
ANANANEEEMMMIII E E E EEE … · buire a Dressler che, nel 1854, riportò un caso di ... me, che...
36
pagina 1 Anemie Emolitic Anemie Emolitic Anemie Emolitic Anemie Emolitic Anemie Emolitiche Autoimmuni he Autoimmuni he Autoimmuni he Autoimmuni he Autoimmuni AN AN AN AN ANEMIE E E E E EMOLITI ITI ITI ITI ITICHE AUT UT UT UT UTOIMMUNI: I: I: I: I: diagnosi e terapia Wilma Barcellini 1 , Maria Antonietta Villa 2 , Nicoletta Revelli 2 , Alberto Zanella 1 1. U.O. Ematologia 2, Dipartimento di Medicina e Specialità Mediche, 2. Centro Trasfusionale e di Immunoematologia, Dipartimento di Medicina Rigenerativa IRCCS Fondazione Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena, Milano. Domande e commenti? Clicca qui
-
Upload
truongliem -
Category
Documents
-
view
217 -
download
0
Transcript of ANANANEEEMMMIII E E E EEE … · buire a Dressler che, nel 1854, riportò un caso di ... me, che...

pagina 1
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
A NA NA NA NA N EEEEE MMMMM IIIII E E E E E EEEEE MMMMM OOOOO LLLLL I T II T II T II T II T I CCCCC HHHHH EEEEEAAAAA U TU TU TU TU TOOOOO IIIII MMMMM MMMMM UUUUU NNNNN I :I :I :I :I :diagnosi e terapia
Wilma Barcellini1, Maria Antonietta Villa2, Nicoletta Revelli2, Alberto Zanella1
1. U.O. Ematologia 2, Dipartimento di Medicina e Specialità Mediche,2. Centro Trasfusionale e di Immunoematologia, Dipartimento di Medicina RigenerativaIRCCS Fondazione Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena, Milano.
Domande e commenti? Clicca qui

pagina 2
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni

pagina 3
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
CENNI STORICI pag 5
CLASSIFICAZIONE pag 7
MECCANISMI PATOGENETICI pag 9
QUADRO CLINICO pag 13
DIAGNOSI pag 15
ANEMIE IMMUNOEMOLITICHE DAFARMACI pag 19
TERAPIA pag 21
ASPETTI PARTICOLARI DELLEANEMIE EMOLITICHE AUTOIMMUNI pag 27
CONCLUSIONI pag 29
BIBLIOGRAFIA pag 31
IIIIINNNNNDDDDDIIIIICCCCCEEEEE

pagina 4
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni

pagina 5
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
CCCCCEEEEENNNNNNNNNNI SI SI SI SI STTTTTOOOOORRRRRIIIIICCCCCIIIII
Circa quindici secoli prima della scoperta deglieritrociti da parte di Malpighi nel 1661, Galeno de-scrisse nel 150 d.c. quello che verosimilmente eraun episodio di anemia emolitica acquisita (1): ri-portò di una persona morsa da una vipera, la cuicute diventò rapidamente pallida. Lo stesso Autoresi spinse poi ad ipotizzare che vi fosse in tutto ciòun’implicazione della milza e, così facendo, antici-pò di circa due millenni Widal, Abrami e Brulè che,nel 1911 al Congrés Français de Médecine di Lio-ne, definirono il ruolo emocateretico della milza nellecrisi emolitiche (2). Alle considerazioni redatte inquesto congresso, sempre nel 1911, Micheli fece se-guire la prima splenectomia terapeutica per l’ane-mia emolitica (3).La prima descrizione di quello che era certamenteun caso di anemia emolitica autoimmune è da attri-buire a Dressler che, nel 1854, riportò un caso diquella forma di emolisi autoimmune che sarebbe sta-ta in seguito definita emoglobinuria parossistica afrigore (4). Il paziente, un bambino di 10 anni vero-similmente affetto da sifilide congenita, sviluppavamacroematuria ogni qualvolta si esponesse alle bas-se temperature.Il lavoro di Dressler anticipò di 15 anni la primadescrizione dell’agglutinazione eritrocitaria, il cuimerito spetta ad Adolf Creite che, nel 1869, nel suoVersuche uber die Wirkung des Serumweisses nachInjection in das Blut, riferì di come proteine sierichedi varie specie animali fossero in grado, una voltainiettate nel torrente circolatorio di conigli, di de-terminare il dissolvimento (lisi) e/o l’aggregazione(agglutinazione) delle emazie della cavia (5). Creitedescrisse inoltre che a queste reazioni seguiva l’emis-sione di urine scure, proprio come il caso del pa-ziente segnalato da Dressler. Reazioni simili furonoosservate anche utilizzando sangue umano e venne-ro descritte da Landois (6).Sebbene Mackenzie nel 1879 ipotizzasse che la di-struzione dei globuli rossi avveniva nel rene (7),Kuessner dichiarò che l’emolisi aveva luogo nel tor-rente circolatorio dopo aver osservato che, duranteuna crisi di emoglobinuria, il siero acquisiva un co-lore rosso proprio per la presenza di emoglobinalibera (8). Vanlair e Masius furono i primi, nel 1871,
a descrivere una crisi emolitica in un paziente ane-mico, non epatopatico (9); spetta però a Minkowski,nei primi anni del ventesimo secolo, il merito di averparlato di ittero acolurico ereditario cronico, sepa-rato dall’ittero ostruttivo (10).Furono Donath e Landsteiner nel 1904 ad identifi-care in un anticorpo anti-emazie la causadell’emoglobinuria parossistica a frigore e a descri-verne il procedimento necessario all’identificazio-ne, definito in seguito proprio test di Donath-Landsteiner (11). I lavori di Karl Landsteiner per-misero di comprendere i fenomeni alla base dellacompatibilità e delle reazioni trasfusionali e gli val-sero il premio Nobel nel 1930.Tra gli anni ’30 e ’40 divenne chiaro chel’autoagglutinazione delle emazie “a freddo” rap-presentava un ben definito evento immunologico chepoteva associarsi a varie malattie; rimaneva invecedi più difficile comprensione la natura immunologicadelle forme emolitiche non associate ad autoagglutinine fredde, cioè le forme da anticorpi caldi.Queste per molti anni furono confuse con l’itteroemolitico costituzionale di Minkowski-Chauffard(sferocitosi ereditaria), sino alla introduzione deltest di Coombs (o test dell’antiglobulina diretto,TAD) nel 1945, che rese chiara la differenza tra ledue condizioni (entrambe caratterizzate dalla pre-senza in varia misura di sferociti nello striscio disangue periferico). Nei pochi anni a seguire venne-ro approfonditi gli aspetti sierologici delle malattieimmunoemolitiche nonché la loro associazione convarie patologie. A metà degli anni ’60, si compreseche alcune forme immunoemolitiche potevano esse-re causate da farmaci. Il primo prodotto implicatonello sviluppo di una AIHA farmaco-indotta fu l’alfa-metildopa. Infine, negli ultimi 20 anni, si sono iden-tificate, e sono andate meglio definendosi, le formeatipiche di anemia emolitica autoimmune, quali leforme TAD-negative e quelle sostenute da IgM “cal-de”.Queste osservazioni furono molto importanti perchécostituirono le premesse per la odierna classifica-zione clinica e sierologica delle anemie emoliticheautoimmuni.

pagina 6
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni

pagina 7
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
Le anemie emolitiche autoimmuni (AEA) compren-dono un gruppo eterogeneo di condizioni morbosecaratterizzate dalla presenza di autoanticorpi diretticontro antigeni eritrocitari e da un quadro clinico diemolisi variabile nella sua gravità. Si tratta di ma-lattie relativamente rare con una incidenza di 1-3casi per 100.000 persone/anno, che possono insor-gere sin dalla prima infanzia, ancorché siano più co-muni dopo la III-IV decade (12).Le AEA vengono distinte in base alle proprietà ter-miche dell’anticorpo, in forme da autoanticorpi “cal-di”, che si legano agli eritrociti ad una temperaturaintorno ai 37°C, e AEA da anticorpi “freddi”, chepossiedono un optimum di reazione a 4°C e che com-prendono due entità cliniche distinte, la malattia daagglutinine fredde (CHD; cold hemagglutinindisease) e la emoglobinura parossistica a frigore. LeAEA da anticorpi “caldi” costituiscono la maggio-ranza delle forme (48-70% dei casi), la malattia da
agglutinine fredde rappresenta il 16-32% dei casi,mentre l’emoglobinura parossistica a frigore rappre-senta una rarità (2% dei casi) (12); infine, vanno ri-cordate le forme “miste” (3% dei pazienti) dove siosservano sia anticorpi “caldi” che “freddi”, e le AEAda farmaci, di cui si tratterà in un capitolo a parte(Tabella 1). Da un punto di vista clinico le AEA ven-gono distinte in forme acute e croniche, sulla basedella presentazione, e in forme primitive (idiopa-tiche) e secondarie ad altre condizioni morbose, fracui malattie infettive, autoimmuni e neoplastiche.In particolare, l’AEA da anticorpi “freddi” rappre-senta una complicanza osservata frequentementenelle sindromi linfoproliferative, quali i linfomi nonHodgkin e la leucemia linfatica cronica, con una pre-valenza variabile nelle diverse casistiche dal 10 al40% dei casi (12). Vanno ricordate le forme secon-darie a infezione da Mycoplasma pneumoniae spes-so iperacute ma a buona prognosi.
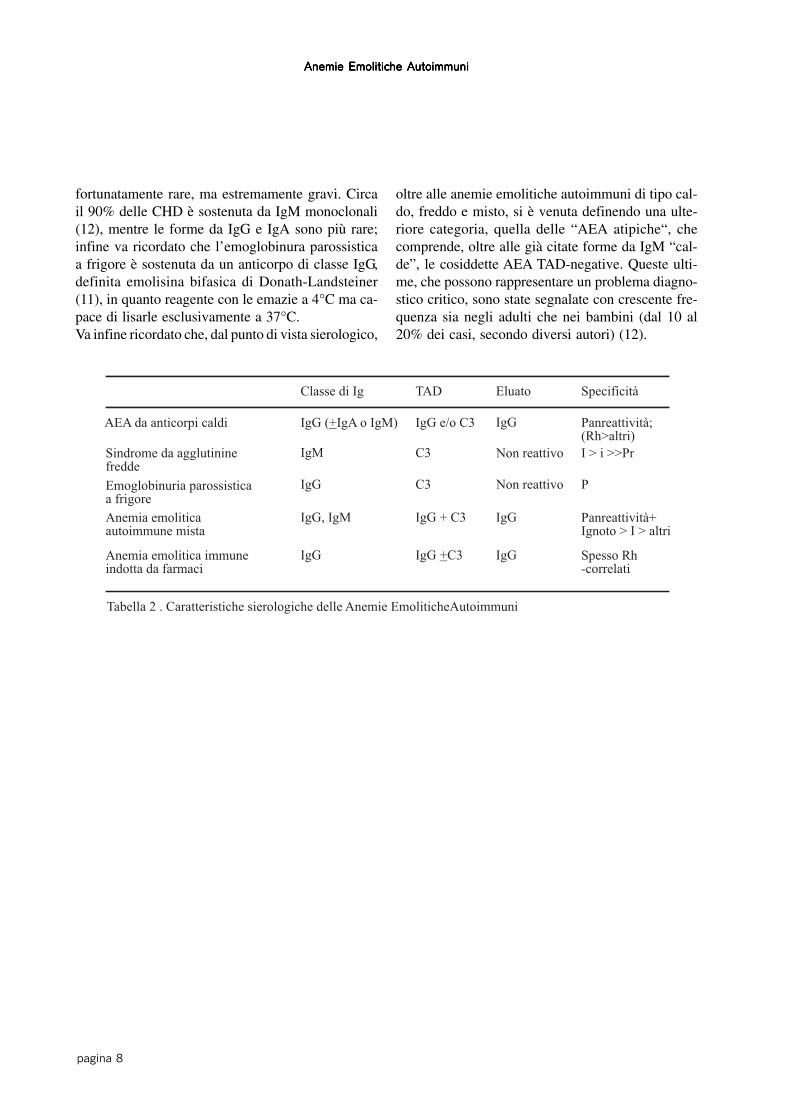
Infine, le AEA si distinguono in base alla classeimmunoglobulinica dell’autoanticorpo (Tabella 2),che più frequentemente è una IgG con attività ter-mica ottimale a 37°C. Un terzo circa delle AEA è
sostenuta da autoanticorpi di classe IgM (12), cheper la loro elevata avidità e capacità di fissare il com-plemento determinano una emolisi più marcata. Ciòè particolarmente vero per le forme da IgM “calde”,
CCCCCLALALALALASSSSSSSSSSIIIIIFFFFFIIIIICCCCCAZAZAZAZAZIIIIIOOOOONNNNNEEEEE
Tabella 1. Classificazione delle Anemie Emolitiche Autoimmuni
Anemia emolitica autoimmune da anticorpi caldiIdiopaticaSecondaria (malattie linfoproliferative, malattie autoimmuni)
Anemia emolitica autoimmune da anticorpi freddi
Sindrome da agglutinine freddeIdiopaticaSecondaria
Acuta temporanea (infezioni)Cronica (malattie linfoproliferative)
Emoglobinuria parossistica da freddoIdiopaticaSecondaria
Acuta temporanea (infezioni)Cronica (sifilide)
Anemia emolitica autoimmune di tipo mistoIdiopaticaSecondaria (malattie linfoproliferative, malattie autoimmuni)
Anemia emolitica immune farmaco-indottaDa adsorbimento di farmaci alla membrana eritrocitariaDa immunocomplessiAutoimmune
pagina 8
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
fortunatamente rare, ma estremamente gravi. Circail 90% delle CHD è sostenuta da IgM monoclonali(12), mentre le forme da IgG e IgA sono più rare;infine va ricordato che l’emoglobinura parossisticaa frigore è sostenuta da un anticorpo di classe IgG,definita emolisina bifasica di Donath-Landsteiner(11), in quanto reagente con le emazie a 4°C ma ca-pace di lisarle esclusivamente a 37°C.Va infine ricordato che, dal punto di vista sierologico,
oltre alle anemie emolitiche autoimmuni di tipo cal-do, freddo e misto, si è venuta definendo una ulte-riore categoria, quella delle “AEA atipiche“, checomprende, oltre alle già citate forme da IgM “cal-de”, le cosiddette AEA TAD-negative. Queste ulti-me, che possono rappresentare un problema diagno-stico critico, sono state segnalate con crescente fre-quenza sia negli adulti che nei bambini (dal 10 al20% dei casi, secondo diversi autori) (12).
I > i >>Pr
P
IgG
Non reattivo
Non reattivo
IgG
IgG
IgG e/o C3
C3
C3
IgG + C3
IgG +C3
IgM
IgG
IgG, IgM
IgG
AEA da anticorpi caldi
Sindrome da agglutininefredde
Emoglobinuria parossisticaa frigore
Anemia emoliticaautoimmune mista
Anemia emolitica immune indotta da farmaci
Eluato TADClasse di Ig
Panreattività;(Rh>altri)
Panreattività+ Ignoto > I > altri
Spesso Rh-correlati
IgG (+IgA o IgM)
Specificità
Tabella 2 . Caratteristiche sierologiche delle Anemie EmoliticheAutoimmuni

pagina 9
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
In generale, alla base dei fenomeni autoimmuni sipossono ipotizzare alcuni meccanismi patogeneticicomuni (13,14). Frequentemente l’autoimmunità puòessere determinata da modificazione degli antigenieritrocitari, ad esempio come conseguenza di agentiinfettivi o farmaci. Sempre in corso di infezioni puòrealizzarsi la cosiddetta reazione crociata, cioè il ri-conoscimento di determinanti del self che “assomi-gliano” a quelli verso i quali si è prodotta la fisiolo-gica risposta immunitaria. Infine, la produzione diautoanticorpi può realizzarsi ad opera dei cosiddetti“cloni proibiti” che compaiono in corso di sindromilinfoproliferative dei linfociti B quali LLC e linfomi.In ogni caso, alla base dei fenomeni autoimmuniesiste una esaltata capacità dell’organismo a produrreautoanticorpi per la rottura della “tolleranza” nei con-fronti del self, che è controllata da complessi mec-canismi cellulari e citochinici non ancora completa-mente conosciuti.
La definizione del tipo e delle caratteristiche termi-che dell’autoanticorpo è di fondamentale importan-za in quanto determina un diverso meccanismopatogenetico alla base dell’emolisi, con conseguen-te diverso quadro clinico e approccio terapeutico (12)
MMMMMECECECECECCCCCCANANANANANIIIIISSSSSMMMMMI PI PI PI PI PAAAAATTTTTOOOOOGGGGGEEEEENNNNNETIETIETIETIETICCCCCIIIII
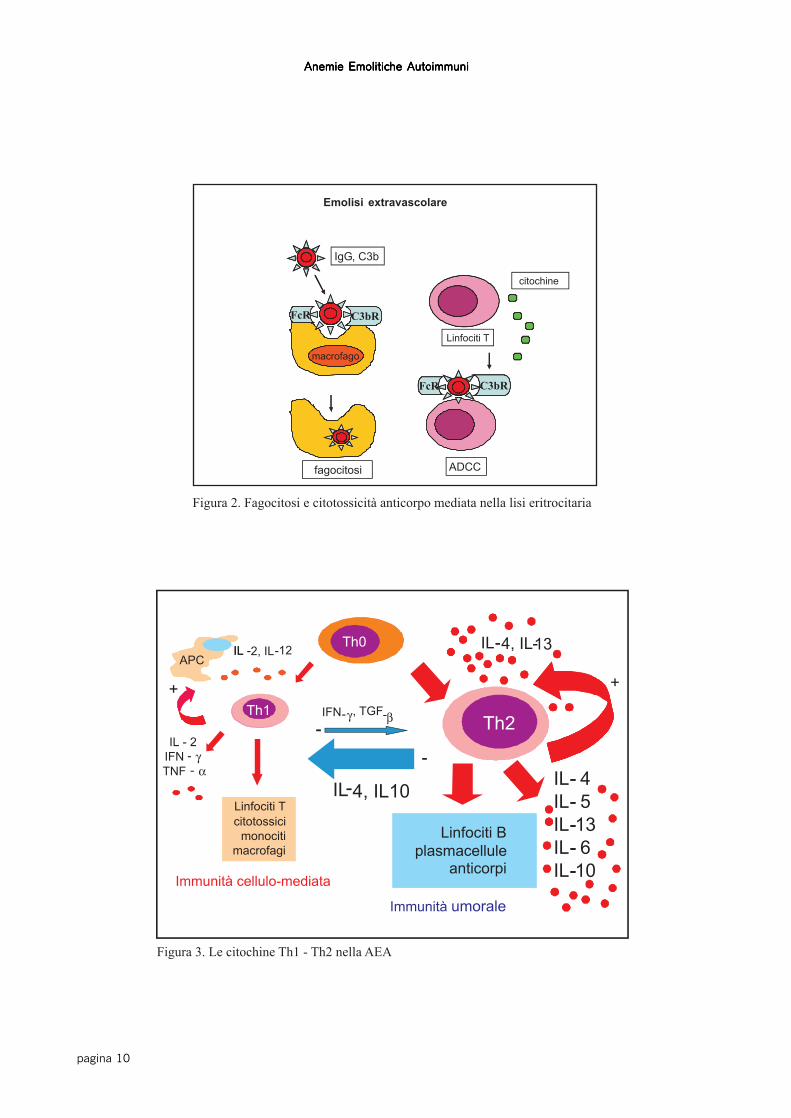
Gli autoanticorpi di classe IgG sono perlopiùmonomeri, in grado di fissare poco il sistema delle
proteine del complemento (Figura 1). La distruzio-ne delle emazie avviene con un meccanismo cosid-detto di ADCC (antibody-dependent cellularcytotoxicity), dove le cellule del sistema monocito-macrofagico fagocitano le emazie attraverso il rico-noscimento del frammento Fc delle IgG (o di frazio-ni complementari quali il C3b). L’emolisi è quindidi tipo extravascolare, ed avviene perlopiù nel fega-to e nella milza. Un meccanismo di tipo ADCC èmediato anche da linfociti attivati da citochine, cheesprimono recettori per il frammento Fc delle IgG eper il C3b (Figura 2). Gli autoanticorpi di classe IgMsono pentameri, dotati di elevata avidità e capacitàdi attivare la cascata complementare fino al com-plesso litico finale (C5-C9), che attraverso l’attiva-zione di “perforine” e altri fattori citotossici, deter-mina la lisi dei globuli rossi direttamente nel torren-te circolatorio (emolisi intravascolare). E’ stato cal-colato che l’emolisi extravascolare comporta la di-struzione di circa 0.25 ml di globuli rossi per kg dipeso corporeo all’ora, che per un uomo del peso di70 kg rappresenta la distruzione di circa 420 ml diemazie nelle 24 ore; viceversa una emolisiintravascolare mediata da IgM determina una poten-ziale distruzione di circa 200 ml di emazie all’ora,con una velocità (e una conseguente gravità clinica)dieci volte maggiore dell’emolisi extravascolare (12).
Il complemento
Complesso litico finale(C5, C6, C7, C8, C9)
Via alterna(Fattore B, D,
properdina)
Via delle lectinemannan binding protein (MBP)
mannanassociated serine-
protease (MASP)
C3-convertasi
Via classica (C1qrs, C2,
C4, C3)
Figura 1. Il sistema di attivazione del complemento
pagina 10
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
Emolisi extravascolare
fagocitosi
macrofago
ADCC
IgG, C3b
Linfociti T
citochine
Figura 2. Fagocitosi e citotossicità anticorpo mediata nella lisi eritrocitaria
FcR C3bR
FcR C3bR
-
-
Th0IL -2, IL-12
Th1
IL - 2IFN γTNF α
IL-4, IL-13
Th2
IL- 4IL- 5IL-13IL- 6IL-10
+
IFN-γ, TGF-β
IL-4, IL10
-
-
Th0IL
APC
Th1Th1
--
Immunità cellulo-mediata
Immunità umorale
+
-
Linfociti Bplasmacellule
anticorpi
Th2
-----
Linfociti T
citotossici monociti
macrofagi
Figura 3. Le citochine Th1 - Th2 nella AEA

pagina 11
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
La regolazione della risposta immune (fisiologicaverso gli antigeni non-self, patologica verso il self)è un processo attivo, regolato da numerose citochine.Classicamente le citochine si distinguono in Th1 eTh2. Le citochine Th1, perlopiù prodotte dai THelper 1 (IL-2, IL-12, Interferon-γ e TNF-α) sonoresponsabili dell’immunità cellulo-mediata attraver-so la generazione dei linfociti T citotossici (CD8+)e dei monociti-macrofagi attivati. Le citochine Th2(IL-4, IL-5, IL-6, IL-10, IL-13) sono responsabilidella immunità umorale, mediante la maturazionedei linfociti B in plasmacellule e la conseguente pro-duzione di anticorpi.Esiste inoltre una regolazione crociata per cui alcu-ne citochine Th1, tra cui l’interferon-γ e il TGF-β(Transforming Grow Factor-β) regolano in sensonegativo la risposta Th2, e viceversa citochine Th2(IL-4 e IL-10) regolano in senso negativo la rispo-sta Th1.Le evidenze di una disregolazione citochinica nellaAEA riportate in letteratura sono contraddittorie e,in modelli animali, riportano a volte una deficienzadi IL-2, oppure una aumentata produzione di IFN-γe di TGF-β (15, 16). Nell’uomo esistono pochi stu-
di: uno dimostra un aumento di IL-2, un altro di IL-4 ed IL-5 (17, 18). Noi abbiamo studiato il profilocitochinico di tipo 1 (IL-2, IFN-γ) e 2 (IL-4, IL-6,IL-10, IL-13) in 21 pazienti con AEA idiopaticanonché la produzione di citochine pro-infiammato-rie (TNF-α, IL-1) e inibitorie (TGF-β), dimostran-do una ridotta produzione di IFN-γ e IL-2, più evi-dente nei pazienti in fase attiva/ emolitica della ma-lattia, e una aumentata produzione di IL-4, IL-13 eIL-6 (19). Infine, i pazienti con AEA in fase attivaavevano una aumentata produzione di TGF-β. Inconclusione, nella AEA esiste uno sbilanciamentodella risposta immunitaria in senso Th2, in linea conil meccanismo di distruzione dei GR mediato daanticorpi. Come in altre patologie autoimmuni, an-che l’immunità cellulare è importante, in quanto“driver” dell’immunità umorale, presente soprattut-to nelle fasi iniziali e di attività della malattia. L’au-mento di TGF-β potrebbe rappresentare un mecca-nismo di feed-back volto a smorzare l’immunitàcellulo-mediata presente nelle fasi di attività dellamalattia e che determina lo shift in senso Th2 dellarisposta immune, con conseguente prevalenza del-l’immunità umorale (Figura 3).

pagina 12
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni

pagina 13
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
Il quadro clinico delle AEA è caratterizzato da unanotevole variabilità in termini di esordio, manife-stazioni cliniche e decorso, da insidioso a fulminan-te. Le AEA, indipendentemente dalle caratteristichedell’autoanticorpo, presentano anemia tendenzial-mente macrocitica di grado estremamente variabile(emoglobina da 3 g/dL nelle forme più gravi fino avalori pressoché normali), reticolocitosi, moderataiperbilirubinemia prevalentemente di tipo indiretto,consumo di aptoglobina, possibile rialzo di LDH e,occasionalmente nelle forme iperacute e massive,emoglobinemia, emoglobinuria ed emosiderinuria.Un altro reperto obiettivo di comune riscontro è co-stituito da splenomegalia usualmente modesta edepatomegalia, rilevabili rispettivamente nella metàed in un terzo dei casi.
Le AEA da agglutinine fredde (CHD, coldhemagglutinin disease) sono caratterizzate daagglutinazione ed emolisi delle emazie nelle sedicorporee dove la temperatura raggiunge quella direazione dell’anticorpo e quindi acrocianosi e feno-meni vasomotori nella microcircolazione superficiale(mani, piedi, orecchie, naso, etc.), scatenati soprat-tutto dall’esposizione al freddo.
L’emoglobinuria parossistica a frigore è stata clas-sicamente descritta in passato in associazione allasifilide. Attualmente si osserva frequentemente neibambini a seguito di malattie infettive prevalente-mente virali, ed è caratterizzata da un esordio acuto,saltuariamente grave con anemia severa edemoglobinuria. I sintomi clinici sono dominati dallaemolisi intravascolare determinata dall’emolisinabifasica di Donath-Landsteiner, un anticorpo di clas-se IgG che si lega agli eritrociti a basse temperature(4 °C) e determina emolisi a 37 °C. Il quadro clinicoè caratterizzato da malessere generale, brividi, feb-bre, crampi, dolori lombari e addominali, fenomenivasomotori ed orticaria a seguito di una esposizionea basse temperature.
La casistica “storica” di pazienti con AEA seguitapresso il nostro Centro negli anni 80-90, costituitada 286 casi di forme calde (rappresentanti il 72%delle AEA totali), di età mediana 48 anni, range1-89, mostrava un quadro assai variabile in termini
di esordio, manifestazioni cliniche e decorso: l’ane-mia era presente nel 90% dei casi, l’ittero nel 82%,mentre meno frequenti erano la splenomegalia (51%)e l’epatomegalia (35%), ed estremamente raral’emoglobinuria (5%).All’esordio, i valori di Hb erano estremamente va-riabili sia nelle forme da anticorpi caldi che freddi,rispettivamente, da 3.6 a 16.5 g/dl (mediana 8.7) eda 5 a 13 g/dl (mediana 9.6) (20). Nelle AEA ireticolociti sono usualmente elevati, con consensualeaumento del VGM, che in casi di emolisi compen-sata rappresenta l’unica anormalità ematologica. Inrari casi è presente reticolocitopenia (1-2% dei casi)di incerta interpretazione. Questi casi rappresenta-no una vera emergenza medica, la reticolocitopeniapotendo essere anche di lunga durata nonostante laterapia, e comportare, prima di risolversi, unfabbisogno trasfusionale talora elevatissimo (sino aoltre 80 unità in meno di 6 mesi) (21).
I parametri clinici delle varie forme di AEA, ottenu-ti da una revisione della più recente casistica delnostro Centro (89 pazienti, di età media 55 anni,range 11-94, 50 maschi e 39 femmine), è riportatain Tabella 3: 62 pazienti (69,7%) erano affetti daAEA da anticorpi caldi, 19 pazienti (21,3%) da sin-drome da agglutinine fredde, 1 paziente da AEA daanticorpi misti e 7 pazienti (7,9%) da AEA TAD-negativa.I nostri dati confermano, in tutti i tipi di AEA, l’am-pia variabilità nei livelli di Hb, da valori pressochénormali a marcatamente ridotti; ugualmente varia-bile è l’alterazione degli indici emolitici, con valorinormali in alcuni pazienti e marcata positività in al-tri; il gruppo di AEA TAD-negative presenta gene-ralmente un quadro clinico più grave, probabilmen-te in conseguenza della difficile e ritardata diagno-si. Il 77.5% dei nostri pazienti (69 casi, di cui 57con AEA da anticorpi caldi, 5 da crioagglutinine, 1da anticorpi misti e 6 DAT negativi) è stato sottopo-sto a terapia; i dati clinici dei pazienti trattati rivela-vano anemia e indici emolitici alterati [Hb mediana7,2 g/dl (range 3 – 11,9), LDH 780 U/l (range 201 –8681), bilirubina indiretta 2,8 mg/dl (range 0,2 –8,2)]; inoltre 17 pazienti presentavano spleno-megalia, 8 epatomegalia e 5 emoglobinuria. Ilfollow-up mediano era di 41 mesi.
QUQUQUQUQUADADADADADRRRRRO CO CO CO CO CLLLLLIIIIINNNNNIIIIICCCCCOOOOO

pagina 14
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
E’ interessante notare la bassa percentuale di pazienticon sindrome da agglutinine fredde che è stato sot-toposto a terapia (solo ¼ dei pazienti) rispetto allaquasi totalità dei pazienti con altre forme di AEA,riflettendo la difficoltà terapeutica di questi casi, cheverrà discussa nel capitolo dedicato alla terapia.Per quanto riguarda la prognosi, negli adulti la so-pravvivenza è riportata essere del 91% ad 1 anno,75% a 5 anni e 73% a 10 anni (12). In età pediatricala malattia in circa l’80% dei casi è acuta e transito-ria in quanto prevalentemente associata ad infezio-
ni virali quali EBV, CMV, varicella, morbillo, H.influenzae ed altri (HP B19, E. Coli, stafilococchi).Anche la prognosi delle AEA idiopatiche ad insor-genza in età compresa tra i 2 e i 12 anni è buona,con guarigione piuttosto rapida (12). In tema dimortalità e di gravità, è doveroso ricordare che par-ticolarmente severe sono le forme atipiche associa-te ad autoanticorpi IgM “caldi”, che hanno una pro-babilità di esito letale più alta di tutte le altre AEAper le quali sono riportate diverse segnalazioni fata-li (22-25).
617 (201-3262)708 (201-8681)LDH (U/l)
8,85 (0,2-55)Reticolociti%
8 (3-9,9)10,3 (4,7-15,6)7,8 (3-15,6)
AEA” ”caldeTotale Parametri
3,27 (1,05-3,7)1,55 (0,4-8,1)2,8 (0,2-8,2)2,5 (0,2-8,2)Bil Ind (mg/dl)
1316 (350-8681)770 (313-4523)
11,95 (1-55)4,5 (1-10)10,6 (0,7-30)
7 (3,8-15,3)Hb(g/dl)
TAD negative AEA “fredde”
Tabella 3. Parametri clinici dei pazienti con AEA seguiti presso il nostro Centro nelperiodo 2001- 2008

pagina 15
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
Il cardine diagnostico delle AEA è rappresentato daltest dell’antiglobulina diretto, che tuttavia non puòessere il solo protagonista, ma deve essere seguitoda altre indagini di laboratorio finalizzate a deter-minare la classe e le caratteristiche termichedell’autoanticorpo, confermare la sua presenza nelsiero e/o nell’eluato dalle emazie e per ultimo esclu-dere che l’autoanticorpo mascheri la presenza dialloanticorpi a basso titolo.
Un corretto inquadramento diagnostico di un pazien-te con sospetta AEA può pertanto richiedere la se-guente batteria di test:
1. test dell’antiglobulina indiretto (TAD) o test di Coombs diretto2. autoagglutinazione3. ricerca di anticorpi eritrocitari nel siero (TAI) o test di Coombs indiretto4. identificazione di anticorpi eritrocitari nel siero ed eluato5. tipizzazione eritrocitaria completa6. tests sierologici addizionali (ad esempio l’autoassorbimento, l’alloassorbimento mira- to e i test per le forme farmaco-indotte)
Il TAD in fase liquida viene in genere eseguito inprovetta utilizzando antisieri antiglobuline umanepolispecifici e monospecifici (anti-IgG, anti-C3 e/oanti-C3d, anti-IgA e anti-IgM) di differenti produt-tori, ed emazie sospese al 3%. Le provette contenen-ti gli antisieri polispecifici e monospecifici vanno let-te dopo centrifugazione immediata tranne il comple-mento che deve essere incubato a temperatura am-biente per 5 minuti.
L’autoagglutinazione è un test molto semplice in ge-nere positivo nelle AEA da autoanticorpi “freddi” manon nelle forme da IgG “caldi”. In caso di auto-agglutinazione positiva è necessario lavare le emaziecon fisiologica a caldo prima di eseguire il TAD.
Il TAD in provetta, eseguito mediante agglutinazionecon antisieri polispecifici, è il test di screening piùfrequentemente utilizzato, tuttavia non è esente dalrilevare sia falsi positivi che negativi (12). In caso diTAD negativo, ma di forte sospetto clinico di ane-mia emolitica autoimmune (Figura 4) è necessarioeseguire indagini di II livello per la ricerca dianticorpi eritrocitari (12). Le cause di falsa negativitàdel TAD eseguito con antisieri polispecifici possonoessere:
DIADIADIADIADIAGGGGGNNNNNOSOSOSOSOSIIIII
TAD standard (provetta)(antisieri polispecifici)
Causa dell’emolisi identificata?
STOP
- Antisieri monospecifici
- TAD II livelloMicrocolonnaFase solidaLavaggi a freddo 4 °CLISSIFA
Siero/Eluato:Screening e
identificazione degli autoanticorpi
Altri(MS-DAT)
+-
si no
+-
+-
Figura 4. Algoritmo diagnostico nelle AEA

pagina 16
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
a) la presenza di autoanticorpi di sola classe IgA,che può essere rilevata dall’uso di antisierimonospecifici; in genere gli anticorpi di classe IgAsono riscontrabili in circa il 14% dei pazienti conAEA e sono usualmente associati ad anticorpi IgGe/o IgM, mentre la sola presenza di anticorpi IgA èabbastanza rara; tuttavia, la reale frequenza di MEAda IgA è probabilmente sottostimata in quantoautoanticorpi IgA non vengono sempre rilevati daisieri antiglobulina umana ad ampio spettro;b) la presenza di autoanticorpi a bassa affinità, chepossono essere invece rilevati dall’esecuzione delTAD dopo lavaggio delle emazie a freddo o con so-luzioni a bassa forza ionica (LISS);c) la presenza di un piccolo numero di molecole IgGlegate ai GR al di sotto del limite di rilevazione del-la metodica in agglutinazione.
Da qui l’utilizzo di metodi dotati di maggiore sensi-bilità, fra cui il TAD in microcolonna e fase solida,vari test immuno-radiometrici ed ELISA, il test diconsumo del complemento e la citofluorimetria.Per quanto riguarda la sensibilità dei vari metodi, ènoto che il classico TAD in provetta diagnostica ef-ficacemente una AEA se almeno 500 molecole diautoanticorpi sono legati ai GR (26), il test inmicrocolonna richiede circa 200-300 molecole/GRper dare un risultato positivo (27), e la cito-fluorimetria, essendo la tecnica in assoluto più sen-
sibile, è in grado di rilevare anche 30-40 molecoledi IgG/GR (28).
Nonostante l’utilizzo di tutti i metodi sopra elencatiè noto che il 5-10% delle AEA continua a presenta-re un TAD negativo. In questi casi la diagnosi è diesclusione e spesso sulla base di una risposta clini-ca alla terapia steroidea.Particolarmente utile nello studio di tali casi si è ri-velato il test di Coombs dopo stimolazionemitogenica in coltura, denominato MS-DAT(mitogen-stimulated direct antiglobulin test). Il testviene eseguito su sangue intero dopo stimolazioneper 48hr con mitogeni quali fitoemagglutinina(PHA), esteri del forbolo (PMA), e fitolacca ameri-cana o pokeweed (PWM); la stimolazionemitogenica determina la produzione in vitro diautoanticorpi da parte dei linfociti B, e il loro suc-cessivo legame alle emazie autologhe; la quantità diautoanticorpi adesi viene valutata mediante un testELISA competitivo in fase solida. Come mostratoin Tabella 4, nei pazienti con AEA, la stimolazionemitogenica aumenta la quantità di IgG legate alleemazie autologhe, rispetto alle colture non stimola-te (19); un analogo aumento, anche se di minoreentità, si osserva in soggetti con leucemia linfaticacronica, condizione nella quale è nota una elevataprevalenza di fenomeni autoimmuni diretti controla serie eritrocitaria (29).
Controlli sani(n=81)
Pazienti con LLC(n=69)
Colture stimolate con PWM
Colture stimolate con PMA
Colture stimolate con PHA
Colture nonstimolate
76+1470+675+975+7
635+134**
183+25***
465+55*
182+37**
623+122**
207+29***
322+49***
134+15**
Pazienti con AEA(n=33)
Tabella 4. - MS-DAT in pazienti con AEA e leucemia linfatica cronica (LLC). I valori sono espressi come ng/ml di IgG legate a GR autologhi (media+*p< 0.05, **p< 0.01 e ***p< 0.001, pazienti versus controlli
ES);
pagina 17
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
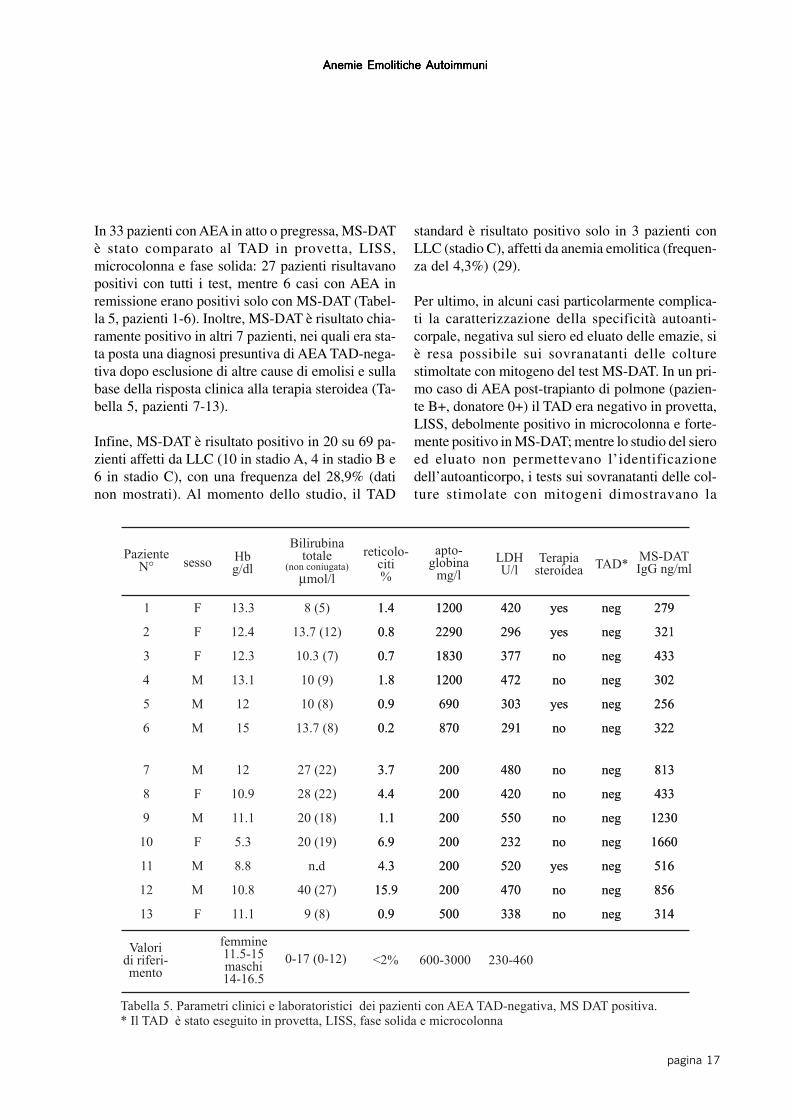
In 33 pazienti con AEA in atto o pregressa, MS-DATè stato comparato al TAD in provetta, LISS,microcolonna e fase solida: 27 pazienti risultavanopositivi con tutti i test, mentre 6 casi con AEA inremissione erano positivi solo con MS-DAT (Tabel-la 5, pazienti 1-6). Inoltre, MS-DAT è risultato chia-ramente positivo in altri 7 pazienti, nei quali era sta-ta posta una diagnosi presuntiva di AEA TAD-nega-tiva dopo esclusione di altre cause di emolisi e sullabase della risposta clinica alla terapia steroidea (Ta-bella 5, pazienti 7-13).
Infine, MS-DAT è risultato positivo in 20 su 69 pa-zienti affetti da LLC (10 in stadio A, 4 in stadio B e6 in stadio C), con una frequenza del 28,9% (datinon mostrati). Al momento dello studio, il TAD
standard è risultato positivo solo in 3 pazienti conLLC (stadio C), affetti da anemia emolitica (frequen-za del 4,3%) (29).
Per ultimo, in alcuni casi particolarmente complica-ti la caratterizzazione della specificità autoanti-corpale, negativa sul siero ed eluato delle emazie, siè resa possibile sui sovranatanti delle colturestimoltate con mitogeno del test MS-DAT. In un pri-mo caso di AEA post-trapianto di polmone (pazien-te B+, donatore 0+) il TAD era negativo in provetta,LISS, debolmente positivo in microcolonna e forte-mente positivo in MS-DAT; mentre lo studio del sieroed eluato non permettevano l’identificazionedell’autoanticorpo, i tests sui sovranatanti delle col-ture stimolate con mitogeni dimostravano la
femmine11.5-15maschi14-16.5
Valoridi riferi-mento
314negno3385000.99 (8)11.1F13
856negno47020015.940 (27)10.8M12
516negyes5202004.3n.d.8.8M11
1660negno2322006.920 (19)5.3F10
1230negno5502001.120 (18)11.1M9
433negno4202004.428 (22)10.9F8
813negno4802003.727 (22)12M7
322negno2918700.213.7 (8)15M6
256negyes3036900.910 (8)12M5
302negno47212001.810 (9)13.1M4
433negno37718300.710.3 (7)12.3F3
321negyes29622900.813.7 (12)12.4F2
279negyes42012001.48 (5)13.3F1
TAD*Terapiasteroidea
LDHU/l
apto-globina
mg/l
reticolo-citi%
sessoPaziente
N°
230-460600 3000-<2%0-17 (0-12)
314negno3385000.9
856negno47020015.9
516negyes5202004.3.
1660negno2322006.9
1230negno5502001.1
433negno4202004.4
813negno4802003.7
322negno2918700.2
256negyes3036900.9
302negno47212001.8
433negno37718300.7
321negyes29622900.8
279negyes42012001.4
MS-DATIgG ng/ml
Bilirubinatotale
(non coniugata)
µmol/l
Hbg/dl
Tabella 5. Parametri clinici e laboratoristici dei pazienti con AEA TAD-negativa, MS DAT positiva.* Il TAD è stato eseguito in provetta, LISS, fase solida e microcolonna

pagina 18
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
positività per anticorpi anti-B, suggerendo la dia-gnosi di “passenger lymphocyte syndrome”, dovutaad anticorpi prodotti dai linfociti del donatore pas-sivamente trasferiti durante il trapianto. In un se-condo caso il TAD era negativo in provetta e LISS,debolmente positivo in microcolonna e fase solida echiaramente positivo in MS-DAT; anche in questocaso lo studio del siero ed eluato era negativo, men-tre le indagini sui sovranatanti delle colture stimo-late con mitogeni dimostravano la positività perautoanticorpi panreattivi in fase solida. In conclu-sione, la diagnosi di AEA deve avvalersi di diversitests dell’antiglobulina diretta, particolarmente neicasi negativi allo standard TAD in provetta. Il testMS-DAT potrebbe essere proposto come un metodoaggiuntivo per la diagnosi dei casi difficili e per lacaratterizzazione della specificità autoanticorpale.Infatti, la stimolazione mitogenica determina un au-mento della produzione di autoanticorpi in vitro esembra quindi in grado di evidenziare unaautoimmunità anti-eritrocitaria latente, includendole AEA TAD-negative, le forme in fase di remissio-ne clinica e quelle condizioni patologiche, quali laLLC, associate ad autoimmunità anti-eritrocitaria.Va ribadita in questa sede l’importanza della
tipizzazione eritrocitaria completa in tutti i pazienticon AEA prima di eventuali trasfusioni. Nei pazien-ti con TAD positivo le emazie vanno tipizzate se pos-sibile con antisieri monoclonali; quando questi nonsono disponibili è necessario trattare i globuli rossicon Clorochina o ZZAP per liberare i siti antigenicidagli anticorpi.
Nei pazienti politrasfusi è invece impossibile avereuna tipizzazione eritrocitaria sierologica attendibileed è pertanto necessario ricorrere alla tipizzazionecon tecniche genomiche, effettuabile con kit com-merciali che utilizzano la tecnica Polymerase ChainReaction-Sequence-Specific Primers (PCR-SSP) suDNA ottenuto da sangue periferico (ABO-Type, Rh-Type, KKDType, Weak D-Type, HPA-Type BAGene,Germany; ABO-SSP, CDE-SSP, weak D-SSP,KKD-SSP, MNSs-SSP, HPA-SSP, Inno-Train DiagnostikGmbH, Germany e BAG Health Care GmbH,Germany) (30, 31).Infine, problematiche diagnostiche particolari ven-gono poste dalle anemie immunoemolitiche da far-maci, che vengono di seguito separatamente consi-derate.

pagina 19
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
I farmaci possono produrre emolisi attraverso mec-canismi sia immuni che non-immuni. Storicamente,l’�-metildopa ed elevate dosi di penicillina sono re-sponsabili della maggior parte dei casi di anemiaemolitica indotta da farmaci con meccanismo im-mune. Studi dei primi anni ’80, quando questi dueagenti erano più comunemente utilizzati, mostrava-no che 12-18% delle AEA erano indotte da farmaci(32, 33). Mentre l’incidenza di anemia emolitica in-dotta da questi farmaci è attualmente molto ridotta,sono sempre di più frequente osservazione casi as-sociati all’assunzione di cefalosporine di seconda eterza generazione, soprattutto cefotexano e ceftri-axone (34, 35). Johnson e coll (36), del Immuno-hematology Reference Lab di Milwaukee ha descrit-to 71 casi di anemia immunoemolitica da farmaci,con 73 anticorpi diretti contro 23 differenti farmaci,tra i quali predominano le ciclosporine, le penicillineed i loro derivati, ed i farmaci infiammatori nonsteroidei. E’ interessante notare che degli 11 anticorpidiretti contro FANS, in 4 casi la diagnosi ha richie-sto indagini su metaboliti urinari del farmaco. Damemorizzare le conclusioni degli Autori, che debbasempre essere presa in considerazione l’eventualitàdi una forma immunoemolitica da farmaco ogniqualvolta un paziente si presenti con una AEA siada anticorpi caldi che freddi. E’ importante sottoli-neare che una corretta diagnosi delle anemieimmunoemolitche da farmaci presuppone un fortesinergismo collaborativo tra clinico e laboratorista
ANANANANANEEEEEMMMMMIIIIIE IE IE IE IE IMMMMMMMMMMUUUUUNNNNNOOOOOEEEEEMMMMMOOOOOLLLLLITIITIITIITIITICCCCCHHHHHE IE IE IE IE INNNNNDDDDDOOOOOTTE DTTE DTTE DTTE DTTE DA FA FA FA FA FARARARARARMAMAMAMAMACCCCCIIIII
dovendo essere utilizzate metodiche complesse nondi uso routinario.
Le anemie immunoemolitiche indotte da farmacisono dovute a diversi tipi di interazione tra il farma-co, l’anticorpo e i componenti della membrana delleemazie. I tre principali meccanismi di induzione in-cludono l’adsorbimento tenace del farmaco allamembrana eritrocitaria, la formazione di immuno-complessi e la produzione di autoanticorpi (Figura5).
Le anemie immunoemolitiche del primo tipo sonosostenute da anticorpi, generalmente di classe IgG,che reagiscono con un complesso farmaco-membra-na formatosi a seguito del tenace legame del farma-co alla membrana stessa. In questi casi il TAD è po-sitivo per IgG e l’emolisi è di tipo extravascolare;farmaci che tipicamente danno questo tipo di emolisisono la penicillina, l’ampicillina, la meticillina, lacarbenicillina e le cefalosporine (cefalotina ecefaloridina).Le forme del secondo tipo sono sostenute dallainterazione fra farmaco e anticorpo anti-farmaco (so-prattutto IgM), con formazione di immunocomplessiche si attaccano alla membrana eritrocitaria, deter-minando emolisi intravascolare e un TAD positivosolo per frazioni complementari; moltissimi farma-ci possono causare anemia immunoemolitica conquesto meccanismo, tra i più frequenti si ricordano
1. Adsorbimento e legame tenace del farmaco alla membrana con produzione di IgG che si legano al complesso farmaco-membrana; emolisi extravascolare; TAD+IgG(esempio penicillina)
2. L’immunocomplesso farmaco-anticorpo anti farmaco (perlopiù IgM) si attacca alla membrana; emolisi intravascolare;TAD + C (esempio chinidina)
3. Il farmaco modifica la membrana del GR rendendola antigenica, induce la produzione di autoanticorpi IgG
emolisi extravascolare; TAD+IgG
(esempio alfa metildopa)
Y
YFigura 5. Meccanismi di induzione delle anemie emolitiche da farmaci
Y
YYY
YY

pagina 20
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
chinidina, fenacetina, idroclorotiazide, rifampicina,sulfamidici, isoniazide, chinina, tetracicline,idralazina, cloropromazina, streptomicina, fluorouracile e molti altri.Infine, le anemie emolitiche secondarie alla produ-zione di autoanticorpi si verificano quando il farma-co interagisce modificando un normale componentedella membrana eritrocitaria e il sistema immunitarioriconosce questo componente dei globuli rossi “al-terato” come non-self; quest’ultimo meccanismo èquello che più si avvicina alle forme autoimmuniclassiche. L’α-metildopa è il tipico farmaco che agi-sce inducendo la formazione di autoanticorpi, e de-terminando la positività del TAD nel 11-36% deipazienti entro 3-6 mesi dalla prima somministra-zione, con una dose-dipendenza (37, 38). Fra gli al-tri farmaci che possono provocare una anemiaimmunoemolitica da autoanticorpi si ricordanocefalosporine, acido mefenamico, procainamide,ibuprofen, diclofenac e interferon-α (39-46). In que-sti casi il TAD è positivo per IgG e l’emolisi è ditipo extravascolare. Infine, va ricordato che i tremeccanismi non sono mutualmente esclusivi. Peresempio, le cefalosporine, che normalmente indu-cono anemia emolitica col meccanismo diadsorbimento del farmaco alla membrana e/o for-mazione di immunocomplessi, possono indurre an-che la formazione di autoanticorpi (39-46).
In Tabella 6 vengono riassunte le caratteristichesierologiche delle tre forme di anemia immuno-
emolitica indotta da farmaci. Oltre alla diversapositività del TAD già accennata in precedenza, vasottolineato che normalmente la ricerca deglianticorpi nel siero e nell’eluato dalle emazie è ne-gativa nelle forme da adsorbimento e da immuno-complessi mentre può essere positiva nel caso di far-maci in grado di indurre la produzione diautoanticorpi, forma che è sierologicamenteindistinguibile da una classica AEA “calda”(anticorpi perlopiù panreattivi, sebbene siano docu-mentate specificità c, e, Wrb, Jka, e U) (47). Tutta-via, il pretrattamento delle emazie con farmaco, per-mette di rilevare la positività per anticorpi nel sieroe/o eluato nelle forme dovute ad adsorbimento; ana-logamente, nelle anemie emolitiche sostenute dallaformazione di immuno-complessi, l’aggiunta del far-maco in vitro e/o il pretrattamento delle emazie confarmaco permette la rilevazione di anticorpi nel sie-ro.Infine, va ricordato che una diagnosi presuntiva dianemia immunoemoltica farmaco-indotta può esse-re confermata solo dalla risposta alla sospensionedel farmaco stesso.
Da un punto di vista terapeutico la sospensione delfarmaco, soprattutto quando esistono terapie alter-native, determina normalmente una risoluzionedell’emolisi dopo alcuni giorni, anche se talvoltasono necessari mesi affinché l’anemia emolitica siesaurisca completamente (47). In casi con emolisisevera si può ricorrere alla terapia steroidea.
++
Normalmente
+++
+++
+Normalmente
+
–
++
–––
++
Normalmente
––
+
––
+
TADPolispecificoIgG
C3Anticorpi nel siero
RoutineFarmaco solubileGR trattati con farmaco
RoutineFarmaco solubileGR trattati con farmaco
Formazione diautoanticorpi
Formazione diimmunocomplessi
Adsorbimentodel farmaco
Anticorpi nell’eluato da GR
Tabella 6. Caratteristiche sierologiche della anemie emolitiche indotte da farmaco. Modificato da Ghers BC & Friedberg RC. AJH, 2002.

pagina 21
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
La terapia delle AEA deve tenere conto della gravi-tà dell’anemia e delle relative conseguenzefisiopatologiche sull’organismo, nonché della rapi-dità di comparsa dello stato anemico. Pertanto, èpossibile identificare tre livelli di anemia e quindidi rischio, con conseguenti diversi approcciterapeutici. Se l’ematocrito è compreso fra 30 e 36%e/o i livelli di emoglobina fra 8 e 12 g/dl vi è l’indi-cazione all’astensione terapeutica fino a inquadra-mento diagnostico completo; se l’ematocrito è com-preso fra 14 e 30% e/o i livelli di emoglobina fra 4,5e 8 g/dl si deve agire a seconda del quadro clinico,preferibilmente però dopo inquadramento diagno-stico; infine, se l’ematocrito è inferiore al 14 % e/oi livelli di emoglobina inferiori a 4,5 g/dl vi è l’indi-cazione all’intervento terapeutico immediato. A pre-scindere dalle forme di AEA secondarie, in cui laterapia è quella della malattia di base, va sottolinea-to che la definizione del tipo e delle caratteristichetermiche dell’autoanticorpo è di fondamentale im-portanza in quanto determina un diverso approccioterapeutico alla AEA.
TERAPIA DELLE ANEMIE EMOLITICHEAUTOIMMUNI DA ANTICORPI “CALDI”
Nelle forme idiopatiche da anticorpi caldi la terapiadi prima scelta è rappresentata dai corticosteroidi.In soggetti adulti una somministrazione giornalierainiziale di 1 mg/kg/die (o 40 mg/m2) di prednisoneper os per circa 3-4 settimane è sufficiente a con-trollare l’emolisi nel 70-85% dei casi. I benefici cli-nici della terapia steroidea compaiono in genere en-tro 10 giorni. Alcuni Autori suggeriscono di incre-mentare la dose a 60 mg/m2 in caso di mancata ri-sposta entro la prima settimana. Quando i livelli diemoglobina ed ematocrito si stabilizzano su valorinormali e si osserva una tendenza allanormalizzazione degli indici emolitici (reticolociti,LDH, bilirubina, aptoglobina), il dosaggio dellosteroide va gradualmente ridotto. Indicativamentetale riduzione deve essere di 10-15 mg/die ogni set-timana fino ad un dosaggio giornaliero di 20-30 mg,quindi di 5 mg/die ogni settimana e, infine, di 2.5mg/die ogni settimana sino alla sospensione del far-maco o al raggiungimento del dosaggio minimo ef-ficace. Questo schema è indicativo, in quanto la ri-duzione dello steroide va adattata ad ogni singolo
paziente con un attento monitoraggio dell’emocromoe degli indici emolitici, in quanto una rapida ridu-zione determina assai frequentemente recidiveemolitiche. Vanno ricordati, perché spessosottostimati, i numerosi effetti collaterali della tera-pia steroidea, usualmente correlati alla dose e alladurata della terapia: ipertensione, diabete, ulcerapeptica, diatesi infettiva, miopatia, cataratta,osteoporosi, irritabilità, insonnia, aumento di pesoe habitus cushingoide. Fra questi una particolare at-tenzione va posta all’osteoporosi, che contrariamentea quanto ritenuto, compare insidiosamente in circail 10-20% dei pazienti che assumono anche piccoledosi di steroide (5-10 mg die di prednisone) per lun-ghi periodi.
Come accennato in precedenza, la terapia steroideaben condotta dimostra una buona efficacia nel 70-85% dei casi; tuttavia solo un terzo di questi rimanein remissione clinica a lungo termine dopo la so-spensione della terapia, mentre circa la metà dei pa-zienti richiede piccole dosi di “mantenimento” disteroide; se tale dose è superiore a 10-15 mg/die, viè l’indicazione a considerare una terapia di secondalinea, che prevede la scelta fra farmaciimmunosoppressori e splenectomia; ciò avviene al-l’incirca nel 20-30% dei casi (12).
La splenectomia induce remissione completa in ol-tre il 50% dei pazienti e nei restanti il controllodell’emolisi richiede comunque una dose inferioredi steroide. Va ricordato che la splenectomia deveessere preceduta da adeguata profilassi vaccinaleanti-pneumococcica, anti-meningococcica e anti-Haemophilus, e che rappresenta comunque un in-tervento a maggior rischio nel soggetto anziano. Inol-tre, molti Autori suggeriscono una terapia antibioti-ca profilattica per 3 anni post-splenectomia per evi-tare le possibili complicanze infettive. Altre possi-bili gravi complicanze descritte, oltre a quelle setti-che, anche generalizzate, sono embolia polmonare,sanguinamento, ascesso ed ematoma intra-addominale. Va infine ricordata la possibile esisten-za di tessuto splenico ectopico/milze accessorie, cheva ricercato soprattutto nei pazienti che recidivanodopo splenectomia (48).
Il trattamento con farmaci citotossici e
TETETETETERRRRRAPAPAPAPAPIAIAIAIAIA

pagina 22
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
immunosoppressori dovrebbe essere preso in consi-derazione solo per pazienti sintomatici che non pos-sono essere sottoposti alla splenectomia o che nonabbiano risposto ad essa. Dei farmaci impiegati nel-le AEA da autoanticorpi caldi (tra i quali azatioprina,ciclofosfamide, 6-mercaptopurina e 6-tioguanina),il più usato è l’azatioprina che, alla dose media gior-naliera di 80 mg/m2/die per almeno 2-3 mesi, dà ri-sultati soddisfacenti in circa i 2/3 dei casi insensi-bili ai trattamenti precedenti. Una buona risposta èstata ottenuta anche con la ciclofosfamide aldosaggio di 60 mg/m2 die al giorno per almeno 6mesi e più recentemente con la ciclosporina alla dosedi 5 mg/kg/die (12, 49-51). Le gammaglobuline pervia endovenosa al dosaggio di 400 mg/kg/die per 5giorni hanno dimostrato una efficacia globale del40%, tendenzialmente migliore nei casi pediatrici(54,5%) rispetto agli adulti (37%) (52). Alcuni ri-sultati terapeutici in casistiche molto limitate sonostati riportati con la somministrazione di danazoloalla dose di 600-800 mg/die, con una efficacia nel60-70% dei casi, di durata tuttavia non specificata(53, 54).
TERAPIA DELLE ANEMIE EMOLITICHEAUTOIMMUNI DA ANTICORPI “FREDDI”
La misura terapeutica più efficace nella sindromeda agglutinine fredde idiopatica è la protezione dalfreddo, che, in considerazione della frequente beni-gnità del quadro clinico, è usualmente sufficiente alcontrollo della sintomatologia. Nei pazienti più gra-vi, buoni risultati sono stati ottenuti con clorambucila basse dosi a regime continuativo (0,08-0,1 mg/kg/die), o intermittente ad alte dosi (0.2 mg/kg/die per14 giorni). Una alternativa terapeutica è rappresen-tata dalla ciclofosfamide alla dose di 60 mg/m2/dieper os per almeno 6 mesi (12). Nei casi iperacuti, ilplasma-exchange può rappresentare una misuraterapeutica efficace per ridurre il titolo delleagglutinine fredde, ma non può essere consideratoun trattamento a lungo termine. Recentemente l’ef-ficacia della plasmaferesi è stata rivalutata secondoi criteri della Evidence Based Medicine, che ha mes-so in luce come non esistano studi clinici controllatima solo segnalazioni aneddotiche di efficacia (55).Corticosteroidi e splenectomia sono quasi sempreinefficaci. La trasfusione di sangue si rende neces-
saria solo occasionalmente.La terapia della emoglobinuria parossistica a“frigore” cronica consiste nella protezione dal fred-do, dal momento che steroidi e splenectomia nontrovano usualmente indicazione pratica. Solo nellametà circa dei casi si rende necessaria la trasfusionedi sangue. Nelle forme secondarie a lue si impone iltrattamento antiluetico che di solito comporta ridu-zione o scomparsa degli attacchi emoglobinurici.Negli altri casi acuti post-infettivi l’emoglobinuriaparossistica a “frigore” si risolve spontaneamenteparallelamente alla guarigione dell’episodio infetti-vo.Negli anni più recenti l’introduzione degli anticorpimonoclonali (vedi capitolo successivo) ha modifi-cato le prospettive terapeutiche delle forme daautoanticorpi freddi e, più in generale, di tutte leAEA.
TERAPIA DELLE AEA “REFRATTARIE”
Le forme refrattarie alla terapia di prima e secondalinea (circa il 5-10%) rappresentano spesso un pro-blema medico critico. In particolare vanno ricorda-te le rare forme con reticolocitopenia e quelle daanticorpi di classe IgM “caldi”, per le quali sonoriportate diverse segnalazioni fatali (20, 22-25).In generale, le opzioni terapeutiche delle AEA “re-frattarie” sono tuttora oggetto di dibattito e non visono Linee Guida o raccomandazioni secondo i cri-teri della Evidence Based Medicine. Trovano indi-cazione farmaci per via parenterale fra cui boli disteroide (prednisolone 10 mg/kg die per 3-5 giorni),ciclofosfamide (800 mg/m2 ogni 2-3 settimane) as-sociato o meno a vincristina (2 mg). Anche in questicasi l’uso di gammaglobuline per via endovenosa ela plasmaferesi vengono associate alla terapiaimmunosoppressiva. Infine, se l’anemia è molto se-vera e clinicamente non sopportata dal paziente, viè l’indicazione alla trasfusione di sangue, che deveessere comunque rimandata quanto più possibile.
Fra le opzioni terapeutiche più recenti e maggior-mente promettenti vi sono gli anticorpi monoclonali,fra cui rituximab (anti-CD20) e alemtuzumab (anti-CD52). Per quanto riguarda il primo, esistono or-mai diverse segnalazioni in letteratura a partire dal-la fine degli anni ’90, perlopiù costituite da
pagina 23
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
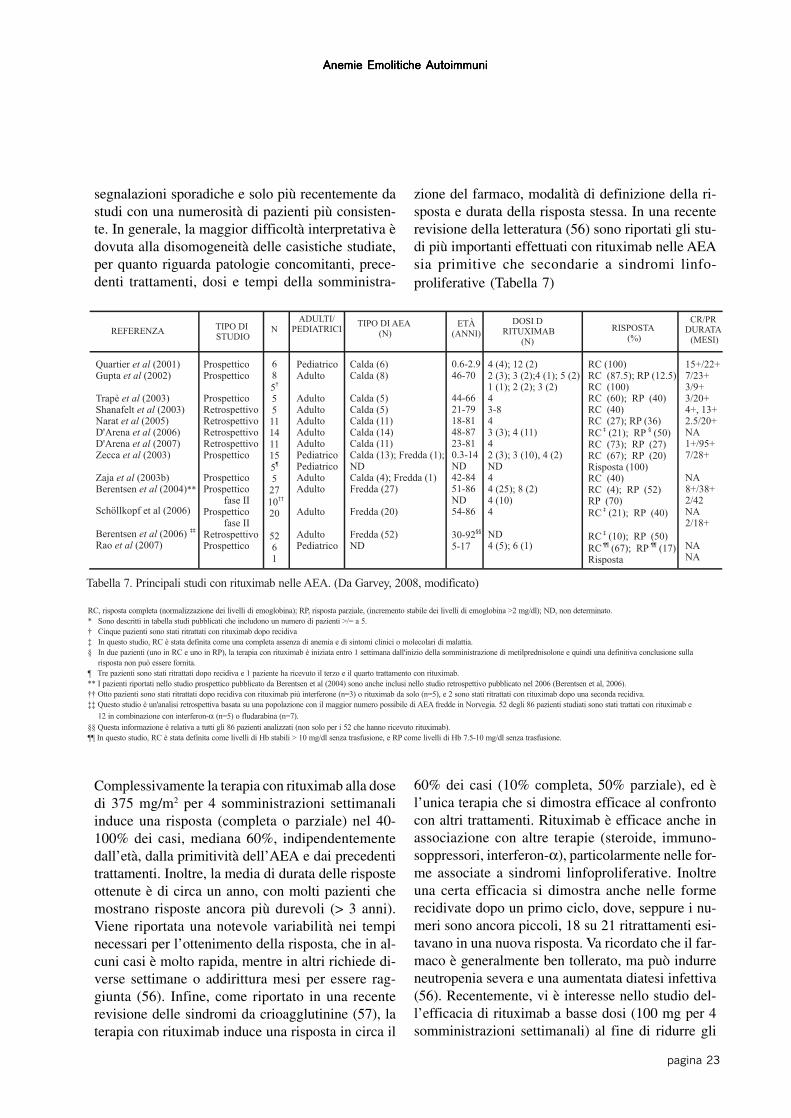
segnalazioni sporadiche e solo più recentemente dastudi con una numerosità di pazienti più consisten-te. In generale, la maggior difficoltà interpretativa èdovuta alla disomogeneità delle casistiche studiate,per quanto riguarda patologie concomitanti, prece-denti trattamenti, dosi e tempi della somministra-
Complessivamente la terapia con rituximab alla dosedi 375 mg/m2 per 4 somministrazioni settimanaliinduce una risposta (completa o parziale) nel 40-100% dei casi, mediana 60%, indipendentementedall’età, dalla primitività dell’AEA e dai precedentitrattamenti. Inoltre, la media di durata delle risposteottenute è di circa un anno, con molti pazienti chemostrano risposte ancora più durevoli (> 3 anni).Viene riportata una notevole variabilità nei tempinecessari per l’ottenimento della risposta, che in al-cuni casi è molto rapida, mentre in altri richiede di-verse settimane o addirittura mesi per essere rag-giunta (56). Infine, come riportato in una recenterevisione delle sindromi da crioagglutinine (57), laterapia con rituximab induce una risposta in circa il
60% dei casi (10% completa, 50% parziale), ed èl’unica terapia che si dimostra efficace al confrontocon altri trattamenti. Rituximab è efficace anche inassociazione con altre terapie (steroide, immuno-soppressori, interferon-α), particolarmente nelle for-me associate a sindromi linfoproliferative. Inoltreuna certa efficacia si dimostra anche nelle formerecidivate dopo un primo ciclo, dove, seppure i nu-meri sono ancora piccoli, 18 su 21 ritrattamenti esi-tavano in una nuova risposta. Va ricordato che il far-maco è generalmente ben tollerato, ma può indurreneutropenia severa e una aumentata diatesi infettiva(56). Recentemente, vi è interesse nello studio del-l’efficacia di rituximab a basse dosi (100 mg per 4somministrazioni settimanali) al fine di ridurre gli
zione del farmaco, modalità di definizione della ri-sposta e durata della risposta stessa. In una recenterevisione della letteratura (56) sono riportati gli stu-di più importanti effettuati con rituximab nelle AEAsia primitive che secondarie a sindromi linfo-proliferative (Tabella 7)
CR/PR DURATA (MESI)
RISPOSTA (%)
DOSI D RITUXIMAB
(N)
ETÀ(ANNI)
TIPO DI AEA (N)
ADULTI/PEDIATRICINTIPO DI
STUDIOREFERENZA
RC, risposta completa (normalizzazione dei livelli di emoglobina); RP, risposta parziale, (incremento stabile dei livelli di emoglobina >2 mg/dl); ND, non determinato.
* Sono descritti in tabella studi pubblicati che includono un numero di pazienti >/= a 5.
† Cinque pazienti sono stati ritrattati con rituximab dopo recidiva
‡ In questo studio, RC è stata definita come una completa assenza di anemia e di sintomi clinici o molecolari di malattia.
§ In due pazienti (uno in RC e uno in RP), la terapia con rituximab è iniziata entro 1 settimana dall'inizio della somministrazione di metilprednisolone e quindi una definitiva conclusione sulla
risposta non può essere fornita.
¶ Tre pazienti sono stati ritrattati dopo recidiva e 1 paziente ha ricevuto il terzo e il quarto trattamento con rituximab.
** I pazienti riportati nello studio prospettico pubblicato da Berentsen et al (2004) sono anche inclusi nello studio retrospettivo pubblicato nel 2006 (Berentsen et al, 2006).
†† Otto pazienti sono stati ritrattati dopo recidiva con rituximab più interferone (n=3) o rituximab da solo (n=5), e 2 sono stati ritrattati con rituximab dopo una seconda recidiva.
‡‡ Questo studio è un'analisi retrospettiva basata su una popolazione con il maggior numero possibile di AEA fredde in Norvegia. 52 degli 86 pazienti studiati sono stati trattati con rituximab e
12 in combinazione con interferon-a (n=5) o fludarabina (n=7).
§§ Questa informazione è relativa a tutti gli 86 pazienti analizzati (non solo per i 52 che hanno ricevuto rituximab).
¶¶ In questo studio, RC è stata definita come livelli di Hb stabili > 10 mg/dl senza trasfusione, e RP come livelli di Hb 7.5-10 mg/dl senza trasfusione.
Quartier et al (2001)Gupta et al (2002)
Trapè et al (2003)Shanafelt et al (2003)Narat et al (2005)D'Arena et al (2006)D'Arena et al (2007)Zecca et al (2003)
Zaja et al (2003b)Berentsen et al (2004)**
Schöllkopf et al (2006)
‡‡Berentsen et al (2006) Rao et al (2007)
ProspetticoProspettico
ProspetticoRetrospettivoRetrospettivoRetrospettivoRetrospettivoProspettico
ProspetticoProspettico fase II Prospettico fase IIRetrospettivoProspettico
68
†55511141115
¶5527
†† 1020
5261
PediatricoAdulto
AdultoAdultoAdultoAdultoAdultoPediatricoPediatricoAdultoAdulto
Adulto
AdultoPediatrico
Calda (6)Calda (8)
Calda (5)Calda (5)Calda (11)Calda (14)Calda (11)Calda (13); Fredda (1);NDCalda (4); Fredda (1)Fredda (27)
Fredda (20)
Fredda (52)ND
0.6-2.946-70
44-6621-7918-8148-8723-810.3-14ND42-8451-86ND54-86
§§30-925-17
4 (4); 12 (2)2 (3); 3 (2);4 (1); 5 (2)1 (1); 2 (2); 3 (2)43-843 (3); 4 (11)42 (3); 3 (10), 4 (2)ND44 (25); 8 (2)4 (10)4
ND4 (5); 6 (1)
RC (100)RC (87.5); RP (12.5)RC (100)RC (60); RP (40)RC (40)RC (27); RP (36)
‡ §RC (21); RP (50)RC (73); RP (27)RC (67); RP (20)Risposta (100)RC (40)RC (4); RP (52)RP (70)
‡RC (21); RP (40)
‡RC (10); RP (50) ¶¶ ¶¶RC (67); RP (17)
Risposta
15+/22+7/23+3/9+3/20+4+, 13+2.5/20+NA1+/95+7/28+
NA8+/38+2/42NA2/18+
NANA
Tabella 7. Principali studi con rituximab nelle AEA. (Da Garvey, 2008, modificato)

pagina 24
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
effetti collaterali e poter proporre questa terapia nonsolo nelle forme “refrattarie” ma anche in prima oseconda linea, prima della splenectomia e dei far-maci immunosoppressori classici (58).
Per quanto riguarda l’uso di alemtuzumab, l’espe-rienza nelle AEA è ancora limitata anche se alcunirisultati vengono riportati, soprattutto nelle formeassociate a LLC. I case reports e le piccole casistichefinora presenti in letteratura per un totale di una ven-tina di casi dimostrano una risposta nei 2/3 dei pa-zienti, di durata non specificata, ma con concomitantigravi effetti collaterali di natura infettiva, in partelegati alle patologie concomitanti e alla lunga storiaclinica dei pazienti (59, 60).
Infine, vi sono alcune recenti segnalazioni in lette-ratura di efficacia della terapia con micofenolato,dove la ottima risposta osservata in un numero esi-guo di pazienti (totale 10) necessita chiaramente diulteriore conferma (61-63).
Per ultimo, va ricordato che alcune forme partico-larmente gravi e refrattarie di AEA sono state sotto-poste a trapianto di midollo autologo o allogenico.Si tratta di segnalazioni sporadiche e di forme asso-ciate ad altre malattie quali sindrome di Evans (ane-mia e piastrinopenia autoimmune), talassemia inter-media e LLC. La valutazione della risposta è im-possibile per l’esiguità dei casi, l’arco temporale am-pio delle segnalazioni (i primi casi sono della fineanni ’90), l’eterogeneità dei regimi trapiantologiciutilizzati e le patologie concomitanti.
LA TRASFUSIONE NELLE AEA
La trasfusione nelle AEA viene rimandata quantopiù possibile e viene indicata nei casi di anemia se-vera e clinicamente non sopportata dal paziente. Inquesti casi i tests pretrasfusionali e le prove di com-patibilità hanno spesso un risultato positivo ed è dif-ficile identificare la presenza di eventualialloanticorpi anti-eritrocitari che potrebbero essereresponsabili di severe reazioni trasfusionaliemolitiche. L’esperienza indica che quando l’incom-patibilità è dovuta alla sola presenza dell’auto-anticorpo, la sopravvivenza dei globuli rossi trasfusiè generalmente buona e dalla trasfusione ci si può
aspettare un significativo e temporaneo beneficio.La decisione di trasfondere o meno questi pazientinon dipende tanto dai risultati delle prove di com-patibilità ma essenzialmente da una valutazione dellenecessità cliniche del paziente (12). In caso di im-possibilità a reperire sangue compatibile, è megliotrasfondere emazie con la specificità antigenicadell’autoanticorpo piuttosto che rischiare unaalloimmunizzazione eritrocitaria. Infatti, i pazienticon autoanticorpi ‘caldi’, quando trasfusi, devonoessere considerati a rischio di alloimmunizzazionee di reazioni post-trasfusionali emolitiche dovute allacoesistenza di alloanticorpi; dati pubblicati indica-no che frequenza di allo immunizzazione nei pazienticon AEA è del 32% (64).
In passato la strategia trasfusionale più utilizzata eraquella di trasfondere le unità che risultavano meno‘incompatibili’ cioè che presentavano i risultati menopositivi nella prova di compatibilità. Si tratta di untipo di approccio piuttosto pericoloso che dovrebbeessere abbandonato, ad eccezione di condizioni estre-mamente urgenti nelle quali non c’è il tempo neces-sario per l’esecuzione dei tests sierologici, in quan-to può essere interpretato in modo diverso dai di-versi operatori sanitari e non è sempre informativonella discussione con i clinici (65).
Una tra le strategie più sicure è quella che prevedel’esecuzione dell’autoassorbimento, che permette dirimuovere l’autoanticorpo dal siero del paziente equindi di identificare nel siero autoassorbito gli even-tuali alloanticorpi. Questa procedura può essere ap-plicata solo se il paziente non è stato trasfuso negliultimi tre mesi, poiché una piccola percentuale dicellule trasfuse potrebbe assorbire gli alloanticorpi,invalidando i risultati (66). L’autoassorbimento, nelcaso di autoanticorpi caldi, si esegue incubando leemazie e il siero autologo con additivo Polyethyleneglycol (PEG) per 30 minuti a 37°C, in modo tale chenel siero autoassorbito residuino solo anticorpi di-retti contro antigeni non presenti sulle cellule delpaziente. I limiti di tale procedura sono i tempi tec-nici, tali da non permetterne l’utilizzo in caso di tra-sfusioni urgenti, e come già ricordato in preceden-za, la non applicabilità nei pazienti recentementetrasfusi.

pagina 25
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
In alternativa, se il paziente è stato recentementetrasfuso, si può ricorrere agli alloassorbimenti mi-rati con emazie selezionate da soggetti a diversofenotipo (R1R1, R2R2, rr, tutti K negativi, unoJk(a-), uno Jk(b-) ed uno Fy(a-), che permettono dirimuovere l’autoanticorpo dal siero del paziente.Come nel caso dell’autoassorbimento i limiti dellaprocedura sono legati ai tempi di esecuzione, nonadatti in caso di urgenza, e la necessità, nei pazientipolitrasfusi, di ripetere la procedura ad ogni eventotrasfusionale. Per tale ragione alcuni autori hannoproposto l’utilizzo di additivi commerciali a bassaforza ionica come il PEG e il LISS, che permettonodi eliminare il pre-trattamento delle emazie conproteasi e quindi di ottenere un risparmio di tempotecnico, con tuttavia lo svantaggio della possibileperdita di alcuni deboli anticorpi (come quelli delsistema KEL e JK) (67, 68).
La trasfusione di globuli rossi selezionati sulla basedella tipizzazione estesa del paziente può determi-nare una significativa misura di sicurezza oltre adovviare al problema di effettuare alloassorbimentiad ogni evento trasfusionale (69). Per assicurare unatrasfusione sicura bisognerebbe tipizzare il pazien-te estensivamente per il fenotipo RH e per gli antigeniclinicamente significativi dei sistemi KEL, JK, FY,MNS e selezionare unità identiche per la trasfusio-ne. Nei casi in cui non fosse possibile eseguire unatipizzazione estesa alcuni autori suggeriscono ditipizzare il paziente con antisieri monoclonali (dovepossibile) almeno per gli antigeni dei sistemi RH,KEL e JK, selezionando unità negative per taliantigeni o almeno negative per gli antigeni E- e K-.In questi due ultimi casi è comunque necessario ri-petere gli alloassorbimenti prima di ogni eventotrasfusionale.
L’approccio utilizzato nal nostro Ospedale prevedel’assegnazione a tutti i pazienti con AEA di unità afenotipo Rh compatibile con il paziente e K negati-ve. Inoltre se la ricerca di alloanticorpi eritrocitari
risulta positiva con tutte e tre le cellule test vengonoassegnate unità anche negative per gli antigeni cli-nicamente significativi dei sistemi KEL, JK, FY,MNS che forniscano risultati negativi nel testdell’antiglobulina indiretto con la metodica standardin microcolonna o in provetta senza additivo.
Nei pazienti con AEA da autoanticorpi ‘caldi’ nonrecentemente trasfusi, viene sempre effettuato unautoassorbimento a +37°C con additivo PEG, segui-to dalla ricerca di alloanticorpi eritrocitari e dalleprove di compatibilità sulle unità da trasfondere.Questi tests pretrasfusionali dovrebbero fornire ri-sultati negativi. Le unità devono essere fresche (nonpiù di 7 giorni), filtrate bed-side ed infuse lentamentesotto stretto monitoraggio clinico.
Nei pazienti con AEA trasfusi negli ultimi tre mesisi esegue invece una tipizzazione eritrocitaria este-sa in biologia molecolare per poter poi eseguire unalloassorbimento mirato con una cellula identica alpaziente. I tests pretrasfusionali nei pazienti con AEAda autoanticorpi ‘freddi’ richiedono meno lavoro tec-nico, in quanto l’autoanticorpo reagisce ad una tem-peratura inferiore a +37°C e pertanto non è necessa-rio proseguire con la procedura dell’assorbimento.Nel caso in cui sia necessario, l’autoassorbimentoviene eseguito a +4°C e il siero autoassorbito utiliz-zato per la ricerca di alloanticorpi eritrocitari, le pro-ve di compatibilità e la determinazione indiretta digruppo AB0. Una corretta gestione della trasfusio-ne in caso di AEA presuppone una buona comunica-zione fra il clinico e lo specialista in medicinatrasfusionale: il primo dovrebbe avvisare tempesti-vamente il Centro Trasfusionale della possibile tra-sfusione di un paziente con AEA e della sua even-tuale urgenza; il secondo dovrebbe illustrare i testspretrasfusionali eseguiti per assicurare una trasfu-sione efficace e sicura, anche se i globuli rossitrasfusi potrebbero non sopravvivere normalmenteper la presenza dell’autoanticorpo.

pagina 26
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni

pagina 27
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
ASSOCIAZIONE CON LA TERAPIA CONANALOGHI DELLE PURINE
E’ ben noto dalla letteratura che circa il 5-10% deipazienti con leucemia linfatica cronica e altre sin-dromi linfoproliferative presenta una AEA, e in mi-nor misura altre citopenie o complicanze autoimmuni(70-72). L’AEA è stata osservata soprattutto nellefasi avanzate della malattia e in pazienti trattati conanaloghi delle purine quali fludarabina e cladribina,suggerendo una associazione fra la terapia con que-sti farmaci e l’insorgenza di AEA (73-75). In parti-colare, è stato ipotizzato che la immunodepressionefarmaco-indotta e la disregolazione del comparto Tlinfocitario possano giocare un ruolo nella genesidell’autoimmunità anti-eritrocitaria. Tuttavia, nonesiste al momento accordo sul ruolo della terapiacon analoghi delle purine come unico fattore di ri-schio per l’insorgenza di AEA, in quanto anche laprogressione della malattia e le precedenti lineeterapeutiche sono stati dimostrati fattori di rischioin analisi multivariata, indipendentemente dalla pre-senza di analoghi delle purine nelle terapie succes-sive alla prima linea con steroide e alchilante (Figu-ra 6) (72). Indipendentemente da queste considera-zioni, la possibile insorgenza di AEA va sempre te-nuta presente nel paziente con leucemia linfatica cro-nica e altre sindromi linfoproliferative, come gravee temibile complicanza, che può essere causa dimorte nel 10-15% dei casi (72).
ASASASASASPPPPPETTI PETTI PETTI PETTI PETTI PARTIARTIARTIARTIARTICCCCCOOOOOLARLARLARLARLARI DI DI DI DI DEEEEELLLLLLLLLLE AEAE AEAE AEAE AEAE AEA
ASSOCIAZIONE DELLA AEA CON LA TRA-SFUSIONE DI SANGUE
Esistono evidenze di una associazione tra AEA e tra-sfusione nel senso che la trasfusione stessa possaessere causa di AEA (12). Se infatti è vero che laprimaria complicanza della trasfusione di sangue èla formazione di alloanticorpi, è però vero che unapiccola percentuale (1-2%) di pazienti sviluppa an-che autoanticorpi, in stretta correlazione temporalecon l’alloimmunizzazione (76). Sono molti i reportscirca la produzione di allo ed autoanticorpi in pa-zienti multitrasfusi pediatrici affetti da talassemia eanemia falciforme: il 7,6% di 184 pazienti con ane-mia falciforme sviluppa autoanticorpi IgG dopo unamedia di 24 trasfusioni (79). Inoltre, su 64 pazientitalassemici, 25% hanno sviluppato positività delTAD ancorché con infrequente emolisi (80). Duesono le ipotesi postulate:a) la persistenza nel ricevente di cellule immuni ori-ginarie del donatore che causano la produzione dianticorpi contro gli eritrociti del ricevente;b) gli alloanticorpi inducono cambiamenticonformazionali negli epitopi antigenici che stimo-lano la produzione di autoanticorpi.
ASSOCIAZIONE DI EMOLISI IMMUNO-ME-DIATA CON IL TRAPIANTO DI MIDOLLOALLOGENICO
Una emolisi immuno-mediata può essere una com-plicanza del trapianto di midollo quando esiste una
0
10
20
30
40
50
60
70
80
%
LLC LLC+AEA
no terapia
I linea
II linea
Chi quadro P<0.001
19.28 -
432.5291.328 (25)Terapiadi II lineacon fludarabina(%)
20.57-479.8199.353 (47)
Terapiadi II lineasenzafludarabina(%)
11.31-202.6847.858 (51)Terapia I linea (%)
Ref12 (2)No terapia (%)
3.43-12.786.561(54)Stadio C (%)
1.13- 4.012.126(23)Stadio B (%)
Ref126(23)Stadio A (%)
CLOD
Figura 6. Leucemia linfatica cronica e AEA: relazione con lo stadio di malattia ela terapia.

pagina 28
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
incompatibilità ABO minore fra il donatore e il ri-cevente, perlopiù quando il primo è di gruppo 0 e ilsecondo di gruppo A, con una frequenza di circa il10-15% dei casi (81, 82). L’emolisi inizia usualmentedue settimane dopo il trapianto, e può avere esordioacuto e grave, con lisi intravascolare accompagnataanche da insufficienza renale. La genesi dell’emolisiimmuno-mediata è stata attribuita alla cosiddetta“passenger lymphocyte syndrome”, già accennata nelcapitolo dedicato alla diagnosi delle AEA, dovutaalla produzione di anticorpi da parte dei linfociti deldonatore passivamente trasferiti al ricevente eimmunologicamente attivi nei confronti delle suecellule eritrocitarie. Oltre all’incompatibilità ABO,gli autoanticorpi sono stati descritti anche nei con-fronti degli antigeni D, E, s, JKb e JKa. Normalmen-te l’emolisi si risolve nel giro di 15 giorni, con lalisi delle residue cellule incompatibili del riceventee con l’attecchimento del trapianto.
In pazienti sottoposti a trapianto di midollo è statadescritta anche una classica AEA, dovuta alla pro-duzione di autoanticorpi da parte dei linfociti deldonatore contro cellule del donatore; in casistichepediatriche si è stimata una incidenza del 6%, conun tempo medio di insorgenza di 4 mesi dal trapian-to, e con una mortalità piuttosto elevata, condizio-nata anche dalla ulteriore terapia immunosoppressivarichiesta per trattare la AEA (83-85). Anche in que-sti casi, vi sono segnalazioni di efficacia della tera-pia con rituximab (86, 87).
ASSOCIAZIONE DI EMOLISI IMMUNO-ME-DIATA CON TRAPIANTO D’ORGANI SOLI-DI
Una emolisi immuno-mediata è stata osservata an-che in corso di trapianti di organi solidi, ed è dovutaal meccanismo del “passenger lymphocytesyndrome”, in quanto linfociti del donatore possonoessere contenuti negli organi trapiantati (88-90). Ilrischio e l’entità dell’emolisi sono proporzionali allamassa linfocitaria contenuta nell’organo trapianta-to: minore nel trapianto di rene (anticorpi positivi
nel 17% dei casi con emolisi solo nel 9%), interme-dio dopo trapianto di fegato (anticorpi positivi nel40 % dei casi con emolisi nel 29%), e maggiore dopotrapianto di cuore e polmoni (anticorpi positivi conassociata emolisi nel 70% dei casi). L’anticorpo èusualmente diretto contro antigeni del sistema Rh.Il quadro clinico insorge da 3 a 24 giorni dopo iltrapianto, ed è generalmente transitorio in quanto laproduzione di autoanticorpi viene meno con la scom-parsa dei linfociti del donatore che, a differenza deltrapianto di midollo, non vanno incontro adattecchimento definitivo. Particolare attenzione vaposta, in questi casi, nella scelta delle trasfusioni,che devono prevedere l’uso di emazie di gruppo 0ed evitare prodotti derivati dal plasma ABO incom-patibili. Le emazie trasfuse devono essere ABO-identiche o compatibili con il siero del ricevente,indipendentemente dal gruppo del donatore d’orga-no; tuttavia, se il donatore e ricevente hanno gruppidiversi, la trasfusione deve essere ABO-compatibi-le con gli eritrociti sia del donatore che del riceven-te.
AEA E TROMBOEMBOLISMO
In una storica revisione del 1967, l’emboliapolmonare è stata riportata come la più frequentecausa di morte (4 casi) su 47 con AEA sottoposti asplenectomia (91). Più recentemente, diversi revi-sioni di casistiche di AEA hanno dimostrato un epi-sodio di tromboembolismo venoso nel 20-30% deipazienti con AEA, spesso associato alla positivitàper anticorpi anti-cardiolipina o anticoagulantelupico (92, 93). Fra i meccanismi responsabili delladiatesi trombofilica si ipotizza un danno della mem-brana eritrocitaria con conseguente esposizione difosfolopidi pro-coagulanti quali fosfatidilserina.Altre ipotesi vedono un ruolo dell’attivazionemonocitaria ed endoteliale da parte di citochine pro-infiammatorie prodotte nel corso delle reazioniautoimmuni. Al momento non esiste un consensocondiviso sull’uso di una terapia profilatticaanticoagulante, se non in quei casi associati a sin-drome da antifosfolipidi.

pagina 29
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
Le AEA sono comunemente una patologia di relati-va facile diagnosi e terapia, con una prognosi favo-revole. Esistono tuttavia casi, fortunatamente rari,di difficile diagnosi o di refrattarietà alle terapiestandard che costituiscono una emergenza medica.Fra questi vanno ricordate le forme TAD negative,quelle associate a farmaci, le AEA da IgM “calde” ele forme con reticolocitopenia. Sono oggi disponi-bili nuovi farmaci, fra cui anticorpi monoclonali enuovi immunosoppressori, il cui utilizzo è stato fi-
COCOCOCOCONNNNNCCCCCLLLLLUSUSUSUSUSIIIIIOOOOONNNNNIIIII
nora limitato alle forme gravi e refrattarie alla tera-pia convenzionale. Sono auspicabili protocollidiagnostici e terapeutici, soprattutto per le forme didifficile diagnosi o refrattarie, nonché trials clinicicontrollati con i nuovi anticorpi monoclonali nonsolo per le AEA più “difficili”, ma anche, come li-nea terapeutica “precoce”, per le forme recidivatedopo terapia steroidea, al fine di evitare gli effetticollaterali della terapia immunosoppressiva classi-ca o della splenectomia.

pagina 30
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni

pagina 31
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
1. Galen C. Volume VIII. In Kuhn DCG (ed):Opera Omnia. Lipsiae 1824:356.
2. Du Role des Hemolysines en Pathologie.Deuxiemie Question du XII Session du CongresFrançais de Médecine, Lyon, 1911.
3. Micheli F. Unmittelbare effekte dersplenektomie bei einem fall von erworbenemhamolytischen splenomegalischen ikterus typusHayem-Widal spleno-hamolytischer ikterus.Weiner Klinische Wochenschr, 1911;24:1269-1274.
4. Dressler. A case of intermittent albuminu-ria and chromaturia. In Major RH (ed): Classic de-scriptions of disease. Springfield, II: Charles CThomas, 1939;590-592.
5. Creite A. Versuche uber die Wirkung desSerumeiweisses nach injection in das Blut. Zeit-schrift für Rationelle Medicin, 1869;36:90-108.
6. Landois L. Die Trasfusion des Blutes.Leipzig, 1875.
7. Mackenzie S. Paroxysmal haemaglobi-nuria with remarks on its nature. Lancet, 1879;2:116,155-117,157.
8. Kuessner B. Paroxysmale hamaglobinurie.Dtsch Med Wochenschr, 1879;5:475-478.
9. Vanlair CF, Masius JR. De lamicrocythemie. Bull Acad R Med Belg 3e Ser,1871;5:515-613.
10. Minkowski O. Ueber eine hereditare, un-ter dem bilde eines chronichen icterus mit urobili-nurie, splenomegalie und nierensiderosis verlau-fende affection. Verhandl Krong Inn Med, 1900;18:316-321.
11. Donath J, Landsteiner K. Ueber paroxys-male Hämoglobinurie. München Med Wschr,1904;51:1590.
12. Petz LD, Garratty G. Immune HemolyticAnemias. Second ed. Philadelphia: ChurchillLivingstone, 2004.
13. Barker RN, Vickers MA, Ward FJ. Con-trolling autoimmunity-Lessons from the study ofred blood cells as model antigens. Immunol Lett,2007;108:20-26.
14. Rose NR. Predictors of autoimmune dis-ease: autoantibodies and beyond. Autoimmunity,2008;41:419-428.
15. Sadlack B, Löhler J, Schorle H, Klebb G,Haber H, Sickel E, Noelle RJ, Horak I. General-ized autoimmune disease in interleukin-2-deficientmice is triggered by an uncontrolled activation andproliferation of CD4+ T cells. Eur J Immunol,1995;25(11):3053-3059.
16. Shen CR, Mazza G, Perry FE, Beech JT,Thompson SJ, Corato A, Newton S, Barker RN,Elson CJ. T-helper 1 dominated responses to eryth-rocyte Band 3 in NZB mice. Immunology, 1996;89:195-199.
17. Fagiolo E, Abenante L. Lymphocyte acti-vation and cytokine production in autoimmunehemolytic anaemia (AIHA). Autoimmunity, 1996;24:147-156.
18. Toriani-Terenzi C, Pozzetto U, Bianchi M,Fagiolo E. Cytokine network in autoimmunehaemolytic anaemia: new probable targets fortherapy. Cancer Detect Prev, 2002; 26:292-298.
19. Barcellini W, Clerici G, Montesano R,Taioli E, Morelati F, Rebulla P, Zanella A. In vitroquantification of anti-red blood cell antibody pro-duction in idiopathic autoimmune haemolyticanaemia: effect of mitogen and cytokine stimula-tion. Br J Haematol, 2000;111:452-460.
20. Zanella A, Rossi F: Autoimmune hemo-lytic anemias: diagnosis and treatment. ForumTrends in Experimental and Clinical medicine,1994;4:39-53.
21. Conley CL, Lippman SM, Ness P.Autoimmune hemolytic anemia with reticulocyto-penia. A medical emergency. JAMA, 1980;244:1688-1690.
22. Garratty G, Arndt P, Domen R, Clarke A,Sutphen-Shaw D, Clear J, Groncy P. Severe
BIBIBIBIBIBBBBBLLLLLIIIIIOOOOOGGGGGRRRRRAFAFAFAFAFIAIAIAIAIA

pagina 32
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
autoimmune hemolytic anemia associated withIgM warm autoantibodies directed against deter-minants on or associated with glycophorin A. VoxSang, 1997;72:124-130.
23. McCann EL, Shirey RS, Kickler TS, NessPM. IgM autoagglutinins in warm autoimmunehemolytic anemia: a poor prognostic feature. ActaHaematol, 1992;88:120-125.
24. Friedmann AM, King KE, Shirey RS,Resar LM, Casella JF. Fatal autoimmune hemo-lytic anemia in a child due to warm-reactiveimmunoglobulin M antibody. J Pediatr HematolOncol, 1998;20:502-505.
25. Nowak-Wegrzyn A, King KE, Shirey RS,Chen AR, McDonough C, Lederman HM. Fatalwarm autoimmune hemolytic anemia resultingfrom IgM autoagglutinins in an infant with severecombined immunodeficiency. J Pediatr HematolOncol, 2001;23:250-252.
26. Issitt P. Serological diagnosis and charac-terization of causative antibody. In: Chaplin H,editor. Methods in haematology. Immune haemo-lytic anaemia. London, USA: ChurchillLivingston; 1985.
27. Dubarry M, Charron C, Habibi B,Bretagne Y, Lambin P. Quantitation of immuno-globulin classes and subclasses of autoantibodiesbound to red cells in patients with and withouthemolysis. Transfusion, 1993;33:466-471.
28. Garratty G, Arndt PA. Applications of flowcytofluorimetry to red blood cell immunology.Cytometry, 1999;38:259-267.
29. Barcellini W, Montesano R, Clerici G,Zaninoni A, Imperiali FG, Calori R, Cortelezzi A,Zanella A. In vitro production of anti-RBCantibodies and cytokines in chronic lymphocyticleukemia. Am J Hematol, 2002;71:177-183.
30. Prager M. Molecular genetic blood grouptyping by the use of PCR-SSP technique.Transfusion, 2007;47:54S-59S.
31. Reid EM. Overview of molecular methodsin immunohematology. Transfusion, 2007;47:10S-16S.
32. Sokol RJ, Hewitt S, Stamps BK.Autoimmune haemolysis: an 18-year study of 865cases referred to a regional transfusion centre. BrMed J (Clin Res Ed), 1981;282:2023-2027.
33. Petz LD, Garratty G. Acquired immunehaemolytic anemias. New York: ChurchillLivingstone;1980.
34. Shammo JM, Calhoun B, Mauer AM,Hoffman PC, Baron JM, Baron BW. First twocases of immune hemolytic anemia associated withceftizoxime. Transfusion, 1999;39:838-844.
35. Arndt PA, Leger RM, Garratty G. Serologyof antibodies to second- and third-generationcephalosporins associated with immune hemolyticanemia and/or positive direct antiglobulin tests.Transfusion, 1999;39:1239-1246.
36. Johnson ST, Fueger JT, Gottschall JL. Onecenter’s experience: the serology and drugs associ-ated with drug-induced immune hemolytic anemia-a new paradigm. Transfusion, 2007;47:697-702.
37. Carstairs KC, Breckenridge A, DolleryCT, Worlledge SM. Incidence of a positive directCoombs test in patients on alpha-methyldopa.Lancet, 1966;2:133-135.
38. Worlledge SM, Carstairs KC, Dacie JV.Autoimmune haemolytic anaemia associated withalpha-methyldopa therapy. Lancet, 1966;2:135-139.
39. Lindstrom FD, Lieden G, Engstrom MS.Dose-related levodopa-induced haemolytic anae-mia. Ann Intern Med, 1977;86:298-300.
40. Scott GL, Myles AB, Bacon PA.Autoimmune haemolytic anaemia and mefenamicacid therapy. Br Med J, 1968;3:534-535.
41. Kleinman S, Nelson R, Smith L,Goldfinger D. Positive direct antiglobulin testsand immune hemolytic anemia in patients receiv-ing procainamide. N Engl J Med, 1984;311:809-812.
42. Salama A, Kroll H, Wittmann G, Mueller-Eckhardt C. Diclofenac-induced immune haemo-lytic anaemia: simultaneous occurrence of red

pagina 33
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
blood cell autoantibodies and drug-dependent anti-bodies. Br J Haematol, 1996;95:640-644.
43. Chenoweth CE, Judd WJ, Steiner EA,Kauffman CA. Cefotetan-induced immunehemolytic anemia. Clin Infect Dis, 1992;15:863-865.
44. LoBuglio AF, Jandl JH. The nature of thealpha-methyldopa red-cell antibody. N Engl JMed, 1967;276:658-665.
45. Bakemeier RF, Leddy JP. Erythrocyteautoantibody associated with alpha-methyldopa:heterogeneity of structure and specificity. Blood,1968;32:1-14.
46. Patten E, Beck CE, Scholl C, Stroope RA,Wukasch C. Autoimmune hemolytic anemia withanti Jka specificity in a patient taking aldomet.Transfusion, 1977;17:517-520.
47. Ewing DJ, Hughes CJ, Wardle DF. Meth-yldopa-induced auto-immune haemolytic anaemia-a report of two further cases. Guys Hosp Rep,1968;117:111-118.
48. Guidelines for the investigation and man-agement of idiopathic thrombocytopenic purpurain adults, children and in pregnancy. British Com-mittee for Standards in Haematology General Hae-matology Task Force. Br J Haematol, 2003;120:574-596.
49. Emilia G, Messora C, Longo G, Bertesi M.Long-term salvage treatment by cyclosporin in re-fractory autoimmune haematological disorders. BrJ Haematol, 1996;93:341-344.
50. Liu H, Shao Z, Jing L. The effectivenessof cyclosporin A in the treatment of autoimmunehaemolytic anemia and Evans syndrome.Zhonghua Xue Ye Xue Za Zhi, 2001;22:581-583.
51. Zhang Y, Chu Y, Chen G. Clinical analysisof 164 cases Coombs test positive autoimmunehemolytic anemia. Zhonghua Xue Ye Xue Za Zhi,1998;19:573-575.
52. Flores G, Cunningham-Rundles C,Newland AC, Bussel JB. Efficacy of intravenous
immunoglobulin in the treatment of autoimmunehaemolytic anemia: results in 73 patients. Am JHematol, 1993;44:237-242.
53. Ahn YS. Danazol in autoimmune haemo-lytic anaemia. Acta Haematol, 1990;84:122-129.
54. Pignon JM, Poirson E, Rochant H. Effi-cacy of danazol in hematologic disorders Br JHaematol, 1993;83:343-345.
55. Von Baeyer H. Plasmapheresis in immunehematology: review of clinical outcome data withrespect to evidence-based medicine and clinicalexperience. Therap Apher Dial, 2003;7:127-140.
56. Garvey B. Rituximab in the treatment ofautoimmune haematological disorders. Br JHaematol, 2008;141:149-169.
57. Berentsen S, Beiske K, Tjønnfjord GE.Primary chronic cold agglutinin disease: an updateon pathogenesis, clinical features and therapy.Hematology, 2007;12:361-370.
58. Provan D, Butler T, Evangelista ML,Amadori S, Newland AC, Stasi R. Activity andsafety profile of low-dose rituximab for the treat-ment of autoimmune cytopenias in adultsHaematologica, 2007;92:1695-1698.
59. Willis F, Marsh JC, Bevan DH, KillickSB, Lucas G, Griffiths R, Ouwehand W, Hale G,Waldmann H, Gordon-Smith EC. The effect oftreatment with Campath-1H in patients withautoimmune cytopenias. Br J Haematol, 2001;114:891-898.
60. Karlsson C, Hansson L, Celsing F, LundinJ. Treatment of severe refractory autoimmunehemolytic anemia in B-cell chronic lymphocyticleukemia with alemtuzumab (humanized CD52monoclonal antibody). Leukemia, 2007;21:511-514.
61. Howard J, Hoffbrand AV, Prentice HG,Mehta A. Mycophenolate mofetil for the treatmentof refractory auto-immune haemolytic anaemia andauto-immune thrombocytopenia purpura. Br JHaematol, 2002;117:712-715.

pagina 34
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
62. Alba P, Karim MY, Hunt BJ.Mycophenolate mofetil as a treatment for auto-immune haemolytic anaemia in patients with sys-temic lupus erythematosus and antiphospholipidsyndrome. Lupus, 2003;12:633-635.
63. Kotb R, Pinganaud C, Trichet C, LambotteO, Dreyfus M, Delfraissy JF, Tchernia G, GoujardC. Efficacy of mycophenolate mofetil in adultrefractory auto-immune cytopenias: a single centerpreliminary study. Eur J Haematol, 2005;75:60-64.
64. Branch DR, Petz LD. Detecting alloanti-bodies in patients with autoantibodies.Transfusion, 1999;39:6-10.
65. Petz LD. ‘Least incompatible’ units fortransfusion in autoimmune haemolytic anemia:should we eliminate this meaningless term? Acommentary for clinicians and transfusion medi-cine professionals. Transfusion, 2003;43:1503-1507.
66. Laine EP, Leger RM, Arndt PA. In vitrostudies of the impact of transfusion on thedetection of alloantibodies after autoadsorption.Transfusion, 2000; 40:1384-1387.
67. Chiaroni J, Touinssi M, Mazet M, DeMicco P, Ferrera V. Adsorption of autoantibodiesin the presence of LISS to detect alloantibodiesunderlying warm autoantibodies. Transfusion,2003;43:651-655.
68. Cheng CK, Wong ML, Lee AW. PEG ad-sorption of autoantibodies and detection ofalloantibodies in warm autoimmune haemolyticanemia. Transfusion, 2001;41:13-17.
69. Shirey RS, Boyd JS, Parwani AV, TanzWS, Ness PM, King KE. Prophylactically antigen-matched donor blood for patients with warmautoantibodies: an algorithm for transfusion man-agement. Transfusion, 2002,42:1435-1441.
70. Mauro FR, Foa R, Cerretti R, GiannarelliD, Coluzzi S, Mandelli F, Girelli G. Autoimmunehaemolytic anaemia in chronic lymphocytic leu-kaemia: clinical, therapeutic, and prognostic fea-tures. Blood, 2000;95:2786-92.
71. Kyasa MJ, Parrish RS, Schichman SA,
Zent CS. Autoimmune cytopenia does not predictpoor prognosis in chronic lymphocytic leukaemia/small lymphocytic lymphoma. Am J Hematol,2003;74:1-8.
72. Barcellini W, Capalbo S, Agostinelli RM,Mauro FM, Ambrosetti A, Calori R, Cortelezzi A,Laurenti L, Pogliani EM, PPedotti P, Liso V,Mandelli F, Zanella A: GIMEMA ChronicLymphocytic Leukemia group. Relationshipbetween autoimmune phenomena and diseasestage and therapy in B-cell chronic lymphocyticleukaemia. Haematologica, 2006;91:1689-92.
73. Myint H, Copplestone JA, Orchard J.Fludarabinerelated autoimmune haemolyticanaemia in patients with chronic lymphocyticleukaemia. Br J Haematol, 1995;91:341-344.
74. Weiss RB, Freiman J, Kweder SL, DiehlLF, Byrd JC. Hemolytic anemia after fludarabinetherapy for chronic lymphocytic leukemia. J ClinOncol, 1998;16:1885-1889.
75. Fleischman RA, Croy D. Acute onset ofsevere autoimmune hemolytic anemia aftertreatment with 2-chlorodeoxyadenosine forchronic lymphocytic leukemia. Am J Hematol,1995;48:293.
76. Young PP, Uzieblo A, Trulock E, LublinDM, Goodnough LT. Autoantibody formation afteralloimmunization: are blood transfusions a riskfactor for autoimmune hemolytic anemia?Transfusion, 2004;44:67-72.
77. Castellino SM, Combs MR, ZimmermanSA, Issitt PD, Ware RE. Erythrocyte autoanti-bodies in paediatric patients with sickle celldisease receiving transfusion therapy: frequency,characteristics and significance. Br J Haematol,1999;104:189-194.
78. Singer ST, Wu V, Mignacca R, KuypersFA, Morel P, Vichinsky EP. Alloimmunization anderythrocyte autoimmunization in transfusion-de-pendent thalassemia patients of predominantlyAsian descent. Blood, 2000;96:3369-3373.
79. Bolan CD, Childs RW, Procter JL, BarrettAJ, Leitman SF. Massive immune haemolysis afterallogeneic peripheral blood stem cell transplanta-

pagina 35
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni
tion with minor ABO incompatibility. Br JHaematol, 2001;112:787-795.
80. Worel N, Greinix HT, Keil F, MitterbauerM, Lechner K, Fischer G, Mayr W, Hocker P,Kalhs P. Severe immune hemolysis after minorABO-mismatched allogeneic peripheral blood pro-genitor cell transplantation occurs more frequentlyafter nonmyeloablative than myeloablativeconditioning. Transfusion, 2002;42:1293-1301.
81. O’Brien TA, Eastlund T, Peters C, NegliaJP, Defor T, Ramsay NK, Scott Baker K.Autoimmune haemolytic anaemia complicatinghaematopoietic cell transplantation in paediatricpatients: high incidence and significant mortalityin unrelated donor transplants for nonmalignantdiseases. Br J Haematol, 2004;127:67-75.
82. Drobyski WR, Potluri J, Sauer D, Gottsch-all JL. Autoimmune hemolytic anemia following Tcell-depleted allogeneic bone marrow transplanta-tion. Bone Marrow Transplant, 1996;17:1093-1099.
83. Chen FE, Owen I, Savage D, Roberts I,Apperley J, Goldman JM, Laffan M. Late onsethaemolysis and red cell autoimmunisation afterallogeneic bone marrow transplant. Bone MarrowTransplant, 1997;19:491-495.
84. Corti P, Bonanomi S, Vallinoto C,Balduzzi A, Uderzo C, Cazzaniga G, Gaipa G,Dassi M, Perseghin P, Rovelli A. Rituximab forimmune hemolytic anemia following T- and B-Cell-depleted hematopoietic stem cell transplanta-tion. Acta Haematol, 2003;109:43-45.
85. Ship A, May W, Lucas K. Anti-CD20monoclonal antibody therapy for autoimmunehemolytic anemia following T celldepleted, haplo-identical stem cell transplantation. Bone MarrowTransplant, 2002;29:365-366.
86. Ramsey G. Red cell antibodies arisingfrom solid organ transplants. Transfusion,1991;31:76-86.
87. Salerno CT, Burdine J, Perry EH, KshettryVR, Hertz MI, Bolman RM, 3rd. Donor-derivedantibodies and hemolysis after ABO-compatiblebut nonidentical heart-lung and lung transplanta-tion. Transplantation, 1998;65:261-264.
88. Triulzi DJ, Shirey RS, Ness PM, Klein AS.Immunohematologic complications of ABO-un-matched liver transplants.Transfusion, 1992;32:829-833.
89. Allgood JW, Chaplin H, Jr. Idiopathic ac-quired autoimmune hemolytic anemia. A review offorty-seven cases treated from 1955 through 1965.Am J Med, 1967;43:254-273.
90. Pullarkat V, Ngo M, Iqbal S, Espina B,Liebman HA. Detection of lupus anticoagulantidentifies patients with autoimmune haemolyticanaemia at increased risk for venous thromboem-bolism. Br J Haematol, 2002;118:1166-1169.
91. Hendrick AM. Auto-immune haemolyticanaemia - a highrisk disorder for thromboembo-lism? Hematology, 2003;8:53-56.

pagina 36
Anemie EmoliticAnemie EmoliticAnemie EmoliticAnemie EmoliticAnemie Emolitiche Autoimmunihe Autoimmunihe Autoimmunihe Autoimmunihe Autoimmuni


















