
Universita degli Studi di Trieste - df.units.it Springolo.pdf · di un sistema quantistico sia...
49
Universit ` a degli Studi di Trieste Dipartimento di Fisica Corso di Studi in Fisica Tesi di Laurea Triennale Polarizzazione di bordo e cariche di vertice Laureando: Matteo Springolo Relatore: Chiar.mo prof. Raffaele Resta ANNO ACCADEMICO 2014-2015
Transcript of Universita degli Studi di Trieste - df.units.it Springolo.pdf · di un sistema quantistico sia...

Universita degli Studi di Trieste
Dipartimento di Fisica
Corso di Studi in Fisica
Tesi di Laurea Triennale
Polarizzazione di bordo e cariche di vertice
Laureando:Matteo Springolo
Relatore:Chiar.mo prof. Raffaele Resta
ANNO ACCADEMICO 2014-2015

a mamma Lucia e papa Valter

Indice
1 Introduzione 2
2 Obiettivi 4
3 Descrizione del sistema fisico 5
4 Appunti Preliminari 74.1 Condizioni al contorno periodiche . . . . . . . . . . . . . . . . . . . 74.2 Elettroni in un potenziale periodico . . . . . . . . . . . . . . . . . . 84.3 Modello a Bande . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84.4 Teorema di Bloch . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
5 Modello Tight-Binding 12
6 Descrizione in modello OBC 24
7 Confronto tra modello OBC e modello TB 38
8 Polarizzazione macroscopica e suo limite termodinamico 41
9 Conclusioni 46
1

Capitolo 1
Introduzione
Questa tesi e ispirata da un articolo di Yuanjun Zhou, Karin Rabe e David Van-derbilt apparso nel Maggio 2015, e in seguito pubblicato come Phys. Rev. B 92,041102(R) (2015), Ref.[1]. Citeremo questo nel seguito come ZRV.
Il messaggio di ZRV e che oltre all’usuale polarizzazione “bulk” (di volume),puo anche esistere una polarizzazione di superficie, parallela alla superficie stessa.Secondo l’elettrostatica elementare se un materiale ha una polarizzazione bulk P,alla sua superficie c’e un accumulo di carica con densita superficiale proporzionalealla componente di P normale alla superficie stessa. La polarizzazione di superficieinvece e parallela a quest’ultima, e in analogia alla polarizzazione di bulk possiamousare questa per calcolare la densita di carica lineare sugli spigoli del cristallite.
Nel lavoro ZRV il problema viene affrontato e discusso con gli strumenti dellamoderna teoria della polarizzazione (fase di Berry e concetti analoghi), ma anchecon simulazioni su cristalliti finiti con Hamiltoniane modello, in 2 e 3 dimensio-ni. In 2 dimensioni la polarizzazione di superficie corrisponde in realta ad unapolarizzazione di bordo (1-dimensionale) e questa permette dunque il calcolo dellecariche di vertice.
In questa tesi abbiamo scelto una Hamiltoniana modello 2d piu semplice diquella scelta da ZRV, ma dove l’effetto nuovo dovrebbe essere altrettanto visibile,non essendo proibito per simmetria. Il nostro sistema modello e il Nitruro di Boro(BN) esagonale, che si ottiene dal grafene introducendo una diversa ionicita per idue atomi nella cella, e di conseguenza aprendo un gap. I nostri cristalliti (flakes)sono rettangolari e di dimensioni variabili.
Poiche il BN ha simmetria trigonale, ci si aspetta che la polarizzazione bulk(ovvero 2d in questo caso) in un flake sia nulla. La teoria moderna della polariz-zazione precisa meglio questo concetto. Nel limite di grandi flakes, il dipolo divisoper l’area (polarizzazione appunto) deve tendere a un “quanto” di polarizzazione,il cui valore puo dipendere dalla terminazione scelta. Zero e il valore piu banaledi questo quanto.
2

CAPITOLO 1. INTRODUZIONE 3
Ci aspettiamo che il nostro flake abbia i vertici carichi, con una carica chetende ad un limite finito per flakes grandi. Questa carica e dovuta alla polarizza-zione di bordo. Le nostre simulazioni confermano quanto scoperto da ZRV, anchenel caso della nostra Hamiltoniana.
Abbiamo anche calcolato la polarizzazione bulk come il dipolo dell’intero flakediviso per la sua superficie e qui abbiamo avuto sorprese. Per grandi flakes il valoredel dipolo diviso l’area non tende a zero, e neppure a uno dei valori quantizzatipermessi. Questo risultato paradossale e in contrasto con la moderna teoria dellapolarizzazione. La soluzione del paradosso rimane un problema aperto che va oltrelo scopo di questa tesi.

Capitolo 2
Obiettivi
Obiettivo di questo lavoro e verificare la presenza di cariche nette ai bordi e aivertici di un cristallo bidimensionale di Nitruro di Boro, come previsto da ZRV. Cisi propone quindi di verificare l’esistenza di tali cariche e la conseguente possibileesistenza della cosiddetta polarizzazione di bordo. Tutto cio viene svolto prendendoin considerazione il cristallo come una macromolecola.
4

Capitolo 3
Descrizione del sistema fisico
Come detto sopra, andremo a studiare un cristallo bidimensionale di Nitruro diBoro. Per fare cio passeremo anche attraverso la descrizione del grafene, solido cri-stallino che presenta la stessa struttura reticolare del Nitruro di Boro. Descriviamoora le cose piu nello specifico.
Grafene e Nitruro di Boro presentano una struttura reticolare (bidimensionalenel caso proposto) di tipo esagonale. Nel caso del grafene i siti del reticolo esagonalesono occupati da atomi tutti uguali fra loro, nella fattispecie da atomi di carboniodistanziati fra loro di un lunghezza pari circa a 1.43 A◦ (distanza di legame). Nelcaso del Nitruro di Boro invece i siti sono occupati alternatamente da atomi diAzoto e da atomi di Boro, distanziati fra di loro di una lunghezza (di legame) pari1.446 A◦.
Come gia detto, abbiamo a che fare con un cristallo, oggetto che per definizionepresenta una struttura spaziale regolare periodica. Tale periodicita viene definitaattraverso i cosiddetti vettori di reticolo diretto. In cristallografia si definisconovettori di reticolo diretto i vettori che definiscono la periodicita spaziale (cioela periodicita nello spazio reale) del cristallo. La porzione di spazio definita elimitata da tali vettori (e con la cui ripetizione si puo quindi ricoprire l’interospazio occupato dal cristallo stesso) viene chiamata cella. I vettori di reticolodiretto piu piccoli capaci di descrivere la periodicita del cristallo vengono dettivettori di reticolo primitivi. Infine, l’insieme di atomi che risiedono nella cellaprende il nome di base.
La struttura esagonale puo essere descritta con una cella primitiva contenenteuna base costituita da due atomi. Come vedremo fra poco, nel lavoro proposto, siusera invece una supercella di forma rettangolare con una base di quattro atomi(quindi una cella ovviamente piu grande della cella primitiva, precisamente grandeil doppio di questa).
I vettori di reticolo diretto scelti sono i vettori
5

CAPITOLO 3. DESCRIZIONE DEL SISTEMA FISICO 6
a1 =(√
3a 0)
a2 = (0 3a)
che definiscono appunto una cella rettangolare nello spazio diretto. Le posi-zioni dei quattro atomi all’interno di questa sono
d1 =(0 1
2a)
d2 =(√
32a 0
)d3 =
(0 3
2a)
d4 =(√
32a 2a
)Il cristallo quindi e rappresentato dalla ripetizione di tali celle e degli atomi
contenuti al suo interno. Chiaramente il numero di celle definisce le dimensionidel cristallo.
Tali asserzioni fanno parte della modellizzazione usata e quindi dell’interoseguente lavoro.

Capitolo 4
Appunti Preliminari
4.1 Condizioni al contorno periodiche
In fisica della materia condensata le quantita e le grandezze piu interessanti siottengono calcolando quest’ultime nel cosiddetto limite termodinamico. Questoconsiste nel considerare il sistema come composto da un numero molto grande diatomi (N −→ ∞) contenuti in un volume sempre piu grande (V −→ ∞), ma inmodo tale che il loro rapporto (ossia la densita volumica di particelle) rimangacostante. Tutto cio viene fatto al fine di far emergere le proprieta di bulk del siste-ma, ovvero le proprieta che non dipendono dalla superficie del sistema, proprietaquest’ultime presenti invece nei sistemi finiti (come ad esempio le macromolecoleche stiamo per studiare). Infatti al limite termodinamico il rapporto tra superfi-cie e volume del sistema tende a zero, e questo equivale appunto a trascurare lastessa superficie e gli effetti dovuti alla sua eventuale presenza. Per eseguire taleoperazione, dal punto di vista pratico, si usano le cosiddette condizioni periodichedi Born-von-Karman (BvK). Queste condizioni impongono che la funzione d’ondadi un sistema quantistico sia periodica su una determinata porzione del reticolocristallino, porzione che viene detta appunto cella di BvK. Questa condizione equi-vale a dire che l’elettrone che si muove in tale cella non viene riflesso all’indietroquando arriva in prossimita dei limiti di questa, ma abbandona la cella rientrandodal lato opposto. Questo equivale a non avere superficie e permette di descriverel’intero sistema limitandosi alla descrizione delle proprieta della cella di BvK, datoche l’intero sistema risulta essere una semplice ripetizione di questa, e quindi ogniproprieta del sistema risulta periodica sulla cella di BvK. In questo modo non siconsidera piu il sistema come una macromolecola dotata di superficie, ma comeun sistema infinito privo di superfici. Inoltre le condizioni di BvK sono necessarieper poter usare il Teorema di Bloch.
7

CAPITOLO 4. APPUNTI PRELIMINARI 8
4.2 Elettroni in un potenziale periodico
Il cristallo preso in considerazione e un sistema fisico a piu corpi. La nostra con-centrazione e posta ora sul comportamento degli elettroni di questo sistema. Ognielettrone interagisce sia con gli ioni del reticolo (considerati ora fissi) sia con gli altrielettroni. Cioe, nell’Hamiltoniana corretta rientrano sia le interazioni elettrone-ione che le interazioni elettrone-elettrone. Nel nostro modello pero facciamo l’ap-prossimazione che gli elettroni siano particelle indipendenti fra loro (quindi senzainterazione elettrone-elettrone). Tale considerazione ci porta a considerare il po-tenziale nel quale sono immersi gli elettroni come periodico nello spazio. Infattila configurazione spaziale degli ioni e periodica nello spazio (per definizione stessadi cristallo) e periodico risulta quindi il potenziale, con medesima periodicita delcristallo. L’Hamiltoniana di singola particella (elettrone) diventa quindi
H = − ~22m∇2 + U(r)
dove U(r) e il potenziale che soddisfa quindi la relazione
U(r) = U(r + R) , ∀r
dove R e il vettore di traslazione che definisce la periodicita del cristallo.
4.3 Modello a Bande
Il modello a bande si applica a quei materiali cristallini per i quali e possibile adot-tare un’approssimazione a particelle indipendenti. Secondo il modello a bande glielettroni in un cristallo sono organizzati in bande di energia, le quali possono essereseparate da regioni energetiche per le quali non esiste nessun orbitale elettronico.Queste regioni proibite sono chiamate “energy gaps” o “band gaps”.
E proprio tale struttura a bande che definisce il comportamento di un mate-riale quando questo viene sottoposto ad un campo elettrico. Infatti il materialerisulta essere un isolante se esiste un consistente gap energetico fra le bande per-messe. In tal caso infatti gli elettroni che riempiono la banda di valenza, anchesotto la sollecitazione del campo elettrico, non possono passare alla bande di con-duzione dove assumerebbero una mobilita maggiore. Se il gap non e troppo grandepero, attraverso un campo elettrico sufficientemente elevato, e possibile promuo-vere alcuni elettroni della banda di valenza a quella di conduzione. In questo casoil materiale viene definito semiconduttore.
Se le bande risultano invece parzialmente sovrapposte o adiacenti il materialeconsiderato si definisce rispettivamente metallo o semimetallo. In tal caso, infat-ti, essendo i livelli energetici permessi distanziati fra loro da valori infinitesimi,

CAPITOLO 4. APPUNTI PRELIMINARI 9
gli elettroni possono passare senza difficolta dalla banda di valenza a quella diconduzione, assumendo quindi una grande mobilita e motivando quindi l’elevataconduzione elettrica del materiale sotto l’azione di un campo elettrico applicato.
4.4 Teorema di Bloch
Il teorema di Bloch afferma che le autofunzioni di un sistema la cui Hamiltonianapresenti un potenziale periodico nelle coordinate spaziali sono della forma:
|ψj,k〉 = eik·r |uj,k〉
che riscritta in rappresentazione delle coordinate diventa:
ψj,k(r) = eik·ruj,k(r)
Cioe le autofunzioni sono uguali a una funzione (dipendente dal vettore k) chepresenta la stessa periodicita del potenziale modulata da un esponenziale complessonel quale rientra il prodotto scalare tra il vettore di Bloch e la posizione dellaparticella.
Quindi vale
uj,k(r) = uj,k(r + R)
dove R e il vettore che definisce la periodicita del potenziale (nel nostro casospecifico sara il vettore di traslazione del reticolo nello spazio diretto).
Le funzioni di Bloch presentano interessanti proprieta:• Se si esegue una traslazione nello spazio pari al vettore R che definisce la
periodicita del potenziale, le funzioni di Bloch vengono moltiplicate per un fattoredi fase, cioe
ψj,k(r + R) = eik·(r+R)uj,k(r + R) = eik·Reik·ruj,k(r + R)
e ricordando che
uj,k(r) = uj,k(r + R)
segue che
ψj,k(r + R) = eik·Reik·ruj,k(r) = eik·Rψj,k(r)

CAPITOLO 4. APPUNTI PRELIMINARI 10
In parole semplici, esse sono autustati degli operatori di traslazione di unvettore R per ogni vettore del reticolo diretto i quali, per la simmetria traslazionaledel cristallo, commutano con l’Hamiltoniana del sistema.
• Imponendo le condizioni al contorno sulla cella di BvK alle funzioni di Bloch,otteniamo una discretizzazione dei vettori di Bloch permessi nello spazio reciproco.Infatti, applicando le condizioni di BvK su una cella di volume finito formata daM1×M2×M3 celle (dove M1,M2,M3 sono il numero di celle nelle direzioni x,y, ez ), indicando con ai i vettori primitivi di traslazione del reticolo diretto e con bii vettori primitivi del reticolo reciproco, dalla relazione ai ·bj = 2πδij, si ha che lafunzione d’onda deve soddisfare, in ogni direzione, la relazione
ψj,k(r) = ψj,k(r + Ri)
e quindi che (dopo qualche manipolazione e ricordando la forma delle funzionidi Bloch)
eik·Ri = 1
cioe k ·Ri = 2πmi
Ossia, ricordando che k =∑3
j=1 vjbj e le relazioni di ortogonalita tra i vettori
del reticolo diretto e quelli del reticolo reciproco, si trova che vj =mj
Mje che quindi
i k sono della forma:
k =∑3
j=1mj
Mjbj
dove gli Mj sono il numero di celle del reticolo nelle rispettive direzioni e glimj sono degli interi con valori rispettivamente 0 ≤ mj ≤ (Mj − 1). Vediamoquindi che, grazie alle condizioni periodiche al contorno di BvK, i vettori di Blochvengono discretizzati e, inoltre, i vettori di Bloch permessi sono solo Mj nellerispettive direzioni, e quindi in un cristallo tridimensionale i soli vettori di Blochpermessi sono M1 ×M2 ×M3
• Il vettore di Bloch k che identifica una funzione di Bloch puo essere semprescelto nella prima zona di Brillouin, poiche ogni k′ che non e contenuto nella primazona di Brillouin puo sempre essere scritto come k′ = k + G , dove k appartienealla prima zona di Brillouin e G e un dato vettore del reticolo reciproco. EssendoeiG·R = 1, il vettore di Bloch e lo stesso sia per k che per k′.
• Se poniamo il potenziale del reticolo uguale a zero, otteniamo come soluzionile onde piane.
Notiamo inoltre che che le funzioni uj,k(r) risultano autofunzioni della matrice
H(k) = e−ik·rHeik·r. Infatti:
H(k) |ψj,k〉 = Ej(k) |ψj,k〉

CAPITOLO 4. APPUNTI PRELIMINARI 11
che possiamo riscrivere come
Heik·r |uj,k〉 = Ejeik·r |uj,k〉
Infine, una semplice manipolazione ci permette di scrivere
e−ik·rHeik·r |uj,k〉 = Ej |uj,k〉
e che quindi |uj,k〉 e autostato dell’Hamiltoniana H(k) = e−ik·rHeik·r , percui:
H(k) |uj,k〉 = Ej(k) |uj,k〉

Capitolo 5
Modello Tight-Binding
Il modello Tight-Binding e un metodo di calcolo usato in fisica dei solidi per ilcalcolo della struttura delle bande elettroniche di un solido attraverso un set difunzioni d’onda approssimate. Tali funzioni d’onda approssimate vengono calcolatecome sovrapposizione delle autofunzioni degli atomi isolati centrate nei siti atomici.Il metodo e quindi strettamente legato al classico metodo LCAO. Cio che cambiada tale modello e il fatto che il set di autofunzioni centrate nei singoli siti atomiciusate come base per la costruzione degli autostati sono leggermente diverse. Infattila base usata viene leggermente modificata. Ogni elemento della base LCAO vienemodulato con un esponenziale complesso nel cui argomento rientra il prodottoscalare tra il cosiddetto vettore di Bloch e la posizione del rispettivo sito atomiconel quale e centrato l’atomo e l’elemento della base riferito a questo. La base vienescelta di questo tipo al fine di rispettare il teorema di Bloch.
La forma dello stato di Bloch risulta quindi essere :
|ψj,k〉 = 1√M
∑R
∑s=1 cj,s(k)eik·(R+ds) |χR,s〉
Dove gli elementi |χR,s〉 corrispondono nel nostro caso agli orbitali a simmetriaπ centrati nei rispettivi siti atomici individuati dai vettori (R + ds).
Il vettore di Bloch e un vettore appartenente ad Rd , dove d e la dimensionedel reticolo reciproco (in questo caso, ad esempio, 2) ed ogni sua componente ha ledimensioni di L−1, dove L e una lunghezza. Tale vettore e un parametro che nonha significato fisico, ma e un parametro che serve solo per descrivere le proprietadi periodicita del cristallo. E quindi un parametro geometrico. Come vedremo frapoco, i valori di tale vettore sono direttamente correlati con i vettori di reticolodiretto (proprio perche vengono costruiti per descrivere le simmetrie del sistema)e lo spazio nel quale risiedono i vettori di Bloch e detto spazio reciproco.
Ricordiamo quali sono i vettori di reticolo diretto scelti per descrivere ilsistema:
12

CAPITOLO 5. MODELLO TIGHT-BINDING 13
a1 =(√
3a 0)
a2 = (0 3a)
Come scritto sopra, i vettori di Bloch devono avere la forma:
k =∑2
j=1mj
Mjbj
dove Mj = M1,M2
(quindi, come detto sopra, avremo M1 ×M2 possibili vettori di Bloch indi-pendenti)
Da quest’ultima segue quindi che i vettori di Bloch sono discretizzati.Ricordando quali sono i vettori di reticolo diretto e le relazioni di ortogonalita
viste sopra, otteniamo i vettori che definiscono la cella (rettangolare) nello spazioreciproco, e questi sono:
b1 = 2π√3a
b2 = 2π3a
E i vettori di Bloch sono quindi:
k = 2πa
(m1√3M1
m2
3M2
)Si vede il perche delle dimensioni dei vettori di Bloch e il perche si parli di
spazio reciproco. Si vede inoltre come i vettori di Bloch permessi formino unagriglia rettangolare nello spazio reciproco, con passi ∆kx = 2π√
3M1ae ∆ky = 2π
3M2a.
La griglia risulta quindi sempre piu fitta all’aumentare di M1 ed M2. Notiamoinoltre come, in analogia con lo spazio diretto, anche i vettori nello spazio reciprocoformino delle celle che ricoprono tutto lo spazio reciproco considerato e il “volume”(nel nostro caso una superficie) di tali celle risulta inversamente proporzionalea quello della cella nello spazio diretto. Tale osservazione tornera utile dopo.Specifichiamo di nuovo inoltre che i possibili valori indipendenti di kx sono M1 equelli di ky sono M2, per un totale quindi di M = M1×M2 possibili vettori k nellospazio reciproco.
Torniamo ora per un attimo all’analisi dell’autostato di Bloch e vediamo poiquindi come questo viene calcolato. Ricordiamo che la sua forma e :
|ψk〉 = 1√M
∑R
∑s=1 cs(k)eik·(R+ds) |χR,s〉
dove M = M1 ×M2, cioe il numero di celle del cristallo. L’introduzione dellasua radice nell’espressione dell’autostato di Bloch serve per ottenere la correttanormalizzazione di questo. E facile verificare infatti tale normalizzzione e come lastessa condizione di normalizzazione si riduca a:

CAPITOLO 5. MODELLO TIGHT-BINDING 14
∑s |cs(k)|2 = 1
I coefficienti non dipendono dalla cella considerata, ma solo dal sito atomicoconsiderato, cioe i coefficienti sono gli stessi per ogni cella. Questi coefficien-ti presentano inoltre una dipendenza dal vettore di Bloch (che fa conservare ladimensione dello spazio degli autostati).
Ricordiamo che il numero di stati usati come base sono in totale 4×M1×M2.Come per il metodo LCAO costruiremo ora la matrice Hamiltoniana su tale
base, per poi diagonalizzarla e ottenere quindi autovalori (le energie) e autovettori(gli autostati di Bloch appunto). Vogliamo cioe risolvere l’equazione agli autovalori
H(k) |ψj,k〉 = Ej(k) |ψj,k〉
Come prima, si usa l’approssimazione ai primi vicini. Gli elementi di matricescritti sulla base considerata risultano quindi:
Hss′,RR′ = αsδss′δRR′ + βδp.v.
Calcolando il valore di aspettazione dell’energia si ottiene:
〈ψk| H |ψk〉 = 1M
∑ss′ c
∗s(k)cs′(k)
∑RR′ eik·(R
′+ds′−R−ds) 〈χR,s| H |χR′,s′〉 =∑s αsc
2s(k) + β
M
∑ss′ c
∗s(k)cs′(k)
∑RR′ eik·(R
′+ds′−R−ds)δp.v.
Si ottiene quindi una forma quadratica dei soli coefficienti e la matrice Hamil-toniana quindi, grazie all’approssimazione ai primi vicini e grazie alla simmetriadel sistema, si riduce ad una matrice 4 × 4. La matrice si riduce quindi ad unamatrice 4 × 4 dipendente dal vettore k , i cui autovalori corrispondono ai livel-li energetici permessi per ogni k, e i cui autovettori permettono di costruire gliautostati di Bloch.
La matrice risulta avere tale formaα1 βγ12 0 βγ14βγ21 α2 βγ23 0
0 βγ32 α1 βγ34βγ41 0 βγ43 α2
dove γij =
∑j e
ik·(dj−di) e il termine che tiene conto dell’interazione di ogni
atomo i con gli atomi vicini (labellati in questo caso con j ). E facile vederecome γij = γ∗ji e come dunque la matrice sia una matrice complessa simmetrica,e come questa sia quindi Hermitiana. Specifico che per costruire questa matricee stata usata la stessa cella, ma una base leggermente diversa, come si vede dalleinterazioni ai primi vicini. I risultati non cambiano.

CAPITOLO 5. MODELLO TIGHT-BINDING 15
Diagonalizzando suddetta matrice dipendente da k si ottengono quattro pos-sibili autovalori per ogni valore del vettore k e quattro possibili autovettori perogni k. Ricordando che i vettori di Bloch permessi sono M1 × M2 abbiamo intutto 4 ×M1 ×M2 possibili energie e 4 ×M1 ×M2 possibili autostati, entrambidipendenti da k. Si verifica cosı che, come dev’essere, il numero di livelli energeticie di autostati rimane uguale e quindi consistente col metodo LCAO.
Riassumendo, abbiamo quattro livelli energetici permessi per ogni vettore k(che labelliamo con la lettera j) e dipendenti dal valore di quest’ultimo. Il set ditali energie in funzione di k va proprio a definire le bande energetiche di valenza edi conduzione del cristallo.
Vediamo in figura 5.1 la forma di tali bande in funzione del vettore bidimen-sionale k.
Figura 5.1: Le quattro bande del grafene in funzione dei vettori k nella prima celladello spazio reciproco
Il fatto di avere quattro possibili bande deriva dal fatto che abbiamo sceltouna supercella rettangolare contenente quattro atomi, invece della cella primitivacontenente due atomi. Se avessimo usato quest’ultima, infatti, avremmo avutodue valori energetici per ogni k, e quindi due bande, una di valenza e l’altra diconduzione. Nel nostro caso invece abbiamo ancora due bande, ma queste sonoripiegate su se stesse nella prima cella dello spazio reciproco. Questo e dovutoproprio alla diversita della cella usata. Questa infatti risulta avere, come gia detto,il doppio degli atomi e il “volume” (una superficie in questo caso) della cella nellospazio reciproco, essendo quest’ultimo inversamente proporzionale al “volume”
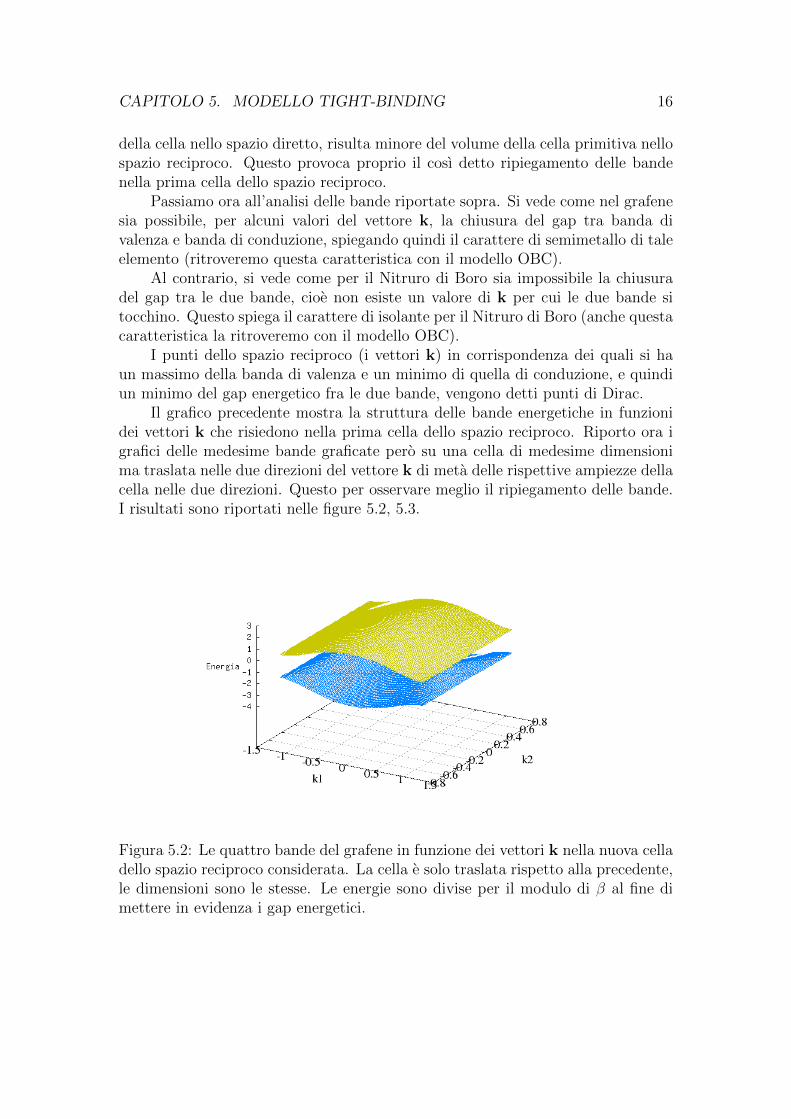
CAPITOLO 5. MODELLO TIGHT-BINDING 16
della cella nello spazio diretto, risulta minore del volume della cella primitiva nellospazio reciproco. Questo provoca proprio il cosı detto ripiegamento delle bandenella prima cella dello spazio reciproco.
Passiamo ora all’analisi delle bande riportate sopra. Si vede come nel grafenesia possibile, per alcuni valori del vettore k, la chiusura del gap tra banda divalenza e banda di conduzione, spiegando quindi il carattere di semimetallo di taleelemento (ritroveremo questa caratteristica con il modello OBC).
Al contrario, si vede come per il Nitruro di Boro sia impossibile la chiusuradel gap tra le due bande, cioe non esiste un valore di k per cui le due bande sitocchino. Questo spiega il carattere di isolante per il Nitruro di Boro (anche questacaratteristica la ritroveremo con il modello OBC).
I punti dello spazio reciproco (i vettori k) in corrispondenza dei quali si haun massimo della banda di valenza e un minimo di quella di conduzione, e quindiun minimo del gap energetico fra le due bande, vengono detti punti di Dirac.
Il grafico precedente mostra la struttura delle bande energetiche in funzionidei vettori k che risiedono nella prima cella dello spazio reciproco. Riporto ora igrafici delle medesime bande graficate pero su una cella di medesime dimensionima traslata nelle due direzioni del vettore k di meta delle rispettive ampiezze dellacella nelle due direzioni. Questo per osservare meglio il ripiegamento delle bande.I risultati sono riportati nelle figure 5.2, 5.3.
Figura 5.2: Le quattro bande del grafene in funzione dei vettori k nella nuova celladello spazio reciproco considerata. La cella e solo traslata rispetto alla precedente,le dimensioni sono le stesse. Le energie sono divise per il modulo di β al fine dimettere in evidenza i gap energetici.

CAPITOLO 5. MODELLO TIGHT-BINDING 17
Figura 5.3: Le quattro bande del Nitruro di Boro in funzione dei vettori k nellanuova cella dello spazio reciproco considerata. La cella e solo traslata rispetto allaprecedente, le dimensioni sono le stesse. Le energie sono divise per il modulo di βal fine di mettere in evidenza i gap energetici.
Una rotazione delle figure metterebbe in evidenza come i gap energetici minimisiano uguali al modulo della differenza tra i valori dei parametri α1 e α2 scelti, equindi che il gap si puo azzerare nel grafene (α1 = α2, quindi semimetallo) e nonpuo azzerarsi nel Nitruro di Boro ( α1 6= α2, quindi isolante) .
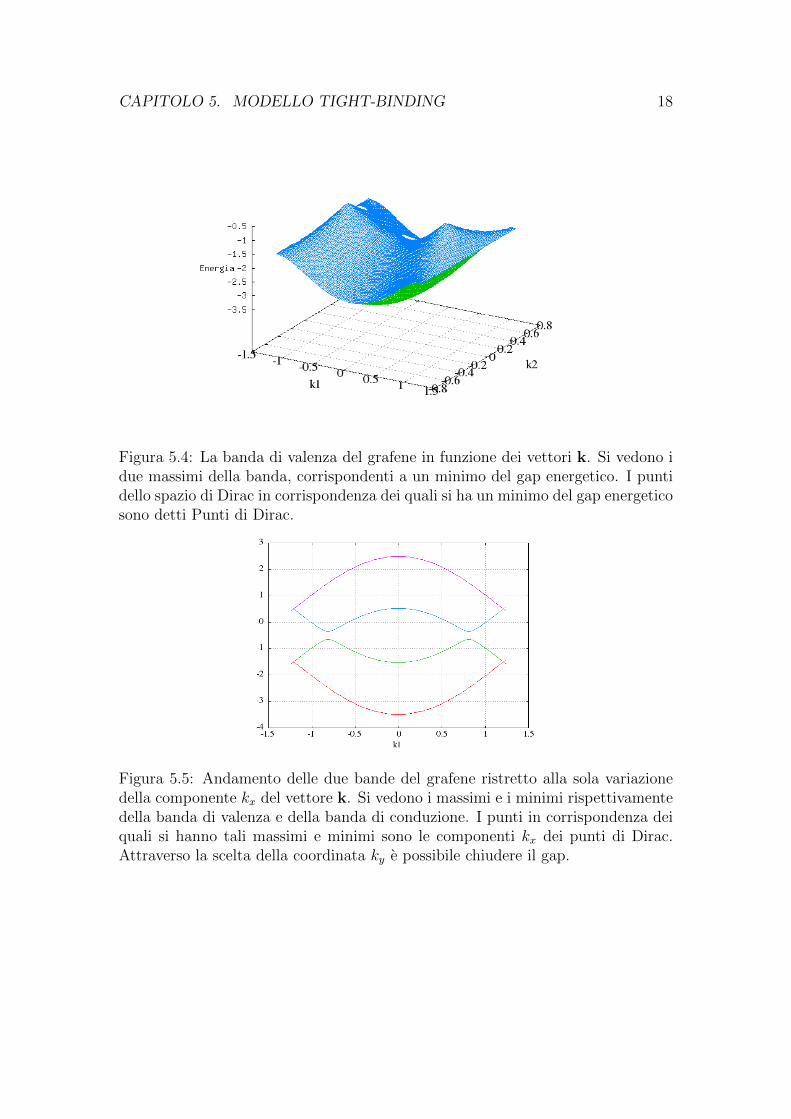
Per mettere in evidenza il fatto che esistono punti dello spazio reciproco incorrispondenza dei quali si ha un massimo della banda di valenza con conseguenteminimo del gap energetico riporto nella figura 5.4 la sola banda di valenza chemette in evidenza il massimo di questa e quindi i cosiddetti punti di Dirac.
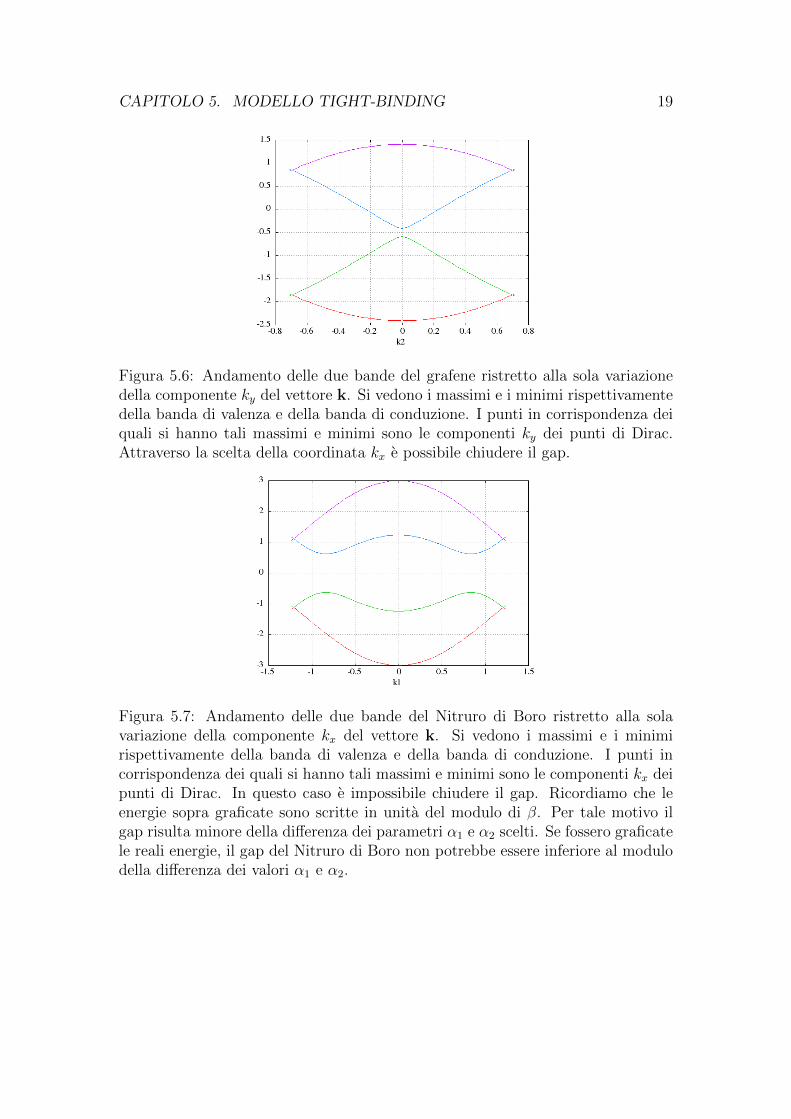
Interessante, per vedere l’effettivo andamento delle bande energetiche in fun-zione di k e ancora per vedere i punti i Dirac, e graficare l’andamento delle bandein funzione prima dei soli valori della componente kx e poi della sola componenteky. Tali grafici sono quelli riportati in figure 5.5, 5.6, 5.7, 5.8.
Abbiamo quindi cosı ottenuto e analizzato la struttura delle bande elettronichedel grafene e del Nitruro di Boro. Le bande trovate, comunque, corrispondono ailivelli energetici permessi di singola particella. Per ottenere l’energia totale diground-state del sistema costituito dai 4 ×M1 ×M2 elettroni, bisogna sommarele energie relative agli stati di singola particella occupati degli elettroni, mentre lostato di ground-state e uguale al determinante di Slater degli autostati di singolaparticella occupati (gli autostati di Bloch).
CAPITOLO 5. MODELLO TIGHT-BINDING 18
Figura 5.4: La banda di valenza del grafene in funzione dei vettori k. Si vedono idue massimi della banda, corrispondenti a un minimo del gap energetico. I puntidello spazio di Dirac in corrispondenza dei quali si ha un minimo del gap energeticosono detti Punti di Dirac.
Figura 5.5: Andamento delle due bande del grafene ristretto alla sola variazionedella componente kx del vettore k. Si vedono i massimi e i minimi rispettivamentedella banda di valenza e della banda di conduzione. I punti in corrispondenza deiquali si hanno tali massimi e minimi sono le componenti kx dei punti di Dirac.Attraverso la scelta della coordinata ky e possibile chiudere il gap.
CAPITOLO 5. MODELLO TIGHT-BINDING 19
Figura 5.6: Andamento delle due bande del grafene ristretto alla sola variazionedella componente ky del vettore k. Si vedono i massimi e i minimi rispettivamentedella banda di valenza e della banda di conduzione. I punti in corrispondenza deiquali si hanno tali massimi e minimi sono le componenti ky dei punti di Dirac.Attraverso la scelta della coordinata kx e possibile chiudere il gap.
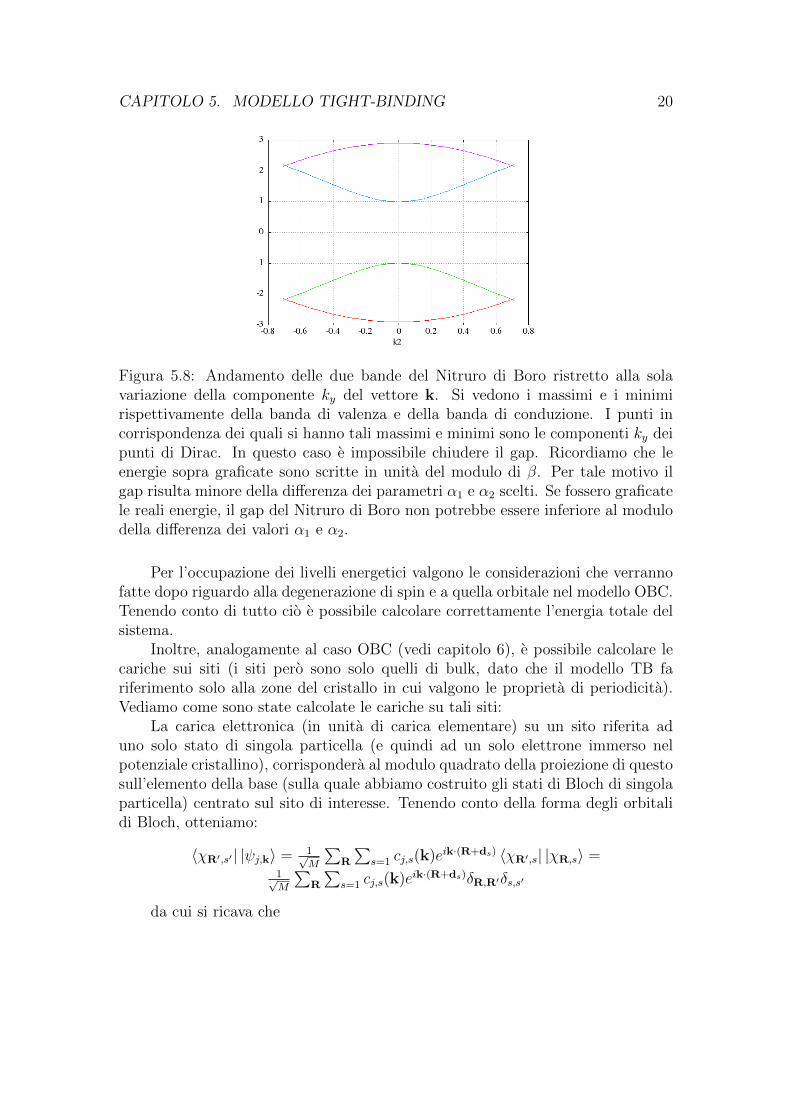
Figura 5.7: Andamento delle due bande del Nitruro di Boro ristretto alla solavariazione della componente kx del vettore k. Si vedono i massimi e i minimirispettivamente della banda di valenza e della banda di conduzione. I punti incorrispondenza dei quali si hanno tali massimi e minimi sono le componenti kx deipunti di Dirac. In questo caso e impossibile chiudere il gap. Ricordiamo che leenergie sopra graficate sono scritte in unita del modulo di β. Per tale motivo ilgap risulta minore della differenza dei parametri α1 e α2 scelti. Se fossero graficatele reali energie, il gap del Nitruro di Boro non potrebbe essere inferiore al modulodella differenza dei valori α1 e α2.
CAPITOLO 5. MODELLO TIGHT-BINDING 20
Figura 5.8: Andamento delle due bande del Nitruro di Boro ristretto alla solavariazione della componente ky del vettore k. Si vedono i massimi e i minimirispettivamente della banda di valenza e della banda di conduzione. I punti incorrispondenza dei quali si hanno tali massimi e minimi sono le componenti ky deipunti di Dirac. In questo caso e impossibile chiudere il gap. Ricordiamo che leenergie sopra graficate sono scritte in unita del modulo di β. Per tale motivo ilgap risulta minore della differenza dei parametri α1 e α2 scelti. Se fossero graficatele reali energie, il gap del Nitruro di Boro non potrebbe essere inferiore al modulodella differenza dei valori α1 e α2.
Per l’occupazione dei livelli energetici valgono le considerazioni che verrannofatte dopo riguardo alla degenerazione di spin e a quella orbitale nel modello OBC.Tenendo conto di tutto cio e possibile calcolare correttamente l’energia totale delsistema.
Inoltre, analogamente al caso OBC (vedi capitolo 6), e possibile calcolare lecariche sui siti (i siti pero sono solo quelli di bulk, dato che il modello TB fariferimento solo alla zone del cristallo in cui valgono le proprieta di periodicita).Vediamo come sono state calcolate le cariche su tali siti:
La carica elettronica (in unita di carica elementare) su un sito riferita aduno solo stato di singola particella (e quindi ad un solo elettrone immerso nelpotenziale cristallino), corrispondera al modulo quadrato della proiezione di questosull’elemento della base (sulla quale abbiamo costruito gli stati di Bloch di singolaparticella) centrato sul sito di interesse. Tenendo conto della forma degli orbitalidi Bloch, otteniamo:
〈χR′,s′ | |ψj,k〉 = 1√M
∑R
∑s=1 cj,s(k)eik·(R+ds) 〈χR′,s| |χR,s〉 =
1√M
∑R
∑s=1 cj,s(k)eik·(R+ds)δR,R′δs,s′
da cui si ricava che

CAPITOLO 5. MODELLO TIGHT-BINDING 21
〈χR′,s′ | |ψj,k〉 = 1√Mcj,s′(k)eik·(R
′+ds′ )
e dunque la carica elettronica su tale sito riferita ad un solo stato di Blochsingolarmente occupato e
qs′ = 1M|cj,s(k)|2
Dove qs′ e la carica elettronica sul sito individuato del vettore (R′ + ds′) nelcaso in cui un solo elettrone sia presente nel reticolo (singola particella immersanel potenziale cristallino).
Tenendo pero conto che ci sono N = 4 ×M1 ×M2 elettroni nel sistema, eche per il BN c’e un gap energetico (quindi non c’e degenerazione) e consideran-do la degenerazione di Spin, il determinante di Slater e uguale al prodotto degliSpin-Orbitali riferiti alle prime due bande e a ogni possibile valore di k. Quindi,ricordando che la carica e estensiva, per avere la carica elettronica totale su un sitodovremo sommare le cariche su tale sito riferite agli spin-orbitali occupati. Cioe:
Qs′ = 2M
∑k
∑j=1,2 |cj,s(k)|2
dove Qs e la carica elettronica totale sul sito individuato dal vettore (R′+ds′),il 2 deriva dalla degenerazione di spin e il j e l’indice che labella le bande (che nellasommatoria va quindi da 1 a 2, data la degenerazione di spin). Una sommatoriaviene fatta sui possibili vettori k dello spazio reciproco, l’altra sulle prime duebande. Quindi la somma totale viene fatta su 2×M1 ×M2 elementi, moltiplicatiper due a causa della degenerazione di spin.
Una veloce analisi dei risultati ottenuti per le cariche sui siti del Nitruro diBoro implementando la formula appena vista mostra che le cariche elettronichesui siti, come ci attendiamo, sono uguali a coppie nella cella (con una precisione di10−16), cioe uguali per gli atomi uguali. Inoltre la media aritmetica delle caricheelettroniche di due siti diversi fa uno (con una precisione di 10−16). Aggiungendoquindi ad ogni sito le cariche protoniche (una per sito) , ogni sito risulta avere unacarica netta e tali cariche risultano essere tutte uguali in modulo, e a segni alternati(segni uguali su siti uguali). Inoltre, come ci si attende, tali cariche risultano esserequasi indipendenti dalle dimensioni del cristallo, cioe dopo pochissime celle il valoredi tali cariche nette converge ad un determinato valore. Ogni cella risulta quindiavere due anioni e due cationi e ogni cella risulta complessivamente neutra (il che cida la conservazione della carica). Vediamo, nei grafici 5.9, 5.10, 5.11, 5.12, valori eandamento delle cariche nette sui siti di bulk rispetto al numero di celle costituentiil cristallo di Nitruro di Boro, calcolate con metodo TB.
Dai grafici riportati si vede subito come le cariche nette sui siti tendano tutteallo stesso valore (assoluto), e come gli andamenti e i segni delle cariche sianouguali a coppie. Gli andamenti sono cioe uguali sui siti uguali. Abbiamo quindidue anioni e due cationi per cella, con cariche nette uguali in modulo ed oppostein segno.
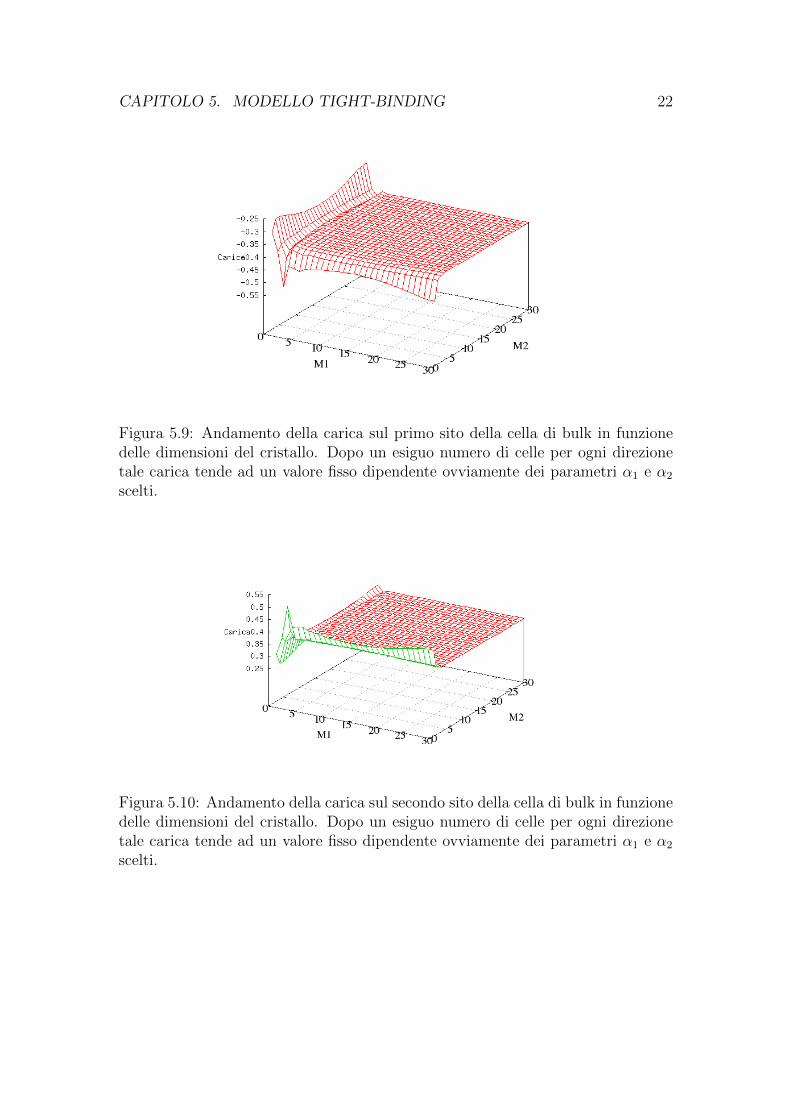
CAPITOLO 5. MODELLO TIGHT-BINDING 22
Figura 5.9: Andamento della carica sul primo sito della cella di bulk in funzionedelle dimensioni del cristallo. Dopo un esiguo numero di celle per ogni direzionetale carica tende ad un valore fisso dipendente ovviamente dei parametri α1 e α2
scelti.
Figura 5.10: Andamento della carica sul secondo sito della cella di bulk in funzionedelle dimensioni del cristallo. Dopo un esiguo numero di celle per ogni direzionetale carica tende ad un valore fisso dipendente ovviamente dei parametri α1 e α2
scelti.

CAPITOLO 5. MODELLO TIGHT-BINDING 23
Figura 5.11: Andamento della carica sul terzo sito della cella di bulk in funzionedelle dimensioni del cristallo. Dopo un esiguo numero di celle per ogni direzionetale carica tende ad un valore fisso dipendente ovviamente dei parametri α1 e α2
scelti. Si nota subito come l’andamento sia identico a quello del primo sito.
Figura 5.12: Andamento della carica sul quarto sito della cella di bulk in funzionedelle dimensioni del cristallo. Dopo un esiguo numero di celle per ogni direzionetale carica tende ad un valore fisso dipendente ovviamente dei parametri α1 e α2
scelti. Si nota immediatamente come l’andamento sia identico a quello del secondosito.

Capitolo 6
Descrizione in modello OBC
Consideriamo ora il cristallo come una macromolecola. Questo equivale a impor-re al sistema le cosiddette condizioni al contorno di Open Boundary Condition(OBC), le quali richiedono che gli stati legati siano a quadrato sommabile su R3,cioe che il valore del loro integrale sullo spazio reale sia finito.
Useremo tale modello per due macromolecole di Nitruro di Boro leggermentediverse fra loro, considereremo cioe due macromolecole quasi identiche, se non perla differenza di un bordo di queste.
Cercheremo quindi di descrivere il sistema attraverso il metodo LCAO (comesi fa con le molecole appunto), per il quale non si tiene conto della periodicita dellastruttura. Per facilitare il tutto pero descriveremo la struttura con il formalismodella cristallografia, cosa che ci aiutera a scrivere la matrice Hamiltoniana e acalcolarne quindi autovalori e autovettori, nostro primissimo obiettivo per giungereal fine ultimo.
Nella descrizione assumiamo che ogni atomo contribuisca con un elettrone π eusiamo come base l’insieme di autostati π di singola particella riferiti ad ogni singo-lo atomo, che considereremo ortonormali fra loro ( e costituenti una base appunto).Indichiamo tale base con la notazione {|χi〉}. La matrice di sovrapposizione fratali stati risulta quindi essere la matrice unita. Dobbiamo ora costruire la matriceHamiltoniana su tali stati. Per fare cio si usa l’approssimazione ai primi vicini,approssimazione per cui le uniche interazioni reciproche tra i siti considerati sonoquelle tra i medesimi siti e i siti adiacenti. Esprimendo tutto cio matematicamente,si ottiene:
〈χi| H |χi′〉 = αiδii′ + βδp.v
e quindi le matrici che descrivono le macromolecole rispettivamente di grafenee Nitruro di Boro sono della forma:
24

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 25α β 0 · · · · · · · · ·β α β · · · · · · · · ·0 β α β · · ·...
......
. . .
0 · · · · · · β α
e
α1 β′ 0 · · · · · · · · ·β′ α2 β′ · · · · · · · · ·0 β′ α1 β′ · · ·...
......
. . .
0 · · · · · · β′ α2
Vediamo ora le differenze fra le due configurazioni di macromolecola consi-
derate e analizziamole quindi distintamente. (Il procedimento generale sara con-cettualmente uguale per le due macromolecole, ci saranno solo alcune differenzequantitative.)
La prima configurazione e quella di una macromolecola formata da (M1×M2)celle, ognuna contenente quattro atomi individuati dai vettori di base visti prima.Ricordiamo che M1 e M2 sono rispettivamente il numero di celle nella direzione xe nella direzione y . La macromolecola risulta quindi essere della seguente forma :
Figura 6.1: Macromolecola di BN asimmetrica. I siti sono colorati di diversi coloriper metterne in evidenza la diversita fra di essi. Quelli in blu vedremo essere glianioni, quelli in rosso i cationi. Il cristallo di grafene e identico, con la differenzache i siti sono tutti uguali fra loro.
E stata quindi calcolata la matrice Hamiltoniana sui siti appartenenti a taleconfigurazione ed e quindi stata eseguita la diagonalizzazione di questa (la forma

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 26
della matrice e quella vista prima) al fine di trovare gli autovalori e gli autovettori.Tutto cio e stato fatto sia per il caso del grafene che per quello del BN (Nitru-ro di Boro). Nel modello LCAO, l’insieme degli autovalori corrispondono al setdi energie di singola particella concesse, quindi lo spettro energetico di singolaparticella, mentre gli autovettori corrispondono di conseguenza agli autostati disingola particella riferiti ai rispettivi valori energetici (gli autovalori). Il sistemanon e pero costituito da una singola particella (elettrone in questo caso), ma dapiu particelle. Piu precisamente, ricordando che per ogni cella abbiamo quattroatomi e che ognuno di questi contribuisce con un elettrone, abbiamo un totale diN = (4 ×M1 ×M2) elettroni. Precisiamo ora che siamo interessanti al ground-state del sistema, cioe allo stato di minima energia. Avendo a che fare con unsistema a piu corpi, per descrivere quest’ultimo dobbiamo costruire lo stato diground-state come determinante di Slater degli autostati di singola particella delsistema occupati. Per fare cio dobbiamo capire prima quali siano gli stati effetti-vamente occupati. Inoltre dobbiamo tenere conto che, essendo il livello energeticodi ground-state pari alla somma dei livelli energetici occupati dalle singole parti-celle, per avere il livello piu basso dobbiamo riempire gli autostati riferiti ai livellienergetici di singola particella piu bassi. Bisogna pero tenere conto anche dellamolteplicita di spin, ossia che ogni autostato di tipo spaziale puo essere doppia-mente occupato, con un elettrone avente spin up e con un elettrone avente spindown. Fatte tali considerazioni, traiamo quindi la conclusione che, nel caso incui i livelli di singola particella non siano degeneri, gli stati di singola particellaoccupati sono i primi N/2 = (2 ×M1 ×M2). Se invece esistono livelli degeneri,bisogna riempire i livelli con occupazione frazionaria. Cioe i livelli si riempionocon occupazione 2 se non sono degeneri, 1 se sono doppiamente degeneri, 1
2se sono
quattro volte degeneri e cosı via.Queste osservazioni saranno utili per riempire correttamente i livelli energetici
di singola particella e quindi gli stati al fine di un corretto calcolo dell’energiatotale, sia nel grafene che nel Nitruro di Boro. Come vedremo sono necessarie diconseguenza anche per il corretto calcolo delle cariche sui siti, obiettivo principaledella simulazione proposta.
Analizziamo quindi i due spettri energetici di singola particella, quello delgrafene e quello del BN.
Ricordiamo che si definisce livello di Fermi il livello energetico di singolaparticella piu alto occupato in un sistema di fermioni (elettroni in questo caso).
Una veloce analisi dello spettro energetico del grafene mostra come i livellisiano densi fra loro. I livelli energetici risultano non degeneri fino a che non sigiunge in prossimita del livello di Fermi. In corrispondenza di questi esistonolivelli degeneri. Tali livelli vanno riempiti quindi, come gia detto, con occupazionefrazionaria. Si vede inoltre come non esista un gap energetico tra i livelli precedenti

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 27
a quello di Fermi e quelli successivi. Questa mancanza di gap energetico motivail carattere di semimetallo del grafene. Riotteniamo quindi quanto visto con ilmodello Tight-Binding. Per il corretto calcolo dell’energia di ground-state di talesistema si e quindi tenuto conto della degenerazione per occupare i livelli.
Nello spettro del Nitruro di Boro, invece, risultano assenti livelli degeneri.Quindi ogni livello e occupato con occupazione 2, e l’energia totale viene calcolatacome somma dei primi (2×M1×M2) livelli energetici di singola particella (ognunomoltiplicato per 2, data la degenerazione di spin). Inoltre e interessante notarecome esista un gap energetico tra il livello di Fermi e il livello energetico successivo.Cioe il modulo della differenza tra il valore energetico del livello di Fermi e quellosuccessivo e finita, e per la precisione vale esattamente quanto il modulo delladifferenza tra i valori α1 e α2 (con una precisione di 10−15 nella nostra simulazione).Questo spiega il carattere isolante del BN. Confermiamo dunque, anche in questocaso, quanto visto con il modello TB.
Passiamo ora al calcolo delle cariche sui siti di nostro interesse. Ricordiamoche ogni autovettore (autostato di singola particella) e la combinazione linearedegli elementi della base {|χi〉}, citati in precedenza, cioe:
|ψ〉 =∑N
i=1 ci |χi〉
Dove N e il numero di elementi della base usata per costruire la matrice equindi gli autovettori. N corrisponde al numero totale di particelle (elettroni). Ici corrispondono alle proiezioni dell’autostato sui rispettivi elementi della base acui si riferiscono.
Ricordiamo inoltre che ogni elemento della base corrisponde a un autostatorelativo al rispettivo atomo isolato, e che quindi risulta essere centrato su questo(Stiamo vedendo quindi queste funzioni corrispondenti agli elementi della basecome delle delta di Dirac centrate sui siti corrispondenti ai nuclei degli atomi).
Quindi se noi proiettiamo il singolo autostato (quindi di singola particella)dell’Hamiltoniana riferita alla macromolecola considerata sui singoli elementi del-la base, per definizione di base ortonormale, otteniamo i rispettivi coefficienti diquesti nella rappresentazione dell’autovettore vista prima. Otteniamo cioe le pro-iezioni dell’autovettore sui singoli elementi della base. Come noto dalla teoriaquantistica, il modulo quadro del singolo coefficiente relativo a un determinatoelemento della base, equivale alla probabilita che il sistema fisico preso in consi-derazione si trovi esattamente in quello stato quantistico (l’elemento della baseconsiderato in questo caso). Dato poi che in questo caso gli elementi della base,come detto, risultano essere funzioni centrate nei rispettivi siti del cristallo, taleprobabilita corrisponde alla probabilita che la singola particella (elettrone) si troviin corrispondenza del sito stesso considerato. (Ricordiamo che gli elementi dellabase sono esattamente quanti i siti)

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 28
Non va dimenticato pero che noi dobbiamo considerare tutti gli N elettronipresenti nel cristallo, e che lo stato che descrive tale sistema a piu corpi e ugualeal determinante di Slater degli autostati di singola particella occupati. La carica euna grandezza estensiva, quindi per ottenere la carica presente sul singolo sito in unsistema di N elettroni bisogna sommare i moduli quadri dei coefficienti (le cariche)relativi a tale sito degli N spin-orbitali di singola particella occupati. Come prima,per fare il calcolo corretto, bisogna tenere bene conto di quali siano effettivamentegli N stati occupati. Occupando quindi correttamente gli stati come fatto prima,otteniamo i seguenti risultati.
Per il grafene, se il cristallo e sufficientemente grande (quando cioe risultanodiventare trascurabili gli effetti di bordo nelle proprieta di bulk) le cariche elet-troniche all’interno del cristallo (bulk) risultano essere unitarie (in unita di caricaelementare) in ogni sito, con una precisione di 10−16. Tenendo conto anche dellacariche nucleari (una carica elementare positiva per atomo) ogni sito risulta essereelettricamente neutro. Di conseguenza risulta essere neutra ogni cella all’internodel cristallo. Anche ai bordi, per il grafene, le cariche elettriche sui siti risultanoessere unitarie, sempre in unita di carica elementare. Lo stesso accade ai vertici.Quindi, prendendo di nuovo in considerazione anche le cariche nucleari, l’interocristallo (bulk, bordi e vertici) risulta essere neutro in ogni suo sito. Non esistonoquindi cariche nette sui siti del cristallo. La nostra concentrazione va in particolarmodo ai vertici a ai bordi. Come appena visto, nel grafene, ogni sito atomicorisulta essere neutro e non esistono quindi cariche nette. Questo ci permette diconcludere che per il grafene non e permessa ne polarizzazione interna (essendoogni sito neutro) ne polarizzazione di bordo (o polarizzazione di superficie, ma nelnostro caso, essendo il cristallo bidimensionale, la “superficie” corrisponde a unlato, una lunghezza, cioe il bordo del cristallo).
Molto diversi, invece, sono i risultati nel caso del Nitruro di Boro. Nella zonadi bulk del cristallo, i siti risultano avere carica elettrica (in unita di carica ele-mentare) diversa da uno. Piu precisamente le cariche elettroniche risultano ugualia coppie (prevedibile avendo quattro atomi per cella, uguali a coppie, e che pre-sentano lo stesso numero di atomi vicini), con valori dipendenti dai parametri α1 eα2 scelti. Facendo la media aritmetica delle cariche elettroniche su due siti diversi,si vede che questa fa uno (con una precisione di 10−7 almeno), per entrambe lemedie fatte. Quindi, se sommiamo ad ogni sito la carica protonica (del nucleo)otteniamo che in ogni cella sono presenti, in modo alternato, cariche positive e ne-gative uguali in modulo, cioe due anioni e due cationi. Ogni sito non risulta quindipiu neutro, ma ci troviamo in presenza di anioni e cationi. Rimane pero neutraogni singola cella (ricordiamo che le medie aritmetiche delle cariche elettroniche didue siti diversi fa uno e che la somma delle cariche elettroniche sui quattro siti faquattro), nel bulk del cristallo. E possibile inoltre vedere come evolve il valore delle

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 29
cariche nette sui siti nel bulk del cristallo in funzione del numero di celle (e quindidelle dimensioni del cristallo). Queste convergono rapidamente ad un determinatovalore. Le differenze si hanno solo per piccole dimensioni del cristallo. Possiamoquindi concludere che, per cristalli sufficientemente grandi, le cariche presenti suisiti di bulk del cristallo sono indipendenti dalle dimensioni del cristallo stesso.
La cosa interessante pero avviene ai bordi e ai vertici. Infatti, sui siti relativial bordo e ai quattro vertici del cristallo, il valore delle cariche elettroniche presentisu tali siti subiscono delle deviazioni rispetto ai valori di bulk. Si hanno deviazionidell’ordine di 10−1. (tali deviazioni del valore della carica sui siti calano moltorapidamente spostandosi dai bordi verso il centro del cristallo, facendoci sapereche gli effetti di bordo diventano subito trascurabili sulle cariche sui siti interni,come visto sopra).
Tali deviazioni sono quindi significative. Si hanno inoltre quindi, tenendoconto anche delle cariche nucleari, delle cariche nette ai bordi e ai vertici, diverseda quelle nel bulk del cristallo.
E pero importantissimo notare un’altra cosa. La media aritmetica fra le cari-che elettroniche sui siti dei vertici inferiore sinistro e superiore sinistro fa uno (conprecisione di 10−15 se il cristallo e sufficientemente grande), e lo stesso accade perla media aritmetica fra le cariche dei siti dei vertici inferiore destro e superioredestro. Di conseguenza, se aggiungiamo ad ogni sito la carica nucleare, e facilevedere come le cariche nette sui vertici inferiore sinistro e superiore sinistro sianouguali in modulo (con la stessa precisione delle medie) ed opposte in segno. Diconseguenza medesima cosa accade fra le cariche nette dei vertici inferiore destroe superiore destro. Le due coppie di cariche sono pero diverse fra loro in modulo.
Importante e vedere l’andamento di tali cariche ai vertici in funzione delledimensioni del cristallo, cioe in funzione del numero di celle che costituisce ilcristallo stesso. Questo si vede nei grafici 6.2, 6.3, 6.4, 6.5 riportati.
Si nota che dopo poche celle le cariche ai vertici sembrano convergere ad undeterminato valore ed essere quindi poi indipendenti dalle dimensioni del cristallo.
Inoltre si vede come le cariche nette siano in modulo uguali a coppie e oppostein segno. Si nota cioe come la carica netta del vertice inferiore sinistro sia uguale inmodulo ed opposta in segno alla carica netta presente sul vertice superiore sinistroe come la stessa cosa accade fra le cariche dei vertici inferiore destro e superioredestro.
Nella figura 6.6 si vede meglio la configurazione elettrica che risulta avere ilNitruro di Boro.
In definitiva, possiamo trarre la conclusione che, a differenza del Grafene, ilNitruro di Boro presenta delle cariche nette ai bordi e ai vertici del cristallo, quasiindipendenti dalle dimensioni del cristallo, e che quindi puo presentare una pola-rizzazione di bordo indipendentemente dalle dimensioni del cristallo. Tali cariche
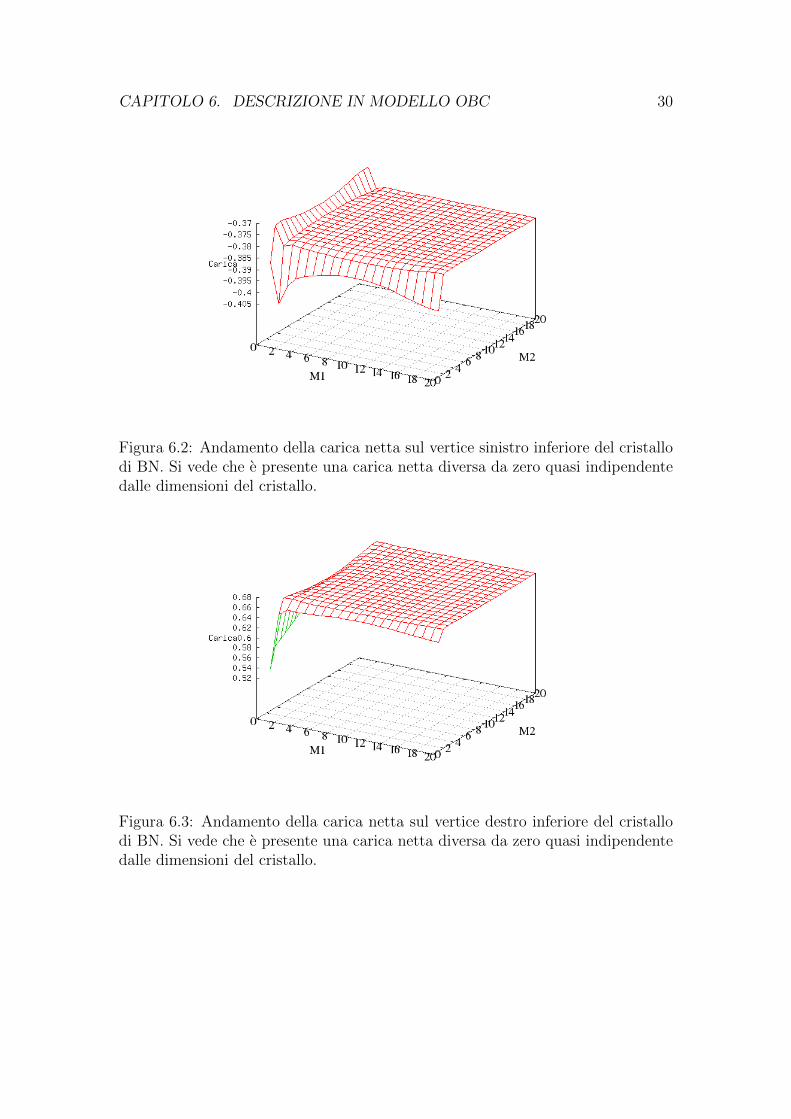
CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 30
Figura 6.2: Andamento della carica netta sul vertice sinistro inferiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo.
Figura 6.3: Andamento della carica netta sul vertice destro inferiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo.
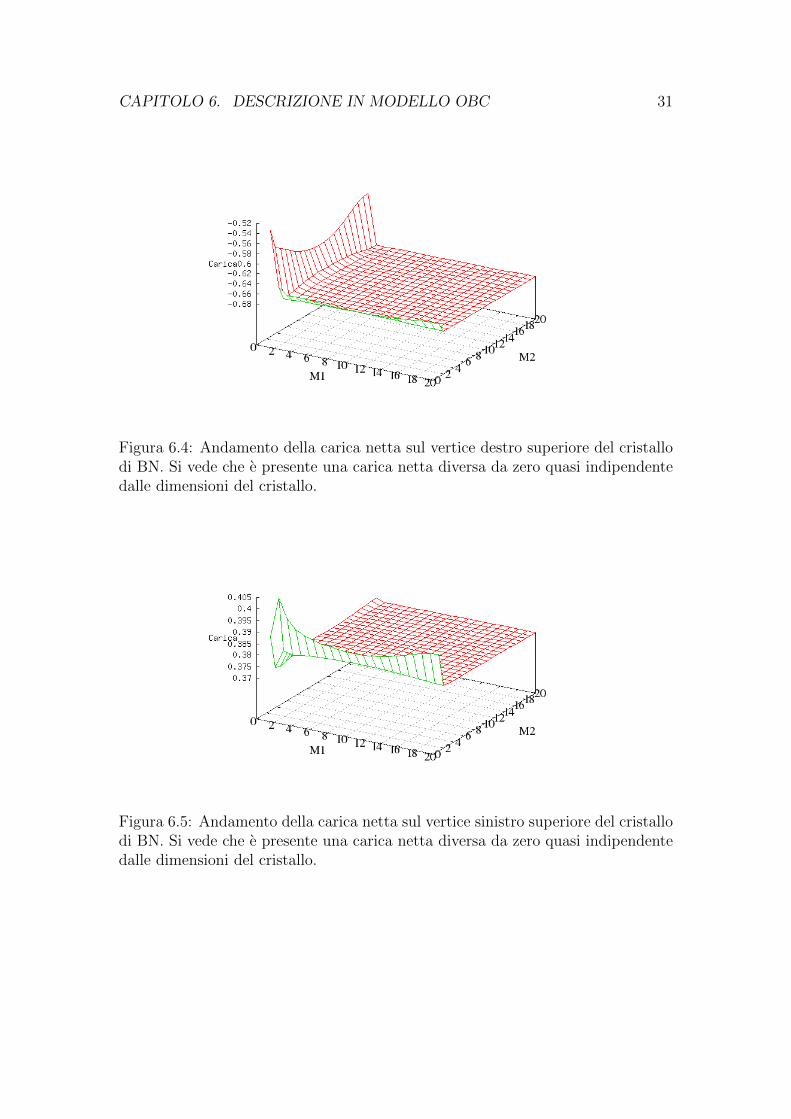
CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 31
Figura 6.4: Andamento della carica netta sul vertice destro superiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo.
Figura 6.5: Andamento della carica netta sul vertice sinistro superiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo.

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 32
Figura 6.6: Macromolecola di BN asimmetrica con rappresentazione delle carichenette presenti ai vertici di questa.
e la possibile polarizzazione si presenta dunque anche nel limite termodinamico.Consideriamo ora la seconda macromolecola e andiamo ad analizzare le me-
desime grandezze viste per la macromolecola precedente.In figura 6.7 e riportata la forma di tale macromolecola.Come si nota subito, questa nuova macromolecola risulta essere uguale alla
precedente con l’aggiunta di un’ulteriore catena di atomi a destra. L’aggiunta ditale catena rende la macromolecola simmetrica in direzione x.
Come prima dobbiamo costruire la matrice Hamiltoniana. I siti non sono piu(4 ×M1 ×M2), ma bensı (4 × (M1 ×M2) + 2 ×M2). Queste saranno quindi lenuove dimensioni della matrice Hamiltoniana.
Procediamo quindi come prima. Costruiamo cioe la matrice Hamiltonina ediagonalizziamola, ottenendo cosı autovalori (energie) e autovettori di singola par-ticella. Una veloce analisi degli spettri mostra, come nel caso precedente, l’assenzadi un gap energetico nello spettro di singola particella del grafene (semimetallodunque), e l’esistenza invece di un gap energetico nello spettro del Nitruro di Boro(isolante quindi).
Analogamente al caso precedente, nel grafene, il livello di Fermi risulta esseredegenere con quelli successivi. Per fare quindi i calcoli giusti va considerata lacorretta occupazione frazionaria di ogni livello. Per il Nitruro di Boro invece, comedetto, c’e il gap, di entita pari al modulo della differenza dei parametri α1 e α2, eil livello di Fermi non e quindi degenere con i successivi. Di conseguenza i livellioccupati nel BN sono i primi (2× (M1×M2) +M2), ognuno con occupazione 2 (ladegenerazione di spin). Tali considerazioni sulla diversa occupazione vanno tenute

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 33
Figura 6.7: Macromolecola di BN simmetrica. I siti sono colorati di diversi coloriper metterne in evidenza la diversita fra di essi. Quelli in blu vedremo essere glianioni, quelli in rosso i cationi. Il cristallo di grafene e identico, con la differenzache i siti sono tutti uguali fra loro.
conto, come gia detto, anche per il corretto calcolo delle cariche elettroniche (e poidi quelle totali) su ogni sito del cristallo. Il metodo di calcolo e identico a quellodescritto per la precedente macromolecola, cambia solo il numero di particelle(elettroni) presenti (4× (M1×M2) + 2×M2 al posto di 4×M1×M2) nel cristalloe quindi il numero di diversi livelli energetici occupati.
I risultati sono i seguenti:• per il grafene le cariche elettroniche su ogni sito (bulk, bordi e vertici)
risultano essere unitarie (con una precisione che va da 10−11 a 10−15). Quinditenendo conto anche delle cariche nucleari, ogni sito risulta essere neutro, cosı comerisulta quindi essere neutro l’intero cristallo. Come per la molecola precedente none quindi ammessa ne polarizzazione interna ne polarizzazione di bordo.• per il Nitruro di Boro, nel bulk le cariche elettroniche risultano essere uguali
a coppie, cioe atomi uguali presentano cariche elettroniche uguali. Inoltre le mediearitmetiche fra le cariche elettroniche su siti diversi fa uno, con una precisionedi 10−7 o 10−8 se il cristallo e sufficientemente grande. Sommando ad ogni sitoanche le cariche protoniche dei nuclei, concludiamo che al centro del cristallo sonopresenti cariche nette diverse da zero e precisamente ci sono anioni e cationi, cioeioni con cariche uguali in modulo e opposte in segno. Ogni sito risulta quindicarico, ma ogni cella del bulk risulta complessivamente neutra. Ai vertici e aibordi avviene qualcosa di diverso e molto interessante. Le cariche ai bordi sidiscostano da quelle di bulk, di una quantita dell’ordine di 10−1. Mentre ai vertici

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 34
le cariche elettroniche si discostano di un valore dell’ordine di 10−2. Tali caricheelettroniche ai vertici risultano essere uguali a coppie. Risultano essere uguali fraloro le cariche elettroniche presenti sui due vertici inferiori, e risultano uguali fraloro le cariche elettroniche presenti sui vertici superiori del cristallo. Inoltre, lamedia aritmetica fra le cariche elettroniche su un vertice inferiore e su un verticesuperiore (per entrambe le coppie di vertici, dato che poi c’e simmetria) fannouno con una precisione di 10−16. Di conseguenza, sommando ancora una volta lecariche nucleari, le cariche nette su un vertice inferiore e su un vertice superiore,sono uguali in modulo ed opposte in segno.
Concludiamo quindi che ai vertici di questa macromolecola sono presenti dellecariche nette, e queste sono tutte uguali fra loro in modulo ed opposte in segno.Precisamente sono presenti due cariche negative ai vertici inferiori e due carichepositive ai vertici superiori.
Preciso che il segno delle cariche si inverte (ma il modulo rimane uguale) seinvertiamo fra loro i valori dei parametri α1 e α2. Inoltre notiamo che l’entita dellecariche elettroniche, e quindi delle cariche nette su ogni sito (cioe la carica degliioni) e dipendente dagli effettivi valori numerici dei parametri α1 e α2 scelti.
Anche in questa configurazione e quindi ammessa polarizzazione di bordo.Anche in questo caso e interessante vedere come l’entita di tali cariche presenti
ai vertici dipenda dalle dimensioni del cristallo (le cariche nel bulk, se il cristallo esufficientemente grande, sono praticamente indipendenti dalle dimensioni di que-sto, quindi per cristalli sufficientemente grandi diventano trascurabili gli effetti dibordo sulle cariche nel bulk). Si scopre che, dopo un esiguo numero di celle, lecariche elettroniche (e quindi le cariche nette) ai vertici tendono ad un valore fisso.
La carica ai vertici quindi, se il cristallo e sufficientemente grande, risul-ta essere indipendente dalle dimensioni del cristallo stesso. Dunque, nel limitetermodinamico sono presenti cariche nette ai vertici del cristallo.
Nelle figura 6.8, 6.9, 6.10 e 6.11 sono illustrati gli andamenti appena discussi.Dai grafici e quindi visibile quanto detto in precedenza. Le cariche nette ai
vertici del cristallo risultano essere tutte uguali in modulo ed avere segno negativoai vertici inferiori e positivo ai vertici superiori. Inoltre tali cariche sono presentianche nel limite termodinamico. La configurazione di carica netta ai vertici delcristallo risulta essere quella di figura 6.12
Traiamo quindi la conclusione che esistono cariche nette ai vertici del cri-stallo, indipendentemente dalle dimensioni di questo. Dunque e ammissibile unapolarizzazione di bordo, anche nel limite termodinamico.
Precisiamo inoltre che che le cariche nette presenti sui siti del bordo sinistrosono uguali a quelle sui siti del bordo destro, mentre le cariche nette presenti suisiti del bordo inferiori sono uguali in modulo ed opposte in segno alle cariche nettepresenti sui siti del bordo superiore (tutto cio con una precisione maggiore di 10−8).

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 35
Figura 6.8: Andamento della carica netta sul vertice sinistro inferiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo.
Figura 6.9: Andamento della carica netta sul vertice destro inferiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo. Si vede inoltre come tale andamento sia identico aquello della carica netta sul vertice sinistro inferiore.
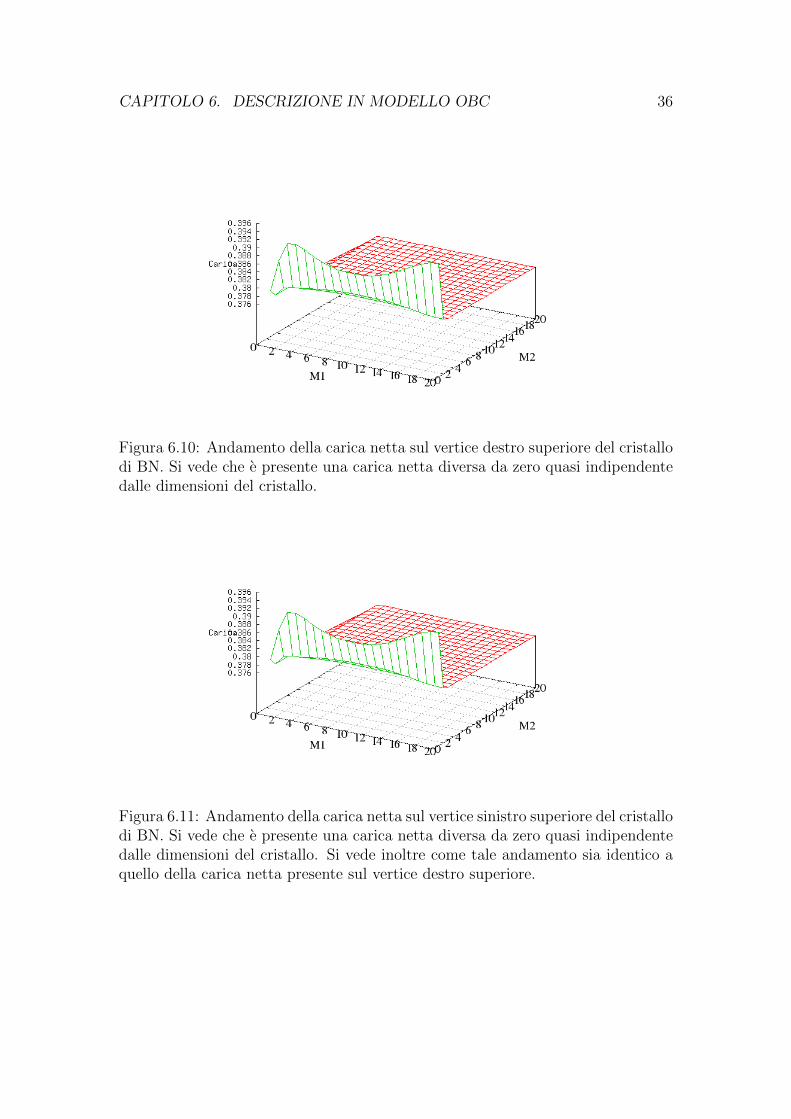
CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 36
Figura 6.10: Andamento della carica netta sul vertice destro superiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo.
Figura 6.11: Andamento della carica netta sul vertice sinistro superiore del cristallodi BN. Si vede che e presente una carica netta diversa da zero quasi indipendentedalle dimensioni del cristallo. Si vede inoltre come tale andamento sia identico aquello della carica netta presente sul vertice destro superiore.

CAPITOLO 6. DESCRIZIONE IN MODELLO OBC 37
Figura 6.12: Macromolecola di BN simmetrica con rappresentazione delle carichenette presenti ai vertici di questa.

Capitolo 7
Confronto tra modello OBC emodello TB
Cerchiamo ora di confrontare i risultati ottenuti per il modello OBC con quelliottenuti con il modello TB. Analizziamo ora solo il caso del Nitruro di Boro, chee quello che ora ci interessa maggiormente.
Iniziamo col confrontare gli spettri energetici. Ci interessa ora vedere se glispettri energetici dello stesso sistema calcolati con i due metodi ( OBC e TB)possano convergere uno all’altro e quindi equivalersi in qualche limite. Per la pre-cisione andiamo a vedere se i due modelli si equivalgono nel limite termodinamico.Per limite termodinamico si intende il limite in cui il numero N di particelle di unsistema fisico tenda ad infinito. Nel nostro caso, avendo il vincolo che ogni cellacontenga quattro elettroni, il limite equivale a fare tendere all’infinito le dimensio-ni del cristallo, e quindi il numero di celle. Facciamo cio perche vogliamo andarea vedere quali sono gli effetti di bordo sulle proprieta del cristallo, e se esiste unlimite in cui questi siano trascurabili.
Per fare questo studio facciamo innanzitutto un’osservazione. Ricordiamo chel’energia totale del sistema equivale alla somme delle energie di singola particellacorrispondenti agli stati di singola particella occupati. Se gli spettri energeticidi singola particella calcolati nei due modelli fossero uguali fra loro, dovrebberoessere uguali anche le energie totali, essendo somma delle energie di singola parti-cella. Quindi, per andare a studiare le differenze dei due spettri, possiamo usarele energie totali dei due sistemi: se in qualche limite queste sono uguali, allorapresumibilmente (in tale limite), lo saranno anche le energie di singola particella.
Ho quindi usato le energie totali e, per mettere in risalto i possibili effetti dibordo, ho considerato un’ulteriore grandezza, l’energia totale per cella. Ho cioediviso l’energia totale per il numero di celle M = M1×M2 del cristallo e analizzatoil suo andamento in funzione proprio del numero di celle.
38
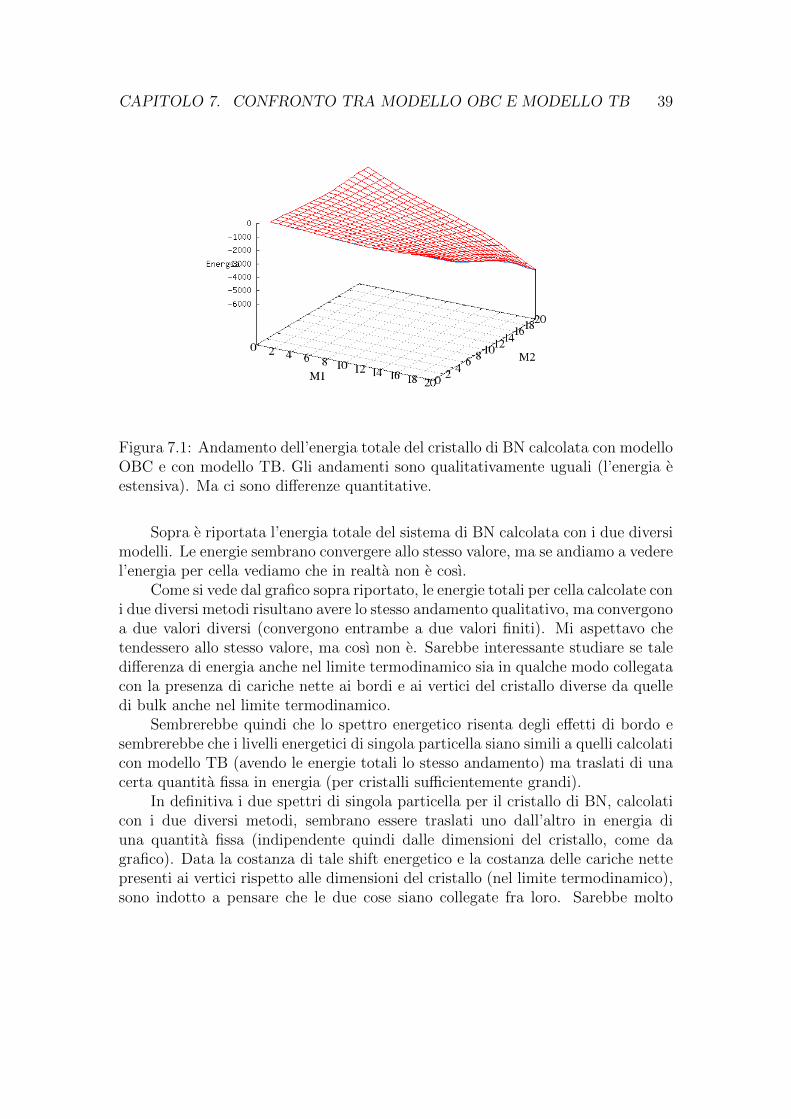
CAPITOLO 7. CONFRONTO TRA MODELLO OBC E MODELLO TB 39
Figura 7.1: Andamento dell’energia totale del cristallo di BN calcolata con modelloOBC e con modello TB. Gli andamenti sono qualitativamente uguali (l’energia eestensiva). Ma ci sono differenze quantitative.
Sopra e riportata l’energia totale del sistema di BN calcolata con i due diversimodelli. Le energie sembrano convergere allo stesso valore, ma se andiamo a vederel’energia per cella vediamo che in realta non e cosı.
Come si vede dal grafico sopra riportato, le energie totali per cella calcolate coni due diversi metodi risultano avere lo stesso andamento qualitativo, ma convergonoa due valori diversi (convergono entrambe a due valori finiti). Mi aspettavo chetendessero allo stesso valore, ma cosı non e. Sarebbe interessante studiare se taledifferenza di energia anche nel limite termodinamico sia in qualche modo collegatacon la presenza di cariche nette ai bordi e ai vertici del cristallo diverse da quelledi bulk anche nel limite termodinamico.
Sembrerebbe quindi che lo spettro energetico risenta degli effetti di bordo esembrerebbe che i livelli energetici di singola particella siano simili a quelli calcolaticon modello TB (avendo le energie totali lo stesso andamento) ma traslati di unacerta quantita fissa in energia (per cristalli sufficientemente grandi).
In definitiva i due spettri di singola particella per il cristallo di BN, calcolaticon i due diversi metodi, sembrano essere traslati uno dall’altro in energia diuna quantita fissa (indipendente quindi dalle dimensioni del cristallo, come dagrafico). Data la costanza di tale shift energetico e la costanza delle cariche nettepresenti ai vertici rispetto alle dimensioni del cristallo (nel limite termodinamico),sono indotto a pensare che le due cose siano collegate fra loro. Sarebbe molto
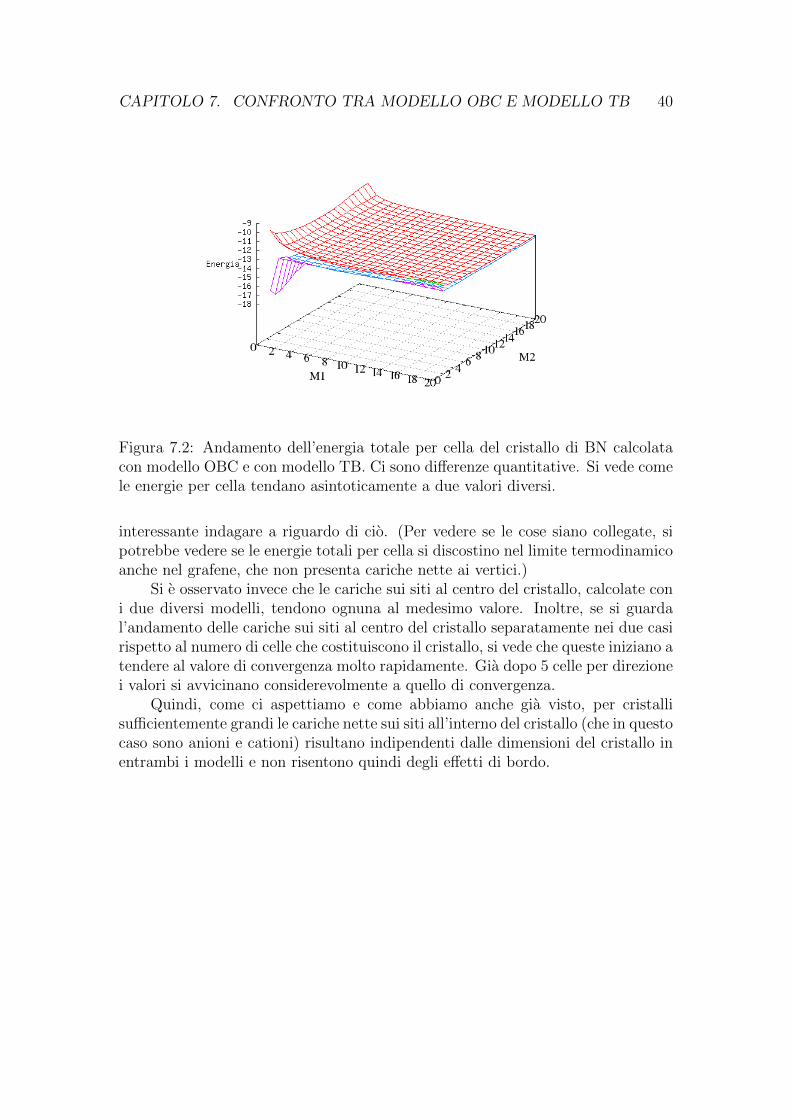
CAPITOLO 7. CONFRONTO TRA MODELLO OBC E MODELLO TB 40
Figura 7.2: Andamento dell’energia totale per cella del cristallo di BN calcolatacon modello OBC e con modello TB. Ci sono differenze quantitative. Si vede comele energie per cella tendano asintoticamente a due valori diversi.
interessante indagare a riguardo di cio. (Per vedere se le cose siano collegate, sipotrebbe vedere se le energie totali per cella si discostino nel limite termodinamicoanche nel grafene, che non presenta cariche nette ai vertici.)
Si e osservato invece che le cariche sui siti al centro del cristallo, calcolate coni due diversi modelli, tendono ognuna al medesimo valore. Inoltre, se si guardal’andamento delle cariche sui siti al centro del cristallo separatamente nei due casirispetto al numero di celle che costituiscono il cristallo, si vede che queste iniziano atendere al valore di convergenza molto rapidamente. Gia dopo 5 celle per direzionei valori si avvicinano considerevolmente a quello di convergenza.
Quindi, come ci aspettiamo e come abbiamo anche gia visto, per cristallisufficientemente grandi le cariche nette sui siti all’interno del cristallo (che in questocaso sono anioni e cationi) risultano indipendenti dalle dimensioni del cristallo inentrambi i modelli e non risentono quindi degli effetti di bordo.

Capitolo 8
Polarizzazione macroscopica e suolimite termodinamico
Andremo ora a studiare la polarizzazione macroscopica del cristallo, al fine distudiarne l’andamento al limite termodinamico e cercare eventuali collegamenti conla densita e quindi le cariche di bordo trovate in precedenza. Facciamo questo conla conoscenza preliminare che polarizzazione macroscopica e densita superficiale(nel nostro caso la densita e una densita lineare, essendo la superficie in realta unbordo) sono correlate fra esse. Piu precisamente sappiamo che vale la relazione:
σ = P · n
dove σ e la densita superficiale del solido, P la polarizzazione e n e il versorenormale alla superficie. Nel nostro caso specifico tale relazione si riduce a
λ = P · n
dove λ e la densita di carica lineare, P la polarizzazione (bidimensionale ora,che giace sul piano del flake) e n e il versore normale allo spigolo considerato. Siconstata subito che le quantita equivalgono dimensionalmente.
Come detto, vogliamo per prima cosa indagare sull’andamento della polariz-zazione rispetto alle dimensioni del cristallo. Facciamo prima delle considerazionipreliminari. Premettiamo intanto che facciamo questo calcolo solo per la secondamacromolecola vista prima, quella simmetrica in direzione x.
Noi calcoleremo la polarizzazione macroscopica del cristallo in modo semi-classico. Moltiplichiamo cioe le cariche nette trovate prima sui siti per le posizionidei rispettivi siti sui quali le cariche risultano localizzate. Otteniamo cosı il vettorebidimensionale richiesto. Prima di eseguire il calcolo ed analizzare i risultati,vediamo cosa ci aspettiamo a priori.
41

CAPITOLO 8. POLARIZZAZIONEMACROSCOPICA E SUO LIMITE TERMODINAMICO42
Nella direzione x il cristallo e simmetrico. Sapendo quindi che la polarizza-zione di un sistema globalmente neutro (quale e il nostro) e indipendente dall’ori-gine scelta, si trae la conclusione che la polarizzazione nella direzione x dovrebberisultare nulla.
Nella direzione y invece il cristallo non risulta totalmente simmetrico.Il cristallo risulta invece simmetrico in entrambe le direzioni, e quindi centro-
simmetrico, nel bulk. Quest’ultima considerazione ci induce a pensare che anchein direzione y, nel limite termodinamico, la polarizzazione tenda a zero. Vedremofra poco che si ottengono delle inaspettate sorprese.
Vediamo intanto cosa accade in direzione x. Come ci si aspetta, la componentein x della polarizzazione risulta nulla (con deviazioni dell’ordine di 10−15 ) perqualsiasi dimensione del cristallo. Si ottengono quindi, per tale componente, irisultati attesi.
Vediamo ora invece cosa si ottiene per la componente in y della polarizzazione.Le simulazioni mostrano come tale componente tenda ad un valore finito di-
verso da zero, con un andamento proporzionale a 1W
e a 1L
, dove W e L sonorispettivamente la larghezza e l’altezza del cristallo. La finitezza di tale valorenel limite termodinamico concorda in realta con quanto trovato in precedenza perle cariche nette ai bordi. Infatti, ricordiamo che le cariche nette trovate sui sitiai bordi risultano essere presenti e diverse da zero anche nel limite termodinami-co. Inoltre a differenza dei bordi sinistro e destro, le cariche sui bordi inferioree superiore sono opposte in segno fra loro, benche uguali in modulo. Ricordandoquindi la relazione fra polarizzazione e densita di carica vista sopra, concludiamoche anche la polarizzazione nella direzione considerata non possa convergere a zeronel limite termodinamico, ma debba convergere invece ad un valore finito cometroviamo. Tale valore dovrebbe essere ovviamente compatibile con la densita dicarica ai bordi e quindi con le cariche nette trovate prima.
Nelle figure 8.1, 8.2 e 8.3 sono riportati gli andamenti della componente iny di P. Notiamo inoltre che invertendo i parametri α1 e α2 (trasformazione chelascia inalterata comunque la simmetria del bulk) la polarizzazione risulta ugualein modulo ed opposta in segno (con una precisione di 10−15). Questo lo si aspettaanche intuitivamente. Infatti invertire i due parametri equivale a invertire gliatomi, e quindi a invertire le cariche sui bordi. Questo induce ovviamente solo uncambio di segno nella polarizzazione.
Vediamo ora quali sono le sorprese che portano con se i risultati ottenuti.Facendo riferimento all’ articolo [3], ci si aspettava che, essendo il bulk cen-
trosimmetrico, il valore a cui converge la componente in y di P nel limite termodi-namico fosse simile a 1
2del cosiddetto quanto di polarizzazione e che la differenza
fra le polarizzazioni ottenute invertendo i parametri α1 e α2 fosse pari esattamen-te al quanto di polarizzazione. Questo deriva dal Teorema di Quantizzazione, il

CAPITOLO 8. POLARIZZAZIONEMACROSCOPICA E SUO LIMITE TERMODINAMICO43
quale afferma che la polarizzazione e quantizzata, e cosı quindi anche la caricasulla superficie di un solido. I valori della polarizzazione permessi differiscono delcosiddetto quanto di polarizzazione, vettore le cui componenti sono:
2eVcell
Ri
dove Vcell e il volume della cella che descrive il reticolo ed Ri il vettore ditraslazione nella direzione i. Il quanto quindi e un vettore con le stesso numerodi componenti dello spazio occupato dal cristallo, nel nostro caso e dunque bidi-mensionale. Il 2 deriva dalla molteplicita di spin. Nell’articolo citato si calcolale cariche agli estremi di una catena quasi-monodimensionale e si verifica che neisistemi con bulk centrosimmetrici la carica netta su tali estremita nel limite termo-dinamico vale meta del quanto di polarizzazione e che i valori permessi differisconofra loro di una quantita pari proprio al quanto di polarizzazione. Se invece il bulknon e centrosimmetrico, nell’articolo si scopre che la polarizzazione puo assumerequalsiasi valore, ma comunque i possibili valori devono differire di una quantitapari a un multiplo intero del quanto di polarizzazione.
Come visto, la simulazione proposta mostra che questo non avviene. La den-sita di carica ai bordi (che e proporzionale alla componente della polarizzazionenormale a tale bordo) risulta essere diversa da meta del quanto. Sembrerebbequindi che la carica netta ai bordi fosse simile a quella prevista considerando solole catene verticali interne al cristallo, le quali non sono simmetriche. Pero, comedetto sopra, ci aspettiamo almeno che le polarizzazioni differiscano almeno di unmultiplo intero del quanto.
Cosı non e.Questo e il risultato inaspettato che e stato trovato. Simulazioni avanzate e
analisi quantitative piu approfondite sono interessanti per indagare il fenomenovisto. Questo va oltre pero all’obiettivo di questa tesi.

CAPITOLO 8. POLARIZZAZIONEMACROSCOPICA E SUO LIMITE TERMODINAMICO44
Figura 8.1: Andamento della componente Py in funzione della larghezzza delcristallo. Si vede che diminuisce in modo proporzionale a 1
We tende ad un valore
finito.
Figura 8.2: Andamento della componente Py in funzione dell’altezza del cristallo.Si vede che diminuisce in modo proporzionale a 1
Le tende ad un valore finito.

CAPITOLO 8. POLARIZZAZIONEMACROSCOPICA E SUO LIMITE TERMODINAMICO45
Figura 8.3: Andamenti della componente Py in funzione dell’altezza del cristallodel cristallo riferiti ai parametri α1 e α2 invertiti. Si vede chiaramente come sianouguali in modulo ed opposti in segno

Capitolo 9
Conclusioni
Riassumiamo quindi i risultati del lavoro fatto. Abbiamo constatato la presenzadi cariche nette ai bordi e ai vertici di un cristallo di Nitruro di Boro. Cosı facen-do abbiamo confermato quanto ottenuto da ZRV, anche usando un’Hamiltonianamodello piu semplice. L’analisi dell’entita di tali cariche mostra inoltre che questesono presenti per qualsiasi dimensione del cristallo considerato, e i valori di talicariche nette convergono a valori finiti indipendenti dalle dimensioni del cristallostesso.
Abbiamo dunque visto il modulo e il segno delle cariche ai vertici, confermandointeressanti simmetrie.
Infine abbiamo studiato anche la polarizzazione macroscopica del cristallo,ottenendo relazioni che confermano i risultati precedenti, ma anche risultati ina-spettati. Ci si propone quindi in un lavoro successivo di analizzare meglio e tentaredi motivare tali fenomeni.
46

Bibliografia
[1] Y. Zhou, K. Rabe, and D. Vanderbilt, Surface polarization and edge charges,Phys. Rev. B (RC) 92, 041102 (2015)
[2] C. Sgiarovello, M. Peressi, and R. Resta, Electron localization in the insulatingstate: Application to crystalline semiconductors, Phys. Rev. B.64. 115202(2001)
[3] K. N. Kudin, R. Car, and R. Resta, Quantization of the dipole moment andof the end charges in push-pull polymers, Chem. Phys.127, 194902 (2007)
47


















