
CARATTERIZZAZIONE BIO-MOLECOLARE DEL CARCINOMA … · Menarca anticipato e menopausa ritardata...
97
CANDIDATO RELATORE Dr. Michele Menicagli Chiar.mo Prof. Antonio Giuseppe Naccarato Anno accademico 2011-2012 CARATTERIZZAZIONE BIO-MOLECOLARE DEL CARCINOMA DELLA MAMMELLA TESI DI SPECIALIZZAZIONE IN PATOLOGIA CLINICA
Transcript of CARATTERIZZAZIONE BIO-MOLECOLARE DEL CARCINOMA … · Menarca anticipato e menopausa ritardata...

CANDIDATO RELATORE
Dr. Michele Menicagli Chiar.mo Prof. Antonio Giuseppe Naccarato
Anno accademico 2011-2012
CARATTERIZZAZIONE BIO-MOLECOLARE DEL CARCINOMA
DELLA MAMMELLA
TESI DI SPECIALIZZAZIONE IN PATOLOGIA CLINICA

2
INDICE
INDICE Pag. 2
1. INTRODUZIONE Pag. 3
2. EPIDEMIOLOGIA Pag. 4
2.1 FATTORI DI RISCHIO Pag. 5
3. MALATTIE BENIGNE DELLA MAMMELLA Pag. 7
4. CARCINOGENESI E PROGRESSIONE TUMORALE Pag. 8
5. FATTORI PROGNOSTICI E PREDITTIVI Pag. 10
6. FATTORI GENETICI E FAMILIARI Pag. 13
6.1 GENI DI SUSCETTIBILITA’ PER IL TUMORE MAMMARIO Pag. 14
6.2 IL CANCRO DELLA MAMMELLA FAMILIARE: BRCA1/BRCA2 Pag. 15
6.3 TP53 Pag. 16
6.4 7.4 ORMONI ENDOGENI Pag. 17
6.5 HER-2 Pag. 20
6.6 TOPOISOMERASI IIα Pag. 22
6.7 EGFR Pag. 23
6.8 VEGF Pag. 23
7. CLASSIFICAZIONE MOLECOLARE DEL CARCINOMA DELLA MAMMELLA Pag. 25
8. ANEUPLOIDIE E CARCINOMA DELLA MAMMELLA Pag. 28
9. CELLULE STAMINALI Pag. 29
10. DETERMINAZIONE DELLO STATO DI HER2 Pag. 34
10.1 TECNICA FISH Pag. 34
10.2 TECNICHE INNOVATIVE DI IBRIDAZIONE IN SITU: CISH E SISH Pag. 40
11. POLISOMIA DEL CROMOSOMA 17 Pag. 44
12. ETEROGENEITA’ TUMORALE Pag. 45
13. TRATTAMENTO DEL CARCINOMA DELLA MAMMELLA Pag. 47
13.1 TARGET THERAPY E BERSAGLI MOLECOLARI Pag. 47
13.2 ANTICORPI MONOCLONALI UMANIZZATI Pag. 48
13.3 TRASTUZUMAB E MECCANISMO D’AZIONE Pag. 49
13.4 TRASTUZUMAB: MECCANSIMI DI RESISTENZA Pag. 53
13.5 STRATEGIE TERAPEUTICHE PER SUPERARE LA RESISTENZA
A TRASTUZUMAB Pag. 61
14. MICRORNA (miRNA) Pag. 65
15. DNA REPAIR COME BERSAGLIO FARMACOLOGICO: SYNTHETIC LETHALITY
STRATEGY Pag. 69
16. CONCLUSIONI Pag. 72
BIBLIOGRAFIA Pag. 73

3
1.0 INTRODUZIONE
Il carcinoma della mammella, rappresenta la neoplasia più frequente nel sesso femminile e la
seconda causa di morte dopo il tumore del polmone. Miglioramenti in termini di sopravvivenza
sono stati ottenuti grazie a vari programmi di screening. Il National Cancer Institute (NCI)
Surveillance, Epidemiology and End Results (SEER) program tra il 1977 e il 1982, ha evidenziato
per neoplasie di dimensioni inferiori ad 1 cm, una frequenza del 5,4%, mentre fra il 1983 e il 1987,
raggiungeva il 14,4% (Gloecker-Ries LA et al., 1994). Anche il tasso di carcinomi in situ è
progressivamente aumentato rispettivamente di 7,2 volte per le forme duttali e di 2,6 volte per le
forme lobulari, nel periodo tra il 1980 e il 2001 (Li CI et al., 2005). Ogni anno in Italia si registrano
circa 35000 nuovi casi e 11000 decessi; in Toscana sono state stimate nel 2010 circa 4100 nuove
diagnosi di tumore della mammella e 850 decessi. L’incidenza della malattia presenta un netto
gradiente tra Nord, Centro e Sud con rischi superiori del 40% al Nord. Nel recente quinquennio,
l’incidenza ha mostrato un aumento variabile dal 2 al 17% (Linee guida AIOM) a fronte di una
netta diminuzione in mortalità grazie agli avanzamenti ottenuti nella diagnosi precoce e nella
strategia terapeutica adiuvante. Tuttavia circa il 20-30% delle pazienti con linfonodi negativi e il
50% di quelle con linfonodi positivi alla diagnosi svilupperà metastasi a distanza, mentre il 7-10%
presenta uno stadio avanzato già al momento della diagnosi. Ad eccezione di casi sporadici (2-3%)
in cui è stata osservata una sopravvivenza di lunga durata, il tumore mammario metastatico rimane
ad oggi una malattia non guaribile, con una sopravvivenza mediana nelle pazienti non
precedentemente trattate di 18-24 mesi, variabile in base all’aggressività biologica, alla sede e
all’estensione della malattia (Cnossen JA et al., 2008). Studi recenti descrivono comunque un
guadagno in termini di sopravvivenza globale pari a 12,5 mesi, grazie all’introduzione di nuovi
agenti terapeutici (Mauri D et al., 2008).
Accanto alle strategie terapeutiche convenzionali, basate sulla chirurgia, linfonodo sentinella,
chemioterapia sistemica ed endocrino-terapia, recentemente sono state introdotte le “terapie target”
basate sull’impiego di anticorpi monoclonali diretti esclusivamente contro le cellule che esprimono
determinati recettori di membrana, bloccando in questo modo la cascata di eventi molecolari che
porta all’attivazione dei fattori di trascrizione e quindi alla crescita incontrollata della neoplasia.
L’amplificazione di determinati geni, è stata particolarmente studiata, in quanto si è dimostrata
avere un significato ai fini prognostici e predittivi di risposta ad alcune terapie. In particolare il 25-
30% dei carcinomi della mammella mostra amplificazione del gene HER-2/neu che codifica per un
recettore coinvolto nella regolazione della crescita cellulare (Seth RM et al., 2006).
Nei carcinomi della mammella in fase avanzata, è indicata la terapia neo-adiuvante, che consiste
nell’uso della chemioterapia in fase pre-operatoria, con l’obiettivo di ridurre la massa del tumore

4
primitivo (riduzione dello stadio T), al fine di rendere operabile, con intento radicale, una neoplasia
localmente avanzata. L’approccio secondario, consiste nella valutazione in vivo della responsività
ai trattamenti e l’eradicazione di eventuali micrometastasi responsabili della ripresa della malattia
(Ruco L et al. 2007).
Per l’individuazione dei fattori predittivi di risposta alle terapie convenzionali (endocrino-terapia) e
target (trastuzumab) in fase pre-operatoria, è possibile ricorrere all’uso dell’ago-biopsia che espone
la paziente ad inconvenienti come l’anestesia locale e il rischio di disseminazione in circolo delle
cellule tumorali. Il prelievo del tessuto permette un’analisi di tipo istologico della lesione, la
conoscenza della sua eventuale invasività e dei parametri biologici. Ai fini della corretta
pianificazione terapeutica si deve tener presente che nel 10-30% dei casi con diagnosi
microistologica di carcinoma in situ, la successiva exeresi chirurgica, rivela la presenza di un
carcinoma invasivo (Estratto Linee Guida F.O.N.Ca.M. 2008).
2.0 EPIDEMIOLOGIA
Il carcinoma della mammella è la neoplasia più frequente nelle donne nei paesi industrializzati, ed è
la maggiore causa di morbilità e mortalità oncologiche. L’incidenza del cancro della mammella
presenta un’ampia variabilità geografica. È quasi 10 volte più frequente nelle popolazioni ricche
dell’occidente rispetto alle aree del terzo mondo. In Italia l’incidenza è di circa 40000 nuovi casi
l’anno, in media con i valori europei (Jemal A et al., 2008) (Fig 1).
I tassi di incidenza aumentano esponenzialmente con l’età, ma intorno ai 50-55 anni, a differenza di
altri tumori epiteliali non dipendenti da fattori ormonali e riproduttivi, l’incremento cessa per poi
riprendere meno pronunciato dopo i 60 anni (Key TJ et al., 2001; Jemal A et al., 2008). In realtà la
tendenza all’incremento dell’incidenza in Italia nelle ultime decadi è riconducibile anche alla
tempestività delle diagnosi in relazione a campagne di screening di prevenzione secondaria.
La mortalità per carcinoma della mammella è rimasta sostanzialmente invariata dal 1930 al 1990
per poi mostrare una leggere riduzione grazie probabilmente alla diagnosi precoce e all’utilizzo di
terapie mirate su base ormonale e genetica. Ad eccezione di casi sporadici (2-3%) in cui si è
osservata una sopravvivenza di lunga durata, il tumore mammario metastatico rimane ad oggi una
malattia non guaribile, con una sopravvivenza mediana nelle pazienti non precedentemente trattate
di 18-24 mesi, variabile in base all’aggressività biologica, alla sede e all’estensione della malattia.
Studi recenti descrivono un guadagno crescente, in termini di sopravvivenza globale pari a 12,5
mesi per l’introduzione di nuovi agenti terapeutici (Mauri D et al., 2008).
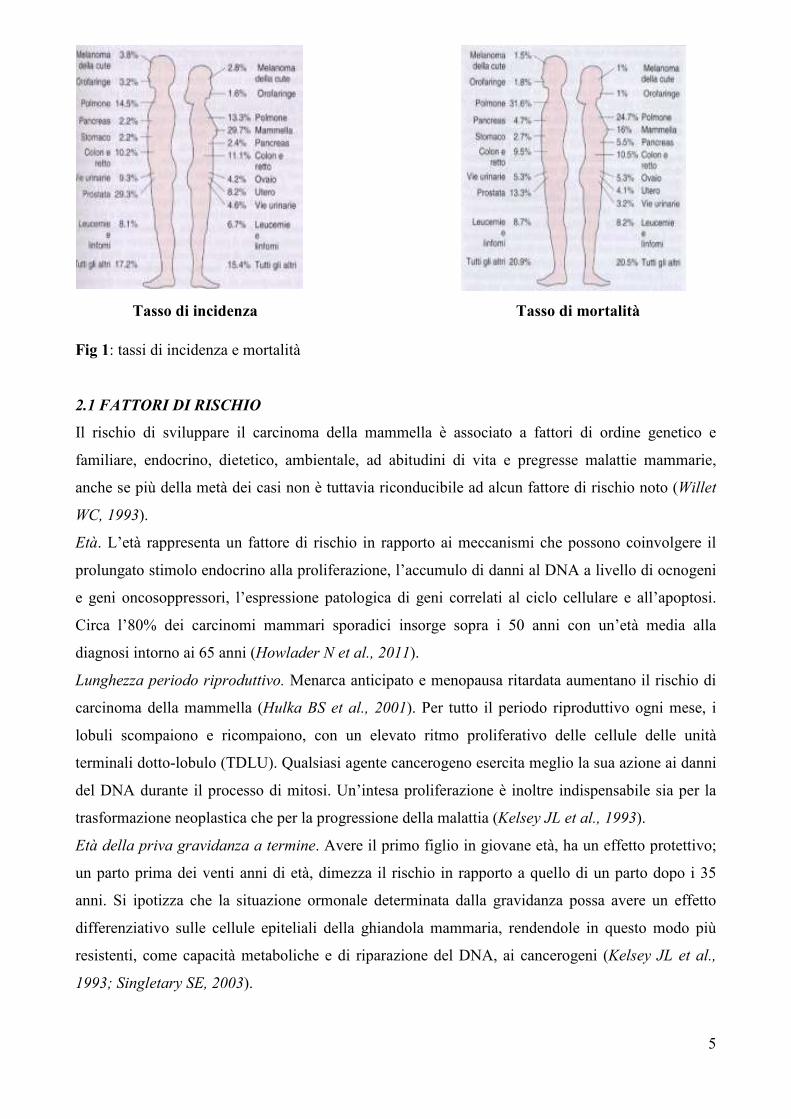
5
Fig 1: tassi di incidenza e mortalità
2.1 FATTORI DI RISCHIO
Il rischio di sviluppare il carcinoma della mammella è associato a fattori di ordine genetico e
familiare, endocrino, dietetico, ambientale, ad abitudini di vita e pregresse malattie mammarie,
anche se più della metà dei casi non è tuttavia riconducibile ad alcun fattore di rischio noto (Willet
WC, 1993).
Età. L’età rappresenta un fattore di rischio in rapporto ai meccanismi che possono coinvolgere il
prolungato stimolo endocrino alla proliferazione, l’accumulo di danni al DNA a livello di ocnogeni
e geni oncosoppressori, l’espressione patologica di geni correlati al ciclo cellulare e all’apoptosi.
Circa l’80% dei carcinomi mammari sporadici insorge sopra i 50 anni con un’età media alla
diagnosi intorno ai 65 anni (Howlader N et al., 2011).
Lunghezza periodo riproduttivo. Menarca anticipato e menopausa ritardata aumentano il rischio di
carcinoma della mammella (Hulka BS et al., 2001). Per tutto il periodo riproduttivo ogni mese, i
lobuli scompaiono e ricompaiono, con un elevato ritmo proliferativo delle cellule delle unità
terminali dotto-lobulo (TDLU). Qualsiasi agente cancerogeno esercita meglio la sua azione ai danni
del DNA durante il processo di mitosi. Un’intesa proliferazione è inoltre indispensabile sia per la
trasformazione neoplastica che per la progressione della malattia (Kelsey JL et al., 1993).
Età della priva gravidanza a termine. Avere il primo figlio in giovane età, ha un effetto protettivo;
un parto prima dei venti anni di età, dimezza il rischio in rapporto a quello di un parto dopo i 35
anni. Si ipotizza che la situazione ormonale determinata dalla gravidanza possa avere un effetto
differenziativo sulle cellule epiteliali della ghiandola mammaria, rendendole in questo modo più
resistenti, come capacità metaboliche e di riparazione del DNA, ai cancerogeni (Kelsey JL et al.,
1993; Singletary SE, 2003).
Tasso di incidenza Tasso di mortalità

6
Precedenti biopsie mammarie con diagnosi di iperplasia epiteliale atipica e carcinoma in situ. Una
spiegazione plausibile si basa sul concetto di “field cancerogenesis” o cancerogenesi a campo;
l’intera popolazione cellulare di un tessuto o di un organo sarebbe esposta all’agente cancerogeno
con possibilità di insorgenza della neoplasia in più zone dello stesso albero ghiandolare. Quindi
sviluppare un’iperplasia tipica o un carcinoma in situ, rappresenta un allarme, per la possibilità di
sviluppare neoplasie infiltranti. Infatti queste lesioni rappresentano le fasi iniziali del processo
neoplastico (Singletary SE, 2003).
Estrogeni endogeni ed esogeni. L’aumentata esposizione ad estrogeni può aumentare il rischio di
carcinoma della mammella. L’obesità è un fattore di rischio nelle donne in post-menopausa, in
quanto nei depositi di grasso si ha produzione endogena di estrogeni (Eliassen AH et al., 2006;
Nichols HB et al., 2009). Il rischio conferito dalla terapia ormonale sostitutiva sembra essere
modesto (Rosemberg LU et al., 2006). Il rischio ambientale (fitoestrogeni, pesticidi) è ancora
oggetto di studio (Rice S et al., 2006).
Esposizione a radiazioni. Il rischio sembra essere importante nelle donne giovani (non oltre i 30
anni) sottoposte a terapia radiante per neoplasia tipo la malattia di Hodgkin (Preston DL et al.,
2002).
Allattamento al seno. Un prolungato allattamento riduce il rischio di carcinoma; durante questo non
si hanno cambiamenti nella struttura della mammella (Singletary SE, 2003).
Dieta. Per quanto riguarda il ruolo della dieta non vi sono ancora studi scientifici certi; un fattore di
rischio su cui sembra esservi consenso è il consumo di alcool (Singletary KW et al., 2001; Baan R
et al., 2007).
Influenza geografica. L’incidenza di carcinoma della mammella negli Stati Uniti e in Europa e 4-7
volte maggiore rispetto ad altri paesi; questo potrebbe essere dovuto all’esposizione a cancerogeni
ambientali e a diverse abitudini come allattamento e dieta (Newman LA et al., 2006).
Familiarità. La presenza di un parente di primo grado (madre o sorella) con carcinoma mammario
rende doppio il rischio di sviluppare un carcinoma della mammella rispetto alla popolazione
generale. Solitamente nei soggetti con predisposizione familiare, il carcinoma compare in età
giovanile (prima dei 40 anni) ed è più frequentemente bilaterale (Lakhani SR et al., 2000;
Singletary SE, 2003). Lo studio genetico di una famiglia basato sulla ricostruzione dell’albero
genealogico, corredato da tutti gli eventi patologici, permette di stabilire se una patologia è del tipo
eredo-familiare. La presenza di una mutazione germinale può essere determinata attraverso un test
genetico che consiste nell’esaminare il DNA di un individuo estratto da cellule di un campione di
sangue o da altri liquidi o tessuti corporei, nella ricerca di alterazioni correlata alla malattia. Le
alterazioni del DNA possono essere numerose, come aberrazioni cromosomiche rilevabili

7
dall’esame del cariotipo, o alterazioni di singoli geni attraverso delezioni, mutazioni puntiformi,
mutazioni frame-shift (inserimento o delezione di singole basi in un esone), amplificazione genica. I
soggetti sani a rischio eredo-familiare possono essere avviati a specifici programmi di sorveglianza
al fine di una diagnosi precoce (Jaffrey SS et al., 2005).
Predisposizione genetica. È stato dimostrato che pazienti affetti da mutazioni a livello dei geni
BRCA1 (17q) e BRCA2 (12q), hanno un aumentato rischio di sviluppare un carcinoma della
mammella entro i 70 anni, di circa il 56% (Allain DC et al., 2007). Le mutazioni a livello di questi
geni, sono responsabili di circa 1/3 dei casi familiari e complessivamente del 10% dei casi di
carcinoma mammario (Walsh T et al., 2006). Le mutazioni del gene BRCA1 sono inoltre coinvolte
nella predisposizione allo sviluppo di carcinomi all’apparato riproduttivo femminile (ovaio e tube).
3.0 MALATTIE BENIGNE DELLA MAMMELLA
Le malattie benigne della mammella, costituiscono un gruppo vasto ed eterogeneo di lesioni la cui
frequenza è maggiore rispetto alle lesioni maligne. La loro importanza deriva dal fatto che alcune
possono simulare clinicamente il cancro della mammella, mentre altre rappresentano fattori di
rischio per lo sviluppo del successivo carcinoma. Solo una minima parte rientra in quest’ultima
categoria, trattandosi di poche lesioni che fanno parte del gruppo delle malattie benigne a carattere
proliferativo. A carico della mammella possono riscontarsi vari tumori benigni fra cui lipomi,
adenomi ed emangiomi; l’asportazione completa di queste lesioni rappresenta un trattamento
adeguato e definitivo. Una delle più note lesioni proliferative della mammella è rappresentata dal
fibroadenoma che si riscontra più frequentemente fra i 25 ed i 35 anni di età, aumenta di dimensioni
durante la gravidanza e tende a regredire con l’avanzare dell’età. Generalmente è una lesione
singola, ma nel 20% dei casi può essere multipla o bilaterale. Microscopicamente sono costituti da
tessuto connettivo e ghiandolare; una loro trasformazione maligna è riportata solo raramente (0,1%
dei casi) per la componente epiteliale in genere sottoforma di carcinoma in situ. Il rischio di
trasformazione è aumentato se è presente iperplasia duttale o una storia familiare di carcinoma
mammario. Altra lesione benigna della mammella è il papilloma intraduttale che nel 90% dei casi è
solitario e costituito da una lesione polipoide all’interno dei dotti che può essere causa di secrezione
ematica del capezzolo. L’escissione chirurgica guarisce il tumore e non c’è in queste condizioni un
aumentato rischio di cancro mammario a lungo termine. Il rischio aumenta in caso di pipillomi
multipli microscopicamente evidenti.
La malattia fibrocistica è la condizione benigna più comune; si riscontra nel 50-90% delle donne in
un’età compresa fra i 25 ed i 45 anni. Generalmente è bilaterale. È rappresentata da un insieme di
alterazioni mammarie tra cui, cisti, fibrosi, infiammazione cronica, iperplasia epiteliale. Questo tipo

8
di patologia di per se non aumenta il rischio di cancro che è invece correlato alla presenza di
iperplasia duttale o lobulare soprattutto se atipica ed associata a storia familiare di carcinoma della
mammella (Bonadonna G et al., 2007).
4.0 CARCINOGENESI E PROGRESSIONE TUMORALE
Il carcinoma mammario si sviluppa dalle cellule epiteliali dell’albero ghiandolare e può originare
diversi istotipi fra i quali i più frequenti sono il carcinoma duttale e lobulare. I due termini sono stati
introdotti ritenendo che la prima forma derivasse dai dotti principali e la seconda dai lobuli.
Fig 2: struttura normale della mammella Tuttavia la maggior parte dei carcinomi insorge a livello delle unità terminali dotto lobulari (TDLU)
e successivamente per meccanismi non del tutto ancora conosciuti da luogo a tumori diversi sia dal
punto di vista morfologico che per il comportamento biologico come ad esempio l’espressione o
meno della proteina di adesione intracellulare E-caderina (Lumachi F et al., 1997) (Fig 2).
Per entrambi i tipi si riconoscono una forma in situ ed una forma infiltrante. Il tipo duttale
rappresenta il 75% dei tumori infiltranti, mentre il lobulare solo il 5%; istotipi infiltranti meno
frequenti sono il carcinoma midollare (15%), il colloide o mucinoso (2%), il tubulare (1-2%) ai
quali si aggiungono altre forme rare. Alle volte le cellule di un carcinoma in situ dei dotti principali
possono migrare fino a raggiungere l’epidermide del capezzolo o dell’areola causando una flogosi
della cute. Tale quadro clinico è noto come malattia di Paget ed è importante per il fatto che con il
tempo il tumore può divenire invasivo.
Il passaggio dalle strutture normali al carcinoma in situ avviene mediante la formazione di lesioni
intermedie, diverse per i due tipi principali di tumore e indicate come lesioni preneoplastiche (Fig
3). Analisi di genetica molecolare e citogenetica hanno infatti dimostrato come il carcinoma della

9
mammella in modo simile ad altre neoplasie derivi da un processo multistep caratterizzato
dall’accumulo di varie alterazioni genetiche (Bombonati A et al., 2011). Uno dei modelli più
accettati è quello di Wellings e Jansen per il quale a partire dall’epitelio normale si può avere lo
sviluppo di un’iperplasia che successivamente può diventare una lesione pre-maligna, carcinoma in
situ ed infine carcinoma invasivo capace di metastatizzare (Wellings GR et al., 1973; Welling GR et
al., 1975) . Determinate alterazioni genetiche si mantengono durante l’evoluzione della lesione ed
esiste una forte correlazione tra il rischio di sviluppare un carcinoma invasivo e la tipologia della
lesione precursore. Pertanto tutti i carcinomi invasivi originano inizialmente da lesioni in situ che
hanno subito alterazioni genetiche ed epigenetiche che influenzano la morfologia e le funzioni
cellulari.
Fig 3: progressione del carcinoma della mammella Il carcinoma in situ, generalmente intraduttale (DCIS), per definizione non supera la membrana
basale del dotto ed è formato da cellule dell’epitelio ghiandolare proliferanti e con caratteristiche di
malignità. In era pre-mammografiica, il carcinoma intraduttale clinicamente evidente non era
frequentemente riscontrabile, essendo meno del 5% di tutti i tumori palpabili. La diffusione dello
screening mammografico ha modificato sensibilmente la sua incidenza, permettendo
l’identificazione di tali lesioni allo stadio preclinico. L’incidenza attuale dei DCIS è circa il 15-30%
di tutti i carcinomi della mammella. Quando il tumore supera la membrana basale diventa micro
infiltrante ed invasivo acquistando la capacità di diffondersi attraverso il sistema emolinfatico ad
altri organi (Fig 4).

10
Fig 4: carcinoma duttale in situ ed infiltrante La cellula da cui originano i carcinomi della mammella è importante per le sue implicazioni
nell’eziologia e nel trattamento. L’ipotesi delle cellule staminali neoplastiche, suggerisce che le
modificazioni maligne avvengano in una popolazione di cellule staminali con proprietà uniche che
le distinguono dalle cellule differenziate (Campbell LL et al., 2007); solo queste contribuirebbero
alla progressione o recidiva tumorale. Il tipo di cellula che più probabilmente è all’origine della
maggior parte dei carcinomi è la cellula luminale che esprime ER; la maggioranza dei carcinomi è
infatti ER positiva e le lesioni pre-cancerogene come le iperplasie atipiche sono simili a questo tipo
di cellule. I carcinomi ER negativi, potrebbero invece derivare da cellule mioepiteliali ER-negative
(Shipitisin M et al., 2007); questo spiegherebbe il fatto di come molte proteine presenti nelle cellule
mioepiteliali siano comuni nei tumori tripli negativi o basiloidi. L’ultima fase nella progressione del
tumore della mammella è la transizione da carcinoma in situ a carcinoma invasivo e rappresenta
purtroppo la fase meno conosciuta.
5.0 FATTORI PROGNOSTICI E PREDITTIVI I criteri prognostici di maggiore importanza sono il grado istologico e lo stadio della neoplasia. La
variabilità prognostica registrata all’interno di categorie di pazienti omogenee per stadio anatomo-
clinico, ha indotto ad una più estesa caratterizzazione del tumore dal punto di vista morfologico e
biofunzionale (Andreopoulou E et al., 2008).
Il sistema di attribuzione del grado più usato considera il pleomorfismo nucleare, la formazione di
tubuli e l’indice mitotico per classificare i carcinomi invasivi in tre gruppi che sono strettamente
correlati alla sopravvivenza. La sopravvivenza a 10 anni dall’85% del grado I scende al 60% nel
grado II e al 15% nel grado III. La riproducibilità del grado è alquanto bassa risentendo della
soggettività dell’osservatore (Kumar V, 2005; Rakha EA et al., 2010) (Fig 5)
Carcinoma duttale in situ Carcinoma duttale infiltrante

11
GRADO I GRADO II GRADO III
Fig 5: grado tumorale
La determinazione dello stadio raggiunto dalla neoplasia alla presentazione è importante per
importare i programmi terapeutici sia chirurgici che radio e/o chemioterapici. La stadiazione viene
eseguita seguendo protocolli che tengono conto delle più recenti acquisizioni scientifiche.
Attualmente si fa riferimento al sistema TNM adottato dall’American Joint Committe on Cancer
(AJCC) nel 2002 e tale sistema si basa su:
• dimensioni della neoplasia (T)
• presenza ed estensione di metastasi ai linfonodi regionali (N)
• presenza di eventuali metastasi a distanza (M)
Dimensioni della neoplasia. Le dimensioni del tumore costituiscono un fattore prognostico
indipendente molto importante. Il vantaggio dello screening mammografico consiste nella
possibilità di riconoscere ed identificare lesioni di dimensioni inferiori a quelli diagnosticati con la
sola clinica. Un aspetto che esemplifica l’importanza delle dimensioni del tumore è il dato,
confermato dalla letteratura, secondo il quale la sopravvivenza a 20 anni per carcinomi di
dimensioni inferiori ad 1 cm e linfonodi negativi è del 90%, mentre per neoplasie di dimensioni
inferiori a 2 cm, la sopravvivenza scende a 2 cm. Alle dimensioni della neoplasia è correlata anche
l’incidenza di metastasi linfonodali.
Metastasi linfonodali. Lo stato dei linfonodi ascellari rappresenta il più importante fattore
prognostico per il carcinoma invasivo della mammella in assenza di metastasi a distanza. La
valutazione clinica del coinvolgimento linfonodale è inaccurata sia per i falsi positivi che per i falsi
negativi, pertanto la biopsia si rende necessaria per una valutazione accurata. In assenza di
interessamento dei linfonodi, la sopravvivenza libera da malattia a 10 anni è vicina al 70-80%, con
un numero di linfonodi interessati da 1 a 3 la percentuale scende al 35-40%, mentre in presenza di
più di 10 linfonodi positivi, la percentuale di sopravvivenza è del 10-15%. Nella stadiazione e
terapia chirurgica del tumore mammario la tecnica del “linfonodo sentinella” sta acquisendo un
ruolo sempre più importante. Si tratta di una tecnica messa a punto nel 1996 da studiosi del National
Cancer Institute of Bethesda, rapidamente diffusasi a livello internazionale e ormai considerata un
cardine della terapia chirurgica conservativa senologica. La tecnica trova la sua giustificazione

12
fisiopatologica nell’osservazione che la diffusione metastatica delle cellule neoplastiche, dal
focolaio tumorale primitivo ai linfonodi ascellari, avviene in modo regolare e progressivo senza
salti di livello. La negatività istologica del primo linfonodo di drenaggio, che riceve il flusso
linfatico proveniente dall’area di mammella interessata dalla neoplasia (linfonodo sentinella
identificato con tecniche radioisotopiche), permette di escludere con ragionevole sicurezza
l’interessamento metastatico dell’intera catena linfonodale ascellare, evitando in questo modo
l’inutile dissezione ascellare completa (valore predittivo negativo maggiore al 96%) (Cserni G,
2002; Cserni G et al., 2003; Cserni G et al., 2004; Turner RR et al., 2008). La positività istologica
del linfonodo sentinella, indica la diffusione regionale della neoplasia con linfoadectomia ascellare
totale. Con l’introduzione di metodologie più sensibili come sezioni seriali dei linfonodi,
immunoistochimica per le cheratine, rilevazione dell’mRNA carcinoma specifico tramite RT-PCR è
stata possibile l’individuazione di micrometastasi (dimensioni inferiori a 0,2 cm), in un numero
maggiore di pazienti. Il 10-20% di pazienti prive di metastasi linfonodali ascellari, presenta tuttavia
recidiva extramammaria e circa lo stesso numero muore per carcinoma della mammella. In queste
pazienti il processo metastatico avviene tramite linfonodi mammari interni o per via ematica
(Fenaroli P et al., 2000; Mabri H et al., 2007). Le procedure per l’allestimento dei preparati del
linfonodo sentinella mancano di standardizzazione e variano in riferimento alle modalità di taglio e
preparazione macroscopica, al numero di sezioni istologiche da allestire ed esaminare e all’uso di
colorazione immunoistochimiche ancillari. Recentemente sono stati pubblicati studi per l’analsi del
linfonodo sentinella con la procedura “One Step Nucleic Acid Amplification” (OSNA), che prevede
l’uso di uno strumento dedicato per RT-PCR e di kit specifici per l’analisi quantitativa di RNA per
la citocheratina 19 (CK19), marcatore di cellule di carcinoma mammario. Questi studi hanno
dimostrato come la procedura OSNA abbia una sensibilità del 95,3% ed una specificità del 94-97%
rispetto alla procedura classica istologica (Pietrabiasi F et al., 2006; Tsujimoto M et al., 2007;
Tamaki Y et al., 2009).
Metastasi a distanza. Le sedi preferenziali di metastasi a distanza sono i segmenti ossei (70-80%),
soprattutto vertebrali, costali, pelvici e della volta cranica. Le metastasi polmonari rappresentano il
60-65%; una volta raggiunto il polmone, attraverso la circolazione arteriosa produce metastasi
epatiche (60%) e cerebrali (25%).
I fattori prognostici indicati nel TNM sono utili per prevedere la prognosi delle singole neoplasie.
Accanto a questi vengono utilizzati altri fattori utili come indicatori di risposta alla terapia.
Sottotipo istologico. La sopravvivenza a 30 anni nelle donne con carcinomi invasivi di tipo speciale
(tubulare, mucinoso, lobulare, papillare) è maggiore del 60%, rispetto a meno del 20% in pazienti
con carcinomi invasivi di tipo non speciale.

13
Invasione vascolare. Le cellule tumorali si trovano all’interno degli spazi vascolari in circa metà dei
carcinomi invasivi; tale reperto è fortemente associato con la presenza di metastasi linfonodali ed è
quindi indice di prognosi sfavorevole (Kumar V 2005).
Indice proliferativo. La proliferazione cellulare può essere misurata sia mediante conta mitotica sia
mediante la rilevazione immunoistochimica di proteine cellulari prodotte durante il ciclo (Ki67) o
mediante citometria a flusso (frazione di cellule in fase S). Numerose evidenze sembrano suggerire
come livelli di espressione di Ki67, compresi fra il 10 e 14% sia in grado di definire gruppi di
pazienti ad alto rischio in termini di prognosi peggiore (de Azambura E et al., 2007). Alti livelli di
proliferazione cellulare sono inoltre stati trovati essere correlati con la negatività per ER e la
positività per HER2 (Viale G et al., 2008). Ki67 avrebbe anche un ruolo predittivo; carcinomi con
alto indice di proliferazione cellulare possono rispondere meglio alla chemioterapia (Paik S et al.,
2004; Yerushalmi R et al., 2010).
Ploidia. La quantità di DNA per cellula tumorale può essere determinata mediante citometria a
flusso; la presenza di cellule aneuploidi, con contenuto anomalo di DNA, è indicativa di precoce
ripresa di malattia (Munteanu D et al., 2004).
Risposta alla terapia neoadiuvante: dopo intervento chirurgico, la maggior parte delle pazienti
riceve un trattamento sistemico o “terapia adiuvante”. La terapia neoadiuvante è un approccio
alternativo in cui il paziente è trattato prima dell’intervento. Tale approccio non aumenta la
sopravvivenza, ma il grado di risposta alla chemioterapia rappresenta un forte fattore prognostico.
Le neoplasie con maggiore probabilità di rispondere meglio sono quelle poco differenziate, ER-
negative ed associate ad aree di necrosi (Gralow JR et al., 2008).
Di recente introduzione è la ricerca di cellule neoplastiche nel midollo osseo o nel sangue con
tecniche di biologia molecolare; la loro presenza è un indice affidabile di diffusione della malattia.
6.0 FATTORI GENETICI E FAMILIARI
Geni e cancro
Il cancro è una malattia genetica dovuta all’alterazione di più geni. Sono interessati geni la cui
funzione è quella di controllare che la moltiplicazione delle cellule avvenga in modo ordinato. Se è
presente un’alterazione, le cellule possono riprodursi in modo disordinato, infiltrare i tessuti vicini e
diffondersi in tutto il corpo. I geni più frequentemente coinvolti sono gli oncogeni che normalmente
stimolano la proliferazione cellulare, i geni oncosoppressori i quali normalmente frenano la
moltiplicazione delle cellule ed i geni riparatori del danno del DNA che favoriscono l’insorgenza
del cancro non correggendo gli errori che si verificano quando il DNA viene duplicato e
consentendo pertanto, l’accumulo di mutazioni. Particolare interesse hanno assunto i test genetici

14
predittivi in grado di individuare soggetti a rischio di sviluppare una neoplasia per il fatto di avere
ereditato un gene mutato, prima della comparsa di segni o sintomi. Un test genetico predittivo dirà
se è presente o meno una mutazione correlata ad una determinata neoplasia. se la mutazione è
presente, lo sviluppo successivo della neoplasia dipende dalla penetranza del gene (Jeffrey SS et al.,
2005).
6.1 GENI DI SUSCETTIBILITA’ PER IL TUMORE MAMMARIO
I geni ad elevata penetranza predisponenti al tumore mammario finora identificati sono: BRCA1 e
BRCA2 (Breast cancer susceptibility gene 1 o 2) (Walsh T et al., 2006), p53 (sindrome di Li-
Fraumeni), la fosfatasi PTEN (malattia di Cowden) (Steck PA et al., 1997), la serin treonin chinasi
STK11/LKB1 (sindrome di Peutz-Jeghers) (Thull DL et al., 2004; Cuatrecasas M et al., 2006;
Sabate JM et al., 2006) e la chinasi ATM (ataxia-telangiectasia) (Chenevix-Trench G et al., 2002)
(Tab 1).
Tab 1: principali geni predisponenti al tumore della mammella
La frequenza di mutazioni varia da 1/1000 nel caso di BRCA1 a circa 1/10000 nelle sindromi più
rare.
Mutazioni relativamente comuni a livello dei geni a bassa penetranza agiscono assieme a fattori
endogeni e condizioni legate allo stile di vita nell’insorgenza di neoplasie sporadiche, che
rappresentano la maggior parte dei carcinomi mammari.

15
6.2 IL CANCRO DELLA MAMMELLA FAMILIARE: BRCA1/BRCA2
Non esiste una mutazione genetica univocamente associata al rischio. L’anamnesi familiare positiva
per carcinoma della mammella, aumenta il rischio di sviluppare la malattia; studi epidemiologici
hanno dimostrato che circa il 12% dei pazienti con carcinoma della mammella hanno almeno un
familiare affetto da malattia e come il rischio aumenta con il numero dei familiari affetti (Jeffrey SS
et al., 2005; Jemal A et al., 2008).
La probabilità che sia presente una mutazione genetica aumenta se la storia familiare include la
giovane età alla diagnosi, raggruppamenti di carcinomi mammari ed ovarici (80% BRCA1),
carcinoma della mammella maschile (66% BRCA2) e altre rare neoplasie come sarcomi e tumori
della corticale del surrene (70% p53).
Dal 5-10% dei carcinomi della mammella compaiono come risultato di specifiche mutazioni in geni
ad alta penetranza (Walsh T et al., 2006). Tali mutazioni, trasmesse con ereditarietà autosomica
dominante sono relative ai due geni oncosoppressori BRCA1 (17q21) e BRCA2 (13q12.3); essi
sono responsabili dell’80-90% delle neoplasie geneticamente determinate, codificando per proteine
nucleari coinvolte nei meccanismi biochimici che controllano l’integrità del genoma (Wooster R et
al., 2003; Allain DC et al., 2007). Rappresentano forti predittori di rischio e conferiscono un
aumentato rischio di neoplasia mammaria, ad esordio spesso bilaterale ed in età precoce (Fig 6).
Mutazioni a carico del gene BRCA1 comportano un rischio del 65-85% di sviluppare un cancro
della mammella nel corso della vita ed un rischio del 39-46% di sviluppare un cancro dell’ovaio
(King MC et al., 2003). Nel sesso maschile mutazioni di BRCA1 non comportano un incremento di
rischio di carcinoma mammario, ma probabilmente di cancro della prostata e del colon
Nel caso di mutazioni di BRCA2, il rischio di sviluppare nel corso della vita, un tumore della
mammella è di circa il 7,5% per gli uomini e del 45-85% per le donne, mentre per il tumore
dell’ovaio è del 10-27% (King MC et al., 2003). Nella popolazione generale una mutazione di
BRCA1 è presente in 1 ogni 500-800 individui; mutazioni a carico di BRCA2 sono meno frequenti.
Negli ebrei Askenazi (dell’Europa occidentale e USA), una mutazione di BRCA2 si riscontra in un
individuo ogni 40 (Chen S et al., 2007); in questa popolazione un’alta percentuale di famiglie sono
associate a poche mutazioni nei due geni denotando per quest’ultime un effetto fondatore.
Le neoplasie in pazienti con mutazioni di BRCA2 non hanno un fenotipo distinto dalle neoplasie
sporadiche a differenza dei carcinomi associati a mutazioni di BRCA1 che presentano un fenotipo
duttale, elevato grado istologico, consistente infiltrato linfocitario, negatività per recettori ormonali
e HER2 ed iperespressione di p53 (Lakhani SR et al., 2002; Tutt A et al., 2008).
Di un terzo gene BRCA3 ne è stata ipotizzata solo l’esistenza, sebbene alcuni studi ne hanno
individuato un possibile locus a livello del braccio corto del cromosoma 8 (Rahman N et al., 2000)

16
Recentemente sono stati identificati altri geni responsabili della suscettibilità al cancro della
mammella come FGFR2 , TNRC9, LSP1, MAP3K1 (Easton DF et al., 2007).
Fig 6: geni BRCA1 e BRCA2
6.3 TP53
Il gene p53 definito “guardiano del genoma” è localizzato sul cromosoma 17p14 ed è un gene
regolatore della trascrizione, stabilizzatore genomico, ed inibitore della progressione del ciclo
cellulare; tramite p21 determina l’arresto del ciclo cellulare per permettere al sistema di riparazione
del DNA di intervenire e correggere eventuali lesioni; tramite l’induzione di BAX determina
l’apoptosi delle cellule nel caso di un danno più esteso. Le anormalità del gene oncosoppressore p53
sono quelle di più comune riscontro nelle neoplasie umane in genere e le sue mutazioni, portando
alla perdita della regolazione del ciclo cellulare sono legate ad una prognosi peggiore anche nel
carcinoma della mammella. Per determinare le mutazioni di p53 sono stati condotti studi sia della
proteina (con tecniche immunoistochimiche), sia del gene stesso (mediante sequenziamento del
DNA). Circa il 30-50% dei carcinomi della mammella presenta una mutazione somatica di p53; le
mutazioni variano secondo popolazione e stadio del tumore. Hanno una maggiore incidenza nei casi
con linfonodi positivi e nei tumori di maggiori dimensioni con malattia a distanza. Inoltre la
frequenza di mutazioni di tale gene è maggiore nei casi di recidiva di malattia rispetto al carcinoma
primitivo ed in pazienti giovani (Marchetti P et al., 2003; Ricevuto E et al., 2003). La presenza di
mutazioni si correla inoltre con prognosi e sopravvivenza peggiore rispetto ai casi wild-type.
Pertanto, la caratterizzazione del genotipo p53 (wild-type e mutato) rappresenta, nel carcinoma
della mammella, uno dei principali fattori prognostici e predittivi della sensibilità a farmaci
genotossici. Il genotipo p53 mutato caratterizza un sottogruppo di pazienti con prognosi meno
BRCA1
BRCA2

17
favorevole e meno sensibili all’azione di farmaci genotossici (alchilanti, cisplatino, antracicline)
(Rodier F et al., 2007). Il 50-70% dei tumori della mammella in donne con mutazioni di BRCA1
presentano anche mutazioni in TP53 suggerendo un’interazione di questi due geni nello sviluppo
della malattia (Arizti P et al., 2000). Molte donne con mutazioni di TP53 nella linea germinale
(sindrome di Li-Fraumeni), che sopravvivono a tumori in età giovanile, sviluppano un cancro in età
adulta.
6.4 ORMONI ENDOGENI
Estrogeno e progesterone nel tessuto mammario influenzano la normale proliferazione, la
differenziazione e la fisiologia cellulare ma possiedono un ruolo primario anche nello sviluppo e
progressione della neoplasia. La prima dimostrazione che il carcinoma mammario è dipendente
dalla stimolazione estrogenica risale alla fine del XIX secolo quando Beatson dimostrò che era
possibile indurre la regressione del tumore della mammella in donne in premenopausa con
ooforectomia (Beatson GT, 1896). Gli effetti fisiologici degli estrogeni sono mediati da un fattore di
trascrizione a localizzazione nucleare, ligando-inducibile, conosciuto come recettore estrogenico
(ER) (Jensen EV et al., 1971). Il legame ormone-recettore promuove una cascata di eventi che
culminano nell’attivazione o repressione di geni specifici fra i quali il gene che codifica per il
recettore progestinico (PgR). A sua volta l’attività di ER è modulata da numerosi co-regolatori
nucleari che possono produrre effetti sia positivi che negativi. Il recettore ER può esistere sia in
forma omodimerica che eterodimerica per la presenza di due isoforme ERα e ERβ.
Il gene del recettore α è localizzato sul cromosoma 6q25.1 e codifica per una proteina si 595
aminoacidi organizzati in 6 domini indicati con le lettere A/B, C, D, E, F (Pavao M et al., 2001). Il
dominio A/B costituisce la porzione amino terminale ed è sede dell’attività transattivante. Il
dominio C è deputato al legame con il DNA; la regione D funge da cerniera e consente
modificazioni conformazionali durante l’attivazione; il dominio E, situato vicino all’estremità
carbossiterminale è deputato al legame con l’ormone steroideo.
Il gene del recettore β è localizzato sul cromosoma 14q22-24. Presenta un elevato grado di
omologia con l’isoforma α che raggiunge il 96% a livello del dominio C e lega l’estradiolo con la
stessa affinità di ERα (Mosselman S et al., 1996). La maggiore diversità fra i due recettori risiede
nella porzione trans attivante in grado di innescare processi di trascrizione diversi per i due
recettori. La forma β inoltre possiede multiple isoforme. Poco si conosce circa il significato clinico
di ERβ; l’espressione immunoistochimica di ERβ appare correlata a quella di ERα (Skliris GP et
al., 2001), ma circa la metà dei casi ERα-negativi, sono immunoreattivi per ERβ (Mann S et al.,
2001). Secondo alcuni autori l’espressione di ERβ rappresenterebbe un indice di prognosi migliore

18
(Omoto Y et al., 2001) e di risposta alla terapia con tamoxifene (Mann S et al., 2001). E’ ipotizzato
che una quota di casi attualmente considerati ERα siano ERβ positivi e quindi potenzialmente
responsivi alla terapia ormonale (Swain SM, 2001). Altri studi riportano come casi negativi per ERα
e positivi per ERβ siano associati ad una quota proliferativa più elevata e non suscettibili alla
terapia con antiestrogeni (Jensen EV et al., 2001), in accordo con quanto dimostrato per l’mRNA di
ERβ che appare essere maggiormente espresso nei casi resistenti al tamoxifene (Speirs V et al.
,1999). Gli studi su ERβ sono ancora in una fase preliminare ma sono importanti per una migliore
definizione del profilo recettoriale di singoli casi nel modulare in modo selettivo i diversi tipi di
recettore con farmaci appropriati (Barkhem T et a., 1998).
I carcinomi della mammella ERα positivi sono associati con un basso indice di proliferazione, basso
grado istologico, diploidia del DNA e quindi sono associati con una buona prognosi (Ross JS et al.,
2005). Più del 90% dei carcinomi lobulari sono ER-positivi mentre, i carcinomi midollari ed
infiammatori sono più frequentemente ER-negativi. Tumori negativi per entrambi i recettori
ormonali sono spesso associati ad un comportamento più aggressivo e presentano amplificazione
dei geni HER-2/neu, c-myc e mutazioni nel gene p53 (Taneja P et al., 2010).
La determinazione del recettore progestinico PgR, ulteriore proteina regolata dagli estrogeni (Jensen
EV et al., 2003; Fournier A et al., 2008), migliora la discriminazione tra neoplasie mammarie
sensibili e non alla terapia ormonale. Anche per il recettore per il progesterone esistono almeno due
diverse isoforme con perso molecolare diverso A e B, codificate dallo stesso gene (Kraus WL et al.,
1993). Le due proteine sono identiche eccetto che per un’ estensione di 164 aminoacidi in sede N-
terminale presente nel recettore B. nonostante la stessa similarità strutturale, le due isoforme hanno
proprietà funzionali diverse L’espressione di PgR è strettamente dipendente da quella di ER.
Tumori che esprimono PgR ma non ER, rappresentano meno dell’1% di tutti i casi di carcinoma
della mammella (Viale G et al., 2007). Esistono evidenze di come nei carcinomi metastatici della
mammella, la risposta al trattamento con farmaci anti-estrogenici sia migliore in pazienti che co-
esprimono ER e PgR, rispetto a quelli che esprimono solo ER (Elldge RM et al., 2000; Liu S et al.,
2010). PgR ha quindi un valore predittivo di risposta alla terapia ormonale e prognostico nei casi
positivi per il recettore estrogenico (Liu S et al., 2010); alti livelli di PgR risultano correlati
negativamente con le dimensioni del tumore ed il grado (Weigel MT et al., 2010).
ER e PgR modulano l’espressione di diversi geni fra i quali AIB-1, c-myc e ciclina D1; in
particolare la ciclina D1 interagisce con ER-α nel promuovere l’attività trascrizionale di
quest’ultima (Neuman E et al., 1997).

19
Un’ elevata concentrazione di ER e PgR non è solo altamente predittiva della risposta alla terapia
ormonale, ma, dato che in genere la loro presenza si accompagna ad una maggiore differenziazione,
può essere utile anche nella stima della sopravvivenza libera da malattia (Bast RC et al, 2001).
Gli estrogeni possono intervenire in tutte le fasi del processo di cancerogenesi mammaria:
iniziazione/trasformazione, promozione e progressione. I meccanismi possibili sono:
- l’induzione di una proliferazione cellulare abnorme attraverso lo stimolo degli ER: questo può
avvenire sia per aumento della quantità di ormone presente (come in caso di obesità), sia per
alterazioni strutturali dei recettori che diventano sensibili a minime quantità di estrogeni come
quelle della donna in post-menopausa.
- effetto genotossico diretto mediato dal citocromo P450; i complessi del citocromo P450 hanno un
ruolo nel catabolismo ossidativo degli estrogeni che porta alla produzione di radicali liberi, che a
loro volta sono causa di stress ossidativo e di danno genomico;
- azione diretta sul genoma, con induzione di uno stato di aneuploidia.
Interessante è il possibile ruolo degli estrogeni ambientali come i fitoestrogeni, pesticidi organo
clorurati.
La valutazione dell’espressione dei recettori ormonali viene routinariamente condotta mediante
analisi immunoistochimica, indicando il risultato come percentuale di cellule positive (Fig 7). La
risposta terapeutica è migliore quando sono presenti entrambi i recettori. Attualmente viene valutata
la positività nei nuclei, in quanto la localizzazione nucleare dei recettori è ben nota. Tuttavia,
informazioni sempre maggiori sulla presenza di recettori in sede extranucleare suggeriscono di
valutare anche tale parametro.
I farmaci impiegati nella terapia ormonale appartengono a diverse categorie; fra esse i SERM (anti-
estrogeni) e gli inibitori delle aromatasi. Gli anti-estrogeni (tamoxifene) competono con gli
estrogeni stessi per il legame ad ER inducendo un effetto citostatico. Gli inibitori delle aromatasi
(letrozolo, anastrozolo) bloccano la formazione degli estrogeni
Fig 7: positività per recettori estro-progestinici

20
6.5 HER-2
Il valore prognostico di HER-2 neu è stato recentemente considerato rilevante nella definizione del
rischio e pertanto deve essere tenuto in considerazione. Le alterazioni di HER-2 sono associate ad
un maggiore rischio di metastasi linfonodali, all’elevato grado istologico, alla negatività per i
recettori steroidei, alla più giovane età d’insorgenza e, più in generale, ad una peggiore prognosi
nelle pazienti affette da carcinoma della mammella (Allen MG, 2008). HER-2 viene attualmente
studiato soprattutto per il suo valore predittivo, in quanto un aumento dell’espressione di questo
oncogene, quando determinato con metodiche immunoistochimiche o con tecnica FISH, è in grado
di predire la risposta a farmaci basati su anticorpi monoclonali come l’herceptin.
Il proto-oncogene ErbB2 (HER2/neu) è situato sul braccio lungo del cromosoma 17 (17q11.2-q12).
Codifica per un mRNA di 4,6 Kb, tradotto in una proteina di 185 kDa, chiamata p185 la quale ha
funzione recettoriale ad attività tirosin-chinasica (Coussens L et al., 1985).
ErbB2 appartiene ad una famiglia di recettori per fattori di crescita che comprende ErbB1, meglio
conosciuto come EGFR, ErbB3 (HER3) ed ErbB4 (HER4).
La struttura monomerica di questi recettori è formata da un dominio trans membrana di 25-30
aminoacidi, da un dominio extracellulare N-terminale di 620 aminoacidi, che lega i fattori di
crescita e strutturato in quattro sottodomini denominati L1, L2 (leucine-rich), CR1, CR2 (cysteine-
rich) ed infine un dominio intracellulare C-terminale, responsabile dell’attività tirosin-chinasica. La
fosforilazione della tirosina nel recettore produce siti di legame per proteine che contengono domini
SH2 (Src homology 2) e PTB (Phosphotyrosine binding). Fanno parte di questo gruppo di proteine
Grb2, Grb7, Crk e Gab1, proteine e lipidi-chinasi come fosfatidilinositolo-3-chinasi, e fosfolipasi
Cγ e proteine fosfatasi come SHP1 e SHP2 (Hynes NE et al., 2005). Dopo attivazione si innescano
meccanismi di trasduzione del segnale che portano a divisione cellulare. L’integrità del segnale di
ErbB richiede l’unione, indipendente dalla fosforilazione, con proteine che regolano l’attività
recettoriale e la corretta localizzazione in membrana. I fattori di crescita che legano questi recettori
sono conosciuti come “hereguline” o “neureguline” ed il loro legame con ErbB3 ed ErbB4 induce
un’eterodimerizzazione con ErbB2 e successiva trasduzione a valle del segnale (Fig 8).

21
Fig 8: segnali cellulari di ErbB2 (HER2/neu)
ErbB2 non è in grado di legarsi a fattori endogeni, ma forma dimeri con altri recettori della
famiglia, già legati, stabilizzandoli ed innescando la trasduzione del segnale chinasi mediata. Tutte
le combinazioni dei quattro recettori possono essere indotte da 10 specifici ligandi di ErbB,
generando segnali molto diversi fra di loro. Alternativamente, l’iperespressione di recettori, che può
essere osservata in alcuni tumori, incluso quello della mammella, promuove la dimerizzazione
spontanea in assenza di ligando e quindi l’attivazione costitutiva.
L’amplificazione di ErbB2 e l’iperespressione della relativa proteina sono riscontrati nel 15-20%
dei carcinomi della mammella (Owens MA et al., 2004) e sono associati alle metastasi linfonodali,
all’elevato grado istologico, alla negatività per i recettori steroidei, alla più giovane età e più in
generale ad una prognosi peggiore (Slamon DJ et al., 1987; Slamon DJ et al., 1989). Dati recenti
suggeriscono come anche l’iperespressione di ErbB3 contribuisca al fenotipo maligno attraverso
SOPRAVVIVENZA
PROLIFERAZIONE
ONCOGENESI
ANGIOGENESI
TUMORIGENESI
INIBIZIONE APOPTOSI
MOTILITA’ CELLULARE
ESPRESSIONE GENICA
PROGRESSIONE NEL CICLO
TRASFORMAZIONE
DIFFERENZIAZIONE
APOPTOSI
FATTORI DI CRESCITA
EGF, TGFα, AMFIREGULINA
NUCLEO
CITOPLASMA
MEMBRANA CELLULARE
TRASCRIZIONE DI BCL-XL, MYC, CCND1, CDKN1A
DIFFERENZIAZIONE, PROLIFERAZIONE, SOPRAVVIVENZA, ONCOGENESI,
ANGIOGENESI

22
l’aumento della motilità cellulare, con induzione di potenziali metastasi e trasduzione di segnali
anti-apoptotici che prolungherebbero la sopravvivenza cellulare e contribuirebbero all’insorgenza di
instabilità genetica e di resistenza farmacologica. Le hereguline sono importanti fattori migratori
delle cellule neoplastiche nel cancro della mammella, inducendo la riorganizzazione dell’actina e la
formazione di strutture citoscheletriche mobili. Inoltre le hereguline stimolano anche PAK1 (p21-
activate-kinase), chinasi implicata nella promozione della migrazione cellulare. Questo può avere
rilevanza nello sviluppo della malattia metastatica che nel carcinoma della mammella è infatti
associata ad iperespressione di ErbB2. questa può anche essere causa di resistenza farmacologica
per induzione della riparazione delle rotture del DNA determinate da chemioterapici come ad
esempio la doxorubicina. L’iperespressione di recettori tirosin chinasici della famiglia ErbB
contribuisce al prolungamento del ciclo cellulare, permettendo la riparazione del DNA e
sovraregolando membri antiapoptotici della famiglia Bcl2.
L’espressione di ErbB2 risulta associata ad una sottoregolazione di BAX e ad una sovraregolazione
di molecole antiapoptotiche come Bcl2 e Bcl-XL; il segnale apoptotico risulta quindi ridotto,
venendo favorita l’instabilità genomica con comparsa della resistenza ad agenti chemioterapici.
Questo vale per farmaci che agiscono producendo danni al DNA come analoghi nucleosidici (5-
fluorouracile, citarabina, fludarabina), antibiotici intercalanti del DNA (adriamicina), agenti
alchilanti che formano legami tra i due filamenti (ciclofosfmide) (Ross JS et al., 1998).
6.6 TOPOISOMERASI IIα
L’isoforma α della topoisomerasi (TOP2A) rappresenta un enzima chiave nella replicazione e
riparazione del DNA e il bersaglio principale di vari agenti chemioterapici fra i quali le antracicline.
Il gene che codifica per tale proteina è localizzato sul cromosoma 17q21 in prossimità del gene
HER2/neu; l’enzima catalizza la rottura e la riunione del DNA a doppia elica con rilassamento della
superelica del DNA. Le antracicline esercitano il loro effetto stabilizzando i tagli del DNA ed
inibendo la replicazione cellulare bloccando l’attività della TOP2A (Kellner U et al., 2002). Diversi
studi hanno dimostrato come aberrazioni del gene TOP2A, in particolare le amplificazioni, siano
responsabili della maggiore sensibilità delle cellule neoplastiche alle antracicline (Faratian D et al.,
2008). TOP2A è risultato essere frequentemente coamplificato con HER2/neu (Di Leo A et al.,
2002), suggerendo pertanto che la sensibilità alle antracicline dei tumori esprimenti HER2 sia in
realtà dovuta alla coamplificazione di TOP2A (Jarvinen TAH et al., 2003; Beser AR et al., 2007). I
dati riportati in letteratura sullo stato di espressione delle due proteine sono contrastanti; in alcuni
studi è stata descritta un’alta correlazione fra l’espressione delle due proteine, mentre in altri
l’iperespressione della proteina TOP2A è stata riportata in meno del 10% dei casi con

23
amplificazione del rispettivo gene (Durbecq V et al., 2004) e nel 30% dei casi non amplificati per
HER-2/neu (Sotiriou C et al., 2003). Quindi l’amplificazione genica sarebbe solo uno dei possibili
meccanismi determinanti l’iperespressione di TOP2A, essendo quest’ultima regolata a diversi livelli
(Bakshi RP et al., 2001). Recentemente è stato dimostrato come l’RNA di TOP2A, quantificabile
mediante la metodica RT-PCR, rappresenti un più utile marcatore prognostico associato alla
risposta alle antacicline (Brase JC et al., 2010).
6.7 EGFR
EGFR è codificato dal gene ErbB1, localizzato sul cromosoma 7q12. EGFR risulta essere
iperespresso nel 15-60% dei carcinomi della mammella (Nicholson RJ et al., 2001) ed il suo reale
significato prognostico rimane ancora da valutare. Tuttavia sembra certo che la sua presenza sia
associata con una perdita di espressione dei recettori estrogenici e una cattiva prognosi (Chan SK et
al., 2006). È stata inoltra osservata una correlazione positiva fra l’espressione di EGFR e
l’iperespressione di HER2 (Rimawi MF et al., 2010). Studi di espressione genica e
immunoistochimici hanno dimostrato che il 50-70% dei tumori della mammella di tipo basale,
esprimono EGFR (Burness ML et al., 2010). Inoltre risulta essere over-espresso nel 30% dei
carcinomi infiammatori della mammella (Yamauchi H et al., 2010). La coespressione di EGFR ed
HER2 è stata osservata nel 10-36% dei carcinomi della mammella ed è associata ad una peggiore
prognosi rispetto ai casi che iperesprimono uno solo dei due recettori (D’Alessio A et al., 2010;
Rimawi MF et al., 2010).
6.8 VEGF
La famiglia del Vascular Endothelial Growth Factor è composta da 5 isoforme (VEGFA, VEGFB,
VEGFC, VEGFD e PLGF) le quali sono ligandi per recettori ad attività tirosin chinasica (VEGFR).
Dopo il legame al rispettivo recettore (soprattutto VEGFR2), vengono attivate vie di segnale
intracellulari fra le quali MEK-ERK e PI3K-Akt, che mediano il processo di angiogenesi.
L’attivazione dell’angiogenesi sia nel tessuto normale che in quello tumorale dipende dalla
proliferazione ed invasione delle cellule endoteliali, dall’incrementata permeabilità vascolare e dal
reclutamento di cellule di supporto come i periciti. VEGF e l’angiogenesi sono fondamentali per la
crescita del tumore e per il processo metastatico in molti tipi di tumore solido (Sakakibara S et al.,
2009). VEGF ha un ruolo determinate nell’angiogenesi del carcinoma della mammella ed è
associato ad una peggiore prognosi (Relf M et al., 1997). Il più importante fattore che determina la
sopravvivenza dei pazienti affetti da carcinoma della mammella è la disseminazione a distanza delle
cellule neoplastiche; lo studio dell’espressione genica nei tumori primitivi, nelle metastasi regionali

24
e in quelle a distanza ha indicato come il gene per VEGF, sia il solo ad essere iperespresso nelle
sedi metastatiche ed associato ad una prognosi sfavorevole, inoltre in pazienti metastatici sono
osservabili incrementati livelli serici di VEGF, i quali risultano essere direttamente correlati al
grado tumorale (Shivakumar S et al., 2009). In uno studio retrospettivo è stato dimostrato un
significativo incremento dei livelli intratumorali di VEGF in pazienti con carcinoma della
mammella triplo negativo rispetto agli altri fenotipi (Linderholm BK et al., 2009; Toft DJ et al.,
2011). Un maggiore rischio di carcinoma invasivo della mammella sarebbe correlato con due
polimorfismi genici di VEGF (VEGF-2578C e VEGF-1154G) capaci di incrementare l’espressione
della relativa proteina (Jacobs EJ et al., 2006). Un’intensa attività angiogenetica è stata osservata
nel carcinoma infiammatorio della mammella in accordo con l’elevato potenziale metastatico di
questa forma di tumore (van der Auwera I et al., 2004).
VEGF per il ruolo centrale nell’angiogenesi, per la sua specificità e la sua correlazione con la
prognosi, è diventato un attraente bersaglio della terapia antitumorale. Sono stati sviluppati
anticorpi (bevacizumab) ed inibitori tirosin chinasici (TKI) (sunitinib) che legano VEGFR (Hayes
DF et al., 2005; Nielsen DL et al., 2010). L’efficacia dei farmaci antitumorali è spesso ridotta dalla
difficoltà di veicolazione delle sostanze, dalla loro mancanza di specificità e dalla resistenza che il
carcinoma sviluppa verso essi. La crescita tumorale si basa su un adeguato apporto sanguigno e
quindi sviluppo di basi al suo interno. I vasi anomali sono un bersaglio evidente per la terapia
farmacologica, attraverso l’uso di sostanze antiangiogeniche, cercando si superare anche il
problema della resistenza dal momento che le cellule endoteliali sono geneticamente stabili,
omogenee e hanno un basso livello mutazionale (Kerbel RS, 1997; Gasparini G et al., 2005).
L’angiogenesi tumorale può essere inibita da sostanze che hanno un effetto diretto sulle cellule
endoteliali e da sostanze che agiscono su cellule che stimolano l’angiogenesi indirettamente (cellule
tumorali e cellule dello stroma). Questa classe di molecole non solo arresta la proliferazione delle
cellule endoteliali, ma ne aumenta la morte per apoptosi inducendo la regressione dei vasi formatisi
(Filho AL et al., 2010). Bevacizumab sarebbe particolarmente indicato per il trattamento dei tumori
della mammella tripli negativi (Greenberg S et al., 2010).
25
7.0 CLASSIFICAZIONE MOLECOLARE DEL CARCINOMA DELLA MAMMELLA
I carcinomi mammari rappresentano un gruppo molto eterogeneo di neoplasie dal punto di vista
morfologico, prognostico e di risposta alla terapia. Recentemente la spiccata eterogeneità del
carcinoma mammario è stata confermata dallo studio del profilo dell’espressione genica che ha
rivelato come ogni singolo carcinoma mammario abbia un suo preciso ed unico assetto molecolare
(Perou CM et al., 2000; Sorlie T et al., 2006; Marchiò C et al., 2008; Tan BK et al., 2008; Sandhu
R et al., 2010; Schnitt SJ, 2010; Goldhirsch A et al., 2011). Nonostante questa grande variabilità, le
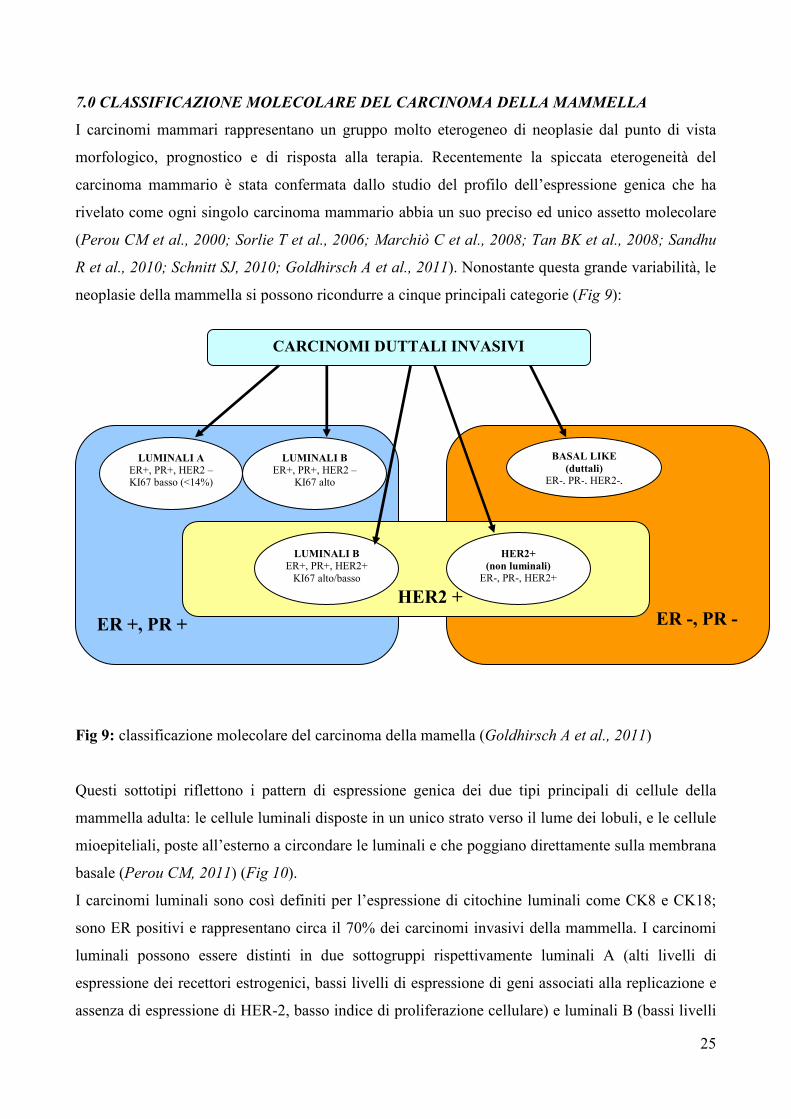
neoplasie della mammella si possono ricondurre a cinque principali categorie (Fig 9):
Fig 9: classificazione molecolare del carcinoma della mamella (Goldhirsch A et al., 2011)
Questi sottotipi riflettono i pattern di espressione genica dei due tipi principali di cellule della
mammella adulta: le cellule luminali disposte in un unico strato verso il lume dei lobuli, e le cellule
mioepiteliali, poste all’esterno a circondare le luminali e che poggiano direttamente sulla membrana
basale (Perou CM, 2011) (Fig 10).
I carcinomi luminali sono così definiti per l’espressione di citochine luminali come CK8 e CK18;
sono ER positivi e rappresentano circa il 70% dei carcinomi invasivi della mammella. I carcinomi
luminali possono essere distinti in due sottogruppi rispettivamente luminali A (alti livelli di
espressione dei recettori estrogenici, bassi livelli di espressione di geni associati alla replicazione e
assenza di espressione di HER-2, basso indice di proliferazione cellulare) e luminali B (bassi livelli
ER +, PR +
LUMINALI A
ER+, PR+, HER2 – KI67 basso (<14%)
ER -, PR -
BASAL LIKE
(duttali) ER-, PR-, HER2-,
HER2 +
LUMINALI B
ER+, PR+, HER2+ KI67 alto/basso
HER2+
(non luminali) ER-, PR-, HER2+
LUMINALI B
ER+, PR+, HER2 – KI67 alto
CARCINOMI DUTTALI INVASIVI

26
di espressione dei recettori estrogenici e alti livelli di espressione di geni coinvolti nella
proliferazione cellulare, iperespressione di HER2) (Perou CM et al., 2000; Sorlie T et al., 2001;
Cheang MCU et al., 2009). Rispondono alla terapia ormonale, mentre la risposta alla chemioterapia
è variabile con il fenotipo B che risponde meglio rispetto a quello A. La prognosi risulta migliore
per il fenotipo A (Schnitt SJ, 2010).
I tumori HER2 comprendono circa il 10-15% dei carcinomi invasivi; sono rappresentati da
neoplasie di alto grado, linfonodo positive, caratterizzate dall’espressione di HER2 e di altri geni
come GRB7 e GATA4 (Sorlie T et al., 2001). Rispondono alla terapia con trastuzumab e alla
chemioterapia con antracicline; tuttavia hanno una cattiva prognosi (Hu Z et al., 2006).
I carcinomi basali detti anche basaloidi, basal-like, o carcinomi a fenotipo basale, costituiscono il
10-20% dei carcinomi invasivi della mammella e sono caratterizzati dall’espressione di
citocheratine come CK5, CK14, CK17, presenti nello strato basale/miopeteliale della ghiandola
normale (Maggie CU et al., 2008; Rakha EA et al., 2008; Choo JR et al., 2010). Frequentemente i
carcinomi basali hanno un assetto sovrapponibile ai carcinomi cosidetti “tripli negativi” per la
perdita di espressione dei recettori estrogenici e progestinici e assenza di iper-espressione di HER-2.
Il fenotipo a cellule basali, triplo negativo è caratterizzato da un’alta probabilità di recidive
(metastasi polmonari e cerebrali) e da una sopravvivenza totale e libera da malattia
significativamente bassa. Altri studi tuttavia hanno attribuito una prognosi peggiore ai sottogruppi
HER-2 positivi (Fulford LG et al., 2007). Inoltre il fenotipo a cellule basali si può associare alla
presenza di mutazioni in BRCA1 (Turner NC et al., 2006; Rakha EA et al., 2008), ed è più
frequente nelle giovani donne di colore. Il riconoscimento di questo sottogruppo di neoplasie è
importante sia per le implicazioni prognostiche che per la loro associazione con i carcinomi eredo-
familiari. I carcinomi “basal-like” sono generalmente neoplasie duttali infiltranti di grado 3; sono
altamente cellularizzate, a margini tondeggianti con crescita di tipo espansivo e sclerosi centrale.
Hanno architettura solida, senza formazione di tubuli e scarso stroma intercellulare. Alcuni
presentano caratterisitiche “simil-midollari” con infiltrato linfocitario alla periferia ed aspetti di tipo
sinciziale della popolazione neoplastica; recentemente è stato dimostrato come il 50% dei carcinomi
midollari veri, presenta immunofenotipo basale e/o mioepiteliale (Jacquemier J et al., 2005). In altri
casi si possono osservare strutture nastriformi associate ad aree di necrosi; le cellule neoplastiche
hanno scarso citoplasma, nucleo tondo-ovale con alto rapporto nucleo/citoplasma e nucleoli
evidenti. Spesso è evidenziabile una componente a cellule fusate, a cellule chiare, a cellule basiloidi
e una metaplasia squamo-cellulare. Presentano un’elevata attività mitotica. Dal punto di vista
immunofenotipico i carcinomi basali sono positivi per vimentina, EGFR, c-Kit, p53, possono
esprimere marcatori del mioepitelio come l’actina muscolo liscio, p63, e CD10 (Reis-Filho JS et al.,

27
2006; Kim MJ et al., 2006) e presentano un’alta espressione della via di segnale di PI3K e bassi
livelli di PTEN (Marty B et al., 2008). Si ritiene che il fenotipo miopeteliale rappresenti un
sottogruppo dei basali. Pertanto di fronte ad una neoplasia duttale NOS di grado 3, triplo negativa è
opportuno procedere ad una caratterizzazione immunofenotipica con vimentina, CK5/6, EGFR che
se positive confermano il fenotipo basal-like (Nielsen TO et al., 2004; Cheang MCU et al., 2008).
La cattiva prognosi che caratterizza questo gruppo di tumori può essere associata solo alle neoplasie
duttali NOS, in quanto l’immunofenotipo basale è comune anche ad altre neoplasie con
differenziazione mioepiteliale che invece hanno una migliore prognosi (Rouzier R et al., 2005).
L’evoluzione peggiore si avrebbe per le neoplasie che co-esprimono il fenotipo basale e quello
mioepiteliale (Fadare O et al., 2008, Maggie CU et al., 2008; Hudis CA et al., 2011).
Fig 10: classificazione molecolare del carcinoma della mammella
Altro sottotipo di carcinoma della mammella è rappresentato dai cosidetti “normal breast”, sulla cui
reale esistenza ci sono tuttavia ancora dei dubbi. Comprenderebbero il 5-10% di tutti i carcinomi
della mammella; esprimono geni caratteristici del tessuto adiposo e presentano una prognosi
intermedia fra i luminali ed i basali, non rispondendo alla chemioterapia neo-adiuvante.
Recentemente è stato identificato un nuovo sottotipo di carcinoma della mammella (Herschkowitz
JI et al., 2007; Eroles P et al., 2012). Comprende il 12-14% dei carcinomi mammari ed è
caratterizzato da una bassa espressione di geni coinvolti nelle adesioni intercellulari fra i quali

28
claudina-3, -4, 7, ed E-caderina. Tale istotipo (cludin-low) è sovrapponibile per alcuni aspetti con i
tumori basal-like; istologicamente sono neoplasie di alto grado con differenziazione metaplastica o
midollare e prognosi sfavorevole (Prat A et al., 2010).
La caratterizzazione del fenotipo molecolare comincia ad essere utilizzata per selezionare terapie
mirate contro bersagli molecolari (target therapy) come ad esempio per quanto riguarda le
indicazioni all’ormonoterapia e alla terapia anti-HER2 (Goldhirsch A et al., 2011). In quest’ottica
numerosi inibitori della via di trasduzione del segnale dell’EGFR (TKI, inibitori della
dimerizzazione, inibitori farnesiltransferasi, anti-RAF, inibitori MAPK, inibitori mTOR) sono
attualmente in fase di studio (Crown J et al., 2012; Gelmon K et al., 2012). L’attivazione della via
di trasduzione di HER2 rappresenta, uno dei meccanismi principali che caratterizzano la
proliferazione delle cellule di carcinoma mammario. Il controllo di tale via a livello del passaggio
PI3K/AKT/mTOR, è mediata da una fosfatasi codificata dal gene PTEN che risulta essere inattivato
in molte neoplasie umane fra le quali il carcinoma della mammella (Daniele L et al., 2009).
8.0 ANEUPLOIDIE E CARCINOMA DELLA MAMMELLA
Nel carcinoma della mammella la frequenza di tumori aneuploidi è piuttosto elevata variando dal 45
al 70% dei casi. L’aneuploidia è direttamente correlata alla scarsa differenziazione, alla
proliferazione cellulare e alla perdita di espressione dei recettori steroidei (Silvestrini R, 2000).
Aneusomie cromosoma specifiche sono spesso associate a fattori prognostici e alla progressione
della malattia (Persons DL et al., 1996); ad esempio l’amplificazione genica è un importante e
frequente meccanismo di iperespressione di determinati oncogeni giocando un ruolo fondamentale
nella crescita e sopravvivenza cellulare. Analisi di Comparative Genomic Hybridization (CGH) su
DCIS hanno evidenziato numerose amplificazioni (1q, 5p, 6q, 8q, 17q, 19q, 20p, 20q) e delezioni
(2q, 5q, 6q, 8p, 9p, 11q, 13q, 1q, 17p, 22q) simili a quelli identificati nei carcinomi invasivi a
conferma di come i carcinomi in situ siano la lesione precursore (Reis-Filho JS et al, 2003). Studi
sono stati focalizzati sulle alterazioni strutturali e/o aneuploidie del cromosoma 20, frequentemente
alterato in altri tumori (ovaio, prostata, vescica, colon, pancreas) e associato con i meccanismi di
iniziazione e progressione del tumore della mammella. La polisomia di questo cromosoma è stata
riscontrata in circa il 90% dei carcinomi duttali, mentre la monosomia e disomia solo

29
rispettivamente nel 4% e 6% dei casi (Nakopoulou L et al., 2002). Alti livelli di polisomia sono
inoltre correlati con una minore sopravvivenza rappresentando un buon fattore prognostico.
9.0 CELLULE STAMINALI
Nei tessuti normali il mantenimento della popolazione cellulare si deve ad una piccola frazione di
cellule, le cellule staminali, che si dividono in modo asimmetrico, autorinnovandosi e dando origine
a progenitori cellulari che, dopo un certo numero di divisioni cellulari, producono cellule
differenziate tessuto-specifiche, non più in grado di dividersi. I tumori sono delle entità eterogenee
per quanto riguarda il potenziale proliferativo, fenotipico e tumorigenico e recentemente è stato
dimostrato come all’interno della massa neoplastica, esista una frazione numericamente esigua di
cellule (1-2% della popolazione tumorale) in grado di rigenerare il tumore in vivo, mentre
popolazioni di cellule più differenziate mancano di questa proprietà. Queste cellule dette anche
Cancer Stem Cells/CSCs) sono simili alle cellule staminali per alcune proprietà come
l’autorinnovamento, lo stato indifferenziato e la multi potenzialità. In base a tali evidenze è
possibile ritenere che il tumore origini da una cellula staminale adulta o da una cellula che
riacquista le caratteristiche di staminalità. La dimostrazione della presenza di una frazione esigua di
cellule autorinnovantesi in grado di rigenerare il tumore, si è avuta nella leucemia mieloide acuta,
con l’osservazione che una sottopopolazione di cellule leucemiche a fenotipo CD34+/CD38- era
responsabile dell’insorgenza della leucemia nei topi (Bonnett D et al., 1997). Recentemente sono
state messe a punto condizioni sperimentali per l’isolamento e propagazione di cellule staminali
oltre che nei mielomi anche da neoplasie solide. Mediante tecniche per l’isolamento di cellule
staminali da tessuti normali, si è dimostrata l’esistenza di cellule che presentano alcuni caratteri di
staminalità nei tumori del sistema nervoso centrale, della prostata, del polmone, dell’ovaio, del
colon, del pancreas, negli epatocarcinomi nei melanomi maligni e nei carcinomi della mammella,
dove sono state descritte cellule staminali con fenotipo CD44+/CD24- (rappresentano lo 0,1-1%
dell’intera popolazione tumorale). La componente tumorigenica è stata distinta da quella non
tumorigenica sulla base dell’espressione di CD44 e CD24; inoculando in un topo 100 cellule a
fenotipo CD44+/CD24 low/Lineage si osserva la formazione del tumore mentre molte più cellule
con fenotipo diverso non sono in grado di formare neoplasia (Al-Hajj M et al., 2003; Nakshatri H et
al., 2009).Altro approccio per la caratterizzazione delle cellule staminali sfrutta alcune
caratteristiche funzionali delle cellule come la capacità di colorarsi con coloranti specifici. Questo
metodo è usato, per l’identificazione tramite citometria a flusso, della cosidetta “side population”
(SP) distinta sulla base di queste cellule di estrudere il colorante vitale Hoechst 33342 (Goodell MA
et al., 1996). Queste cellule rappresentano solo una piccola frazione di tutte le cellule e per la loro

30
capacità di estrudere farmaci, rappresentano la frazione chemioresistente all’interno del tumore
(Alvi AJ et al., 2003; Hirschmann-Jax C et al. 2004). Inoltre le SP sembrerebbero rappresentare una
fonte arricchita di cellule staminali (Challen GA et al. 2006). Le cellule staminali tumorali quindi
potrebbero derivare dalle stesse cellule staminali normali in seguito ad una mutazione, mentre altre
linee di ricerca sembrano sostenere come le cellule staminali tumorali abbiano origine da cellule
progenitrici mutate; questi progenitori (transit-amplyfing cells) possiedono una certa attività
replicativa, ma non sarebbero in grado di autorinnovarsi se non in seguito ad una mutazione. Tre
sono le situazioni che possono verificarsi e che riguardano le potenzialità delle cellule staminali
tumorali: 1) le cellule staminali tumorali, in seguito ad una mutazione di una cellula staminale
normale potrebbero dare origine ad altre cellule staminali tumorali e alla formazione del tumore; 2)
potrebbero rappresentare una piccola riserva di cellule resistenti alla terapia e responsabile di
ricadute; 3) potrebbero dare origine a metastasi in siti distanti rispetto al tumore primario (Fig 13).
Fig 13: trasformazione della cellula staminale normale in cellula staminale tumorale
Mentre le cellule staminali normali e tumorali condividono molte caratteristiche molecolari e
funzionali, la regolazione dell’autorinnovamento cellulare attraverso i pathways di Wnt, BMI-1,
Notch, Hedgehog, operativo e strettamente controllato nelle cellule staminali normali, è del tutto
alterata nelle cellule staminali tumorali e la conoscenza delle alterazioni molecolari che la
sostengono è cruciale per l’identificazione dei bersagli molecolari per interventi terapeutici
potenzialmente altamente selettivi (Liu S et al., 2010; Jain P et al., 2011) (Fig 14).
CELLULA STAMINALE
CELLULA STAMINALE
TUMORALE
PROGENITORE
CELLULARE
PROGENITORE
CELLULARE
TUMORALE
CELLULA
DIFFERENZIATA
CELLULA
TUMORALE

31
In particolare Wnt nel carcinoma della mammella sembra essere coinvolto in un cross-talk con i
recettori per gli ormoni steroidei attraverso pathways comuni; è stato anche dimostrato che
l’interferenza della sua funzionalità attraverso antagonisti specifici aumenta l’espressione di
marcatori di differenziamento. Un significato prognostico sembra avere la beta-catenina
(Nakapoulou L et al., 2006), la cui localizzazione intracellulare è differentemente associata con il
decorso della malattia (favorevole per l’espressione citoplasmatica e sfavorevole per quella
nucleare) e SFRP-1 (secreted frizzled-related protein-1) la cui metilazione del promotore è un
fattore sfavorevole (Veeck J et al., 2006).
Alterazioni della via Hedgehog sono implicate nello sviluppo di differenti patologie neoplastiche,
inclusa la patologia mammaria. In particolare Sonic Hedgehog identifica carcinomi della mammella
infiammatori a diversa aggressività biologica (Bièche I, et al, 2004) mentre la ciclopamina,
alcaloide che interferisce in modo specifico con la via di Hedgehog riduce l’espressione di Gli1 e
rallenta la crescita tumorale (Kubo M et al., 2004). Bmi1 è uno dei componenti del complesso
Polycomb indotto attraverso la vaia di segnalazione di hedgehog, responsabile
dell’autorinnovamento di cellule staminali normali e leucemiche, reprime geni che inducono
senescenza e morte cellulare e immortalizza cellule dell’epitelio umano. Un profilo molecolare
basato sull’espressione di 11 geni presenti nel pathway di BMI-1 sembrerebbe essere un potenziale
indicatore prognostico sia in termini di ripresa globale che di metastasi a distanza e di morte sia in
neoplasie epiteliali che non compreso il carcinoma della mammella (Glinsky GV et al., 2005;
Lawson JC et al., 2009). Altra interessante osservazione riguardo le cellule staminali di carcinoma
della mammella, è l’iperespressione di HER2 e la loro sensibilità a farmaci specifici come
trastuzumab e lapatinib (Magnifico A et al., 2009). È stato inoltre dimostrato una correlazione
diretta fra l’espressione di HER2 e la “side popultion”: il numero di cellule di questa popolazione
diminuisce in presenza di inibitori di HER2 (Nakanishi T et al., 2010).
Un terzo dei tumori della mammella si sono dimostrati non responsivi al trattamento con inibitori
specifici di HER2/neu. Evidenze sperimentali indicano come tale resistenza sia associata a
mutazioni del gene oncosoppressore PTEN, a mutazioni di PI3K o a forme tronche del dominio
extracellulare di HER2, con conseguente aberrante attivazione della via di segnale PI3K/AKT
(Nagata Y et al., 2004). Tale via di segnale avrebbe un ruolo importante nella regolazione delle
cellule tumorali staminali di mammella che, come dimostrato da studi in vivo, sarebbero potenziali
bersagli di inibitori selettivi di AKT (Korkaya H et al., 2007).

32
Fig 14: vie di trasduzione del segnale in cellule staminali del carcinoma della mammella
Recenti evidenze sulle cellule staminali della ghiandola mammaria depongono per una sua
negatività per i recettori estrogenici (ER) nei progenitori e nelle cellule più differenziate (Liu S et
al., 2010). Il momento dello stadio differenziativo del lineage cellulare al quale corrisponde la
trasformazione neoplastica è determinante nella storia clinica della malattia. Se la trasformazione
neoplastica insorge nella cellula staminale senza recettori per gli estrogeni, verosimilmente il
tumore sarà di tipo basale, indifferenziato, a prognosi sfavorevole e poco responsivo alle terapie
ormonali. Un tumore generato da cellule ben differenziate apparterrà al fenotipo luminale A,
caratterizzato da una migliore prognosi ed elevata probabilità di risposta al trattamento ormonale.
Al progenitore parzialmente differenziato corrisponderà un tumore con un comportamento clinico e
biologico eterogeneo (Fig 15).
Fig 15: relazione tra stadio di differenziazione e tipo di tumore

33
Esistono evidenze sperimentali che dimostrano l’efficacia di nuovi agenti terapeutici
sull’autorinnovamento di cellule staminali/progenitori normali (Liu S et al., 2006). Tuttavia esistono
difficoltà a livello dell’identificazione sia di marcatori di staminalità che di nuovi bersagli per
farmaci innovativi (Hill RP, 2006).
Gli attuali trattamenti, indirizzati contro le cellule proliferanti hanno come scopo la riduzione
dell’intera massa tumorale e non necessariamente l’uccisione della frazione staminale/tumorigenica,
caratterizzata dall’elevata espressione di proteine appartenenti alla famiglia dei trasportatori di
membrana ABC (favoriscono l’efflusso cellulare di farmaci e sono coinvolti nella resistenza ad
agenti terapeutici come paclitaxel, cisplatino, 5-fluoruracile, antracicline) (Dean M et al., 2005), da
alterazioni nei meccanismi di riparazione del DNA, dalla presenza di fattori citoprotettivi, e da una
relativamente modesta velocità di crescita a fronte di un elevato potenziale proliferativo. La Cancer
stem cell hypothesis, sostiene che all’interno del tumore, non tutte le cellule abbiano la stessa
capacità di auto rinnovarsi e proliferare ma solo una piccola frazione di cellule (Tumor initiating
cells o TICs) siano in grado di rinnovarsi in maniera illimitata, mentre il resto del tumore è
composto da cellule progenitrici con limitata capacità proliferativa o da cellule totalmente
differenziate. Sono diverse le strategie terapeutiche che potrebbero essere applicate nel tentativo di
colpire in modo specifico le TICs: 1) inibire le vie di trasduzione del segnale maggiormente
coinvolte nel mantenimento della popolazione cellulare in particolare le vie Wnt (You L et al.,
2004), Notch (Fan X et al., 2006) e PI3K/Akt/mTOR (Hambardzumyan D et al., 2008); 2)
sensibilizzare le cellule ad agenti chemioterapici usando inibitori delle chinasi che controllano i
punti di ingresso nel ciclo cellulare (Bao S et al., 2006); 3) indurre la differenziazione delle TICs
usando proteine come le BMPs (Bone Morphogenetic proteins) (Piccirillo SG et al., 2006) o
anticorpi monoclonali CD44 specifici (Marangoni E et al., 2009); 4) terapia anti-angiogenetica
privando le cellule staminali dell’ambiente ideale per la loro sopravvivenza (Calabrese C et al.,
2007).
Le cellule staminali cancerose offrono pertanto prospettive per il miglioramento della prognosi e del
trattamento del tumore. La loro caratterizzazione molecolare attraverso l’analisi del profilo genetico
e proteico è importante per la comprensione dei meccanismi biologici alla base della formazione dei
tumori solidi e nell’individuazione di nuovi obiettivi terapeutici e diagnostici.

34
10.0 DETERMINAZIONE DELLO STATO DI HER-2
Nonostante il significato prognostico, è soprattutto per il valore predittivo che HER-2 viene
attualmente studiato. In particolare ci sono diversi farmaci per i quali i dati clinici e sperimentali
indicano un possibile valore predittivo tra i quali tamoxifene, taxani, herceptin e trastuzumab. Nello
specifico l’amplificazione e l’aumentata espressione di HER2 forniscono dati prognostici negativi e
sembrano essere predittivi di una risposta a regimi chemioterapici intensivi.
La rivelazione dell’espressione di HER2 /neu nei tessuti è stata oggetto di un’ampia gamma di
metodologie di analisi. Recentemente la FDA ha approvato sia l’impiego della tecnica FISH, per la
determinazione dell’amplificazione genica sia l’impiego dell’immunoistochimica per la valutazione
dell’espressione proteica per una definizione pratica delle opzioni di trattamento in pazienti con
carcinoma della mammella HER2 positivo.
10.1 TECNICA FISH
Fra le nuove e ormai consolidate tecniche di biologia molecolare la FISH (Fluorescente In Situ
Hybridization) fa parte del gruppo di quelle che si basano sul principio dell’ibridazione degli acidi
nucleici e si è rivelata capace di potenti sinergie con il campo della diagnostica istopatologica
(Anguiano A, 2000). Questa metodica ha ampliato le applicazioni di citogenetica cellulare e
tissutale integrando i dati della citogenetica convenzionale, che ha come oggetto di studio la
morfologia dei cromosomi metafisici, con quelli forniti da sonde marcate, che hanno come
bersaglio il DNA del nucleo interfasico. I settori di maggiore interesse nella diagnostica
istopatologica sono quelli dell’oncoematologia e di alcune neoplasie solide.
Come primi esperimenti di ibridazione in situ sono state eseguite metodiche che sfruttavano
marcatori radioattivi (Gall JC et al., 1969). L’impiego della FISH per la marcatura dei geni, quindi
per la mappatura dei cromosomi risale ai primi anni ’90 (Ferguson-Smith, 1991).
L’ibridazione in situ in fluorescenza (FISH) è una tecnica che permette la localizzazione di una
specifica sequenza di DNA su preparati fissati di cromosomi in metafase, nuclei interfasici e sezioni
di tessuto ottenute da qualsiasi tipo di materiale biologico (sangue, biopsie, liquido amniotico,
gameti), sia esso fresco, conservato o paraffinato. Come primi esperimenti di ibridazione in situ
sono state eseguite metodiche che sfruttavano marcatori radioattivi (Gall JC et al., 1969).
L’impiego della FISH per la marcatura dei geni e quindi la mappatura dei cromosomi risale agli
anni ’90 (Ferguson-Smith, 1991). La tecnica prevede l’utilizzo di sonde di DNA in grado di
ibridare con l’intero cromosoma o con singole sequenze e costituisce un potente strumento in
aggiunta alle tecniche citogenetiche classiche. Generalmente la morfologia del cromosoma, delle
cellule o del tessuto viene conservata per permettere una precisa ed inequivocabile interpretazione

35
del target (Bentz et al., 1993). La FISH si basa sulla proprietà del DNA di fondersi in modo
reversibile e prevede il legame tra una sonda molecolare (frammento di DNA specifico per la
regione di interesse marcato con composti fluorescenti) e la sequenza di DNA complementare del
preparato che è stato fissato e montato su un vetrino portaoggetti. Il DNA bersaglio dopo fissazione
viene sottoposto a denaturazione al calore in presenza di formamide. In questo modo il DNA
bersaglio diventa disponibili per l’annealing con una sonda di DNA a singola elica e sequenza
complementare marcata con una sostanza fluorescente. Terminata l’ibridazione, la sonda di DNA
non legata o legata in modo aspecifico è rimossa per mezzo di lavaggi di stringenza e il DNA viene
in seguito colorato con un colorante di contrasto come il DAPI (4,6-diamidino-2-fenilindole).
L’ibridazione della sonda viene quindi analizzata con un microscopio a fluorescenza (Fig 16).
Fig 16: tecnica FISH La sonda o probe può essere rappresentato da un frammento di DNA o RNA che viene marcato; è
rappresentata da sequenze di DNA clonato, prodotti di PCR ed oligonucleotidi sintetici; la sua
lunghezza ideale varia dalle 50 alle 150 paia di basi.
I principali tipi di sonda utilizzate nella FISH sono (Fig 17):
CEP (Chromosome Enumeration Probe): sequenze di DNA ripetute in tandem (DNA satellite)
cromosoma specifiche. Sono sonde che individuano la regione centromerica del cromosoma e
consentono di valutare la ploidia (esempio CEP17);
TEL (Telomeric Probe): sonde telomeriche utili per individuare delezioni e riarrangiamenti
cromosomici (Fig 17B)
LSI (Locus Specific Identifier): sonde a DNA date da sequenze specifiche omologhe di regioni di
DNA o loci e direttamente marcate con fluorocromi;
WCP (Whole Chromosome Painting): sonde utili per l’identificazione di traslocazioni,
riarrangiamenti ed ibridi umani/animali;

36
Sequenze genomiche in YAC (Yeast Artificial Chromosome), BAC (Bacterial Artificial
Chromosome) e PAC (Plasmid Artificial Chromosome) per l’analisi di regioni specifiche.
Le sonde possono essere utilizzate sia singolarmente che in co-ibridazione.
Fig 17: tipi di sonde Le sonde possono essere caratterizzate da un frammento di DNA conosciuto marcato inserendo
direttamente nella catena un gruppo fluorescente (FITC, Phicoeritrina…) od un gruppo chimico che
sarà successivamente riconosciuto da un sistema di rivelazione antigene anticorpo fluorescente
(biotina, digossigenina…). Lunghezza e composizione delle sonde sono variabili a seconda del
bersaglio (Fig 18).
Fig 18: marcatura sonda
Le condizioni che possono destabilizzare la doppia elica provocando la separazione delle due catene
sono: le alte temperature, un pH alcalino estremo (maggiore di 13); la bassa forza ionica
(concentrazione di ioni sodio); presenza in soluzione di sostanze che rompono i ponti ad idrogeno
(urea, formamide). La denaturatone della doppia elica si accompagna a grosse variazioni delle
proprietà fisiche di soluzioni di DNA come la diminuzione della viscosità e l’aumento
MARCATURA TELOMERO
MARCATURA TELOMERO
MARCATURA CENTROMERO
GENE
MARCATURA DEL GENE
E AMPLIFICAZIONE DEL
SEGNALE
WHOLE CHROMOSOME
PAINTING
BERSAGLIO
SONDA MARCATA
CON BIOTINA
Ab LINK: FITC LABELED ANTI-
BIOTINA
AMPLIFICATION-Ab: FITC-LABELED
ANTI-MOUSE-IgG
SONDA DIRETTA SONDA INDIRETTA

37
dell’assorbanza a 260 nm. La denaturazione del DNA non è un processo irreversibile in quanto se
dopo la separazione delle due eliche la temperatura viene fatta scendere in modo graduale, le
singole eliche complementari si possono riappaiare con riformazione della doppia elica
(ibridazione). La discesa graduale della temperatura e la permanenza delle molecole per un certo
tempo pochi gradi sotto la temperatura di denaturazione, sono condizioni fondamentali affinché si
possa avere ibridazione, processo dipendente dai moti di agitazione termica. La temperatura di
melting (Tm) definita come temperatura alla quale metà del DNA si presenta sottoforma denaturata
caratterizza la stabilità dell’ibrido che di forma tra la sonda ed il suo filamento complementare; è
critica nel determinare la temperatura ottimale per usare sonde oliginucleotidiche. La Tm dipende
dalla lunghezza della sonda, dalla composizione in basi G+G e A+T, dal grado di omologia, dalla
concentrazione della sonda e del bersaglio e dall’ambiente chimico (i cationi monovalenti come il
sodio stabilizzano la doppia elica mentre i denaturanti chimici come l’urea e la formamide la
destabilizzano). Il buon esito di una reazione di ibridazione dipende anche dalla stringenza che è
definita come la specificità con cui una sonda si lega ad una determinata sequenza bersaglio. In
condizioni di alta stringenza la sonda può ibridare solo una sequenza esattamente complementare ad
essa, mentre in condizioni di bassa stringenza l’ibridazione può avvenire anche tra la sonda e
sequenze solo parzialmente complementari. Condizioni di ibridazione ad alta stringenza che
richiedono un’alta percentuale di omologia tra sonda e bersaglio prevedono alta temperatura, bassa
concentrazione salina e presenza di denaturanti chimici (formamide). Condizioni di bassa stringenza
che richiedono di una minore omologia fra sonda e bersaglio prevedono una bassa temperatura,
un’alta concentrazione salina e l’assenza di denaturanti chimici (Wang S et al., 2000).
La FISH permette la localizzazione di geni e il rilevamento morfologico diretto di difetti o anomalie
genetiche in patologie ereditarie, l’analisi pre- e post-natale sia su campioni ematologici che su
tumori solidi, la verifica di aneuploidie importanti in nuclei interfasici (trisomia 21, neoplasie), lo
studio della metafase tramite la colorazione di cromosomi (cariotipizzazione). Amplificazioni,
traslocazioni e delezioni sono le principali alterazioni che possono essere studiate mediante FISH.
La FISH è in grado di valutare il numero di copie del proto-oncogene c-erbB-2 (sonda LSI-
HER2/neu), in relazione al numero di copie del cromosoma 17 (sonda CEP17) per distinguere
l’amplificazione genica dall’aneuploidia cromosomica. Esiste amplificazione genica se il rapporto
fra il numero di segnali c-erbB2 (rosso)/CEP17 (verde) risulta essere maggiore di due (Fig 19).

38
Fig 19: sonda LSI HER-2/neu
L’algoritmo proposto nella valutazione dello status di HER-2 nel carcinoma della mammella
prevede una prima fase di screening con la tecnica immunoistochimica (IHC), metodo pratico e
affidabile, a basso costo e di facile esecuzione. I risultati dell’immunoistochimica vengono espressi
attraverso uno schema di classificazione standard basato su tre parametri: percentuale delle cellule
colorate, completezza o meno della colorazione di membrana ed intensità di colorazione. Il
punteggio ottenibile varia da 0 a 3+; punteggio 0 (nessuna colorazione), punteggio 1+ (debole
parziale colorazione di membrana in più del 10% delle cellule tumorali), punteggio 2+ (moderata
colorazione di tutta la membrana tra il 10 ed il 30% delle cellule tumorali), 3+ (intensa colorazione
di tutta la membrana in più del 30% delle cellule tumorali. I risultati 0 ed 1+ sono considerati
negativi, mentre 2+ e 3+ sono considerati positivi. I pazienti con risultato positivo è un buon
candidato alla terapia con trastuzumab in grado di bloccare i recettori ed inibendo la replicazione e
la crescita del tumore (Wolf AC et al., 2007; Sauter G et al., 2009) (Fig 20).
Fig 20: espressione immunoistochimica di HER2
La FISH ed immunoistochimica sono due tecniche complementari fondamentali nel trattamento del
carcinoma della mammella (Hicks DG etal., 2008). Recentemente è stata dimostrata la tendenza
0 1+ 2+ 3+

39
dell’immunoistochimica a dare risultati falsi positivi; la concordanza fra le due metodiche è
dell’88% e tale correlazione aumenta per i casi 3+. Spesso i casi negativi alla FISH risultano
erroneamente positivi all’immunoistochimica soprattutto quelli con score +2. quindi lo score +2 non
dovrebbe mai essere utilizzato come criterio per la terapia senza la conferma per mezzo della FISH
(Tubbs RR et al., 2001). La FISH viene considerata principalmente come metodica di valutazione di
secondo livello, in accordo con le linee guida internazionali e nazionali (Sauter G et al., 2009), da
usare dopo l’immunoistochimica per dirimere quei casi in cui la determinazione
immunoistochimica abbia fornito risultati equivoci (2+) o per contribuire a risolvere casi clinici
specifici (Fig 21). Tuttavia l’impiego di tecniche di ibridazione in situ è comunque possibile anche
per la valutazione di primo livello dello stato di HER2 nel carcinoma della mammella. Deve essere
privilegiata l’adozione di tecniche dual color che utilizzano due fluorocromi diversi per visualizzare
sullo stesso preparato istologico o citologico la regione centromerica del cromosoma 17 (sonda
CEP17) ed il numero di copie del gene HER2 (sonda locus specifica).
La valutazione deve essere fatta su almeno 20 cellule neoplastiche e in almeno due campi diversi
della componente invasiva per rendere ragione dell’eterogeneità tumorale. Deve comprendere il
rapporto tra i segnali del gene HER2 e i segnali centromerici; se questo è maggiore di 2,2 si ha
amplificazione; un rapporto compreso fra 1,8 e 2,2 è considerato equivoco o bordeline dalle linee
guida ASCO/CAP e in questo caso si deve ripetere la reazione o la lettura; se il rapporto
HER2/CEP17 è inferiore di 1,8 allora il caso sarà non amplificato. È necessario segnalare la
percentuale di cellule neoplastiche in cui il gene HER2 risulta amplificato, il valore di cut-off
applicato per lo score finale e la media dei segnali CEP17 per fornire indicazioni sulla polisomia.
La FISH pur rimanendo la metodica di riferimento per la predittività di risposta al trattamento con
trastuzumab, presenta alcune criticità: la necessità di attrezzature costose, specifiche e dedicate,
impossibilità di conservare a lungo i preparati per decadimento del segnale fluorescente e la
mancanza del dettaglio morfologico che non permette un confronto con l’immunoistochimica.

40
Fig 21: Iter diagnostico per la valutazione dello stato di amplificazione del gene
10.2 TECNICHE INNOVATIVE DI IBRIDAZIONE IN SITU: CISH e SISH
Recentemente sono stati sviluppati nuovi metodi di ibridazione in situ in campo chiaro (ISH cromo
genica o CISH) (Shah S et al., 2010). Studi preliminari hanno inoltre proposto un’ulteriore tecnica
di ISH, molto sensibile basata sulla metallografia enzimatica e conseguente deposizione di argento
metallico (SISH) usata per la tipizzazione di HER2 nel carcinoma della mammella. I risultati di uno
studio multicentrico in Italia (Carbone A et al., 2008), hanno dimostrato un’alta riproducibilità
interlaboratorio e fra i diversi osservatori per quanto riguarda la SISH applicato allo studio di HER2
nel carcinoma della mammella. Inoltre è stata dimostrata una forte concordanza fra FISH, CISH e
SISH. Discordanze fra FISH e SISH sono state risportate nei casi di polisomia del cromosoma 17,
casi in cui la valutazione dei preparati allestiti con la SISH indicava un’amplificazione di basso
Analisi IHC
HER2
Carcinoma mammella
(componente invasiva)
IHC
3+
(Overespressione di
HER2positivo)
IHC
2+
(borderline)
IHC
0/+1
(negativo)
Analisi FISH
HER2
FISH (+) per
amplificazione di HER2
FISH (–) per
amplificazione di HER2
FISH
(borderline)

41
livello/equivoco mentre i risultati corrispondenti in FISH non indicavano amplificazione (Carbone
A et al., 2008). È auspicabile che i metodi di ISH in campo chiaro siano implementati nei laboratori
di anatomia patologica. Queste metodiche offrono alcuni vantaggi come la stabilità del segnale, la
preservazione della morfologia cellulare e tissutale, la possibilità di archiviare in modo permanente
i preparati.
La CISH (Chromogenic In Situ Hibridization) è una metodica che permette l’osservazione in campo
chiaro di specifiche sequenze geniche mediante una reazione perossidasica. A differenza della
FISH, i preparati allestiti con la metodica CISH possono essere osservati con un microscopio ottico
tradizionale, la valutazione può avvalersi del correlato morfologico ed il segnale è permanente. Le
amplificazioni vengono evidenziate mediante sonde marcate con digossigenina che vengono
rivelate mediante applicazione sequenziale di un anticorpo mouse anti-digossigenina, di un
anticorpo rabbit anti-mouse, un polimero anti-rabbit ed evidenziate mediante precipitazione del
cromogeno DAB (Tanner et al., 2000) (Fig 22).
Fig 22: principio della CISH
Lo stato di HER2 viene valutato secondo il seguente score: alti livelli di amplificazione se sono
presenti più di 10 segnali per nucleo in più di 50 cellule neoplastiche; bassi livelli di espressione se
il numero di segnali varia da 6 a 10 per nucleo in più di 50 cellule neoplastiche; nessuna
amplificazione se il numero di segnali varia da 1 a 5 per nucleo. È stata riportata una concordanza
fra FISH e CISH variabile dall’85 al 100% (Arnould L et al., 2003; Gupta D et al., 2003; Isola A et
al., 2004; Gong Y et al., 2005; Penault-Llorca F et al., 2009) (Fig 23)

42
Fig 23: visualizzazione del segnale con tecnica CISH
Oltre alla CISH standard esiste anche la CISH dual-color (dc-CISH) in cui si usano due sonde: una
sonda HER2 marcata con digossigenina (segnale verde) ed una sonda centromerica marcata con
biotina (segnale rosso) (Laakso M et al., 2006) (Fig 20). Un valore ≥2 del rapporto HER2/CEP17 è
indicativo di amplificazione (Caballero-Garcia T et al., 2010). La doppia ISH in campo chiaro ha
mostrato una concordanza del 98,9% con la FISH tradizionale (Nitta H et al., 2008; Hoff K et al.,
2010; Mollerup J et al., 2012) (Fig 24).
Fig 24: CISH dual color
La SISH (Silver In Situ Hybridization) sfrutta il principio della metallografia enzimatica mediante
la quale un enzima, la perossidasi, catalizza la riduzione di ioni argento in argento metallico con
deposizione di particelle metalliche nella sede del target ibridizzato al probe (Tubbs R et al., 2004).
I metodi di marcatura con oro colloidale di anticorpi e altre proteine è stata introdotta a partire dagli
anni ’70 in metodi di immunoelettronmicroscopia. Successivamente è stato dimostrato come
microparticelle d’oro siano in grado di catalizzare la deposizione di argento da una miscela di sali di
argento e agenti riducenti (autometallografia o silver enhancement). Risultati migliori sono stati
ottenuti con particelle d’oro ancora più piccole (1,4 nm) o Nanogold che possono essere legate ad
anticorpi, proteine o peptidi. Infine sono state sviluppate tecniche di ISH che abbinano l’uso di
sonde marcate con Nanogold con l’autometallografia. La metallografia enzimatica è stata descritta
da Hainfeld nel 2002; la perossidasi catalizza la riduzione di ioni argento ad argento metallico con
NO AMPLIFICATO AMPLIFICATO

43
le nanoparticelle che si depositano nella sede in cui il target si è ibridizzato con il probe. La SISH,
messa a punto per lo studio dello stato del gene HER2 e per la verifica della ploidia del cromosoma
17, opera in automatismo in un sistema chiuso; il precipitato d’argento è depositato sui nuclei ed
una singola copia del gene HER-2 o del cromosoma 17 è visualizzata come punti grigio-neri che
risaltano nettamente sull’ematossilina usata come controcolorante (Tubbs R et al., 2002; Carbone A
et al., 2008) (Fig 25).
Fig 25: principio tecnica SISH
Affinché il preparato possa essere considerato adeguato i segnali devono essere netti e facilmente
visibili anche con un obiettivo di 20x. Si deve controllare che le cellule normali (linfociti, cellule
endoteliali) abbiano da uno a due segnali. L’osservazione dei segnali sulle cellule normali è
fondamentale, in quanto la dimensione dei segnali è il riferimento per il conteggio dei segnali sulle
cellule neoplastiche. Un punto discreto viene considerato come copia singola di HER-2 o del
cromosoma 17 (Fig 26).
Fig 26: copia singola di HER2
Un grande cluster è costituito da segnali multipli aggregati non contabili; il grande cluster viene
conteggiato, per definizione, come costituito da 12 copie, mentre un piccolo cluster viene
conteggiato come costituito da 6 copie (Fig 27)

44
Fig 27: Cluster di segnali
Il segnale non amplificato è rappresentato da uno, due dot per cellula. Con l’amplificazione è
possibile osservare due tipi di pattern: due grandi cluster indicano la presenza di copie ripetute di
HER2 su un singolo cromosoma (regioni cromosomiche espanse); un pattern più distribuito
permette il conteggio dei singoli granuli ed è indicativo di un’amplificazione tipo double minutes
chromosomes (strutture cromosomiche circolari privi di centromero) (Tab 3).
Tab 3: algoritmo SISH
Numerosi studi hanno dimostrato una forte concordanza fra i risultati ottenuti mediante FISH e
SISH nella determinazione dello stato di HER2 nel carcinoma della mammella (Papouchado BG et
al., 2010).
11.0 POLISOMIA DEL CROMOSOMA 17
Sulla base delle linee guida FDA e più recentemente delle raccomandazioni ASCO/CAP,
l’amplificazione del gene HER2, quando si utilizzino tecniche dual color, viene indicata come
rapporto >2 o >2,2 tra numero di copie del gene HER2 ed i segnali del centromero 17. Tumori con
aumento del numero di segnali sia per il gene HER2 sia per CEP17, ma con un rapporto <2 (1,8 per
ASCO/CAP) sono considerati polisomici (Hyun CL et al., 2008; Vanden Bempt I et al., 2008). La
polisomia, definita come presenza di 3 o più copie del centromero 17, in assenza di amplificazione
genica può essere associata ad un’intensità di colorazione all’immunoistochimica 2+ e più
raramente 3+; solo i casi polisomici con iperespressione della proteina (3+) sono eleggibili al
GRANDI CLUSTER PICCOLI CLUSTER

45
trattamento terapeutico. Quindi diventa importante verificare la reale incidenza della polisomia 17
in assenza di amplificazione genica e il meccanismo biologico responsabile dell’aumento dei
segnali centromerici (Fig 28). L’incidenza dei casi polisomici è ancora oggetto di discussione; i dati
in letteratura oscillano tra il 4 ed il 30% in funzione dei criteri usati per la diagnosi. Un recente
studio, attraverso l’utilizzo della tecnica CGH (microarray based comparative genomic
hybridization) ha suggerito come la polisomia possa essere considerata un evento molto raro; infatti
solo il 5% delle polisomie individuate con la FISH sono state confermate dalla CGH; ciò che
inizialmente era stato definito polisomico con la FISH si è dimostrato un aumento (gain) o
amplificazione della sola zona del centromero e non un aumento del numero di cromosomi 17; il
20% delle pazienti, considerate polisomiche alla FISH e con un rapporto HER2/CEP17 <2,
mostravano con la tecnica CGH amplificazione del gene HER2 e quindi erano eleggibili alla terapia
con trastuzumab. I casi di carcinoma della mammella con vera polisomia del cromosoma 17
sarebbero rari e dovrebbero essere considerati amplificati tutti quei casi con più di 6 segnali di
HER2 per cellula indipendentemente dal numero dei segnali CEP17 (Marchiò C et al., 2009; Yeh IT
et al., 2009; Gunn S et al., 2010; Oakman C et al, 2010).
Fig 28: polisomia del cromosoma 17
12.0 ETEROGENEITA TUMORALE
Alcuni tumori mammari appaiono eterogenei sia per quanto riguarda la morfologia che le
caratteristiche genetiche e si possono avere discordanze tra le metodiche di FISH ed
immunoistochimica. L’eterogeneità genetica, rilevata all’immunoistochimica, viene definita focale
quando localizzata in cloni definiti di cellule HER2 positive, o diffusa quando ci sono aree 3+
frammiste ad aree 2+ o negative. Secondo le raccomandazioni ASCO/CAP, viene definito
eterogeneo, un tumore che, mediante tecnica FISH, presenta amplificazione genica in più del 5%
ma in meno del 50% delle cellule neoplastiche infiltranti, con un rapporto HER2/CEP17 ≥ 2. In

46
base a questa definizione, l’incidenza di eterogeneità intratumorale varia dal 5 al 30% (Fig 29). Tale
eterogeneità potrebbe essere responsabile della diversità nell’amplificazione del gene o
iperespressione di HER2 tra carcinoma primario e metastasi, evidente in circa il 14% dei casi
(Tapia C et al. 2007). L’impatto dell’eterogeneità genetica sulla storia naturale della neoplasia e
sulle scelte terapeutiche è ancora oggetto di studio. Recentemente è emerso il beneficio della terapia
con trastuzumab in casi eterogenei con amplificazione focale. Inoltre prima di escludere pazienti
con tumori eterogenei dal possibile beneficio del trattamento, è raccomandabile una rivalutazione
dello stato di HER2 con ulteriori analisi immunoistochimiche e/o tecniche di ibridazione in situ da
eseguirsi su ulteriori inclusioni della neoplasia primitiva o sulle eventuali metastasi linfonodali
(Lewis JT et al., 2005; Hanna W et al., 2007; Chivukula M et al., 2008; Brunelli M et al., 2009
Vance GH et al., 2009; Oakman C et al., 2010, Lee S et al. 2011).
Fig 29: esempio di eterogeneità tumorale
IHC FISH

47
13.0 TRATTAMENTO DEL CARCINOMA DELLA MAMMELLA
I tipi di trattamento per il carcinoma della mammella possono essere sistemici o locali. Le terapie
locali mirano ad asportare, distruggere o controllare le cellule neoplastiche di una determinata zona.
Le terapie sistemiche distruggono e controllano le cellule tumorali diffuse in tutto l’organismo.
L’intervento chirurgico rappresenta il trattamento più diffuso nel caso di tumore della mammella.
Altro tipo di trattamento è rappresentato dalla terapia radiante che consiste nell’uso di radiazioni ad
alta energia per distruggere le cellule neoplastiche ed impedirne la crescita. La chemioterapia
consiste nella somministrazione di una combinazione di farmaci anti-tumorali in cicli. L’endocrino
terapia è mirata a bloccare l’azione degli ormoni sulle cellule neoplastiche. Può prevedere l’uso di
farmaci che modificano il funzionamento ormonale oppure l’asportazione chirurgica delle ovaie,
produttrici degli ormoni femminili.
Altra terapia è la chemioterapia adiuvante che ha lo scopo di eliminare o ridurre la massa tumorale;
si tratta di un trattamento che precede l’intervento chirurgico in pazienti affetti da carcinoma al fine
di ridurre lo stadio T della neoplasia in modo da consentirne l’asportazione chirurgica. La terapia
pre-chirurgica è in genere meglio tollerata dal paziente rispetto alla sua somministrazione dopo
chirurgia e riduce la probabilità di diffusione delle cellule neoplastiche in corso di intervento
chirurgico.
13.1 TARGET THERAPY E BERSAGLI MOLECOLARI
Negli ultimi anni sono stati condotti progressi importanti nella scoperta di lesioni genetiche alla
base delle neoplasie. Queste scoperte hanno aperto nuovi scenari per lo sviluppo di tageted therapy,
terapie molecolari indirizzate selettivamente contro le proteine responsabili della formazione di
tumori. Le cellule tumorali dipendono in maniera assoluta da specifiche vie di trasduzione del
segnale e l’inibizione di queste può portare ad un arresto irreversibile della crescita o a morte delle
cellule neoplastiche stesse. L’uso di targeted therapy richiede di una corretta definizione del target.
Un bersaglio terapeutico ideale deve:
essere esclusivo del tumore;
essere assente o irrilevante nel tessuto sano;
rivestire un ruolo patogenetico importante nella progressione del tumore;
poter essere bersagliabile farmacologicamente;
poter essere misurabile clinicamente;
Le proteine codificate da oncogeni e geni oncosoppressori rappresentano dei bersagli elettivi per la
messa a punto di terapie molecolari; colpire bersagli molecolari specifici rappresenta un approccio
potenzialmente più efficace e meno tossico rispetto alle terapie tradizionali.

48
Bersagli molto promettenti sono le proteine RAS; esse rappresentano le oncoproteine più spesso
coinvolte nei tumori dell’uomo. Per poter funzionare le proteine RAS devono essere coniugate a
catene lipidiche di enzimi (farnesiltransferasi o geraniltransferasi). Inibitori di questi enzimi
possono avere un’efficacia terapeutica grazie al blocco di RAS o di proteine della stessa famiglia
(Baselga J et al., 2005; Carlomagno F et al., 2005)
Altri bersagli terapeutici promettenti sono le oncoproteine con funzione anti-apoptotica. La loro
inibizione potrebbe favorire la morte delle cellule tumorali. Sono in sperimentazione molecole in
grado di inibire le proteine IAP (Inhibitor of Apoptosis) o Bcl2, altra proteina con funzione
antiapoptotica. Esistono strategie terapeutiche che hanno come obiettivo l’attivazione di
oncosoppressori. La proteina p53 ha potenti effetti antitumorali, in gran parte mediati dalla capacità
di indurre apoptosi o arresto del ciclo cellulare. Recentemente sono stati identificati composti che
hanno la capacità di bloccare la degradazione di p53, aumentandone i livelli intracellulari (Baselga
J et al., 2005; Carlomagno F et al., 2005). Altra classe di composti antitumorali sono gli inibitori
delle istonedeacetilasi (HDAC), enzimi che favoriscono l’acetilazione degli istoni ed il loro distacco
dalla cromatina. Questi composti sono in grado di indurre la trascrizione genica ed il loro effetto
terapeutico è legato alla capacità di riattivare l’espressione di proteine con funzione di soppressione
tumorale (Minucci S et al., 2006).
13.2 ANTICORPI MONOCLONALI UMANIZZATI
La terapia con anticorpi monoclonali (mAb) è basata sulla scoperta che mAb specifici per un
antigene possono essere ottenuti fondendo una cellula B che produce anticorpi con una cellula
immortalizzata di mieloma. Provocando la fusione cellulare di linfociti B provenienti da un topo
con una linea cellulare proveniente da un mieloma, si ottiene un ibridoma; facendo crescere i singoli
ibridomi si ottengono cloni cellulari ognuno dei quali sintetizza uno specifico anticorpo
monoclonale con struttura chimica definita e con la capacità di riconoscere un singolo epitopo
(Köhler G et al., 1975). Nel 1975 sono stati prodotti i primi anticorpi murini mediante la tecnica
degli ibridomi; tuttavia questi anticorpi sono immunogeni per l’uomo inducendo risposte anticorpali
anti-immunoglobulina di topo. Grazie allo sviluppo di tecniche di ingegneria genetica è stato
possibile produrre anticorpi più simili a quelli umani e quindi meno immunogeni (Orlandi R et al.,
1989). Gli anticorpi chimerici sono stati prodotti da geni ibridi derivanti dalla fusione di geni
codificanti per le regioni variabili (VH e VL) delle Ig di un mAb di topo con corrispondenti regioni
H e L di anticorpi umani. Questi geni ibridi devono essere inseriti mediante trasfezione genica in
una cellula (trasfettoma) che diventerà in questo modo produttrice di anticorpi chimerici. Questi
anticorpi pur mantenendo la specificità di riconoscimento dell’anticorpo originale murino, sono

49
meno immunogenici rispetto alle Ig murine ed inducono meno anticorpi umani anti-topo (human
anti-mouse antibody, HAMA) (Brüggemann M et al., 1989). Dal momento che la regione variabile
che riconosce l’antigene è ancora murina, possono essere ancora prodotti anticorpi neutralizzanti.
Gli anticorpi primatizzati, hanno la regione variabile codificata da geni di primati; la loro
immunogenicità è ridotta ma tuttavia sono ancora possibili reazioni da parte dell’ospite. Negli
anticorpi umanizzati, tutte le porzioni che sono coinvolte nel legame con l’antigene, escluse le
regioni ipervariabili, sono ottenute da sequenze geniche umane. Questi anticorpi sono ottenuti
mediante la tecnica del CDR-grafting, attraverso l’isolamento di cDNA delle catene L e H da
ibridomi prodotti in topi transgenici in cui vengono inseriti geni umani. Con la PCR vengono
amplificate le regioni variabili VH e VL degli anticorpi murini (CDR). Attraverso cicli di
mutagenesi sono sostituiti i frammenti CDR. I geni ricombinanti vengono clonati in vettori di
espressione e si introducono in cellule ospiti idonee (E coli o mammifero) per la produzione di
anticorpi. Meno del 10% dell’anticorpo monoclonale umanizzato ha sequenze geniche murine.
Gli anticorpi monoclonali possono essere utilizzati a scopo terapeutico come mAb modificati o
mAb coniugati. Gli anticorpi non coniugati di classe IgG1 possono agire direttamente a livello
cellulare provocando apoptosi, citotossicità complemento-mediata (CDC) o citotossicità cellulare
anticorpo-mediata (ADCC). Gli anticorpi possono anche agire tramite blocco dei recettori per
fattori di crescita con inibizione della crescita ed alterazioni del ciclo cellulare.
Gli anticorpi coniugati non sfruttano l’attività immunologica intrinseca, ma servono a dirigere in
modo specifico il composto tossico a cui sono coniugati, come agenti chemioterapici, tossine o
radioisotopi (Slamon DJ et al., 2001).
13.3 TRASTUZUMAB E MECCANISMO DI AZIONE
Herceptin (trastuzumab) è un anticorpo monoclonale umanizzato ricombinante (mAb) diretto contro
il dominio extracellulare della proteina HER2; è costituito da una regione complementare
determinante di un anticorpo murino (clone 4B5) integrata nella struttura di una IgG1 umana
(Carter P et al., 1992).
Il razionale per iniziare lo studio di trastuzumab nel trattamento del carcinoma mammario operabile
si è basato su diversi fattori:
• la malattia HER2 positiva è associata a prognosi sfavorevole; è una malattia aggressiva con
alto rischio di recidive e metastasi
• la positività di HER2 è un evento precoce nello sviluppo del carcinoma della mammella
• trastuzumab è diretto in modo specifico contro il carcinoma mammario positivo per HER2 e
nella malattia metastatica è associato ad un beneficio clinico significativo

50
• trastuzumab ha dimostrato di indurre scarsi effetti collaterali e di essere dotato di un profilo
di tossicità favorevole.
Sebbene il meccanismo attraverso il quale herceptin induca una regressione dei tumori con
iperespressione di HER-2 non sia completamente conosciuto, diversi effetti molecolari e cellulari
sono stati osservati in sperimentazioni con modelli in vivo e vitro (Valabrega G et al., 2007;
Daniele L et al., 2009) (Tab 4).
HER2 attiva molteplici vie di segnale intracellulari comprese quelle che coinvolgono fosfatidil-
inositolo 3,4,5 trifosfato chinasi (PI3K), e MAP chinasi (MAPK). Herceptin riduce la trasmissione
del segnale attraverso queste vie, promuovendo l’arresto del ciclo cellulare e l’apoptosi. La
riduzione del segnale attivato dal recettore può condurre ad un'internalizzazione dello stesso
mediata da herceptin e ad una sua successiva degradazione (Sliwkowsky MX et al., 1999; Baselga J
et al., 2001; Gabriel N et al., 2005). Non è tuttavia chiaro se herceptin determini una riduzione
dell’espressione di HER2 sulla superficie cellulare in quanto è stato dimostrato che i livelli del
recettore non si modificano in risposta al trattamento con l’anticorpo monoclonale (Le XF et al.,
2003). Un meccanismo attraverso il quale il farmaco può bloccare la via di segnale di PI3K è stato
recentemente descritto da Nagata et al. (Nagata Y et al., 2004); PTEN (MMAC1/TEP), è una
fosfatasi che de fosforila in posizione D3 di membrana il PI3 sito di ancoraggio del dominio
plecstrina-omologo di AKT alla membrana cellulare (Parson R et al., 2003). Dal momento che
PI3K catalizza la produzione di PI3, PTEN antagonizza l’azione di PI3K e regola negativamente
l’attività di AKT. Herceptin attiva la fosfatasi PTEN con rapida defosforilazione di AKT e
conseguente inibizione del ciclo cellulare (Nagata Y et al., 2004). Le cellule trattate con herceptin,
vanno quindi incontro all’arresto del ciclo cellulare in G1 con riduzione della proliferazione.
L’arresto del ciclo è accompagnato dalla riduzione dell’espressione di proteine coinvolte nel
sequestro dell’inibitore delle chinasi ciclino-dipendenti (cdk), p27kip1, inclusa la ciclina D1; questo
comporta il rilascio di p27kip1 permettendo a questa di legare ed inibire il complesso ciclina E/cdk2
(Lane HA et al., 2000). Il trattamento determina inoltre un accumulo intracellulare di p27kip1 per
ridotto sequestro da parte del proteo soma (Lane HA et al., 2001). Come singolo agente, herceptin
può ridurre drasticamente le dimensioni del tumore in pazienti con carcinoma della mammella
metastatico che iperesprime HER2. Rimane da chiarire se questo sia dovuto ad un effetto
citotossico diretto sulle cellule tumorali o ad un meccanismo indiretto (attività anti-angiogenetica o
stimolazione della risposta immunitaria). Studi in vivo hanno dimostrato come herceptin possa
indurre apoptosi in cellule di carcinoma della mammella (Chang JC et al., 2003).
L’iperespressione di HER2 è associata ad un incremento nel processo di angiogenesi e
dell’espressione di VEGF (Laughner E et al., 2001); il trattamento di tumori mammari HER2+

51
comporta la riduzione delle dimensioni del tumore, della densità dei piccoli vasi in vivo (Izumi Y et
al., 2002) e la migrazione di cellule neoplastiche in vitro (Klos KS et al., 2003). È stata inoltre
dimostrata una riduzione nella sintesi di fattori pro-angiogenici e un aumento nella produzione di
fattori anti-angiogenici (Izumi Y et al., 2002; Klos KS et al., 2003). La combinazione di herceptin e
paclitaxel avrebbe la capacità di inibire gli effetti correlati all’angiogenesi in modo più efficace
rispetto al solo anticorpo monoclonale (Klos KS et al., 2003; Gutierrez C et al., 2011) (Fig 30).
Tab 4: meccanismi di azione di trastuzumab
Fig 30: vie di segnale di HER2 e target therapy
Modelli in vivo e studi clinici hanno dimostrato per trastuzumab un effetto citotossico in parte
correlato alla stimolazione della risposta immunitaria. Herceptin ha la capacità di attivare la
citotossicità cellulo-mediata anticorpo dipendente (ADCC) in diverse linee cellulari di carcinoma
della mammella (Cooley S et al., 1999; Slamon DJ et al., 2001). Le cellule NK, coinvolte
CITOTOSSICITA CELLULARE ANTICORPO MEDIATA (ADCC)
INIBIZIONE DEL TAGLIO PROTEOLITICO DI HER2
INTERNALIZZAZIONE E DEGRADAZIONE DI HER-2
ATTENUAZIONE DEI SEGNALI CELLULARI; ARRESTO IN FASE G1
INIBIZIONE DELLA VIA DI SEGNALE PI3K-AKT
INIBIZIONE DELL’ANGIOGENESI TUMORALE
INIBIZIONE DELLA RIAPARAZIONE DEL DNA
INDUZIONE DI p27Kip1 E DELL’INTERAZIONE p27Kip1- CDK2
Dominio di legame
per il ligando
Dominio tirosin
chinasico
Trascrizione
• Proliferazione
• Motilità
• Invasione
• Resistenza all’apoptosi

52
nell’ADCC esprimono il recettore Fcγ attraverso il quale si legano alla componente Fc del farmaco
con conseguente lisi cellulare.
Studi in vitro hanno dimostrato una sinergia fra herceptin e diversi farmaci chemioterapici (Pegram
M.D et al., 2004). Esisterebbe una correlazione fra il danno causato al DNA dai farmaci
chemioterapici e il blocco della riparazione da parte di trastuzumab; inoltre l’anticorpo monoclonale
sarebbe in grado di promuovere la rottura del legamento di DNA nelle cellule che iperesprimono
HER2 (Mayfield S et al., 2001) e di modulare la trascrizione di geni i cui prodotti sono coinvolti
nella riparazione del DNA (Kauraniemi P et al., 2004). Attualmente herceptin viene somministrato
in associazione a chemioterapici come paclitaxel e docetaxel che migliorano la risposta alla terapia,
il tempo alla progressione e la sopravvivenza globale rispetto alla ionoterapia con solo trastuzumab
(Esteva FJ et al., 2002; Baselga J et al., 2004; Gabriel N et al., 2005; Romond EH et al., 2005;
Jackisch C, 2006).
Esisterebbe un’interazione tra i segnali di trasduzione di HER2 e quelli di ER. HER2 è in grado di
attivare la via Ras/MAPK che a sua volta determina la fosforilazione e attivazione del recettore
degli estrogeni. Nei tumori con iperespressione di HER2, la fosforilazione e attivazione della
proteina AIB1 determinano un potenziamento dell’attività trascrizionale di ER ed un aumento
dell’attività agonista del tamoxifene. Attraverso MAPK vengono fosforilati sia il recettore che i
coattivatori, promuovendo la proliferazione, mentre da parte del recettore viene favorita la
produzione di fattori di crescita che potenziano ulteriormente la via MAPK. Queste vie possono
essere inibite somministrando farmaci come il trastuzumab, in grado di bloccare la via Ras e di
evitare la fosforilazione del coattivatore a livello di ER. Gli inibitori dell’aromatasi, riducendo la
sintesi degli estrogeni, riducono l’attivazione di ER; in questo modo ER non è più un target per la
cascata delle MAPK indotta da HER2 e viene bloccato il meccanismo di resistenza ipotizzato per il
tamoxifene (Baselga J et al., 1998; Arpino G et al., 2008; Giuliano M et al., 2011). Il promotore di
HER2 contiene inoltre un elemento di risposta all’estrogeno che può inibire la trascrizione del gene;
questo è stato dimostrato in linee cellulari di carcinoma della mammella MCF-7 dove l’estrogeno
induce una diminuzione di HER2, mentre la presenza dell’antiestrogeno determina una parziale
inversione dell’effetto (Dati C et al., 1990). Un mediatore del segnale di HER2 stimola la
fosforilazione di ER portando ad un aumento della trascrizione e proliferazione; in questa situazione
l’effetto agonista del tamoxifene supera quello antagonista convertendolo da soppressore a
stimolatore delle cellule di tumore mammario. Questo fenomeno spiegherebbe in parte la relativa
resistenza che tumori ER e HER2 positivi mostrano al trattamento con tamoxifene (Dati C et al.,
1990) (Fig 31). Una strategia promettente nel superare o prevenire la resistenza alla terapia

53
endocrina potrebbe essere la combinazione di agenti ormonali e farmaci diretti contro effettori a
valle dei recettori tirosin-chinasici quali m-TOR (Giuliano M et al., 2011; Baselga J et al., 2012).
Fig 31: interazione di HER2 con ER
13.4 TRASTUZUMAB: MECCANSIMI DI RESISTENZA
Sebbene il trastuzumab venga considerato il trattamento standard di prima linea in pazienti con
diagnosi di carcinoma della mammella HER-2 positivo, un numero significativo di pazienti (circa il
40%) con diagnosi di tumore mammario HER-2 positivo non beneficia di questa terapia in quanto
ha sviluppato una resistenza primaria o secondaria al farmaco (Scaltriti M et al., 2007) (Fig 32).
I meccanismi di resistenza al trastuzumab maggiormente studiati sono (Nahta R et al., 2006;
Pohlmann PR et al., 2009; Fiszman GGL et al., 2011):
• accumulo della forma tronca del recettore (p95HER2);
• perdita di espressione della proteina PTEN;
• ostacolo al legame dell’anticorpo monoclonale al recettore;
• la sovra regolazione delle vie di segnale a valle di HER2;
• vie di segnale alternative (IGF-1R);
• incapacità di determinare la citotossicità cellulare anticorpo mediata.
CRESCITA
CRESCITA

54
Fig 32: meccanismi di resistenza a trastuzumab
Mutazioni somatiche nella regione del gene egfr codificante per il dominio TK, correlano con la
risposta all’inibitore tirosin-chinasico gefitinib in pazienti affetti da neoplasia polmonare (Paez JG
et al., 2004). In modo similare, mutazioni nella regione codificante per il dominio extracellulare
(ECD) possono essere presenti nel gene HER2 impedendo il legame di trastuzumab al recettore.
Inoltre l’interazione fra anticorpo e recettore può venir meno se i livelli di HER2 si riducono nel
tempo. Studi immunoistochimici hanno tuttavia dimostrato come l’iperespressione del recettore sia
mantenuta in cellule neoplastiche mammarie ottenute da pazienti recidivati dopo completa risposta
al trattamento con trastuzumab (Gennari R et al., 2004). Recenti studi hanno dimostrato come
l’amplificazione del segnale mediato da PI3K contribuisce ad indurre resistenza alla terapia. PTEN
(Phosphatase and TENsin homologue deleted on chromosome 10) agisce come antagonista della
funzione di PI3K, inibisce l’attività di AKT e la crescita tumorale (Lu Y et al., 1999). La riduzione
dell’espressione di PTEN blocca l’inibizione della proliferazione cellulare mediata da herceptin in
cellule di carcinoma della mammella (Nagata Y et al., 2004; Pandolfi PP, 2004) che iperesprimono
HER2, con aumento del segnale attivato da PI3K. Recenti studi, condotti su una serie di carcinomi
della mammella HER-2 positivi, hanno dimostrato come il trastuzumab possa bloccare la via PI3K
incrementando l’attività fosfatasica di PTEN e la sua localizzazione a livello di membrana
(Valabrega G et al., 2007; Nagata Y et al., 2004) e come la perdita di PTEN sia responsabile della
resistenza al trattamento (Pandolfi PP, 2004). La perdita di funzione di PTEN può essere
determinata nel 5-10% dei tumori della mammella da mutazioni. L’aploinsuffucuenza di PTEN, per
perdita di eterozigosi a livello del locus genico è stata osservata nel 50% dei tumori della mammella
(Li J et al., 1997); inoltre è stata descritta una mutazione epigenetica di PTEN (Mutter GL et al.,
2000). In pazienti resistenti al trastuzumab, per una ridotta espressione di PTEN, potrebbe essere
vantaggioso l’impiego di farmaci in grado di inibire PI3K.

55
Mutazioni attive a livello del gene codificante la sub unità catalitica della chinasi PI3K sono state
identificate in circa io 25% dei carcinomi primitivi della mammella, principalmente a livello
dell’esone 9 e 20, determinando un aumento costitutivo della trasmissione intracellulare del segnale
mediato dalla stessa chinasi (Isakoff SJ et al., 2005). È stato pertanto valutato il ruolo della perdita
di espressione di PTEN e della presenza di mutazioni attivanti di PI3K in pazienti affette da
carcinoma della mammella HER2 positivo e trattate con trastuzumb in monoterapia o in
associazione con chemioterapici. Né la perdita di espressione di PTEN, né mutazioni di PI3K
considerate singolarmente risultavano essere correlate con una peggiore prognosi. Valutando
simultaneamente il ruolo prognostico della perdita di espressione di PTEN e delle mutazioni
attivanti di PI3K, è stata osservata una differenza significativa in termini di progressione tra la
popolazione definita con “pathway PI3K attivo” (perdita di espressione di PTEN/PI3K mutato) e la
popolazione definita con “pathway PI3K non attivo” (alta espressione di PTEN/PI3K non mutato).
Recentemente uno studio pubblicato da Esteva e coll (Esteva FJ et al., 2010) su pazienti affette da
carcinoma della mammella metastatico HER2 positivo trattate con trastuzumab da solo o associato
a regimi chemioterapici, ha valutato la correlazione fra la perdita di espressione di PTEN, lo stato
mutazionale di PI3K e l’iperespressione di pAKT e p-p70S6K, con la risposta al trattamento e
l’autcome clinico. I risultati hanno evidenziato come non esista nessuna correlazione tra lo stato dei
4 determinati e la risposta al trattamento o l’outcome clinico quando ciascun determinate è stato
valutato singolarmente. La valutazione dello stato di attivazione del pathway PI3K come perdita di
espressione di PTEN/presenza di mutazioni a livello di PI3K, veniva osservata una correlazione
statisticamente significativa sia per la risposta al trattamento che per la sopravvivenza globale.
Herceptin aumenta l’emività di p27kip1 riducendone la fosforilazione mediata dal complesso ciclina
E/cdk2 e bloccando di conseguenza la sua degradazione ubiquitino-dipendente (Le XF et al., 2003).
Il farmaco inoltre aumenta l’associazione di p27kip1 e cdk2 con arresto del ciclo cellulare in G1
(Lane HA et al., 2000). Linee cellulari SKBR3 resistenti ad herceptin, dopo esposizione continua al
farmaco esprimono livelli ridotti di p27kip1 con elevata attività di cdk2; la trasfezione di p27kip1
comporta di nuovo la sensibilità cellulare al trastuzumab confermando come la proteina sia un
mediatore critico nella risposta ad herceptin (Natha R et al., 2003), potendo servire come marcatore
di risposta al trastuzumab o come target terapeutico nei sottogruppi di pazienti affetti da tumore
della mammella progrediti durante la terapia con l’anticorpo monoclonale.
La via di trasduzione del segnale PI3K/AKT/mTOR ha una funzione critica nella proliferazione
cellulare, nella progressione del ciclo cellulare, nell’apoptosi e nel metabolismo cellulare. AKT è la
serin treonin chinasi che viene direttamente attivata in risposta a PI3K e rappresenta il suo più
importante effettore (Engelman JA et al., 2006; Gonzales-Angulo AM et al., 2011). Nonostante

56
AKT sia l’effettore di PI3K maggiormente coinvolto nell’insorgenza e sviluppo di neoplasie, vi
sono altre vie indipendenti da AKT, ma che vengono attivate da PI3K, implicate nella polarità e
nella migrazione cellulare (Cain RJ et al., 2009). Il principale effettore a valle di AKT è il
complesso mTORC1: esso è capace di integrare segnali provenienti dall’esterno ed interno della
cellula come ad esempio segnali legati allo stato energetico, alla presenza di nutrienti e di fattori di
crescita. Allo stato attuale vi è uno sviluppo clinico di molti farmaci che hanno come bersaglio
l’asse di sopravvivenza PI3K/AKT/mTOR; in particolare sono state sviluppate molecole che vanno
ad inibire PI3K, AKT ed inibitori di mTOR sia allosterici che del sito catalitico. Importante è la
complessa regolazione di mTORC1 che gioca un ruolo fondamentale dal punto di vista terapeutico;
infatti si stanno attualmente sviluppando farmaci doppi inibitori di PI3K e mTOR. I doppi inibitori
potrebbero offrire un vantaggio terapeutico in quanto come noto PI3K non è l’unico regolatore di
mTOR (Engelman JA et al., 2009). La via principale attraverso la quale l’asse PI3K/AKT/mTOR
viene inattivato è la defosforilazione dei prodotti delle PI3K che fungono da secondi messaggeri per
l’attivazione di AKT. Le molecole di PtdIns(3,4,5)P3, non sono il substrato di nessuna delle
fosfolipasi C conosciute ma sono defosforilate dalle fosfatasi PTEN e SHIP (SH domain-containing
Inositol Phosphatases). PTEN è una fosfatasi dotata di doppia specificità in quanto capace di
defosforilare sia proteine che lipidi ed è in grado di rimuovere il gruppo fosfato in posizione 3
(Stambolic V et al., 1998). SHP1 e SHP2 defosforilano PtdIns(3,4,5)P3 rimuovendo il gruppo
fosfato in posizione 5 (Choi Y et al., 2002). Quindi PTEN agisce come antagonista della funzione di
PI3K inibisce l’attività di AKT e la crescita tumorale (Lu Y et al., 1999; Gori S et al., 2009) (Fig
33).
Fig 33: iperegolazione delle vie di segnale a valle di HER2
Altro potenziale meccanismo di resistenza al trastuzumab è l’accumulo a livello cellulare della
forma tronca di HER2, conosciuta come p95HER2 o frammento C-terminale. Questi frammenti
sono frequentemente ritrovati in carcinomi della mammella esprimenti HER2 (Molina, MA et al.,

57
2002) ed originano dalla proteolisi, ad opera di una metalloproteinasi non ancora ben definita, del
dominio extracellulare di HER2 (Christianson TA et al., 1998; Codony-Servat J et al., 1999) o da
un inizio alternativo di traduzione a partire da due residui di metionina (611 e 687) che sono
localizzati rispettivamente prima e dopo il dominio transmembrana (Anido J et al., 2006). La
funzione biologica di p95HER2 non è completamente conosciuta, nonostante sia stato dimostrato
come la sua iperespressione determini la crescita di tumori trapiantati in topi nudi (Anido J et al.,
2006). Il frammento C-terminale possiede attività TK richiesta per la crescita tumorale. Il fatto che
la forma tronca del recettore possieda attività tirosin-chinasica in assenza del legame tra
trastuzumab e il dominio extracellulare, ha portato ad ipotizzare come i tumori esprimenti p95HER2
siano resistenti ad herceptin ma sensibili ad altre molecole in grado di inibire l’attività tirosin-
chinasica di HER2 come il lapatinib, inibitore tirosin-chinasico (TKI) a basso peso molecolare
(Wood ER et al., 2004). Il potenziale ruolo predittivo di resistenza al trattamento con trastuzumab è
stato studiato in primis da Scaltriti e coll, sia in vitro che in vivo. Linee cellulari di carcinoma della
mammella (MCF-7 e T47D), precedentemente transfettate con vettori contenenti la forma integra o
quella tronca di HER2, sono state studiate sia prima che dopo esposizione a trastuzumab e lapatinib.
I risultati hanno evidenziato come il trattamento con lapatinib inibisce la fosforilazione di entrambe
le forme di HER2 e delle chinasi a valle (AKT e MPAKs); contrariamente trastuzumab non ha
dimostrato avere nessun effetto sulle cellule che esprimevano p95HER2. Per confermare tali
risultati in vivo, la linea cellulare MCF7, transfettata con le due forme del recettore HER2, è stata
inoculata in cavie atimiche, successivamente suddivise in tre gruppi ed esposti a trastuzumab,
lapatinib o placebo rispettivamente. Le cavie il cui tumore esprimeva la full-lenght HER2 hanno
risposto a trastuzumab e lapatinib, mentre quelle che esprimevano p95HER2 sono risultate
resistenti a trastuzumab e sensibili a lapatinib (Scaltriti M et al., 2007) (Fig 34).
Fig 34: Forma tronca di HER2

58
Il mascheramento dell’epitopo riconosciuto da trastuzumab è stato investigato come possibile
meccanismo di resistenza al farmaco. In un modello preclinico con linee cellulari umane JIMT-1
HER2 positive, resistenti al trastuzumab, è stato osservato come la resistenza sia associata alla
presenza di mucina 4 (MUC-4), glicoproteina associata alla membrana, in grado di impedire il
legame dell’anticorpo monoclonale allo specifico epitopo di HER2 (Nagy P et al., 2005).
CD44 e l’acido ialuronico possono ridurre il legame di trastuzumab allo specifico epitopo di HER2.
CD44 è un recettore transmebrana specifico per l’acido ialuronico; è stato osservato come il legame
ligando-recettore, in cellule di carcinoma ovarico, attiva segnali di trasduzione fra i quali RAS e
PI3K (Bourguignon LY et al., 2001). Linee cellulari di carcinoma della mammella JIMT-1 trattate
con inibitori della sintesi di acido ialuronico, hanno mostrato una riduzione nei livelli di acido
ialuronico sia in vivo che in vitro, con un incremento del legame di trastuzumab ad HER2 e
conseguente effetto antitumorale (Palyi-Krekk Z et al., 2007). È stato dimostrato come il legame di
polimeri di acido ialuronico a CD44 possa contribuire all’attivazione della via di segnale
PI3K/AKT; il blocco di questo legame attraverso anticorpi anti-CD44 potrebbe sopprimere questa
via di segnale con inibizione della crescita cellulare indipendente dall’ancoraggio sia in vitro che in
vivo (Ghatak S et al., 2002) (Fig 35).
Fig 35: Mascheramento epitopo
Il fattore di crescita insulino simile (IGF) ed il suo recettore (IGF-IR) risultano essere
frequentemente iperespressi e coinvolti nella proliferazione cellulare, trasformazione e
metastatizzazione (Pandini G et al., 1999; Yee D, 2002). Alti livelli di IGF prevengono l’apoptosi
indotta da chemioterapici e radiazioni. L’iperattivazione della via di segnale coordinata da IGF-IR è
associata a resistenza a trastuzumab in cellule neoplastiche HER2+. E’ stato dimostrato come
l’arresto della crescita cellulare mediato da herceptin, è perso in cellule che iperesprimono IGF-IR e
tale effetto può essere annullato se al mezzo di coltura viene aggiunta la proteina 3 in grado di

59
inibire IGF (Lu YH et al., 2001). Pertanto IGF-IR potrebbe rappresentare un possibile mediatore
target terapeutico in pazienti con carcinoma della mammella resistente al trastuzumab (Nahta R et
al., 2005; Valabrega G et al., 2007) (Fig 36).
Dei quattro membri della famiglia EGFR, HER2 è l’unico a non avere un ligando naturale. Tuttavia,
l’eterodimerizzazione di HER-2 con altri membri della famiglia può essere indotta dai ligandi di
HER1, HER3 e HER4. In presenza di un eccesso di ligando, HER2 o di entrambi, i risultanti
eterodimeri possono indurre la proliferazione cellulare e bloccare l’apoptosi, quindi interferire con
trastuzumab (Diermeier S et al. 2005). Fra tutti i ligandi TGF-α sembra avere un ruolo potenziale
nel meccanismo di resistenza (Valabrega G et al., 2005).
Il recettore c-Met ed il suo ligando HGF sono frequentemente overespressi nel carcinoma della
mammella e correlati con un decremento dell’intervallo libero da malattia e della sopravvivenza
totale (Kang JY et al., 2003). c-Met è risultato essere frequentemente co-espresso con HER2 in linee
cellulari e questo potrebbe contribuire alla resistenza al trastuzumab, garantendo l’attivazione di
Akt. Cellule che iperesprimono HER2 rispondono alla terapia con trastuzumab con una rapida
sovraregolazione dell’espressione di c-Met; tuttavia l’attivazione di c-Met avrebbe l’effetto di
rendere le cellule resistenti al trattamento con trastuzumab (Shattuck DL et al. 2008) (Fig 35).
Fig 36: vie recettoriali alternative
Un polimorfismo genetico con espressione di una valina (V) o fenilalanina (F) in posizione 158 di
FcγRIIIa influenza in modo significativo la sua affinità per IgG1 (Koene HR et al., 1997). Cellule
dell’immunità che esprimono la variante recettoriale FcγRIIIa V/V mediano l’azione ADCC di
trastuzumab in modo più efficace rispetto alla variante allelica FcγRIIIa V/F (Koene HR et al.,
1997; Shields RL et al., 2001). Ulteriori evidenze hanno confermato l’importanza del meccanismo
di ADCC nel trattamento di tumori che iperesprimono HER2; in linee cellulari di carcinoma della
mammella MCF7 e SKBR3 trattate con lapatinib da solo o in combinazione con trastuzumab si
osservava inibizione della fosforilazione di HER2 con prevenzione della sua ubiquitinazione e

60
conseguente accumulo della forma inattiva del recettore sulla superficie cellulare. Trastuzumab da
solo causava invece una degradazione di HER2. L’accumulo di HER2 indotto da lapatinib
amplificherebbe l’effetto citotossico immuno-mediato di trastuzumab (Scaltriti M et al., 2009) (Fig
37).
Fig 37: effetto ADCC di trastuzumab

61
13.5 STRATEGIE TERAPEUTICHE PER SUPERARE LA RESISTENZA A
TRASTUZUMAB
La resistenza al trastuzumab, rappresenta uno dei maggiori problemi clinici che ha indirizzato
l’attenzione verso nuovi farmaci che hanno come bersaglio HER2 o vie di segnale alternative.
Poiché EGFR ed HER-2 risultano essere co-espressi in circa il 30% dei carcinomi della mammella
il blocco di entrambi potrebbe rappresentare una strategia terapeutica per superare la resistenza al
trastuzumab (Normanno N et al., 2009; Tsang RY et al., 2012).
Lapatinib (GW572016)
Lapatinib è un inibitore tirosin chinasico diretto verso EGFR ed HER2. A differenza di altri inibitori
tirosin chinasici, l’interazione di lapatinib con EGFR ed HER2 è reversibile ma la dissociazione da
essi è più lenta. Lapatinib si lega al dominio citoplasmatico che lega ATP bloccando la
fosforilazione ed attivazione del recettore prevenendo l’innesco di vie di segnalazione a valle come
ERK1/2 e PI3K/Akt (Amundadottir LT et al., 1998; Wood ER et al., 2004; Gutierrez C et al.,
2011). Studi in vitro hanno dimostrato coma la combinazione di lapatinib con anticorpi anti-HER2
sia in grado di amplificare il segnale apoptotico in cellule di carcinoma della mammella che
iperesprimono HER2 e come tale effetto sia mediato dalla down-regolazione della survivina (Xia W
et al., 2005). Interessante è stata l’osservazione di come la resistenza a lapatinib possa essere
mediata da un incremento del segnale mediato da ER in tumori della mammella positivi per ER e
con aumentati livelli di espressione di HER2 (Xia W et al., 2006). Lapatinib avrebbe l’effetto di
indurre un significativo segnale apoptotico in cellule resistenti a trastuzumab ed effetti inibitori sul
segnale che deriva dall’interazione IGF-I/IGF-IR (Nahta R et al, 2006) (Fig 38).
Fig 38: meccanismo d’azione di lapatin
Cascata di segnali

62
Teoricamente, piccole molecole in grado di inibire l’attività tirosin chinasica di EGFR e HER2
hanno diversi vantaggi rispetto ad anticorpi monoclonali diretti contro HER2. L’inibitore di un solo
recettore può non essere efficace verso eterodimeri contenenti sia ErbB1 che ErbB2 (Rusnak DW et
al., 2001; Zhou Y et al., 2006). Altro vantaggio di lapatinib rispetto a trastuzumab è rappresentato
dalla sua capacità di inibire le forme tronche dei recettori ErbB1 ed ErbB2, che mancano del
dominio extracellulare (Xia W et al., 2004; Saez R et al., 2006; Scaltriti M et al., 2010).
Trastuzumab e lapatinib hanno meccanimi di azione complementari e attività antitumorale sinergica
in modelli si tumore della mammella con aumentata espressione di HER2. La doppia inibizione del
recettore potrebbe pertanto rappresentare un valido approccio al trattamento del tumore della
mammella positivo per HER2 nel contesto neoadiuvante (Baselga J et al., 2012).
Pertuzumab (2C4)
L’anticorpo monoclonale umanizzato ricombinante pertuzumab rappresenta una nuova classe di
farmaci denominati inibitori della dimerizzazione. Questi farmaci hanno la capacità di bloccare il
segnale derivante da latri membri della famiglia HER e inibire segnali in cellule che esprimono
normali livelli di HER2; in particolare pertuzumab blocca la dimerizzazione di HER2 con EGFR e
HER3 inibendo in questo modo la cascata di segnali che derivano dalla formazione degli
eterodimeri HER2/EGFR e HER2/HER3 (Agus DB et al., 2002). Pertuzumab possiede inoltre la
capacità di inibire l’interazione fra IGF-IR ed HER2 in cellule resistenti al trastuzumab (Nahta R et
al., 2005). Trastuzumab e pertuzumab, si legano ad epitopi differenti del dominio extracellulare di
HER2; trastuzumab si lega al dominio IV mentre pertuzumab si lega vicino ai domini I, II e III
(Franklin MC et al., 2004) Pertanto, teoricamente pertuzumab potrebbe essere efficace in tumori
resistenti al trastuzumab. Tuttavia mentre la combinazione trastuzumab-pertuzumab produce un
effetto apoptotico sinergico su cellule di carcinoma della mammella responsive ad herceptin, non si
osservano evidenze significative sulla vitalità delle cellule trastuzumab-resistenti (Tanner M et al.,
2004; Nahta R et al., 2005). Questo comportamento potrebbe riflettere aberrazioni nelle vie di
segnale a valle che conferiscono resistenza ad una varietà di farmaci che hanno come bersaglio
HER2 (Fig 39).

63
Fig 39: meccanismo di azione di pertuzumab Inibizione di IGF-IR
In base ad evidenze precliniche che suggeriscono un ruolo di IGF-IR nello sviluppo della resistenza
al trastuzumab (Lu Y et al., 2001; Nahta R et al., 2005), agenti diretti contro tale recettore sono stati
testati in modelli sperimentali. In modelli cellulari tumorali umani di carcinoma della mammella
con elevati livelli di espressione di EGF-IR e bassi livelli di HER2 (linee cellulari MCF7) o
viceversa con bassi livelli di IGF-IR ed alti livelli di HER2 (linee cellulari BT474) è stato
dimostrato come il trattamento combinato con farmaci, sia anticorpi monoclonali che piccole
molecole, inibitori di HER2 ed IGF-IR, causa inibizione della proliferazione in modo sinergico
anche in situazioni di scarsa sensibilità al singolo inibitore per mancanza del bersaglio specifico.
Pertanto il trattamento combinato determina una perturbazione del ciclo cellulare con induzione di
apoptosi ed inibizione di trasduttori del segnale a valle inclusi MAPK e AKT (Camirand A et al.,
2002; Di Giovanna MP et al., 2006; Chakraborty AK et al., 2008; Bhargava R et al., 2011). Inoltre
è stato dimostrato come lapatinib in combinazione con l’anticorpo monoclonale α-IR3 diretto
contro IGF-IR, incrementi il segnale apoptotico in cellule resistenti al trastuzumab (Nahta R et al.,
2006).
T-DM1
Un trattamento nuovo, attualmente in fase di sviluppo per il carcinoma della mammella HER2
positivo in stadio avanzato è rappresentato dall’utilizzo di farmaci anticorpo-coniugati (ADC) o
“anticorpi armati”. Gli ADC per la loro natura di farmaci mirati, permettono la somministrazione di
alte dosi di chemioterapici altrimenti intollerabili. T-DM1 (trastuzumab-emtansine) è stato
concepito per combinare i benefici clinici di trastuzumab con la chemioterapia a base di DM1
(potente inibitore dei microtubuli); DM1 si lega al trastuzumab mediante un legame stabile che
mantiene intatto il coniugato T-DM1 fino a quando questo non raggiunge le cellule tumorali HER2-
positive (Burris HA, 2011) (Fig 40). Trasruzumab, si lega alle cellule tumorali positive per HER2 e
blocca i segnali che contribuiscono alla crescita e sopravvivenza del tumore, attivando nello stesso
Nessun segnale
Arresto della crescita
Nessun segnale
Arresto della crescita
Segale di crescita

64
tempo il sistema immunitario dell’organismo contro queste cellule. DM1 viene internalizzato nelle
cellule tumorali e rilasciato (Lewis Phillips GD et al., 2008; Junttila TT et al., 2010).
Fig 40: meccanismo di azione del coniugato T-DM1
Stategie future
Terapie innovative per by-passare la resistenza a trastuzumab sono rappresentate dall’inibizione di
PI3K/AKT (Van Ummersen L et al., 2004; Leighl N et al., 2008), e mTOR (Chan S et al., 2005;
O’Regan R et al., 2009; Pohlmann PR et al, 2009; Jones KL et al., 2010), critici per la crescita
cellulare, la proliferazione e l’angiogenesi ed implicati in diversi tipi di tumore (Fig 41). Fra i
pazienti con carcinoma della mammella positivo per HER2, quelli con alti livelli di AKT
fosforilato, mutazioni attivanti di PI3CKA e perdita di PTEN, presentano una peggiore prognosi
dopo terapia con trastuzumab, rispetto ai pazienti senza tali alterazioni (Wang L et al., 2011). Negli
ultimi anni sono stati sviluppati composti in grado di colpire molteplici recettori
contemporaneamente (targeted multitherapy). Questi composti multifunzionali sono stati progettati
al fine di sfruttare interazioni tra vari target terapeutici e per essere più resistenti ai meccanismi
compensativi (Faive S et al., 2006; Le Tourneau C et al., 2008; Mohd Sharial MSN et al., 2012).
Fig 41: Inibizione di PI3K e mTOR

65
14.0 MicroRNA (miRNA)
Negli ultimi anni la lista delle classi di geni classicamente considerati come oncogeni e
oncosoppressori è stata rivisitata, essendosi resa necessaria l’espansione di quest’ultima per
includere anche la famiglia di miRNA. I miRNA sono piccole molecole di RNA non codificanti, in
grado di regolare l’espressione genica agendo a livello post-trascrizionale (He L et al., 2004). La
loro funzione è quella di legare molecole di RNA bersaglio, inducendo l’inibizione della traduzione
o la degradazione dell’mRNA, modulando in tal modo un ampio spettro di processi fisiologici,
quali il controllo del ciclo cellulare, il differenziamento e l’apoptosi. L’espressione dei miRNA è in
genere tessuto specifica e dipendente dai livelli di differenziazione della cellula per cui la presenza
di significative alterazioni nei livelli di espressione dei miRNA in un tessuto tumorale potrebbe
essere secondaria ai processi più direttamente implicati nella tumorigenesi piuttosto che un
elemento causale direttamente associato alla trasformazione neoplastica. Un significativo numero di
geni di miRNA sono localizzati in regioni genomiche frequentemente riarrangiate nei tumori come i
siti fragili, regioni delete (minimal region of loss of heterozigosity, LOH) o amplificate (minimal
amplicons) e regioni comuni di break-point, fornendo un’evidenza del loro ruolo nella patogenesi
del cancro (Gaur A et al., 2007). Alterazioni nei livelli di espressione dei miRNA sono state
documentate in diversi tipi di tumori e fra questi i miRNA che hanno come target geni regolatori
dello sviluppo tumorale come E2F e RAS (Gaur A et al., 2007). A seconda del tipo di funzione
biologica del miRNA, determinata dalle proteine che regola, si possono distinguere miRNA
oncogeni (oncomiRNA) e miRNA oncosoppressori (Croce CM, 2009) (Fig 11). I primi si trovano
in regioni amplificate o sovra espresse nel tumore, con aumento della proliferazione cellulare,
dell’angiogenesi, dell’invasività e riduzione dell’apoptosi. I secondi si trovano in regioni delete o
silenziate nel tumore ed hanno un effetto biologico opposto. Esempi di oncomiRNA sono il miR-
155 amplificato in molti tumori ematologici, nel tumore del polmone e della mammella e il cluster
miR-17-92, che comprende 6 geni diversi per miRNA sovra espressi in tumori solidi e nel linfoma
diffuso a grandi cellule B. Tra i miRNA oncosoppressori troviamo miR-15a e miR-16-1 deleti nella
leucemia linfatica cronica e che ha come bersaglio la proteina antiapoptotica Bcl-2 e la famiglia let-
7 che reprime l’espressione dell’oncogene RAS (Heneghan HM et al., 2009; Findlay VJ, 2010).

66
Fig 11: miRNA oncogeni e oncosoppressori
Tra i miRNA potenzialmente rilevanti nel carcinoma della mammella, è stato identificato il miR-
205 che ha come bersaglio il recettore tirosin chinasico HER3 (Tab 2). HER3, esercita un ruolo
importante nella tumorigenesi mediata dall’oncogene HER2; la co-espressione dei due membri
della stessa famiglia recettoriale, rappresenta un fattore prognostico negativo essendo responsabile
dell’attivazione del meccanismo di sopravvivenza mediato dalle chinasi PI3K ed AKT. In vitro è
stato dimostrato come miR-205 regola negativamente l’espressione di HER3; l’espressione ectopica
di questo miRNA in una linea cellulare di carcinoma della mammella caratterizzata da bassi livelli
di espressione di miR-205, causa una riduzione dell’espressione di HER3. La capacità di questo
miRNA di inibire la proliferazione, è stata confermata in un modello cellulare di carcinoma
mammario; l’espressione ectopica di miR-205, determina una riduzione dei livelli della forma attiva
(fosforilata) del mediatore AKT suggerendo un suo possibile ruolo nel modulare la responsività ad
inibitori tirosin chinasici come Gefitinib e Lapatinib. Pertanto l’introduzione di miR-205 in cellule
di carcinoma mammario, modulando l’espressione di HER3 e spegnendo di conseguenza la via
mediata da AKT è capace di contrastare i meccanismi di resistenza al trattamento mediati dal
recettore, ristabilendo l’effetto pro-apoptotico dei farmaci utilizzati (Iorio MV et al., 2009). Il ruolo
oncosoppressivo di miR-205 è stato inoltre dimostrato in un modello di carcinoma della mammella
triplo negativo, sottogruppo caratterizzato da un fenotipo estremamente indifferenziato e
clinicamente molto aggressivo. Non essendo stati ancora identificati specifici marcatori non esiste,
per tali tumori una specifica e mirata terapia. I miRNA potrebbero fornire informazioni biologiche
aggiuntive necessarie per spiegare il comportamento di tale sottogruppo e rappresentare un possibile
strumento o bersaglio per una terapia specifica. È stato dimostrato come miR-205 sia in grado di
inibire sia la proliferazione che la migrazione in modelli in vitro di tumori tripli negativi, sia la
crescita in vivo in modelli murini, attraverso la modulazione di molecole coinvolte nei processi di
proliferazione, adesione, migrazione ed invasione (Piovan C et al., 2012).

67
Tab 2: alterata espressione dei miRNA nel carcinoma della mammella
Interessante è il possibile ruolo dei miRNA nella regolazione della transizione da carcinoma duttale
in situ a carcinoma invasivo; la possibilità di inibire i miRNA associati con l’invasività del tumore,
potrebbe arrestare la progressione allo stadio pre-invasivo (Volinia S et al., 2012) (Fig 12).
Fig 12: alterata espressione di miRNA durante la progressione tumorale
Ad oggi si sta valutando ed investigando il possibile utilizzo di queste piccole molecole ad RNA
come terapia innovativa, nel tentativo di modulare la loro espressione nei tumori. In particolare si
Mammell
a normale
Carcinoma
duttale in
situ
Carcinoma
duttale
invasivo
Espressione e ruolo miRNA Target

68
stanno elaborando approcci per reintrodurre microRNA la cui espressione è persa nel tumore, o
viceversa inibire microRNA oncogenici utilizzando specifiche molecole antisenso (antagomiR)
(Hota J et al., 2009; Iorio MV et al., 2011).
I miRNA potrebbero pertanto essere anche considerati buoni target per la terapia antitumorale.
L’introduzione di miRNA o antagomiR in tumori in cui questo è stato perso o risulta essere
sovraespresso può portare all’arresto del ciclo cellulare e/o apoptosi (Kota J et al., 2009). Per
tumori in cui si assiste ad un silenziamento epigenetico di geni oncosoppressori si potrebbe
procedere con una terapia basata sull’introduzione di miRNA che controllano la DNA metil-
transferasi (DNMT), come i membri della famiglia del miR-29 (Fabbri M et al., 2007). Da studi di
profiling su pazienti tumorali si è evidenziato come specifici set di miRNA sono associati al tessuto
di origine (Rosenfeld N et al., 2007) o a determinate caratteristiche istopatologiche e di aggressività
(Blenkiron C et al., 2007). Pertanto alcuni miRNA potrebbero considerarsi marcatori importanti
nella stratificazione di pazienti sia in fase diagnostica che prognostica.
Tre sono le metodologie che permettono di analizzare i livelli di espressione di miRNA; un classico
northern blot con una sonda diretta verso il miRNA di interesse, l’uso di microarray o l’analisi
mediante la tecnica MIRMasa. Quest’ultima tecnica consiste nell’incubare l’RNA estratto con due
sonde di cui una complementare ad una metà del miRNA di interesse marcata con un fluoroforo
(capture oligo), l’altra complementare all’altra metà di miRNA e marcata con biotina (detection
oligo). Dopo incubazione si esegue una purificazione con sfere ricoperte di streptavidina e si
catturano i detection oligo. Se nell’estratto non sono presenti i miRNA di interesse si purificano
solo i detection oligo e pertanto la frazione purificata non emette luce se stimolata con lunghezze
d’onda appropriate. Se i miRNA sono presenti, questi ibrideranno contemporaneamente le due
sonde e la purificazione con le biglie selezionerà sia i detection oligo, sia i miRNA che i capture
oligo per cui il purificato sarà fluorescente e l’intensità del segnale è direttamente proporzionale alla
quantità di miRNA presente nell’estratto totale.
Recentemente è stata osservata la presenza di miRNA nel siero e nel plasma umano in diversi tipi di
tumore come quelli del colon, della mammella, della prostata, del polmone e dell’ovaio (Chen X et
al., 2008). Questi possono essere utilizzati come marcatori descriminanti non invasivi per la
diagnosi del fenotipo tumorale. I livelli di miRNA nel siero sono molto stabili (Xi Y et al., 2007) e
possono essere individuati mediante semplici prelievi di sangue. È stato dimostrato per alcuni
tumori, come i miRNA nel siero hanno un’origine tumore-specifica (Lu J et al., 2005) e come
possono essere usati nella diagnosi precoce del cancro (Mitchell PS et al., 2008).

69
15.0 DNA REPAIR COME BERSAGLIO FARMACOLOGICO: SYNTHETIC LETHALITY
STRATEGY
La “synthetic lethality” si riferisce a quella condizione in cui due o più mutazioni non alleliche e
non essenziali, quindi di per se non letali, lo diventano se presenti all’interno di una stessa cellula.
Un esempio tipico di farmaci che possono funzionare in questo modo è rappresentato dagli inibitori
di PARP-1 (Ashworth A, 2008). Gli enzimi PARP (PolyAdenosine diphosphate-Ribose Polymerase)
costituiscono una famiglia di 17 enzimi nucleari coinvolta in una serie di processi cellulari quali la
riparazione del DNA e l’apoptosi (Schreiber V et al., 2006). PARP-1 in particolare interviene nel
riparo di danni al DNA (basi alchilate) che possono essere indotti dal trattamento con agenti
chemioterapici alchilanti. L’attivazione di PARP è alla base del fenomeno della resistenza dei
tumori alla chemioterapia. Inibendo PARP, si attenua nei tumori la capacità di resistere agli agenti
alchilanti e si ripristina la loro sensibilità alla chemioterapia. Inattivando PARP in cellule con
deficit di riparo come quelle BRCA-mutate, nel nucleo si accumulano frammenti danneggiati di
DNA a singolo e doppio filamento, con arresto della crescita cellulare, della sua divisione fino alla
morte delle cellule tumorali (Bryant et al. 2005; Farmer et al. 2005; Aly A et al., 2011) (Fig 42).
Studi preclinici e clinici hanno dimostrato come la combinazione di inibitori di PARP-1 (olaparib)
con agenti chemioterapici derivati dal platino, che inducono danni al DNA attraverso addotti e
cross-linking, potenzia la loro citotossicità (Annunziata CM et al., 2010; Hastak K et al., 2010;
Underhill C et al., 2011).
Di particolare interesse sono alcune evidenze precliniche che suggeriscono un potenziale più ampio
di utilizzo degli inibitori PARP. È stato infatti osservato che oltre BRCA1/2, la perdita di funzione
di varie proteine coinvolte nei meccanismi di riparo del DNA, generi sinteticamente un fenotipo
letale quando PARP viene inibito. Ad esempio cellule con geni PTEN difettosi, sono più sensibili ai
PARP inibitori rispetto alle cellule normali (Mendes-Pereira AM et al., 2009; Dededs KJ et al.,
2011). Recenti studi hanno dimostrato come PTEN avrebbe un ruolo importante nel mantenimento
dell’integrità cromosomica e nella riparazione dei danni al DNA (Meyn RE, 2009). L’inattivazione
di questi geni è stata riportata in un gruppo di tumori umani e potrebbe quindi costituire un
biomarker predittivo per selezionare pazienti da trattare con inibitori di PARP. Interessante è la
possibile applicazione di inibitori di PARP in tumori tripli negativi della mammella (TNBC). Questi
tumori non sono spesso mutati in BRCA1 (15-20%) ma presentano delle caratteristiche fenotipiche
e cliniche che ricordano i tumori BRCA1 mutati; tale fenotipo viene definito BRCAness (Turner N
et al., 2004; Anders CK et al., 2010; Kruse V et al., 2011). Questi tumori sono frequentemente di
alto grado, mutati in p53, esprimono EGFR e sono sensibili alla chemioterapia. I possibili
meccanismi responsabili del fenotipo BRCAness includono: la metilazione del promotore del gene

70
per BRCA1 (Turner N et al., 2004), la ridotta espressione dell’mRNA di BRCA1 (Natrajan R et al.,
2010), l’iper-regolazione di soppressori trascrizionali di BRCA1 come ID4 (Turner NC et al., 2007)
e HMG1 (Baldassarre G et al., 2003), l’iper-regolazione di miR-182 (Moskwa P et al., 2011),
mutazioni in geni che codificano per proteine che interagiscono con BRCA1/2 o che agiscono nella
stessa via di riparazione del DNA come CHK2 (checkpoint homolog kinase 2), PALB2 (Partner and
localizer of BRCA2), BRIP1 (BRCA1 interacting protein C-terminal helicase), RAD50 e RAD51 e
BARD1 (BRCA1-associated RING domain protein 1) (Kuusito KM et al., 2011; Wang XZ et
al.,2011). Quindi la migliore comprensione della rete biologica e molecolare delle proteine del
pathway di BRCA nella risposta al danno al DNA potrebbe aiutare nella identificazione di pazienti
con TNBC da indirizzare a terapie non convenzionali maggiormente mirate a colpire cellule con
deficit di riparo del DNA (Pal Sk et al., 2009; Carey LA et al., 2010; O'Shaughnessy J, et al., 2011;
Weill MK et al., 2011).
Fig 42: inibizione di PARP

71
16. CONCLUSIONI
Il carcinoma della mammella rimane ad oggi la principale causa di morte per cancro nelle donne
nonostante i notevoli progressi ottenuti sia nella diagnosi che nella terapia. Nonostante lo studio dei
profili di espressione genica, che ha contribuito ad identificare le alterazioni molecolari
caratteristiche dei tumori umani, abbia fornito la maggior parte dei nuovi bio-marcatori con
possibili applicazioni nell’ambito diagnostico, di sviluppo di nuovi farmaci e di nuove terapie
mirate, non ha ancora elucidato in modo esaustivo tutti i meccanismi all’origine dei tumori, incluso
il tumore della mammella. Questo suggerisce come il processo di tumorigenesi possa avvenire
attraverso meccanismi nuovi non ancora del tutto caratterizzati.
Il tumore della mammella è una malattia eterogenea caratterizzata da un ampio spettro di variabilità
cliniche, patologiche e molecolari che può rendere conto delle diverse risposte alle terapie. Per
molti anni la pratica clinica è stata guidata dai risultati delle metanalisi e da una classificazione
esclusivamente morfologica del carcinoma della mammella.
Le pazienti affette da carcinoma mammario HER2 positivo hanno in genere una peggiore prognosi
e un andamento della malattia maggiormente aggressivo rispetto alle pazienti i cui tumori non
esprimono la proteina di membrana. Attualmente il trattamento con trastuzumab, in associazione
con la chemioterapia, rappresenta il trattamento standard di prima linea nelle pazienti con
carcinoma mammario metastatico che iperesprime HER2. Tuttavia il 40% delle pazienti con
diagnosi di tumore metastatico HER2 positivo, trattate con trastuzumab, non presenta alcun
beneficio per lo sviluppo di una resistenza primaria o secondaria al farmaco. Lapatinib in
associazione alla capecitabina rappresenterebbe una valida opzione terapeutica nel caso di
resistenza a trastuzumab. Ad oggi sono stati individuati diversi determinanti molecolari che
sembrano essere implicati nei meccanismi di resistenza al trastuzumab, sebbene nessuno sia stato
ancora validato nella pratica clinica. Analisi precliniche e retrospettive hanno dimostrato come la
forma recettoriale tronca di HER2, la perdita di espressione di PTEN e lo stato mutazionale di
PI3K/AKT risultino essere associate ad una resistenza al trastuzumab.
Ricerche di “gene espression profiling” hanno permesso di individuare alcuni sottogruppi
molecolari di carcinoma della mammella con implicazioni clinico-prognostiche e terapeutiche. In
particolari i tumori tripli negativi rappresentano un tipo particolarmente aggressivo di carcinoma
della mammella, tipicamente caratterizzato da una prognosi infausta. Queste neoplasie
rappresentano il 15-20% di tutti i tumori della mammella e si distinguono in quanto a differenza
delle forme più comuni non esprimono il recettore per gli estrogeni, per il progesterone e non
presentano amplificazione del gene codificante per il fattore di crescita epidermico. La loro crescita
è pertanto guidata da meccanismi molecolari distinti e per questo sono resistenti ai trattamenti

72
standard. Questi tumori sono caratterizzati da un’estrema variabilità genotipica e presentano un
carattere fortemente evolutivo legato principalmente ad una forte instabilità del loro genoma.
L’unica arma oggi a disposizione per questo tipo di tumori rimane la chemioterapia. Numerosi
target terapeutici stanno emergendo come potenziali opzioni nel trattamento di questo gruppo di
neoplasie fra cui inibitori di EGFFR, PARP-inibitori, inibitori di c-KIT ed inibitori della via di
segnale PI3K/AKT.
Recentemente una nuova classe di molecole di RNA non codificanti, noti come microRNA
(miRNA), capaci di regolare l’espressione genica attraverso il legame con sequenze regolatorie
(3’UTR) su mRNA, è stata associata a diverse malattie umane incluso il tumore della mammella.
Un crescente numero di evidenze sperimentali ha mostrato come i miRNA abbiano un ruolo causale
nel processo di tumorigenesi, agendo come una nuova classe di oncogeni od oncosoppressori. Dopo
le iniziali osservazioni dell’associazione fra miRNA e tumori umani, questo campo di ricerca sta
crescendo incredibilmente. Attualmente si sta cercando di elucidare i meccanismi molecolari in cui i
miRNA sono coinvolti e di validare il loro potenziale ruolo sia come biomarcatori che come
strumenti o bersagli di terapie innovative. Tra i miRNA potenzialmente rilevanti nel carcinoma
della mammella è stato identificato miR-205 capace di interferire con la proliferazione cellulare
mediata dai recettori della famiglia HER ed in grado di aumentare la responsività al trattamento con
inibitori tirosin-chinasici (TKI). I miRNA potrebbero fornire informazioni biologiche necessarie per
spiegare il comportamento dei tumori triplo negativi e rappresentare un possibile strumento o
bersaglio per la terapia specifica.
Pertanto la caratterizzazione genotipica dei tumori della mammella è indispensabile per
comprendere i meccanismi biologici che ne guidano la crescita e che determinano la risposta ai
trattamenti. Tale procedura si rende ancora più necessaria per i tumori tipli negativi per i quali gli
approcci terapeutici standard si rivelano spesso inefficaci e altre strategie devono essere sviluppate
e sperimentate.

73
BIBLIOGRAFIA
- Ahmed I et al. Mouse mammary tumor virus: a cause of breast cancer in humans? Cancer
ther 6: 537-44, 2008.
AIMO. - www.aiom.it/Attività Scientifica/Linee guida/Neoplasie della mammella
- AJCC. Cancer staging manual. 6. New York, Springer, 2002.
- Al-Hajj M et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci
USA: 100, 3983-88, 2003
- Allain DC et al. Management options after prophylactic surgieries in women with BRCA
mutations: a review. Cancer Control 14: 330-37, 2007.
- Allen MG. Current issues in ER and HER testing by IHC in breast cancer. Mod Pathol 21: S8-
S15, 2008.
- Allred DC et al. Ductal carcinoma in situ and the emergence of diversity during breast cancer
evolution. Clin Cancer Res 14: 370, 2008.
- Alvi AJ et al. Functional and molecular characterisation of mammary side population cells.
Breast Cancer Res 5: R1-R8, 2003.
- Amundadottir LT et al. Signal transduction pathways activated and required for mammary
carcinogenesis in response to specific oncogenes. Oncogene 16: 737–46. 1998.
- Anders CK et al. Poly(ADP-Ribose) polymerase inhibition: “targeted therapy for triple
negative breast cancer. Clin Cancer Res v16: 4702-10, 2010.
- Andreopoulou E et al. Prognostic factors in metastatic breast cancer: successes and challenges
toward individualized therapy. J Clin Oncol 26: 3660-62, 2008.
- Anguiano A. L’ibridazione in situ in fluorescenza (FISH); principi generali ed applicazioni
cliniche. Journal of Clinical Ligand Assay 23: 33-42, 2000.
- Anido J et al. Biosynthesis of tumorigenic HER-2 C-terminal fragments by alternative initiation
of translation. EMBO J 25: 3234-44, 2006.
- Annunziata CM et al. Poly(ADP-Ribose) polymerase as a novel therapeutic target in cancer. Clin
Cancer Res 16: 4517, 2010.
- Arizti P et al. Tumor suppressor p53 is required to modulate BRCA1 expression. Mol Cell Biol
20: 7450-59, 2000.
- Arnould L et al. Agreement between chromogenic in situ hybridisation (CISH) and FISH in the
determination of HER2 status in breast cancer. Br J Canc 88: 1587-91, 2003.
- Arpino G et al. Crosstaltk between the estrogen receptor and HER tyrosine kinase receptor
family: molecular mechanism and clinical implications for endocrine therapy resistence. Endocr
Rev 29: 217-33, 2008.

74
- Ashworth A et al. A synthetic lethal therapeutic approach: Poly (ADP-ribose) polymerase
inhibitors for the treatment of cancer deficient in DNA double-strand break reapir. J Clin Oncol 26:
3785-90, 2008.
- Baan R et al. Carcinogenicity of alcoholic bever-ages. Lancet Oncol 8:292-93, 2007.
- Baldassarre G et al. Negative regulation of BRCA1 gene expression by HMGA1 proteins
accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol
23:2225-38, 2003.
- Bakshi RP et al. Functional and regulatory characteristics of eukaryotic type II DNA
topoisomerase. Crit Rev Biochem Mol Biol 36: 1-37, 2001.
- Barkhem T et al. Differential response to estrogen receptor alpha and estrogen receptor beta to
partial estrogen agonists/antagonists. Mol Pharmacol 54: 105-12, 1998.
- Baselga J et al. Recombinant humanized anti Her2 antibody (Herceptin™) enhances the
antitumor activity of paclitaxel and doxorubicin against Her2/neu overexpressing human breast
cancer xenografts. Cancer Res 58: 2825-31, 1998.
- Baselga J et al. Mechanism of action of trastuzumab and scientific update. Semin Oncol 28: 4-11,
2001.
- Baselga J et al. Future options with Trastuzumab for Primary Systemic and Adjuvant Therapy.
Semin Oncol 31: 51-57, 2004.
- Baselga J et al. Critical update and emerging trends in epidermal growth factor receptor targeting
in cancer. J Clin Oncol 23: 2445-59, 2005.
- Baselga J et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO) :
a randomised, open-label, multicentre, phase 3 trial. Lancet 379: 633-40, 2012.
- Baselga J et al. Everollimus in postmenopausal hormnone receptor-positive advanced breast
cancer. N Engl J Med 366: 520-29, 2012.
- Bast RC et al. American Society of Clinical Oncology Tumor Markers Expert Panel 2000 update
of recommendations for the use of tumor markers in breast and colorectal cancer: clinical practice
guidelines of the American Society of Clinical Oncology. J Clin Oncol 19: 1865-78, 2001.
- Beatson GT. On the treatment of inoperable cases of carcinoma of the mamma: suggestion for a
new method of treatment with illustrative cases. Lancet 2: 104-07, 1896.
- Bempt I et al. Polysomy 17 in breast cancer: clinicopathologic significance on HER-2 testing. J
Clin Oncol 26: 4869-74, 2008.
- Besere AR et al. HER-2, TOP2A and chromosome 17 alterations in breast cancer. Pathol Oncol
Res 13: 180-85, 2007.

75
- Bhargava R et al. Insuline-like growth factor receptor-1 (IGF-1R) expression in normal breast ,
proliferative breast lesions, and breast carcinoma. Appl Immun Mol Morphol 19: 218-25, 2011.
- Bieche I et al. Molecular profiling of inflammatory breast cancer: identification of a poor-
prognosis gene expression signature. Clin Cancer Res 10: 6879-95, 2004.
- Bieche I et al. Molecular profiling of inflammatory breast cancer: a hyperproliferative phenotype.
Clin Cancer Res 12: 5047-54, 2006.
- Bittner JJ. Some possible effects of nursing on the mammary gland tumor incidence in mice.
Science 84: 162-68, 1936.
- Blenkiron C et al. MicroRNA expression profiling of human breast cancer identifies new
markers of tumor subtype. Genome Biol 8: R214, 2007.
- Bonadonna G et al. Medicina Oncologica. Vol 2: 800-64, 2007.
- Bonnet D et al. Human acute myeloid leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med 3: 730-37, 1997.
- Bombonati A et al. The molecular pathology of breast cancer. J Pathol 223: 307-17, 2011.
- Bourguignon LY et al. Hyaluronan promotes CD44v3–2 interaction with Grb2-185(HER2)
and induces Rac1 and Ras signaling during ovarian tumor cell migration and growth. J Biol
Chem 276: 48679–92, 2001.
- Boussen H et al. Inflammatory breast cancer in Tunisia: epidemiological and clinical trends.
Cancer 116: 2730-35, 2010.
- Brase JC et al. ERBB2 and TOP2A in Breast Cancer: A comprehensive analysis of gene
amplification, RNA levels, and protein expression and their influence on prognosis and prediction.
Clin Cancer Res 16: 2391-401, 2010.
- Brinda A et al. Search for DNA of exogenous mouse mammary tumor virus-related virus in
human breast cancer samples. J Gen Virol 88: 1806-09, 2007.
- Brown AMC. Wnt signalling in breast cancer: have we come full circle? Breast Cancer Res
3: 351-55, 2001.
- Brüggemann M et al. The immunogenicity of chimeric antibodies. J Exp Med 170: 2153-57,
1989.
- Brunelli M et al. Gentotypic intratumoral heterogeneity in breast carcinoma with HER2/neu
amplification. Am J Patthol 131: 678-82, 2009.
- Jackisch C. Her2 positive metastatic breast cancer: optimizing Trastuzumab based therapy. The
Oncologist 11: 34-41, 2006.
- Burris HA. Trastuzumab emtansine a novel antibody-drug conjugate for HER2 positive breast
cancer. Exp Opin Bio Ther 11: 807-19, 2011.

76
- Burness ML et al. Epidermal growth factor receptor in triple negative and basal like breast
cancer: promising clinical target on only marker? Cancer J 16: 23-32, 2010.
- Caballero-Garcia T et al. Determination of HER2 amplification in primary breast cancer using
dual-colour chromogenic in situ hybridization is comparable to fluorescence insitu
hybridization:aEuropean multicentre study involving 168 specimens. Hystopatology 56: 472-80,
2010.
- Cain RJ et al. Phosphoinositide 3-kinases in cell migration. Biol Cell 101:13-29, 2009.
- Calabrese C et al. A perivascular niche for brain tumor stem cells. Cancer Cell 11: 69-82, 2007.
- Callahan R et al. MMTV-induced mammary tumorigenesis: gene discovery, progression to
malignancy and cellular pathaways. Oncogene 19: 992-1001, 2001.
- Camirand A et al. Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes
synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit 8:
BR521-BR526, 2002.
- Campbell LL et al. Breast tumor heterogeneity. Cancer stem cells or clonal evolution? Cell Cycle
6: 2332, 2007.
- Carbone A et al. High interlaboratory reproducibility of the Silver in Situ Hybridization Assay
(SISH) for the detection of HER2 gene status in breast carcinoma: a study reporting results from
five cancer institutes. USCAP Annual Meeting, Denver, March 1-7, 2008.
- Carey LA. Directed theraphy of subtypes of triple negative breast cancer. The Oncologist 15: 49-
56, 2010.
- Carlomagno F et al. Receptor tyrosine kinases as targets for anticancer therapeutics. Curr Med
Chem 12: 1773-81, 2005.
- Carter P et al. Humanization of an anti-p185 Her2 antibody for human cancer therapy. Proc Natl
Acad Sci USA 89: 4285-89, 1992.
- Chakraborty AK et al. Co-targeting Insuline Like Growth Factor I Receptor and HER2: dramatic
effects of HER2 inhibitors of nonoverexpressing breast cancer. Cancer Res 68: 1538-45, 2008.
- Challen GA et al. A side order of stem cells: the SP phenotype. Stem Cells 24: 3–12, 2006.
- Chan S et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily
pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 23: 5314-22,
2005.
- Chan SK et al. The role of epidermal growth factor receptor in breast cancer. J Mamm Gland Biol
Neopl 11: 3-11, 2006.
- Chang JC et al. Induction apoptosis without change in cell proliferation in primary breast cancer
with neoadjuvant trastuzumab. Breast Cancer Res Treatm 82: 24, 2003.

77
- Cheang MCU et al. Basal-Like breast carcinoma defined by five biomarkers has superior
prognostic value thar triple negative phenotype. Clin Cancer Res 14: 1368-76, 2008.
- Cheang MCU et al. HER2 status, and prognosi of patients with luminal B breast cancer. J Natl
Cancer Inst 101: 736-50, 2009.
- Chen S et al. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 25: 1329-33,
2007.
- Chen X et al. Characterization of microRNAs in serum: a novel class of biomarkers for
diagnosis of cancer and other diseases. Cell Res 8: 997-1006, 2008.
- Chenevix-Trench G et al. Dominant negative ATM mutations in breast cancer families. J
Nat Cancer Inst 6: 205-215, 2002.
- Chivukula M et al. Clinical importance of HER2 immunohistologic heterogeneous expression in
core-needle biopsies vs resections pecimens for equivocal (immunohistochemicalscore 2+) cases.
Mod Pathol 21: 363-68, 2008.
- Choi Y et al. PTEN, but not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces
growth inhibition and apoptosis of myeloma cells. Oncogene 21: 5289-300, 2002.
- Choo JR et al. Biomarkers for Basal-like Breast Cancer. Cancer 2: 1040-65, 2010.
- Christianson TA et al. NH2-terminally truncated HER-2/neu protein: relationship with shedding
of the extracellular domain and with prognostic factors in breast cancer. Cancer Res 58: 5123-29,
1998.
- Clark MF et al. Cancer stem cells perspective on current status and future directions: AACR
workshop on cancer stem cells. Cancer Res 66: 9339-44, 2006.
- Cnossen JA et al. Long term surviva lwith metastatic breast cancer (MBC): results of a
retrospective, single-centre analysis from 2000-2005. J Clin Oncol 26 (Suppl.): 1128, 2008.
- Codony-Servat J et al. Cleavage of the HER-2 ectodomain is a pervanadate-activable process
that is inhibited by the tissue inhibitor of metalloproteinases-1 in breast cancer cells. Cancer Res 59:
1196-201, 1999.
- Collins AT et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res
65: 10946-51, 2005.
- Cooley S et al. Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct
mechanisms of antibody dependent cellular cytotoxicity against LFA-3 and HER-2/neu. Exp
Hematol 27: 1533-41, 1999.
- Coussens L et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares
chromosomal location with neu oncogene. Science 230: 1132-39, 1985.

78
- Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 10:
704-14, 2009.
- Crown J et al. Emerging target therapies in triple-negative breast cancer. Ann Oncol 23: 56-65,
2012.
- Cserni G. Complete sectioning of axillary sentinel lymph nodes in patients with breast. Analysis
of two different step sectioning and immunohistochemistry protocols in 246 patients. J Clin Pathol
55: 926-31, 2002.
- Cserni G et al. Pathological work-up of sentinel lymph nodes in breast cancer. Review of current
data to be considered for the formulation of guidelines. Eur J Cancer 39: 1654-67, 2003.
- Cserni G et al. Discrepancies in current practice of pathological evaluation of lymph nodes in
breast cancer. Results of questionnarie-based survey by the European Working Group for breast
screening pathology. J Clin Pathol 57: 695-701, 2004.
- Cuatrecasas M et al. ATM gene espression is associated with differentiation and angiogenesis in
infiltrating breast carcinomas. Hist Histopathology 21: 149-56, 2006.
- Daniele L et al. Anti-HER2 Treatment and Breast Cancer: State of the Art, Recent Patents, and
New Strategies. Recent Patents on Anti-Cancer Drug Discovery 4: 9-18, 2009.
- D’Alessio A et al. Effects of the combined blockade of EGFR and ErbB-2 on signal transduction
and regulation of cell cycle regulatory proteins in breast cancer cells. Breast Cancer Res Treat 123:
387-89, 2010.
- Dati C et al. Inhibition of c-erbB-2 oncogene expression by estrogens in human breast cancer
cells. Oncogene 5: 1001-06, 1990.
- de Azambura E et al. Ki67 as prognostic marker in early breast cancer: a meta-analysis of
published studies involving 12,155 patients. Br J Cancer 96: 1504–13, 2007.
- Dean M et al. Tumour stem cells and drug resistance. Nature Rev Cancer 5: 275- 84, 2005.
- Dedes KJ et al. Synthetic lethality of PARP inhibition in cancer lacking BRCA1 and BRCA2
mutations. Cell Cycle 10: 1192-99, 2011.
- Diermeier S et al. Epidermal growth factor receptor coexpression modulates susceptibility to
herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and
activation. Exp Cell Res 304: 604-19, 2005
- DiGiovanna MP et al. Combinations of HER2, estrogen receptor (ER) and IGF-I receptor
(IGF1R) inhibitors induce apoptosis in breast cancer cells: Dramatic effects of HER2 inhibitors on
non-overexpressing cells. Proc Am Assoc Cancer Res 47: 1226, 2006.
- Durbecq V et al. Correlation between topoisomerase IIalpha gene amplification and protein
expression in Her-2 amplified breast cancer. Int J Oncol 25: 1473-79, 2004.

79
- Edgar R et al. Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016
(Lapatinib). Cancer Res 64: 6652-59, 2004.
- Eliassen AH et al. Adult weight change and risk of postmenopausal breast cancer. JAMA 296:
193-201, 2006.
- Elledge RM et al. Estrogen receptor (ER) and progesterone receptor (PgR), by ligand-binding
assay compared with ER, PgR and pS2, by immuno-histochemistryin predicting response to
tamoxifen in metastatic breast cancer: a Southwest Oncology Group Study. Int J Cancer 89: 111-
17, 2000.
- Engelman JA et al. The evolution of phosphatidylinositol 3-kinases as regulators of growth and
metabolism. Nat Rev Genet 7: 606-19, 2006.
- Engelman JA et al. Targeting PI3K signalling in cancer: opportunities, challenges and
limitations. Nat Rev Cancer 9: 550-62, 2009.
- Esteva FJ et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-
2 overexpressing metastatic breast cancer. J Clin Oncol 20: 1800-08, 2002.
- Eroles P et al. Molecular biology in breast cancer:Intrinsic subtypes and signaling
pathways. Cancer Treatm Rev 38: 698-707, 2012.
- Esteva FJ et al. PTEN, PIK3CA, p-AKT and p-p70S6K status: association with
trastuzumab response and survival in patients with HER2-positive metastatic breast cancer.
Am J Pathol 177: 1647-56, 2010.
- Estratto Linee Guida F.O.N.Ca.M. 2008 Cap. 5 Diagnosi
- Etkind PR et al. Clonal isolation of different strains of mouse mammary tumor virus like
DNA sequences from both the breast tumors and non-Hodgkin’s lymphomas of individual
patients diagnosed with both malignancies. Clin Cancer Res 10: 5656-64, 2004.
- Fabbri M et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by
targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci 104:15805-10, 2007.
- Fadare O et al. Clinical and pathologic aspects of basal-like breast cancers. Nat Clin Prac Oncol.
25: 149–159, 2008.
- Faedo M et al. Mouse mammary tumor-like virus is associated with p53 nuclear accumulation
and progesterone receptor positivity but not estrogen positivity in human female breast cancer. Clin
Cancer Res 10: 4417-19, 2004.
- Faivre S et al. New paradigm in anticancer therapy: targeting multiple signalling pathways with
kinase inhibitors. Sem Oncol 4: 407-20, 2006.
- Fan X et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in
embryonal brain tumors. Cancer Res 66: 7445- 52, 2006.

80
- Fang D et al. A tumorigenic subpopulation with stem cell properties in melanoma. Cancer Res
65: 9328-37, 2005.
- Farnie G et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Nocth
and epidermal growth factor receptor signalling pathway. J Natl Cancer Inst 99: 616-27, 2007.
- Faschinger A et al. Mouse mammary tumor virus integration site selection in human and mouse
genomes. J Virol 82: 1360-67, 2008.
- Fenaroli P et al. Axillary sentinel node biopsy under local anaesthesia in early breast cancer. Ann
Oncol 11: 1617-18, 2007.
- Findlay VJ et al. MicroRNAs and breast cancer. Open Cancer J 3: 55-61, 2010.
- Filho AL et al. Angiogenesis and breast cancer. J Oncol 2010: 1-7, 2010.
- Fiszman GL et al. Molecular mechanisms of trastuzumab resistance in HER2 overexpressing
breast cancer. Int J Breast Cancer 2011: 1-11, 2011.
- Ford CE et al. MMTV virus-like RNA transcripts and DNA are found in affected cells of human
breast cancer. Clin Cancer Res 10: 7284-89, 2004.
- Fournier A et al. Use of different hormone therapies and risk of histology- and hormone receptor-
defined invasive breast cancer. J Clin Oncol 26: 1260-68, 2008.
- Fulford LG et al. Basal-like grade III invasive ductal carcinoma of the breast: Patterns of
metastasis and long-term survival. Breast Cancer Res 9: R4, 2007.
- Gabriel N et al. Trastuzumab in the treatment of breast cancer. N Engl J Med 16: 1734-36, 2005.
- Gall JC et al. The formation and detections of RNA_DNA hybrid molecules in cytological
preparations. Proc Natl Acad Sci USA 63: 378-83, 1969.
- Galli R et al. Isolation and characterization of tumorigenic stem like neural precursor from human
glioblastoma. Cancer Res 64: 7011-21, 2004.
- Gasparini G et al. VEGF e angiogenesi tumorale. Le prospettive della terapia antiangiogenica
pp.1-12,19-21, 2005.
- Gaur A et al. Characterization of microRNA expression levels and their biological correlates in
human cancer cell lines. Cancer Res 67: 2456-68, 2007.
- Gelmon K et al. Targenting triple-negative breast cancer: optimising therapeutic outcomes. Ann
Oncol 23: 2223-34, 2012.
- Gennari R et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients
with primary operable breast tumors overexpressing HER2. Clin Cancer Res 10: 5650-55, 2004.
- Ghatak S et al. Hyaluronan oligosaccharides inhibit anchorage-independent growth of
tumor cells by suppressing the phosphoinositide3-kinase/Akt cell survival pathway.J Biol
Chem 277: 38013–20, 2002.

81
- Giuliano M et al. Biological mechanisms and clinical implications of endocrine resistence
in breast cancer. The Breast 20: S42-S49, 2011.
- Glinsky GV et al. Microarray analysis identifies a death-from-cancer signature predicting therapy
failure in patients with multiple types of cancer. J. Clin. Invest 115:1503–21, 2005.
- Gloecker-Ries LA et al. Survival from breast cancer according tot umor size and nodal
status. Surg Oncol Clin N Am. 3:35-53, 1994.
- Goodell MA et al. Isolation and functional properties of murine hematopoietic stem cells
that are replicating in vivo. J Exp Med Vol. 183: 1797–06, 1996
- Goldhirsch A et al. Stategies for subtypes-dealing with the diversity of breast cancer:
highlights of the St Gallen International Expert Consensus of tre primary therapy of early
breast cancer 2011. Ann of Oncology 22: 1736-47, 2011.
- Gong Y et al. Reliability of chromogenic in situ hybridization for detecting HER-2 gene status in
breast cancer: comparison with fluorescence in situ hybridization and assessment of interobserver
reproducibility. Mod Pathol 18: 1015-21, 2005.
- Gonzales-Angulo AM et al. PI3K Pathway mutations and PTEN levels in primary and metastatic
breast cancer. Mol Cancer Ther 10: 1093-101, 2011.
Gori S et al. EGFR, pMAPK, pAKT and PTEN status by immunohistochemistry: correlation with
clinical outcome in HER-2 positive metastatic breast cancer patients, treated with trastuzumab. Ann
Oncol 20: 648-54, 2009.
- Gralow JR et al. Preoperative therapy in invasive breast cancer: pathologic assessment and
systemic therapy issues in operable disease. J Clin Oncol 26: 814-19, 2008.
- Greenberg S et al. Triple negative breast cancer. Cancer Immunon, Immunother 55: 1565-74,
2010.
- Gunn S et al. Clinical array-based karyotyping of breast cancer with equivocal lHER2 status
resolves gene copy number and reveals chromosome 17 complexity. BMS Cancer 10: 396-403,
2010.
- Gupta D et al. Comparison of fluorescence and chromogenic in situ hybridization for detection of
HER-2/neu oncogene in breast cancer. Am J Clin Pathol 19: 381-87, 2003.
- Gutierrez C et al. HER2: biology, detection, and clinical implications. Arch Pat Lab Med 135:
55-62, 2011.
- Hambardzumyan D et al. PI3K pathway regulates survival of cancer stem cells residing in the
perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 22: 436–48, 2008.
- Hanna W et al. Intratumoral heterogeneity of HER2/neu in Breast Cancer-a rare event. Breast
Journal 13: 122-29, 2007.

82
- Hastak K et al. Synergistic chemosensitivity of triple-negative breast cancer cell lines to
poly(ADP-Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res 70: 7970-80,
2010.
- Hayes DF et al. Angiogenesis as targeted breast cancer therapy. Breast 16: 17-19, 2007.
- He L et al. MicroRNAs: small RNAs with big role in gene regulation. Nat Rev Genet 5: 522-31,
2004.
- Heneghan HM et al. MicroRNAs as novel biomarkers for breast cancer. J Oncol 2010: 1-7, 2009.
- Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res 65: 1891-96,
2006.
- Herschkowitz JI et al. Identification of conserved gene expression features between murine
mammary carcinoma models and human breast tumors.GenomeBiol 8:R76, 2007.
- Hicks DG et al. HER2+ breast cancer: review of biological relevance and ptimal use of diagnostic
tools. Am J Clin Pathol 129: 263-73, 2008.
- Hirschmann Jax C et al. A distinct “side population” of cells with high drug efflux capacity in
human tumor cells. Proc Natl Acad Sci USA 101: 14228-33, 2004.
- Hoff K et al. Visualization of FISH probes by dual-color Chromogenic In Situ Hybridization. Am
J Clin Pathol 133: 205-11, 2010.
- Howlader N et al. SEER Cancer Statistics Review, 1975-2008. Bethesda, MD: National Cancer
Institute 2011
- Hynes NE et al. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat Rev
Cancer 5: 341-54, 2005.
- Hyun CL et al. The effect of chromosome 17 polysomy on HER-2/neu status in breast cancer. J
Clin Pathol 61: 317-21, 2008.
- Hu Z et al. The molecular portraits of breast tumors are conserved across microarray platforms.
BMC Genomics 7: 96, 2006.
- Hudis CA et al. Triple negative breast cancer: an unmet medical need. The Oncologist 16: 1-11,
2011.
- Hulka BS et al. Breast cancer: hormones and other risk factors. Maturitas 38: 103-13, 2001.
- Iorio MV et al. MicroRNA-205 regulates HER3 in human breast cancer. Cancer Res 69: 2195-
2200, 2009.
- Iorio MV et al. Breast cancer and microRNAs: therapeutic impact. Breast 20: S63-S70, 2011.
- Isakoff SJ et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary
epithelial cells. Cancer Res 65: 10992-11000, 2005.

83
- Isola J et al. Interlaboratory comparison of HER-2 oncogene amplification as detected by
chromogenic and fluorescence in situ hybridization. Clin Canc Res 10: 4793-98, 2003.
- Izumi Y et al. Tumor biology: Herceptin acts as an anti-angiogenic cocktail. Nature 416: 279-80,
2002.
- Jackisch C. Her2 positive metastatic breast cancer: optimizing Trastuzumab based therapy. The
Oncologist 11: 34-41, 2006.
- Jacobs EJ et al. Polymorphisms in the vascular endothelial growth factor gene and breast cancer
in the Cancer Prevention Study II cohort. Breast Cancer Res 8: R22, 2006.
- Jacquemier J et al. Typical medullary breast carcinomas have a basal/myoepithe-lial phenotype.
J Pathol 207: 260-68, 2005.
Jain P et al. Breast cancer stem cells: a new challenge for breast cancer treatment. Front Bios 16:
1824-32, 2011.
- Jarvinen TAH et al. HER-2/neu and Topoisomerase IIa simultaneous drug targets in cancer.
Comb Chem High Throughput Screen 6: 79-99, 2003.
- Jeffrey SS et al. Genoma-based prognosis and therapeutic prediction in breast cancer. J Natl
Compr Canc Netw 3: 291-300, 2005.
- Jensen EV et al. Estrogen receptor and breast cancer response to adrenalectomy. Natl Cancer Inst
Monogr 34: 55-70, 1971.
- Jensen EV et al. Estrogen receptor and proliferation markers in primary and recurrent breast
cancer. Proc Natl Acad Sci USA 98: 15197-202, 2001.
- Jensen EV et al. The estrogen receptor: a model for molecular medicine. Clin Cancer Res 9:
1980, 2003.
- Johal H et al. .Mouse mammary tumour virus-like virus (MMTV-LV) is present with in the liver
in a wide range of hepatic disorders and unrelated to nuclear p53 expression or
hepatocarcinogenesis. J Hepatol 50: 548–54, 2009.
- Jones KL et al. Evolving novel anti-HER2 strategies. Lancet Oncol 10: 1179-87, 2010.
- JunttilaTT et al. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of
trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res
Treat 128: 347–56, 2010.
- Kang YJ et al. Tissue microarray analysis of hepatocyte growth factor/Met pathway components
reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the
progression of node-negative breast cancer. Cancer Res 63: 1101–05, 2003.
- Kauraniemi P et al. Effects of Herceptin treatment on global gene expression patterns in
HER2-amplified and nonamplified breast cancer cell lines. Oncogene 23: 1010-13, 2004.

84
- Kei TJ et al. Epidemiology of breast cancer. Lancet Oncol 2: 133-40, 2001.
- Kim MJ et al. Clinicopathologic significance of the basal-like subtype of breast cancer: a
comparison with hormone receptor and Her2/neu –overexpressing phenotypes. Hum Pathol 37:
1217-26, 2006.
- Kellner U et al. Culprit and victim DNA topoisomerase II. Lancet Oncol 3: 235-43, 2002.
- Kelsey JL, et al. Reproductive factors and breast cancer. Epidemiol Rev. 15: 36-47, 1993.
- Kerbel RS. “A cancer therapy resistant to resistance.” Nature 390: 335-36, 1997.
- King MC et al. New York Breast Cancer Study Group. Breast and ovarian cancers risks due to
inherited mutations in BRCA1 and BRCA2. Science 302: 643-46, 2003.
- Klos KS et al. Combined trastuzumab and paclitaxel treatment better inhibits ErbB-2-mediated
angiogenesis in breast carcinoma through a more effective inhibition of akt than either treatment
alone. Cancer 98: 1377-85, 2003.
- Knudsen KE. The cyclin D1b splice variant: an old oncogene learns new tricks. Cell Div 1:15,
2006.
- Koene HR et al. Fc γRIIIa-158V/Fpolymorphism influences the binding of IgG by natural killer
cell Fc γRIIIa, independently of the FcγRIIIa-48L/R/H phenotype. Blood: 90: 1109–14, 1997.
- Köhler G et al. Continuous cultures of fused cells secreting antibody of predefined specificity.
Nature 256: 495-97, 1975.
- Korkaya H et al. Regulation of mammary stem/progenitor cells by PTEN/AKT/beta-catenin
signaling. PLoS Biol 7: 139-47, 2007.
- Korkaya H et al. HER2 regulates the mammary stem/progenitors cell population driving
tumorigenesis and invasion. Oncogene 27: 6120-30, 2009.
- Kota J et al. herapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer
model. Cell 137: 1005-17, 2009.
- Kraus WL et al. Cloning of the rat progesterone receptor gene 5’-region and identification of two
functionally distinct promoters.Mol Endocrinol 7: 1603-16, 1993.
- Krus V et al. PARP-inhibitors in breast and ovarian cancer. Belg J Med Oncol 5: 62-69, 2011.
- Kubo M et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast
cancer. Cancer Res 64: 6071-74, 2004.
- KumarV, Cotran RS, Robbins LS. Cap. 19 Anatomia Patologica 7° edizione EMSI
Edizioni Mediche Scientifiche Internazionali-Roma 2005.
- Kuusisto KM et al. Screening for BRCA1, BRCA2, CHEk2, PALB2, BRIP1, RDA50 e
CDH1 mutations in high-risk Finnish BRCA1/2 –founder mutation negative breast and/or
ovarian cancer individuals. Breast Cancer Res 13: R20, 2011.

85
- Laakso M et al. Dual-colour chromogenic in situ hybridization for testing of HER-2 oncogene
amplification in archival breast tumours. J Pathol 210: 3-9, 2006.
- Lakhani SR et al. The pathology of familial breast cancer: histological features of cancers in
families not attributable to mutations in BRCA1 and BRCA2. Clin Cancer Res 6: 782-89, 2000.
- Lakhani SR et al. The pathology of familial breast cancer: predictive value of immu-
nohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with
mutations in BRCA1 and BRCA2. J Clin Oncol 20: 2310-18, 2002.
- Lane HA et al. ErbB2 potentiates breast tumor proliferation through modulation of p27(Kip 1)-
Cdk2 complex formation: receptor overexpression does not determine growth dependency. Mol cell
Biol 20: 3210-23, 2000.
- Lane HA et al. Modulation of p27(Kip 1)-Cdk2 complex formation through 4D5 mediated
inhibition of HER2 receptor signalling. Ann oncol 12: 21-22, 2001.
- Laughner E et al. HER2 (neu) signaling increase the rate of hypoxia-inducibile factor 1 alpha
(HIF-1 alpha) synthesis: novel mechanism for HIF-1-mediated vascular endotelial growth factor
expression. Mol Cell Biol 21: 3995-4004, 2001.
- Lawson JC et al. Cancer stem cells in breast cancer and metastasis. Breast Cancer Res Treatm
118: 241-54, 2009.
- Lawson JS et al. Breast Cancer like Sequences in Human − Mouse mammary tumor virus.
Cancer Res 70: 3576-85, 2010.
- Le XF et al. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-
induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem 278: 23441-50, 2003.
- Lee S et al. evaluation of intratumoral HER-2 Heterogeneity by Fluorescence In Situ
Hybridization in invasive breast cancer: A single insitution study. J Korean Med Sci 26: 1001-06,
2011.
- Leighl N et al. A Phase 2 study of perifosine in advanced or metastatic breast cancer. Breast
Cancer Res Treat 108: 87–92, 2008.
- Le Tourneau C et al. New development in multitarget therapy for patients with solid tumours.
Cancer Treatm Rev 34: 37-48, 2008.
- Levine PH et al. Increased detection of breast cancer virus sequences in inflammatory breast
cancer. Adv Tumor Virol 1: 3-7, 2009.
- Lewis JT et al. Analysis o fintramural heterogeneity and amplification status in breast carcinomas
with quivocal (2+ ) Her2 immunostaining. Am J Clin Pathol 124: 273-81, 2005.
- Lewis Phillips GD et al. HER-2 positive breast cancer with trastuzumab-DM1, an antibody
cytotoxic drug conjugate. Cancer Res 68: 9280-90, 2008.

86
- Li CI et al. Risk of invasive breast carcinoma among women diagnosed with ductal carcinoma in
situ and lobular carcinoma in situ, 1988-2001. Cancer 106: 1197, 1995.
- LI C et al. Identification of pancreatic cancer stem cells. Cancer Res 67: 1030-37, 2007.
- Li J et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast,
and prostate cancer. Science 275: 1943-47, 1997.
- Linderholm BK et al. Signifcantly higher levels of vascular endothelial growth factor (VEGF)
and shorter survival times for patients with primary operable triple-negative breast cancer. Ann
Oncol 10: 1639-46, 2009.
- Liu S et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human
mammary stem cells. Cancer Res 66: 6063–71, 2006.
- Liu S et al. Progesterone receptor is a significant factor associated with clinical outcomes and
effect of adjuvant tamoxifen therapy in breast cancer patients. Breast Cancer Res Treat 119: 53-61,
2010.
- Liu S et al. Targeting breast cancer stem cells. J Clin Oncol 28: 4006-12, 2010.
- Lu J et al. MicroRNA expression profiles classify human cancers. Nature 435 : 834–838, 2005.
- Lu Y et al. The PTEN/MMACA1/TEP tumor suppressor gene decreases cell growth and induces
apoptosis and anoikis in breast cancer cells. Oncogene 18: 7034-45, 1999.
- Lu Y et al. Insulin-like growth factor-I receptor signalling and resistance to trastuzumab
(Herceptin) J Natl Cancer Inst 93: 1852–57, 2001.
- Lumachi F et al. I tumori della mammella. Unipress Padova, 8-10, 1997.
- Mabry H et al. Sentinel node mapping for breast cancer: progress to date and prospects for the
future. Surg Oncol Clin N Am 16: 55, 2007.
- Magnifico A et al. Tumor-initiating cells of HER2 positive carcinoma cell lines express the
highest oncoprotein levels and are sensitive to trastuzumab. Clin Cancer Res 15: 2010-21, 2009.
- Maggie CU et al. Basal-like breast cancer defined by five biomarkers has superior prognostic
value than triple-negative phenotype. Clin Cancer Res 14: 1368-76, 2008.
- Mann S et al. Estrogen receptor beta expression in invasive breast cancer. Human Pathol 32: 113-
18, 2001.
- Marchetti P et al. Prognostic value of p53 molecular status in high-risk primary breast cancer.
Ann Oncol 14: 704-08, 2003.
- Marchiò C et al. Molecular diagnosis in breast cancer. Diagn Histopath 14: 202-13, 2008.
- Marchiò C et al. Does chromosome17 centromere copy number predict polysomy in breast
cancer? A fluorescence in situ hybridization and microarray-based CGH analysis. J Pathol 219: 16-
24, 2009.

87
- MartY B et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase
pathway in basal-like breast cancer cells. Breast Cancer Res 10: R101, 2008
- Mauri D et al. Multiple-treatments meta-analysis of chemotherapy and targeted therapies in
advanced breast cancer. J Natl Cancer Inst 100: 1780-91, 2008.
- Mayfield S et al. DNA strand breaks and cell cycle perturbation in Herceptin treated breast
cancer cell lines. Breast Cancer Res Treatm 70: 123-29, 2001.
- Melana SM et al. Characterization of viral particles isolated from primary cultures of
human breast cancer cells. Cancer Res 67: 8960-65, 2007.
- Mendes-Pereira AM et al. Synthetic lethal targeting of PTEN mutant cells with PARP
inhibitors. EMBO Mol Med 1: 315-22, 2009.
- Meyn RE. Linking PTEN with genomic instability and DNA repair. Cell Cycle 8: 2198-
210, 2009.
- Mitchell PS et al. Circulating microRNAs as stable blood-based markers for cancer
detection. Proc Natl Acad Sci 105: 10513-18, 2008.
- Minucci S et al. Hystone deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer 6: 38-51, 2006.
- Mohd Sharial MSN et al. Overcoming resistence and restoring sensitivity to HER-2 targeted
therapies in breast cancer. Ann Oncol 0: 1-12, 2012.
- Molina MA et al. NH(2)-terminal truncated HER-2 protein but not full-lenght receptor is
associated with nodal metastasis in human breast cancer. Clin Cancer Res 8: 347-53, 2002.
- Mollerup J et al. Dual color chromogenic in situ hybridization for determination of HER2 status
in breast cancer: a large comparative study to current state of the art fluorescence in situ
hybridization. BMC Clin Pathol 12: 3, 2012.
- Moskwa P et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and
sensitivity to PARP inhibitors. Mol Cell 41:210-20, 2011.
- Mosselman S et al. ER beta: identification and characterization of novel human oestrogen
receptor. FEBS Lett 392: 49-53, 1996.
- Moulder SL et al. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839
(Iressa) inhibits HER2/neu (erbB2)- overexpressing breast cancer cell in vitro and in vivo. Cancer
Res 61: 8887-95, 2001.
- Moy B et al. Lapatinib: Current Status and Future Directions in Breast Cancer. The Oncologist
11: 1047,57, 2006.
- Munteanu D et al. Flowcytometric evidence of DNA ploidy in human breast cancer. J Prev Med
12: 59-65, 2004.

88
- Mutter GL et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial
precancers. J Natl Cancer Inst 92: 924-30, 2000.
- Nagata Y et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of
PTEN predicts trastuzumab resistance in patients. Cancer Cell 6: 117-27, 2004.
- Nagy P et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1,a herceptin-
resistant, MUC4-expressing breast cancer cell line. Cancer Res 65: 473–82, 2005.
- Nakanishi T et al. Side population cells in luminal type breast cancer have tumor-initiating cell
propiertes, and are regulated by HER2 expression and signalling. Br J Cancer 102: 815-26, 2010.
- Nakopoulou L et al. Aneuploidy of chromosome 20 in invasive breast cancer correlates with
poor outcome. Cancer Genet Cytogenet 134: 127-32, 2002.
- Nakopoulou L et al. Study of phospho-beta-catenin subcellular distribution in invasive breast
carcinomas in relation to their phenotype and the clinical outcome. Mod Pathol 19: 556–63, 2006.
-Nakshatri M et al. Breast cancer stem cells and intrinsic subtypes: controversies rage on. Current
Stem Cell Res&Therapy 4: 50-60, 2009.
- Natha R et al. P27(kip1) down-regulation is associated with Trastuzumab resistance in breast
cancer cells. Cancer Res 64: 3981-86, 2003.
- Natha R et al. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2
heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 65:
11118-28, 2005.
- Natrajan R et al. An integrative genomic and transcriptomic analysis reveals molecular pathways
and networks regulated by copy number aberrations in basal-like HER2 and luminal cancers. Breast
Cancer Res Treatm 121: 575-89, 2010.
- Neuman E et al. Cyclin D1 stimulation of estro-gen receptor transcriptional activity independent
of cdk4. Mol Cell Biol 17: 5338-47, 1997.
- Newman LA et al. Meta-analysis of survival in African American and white American patients
with breast cancer: ethnicity compared with socioeconomic status. J Clin Oncol 24: 1342-49, 2006.
- Nichols HB et al. Body mass index before and after breast cancer diagnosis: associations with all-
cause, breast cancer, and cardiovascular disease mortality. Cancer Epidemiol Biomarkers Prev18:
1403-09, 2009.
- Nicholson RJ et al. EGFR and cancer prognosis. Eur J Cancer 37: 9-15, 2001.
- Nielsen TO et al. Immunohistochemical and clinical characterization of the basal-like subtype of
invasive breas tcarcinoma. Clin Cancer Res 10: 5367–74, 2004.
- Nielsen DL et al. Antiangiogenic therapy for breast cancer. Breast Cancer Res 12: 209-24, 2010.

89
- Nitta H et al. Development of automated brightfield double in Situ hybridization (BDISH)
application for HER2 gene and chromosome17 centromere (CEN17) for breast carcinomas and an
assay performance comparison to manual dual color HER2 fluorescence in Situ hybridization
(FISH). Diagn Pathol 3: 41-52, 2008.
- Normanno N et al. Target-based therapies in breast cancer: current status and future perspective.
End Rel Cancer 16: 675-702, 2009.
- Nuss R et al. Many tumors induced by the mouse mammary tumor virus contain a provirus
integrated in the same region of the host genome. Cell 31: 99-109, 1982.
- Oakman C et al. Chemotherapy with or without trastuzumab. Ann of Oncology 21: 112-19, 2010.
- O’Brien CA et al. A human colon cancer cell capable of initiating tumor growth in
immunodeficient mice. Nature 445: 106-10, 2007.
- Omoto Y et al. Clinical value of the wild-type estrogen receptor beta expression in breast cancer.
Cancer Lett 163: 207-12, 2001.
- O’Regan R et al. RAD001 (everolimus) in combination with weekly paclitaxel and trastuzumab
in patients with HER-2-overexpressing metastatic breast cancer with prior resistance to
trastuzumab:a multicenter phase I clinical trial. Cancer Res 69: 3119, 2009.
- Orlandi R et al. Cloning immunoglobulin variable domains for expression by the polymerase
chain reaction. Proc Natl Acad Sci USA 86: 3833-37, 1989.
- O'Shaughnessy J, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N
Engl J Med 364: 205-14, 2011.
- Owen MA et al. HER2 amplification ratios by fluorescence in situ hybridization and correlation
with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin Breast Cancer 5: 63-69,
2004.
- Paez JG et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib
therapy. Science 304: 1497-500, 2004.
- Paik S et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast
cancer. N Engl J Med 351: 2817–26, 2004.
- Pal SK et al. Triple negative breast cancer: novel therapy and new directions. Maturitas 63: 269-
74, 2009.
- Palyi-Krekk Z et al. Hyaluronan-induced masking of ErbB2 and CD44 enhanced
trastuzumab internalisation in trastuzumab resistant breast cancer.Eur J Cancer 43: 2423–33,
2007.

90
- Pandini G et al. Insulin and insulin-like growth factor (IGF-I) receptor overexpression in
breast cancer, leads to insulin/IGF-I hybrid receptor overespression.: evidence for a second
mechanism of IGF-I signaling. Clin Cancer Res 5: 1935-44, 1999.
- Pandolfi PP. Breast cancer-loss of PTEN predict resistance to treatment. N Engl J Med 351:
2337-38, 2004.
- Papouchado BG et al. “Silver in situ hybridization (SISH) for determination of HER2 gene status
in breast carcinoma: comparison with fish and assessment of interobserver reproducibility. Am J
Surg Pathol 34: 767-76, 2010.
- Parson R et al. PTEN and cancer. Methods Mol Biol 222: 147-66, 2003.
- Pavao M et al. Estrogen receptor antibodies: specificity and utility in detection, localization and
analysis of estrogen receptor alpha and beta. Steroids 66: 1-16, 2001.
- Penault-Llorca F et al. Emerging technologies for assessing HER2 amplification. Am J Clin
Pathol 132: 539-48, 2009.
- Perou CM et al. Molecular portraits of human breast tumors. Nature 406: 747-52, 2000.
- Perou CM. Molecular stratification of triple-negative breast cancer. Oncologist 16 Suppl 1: 61-
70, 2011,
- Persons DL et al. Chromosome-specific aneusomy in carcinoma of the breast. Clin Cancer Res
2: 883-88, 1996.
- Pietribiasi F et al. Protocol for diagnostic assessment of sentinel lymph nodo in breast pathology:
a proposal of SIAPEC-IAP, Piemonte Region Italy. Pathologica 98: 167-70, 2007.
- Piovan C et al. Oncosuppressive role of p53-induced miR-205 in triple negative breast cancer.
Mol Oncol 6: 458-72, 2012.
- Piccirillo, SG et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain
tumour!initiating cells. Nature 444: 761–65, 2006.
- Pogo BG et al. Sequences homologous to the MMTV env gene in human breast carcinoma
correlate with overexpression of laminin receptor. Clin Cancer Res 5: 2108-11. 1999.
- Pogo BG et al. Human Mammary Tumor Virus in inflammatory breast cancer. Cancer 116: 2741-
44, 2010.
- Pohlmann PR et al. Resistance to Trastuzumab in Breast Cancer. Clin Cancer Res 15: 7479-91,
2009.
- Prat A et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of
breast cancer. Breast Cancer Res 12: R68, 2010.
- Preston DL el al. Radiation effects on breast cancer risk: a pooled analysis of eight cohorts.
Radiat Res 158: 220-35, 2002.

91
- Racila E et al. Detection and characterization of carcinoma cells in the blood. Proc Natl
Acad Sci USA 95: 4589-94, 1998.
- Rahman N et al. Absence of evidence for a familial breast cancer susceptibility gene at
chromosome 8p12-p22. Oncogene 19: 4170-73, 2000.
- Rakha EA et al. Basal-like breast cancer: a critical review. J Clin Oncol 26: 2568-81, 2008.
- Rakha EA et al Breast cancer prognostic classification in the molecular era: the role of
histological grade. Breast Cancer Res 12: 207-18, 2010.
- Reis-Filho JS et al. The diagnosis and management of pre-invasive breast disease: genetic
alterations in pre-invasive lesions. Breast Cancer Res 5: 313-19, 2003.
- Ramawi MF et al. EGFR expression in breast cancer association with biological phenotype and
clinical outcomes. Cancer 116: 1234-42, 2010.
- Reis-Filho JS et al. Distribution and significance of nerve growth factor receptor
(NGFR/p75NTR) in normal, benign and malignant breast tissue. Mod Pathol 19:307-19, 2006.
- Relf M et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic
and basic fbroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell
growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its
relation to angiogenesis. Cancer Res 57: 963-69, 1997.
- Reya T et al. Stem cells, cancer, and cancer stem cells. Nature 414: 105-11, 2001.
- Rice S et al. Phytoestrogens and breast cancer-promoters or protectors? End Rel Cancer 13: 995-
1005, 2006.
- Ricevuto E et al. Can analysis of the molecular status of the p53 gene contribute to improving the
therapeutic stategy for breast carcinoma? Tumori 89: 197-99, 2003.
- Rodier F et al. Two faces of p53: aging and tumor suppression. Nucleic Acid Res 35: 7475-82,
2007.
- Romond EH et al. Trastuzumab plus Adjiuvant Chemoterapy for operable Her2 positive breast
cancer. N Engl J Med 353: 1673-84, 2005.
- Rosembreg LU et al. Menopausal hormone therapy and other breast cancer risk factor in
relation of risk of different histological subtypes of breast cancer: a case control study. Breast
Cancer Research 8: R11, 2006.
- Rosenfeld N et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 26:
462-69, 2008.
- Ross JS et al. Molecular Oncology of Breast Cancer Jones and Bartlett Publishers, Sudsbury,
Massachusetts. 2005.

92
- Ross JS et al. The HER-2/neu oncogene in breast cancer. Prognostic factor, predictive
factor, and target for therapy. Stem Cell 16: 413-28, 1998.
- Rouzier R et al. Breast cancer molecular subtypes respond differently to preoperative
chemotherapy. Clin Cancer Res 11: 5678-85, 2005.
- Ruco L et al. Anatomia Patologica Le Basi Cap. 9 Utet 2007.
- Rusnak DW et al. The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase
inhibitors: potential therapy for cancer. Cancer Res 61: 7196–203, 2001.
- Sabate JM et al. Evaluation of breast involvement in relation of Cowden syndrome; a
radiological and clinicopathological study of patients with PTEN germ-line mutations. Eur
Radiol 16: 702-06, 2006.
- Saez R et al. p95HER-2 predicts worse outcome in patients with HER-2-positive breast
cancer. Clin Cancer Res 12: 424–31, 2006.
- Sakakibara S et al. Regulation of angiogenesis in malignancies associated with Epstein-Barr
virus and Kaposi’s sarcoma-associated herpes virus. Fut Microb 4: 903-17, 2009.
- Sandhu R et al. Microarray based gene expression profiling for molecular classification of breast
cancer and identification of new thargets for therapy. Lab Med 41: 364-72, 2010.
- Sash S et al. Testing for HER2 in Breast Cancer: A Continuing Evolution. Path Res Int 22: 1-16,
2010.
- Sauter G et al. Guidelines for human epidermal growth factor receptor2 testing: biologic and
methodologic considerations. J Clin Oncol 27: 1323-33, 2009.
- Scaltriti M et al. Expression of p95HER-2, a truncated form of the HER-2 receptor, and response
to anti-HER-2 therapies in breast cancer. J Natl Cancer Inst 99: 628-38, 2007.
- Scaltriti M et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and
accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 28:
803–14, 2009.
- Scaltriti M et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal
growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin
Cancer Res 16: 2688-95, 2010.
- Schnitt SJ et al. Classification and prognosis of invasive breast cancer: from morphology to
molecular taxonomy. Mod Pathol 23: S60-64, 2010.
- Seth RM et al. Analysis of the HER2/neu gene amplification in microdissected breast
cancer tumour samples. Anticancer Res 26(2A): 927-31, 2006.
- Shattuck DL et al. Met receptor contributes to trastuzumab resistance of Her2-overexpressing

93
- Shields RL et al. High resolution mapping of the binding site on human IgG1 for Fc γRI, Fc γRII,
Fc γRIII, FcRn and design of IgG1variants with improved binding to theFc γ R .J Biol Chem 276:
6591–604, 2001.
- Shipitisin M et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 11: 259, 2007.
- Shivakumar S et al. Evaluation of serum vascular endothelial growth factor (VEGF) and
microvessel density (MVD) as prognostic indicators in carcinoma breast. J Cancer Res Clin Oncol
135: 627-36, 2009.
- Schreiber V et al. Poly(ADP-ribose): novel functions for old molecule. Nat Rev Mol Cell Biol 7:
517-28, 2006.
- Skliris GP et al. Immunohistochemical detection of ERbeta in breast cancer: towards more
detailed receptor profiling. Br J Cancer 84: 1095-98, 2001.
- Singletary KW et al. Alcohol and breast cancer: review of epidemiologic and experimental
evidence and potential mechanisms. JAMA 86: 2143-51, 2001.
- Silvestrini R. Relevance of DNA-ploidy as a prognostic instrument for solid tumors. Ann Oncol:
259-61, 2000.
- Singletary SE. Rating the risk factors for breast cancer. Ann Surg 237: 474-82, 2003.
- Slamon DJ et al. Human breast cancer: correlation of relapse and survival with amplification of
the HER-2/neu oncogene. Science. 235: 177-82, 1987.
- Slamon DJ et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer.
Science 244: 707-12, 1989.
- Slamon DJ et al. Use of chemoteraphy plus a monoclonal antibody against HER2 for
metastatic breast cancer that overespress HER2. N Engl Journal Med 344: 783-92, 2001.
- Sliwkowsky MX et al. Nonclinical studies addressing the mechanism of action of trastuzumab
(Herceptin). Semin Oncol 26: 60-70, 1999.
- Sorlie T et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with
clinical implications. Proc Natl Acad Sci U S A 98: 10869–77, 2001.
- Sorlie T et al. Distinct molecular mechanisms underlying clinically relevant subtypes of breast
cancer: gene expression analyses across three different platforms. BBMC Genomics 26: 127, 2006.
- Sotiriou C et al. Breast cancer classificationand prognosis based on gene expression profiles from
a population-based study. Proc Natl Acad Sci USA 100: 10393-98, 2003.
- Speirs V et al. Increased expression of estrogen receptor beta mRNA in tamoxifen-resistant breast
cancer patients. Cancer Res 59: 5421-24, 1999.
- Speirs V et al. Prognostic signifcance of oestrogen receptor beta in breast cancer. Br J Cancer 87:
405-09, 2008.

94
- Stambolic V et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor
suppressor PTEN. Cell 95: 29-39, 1998.
- Steck PA et al. Identification of a candidate tumor suppressor gene, MMAC1, at chromosome
10q23.3 that is mutated in multiple advanced cancers. Nat Genetics 15: 356-62, 1997.
- Stewart TH et al. Breast cancer incidence highest in the range of one species of house mouse,
Mus domesticus. Br J Cancer 82: 446-51, 2000.
- Sugiyama T et al. Signal transduction involving the Dmp1 transcription factor and its alteration
in human cancer. Clin Med Oncology 2: 209-19, 2008.
- Szabo S et al. Of mice, cats, men: is human breast cancer a zoonosis? Microsc Res Tech 68: 197-
208, 2005.
- Tamaki Y et al. Molecular detection of lymph node metastases in breast cancer patients: results
of multicentric trial using One-Step Nucleic Acid Amplification Assay. Clin Cancer Res 15: 2879-
84, 2009.
- Tan BK et al. Clinical validation of a customized multiple signature microarray for breast cancer.
Clin Cancer Res 15: 461-69, 2008.
- Taneja P et al. Classical and novel prognostic markers for breast cancer and their clinical
signifcance. Oncology 4: 15-34, 2010.
- Tanner A et al. A practical alternative for Fluorescence in Situ Hybridization to detect HER-
2/neu oncogene amplification in archival breast cancer samples. Am J Pathol 157: 1467-72, 2000.
- Tapia C et al. HER2 gene status in primary breast cancers and matched distant metastases. Breast
Cancer Res 9: R31-R38, 2007.
- Teneja P et al. Classical and novel prognostic Markers for breast cancer and their clinical
signifcance. Oncology 4: 15-34, 2010.
- Thull DL et al. Recognition and management of hereditary breast cancer syndromes. Oncologist
9: 13-24, 2004.
- Tian H et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic
carcinogenesis. Proc Natl Acad Sci USA 106: 4254-59, 2009.
Toft DJ et al. Minireview: Basal-like breast cancer: from molecular profiles to target therapies. Mol
Endocrinol 25: 199-211, 2011.
- Tsujimoto M et al. One-step nucleic acid amplification for intraoperative detection of lymph
node metastasis in breast cancer patients. Clin Cancer Res 15: 4807-16, 2007.
- Tsang RY et al. Beyond trastuzumab: novel therapeutic stategies in HER2-positive metastatic
breast cancer. Br J Cancer 106: 6-13, 2012.

95
- Tubbs RR et al. Discrepancies in clinical laboratory testing of eligibility for trastuzumab therapy:
apparent immunohistochemical false-positive do not get the message. J Clin Oncol 15: 2714-21,
2001.
- Tubbs R et al. Gold-facilitated in situ hybridization: a bright-field autometallographic alternative
to fluorescence in situ hybridization for detection of HER-2/neu gene amplification. Am J Pathol
160: 1589-95, 202.
- Tubbs R et al. Novel bright field molecular morphology methods for detection of HER2 gene
amplification. J Mol Histol 35: 589-94, 2004.
- Turner N et al. Hallmarks of BRCAness in sporaidc cancer. Nat Rev Cancer 4: 814-19, 2004.
- Turner NC et al. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 25: 5846-53,
2006.
- Turner NC et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 26: 2126-
32; 2007.
-Turner RR et al. Nodal stage classification for breast carcinoma: improving interobserver
reproducibility through standardized histological criteria and image-based training. J Clin Oncol 26:
258-63, 2008.
- Tutt A et al. Can genetic testing guide treatment in breast cancer. Eur J Cancer 44: 2774-80,
2008.
- Underhill C et al. A review of PARP inhibitors: form bench to bedside. Ann Oncol 22: 268-79,
2011
- Valabrega G et al. Trastuzumab: mechanism of action, resistance and future perspectives in
HER-2 overexpressing breast cancer. Ann Oncol 18: 977-84, 2007.
-Vance GH et al. Genetic heterogeneity in HER2 testing in breast cancer. Arch Pathol Lab Med
133: 611-12, 2009.
- Van der Auwera I et al. Increased angiogenesis and lymphangiogenesis in infammatory versus
non infammatory breast cancer by real-time reverse transcriptase-PCR gene expression
quantification. Clin Cancer Res 10: 7965-71, 2004.
- Vanden Bemp I et al. Polysomy 17 in breast cancer: clinico pathologic significance and impact. J
Clin Oncol 26: 4869-74, 2008.
- Van Ummersen L et al. A phase I trial of perifosine (NSC 639966) on a loading
dose/maintenancedose schedule in patients with advanced cancer. Clin Cancer Res 10: 7450-56.
2004.
- Veeck J et al. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated
with unfavourable prognosis. Oncogene 25: 3479-88, 2006.

96
- Viale G et al. Prognostic and predictive value of centrally reviewed expression of estrogen and
pro-gesterone receptors in arandomized trial comparing letrozole and tamoxifen adjuvant therapy
for postmenopausal early breast cancer: BIG1-98. J Oncol 25: 3846-52, 2007.
- Viale G et al. Predictive value of tumor Ki67 expression in two randomized trials of adjuvant
chemoendocrine therapy for node-negative breast cancer. J Natl Cancer Inst 100: 207–12, 2008.
- Volinia S et al. Breast cancer signatures for invasiveness and prognosis defined by deep
sequencing of microRNA. Prot Natl Acad Sci USA 21: 3024-29, 2012.
- Walsh T et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2 and TP53 in families at high
risk of breast cancer. JAMA 295: 1379-88, 2006.
- Wang L et al. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib.
BMC Cancer 11: 248, 2011.
- Wang S et al. Laboratory assessment of the status of HER-2/neu protein and ocnogene in the
breast cancer specimens: comparasion of immunohistochemistry assay with fluorescence in situ
hybridization assays. J Clin Pathol 53: 374-81, 2000.
- Wang XZ et al. The ups and downs of DNA repair biomarkers for PARP inhibitor therapies. Am
J Cancer Res 1: 301-27, 2011.
- Wang Y et al. Presence of MMTV-like env gene squences in gestational breast cancer. Med
Oncol.20: 233-36, 2003.
- Weigel MT et al. Current and emerging biomarkers in breast cancer: prognosis and
prediction. End Rel Cancer 17: R245-62, 2010.
- Weill KM et al. PARP inhibitor treatment in ovarian and breast carcinoma. Curr Probl
Cancer 35: 7-50, 2011.
- Wellings SR et al. On the origin and progression of ductal carcinoma in the human breast. J
Natl Cancer Inst 50: 1111-18, 1973.
- Wellings SR et al. An atlas of subgross pathology of the human breast with special
reference to possible precancerosu lesions. J Natl Cancer Inst 55: 231-73, 1975.
- Wieczorek M et al. Silencing of Wnt-1 by siRNA induces apoptosis of MCF7 human breast
cancer cells. Cancer Biol Ther 7: 268–74, 2008.
- Willet WC. Recent findings regarding lifestyle risk factors in the epidemiology of breast and
colon cancer. Philadelphia, J.B. Lippincott Co 164-177, 1993.
- Wolff AC et al. American Society of Clinical Oncology/College of American Pathologists
guideline Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast
cancer. J Clin Oncology 25: 118-45, 2007.

97
- Wood ER et al. A unique structure for epidermal growth factor receptor bound to GW572016
(Lapatinib): relationships among protein conformation inhibitor off-rate, and receptor activity in
tumor cells. Cancer Res 64: 6652-59, 2004.
- Wooster R et al. Breast and ovarian cancer. N Engl J Med 348: 2339-47, 2003.
- Xi Y et al. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and
formalin-fixed paraffin-embedded samples. RNA, 13: 1668–74, 2007.
- Xia W et al. Antitumor activity of GW572016: A dual tyrosine kinase inhibitor blocks EGF
activation of EGFR/erbB2 and downstream Erk 1/2 and Akt pathways. Oncogene 21: 6255-63,
2002.
- Xia W et al. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through
heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase
inhibitor GW572016. Oncogene 23: 646–53, 2004.
- Xia W et al. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2
tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpress-
ing breast cancer cells. Oncogene 24: 6213-21, 2005.
- Xia W et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a
therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA 103: 7795-800,
2006.
- Yamauchi H et al. Targeted therapy in inflammatory breast cancer. Cancer 116: 2758-59, 2010.
- Yee D. The insuline-like growth factor system as treatment target in breast cancer. Semin Oncol
29: 86-95, 2002.
- Yie IT et al. Clinical validation of an array CGH test for HER2 status in breast cancer reveals that
polisomy 17is a rare event. Modern Pathol 22: 1169-75, 2009.
- You L et al. An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant melanoma cells
and inhibits tumor growth. Cancer Res. 6: 5385–89, 2004.
- Yurushalmi R et al. Ki67 in breast cancer: prognostic and predictive potential. Lancet Oncology
11: 174-83, 2010.
- Zhou Y et al. Blockade of EGFR and ErbB2 by novel dual EGFR and ErbB2 tyrosine kinase
inhibitor GW572016 sensitizes human colon carcinoma GEO cells to Apoptosis. Cancer Res 66:
404-11, 2006.

















