Chimica Fisica dei Materiali - unibas.it · elettrodo inerte (Pt) i livelli energetici sono...
33
Chimica Fisica dei Materiali Dr. Sergio Brutti Tecniche voltammetriche
Transcript of Chimica Fisica dei Materiali - unibas.it · elettrodo inerte (Pt) i livelli energetici sono...

Chimica Fisica dei Materiali
Dr. Sergio Brutti
Tecniche voltammetriche

Una tecnica elettrochimica è una metodo sperimentale di analisi di
proprietà funzionali e prestazioni di un sistema chimico-fisico mediante
somministrazione di un segnale elettrico (potenziale, corrente) e/o una
sua rilevazione (potenziale, corrente).
Tecniche elettrochimiche
Sistema chimico fisico
(e.g.
materiale/composito,
cella elettrochimica, fase
liquida)
Segnale elettrico Risposta elettrica
Potenziale cost.
Potenziale altern.
Corrente DC
Corrente AC
Corrente
Potenziale
Fase (segnali AC/AV)
Dipendenza dal tempo

Tecniche Voltammetriche
Una tecnica voltammetrica prevede l’applicazione di un potenziale
variabile con il tempo (usualmente con dipendenza lineare). La risposta
del sistema elettrochimico è data dalla corrente continua erogata (o
assorbita) in coincidenza temporale con il segnale di potenziale.
Cella elettrochimica
(galvanica o elettrolitica) Segnale elettrico Risposta elettrica
Potenziale
variabile con il
tempo
Corrente di risposta
in coincidenza
temporale col
potenziale

Linear sweep voltammetry / LSV
Cella elettrochimica
(galvanica o elettrolitica) Segnale elettrico Risposta elettrica
Potenziale variabile
linearmente con il tempo Corrente di risposta in
coincidenza temporale col
potenziale 𝐄 𝐭 = 𝑬𝟏 + 𝒗𝒔 ∙ 𝒕
Tempo / s
Po
ten
zia
le / V
E1
E2
Potenziale / V
Co
rrente
/ A
Processo redox
(ossidazione)
Processo redox
(riduzione)

Voltammetria e f.e.m.
Specie in
soluzione
En
erg
ia / e
V
Ox+
Red-
In voltammetria i segnali i riduzione e ossidazione cadono a potenziali prossimi al
potenziale redox termodinamico della coppia red/ox considerata.
La fem è correlata alla distanza in energia tra gli stati ossidati e ridotti::
Potenziale / V
Co
rrente
/ A
Eox>fem
Ered<fem
fem
Ox++ne-→Red-
DG°=Ered-Eox
Ox++ne-→Red- 𝜇 𝑂𝑥+(𝑠𝑜𝑙) + 𝑛𝜇𝑒− 𝑒𝑙 = 𝜇 𝑅𝑒𝑑− 𝑠𝑜𝑙
𝐸 = ∆Φ −𝜇°𝑒− 𝑒𝑙
𝐹=
𝜇°𝑂𝑥+ 𝑠𝑜𝑙 − 𝜇°𝑅𝑒𝑑− 𝑠𝑜𝑙
𝑛𝐹+
𝑅𝑇
𝑛𝐹∙ 𝑙𝑛
𝑎𝑂𝑥+ 𝑠𝑜𝑙
𝑎𝑅𝑒𝑑− 𝑠𝑜𝑙
𝐸𝑜 =𝜇°𝑂𝑥+ 𝑠𝑜𝑙 − 𝜇°𝑅𝑒𝑑− 𝑠𝑜𝑙
𝑛𝐹=
−∆𝐺𝑜
𝑛𝐹

Equilibrio dinamico in soluzione ed allineamento di
banda
Specie in
soluzione
En
erg
ia / e
V
Pt
Banda di
valenza
Banda di
Conduzio
ne Fermi
level
Ox+
Red-
Se ho in soluzione una specie chimica nei suoi stati redox ox e red ed immergo un
elettrodo inerte (Pt) i livelli energetici sono descritti dal seguente grafico (qualitativo)
Specie in soluzione
Energ
ia / e
V
Pt
Banda di Conduzio
ne Fermi level
Ox+
Red-
Banda di valenza
Specie in soluzione
Energ
ia / e
V
Pt
Banda di Conduzio
ne
Fermi level
Ox+
Red-
Banda di valenza
Polarizzare un elettrodo altera gli allineamenti di banda tra la soluzione e il Pt
modificando il livello di fermi di quest’ultimo.

Ossidazioni irreversibili Consideriamo la cella descritta.
La cella ha OCV=3.0V ma non essendoci
alcuna reazione redox attiva questo
valore non rappresenta nessuna fem.
Polarizzando in anodica (ovvero fissando
il potenziale applicato alla cella
dall’esterno con un valore di DV>OCV)
nessuna redox a carico del Pt è possibile
fino a quando a DV>5.5V circa comincia
un processo redox attivo non legato ad
intercalazioni/deintercalazioni.
A questo potenziale il Pt agisce come
elettrodo cosiddetto di terzo tipo ovvero
inerte rispetto alla redox.
DM
C L
iPF
6
Ele
ttrolita
liquid
o
non a
cquoso
Pt - c
ato
do L
itio
- a
nodo
Potenziale / V
Corr
ente
/ A
Che succede?

Ossidazione del DMC Al (Pt) stanno avvenendo reazioni di
ossidazione irreversibili del DMC.
DM
C L
iPF
6
Ele
ttrolita
liquid
o
non a
cquoso
Pt - c
ato
do L
itio
- a
nodo
Potenziale / V
Corr
ente
/ A
Questa reazione rilascia radicali e CO2.
Perché si è attivata a quel determinato
potenziale e non ad un potenziale minore
(o maggiore)?

Energia di ionizzazione del DMC La reazione di ossidazione del DMC è un processo a più stadi. Il primo è la
ionizzazione del DMC a DMC+.
-e-
+
Questa reazione elementare non è spontanea ed è proprio il processo
governato dall’energia di prima ionizzazione della molecola DMC (Eion).
L’energia Eion(DMC) di ionizzazione del DMC è la distanza in energia tra
l’HOMO e il LUMO della molecola stessa
Poiché l’Eion(Li)= 1.48eV la differenza:
∆𝐄 = 𝑬𝒊𝒐𝒏 𝑫𝑴𝑪 − 𝟏. 𝟒𝟖 𝒊𝒏 𝑽𝒐𝒍𝒕
E’ il potenziale redox standard rispetto al litio al quale avviene la
ionizzazione (ossidazione) del DMC.
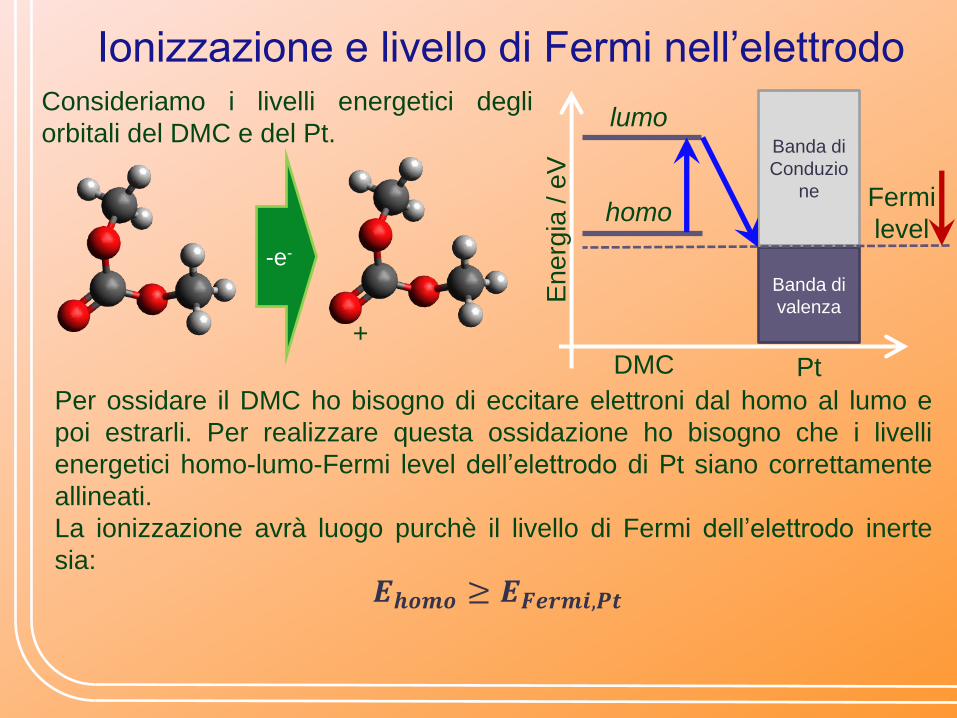
Ionizzazione e livello di Fermi nell’elettrodo Consideriamo i livelli energetici degli
orbitali del DMC e del Pt.
-e-
+
DMC
En
erg
ia / e
V
Pt
Banda di
valenza
Banda di
Conduzio
ne Fermi
level homo
lumo
Per ossidare il DMC ho bisogno di eccitare elettroni dal homo al lumo e
poi estrarli. Per realizzare questa ossidazione ho bisogno che i livelli
energetici homo-lumo-Fermi level dell’elettrodo di Pt siano correttamente
allineati.
La ionizzazione avrà luogo purchè il livello di Fermi dell’elettrodo inerte
sia:
𝑬𝒉𝒐𝒎𝒐 ≥ 𝑬𝑭𝒆𝒓𝒎𝒊,𝑷𝒕

Ionizzazione e livello di Fermi nell’elettrodo Consideriamo i livelli energetici degli orbitali del DMC e del Pt.
-e-
+
DMC Energ
ia / e
V
CO2
homo
lumo
Banda di
valenza
Banda di
Conduzio
ne
Fermi
level
Pt
DMC
Ox++ne-→Red-
-nFE=DG°=Ered-Eox
DE’=e•ηan

LSV anodica del DMC Dalle precedenti è quindi possibile interpretare l’andamento della risposta
in corrente della LSV del DMC.
Potenziale / V
Corr
ente
/ A
Fino a che la ddp<fem della ionizzazione
nessuna corrente passerà nel circuito
esterno. Appena la ddp>fem di
ionizzazione si osserverà un segnale in
corrente positivo perché il sistema
fornisce elettroni (sto ossidando il DMC).
-e-
+ fem
E=fem+DE’

Riduzione del DMC Se polarizzo in catodica (a potenziali via via minori
rispetto al riferimento o al controelettrodo) l’elettrodo
(Pt) ad un dato potenziale di attiveranno i processi di
riduzione del DMC DMC LiPF6
Elettrolita liquido
non acquoso
Pt - catodo
Litio - anodo
+e-
Questa reazione rilascia radicali e anioni metossido.
Perché si è attivata a quel determinato potenziale e non ad un
potenziale minore (o maggiore)?
Possiamo ragionare in modo analogo a prima ma rovesciato.
In questo caso l’energetica elementare che devo considerare sarà
l’affinità elettronica del DMC.
− ●

Affinità elettronica e livello di Fermi nell’elettrodo
Consideriamo i livelli energetici degli
orbitali del DMC e del Pt.
+e-
-
DMC
En
erg
ia / e
V
Pt
Banda di
valenza
Banda di
Conduzio
ne
Fermi
level
homo
lumo
Per ridurre il DMC ho bisogno di spostare il livello di Fermi dell’elettrodo
inerte di Pt al di sopra dell’energia del lumo. Per realizzare questa
riduzione ho bisogno che i livelli energetici lumo-Fermi level dell’elettrodo
di Pt siano correttamente allineati.
Il sistema quindi accetterà elettroni purchè il livello di Fermi dell’elettrodo
inerte sia: 𝑬𝒍𝒖𝒎𝒐 ≤ 𝑬𝑭𝒆𝒓𝒎𝒊,𝑷𝒕

Affinità elettronica e livello di Fermi nell’elettrodo
Consideriamo i livelli energetici degli orbitali del DMC e del Pt.
+e-
-
DMC En
erg
ia / e
V
MeO- & radicals homo
lumo
Banda di
valenza
Banda di
Conduzio
ne
Fermi
level
− ●
Pt
DMC-
Ox++ne-→Red-
-nFE=DG°=Ered-Eox
DE’’=e•ηcat
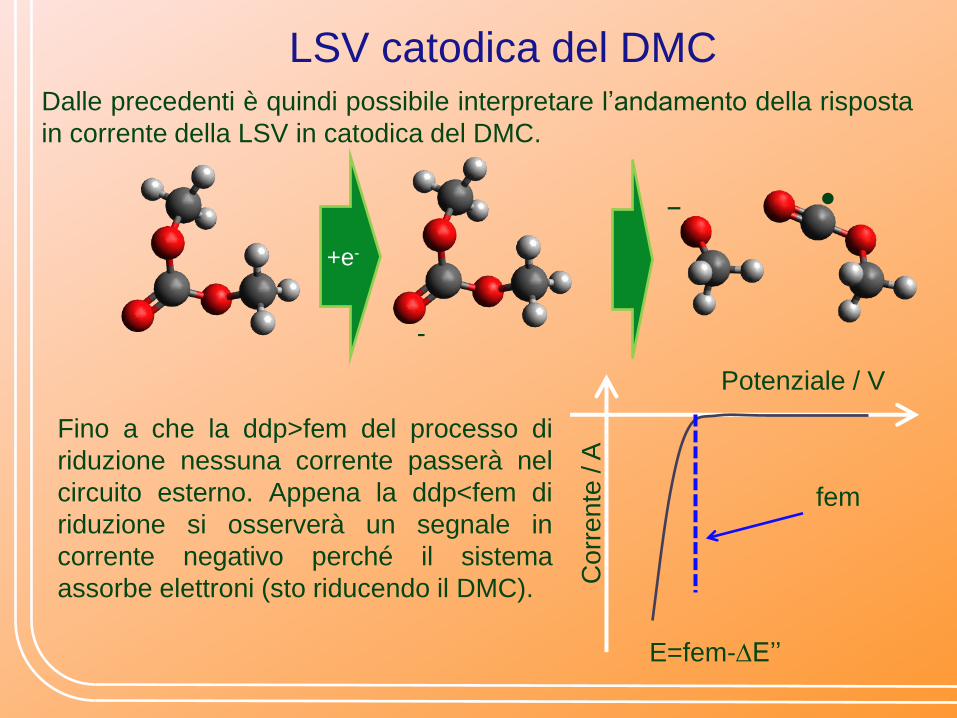
LSV catodica del DMC Dalle precedenti è quindi possibile interpretare l’andamento della risposta
in corrente della LSV in catodica del DMC.
Potenziale / V
Corr
ente
/ A
Fino a che la ddp>fem del processo di
riduzione nessuna corrente passerà nel
circuito esterno. Appena la ddp<fem di
riduzione si osserverà un segnale in
corrente negativo perché il sistema
assorbe elettroni (sto riducendo il DMC).
+e-
-
− ●
fem
E=fem-DE’’

Picco voltammetrico I segnali voltammetrici sono caratterizzati quindi da un potenziale redox di onset
maggiore/minore della fem del corrispondente processo a causa delle
sovratensioni.
Oltre il potenziale di onset il segnale voltammetrico della corrente cresce
esponenzialmente per poi raggiungere un massimo e decrescere. Possiamo quindi
distinguere 3 parti del segnale voltammetrico:
1. Onset
2. Crescita
3. Massimo & decrescita
Specie in
soluzione
En
erg
ia / e
V
Pt
Banda di
valenza
Banda di
Conduzio
ne Fermi
level
Ox+
Red-
Potenziale / V
Co
rre
nte
/ A
Eox>fem
Ered<fem
fem

Parte crescente del picco voltammetrico Subito dopo il potenziale di onset il segnale voltammetrico in corrente cresce
esponenzialmente al crescere del potenziale imposto (ddp).
Questo andamento è legato alla
variazione della costante cinetica del
processo redox in funzione del potenziale.
La costante cinetica di un semiprocesso di
riduzione generico:
An+ + e- → A(n-1)+
𝑘𝑟𝑒𝑑 = 𝑘𝑟𝑒𝑑𝑜 ∙ 𝑒
−𝛼𝐹η𝑅𝑇
In cui α e η hanno lo stesso significato che
nella eq.di Tafel (coefficiente di
trasferimento di carica e sovratensione).
Potenziale / V
Co
rre
nte
/ A
Eox>fem
Ered<fem
fem
Sicchè all’aumentare della sovratensione in riduzione, la costante cinetica aumenta
e quindi il segnale i corrente aumenta anch’esso.

Parte decrescente del picco voltammetrico La porzione esponenziale crescente del picco in corrente voltammetrico si conclude in un
massimo ed in una porzione discendente.
Da un punto di vista algebrico questo andamento è paradossale
perché le equazioni prevedono una crescita indefinita delle
costanti cinetiche.
An+ + e- → A(n-1)+
𝒌𝒐𝒙 = 𝒌𝒐𝒙𝒐 ∙ 𝒆
𝟏−𝜶 ∙𝑭𝜼𝑹𝑻
Ma la velocità effettive di reazione è data da:
𝒅 𝑨(𝒏−𝟏)+
𝒅𝒕= 𝒌𝒐𝒙 ∙ 𝑨𝒏+
L’aumento della corrente è parallelo all’aumento della velocità.
Esso è parallelo all’aumento della costante cinetica solo se la
concentrazione [An+] dei reagenti resta costante all’elettrodo.
Potenziale / V
Co
rre
nte
/ A
Eox>fem
Ered<fem
fem
Ele
ttro
do -
pla
tino
An+ An+
An-1+
An+
An+
An+
An+
An+
H2O
H2O
H2O
H2O An-1+
An-1+
An-1+
An+
e-
An+ An
+ An
+ An-1+
An+
Diffusione lungo grandiente di concentrazione di A(n-1)+
Diffusione lungo gradiente di campo elettrico di An+

Parte decrescente del picco voltammetrico
Ele
ttro
do
- p
latino
An+ An+
An-1+
An+
An+
An+
An+
An+
H2O
H2O
H2O
H2O An-1+
An-1+
An-1+
An+
e-
An+ An
+ An
+ An-1+
An+ Diffusione lungo grandiente di concentrazione di A(n-1)+
Diffusione lungo gradiente di campo elettrico di An+
H2O
H2O
H2O
H2O
Distanza dall’elettrodo
Co
nc. [A
n+]
ddp=OCV i=0
Adsorbimento
sull’elettrodo
Distanza dall’elettrodo
Co
nc. [A
n+]
ddp>fem i>0 Diffusion ≈ Rate
Consumo (Rate)
elettrochimico
Distanza dall’elettrodo
Conc. [A
n+]
ddp>>fem i>>0
Diffusion < Rate
Consumo (rate)
elettrochimico

Descrizione del picco voltammetrico
Distanza dall’elettrodo
Co
nc. [A
n+]
ddp>fem i>0
Diffusion ≈ Rate
Consumo (Rate)
elettrochimico
Distanza dall’elettrodo
Co
nc. [A
n+]
ddp>>fem i>>0
Diffusion < Rate
Massiccio consumo
(rate) elettrochimico
Distanza dall’elettrodo
Co
nc. [A
n+]
ddp>fem i>0
Diffusion > Rate
Modesto
consumo (Rate)
elettrochimico
Potenziale / V
Co
rre
nte
/ A
fem
Corrente di picco
An+ + e- → A(n-1)+
𝒅 𝑨(𝒏−𝟏)+
𝒅𝒕= 𝒌𝒐𝒙 ∙ 𝑨𝒏+
𝒌𝒐𝒙 = 𝒌𝒐𝒙𝒐 ∙ 𝒆
𝟏−𝜶 ∙𝑭𝜼𝑹𝑻

Reversible vs irreversible e-trasfer
Potenziale / V C
orr
en
te / A
fem
Potenziale / V
Co
rre
nte
/ A
fem
Ep Ep/2
ip
Ip/2
ELECTRON
TRASNFER
REVERSIBILE
ELECTRON
TRASNFER
IRREVERSIBILE
α=1
Ep indipendente da vs
𝑬𝒑 − 𝑬𝒑/𝟐 = 𝟐. 𝟐𝟎 ∙𝑹𝑻
𝑭
𝒊𝒑 = 𝟎. 𝟑𝟏𝟖𝑨𝑭 𝑫𝒗𝒔 𝑨 𝒃𝒖𝒍𝒌
𝑭
𝑹𝑻
α<1
Ep dipende da vs
𝒗𝒔𝟏 𝒗𝒔𝟐 = 𝟏𝟎 → ∆ 𝑬𝒑 ≅ 𝟏. 𝟏𝟔𝑹𝑻
𝜶𝑭
𝑬𝒑 − 𝑬𝒑/𝟐 = 𝟏. 𝟖𝟔 ∙𝑹𝑻
𝑭
𝒊𝒑 = 𝟎. 𝟒𝟗𝟔 𝜶𝑨𝑭 𝑫𝒗𝒔 𝑨 𝒃𝒖𝒍𝒌
𝑭
𝑹𝑻

Reversible vs irreversible e-trasfer
Picchi simmetrici e
indipendenti come
posizione in E dalla
velocità di scansione.
Correnti di picco crescenti
e picchi più stretti al
crescere della velocità di
scansione
E di picco distante dalla
fem
Picchi asimmetrici e
dipendenti come posizione
in E dalla velocità di
scansione.
Correnti di picco crescenti
e picchi più stretti al
crescere della velocità di
scansione
Potenziale / V
Co
rre
nte
/ A
fem
ELECTRON
TRASNFER
REVERSIBILE
Vs c
rescenti
Potenziale / V
Co
rre
nte
/ A
fem
ELECTRON
TRASNFER
IRREVERSIBILE

Voltammetria ciclica – processo reversibile
Potenziale / V
Co
rre
nte
/ A
fem
ELECTRON
TRASNFER
REVERSIBILE
Tempo / V
Dd
p / V
E2
E1
𝑬𝒑𝒐𝒙 − 𝑬𝒑
𝒓𝒆𝒅 = 𝟐. 𝟐𝟎 ∙𝑹𝑻
𝒏𝑭
𝒊𝒐𝒙𝒅𝒕 =𝒕𝒊𝒏𝒗
𝟎 𝒊𝒓𝒆𝒅𝒅𝒕
𝒕𝒆𝒏𝒅
𝒕𝒊𝒏𝒗
Epox
Epred
tinv tend

Voltammetria ciclica – processi irreversibili
Potenziale / V
Co
rre
nte
/ A
fem
ELECTRON
TRASNFER
parzialmente
reversibile
𝒊𝒐𝒙𝒅𝒕 ≠𝒕𝒊𝒏𝒗
𝟎 𝒊𝒓𝒆𝒅𝒅𝒕
𝒕𝒆𝒏𝒅
𝒕𝒊𝒏𝒗
Epox
Epred
Co
rre
nte
/ A
ELECTRON
TRASNFER
irreversibile

Batterie Litio-ione: CG vs CV Processi redox differenti hanno CG e CV di scarica/carica differenti.
Catodo: FePO4 + Li+ + e- = LiFePO4
Anodo: Li+ + e- = Li
CG – profilo caratterizzato da
un plateau di potenziale.
CV – profilo caratterizzato da
un picco singolo stretto di
corrente.
DGR=DGf(LiFePO4)- DGf(FePO4)
Potenziale / V C
orr
en
te / A
Eox>fem
Ered<fem
fem
CC
V /
V
Tempo / s
Corrente
costante
En
d o
f ch
arg
e
LiFePO4 → FePO4+Li
Li + FePO4 → LiFePO4

Batterie Litio-ione: CG vs CV Processi redox differenti hanno CG e CV di scarica/carica differenti.
CG – profilo caratterizzato da
due plateaux consecutivi di
potenziale.
CV – profilo caratterizzato da
due picchi separati di corrente.
Potenziale / V C
orr
en
te / A
Eox>fem
Ered<fem
fem
CC
V /
V
Tempo / s
Corrente costante
En
d o
f ch
arg
e
LiCoPO4 → Li0.7CoPO4+0.3Li → CoPO4+Li
Li+CoPO4 → Li0.7CoPO4+0.3Li → LiCoPO4
Catodo: CoPO4 + 0.7 Li+ + 0.7 e- =Li0.7CoPO4
Catodo: Li0.7CoPO4+0.3 Li++0.3e- =LiCoPO4
Anodo: Li+ + e- = Li

Batterie Litio-ione: CG vs CV Processi redox differenti hanno CG e CV di scarica/carica differenti.
CG – profilo caratterizzato da
una «slope» di potenziale.
CV – profilo caratterizzato da
un largo picco (banda) di
corrente.
Potenziale / V C
orr
en
te / A
Eox>fem
Ered<fem
fem
CC
V /
V
Tempo / s
Corrente costante
En
d o
f d
isch
arg
e
b-LiTiO2 → b-TiO2+Li
Li+b-TiO2 → b-LiTiO2
Catodo: b-TiO2 + xLi+ + xe- = b-LixTiO2
Anodo: Li+ + e- = Li
DGR=DGf(b-LixTiO2)-DGf(b-TiO2)
DGf(b-LixTiO2) = f(x)

Batterie Litio-ione: CG vs CV Processi redox differenti hanno CG e CV di scarica/carica differenti.
CG – profilo caratterizzato da
un plateau e una «slope» di
potenziale.
CV – profilo caratterizzato da
un largo picco (banda) di
corrente sovrapposto ad un
picco stretto.
Potenziale / V C
orr
en
te / A
Eox>fem
Ered<fem
fem
CC
V /
V
Tempo / s
Corrente costante
En
d o
f d
isch
arg
e
Li0.5+xTiO2 → Li0.5TiO2+xLi→ TiO2+(0.5+x)Li
Catodo: TiO2 + 0.5Li+ + 0.5e- = Li0.5TiO2
Catodo: Li0.5TiO2 + xLi+ + xe- = Li0.5+xTiO2
Anodo: Li+ + e- = Li
Li0.5+xTiO2 → Li0.5TiO2+xLi→ TiO2+(0.5+x)Li

Cronoamperometria a potenziale costante
fem Po
ten
zia
le / V
Tempo / s Tempo / s C
orr
ente
/ A
𝒊 =𝒏𝑭𝑨 𝑫 𝑨 𝒃𝒖𝒍𝒌
𝝅𝒕 Eq. Di Cottrell
Il best-fitting non lineare (o linearizzando l’andamento con una relazione tipo i vs. 1/t)
della porzione ad andamento regolare consente di ricavare il coefficiente di
diffusione.
Sqrt(1/t) / s-1/2
Co
rre
nte
/ A
Carica nello step
di potenziale 𝑸 = 𝒊 𝒅𝒕
𝒕𝒆𝒏𝒅
𝟎
La carica scambiata
nell’intervallo di tempo è
proporzionale all’avanzamento
della reazione redox

Voltammetria a step (potential stepping test)
Questa tecnica prevede di applicare step di potenziale crescenti/decrescenti
(senza transienti) mentre viene registrato l’andamento della corrente
corrispondente.
Caratteristiche proprie di ogni esperimento sono:
1. Ampiezza dello step di potenziale (5 mV, 10 mV, etc.)
2. Durata dello step di potenziale
i. Cutoff in tempo
ii. Cutoff in corrente (quanto la corrente scende sotto una soglia minima)
iii. Cutoff misto tempo/corrente (OR)
Tempo / s
Corr
ente
/ A
𝑸𝒔𝒕𝒆𝒑 = 𝒊 𝒅𝒕𝒕𝒆𝒏𝒅
𝒕𝟎
Po
ten
zia
le / V
Tempo / s
𝑽𝒔𝒕𝒆𝒑

Voltammetria a step (potential stepping test) P
ote
nzia
le / V
Tempo / s Tempo / s C
orr
ente
/ A
Curva della capacità
differenziale
Carica / mAh
Po
ten
zia
le / V
Curva di
caricamento
𝑸𝒔𝒕𝒆𝒑 = 𝒊 𝒅𝒕𝒕𝒆𝒏𝒅
𝒕𝟎
L’aspetto della curva
di caricamento
dipende dalla
termodinamica del
processo redox
analogamente a
quanto accade per le
CG e le CV.
𝑽𝒔𝒕𝒆𝒑
dQ/dV
Potenziale / V
dQ
/dV
«pseudo
termodinamica» No sovratensioni!!
«pseudo
termodinamica» No sovratensioni!!

PITT (potentiostatic intermittent titration test)
Lo studio degli andamenti delle correnti negli step di potenziale e del potenziale negli intervalli di
rilassamento ad OCV consente di ricavare il coefficiente di diffusione nel materiale solido ad un
dato potenziale termodinamico utilizzando una versione semplificata dell’equazione di Cottrell:
Tempo / s
Corr
ente
/ A
𝑸𝒔𝒕𝒆𝒑 = 𝒊 𝒅𝒕𝒕𝒆𝒏𝒅
𝒕𝟎
P
ote
nzia
le / V
Tempo / s
𝑽𝒔𝒕𝒆𝒑
OCV
𝒊 =𝒏𝑭𝑨 𝑫 𝑨 𝒃𝒖𝒍𝒌
𝝅𝒕 Eq. Di Cottrell
𝑫 =𝒊 𝒕𝟏/𝟐
𝑪𝒐𝒕𝒕𝒓𝒆𝒍𝒍∙ 𝑳 𝝅
𝑸𝒔𝒕𝒆𝒑
𝟐
Diffusion
coefficient from
PITT
OCV
In cui 𝒊 𝒕𝟏/𝟐𝑪𝒐𝒕𝒕𝒓𝒆𝒍𝒍
è il valore del prodotto nell’intervallo di linearità di Cottrell e L lo spessore
dell’elettrodo