C. I. Di Patologia e Fisiopatologia Generalijavadevil.altervista.org/sbob/3a1sC/patologia/Patologia...
105
C. I. Di Patologia e Fisiopatologia Generali a. a. 2013-2014 Prof.ssa Teti 1
Transcript of C. I. Di Patologia e Fisiopatologia Generalijavadevil.altervista.org/sbob/3a1sC/patologia/Patologia...

C. I. Di Patologia e Fisiopatologia Generali
a. a. 2013-2014
Prof.ssa Teti
1

09/10/2013 Prof.ssa Teti
1Introduzione allaIntroduzione alla
Patologia GeneralePatologia GeneraleLa patologia generale studia “la malattia” e non “le malattie”. La malattia nel senso di una condizione che può avere riscontri diversi, ovviamente, su diversi organi o apparati in una condizione fisiopatologica. Quindi capire le cause, i meccanismi, il come e il perché si instaura la malattia. Quindi gli studi relativi ai sintomi sono attinenti ad altre discipline che studiano i “malati”, noi, invece, studiamo la “malattia”. Ora dobbiamo capire cosa sono i “fenomeni biologici”. I fenomeni biologici sono intesi, in base ad una visione teleonomica della medicina, nella loro integrità, nel loro coinvolgimento dell’intera persona non di un organo singolo o di un apparato, non solo dal punto di vista fisico, ma anche mentale, spirituale, psicologico e di considerarli come il risultato della risposta che ogni persona riesce a dare agli eventi che incidono sulla sua salute. Quindi i
fenomeni biologici, sia che siano fisiologici sia che siano patologici, sono relativi alla cosiddetta qualità del coping che può avere diversi valori alto, medio e basso, che è la capacità maggiore o minore di rispondere agli eventi della vita. Quindi al modo con cui vengono affrontati gli eventi della vita sia all’interno che all’esterno. La malattia, come del resto anche la salute, è limitata da un lato dalla condizione che noi abbiamo di rispondere a questi eventi biologici, dall’altro dai limiti che noi abbiamo di rispondere. Se avessimo possibilità illimitate di rispondere ci sarebbe la malattia. Quindi la malattia è condizionata da un lato dai limiti di risposta, dall’altro dalla capacità di rispondere adeguatamente a questi stimoli e quindi di realizzare noi stessi la nostra condizione di salute.Dobbiamo rifarci al concetto di salute. Diamo diverse definizioni di salute. La condizione di salute è il risultato di quel
2

lavoro di costruzione di quei fattori che riescono a proteggere la persona da questi eventi dannosi. E questi fattori di protezione non riguardano solo una condizione di tipo fisico, ma anche mentale, spirituale ecc… Difatti la medicina teleonomica, quella attuale, non si preoccupa soltanto di valutare le patologie a livello degli organi o dei punti riflessi relativi a questi organi, ma di valutare , di prendersi cura, di curare l’individuo inteso nel suo insieme di corpo, di mente, di spirito. Per cui noi possiamo considerare che la condizione di salute dipende molto dall’entità e dalla qualità del coping, cioè dalla qualità di come noi riusciamo a rispondere agli eventi esterni dannosi. La medicina quale noi vogliamo intendere, che voi dovreste praticare nel vostro futuro, è quell’attività, parliamo proprio di “missione della medicina”.E’ diverso parlare di “prendersi cura” e “curare”, non sono sinonimi, sono due concetti completamente diversi: curare è predisporre una terapia, prendersi cura è qualcosa di più, è proprio prendersi in carico il malato, curare e valutare le condizioni della persona malata fin dal concepimento, non dalla nascita, per poter conseguire e conservare le condizioni di salute. E’ importante che la medicina, prima di intervenire terapeuticamente, si occupi di individuare tutte quelle risorse che sono necessarie per realizzare la condizione di salute. Prima della prevenzione noi dobbiamo fare in modo di stimolare, di indurre tutte quelle condizioni che possono proteggere o far rispondere a stimoli nocivi e quindi a realizzare la condizione di salute. Dovremmo cercare, noi docenti, di insegnare che appunto la prevenzione e la
cura del malato devono essere azioni che ogni medico deve realizzare per sviluppare, accrescere, migliorare le capacità e le risorse personali per poter aumentare le difese contro il rischio , contro tutte le condizioni che minano alla salute. Quindi, si è passati dalla medicina che si basa sulle evidenze alla medicina basata sulle risorse. Se i fatti biologici sono determinati dagli eventi nocivi, dannosi e dai limiti delle nostre risposte, questi fatti biologici devono essere studiati tenendo conto del fatto che sono determinati da due eventi: dalle cause cosiddette “prossime” e dalle cause “remote” o “evolutive”. -Per esempio le discipline che rispondono alla domanda del “come” sono discipline che studiano le cause prossime (come si è realizzato questo evento? In che modo si svolge questa funzione?) quindi una biologia di tipo funzionale.-Poi ci sono le discipline che valutano le cause remote cioè quelle che si sono realizzate nel corso dell’evoluzione, tanto per intenderci per quanto riguarda il dimorfismo sessuale la causa “prossima” è l’influenza degli ormoni nel determinare il sesso, quella “evolutiva” è l’instaurarsi di questo dimorfismo ha sicuramente realizzato una condizione di vantaggio rispetto all’altra situazione. Allora in questi termini come si pone la patologia generale? Come una disciplina che si sforza di rispondere a entrambe le domande: del come e del perché. Studia le cause prossime, quali sono le cause della malattia. La patologia generale studia le cause della malattia e i meccanismi che si sono evoluti nel tempo che hanno condotto poi a quella condizione per cui quella malattia dava quella causa si determina in un determinato modo. Quindi la patologia
3

generale studia l’eziopatogenesi, studia le cause e i meccanismi con cui le cause determinano le malattie. L’oggetto di studio della patologia è la malattia considerata come condizione “fisiopatologica”. “Fisiologia” e “patologia” sembrano due termini antitetici, ma in realtà non è così. Innanzitutto se non ci fosse la fisiologia non ci sarebbe la patologia. Il fatto che non sia un antitetico sta nella condizione in cui la fisiologia può impercettibilmente allontanarsi dalle condizioni di salute e impercettibilmente arrivare a quelle di patologia. Non c’è uno stacco netto, una scissura tra condizione di salute e condizione di patologia. C’è uno spettro infinito di situazioni sfumate che gradualmente e impercettibilmente possono andare da una situazione di normalità a una di patologia.La condizione fisiopatologica sta a significare proprio questa deviazione maggiore o minore, a seconda della condizione di cui si parla, dalla fisiologia. Quindi la condizione fisiopatologica significa che c’è un discostarsi, con uno spettro continuo di condizioni di situazione, da una realtà che si pensa sia quella normale. Questo sta a significare che la malattia è considerata in modo dinamico, in continua evoluzione, in continuo divenire. Cioè una malattia ha un’espressione in un determinato momento, dell’evoluzione della malattia ci può essere un’altra condizione. Non è un fatto statico che è determinato da una situazione che nasce in un certo modo e rimane costante nel tempo. La malattia è una situazione che evolve nel tempo, è una condizione altamente dinamica. Quindi la patologia è una disciplina di natura dinamica perché studia un evento
dinamico, ma non solo, ma è anche la disciplina che ha carattere olistico, viene da “olos” che vuol dire “tutto”, insieme. Perciò si chiama patologia generale, non perché è generica, ma considera la malattia dal punto di vista integrale, cioè tutti i suoi aspetti. La patologia generale non studia un evento che si realizza a livello di un determinato organo o apparato e si limita lì lo studio, la patologia generale studia i rapporti, perciò è importante che voi apprendiate questo studio. Che quell’evento che sembra colpisca quel determinato organo o apparato ha rapporto con altri organi o apparati. Questo vuol dire condizione fisiopatologica, perché se c’è l’alterazione della fisiologia di un organo o apparato, questo di necessità comporta l’alterazione della fisiologia di altri organi o apparati non rimane limitata lì. Q uesto è il carattere olistico e dinamico. L’alterazione che l’anatomopatologo va a studiare, non è la causa della malattia, ma è la conseguenza della malattia. La malattia ha causato quella lesione a quel determinato livello. Quindi la malattia non deve essere studiata come aveva proposto Morgagni nel suo libro famosissimo “De sedibus et causis morborum per anatomen indagatis” cioè andare a studiare le cause della malattia attraverso l’indagine anatomica. Questo modello di studio anatomopatologico attiene a un concetto di tipo riduzionistico, cioè le malattie sono ridotte, limitate a quella lesione di quel determinato distretto. Come se l’organismo umano fosse una macchina e quindi andare a vedere dove è il guasto. Questo approccio è stato, con la nascita della patologia generale, abbandonato. È rimasto invece a livello degli anatomopatologi e soprattutto con questo
4

libro di Bernardi, “L’introduzione allo studio della medicina sperimentale”, che si può considerare come l’inizio della patologia generale perché la patologia generale tutto il suo contenuto lo ricava da un’indagine sperimentale. Perché tutte le conoscenze della patologia generale derivano dall’attività di ricerca sperimentale. L’approccio riduzionistico può sembrare una condizione di buon senso, però non può essere accettato perché intanto dobbiamo capire che cosa significa disfunzione, che cosa significa guasto, che cosa significa alterazione. E hanno significato soltanto se noi definiamo bene che cose significa normalità, che cosa significa funzionalità. La normalità quando parliamo di un soggetto umano è relativa alla salute e quindi dobbiamo capire che questa definizione che era stata data da Boorst che “la vita o la salute è una gerarchia tipica di sistemi funzionali che mantengono la vita”, queste sono parole interconnesse non è accettabile perché non si capisce cosa è questa gerarchia di questi sistemi interconnessi e che cos’è la funzionalità nomale. Non lo possiamo ricavare per via statistica perché se ci dovesse essere una situazione in cui una condizione patologica è presente nella stragrande maggioranza della popolazione alla fine noi diremo che questa è la normalità, quindi non è questa la via per definire la condizione di salute; quindi la via statistica non può essere utilizzata perché c’è una grande variabilità dei fenomeni biologici. Invece partendo dagli stessi concetti dalla patologia generale, che abbiamo detto che ha un carattere olistico, la patologia generale è importante per poter definire bene la condizione di salute assieme agli altri saperi. L’olismo della patologia
generale è realizzato attraverso l’apporto di tutte queste materie, sono importanti le nozioni di base delle altre materie.Se i fenomeni biologici sono complessi e tra di loro correlati, immaginate quanta maggiore complessità esiste tra i fenomeni patologici. Anche quelli elementari sono molto complessi perché c’è un’alta integrazione tra i sistemi biologici.Abbiamo detto che non sono antitetiche fisiologia e patologia cioè non c’è contrapposizione tra stato di salute e stato di malattia. Dobbiamo quindi capire bene cosa sia la condizione di salute o di normalità. Intanto potremo rivolgerci a un modello astratto in cui noi possiamo dire che la “salute” è quella condizione in cui non ci sono grosse variazioni rispetto a un modello astratto di salute che ci possiamo dare, si può decidere quali sono le condizioni di normalità e magari, in maniera astratta, poi stabilire che la condizione di salute è quella condizione da cui non ci si allontana molto da questa definizione di salute data in maniera astratta e potrebbe essere un sistema. Però dobbiamo considerare che la situazione è un po’ più complessa perché la vita in realtà, la sopravvivenza del nostro organismo, è assicurata dall’integrazione tra diversi stati stazionari. Lo “stato stazionario” è la condizione di equilibrio dinamico, ogni specifica determinata piccolissima funzione del nostro organismo è da considerare come un equilibrio altamente dinamico, in continuo riaggiustamento perché continuamente sottoposto a variazioni da parte di stimoli interni ed esterni, come visto all’inizio della lezione, e quindi di conseguenza se noi per esempio consideriamo la temperatura corporea in condizioni normali, siamo in grado di mantenere
5

costante la nostra temperatura corporea, noi riusciamo a mantenere costante la nostra temperatura corporea nonostante la variazione della temperatura ambientale entro certi limiti con meccanismi di aggiustamento e di compenso che assicurano la costanza della temperatura corporea, che è 37°, sia in inverno che estate e quando c’è una variazione all’esterno entro certi limiti, perché quando si superano questi limiti o in altre condizioni andiamo in una situazione patologica. Stiamo parlando dal punto di vista della condizione di salute, di normalità. La glicemia, uguale, noi la manteniamo costante in condizioni di normalità, perché c’è un mirabile equilibrio, per esempio, tra ormoni che abbassano la glicemia e ormoni che sono più numerosi di quelli che la abbassano che invece la innalzano. L’organismo è in grado di mantenere costanti molte funzioni. Però questa costanza non vuol dire che queste funzioni rimangono fisse a un determinato parametro nel tempo. Il parametro è uguale nel tempo, però questa uguaglianza, questa costanza del parametro è il risultato di un continuo aggiustamento, non è un dato che si ha e si mantiene così, è un dato che viene mantenuto costante per l’intervento di diversi sistemi, meccanismi di regolazione, che sono meccanismi automatici di controllo, cioè che ogni funzione viene mantenuta in questi parametri per l’intervento di meccanismi omeostatici di controllo che ne regolano e ne assicurano la costanza. E questo si può fare perché abbiamo bisogno di enzimi, abbiamo bisogno di energia, abbiamo bisogno dell’apporto di molecole necessarie per la costituzione degli enzimi, ma la costanza delle funzioni viene assicurata dal
coordinamento esercitato da meccanismi omeostatici di controllo che determinano la stazionarietà dell’intero organismo. Che vuol dire “stazionarietà”? Che mantengono la costanza delle funzioni però la costanza che non è stabile, cioè non è il risultato di una regolazione di tipo statico, ma sempre altamente dinamico e questo coordinamento è determinato da questi meccanismi di controllo. La caratteristica fondamentale è la costanza di ogni funzione ad opera di questi “servomeccanismi”: sono meccanismi automatici che entrano automaticamente in funzione quando una funzione è alterata. Sono automatici però devono avere una certa “elasticità”. La salute è data non dal buon funzionamento del sistema immunitario o del sistema endocrino separatamente, c’è un overlucking, una sovrapposizione tra i vari sistemi però questo vale per tutti i sistemi del nostro organismo che sono tra di loro in equilibrio omeostatico. È questo che assicura la condizione di salute, che quindi forse stiamo arrivando a una definizione di salute un po’ più concreta, meno astratta di quella che abbiamo detto prima. Cioè la condizione di salute la possiamo ritenere quella in cui viene mantenuta mediante l’omeostasi la costanza della funzione come in un equilibrio che è il migliore possibile. Ogni funzione ha un equilibrio che è il migliore possibile, il più idoneo alla sopravvivenza. Perché non è che ci sono due possibilità, o una funzione è in equilibrio o non è in equilibrio, perché se è in equilibrio c’è la condizione di salute, se non è in equilibrio c’è la morte. Ci sono vari stadi intermedi come dicevo poco fa, in cui quella funzione è in equilibrio però può raggiungere un equilibrio che non è il migliore possibile ma non è il più idoneo
6

alla sopravvivenza, e queste sono le condizioni patologiche.Allora per definire la condizione di salute: è quella condizione in cui tutte le funzioni dell’organismo si trovano in uno stadio stazionario che è il migliore possibile, che è il più idoneo alla sopravvivenza. Poi ci sono condizioni in cui ci si allontana da questo che è lo stato stazionario migliore possibile e piano piano l’organismo adotta vari altri equilibri dinamici che sono meno idonei alla sopravvivenza. Ci può essere quindi la condizione di normalità o gli stessi stimoli hanno trovato una possibilità di risposta adeguata da parte dell’organismo che non hanno alterato la condizione di salute e qua invece lo stesso stimolo nocivo che da l’infiammazione è eccessivo o più virulento, allora vedete che invece si eccede, si instaura un equilibrio diverso da quello della salute e si va nella situazione di malattia.Vediamo coma da un equilibrio dinamico di 37° al variare della temperatura esterna si può passare alla patologia, non possiamo fare l’aggancio alla patologia. Per mantenere costante la temperatura corporea ci vuole un equilibrio tra fattori termogenici e fattori termodispersivi. Questo equilibrio viene raggiunto a 37°, condizione di normalità, in base a quanto detta il set point ipotalamico. Però ci saranno delle condizioni in cui questa funzione non è mantenuta costante, in cui questa temperatura corporea si può elevare. Le due principali patologie relative all’alterazione della riduzione del controllo della temperatura corporea sono: le ipertermie cosiddette “non febbrili” (colpi di calore e colpi di sole) e le ipertermie “febbrili” (febbre). Tutte e due queste condizioni patologiche sono espressione di un’alterazione di quel
meccanismo omeostatico di controllo che regola la temperatura corporea. Però si realizzano in un modo diverso:• Nel caso delle ipertermie NON FEBBRILI succede che il guasto dell’azione di questo meccanismo omeostatico sta nel fatto che i fattori che producono calore superano quelli che disperdono calore. Quelli che disperdono calore sono il brivido, gli ormoni che disperdono calore sono soprattutto gli ormoni tiroidei. Ci possono essere condizioni ambientali proibitive: aumento della temperatura esterna oltre determinati valori, ci può essere un’alterazione dell’ambiente data dall’aumento dell’umidità. Tutti questi fattori nella bilancia tra quelli che disperdono calore e quelli che lo producono fa pendere il piatto da parte dei fattori che “producono calore”, e alleggeriscono il piatto di quelli che disperdono il calore. Perché con l’aumento della temperatura si capisce che l’organismo deve sopperire a un aumento esterno abbastanza significativo però con l’aumento dell’umidità c’è anche un impedimento della messa in funzione di un meccanismo di termodispersione, la sudorazione è uno di questi. Quindi da un lato si raccolgono più intensi gli stimoli termogenici e dall’altro meno validi impediti dall’aumento dell’umidità, la quale fa avvertire una temperatura superiore. Fa avvertire una temperatura superiore perché impedisce la messa in azione di un meccanismo di dispersione del calore. Allora in questo caso noi abbiamo una situazione in cui il livello dell’equilibrio dinamico dei due fattori rimane costante a 37°, ma quelli che sono alterati sono i fattori che entrano in equilibrio tra di loro per arrivare a quel determinato valore. Quindi essendo
7

preponderanti i fattori che producono il calore e meno attivi quelli che lo disperdono, si alza la temperatura corporea. Questo è il “colpo di calore”. Però può capitare anche un’altra cosa: che anche se non ci sono condizioni ambientali esterne così proibitive, ci può essere un colpo di calore. Allorché questo accade, si verifica nelle persone anziane o nei bambini molto piccoli, cioè in persone che hanno o un’ immaturità (i bambini piccoli) o un’alterazione dell’elasticità di questi meccanismi omeostatici di controllo che regolano la temperatura corporea (soprattutto nelle nostre regioni, i colpi di calore colpiscono le persone anziane perché l’elasticità è meno valida perché c’è un progressivo deterioramento dei meccanismi omeostatici di controllo che, magari, in condizioni normali, fanno bene il loro lavoro, ma che se sono sottoposti all’azione di agenti un po’ eccessivi che superano le capacità di risposta non sono in grado di far fronte a queste condizioni diverse). Quindi possiamo avere un’ipertermia non febbrile o perché c’è un eccesso di umidità o di calore all’esterno in presenza di una perfetta integrità dei sistemi automatici di controllo oppure in presenza di un leggero aumento di calore, di umidità nell’ambiente, c’è una ridotta capacità di far fronte di rispondere alla temperatura esterna. Questa è la situazione, le cause del colpo di calore. Un colpo di sole è dovuto ai raggi solari sui vasi con ustioni e può portare anche alla morte.
• Nella febbre, ipertermia FEBBRILE, si ha un aumento della temperatura corporea, ma con un meccanismo totalmente diverso perché nella febbre si realizza un perfetto equilibrio tra fattori
che producono calore e fattori che disperdono calore soltanto che questo equilibrio viene raggiunto ad un livello superiore rispetto a 37° cioè nella febbre è il valore di regolazione del set point ipotalamico che viene modificato, ma l’equilibrio dinamico è perfetto tra i vari fattori solo che questo equilibrio si realizza ad un livello superiore . Questo perché durante la febbre si producono “pirogeni endogeni” rappresentati da citochine proinfiammatorie e anche da prostaglandine2 che sono in grado di agire direttamente nell’omeostasi regolando il set point ipotalamico determinando un innalzamento del punto di regolazione della temperatura corporea. Comunque le due condizioni sono l’espressione di un’alterata omeostasi nel primo caso per uno squilibrio tra i fattori che regolano la temperatura corporea, nel secondo caso per un’alterazione del livello di equilibrio.
La stessa cosa si può dire della riparazione dai danni delle radiazioni ultraviolette. I danni sono la dimerizzazione della timina che può determinare anche dei tumori, si può passare dall’ustione all’epitelioma che è un tumore maligno degli epiteli di rivestimento oppure xeroderma pigmentoso. Anche qui ci sono due condizioni da considerare perché nel caso dell’ustione è un’infiammazione. Se andiamo a vedere il discorso relativo all’epitelioma e tumore causata da un’eccessiva esposizione al sole. Queste condizioni sono dovute al fatto che magari l’organismo ha un corredato di buoni sistemi di difesa nei confronti dei sistemi correlati con le radiazioni. Questi sistemi sono: quei meccanismi di “taglia e cuci”, di enzimi di riparazione del dna che ci legano la catena di DNA alterata, ricostituiscono
8

la porzione escissa o viceversa a seconda se le timine dimerizzate sono vicine o lontane. L’epitelioma si può realizzare in persone che hanno un normale corredo di questi enzimi solo che l’esposizione eccessiva ha superato le capacità riparative di questi enzimi.Lo xeroderma pigmentoso è una condizione che si realizza nell’età infantile, sempre per un’eccessiva esposizione alle radiazioni ultraviolette però anche un’esposizione se non eccessiva prolungata anche se meno prolungata di quella dell’epitelioma, si ha in età infantile perché si realizza nei soggetti che hanno una carenza di questi fattori di protezione cioè di questi enzimi di riparazione delle mutazioni, delle alterazioni del DNA che ci sono in età infantile perché appunto congenite e quindi determinano una situazione che si chiama xeroderma pigmentoso, che è una lesione precancerosa, alcuni lo considerano un tumore in situ in cui si ha un ispessimento, un’ipercheratosi pigmentata rispetto al normale ciclo dei melanociti.Vedete che queste alterazioni sono dovute o ad un eccesso di esposizione o ad una carenza di fattori protettivi come gli enzimi di riparazione .L’autoregolazione endocrina è un altro esempio di come ci sia questo equilibrio omeostatico che regola il livello di tutti gli ormoni. E’ importante soffermarci sull’alterazione riguardante i meccanismi della proliferazione perché anche qua ci possono essere varie patologie. Anche la proliferazione è un evento che è sotto controllo dei meccanismi omeostatici.Le patologie della proliferazione sono essenzialmente tre: iperplasia, lo sviluppo di tumori benigni, lo sviluppo di tumori maligni. Queste sono le tre condizioni
principali in cui noi riscontriamo l’alterazione della proliferazione. Anche qui per la proliferazione ci sono dei meccanismi omeostatici di controllo che sono alterati in maniera diversa a seconda che parliamo di iperplasia , di tumori benigni o di tumori maligni.Per quanto riguarda la prima patologia: iperplasia. E’ quella condizione in cui noi abbiamo un aumento del volume di un organo per l’aumento del numero delle cellule che lo compongono. Nelle condizioni di normalità per un lungo periodo di tempo ogni organo ed apparato mantiene il suo volume costante . Ma questa costanza nel tempo di queste dimensioni negli organi è dovuta a un fatto dinamico perché c’è un continuo aggiustamento tra produzione di nuove cellule e morte delle cellule e c’è un terzo evento che interviene nel mantenimento della costanza che è la “differenzazione”. Sono tre fattori che incidono sulla costanza delle dimensioni dell’organismo:Iperplasia: o aumenta il numero di cellule perché aumenta la proliferazione o per una ridotta morte cellulare, c’è una maggiore sopravvivenza delle cellule muoiono di meno .C’è questo equilibrio per cui le dimensioni sono costanti. Ammettiamo di entrare nel campo dell’iperplasia. Quindi l’iperplasia è quella condizione in cui il numero delle cellule aumenta perché c’è o uno stimolo ormonale o uno stimolo funzionale, cioè quel tessuto o quell’organo è sottoposto ad un eccesso di ormoni oppure ad una richiesta maggiore di lavoro e allora il tessuto risponde a queste sollecitazioni aumentando il numero delle cellule. Cessato lo stimolo, finita la sollecitazione ormonale e la richiesta funzionale, l’organo
9

ritorna alle condizioni precedenti e quindi ritorna alle dimensioni che aveva prima. Nel caso dell’iperplasia dovuta a tumori benigni noi non abbiamo una risposta a degli stimoli ad una sollecitazione di un’attività funzionale o meno, ma c’è un maggiore aumento della proliferazione perché c’è una mutazione a livello di geni che sono responsabili della proliferazione, che attivano la proliferazione. Se abbiamo una mutazione a livello di geni che hanno una funzione positiva, che aumentano l’attività proliferativa, è importante l’equilibrio tra proliferazione e morte cellulare, ma anche per quanto riguarda la stessa proliferazione ci sono fattori che la aumentano e fattori che la riducono. Ogni evento è sottoposto a sistemi di controllo, non solo la proliferazione con la morte cellulare, ma anche della stessa proliferazione tra eventi che la attivano ed eventi che la inibiscono.Se abbiamo una mutazione di geni che codificano per proteine che aumentano la proliferazione avremo l’iperplasia del tumore benigno. Se la mutazione riguarda geni che non solo codificano per proteine che aumentano la proliferazione, ma anche per proteine relative alla differenziazione, per cui questi geni codificano per proteine con differenzazioni patologiche, anomale, atipiche, non funzionanti noi avremo un tumore maligno. L’iperplasia da tumore maligno è determinata da mutazioni di geni che aumentano la proliferazione cellulare o mutazioni di geni che diminuiscono la proliferazione cellulare perché l’aumento della proliferazione cellulare ce la possiamo avere perché o aumenta la funzione dei geni che attivano la proliferazione o perché abbiamo un’ipofunzione per delezione, per mutazione di geni che inibiscono la
proliferazione di geni che inibiscono la proliferazione cellulare. Però nel tumore maligno i geni coinvolti sono geni che non sono importanti solo nel regolare la proliferazione, sono geni anche che regolano la differenziazione perché noi nei tumori maligni riscontriamo non solo un aumento della proliferazione, ma anche un’alterazione della differenzazione, cosa che non riscontriamo nel tumore benigno dove la vera differenza da dove poi discendono tutte le altre, che poi sono conseguenze e non cause, è che nei tumori maligni c’è una mutazione di geni che regolano la proliferazione e la differenzazione per cui la cellula neoplastica è una cellula che non solo attiva la proliferazione, ma anche una differenzazione alterata e invece nei tumori benigni si ha solo una mutazione dei geni che regolano la proliferazione. Se si fa una biopsia di un tumore benigno come un adenoma della mammella è più facile scoprirlo rispetto a un adeno-carcinoma dove si perdono i caratteri della differenzazione. Il concetto di tutto ciò è che nell’iperplasia si ha un’alterazione dell’equilibrio dinamico della costanza che regola la proliferazione. Finito lo stimolo, la capacità dei meccanismi di controllo, fanno ripristinare la condizione iniziale . Nell’iperplasia del tumore benigno e maligno questo non si realizza. Eliminato l’agente cancerogeno, la cellula non torna a proliferare perché l’agente eziologico ha determinato la mutazione a livello genico che rimane in maniera costante e perenne nel tumore. Quindi la gravità e complessità dei tumori sta nel fatto che sono mutati e alterati proprio i meccanismi di controllo che sono i geni, una volta che sono mutati, questo non è reversibile. Per il fatto che nei
10

tumori è alterata anche la differenzazione sono alterate anche le proteine di membrana quelli che costituiscono i sistemi di funzione, sono i desmosomi soprattutto che sono alla base del distacco delle cellule neoplastiche del tumore primario e sono alla base del fatto che le cellule neoplastiche non sono più in grado di cedere i segnali di inibizione della crescita provenienti dalle altre cellule perché la proliferazione è regolata dai geni, ma anche dal contatto cellula-cellula, sappiamo che quando le cellule normali vengono messe a confluire su una piastra di coltura appena entrano in contatto l’uno con l’altra non producono e non si muovono e quindi formano un monostrato . Se mettiamo in coltura cellule neoplastiche non si forma il monostrato, ma si formano tanti multistrati discontinui tra di loro, questo prende il nome di inibizione da contatto. L’inibizione da contatto è mediata da molecole trasmesse tra cellule e cellule che comunicano tra di loro. Questa comunicazione nelle cellule neoplastiche viene alterata, viene soppressa perché vengono alterati i geni che codificano per queste proteine che fanno parte delle giunzioni comunicanti, dei desmosomi e perché sono alterati, modificati i geni che regolano proliferazione e differenziazione. Quindi tanto vero che la cellula neoplastica è insensibile a ogni regolazione e questo sta alla base della definizione di tumore. Si basa sul fatto che la comunicazione cellula-cellula nelle cellule neoplastiche viene persa. Abbiamo detto finora che quando c’è un’alterazione entrano in funzione i meccanismi omeostatici di controllo che mantengono costante la funzione. Abbiamo visto che l’attività di questi
sistemi di controllo può essere annullata da fattori ambientali o da alterazione dei geni ecc. Però ci sono delle condizioni in cui l’organismo cerca di evitare di arrivare d’ambrè (??????) nella patologia, cerca di mettere in atto dei meccanismi omeostatici che assicurino la sopravvivenza, quanto più possibile una situazione di salute anche se possono mettere in atto solo meccanismi, equilibri diversi, stati stazionari meno idonei alla sopravvivenza, diversi da quello iniziale, da quello che è il migliore. Quindi quando lo stimolo è troppo intenso e non può essere compensata dal meccanismo omeostatico fisiologico cioè preposto a mantenere costante quella funzione, l’organismo mette in atto altri meccanismi che ovviamente sono meno fisiologici del precedente, meno idonei del precedente, ma che però assicurano all’organismo il mantenimento dello stato stazionario, quindi salute, sopravvivenza, uno stato stazionario che, ovviamente, non è il precedente, è meno idoneo del precedente alla sopravvivenza . Vi potrà sembrare strano, ma anche la pigmentazione, la cosiddetta “tintarella” che noi tanto bramiamo durante l’estate è l’espressione della messa in atto di un meccanismo omeostatico di controllo, di riparazione dai raggi ultravioletti che non è il più idoneo alla sopravvivenza, perché la pigmentazione nasce come evento di difesa perché non è uguale al fenotipo iniziale che non è pigmentato. Prima di arrivare alle ustioni se si può, all’epitelioma, al melanoma, patologie per l’alterazione, per l’impossibilità di mettere in atto meccanismi omeostatici di controllo per l’azione delle radiazioni ultraviolette, l’organismo cerca di adottare un equilibrio dinamico diverso, che è quello della
11

pigmentazione della cute che si ha con l’aumento della sintesi di melanina che è alla base dell’abbronzatura ed è l’espressione di un adattamento, di una messa in atto di un meccanismo omeostatico non idoneo, meno idoneo al precedente per riparare i tessuti sottostanti alla cute dall’azione lesiva delle radiazioni ultraviolette, perché la pigmentazione di colore scuro, come sapete, è in grado di assorbire le radiazioni ultraviolette e quindi comporta una minor esposizione degli organi meno all’azione lesiva dei raggi. Quindi l’abbronzatura è l’espressione visibile, appariscente, della messa in atto di un equilibrio dinamico che si discosta da quello iniziale. Più patologica è la messa in azione di altri meccanismi omeostatici come quello della tachicardia in alcune situazioni, la tachicardia non è un evento paragonabile a quello della tintarella, però è un evento che può essere sovrapponibile a quello della tintarella perché è un’altra espressione di come l’organismo è in grado di attivare dei meccanismi omeostatici meno idonei di quelli normali, fisiologici quando con quelli fisiologici non è in grado di far fronte a determinate situazioni. L’esempio della tachicardia in queste condizioni patologiche è calzante, cioè nella febbre noi tutti sappiamo che c’è un aumento della frequenza del battito cardiaco perché la temperatura aumenta il metabolismo di base che richiede un maggiore afflusso di ossigeno e quindi di sangue, e il cuore come può far fronte a questa maggiore richiesta? Aumentando la frequenza. Dovete fare appello alle vostre conoscenze di base per rispondere alle domande che la patologia vi pone. Tachicardia nell’ipertensione arteriosa, anche qua ci viene in soccorso la fisiologia. Perché c’è la
tachicardia nell’ipertensione arteriosa? Nell’ipertensione arteriosa si ha un aumento delle resistenze periferiche quindi il cuore deve fare maggiore sforzo per pompare il sangue, e questa sollecitazione può rispondere aumentando il numero dei battiti. Nella stenosi, anche di alcune valvole, oppure anche nell’ipertiroidismo noi abbiamo una situazione in cui gli ormoni tiroidei aumentano tutte le funzioni del cuore che riguardano la frequenza del battito, la conduzione, la forza contrattile. Tutte queste situazioni si ripercuotono a livello cardiaco con l’aumento della frequenza. Quindi noi potremmo considerarci salvi dal momento che i meccanismi omeostatici risolvono tutti i problemi, però i meccanismi omeostatici sono importantissimi, ma non tanto, nel senso che se noi riflettiamo su quello che causano l’infiammazione e l’immunità, ci rendiamo conto che questi meccanismi omeostatici talvolta possono essere dannosi, difatti non è un caso che l’infiammazione la studiate in patologia generale. Infiammazione e immunità sono dei fenomeni, degli eventi difensivi, perché alla base dell’infiammazione non c’è altro che la fagocitosi cioè quell’evento per cui i fagociti (macrofagi e neutrofili) inglobano agenti estranei, batterici, li distruggono e quindi ci difendono dall’infezione. Il problema sta nel fatto che in alcune condizioni particolari di difesa, a un aumento del numero dei fagociti o un aumento della carica batterica e altre situazioni concomitanti alla fagocitosi consegue un danno del tessuto a livello del quale si è verificata la fagocitosi con la fuoriuscita degli enzimi lisosomiali, perché non sempre succede che il fagocita ingloba la particella e la digerisce, altre volte per
12

l’eccesso di carica batterica o per un particolare agente patogeno difficile a essere distrutto all’interno delle cellule, o per deficit del sistema immunitario che può provocare patologie, si può realizzare un danno a livello del tessuto nel quale si è realizzata la fagocitosi e abbiamo l’infiammazione. Spesso i radicali liberi che si producono durante la fagocitosi distruggono anche le cellule circostanti. Gli enzimi idrolitici che liberano i lisosomi non rimangono confinati nell’ambito del fagolisosoma, ma vengono riversati all’esterno e quindi abbiamo un danno, una necrosi, una morte cellulare di tessuto dove avviene l’infiammazione e quindi abbiamo un evento patologico. L’immunità: se non avessimo l’immunità noi non saremmo stati qui oggi, fondamentale nella difesa contro oggetti estranei e però quanti eventi immunologici possono causare patologia? L’autoimmunità non può essere soppressa perché è diretta contro gli antigeni del nostro organismo. Un altro esempio è anche l’ipersensibilità: le IgE erano nate come difesa nelle infezioni parassitarie, però poi soprattutto nei paesi più industrializzati, più inquinati queste IgE sono causa di vere e proprie patologie senza dare una infiammazione, perché sono dirette contro sostanze che non sono patogene, che non sono dannose e però sono causa di patologie. Addirittura in altri casi può essere vantaggioso che un meccanismo omeostatico vada fuori funzione, che non intervenga proprio, come per esempio nell’insufficienza cardiaca di tipo congestizio in cui noi abbiamo un difetto, un’ alterazione del ritorno venoso per cui si ha un aumento della pressione idrostatica quindi un’alterazione del meccanismo
omeostatico che regola lo scambio di liquidi tra vasi e tessuti, dato dall’aumento dello squilibrio tra la pressione idrostatica in aumento e quella colloidosmotica che si mantiene uguale, costante e quindi fuoriuscita di liquido, formazione di edemi. In questa situazione gli edemi sono utili al cuore, perché il cuore in una condizione di sofferenza non riesce ad assicurare un ottimale ritorno venoso per cui se si riduce la volemia (perché gli edemi fanno ridurre la volemia) è un fatto vantaggioso per il cuore, perché deve lavorare meno. Tuttavia, l’ipovolemia non è altrettanto vantaggiosa per il rene che quindi appena si accorge che c’è ipovolemia, attiva il sistema renina-angiotensina-aldosterone per riportare la volemia alle condizioni precedenti. E questo risulta utile per il rene, ma non altrettanto per il cuore. Quindi per il cuore questa messa in atto di questo meccanismo omeostatico di controllo non è certamente vantaggiosa, sarebbe stato meglio non averlo. Da quello che abbiamo detto finora risulta che noi dobbiamo considerare i processi biologici come eventi altamente integrati tra di loro e ciò vuol dire che l’alterazione di uno di essi può sbilanciare l’azione di molti altri distretti biologici. Quindi sono da considerare come lo spostamento non di un equilibrio omeostatico, ma di più equilibri omeostatici e questo vale per tutte le malattie sia genetiche sia acquisite, perché nelle malattie genetiche c’è l’alterazione di un meccanismo omeostatico perché è alterato quel gene che determina il malfunzionamento di quel meccanismo omeostatico o lo mette fuori funzione o lo riduce o lo altera, perché viene alterato una qualunque proteina o fattore codificato da quel gene, quel meccanismo omeostatico è
13

alterato, ovviamente. Però il discorso è che quell’alterazione di quel meccanismo omeostatico per l’alterazione di quel gene non rimane limitato a quel meccanismo omeostatico, ma sbilancia e altera altri meccanismi omeostatici che sono alterati solo sotto il profilo funzionale, mentre quello determinato dalla mutazione del gene è alterato in maniera strutturale. Però anche nella malattia genetica, che poi è alterato quel gene, quel prodotto genico, anche in questo caso la patologia è correlata all’alterazione di quello più altri meccanismi omeostatici. Nelle malattie acquisite la situazione è più complessa perché lo stimolo patologico può bloccare un meccanismo omeostatico e quindi la malattia si può realizzare perché lo stimolo patologico, la sostanza patogena blocca un meccanismo omeostatico, impedisce che venga messo in azione o ne altera il suo livello di equilibrio. È il caso della febbre che è un evento sistemico in cui il meccanismo omeostatico esiste, va bene, è perfetto soltanto che il suo livello di equilibrio è spostato verso l’eccesso. In altre condizioni il livello di equilibrio può essere anche in difetto. O ancora si può realizzare la situazione in cui l’agente patogeno inganna un meccanismo omeostatico e lo induce a entrare in funzione quando non è necessario e viene a capo la patologia. Un esempio di come uno spostamento di un equilibrio portato allo stato iniziale molto piccolo, limitato a una piccola sostituzione, di un solo amminoacido nella catena dell’emoglobina, come accade nell’anemia falciforme, può causare la morte se non viene diagnosticata in tempo, perché la sostituzione dell’acido glutammico con la valina in una determinata posizione della catena
globinica (posizione 6), perché se fosse un’altra posizione non ci sarebbe questo, determina un’ alterazione della solubilità in una condizione di ipossia, quindi la precipitazione dell’ emoglobina, formazione delle emazie falciformi che si impilano nel microcircolo, determina il blocco dei vasi che può portare a necrosi e anche morte. Quindi questo viene portato come esempio di amplificazione biochimica dell’anemia falciforme. Perché in questa condizione un piccolo evento iniziale, una piccola mutazione può portare a conseguenze molto gravi. Come vi dicevo poco fa, nella vecchiaia, si ha un progressivo deterioramento dei meccanismi di regolazione che sbilancia l’omeostasi e se la vecchiaia è questa, la morte si verifica allorché il sistema aperto che è rappresentato dal nostro organismo non riesce più a mantenere lo stato stazionario e quindi a mantenersi in equilibrio con l’ambiente circostante, quindi le condizioni sono tali che l’organismo non riesce più ad assicurare l’omeostasi e quindi a entrare in equilibrio con l’ambiente circostante. Questi sono gli eventi che si verificano dalla nascita all’età adulta in cui progressivamente i fattori ambientali ossidanti, fattori inquinanti, l’obesità, il diabete, alcune malattie che si incrementano con l’avanzare dell’età. Sono le cosiddette “malattie della terza età” in cui si ha l’accumulo e l’intervento di molti fattori esterni che non sono però validamente controbilanciati dai malati dai meccanismi omeostatici che sono andati incontro a deterioramento. Quindi i fenomeni patologici vi invito a considerarli in questi termini di alterazione dell’omeostasi, prendiamo tutto dalla fisiologia. Vediamo i fenomeni patologici come un’alterazione dei fenomeni di
14

omeostasi e dell’alterazione dei meccanismi omeostatici preposti al mantenimento della costanza delle funzioni e dobbiamo assolutamente ricercare una sintesi nella molteplicità degli eventi patologici e dovete considerare la malattia non più come un evento localizzato ma come un processo in continuo divenire che coinvolge l’intero organismo. Quindi quando voi studiate la patologia generale, non studiate gli
argomenti non in maniera staccata e separata tra di loro, ma integrati tra di loro, riferendo alcuni dati che avete appreso nelle nozioni di base della patologia generale ad altri fenomeni patologici, trovare somiglianze,interazioni, connessioni tra i vari eventi. Non studiate in maniera artificiosa, mnemonica, banale. Dovete studiare riflettendoci, capendoli e correlandoli tra di loro.
Francesca Mazzeo
15

14/10/2013 Prof.ssa Teti
2Risposte cellulari alRisposte cellulari al
dannodannoContinuiamo sulla falsa riga di quello che abbiamo detto nella scorsa lezione cioè che la patologia è il risultato di un’incapacità, impossibilità, inadeguatezza dei meccanismi omeostatici di controllo fisiologici a rispondere ad un danno, ad una noxa patogena e quindi dobbiamo vedere quali sono le risposte della cellula a questi agenti patogeni. Cioè la cellula abbiamo visto l’altra volta come ha la possibilità di regolare le proprie funzioni, i meccanismi omeostatici che regolano la costanza di funzione e che possono essere di danno, possono essere quelli che poi non sono i più idonei; vediamo quindi come una cellula sottoposta a un danno risponde a questo danno. In condizioni fisiologiche, di normale funzione della cellula, la cellula è sottoposta a dei limiti. È confinata, è limitata da diversi elementi, diversi fattori. Questi elementi che limitano le funzioni, le attività della cellula sono rappresentati ovviamente dai suoi programmi genetici
di metabolismo, di funzione, di specializzazione, di differenziazione: cioè ogni cellula è differenziata in base all’espressione dei propri geni. Ho dei programmi genetici (le cellule hanno tutte gli stessi geni, però nelle diverse tipologie di cellule, questi geni sono diversamente espressi e da questo dipende la differenziazione, dipende il tipo di proteine quindi le funzioni che una cellula svolge) quindi le cellule hanno dei limiti che derivano dai propri programmi di specializzazione, perché la specializzazione è un caso ulteriore rispetto alla differenziazione, quindi ogni cellula è limitata al suo grado di differenziazione e di specializzazione nello svolgere una determinata funzione. È limitata da altri fattori, per esempio (l’abbiamo accennato la volta scorsa) dall’inibizione esercitata dalle cellule vicine: abbiamo parlato del fatto che le cellule, quando sono a contatto, sono vicine l’una all’altra e si scambiano delle
16

informazioni rappresentate da piccole molecole, ioni e queste molecole trasmettono dei messaggi che inibiscono alcune attività della cellula come per esempio la proliferazione. Uno dei meccanismi con cui la proliferazione viene contenuta entro certi limiti è proprio dato dai messaggi di inibizione delle cellule vicine attraverso le giunzioni comunicanti. Ovviamente dalla disponibilità di substrato, di sostanze nutritive. Quindi, ha diversi limiti la cellula, però nonostante questi limiti, se la cellula è sottoposta a determinati stimoli, a determinati agenti patogeni, la cellula può rispondere in maniera diversa da quella che è la normalità: o con un eccesso, un aumento di funzione, o con un difetto di funzione, perché la cellula può rispondere anche riducendo la propria attività. La cellula è in grado di rispondere a queste richieste, a queste attività patologiche (ora vedremo il danno), ma anche una risposta a queste richieste fisiologiche mediante una messa in opera di quei meccanismi che abbiamo detto essere meccanismi omeostatici di controllo, però appunto quando gli stress, le richieste fisiologiche sono eccessive, tanto da diventare stress, o quando la cellula è addirittura sottoposta all’azione di questi agenti patogeni, le cellule vanno incontro a determinate condizioni di adattamento che possono essere diverse, ovviamente riguardano sia la struttura sia la morfologia. In questa maniera, quando le cellule rispondono adattandosi a uno stress eccessivo fisiologico o a una noxa patogena, le cellule (di necessità) devono costituire, fondare un nuovo sistema omeostatico, un nuovo equilibrio, diverso dal precedente. Perché devono stabilire un nuovo equilibrio? Per poter far fronte a queste
mutate esigenze e quindi le cellule modificano la loro attività, la loro funzione perché arrivano a stabilire un equilibrio dinamico diverso da quello che precedeva l’azione degli agenti patogeni o dell’eccesso di stimoli fisiologici. Le cellule raggiungono un diverso equilibrio dinamico, lo raggiungono attraverso questa risposta adattativa che è una risposta di adattamento alle nuove condizioni. Tuttavia succede che anche la risposta adattativa ha dei limiti, non è che la cellula possa rispondere in maniera adeguandosi a qualunque condizione. Quando la risposta adattiva non riesce più a compensare, a dare adeguate risposte agli stimoli eccessivi o patologici, allora la cellula va incontro al danno. La cellula, prima di andare incontro a un danno, cerca di adattarsi alle nuove condizioni, ma se questo non è possibile, la cellula subisce un danno; danno che, in prima istanza, è un danno reversibile, cioè un danno che può regredire se lo stimolo patologico viene allontanato (può modificare la propria struttura, la propria funzione per via di un danno ai suoi organuli cellulari). Un danno che però, se lo stimolo dura poco o non è eccessivo, può essere reversibile. Se lo stimolo patologico è eccessivo o dura a lungo nel tempo, il danno che prima era reversibile diventa irreversibile e ovviamente è uno step che porta poi alla morte cellulare. Ecco le risposte della cellula agli agenti lesivi, quindi la morte cellulare non è un fatto repentino, ma una conseguenza di tutta una serie di risposte cellulari. A seconda della natura, della tipologia dello stimolo patologico, la cellula può adattarsi e mettere in atto delle risposte adattative che possono o aumentare l’attività policlonale o la possono ridurre.
17

Vedete in questo diagramma che c’è un danno, si può avere anche una ridotta attività funzionale della cellula fino alla morte cellulare che si estrinseca nella condizione di necrosi. La necrosi è quella condizione di morte cellulare che riguarda un certo numero di cellule per l’azione di agenti lesivi rappresentati soprattutto da tossine, sostanze tossiche o soprattutto da una ridotta pressione di ossigeno, quindi ipossia. In condizioni di ridotto apporto di ossigeno, la cellula riduce la propria attività fino ad andare incontro a morte, la necrosi è una condizione di morte cellulare che riguarda un consistente gruppo di cellule, quasi tutto un tessuto.
2.1 Cause di danno cellulare2.1 Cause di danno cellulareVediamo quali sono le risposte cellulari al danno [che sono limitate nonostante gli agenti lesivi possano essere numerosi]. Vediamo intanto quali sono questi agenti lesivi che possono portare al danno.
• AGENTI FISICIo Traumi meccanicio Radiazionio Temperatura (ustioni, congelamenti)
• AGENTI CHIMICI• AGENTI INFETTIVI/BIOLOGICI• IPOSSIA• REAZIONI IMMUNOLOGICHEo Ipersensibilità
o Autoimmunità• ANOMALIE GENETICHE• DISTURBI NUTRIZIONALIo Carenza alimentareo Carenza vitaminicao Obesità
Quali sono le risposte cellulari a questi danni? La prima fase è rappresentata dall’adattamento: la cellula cerca di adattarsi. La flessibilità è un processo che è importante sia sotto il profilo umano e sociale, sia sotto il profilo anche biologico, anche le cellule cercano di adattarsi alle mutate condizioni.
18

2.2.Esempi di risposte cellulari2.2.Esempi di risposte cellulariQuali sono le condizioni principali di adattamento? Non dobbiamo considerarle come patologie, ma come processo di adattamento, messa in atto di meccanismi omeostatici diversi da quello iniziale che è migliore per la sopravvivenza.
2.2.1 ADATTAMENTI CELLULARIa. Atrofiab. Ipertrofiac. Iperplasiad. Metaplasiae. Displasia
Come vi dicevo le risposte adattative possono portare a un aumento della funzione od ad una diminuzione della funzione ed in questo caso, nel caso della ridotta funzione, ovviamente l’atrofia che è la riduzione del volume di un organo per riduzione del volume delle cellule è una risposta. Le cellule, in determinate condizioni, possono andare incontro ad atrofia. La cellula come fa a svolgere le proprie funzioni in carenza di ossigeno? LIMITA la propria attività funzionale alla semplice sopravvivenza, cioè cerca di limitare le proprie funzioni a quelle necessarie per sopravvivere. Per fare questo, le cellule degradano i loro componenti cellulari, i loro organuli: c’è una riduzione del numero dei mitocondri, dei ribosomi, riduzione proprio delle attività degli organuli che sono preposti alle attività funzionali della cellula. Quindi facendo questo, la cellula riduce il proprio volume degradando, non attraverso gli enzimi lisosomiali, ma attraverso un altro processo che coinvolge il proteosoma, le
cellule riducono i propri organuli vitali, quelli che svolgono le attività funzionali della cellula e le cellule vanno in contro alla riduzione del proprio volume, mantengono le attività funzionali strettamente necessarie alla sopravvivenza. Un altro tipo di risposta adattativa è proprio l’esatto opposto: l’ipertrofia. Quella condizione in cui un organo o un tessuto aumenta il proprio volume per aumento del volume delle cellule. Le cellule diventano più grosse perché aumentano il loro numero di organuli vitali, ribosomi, mitocondri e quindi aumenta il loro volume, non aumenta solo il liquido citoplasmatico, aumentano gli organuli e questo perché la cellula deve svolgere un’attività funzionale maggiore, viene richiesta un’attività maggiore alle cellule e queste, per rispondere a questa richiesta di lavoro, aumentano il loro numero di organuli e quindi aumenta il loro volume. Il classico esempio è quello muscolare: il muscolo risponde a una maggiore richiesta funzionale con l’allungamento delle sue fibre, con un aumento del volume delle fibre perché le cellule devono svolgere una maggiore attività e quindi aumentano gli organuli cellulari. Un’altra risposta adattativa è l’iperplasia. Mi sembra di avervi detto che è la risposta delle cellule a uno stimolo, ad una richiesta funzionale maggiore, ad uno stimolo ormonale. Quando le cellule sono sottoposte a questi stimoli e sono cellule che hanno mantenuto la capacità di proliferare, quelle muscolari non l’hanno mantenuta. C’è un solo caso in
19

cui le cellule muscolari aumentano la capacità proliferativa, quale?- La lesione muscolare.Sì, infatti, la vedremo tra poco, è stata studiata recentemente questa situazione dell’iperplasia, cioè l’aumento del numero delle cellule che però è una condizione abbastanza ridotta, un evento limitato rispetto alla risposta dell’ipertrofia. Le cellule muscolari, ad esempio nell’aterosclerosi dove c’è un ispessimento della tonaca muscolare per azione di una citochina prodotta dalle cellule endoteliali (platelet derived growth factor, PDGF) che è l’unico in grado di dare proliferazione. Con l’ispessimento della tonaca muscolare a livello dei vasi sanguigni, questi vanno incontro ad aterosclerosi per l’azione di questa citochina prodotta dalle piastrine e si chiama proprio PDGF, uno dei più potenti fattori mitogeni. Quella è la condizione unica, patologica. C’è comunque un’iperplasia di risposta che accompagna l’ipertrofia di cellule muscolari, ma diciamo questa componente iperplastica è molto ridotta rispetto alla componente ipetrofica nella risposta adattativa delle cellule muscolari. Ci sono invece cellule che hanno mantenuto intatta la loro capacità di proliferare e quindi, a maggiori richieste funzionali, rispondono con un aumento. Aumentano le schiere. È una condizione di risposta adattativa a degli stimoli, ma, una volta eliminati, le cellule ritornano alla condizione precedente, cioè riducono il loro rave proliferativo e quindi il tessuto ritorna alle condizioni iniziali come dimensioni. Un’altra situazione di risposta adattativa è rappresentata dalla metaplasia. La metaplasia indica un passaggio, una costituzione di un tipo di tessuto con un altro tipo di tessuto ugualmente
differenziato, ben differenziato come il tessuto che viene sostituito. In condizioni molto critiche, le cellule pur di assicurare la sopravvivenza del tessuto, sostituiscono il tipo di differenziazione. Non è che cambiano il livello di differenziazione, viene sostituito un tessuto ben differenziato, con un altro tessuto, altrettanto ben differenziato, non ugualmente differenziato. Questa sostituzione consente alle cellule di sopravvivere e far fronte a queste mutate condizioni ambientali. Molte volte c’è una riduzione della specializzazione delle cellule che rende le cellule più fragili, più sensibili all’azione degli agenti lesivi, con cellule un po’ meno specializzate, ma più resistenti. E questa è la metaplasia.
2.2.2 DANNO CELLULARE ACUTOf. Danno reversibileg. Morte cellulare:I NecrosiII Apoptosi
Dopo l’attivazione di queste risposte adattative, se gli stimoli persistono, la cellula va incontro al danno. Si passa attraverso varie fasi: un danno acuto che prima è reversibile e poi diventa irreversibile, poi la morte cellulare che può avvenire con due meccanismi: necrosi e apoptosi. L’apoptosi riguarda una morte cellulare che avviene con un meccanismo completamente diverso rispetto alla necrosi e perché è diverso? Lo vedremo quando parleremo più approfonditamente dell’apoptosi, perché l’apoptosi interviene nei meccanismi di regolazioni delle dimensioni del tessuto. Una delle costanti in un organismo è la dimensione di un tessuto, tutto è dato da
20

un equilibrio tra proliferazione e morte cellulare e differenziamento. La morte cellulare che interviene nella regolazione delle dimensioni di un tessuto, è la morte per apoptosi, non per necrosi. La necrosi è sempre l’espressione di una morte cellulare patogena, magari una noxa patogena che ha causato un danno irreversibile ed ha portato a morte la cellula. Poi ci sono altre differenze e lo vedremo, in questo momento mi interessa che abbiate chiaro questo concetto. La morte cellulare che regola le dimensioni di un tessuto, che fa parte, che entra in gioco con la proliferazione e la differenziazione, è la morte per apoptosi. Voi potreste dire “L’apoptosi è una morte per così dire fisiologica.” Interviene nel mantenimento dell’omeostasi. Ed infatti l’apoptosi ha questo ruolo, ma non ha solo questo ruolo, nel senso che l’apoptosi è un evento, sia che sia ridotta, sia che sia aumentata, che può avere anche caratteri patologici. Ha una doppia faccia! Strumento, meccanismo di regolazione delle normali dimensioni di un tessuto, ma anche processo nettamente patologico, sia che sia in eccesso (cioè le cellule vanno incontro ad un eccesso di morte cellulare per apoptosi: Parkinson) oppure una riduzione dell’apoptosi (come nel cancro, in molti tipi di cancro). L’apoptosi ha questa duplice veste che interviene nei normali meccanismi omeostatici, ma che può anche essere causa di patologie. La necrosi è un evento sempre patologico, dovuto a danno irreversibile della cellula.
2.2.3 ALTERAZIONI SUBCELLULARI ED INCLUSIONI
Altre risposte cellulari sono alcune
alterazioni che riguardano proprio gli organuli subcellulari, proprio perché gli organuli subcellulari sono quelli preposti alle normali funzioni delle cellule. Potremmo avere un danno a livello mitocondriale, se c’è un eccesso di stimolazione da parte degli ormoni della tiroide, noi abbiamo delle alterazioni funzionali a livello dei mitocondri, fino ad arrivare alla dissociazione tra fosforilazione e ossidazione. È ovvio che gli organuli cellulari siano coinvolti, come anche le inclusioni.
2.2.4 ACCUMULI INTRACELLULARI
Altre risposte adattative sono gli accumuli intracellulari, le cosiddette degenerazioni. Le degenerazioni sono delle condizioni patologiche in cui c’è un accumulo di sostanze in cellule che normalmente non le contengono. Per esempio i trigliceridi che sono presenti a livello degli adipociti, non costituiscono causa di degenerazioni, di obesità sì, ma non di degenerazione. Se i trigliceridi sono presenti a livello degli epatociti, allora noi abbiamo la steatosi, una grave condizione degenerativa. Le lipidosi riguardano invece i lipidi complessi e carenze enzimatiche, ci sono le glicogenosi, diversi casi di accumuli.
2.2.5 CALCIFICAZIONI PATOLOGICHE
L’altro capitolo sono le calcificazioni patologiche che fanno parte delle degenerazioni perché sono patologie in cui si ha accumulo di sali di calcio, di pirofosfato, deposizione non a livello dell’osteone come nella calcificazione fisiologica, ma a livello di vari organi e tessuti: questa è degenerazione. Possono essere la conseguenza o di una
21

ipercalcemia, cioè le cosiddette calcificazioni metastatiche, dovute all’aumento, all’alterazione a livello degli ormoni che regolano il metabolismo del calcio e del fosforo con aumento della calcemia che supera di 11 mg x 100 ml. Dobbiamo sempre chiederci quali sono i valori normali, perché poi viene spontaneo agli esami che ve lo chiediamo, a parte questo è importante che se voi studiate una patologia in cui c’è un aumento di qualcosa, è naturale sapere l’aumento rispetto a cosa, a quale valore. Chiedetevi sempre il perché. Il valore di calcemia è molto limitato, tra 9 e 11 mg x 100 ml. Un range molto basso. In particolari condizioni patologiche come l’iperparatiroidismo, lo pseudoiperparatiroidismo, altre patologie che determinano un’ipercalcemia, possono favorire, in presenza di altre sostanze che sono le sostanze scatenanti, la precipitazione di fosfato di calcio, ma le calcificazioni eterotopiche hanno un luogo diverso di deposizione di calcio rispetto al normale luogo di deposizione. L’altra forma di calcificazione eterotopica è la calcificazione distrofica che è dovuta a una condizione di necrosi cellulare. In molte condizioni, un’evoluzione della necrosi è la calcificazione. In un ambito di cellule che sono andate incontro a necrosi, è più facile che ci sia la precipitazione di Sali di calcio. Il motivo sta nel fatto che, in corso di necrosi, si ha la liberazione di enzimi lisosomiali da parte di cellule necrotiche che danneggiano anche le cellule vicine e, tramite gli enzimi lisosomiali, viene attivata la fosfatasi acida, che avrebbe una funzione simile a quella della fostafasi alcalina che è quella fisiologica che fa depositare i sali di calcio correttamente. In questa condizione in cui c’è questa
liberazione massiccia di fosfatasi acida, i sali di calcio in una situazione normale di calcemia in cui non c’è un’alterazione della funzionalità degli ormoni che regolano questo metabolismo, si ha precipitazione nel luogo in cui il tessuto è andato incontro a necrosi e sono moltissime le condizioni: il granuloma tubercolare che voi studierete nell’infiammazione cronica che porta ad un particolare tipo di necrosi e poi quello che noi vediamo tante volte anche in un reperto radiografico: una piccola calcificazione a livello dell’ilo polmonare, è l’esempio di una calcificazione che si è potuta realizzare a livello di un tubercolo che è andato incontro a necrosi. C’è appunto la liberazione degli enzimi lisosomiali, della fosfatasi acida. C’è la calcificazione a livello dei vasi sanguigni, una placca ateromatosica, una lesione infartuata… ci sono varie condizioni in cui c’è questo accumulo di calcio e sempre fa parte delle risposte cellulari al danno. Quelle adattative sono queste, ma queste sono risposte cellulari diverse rispetto ad un evento dannoso. Atrofia: parlavamo dell’atrofia e della riduzione della grandezza e della funzione di una cellula. Se qualcuno vi dovesse domandare di discutere sull’atrofia, voi dovreste rispondere con maggiore correttezza: “Non è solo una riduzione della grandezza, ma anche della funzione. La riduzione della grandezza è in funzione della riduzione della funzione”. Tutti voi sanno come può ridursi un arto dopo tanto tempo che è stato per esempio ingessato. Una delle possibili cause è la ridotta utilizzazione da immobilizzazione di un arto in seguito ad una frattura. Tuttavia siccome le cause di atrofia non
22

sono dovute soltanto ad una ridotta utilizzazione di un arto, ma può esserci l’implicazione di stimoli da parte delle terminazioni nervose che hanno una funzione trofica sulle cellule muscolari. Un altro esempio di atrofia muscolare è quella che riguarda l’infezione da virus della poliomielite, non è dovuta a immobilizzazione ma è una atrofia di tipo neurogeno. Ipetrofia: Aumento anche della funzione di un organo in seguito ad uno stimolo ormonale o in seguito ad una maggiore richiesta di lavoro, per esempio l’ipertrofia cardiaca in seguito ad ipertensione arteriosa, in seguito ad una alterazione delle valvole cardiache, aortica. In particolare in queste condizioni, l’ipertrofia ventricolare, le miofibrille implicate sono quelle che riguardano la contrazione rapida. Un altro esempio di ipertrofia è rappresentata dall’aumento del volume della prostata. Di solito l’ipertrofia prostatica aumenta con l’età, è quasi fisiologico che ci sia un aumento del volume della prostata, questo per eccesso di ormoni di tipo estrogeno, può aumentare la produzione di estrogeni e siccome ci sono dei recettori per gli estrogeni sulla prostata, allora si può avere l’ipertrofia prostatica a causa degli estrogeni o per azione dei fattori di crescita. Per quanto riguarda l’ipertrofia muscolare abbiamo il caso in cui aumentano il numero delle fibrille come in questo caso, oppure aumenta il sarcoplasma e si parla di ipertrofia sarcoplasmatica, oppure in particolari condizioni può esserci un’iperplasia con un aumento dell’aumento delle fibre muscolari. Intervengono vari fattori: l’aumento delle
miofibrille (aumentano le proteine contrattili, actina e miosina), aumento del connettivo, della vascolarizzazione e l’aumento delle fibre (è ancora controverso questo discorso perché appunto l’ipertrofia muscolare è un processo a più livelli, multidimensionale, perché ci sono molti fattori coinvolti, c’è coinvolto anche il processo iperplasico che è sostenuto dalle cellule satelliti). C’è un intervento della proliferazione delle cellule satelliti, un intervento dei meccanismi che la stimolano, del sistema immunitario, di fattori di crescita ed anche degli ormoni. Ci sono segnali all’interno delle cellule che attivano l’attività contrattile, come quelle calcio-dipendenti attraverso le protein-chinasi, le MAP-chinasi. Questo sistema della Rapamicina (Mamalian target of rapamicin) che è una protein-chinasi che fosforila serina e treonina e regola la crescita, la proliferazione, la motilità delle cellule muscolari. Per cui le cellule possono essere indotte a modificare la propria condizione di sintesi proteica e quindi ad aumentare la loro attività di sintesi proteica e ad inibire il catabolismo perché aumentare gli organuli cellulari porta ad aumentare la sintesi e ridurre il catabolismo. L’inverso dell’atrofia. S’è visto che nell’ipertrofia muscolare intervengono diverse citochine, proprio perché sono in grado di interagire con recettori specifici presenti a livello muscolare e poi ci sono proprio ormoni anabolici come l’IGF-1 (uno dei più potenti fattori mitogenici), il testosterone e l’ormone della crescita che hanno un ruolo molto importante nel determinare l’ipertofia. Come vedete l’ipertrofia muscolare non è un evento banale, ma un
23

evento complesso in cui intervengono molteplici fattori e molteplici vie di trasduzione del segnale, attivazione di vie di trasduzione del segnale. Iperplasia: Abbiamo detto che l’iperplasia è un aumento del numero delle cellule, sempre per stimolazione ormonale, per una maggiore richiesta funzionale e questa iperplasia abbiamo visto può riguardare anche l’iperplasia a livello del muscolo. Le cellule satelliti che rivestono la fibra muscolari, in seguito al trauma, vengono attivate e vengono indotte a proliferare. Se il trauma è breve, non è di grande entità, la cellula satellite torna allo stato di quiescenza, ma se lo stimolo persiste a lungo, le cellule satelliti vanno incontro a proliferazione e migrano verso la fibra danneggiata. Ci sono due vie: Si fondono con la fibra danneggiata e questo dà luogo all’ipertrofia, la fibra aumenta di volume. Oppure le cellule satellite vanno incontro a un processo di proliferazione e si fondono tra di loro dando luogo a nuove fibre muscolari e questa è l’iperplasia. Se si fondono con la fibra danneggiata, questa aumenta di volume, se si fondono tra di loro dando origine a nuove fibre muscolari, abbiamo l’iperplasia. Metaplasia: è la conversione, sostituzione di un tipo di tessuto ben differenziato con un tipo di tessuto altrettanto ben differenziato. Avviene per alterazione della maturazione delle cellule, ancora non è chiaro se è dovuto alla stimolazione delle cellule staminali che vengono colpite con il segnale a differenziarsi in altro modo o delle cellule mesenchimali.
Displasia: Possiamo considerare anche questa una risposta adattativa, in cui abbiamo delle alterazioni della grandezza, della morfologia e della disposizione delle cellule nell’ambito dei tessuti. Nella displasia l’architettura generale di un tessuto viene alterata. Per esempio la cheratosi attinica che avviene a livello dell’epidermide. È un’alterazione della morfologia, della grandezza e soprattutto del posizionamento, dell’organizzazione delle cellule nell’ambito di un tessuto la cui architettura viene modificata. A livello della mammella vi sono varie forme di displasia. Abbiamo visto i cambiamenti possibili che seguono a un danno cellulare: abbiamo visto la cellula normale subisce un danno in base all’intensità, alla durata, all’entità del danno, la cellula può andare incontro a cambiamenti adattativi, oppure può andare incontro a danno o a cambiamenti reversibili (per esempio le degenerazioni) oppure ancora può andare incontro a cambiamenti irreversibili che sono l’anticamera della morte, della necrosi. Volevo solo farvi notare che il danno che può dare luogo a uno di questi tre cambiamenti cellulari, ma che anche questi cambiamenti cellulari sono tra di loro connessi. O il danno da degenerazione od un guasto irreversibile che porta direttamente alla morte, oppure si può arrivare tramite queste varie fasi di adattamento, di danno acuto reversibile, di danno acuto irreversibile e poi quindi morte. Il danno può dare luogo ad una delle tre risposte, oppure l’ultima risposta può essere conseguenza della prima.
24

2.3 Come si presenta la cellula2.3 Come si presenta la cellula danneggiata danneggiataLa cellula che va incontro a un danno [figura; nota del correttore: la figura fa riferimento alla differenza tra necrosi e apoptosi, ma è l’unica che ho trovato che mostra i blebs] questa è una cellula normale, questo è un danno reversibile. Qui però già vedete che ci sono delle alterazioni a livello della membrana: queste estroflessioni che si chiamano blebs. Vedete come ci sia una dilatazione, un rigonfiamento del reticolo
endoplasmico, un rigonfiamento un po’ generalizzato di tutta la cellula. Incominciano i processi di autofagia perché? C’è una dilatazione, un aumento di permeabilità della membrana lisosomiale con liberazione di enzimi lisosomiali che determinano distruzione degli organuli cellulari, quindi il processo di autofagia; ed il rigonfiamento dei mitocondri, presenza di corpi densi.
Nel danno reversibile, si ha perdita della funzione cellulare e si determina un danno strutturale, però come vedete da questa diapositiva, la cellula può cambiare la sua condizione e ritornare alla situazione precedente. Quindi, una cellula danneggiata con danno reversibile, ritorna alla sua condizione precedente (vedete che c’è una freccia bidirezionale). Nel danno irreversibile le cellule quasi si dissolvono, si ha una completa alterazione profonda, massiccia del reticolo endoplasmico rugoso, il nucleo va incontro a danno profondo o per picnosi o per
cariolisi, si ha un rigonfiamento mitocondriale con alterazione della fosforilazione ossidativa e soprattutto si hanno dei gravi danni a livello della membrana; il guasto principale è l’adesione della membrana perché si hanno soluzioni di continuità e quindi alterazione soprattutto della pompa sodio-potassio e quindi ingresso di sodio e di acqua all’interno della cellula che comportano maggiore rigonfiamento della cellula e di tutti gli organuli con perdita di funzione e in questo caso il passaggio dal danno reversibile a irreversibile è a unico senso
25

(non c’è la freccia bidirezionale) dove la cellula va incontro a morte. Qua sono rappresentate grossolanamente le due condizioni di morte cellulare. Vedete quella per necrosi in cui si ha una cellula che è rigonfia, ingrandita, con lesione dei granuli cellulari e con estroflessioni della membrana, non so se lo notate, queste estroflessioni contengono solo il citoplasma non ha gli organuli cellulari, mentre invece quando la cellula va incontro ad apoptosi, vedete che si formano queste grosse estroflessioni della membrana profonde, vediamo però che noi ritroviamo gli organuli cellulari che
sono ancora perfettamente funzionali e la cellula, anziché andare incontro ad un processo di rigonfiamento, nell’apoptosi va incontro ad un processo di raggrinzimento cellulare. Vedete che poi, a livello di queste estroflessioni, si ha la rottura e vedete questo pezzo che si stacca dalla cellula e questi corpi che si distaccano dalla cellula che contengono gli organuli cellulari funzionanti si chiamo apoptosomi. Mentre, invece, la cellula che va incontro a necrosi, vedete qui non ha nessun organulo ha solo citoplasma e poi va incontro prima a rigonfiamento e poi a lisi.
2.4 Radiazioni ionizzanti2.4 Radiazioni ionizzanti
Un esempio di morte cellulare a livello di cellule proliferanti o non proliferanti è dato dall’azione degli agenti fisici, delle radiazioni ionizzanti che determinano, in presenza di acqua, una radiolisi cioè una rottura delle catene del DNA. A livello della rottura si formano i radicali liberi, in presenza di acqua, e queste possono portare, nelle cellule proliferanti, a danno del DNA (quindi non possono rispondere con l’aumento della replicazione perché sono incapaci di proliferare e quindi si ha la morte cellulare) oppure a livello delle cellule non proliferanti, il danno maggiore che i radicali liberi possono produrre è a livello degli acidi grassi insaturi dei lipidi di membrana (sono particolarmente sensibili all’azione dei radicali liberi dell’ossigeno) quindi perdita dell’integrità di membrana [per ossidazione dei lipidi di membrana] e la morte cellulare. Abbiamo detto che questi agenti lesivi
gravi, persistenti, inducono tutti questi danni alle cellule, però guardate che la cellula è dura a morire. Nello stesso momento in cui la cellula subisce un danno, è impressionante il fatto che la cellula metta in atto dei sistemi di sopravvivenza che cercano di ridurre il danno e cercano anche le cellule di attivare quei processi di proliferazione cellulare se le cellule non sono gravemente danneggiate. Cioè le cellule hanno la capacità di stabilire quali sono quelle che devono proliferare e invece quelle che è meglio vadano incontro a morte. Perché? Se le cellule subiscono un profondo danno a livello del DNA, queste cellule sono indotte, attivano i programmi di morte cellulare che le porta alla morte per apoptosi (detta anche morte altruista) per evitare di trasmettere alle cellule figlie, ove mai avessero mantenuto la capacità di replicarsi, il danno ricevuto. Ci può essere
26

il caso che abbiano ricevuto un danno grave al DNA, ma abbiano mantenuto le cellule la capacità di proliferare. In questo caso le cellule sicuramente trasmetterebbero un danno alle cellule figlie, quindi avremmo un tessuto ricco di cellule danneggiate che non possono svolgere la loro funzione. Le cellule che hanno ricevuto questo danno profondo, prima disattivano i programmi di riparazione del danno al DNA (perché nell’apoptosi succede questo: c’è la disattivazione). Le cellule hanno tanti programmi di riparazione di un danno al DNA, però quando il danno è serio ci vuole tanto tempo, nel frattempo le cellule possono proliferare. Prima che la replicazione avvenga, le cellule disattivano il loro programma di riparazione e vanno incontro a morte. O rispondono in questo modo, oppure le cellule che invece non hanno subito un danno mortale, vengono indotte a proliferare. Noi dobbiamo intendere le cellule non come entità a sé stanti, ma come parte di un tutto che è il tessuto, cioè la cellula attiva questi programmi di morte e di replicazione cellulare per il mantenimento dell’omeostasi del tessuto, non della
singola cellula, ecco la socialità delle cellule che è persa nel caso di una cellula trasformata. Le cellule danno una risposta, più che per la singola cellula, per la visione di un insieme, di un’entità superiore della cellula che è l’integrità del tessuto. - Ma è l’agente nocivo che stimola la cellula a proliferare o è la cellula che subisce il danno?È la cellula. Allora, l’agente nocivo determina un danno X. Se il danno è riparabile entro breve tempo, prima che la cellula si replichi, vengono attivati tutti quei meccanismi (come la PAP) di riparazione del DNA e quindi poi la cellula va incontro a replicazione. Tuttavia se l’agente lesivo è tale che ha danneggiato gravemente il DNA, per cui la cellula impiegherebbe troppo tempo per ripararsi e nel frattempo la cellula va incontro a proliferazione, la cellula invece attiva il programma di apoptosi, viene disattivata la riparazione del DNA e poi vengono attivati i meccanismi di morte cellulare. È sempre una lama a doppio taglio. Agente lesivo e la risposta a seconda dell’entità dello stimolo e del danno.
2.5 Come risponde la cellula?2.5 Come risponde la cellula?Un tipo di risposta può essere la heat shock response. È stata studiata per la prima volta in cellule sottoposte a danno termico. Viene attivato un certo tipo di risposta dovuta allo shock termico. Viene adottata non solo in seguito allo shock termico, ma anche in seguito a stimoli
molto gravi.La risposta in che cosa consiste? Nell’attivazione di alcuni geni che normalmente sono repressi. Quando c’è questo shock termico o questi stimoli altamente lesivi, alcuni geni vengono attivati.
27

I geni ci sono, alcuni sono espressi altri no. Anche condizioni non solo della differenziazione, ma anche la risposta a stimoli nocivi. Quali sono geni che andremo ad attivare? Un gene che si chiama GADD 45 che significa Growth Arrest of DNA Damage. Un gene che produce delle proteine che inducono a resto della crescita che cercano di riparare il danno nel DNA. Geni che sono espressi in una cellula sottoposto non dico solo a shock termico, ma anche ad altri tipi di agenti lesivi che inducono alla riparazione del DNA. Oppure altri geni che codificano per proteine come l’ubiquitina che sono proteine che vengono chiamate proteine chaperon. Lo shock termico e gli altri stimoli, alterano, modificano la struttura terziara-quaternaria delle proteine. Le proteine perdono la loro struttura tridimensionale e si allungano “unfolded proteins” cioè non sono più impacchettate come lo sono in condizione fisiologica. Queste proteine creano un grave danno alla cellula e allora vengono eliminate attraverso un inglobamento in proteine più grosse come l’ubiquitina (è una proteina molto grossa, si chiama così perché è presente in tutto l’organismo) e questa ubiquitinazione comporta l’attivazione di un processo complesso (la formazione del cosiddetto proteosoma) e della degradazione di queste proteine senza che si danneggi la cellula. Al contrario di quello che accade durante la necrosi: c’è l’aumento della permeabilità dei lisosomi e vengono liberati gli enzimi lisosomiali che danneggiano la cellula. Questi enzimi che degradano proteine unfolded che sono di danno per la cellula, sono dei processi silenti che non danneggiano la cellula. Sono vari tipi di risposta che la cellula può
dare in seguito allo shock termico o ad un danno consistente e questo è un esempio.L’apoptosi è il programma per cui viene disattivato di riparazione perché troppo lungo ed il danno è troppo complesso e nel frattempo la cellula potrebbe proliferare e trasmettere il proprio danno alle cellule figlie e poi d’altra parte vi avevo detto che le cellule che non sono state danneggiate, se quelle vanno incontro a morte, per mantenere costante l’omeostasi è necessario che si formino altre cellule. Nello stesso momento (questa è la cosa secondo me mirabile) in cui alcune cellule vanno incontro a morte, lo stesso danno induce le cellule che non sono state gravemente danneggiate ad attivare il loro programma di riparazione con l’attivazione, l’espressione di geni (se la cellula fa una cosa, è perché ha delle proteine che le fanno fare determinate cose). I geni che codificano per alcuni fattori di trascrizione che si legano a siti di consenso del DNA ed alcuni geni determinano la proliferazione come c-fos, c-jun, c-myc. La grandezza della popolazione cellulare di un tessuto è determinata da tre eventi: proliferazione, apoptosi e differenziazione. Queste sono le patologie della proliferazione cellulare. A partire da una situazione di normalità, si può avere un accumulo di cellule in seguito ad un processo iperplastico o tumorale, le cellule si accumulano, sono superiori a quelle di partenza ed invece questa è la condizione del numero cellulare per eccesso di perdita cellulare. È chiaro che l’entità della popolazione cellulare dipende da questi tre fattori ed ovviamente se c’è uno stimolo della proliferazione, la popolazione aumenta ecc…diverso è per l’apoptosi.
28

2.6 Ciclo cellulare2.6 Ciclo cellulareBrevemente vi volevo far ricordare la distinzione tra le cellule che mantengono il loro grado di proliferazione o le cellule che sono dette labili. Quelle permanenti sono quelle che non si dividono più, quelle quiescenti si dividono in particolari condizioni (di normalità ma in seguito a determinati stimoli). Le cellule quiescenti si trovano in una condizione G0, mentre quelle permanenti escono completamente dal ciclo proliferativo.
Vi sto parlando della patologia del processo proliferativo, stiamo entrando nel discorso dei fattori che intervengono nel mantenimento dell’omeostasi del numero di cellule in un tessuto che sono rappresentate dalla proliferazione, dall’apoptosi e dalla differenziazione. Vediamo come avviene la proliferazione, quali sono i fattori responsabili, vediamo poi come si estrinseca l’apoptosi. La proliferazione avviene attraverso queste quattro fasi.
Non so se vi ricordate che la fase G1 prende più della metà di tutto il ciclo mitotico. Perché?S.: "Avviene l’accrescimento cellulare."P.: "In che senso? L’aumento della proliferazione? Ma quello si ha nella fase M."S.: "No, nel senso che avviene l’accrescimento degli organuli."P.: "E ci vuole tutto questo tempo? È un poco lento."
Può essere giusta anche la vostra idea. Io vedo tutto in termini di regolazione degli eventi biologici.
Quando la cellula va incontro a proliferazione, è un evento molto importante. La cellula prima di andare incontro a proliferazione, ci pensa due volte. Vi ho detto prima che ha la capacità di resistere, di affrontare tutto quello che può, però è anche una cellula che prima di prendere una decisione, dev’essere sicura che quella decisione sia giusta. E l’allungamento, questo maggior tempo che la cellula impiega in fase G1 dà conto di questa necessità per la cellula, perché? Voi lo sapete che una volta che si supera la fase G1, il cosiddetto checkpoint, non c’è più niente da fare: la cellula prolifera.
29

E allora la cellula ci pensa bene prima di superare il checkpoint! Deve valutare se questo è un evento utile per il tessuto di cui la cellula fa parte! Secondo me dev’essere ben sicura la cellula, come fa la cellula a saperlo? Perché a livello della fase G1 intervengono molti se non tutti i meccanismi di regolazione della proliferazione. Quali sono? Agenti che attivano la proliferazione: abbiamo i cosiddetti fattori di crescita. Se non ci sono fattori di crescita, la cellula non prolifera. Secondo voi i fattori di crescita, in quale momento della vita della cellula intervengono? Nella fase G1! Nella fase G1 noi possiamo distinguere due sottofasi, due metà.
Nella prima metà della fase G1, intervengono i cosiddetti fattori di competenza. Le cellule, dalla fase G0 sono indotte a proliferare quindi a entrare nella fase G1 ed entrano nella prima metà per azione dei fattori di competenza che sono rappresentati dall’FGF (Fibroblast Growth Factor) e da quel fattore di crescita di cui vi parlavo poco fa PDGF (Platelet Derived Growth Factor). La cellula in fase G0, viene indotta ad entrare in fase G1. Ammettiamo che questa cellula non riceva ulteriori stimoli: ha avuto lo stimolo a passare nella fase G1. Se noi non diamo altri stimoli, la cellula ritorna nella fase G0. Quindi non è sufficiente che lei passi dalla G0 alla G1 per andare a proliferare: ha bisogno di ulteriori stimoli per andare
avanti nella fase G1 ed andare incontro alla proliferazione. Come vedete qua c’è l’ingresso ma anche l’uscita, ritorna alla fase G0.
Ammettiamo che la cellula vada avanti e vada nella seconda metà della fase G1 nella fase di progressione in cui intervengono altri fattori di crescita, per esempio l’IGF-1 che inducono la cellula a inoltrarsi nella fase G1 ed a progredire fino ad arrivare al checkpoint e sono questi fattori che inducono le cellule ad andare verso la fase S. Tuttavia se le cellule arrivano in questa fase di progressione e poi non diamo stimoli ulteriori, la cellula rimane in fase G1 e sapete che fa la cellula se rimane in fase G1 per un po’ di tempo? Va incontro ad apoptosi. Nella fase G1 si possono creare diversi eventi, perciò è questo il motivo secondo me.
Se ci sono gli stimoli adeguati, i fattori di competenza, i fattori di progressione, superano il checkpoint e poi il destino è quello, ma se non ricevono stimoli adeguati, se sono cellule stabili, tornano in fase G0, altrimenti vanno incontro ad apoptosi. Vedete che nella fase G1 si gioca il destino della cellula, o sopravvive o muore. Non è detto che per forza vada a proliferare, è una fase molto importante della fase della proliferazione e questo è il ruolo dei fattori di crescita, credo lo conosciate questo meccanismo.
30

2.7 Oncogèni2.7 OncogèniPerché i fattori di crescita agiscano, è necessario l’intervento di due fattore di crescita, io mi soffermo su questo perché tutti i geni che codificano per fattori di crescita, per i recettori dei fattori di crescita, per proteine che sono a ridosso della membrana, sono tutti geni coinvolti nei tumori. Se questi geni sono mutati con guadagno di funzione, questi geni sono la causa di molti tumori, i cosiddetti oncogéni. Non fate la confusione con oncògeni. Oncògeni: Generatori di tumori, sono le cause chimiche, fisiche, biologiche che possono portare al tumore. Oncogèni: Geni mutati nel tumore
I geni che codificano per fattori che agiscono in maniera positiva nella proliferazione si chiamano proto-oncogèni, sono geni che funzionano regolarmente perché la cellula deve proliferare in maniera regolata ed adeguata. Questi geni che attivano la proliferazione si chiamano proto-oncogeni. Quando i proto-oncogeni subiscono delle mutazioni con guadagno di funzione, si chiamano ongogèni. Per contro, ci sono altri geni che producono fattori che inibiscono la proliferazione, perché la proliferazione è uno stato dell’equilibrio tra geni che attivano e geni che inibiscono. I geni che codificano per proteine che inibiscono la proliferazione si chiamano geni oncosoppressori o anti oncogèni. Non è che agiscono sugli oncogèni, agiscono per conto loro in maniera autonoma nell’inibire della proliferazione.
Tutte queste cose che stiamo facendo adesso fanno parte dei proto-oncogèni. I fattori di trascrizione sono codificati da proto-oncogèni. I fattori di crescita devono interagire con due recettori per lo meno, due molecole. Vedete questo è necessario perché questi recettori che prima sono distanti l’uno dall’altro, modificano la loro struttura ed entrano in contatto. Il contatto attiva un’attività enzimatica, presente nella coda del recettore del fattore di crescita che è un’attività di protein-chinasi a livello di tirosina: una tirosin-chinasi. Le tirosine presenti all’interno della parte intracitoplasmatica dei recettori dei fattori di crescita si autofosforilano. La autofosforilazione determina un’attivazione e fosforilazione di alcune proteine che si trovano proprio a ridosso della parte interna della membrana, sono delle proteine particolari che hanno un modulo che si chiama SH2 che è un modulo di omologia, sono moduli di omologia (H sta per homology) col modulo presente nella proteina principe studiata in un tipo di tumore (sarcoma) che si chiama SRC [Pronuncia: SARC].La proteina SRC, codificata da un proto-oncogéne studiato nel sarcoma, è una proteina che ha un modulo. Questo modulo è presente, anche se con conformazione diversa, nelle altre proteine che si trovano a livello della parte interna della membrana plasmatica, che sono proteine che hanno un modulo simile al modulo presente sulla proteina SRC. Questo modulo viene chiamato SH2. Perché ve lo sto dicendo? Con questo
31

modulo SH2, queste proteine che si trovano a livello della membrana, interagiscono con le tirosine fosforilate presenti nella coda citoplasmatica del recettore e vengono a loro volta attivate. L’attivazione di queste proteine determina l’attivazione di altre proteine a livello delle G-Protein (sono codificate dal gene ras, si chiamano G perché legano il GTP) a livello di altri che poi attivano raf che a sua volta attiva le MAP-chinasi con un processo di fosforilazione a cascata (MAP-k, MAP-k-k, MAP-k-k-k, ecc…) ci sono varie MAP-chinasi. Alla fine che fanno? Fosforilano i fattori di trascrizione, perché i fattori di trascrizione non si possono legare ai loro siti di consenso se non sono fosforilati. La fosforilazione ad opera delle MAP-chinasi rende questi fattori di trascrizione c-fos, c-jun, c-myc, fosforilati e vanno a legarsi ai siti di consenso attivando i geni con attivazione della proliferazione. [Disegno di una semiluna con trattino per indicare il modulo SH2. Se dovessi reperire l’immagine sull’Idelson, aggiornerò la lezione.]Questo modulo è presente in tutte queste proteine che si trovano a ridosso della membrana. Ogni proteina si lega a un residuo di tirosina fosforilata, ci potrebbe essere un po’ di confusione, tutte hanno il modulo SH2. Lungo il recettore del PDGF ci sono tante tirosine fosforilate, a seconda della posizione avremo un diverso numero [Y624
ad esempio]. Accanto ci sono altre sigle che riguardano gli amminoacidi che sono successivi alla tirosina. Ogni tirosina fosforilata è legata ad un certo tipo di amminoacido. Il modulo SH2 di queste proteine di membrana, si lega ad un solo tipo di tirosina fosforilata, perché questo modulo
SH2 è simile ma ha una diversa conformazione nelle diverse proteine e quindi questo modulo SH2 si lega ad un solo tipo di tirosina fosforilata che ha quella sequenza di amminoacidi precisa. Il modulo SH2 della proteina SRC si lega solo e soltanto a questa tirosina perché a questa tirosina seguono questi amminoacidi (la glicina, la lisina, la valina, l’acido aspartico) che sono successivi alla tirosina fosforilata. Quest’altro si può legare solo a questa tirosina perché è seguita solo da questi amminoacidi e non da altri. Questa è la spiegazione di quello che vi sto dicendo io a parole: il modulo SH2 di SRC si lega a quella tirosina in quella posizione perché è formato da due tasche.
Una tasca è quella che contiene la tirosina fosforilata e questa tasca è comune a tutti i moduli SH2. Tutti i moduli SH2 hanno la prima tasca identica, perché è la prima tasca che lega la tirosina fosforilata.
La diversità sta nella seconda tasca. La seconda tasca del modello SH2 di SRC è in grado di legare un amminoacido che è presente in posizione 3 rispetto alla tirosina fosforilata. Vedete che c’è una distanza tra la prima tasca e la seconda tasca, questa distanza è data da due amminoacidi. La seconda tasca di SH2 di SRC può legare solo l’isoleucina.
L’altro modulo SH2 della fosfolipasi C ha il primo modulo identico, il secondo modulo lega addirittura subito l’amminoacido immediatamente successivo alla tirosina fosforilata ed è una tasca grande, quindi prende 5 amminoacidi immediatamente successivi alla tirosina fosforilata perché ha questa conformazione.
32

Mentre il modulo SH2 di SRC ha la conformazione piccola, prende solo un amminoacido, la seconda tasca che prende un solo amminoacido [..] e dista due amminoacidi dalla tirosina fosforilata e quindi piglia il terzo amminoacido. Ecco
perché e come queste proteine che mediano la trasduzione del segnale delle MAP-chinasi, legano in maniera diversa le tirosine presenti nella coda citoplasmatica del recettore del fattore di crescita e quindi inducono la proliferazione.
Antonio De Maria
33

21/10/2013 Prof.ssa Teti
3Risposta al dannoRisposta al danno
cellularecellulare
Ricordiamo che l'infiammazione è quel processo di difesa che nasce nei confronti di un'infezione che però in alcune circostanze può diventare causa di danno al tessuto circostante e di necrosi cellulare. Noi invece stiamo seguendo un'altra linea direttiva che è quella delle risposte cellulari al danno, infatti, se ricordate, vi ho detto che le cellule rispondono in maniera diversa a un danno causato da agenti patogeni e la risposta è caratterizzata in primo luogo da risposte di tipo adattativo tra cui c'è l'iperplasia, l'ipertrofia, la metaplasia e successivamente da un danno cellulare che prima può essere reversibile e poi irreversibile. Stavamo parlando, la volta scorsa, della patologia e delle alterazioni che riguardano l'equilibrio tra proliferazione e morte cellulare, che interviene nei meccanismi di regolazione dell'omeostasi nei tessuti, cioè, nell'equilibrio tra cellule che vengono prodotte e cellule che vengono perse, dato
dall'apoptosi e dal differenziamento. Parlavamo anche dei meccanismi che attivano e che regolano la proliferazione, perché indipendentemente dall'apoptosi ci sono meccanismi che attivano e inibiscono il processo proliferativo. In particolare abbiamo accennato ai moduli SH2 che sono specifici per ogni proteina citoplasmatica e che sono specifici nel legame con una particolare proteina fosforilata per esempio nella coda citoplasmatica dei recettori dei fattori di crescita.
Fig. 3.1 Espressione dei complessi delle cicline con leCDK.
34
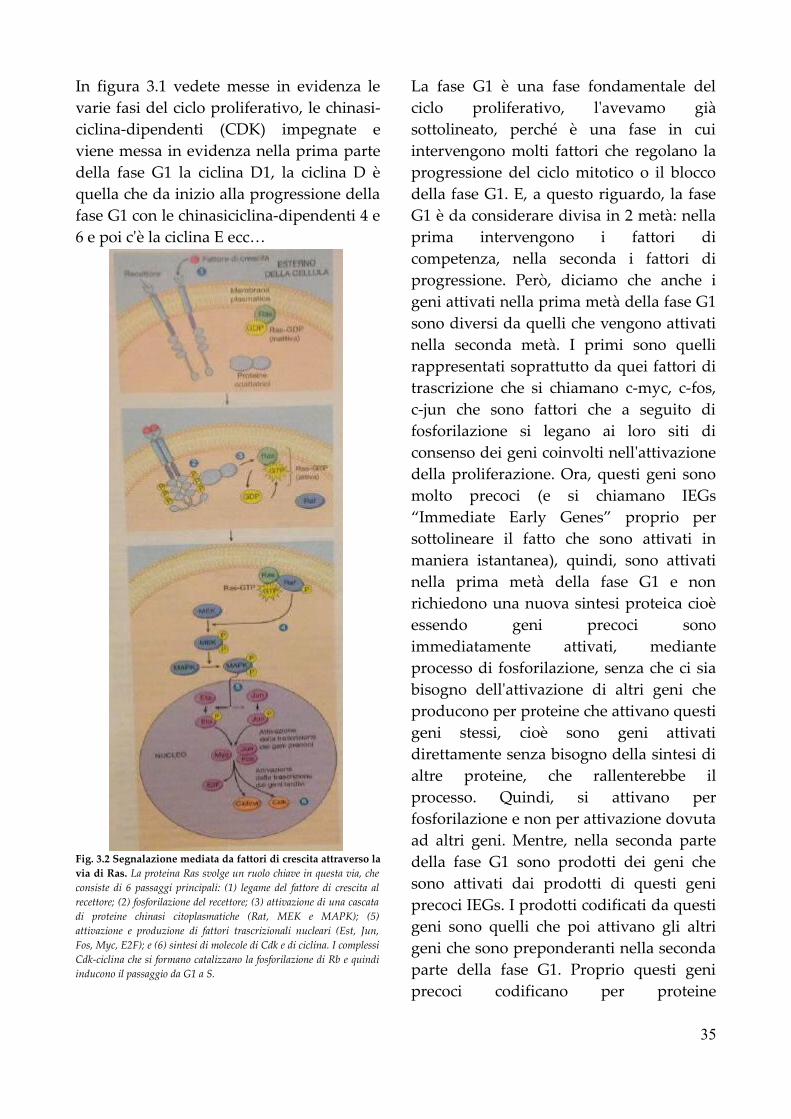
In figura 3.1 vedete messe in evidenza le varie fasi del ciclo proliferativo, le chinasi-ciclina-dipendenti (CDK) impegnate e viene messa in evidenza nella prima parte della fase G1 la ciclina D1, la ciclina D è quella che da inizio alla progressione della fase G1 con le chinasiciclina-dipendenti 4 e 6 e poi c'è la ciclina E ecc…
Fig. 3.2 Segnalazione mediata da fattori di crescita attraverso la via di Ras. La proteina Ras svolge un ruolo chiave in questa via, che consiste di 6 passaggi principali: (1) legame del fattore di crescita al recettore; (2) fosforilazione del recettore; (3) attivazione di una cascata di proteine chinasi citoplasmatiche (Rat, MEK e MAPK); (5) attivazione e produzione di fattori trascrizionali nucleari (Est, Jun, Fos, Myc, E2F); e (6) sintesi di molecole di Cdk e di ciclina. I complessi Cdk-ciclina che si formano catalizzano la fosforilazione di Rb e quindi inducono il passaggio da G1 a S.
La fase G1 è una fase fondamentale del ciclo proliferativo, l'avevamo già sottolineato, perché è una fase in cui intervengono molti fattori che regolano la progressione del ciclo mitotico o il blocco della fase G1. E, a questo riguardo, la fase G1 è da considerare divisa in 2 metà: nella prima intervengono i fattori di competenza, nella seconda i fattori di progressione. Però, diciamo che anche i geni attivati nella prima metà della fase G1 sono diversi da quelli che vengono attivati nella seconda metà. I primi sono quelli rappresentati soprattutto da quei fattori di trascrizione che si chiamano c-myc, c-fos, c-jun che sono fattori che a seguito di fosforilazione si legano ai loro siti di consenso dei geni coinvolti nell'attivazione della proliferazione. Ora, questi geni sono molto precoci (e si chiamano IEGs “Immediate Early Genes” proprio per sottolineare il fatto che sono attivati in maniera istantanea), quindi, sono attivati nella prima metà della fase G1 e non richiedono una nuova sintesi proteica cioè essendo geni precoci sono immediatamente attivati, mediante processo di fosforilazione, senza che ci sia bisogno dell'attivazione di altri geni che producono per proteine che attivano questi geni stessi, cioè sono geni attivati direttamente senza bisogno della sintesi di altre proteine, che rallenterebbe il processo. Quindi, si attivano per fosforilazione e non per attivazione dovuta ad altri geni. Mentre, nella seconda parte della fase G1 sono prodotti dei geni che sono attivati dai prodotti di questi geni precoci IEGs. I prodotti codificati da questi geni sono quelli che poi attivano gli altri geni che sono preponderanti nella seconda parte della fase G1. Proprio questi geni precoci codificano per proteine
35

fondamentali per attivare altri geni coinvolti positivamente nella proliferazione. Quindi vedete è un processo complesso, articolato, tra geni IEGs ( della prima metà della fase G1) e “Late Response Genes” o geni attivati tardivamente, proprio dai prodotti degli IEGs. Gli IEGs vengono chiamati anche “gateway to the genomic response” perché rappresentano l’ingresso alla risposta genomica per la proliferazione e sono attivati direttamente dai fattori di crescita o dalle proteine virali.[Esempio di via di trasduzione del segnale dove sono coinvolti i geni precoci e tardivi. L’eterodimero formato da Fos e Jun è un complesso noto come AP-1] Figuraa) geni precoci o IGEs, quali c-myc (codifica per il fattore di trascrizione Myc), c-fos (codifica per il fattore di trascrizione Fos), c-jun (codifica per il fattore di trascrizione Jun) il cui RNA aumenta molto prima della metà della fase G1 e la cui induzione non necessità di sintesi proteica;b) geni tardivi, il cui RNA aumenta dopo la metà della fase G1 e che dipendono dalla sintesi proteica dei prodotti codificati dagli immediate early genes.
Anche la differenziazione entra in gioco nell'equilibrio omeostatico che regola le dimensioni di un tessuto e interviene in maniera diversa a seconda del tipo di tessuto. Per esempio la differenziazione ha un certo ruolo per quanto riguarda le cellule del midollo osseo perché le cellule del midollo osseo una volta che si differenziano non si dividono più. Voi lo sapete bene, per quanto riguarda il sangue, che è l'unico tessuto umano in cui è nettamente distinguibile il compartimento di proliferazione rispetto al compartimento
di differenziazione. Perché il primo si trova a livello del midollo osseo, il secondo nel sangue periferico. Perciò rispetto agli altri tessuti i due compartimenti sono fisicamente distinguibili. Quando le cellule del midollo osseo vanno incontro ad un processo di differenziazione (il midollo osseo ha la peculiarità che le cellule che qui si dividono non solo si differenziano ma anche maturano -compartimento di espansione-, le cellule che proliferano man mano maturano e poi differenziano) lasciano il midollo osseo, durante la cosiddetta fase di emersione e vanno nel torrente circolatorio. I compartimenti sono talmente distinti che le cellule una volta differenziate sicuramente non proliferano più e quindi la differenziazione gioca un ruolo importante nel mantenimento dell'omeostasi proprio perché impedisce che poi queste cellule proliferino ancora, interviene in senso negativo quindi nella proliferazione. Mentre invece le cellule epatiche, essendo cellule stabili, anche se si differenziano non perdono completamente la capacità di proliferare e se opportunamente stimolate comporta un ritorno dalla fase G0 alla fase G1. La differenziazione assume questo ruolo importante, nel tessuto ematico, nel regolare negativamente la proliferazione: su questa base è comprensibile il meccanismo per cui nelle patologie del midollo, le leucemie, l'aumento della massa dei blasti leucemici non è dovuto, come tutti potreste credere, ad un aumento della loro attività proliferativa rispetto ai blasti normali (ci sono vari parametri che valutano il grado di proliferazione: il tempo di permanenza nelle varie fasi del ciclo mitotico, il numero di cellule che una volta lasciato il ciclo mitotico va verso la
36

differenziazione, presi in considerazione per valutare l'entità della proliferazione) ma è stato visto che aumenta il pool delle cellule differenziate per una maggiore permanenza nel compartimento di differenziazione. Nelle leucemie è alterato quindi il processo apoptotico, poiché, come vedremo meglio, una delle caratteristiche dell'apoptosi è che è un processo fisiologico ma le sue alterazioni sia in senso positivo che in senso negativo
sono causa di patologia, nel caso dei tumori in genere ma soprattutto delle leucemie dove c'è questo aumento del tempo di differenziazione: queste cellule stanno a lungo nel compartimento di differenziazione perché c'è una ridotta apoptosi. Questo per spiegare meglio nelle patologie neoplastiche il ruolo della differenziazione.
3.1 Meccanismi che limitano la3.1 Meccanismi che limitano la proliferazione proliferazione
Una delle proteine normali che limitano il processo proliferativo è la proteina p53. Essa è codificata da un gene onco-soppressore, è coinvolta nella regolazione del controllo del ciclo cellulare perché va a determinare o un blocco di esso se c'è un danno al DNA in fase G1 oppure se il danno non è riparabile manda in apoptosi la cellula (definizione correttamente esposta dalla collega A, nda).È un fattore di trascrizione perché si lega a siti di consenso di vari geni che sono di diverso genere. Dovete sapere che in più del 50% di tutti i tumori del mondo la p53 è mutata. Nel contempo è rarissimo, forse non succede mai, che in un tumore sia mutato un solo gene infatti un tumore è la risultante di una serie di mutazioni a livello di più geni che possono trovarsi anche su cromosomi diversi; noi parliamo tra l'altro di
cancerogenesi cioè esiste un processo dinamico in cui si passa attraverso vari stadi in cui sono coinvolti vari geni; di solito la mutazione di p53 è un evento abbastanza precoce che precede le mutazioni di altri geni.Proprio a causa di questo importantissimo ruolo essa in condizioni normali è presente in piccolissime quantità. D'altronde il punto è sempre là: ci tengo che voi abbiate chiaro il concetto che tutte le risposte cellulari sono sotto il controllo di più fattori che tendono a tenere in equilibrio omeostatico le varie funzioni della cellula. Per cui la p53 se fosse presente in grandi quantità attiverebbe la morte cellulare, bloccherebbe la proliferazione in maniera molto significativa, per cui in condizioni fisiologiche le quantità di p53 hanno livelli così bassi proprio perché servono esclusivamente ad entrare in gioco con
37

altri fattori che attivano la proliferazione. Però se c'è una situazione in cui si ha un grave danno al DNA la p53 aumenta la sua concentrazione perché non viene degradata. Essa infatti viene degradata quasi dopo la sua sintesi, ma in una cellula sottoposta a stress si accumula. Tutto questo perché viene fosforilata: viene attivata una protein-chinasi DNA-dipendente dal danno del DNA (le cellule devono rispondere adeguatamente agli stress, per cui lo stesso stress che altera il DNA è quello che attiva la p53) che fosforila p53 a livello di due serine chiave. Per cui, una volta fosforilata, essa si accumula e può esercitare la sua funzione, maggiore che in una cellula non danneggiata. Parallelamente a questo meccanismo, ne interviene un altro, in quanto il livello di p53 è fondamentale per la cellula, ne va della sua vita o morte: la p53 è regolata da un'altra proteina che si chiama Mdm2 (Mouse double minute 2 homolog, conosciuta anche come E3 ubiquitin-protein ligase Mdm2), codificata dallo stesso gene (gene MDM2), che inibisce la p53 con due meccanismi (vedete quant'è importante la regolazione dei livelli di p53): da un lato agisce sul gene di p53 bloccando la sua produzione (fattore di trascrizione negativo per p53), inoltre idrolizza la p53 non fosforilata, per cui quando viene fosforilata la p53 da questa protein-chinasi DNA-dipendente, la stessa p53 si copre di gruppi fosfato, si protegge dall'azione idrolitica di Mdm2. Per cui Mdm2 agisce a due livelli: sul gene di p53 bloccandolo e sulla proteina degradandola sempre con il sistema dell'ubiquitinazione. La regolazione dei livelli delle proteine intracellulari sia nell'atrofia, sia in questo caso della p53 è mediato da un processo di distruzione non idrolitica, non dovuta
all'azione di enzimi idrolitici lisosomiali sennò la cellula va incontro ad autofagia come succede in alcune condizione patologiche. In questo caso vengono degradate singole proteine in maniera silente, in maniera che non ci siano mutazioni cellulari, in base a un processo di ubiquitinazione. L'ubiquitina, una grossa molecola, ricopre le proteine che devono essere degradate e le inserisce in un complesso più ampio che si chiama proteasoma nell'ambito del quale le proteine vengono degradate.In cellule normali non sottoposte a stress p53 è una proteina altamente instabile che in genere viene degradata subito per proteolisi dopo la sua stessa sintesi. Ha un'emivita di circa 20 minuti. Allo stato stazionario la concentrazione di p53 in queste cellule è molto bassa. Essa si lega ai siti di consenso presenti in vari geni; in seguito ad alcuni stress o danni al genoma, p53, può essere fosforilata nel suo dominio N-terminale da alcune chinasi. La fosforilazione (serina 19 e 37) blocca il legame di Mdm2 e salva p53 da ubiquitinazione e degradazione.1. Stress determina attivazione protein-chinasi-DNA-dipendente;2. Mdm2 idrolizza la p53 non fosforilata.
Il gene Mdm2 viene attivato da p53, la quale si autoregola, cioè attiva il gene di una proteina che la distrugge; e questo è un altro esempio di meccanismo automatico di controllo delle risposte cellulari. La proteina Mdm2 agisce sia come ubiquitina-ligasi (porta l'ubiquitina sulla p53) che riconosce una lisina nel dominio trans-attivante (attiva i geni che sono sotto il suo controllo in posizione trans) sulla p53. Quindi determina il legame dell'ubiquitina proprio a livello di
38

questo dominio trans-attivante e impedisce che la p53 svolga le sue funzioni perché ne attiva la degradazione. Però, d'altra parte, come ho già detto, inibisce la trascrizione di p53.Vi avevo accennato l'altra volta, che ci sono due tipi di geni coinvolti nella oncogènesi:- i protoncogèni che attivano la proliferazione e la cui mutazione comporta un guadagno di funzione; (e)- gli oncosoppressori che invece hanno il ruolo di inibire la proliferazione per cui c'è una riduzione di funzione.
Protoncogène ―› più proliferazione (guadagno di funzione).Oncosoppressori ―› proliferazione inibita (riduzione di funzioni).
Le mutazioni che coinvolgono i protoncogèni sono mutazioni cosiddette dominanti perché basta che ci sia una mutazione in uno dei due alleli per cui il prodotto codificato da quei geni sia mutato.Se c'è un guadagno di funzioni basta un allele per determinare un aumento della proteina. Per cui si dice che è una mutazione dominante. Al contrario per i geni onco-soppressori si dice nella quasi totalità dei casi, tranne che per p53, che le mutazioni sono recessive. Le mutazioni che possono riguardare gli onco-soppressori sono perlopiù delezioni o mutazioni puntiformi che determinano un'alterazione della sequenza della proteina perché questa non è più funzionale; se questo accade a livello di uno dei due alleli, l'altro allele funzionante può sopperire alle carenze dell'allele mutato, si produce una quantità di proteina normale, non patologica, la quale può essere in grado di svolgere le sue
normali funzioni: perché ci sia una conseguenza a livello della proliferazione è necessario che la mutazione riguardi tutti e due gli alleli. Il comportamento del gene TP53 è anomalo rispetto agli altri onco-soppressori, perché si può comportare o come onco-soppressore recessivo o come oncosoppressore dominante.Per cui il gene TP53 può andare, nella trasformazione neoplastica, ad un duplice destino:1) si comporta in maniera recessiva ( ome tutti gli altri onco-soppressori) se manca, è deleto con conseguente impossibilità alla sintesi del prodotto da esso fisiologicamente codificato che, di conseguenza, risulta mancante nella popolazione cellulare di quel tumore. In tal caso, l'altro allele, che è funzionante, è in grado di codificare per la proteina normale, non mutata, fisiologica che può svolgere così la sua funzione. Questo è il comportamento recessivo che la TP53 condivide con tutti gli altri geni oncosoppressori. Il comportamento recessivo quindi è nel caso della delezione: è deleto un allele, l'altro funziona;2) TP53 si può comportare in maniera dominante qualora uno solo dei due alleli non vada incontro ad una mutazione per delezione ma ad una mutazione per sostituzione di basi. In tal caso la copia indenne codifica per una p53 wild-type mentre l’altra copia che è mutata per una p53 abnorme e mutata.
Voi potreste pensare che quella wild-type potrebbe funzionare e invece no perché la p53 per poter agire e funzionare deve essere sotto forma di un omotetramero, formato da 4 subunità di p53. Se in questo tetramero ci sono delle copie mutate il tetramero non funziona, quindi in questo
39

caso p53 si comporta da gene dominante. Quindi se la mutazione del gene p53 è una delezione allora p53 si comporta da recessivo come tutti gli oncosoppressori di questo mondo, se invece la mutazione è a livello strutturale, la struttura del gene è alterata, si producono delle copie wild-type e delle copie mutate che vanno a formare un tetramero inefficiente, ecco quindi che la mutazione è mutante e il tetramero non essendo omotetramero non funziona.
Delezione di p53 ―› si comporta da recessivo. Alterazione di p53 ―› copie wild type e mutate, formazione di tetramero inefficiente (mutazione dominante).La p53 è una fosfoproteina molto versatile perché innesca reazioni che determinano la comparsa di varie risposte cellulari. Essa è dotata di 4 domini:a) il dominio trans-attivante, responsabile dell'attivazione o della repressione del gene, perchè questo dominio può essere sia in senso positivo che negativo, quindi agisce sull'espressione del gene target;b) il dominio legante il DNA (DNA-binding);c) il dominio oligomerizzante, perchè la p53 agisce come tetramero;d)il dominio responsabile della auto inibizione della sua attività trans-attivante
(la p53 inibisce se stessa grazie a questo dominio).
Questo ci fa vedere come le cellule mettano in atto molti meccanismi che regolano le risposte cellulari, che intervengono in ogni fase dell'azione della proteina. Come avviene per p53 che attiva il gene che codifica per una proteina che la inibisce e che si auto inibisce da sola, inibendo la sua attività trans-attivante.I segnali di sopravvivenza (soprattutto fattori di crescita che attivano Mdm2) e segnali mitogeni (attivando altri fattori di crescita come MAP chinasi che fosforilano soprattutto in tyr) causano la rapida degradazione di p53 in quanto incrementano il livello della proteina Mdm2. Gli stimoli mitogeni agiscono attraverso fattori di trascrizione che, come p53, attivano la loro trascrizione del gene Mdm2.La p53 agisce come fattore di trascrizione su un gene chiave della proliferazione, ovvero, p21. Questa proteina agisce a due livelli: da un lato blocca il PCNA (Antigene nucleare di cellule in proliferazione che, consentendo alla DNA-polimerasi delta il contatto con il filamento danneggiato, svolge un ruolo importante sia per la sintesi di DNA che per la sua riparazione) e dall'altro agisce anche a livello del complesso Ciclina-Chinasi-dipendente (CDK), legandosi al complesso già formato e, bloccando la fosforilazione delle cicline, causa un blocco del ciclo proliferativo. Quando non c'è p21
40

il complesso chinasi-ciclina-dipendente attiva la proliferazione. C'è un altro gene che codifica per p15, un oncosoppressore perchè blocca la proliferazione sempre a livello delle Chinasi-Ciclinadipendente, però a un diverso livello, in quanto, in questo caso, la p15 si lega alla Chinasi-Ciclinadipendente impedendo che sia attivata dal legame con la Ciclina. Quindi, p15 si lega alla chinasi e impedisce che la ciclina si leghi a quest’ultima. Però la p53 agisce a più livelli, come fattore di trascrizione particolarmente attivo. Poi c'è un altro gene presente in 3 isoforme, ovvero il GADD45 (Growth Arrest and DNA Damage), il cui prodotto si lega anch'esso al DNA e attiva la riparazione di esso. Il suo legame col DNA è mediato dalla p45 che viene ad essere espressa in condizioni di stress.
Inoltre p53 attiva l'apoptosi. Il gene BAX se prodotto attiva l'apoptosi non è l'unico ad essere attivato da p53 ma ce ne sono altri. Per sommi capi possiamo dire che p53 attiva alcuni geni chiave che facilitano il processo apoptotico ed ha un effetto negativo su un membro delle famiglia di BCL che attiva la proliferazione. BCL la possiamo definire una sorta di famiglia allargata perché contiene geni che attivano la proliferazione e geni che attivano
l'apoptosi. In particolare BCL-2 è un membro di questa famiglia che attiva in maniera significativa la proliferazione, tanto è vero che il suo gene è un proto-oncogene attivato in alcune leucemie. Altri membri di questa famiglia come BAX invece attivano l'apoptosi.La cellula risponde alle condizione di stress attivando le 3 isoforme del gene GADD45 che da un lato può determinare la sopravvivenza cellulare dall'altro attivare le vie della morte e della senescenza cellulare, perché nel caso in cui il GADD45 si lega a PCNA e questo DNA non è eccessivamente danneggiato allora la cellula preferisce ripararlo, e le 3 isoforme codificate da esso determineranno dunque la riparazione del DNA. Anche sotto questo aspetto la fase G1 è fondamentale perché l'allungamento di essa dà un po' di tempo alla cellula per riparare i danni al DNA in modo che esso possa andare incontro alla sintesi e quindi di trasmettere alle cellule figlie il DNA ricevuto. Quindi l'interazione con le chinasi-ciclina-dipendenti e la p21 determina un arresto del ciclo cellulare alla fase G1 e permettere dunque la sopravvivenza della cellula e può influire anche sul blocco del ciclo cellulare anche dopo la fase G0. D'altra parte, con l'attivazione di determinate vie di trasduzione del segnale di MAP chinasi, questo gene GADD45 può attivare la p28 e la Jun-chinasi (JNK) che sono le vie dello stress cellulare che danno luogo all'apoptosi e alla senescenza. Tra le altre proteine che inibiscono la proliferazione ce ne sono alcune fondamentali come la p21 che è indotta dalla p53 e determina il legame con la chinasi-ciclina-dipendente e il DNA. Ci sono poi p16, p15, p14 che sono proteine che bloccano il ciclo mitotico che vanno ad agire sul complesso
41

CiclinaD/Cdk 4-6, lo stesso attivo nella fase G1, agiscono quindi tutte allo stesso livello. Essendo onco-soppressori spesso nelle neoplasie sono deleti.Tutti questi fattori se son regolati da geni
sono coinvolti nella trasformazione neoplastica, per cui io faccio sempre il riferimento ai tumori per abituarvi a fare i collegamenti.
[nota del correttore: Immagine presa da una diapositiva riassuntiva della professoressa. I simboli con i rettangolini sono i geni]
42

3.2 Caratteristiche principali degli3.2 Caratteristiche principali degli inibitori delle chinasi inibitori delle chinasi ciclino-dipendenti ciclino-dipendentip21 ―› la sua sintesi è indotta dall'anti-oncoproteina p53; agisce sui vari complessi Cdk tramite il dominio N-terminale, sul PCNA tramite il dominio C-terminale.p16 ―› modificato dal gene onco soppressore MTS1 che è deleto, ipermetilato o mutato in numerose neoplasie umanep15 ―› codificato dal gene onco soppressore MTS2 che è deleto o ipermetilato in alcune neoplasie umane; la sua sintesi è indotta da TGF-β.p19 ―› la sua sequenza amminoacidica è parzialmente omologa (48%) a quella dell'inibitore p16.p27 ―› è degradato per proteolisi da ubiquitinazione quando la cellula deve entrare in fase S; il suo rapporto stechiometrico con i complessi Cdk è modificato dal TGF-β.p57 ―› si presume che il gene codificatore sia un onco soppressore.p18 ―› debolmente attivo sulla proteina bersaglio Cdk6-D.
Finora abbiamo detto che gli oncosoppressori posso essere mutati o per delezione o per mutazione puntiforme, però negli ultimi tempi si è messo in luce un meccanismo di alterazione dei geni soprattutto geni oncosoppressori che non è dovuto ad alterazioni strutturali del gene, cioè delezioni o sostituzioni di basi ma è dovuto a modificazioni chimiche delle basi dei geni; modificazioni che hanno aperto
un campo, per ora oggetto di molti studi ancora in progresso che però stanno dando ottimi risultati, che si chiama epigenetica, che quindi si occupa non delle alterazioni strutturali ma delle modificazioni chimiche delle basi azotate e delle proteine istoniche.Si è visto che molto geni oncosoppressori non vengono espressi a causa proprio di modificazioni epigenetiche. Il gene non è alterato quelle che spesso risultano alterate sono le citosine che sono metilate, non citosine qualunque, ma citosine che fanno parte di alcune regioni del promoter che si chiamano CpG Islands (P sta per fosforo). Esse sono delle regioni con elevata frequenza di citosine seguite da guanina. Di solito queste citosine metilate sono relative a citosine presenti in queste isole però qualche volta, a livello del gene dell'interleuchina 8, che come studierete nell'infiammazione è una chemochina (una citochina che attira i neutrofili nella sede dell'infezione), in realtà, sono solo 6 CpG nel promoter e la metilazione determina un silenziamento di IL-8. Essa è prodotta in grande quantità non solo dai neutrofili ma anche in molti tumori perché in molte condizioni neoplastiche c'è una iperattività a livello infiammatorio, c'è una stretta relazione tra infiammazione cronica e neoplasie. Quindi la metilazione di queste citosine seguite da guanina può reprimere l'espressione del gene. Molti geni oncosoppressori hanno questa alterazione
43

epigenetica e questo è un evento molto importante perché è reversibile. Io vi ho detto l'altra volta che nei tumori ci sono delle alterazioni strutturali dei geni coinvolti nella proliferazione che, anche se viene allontanata la causa che le ha prodotte, ossia l'agente cancerogeno, rimangono e la cellula, ovviamente, rimane una cellula trasformata. Ma se l'alterazione dei geni oncosoppressori è dovuta ad un'alterazione epigenetica allora si può intervenire chimicamente e cercare di demetilare queste citosine e fare riesprimere i geni oncosoppressori. Un’ altra modificazione epigenetica molto importante nello sviluppo dei tumori è, per esempio, il grado di acetilazione degli istoni, voi sapete che, affinché ci sia una espressione genica, è necessario che i geni siano deacetilati. Vi sono molti agenti
utilizzati dal punto di vista terapeutico che sono inibitori delle deacetilasi, mutati in diversi primers dei tumori, che agiscono a vari livelli soprattutto a livello dell'istone H3/H4. Questa nuova branca dello studio dei tumori, l'epigenetica, si sta sviluppando perché in numerose neoplasie umane questi geni oncosoppressori possono essere mutati dal punti di vista epigenetico, per esempio ipermetilati. Poi ci sono altri geni coinvolti nella regolazione degli inibitori delle chinasi-ciclina-dipendenti, tra cui il p19 omologo al p16, p27 che al momento dell'ingresso della fase S viene degradato per ubiquitinazione ed è sotto il controllo del TGF-β che è un inibitore della proliferazione, e poi altri geni un po' meno determinanti.
3.3 Metaplasia3.3 MetaplasiaUn caso di risposta adattativa della cellula alle condizioni patologiche è costituito dalla metaplasia, cioè da un cambiamento reversibile in cui un tipo cellulare adulto, già differenziato, è sostituito da un altro tipo cellulare adulto altrettanto ben differenziato, nell’ambito della stessa ontogenesi. In tal modo, cellule più sensibili allo stress sono sostituite con cellule più capaci di affrontare l'ambiente avverso. Esso è un evento reversibile, che può evolvere nelle condizioni precedenti, se lo stimolo che lo ha attivato, cessa. Perché le cellule di un tessuto si trasformano in cellule di un altro tessuto? Di solito, il principio, ritenuto
comunemente valido, è che le cellule in seguito all'azione di stimoli nocivi cercano di opporre a questi delle cellule più resistenti, meno sensibili al danno, che sicuramente sono ben differenziate ma che sono meno specializzate, un po' più rozze, che sappiano affrontare più adeguatamente questi agenti nocivi.Un esempio è dato dalla metaplasia squamosa del tratto respiratorio in seguito all'azione irritativa cronica esercitata dal fumo di sigaretta. Vi è un tessuto epiteliale colonnare cilindrico sostituito con l'epitelio piatto, ben differenziato anche, ma meno specializzato. Il primo svolge funzioni che il secondo non svolge: intanto ha le ciglia e
44

poi secerne muco, capacità fondamentali nel tratto bronchiale. Però se la nostra mucosa bronchiale è sottoposta ad inquinamento ambientale o a fumo di sigaretta (contiene idrocarburi policiclici) ad azione continua, questi agenti possono determinare la metaplasia squamosa. Sostituzione di cellule funzionali e protettive (produzione di muco, importante nella difesa contro le infezioni e contro altri agenti) con cellule funzionalmente meno valide. Se lo stimolo persiste, la metaplasia può evolvere in displasia, proliferazione cellulare eccessiva e a lungo andare possono sfociare in tumori bronchiogeni (spesso carcinomi in cellule squamose, segno che quella metaplasia è insorta su una metaplasia squamosa). Possono iniziare delle risposte cellulari ad altri stimoli a cui le cellule possono essere sottoposte che possono determinare appunto la displasia.Le cellule epiteliali colonnari della trachea e dei bronchi sono sostituite da cellule epiteliali squamose stratificate. Condizioni in cui ci sono stimoli cronici come la presenza di calcoli, processi infiammatori irritativi che inducono la sostituzione di cellule cilindriche con quelle epiteliali piatte.Calcoli nei dotti escretori delle ghiandole salivari maggiori e del pancreas e nei dotti biliari possono indurre la sostituzione dell'epitelio colonnare in quello squamoso.La deficienza di vitamina A (la quale interviene nel processo di cheratinizzazione) induce metaplasia squamosa delle cellule epiteliali con comparsa iniziale di ipercheratosi generalizzata che non è quindi solo follicolare (corneificazione a livello dei follicoli piliferi) ma avviene anche a livello del cute a cui segue frinoderma (pelli di
rospo) con perdita di cute sotto forma di grosse scaglie. C'è la cheratinizzazione a livello delle mucose bronchiolari, quindi brochiolite, a livello delle bacinetto renale con pielonefriti e pieliti, a livello della congiuntiva e della cornea con xeroftalmia (ispessimento e quindi secchezza a livello dell'occhio, con conseguente perdita di lucentezza). L'eccesso di vitamina A sopprime la cheratinizzazione. Il problema della metaplasia squamosa è che si rimpiazzano le cellule normali con cellule più resistenti al danno ma che non svolgono nessuna funzione utile.Si può comunque verificare una metaplasia da epitelio squamoso (piatto) a epitelio colonnare (cilindrico) come nell'esofagite di Barrett (non vi è infiammazione) in cui l'epitelio squamoso esofageo è sostituito da cellule colonnari gastriche. È da tenere sotto controllo perché qualsiasi forma di metaplasia, che comunque è reversibile, se persiste nel tempo può dare luogo a trasformazioni tanto che per alcuni testi l’esofagite di Barrett si tratta di lesione precancerosa che può regredire o evolvere verso una neoplasia. Con questo termine si indica una lesione a livello della quale più facilmente, da un punto di vista statistico, più frequentemente, c'è il passaggio a una condizione neoplastica, cioè rispetto a un tessuto che non la presenta. Tutto questo, però, non ci dice che cos'è una lesione precancerosa, per cui, secondo me una definizione più adeguata è quella di una lesione in cui riscontriamo sia una metaplasia che un aumento dell'attività proliferativa. Vedete quindi, come si può passare, attraverso tanti passaggi, da una condizione di difesa, di adattamento, a una condizione patologica, neoplastica. È una lesione che non necessariamente va
45

incontro a neoplasia però a nessuno sfugge il fatto che cellule metaplastiche e in attiva proliferazione possono essere più sensibili all'azione degli agenti oncogeni e così andranno incontro a tumori più facilmente di cellule non proliferanti. Cellule metaplastiche possono essere più sensibili ad agenti di natura oncogena.Per esempio la leucoplachia a livello della mucosa orale è anch'essa una lesione precancerosa perché abbiamo una prosoplasia, cioè metaplasia in cui il tessuto che sostituisce l'altro ha un più altro grado di differenziazione (perché c'è cheratinizzazione) e c'è anche aumento della proliferazione. Durante la prima lezione abbiamo parlato dello Xeroderma pigmentosum che è anche una lesione precancerosa perché abbiamo una metaplasia in quanto anche qui abbiamo una cheratinizzazione eccessiva e aumentano anche la melanine perché aumenta il numero di melanociti (per questo pigmentosum).Per quanto riguarda il carcinoma cervicale in situ ci sono due diverse tendenze se considerarla tumore o lesione precancerosa. Si dice in situ in quanto il carcinoma non ha invaso le strutture circostanti, cioè la membrana basale non è andata ad invadere altri tessuti. Quindi, una caratteristica dei tumori maligni è l'invasività e deriva sempre dalla vera differenza tra tumori maligni e tumori benigni che è quella dell'alterazione dei geni che regolano la proliferazione e la differenziazione.L'invasività è conseguenza dell’alterazione dei geni che codificano la differenziazione perché verranno alterate: le strutture di membrana, le giunzioni intercellulari che determinano una ridotta adesione omotipica (cioè tra cellule dello stesso
tumore) così che le cellule tumorali si distaccano, danno metastasi e invadono altri tessuti. Il tumore è la conseguenza di mutazioni strutturali o epigenetiche che coinvolgono geni per la differenziazione e la proliferazione; quindi, per me, il carcinoma cervicale in situ è un tumore e non una lesione precancerosa perchè la lesione precancerosa, secondo quello che abbiamo detto prima, è una lesione in cui noi abbiamo un aumento della proliferazione ma non per alterazione genica bensì per risposta a determinati stimoli.È, quindi, un processo di metaplasia. Il carcinoma in situ può essere, ad esempio, un momento dello sviluppo del tumore cioè può essere il tumore che è radicato senza però compromettere la salute dell'individuo in quanto non ha invaso strutture vicine e non ha dato luogo a metastasi. In questo caso, l'eliminazione del tumore può essere di valido aiuto, ma non perché non si tratti di tumore, ma perchè il tumore è in uno stadio precedente in cui non è entrato nella fase di progressione neoplastica (metastasi) in cui entra ogni tumore una volta che si è instaurato. Per cui, anche se molti libri di testo ne parlano come lesione precancerosa, per me si tratta di tumore; per me lesione precancerosa è laddove non c'è alterazione genetica o epigenetica ma c'è metaplasia con aumento della proliferazione.Anche le cellule del tessuto connettivo fibroso possono trasformarsi in osteoblasti (metaplasia ossea) e condroblasti (metaplasia cartilaginea) soprattutto a livello dei foci alterati in seguito a trauma.La metaplasia potrebbe essere il risultato di una riprogrammazione genetica di:a) cellule staminali, per esempio in un
46

tessuto per cui esse sono programmate a evolversi verso un tipo di cellula anziché verso un altro; (e)b) di cellule mesenchimali indifferenziate presenti nel tessuto connettivo.
Ci sono altre sostanze che possono indurre l'insorgere di una metaplasia, oltre al fumo di sigaretta, quali sostanze chimiche, vitamine, fattori di crescita, derivati dall'acido retinoico e citochine che regolano crescita e differenziazione.Fattori morfogenetici dell'osso, appartenenti alla superfamiglia TGF-#, inducono nelle cellule staminali l’espressione di caratteristiche condrogeniche od osteogeniche, mentre ne sopprimono il differenziamento in senso muscolare o adiposo. Alcuni farmaci citostatici, molto utilizzati nella terapia dei tumori, possono alterare la differenziazione perché alterano i quadri
di metilazione delle citosine e possono trasformare le cellule mesenchimali da un tipo (fibroblasto) a un altro (muscolo, cartilagine). La differenziazione è dovuta all'espressione di alcuni geni e al silenziamento di altri che è diversa nelle varie cellule, se sono presenti fattori come questi farmaci citostatici che alterano i quadri di metilazione del DNA ovviamente modifica il quadro di espressione genica, cioè alcuni geni vengono repressi e altri no. Tutti fattori che modificano l'entità di espressione dei geni, modificano anche il grado di differenziazione, il tipo di differenziazione. Per cui anche questi farmaci possono intervenire per determinare la metaplasia, come altri agenti metilanti che sono in grado di modificare l'espressione degli onco-soppressori e quindi indurre i tumori.
Antonio Ferlito
47

28/10/2013 Prof.ssa Teti
4Apoptosi e necrosiApoptosi e necrosi
La metaplasia è un processo del tutto reversibile, contrariamente a quello che succede nei tumori, dove il processo è irreversibile quando è dovuto a una modificazione strutturale dei geni, non a un'alterazione epigenetica. Mentre, abbiamo detto, la metaplasia è un processo reversibile perché, se si allontana lo stimolo nocivo o la stimolazione è di breve intensità e durata, l'alterazione è soltanto fenotipica (quindi alterazione morfologica della differenziazione) e di tipo reversibile, proprio perché il genoma cellulare è integro e quindi l'espressione dei geni prima repressi, o diversamente espressi, può ritornare nella norma. Nonostante ciò, la metaplasia, come vi accennavo la volta scorsa, è un processo che, nonostante sia reversibile e nonostante sia un processo di adattamento delle cellule in determinate condizioni ambientali, può costituire il punto di partenza per l'instaurarsi di altri processi patologici. Vi ricordavo la volta scorsa che, quando si ritrova un carcinoma polmonare [...], è logico dedurre che questo sia un' evoluzione di una metaplasia squamosa, infatti ci sono
diversi passaggi tra un processo metaplastico e uno neoplastico, in seguito all'instaurarsi di stimoli nocivi che possono indurre un'iperplasia. Già vi ho detto la volta scorsa che, nelle condizioni in cui c'è un processo metaplastico con un aumento della proliferazione, noi possiamo definire delle reazioni precancerose e quindi è molto più facile l'instaurarsi di un processo iperplastico in un tessuto metaplastico, piuttosto che in un tessuto normale. Le cellule andate incontro a metaplasia, sotto stimolo ormonale, funzionale, chimico, fisico, traumatico, che può indurre un aumento della proliferazione, possono andare incontro a quelle lesioni che abbiamo definito precancerose, come per esempio i polipi intestinali. Successivamente, sempre a livello di queste lesioni, che sono reversibili e non neoplastiche, se ci sono altri fattori, come per esempio esposizione a sostanze chimiche o diciamo condizioni che possono determinare più facilmente un danno a livello della struttura dei geni(quindi delezione, mutazione ecc), si passa a una condizione neoplastica vera e
48

propria. Per esempio non è difficile la transizione tra polipo intestinale a cancro del colon. Quindi sono tutti passaggi in cui queste condizioni di adattamento, come la metaplasia, in risposta a stimoli, danni o agenti lesivi, similmente all'iperplasia, sono maggiormente esposti e sensibili, a lungo andare, all'azione di agenti cancerogeni. Questi agenti, che danno lesioni a livello genico, sono agenti cancerogeni veri e propri e queste cellule sono maggiormente esposte agli agenti oncogeni rispetto alle cellule normali e, di conseguenza, può essere il punto di partenza per una condizione neoplastica.Abbiamo detto che l'iperplasia è l'altra risposta adattativa delle cellule che hanno mantenuto la loro attività proliferativa e l'aumento della proliferazione. L'iperplasia è dovuta all'attivazione dei geni che codificano per i fattori di trascrizione coinvolti nell'attivazione genica che sono: C- Fos , C-Jun , C-Myc , coinvolti nella cosiddetta risposta precoce, quindi sono espressi nella prima metà della fase G1. Per quanto riguarda le cellule cosiddette stabili, diciamo che queste possono andare incontro a proliferazione, se sottoposte a determinati stimoli. Un esempio è l'epatectomia parziale che dà luogo a un aumento della proliferazione per l'azione di agenti ossidanti, di citochine, di carico metabolico e può dare luogo a un aumento della proliferazione perché sono preponderanti i fattori di crescita, nel topo sopratutto EGF (epidermal Growth factor), ma anche FGF (fibroblast growth factor), e sono poi in equilibrio con sostanze che possono intervenire successivamente e che inibiscono la proliferazione,per cui abbiamo un aumento di questi fattori che inibiscono la crescita, in particolare per l'epitelio il TGF-Beta (trasforming growth
factor), e la riduzione dei fattori di crescita,i cosiddetti fattori adiuvanti,di cui abbiamo parlato l'altra volta (sono i fattori di promozione e progressione del ciclo mitotico)L'ipertrofia è l'aumento del volume di un tessuto per aumento dei componenti strutturali perché è espressione di un aumento della richiesta funzionale, di una richiesta lavorativa aumentata, di stimoli ormonali, che comportano un maggior impegno metabolico e funzionale, per cui aumentano gli organuli preposti alla sintesi proteica, alla respirazione ecc e quindi le cellule aumentano di volume. Dobbiamo tenere presente che, sebbene ipertrofia e iperplasia siano processi del tutto diversi tra di loro (uno comporta l'aumento del numero di cellule, l'altro del volume), non si escludono a vicenda, possono anche verificarsi frequentemente e, come abbiamo visto nelle lezioni che abbiamo fatto, sono innescate dagli stessi meccanismi, cioè richiesta funzionale o stimolo ormonale. Per esempio un aumento del volume dell'utero può essere dovuto a entrambi i processi(iperplasia e ipertrofia). In altri casi anche se le cellule possono dividersi, magari possono non rispondere a un processo di stimolo funzionale, non vanno incontro a iperplasia, ma vanno incontro a ipertrofia, se sono presenti dei fattori che inibiscono la proliferazione. Dobbiamo sempre tener conto che ogni processo biologico, fisiologico e sopratutto patologico è sottoposto al controllo di fattori che lo attivano e lo inibiscono e il risultato dipende dal prevalere degli uni o degli altri, quindi anche cellule capaci di proliferare, se sottoposte a determinati stimoli possono non proliferare e andare incontro a ipertrofia se sono presenti
49

fattori che inibiscono la proliferazione,in particolar modo il TGF-beta.Questa [slide] è la risposta del miocardio a un aumentato stretching meccanico, all'azione di alfa agonisti e fattori di crescita. Vediamo che le cellule miocardiche rispondono con un'ipertrofia, però c'è sempre un'attivazione dei geni target dei fattori di trascrizione, C-Fos, C-jun e C- Myc, che non portano all'iperplasia, ma che portano a un aumento dell'attività contrattile,aumenta la risposta allo stress, quindi aumentano le cosiddette proteine chaperon, ad esempio heat shock protein, c'è uno switch delle isoforme dell'actina e della miosina (l'actina diventa Alfa-actina simile a quella della muscolatura scheletrica) , c'è il cambiamento di isoforma dell'ATPasi Na\K dipendente e riduzione dei beta recttori. Importante da un punto di vista funzionale è l'aumento del fattore natriuretico atriale (ANP), che agisce eliminando liquidi e sodio, quindi è utile per l'attività funzionale cardiaca e, di conseguenza, anche questo è un meccanismo di adattamento e di risposta a una maggiore richiesta funzionale.L'atrofia è il contrario dell'ipertrofia, in cui si ha una riduzione del volume per riduzione delle attività metaboliche, cioè le cellule riducono le loro funzioni a quelle minime necessarie per la loro sopravvivenza. Come fanno a ridurre le loro funzioni? Riducendo i loro organuli cellulari e questo comporta proprio una distruzione di questi organuli e quindi una riduzione del volume della cellula, che diventa più piccola perché contiene un minor numero di organuli. Queste condizioni di atrofia sono diverse e ci sono varie cause. Le principali sono:
• Diminuito carico di lavoro a livello
muscolare,• Perdita di innervazione, ad esempio
nella poliomielite l'atrofia muscolare è proprio dovuta a una perdita di innervazione, quindi perdita di attività e trofismo da parte dei nervi;
• Diminuito apporto di sangue, che determina una riduzione dell'attività funzionale delle cellule,
• Inadeguata nutrizione,• Riduzione stimolazione endocrina,• Invecchiamento.
Con questo sistema le cellule cercano di stabilire un equilibrio dinamico diverso dal precedente e ne stabiliscono uno meno valido,però utile per la condizione in cui si trovano. Questo nuovo equilibrio comporta solo la sopravvivenza e non altre funzioni che la cellula non può più assolvere. Le cellule che vanno incontro ad atrofia, se lo stimolo permane, vanno in apoptosi, cioè non è una condizione che può perdurare a lungo per cui o finisce lo stimolo che ha indotto l'atrofia, e in tal caso si ritorna alla condizione precedente, oppure, se lo stimolo perdura, le cellule vanno incontro a morte programmata, l'apoptosi. Come arriva la cellula a questo risultato?
• Mediante il blocco della sintesi di nuovi organuli, infatti sapete che gli organuli sono sottoposti a un continuo turnover.
• Mediante la degradazione degli organuli già presenti.
Questo turnover è sotto il controllo di alcuni ormoni, per esempio l'insulina, gli ormoni tiroidei, i glicocorticoidi, le prostaglandine, che sono dunque coinvolti nel controllo degli organuli all'interno
50

delle cellule. Se c'è un'atrofia prolungata, quindi una condizione di degradazione proteica che può anche essere lieve, ma che è continua nel tempo, si incorre in una condizione di morte cellulare, come accade per esempio nelle distrofie muscolari. La distrofia muscolare è l'esito di questo lungo periodo di atrofia, causato da varie condizioni, che portano poi alla morte delle cellule muscolari. Ovviamente, in questo turnover di degradazione degli organuli sono molto importanti le Heat Shock Protein, che ricoprono le proteine alterate e le avviano alla distruzione a livello del proteasoma. L'atrofia, però, non sempre si realizza per attivazione del proteasoma, ma, a un certo punto, può coinvolgere anche le idrolasi lisosomiali e questo porta a un processo che si chiama autofagia, cioè la cellula mangia se stessa e questo si manifesta con la formazione di vacuoli, si perde citoplasma, si perdono gli organuli e si realizzano questi vacuoli autofagici, che sono espressione di questa attività idrolitica lisosomiale dell'atrofia. C'è un particolare tipo di atrofia, chiamata atrofia bruna, che è evidente nella cute delle persone anziane ed è causata
dall'invecchiamento e porta proprio alla formazione di questi vacuoli e alla presenza, all'interno di questi vacuoli, di alcuni granuli di lipofuscina, che danno una colorazione bruna e che sono l'espressione dei prodotti di digestione della cellula parzialmente digeriti.Tutti i comportamenti della cellula analizzati finora sono tesi al mantenimento dell'equilibrio omeostatico delle dimensioni di un tessuto. L'altra condizione che entra in gioco nel mantenimento dell'equilibrio omeostatico è la morte cellulare, prevalentemente per apoptosi. L'apoptosi è in equilibrio con i fattori che favoriscono la proliferazione per mantenere costante le dimensioni di un tessuto. Ci sono due possibilità di morte cellulare:2. Apoptosi3. NecrosiVedremo che anche in questo caso una non esclude l'altra, cioè una cellula che va incontro ad apoptosi potrebbe andare incontro anche a necrosi. Vediamo quali sono le differenze più importanti tra i due processi.
51

Partiamo dalle differenze morfologiche.Dobbiamo considerare differenze che riguardano la plasmamembrana, cioè il punto chiave che regola tutte le funzioni della cellula, non a caso nei tumori la prima ripercussione dell'alterazione dei geni è a livello della plasmamembrana. Nella necrosi la plasmamembrana è alterata, perché la necrosi è dovuta essenzialmente all'azione di tossine o a condizioni di ipossia, quindi ridotta disponibilità di ossigeno, che riduce la capacità di produrre molecole come l'ATP e questo altera la possibilità da parte della plasmamembrana di mantenere in attività la pompa Na\K e quindi si ha un accumulo di sodio all'interno della cellula e un richiamo di liquidi e questo comporta un rigonfiamento, non solo della cellula, ma anche degli organuli cellulari, compresi i lisosomi, il che porta alla liberazione di enzimi idrolitici. Sia nella necrosi sia nell'apoptosi si hanno delle alterazioni a livello della membrana plasmatica, si formano infatti queste estroflessioni che si chiamano BLEBS. Inizialmente si formano piccole estroflessioni, successivamente iniziano a esserci alterazioni a livello nucleare.Le BLEBS della necrosi si fondono una con l'altra, diventando più grandi. Un'altra cosa da notare è che all'interno di queste estroflessioni in una cellula in necrosi NON ci sono gli organuli cellulari, che rimangono al centro della cellula. Il contrario avviene nell'apoptosi, in quanto le estroflessioni NON si fondono tra di loro e in queste estroflessioni ci sono organuli cellulari ancora vitali e funzionantiAltre differenze sono a livello nucleare. Per quanto riguarda l'apoptosi a livello del nucleo si può notare la frammentazione del
DNA in piccoli pezzi di lunghezza uguale tra di loro e molto regolari, non a caso un test per vedere se la cellula è in apoptosi è proprio lo studio della frammentazione del DNA, mentre a livello della cellula in necrosi il nucleo va incontro a lisi, picnosi o caveolisi, in quanto il DNA viene frammentato in maniera disorganica e caotica dagli enzimi lisosomiali, mentre la frammentazione del DNA nell'apoptosi è dovuta ad altri enzimi che si chiamano CASPASI, che agiscono in punti precisi del DNA, cioè hanno un'azione specifica su alcuni punti della struttura del DNA, in quanto vanno a distruggere il DNA a livello dei residui di acido aspartico, presenti a livello internucleosomale. Quindi la rottura del DNA avviene a livello internucleosomale, per azione di enzimi specifici che sono le CASPASI. Nel caso della necrosi, invece, il DNA viene distrutto da enzimi lisosomiali che non hanno alcuna attività specifica. La cellula, che va incontro a necrosi, presenta poi delle soluzioni di continuità a livello della plasmamembrana, si ha, infatti, la rottura della plasmamembrana e la fuoriuscita di tutto il contenuto cellulare al di fuori della cellula. Gli organuli hanno perso la loro vitalità e non sono più funzionanti. La conseguenza della fuoriuscita del contenuto intracellulare è l'INFIAMMAZIONE. Quindi, possiamo dire che una necrosi è causa di INFIAMMAZIONE. La morte per necrosi distrugge le cellule circostanti determinando il processo infiammatorio, mentre l'apoptosi non crea nessuna risposta infiammatoria perché non c'è rottura della membrana e le BLEBS si approfondiscono sempre di più fino a formare i cosiddetti corpi apoptotici, che contengono ognuno di essi un numero di
52

organuli cellulari ancora vitali e funzionanti. Questi corpi apoptotici a loro volta vengono fagocitati, però, dato che sono vitali, hanno dei recettori sulla superficie e questo è importante perché vengono impegnati dei recettori specifici dei macrofagi, che riconoscono delle strutture presenti sui corpi apoptotici, non determinando di conseguenza la risposta da parte di geni proinfiammatori. I macrofagi hanno molti recettori sulla loro membrana, alcuni di questi attivano la via di trasduzione del segnale che porta all'attivazione di geni proinfiammatori e abbiamo l'infiammazione, altri, invece, non sono collegati e non attivano la via di trasduzione del segnale che attiva i geni poinfiammatori, ma attivano altre vie di trasduzione del segnale che determinano poi la distruzione e la formazione di corpi apoptotici, senza implicare geni proinfiammatori. I corpi apoptotici, avendo dei ligandi ben precisi, interagiscono con i macrofagi dotati dei recettori che non attivano i geni proinfiammatori, infatti la morte per apoptosi non determina infiammazione per questo motivo. Bisogna, inoltre, sottolineare che le cellule apoptotiche possono essere fagocitate non solo dai macrofagi, ma anche dalle cellule parenchimali vicine. Le cellule parenchimali, infatti, presentano funzioni fagocitiche solo ed esclusivamente nei confronti di questi corpi apoptotici. Quindi, il risultato della morte per necrosi o apoptosi è completamente diverso. L'apoptosi proprio per le sue caratteristiche e per la non produzione di infiammazione è stata paragonata alla caduta delle foglie. Si distingue, dunque, dalla necrosi, perché SOLO con l'apoptosi le cellule contribuiscono all'equilibrio
omeostatico di un tessuto ed è inoltre fondamentale per bloccare lo sviluppo delle cellule cancerose e per distruggerle. Ci sono molti tumori in cui l'apoptosi gioca un ruolo fondamentale, in quanto l'alterazione dell'apoptosi stessa è un meccanismo fondamentale attraverso il quale il tumore si sviluppa. Le leucemie sono un esempio eclatante di come la riduzione dell'apoptosi possa essere un presupposto fondamentale per lo sviluppo della malattia e per la presenza di un numero elevato di cellule cancerose. Il malfunzionamento dell'apoptosi è implicato anche in altre condizioni patologiche che non sono solo quelle neoplastiche, per esempio nelle malattie neurodegenerative, in cui l'apoptosi sia in eccesso. La morte eccessiva di cellule del sistema nervoso può per esempio determinare il Prakinson. L'AIDS è un'altra condizione in cui si ha attivazione dell'apoptosi, determinata dall'interazione della glicoproteina del virus con il recettore CD4 dei linfociti T. Quindi l'eccesso dell'apoptosi è un danno per l'organismo, per esempio nel Parkinson, nell' AIDS, nello stroke ischemico. Allo stesso modo un difetto dell'apoptosi può essere causa di danno come per esempio nei tumori. Quindi non è l'evento causa di malattia, ma l'equilibrio nel quale questo evento si inserisce, alterandolo. L'apoptosi si confronta con altri fattori, come la proliferazione. Qui ho riassunto le differenze in relazione agli stimoli, in quanto le cause sono diverse. La necrosi è dovuta essenzialmente all'azione di sostanze tossiche o a condizioni di ipossia, che alterano la capacità della cellula di produrre ATP. Diciamo quindi che una condizione fondamentale della necrosi è la
53

riduzione della produzione di ATP, cosa che non avviene nell'apoptosi, anzi la produzione di quantità sufficienti di ATP è condizione affinché si verifichi l'apoptosi: infatti, in parole povere, l'apoptosi è un processo ATTIVO (In biologia si definisce un processo attivo o passivo a seconda che si utilizzi ATP). In questo caso l'apoptosi è un processo attivo perché ha bisogno dell'ATP perché si realizzi, mentre la necrosi si realizza perché manca l'ATP. Per l'apoptosi ci sono stimoli patologici e fisiologici, quindi è innescata sicuramente da eventi patologici, ma anche da eventi fisiologici. Da un punto di vista morfologico come si presenta la cellula in necrosi e come si presenta in apoptosi? La cellula in necrosi si presenta rigonfia perché al suo interno ha liquidi, quindi sodio e acqua. La cellula che va incontro ad apoptosi è invece una cellula raggrinzita. E' come se la cellula si raggrinzisse (Si dice shrinked = raggrinzito). La cellula può entrare in apoptosi anche senza andare incontro ad atrofia. Nell'apoptosi si attivano questi enzimi che vanno a frammentare il DNA, ma vanno a frammentare anche altri substrati, per cui la cellula si raggrinzisce. Certo è che la cellula non si rigonfia perché l'ATP è bello e funzionante, però c'è proprio il raggrinzimento perché questi enzimi, che si chiamano CASPASI, idrolizzano molti substrati, per cui la cellula si raggrinzisce. Dunque qui non c'entra il discorso dell'osmosi, qui c'entra l'attivazione di enzimi che distruggono strutture cellulari, per cui la cellula va incontro a raggrinzimento.Un altro concetto importante, proprio per le cause che riguardano la necrosi e l'apoptosi, è il coinvolgimento delle cellule, cioè nella necrosi sono coinvolte molte
cellule, si può dire che sia coinvolto tutto il tessuto, perché è inverosimile che una o due cellule siano sottoposte all'azione di tossine o a ipossia, in quanto, in una possibile condizione di ipossia, è coinvolto tutto il tessuto o se ci sono delle tossine, queste riguardano un grande numero di cellule. Invece, l'apoptosi riguarda la singola cellula o al massimo due cellule, in quanto è un processo attivato dalla stessa cellula esposta a un determinato stimolo fisiologico o patologico. L'apoptosi dunque si realizza o per un’interazione tra ligandi e recettori presenti sulla cellula o perché la cellula ha subito un grave danno al DNA e decide di attivare il programma di morte per evitare di trasmettere il danno alle cellule figlie, quindi è una decisione presa da una cellula di fronte a un determinato stimolo. Quindi, è l'attivazione di un programma (non a caso si parla di morte programmata) che si trova all'interno della cellula, dunque è un meccanismo volontario della cellula, che decide di attivarlo. Nella necrosi è coinvolta una massa cellulare, perché è la massa esposta all'ipossia o all'azione di tossine, quindi è un evento passivo, cioè le cellule sono danneggiate o dalla carenza di ossigeno o dalla presenza di tossine.Nell'apoptosi la cromatina viene condensata, quindi la frammentazione del DNA determina la condensazione della cromatina, cosa che non avviene invece nella necrosi. Nella necrosi abbiamo la distruzione degli organelli in seguito all'ingresso di sodio e di acqua, proprio per questo gradiente di osmolarità che si realizza nella necrosi; organelli che si mantengono vitali all'interno dei corpi apoptotici, anche se ridotti in seguito ad atrofia. Il meccanismo che porta alla necrosi è
54

quello mediato dalla deplezione di ATP, in quanto le cause che inducono la necrosi sono tutte cause che determinano una diminuzione della produzione di ATP, cosa che non si verifica nell'apoptosi. Quindi, qual è il meccanismo dell'apoptosi? L'attivazione genica, cioè il "programma" è scritto sui cosiddetti geni della morte (è brutto dirlo, ma è così), che vengono attivati. Quindi si ha un processo di attivazione genica, quindi un processo attivo, che richiede ATP e che determina l'attivazione di geni prima poco espressi o non espressi o espressi a livello [uno starnuto non mi permette di capire bene la parola]. Questa attivazione genica comporta a sua volta l'attivazione di alcuni enzimi, tra cui le ENDONUCLEASI, ma anche di altri enzimi che intervengono nell'apoptosi. Nell'apoptosi non c'è danno di membrana che invece è presente nella necrosi e che è dovuto a un'alterazione della pompa Na\K per deplezione dell'ATP e, inoltre,a causa di un aumento di acqua c'è anche un'attivazione dei radicali liberi: infatti c'è un danno da radicali liberi, che non c'è nell'apoptosi.La reazione tissutale è diversa perché nella necrosi si ha l'infiammazione, nell'apoptosi non si ha infiammazione perché i corpi apoptotici vengono fagocitati. Voi sicuramente sapete che la fagocitosi è alla base dell'infiammazione, non c'è infiammazione senza fagocitosi, per cui quando vi dico che i corpi apoptotici vengono fagocitati potete non ritrovarvi. Però vi ho spiegato che i corpi apoptotici all'interno contengono organuli perfettamente vitali e sulla loro membrana, perfettamente integra, presentano dei ligandi che interagiscono con dei recettori sui macrofagi che non attivano i geni proinfiammatori, ecco perché non c'è
infiammazione nonostante ci sia la fagocitosi.Se non ci fosse l'apoptosi, non si avrebbe uno sviluppo armonico e ordinato durante lo sviluppo embrionale, in quanto durante lo sviluppo embrionale c'è una tumultuosa proliferazione e replicazione cellulare, che porta a un eccesso di cellule che poi devono essere eliminate proprio attraverso l'apoptosi.Caratteri fondamentali dell'apoptosi sono dunque lo shrinkage, quindi questo raggrinzimento, che rende la cellula più piccola, per cui il citoplasma risulta più denso e gli organelli sono ammassati. La cromatina si addensa alla periferia, subito sotto la membrana nucleare e queste masse sono ben definite, proprio perché intervengono questi enzimi che agisco a livello internucleosomale, quindi sono ben definiti, anche se hanno varie forme di grandezza. Anche il nucleo si può frammentare. A livello della membrana si formano queste BLEBS, che poi vanno incontro a un approfondimento maggiore fino a quando si frammenta la cellula e si formano i corpi apoptotici, rivestiti da membrana integra e formati da citoplasma e da organelli impacchettati, ma ancora vitali. La fagocitosi si realizza anche da parte delle cellule parenchimali adiacenti, oltre che da parte dei macrofagi. La cosa sorprendente è che quando una cellula andata incontro ad apoptosi viene fagocitata, nell'uno o nell'altro modo, immediatamente viene sostituita da una cellula nuova, che va a ricoprire il vuoto lasciato dalla cellula apoptotica, senza che vi sia danno a livello tissutale e questo è alla base del mantenimento dell'equilibrio omeostatico. L'importanza dell'apoptosi nello sviluppo embrionale è data dal comportamento di questa molecola di
55

segnale morfogenetico, che si chiama SHH ( Sonic Hedgehog Homolog), molecola connessa con l'apoptosi e fondamentale nello sviluppo degli organi dell'embrione di drosophila perché agisce secondo il proprio gradiente di concentrazione, cioè viene prodotta dalla notocorda e poi si diffonde in base a un gradiente di concentrazione e in base al gradiente di concentrazione stesso si ha lo sviluppo di vari organi e apparati. Questa molecola interagisce con un suo recettore che si chiama PTC1 (Patched One) e questo legame induce le cellule a proliferare e a sopravvivere. Se però questo recettore rimane privo del suo ligando, perché l'SHH viene prodotta in minore quantità o perché c'è un gradiente in cui la concentrazione di questo fattore morfogenetico è bassa, il recettore stesso perde la sua porzione intracitoplasmatica, che viene idrolizzata, e espone un dominio che attiva l'apoptosi. Quindi lo stesso processo può indurre o proliferazione e sopravvivenza o morte cellulare a seconda della concentrazione di questo fattore SHH, per cui se questo manca o è ridotta la sua concentrazione, si ha l'attivazione dell'apoptosi perché viene degradata la porzione intracitoplasmatica del recettore Patched 1 e quindi vengono attivate le CASPASI. Il gene si chiama HH e controlla la segmentazione dell'embrione di Drosophila e si è visto che nei moscerini in cui era alterato, mutato o deleto questo gene e quindi il fattore morfogenetico non veniva prodotto, il moscerino in questione assumeva un aspetto genetico molto strano caratterizzato da estroflessioni che somigliavano alle spine del riccio o dell'istrice, da questo deriva il nome Hedgehog (= istrice,riccio). Vi sto parlando
di questa molecola non solo per farvi capire che nei processi proliferativi si possono innescare, in determinate condizioni, processi apoptotici, ma anche perché questa molecola è importante per la sopravvivenza e per la proliferazione cellulare in età adulta, non solo nel periodo embrionale, perché è coinvolta nella divisione della cellula staminale e può essere coinvolta anche nella patogenesi di alcuni tipi di cancro.L'apoptosi è fondamentale anche nella risposta immunitaria, in quanto durante il processo di sviluppo dei linfociti T si possono formare anche dei cloni di cellule autoreattive che devono essere eliminate mediante l'apoptosi: infatti, se non ci fosse un adeguato processo apoptotico, noi andremmo incontro a malattie autoimmuni. Anche l'attività citotossica dei CD8+ è mediata da un processo apoptotico un po' particolare perché, interagendo con la cellula target, liberano perforina che determina la formazioni di pori a livello della membrana e così inoculano Granzyme che è una serin-proteasi. Le CASPASI si chiamano così perchè C= cisteina ASP = acido aspartico, cioè il sito attivo è dato dalla cisteina e idrolizzano le proteine a livello dell'acido aspartico. Ora il Granzyme va sempre ad agire sull'acido aspartico, ma non è una cistein-proteasi, bensì una serin-proteasi, quindi il suo sito attivo è dato dalla serina, non dalla cisteina per cui è una CASPASI-simile. Dell'apoptosi si dice che è una morte pulita, perché non determina infiammazione, e programmata, perché determina l'attivazione dei geni della morte e altruista. Noi non abbiamo le mani palmate perché, per fortuna, abbiamo avuto l'apoptosi che ci ha distrutto le membrane interdigitali.
56
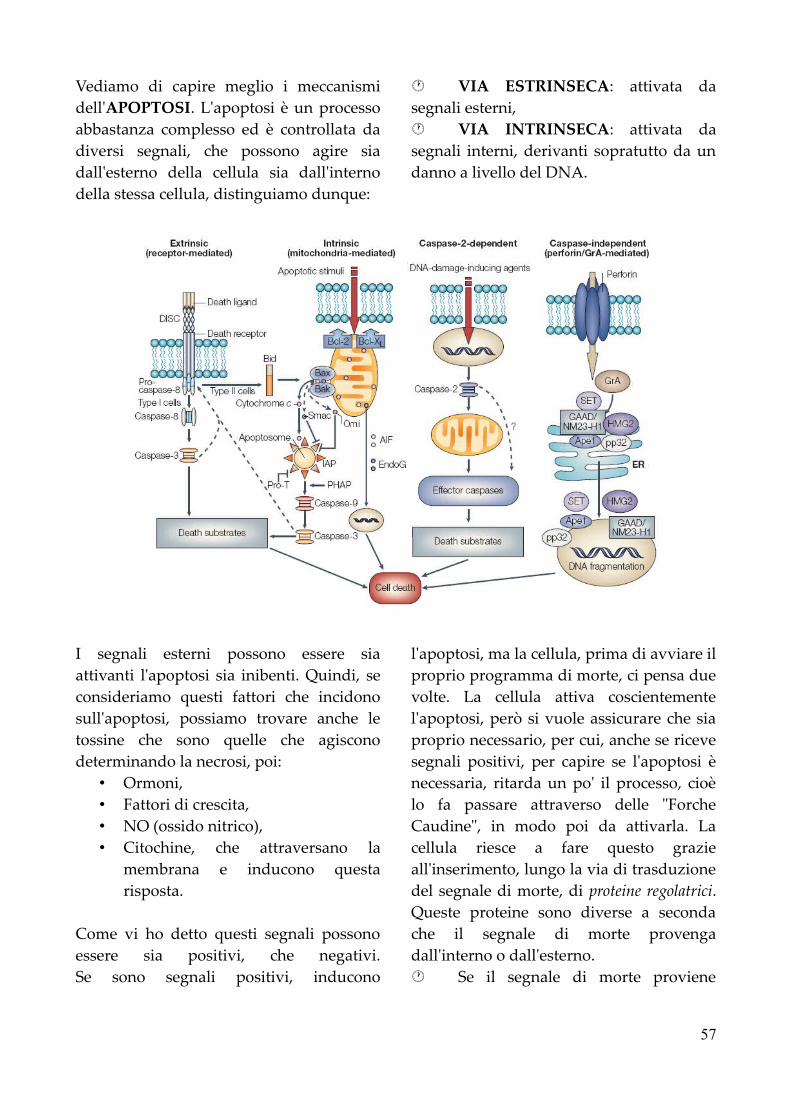
Vediamo di capire meglio i meccanismi dell'APOPTOSI. L'apoptosi è un processo abbastanza complesso ed è controllata da diversi segnali, che possono agire sia dall'esterno della cellula sia dall'interno della stessa cellula, distinguiamo dunque:
VIA ESTRINSECA: attivata da segnali esterni, VIA INTRINSECA: attivata da segnali interni, derivanti sopratutto da un danno a livello del DNA.
I segnali esterni possono essere sia attivanti l'apoptosi sia inibenti. Quindi, se consideriamo questi fattori che incidono sull'apoptosi, possiamo trovare anche le tossine che sono quelle che agiscono determinando la necrosi, poi:
• Ormoni,• Fattori di crescita,• NO (ossido nitrico),• Citochine, che attraversano la
membrana e inducono questa risposta.
Come vi ho detto questi segnali possono essere sia positivi, che negativi. Se sono segnali positivi, inducono
l'apoptosi, ma la cellula, prima di avviare il proprio programma di morte, ci pensa due volte. La cellula attiva coscientemente l'apoptosi, però si vuole assicurare che sia proprio necessario, per cui, anche se riceve segnali positivi, per capire se l'apoptosi è necessaria, ritarda un po' il processo, cioè lo fa passare attraverso delle "Forche Caudine", in modo poi da attivarla. La cellula riesce a fare questo grazie all'inserimento, lungo la via di trasduzione del segnale di morte, di proteine regolatrici. Queste proteine sono diverse a seconda che il segnale di morte provenga dall'interno o dall'esterno. Se il segnale di morte proviene
57

dall'interno queste proteine regolatrici sono coinvolte nella funzionalità mitocondriale, quindi si tratta di proteine della famiglia BCL, coinvolte nella regolazione della permeabilità mitocondriale. Sono proteine regolatrici perché una volta che viene alterata la permeabilità mitocondriale, viene attivata la via apoptotica intrinseca. Se il segnale di morte proviene dall'esterno le proteine regolatrici sono caratterizzate da proteine adattatrici,cioè proteine che si inseriscono nella porzione intracitoplasmatica del recettore della morte. Infatti, i segnali esterni vanno a interagire con dei recettori della morte presenti sulle cellule. Una volta che è avvenuta l'interazione, non si ha subito l'attivazione dell'apoptosi, ma a livello del recettore, impegnato con il ligando della morte, vanno ad inserirsi delle proteine cosiddette adattatrici,che a loro volta legano le CASPASI, che così si attivano. Ci sono, dunque, degli intermediari tra il recettore, che interagisce con il ligando della morte, e le CASPASI, che poi sono quelle responsabili dell'apoptosi. Quindi l'apoptosi si attiva, se per esempio il segnale perdura,ma se la cellula si rende conto che è possibile evitare l'apoptosi, allora in questo modo può non attivare il programma di morte cellulare.
I fattori che inducono i segnali apoptotici esterni possono essere rappresentati dal ligando del TNFR, sono recettori della famiglia del TNF (Tumor Necrosis Factor) e ve ne sono di due tipi:
• TNFR 1;• TNFR 2.
Un altro recettore è il FAS (anche chiamato CD95), che interagisce con un ligando che si chiama Fas-ligand. Ricapitolando
abbiamo due tipi recettori: TNFR (1 e 2), recettore per il TNF, FAS (CD95), recettore per il FAS-ligando.
In realtà c'è un terzo tipo di recettore che si chiama DR4- DR5Questi sono i principali recettori che legano i ligandi che attivano l'apoptosi, ma ci sono anche altre condizioni che attivano l'apoptosi, come il calore, le radiazioni, la carenza di sostanze nutritive che portano prima all'atrofia, poi all'apoptosi. Ancora possiamo menzionare i glicocorticoidi: infatti, un eccesso di terapia cortisonica può portare a una deplezione di linfociti. Prima si ha una spremitura del midollo e quindi una maggiore presenza di linfociti T in circolo,ma una prolungata terapia cortisonica può proprio portare a una deplezione centrale dei linfociti, per questo non bisogna mai esagerare con la terapia cortisonica, se non strettamente necessario.Una delle risposte più rapide quando viene attivata l'apoptosi è la degradazione di alcuni enzimi chiave della riparazione del DNA. Cioè alcuni enzimi implicati nella riparazione del DNA vengono subito eliminati nel momento in cui viene attivata l'apoptosi, per evitare, in caso di danno eccessivo che richieda troppo tempo per la riparazione, che lo stesso danno venga trasmesso alle cellule figlie. Viene quindi disattivato, per prima cosa, un enzima importante nella riparazione del DNA che è l'enzima PARP (Poli-ADP-ribosio polimerasi), che fa parte del complesso del riparosoma e che recupera molecole di poliribosio da parte del NAD e li porta in substrati fondamentali per la riparazione del DNA. Quindi è un enzima che depaupera il NAD di molecole di ribosio e li porta a livello del DNA danneggiato:
58

infatti PARP riconosce proprio siti del DNA danneggiato e ha la capacità di polimerizzare e quindi di riparare il DNA. PARP presenta tre domini:
• DNA-binding, che lega il DNA;• Dominio catalitico, che ha questa
attività polimerasica;• Dominio di auto modificazione, in
quanto tutti questi enzimi polifunzionali hanno la capacità di modificare la loro conformazione.
Durante l'apoptosi PARP viene idrolizzata dalle CASPASI, che ne determinano la frammentazione in due porzioni:
• Uno molto piccolo di 24 kD che interagisce con il DNA danneggiato;
• Un'altra parte che contiene la porzione catalitica e che rimane con il riparosoma.
La presenza di questo frammento legato al sito del DNA impedisce il legame delle endonucleasi al DNA stesso, quindi non solo viene impedito il processo di riparazione mediato da PARP, ma viene impedito anche qualsiasi altro processo di riparazione, perché il sito è coperto dal dominio DNA-binding del PARP, quindi si impedisce l'accesso alle endonucleasi e quindi il processo di riparazione (le endonucleasi scindono per poi polimerizzare e riprodurre il nuovo frammento).Ricapitolando: la grossa porzione del PARP rimane legato al riparosoma ed è costituito dal dominio catalitico e dal dominio di auto modificazione, mentre il dominio DNA-binding rimane invece legato al sito danneggiato del DNA, impedendo l'azione di qualsiasi enzima coinvolto nella riparazione, lo lascia danneggiato. Questa scissione di PARP
agisce in due modi: 1. Mediante un impedimento sterico dovuto al legame del dominio DNA-Binding con il DNA danneggiato, 2. Mediante l'altra porzione rimasta legata al riparosoma, che ovviamente non è più efficiente e funzionante, per la mancanza appunto del DNA-binding.
Ci possono essere delle conseguenze diverse se questo processo non è sotto controllo, perché l'attivazione del PARP, in condizione di normalità, può essere utile alla riparazione del DNA, in quanto questo è uno stimolo dato da agonisti di un recettore NMDA di glutammato, che agisce attraverso questo recettore e porta alla produzione di ossido nitrico. Se l'ossido nitrico è a basse concentrazioni (L'ossido nitrico, sapete, è un potentissimo radicale libero, coinvolto anche nell'infiammazione, infatti è un mediatore gassoso dell'infiammazione), determina un danno del DNA e se il danno è breve si ha l'attivazione della PARP e la riparazione, se invece il danno al DNA è eccessivo, si ha un over-attivazione di PARP con deplezione del NAD, perché va a rifornirsi di ribosio a livello del NAD, per cui si ha una ribosilazione del NAD eccessiva, dunque deplezione del NAD, riduzione di ATP e quindi anche morte cellulare. Questo è il meccanismo del duplice effetto dell'attivazione di PARP. Consideriamo l'apoptosi attivata dal meccanismo intrinseco, quindi danno del DNA. Abbiamo detto che ci sono delle proteine regolatrici, che regolano la risposta apoptotica, abbiamo infatti visto poco fa che c'è questo meccanismo intermedio, che cerca di vedere se effettivamente è necessario arrivare all'apoptosi e che regolano la permeabilità
59

della membrana mitocondriale. Per esempio l'ossido nitrico, essendo radicale libero, agisce a livello del potenziale della membrana mitocondriale, aumentando la permeabilità e questo porta a due eventi fondamentali.
• Liberazione del citocromo C,• Liberazione di alcuni prodotti che
attivano l'apoptosi, presenti sempre a livello del mitocondrio e questi prodotti si chiamano SMAC\ DIABLO.
SMAC = Second Mitochondria-derived Activator of Caspases [La prof ha detto "apoptosis",ma credo si sia sbagliata perchè su Wikipedia e sul libro c'è scritto "Caspases"] e sono secondari all'aumento della permeabilità mitocondriale.Il citocromo C attiva, con le molecole adattatrici che si chiamano APAF, le varie CASPASI e quindi meccanismo intrinseco dell'apoptosi. Le altre molecole che si chiamano SMAC attivano anch'esse l'apoptosi, ma con un altro meccanismo.Le SMAC/DIABLO sono a loro volta controbilanciate da alcune proteine che si chiamano IAP ( Inhibitors of Apoptosis protein) e sono proteine inibitrici dell'apoptosi. Quindi ogni evento, in questo caso l'apoptosi, è regolato da fattori che lo inibiscono e fattori che lo attivano. Vediamo le SMAC, che sono bersaglio delle IAP, ma le IAP non agiscono solo a livello delle SMAC, ma anche a livello delle CASPASI, cioè gli enzimi proprio effettori dell'apoptosi. Queste CASPASI sono enzimi specifici che agiscono sull'acido aspartico, hanno come sito attivo la cisteina ed effettuano la loro azione su diversi substrati. A parte la PARP, di cui già abbiamo parlato, agiscono su varie proteine chiave del metabolismo e
dell'attività funzionale della cellula, per esempio le LAMINE, che si trovano a ridosso della membrana nucleare e che legano il DNA, oppure le protein-chinasi DNA dipendenti, quindi tutti quei fattori che attivano i processi di riparazione e attivano un fattore di degradazione del DNA che determina frammentazione del DNA stesso. Dunque le CASPASI attivano proprio questo fattore di frammentazione, che si chiama DFF (DNA fragmentation factor) e questo determina appunto una delle caratteristiche principali dell'apoptosi che è il DNA in frammenti ben ordinati. Le CASPASI possono essere distinte in:- CASPASI iniziatrici,- CASPASI effettrici.Quindi, possiamo dire che anche questo è un processo che procede secondo delle tappe, cioè le CASPASI non vengono subito attivate. Se parliamo della via estrinseca parliamo di ligando ―› recettore ―› proteina adattatrice ―› attivazione di CASPASI iniziatrici ―› attivazione di CASPASI effettrici. Quindi vedete che c'è un pathway abbastanza complesso formato da più step, che porta sicuramente alla morte cellulare, ma è anche vero che a livello di ciascuno stadio il processo si può interrompere, quindi non necessariamente la cellula va incontro ad apoptosi. L'apoptosi è un processo così importante per l'economia del nostro organismo, che si è mantenuto costante nel tempo il meccanismo con cui si realizza, non a caso ci sono proteine omologhe alle proteine apoptotiche dei mammiferi, che sono state riscontrate addirittura in un nematode,nel Caenorhabditis Elegans. In quest'ultimo è stata individuata una proteina, che si chiama CED 3, che ha molte omologie con la CASPASI 1 dei mammiferi, che è anche un enzima che converte l'interleuchina 1
60

(Interleukin-1 converting enzyme). Questo è un altro collegamento con l'infiammazione, in quanto l'interleuchina 1 è una citochina coinvolta in vari processi biologici, prodotta dai fagociti, nel riconoscimento e presentazione dell'antigene ed è coinvolta anche nell'apoptosi, perché appunto la CASPASI idrolizza l'interleuchina 1-beta. Quindi, abbiamo detto, CED 3 di C. Elegans è un omologo di CASPASI 1 dei mammiferi e si chiama ICE (Interleukin-1 converting enzyme). Altre molecole omologhe sono il CED 4 e il CED 9. IL CED 4 è omologo di una proteina coinvolta nella via intrinseca,cioè APAF di cui ho parlato poco fa, mentre CED 9 è una molecola che inibisce l'apoptosi, che attiva la proliferazione e che è omologa a BCL2. Cioè nel C. Elegans vi sono tre proteine coinvolte nell'apoptosi:
• CED3 omologo a CASPASI 1, coinvolta nella via estrinseca
• CED4 omologo della molecola adattatrice APAF, coinvolta nella via intrinseca
• CED9 omologo a BCL2, anti-apoptotica, attiva la proliferazione.
Vediamo appunto come nel C. Elegans CED 9 blocca CED 3 e CED 4, che invece attivano la morte cellulare. Nei mammiferi una cosa simile viene effettuata da BCL 2 che agisce su una CASPASI omologa che attiva la morte cellulare.Nella Drosophila Melanogaster abbiamo un gene simile che ha un meccanismo sovrapponibile a quello dei mammiferi.La via intrinseca è attivata da problemi interni alla cellula, come alterazione del DNA, stress ossidativi, come per esempio il danno causato dall'ossido nitrico o dai radicali liberi dell'ossigeno, da un danno
mitocondriale. La via estrinseca è mediata da ligandi che agiscono con recettori specifici presenti sulla cellula.Nella via intrinseca abbiamo la lesione a livello della membrana mitocondriale, che comporta, oltre alla liberazione di SMAC, anche la librazione di citocromo C, fondamentale nell'attivazione dell'apoptosi per via intrinseca. Il citocromo C, infatti, si lega a una proteina adattatrice che è APAF-1 (apoptotic protein activating factor, cioè fattore di attivazione delle proteine coinvolte nell'apoptosi). L'APAF-1 si ritrova in forma inattiva, ma viene attivata dal legame con il citocromo C, liberato in seguito a danno della membrana mitocondriale. Questo evento porta all'aggregazione di 7 molecole di APAF-1 tra di loro. Queste molecole adattatrici ovviamente sono polifunzionali, cioè hanno vari domini, in quanto devono legare il citocromo C, devono interagire tra di loro e devono reclutare le CASPASI per attivarle, quindi, con un dominio che si chiama CARD (Caspase Recruitment Domain), reclutano la procaspasi 9, che è la forma inattiva e la inserisce in un complesso che si chiama APOPTOSOMA e, a questo livello, la procaspasi 9 viene attivata, mediante un clivaggio, in CASPASI 9. Questa è la CASPASI iniziatrice della via intrinseca, in quanto a sua volta attiva le CASPASI a valle, cioè le CASPASI effettrici, che sono essenzialmente la CASPASI 3 e la CASPASI 7, quindi avviene l'attivazione della cosi detta "Cascata delle CASPASI".A livello dell'apoptosoma abbiamo dunque:1. Dominio CARD, che serve a reclutare la procaspasi 9;2. Dominio simile a CED 4, quindi abbiamo questo dominio che è simile alla
61

proteina CED4 importante nell'apoptosi del nematode (per questo vi ho parlato del C.Elegans);3. Dominio C-terminale, che si chiama WD40. Questo è un dominio che presenta 12 o 13 unità di ripetizione, ogni unità, di queste 12 o 13, è costituita da 40 residui di acido aspartico e di triptofano. Questo è un elemento regolatore negativo, che interagisce con CARD, ma che probabilmente può interagire anche con altre molecole regolatrici, quindi con altre molecole adattatrici dell'apoptosi. Notate come l'apoptosi si autoregoli. E' importante sottolineare come l'espressione di APAF-1 sia sotto il controllo di p53. Quando il citocromo C fuoriesce dal mitocondrio, si lega ad APAF 1 modificandone la conformazione. Infatti, da una conformazione chiusa, grazie al legame con il citocromo C e
successivamente con le molecole di ATP che fuoriescono dal mitocondrio (ecco perché è fondamentale l'ATP), si passa a una conformazione aperta. Quindi sette molecole di APAF-1 si aggregano tra di loro e, attraverso il dominio CARD, reclutano le molecole di procaspasi-9, formano l'apoptosoma e attivano le procaspasi-9, che diventano CASPASI 9. Vedete quindi l'attivazione di procaspasi 3 e procaspasi 7 e l'attivazione dell'apoptosi. Il dominio CED 4 - like è importante per questo cambiamento conformazionale e i domini WD40 interagiscono con il citocromo C.
[Nota: nelle sbob comprate in copisteria la stessa Prof.ssa Teti afferma che la proteina CED3 è omologa alla CASPASI 3, compresa tra le CASPASI effettrici. Nonostante ciò, nella registrazione si sente chiaramente che la prof parla di CASPASI 1 o non di CASPASI 3]
Francesca Galletta
62

06/11/2013 Prof.ssa Teti
5Meccanismi ApoptoticiMeccanismi Apoptotici
Patologia cellulaPatologia cellula neoplasticaneoplastica
(la prof.ssa Teti inizia la lezione riassumendo in breve qualcosa riguardo la lezione precedente)Riprendiamo oggi l’argomento dell’apoptosi trattato nella lezione precedente!!Sono chiari i vari domini del complesso di APAF-1? E le modalità con ci esso modifica la sua conformazione? Ricordiamo che il primo cambiamento conformazionale avviene grazie al legame con il citocromo C mentre il secondo cambiamento conformazionale è dato dal legame con l’ATP. Infatti l’apoptosi è un azione cellulare che RICHIEDE ENERGIA proprio perché l’ATP serve per modificare la conformazione di APAF in modo che si dispieghi la molecola, in modo che possano associarsi 7 molecole di APAF-1 con 7 MOLECOLE DI PRO-CASPASI 9 formando il complesso dell’APOPTOSOMA!…………………………………La prof.ssa proietta una slide nella quale sono rappresentate le due vie INTRINSECA ED ESTRINSECA DELL’APOPTOSI.Come notate dall’immagine, abbiamo una rappresentazione delle due vie dell’apoptosi, intrinseca ed estrinseca.
4. VIA ESTRINSECA è caratterizzate
63

dall’impegno di RECETTORI DI SUPERFICIE. Sono essenzialmente di tre tipi: - RECETTORI FAS/ FAS-L- RECETTORI TNFR1-TNFR2 (famiglia di recettori che lega il TNF alfa)- RECETTORI PER I TRIAL
5. VIA INTRINSECA mediata da alterazioni portate alla cellula soprattutto a livello del DNA.
(LA PROF.ssa inizia a fare esempi per ciascuna via ma questi saranno poi successivamente ripetuti durante la lezione e approfonditi)ESEMPIO VIA ESTRINSECA :Qui è rappresentato principalmente la via estrinseca mediata dal recettore FAS chiamato anche CD95. È il recettore per il LIGANDO FAS (FAS-LIGAND O FAS-L) che è un componente della famiglia del TNF. Il legame del ligando con il suo recettore (lo vedremo meglio più tardi durante la lezione) comporta il fatto che a livello del recettore che si lega con il FAS-L si debba inserire una MOLECOLA ADATTATRICE. La cellula infatti prima di inoltrare il programma apoptotico deve rendersi conto se esso risulta essere necessario o meno. Per cui risulterà rallentato il pathway che porta alla morte cellulare mediante appunto l’inserimento di queste MOLECOLE ADATTATRICI. Quella che interviene legandosi al recettore dopo che esso si è a sua volta legato con il FAS-L si chiama FADD (FAS associated DEATH DOMAIN). Questi Recettori e le molecole adattatrici presentano dei domini particolari chiamati DOMINI DELLA MORTE (DD) perché implicati nella realizzazione del programma di morte cellulare. Per cui FADD è una PROTEINA CHE CONTINE DOMINI DELLA MORTE (DD) , associata al recettore FAS. Essendo una molecola adattatrice ha due regioni diverse:
Regione che si lega a FAS Regione che lega la PRO-CASPASI 8 che è coinvolta appunto nella via di
attivazione estrinseca. In questo caso i domini non saranno più DD ma DE (death effector ) che interagiscono appunto con la pro-caspasi otto che attivata a CASPASI 8 ATTIVERA’ LA PROCASPASI-3 . La pro-caspasi 8 fa parte della caspasi INIZIATRICI mentre la 3 delle EFFETRICI.
64

Notiamo dunque che FADD presenta una doppia faccia come una medaglia: DOMINI DD e domini DE che legano rispettivamente FAS E PRO-CASPASI 8.Tutto questo complesso che si viene a formare ovvero : FAS-L / FAS/ FADD/ PRO-CASPASI 8 è definito come DISC (DEATH INDUCING SIGNALLING COMPLEX). È un complesso di segnalazione che induce la MORTE, che attiva la caspasi 3. MA: PRO-CASPASI 3 è anche attivato da un’altra caspasi iniziatrice quale è la CASPASI 9 che è attivata invece mediante la via INTRINSECA.La via intrinseca si gioca tutta a livello della PERMEABILITA’ MITOCONDRIALE DOVE AVVERRA’ UNA SORTA DI LOTTA.
Sono coinvolte sia la P53 che non agisce direttamente sulla membrana mitocondriale, ma agisce attraverso l’attivazione di alcune proteine che invece modificano la permeabilità della membrana, sia membri della famiglia BCL-2 che è costituita da membri con azioni contrastanti, alcuni dei quali INIBISCONO L’APOPTOSI e altri che invece FAVORISCONO L’APOPTOSI, quindi sono ANTI-PROLIFERATIVI O PRO-APOPTOTICI.
(la prof.ssa si riferisce spesso alla famiglia delle bcl-2 utilizzando la denominazione di “FAMIGLIA DELLE BCL”. Ho controllato su internet ed entrambi le denominazioni sono corrette .)
Ad esempio : BCL-2 a sua volta è il nome di un MEMBRO della stessa famiglia delle BCL , e si tratta del prodotto di un PROTONCOGENE che attiva la proliferazione cellulare e INIBISCE l’apoptosi. Per cui A LIVELLO DELLA MEBRANA MITOCONDRIALE grazie ai membri della famiglia di BCL-2 pro-apoptotici si avrà un aumento della permeabilità mitocondriale, la fuoriuscita di citocromo c, l’assemblamento dell’apoptosoma, reclutamento di pro.-caspasi9 che si attiva a caspasi 9 che avrà come target sempre lo stesso membro attivato anche dalla pro-caspasi 8 che è la PRO-CASPASI 3.
CASPASI E PROCASPASI 3>> L’azione su PRO-CASPASI 3 che diventa CASPASI 3 è mediata anche da un altro composto che prende il nome di PAC-1 (si legge in inglese pac one) che sta per first procaspase activating compound il quale agirà su residui di ACIDO ASPARTICO presenti sulla PRO-CASPASI 3 .PRO-CASPASI 3 nella sua forma lineare presenta: (immagine in basso )
PRO-DOMINIO che viene scisso nella sua attivazione da PRO-CASPASI 3 a CASPASI3
65

Domini P17-P12 che vengono scissi in mezzo da PAC-1 a livello dell’acido aspartico in posizione 175 . Una volta liberati I monomeri p17 e p12 si assembleranno in ETEROTETRAMERI e diventa così la caspasi 3 attiva
SITO SAFETY CATH composto da 3 residui di acido aspartico (TRA 179-181) , livello dei quali agisce sempre PAC-1
È importante ricordare anche la CISTEINA 163 che rappresenta il sito catalitico della PRO-caspasi 3. Ricordiamo che le caspasi sono cistein proteasi pechè appunto hanno il sito attivo costituito dalla cisteina; in questo caso si tratta della cisteina 163 che dà l’azione
QUALI SONO I SUBSTRATI DELLA CASPASI 3?Sono presi qui in considerazione soprattutto quelli che agiscono a livello del DNA:
66

PARP (primo enzima idrolizzato e quindi inattivato)
COMPLESSO CARD- ICAD (proteina chaperon di CAD).L’acronimo CAD sta per CASPASE ACTIVATED
DESOSSIRIBONUCLEASE. Essa frammenta il DNA IN MANIERA REGOLARE ed è ATTIVATA DALLE CASPASI. Infatti la caspasi libera il CAD dalla sua molecola inibitrice, che la blocca ovvero I-CAD (INHIBITOR OF CAD).Per cui soltanto idrolizzando I-CAD, CAD viene liberato e può agire a livello inter nucleosomale. Questo è uno degli enzimi coinvolti nella frammentazione precisa e regolare del Dna in quanto ce ne sono altri.
DFF (FATTORE DI FRAMMENTAZIONE DNA) costituit0 in maniera simile a CAD ed I-CAD da DUE SUBUNITA’ : 45 E 40. Sotto questa forma eterodimerica è inattivo. La caspasi 3 idrolizza il fattore di frammentazione 45, liberando il fattore di frammentazione del DNA 40 che si OLIGOMERIZZA unendosi ad altre molecole di DF40 e da ciò esso sarà in grado di IDROLIZZARE IL DNA.
Anche qui così (come in CAD) abbiamo un discorso di regolazione dovuta al fatto che condizioni NON DI MORTE CELLULARE, le desossiribonucleasi sono INATTIVE: una CAD, bloccata da I-CAD, idrolizzata da caspasi 3 ed il DFF perché sotto forma di eterodimero inattivo.
67

MODELLI FAMIGLIE RECETTORI : VIA ESTRINSECA APOPTOSI
1. RECETTORE FAS/FAS-L :
Quando FAS interagisce con il suo ligando, si assiste ad un processo di TRIMERIZZAZIONE di FAS PRIMA E DI FADD DOPO. Tutti i recettori, anche quelli che hanno come ligandi i fattori di crescita, quando legano il ligando tendono a dimerizzare, questo del FAS è un processo simile solo che invece di una dimerizzazione avremo una TRIMERIZZAZIONE del recettore. A questo punto (come già detto in precedenza) vi sarà il reclutamento di FADD che si legherà al FAS mediante i domini DD (della morte). Vi è un certo numero di molecole FADD che interagiscono con il DEATH DOMAIN POSSEDUTO ANCHE DA FAS.A livello di FADD sono messi in evidenza anche i DOMINI EFFETTORI DELLA MORTE (DE) che reclutano la pro-caspasi 8, formando il complesso DISC.Questo è il modello in cui FADD si lega sulla faccia ESTERNA della coda citoplasmatica di FAS ma potrebbe anche legarsi sulla superficie interna.
PUNTI DI CONTATTO TRA VIA INTRINSECA ED ESTRINSECA
Abbiamo parlato di apoptosi e di via intrinseca ed estrinseca come se fossero due processi separati; in realtà ci sono sempre delle interazioni tra le due. Così come avviene nella coagulazione dove avrò vari punti di contatto tra via intrinseca e via estrinseca, così anche nella cascata dell’apoptosi troveremo diversi punti di contatto. (fare riferimento all’immagine nella pagina seguente)
Quando Caspasi 8 viene attivata dal legame con FADD; abbiamo visto poco fa che attiva PRO-CASPASI 3 IN CASPASI 3 ma, non solo, la CASPASI 8 è anche in grado di agire IDROLIZZANDO UN MEMBRO DELLA FAMIGLIA BCL chiamato BID (bcl-2 interacting domain) che è un DOMINIO DI INTERAZIONE CON BCL-2.
68

Succede che BID idrolizzato da CASPASI 8 diventa una piccola molecola detta T-BID (truncated bid), trattasi della porzione C-TERMINALE, che è in grado di interagire con la plasmamebrana mitocondriale , alterandone la permeabilità e quindi ATTIVANO LA VIA INTRINSECA. Quindi vedete che c’è uno stretto legame tra l’una e l’altra via poichè attraverso caspasi 8 e BID possiamo avere una interazione con la via intrinseca.
A LIVELLO DELLA VIA INTRINSECA agiscono i vari membri della famiglia BCL. Essa è Costituita da membri che ATTIVANO L’APOPTOSI (bax-bad-bim) che dal citosol si Localizzano sulla MEMBRNA MITOCONDRIALE permeabilizzandola (RILOCALIZZAZIONE), e la cui azione è antagonizzata da BCL-2 (un altro membro) che è appunto un ANTI-APOPTOTICO, poiché impedisce la fuoriuscita del citocromo C e quindi la formazione dell’apoptosoma.
L’altro punto di congiunzione tra via intrinseca e estrinseca è la IDROLISI-ATTIVAZIONE DI PRO-CASPASI 3 che può avvenire sia ad opera della CASPASI 9 che della pro-caspasi 8.
Caspasi 3 poi agirà a livello a livello del DFF (fattore di frammentazione del DNA) idrolizzando la subunità 45 e liberando la subunità 40 che entra nel nucleo e cambierà la sua conformazione diventando un OLIGOMERO. Questo cambiamento di conformazione provoca un’Acquisizione dell’attività DNA-asica e quindi è in grado di determinare la frammentazione del DNA. Oltre al recettore FAS, di cui abbiamo appena parlato, esistono altri recettori della via ESTRINSECA OVVERO:
69

2. RECETTORE TNFR-1 e TNFR-2 (recettori per i membri della famiglia del TNF (tumor necrosis factor) :
A livello di questi recettori si possono verificare due risposte cellulari diverse e addirittura OPPOSTE!!!Si può verificare infatti una risposta cellulare che porta alla MORTE (APOPTOSI) ed una che porta alla SOPRAVVIVENZA (PROLIFERAZIONE). Sono quindi due eventi completamente OPPOSTI TRA LORO, OPPOSTI FINO ALL’ESCLUSIONE RECIPROCA!!Questo a dimostrazione di come una cellula prima di andare incontro a MORTE valuti attentamente le condizioni, l’ambiente, la situazione in cui si trova per poter prendere la DECISIONE! COME FA LA CELLULA A PRENDERE UNA DECISIONE?MEDIANTE LE MOLECOLE ADATTATRICI che sono quelle che regolano il DESTINO DELLA CELLULA DOPO IL LEGAME DEI RECETTORI CON IL LIGANDO. A livello di questi recettori si pensava che interagisse direttamente la molecola FADD che appunto era quella molecola adattatrice di FAS (vista sopra). Se fosse così però la cellula non avrebbe altro destino che l’apoptosi, invece:
- A livello di questi recettori (TNFR-1 e TNFR-2) si inserisce una diversa molecola adattatrice chiamata TRADD (TNFR-1 associated death domain protein) che a sua volta può:
Se la cellula deve andare incontro ad Se la cellula NON deve andare incontro APOPTOSI, legarsi a FADD ad apoptosi, legarsi ad un’altra molecola Ovvero RIP – 1 (receptor interacting Protein), una serin-treonin chinasi Contentente DD (death domain) Tutto dipende dalle CONDIZIONI AMBIENTALI, DIPENDE SE CI SONO STIMOLI DI SOPRAVVIVENZA CHE SUPERANO QUELLI DI MORTE E VICEVERSA.Ad esempio nei tumori succede che i segnali di morte cellulare sono meno intensi e forti rispetto a quelli della sopravvivenza (citochine, fattori di crescita) che naturalmente sposteranno l’equilibrio verso la SOPRAVVIVENZA!!!
Quindi tutto ciò (si riferisce alla doppia possibilità di risposta) si può realizzare nella cellula poiché al recettore si vanno a legare appunto queste molecole adattatrici.
In particolare :
Dal legame di TRADD CON FADD avremo che: FADD legherà la PRO-CASPASI 8 E QUINDI SI ATTIVA LA CLASSICA CASCATA CHE PORTA ALL’APOPTOSI.
70
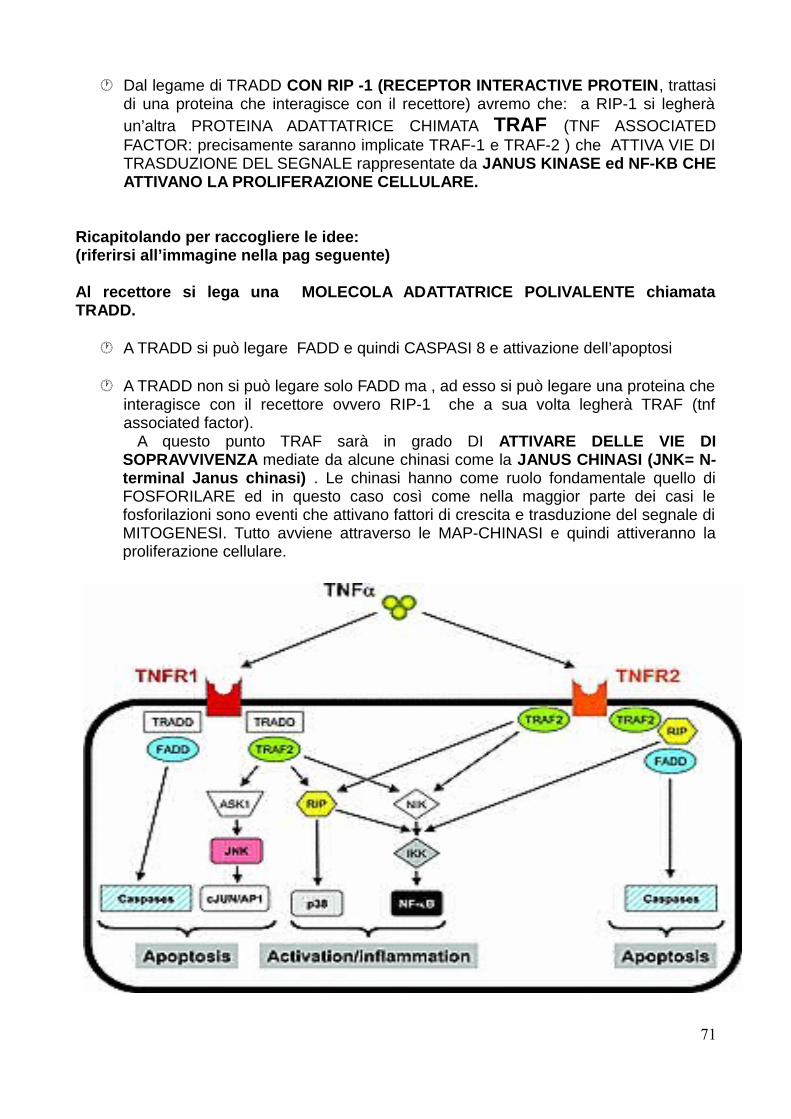
Dal legame di TRADD CON RIP -1 (RECEPTOR INTERACTIVE PROTEIN, trattasi di una proteina che interagisce con il recettore) avremo che: a RIP-1 si legherà un’altra PROTEINA ADATTATRICE CHIMATA TRAF (TNF ASSOCIATED FACTOR: precisamente saranno implicate TRAF-1 e TRAF-2 ) che ATTIVA VIE DI TRASDUZIONE DEL SEGNALE rappresentate da JANUS KINASE ed NF-KB CHE ATTIVANO LA PROLIFERAZIONE CELLULARE.
Ricapitolando per raccogliere le idee:(riferirsi all’immagine nella pag seguente)
Al recettore si lega una MOLECOLA ADATTATRICE POLIVALENTE chiamata TRADD.
A TRADD si può legare FADD e quindi CASPASI 8 e attivazione dell’apoptosi
A TRADD non si può legare solo FADD ma , ad esso si può legare una proteina che interagisce con il recettore ovvero RIP-1 che a sua volta legherà TRAF (tnf associated factor). A questo punto TRAF sarà in grado DI ATTIVARE DELLE VIE DI SOPRAVVIVENZA mediate da alcune chinasi come la JANUS CHINASI (JNK= N-terminal Janus chinasi) . Le chinasi hanno come ruolo fondamentale quello di FOSFORILARE ed in questo caso così come nella maggior parte dei casi le fosforilazioni sono eventi che attivano fattori di crescita e trasduzione del segnale di MITOGENESI. Tutto avviene attraverso le MAP-CHINASI e quindi attiveranno la proliferazione cellulare.
71

Le vie di attivazione della proliferazione si attivano anche mediante l’attivazione di un FATTORE DI CRESCITA NUCLEARE: NF-KB . Trattasi di un fattore di crescita che è stato trovato per la prima volta nei LINFOCITI B dove era necessario per la sintesi delle catene leggere K. Poi si è visto però che si tratta di un FATTORE DI TRASCRIZIONE UBIQUITARIO CHE ATTIVA MOLTI GENI PRO-INFIAMMATORI. (Durante l’infiammazione ad esempio i macrofagi ,per produrre le citochine pro-infiammatorie, sono stimolati mediante i toll like recetor ,ad attivare l’NF-KB che si và a legare a dei particolari SITI DI CONSENSO per esso presenti in quasi tutti i geni PRO-INFIAMMATORI ) .Tutto ciò non basta poiché NF-KB è in grado anche di attivare l’espressione dei geni che regolano la PROLIFERAZIONE CELLULARE. Per quanto concerne sempre NF-KB, trattasi di un sistema che presuppone la presenza di un INIBITORE DI NF-KB che viene FOSORILATO ED INVIATO ALLA DISTRUZIONE NELL’AMBITO DEL PROTEASOMA. Rimarranno le subunità dell’NKFB che migrano verso p50 e p65 che migrano nel nucleo dove agiranno come fattori di trascrizione per geni coinvolti nella proliferazione cellulare.Quindi le vie DI TRASDUZIONE CELLULARE che attivano NF-KB E JNK SONO VIE CHE ATTIVANO LA PROLIFERAZIONE CELLULARE.
Per cui paradossalmente siamo partiti da un LIGANDO DI MORTE con il TNFR-1 e si arriva all’attivazione della proliferazione cellulare. Questo ACCADE PREVALENTEMENTE NEI TUMORI!!!
3. TRAIL-R : RECETTORE PER ligando TRIAL
TRAIL è un ligando: TNF REALATET APOPTOSIS INDUCED LIGAND, che riconosce essenzialmente 4 tipi di RECETTORI però ce ne sono anche di più.
Questi ultimi prendono il nome di TRAIL-R1, TRAIL-R 2, TRAIL-R 3, TRAIL-R-4. Questa nomenclatura però è stata sostituita da una più snella. Infatti conosciamo:
DR4= TRAIL-R 1 e DR5 = TRAIL-R-2. Questi due recettori hanno una coda citoplasmatica costituita dai DEATH DOMAIN (DD) che quindi interagendo con il TRAIL possono trasmettere il segnale di morte cellulare.
DcR1 = TRAIL-R 3 e DcR2= TRAIL-R 4, altri due recettori che o non hanno il DEATH DOMAIN oppure lo hanno molto molto CORTO e
72

quindi sono INCAPACI DI TRASMETTERE IL SEGNALE DELLA MORTE ALL’INTERNO DELLA CELLULA.DcR2 ha una porzione citoplasmatica piccolissima (DD);
DcR1 non ha questa porzione citoplasmatica.
Perché sono stati denominati DcR? Questo acronimo “Dc” riguarda il soprannome che è stato dato a questi recettori ovvero DECOY RECEPTORS, IN QUANTO SI TRATTA DI RECETTORI CHE INGANNANO I LEGANDI DI MORTE CELLULARE QUALI IL TRAIL. Il TRAIL infatti interagisce con DCR1 e DCR2 convinto di indurre l’APOPTOSI. In realtà l’Apoptosi NON VIENE INDOTTA perché la porzione citoplasmatica di questi recettori è appunto piccola o non presente totalmente. Allora la cellula sta ricevendo un segnale di morte cellulare che non può essere trasmesso e allora questi recettori sono stati così paragonati a della TRAPPOLE CHE INGANNANO IL SEGNALE DI MORTE CELLULARE IN QUANTO VI E’ L’INTERAZIONE CON IL SEGNALE DI MORTE CELLULARE MA NON VI POTRA’ ESSERE LA RISPOSTA APOPTOTICA E QUINDI DI MORTE.
NON SI TRATTA DI UN FEED-BACK, poiché il feed back avviene quando qualcosa viene attivato e poi il prodotto stesso andrà ad agire sulla struttura che ha generato la risposta. Qui più che feed-back, si ritorna al concetto di EQUILIBRIO OMEOSTATICO e regolazione dell’apoptosi.
È un processo regolato attraverso lo stesso meccanismo!!! È difficile scappare a questa regolazione poiché il meccanismo è lo stesso ci saranno recettori che trasmetteranno il segnale di apoptosi, mentre altri no.
NON SI TRATTA NEANCHE DI AFFINITA’!!! Non è che DCR1-2 siano più o meno affini al TRAIL. TUTTI E QUATTRO I RECETTORI HANNO LA STESSA IDENTICA AFFINITA’ solo che DcR1 e DcR2 non hanno o hanno una corta coda citoplasmatica e quindi non possono trasmettere il segnale quindi si ritorna ancora al fatto che
“ALCUNI RECETTORI PER LA LORO CONFORMAZIONE TRASMETTONO IL SEGNALE ED ALTRI NO” e ciò comporta UN EQUILIBRIO in quanto anche in presenza di segnali apoptotici viene impedita una TOTALE MORTE CELLULARE poiché appunto sono presenti alcuni recettori che non solo non trasmettono il segnale della morte MA DCR1 E DCR2 INTERAGENDO CON IL TRAIL LO SOTTRAGGONO DAL POSSIBBILE LEGAME CON DR4 E DR5 che indurrebbero apoptosi.!! È PROPRIO QUI CHE SI GIOCA L’EQUILIBRIO ed e’ per questo motivo che SONO CHIAMATI RECETTORI DECOY OVVERO TRAPPOLA!!!
Questo equilibrio è importante si a nelle cellule normali che nelle CELLULE NEOPLASTICHE in quanto in queste troveremo una DIVERSA
73

ESPRESSIONE di questi recettori. A parte il fatto che è molto complicato il discorso poiché anche a livello di questi recettori possono essere attivati vie di sopravvivenza cellulare quali NFKB E JNK come a livello di TNFR.
Quindi DCR1-DCR2 rispetto a DR4-DR5, svolgono un RUOLO OPPOSTO!!! L’ESPRESSIONE di DCR1-DCR2 dovrebbe essere esaltata nei tumori poiché si oppongono all’apoptosi invece, stranamente, nella maggior parte dei casi sono IPO-ESPRESSI IN QUANTO IPERMETILATI. (noi lo stiamo studiando in laboratorio in alcune linee tumorali e anche in alcuni campioni di tumori). Questo non si concilia con un processo di TRASFORMAZIONE NEOPLASTICA ma :
in corso di neoplasie la presenza di fattori di crescita e di attivazione può determinare lo slittamento verso la via di proliferazione piuttosto che verso un’altra. Quindi si può CAPOVOLGERE il ruolo di DcR1- DcR-2 con quello di DR-4, DR-5. Determinante è quindi il diverso ambiente cellulare in cui le cellule si trovano a persistere.
A livello di TRAIL-R in particolare DR4-DR5 non abbiamo parlato di PROTEINA ADATTATRICE; ne abbiamo parlato invece per quanto concerneva TNFR1-2, ovvero TRADD, FADD, RIP-1. Diciamo meglio che la molecola che funge da PROTEINA ADATTATRICE per DR4-5 è PRESENTE MA E’ SCONOSCIUTA e probabilmente si ritiene che a livello di questa proteina che ancora non è stata individuata ma che comunque si va a legare al recettore DR4-DR5, venga ATTIVATA la PRO-CASPASI 10 in caspasi 10 che comunque agisce sempre attivando la PRO-CASPASI 8 in caspasi 8. Da queste cascate iniziatrici poi si attiverà la cascata delle varie caspasi che porterà all’apoptosi e che è regolata in senso negativo dalle proteine della famiglia delle BCL.
IAP protein
Nel meccanismo di regolazione dell’apoptosi sono importanti alcune proteine INIBITRICI DELL’APOPTOSI ovvero le INHIBITORS OF APOPTOSIS o IAPs che antagonizzano l’azione degli attivatori dell’apoptosi liberati secondariamente dai mitocondri ovvero le SMAC/DIABLO (ricordiamo che i mitocondri nella via intrinseca sono coinvolti non solo perché liberano il CITOCROMO C ma anche perché liberano queste SMAC/DIABLO). Ma, le IAPs agiscono anche DIRETTAMENTE, bloccando le CASPASI.
Le IAPs sono delle proteine un po’ particolari in quanto hanno un alto grado di omologia con alcune sequenze di proteine prodotte da alcuni virus chiamati BACULOVIRUS. Queste proteine sintetizzate dai baculovirus , vengono prodotte proprio per evitare che le cellule da essi infettate vadano incontro a
74
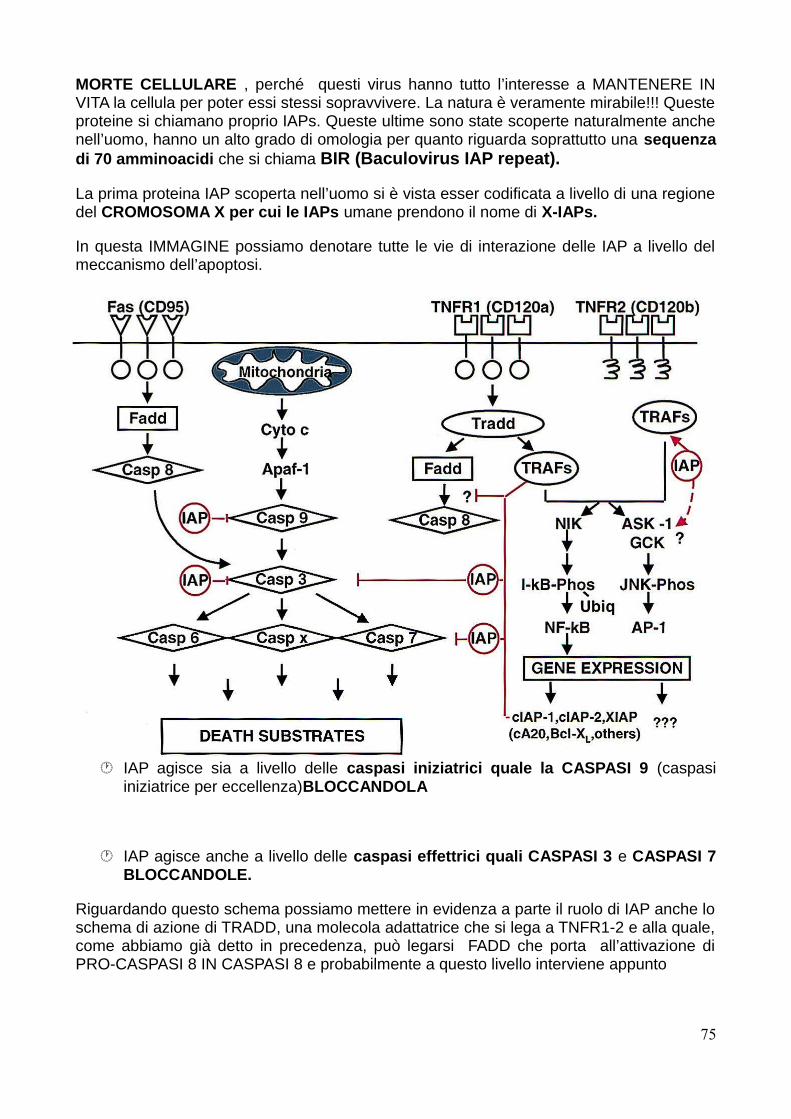
MORTE CELLULARE , perché questi virus hanno tutto l’interesse a MANTENERE IN VITA la cellula per poter essi stessi sopravvivere. La natura è veramente mirabile!!! Queste proteine si chiamano proprio IAPs. Queste ultime sono state scoperte naturalmente anche nell’uomo, hanno un alto grado di omologia per quanto riguarda soprattutto una sequenza di 70 amminoacidi che si chiama BIR (Baculovirus IAP repeat).
La prima proteina IAP scoperta nell’uomo si è vista esser codificata a livello di una regione del CROMOSOMA X per cui le IAPs umane prendono il nome di X-IAPs.
In questa IMMAGINE possiamo denotare tutte le vie di interazione delle IAP a livello del meccanismo dell’apoptosi.
IAP agisce sia a livello delle caspasi iniziatrici quale la CASPASI 9 (caspasi iniziatrice per eccellenza)BLOCCANDOLA
IAP agisce anche a livello delle caspasi effettrici quali CASPASI 3 e CASPASI 7 BLOCCANDOLE.
Riguardando questo schema possiamo mettere in evidenza a parte il ruolo di IAP anche lo schema di azione di TRADD, una molecola adattatrice che si lega a TNFR1-2 e alla quale, come abbiamo già detto in precedenza, può legarsi FADD che porta all’attivazione di PRO-CASPASI 8 IN CASPASI 8 e probabilmente a questo livello interviene appunto
75

IAP che si lega e blocca l’azione della CASPASI 8 (questo fenomeno è ancora in dubbio, più sicura è l’azione sulla CASPASI 9 accennata poco fa)
OPPURE agirà su TRADD. Infatti, mediante l’intervento di RIP-1, può legarsi la molecola TRAF dal quale vengono attivate le vie di fosforilazione mediante delle chinasi , tra le quali JNK (JANUS CHINASI) che portano alla fosforilazione del complesso AP1. Quest’ultimo è appunto un complesso formato da vari fattori di trascrizione quali c-fos, c-jun, Fra 1, Fra 2, Jun B, Jun D che possono formare omodimeri ed eterodimeri. In questo caso non dobbiamo citare c-myc (non dobbiamo fare la cantilena c-fos, c-jun, c-myc in quanto questi tre sono dei fattori di trascrizione attivati nella prima parte della fase G1, quindi sono i geni cosiddetti precoci) poiché si tratta di un prodotto di un proto-oncogene che noi troviamo attivato nel LINFOMA DI burkitt Nel complesso AP1 fosforilato da JANUS CHINASI NON ABBIAMO LA PARTECIPAZIONE DI c-myc.
Ritornando allo schema, TRAF una volta legatosi a TRADD può portare alla fosforilazione dell’INIBITORE DI NF-KB (INF-kB) che sarà ubiquitinato, idrolizzato a livello del proteasoma mentre si libera NF-KB che andrà ad attivare vari geni coinvolti nella proliferazione tra i quali alcuni geni della famiglia BCL-2, ovvero BCL-XL che attiveranno la proliferazione.
Anche a questo livello probabilmente (ancora non se ne ha anche qui la certezza) intervengono le IAPs: non solo esse bloccheranno le caspasi 3, 7, 9 ma
PROBABILMENTE intervengono anche nell’attivazione della via di sopravvivenza ATTIVANDO LA VIA DI TRAF e le JANUS CHINASI implicate dirottando la risposta cellulare verso la sopravvivenza piuttosto che verso l’apoptosi.
MEMBRI DELLA FAMIGLIA BCL-2
Trattasi di una numerosa famiglia, i cui membri non vanno molto d’accordo, poiché alcuni agiscono in un modo ed altri in un’altra.
Vediamo cosa succede a livello di questi membri della famiglia BCL quando una cellula avvia il suo programma di morte cellulare.
Cominciamo però dalla:
Cellula in CONDIZIONI DI RIPOSO.
Tutto di gioca a livello della MEMBRANA MITOCONDRIALE dove sono appunto localizzate le proteine BCL-2 che sono prodotti di un protoncogene che attiva la proliferazione impendendo l’apoptosi impedendo l’azione di un altro membro che si chiama BAK che a sua volta è posizionato a livello della membrana mitocondriale.
76

Per cui a livello della membrana mitocondriale vi sono 2 ANTAGONISTI:
- BAK che facilita e dunque favorisce l’aumento della permeabilità mitocondriale
- BCL-2 che impedisce l’aumento della permeabilità mitocondriale impedendo BAK che quindi sarà TENUTO IN STALLO e BLOCCATO da BCL-2
A livello del citosol ci sono altri membri della famiglia BCL-2:
- BAX che sono tenute a livello del citosol da BCL-2 che quindi avrà doppia funzione, da un lato bloccherà BAK a livello della membrana mitocondriale, dall’altra bloccherà nel citosol le molecole di BAX che fa sempre parte di questa famiglia. Il meccanismo di questo sequestro a livello del citosol ancora è sconosciuto, probabilmente BCL-2 complesserà con BAX così come fa con BAC.
- BAD = mantenuto a livello del citosol da un meccanismo di FOSFORILAZIONE. Se infatti BAD è FOSFORILATO è SEQUESTRATO NEL CITOSOL e non può migrare a livello mitocondriale per alterare la membrana mitocondriale . BAD è fosforilato nel caso in cui la cellula sia esposta a stimoli mitogenici, poiché questi attivano le MAP-KINASI che tra le altre cose fosforilano appunto BAD.
Per cui RIASSUMENDO IN CONDIZIONI DI RIPOSO ABBIAMO:
BAD fosforilato anche da qualche piccolo segnale di sopravvivenza che la cellula riceve
BAX bloccato nel citosol da BCL-2
BAK che sta sempre sulla membrana mitocondriale ma è reso inoffensivo da BCL-2.
Succede che ad un certo punto che la
cellula riceve un SEGNALE DI APOPTOSI
Vengono attivati tutti i membri della famiglia BCL-2 ad attivita’ pro-apoptotica che fanno parte di un gruppo che prende il nome di BH3-ONLY. Si tratta di proteine che hanno una piccola sequenza di 9-13 amminoacidi di omologia con le proteine
77
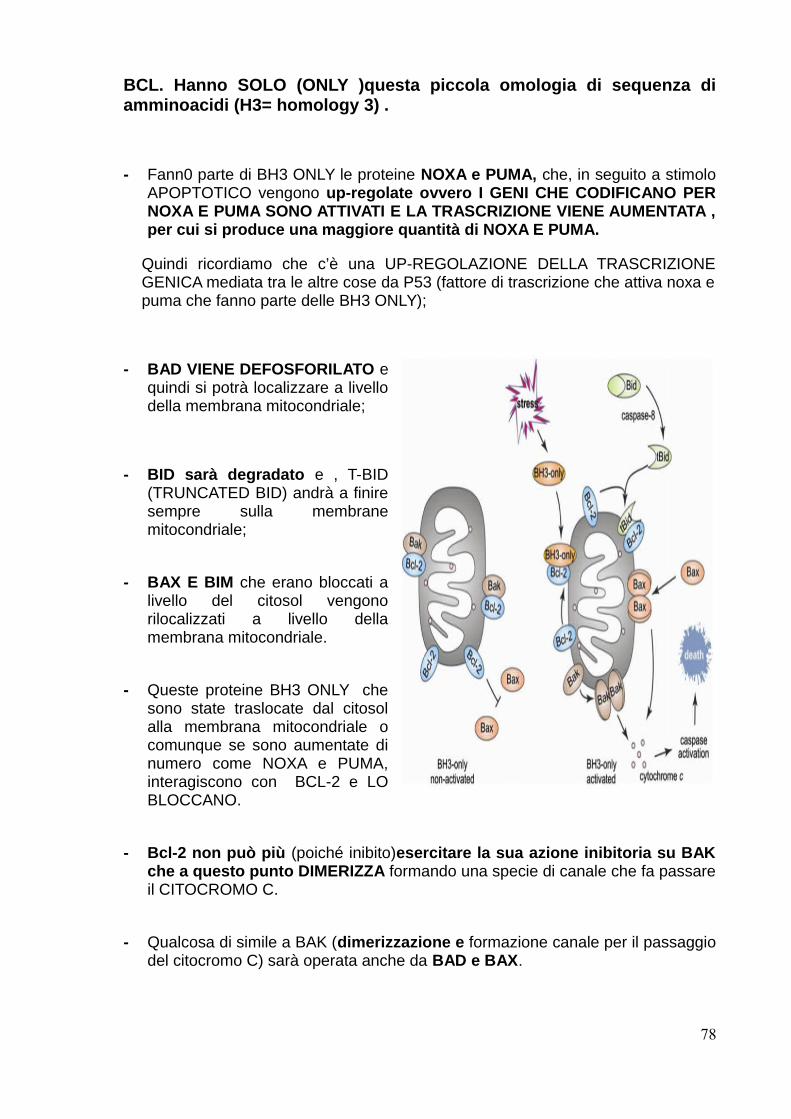
BCL. Hanno SOLO (ONLY )questa piccola omologia di sequenza di amminoacidi (H3= homology 3) .
- Fann0 parte di BH3 ONLY le proteine NOXA e PUMA, che, in seguito a stimolo APOPTOTICO vengono up-regolate ovvero I GENI CHE CODIFICANO PER NOXA E PUMA SONO ATTIVATI E LA TRASCRIZIONE VIENE AUMENTATA , per cui si produce una maggiore quantità di NOXA E PUMA.
Quindi ricordiamo che c’è una UP-REGOLAZIONE DELLA TRASCRIZIONE GENICA mediata tra le altre cose da P53 (fattore di trascrizione che attiva noxa e puma che fanno parte delle BH3 ONLY);
- BAD VIENE DEFOSFORILATO e quindi si potrà localizzare a livello della membrana mitocondriale;
- BID sarà degradato e , T-BID (TRUNCATED BID) andrà a finire sempre sulla membrane mitocondriale;
- BAX E BIM che erano bloccati a livello del citosol vengono rilocalizzati a livello della membrana mitocondriale.
- Queste proteine BH3 ONLY che sono state traslocate dal citosol alla membrana mitocondriale o comunque se sono aumentate di numero come NOXA e PUMA, interagiscono con BCL-2 e LO BLOCCANO.
- Bcl-2 non può più (poiché inibito)esercitare la sua azione inibitoria su BAK che a questo punto DIMERIZZA formando una specie di canale che fa passare il CITOCROMO C.
- Qualcosa di simile a BAK (dimerizzazione e formazione canale per il passaggio del citocromo C) sarà operata anche da BAD e BAX.
78

Ci sono le varie vie di FAS e altri stimoli della morte tramite recettori che non sono il FAS. Però la cellula è sottoposta anche a fattori di sopravvivenza.
Partiamo dalla situazione di SOPRAVVIVENZA perché se la cellula non muore, deve vivere e deve avere dei SEGNALI DI SOPRAVVIVENZA.
(ho inserito in basso l’immagine effettivamente proiettata dalla prof. Quindi basta seguirla nei minimi dettagli per comprendere la seguente spiegazione che ad essa è riferita)
Vedete come tutti i segnali di sopravvivenza : PKC,PKC,ERK1/2 BI3K, sono tutti fattori che ATTIVANO LE MAP KINASI. Ne esiste anche un altro di segnale che è PKB che assieme a PKC dà vie di sopravvivenza e di fosforilazione di tirosin chinasi molto importanti.
BAD in seguito all’azione di queste chinasi VIENE FOSFORILATO su degli specifici residui amminoacidici, indicati con dei numeretti, e quindi RIMANE SEQUESTRATO NEL CITOSOL.
Nel caso in cui NON C’E’ LO STIMOLO DI SOPRAVVIVENZA (riferirsi sempre all’immagine precedente) (Si parte da uno stress genotossico, da un danno al DNA!!! )
BAD NON sara’ FOSFORILATO (senza i numeretti) , dunque si porterà alla membrana mitocondriale dove bloccherà BCL-XL (una isoforma di un membro della famiglia delle BCL ovvero BCL-XL ; l’altra isoforma è BCL-XS). Abbiamo infatti due fattori ANTI-APOPTOTICI : BCL-2 e BCL-XL.
79

Inoltre, se c’è l’attivazione della caspasi 8, BID da BID si ricava T-BID e quest’ultimo si muoverà a livello anch’esso della membra mitocondriale BLOCCANDO BCL-XL.
T-BID facilità la dimerizzazione di BAC e quindi la formazione di un canale a livello della membrana mitocondriale con fuoriuscita di CITOCROMO C.
Grazie all’azione di P53 avremo l’UP-REGOLAZIONE di NOXA E PUMA che vanno anche esse alla membrana mitocondriale dove BLOCCANO BCL-2
BIM blocca BCL-2
BAX è attivato anche da p53 e paradossalmente può essere attivato anche dalle JANUS KINASI che in questo caso hanno azione PRO-APOPTOTICA. Però SE PREVALE BCL-2 allora BAX risulterà bloccato.
Tutto il complesso della risposta in generale NON E’ LINEARE. Parlavo del discorso di recettori DECOY che bloccano l’apoptosi, e invece no, ci sono condizioni che possono comportare un meccanismo completamente opposto.
(La prof.ssa fa una sorta di riepilogo appoggiandosi ad un’immagine)
Come vedete nella cellula apoptotica , BAD non è fosforilato da parte delle chinasi come AKT. Esso andrà a livello della membrana mitocondriale bloccando le due molecole pro-apoptotiche BCL-2 e BCL-XL e anche qui avremo la formazione di un canale formato da Bax che consente la fuoriuscita del citocromo C.
Mentre in una cellula normale, con segnale di sopravvivenza, BAX è fosforilato e quindi BCL-2 e BCL-XL evitano che BAX possa formare il canale in questione.
80

Da tenere presente è ancora il CROSS-TALK tra la via intrinseca e la via estrinseca mediato sia a livello della caspasi 9 su caspasi 3 che a livello della caspasi otto su BID che agirà già a livello mitocondriale.
SINTESI MAGGIORE O MINORE DEI MEMBRI DELLA FAMIGLIA BCL-2 coinvolti nella proliferazione e nell’apoptosi.
Vedete che quando normalmente BAX e BCL-2 sono in equilibrio tra di loro. Quando aumenta la sintesi di BAX per cui si formano degli omodimeri di BAX allora preverrà l’apoptosi, viceversa se abbiamo un aumento dell’espressione del gene che codifica per BCL-2 allora si formeranno degli omodimeri di BCL-2 che avranno un’aizone ANTI-APOPTOTICA. Tutto si gioca su un EQUILIBRIO DI ESPRESSIONE DI GENI.
Ci sarà normalmente anche un equilibrio tra l’espressione delle isoforme del gene BCLX. Dello stesso gene esistono infatti le due isoforme BCL-XS, BCL-XL.
L’isoforma BCL-XL (isoforma large) si comporta come BCL-2 nel BLOCCARE L’APOPTOSI. Se prevale essa infatti avremo un blocco dell’APOPTOSI
L’ISOFORMA BCL-XS (isoforma small) invece se prevale porterà ad
81

un’ATTIVAZIONE DELL’APOPTOSI.
Anche questa è la dimostrazione di COME UNA CELLULA POSSA RISPONDERE ad uno STRESS FISIOLOGICO O AMBIENTALE mediante l’espressione dei GENI DELLO STRESS , quali P38 (CHE è UNA PROTEIN CHINASI), JANUS CHINASI, GADD 45 (growtn arrest and DNA damnage) con le tre isoforme che viene attivato proprio dal danno al DNA.
Le tre isoforme di Gadd 45 ad esempio interagiranno con PCNA , questo ANTIGENE di risposta nucleare favorendo la riparazione del DNA, oppure legando P21 e quindi interviene a livello dell’arresto del ciclo cellulare in G1, oppure a livello del legame con la ciclina B1- con la chinasi ciclino dipendente.
PROF.SSA TETI : Io, di solito, quando facevo queste lezioni parlavo anche delle degenerazioni però ho pensato di completare invece con le alterazioni dei meccanismi che regolano la proliferazione cellulare con qualche accenno riguardo il comportamento della cellula neoplastica. Comunque noi quest’anno purtroppo abbiamo poche lezioni (meno dell’anno scorso) , quindi ho paura che non riusciremo a fare tutto. Le degenerazioni non so se riusciremo a farle, ma sono cose abbastanza codificate in quanto fanno parte della vecchia patologia anche se ci sono questioni relative agli enzimi che sono leggermente più complicate. Intanto ho pensato che fosse utile focalizzarci un attimino su ciò che concerne la trasformazione neoplastica.
82

I tumori, sia che siano maligni, sia che siano benigni, sono conseguenze di alterazioni geniche. Queste alterazioni nella maggior parte dei casi sono dovute a mutazioni geniche , e in un’ altra percentuale dei casi ,che stanno diventando sempre più numerosi, ad alterazioni epigenetiche cioè chimiche delle basi del DNA.
Si tratta comunque di un’alterazione genica che , nel caso delle mutazioni geniche è irreversibile mentre, nel caso delle alterazioni epigenetiche potrebbe diventare reversibile ma non con l’allontanamento dello stimolo che lo ha indotto ma con l’intervento di sostanze e terapie tese a modificare l’alterazione epigenetica mentre invece tutte le altre forme di alterazione della proliferazione sono tutte forme che sono REVERSIBILI IN SEGUITO ALL’ALLONTANAMENTO DELLO
STIMOLO CHE LO HA PRODOTTO. (Questo CONCETTO è fondamentale per capire lo sviluppo dei tumori!!!)Se l’alterazione NEOPLASTICA riguarda i geni e quindi il nucleo ( e da qui si parte, non c’è dubbio) la struttura della cellula che subisce modificazioni significative nel senso della patogenesi della trasformazione neoplastica è la PLASMAMEMBRANA. In realtà TUTTA la cellula subisce delle modificazioni, diventa una cellula ATIPICA (solo nella cellula neoplastica possiamo utilizzare questo aggettivo). Saranno alterate le strutture dal punto di vista funzionale, morfologico della cellula , ma tra tutte le strutture cellulari quella che ha un significato patogenetico da cui derivano altre patologie di alterazione della cellula neoplastica è senza dubbio la MEBRANA PLASMATICA perché tramite essa la cellula comunica con le altre cellule. Quindi l’importanza delle alterazioni della membrana plasmatica sta nel fatto che la cellula neoplastica si comporta in maniera diversa rispetto alle altre cellule proprio per le alterazioni che ha a livello della plasma-membrana.
83

È vero che i geni codificano per proteine coinvolte nella proliferazione ma, la membrana plasmatica REGOLA la proliferazione, si parla di controllo dal CONTATTO CELLULA-CELLULA, in quanto le cellule possono comunicare tra di loro e inviare dei segnali di inibizione della proliferazione e del movimento. Quindi le modifiche a livello della membrana plasmatica sono strettamente correlate, come evidenziano alcuni esperimenti, con l’invasività e la crescita patologica.
Sono coinvolte nella trasformazione neoplastica tutte le giunzioni
cellulari dalle aderenti, alle comunicanti alle occludenti. Esse presenteranno grado variabile a secondo delle caratteristiche del tumore ovvero invasività e malignità.Come sapete le giunzioni intercellulari aderenti formano legami forti tra cellula e cellula, quelle comunicanti consentono passaggio di ioni, di messaggi e molecole che regolano la proliferazioni mentre le occludenti formano una barriera selettiva alla DIFFUSIONE di varie sostanze. Il fatto che siano alterate le giunzioni occludenti vuol dire che la cellula è più esposta all’aggressione di sostanze nocive e lesive che altrimenti non arriverebbero. Quindi tutti e tre i tipi di giunzione intervengono in modo diverso a seconda della loro funzione nella patogenesi della trasformazione.
RUOLO SINGOLE GIUNZIONI NEI TUMORI: GIUNZIONI ADERENTI (maculae adherens - desmosomi)
In questa immagine sono mostrate le principali componenti che partecipano alla formazioni delle giunzioni aderenti. L’Alterazione della CADERINA è frequentissima nelle alterazioni tumorali. L’espressione della E-CADERINA è in quasi tutti i tumori RIDOTTISSIMA. La E- CADERINA è infatti fondamentale nell’adesione CELLULA-CELLULA per cui una delle cause della diminuita adesione cellulare nelle trasformazioni neoplastiche sarà una ridotta espressione della E-CADERINA che fisiologicamente, forma una sorta di CERNIERA MOLECOLARE poiché i dimeri di
84

caderina di una cellula con quelli dell’altra cellula vanno a formare unendosi una struttura che a vista d’occhio somiglia molto ad una cerniera . La cosa interessante non è solo il fatto che la E-CADERINA formi la molecola base per l’adesione cellula- cellula ma è rilevante il fatto che la E-CADERINA svolge un ruolo importante anche nella regolazione della proliferazione cellulare. Attraverso la E-CADERINA avremo il legame con il CITOSCHELETRO quindi con tutte le proprietà motrici e funzionali della cellula. La E-caderina è legata alla BETA.CATENINA ,legata a sua volta alla ALFA-CATENINA che interagisce e si lega ai FILAMENTI DI ACTINA. O ancora la caderina può essere ancorata ad altri componenti come la vinculina o la radexina e così via.
In alcuni tipi cellulari (non in tutti) la E-CADERINA tramite la BETA-CATENINA è legata ad una proteina detta APC, coinvolta nell’adesione cellulare e nella
trasmissione del messaggio all’interno della cellula, che è codificata da un GENE ONCOSOPPRESSORE.
In alcuni tumori soprattutto in quelli del colon poliposici si è visto che c’è un deficit di espressione di questo gene per cui una minor produzione di APC. L’acronimo APC sta infatti per ADENOMATUS POLIPOSIS COLI , gene che è alterato, soppresso, deleto e quindi il suo prodotto non viene prodotto in questo tipo di tumori dove appunto ne rappresenta una causa principale. Si è visto ancora che nei tumori invasivi più che maligni, le giunzioni aderenti ed in particolare le maculae adherens e i desmosomi sono ridotti di numero e di grandezza!!! Vi è una correlazione diretta tra la diminuzione di grandezza e del numero e quindi della quantità delle giunzioni aderenti e l’INVASIVITA’ DEL TUMORE. Meno sono presenti è più invasivo è il tumore. Pare che la correlazione sia più con l’invasività piuttosto che con la malignità del tumore, anche se la malignità è abbinata
all’invasività.
85

Nelle cellule maligne l’adesione tra cellula e cellula risulta quindi essere ridotta. Questa diminuzione dell’adesione è alla base della PRIMA FASE DELLE METASTASI. Noi tra le caratteristiche dei tumori maligni parliamo di TENDENZA ALLA METASTASI intendendola come una conseguenza del fatto vero del tumore maligno che è l’alterazione dei geni che codificano per PRODOTTI DI DIFFERENZIAZIONE e che codificano per prodotti della proliferazione. Questa è una CONSEGUENZA poiché le alterazioni della plasma-membrana sono CONSEGUENZA DEL FATTO CHE NEI TUMORI MALIGNI SONO ALTERATI I GENI CHE SONO RESPONSABILI NON SOLO DELLA PROLIFERAZIONE MA ANCHE DELLA DIFFERENZIAZIONE.
Risulta evidente come le cellule normali siano tra di loro unite mediante le E-CADERINE mentre nelle cellule maligne vi è una riduzione delle E-CADERINE per cui esse non sono più unite tra di loro. In poche parole nelle cellule NEOPLASTICHE viene a mancare L’ADESIONE OMOTIPICA (ADESIONE TRA CELLULE DELLO STESSO TIPO).
Ma, nonostante nelle cellule neoplastiche a causa della diminuzione della E-CADERINA viene a mancare l’adesione OMOTIPICA, dall’altro si ha un AUMENTO DELL’ADESIONE ETEROTIPICA poiché le cellule tumorali subiscono una transizione dal tipo EPITELIALE AL TIPO MESENCHIMALE che ha affinità con cellule non omotipiche; si tratta di una transizione definita TEM (transizione epitelio-mesenchimale).
Le cellule neoplastiche NON SONO PIU’ UNITE TRA DI LORO, sono separate, sono cellule definite ASOCIALI (questo termine è perfetto in quanto riassume quello che è l’aspetto della INCOMUNICABILITA’ delle cellule tra di loro perché non solo mancano le e-caderine ma mancano anche le giunzioni comunicanti).La cellula neoplastica PERO’ DIVENTA CAPACE DI ADERIRE ALLA MATRICE EXTRACELLULARE.
GIUNZIONI COMUNICANTI
86
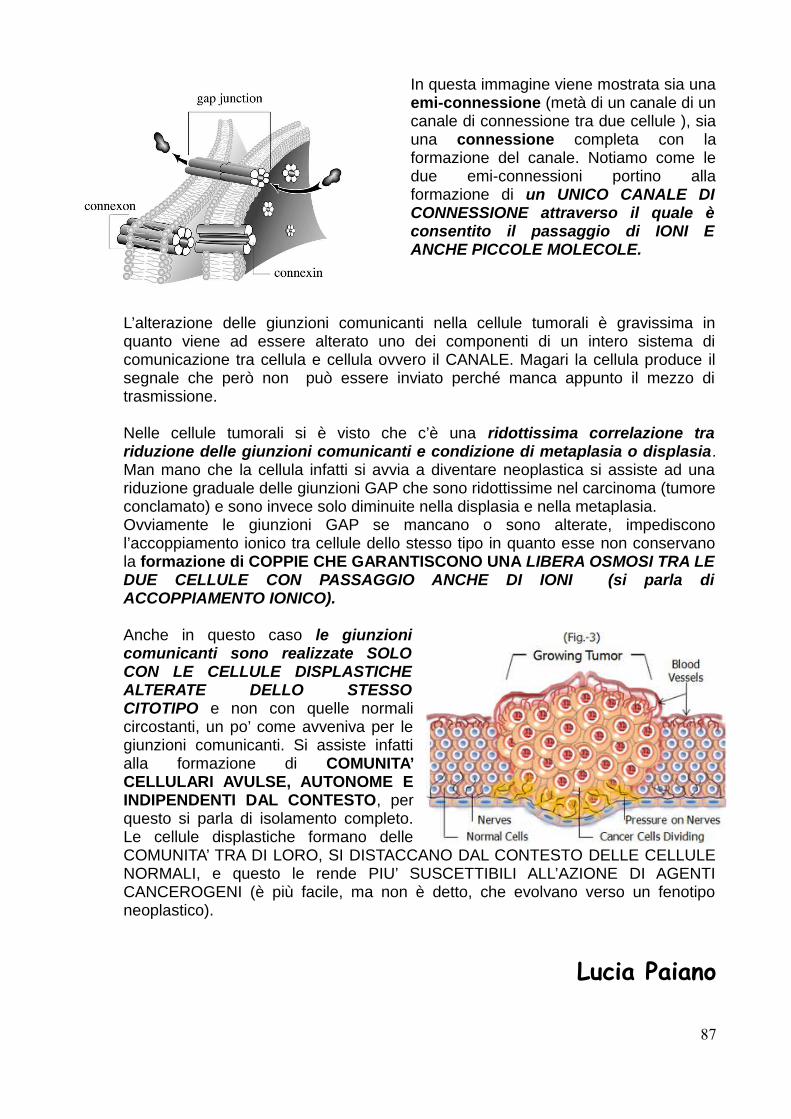
In questa immagine viene mostrata sia una emi-connessione (metà di un canale di un canale di connessione tra due cellule ), sia una connessione completa con la formazione del canale. Notiamo come le due emi-connessioni portino alla formazione di un UNICO CANALE DI CONNESSIONE attraverso il quale è consentito il passaggio di IONI E ANCHE PICCOLE MOLECOLE.
L’alterazione delle giunzioni comunicanti nella cellule tumorali è gravissima in quanto viene ad essere alterato uno dei componenti di un intero sistema di comunicazione tra cellula e cellula ovvero il CANALE. Magari la cellula produce il segnale che però non può essere inviato perché manca appunto il mezzo di trasmissione.
Nelle cellule tumorali si è visto che c’è una ridottissima correlazione tra riduzione delle giunzioni comunicanti e condizione di metaplasia o displasia. Man mano che la cellula infatti si avvia a diventare neoplastica si assiste ad una riduzione graduale delle giunzioni GAP che sono ridottissime nel carcinoma (tumore conclamato) e sono invece solo diminuite nella displasia e nella metaplasia.Ovviamente le giunzioni GAP se mancano o sono alterate, impediscono l’accoppiamento ionico tra cellule dello stesso tipo in quanto esse non conservano la formazione di COPPIE CHE GARANTISCONO UNA LIBERA OSMOSI TRA LE DUE CELLULE CON PASSAGGIO ANCHE DI IONI (si parla di ACCOPPIAMENTO IONICO).
Anche in questo caso le giunzioni comunicanti sono realizzate SOLO CON LE CELLULE DISPLASTICHE ALTERATE DELLO STESSO CITOTIPO e non con quelle normali circostanti, un po’ come avveniva per le giunzioni comunicanti. Si assiste infatti alla formazione di COMUNITA’ CELLULARI AVULSE, AUTONOME E INDIPENDENTI DAL CONTESTO, per questo si parla di isolamento completo. Le cellule displastiche formano delle COMUNITA’ TRA DI LORO, SI DISTACCANO DAL CONTESTO DELLE CELLULE NORMALI, e questo le rende PIU’ SUSCETTIBILI ALL’AZIONE DI AGENTI CANCEROGENI (è più facile, ma non è detto, che evolvano verso un fenotipo neoplastico).
Lucia Paiano
87

14/11/13 Pro.ssa Teti
6Tappe dellaTappe della
cancerogenesicancerogenesi
Vi ricordate che avevamo detto che nei tumori, le giunzioni occludenti, come tutte le giunzioni intercellulari, sono:
• ridotte di numero;• più sottili;• discontinue;• le fibrille sono ridotte;• ridotte di spessore.
Già abbiamo parlato di quelle aderenti, le giunzioni occludenti, quando sono ridotte di numero generano delle comunità cellulari. Le giunzioni comunicanti, come voi sapete, generano delle coppie di cellule; le cellule sono accoppiate dal punto di vista ionico e allora comunicano tra di loro con segnali,soprattutto, di inibizione della crescita. Durante la trasformazione neoplastica, queste giunzioni comunicanti sono ridotte, per cui le cellule non hanno più questo accoppiamento ionico, chimico e soprattutto lo perdono con le cellule
normali dello stesso tessuto; per cui queste cellule già trasformate rimangono come delle isole, come delle zone di cellule che tra loro comunicano, ma che hanno perso i contatti con le cellule normali circostanti. Questo crea una comunità cellulare che diventa più insensibile ai segnali di regolazione e più sensibile ai segnali di trasformazione neoplastica, perché perdono non solo il contatto, ma proprio la comunicazione con le cellule; questo è un evento importante nella trasformazione neoplastica e quindi hanno poi tutto un processo di trasduzione del segnale diverso, modificato, rispetto a quello delle cellule normali.Quelle occlundentes sono quelle zonulae che determinano la formazione di una vera e propria barriera contro la diffusione attraverso gli epiteli e quindi impediscono il transito di molecole, anche di molecole dannose, di molecole oncogene, che possono trasformare le cellule. È quindi
88

una difesa importantissima quella della continuità delle zonulae occludentes. Ed anche nei tumori (le occludentes) sono ridotte di numero e densità.Le zonulae occludentes, quindi, non svolgono solo la funzione di creare questa barriera selettiva e ovviamente quella della adesione intercellulare, ma svolgono anche un’altra funzione, quella di rendere stabile la topografia biochimica della membrana cellulare. Che cosa significa questo? Come voi sapete, molte cellule epiteliali hanno delle funzioni diverse ai diversi poli della cellula. Ad esempio cellule che hanno: un polo basale, un polo volto verso il lume.Questi si chiamano “poli cellulari” perché hanno attività enzimatiche diverse, svolgono funzioni diverse. Cioè a un polo una cellula ha una funzione, ad un altro polo ha un’altra funzione. Questo livello di differenziazione, questa topografia biochimica della membrana, viene mantenuta proprio dalle zone occludenti, perché sapete che impediscono (sapete che il modello di membrana, riconosciuto attualmente, è quello fluido; quindi c’è una riorganizzazione delle proteine di membrana, anche delle proteine integrali di membrana) che ci sia una eccessiva mobilizzazione di queste attività enzimatiche, quindi mantengono la topografia biochimica della membrana cellulare.Difatti in molte alterazioni, anche displastiche, si comincia ad avere anche un’alterazione della topografia biochimica. Proprio la displasia, lo abbiamo detto all’inizio della lezione, è quella condizione in cui proprio si altera l’architettura del tessuto, proprio perché si altera la localizzazione di queste attività enzimatiche.Io sto facendo tutto un discorso iniziale
sull’importanza della membrana plasmatica nella trasformazione neoplastica e sul ruolo che svolge, perché la membrana non solo partecipa alle attività cellulari attraverso le giunzioni di cui abbiamo parlato, ma anche attraverso l’espressione di recettori che sono fondamentali per la crescita neoplastica e che sono recettori per il fattore di crescita.Quindi la membrana della cellula neoplastica, non solo presenta queste modificazioni a livello delle strutture giunzionali, ma ne presenta anche di importanti in relazione ai recettori del fattore di crescita. Ed è un fatto significativo diciamo questo.Perché? Perché la cellula neoplastica è in grado non solo di esprimere il recettore del fattore di crescita, ma di produrre anche il fattore di crescita. Un po’ come i linfociti T attivati che producono interleuchina 2 ed il recettore per l’interleuchina 2. Accade lo stesso nella cellula neoplastica, cioè c’è un’attivazione autocrina della cellula neoplastica. Sono capaci quindi di una regolazione autocrina: la cellula produce sia il fattore di crescita (che interagisce con la stessa cellula che lo ha secreto, perché la cellula è in grado di produrre anche i recettori) che i recettori.Quali sono i peptidi che hanno questa capacità di attivazione autocrina, cioè proliferativa?Sono, in primo luogo:
• il trasforming growth factor-alpha TGF-α (vi ricordo, invece, che l’isoforma beta è un inibitore della proliferazione, soprattutto al livello epiteliale);
• il fattore di crescita legato alle piastrine (PDGF) che è uno dei più potenti fattori legati alla proliferazione, prodotto dalle
89

piastrine, ma anche dalle cellule neoplastiche ed è un fattore ad attività autocrina.È, come vi ho detto l’altra volta, addirittura in grado di attivare la proliferazione delle cellule muscolari;
• bombesina, che è un peptide vasoattivo, prodotto al livello delle cellule gastriche dell’antro pilorico e del duodeno. È in grado di stimolare la gastrina, ma è anche un potente agente mitogeno.
Il TGF-α è prodotto da vari tipi di neoplasie, non da alcune ed è, come dice la stessa parola, correlato con la trasformazione. PDGF ha una produzione un po’ più ristretta, a livello soprattutto dei sarcomi, cioè dei tumori del tessuto connettivo e a livello dei gliomi. La bombesina è ancora più ristretta come attività, perché è prodotta dalle cellule del carcinoma al polmone.A questo punto conviene fare un pochino una riflessione su alcuni punti fondamentali della neoplasia. Allora, il primo punto fondamentale è che la neoplasia è di tipo MONOCLONALE. Cosa vuol dire? Vuol dire che la neoplasia coinvolge una sola cellula e che da questa cellula trasformata, si produce un clone; si producono cellule identiche, uguali alla cellula da cui derivano.Mentre, invece, quando abbiamo un processo IPERPLASTICO, questo coinvolge più cellule perché sono tutte sottoposte all’azione di un agente stimolante. La neoplasia è il frutto, come vi ho sempre detto, di una trasformazione a livello genico. Quell’alterazione a livello genico è di una cellula non può coinvolgere più cellule. La maggior parte delle alterazioni a livello genico, nei
tumori, sono alterazioni di tipo strutturale, cioè alterazioni numeriche dei cromosomi, alterazioni dei geni nel senso di mutazioni (geni sottoposti a mutazioni puntiformi, traslocazioni, delezioni…) però, sempre più evidente, risulta il fatto che in alcuni tumori ci sono anche modificazioni epigenetiche, cioè delle modificazioni chimiche sia delle basi del DNA sia degli istoni.Allora, intanto io vorrei dirvelo questo, perché entrerete in contatto con questa realtà. Ci sono tre tipi di modificazioni epigenetiche:
• dovute ad acetilazione e metilazione degli istoni;
• modificazione dei microRNA;• metilazione del DNA.
Cosa sono i microRNA? Risposta di una collega: “sono RNA che vanno a bloccare dei geni, legandosi a determinate sequenze…” Risposta della professoressa: "devi dire quali sequenze. Il fatto è questo i microRNA non vanno a bloccare i geni. I microRNA interagiscono con le sequenze 3’UTR degli mRNA, quindi a livello trascrizionale non genico, siamo molto più giù. Quindi, sono degli RNA piccoli, che agiscono interagendo con questa porzione 3’ UTR di questi RNA messaggeri, che vengono distrutti, degradati e non posso poi tradurre il messaggio.Ci sono microRNA che agisco in maniera diretta, altri che agiscono in maniera indiretta. Quelli diretti agiscono direttamente sul RNA, bloccando poi la traduzione. E questo vale per tutte le cellule: se un microRNA ha un’azione diretta, questa azione diretta si esplica a livello di tutte le cellule dell’organismo, quindi non c’è specificità tissutale.Ci sono altri microRNA che, invece,
90

sempre bloccano la traduzione di alcuni geni, ma in maniera indiretta cioè attraverso altri microRNA che a loro volta, poi, agiranno a valle. Questa è tessuto-specifica, quindi dipende dal tipo di espressione di differenziazione che ha quella cellula.Quindi, sono queste le alterazioni epigenetiche coinvolte nei tumori. Un altro punto importante è questo: lo sviluppo della maggior parte dei tumori è un processo multifasico, in cui c’è il coinvolgimento di molteplici mutazioni che colpiscono geni diversi, anche su cromosomi diversi. Cioè è raro che ci sia un tumore causato dalla mutazione di un solo gene: mutato questo gene, ci sta questo tumore. È molto raro. Perché ci sia un tumore è necessario che siano mutati più geni. Si chiamano proprio geni precoci, che si trovano nella prima fase della trasformazione e che poi vengono, successivamente, mutati. Questo che cosa comporta? Questo comporta non solo che ci siano più geni mutati anche su cromosomi diversi, ma anche che il processo avvenga in più fasi.Ci sono tre fasi che si chiamano appunto tappe della cancerogenesi e sono:
• induzione;• promozione;• progressione neoplastica.
Per questo lo definiamo un processo multifasico. L’induzione è quella fase in cui agiscono gli agenti oncogeni. Cioè in cui, le sostanze con potere oncogeno (che possono essere agenti fisici, chimici, biologici) inducono una trasformazione, ovvero una mutazione a livello genico. Questo vuol dire indurre una trasformazione.Parliamo, ad esempio, degli agenti
oncogeni chimici. Ci sono agenti oncogeni chimici che sono completi, cioè che inducono sia induzione sia promozione. Attivano non solo, quindi, l’induzione, ma anche la proliferazione neoplastica. Ma ci sono degli agenti oncogeni (la maggior parte) che vengono detti incompleti, cioè sono in grado di modificare la struttura dei geni, però le cellule non manifestano immediatamente questa trasformazione a livello genico. Cioè determinano quello che si chiama stato di induzione ovvero quella condizione in cui la cellula ha subito la mutazione a livello del DNA, ma quella cellula non presenta modificazioni del fenotipo. Ha soltanto modificazioni a livello del genotipo, ma non del fenotipo, e rimane in uno stato detto di quiescenza. La cellula si chiama cellula silente o dormant cell.Questo stato di induzione può durare anche a lungo, fino a quando non intervengono i cosiddetti fattori promuoventi. I fattori promuoventi non sono fattori in grado di modificare il DNA, questo lo dovete avere ben chiaro. Sono agenti in grado di attivare la proliferazione cellulare, quindi sono soprattutto:
• fattori di crescita;• citochine;• ormoni.
Questi agenti promuoventi che non sono assolutamente cancerogeni (i cancerogeni sono quelli che agiscono a livello dell’induzione) determinano la proliferazione soprattutto a livello di queste cellule che avevano già subito una modificazione a livello del DNA, determinando il passaggio dallo stato di induzione a quello di promozione, con attivazione della proliferazione cellulare.Poi una volta avvenuto questo, il
91

successivo passaggio allo stato di progressione è quasi automatico.Questo concetto, che è un processo multifasico, non è importante solo per capire la progressione del tumore, ma anche per capire perché ci sono delle recidive, cioè perché anche in seguito a trattamento chirurgico o chemioterapico, possono rimanere delle cellule che dal punto di vista macroscopico o fenotipico non presentano modificazioni, ma che poi hanno una mutazione a livello del loro genoma e quindi possono essere in grado di entrare nella fase di promozione e progressione in seguito all’azione di sostanze come virus o di agenti oncogeni attivanti, in generale, la proliferazione. Quando passiamo allo stato di progressione, anche questa fase avviene in più tappe.Dovete avere l’idea chiara che, quando c’è un tumore già formato (cioè in fase di promozione e progressione), si generano all’interno della massa tumorale i cosiddetti subcloni. Abbiamo detto che è una massa MONOCLONALE, però all’interno di questo clone di cellule, si realizzano diverse comunità neoplastiche, diverse tra di loro: si formano dei subcloni, perché la cellula neoplastica ha un’instabilità genomica, il genoma è instabile per le modificazioni che ha subito. Questo genera dei sublconi che hanno potenziale metastico, potenziale invasivo, potenziale maligno diverso.E che succede? Si ha la selezione. Cioè tra questi subcloni, si crea una competizione e vengono selezionati quelli più aggressivi.L’induzione viene anche detta iniziazione, quindi iniziazione è un sinonimo di induzione. Una volta avvenuta l’iniziazione, la crescita neoplastica deve essere sostenuta (quindi per andare allo
stadio di promozione e progressione) da fattori che non coinvolgono mutazioni del DNA, lo abbiamo chiarito questo credo.Ecco, questo è un altro fatto interessante secondo me. Noi abbiamo detto mutazione genica, d’accordo. Ci sono però anche i cosiddetti fattori ambientali, che possono essere degli agenti oncogeni, quelli che causano i tumori. L’incidenza dei tumori, di particolari tipi di tumori, varia moltissimo di paese in paese. Ovviamente molto importante è l’alimentazione; purtroppo ieri ho seguito la vicenda della Terra dei Fuochi, quella zona tra Napoli e Caserta in cui si ha un’alta incidenza di tumori, per la presenza di sostanze tossiche. Noi abbiamo Milazzo, secondo me, terribile come inquinamento. In Giappone c’è il consumo frequente di pesce crudo, che può essere la causa della maggiore incidenza di tumori gastrici in Giappone, rispetto che in altri paesi.Il fumo, ve l’ho già detto, vero? Non lo ripeto.Questa è la diapositiva che avevo messo apposta e che vi spiega un pochino il discorso della cancerogenesi, di questo processo multifasico. Queste sono cellule che hanno il genotipo e il fenotipo normale. Questa cellula con la X si trova nella fase di iniziazione ed è una cellula che ha subito un danno a livello del DNA, però vedete che dal punto di vista fenotipico non presenta nessuna modificazione rispetto alle altre cellule, solo dal punto di vista genotipico.Qui (in questa slide) più che parlare di promozione e progressione, si parla di promozione al primo ed al secondo stadio, ma comunque il concetto è lo stesso. Ecco nello stadio di promozione o primo stadio di promozione, la cellula è sottoposta
92

all’azione di agenti promuoventi che attivano la proliferazione. Perché, se vi ricordate, io mi sono prolungata un pochino nel meccanismo di azione dei fattori di crescita.Vi ho detto che attivano le protein-chinasi, le tirosin-chinasi a livello del recettore, a livello delle proteine di membrana e questo determina, appunto, un’attivazione della proliferazione per cui le cellule (prima qualcuna poi le altre), iniziano a modificarsi anche a livello del fenotipo. E questa può essere considerata una condizione pre-neoplastica.Successivamente, un ulteriore agente promuovente, che è un mitogeno che agisce principalmente, specificamente sulle cellule già modificate (abbiamo parlato, se vi ricordate, delle alterazioni pre-neoplastiche, sono cellule più sensibili all’azione di determinati agenti mitogenici, promuoventi) determinando l’espansione quindi la promozione di secondo stadio, quindi la progressione del processo neoplastico. Quali sono i fattori coinvolti nell’invasione? Perché una delle caratteristiche dei tumori maligni, la ripeto, è che presentano un’alterazione sia a livello dei geni che regolano la proliferazione, che dei geni che regolano poi l’espressione di proteine, è l‘invasività.Perché un tumore possa invadere i tessuti vicini o addirittura, diciamo, staccarsi dal tumore primitivo e invadere i tessuti lontani…la metastasi è la riproduzione a distanza di un tumore. Cioè la capacità di un tumore di staccarsi e riprodursi a distanza, questa è la definizione di metastasi. Per riprodursi a distanza, le cellule devono avere un potenziale invasivo abbastanza alto e un notevole apporto ematico. Quindi, una delle caratteristiche della presenza di un tumore
è la formazione di nuovi vasi sanguigni, la neoangiogenesi, perché durante lo sviluppo tumorale, si producono dei fattori di crescita che hanno anche la capacità di attivare la proliferazione delle cellule endoteliali, a livello delle venule preesistenti e quindi si creano, in questo modo, nuovi vasi sanguigni. Per cui: adeguato apporto ematico.Per poter dare luogo alla metastasi, una cellula o due cellule, si devono staccare dal tumore primitivo, quindi la prima condizione è la riduzione della adesione tra cellule. Ecco perché ho iniziato questa lezione con la membrana. L’adesività omotipica viene alterata. C’è un fatto importante in questo processo: le cellule tumorali perdono, riducono l’adesività omotipica, però acquisiscono il carattere di aderire a molecole non omotipiche, in particolare a molecole della matrice extracellulare. Questo fenomeno si spiega con la transizione da carattere di cellula epiteliale a carattere di cellula mesenchimale, il così detto TEM (Transizione Epitelio Mesenchimale). Per cui, le cellule perdono le caderine e le altre molecole di adesione, ma acquisiscono altre molecole di adesione, come le integrine, che gli consentono di aderire ad altre molecole sia della membrana basale sia della matrice extracellulare. Questa è la transizione a cellula mesenchimale.In più le cellule neoplastiche hanno la capacità di produrre degli enzimi in grado di degradare la matrice extracellulare, cioè le cellule per dare luogo alle metastasi, devono camminare in un mare piuttosto ostile, denso, costituito dal collagene della matrice extracellulare, per arrivare ad un vaso linfatico, ad un vaso sanguigno. E come possono fare tutto questo? Se alterano la matrice extracellulare, possono
93

crearsi un passaggio. Quindi, sono in grado di produrre enzimi proteolitici e poi sono capaci di movimento attivo.Ecco, infatti, come si sviluppa l’angiogenesi: c’è una cellula neoplastica che fa già parte di un gruppetto di cellule e perché ci sia questo stato di promozione, di progressione, cioè che diventi una massa neoplastica significativa, necessita di un adeguato apporto di sangue. Quando si forma questo gruppo di cellule, alcune di queste, sono più lontane di altre dal vaso sanguigno per cui si trovano in condizione di ipossia; questa condizione determina l’attivazione dell’espressione di VEGF (Vascular Endothelium Growth Factor). Quindi le cellule neoplastiche producono grosse quantità di VEGF, ma non solo di questo, anche di FGF (Fibroblast Growth Factor) che un fattore proliferativo, mitogenico, ma anche angiogenico; pertanto questi inducono le cellule endoteliali di quel sito a proliferare, con formazione di nuovi vasi sanguigni che ovviamente si inoltrano nella massa tumorale, fornendo il sostegno attraverso sia ossigeno che sostanze nutritive, necessarie per la progressione e quindi poi anche per l’invasività.Il primo problema è che questi vasi sanguigni non sono ben strutturati. Non sono strutturati come i vasi sanguigni normali perché mancano della membrana basale; questi vasi sono soggetti al cosiddetto “ leakage” cioè trasudano liquido e sangue e questo è il motivo (spesso) del sanguinamento di alcuni tumori, che può rappresentare un allarme della presenza di un tumore, di una rottura del vaso perché, appunto, questi vasi sanguigni sono meno strutturati di quelli normali.
Ecco vedete la cellula tumorale, deve assolutamente perdere l’adesività omotipica però acquisisce la capacità di legare alcune molecole della membrana basale, come la laminina, il collagene di tipi IV…e ovviamente perché la cellula neoplastica esprime in maggiore quantità, quando c’è questa transizione verso la cellula mesenchimale, di recettori per la lamininina, che sono rappresentati dalle integrine, le beta1 integrine.Nel libro c’è un errore, ma non l’ho potuto cambiare, sono le beta1 integrine e non le beta2.Sono le VLA (Very Late Antigens) 2, 4, 5 e 6. Anche queste sono coinvolte nell’infiammazione, però per quanto riguarda i neutrofili, del legame non l’endotelio sono responsabili le beta2 integrine, sono CD11 e CD18 a essere coinvolte. Allora, queste integrine (VLA) interagiscono, a seconda che siano la 2, la 4, la 5 o la 6, con vari tipi di molecole della matrice extracellulare come: la laminina, la fibronectina. Con la fibronectina anche il beta4 ed il beta3.Che cosa succede? Succede che attraverso queste integrine beta1, la cellula neoplastica, aderisce alla membrana basale. Voi dovete capire una cosa, può sembrare strano, ma perché la cellula progredisca nel suo cammino (dopo che si è staccata), deve aderire prima aderire e poi può progredire. Perché? Perché aderendo libera questi enzimi proteolitici, distrugge quello che le è intorno progredisce; poi aderisce di nuovo, libera enzimi proteolitici e continua ad avanzare. Quindi, può sembrare un paradosso che la cellula per progredire, debba aderire. Ma l’adesione è necessaria affinché la cellula possa poi liberare questi enzimi proteolitici, che le consentono di degradare
94

la matrice extracellulare e di andare avanti.Fa così anche con la membrana basale, si lega alla laminina e libera soprattutto le “gelatinasi a e b” che sono quelle più specificamente coinvolte nella lisi del collagene di tipo IV, presente nella membrana basale.E vedete cosa succede? 1. Aderisce;2. Si lega alla laminina;3. Libera le gelatinasi;4. Viene interrotta la membrana basale e la cellula si insinua in questa lesione, in questa soluzione di continuità, emettendo degli pseudopodi (come fanno i neutrofili, è lo stesso concetto; la biologia utilizza meccanismi simili, quando li perfeziona, li utilizza in tutte le circostanze).
E poi che cosa succede? Che appena la cellula passa attraverso la membrana basale, si trova a contatto con la matrice extracellulare. E che fa? Immediatamente interagisce, soprattutto, con la fibronectina.Questo è il recettore per la fibronectina, aderisce alla matrice extracellulare. Questi sono gli enzimi proteolitici che, dopo aver aderito alla matrice extracellulare, vengono prodotti; vedete, sono soprattutto:
• metalloproteinasi (MMP);• gelatinasi per il collagene della
matrice extracellulare;• stromelisine;• serinproteasi (che non sono
metalloproteasi);• l’attivatore tissutale (tPA) del
plasminogeno.
Le cellule, abbiamo detto, si legano alla membrana, alla matrice, liberano gli enzimi proteolitici e si muovono. Quindi, devono acquisire anche capacità di movimento. Perché si muovono le cellule
neoplastiche? Perché producono loro stesse e vengono prodotti anche da cellule a loro vicine dei fattori che si chiamano “scattering factors”, cioè fattori di dispersione, prodotti sia dalle cellule neoplastiche sia dai fibroblasti. Quindi, agiscono sia per via autocrina sia per via paracrina.Scatter vuol dire disperdere. Fattore di dispersione che veniva chiamato, prima che lo si identificasse, “hepatocytes growth factor”.Seguiamo l’andamento di queste cellule, seguiamo tutte le tappe dell’invasività di queste cellule: abbiamo il tumore primario e abbiamo visto che c’è una modificazione a livello delle giunzioni intercellulari, questo facilita la formazione di cloni un po’ più isolati, rispetto alle cellule del tessuto. A un certo punto anche l’adesività omotipica si altera e si ha il distacco di cellule dal tumore primario.Le cellule fanno questa strada: membrana basale, matrice extracellulare, finché migrano (con lo scatter factor) e arrivano sino ad un vaso sanguigno. E qui fanno sempre la stessa identica cosa, si legano alla membrana basale vascolare (la precedente era la membrana basale tissutale). Quindi:
• aderiscono;• gelatinasi;• ingresso nel torrente circolatorio.
All’interno del torrente circolatorio, invasione delle venule. All’interno di questo torrente circolatorio succede che…qui c’è scritto trasporto delle cellule sottoforma di microemboli. Perché sottoforma di microemboli? Perché la cosa antipatica è che questa cellula neoplastica, all’interno dei vasi, continua a proliferare.Perché se rimanesse sola, potrebbe anche
95

essere distrutta, avvolta dalla fibrina, potrebbe esserci l’azione dei fattori della coagulazione, ci potrebbe essere l’attivazione a dissolvere questo coagulo, quindi l’azione del fibrinogeno, della trombina.Invece la cellula neoplastica, all’interno dei vasi continua la sua attività proliferativa; per cui da una cellula che entra (dico una, ma potrebbero essere anche due) si forma un microembolo, cioè un gruppetto di cellule e quindi viene trasportato, all’interno del vaso, un gruppetto di cellule che poi, ad un certo punto, diventando grande, si dispone all’esterno ed entra in contatto con la membrana basale del vaso e c’è il processo inverso. Processo inverso, nel senso che ora uscirà del vaso, sempre però:
• adesione;• gelatinasi;• fuoriuscita;• migrazione all’interno del tessuto
anche se non si sa se c’è l’attecchimento o meno.
Perché noi abbiamo tre fasi che sono:• ristagno;• migrazione;• attecchimento.
Non sempre le cellule che fanno tutto questo percorso poi attecchiscono nella sede, nella quale poi fuoriescono dal vaso sanguigno. Ci sono due condizioni per cui le cellule neoplastiche attecchiscano per la terza fase della metastasi e quindi per dare origine alla metastasi vera e propria. Sono due condizioni:
1. che il tessuto a livello del quale si sono liberate le cellule neoplastiche, non abbia grandi difese immunitarie, che non sia ricco di
linfociti, che non abbia capacità di difese immunitarie troppo elevate; la milza, per questo, non è mai sede di metastasi, il fegato sì. Ma il fegato perché? Perché nel fegato si realizza un’altra condizione;
2. perché la seconda condizione di attecchimento è che la cellula neoplastica trovi un metabolismo adeguato alle sue necessità.
Allora nel fegato che metabolismo c’è? Perché il fegato è una sede preferita? Perché nel fegato è abbastanza sostenuta la glicolisi anaerobica. Le cellule neoplastiche utilizzano la glicolisi anche in presenza di ossigeno; nel fegato si verificano anche delle situazioni di ipossia, con attivazione della glicolisi perché le cellule epatiche sono costrette. Le cellule neoplastiche però, pur in presenza di ossigeno, non utilizzano l’ossigeno e quindi attivano la glicolisi. E ci sono molti motivi per cui questo si verifica.Una collega risponde: “ un blocco dei sistemi navetta”. Riprende la Prof.ssa: esatto perché le cellule neoplastiche sono spesso carenti di alfa glicerofosfato deidrogenasi, per cui viene alterato il passaggio per ossidare il NADH. Il NADH non può passare attraverso la membrana mitocondriale e lo fa cedendo i suoi elettroni. Quindi, questo è il motivo principale. Per cui, se trovano questo metabolismo anaerobio più attivo, come accade nel fegato (dove non c’è una enorme attività immunitaria) ecco che le cellule attecchiscono.Vediamo quali sono i geni coinvolti nella trasformazione neoplastica. Abbiamo accennato l’altra volta al fatto che siano geni detti “protoncogeni”, che hanno un’attività positiva sulla proliferazione, perché codificano per proteine che attivano
96

la proliferazione e altri oncogeni che invece sono normali geni, che però (al pari dei protoncogeni) codificano per proteine che inibiscono la proliferazione. Ovviamente, mi pare che lo avevamo anche accennato, quando ci sono mutazioni a carico di questi protoncogeni, queste mutazioni sono di tipo dominante cioè è sufficiente che sia mutato un solo allele perché si abbia la trasformazione.Al contrario per quanto riguarda gli oncosoppressori. Per gli oncosoppressori parliamo di una mutazione recessiva (sono, infatti, per lo più deleti), quindi è necessario che vengano inattivati tutti e due gli alleli; perché quella proteina, anche se codificata in quantità minore dall’allele non mutato, può essere sufficiente a svolgere la funzione.Le proteine codificate dai protooncogeni sono coinvolte nel processare, nel trasdurre il segnale proveniente dagli stimoli mitogenici sino al nucleo. Però che cosa succede? Succede che nelle cellule trasformate, in cui appunto gli oncogeni mutati codificano per queste proteine coinvolte nella trasduzione del segnale, queste proteine si comportano come se fossero in presenza di stimoli mitogenici, anche in assenza di qualunque stimolo mitogenico…non so se è chiaro. E questo perché sono mutate con guadagno di funzione e quindi agiscono come se fossero state attivate da segnali mitogenici, anche in assenza di questi. Ecco perché queste cellule sono caratterizzate da una crescita così detta autonoma.Voi trovate nella definizione di tumore: crescita autonoma, afinalistica…io non vorrei che lo imparaste a memoria… Dovete rendervi conto di cosa significa crescita autonoma, proprio del discorso dell’economia della cellula…cioè la cellula
attiva questi processi di proliferazione anche in assenza di stimoli mitogenici, perché sono proprio alterate le proteiche che trasmettono questi messaggi.Ovviamente le cellule sono in grado di inibire anche le cellule che sono vicine e gli anti-oncogeni agiscono proprio in questo senso. Quindi che fanno questi anti-oncogeni? Codificano per proteine che partecipano alla trasmissione del segnale proliferativo; anche in questo caso, se queste proteine coinvolte nella trasmissione del segnale di inibizione sono modificate o non sono prodotte perché sono deleti i geni, questi segnali di inibizione non vengono attivati anche se la cellula è sottoposta a stimoli di natura inibitoria. Perciò i prodotti degli anti-oncogeni non sono da considerare dei prodotti semplicemente di tipo citostatico, ma sono dei prodotti che agiscono attivamente nella cellula, non in maniera passiva.L’inibizione della proliferazione è un evento attivo, che ha le stesse caratteristiche del processo che invece porta all’attivazione della proliferazione cellulare. E sappiamo che sono proprio queste molecole, a basso peso molecolare, che passano attraverso le giunzioni comunicanti.Come risponde la cellula, se tutto va bene, a questo segnale inibitorio? Può essere indotta a rimanere in fase G1, quindi le viene impedito di superare il checkpoint e di passare in fase S. Quindi viene costretta a rimanere in fase G1. Anche se vi ho detto che, dopo un certo periodo di tempo che la cellula sta in fase G1, senza ricevere alcuno stimolo, muore. Oppure viene stimolata ad andare direttamente nel compartimento di differenziazione.
97

Ora, non so se voi avete fatto il ciclo mitotico della cellula. Oltre alle quattro fasi del ciclo mitotico della cellula, quando si producono in condizioni fisiologiche, due cellule alla fine della fase M, di norma, una cellula ritorno nel ciclo e l’altra prosegue nel compartimento di differenziazione. Quando ci sono stimoli inibitori della proliferazione cellulare, tutte e due le cellule prodotte, magari sono indotte ad entrare nel compartimento di differenziazione e quindi a non proliferare più; in questo modo si riduce il grado di proliferazione. Oppure ancora, la cellula viene indotta ad andare incontro all’apoptosi, muore subito e non se ne parla più. Questi sono i tre meccanismi di risposta al segnale di inibizione della crescita.Abbiamo detto:
• strutturali;• numeriche;• puntiformi.
E tutti i vari tipi di mutazioni penso che li conosciate…Questa (riferendosi ad una slide) è una dimostrazione di quali siano le modificazioni più importanti a livello genico; c’è ad esempio un:
• processo di amplificazione genica (un protoncogene produce una quantità maggiore del suo prodotto, quindi da una copia o due del gene, in seguito alla duplicazione, si hanno più copie di oncogene);
• processo di traslocazione (come avviene nel linfoma di Burkitt, ad esempio) è una traslocazione (la più frequente 8 -14, ma anche altre) che comporta il fatto che un proto oncogene che è il C-Myc (che non è solo un fattore di trascrizione, ma è
anche uno di quei geni precocemente attivati in G1 e che viene regolato da una sua regione regolatrice a livelli basali), viene traslocato in una regione sotto il controllo di una regione regolatrice di una delle catene delle immunoglobuline.
Le immunoglobuline sono prodotte in grandi quantità nelle cellule B, quindi la regione “promoter”, la regione regolatrice delle immunoglobuline, è una regione che attiva l’espressione del gene, la trascrizione del gene. Se C-Myc, quindi, va sotto il controllo di questa regione regolatrice delle immunoglobuline, viene prodotto in maggiori quantità. Appunto nel linfoma di Burkitt ci sono questi tipi di traslocazione, che riguardando il cromosoma 8 che può traslocare non solo con il cromosoma 14 (che sarebbe quello che codifica per le catene pesanti), ma anche con le altre regioni del cromosoma 2 e 22 ( che codificano per le catene “k” e “lambda” delle catene leggere). Quindi può avvenire non solo la traslocazione più frequente che conoscete e questo appunto determina una “traslocazione reciproca” per cui avremo un cromosoma 8 q- a livello del braccio lungo del cromosoma 8, con una porzione del cromosoma 8 che è andata a traslocare su una porzione del cromosoma 14, che diventa appunto +.Un altro tipo di mutazione presente nei tumori è quella che vediamo, ad esempio, nella leucemia mieloide. Si tratta di un altro tipo di traslocazione. Una traslocazione 9 – 22. Sul cromosoma 22 è presente una regione in cui si verificano frequentemente delle rotture di combinazione, questa regione prende il nome di BCR: “breakpoint cluster region”,
98

perché queste zone di frattura sono tutte raggruppate in una regione.Che cosa succede nella traslocazione 9 – 22? Succede che questa regione BCR va a traslocare, sulla regione del cromosoma 9, dove si trova un protoncogene chiamato ABL. E la traslocazione da risultati diversi perché si forma un ibrido BCR-ABL che codifica per una proteina ad attività intensissima tirosino-chinasica, e quando si ha l’attiva di una proteina tirosino-chiansica, si ha l’attivazione della proliferazione cellulare in maniera enorme. Questo ibrido è sotto il controllo trascrizionale del gene BCR.L’intensa attività tirosino-chinasica di questa proteina di fusione (BCR-ABL), porta a sua volta ad attivare (a fosforilare a livello della tirosina) altri enzimi coinvolti nella proliferazione cellulare, soprattutto la via AKT. Quindi via RAS, RAF da queste poi l’attivazione della fosfatidilinotistolo chinasi AKT e quindi questa via…e soprattutto ad attivare una proteina (perché io prima ne ho parlato, perché queste proteine di cui abbiamo parlato nella regolazione della proliferazione, sono tutte coinvolte nei tumori) mediante la via KT della BCL2. Questo evento porta all’attivazione di segnali fortemente positivi per la proliferazione e questo soprattutto se in un primo momento si ha un’attività proliferativa anti-apoptotica (perché BCL2 è antiapoptotico) prima ancora che ci sia un’alterazione della differenziazione, anche se poi questo ci sarà perché nei tumori maligni abbiamo sempre una alterazione della proliferazione e della differenziazione. Ve ne ho parlato sia come esempio, ma anche perché questa proteina è stata una delle prime ad essere stata associata ad una neoplasia umana, in cui si è delucidato
il meccanismo alla base della patologia (leucemia mieloide). È importante che questa proteina abbia attività tirosino-chinasica, per poter essere oncogena (quindi causa di tumore), perché se invece ci sono mutazioni di questa proteina che ne ledono l’attività tirosino-chinasica, non avrà più potere trasformante. C’è anche il coinvolgimento di C-sis.Volevo dirvi un’altra cosa in relazione alla leucemia mieloide cronica…non tutte le cellule neoplastiche sono dovute alla presenta della modificazione di cui abbiamo parlato. Circa il 95% delle cellule presentano quella modificazione, ma c’è una percentuale minore presenta una modificazione a livello del gene Jak2. Jak2 è un gene che fa parte di quelli che codificano per una proteine detta Jak chinasi, che è coinvolto negli eventi proliferativi. In condizioni normali, un agente mitogeno interagisce con il recettore Jak2, che attiva Stat il quale è un fattore di trascrizione che agisce a livello genico. Nella leucemia mieloide cronica, in questi casi, una mutazione (noi la facciamo anche come diagnostica molecolare, perché è una mutazione diciamo già definita del gene Jak2) a livello di Jak2, determina l’attivazione di Jak2 anche in assenza di uno stimolo mitogenico, quindi Jak2 agisce, attiva Stat e questo, a sua volta, attiva la proliferazione, anche in assenza di un fattore mitogenico. Il meccanismo della formazione della leucemia mieloide cronica, indipendentemente dalla formazione del cromosoma a filamento (quello della prima traslocazione di cui abbiamo parlato), è dovuto ad una modifica del gene Jak2 che attiva la via di trasduzione del segnale, indipendentemente dal legame del recettore a cui è legato con il fattore di
99

crescita.Questa è la duplicazione, quindi è il secondo meccanismo, quello numerico. Duplicazione, amplificazione abbiamo già visto. Ci sono anche delle forme di trisomia presenti nelle forme mielodegenerative. E poi la mutazione puntiforme che a livello dei protoncogeni riguarda quasi esclusivamente RAS, perché nelle altre mutazioni dei proto oncogeni, abbiamo la mutazione, la traslocazione ecc…Mentre la mutazione puntiforme riguarda RAS, perché RAS codifica per g-protein che, in condizioni di attivazione, lega GTP.In condizione di riposo lega GDP, in condizione di attivazione lega GTP e trasmette il segnale a RAF.Cosa succede? Appena trasmette il segnale di attivazione a RAF, le g-protein attivano subito la loro attività enzimatica GTPasica che idrolizza il GTP, trasformandolo in GDP.In questo modo RAS e g-protein non trasmettono più il segnale mitogenico.È un processo molto importante che funziona proprio per evitare che ci siano processi di attivazione eccessivi; e la stessa proteina g-protein che trasmette il segnale di attivazione a RAF e poi attiva la sua attività enzimatica GTPasica, per tornare allo stato di riposo. Le proteine G, alterate per mutazioni puntiforme, presentano delle alterazioni nella sequenza di amminoacidi relativi alla loro attività GTPasica, per cui non sono più in grado di idrolizzare il GTP e mantengono così il loro stato di attivazione, anche se lo stimolo mitogenico è stato allontanato. Anche i virus sono agenti oncogeni. La cosa paradossale, diciamo particolare, è che un retrovirus (perché di retrovirus stiamo parlando) anche se non è
trasformante, lo può diventare dopo che infetta la cellula. Perché avendo la capacità di interagire con il genoma cellulare, quando si moltiplica, si duplica il virus, può avere all’interno del suo genoma un gene cellulare, sotto il controllo di una regione regolatrice virale e quindi può diventare, a sua volta, un virus trasformante attraverso questo processo: virus non trasformante, la regione regolatrice regola alcune proteine virali, l’infezione alla cellula animale che ha un proprio oncogene, se si inserisce un protoncogene nella zona vicina ed il retrovirus si riproduce completamente, fuoriescono dalla cellula delle particelle virali che sono ora oncogene, trasformanti. Perché quella porzione del protoncogene della cellula animale si trova ora sotto il controllo della regione regolatrice virale. Quindi, l’infezione della cellula e la cellula che rende trasformante il virus. È un discorso un po’ particolare.Questo è il meccanismo della formazione di queste particelle nuove virali.Questo credo che lo sappiate, tutto il processo di ingresso del genoma virale nella cellula, la formazione del RNA virale, la duplicazione mediante l’azione della trascrittasi inversa, si passa da singolo filamento a doppio filamento di DNA, doppio filamento che si può integrare nel genoma cellula quindi si ha il pro-virus.Trascrizione, traduzione, formazione di nuove particelle e quindi di virus infettante.Allora appunto i retrovirus creano nuovi oncogéni modificando, deregolando i protoncogeni cellulari. I geni oncogeni dei virus a RNA non sono necessari ai virus e la replicazione dei virus non è citotossica, mentre invece è citotossica la replicazione dei virus a DNA, non so se questo ve lo
100

ricordate. Difatti i virus a DNA determinano infiammazione quando provocano lisi cellulare, quando si replicano quindi totalmente. Sono trasformanti e quindi determinano tumori (che sono per lo più, però tumori benigni, mentre i virus a RNA causano tumori maligni), quando non viene riprodotto tutto l’intero virus, ma quando vengono trascritti soltanto quei geni detti “precoci”, cioè quei geni che determinano la sintesi di proteine trasformanti. Che non siano né proteine strutturali né proteine trasformanti proprie del virus, allora si ha la trasformazione cellulare…per quanto riguarda i virus a DNA.Con i virus a RNA, la trasformazione, si ha anche con la replicazione del virus, che però non è citotossica, altrimenti non si avrebbe la trasformazione.I vari protoncogeni partecipano a tutte le fasi della crescita e dello sviluppo della cellula, quindi ad eventi biologici, biochimici e molecolari come:
• interazione ligando-recettore;• gli enzimi coinvolti nella risposta
recettoriale;• negli eventi che sono coinvolti nella
trascrizione e nella traduzione.
Abbiamo quattro classi di oncogeni:• la prima classe di divide in due
sottoclassi: prima A e prima B; la sottoclasse prima B è costituita proprio dalle tirosino-chinasi citoplasmatiche, cioè quelle di cui abbiamo parlato in precedenza, quelle con il modulo SH2: SARC, FOSFOLIPASI C GAMMA ecc. I geni che codificano per queste proteine sono protoncogeni. L’altra sottoclasse è costituita dai recettori per i fattori di crescita;
• la classe seconda è costituita dai fattori di crescita;
• la classe terza è costituita dalle proteine coinvolte nella trasduzione del segnale, in particolare dalle G-protein e da tutte quelle dei processi post recettoriali;
• la classe quarta è costituita dai fattori di trascrizione, in grado di legarsi a sequenze specifiche del DNA.
Nella classe prima B ci sono appunto questi recettori per i fattori di crescita, aderenti proprio alla parte interna della plasmamembrana.Andiamo velocemente. La proteina SARC è così chiamata perché ha molta omologia con un gene del virus che causa il sarcoma di Rous. Il sarcoma di Rous è un tumore che coinvolge il petto di pollo ed è stato studiato nei primi anni del ‘900, quando ancora non si sapeva che c’erano i virus. È stata scoperta una cosa stranissima, che un filtrato che non conteneva cellula, proveniente da un tumore di un pollo (appunto da un sarcoma), era in grado di riprodurre lo stesso tumore all’interno di un pollo non ammalato; questo proprio perché era causato da un virus. I virus all’inizio venivano chiamati “ particelle filtranti”, perché filtravano anche attraverso i filtri più stretti. Questo è l’esperimento di Rous, che ha dimostrato che: da un pollo con il sarcoma a livello del petto, in seguito all’estrazione di questo sarcoma e alla sua “omogeneizzazione” (perché veniva, diciamo, omogeneizzato) e filtrazione (attraverso un filtro molto stretto), inoculando solo il filtrato in un pollo giovane, il tumore si ripresentava.Questi sono tutti tumori che sono stati
101

studiati negli animali. Questa è la leucemia murina di Abelson (studiata nei topi), in cui abbiamo un gene ABL che è omologo a quello che abbiamo trovano coinvolto nella leucemia mieloide cronica. Questo ABL presente nel topo ha molta omologia con l’ABL che troviamo nell’uomo e con i geni omologhi a SARC, quindi fa parte anche di quelle proteine citoplasmatiche.Poi l’interazione dei fattori di crescita con i loro recettori.Parliamo dei recettori per i fattori di crescita. Abbiamo detto una classe prima B. Uno dei recettori più studiati è il recettore per un fattore di crescita che viene detto “Epithelial Growth Factor”, che è un potente agente mitogenico per tutte le cellule, non solo per quelle epiteliali. E allora questo recettore EGF è codificato da un protoncogene che si chiama EV, importante anche nel carcinoma della mammella. Questo recettore, codificato da una mutazione del gene EV, comporta il fatto che manchi della porzione extracitoplasmatica e questo significa una cosa importantissima, cioè che il recettore (carente della porzione extramembrana, extracitoplasmatica) si comporti come un recettore che ha interagito con un fattore di crescita.La cellula percepisce un segnale mitogenico quando la porzione extramembrana del recettore è complessata con il fattore di crescita, se questa porzione extramembrana manca, la cellula percepisce lo stesso segnale andando incontro ad attività proliferativa, indipendentemente dalla presenza del fattore di crescita. E soprattutto è associato al carcinoma epiteliale del polmone.Un altro oncogene è il virus dell’eritoblastosi chiamato EV-A, che
potenzia l’azione di EV-B, ma che ha anche una sua attività autonoma in quanto codifica per il recettore ad alta affinità per l’ormone tiroideo.L’ormone tiroideo si lega al recettore, che ha una localizzazione intranucleare. Si lega a regioni regolatrici, attivando molti geni coinvolti nella proliferazione.Un altro protoncogene importante a livello della proliferazione delle cellule del midollo osseo è il gene M-CSF (Colony stimulating factor), che codifica per il recettore per il Monocyte Colony stimulating factor 1, che è quindi un fattore di proliferazione e differenziazione per i monociti. Quindi, quando è mutato può essere coinvolto in processi leucemici.C-sis ha delle omologie di sequenza con la catena b del PDGF (Platelet- Derived Growth Factor).Un altro fattore di crescita importante è il Fibroblast Growth Factor, che è coinvolto nel Sarcoma di Kaposi e in molti tumori della vescica o dello stomaco. Nel carcinoma mammario, il corrispondente oncogéne prende il nome di IT2. Il FGF non è solo un fattore mitogenico in senso lato, ma anche un potente fattore angiogenico, come il BEGF (Basic Epidermal Growth Factor). Quindi ha fattore mitogenico per i fibroblasti ed angiogenico perché incrementa lo sviluppo di vasi sanguigni e quindi favorisce la crescita dei tumori solidi. Però l’FGF, al contrario dei BEFG, è anche un protoncogene.Questa è un po’ la sequenza che avviene in condizioni normali. Penso che già lo sappiate dalla biochimica, ve l’ho fatto vedere perché lo possiate ricordare.Quando appunto presentano queste mutazioni puntiformi, le proteine G, non sono più in grado di attivare l’attività GTPasica e quindi mantengono sempre il
102

loro stato di attivazione.Vedete come dall’interazione tra recettore e fattore di crescita si ha tutta l’attivazione di una serie di risposte a livello della membrana che portano all’attività di 21RAS. GAP vuol dire GTPase protein, quindi attivazione di GTPasi; e badate che c’è anche un altro tipo di mutazione, molto meno frequente, che non riguarda RAS, ma riguarda la GTPasi. Perché le G-protein hanno attività GTPasica, ma pur avendo questa attività queste proteine, è un’attività che comunque deve essere attivata. La cellula per mettersi al riparo da eccessivi stimoli mitogenici, ha addirittura una proteina che attiva l’attività GTPasica; mutazioni di questa proteina possono determinare la condizione di attività iperproliferativa. Quindi, non soltanto a livello delle G- protein, ma anche nella proteina che attiva la GTPasi. Appunto RAF che viene subito attivata dalle G protein, le fosfolipasi C.E queste sono le vie del segnale attivate dalle Fosfolipasi C, diacilglicerolo e proteichinasi C, inositolo trifosfato che attiva la mobilizzazione del calcio, il DAG che attiva la protein chinasi C che ha anche attività tirosin-chinasica, SARC che ha diversi substrati e la Fosfoinositolo 3 chinasi.Mi ero dimenticata di dirvi che in molti tumori c’è un’amplificazione genica extra cromosomica, cioè molte volte l’amplificazione dei geni avviene a livello di regioni di DNA che sono extracromosomiche, che hanno DNA circolare e che presentano molte copie geniche, soprattutto di proto oncogeni come C-Myc e RAS. Si chiama “double” (?). E poi inserzione di un nuovo promotore trascrizionale, come per esempio la trasformazione virale, come
abbiamo visto poco fa.Vedete, nelle cellule normali si ha la presenza di livelli basali di fattori di crescita, di recettori, di tutti i segnali di trasduzione normali e basali Nelle cellule neoplastiche si ha un’iperattività o dei fattori mitogenici, o dei recettori per i fattori mitogenici, oppure delle vie di trasduzione del segnale o dei fattori che attivano la trascrizione genica.Per quanto riguarda gli oncosoppressori, di P53 ne abbiamo parlato, l’altro oncosoppressore abbastanza importante è quello che codifica per il gene RB del retinoblastoma, che è coinvolto nella proliferazione, perché quando non è fosforilato lega i due fattori che danno il via alla proliferazione cellulare (al passaggio alla fase S) E2F e DP1. Quando invece viene fosforilato, perché vengono attivate le vie di fosforilazione a livello di tirosino-chinasi, “molla” il complesso E2F- Dp1 e l’RB viene fosforilato dai complessi chinasi ciclina dipendenti.Il P53 lo abbiamo visto, soprattutto tramite al P21, blocca pure questi complessi.Una cosa interessante è che alcuni virus, come il papilloma virus, si comportano bloccando i prodotti dei geni oncosoppressori. Cioè i geni oncosoppressori non sono mutati o alterati, codificano il loro prodotto genico, eppure non funzionano, perché sono complessati dalle proteine del papilloma virus. Sono il 16 ed il 18 che producono l’uno la proteina 7 che lega l’RB ed il 18 che produce la proteina E6 che lega P53. Quindi come se questi geni fossero deleti e invece sono complessati dalle proteine del papilloma virus.Qui c’è la metilazione, parlando delle varie mutazioni. Ancora abbiamo qui l’alterazione genetica quindi abbiamo il
103

cambiamento nella sequenza del DNA. Abbiamo avuto la mutazione rispetto a questa sequenza. Questa sequenza presenta una mutazione adenina al posto di una timina, e quindi poi si la divisone cellulare e poi l’amplificazione cellulare e quindi si mantiene la mutazione. Per cui a livello del gene, per esempio si forma l’RNA e questa alterazione che si ritrova nella proteina che poi viene tradotta nell’RNA, non può essere revertita da nessun trattamento farmaceutico.Se invece abbiamo una modificazione come ad esempio una metilazione a livello della citosina (le citosine coinvolte, lo avevamo detto, sono prevalentemente a livello del promoter), il gene viene silenziato.Ci sono molti studi promettenti di trials clinici che utilizzano dei farmaci che sono in grado di demetilare le citosine, oppure ci sono proprio inibitori delle acetilasi, quelli di vecchia generazione e quindi poco specifici e quelli di nuova generazione che invece sono specifici per la classe di istoni coinvolti, che revertono la metilazione della citosina. Con l’intervento di queste sostanze, che riescono ad attuare la deacetilazione, quindi il gene viene espresso.La metilazione del DNA viene riconosciuta ampiamente sia come meccanismo patogenetico di tumori come markers di un tumore e quindi utilizzata a livello diagnostico.In questa diapositiva si vede come la replicazione possa venire regolarmente, in condizioni normali, nella trascrizione di un tumor suppressor g; come invece questa non si verifica quando si ha la metilazione della citosina, quando vengono attivati i geni che codificano per le DNA metiltransferasi. Ci sono tre tipi di DNA
metiltransferasi:• 2 tipi (DMT3A e DMT3B) sono
prodotti nella metilazione de novo, cioè attivano la metilazione di citosine de novo;
• Il DMT1 invece mantiene lo stato di metilazione.
Cioè la metilazione dei geni non è negativa, diventa negativa quando è eccessiva e rivolta a livello del tumor suppressor gene, ma uno stato di metilazione dei geni è necessario per mantenere la trascrizione genica a livello basale. Quindi chi mantiene lo stato di metilazione (metilazione di mantenimento), cioè fa metilare le basi di nuove catene di Dna, in modo similare a quelle del DNA parentale è il DMT1. Per quanto riguarda quello di nuova metilazione, sono DMT3A e DMT3B. E queste si studiano proprio in corso di tumore.L’inattivazione degli oncogéni non avviene solo per metilazione, ma anche per alterazioni strutturali quali possono essere: delezione o mutazione di senso errato. Come per P53, che può essere alterato o per delezione o per mutazione di senso errato. Vedete che ci sono molti tipi di tumore in cui c’è delezione o inativazione soprattutto del tumor suppressor gene come nel:
• retinoblastoma;• tumore di Wilms;• alcuni tipi di carcinoma del colon.
Abbiamo detto che si chiama retinoblastoma perché riguarda soprattutto l’occhio, però non è sempre così. La delezione che riguarda il retinoblastome è sul cromosoma 13. E il tumore di Wilms riguarda il rene, è dovuto
104

ad una delezione su una regione (P13) del cromosoma 11. Sono tumori tessuto-specifici. Questi tumori embrionali (retinoblastoma e nefroblastoma di Wilms) sono tutti tumori che si trovano nella prima infanzia: nei bambini e negli adolescenti. La mutazione riguarda, infatti, la linea germinale, si ha un’alterazione a livello germinale di un allele e poi il tumore si sviluppa quando si ha l’alterazione dell’altro allele. Quindi queste delezioni degli antioncogeni riguarda soprattutto la linea germinale.Nelle cellule predisposte basta che ci sia un solo allele mutato. Però perché ci sia il tumore è necessario che entrambi gli alleli siano mutati, questo è un esempio di quello che avviene nel retinoblastoma, in cui manca la regione codificante per l’RB. È importante che, nel caso del retinoblastoma, si distingua se la mutazione è a livello somatico o della linea germinale. Perché la proteina RB si trova a livello di tutti i tessuti e non solo della retina, abbiamo visto, infatti, che prende parte alla proliferazione cellulare. Le mutazioni dei geni oncosoppressori sono prevalentemente a livello della linea germinale, mentre le mutazioni dei protoncogeni solo a livello somatico. Se il gene che codifica RB1 è mutato e la mutazione riguarda solo la linea germinale, parliamo di retino blastoma. Se invece la mutazione riguarda il proto oncogene (quindi parliamo di mutazioni somatiche), il tumore si verifica anche a livello di altri distretti, di altri organi; quindi non è localizzato solo a livello della retina. Quindi l’alterazione del gene che codifica per RB1 può riguardare sia la linea somatica sia quella germinale.Una mutazione simile a quella di RB si ha con il Papilloma Virus, che complessa l’E7,
che complessa l’RB.Il tumore di Wilms, vi ho detto che è tessuto specifico, nefroblastoma. Codifica per un fattore di trascrizione che ha la zinc finger, che consente il legame (di questo fattore di trascrizione) a sequenze specifiche del DNA. E antagonizza il recettore per l’epithermal growth factor, e questo è anche importante. Poi c’è un’altra mutazione (delezione) che riguarda una regione che coinvolge la codificazione di un gene che codifica per una fosfatasi.Quindi, per un gene che codifica per un enzima che è in equilibrio con una tirosino-chinasi, ecco perché questo è un gene oncosoppressore, perché in questa regione (3, P13, 23) che comporta il tumore in varie zone come polmone ed ovaio, c’è un locus che codifica per la tirosin fosfatasi gamma. Cioè per un enzima fostatasi che idrolizza gli atomi di fosforo dovuti all’attività tirosino-chinasica, che evita quindi il segnale di proliferazione. In questi tumori viene persa questa regione allelica che codifica per questa fosfatasi.Una cosa importante nel tumore del colon non poliposico, viene alterato un altro gene. Questo è importantissimo perché, se si fa uno screening adeguato (perché, in questo tumore, abbiamo un’alterazione dei processi di riparazione del DNA, del mismatch repair), nei soggetti a rischio, si possono sventare molti casi di tumore al colon. Quindi, l’individuazione di questi geni coinvolti nel mismatch repair è di fondamentale importanza anche a livello di prevenzione.Vi volevo lasciare con questo messaggio, dato che voi poi farete i medici, perché questo vi potrà aiutare a salvare molte vite. La ricerca scientifica è quindi importante sia a livello clinico sia preventivo.
Ludovica Crocè
105