![El “Tour” desdeBarcelóna VIENE i.iiÉiai$;0] JOIE MIJRI ...hemeroteca-paginas.mundodeportivo.com/./EMD02/HEM/... · duelo a treCe asaltos. El vence-dor petdibtrá 350.00!) pesetas,](https://static.fdocumenti.com/doc/165x107/5bb768a309d3f2f7768d8485/el-tour-desdebarcelona-viene-iiieiai0-joie-mijri-hemeroteca-.jpg)
Titolo RUOLO DELL’ETERODIMERIZZAZIONE DEI … · L'eterodimerizzazione del MOR con DOR conduce a...
87
UNIVERSITA’ DI PISA FACOLTA’ DI FARMACIA Corso di Laurea Specialistica in Farmacia Tesi di Laurea Titolo RUOLO DELL’ETERODIMERIZZAZIONE DEI RECETTORI A PROTEINA G Candidato TRONFI DAVIDE Relatore Correlatore Prof. Lucacchini Antonio Prof. Giannaccini Gino Anno Accademico 2009-2010
Transcript of Titolo RUOLO DELL’ETERODIMERIZZAZIONE DEI … · L'eterodimerizzazione del MOR con DOR conduce a...

UNIVERSITA’ DI PISA
FACOLTA’ DI FARMACIA
Corso di Laurea Specialistica in Farmacia Tesi di Laurea
Titolo
RUOLO DELL’ETERODIMERIZZAZIONE DEI
RECETTORI A PROTEINA G
Candidato
TRONFI DAVIDE
Relatore Correlatore
Prof. Lucacchini Antonio Prof. Giannaccini Gino
Anno Accademico 2009-2010

Riassunto analitico
I recettori accoppiati alle proteine G (GPCR) costituiscono una grande famiglia di recettori di
membrana che rivestono un ruolo cardine nella trasduzione del messaggio. I GPCR sono dei
componenti fondamentali di molti processi biologici, tra cui le attività del SNC, la regolazione
dell’appetito, della digestione, della pressione, del metabolismo e dell’infiammazione.
Alterazioni dei GPCR sono alla base di condizioni patologiche come l’asma, di malattie
neurodegenerative, il diabete… . L’attivazione dei GPCR e la conseguente trasmissione del
segnale, prevede una serie di eventi scatenati dal legame del farmaco al dominio extracellulare
che determina una modificazione della conformazione del GPCR seguita dall’attivazione della
proteina G e dell’effettore (enzimatico o canale). Viene affrontato il ruolo che
l’eterodimerizzazione gioca nella funzione di alcuni GPCR e viene affrontata l’importanza degli
eterodimeri nella patogenesi di alcune malattie. E’ stato discusso il meccanismo di attivazione
delle proteine G eterodimeriche che prevede la dissociazione del complesso Gβγ dal monomero
Gα, e si avvale dell’attività del canale Ca++ che viene usato come sensore dell’attività della
proteina G. Ciò suggerisce che gli eterodimeri dei recettori a proteina G rappresentano bersagli
farmacologici innovativi per l’identificazione di composti potenzialmente selettivi con meno
effetti collaterali.

INDICE INTRODUZIONE 1
1. ETERODIMERI DEL GPCR : NUOVE PROSPETTIVE DI RECETTORI 2
2. PRIMI STUDI SULL’ETERODIMERIZZAZIONE 7
3. RECETTORE GABA B 9
4. RECETTORI DELLA MODULAZIONE DEL DOLORE 11
5. COMPLESSO A2A/D2 18
6. COMPLESSO D2/D3 36
7. COMPLESSO SSTR5/SSTR1 38
8. COMPLESSO SSTR5/D2 39
9. COMPLESSO A1/D1 41
10. COMPLESSO mGluR1αA1 44
11. COMPLESSO P2Y1/A1 46
12. COMPLESSO mGLuR5/A2A 48
13. COMPLESSO B2/AT1 50
14. COMPLESSO GABA A/D5 52
15. COMPLESSI OLIGOMERICI E RECETTORI TIROSIN-CHINASICI 56
16. COMPLESSI CALCITONINA/RAMP1-3 58
17. COMPLESSO D1/CALCIONE 60
18. COMPLESSO A1/ADENOSINA DEAMMINASE 63
19. COMPLESSO A1/HSC 73 65
20. CONCLUSIONI 67
BIBLIOGRAFIA 69

1
INTRODUZIONE
I recettori accoppiati alle proteine-G (GPCR) sono proteine di membrana che convertono
informazioni extracellulari in segnali intracellulari, sono implicati in più processi fisiologici.
Come tali, questi recettori (GPCR) rappresentano bersagli importanti dei farmaci. Gli
avanzamenti nella conoscenza della biologia del GPCR suggeriscono che un dato recettore a
proteina-G sia implicato solitamente in più di un processo fisiologico. Di conseguenza, i farmaci
che mirano ad un tipo recettoriale dentro un contesto patologico specifico sono probabili ad
agire sullo stesso tipo recettoriale di altri tessuti che non sono implicati nella malattia, e
conducono agli effetti indesiderati del farmaco. Nell'ultimo decennio, parecchi meccanismi che
regolano la funzione e le proprietà dei GPCR sono stati identificati ed hanno mostrato che,
l’eterodimerizzazione [1] appare come processo importante nella specializzazione della funzione
del recettore e regolano eventi patologici. I recettori eterodimeri esistono in particolari tessuti e
rappresentano le entità funzionali che mediano le diverse risposte biologiche. Queste proprietà
suggeriscono che i recettori eteromeri possano essere usati come bersaglio di nuovi farmaci
perché i composti che mirano a specifici eterodimeri sono probabili a migliorare la specificità e a
ridurre gli effetti indesiderati.

2
1. ETERODIMERI DEL GPCR : NUOVE PROSPETTIVE DI RECETTORI
Un concetto emergente nella ricerca degli GPCR è la promozione di cascate di segnali
eterodimerici specifici che permettono la diversificazione degli effetti del recettore in un
contesto dipendente (figura 1). La modulazione delle proprietà di legame
dell’eterodimerizzazione dei GPCR sono state descritte per diverse coppie di recettori [2-5].
Questo è pensato per essere la conseguenza di un'alterazione nelle tasche di legame, rivelando
che qualche ligando potrebbe preferire i recettori nella loro struttura omomerica o
eterodimerica. Per esempio, una diminuzione nell’affinità e nella potenza dei recettori agonisti
selettivi è stata osservata nel caso dei recettori oppioidi δ−κ (DOR-KOR) [4], nel DOR-MOR [6] e
nel recettore della somatostatina sst2A-sst3 [7]. In contrasto, un aumento dell’affinità del
recettore β2 adrenergico (AR) il ligando era stato osservato per coppie di recettori β1−AR-
β2−AR [8]. Questi studi indicano che l'eterodimerizzazione altera il ligando che lega, ma i
cambiamenti nella farmacologia del recettore non sembrano seguire un modello comune
perché ogni coppia del recettore rappresenta un'unica alterazione nella farmacologia del
recettore. Questi cambiamenti potrebbero risultare dalla modulazione allosterica di un
protomero nella legatura del ligando all'altro protomero dentro l'eterodimero [9]. Studi hanno
mostrato che l’eterodimerizzazione del recettore cambia nell'agganciamento della proteina-G.
In molti casi, l'eterodimerizzazione conduce ad alterazioni nell'estensione dell’agganciamento
alla stessa proteina-G ma recenti scoperte mostrano che, in alcuni casi, c'è un cambiamento
nella natura della proteina-G agganciata. Un esempio di studio di un cambiamento
nell'estensione dell’agganciamento della proteina-G è l'eterodimero α2a-AR-MOR che legato
alla morfina inibisce il segnale di norepinefrina mediata a Gαi e a valle la cascata delle MAPK
[10]. Il trasferimento di energia di risonanza di Forster (FRET) ha rivelato un cambiamento di
conformazione che propaga da un recettore all'altro, causando una rapida inattivazione del
secondo recettore con frazioni cinetiche [11]. Queste scoperte suggeriscono che
l’eterodimerizzazione induce un cambiamento nella struttura del recettore che è responsabile
delle nuove proprietà dei recettori. L'eterodimerizzazione può risultare anche in un cambio nel

3
tipo della proteina-G agganciata. L'eterodimerizzazione tra DOR ed il recettore sensoriale
neurone specifico 4 era riportato a cambiamento da un Gαi/o a un Gαq [12].
L'eterodimerizzazione tra i recettori D1-D2 della dopamina sono stati trovati per cambiamento
da un Gαs/olf (D1R) o Gαi (D2R) a un Gαq/11, e l'eterodimerizzazione tra DOR e MOR è stata
mostrata per cambiamento da un Gαi a un Gαz [13-15]. Nuovi studi indicano che l’attivazione
degli eterodimeri conducono a un cambiamento nel reclutamento di diverse molecole di
segnale. Questo conduce alla specializzazione degli effetti a valle come l'attivazione
dell’espressione di fattori di trascrizione e di diversi geni. L'eterodimerizzazione dell’α2a-AR con
l’α2c-AR riduce la fosforilazione GRK e il reclutamento della β-arrestina, agendo a valle la
fosforilazione Akt [16]. L'eterodimerizzazione del MOR con DOR conduce a un cambio da uno
maggior mediato dalla proteina-g a uno mediato dalla b-arrestina, nel giro risultante
nell'attivazione di differenti fattori di trascrizione [17]. Un protomero può far da ponte nel
reclutare delle molecole di segnale che promuovono diversi segnali all'altro protomero, come
proposto nel caso dell’α2a-AR-MOR [10]. Tale meccanismo deve essere ancora esplorato per le
altre coppie del recettore, ma potrebbe rappresentare un mezzo generale per diversificare la
funzione dei GPCR. L'eterodimerizzazione del GPCR conduce a cambiamenti nell’agganciamento
sia qualitativi che quantitativi. Questi cambiamenti possono modulare la forza della risposta di
un agonista ad agganciarsi alle diverse proteine-G e/o di associati complessi di segnale. Queste
nuove proprietà evidenziano l'eterodimerizzazione del recettore come un maggior meccanismo
che allarga i vari effetti del GPCR ed i contributi alla descrizione funzionale dei recettori. Questo
suggerisce che mirare ad un specifico tipo di un recettore dentro un eterodimero potrebbe
consentire l'attivazione di un percorso fisiologico discreto, con gli effetti limitati sugli altri
processi che coinvolgono lo stesso recettore.
L’identificazione dei recettori eteromeri nel vivo e la loro implicazione nei processi
fisiopatologici è critica per il loro riconoscimento come nuovi bersagli di farmaci. Solo poche
tecniche tengono conto della rivelazione dei complessi GPCR nei tessuti nativi (Figura 2). Il
ligando Fluoroforo è stato sviluppato quale potrebbe essere usato per eseguire analisi FRET
interagendo tra i recettori endogeni. Gli eterodimeri del recettore riconoscono anticorpi

4
specifici e rappresentano gli strumenti utili per la rivelazione ed analizzano i complessi GPCR nel
vivo. Il ruolo di un eterodimero può essere facilitato dalla caratterizzazione e dalla mutazione
dei residui o dai domini implicati nell'associazione recettore-recettore. Diversi approcci, per
esempio modellare molecole, cambiando dimeri di recettore, crosslinking , tecniche biofisiche
ed il dominio di transmembrane (TM) sono stati usati per determinare tali domini. Questi studi
hanno dimostrato l'esistenza di interfacce di interazione multiple, compreso TM1 e compreso
TM4 nell’omomero D2R ed identificato come l’interfaccia della dimerizzazione mGluR2 TM 4/5
con 5HT2aR nel complesso 5HT2aR-mGluR2. Le strategie sono mirate a evitare o rompere
l'interazione recettore-recettore usando delle piccole molecole o di peptidi che potrebbero
produrre degli approcci alternativi per mirare all'attività degli eterodimeri.

5
Figura 1. Modulazione della funzione del recettore dall’eterodimerizzazione
(a) L’eterodimerizzazione conduce alla formazione di un alterato sito di legame. Mostrato per DOR-KOR , DOR-MOR, e MOR-sst2A.
(b) L’eterodimerizzazione conduce a una diminuzione o aumento nell’accoppiamento alla proteina G. La diminuzione nell’accoppiamento era mostrata per l’eterodimero DOR-MOR; l’aumento nell’accoppiamento era mostrato per α2−AR-MOR, e AT1R-B2R.
(c) L’eterodimerizzazione conduce all’accoppiamento a una nuova proteina G. Mostrato per l’eterodimero MOR-DOR accoppiato a Gαz; l’eterodimerizzazione di due differenti recettori accoppiati conducono all’accoppiamento preferenziale a una proteina G dopo stimolazione. Mostrato per DOR-SNSR-4.
(d) L’eterodimerizzazione conduce a un cambio nell’accoppiamento del recettore, da proteina G a β-arrestina, e la successiva attivazione di differenti segnali e fattori di trascrizione. Mostrato per l’eterodimero MOR-DOR.

6
Figura 2. Emergenti tecniche degli eterodimeri a scoprire (a-c) e a modulare (d-i) gli eterodimeri GPCR possono essere scoperti usando 3 principali approcci.
(a) Il ligando mediato fluoroforo agganciato FRET. Il ligando fluoroforo agganciato per due recettori può essere analizzato usando FRET solo se i loro due recettori analoghi sono in prossimità vicini in un complesso eterodimerico.
(b) Gli anticorpi eterodimerici selettivi che riconoscono solo l'eterodimero possono essere usati per studiare la localizzazione ed i livelli degli eterodimeri.
(c) Il ligando eterodimerico specifico può legare solo al recettore alterato che legandosi alla tasca ha formato un eterodimero. L'eterodimero del GPCR può essere modulato usando sei principali approcci.
(d) Gli agonisti eterodimerici specifici possono attivare un recettore solo se in una forma eterodimerica. (e) Gli antagonisti eterodimerici specifici possono bloccare un solo recettore se in una forma eterodimerica. (f) Gli anticorpi eterodimerici specifici possono bloccare il legame e/o il segnale del recettore solo se in una forma eterodimerica. (g) L’attivazione degli anticorpi eterodimerici specifici può attivare recettori solo se in una forma eterodimerica. (h) ligandi bivalenti possono legare e attivare recettori solo se i loro recettori sono in una forma eterodimerica. (i) I ligandi di un recettore possono agire come modulatori allosterici negativi o positivi della interazione recettoriale in un
eterodimero. I modulatori allosterici possono alterare le proprietà di legame e/o di segnale della interazione recettoriale.

7
2. PRIMI STUDI SULL’ETERODIMERIZZAZIONE
Gli studi sui recettori a proteina-G (GPCR) hanno inizio nel 1979-1980 alla ricerca di una
spiegazione da dove tutte le recenti scoperte dei neuropeptidi nel cervello potrebbero
integrare i loro messaggi con quelli dei classici trasmettitori come le monoammine. Luigi F.
Agnati e Kjell Fuxe hanno postulato che un'interazione intramembrane tra i recettori dei
neuropeptidi e delle monoammine potrebbero essere coinvolte. Le prime osservazioni sono
state pubblicate nel 1980 [18], mostrando come la sostanza P potrebbe modulare la serotonina
con alta-affinità che lega dei siti nelle preparazioni di membrana del midollo spinale usando le
tecniche di legame biochimiche [19]. Successivamente si è scoperto che non solo i recettori dei
neuropeptidi e dl monoammine sono stati coinvolti nelle interazioni recettore/recettore
intramembrana ma anche in certi tipi di recettori del glutammato e dell’adenosina. Questi
risultati sono stati tutti ottenuti nel luogo di riconoscimento dei recettori, usando esperimenti di
saturazione e di legame competitivo. Un'indicazione di un effetto sull’accoppiamento della
proteina-G e perciò sull'efficacia del recettore modulato potrebbe essere ottenuto studiando
come la modulazione potrebbe controllare la scomparsa GTP-indotta dello stato di alta-affinità
del recettore. Questo implicherebbe un’attivazione della proteina-G con formazione di Gα-GTP
e dimeri βγ associati a regolazione incrociata di GPCR con scomparsa dello stato di alta affinità
del recettore. Gli studi sono stati estesi per mostrare le interazioni multiple recettore/recettore.
Nel 1993 [20], è stato introdotto il meccanismo molecolare per queste interazioni
recettore/recettore intramembrana fra GPCR e potrebbero essere la formazione dei complessi
eteromerici, il più semplice un eterodimero. Questo concetto è stato basato sull'indicazione al
tempo che la dimerizzazione in seguito a attivazione agonista potrebbe essere un fenomeno
generale essenziale per l'attivazione del recettore [21], il migliore esempio è la dimerizzazione
dei recettori tirosin chinasici [22,23,24]. Così, è stato presunto che GPCR esista principalmente
come l'omodimero che interagisce con gli altri tipi di omodimeri a formare gli eterodimeri. Le
proporzioni relative di omo e eterodimeri sarebbero determinate dalle concentrazioni dei due
trasmettitori, la densità dei due recettori ed i loro modelli di distribuzione, e le caratteristiche

8
uniche di ogni interazione recettore/recettore [25]. È stato postulato [26,27] che la formazione
e/o la stabilizzazione dei complessi eteromerici dei GPCR potrebbero essere migliorate
dall’associazione di proteine soprattutto nelle membrane sinaptiche. Deve essere notato che la
maggior parte del GPCR è localizzato nelle membrane extrasinaptiche e quindi i potenziali
complessi eteromerici discussi sopra potrebbero essere anche raggiunti dal segnale di
trasmissione di volume [28]. Dai dati di recenti esperimenti viene confermata
l’eterodimerizzazione (figura 3).
Figura 3. Eterodimerizzazione tramite interazioni recettore-recettore intramembrana

9
3. RECETTORE GABA B
La prima prova che due sottotipi recettoriali del GABA B, GABA B R1 e GABA B R2 subiscono
l'eterodimerizzazione e che questo processo è essenziale per l'espressione della cellula di
superficie del recettore funzionale è stata data in tre studi pubblicati simultaneamente nel
dicembre 1998 da Kaupmann [29], da Jones [30] e da White [31]. Questo è stato anche
dimostrato da Kuner [32] e Ng [33] nel gennaio 1999 [34,35]. La pertinenza fisiologica di queste
scoperte è sostenuta dalla dimostrazione della coimmunoprecipitazione nelle membrane della
corteccia cerebrale delle proteine del GABA B R1 con GABA B R2, della loro localizzazione nelle
spine dorsali dendritiche [29] ed il sostanziale grado di co-espressione dei livelli di m RNA di
GABA B R1 e GABA B R2 in molte popolazioni cellulari nervose [32]. I cambiamenti sono accaduti
anche a livello del luogo di riconoscimento come risultato dell'eterodimerizzazione, poiché la
potenza degli agonisti e degli agonisti parziali è diventata aumentata [29]. Il cambiamento più
drammatico è comunque che l'eteromero recettoriale del GABA B formato, a differenza dei suoi
componenti monomerici, può diventare funzionale e accoppiarsi alle proteina-G portando
internamente alla regolazione della reattività dei canali k+, dei canali Ca++, e dell’adenilato-
ciclasi [36,37]. Così, sembra come se il recettore GABA B predominante nativo sia l'eteromero
del recettore GABA B. E’ stata presentata la prova che il dominio del COOH-terminale è
coinvolto nella formazione di quest'eterodimero da un'interazione tra gli avvolgimenti [32,38].
Nel 2000, è stata fatta l'osservazione importante in cui l'interazione a spirale al dominio del
COOH-terminale blocca una ritenzione al reticolo endoplasmatico del recettore GABA B R1 [25].
Comunque, anche se il mutante GABA B R1 il motivo di ritenzione era stato tolto e questo
recettore mutante potrebbe essere espresso sulla superficie della cellula, è rimasto
funzionalmente inattivo, sottolineando un probabile ruolo cruciale della eterodimerizzazione
del recettore GABA B nella trasmissione del segnale. Queste scoperte importanti danno un
esempio chiaro della importante funzione di interazioni recettore/recettore intramembrane
attraverso l'eteromerizzazione, compreso la maturazione recettoriale cellulare e l’espressione
del recettore sulla superficie cellulare, e nel segnale del recettore, cioè, nell’agganciamento alla

10
proteina-G e in un’aumentata potenza di legame degli agonisti e degli agonisti parziaIi, in linea
con il lavoro precedente sulle interazioni recettore/recettore [20,39,40]. È ben saputo che il
ruolo dei recettori GABA B è distinto nel modulare le reti neuronali e che farmaci agonisti che
agiscono, su questi recettori appaiono per avere le proprietà anticonvulsive e ansiolitiche. È
quindi di interesse sostanziale che il composto anticonvulsivo gabapentin è un agonista seIettivo
all’eteromero GABA B R1a/GABA B R2 [41]. Questo è un esempio di come la composizione
moIecolare dell'eteromero determina il suo profilo farmacoIogico e provoca un nuovo agonista
selettivo recettoriale del GABA B per un certo tipo di recettore eteromero dipendente GABA B
sulla specie variante coinvolta. In questo studio, questi risultati sono stati messi in correlazione
con una capacità selettiva di aumentare il segnale del recettore GABA B postsinaptico senza
alterare la trasmissione del GABA a livello presinaptico [41]. Questo nuovo tipo di selettività
farmacologica basata sull'unico eteromero potrebbe avere quindi una considerevole attenzione
per lo sviluppo di farmaci, e serve per mostrare la rilevanza farmacologica di interazioni
recettore/recettore intramembrana che potrebbe dare origine a nuovi sottotipi recettoriali con
un’unica farmacologia basata sulla composizione dell'eteromero formato per cambiare le
caratteristiche biochimiche della tasca di legame del recettore.

11
4. RECETTORI DELLA MODULAZIONE DEL DOLORE
Recettori oppioidi
I recettori oppioidi agonisti µ e κ sono stati considerati importanti nella modulazione del dolore,
e bersagli per la terapia dell’analgesia. Sfortunatamente, l’analgesia prodotta dal ligando
selettivo è limitata dagli effetti collaterali meccanismo-dipendenti. Più recentemente,
l'upregulation del sottotipo RGS4 della proteina regolatrice di segnale, è stata dimostrata nel
dolore neuropatico, una possibile spiegazione per l'efficacia ridotta della morfina in questa
condizione [42]. Questo suggerisce che l’inibitore sito specifico di RGS4 potrebbe migliorare
l'efficacia della morfina e ridurre forse gli effetti collaterali come la costipazione e la
depressione respiratoria. D'altra parte, il centro d'interesse della ricerca sui bersagli del
recettore degli oppioidi si è rivolto verso i recettori oppioidi δ (DORs), i ligandi a cui hanno la
potenza per l'efficacia analgesica con gli effetti collaterali minimi. Molti studi, compresi quelli
che coinvolgono la riduzione del recettore con l’oligonucleotide antisenso o la soppressione del
gene DOR [43]; hanno sostenuto il concetto che i ligandi selettivi δ, che comprendono agonisti
peptidici e non peptidici produce analgesia senza la notevole depressione respiratoria,
l'inibizione della motilità gastrointestinale o l'azione sedativa. Sebbene l'iperattività sia stata
notata con alcuni composti δ selettivi nei roditori, questo non è stato osservato nell’uomo. In
modo interessante, i ligandi DOR forniscono meno efficacia analgesica della morfina, sebbene
la loro efficacia appaia dipendere dalla natura dello stimolo del dolore e l'ambiente chimico, che
potrebbe essere determinante per l’espressione del recettore. Conseguentemente i ligandi δ
selettivi hanno efficacia analgesica bassa nei modelli di dolore acuto, ma mostrano un’avanzata
attività per somministrazione ripetitiva di morfina e dopo un episodio di infiammazione.
L'aumento di efficacia è stata attribuita a stimolazione indotta del DOR contenuto nelle
vescicole citoplasmatiche dalla loro localizzazione endocellulare, seguita dalla incorporazione
nelle membrane del nervo dopo fusione vescicolare [44]. Allo stesso modo l’aumento di traffico
del DOR accade nei neuroni sensoriali, dove la produzione del segnale calcio-dipendente

12
dall'attivazione del nervo afferente migliora l'espressione della liberazione di GPCR e DOR nella
membrana del nervo [45]. La possibilità dell'eterodimerizzazione dei recettori oppioidi µ, δ e k
potrebbe spiegare l'esistenza di più di tre sottotipi recettoriali degli oppioidi come caratterizzato
farmacologicamente [46]. L'eteromero oppioide δ/K [4,6] è stato mostrato avere una
farmacologia unica con un’alta affinità piuttosto per ligandi non selettivi ma molto piccola
affinità per aumenti di δ e k selettivi. Così, un nuovo sottotipo del recettore oppioide legandosi
potrebbe essere apparso attraverso questa interazione recettore/recettore tramite
l'eterodimerizzazione del recettore δ/κ oppioide. Anche, il δ e k eteromero hanno avuto delle
conseguenze per l'internalizzazione dell’agonista-indotto del recettore oppioide δ, che si è
ridotto. Così, un altro ruolo funzionale di quest'interazione intramembrana del
recettore/recettore attraverso l'eteromerizzazione potrebbe essere il controllo
dell’internalizzazione del recettore. L'eteromero del recettore µ/δ oppioide, dimostrato da
George [6], mostra i cambiamenti mostrati nelle proprietà farmacologiche al sito di
riconoscimento con un'affinità ridotta per gli agonisti selettivi e un aumento di affinità per certi
peptidi encefalici. Può infatti l'eteromero µ/δ essere il bersaglio di distinti peptidi encefalici. Di
interesse speciale era la dimostrazione che l'eterodimero µ/δ, a differenza dei recettori µ e δ
quando espressi soli, potrebbero diventare resistenti alle proteine-G della tossina della
pertosse, come Gz. Così, alla proteina-G agganciata il segnale è diventato alterato
nell'eteromero. Infatti, la maggiore funzione dell’interazione recettore/recettore
intramembrana si basa sull'eteromerizzazione µ/δ, in cui potrebbe esserci un cambiamento
nella selezione dell’agganciamento alla proteina-G che coinvolge un cambiamento di
conformazione nell’interfaccia alla proteina-G del recettore eteromero µ/δ. L'altro
cambiamento della funzione è l’alterazione delle proprietà di legame del sito di riconoscimento
mentre un modello di nuova legatura del ligando è basato sui cambiamenti di affinità; una
nuova tasca di legatura sembra essere apparsa [47].

13
Recettori cannabinoidi
I recettori Cannabinoidi sono molto distribuiti in ogni parte del sistema nervoso dei mammiferi
e i sistemi cannabinoidi sono stati dimostrati nella modulazione del dolore [48]. Nell’estratto
della cannabis e nei ligandi degli cannabinoidi è stata dimostrata l'efficacia clinica in un numero
di condizioni di dolore cronico, e c'è quindi grande fiducia che il recettore dei cannabinoidi (CB1
o CB2) e dei ligandi possa essere identificato con la migliore efficacia ed un ridotto profilo di
effetto collaterale. Comunque, ad oggi, sia l'analgesia e sia gli effetti collaterali sono stati
attribuiti ai recettori CB1 localizzati nel SNC, nel midollo spinale e nei neuroni sensoriali
periferici. Diversi nuovi approcci sono considerati per una migliore terapeuticità, per esempio, lo
sviluppo dei ligandi CB1 produce ad analgesia su un'azione periferica ed evita così gli effetti
collaterali nel SNC. D'altra parte, la localizzazione dei recettori CB2 fuori del SNC mantengono la
promessa dell’analgesia con una migliore sicurezza. Fino ad ora, i recettori CB2 sono stati solo
trovati nei tessuti periferici relativi all’infiammazione ed un numero di ligandi CB2 selettivi è la
dimostrazione degli effetti degli antinocicettivi in una varietà di modelli del dolore [49] senza gli
effetti collaterali nocivi. È attualmente non chiaro se al ligando CB2 selettivo riduca il dolore solo
nella periferia; forse inibendo la liberazione di mediatori infiammatori ed il trophins dai
macrofagi e cellule immuni. Ulteriori studi recenti suggeriscono che l'espressione CB2 accade
anche nel SNC, particolarmente nella microglia spinale seguendo le lesioni dolorose
neuropatiche [50], che alzano la possibilità di azioni addizionali nel SNC senza gli effetti
collaterali dei CB1. Finalmente, ci potrebbero essere delle opportunità nello sviluppare le
combinazioni di agonista oppioide µ e CB1 siccome la sinergia analgesica è stata dimostrata nei
modelli di dolore acuti, suggerendo che le basse dosi delle combinazioni di MU/CB1 forniranno
l'efficacia analgesica adeguata con gli effetti collaterali ridotti. È attualmente non chiaro se la
sinergia sarà trovata nelle condizioni di dolore più croniche e se la riduzione degli effetti
collaterali sarà sufficiente per offrire una finestra terapeutica vantaggiosa.

14
Recettori dei neuroni sensoriali specifici
I recettori dei neuroni sensoriali specifici (SNSRs) sono una famiglia di GPCRs che sono espressi
selettivamente in un sottoinsieme di neuroni sensoriali dei gangli dorsali e del trigemino [51].
Questi recettori costituiscono un sottoinsieme di una più grande famiglia di geni affini che fa
riferimento ai geni Mas (Mrgs) [52]. Ci sono sei sottotipi del SNSR umano derivati dai geni
hMrgX, un solo sottotipo nei ratti e diversi da 50 a 60 nei topi. La diversità filogenetica della
famiglia del recettore Mrg non è attualmente capita. In modo importante, l’SNSRs è localizzato
preferenzialmente a IB4 POSITIVO di piccoli neuroni di diametro. Inoltre, approssimativamente
metà dei neuroni SNSR-POSITIVI contengono pure il dannoso recettore vanilloid 1 (TRPVR1),
indicando che i neuroni SNSR-POSITIVI potrebbero essere coinvolti in una gamma diversa di
funzioni sensoriali. Gli sforzi a identificare ligandi ha prodotto diversi attivatori per la famiglia di
SNSR/Mrg. Han [53] ha descritto peptidi affini RFamide che attivano dei membri di questa
famiglia, mentre Bender [54] ha identificato l'adenina come un attivatore di Mrg del topo. Non è
chiaro se questo ligando sarebbe coinvolto nella segnalazione del dolore. Comunque, di
pertinenza del dolore e la unica distribuzione di SNSRs è il fatto che i peptidi derivano dal gene
pro-encefalina A che è stato identificato e attiva il SNSRs. Così, il peptide della midollare del
surrene del bovino 22 (BAM22), e l'elaborazione endogena del frammentano BAM8-22 , attiva il
SNSRs umano nei test funzionali, ma erano approssimativamente di 10 cavità meno potente allo
SNSR del topo [51]. In modo interessante, BAM8-22 non ha attività su qualsiasi dei recettori
oppioidi, suggerendo che gli stessi prodotti di gene possono entrambi facilitare e possono
inibire dei neuroni sensoriali. Le scoperte inoltre, più recenti hanno dimostrato che γ2-MSH e
CT-γ2-MSH potentemente hanno attivato anche SNSR del topo. La valutazione ulteriore
funzionale di SNSRs con BAM8-22 ha rivelato l'aumento di eccitabilità vertebrale, indicato dalla
potenza di risposte nocicettive evocate dagli stimoli termici e meccanici. Inoltre, la
somministrazione vertebrale di intradermale e intratecale periferica di BAM8-22 e CT-γ2-MSH
ha causato dose dipendente hyperalgesia del compartimento termico. Questi dati sostengono
fortemente un ruolo per SNSRs nella modulazione di dolore ed il bisogno di un analgesico
antagonista.

15
Recettore Neuromedina U
Il recettore Neuromedina U (NMU), un peptide di 23 amminoacidi era all'inizio isolato dal
midollo spinale del porcino, ma è anche trovato in un numero di altri tessuti centrali e periferici.
Un N-terminale più breve, di 8 amminoacidi è stato anche identificato che è apparentemente sia
sufficiente sia necessario per conferire la piena attività biologica del peptide. Due recettori per
NMU sono stati identificati, NMUR1 e NMUR2: NMUR1 mRNA appare per essere più
abbondante nei tessuti periferici ed in modo interessante negli strati di piccoli e medi dei
neuroni sensoriali; NMUR2, d'altra parte, appare per essere più specificatamente limitato al
SNC, sebbene un’ alta densità di mRNA del NMUR2 appare nella lamina vertebrale I e II,
indicando un ruolo possibile per NMU nella trasmissione del dolore [55]. Il sostegno ulteriore
per l'attività dei recettori NMU nella modulazione del dolore è indicato dalla dimostrazione che
la somministrazione vertebrale intratecale del ligando peptide NMU di 23 amminoacido ha
prodotto le risposte meccaniche e termiche di stimoli pro-nocicettive alla periferia. In più, 23
amminoacido del NMU hanno migliorato l'eccitabilità di neuroni del corno dorsale, come pure la
sensibilità agli stimoli meccanici dannosi, nel modello flessore-riflesso del topo [55]. Comunque,
il contributo dei sottotipi del recettore NMU alla modulazione dell’eccitabilità o del dolore
vertebrale è attualmente non chiaro, come i sottotipi agonisti selettivi e gli antagonisti non sono
disponibili.
Recettori della Galanina
C’è la prova dell'implicazione dei recettori galanin e recettori galanin nella trasmissione del
dolore. Galanin è stato localizzato alla radice del midollo spinale e gangli dorsali, e può essere
aumentato in queste aree seguenti queste ferite dolorose del nervo [56]. Negli studi funzionali,
il galanin ha prodotto i cambiamenti dell’eccitabilità nel midollo spinale che erano entrambi
eccitatori ed inibitori. Comunque, sotto le condizioni di ipereccitabiltà vertebrale dopo la ferita
dolorosa del nervo, il galanin ha ridotto l’attività riflessa del flessore C evocata dalla fibra e
l'allodynia indotta da un numero di stimoli sensoriali. Il sostegno ulteriore per un ruolo di

16
inibitore globale del galanin nel midollo spinale viene dal trattamento con un oligonucleotide
antisenso di galanin. Questo ha prodotto un’aumentata autotomia del limbo periferico
seguendo la transazione del nervo sciatico, suggerendo il dolore esagerato in mancanza di
galanin endogeno [57]. Oltre a modulare l'eccitabilità neuronale, è stato suggerito che il galanin
è un peptide trofico che è coinvolto nei processi di maturazione e rigenerazione del nervo. In
rapporto con questo, la soppressione del gene di galanin ha ritardato la rigenerazione di nervo
funzionale dopo le lesioni dolorose periferiche del nervo. Mentre diversi ruoli funzionali sono
stati identificati per il galanin nelle conseguenze di ferita periferica, non è chiaro attraverso cui
questi effetti sono mediati poiché un numero di sottotipi del recettore(Gal-R1, Gal-R2 e Gal-R3)
sono stati identificati. Tutti i tre sottotipi dei recettori sono localizzati nel midollo spinale e nei
gangli sensoriali, sebbene gli strumenti molecolari e i ligandi recettore selettivo sono implicati
Gal-R1 e Gal-R2 poiché essendo più particolarmente coinvolti nella modulazione del dolore.
Così, gli agonisti Gal-R2 hanno causato moderata antihyperalgesia in diversi modelli di dolore
cronico, mentre la riduzione di Gal-R1 seguendo il trattamento di antisense ha migliorato
l'hyperalgesia neuropatico [58], suggerendo che questo recettore inibisce anche l'eccitabilità
vertebrale. Comunque, nel globale c'è l'incertezza considerevole del ruolo specifico dei recettori
di galanin nella modulazione del dolore cronico. Ricerche in quest'area continuano, essere
ostacolate dalla mancanza di biodisponibilità dedli antagonisti selettivi.
Recettore dell’ormone Paratiroideo 2
Due sottotipi recettoriali dell’ormone paratiroide (PTH) sono stati identificati nei mammiferi. Il
recettore PTH1 è attivato dal peptide da PTH e PTH-affine, ed è responsabile principalmente
dell’omeostasi di calcio, e sviluppo di tessuto e rimodellazione. Il recettore PTH2 , che è attivato
da PTH, può essere anche attivato con affinità più grande e alta selettività dal recentemente
identificato ligando peptide di 39 amminoacidi (TIP-39), ottenuto dagli estratti ipotalamici di
bovini, che divide dell'omologia con PTH [59]. La prova di un ruolo per il recettore PTH1 nella
modulazione del dolore è limitato, poiché è distribuito molto nei tessuti periferici. Comunque, il
recettore PTH2 ha una distribuzione limitata del SNC (il talamo e la corteccia sensoriale), ed è

17
espresso fortemente nei tratti di fibra e nelle estremità dentro la lamina superficiale e del
midollo spinale, come pure in una sottopopolazione di neuroni dei gangli dorsali. Veramente, la
somministrazione locale di TIP-39 sia il midollo spinale o in un membro periferico ha prodotto il
comportamento nocefensive, mentre la somministrazione vertebrale di neutralizzare l’anticorpo
TIP-39 risposte causate nei modelli di dolore acuti risposte antinociceptive [60]. Mentre non c'è
attualmente alcuna prova che il recettore PTH2 gioca un ruolo nel dolore cronico,con i dati
ottenuti si sostiene un ruolo per il recettore PTH2 nella modulazione delll'eccitabilità vertebrale
e suggerisce che un antagonista sarebbe essere l'antinociceptive.
In conclusione diversi GPCRs e loro ligandi giocano ruoli importanti nella modulazione del dolore
acuto e cronico. Gli oppioidi e i cannabinoidi nel particolare sono di valore stabilito terapeutico
per la gestione del dolore, e lo sfruttamento ulteriore dei sottotipi di GPCR specifici
(δ-OPPIOIDI,CB1 e CB2) per questi ligandi produrrà potenzialmente degli analgesici più potenti
con i profili di effetto collaterale favorevoli. Più recentemente, l'identificazione di un numero di
GPCRs nei percorsi del dolore, il SNSRs, il NMU, il Gal R1/2 e il PTH2, ed il ligando selettivo che
modula la trasmissione del dolore, hanno evidenziato per continuare le opportunità
terapeutiche. Finalmente, la scoperta e la caratterizzazione di proteine di RGS coinvolte nella
regolazione del GPCR sono una sfida ulteriore alla comprensione degli GPCRs nella modulazione
del dolore che aggiunge una dimensione addizionale dei bersagli per l'analgesia.

18
5. COMPLESSO A2A/D2
Interazioni tra recettori A2A e D2
Nel 1991, l'interazione antagonistica recettore/recettore A2A/D2 è stata dimostrata nelle
preparazioni della membrana dello striatal con recettori A2A riducendo l'affinità dei recettori
D2, soprattutto nello stato di alta affinità, per gli agonisti [61]. Questo ha offerto un
meccanismo alle nuove interazioni antagonistiche adenosina/dopamina trovate nel cervello [62-
66]. L’ interazione recettore/recettore intramembrana A2A/D2 accade in tutti i tipi di cellule ed
in altri recettori. D2L e D2S potrebbero subire la stessa modulazione dall’attivazione del
recettore A2A, almeno a livello del sito di riconoscimento. La specificità è dimostrata dal
fallimento dei recettori agonisti A1 ad alterare l’affinità dei recettori D2 [61]. Hillion [67] ha
recentemente riferito, in cui si è basato sugli esperimenti della coimmunoprecipitazione, che
l'eteromerizzazione del recettore umano A2A ed umano D2L esiste nello stato basaIe delle
cellule del neuroblastoma SH-SY5Y con i recettori D2L contengono i recettori nativi A2A, ed
esistono nelle cellule dei fibroblasti con il recettore umano D2L con etichettato il recettore A2A
del cane. Esiste anche lì un alto grado di localizzazione dei recettori D2 e A2A in queste cellule e
nelle culture primarie dei neuroni dello striatal del topo (figura 4). L'esistenza di monomeri e di
omomeri contro i complessi eteromerici rimane in queste cellule ad essere determinate come
semplice esistenza del complesso eteromerico, l’eterodimero A2A/D2 (figura 5). Di nuovo,
dovrebbe essere accentuato che questo complesso eteromerico esiste in mancanza di agonisti
esogeni e la specificità dell'eteromerizzazione del recettore A2A/D2 è mostrata dall'assenza del
recettore A2A/D1 nelle cellule che esprimono i recettori D1 e etichettati recettori A2A. Un
significato funzionale di quest'interazione intramembrana di recettore/recettore attraverso
l'eteromerizzazione è poi di ridurre l’affinità dello stato di agonista di alta-affinità dei recettori
D2. Altro significato è di neutralizzare il recettore D2 all’aggancio alla proteina-G, poiché
l’agonista A2A neutralizza la scomparsa del recettore D2 analogo indotto dal GTP nello stato di
alta affinità (RH) attraverso un sito di azione indipendente del sito di legame del GTP (figura 6).

19
Così, l’essenza di questa eteromerizzazione del recettore A2A/D2 potrebbe essere nel
convertire il recettore D2 in uno stato di attività funzionale fortemente ridotta (figura 7). In
linea, con quest'attivazione del recettore A2A neutralizzato da risposte dei recettori D2 Ca++
intracellulari indotti [68] e l'inibizione del recettore mediato D2 della formazione di cAMP
[67,69]. E’ stato basato sugli studi delle chimere del recettoreD1/D2 [70-73] dove il dominio
TM 5 e 6 più il terzo loop del recettore D2 è stato sostituito dal dominio corrispondente del
recettore D1, è probabile che questi domini dei recettori D2 sono delle parti dell'interfaccia
A2A/D2, dall'affinità di questa chimera del recettore D1/D2 per la dopamina non può essere
modulata in lungo dall'attivazione di agonista del recettore A2A [73]. Fino ad ora non è stato
possibile per mostrare una reciproca regolazione di affinità del recettore A2A/D2 da cui il
recettore D2 controlla sull'attivazione l'affinità dell’agonista del recettore A2A. L'interazione
intramembrana recettore/recettore del recettore A2A/D2 attraverso l'eteromerizzazione ha
anche un impatto sul traffico del recettore [67]. Così, la coaggregazione dei recettori D2 e A2A
nelle cellule di membrana e nelle cellule del neuroblastoma potrebbero essere dimostrate dopo
trattamento con agonisti del recettore A2A o D2 per 3 h per mezzo della combinazione con
delle cellule non permeabilizzate. Il recettore D2 dell’aggregazione agonista indotto dei
recettori A2A era assente nelle cellule del neuroblastoma dei genitori, con un'espressione molto
ridotta dei recettori D2. L’aumentato di sviluppo dei recettori aggregati A2A/D2 sulla
membrana della cellula dopo trattamento prolungato con A2A o D2 è stato associato con un
agonista recettoriale A2A a incrementare i livelli di cAMP. Così, il recettore A2A/D2 aggregato
sviluppato era associato all'appartenenza di entrambe omologie e desensibilizzazione di
eterologhi del recettore mediato D2 dei recettori A2A. In contrasto, il recettore D2 non
desensibilizza sotto queste condizioni in termini di inibizione dell’accumulazione di cAMP, forse
collegato sostanzialmente alla più alta densità dei recettori D2, di cui potrebbe rappresentare
risparmio dei recettori. Un alto grado di localizzazione dei recettori A2A e D2 sono stati anche
trovati nelle colture dei neuroni dello striatal ed anche qui l'agonista A2A o l’agonista D2 dopo
un'esposizione prolungata potrebbe indurre la coaggregazione dei recettori A2A/D2. La
cointernalizzazione dei recettori A2A/D2 potrebbero essere dimostrati direttamente incubando

20
anche anticorpi fluorescenti dei recettori D2 ed A2A insieme a agonisti recettoriali A2A e D2 a
4°C per 2 h seguita dall'incubazione per 3 h a 37° C, consentendo di etichettare i recettori
A2A/D2 internalizzati sotto l'influenza dei due agonisti. Tale sinergismo per ciò che riguarda la
coaggregazione e la internalizzazione dei recettori A2A/D2 non potrebbero essere dimostrati
nei neuroni dello striatal. È di interesse sostanzioso che negli esperimenti di accumulazione del
cAMP sulle cellule del neuroblastoma, il trattamento combinato di agonista è stato associato
con lo sviluppo di una desensibilizzazione del recettore D2 mentre ha visto l’inibizione ridotta
dall’attivazione del recettore D2 dell'accumulazione di cAMP [67]. Così, il recettore dell’
eteromerizzazione A2A/D2 appare essere coinvolto nell’aggregazione, nell’internalizzazione e
nella desensibilizzazione dei recettori A2A e D2 [67]. Finalmente, quest'interazione
intramembrana recettore/recettore A2A/D2 attraverso l'eteromerizzazione potrebbe aiutarci a
capire la tolleranza trasversale e la sensibilizzazione trasversale trovata in vivo tra gli agonisti di
dopamina e nell’azione dei farmaci che agiscono sui recettori A2A [74,75] e anche la riduzione
dell'attività antiparkinsoniana [72].
Figura 4. Neurone efferente GABAergico dello striatal.
21
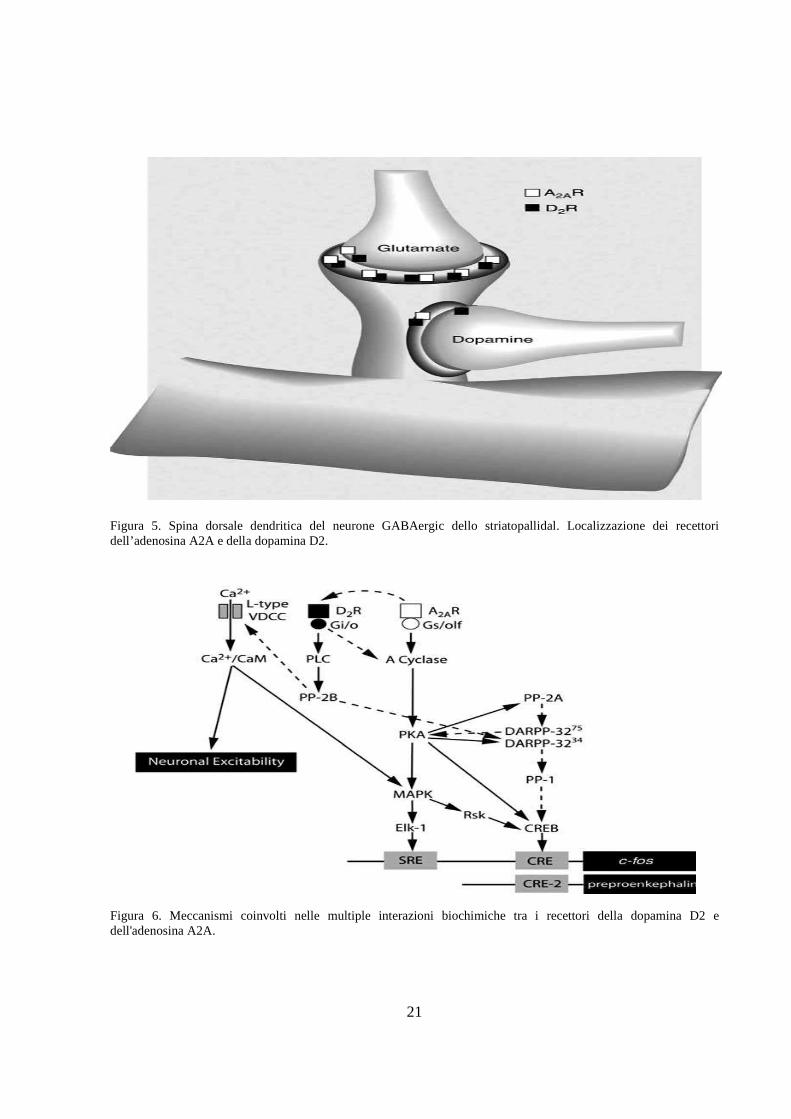
Figura 5. Spina dorsale dendritica del neurone GABAergic dello striatopallidal. Localizzazione dei recettori dell’adenosina A2A e della dopamina D2.
Figura 6. Meccanismi coinvolti nelle multiple interazioni biochimiche tra i recettori della dopamina D2 e dell'adenosina A2A.
22
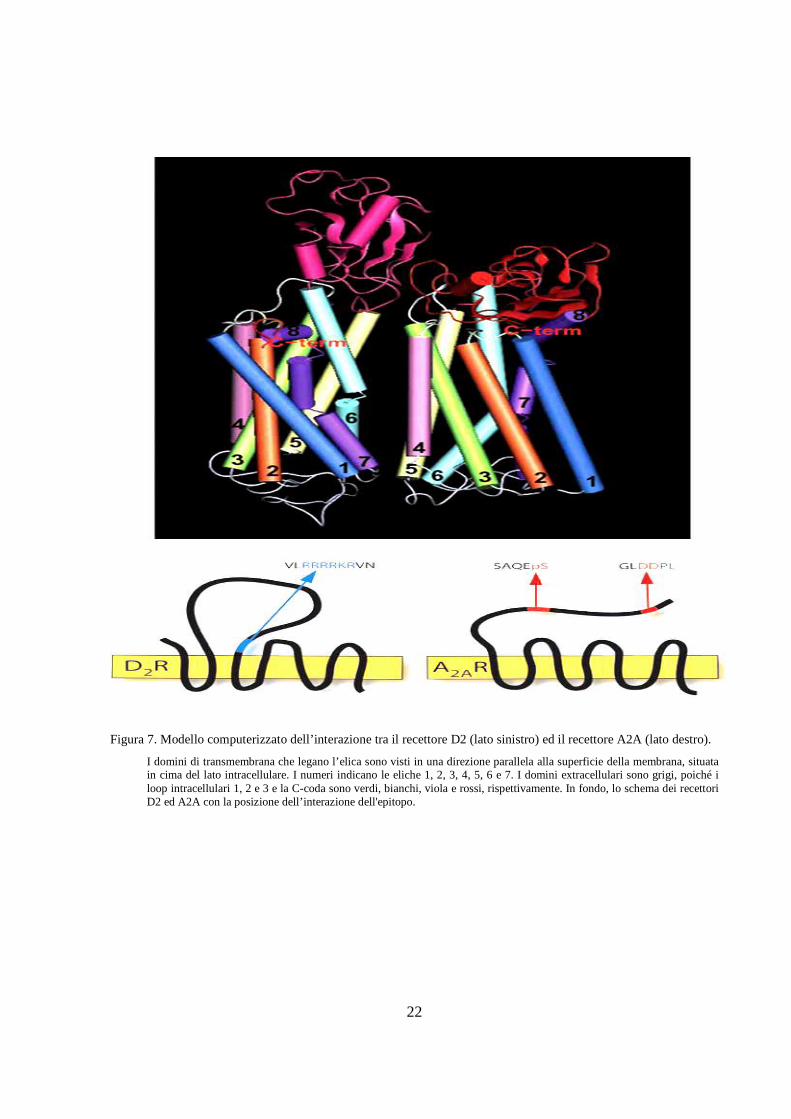
Figura 7. Modello computerizzato dell’interazione tra il recettore D2 (lato sinistro) ed il recettore A2A (lato destro).
I domini di transmembrana che legano l’elica sono visti in una direzione parallela alla superficie della membrana, situata in cima del lato intracellulare. I numeri indicano le eliche 1, 2, 3, 4, 5, 6 e 7. I domini extracellulari sono grigi, poiché i loop intracellulari 1, 2 e 3 e la C-coda sono verdi, bianchi, viola e rossi, rispettivamente. In fondo, lo schema dei recettori D2 ed A2A con la posizione dell’interazione dell'epitopo.

23
Interazioni a livello biochimico
L’interazione tra i recettori A2A e D2 è stata mostrata usando i test della
coimmunoprecipitazione e la tecnica della risonanza di trasferimento di energia di fluorescenza
e bioluminescenza (FRET o BRET) su cervelli di topo sprague-dawley. A livello biochimico, due
tipi di interazioni antagonistiche del recettore A2A/D2 sono state scoperte e ciò potrebbe
spiegare le interazioni del recettore A2A/D2 che hanno descritto entrambi a livello neuronale e
del comportamento [76-80]. Dapprima, per mezzo di un'interazione allosterica nel recettore
eteromero, lo stimolo del recettore A2A diminuisce l'affinità del recettore D2 per i loro agonisti
[81]. Poi, la stimolazione del recettore D2 alla proteina G i/o inibisce l'accumulazione di cAMP
indotta dalla stimolazione del recettore A2A alle proteine G s/olf [76,79,80]. Nella buona
conoscenza del ruolo della dopamina nella malattia del Parkinson, della schizofrenia e
dell’abuso di farmaci, è stato suggerito che le interazioni del recettore A2A/D2 nel sistema
centrale nervoso potrebbero fornire dei nuovi approcci terapeutici per combattere questi
disordini [78,82]. Un'interazione elettrostatica epitopo-epitopo tra un epitope ricco di Arginina
del N terminal del terzo loop endocellulare (3IL) del recettore D2 ed un epitope contenente una
Serina fosforilata localizzata nella parte distale del C terminal del recettore A2A sono coinvolti
nell'interfaccia dell’eteromero del recettore A2A/D2 [83-85]. Lo stesso epitopo ricco di Arginina
del recettore D2 può interagire con la CaM [86-89]. In mancanza di residui fosforilati, adiacenti
gli aspartati o i glutammati, che sono abbondanti nella CaM, possono così formare complessi
non-covalenti con l'epitopo ricco di Arginina [90]. Quindi, la CaM può trasportare
potenzialmente un segnale Ca++ al recettore D2 attraverso un legame diretto al 3IL del
recettore D2 [86]. I dati di spettrometria di massa hanno mostrato che CaM del bovino può
formare dei complessi multipli non-covalenti con un peptide ricco di Arginina corrispondente
alla regione del N-terminaI del 3IL del recettore D2 (VLRRRRKRVN) [88] come pure un peptide
dal prossimale C terminal del recettore A2A [88]. Quest'epitopo, la cui sequenza è RIREFRQTFR
nel recettore umano A2A, contiene anche diversi residui di Arginina. Poiché la sospettata
interazione tra il recettore A2A e CaM attendevano la conferma dai test che usano delle
proteine complete, lo studio attuale è stato intrapreso per dimostrare l'esistenza di interazioni

24
tra il recettore A2A e CaM entrambi in un sistema celIulare di espressione della proteina
ricombinante nel cervello. Un approccio proteomics è stato usato per la scoperta di interazioni
proteina-proteina tra il recettore A2A e CaM nel cervello del topo, poiché BRET nelle cellule del
trasporto ha dimostrato un'interazione diretta tra CaM e questo recettore. Inoltre, dall’uso di
BRET e le conseguenti tecniche di trasferimento di energia di risonanza (SRET) ed analizzando il
segnale MAPK nelle cellule di transporto, la prova è stata ottenuta per l’oligomerizzazione
CaM-A2A-D2 del recettore ed un selettivo Ca++ ha mediato la funzione della modulazione del
recettore A2A e D2 nel recettore eteromero A2A/D2 (figura 8). Dai test fatti c’e la prova per la
legatura della CaM ai recettori dell’adenosina A2A (figura 9). È stato dimostrato che la sequenza
RIREFRQTFR, che è presente nella parte prossimale del C terminale del recettore A2A, è il
dominio di legatura della CaM in questi recettori. Inoltre, formano la prova per
l’oligomerizzazione del recettore CaM-A2A-D2 e per una specifìca modulazione Ca++
dipendente della funzione del recettore eteromero A2A-D2. La legatura di CaM al recettore D2
rompe l'agganciamento di Gi/o e quindi la sua capacità di inibire l'attività dell’adenilato
ciclasico. I test suggeriscono che CaM e il recettore A2A potrebbero interagire, ma l'esistenza di
una terza proteina che costruisce un ponte non può essere esclusa. A questo scopo, i test di
BRET sono stati eseguiti nelle cellule transfected con il cDNAs per le proteine di fusione
A2ARRluc e CaMYFP. L'iperbole ottenuta aumenta il rapporto di YFP/Rluc indica che
un'interazione specifica tra CaM e il recettore A2A può accadere nelle cellule viventi.
Esperimenti simili sono stati eseguiti usando D2RRluc e CaMYFP a confermare che queste due
proteine potrebbero stabilire delle interazioni molecolari dirette. La specificità delle interazioni
CaM e recettore A2A o CaM e recettore D2 nelle ceIlule HEK è stata confermata dal non
specifico segnale di BRET ottenuto quando a testato A1RRluc e CaMYFP. L'immunoIocalizzazione
dei recettori e CaM sono stati eseguiti nelle cellule HEK tranfettate. Quando espresse in
mancanza dei recettori, la CaM ha mostrato una localizzazione citosolica. L’espressione della
CaM e qualsiasi dei due recettori non ha modificato la localizzazione del recettore (figura 10).
D'altra parte, una localizzazione significativa della membrana della CaM è stata solo osservata
quando la proteina era espressa con entrambi i recettori A2A o D2. Gli esperimenti di BRET sono

25
stati eseguiti per dare conoscenza nel ruolo del Ca++ nell'interazione di CaM con entrambi i
recettori A2A o D2 (figura 11). Da una parte, questi dati indicano che Ca++ non ha avuto bisogno
per CaM di poter interagire con i recettori A2A o D2. D'altra parte, l’aumento di ionoforo induce
nella concentrazione intracellulare di Ca++ non rompe l'oligomerizzazione del CaM con i
recettori ma conduce ai cambiamenti di conformazione nella molecola CaM e/o i recettori che
altera la distanza tra Rluc e YFP (figura 12). Per mezzo delle sperimentazioni di competizione di
BRET eseguite con le cellule che esprimono A2A Rluc e CaM YFP, l'aumento dell’espressione del
recettore D2 non ha modificato il segnale di BRET dovuto a A2RRluc e CaMYFP (figura 13).
Analogamente, aumentando l'espressione di CaM non ha modificato il segnale di BRET dovuto a
A2RRluc e D2RYFP. Queste scoperte mettono in relazione la capacità potenziale dei recettori di
CaM ed A2A per legare a qualsiasi dei due epitopi ricchi di Arginina del 3IL del recettore D2L. La
specificità di questi effetti è stata dimostrata dall'indipendenza del segnale di BRET con
l'espressione crescente del recettore A1. Lo studio attuale dimostra l'esistenza delle interazioni
intermolecolari oligomeriche tra i recettori CaM, A2A e D2. Quando non espressi insieme, i
recettori A2A o D2 potrebbero legare potenzialmente la CaM, ma nelle cellule che esprimono gli
eteromeri del recettore A2A-D2, i legami del CaM preferenzialmente al recettore A2A (figura
14). Inoltre, la CaM trasduce i cambiamenti di Ca++ dipendenti delle MAPK che segnalano
nell’oligomero del recettore CaM-A2A-D2 (figura 15). Questo lavoro aggiunge delle nuove
informazioni della funzione degli eteromeri del recettore A2A-D2, che sono considerati come un
bersaglio per lo sviluppo degli agenti anti-parkinsonian.
26
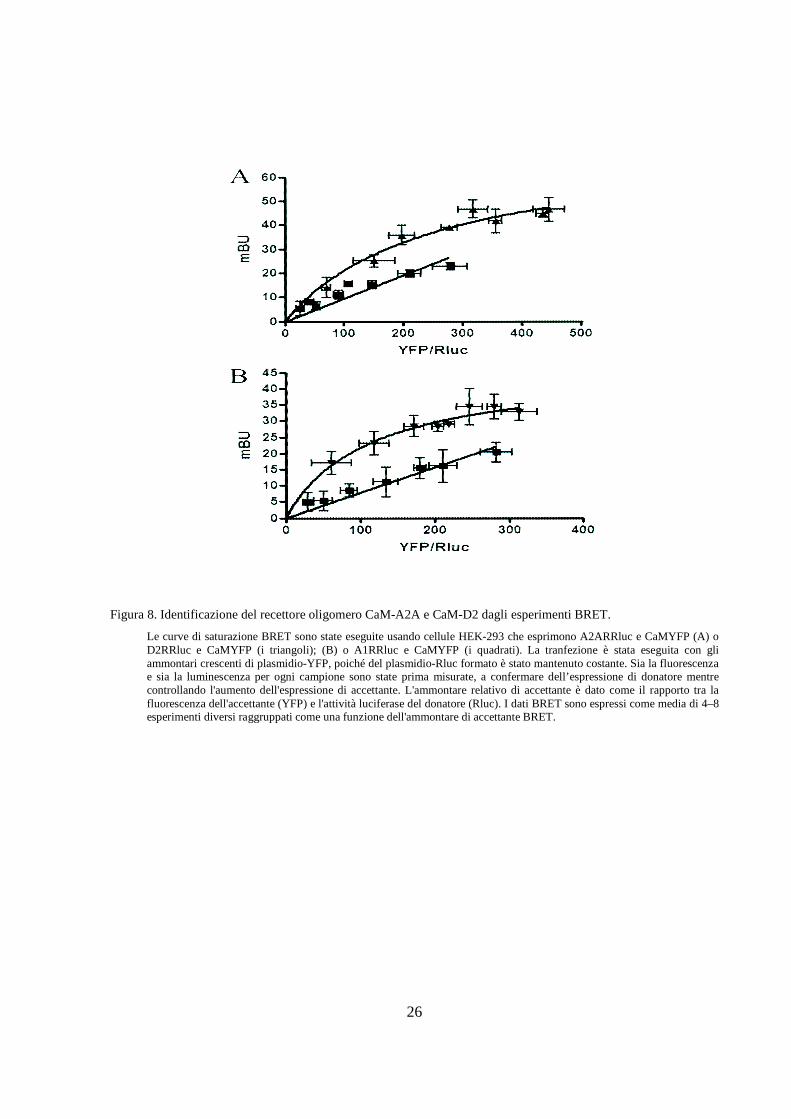
Figura 8. Identificazione del recettore oligomero CaM-A2A e CaM-D2 dagli esperimenti BRET.
Le curve di saturazione BRET sono state eseguite usando cellule HEK-293 che esprimono A2ARRluc e CaMYFP (A) o D2RRluc e CaMYFP (i triangoli); (B) o A1RRluc e CaMYFP (i quadrati). La tranfezione è stata eseguita con gli ammontari crescenti di plasmidio-YFP, poiché del plasmidio-Rluc formato è stato mantenuto costante. Sia la fluorescenza e sia la luminescenza per ogni campione sono state prima misurate, a confermare dell’espressione di donatore mentre controllando l'aumento dell'espressione di accettante. L'ammontare relativo di accettante è dato come il rapporto tra la fluorescenza dell'accettante (YFP) e l'attività luciferase del donatore (Rluc). I dati BRET sono espressi come media di 4–8 esperimenti diversi raggruppati come una funzione dell'ammontare di accettante BRET.
27
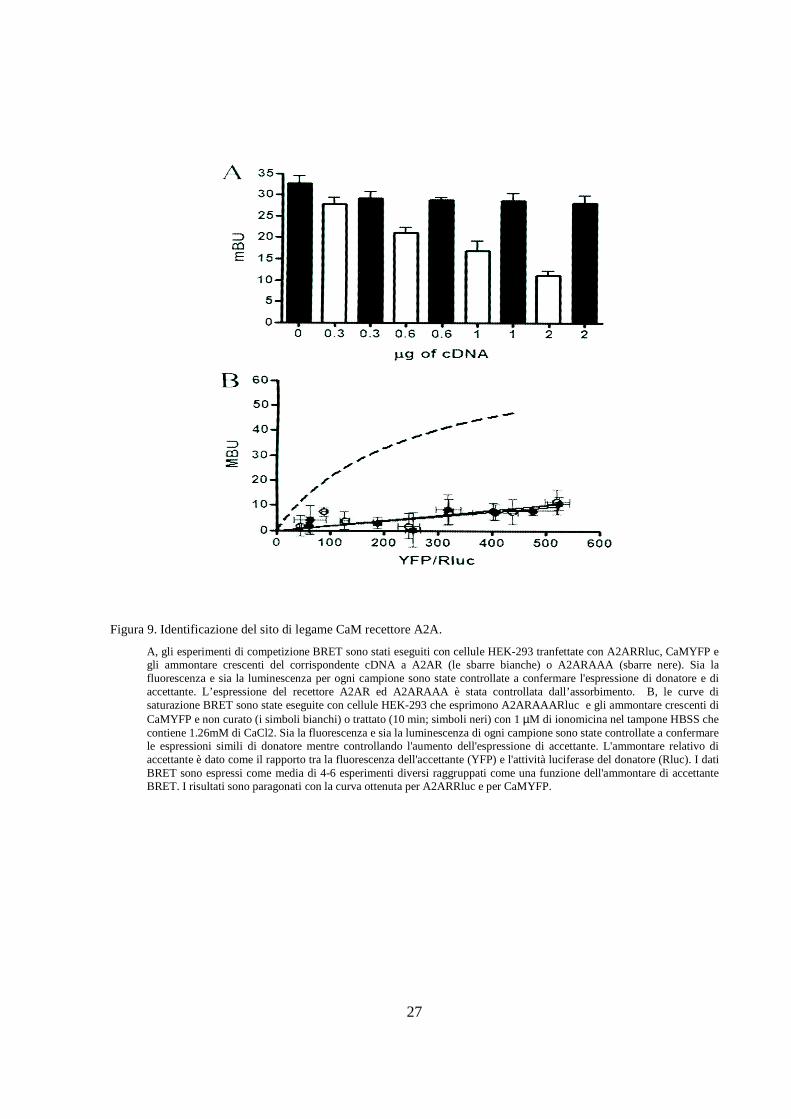
Figura 9. Identificazione del sito di legame CaM recettore A2A.
A, gli esperimenti di competizione BRET sono stati eseguiti con cellule HEK-293 tranfettate con A2ARRluc, CaMYFP e gli ammontare crescenti del corrispondente cDNA a A2AR (le sbarre bianche) o A2ARAAA (sbarre nere). Sia la fluorescenza e sia la luminescenza per ogni campione sono state controllate a confermare l'espressione di donatore e di accettante. L’espressione del recettore A2AR ed A2ARAAA è stata controllata dall’assorbimento. B, le curve di saturazione BRET sono state eseguite con cellule HEK-293 che esprimono A2ARAAARluc e gli ammontare crescenti di CaMYFP e non curato (i simboli bianchi) o trattato (10 min; simboli neri) con 1 µM di ionomicina nel tampone HBSS che contiene 1.26mM di CaCl2. Sia la fluorescenza e sia la luminescenza di ogni campione sono state controllate a confermare le espressioni simili di donatore mentre controllando l'aumento dell'espressione di accettante. L'ammontare relativo di accettante è dato come il rapporto tra la fluorescenza dell'accettante (YFP) e l'attività luciferase del donatore (Rluc). I dati BRET sono espressi come media di 4-6 esperimenti diversi raggruppati come una funzione dell'ammontare di accettante BRET. I risultati sono paragonati con la curva ottenuta per A2ARRluc e per CaMYFP.
28
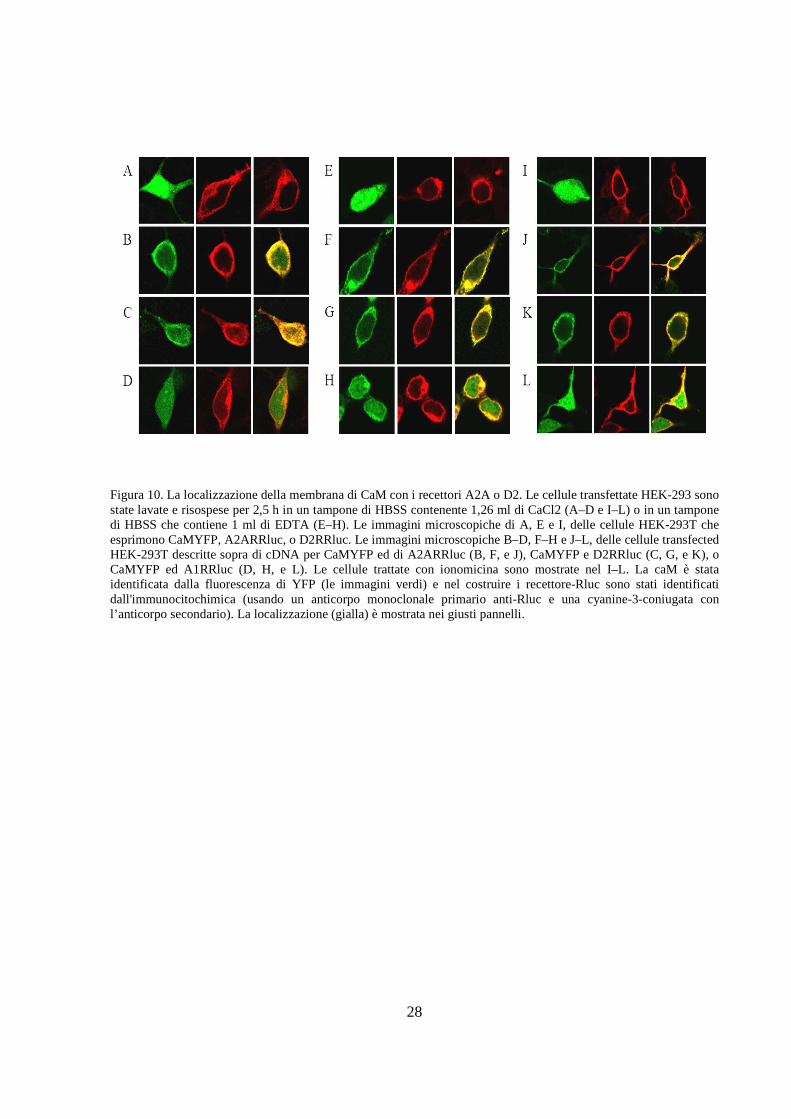
Figura 10. La localizzazione della membrana di CaM con i recettori A2A o D2. Le cellule transfettate HEK-293 sono state lavate e risospese per 2,5 h in un tampone di HBSS contenente 1,26 ml di CaCl2 (A–D e I–L) o in un tampone di HBSS che contiene 1 ml di EDTA (E–H). Le immagini microscopiche di A, E e I, delle cellule HEK-293T che esprimono CaMYFP, A2ARRluc, o D2RRluc. Le immagini microscopiche B–D, F–H e J–L, delle cellule transfected HEK-293T descritte sopra di cDNA per CaMYFP ed di A2ARRluc (B, F, e J), CaMYFP e D2RRluc (C, G, e K), o CaMYFP ed A1RRluc (D, H, e L). Le cellule trattate con ionomicina sono mostrate nel I–L. La caM è stata identificata dalla fluorescenza di YFP (le immagini verdi) e nel costruire i recettore-Rluc sono stati identificati dall'immunocitochimica (usando un anticorpo monoclonale primario anti-Rluc e una cyanine-3-coniugata con l’anticorpo secondario). La localizzazione (gialla) è mostrata nei giusti pannelli.
29
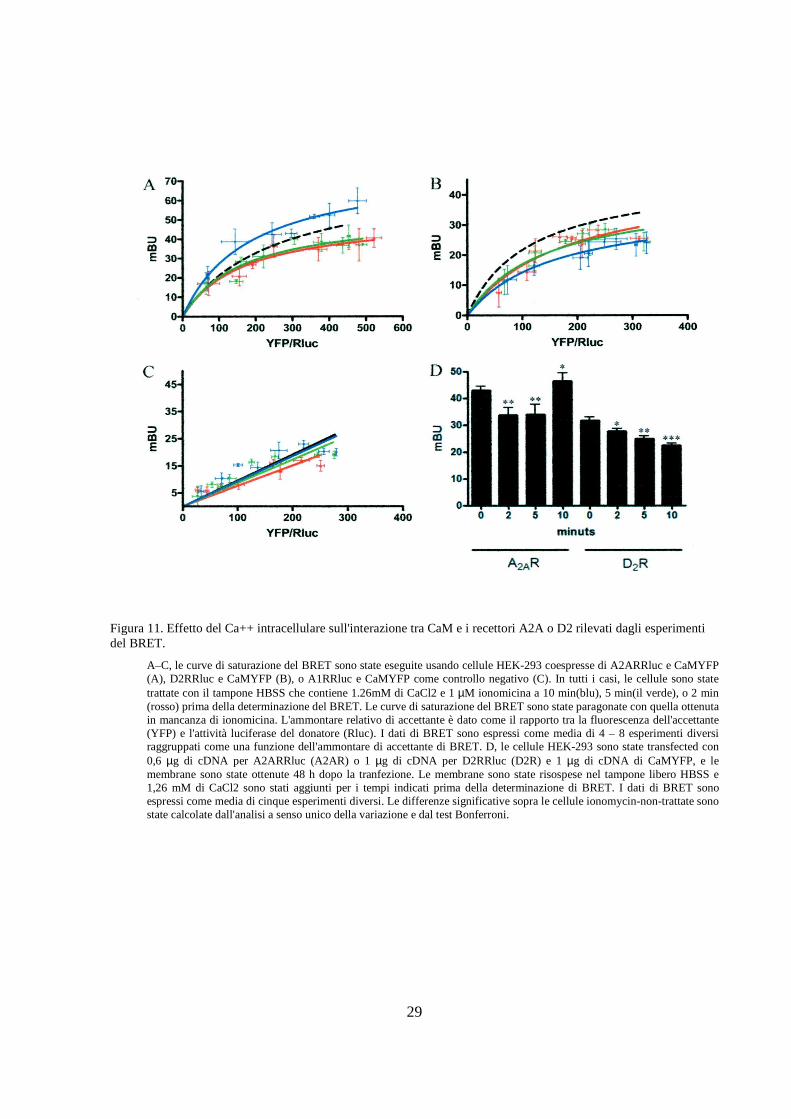
Figura 11. Effetto del Ca++ intracellulare sull'interazione tra CaM e i recettori A2A o D2 rilevati dagli esperimenti del BRET.
A–C, le curve di saturazione del BRET sono state eseguite usando cellule HEK-293 coespresse di A2ARRluc e CaMYFP (A), D2RRluc e CaMYFP (B), o A1RRluc e CaMYFP come controllo negativo (C). In tutti i casi, le cellule sono state trattate con il tampone HBSS che contiene 1.26mM di CaCl2 e 1 µM ionomicina a 10 min(blu), 5 min(il verde), o 2 min (rosso) prima della determinazione del BRET. Le curve di saturazione del BRET sono state paragonate con quella ottenuta in mancanza di ionomicina. L'ammontare relativo di accettante è dato come il rapporto tra la fluorescenza dell'accettante (YFP) e l'attività luciferase del donatore (Rluc). I dati di BRET sono espressi come media di 4 – 8 esperimenti diversi raggruppati come una funzione dell'ammontare di accettante di BRET. D, le cellule HEK-293 sono state transfected con 0,6 µg di cDNA per A2ARRluc (A2AR) o 1 µg di cDNA per D2RRluc (D2R) e 1 µg di cDNA di CaMYFP, e le membrane sono state ottenute 48 h dopo la tranfezione. Le membrane sono state risospese nel tampone libero HBSS e 1,26 mM di CaCl2 sono stati aggiunti per i tempi indicati prima della determinazione di BRET. I dati di BRET sono espressi come media di cinque esperimenti diversi. Le differenze significative sopra le cellule ionomycin-non-trattate sono state calcolate dall'analisi a senso unico della variazione e dal test Bonferroni.
30
Figura 12. L’interazione CaM con i recettori eterodimerici A2A-D2 scoperti dagli esperimenti di competizione BRET.
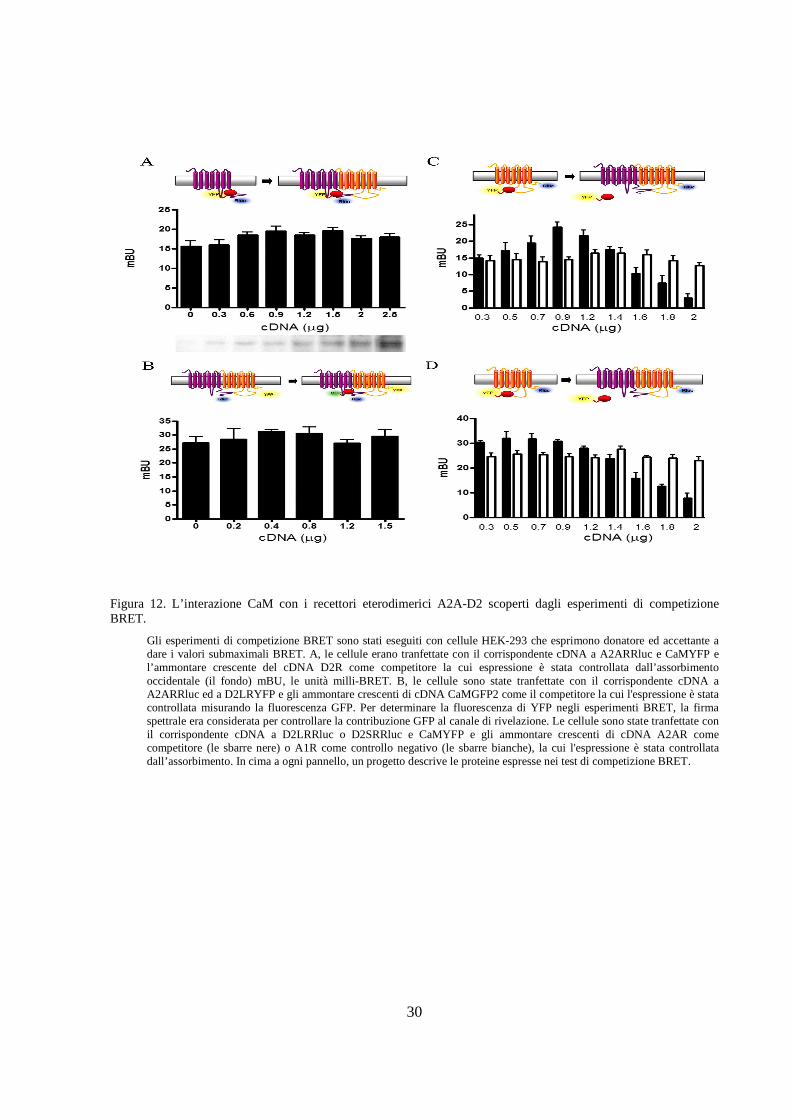
Gli esperimenti di competizione BRET sono stati eseguiti con cellule HEK-293 che esprimono donatore ed accettante a dare i valori submaximali BRET. A, le cellule erano tranfettate con il corrispondente cDNA a A2ARRluc e CaMYFP e l’ammontare crescente del cDNA D2R come competitore la cui espressione è stata controllata dall’assorbimento occidentale (il fondo) mBU, le unità milli-BRET. B, le cellule sono state tranfettate con il corrispondente cDNA a A2ARRluc ed a D2LRYFP e gli ammontare crescenti di cDNA CaMGFP2 come il competitore la cui l'espressione è stata controllata misurando la fluorescenza GFP. Per determinare la fluorescenza di YFP negli esperimenti BRET, la firma spettrale era considerata per controllare la contribuzione GFP al canale di rivelazione. Le cellule sono state tranfettate con il corrispondente cDNA a D2LRRluc o D2SRRluc e CaMYFP e gli ammontare crescenti di cDNA A2AR come competitore (le sbarre nere) o A1R come controllo negativo (le sbarre bianche), la cui l'espressione è stata controllata dall’assorbimento. In cima a ogni pannello, un progetto descrive le proteine espresse nei test di competizione BRET.
31
Figura 13. SRET per CaM, recettore A2A e recettore D2 nelle cellule viventi.
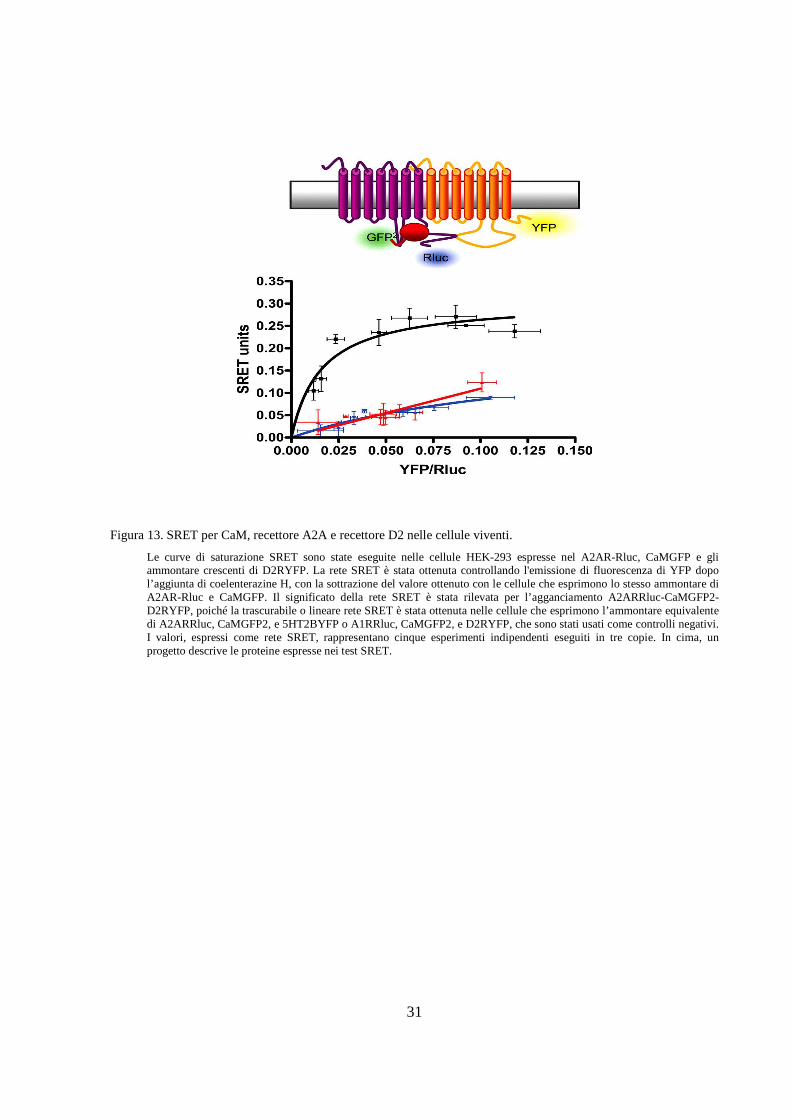
Le curve di saturazione SRET sono state eseguite nelle cellule HEK-293 espresse nel A2AR-Rluc, CaMGFP e gli ammontare crescenti di D2RYFP. La rete SRET è stata ottenuta controllando l'emissione di fluorescenza di YFP dopo l’aggiunta di coelenterazine H, con la sottrazione del valore ottenuto con le cellule che esprimono lo stesso ammontare di A2AR-Rluc e CaMGFP. Il significato della rete SRET è stata rilevata per l’agganciamento A2ARRluc-CaMGFP2-D2RYFP, poiché la trascurabile o lineare rete SRET è stata ottenuta nelle cellule che esprimono l’ammontare equivalente di A2ARRluc, CaMGFP2, e 5HT2BYFP o A1RRluc, CaMGFP2, e D2RYFP, che sono stati usati come controlli negativi. I valori, espressi come rete SRET, rappresentano cinque esperimenti indipendenti eseguiti in tre copie. In cima, un progetto descrive le proteine espresse nei test SRET.
32
Figura 14. Effetto del Ca++ intracellulare sulle interazioni molecolari tra CaM, i recettori A2A, and D2.
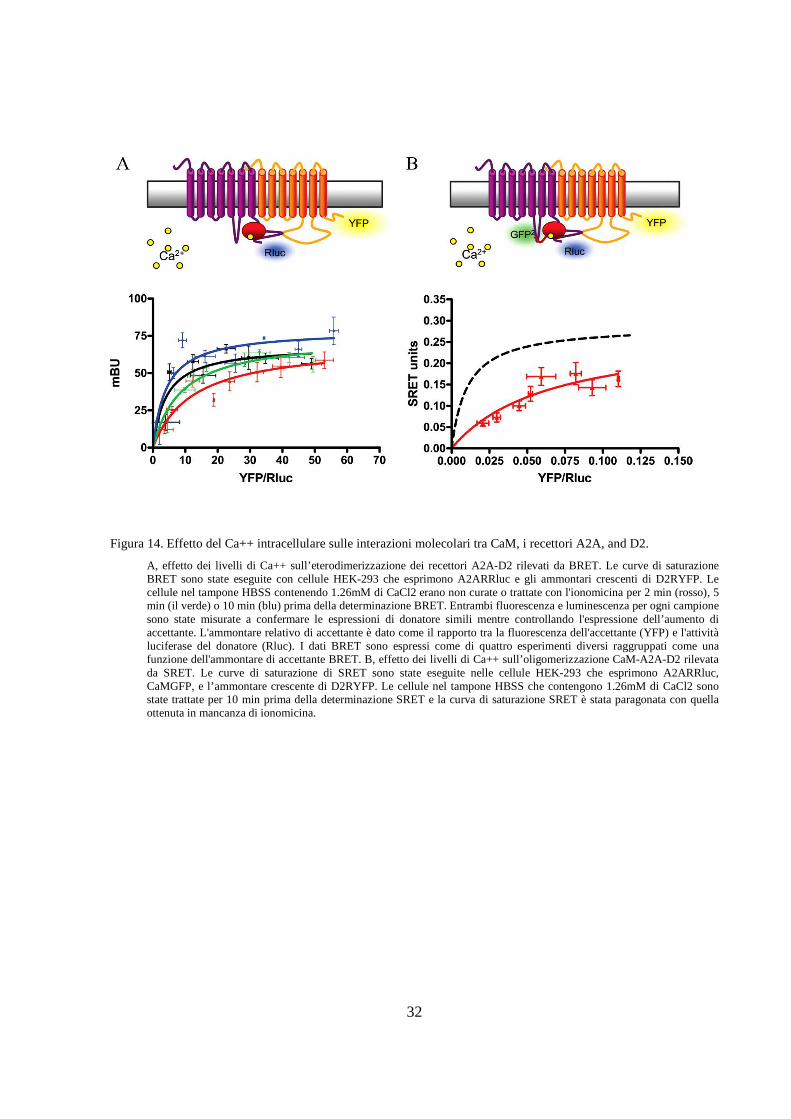
A, effetto dei livelli di Ca++ sull’eterodimerizzazione dei recettori A2A-D2 rilevati da BRET. Le curve di saturazione BRET sono state eseguite con cellule HEK-293 che esprimono A2ARRluc e gli ammontari crescenti di D2RYFP. Le cellule nel tampone HBSS contenendo 1.26mM di CaCl2 erano non curate o trattate con l'ionomicina per 2 min (rosso), 5 min (il verde) o 10 min (blu) prima della determinazione BRET. Entrambi fluorescenza e luminescenza per ogni campione sono state misurate a confermare le espressioni di donatore simili mentre controllando l'espressione dell’aumento di accettante. L'ammontare relativo di accettante è dato come il rapporto tra la fluorescenza dell'accettante (YFP) e l'attività luciferase del donatore (Rluc). I dati BRET sono espressi come di quattro esperimenti diversi raggruppati come una funzione dell'ammontare di accettante BRET. B, effetto dei livelli di Ca++ sull’oligomerizzazione CaM-A2A-D2 rilevata da SRET. Le curve di saturazione di SRET sono state eseguite nelle cellule HEK-293 che esprimono A2ARRluc, CaMGFP, e l’ammontare crescente di D2RYFP. Le cellule nel tampone HBSS che contengono 1.26mM di CaCl2 sono state trattate per 10 min prima della determinazione SRET e la curva di saturazione SRET è stata paragonata con quella ottenuta in mancanza di ionomicina.
33
Figura 15. Effetto del Ca++ intracellulare sulla fosforilazione mediata ERK 1/2 dei recettori eterodimeri A2A-, D2-, e A2A-D2.
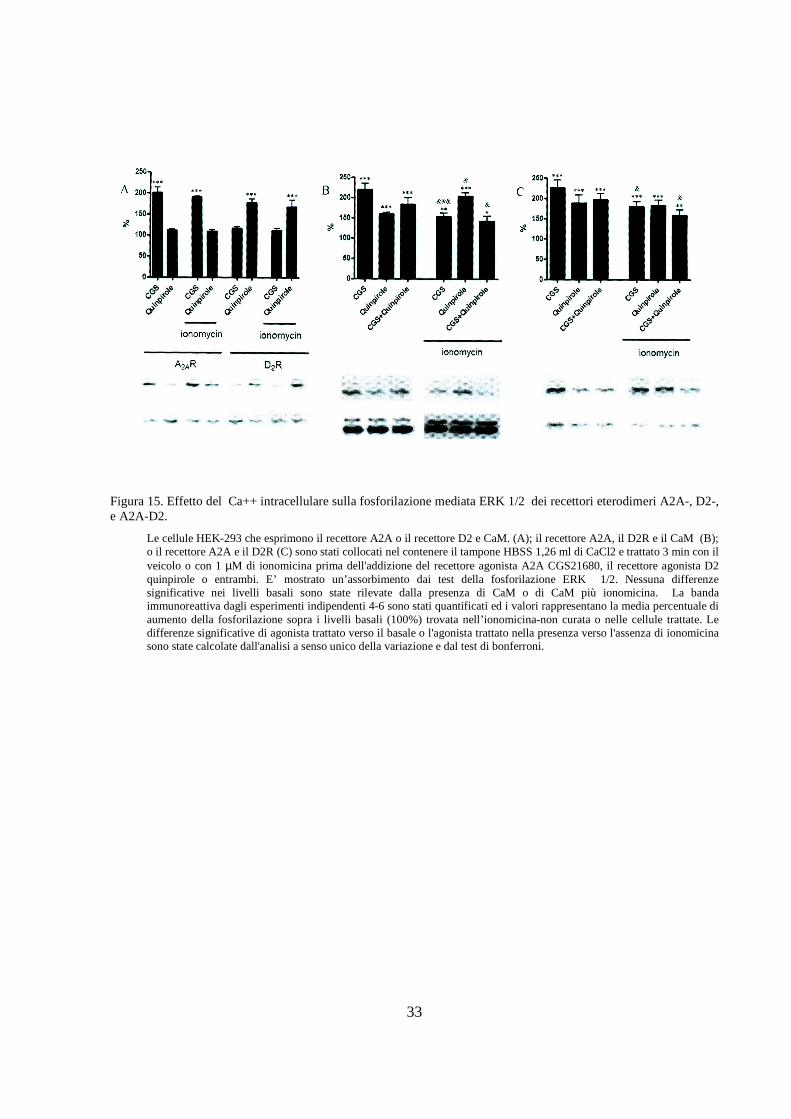
Le cellule HEK-293 che esprimono il recettore A2A o il recettore D2 e CaM. (A); il recettore A2A, il D2R e il CaM (B); o il recettore A2A e il D2R (C) sono stati collocati nel contenere il tampone HBSS 1,26 ml di CaCl2 e trattato 3 min con il veicolo o con 1 µM di ionomicina prima dell'addizione del recettore agonista A2A CGS21680, il recettore agonista D2 quinpirole o entrambi. E’ mostrato un’assorbimento dai test della fosforilazione ERK 1/2. Nessuna differenze significative nei livelli basali sono state rilevate dalla presenza di CaM o di CaM più ionomicina. La banda immunoreattiva dagli esperimenti indipendenti 4-6 sono stati quantificati ed i valori rappresentano la media percentuale di aumento della fosforilazione sopra i livelli basali (100%) trovata nell’ionomicina-non curata o nelle cellule trattate. Le differenze significative di agonista trattato verso il basale o l'agonista trattato nella presenza verso l'assenza di ionomicina sono state calcolate dall'analisi a senso unico della variazione e dal test di bonferroni.

34
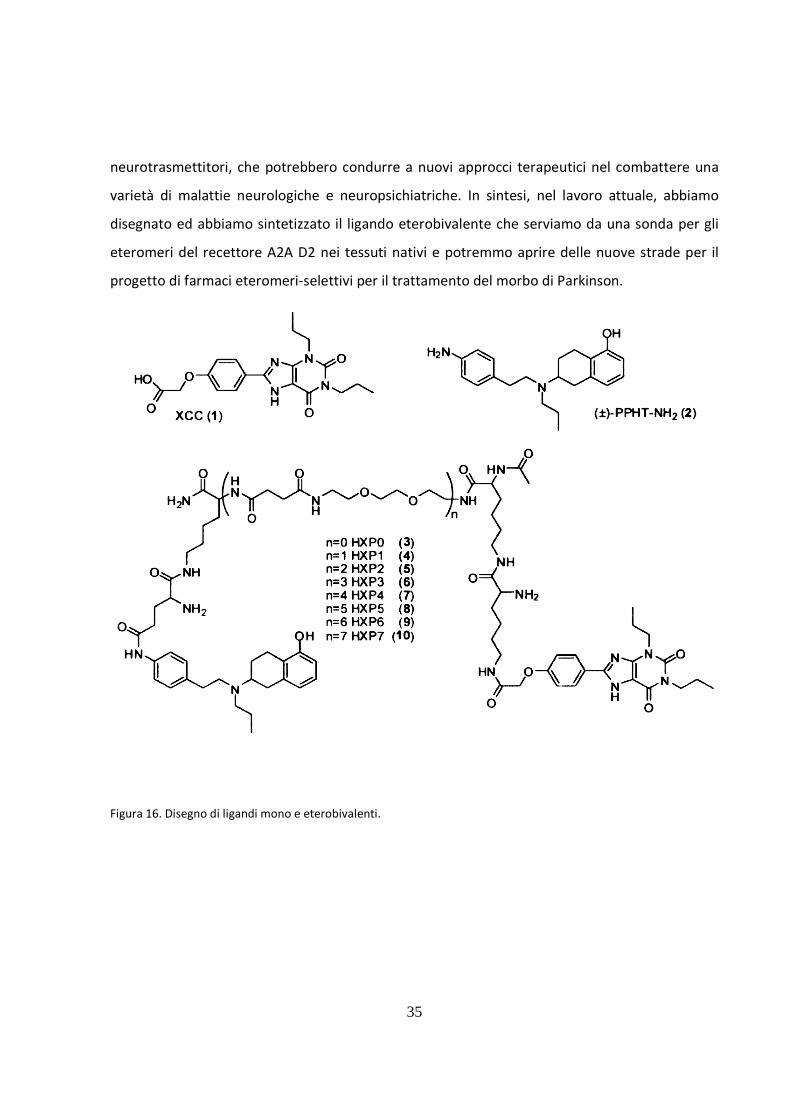
Ligandi bivalenti
E’ stato disegnato ed è stato sintetizzato per studiare gli eteromeri A2A/D2, una famiglia di
ligandi etero bivalenti che contengono collegato un agonista D2 e un antagonista A2A. Queste
scoperte indicano l'esistenza degli eteromeri dei recettori A2A/D2 e di una simultanea
interazione dei ligandi etero bivalente con entrambi i recettori. L'effetto cooperativo derivato
dall'interazione simultanea suggerisce l'evento degli eteromeri A2A/D2 nelle cellule
cotransfected e nello striatum del cervello. L’azione bivalente dopamina/adenosina potrebbe
costituire un nuovo concetto nella farmacoterapia del morbo del Parkinson. Nello studio
attuale, hanno sintetizzato test di ligandi eterobivalenti costituiti da due metà farmacoforiche,
XCC (1) come recettore A2A antagonista e (±)-PPHT-NH2 (2) come recettore D2 agonista, che
sono collegati tramite linkers Lys-Lys-[PEG/polyamide]n-Lys-Glu (figura 16). Questi risultati
indicano che un Iigando eterobivalente legato al D2R può successivamente localizzarsi nella
vicinanza del A2AR con una più alta affinità del corrispondere ligando A2AR monovalente. Allo
stesso modo, un composto eterobivalente al A2AR in seguito legato a D2R localizzato in più
vicino la prossimità con una più alta affinità del suo contrasto D2R monovalente. Questo indica
un fattore di concentrazione significativo esercitato dal Iigando eterobivalente che può essere
solo considerato se entrambi i recettori sono in una prossimità molto vicina. Così, il ligando
eterobivalente di dopamina-adenosina è usato per mostrare l'evento di questo eteromero nello
striatum del cervello e l'affinità aumentata di questo ligando ha rilevato che quando entrambi i
recettori sono presenti è un'indicazione forte di legatura all'eteromero del recettore A2A-D2. Le
potenti tecniche biofisiche come il trasferimento di energia di risonanza di fluorescenza (FRET) e
il trasferimento di energia di risonanza della bioluminescenza (BRET) sono stati decisivi nel
dimostrare la formazione di eteromeri nei sistemi di espressione dei sistemi eterologhi. Il
ligando eterobivalente sono degli strumenti di valore farmacologico che consente la
dimostrazione dell'esistenza degli eteromeri del recettore A2A-D2 anche nel tessuto nativo e
può essere usato per studiare il comportamento di un specifico dimero GPCR senza qualunque
modifica di recettore. Inoltre, le strategie di ligando etero bivalente possono essere sviluppate
per identificare gli eteromeri che coinvolgono gli altri recettori neuromodulatori e/o
35
neurotrasmettitori, che potrebbero condurre a nuovi approcci terapeutici nel combattere una
varietà di malattie neurologiche e neuropsichiatriche. In sintesi, nel lavoro attuale, abbiamo
disegnato ed abbiamo sintetizzato il ligando eterobivalente che serviamo da una sonda per gli
eteromeri del recettore A2A D2 nei tessuti nativi e potremmo aprire delle nuove strade per il
progetto di farmaci eteromeri-selettivi per il trattamento del morbo di Parkinson.
Figura 16. Disegno di ligandi mono e eterobivalenti.

36
6. COMPLESSO D2/D3
I recettori D2 e D3 sono noti esistere come i monomeri e come gli omomeri [91,92]. Nel 2001,
l’ulteriore prova venuta dai recettori D2 e D3 possono formare gli eteromeri con le proprietà
uniche funzionali [93]. Usando la coimmunoprecipitazione si sperimentano differenti recettori
etichettati D2 e D3 ed hanno mostrato che i recettori D2 e D3 nelle cellule HEK-293 possono
formare un complesso eteromerico. Inoltre, potevano dimostrare che i frammenti della coda del
recettore di dopamina potrebbero formare i recettori di dopamina funzionali che limitano gli
agonisti e gli antagonisti della dopamina con un profilo farmacologico diverso paragonato con
recettori nativi D2 e D3, con l'affinità più alta di tutti essendo trovati con la combinazione dei
frammenti della coda. Così, gli eteromeri D2/D3 potrebbero essere formati attraverso il
meccanismo del cambio del dominio come proposto da Gouldson e da Reynolds [94,95] che si
sono basati sulle dimostrazioni della complementazione funzionale tra le chimere dei recettori
α2 adrenergici e M3 muscarinico [96]. In accordo con la formazione degli eteromeri D2/D3 nelle
cellule, questi tipi di frammenti del recettore D2 e D3, quando coespressi con i recettori nativi
D2 e D3, hanno ridotto l'espressione dei recettori della dopamina nativi indicando la complessa
formazione del recettore frammento/nativo. Comunque, non solo sono recettori D2 trunc/D3
tail e D3 trunc/D2 tail capaci di legare i ligandi ma possono agganciare anche in una maniera
inibitoria all’adenilato ciclasico e alla stessa estensione come il recettore nativo D2. È anche di
interesse sostanziale che il recettore D3 sotto le condizioni in cui non può inibire l’adenilato
ciclasico VI può sviluppare tale agganciamento dal trasporto con i recettori D2. È quindi possibile
che nel complesso del recettore eteromerico D2/D3 formato, il recettore D2 può fare il possibile
all’aggancio alla proteina-G del recettore D3 all’adenilato ciclasico VI. Altrimenti, il recettore D3
che lega la tasca sull'attivazione dagli agonisti D3 può, attraverso i cambiamenti di
conformazione, trasferire la tasca D2 dell’eteromero in uno stato attivato, conducendo
all’attivazione di Gi e all’inibizione di adenilato ciclasico VI. Finalmente, nel caso dell’attività
dell’adenilato ciclasico V, la coespressione dei recettori D2 e D3 sono risultati anche in
un’aumentata potenza dell’agonista D3 per inibire questo adenilato ciclasico paragonato con il

37
recettore wild-tipo D2 quando attivato dagli agonisti D2. Una funzione del complesso
recettoriale eteromerico D2/D3 potrebbe essere quindi di consentire un agganciamento
inibitorio più forte dei recettori D3 all’adenilato ciclasico. La localizzazione dei recettori D2 e D3
è stata dimostrata nelle cellule nervose dei gangli basali [97,98] mostrando che c'è il potenziale
a formare l’eteromero funzionale D2/D3 anche nel vivo.

38
7. COMPLESSO SSTR5/SSTR1
Lo studio di Rocheville [99] dà una bell'illustrazione delle interazioni intramembrana
recettore/recettore sulla loro pertinenza funzionale e del rapporto dei monomeri, dei omomeri, e
degli eteromeri fra cinque sottotipi recettoriali di somatostatina. Usando l'analisi FRET, il sottotipo
recettoriale umano della somatostatina SSTR5 è stato mostrato per esistere come un monomero
nello stato basale, e sull'attivazione dell’agonista è stato convertito in un omomero. I dati hanno
suggerito che la dimerizzazione dell’agonista-indotto del recettore SSTR5 erano essenziali per
segnalare. L'eteromerizzazione dell’agonista-indotto dei recettori SSTR5 e SSTR1 e potrebbe
anche essere dimostrata per lo specifico sottotipo. Il suggerimento è stato fatto che l'alto livello
riferito dell’espressione dell’omomero basale del GPCR potrebbe essere dovuto all'over-
espressione del recettore. Le interazioni intramembrana recettore/recettore del SSTR5 e SSTRI
attraverso l'eteromerizzazione sono stati mostrati per avere delle conseguenze funzionali
importanti per i recettori che partecipano. Oltre ai cambiamenti nell'affinità dell’agonista che
sviluppano di solito sui cambiamenti nello stato di oligomerico, le alterazioni marcate
nell'internalizzare e nella regolazione dell’agonista-dipendente dei recettori SSTR1 sono accadute
attraverso la formazione di un eteromero con i recettori SSTR5. Così, il solo recettore SSTR1 ha
subito solo l'internalizzazione agonista-indotto come un eterodimero con il recettore SSTR5.
Inoltre, l'eteromerizzazione ha consentito all’agonista del recettore della somatostatina a indurre
la regolazione di legatura dell’agonista al recettore SSTRI. Il recettore SSTR5 che segnala alla
proteina-G non è coinvolto probabilmente in questa risposta dell'eteromero, poiché la coda del
COOH-terminale del recettore SSTR5 era stato cancellato abolendo la regolazione di adenilato
ciclasico. Questi risultati sono di interesse sostanziale da allora in questa maniera la
desensibilizzazione dei sottotipi recettoriali di somatostatina attivata può essere compensata da
una regolazione dei sottotipi del recettore della somatostatina non attivata, come SSTR1 ha fatto
il possibile attraverso l'eteromerizzazione. Così, un altro significato funzionale delle interazioni
intramembrane recettore/recettore tramite l'eteromerizzazione potrebbe essere la sensazione di
un isorecettore poiché l'altro isorecettore dell'eteromero subisce la desensibilizzazione.

39
8. COMPLESSO SSTR5/D2
La scoperta di quest'interazione intramembrana recettore/recettore attraverso l'etero
oligomerizzazione hanno dato una nuova maniera per capire le interazioni della
somatostatina/dopamina ben conosciute nel cervello, coinvolte per esempio nel controllo
dell’attività motoria [100-108]. In questo caso l'etero-oligomerizzazione ha coinvolto distinti
GPCR e non isorecettori che hanno lo stesso ligando endogeno. Era per mezzo della microscopia
FRET che l’interazione diretta recettore/recettore SSTR5/D2 potrebbe essere determinata e
l'oligomerizzazione era stata appena osservata nello stato basale ma solo dopo il trattamento
con l'altro agonista. Il trattamento simultaneo con i due tipi di agonisti non ha avuto ulteriore
azione insieme [109]. È saputo che recettori omomeri D2 esistono nello stato basale [92] e gli
omomeri della somatostatina sono indotti dagli agonisti del recettore della somatostatina.
Rimane per essere determinata quali eterodimeri sono formati o i più grandi complessi
oligomerici nel caso dell’eteromerizzazione SSTR5/D2. Il significato funzionale di
quest'interazione diretta intramembrana recettore/recettore appare avere diverse pieghe
[109]. Dapprima, la tasca obbligatoria del recettore SSTR5 è stato segnatamente alterato,
poiché un aumento di 30 pieghe nell'affinità è stata trovata sull'attivazione dell’agonista
recettoriale D2, poiché gli antagonisti del recettore D2 hanno ridotto l'affinità dei recettori
SSTR5 per l’agonista della somatostatina SST-14. Così, il differente stato conformazionale del
recettore D2 ha un'azione modulatoria sostanziosa sulla tasca obbligatoria del SSTR5. Il
riconoscimento dell'interazione al luogo di livello aveva anche migliorato l’affinità reciproca
dall'agonista della somatostatina del recettore D2 per gli antagonisti. Secondo, la proteina-G del
recettore SSTR5 è stata migliorata dall’attivazione del recettore D2, poiché la riduzione di
legatura di agonista del recettore SSTR5 dal GTP γ S è stata migliorata dall’agonista D2. Inoltre,
le risposte inibitorie sull'accumulazione di cAMP sono state considerevolmente migliorate dai
trattamenti simultanei di agonista, accentuando il miglioramento dell’attività funzionale
attraverso l'etero oligomero formato ed i cambiamenti di conformazione indotti dagli agonisti in
questo complesso. Infatti, questi cambiamenti funzionali nelle interazioni intramembrane

40
recettore/recettore indotti dagli agonisti SSTR5 e D2 potrebbero spiegare l’aumento di
neurotrasmissione mediato dei recettori della somatostatina e del D2 trovati nel vivo dopo
trattamenti con agonista somatostatina o dopamina. In questo studio, Rocheville [109] ha usato
anche il mutante recettoriale 318-SSTR5, con una soppressione della coda del COOH-terminale.
Questo recettore mutante SSTR5 era stato precedentemente mostrato per legare gli agonisti
della somatostatina con un’affinità immutata, ma diversa del recettore wild-tipo SSTR5, non può
produrre inibizione delll'accumulazione di cAMP indotta forskolin [110]. L’interessante scoperta
era con i recettori D2 e mutante 318-SSTR5 potrebbero restaurare l'agonista della
somatostatina a segnalare adenilato ciclasico fornito al sito di riconoscimento non è stato
bloccato da un antagonista D2. Queste osservazioni possono essere spiegate dalla formazione di
un etero-oligomero nelle cellule che usano CHO-K , in cui il SSTR5 la tasca di legatura quando
attivata dagli agonisti può segnalare tramite un cambiamento di conformazione nella tasca di
legatura della dopamina D2. Questo condurrebbe a un agganciamento del luogo di
riconoscimento D2 al sito della proteina-G seguita dalla sua successiva attivazione e la sua
inibizione di adenilato ciclasico. Questo cambiamento di conformazione non può accadere
quando la tasca di legatura D2 è in uno stato obbligatorio antagonistico. Indica infatti che il
recettore SSTR5 può segnalare tramite un cambiamento di conformazione nel recettore come
alla produzione dell’agonista D2. In altre parole, una cross-attivazione del recettore D2 può
accadere in mancanza di dopamina da un'interazione diretta recettore/recettore nelle
interfacce del recettore etero-oligomero. Così, il recettore SST attivato può non solo modulare
l'attivazione all’agganciamento recettoreD2/proteina G i all’adenilato ciclasico ma produce
anche un'attività costitutiva del recettore D2 quando non è chiuso a chiave in uno stato
antagonistico. Da un'altra prospettiva, rappresenta un esempio di come un GPCR mutante può
regolare il suo segnale attivando un altro recettore agganciato allo stesso tipo di proteina-G.

41
9. COMPLESSO A1/D1
L'articolo sugli eteromeri A1/D1 è stato pubblicato da un paio di mesi dopo la pubblicazione
dell'articolo del recettore oligomero SST5/D2 e dà un altro esempio di eteromerizzazione tra
distinti GPCR [104]. Un numero di osservazioni morfologiche e neurochimiche indicano che le
interazioni recettore/recettore di adenosina A1 e dopamina D1 esistono nei gangli basaIi
[63,65,111,113,114] e la localizzazione dei recettori A1 e D1 esistono nelle culture primarie
corticali [104]. L'articolo di Ginés [104] dà la prima prova che quest'interazione
recettore/recettore può coinvolgere i complessi recettoriali eteromerici A1/D1 poiché tali
complessi potrebbero essere dimostrati nel trasporto A1/D1 dei fibroblasti per mezzo della
coimmunoprecipitazione. Così, precedentemente sono stati trovati dei recettori A1 indotti del
recettore D1, ha dimostrato come la scomparsa del recettore indotto A1 dell'alta affinità del
recettore agonista D1 che si lega ai siti nelle preparazioni della membrana [65,111,112],
potrebbe essere il risultato di un'interazione fisica del recettore A1 con il recettore D1 in questo
complesso eteromerico, conducendo allo spaiare del recettore D1 alla sua proteina G in questa
interazione funzionale di complesso eteromerico. L'analisi della coimmunoprecipitazione
dimostra la sua esistenza già nello stato basale e la sua specificità per mostrare
l'eteromerizzazione del recettore A1/D2 nelle cellule di trasporto dei fibroblasti del recettore
A1/D2. Comunque, l'eteromerizzazione recettoriale A1/D1 nelle cellule di fibroblasti è stata
fortemente ridotta dal trattamento di agonista del recettore D1, mostrando una dipendenza
dell’agonista ed il blocco simultaneo del trattamento dell’agonista recettoriale D1 e A1 con
questa rottura del complesso eteromerico. Così, come lo studio di Rocheville [109], questo
studio mostra come trattamenti di agonisti soli o simultanei ai cambiamenti conformazionali
nelle loro rispettive tasche obbligatorie che sono trasmessi all'interfaccia eteromerica e
risultano nel rafforzare o nella distruzione del complesso. In questo caso, l'interazione fisica è
mantenuta quando il recettore A1 e D1 che lega delle tasche è simultaneamente attivato dagli
agonisti che consentono l'interazione antagonistica intramembrana recettore/recettore, cioè la
proteina-G che spaia con la scomparsa dello stato di alta affinità del recettore D1 per gli

42
agonisti. Un significato funzionale di quest'interazione intramembrana recettore/recettore spaia
quindi il recettore D1 dalla proteina-G s. Questo è nel miglioramento dell’attività funzionale dei
recettori oligomerici SSTR5/D2, soprattutto dopo il trattamento combinato di agonista [109]. Il
complesso del recettore eteromerico A1/D1 potrebbe dare quindi delle basi molecolari ben
documentate per le interazioni antagonistiche recettore/recettore A1/D1 trovate nel cervello
delle reti neuronali [63,65,113,114]. L'eteromerizzazione del recettore A1/D1 appare anche per
avere un impatto sul traffico del recettore [104]. Così, un agonista del recettore A1, dopo 3 h di
esposizione, ha prodotto una coaggregazione dei recettori A1 e D1. D'altra parte, un agonista
del recettore D1 dopo 3 h di esposizione ha prodotto solo un'aggregazione della immuno
reattività recettoriale D1 con una mancanza di coaggregazione in accordo con la capacità
dell'agonista del recettore D1 per rompere l'eteromerizzazione del recettore A1/D1. Il recettore
D1 che segnala e rimasto inalterato dalla formazione di gruppi di recettore D1 o A1/D1, mentre
ha visto in termini un cambio dalla stimolazione del recettore un'accumulazione di cAMP e così
con nessun segno della desensibilizzazione del recettore D1. In contrasto, i trattamenti
combinati dei recettori agonisti A1 e D1, sotto le stesse condizioni non sono risultati nella
formazione di gruppi del recettore A1/D1, ma il diffondere della localizzazione del recettore
A1/D1 è stata mantenuta. Inoltre, ora i segni della desensibilizzazione del recettore D1 hanno
sviluppato riduzioni negli aumenti dei livelli di cAMP del recettore indotto D1. Così, le
caratteristiche essenziali della desensibilizzazione del recettore D1 potrebbero mantenere
l’eteromerizzazione con nessun recettore A1/D1 coaggregato formato dopo esposizione
combinata prolungata al recettore agonista A1 e D1 con nessuna indicazione di internalizzazione
del recettore. Sembra possibile che la desensibilizzazione del recettore D1 potrebbe essere
principalmente causata da un cambiamento prolungato allosterico nel recettore D1 causato
dall'interazione recettore/recettore del A1/D1 dentro il complesso eteromerico, che potrebbe
essere raccontato ai cambiamenti successivi della fosforilazione e/o all'associazione con le
molecole β-arrestine [115,116], conducendo complessivamente a una riduzione del recettore
D1/proteina-G. Così, potrebbe essere suggerito che l’interazione intramembrana
recettore/recettore A1/D1 in questo complesso eteromerico è pertinente non solo per

43
l'antagonismo acuto di segnalazione del recettore D1 ma anche per un antagonismo persistente
di lungo termine che segnala il D1 alla proteina-G. I dettagli della composizione e la
stechiometria del complesso del recettore eteromerico A1/D1 sono sconosciuti, ed i recettori
A1 e D1 sono saputi per esistere come monomeri e come omomeri [92,117]. È sconosciuto se
eteromero è preferito quando i recettori A1 e D1 sono coespressi nelle stesse cellule.

44
10. COMPLESSO MGLUR1αααα////A1
C'è la prova che il gruppo 1 del recettore metabotropico del glutammato mGIuR1α e
dell’adenosina A1 sono localizzati in certi tipi di neuroni cerebrali e che le interazioni funzionali
accadono tra i recettori di adenosina e di glutammato nel cervello [118-120]. Gli esperimenti
della coimmunoprecipitazione sugli estratti solubili dalle sinapsi del cervelletto di topo hanno
mostrato quei recettori del mGIuR1α possono coimmunoprecipitare con gli anticorpi del
recettore anti-A1. Così, i recettori del mGIuR1α e dell’A1 potrebbero esistere come eteromeri in
certe popolazioni cellulari del cervelletto [119]. Successivi studi della coimmunoprecipitazione
sulle cellule di HEK-293 hanno mostrato quei complessi di recettore eteromerico del
mGluR1α/Α1 che esistono anche in queste cellule. La specificità del sottotipo del recettore è
stata mostrata dal fallimento della congiunzione della variante del COOH-terminale, il mGIuR1α
all'immunoprecipitato con il recettore A1, indicando l'implicazione della coda del COOH-
terminale nella formazione di questo complesso eteromerico. È sconosciuto come questo
complesso eteromerico si attacca al dimero del recettore mGIuR1, dove l'interfaccia modula il
sito di legatura del glutammato della regione extracellulare del recettore [121]. Il ruolo
funzionale di questa eteromerizzazione è stata soprattutto studiata nelle cellule HEK-293. E’
stato trovato che l'acido quisqualico ha migliorato sostanzialmente l'aumento di segnalazione
della produzione di Ca++ dall'attivazione del recettore A1, e lo stesso era vero quando la
modulazione della funzione del recettore A1 di mGIuR1α è stata studiata. Così, sembra come se
l'eteromerizzazione ha condotto allo sviluppo di risposte cooperative nell’attivazione simultanea
della segnalazione nel Ca++ sui recettori dentro l'eteromero del recettore di mGluR1α/A1.
Anche nelle culture primarie corticali, dove un’alta localizzazione è stata osservata alle posizioni
dendritiche, un'interazione sinergica è stata trovata in termini di una riduzione di neurotossicità
indotta di NMDA [119,120]. Una ridotta neuro protezione è stata osservata nel recettore
mancante A1 [122]. Sembra possibile che l’eteromerizzazione del recettore mGluR1α/A1 può
avere luogo indirettamente, poiché il COOH-terminale interagisce con le specifiche proteine di
bersaglio. Così, l'omero-1a della proteina con un permesso omologo VASP collega il dominio che

45
lega al COOH-terminale del recettore mGluR1α. L’omero 1-c può collegare insieme proteine
ricche di prolina (PPSPF) [123]. È di interesse sostanzioso che questo o un motivo simile hanno
trovato in entrambi la parte COOH-terminale del mGluR1α e nella parte COOH-terminale del
recettore A1. Così, l'omero può essere una parte importante di questo complesso eteromerico
come una proteina adattatore [124,125]. Esso dovrebbe essere anche notato che l'omero può
anche collegare mGIuR1α alla Canna, poiché contiene anche un motivo simile (PPEEF). La canna
è un’impalcatura multimerica di proteina postsinaptica che potrebbe portare il mGluR1α alla
posizione appropriata sulla superficie cellulare ed è parte del complesso PSD-95 del recettore
associato NMDA [123].

46
11. COMPLESSO P2Y1/A1
Nel 2001, la scoperta è stata fatta negli esperimenti della coimmunoprecipitazione sulle cellule
HEK-293 dei recettori purinocettori P1 e dell’adenosina A1 agganciati alla proteina Gi/o che
possono formare un complesso eteromerico con i recettori purinocettori P1 ATP e purinocettori
P2y1 agganciati alla proteina Gq [126], mostrando meno del 5% l’omoIogia con l'un l'altro [127].
La parte del COOH-terminaI dei recettori A1 non è stata mostrata essere coinvolta in questo tipo
di eteromerizzazione. Studi nell'immunofluorescenza hanno mostrato una localizzazione
marcata dei recettori A1 e P2Y1. Inoltre, la coimmunoprecipitazione dei recettori A1 con P2Y1
indicano che l'eteromerizzazione tra sottotipi recettoriali P1 e P2 potrebbero essere un
meccanismo piuttosto esteso per l’immediato cross-taIk tra, per esempio, i recettori inibitori A1
e recettori eccitatori P2Y1. Un cambiamento marcato nel segnalare è stato trovato nel
complesso eteromerico. Così, è diventato possibile per il recettore agonista P2Y1 ADPβS a
indurre un segnale tramite proteina Gi/o agganciata al recettore A1, un'azione bloccata dalla
tossina della tosse e il recettore antagonista A1 ma non dal recettore antagonista P2Y1. Questi
risultati interessanti sono sembrati essere spiegati dallo sviluppo di una capacità della riduzione
del ADPβS etichettato H N6-phenylisopropyladenosine legato dal complesso eteromerico nella
gamma di alta affinità. È stato quindi suggerito che la tasca del ligando del recettore A1
dell'eteromero aveva cambiato per legare segnatamente l’agonista P2Y1 associato con
l'attivazione della proteina di Gi/o. Così, è stato correttamente suggerito che l'associazione
eteromerica produce un recettore A1 P2Y1. Comunque, è anche possibile per proporre un altro
meccanismo molecolare. Così, potrebbe esistere un’interazione recettore/recettore A1/P2Y1 a
livello del sito di riconoscimento che cambia la farmacologia di altre tasche della legatura del
recettore A1 e P2Y1. Il cambiamento di conformazione dell’agonista indotto del recettore P2Y1
nel recettore P2Y1 che lega la tasca ora non solo comanda a un'attivazione delle proteine Gq ma
anche a un cambiamento nella struttura del recettore A1, convertendo in uno stato di agonista
capace di accendere alla proteina Gi/o. Tale cambiamento di conformazione non può più
accadere quando la tasca di legatura del recettore A1 è occupata dall'antagonista del recettore

47
A1 chiudendo in uno stato di antagonista. Il recettore antagonista P2Y1 non potrebbe bloccare
l'azione dell’agonista del recettore P2Y1 poiché sembra possibile che nel complesso
eteromerico l'antagonista usato non potrebbe avere l'affinità sufficiente per la tasca di legatura
del recettore P2Y1 [128]. Questo complesso di recettore eteromerico P2y1/A1 è di interesse
sostanzioso, poiché consente che l'attivazione eccitatoria del recettore P2Y1 ATP è subito
attivato nel meccanismo dell’inibizione parallela del recettore A1. In questa maniera,
l'eccitazione e l’aumento della spesa di energia sono causate dall'attivazione del recettore P2Y1
ATP e inizia a essere anche neutralizzato a un momento quando l'ATP extracellulare non è stato
guastato all'adenosina, il maggiore ligando per il recettore A1 [129-131].

48
12. COMPLESSO MGLUR5/A2A
Nel 2002, Ferré ha potuto dimostrare nella cellula HEK-293 una sostanziosa sovrapposizione
nella distribuzione dell’A2A e il gruppo 1 recettore metabotropico del glutammato mGluR5.
Inoltre, in queste cellule gli esperimenti della coimmunoprecipitazione hanno mostrato che i
recettori mGluR5 e A2A hanno formato i complessi eteromerici che sono apparsi essere selettivi
tali complessi non sono stati formati tra mGluR5 e mGluR1β. I complessi eteromerici
A2A/MGLUR5 sono stati anche dimostrati nelle preparazioni della membrana dello striatal del
topo con gli esperimenti della coimmunoprecipitazione [132]. Queste scoperte sono di interesse
speciale, poiché nei recettori A2A e MGLUR5 nello striatum sembrano avere un modello di
distribuzione simile nei neuroni del GABA dello striatopallidal con una localizzazione delle
sinapsi periferiche alle presunte sinapsi glutamatergiche postsinaptiche asimmetriche. Inoltre,
negli studi sul comportamento sinergico dei recettori agonisti A2A e mGluR5 nel neutralizzare i
recettori D2 indotti che cambiano il comportamento ai recettori di dopamina ipersensibili [133]
e, negli studi biochimici, sia recettori agonisti A2A e sia mGluR5 hanno ridotto l'affinità dei siti di
legame del recettore agonista D2 ad alta affinità [133-135]. Attualmente, è sconosciuto se il
recettore A2A e il recettore mGluR5 sono collegati insieme tramite l'eteromerizzazione diretta o
se, per esempio, le proteine citosoliche omeriche sono coinvolte e si possono legare alla parte
COOH-terminale del recettore mGluR5 e producono il loro gruppo. Le proteine di shank hanno
un ruolo nella impalcatura con i motivi di interazione multipla proteina/proteina come regioni
ricche di prolina e domini PDZ potrebbero anche essere coinvolte [136], soprattutto partecipano
collegandosi insieme il mGluR5 con i recettori NMDA [137]. Il complesso eteromerico del
recettore A2A/MGLUR5 nelle cellule HEK-293 non ha mostrato il sinergismo nella mobilitazione
del Ca++ e accumulazione di cAMP. Tuttavia, un sostanzioso sinergismo è stato trovato dopo i
trattamenti di agonista in termini di MAPK e l'espressione del c-fos nelle cellule HEK-293 [132]. È
adesso sconosciuto come i segnali dall'eteromero possono causare questo forte sinergismo
funzionale che è stato anche osservato nello striatum nel vivo dopo i trattamenti combinati di
agonista A2A e mGluR5. Così, il recettore mGluR5 e A2A potrebbe mediare il sinergismo

49
dell’adenosina e del glutammato in caso di espressione del c-fos nello striatum che coinvolge il
complesso eteromerico recettore A2A/mGluR 5. C'è una distinta possibilità che la combinata
attivazione dei due recettori del complesso eteromerico A2A/mGluR5 potrebbe condurre alla
ridotta desensibilizzazione del mGluR5 consentendo un incremento della defosforilazione per
sviluppare grazie all'incremento di attivazione e/o grazie alla disponibilità della proteina
phosphatase 2B al recettore mGluR5 [138,139]. Sembra probabile che il sinergismo dimostrato
nell'espressione dello striatal di topo del c-fos ha delle conseguenze funzionali importanti ,
poiché è stato uguagliato da un sinergismo del recettore agonista CHPG e del recettore agonista
CGS 21680 a neutralizzare l’attività motoria delle phencyclidine indotto nei topi, che è una
risposta del comportamento conosciuta per essere estremamente dipendente sulla funzione del
recettore D2. Sembra possibile che l'attivazione combinata dei recettori A2A e mGluR5 nello
striatum potrebbero neutralizzare il forte conosciuto tonico recettore D2 dalll'inibizione del
recettore mediato dall’adenilato ciclasico e l’espressione di immediati primi geni nei neuroni
GABA-ergici dello striatopallidal. Poiché gli immediati primi geni sono coinvolti nel collegamento
alle risposte neuronali adattabili tra breve a lungo termine, i complessi recettore eteromerici,
A2A/mGluR5 potrebbero avere un ruolo nella plasticità della depressione a lungo termine e
potenziare la sensibilizzazione ai psicostimolanti collegati all'espressione del c-fos dopamina-
indipendente. Finalmente, il trattamento cronico ma non acuto con un antagonista mGluR5 può
indietreggiare a un deficit cinetico nel modello 6-0H-dopamina della malattia del Parkinson
[140]. Questo potrebbe essere raccontato a un alterato traffico del recettore eteromero
A2A/mGluR5, conducendo alla sua interiorizzazione e/o alla ridistribuzione che consente un
predominio dei segnali D2.

50
13. COMPLESSO B2/AT1
Le prime indicazioni della possibile esistenza di un’interazione recettore/recettore di una
bradichinina/angiotensina II è stata ottenuta dall'autoradiografia quantitativa del recettore nel
nucleo solitarius tractus del cervello del topo, un centro centrale cardiovascolare [141]. Le
scoperte hanno suggerito che recettori della bradichinina B2 del nucleo solitarius tractus sono
stati coinvolti nel modulare in una differente maniera l'alta e bassa affinità che lega dei siti dei
recettori dell'angiotensina II senza gli effetti sui valori di Bmax dei siti di legatura di agonista
AT1. Così, l'affinità dello stato di agonista di alta-affinità dei recettori AT1 è stata ridotta dalla
bradichinina; mentre la bradichinina aumentava l’affinità dello stato dell’agonista di bassa
affinità che usa i radioligandi di agonista ed antagonista per il recettore AT1. È stato suggerito
che quest'interazione recettore/recettore può contribuire all'attività centrale di vasopressoria
della bradichinina riducendo la crescente trasmissione mediata AT1 agli stati di agonista di alta e
bassa affinità, considerati nell’essere coinvolti rispettivamente nell'attività di vasodepressoria e
vasopressoria [141]. Comunque, un'altra interpretazione dei risultati dagli esperimenti di
competizione con un antagonista del recettore AT1 iodinated contro l'angiotensina II è quella
bradichinina che riduce l’affinità del sito di legame dell'antagonista del recettore AT1,
consentendo una migliorata competizione dall'angiotensina II visto come una riduzione nei
valori IC50. Nel globale potrebbe essere considerato che lo stato di antagonista del recettore
AT1 può essere differenzialmente regolato dall’attivazione del recettore B2 contro lo stato di
agonista. La modulazione del sito di legame del recettore antagonista AT1 della bradichinina
tuttavia rimane ancora a essere determinato. Recentemente la scoperta è stata fatta con
recettori dell'angiotensina AT1 e bradichinina B2 che formano eteromeri nelle cellule dei
muscoli lisci e nelle cellule HEK-293, con espressi i recettori AT1 e B2 [142] che indicano che
questa potrebbe essere la base molecolare per le interazioni intramembrana
recettore/recettore precedentemente osservate tra questi due recettori. La cromatografia
dell’immuno-affinità è stata eseguita sulle proteine dalle cellule muscolari lisce e recettori
dimeri AT1 sono stati attaccati con gli anticorpi del recettore anti-B2. Poiché la bradichinina e

51
l'angiotensina II erano stati cross-linked agli anticorpi del recettore AT1 e B2 prima della
cromatografia di immuno-affinità, i risultati hanno suggerito che i recettori ad alta affinità di
AT1 e B2 formano i complessi eteromerici sulle cellule muscolari lisce. Le cellule HEK-293,
quando espresse solo su uno dei due recettori mostrati, formano un monomerico, ma quando
c’è co-espressione dei recettori AT1 e B2 è stato dimostrato che formano un eteromero,
coesistendo dei recettori AT1 e B2. Lo stabile recettore eteromero AT1/B2 potrebbe essere
dimostrato dalla sds-page e non era dipendente sugli agonisti ma sulla densità dei due recettori.
Così, sembra probabile che le interazioni intramembrana recettore/recettore [141] prima
riportate riflettono i cambiamenti di conformazione nelle tasche di agonista-indotto degli
eteromeri preformati che conducono alle alterazioni nell'affinità del ligando dell’altra tasca di
legatura. La scoperta più impressionante nell'articolo da AbdAlla [143] era l'aumento
dell'agganciamento del recettore AT1/proteina-G nel recettore eteromero AT1/B2. Questo è
stato visto, per esempio, dall’aumentato grado di ridistribuzione del AT-1 stimolato della
proteina Gα nel citosol, dall'aumento marcato di legatura della angiotensina II stimolata che
lega GTPγS e gli aumenti sostanziosi dei fosfati di inositolo. Un'analisi elegante con il recettore
mutante B2 ha dimostrato che l'aumento di segnale del AT1 nell’eteromero dipendeva
dall’interfaccia della proteina-G dei recettori B2 ma non sulla legatura della bradichinina ai
recettori B2. L'eteromero era comunque formato in modo indipendente dell’interferenza con
l'agganciamento della proteina-G e con la legatura della bradichinina. Così, una importante
funzione significativa dell'eteromero in questo caso è il miglioramento dell'agganciamento del
recettore AT1/proteina-G e così del recettore AT1 che segnala [143]. È stato anche mostrato che
la aumentata presenza del recettore eteromero AT1/B2 potrebbe contribuire allo sviluppo
dell’ipersensibilità di angiotensina II nella preeclampsia. Tuttavia un altro significato funzionale
potrebbe essere un cambiamento nel traffico recettoriale, poiché il recettore eteromero AT1/B2
diventa internalizzato attraverso un percorso dinamico dipendente in contrasto quando sono
espressi soli.

52
14. COMPLESSO GABA A/D5
L'interazione diretta tra proteine/proteine dei recettori GABAA e dopamina D5 sono stati
riferiti in una rivista di Liu [144]. Una localizzazione dei recettori del GABAA e D5 è stata
dimostrata nei neuroni dell’ippocampale coltivati per mezzo di studi dell’immuno fluorescenza
nella combinazione con la microscopia di laser. Questo era in linea con il lavoro indicato
precedentemente che i recettori D5 nei neuroni dell’ippocampale esistono sugli alberi dendritici
e nel colle di axon, le regioni ricche nelle sinapsi del GABA [145,146]. Nelle macchie Occidentali,
i recettori del GABAA dell’ippocampale potrebbero essere dimostrati dopo la precipitazione
dell’affinità con il D5 ma non le proteine di fusione GST dei recettori D1 COOH-terminali. Inoltre,
le proteine di fusione GST con il secondo loop intracellulare del recettore GABA A γ2 hanno
precipitato l'ippocampale del D5 ma non i recettori D1. Questi risultati hanno indicato
un'interazione fisica tra la parte del COOH-terminale del recettore D5 e la subunità γ2 del
recettore GABA A, più esattamente il secondo loop intracellulare. L’eteromerizzazione del
recettore GABAA/D5 è stato dimostrato negli esperimenti della coimmunoprecipitazione. Nei
test di copertura della macchia, è stato mostrato che la sds-page ha separato la proteina della
fusione GST della subunità γ2 ma nessun’altra sottounità potrebbe legare direttamente il
peptide tradotto in vitro [S]metionina del D5 COOH-terminale. Similmente, la [S]methionine del
secondo loop intracellulare della subunità γ2 potrebbe legare direttamente alla proteina di
fusione del recettore GST-D5 COOH-terminale. Dovrebbe anche essere considerato che la
proteina di fusione GST del secondo loop intracellulare della subunità β2 non potrebbe legare
alla parte COOH-terminale del recettore D5 nel test di copertura della macchia malgrado il fatto
che questa parte può immunoprecipitare recettori D5. Quest'interazione potrebbe essere quindi
indiretta tramite le proteine associate o coinvolgere altri parti del recettore D5 COOH-terminale.
Comunque, questa parte del recettore GABAA potrebbe avere anche un ruolo nella formazione
del complesso del recettore eteromerico del GABAA/D5. È di interesse che esiste lì una proteina
associata recettore GABAA che può interferire con la capacità della coda COOH-terminale del
recettore D5 nell’interagire con il loop γ2 intracellulare [147]. Gli studi nelle cellule HEK-293

53
hanno dimostrato che la coattivazione degli agonisti dei recettori D5 e GABAA era necessario
per aver luogo la coimmunoprecipitazione. Così, i cambiamenti di agonista-indotto nel secondo
loop intracellulare della subunità γ e nella parte del COOH-terminale del recettore D5 è
essenziale per la formazione di questo complesso eteromerico. Un significato funzionale di
quest'eteromerizzazione appare per essere concessa reciprocamente la cross-talk inibitoria a
prendere posto. Così, nelle cellule HEK-293 la ridotta attivazione del recettore D5 e del GABAA
diminuisce la pendenza della curva di voltaggio. Questi cambiamenti sono stati causati da un
meccanismo cAMP-indipendente ed erano per mezzo delle chimere dei recettori D1/D5 che si è
dimostrato essere completamente dipendenti sull’interazione del recettore D5 COOH-
terminale/γ2 nel complesso eteromerico formato. Così, questi risultati indicano che D5 può
ridurre la forza sinaptica sopra i recettori del GABAA tramite questo complesso. In accordo con
questa veduta, questi studi elettrofisiologici nelle parti dell’ippocampale hanno mostrato che un
agonista D1/D5 potrebbe ridurre l'ampiezza inibitoria della miniatura del recettore mediato
GABAA e le correnti post-sinaptici indipendenti del PKC e PKA. L'iniezione di un peptide GST-
codificato D5 COOH-terminale nel neurone registrato ha evitato quest'azione. Questo
complesso eteromerico potrebbe essere quindi di pertinenza nel vivo, dove potrebbe
controllare la forza sinaptica del recettore GABAA e segnalare come sopra il recettore D5 era nel
giro modulato dal recettore GABAA in una maniera inibitoria. Così, nelle cellule coespresse dai
due recettori, il recettore GABAA ha ridotto la massima attivazione dell’adenilato ciclasico della
dopamina da 45%, che era selettivo per il D5 contro il recettore D1. Quest'azione non ha
coinvolto dei cambiamenti nel luogo di riconoscimento del D5 mentre ha mostrato dall'assenza
dei cambiamenti nell'affinità di agonista e antagonista del recettore di dopamina né avevano
alcun cambiamento nei valori di Bmax dei siti osservati del D5 antagonista. Invece, sembra che
l'attivazione di GABAA di questo complesso eteromerico riduca il recettore D5/proteina-Gs
attraverso l’interazione fisica del secondo loop intracellulare γ2/recettore D5 COOH-terminale.
Così, l'espressione dei minigeni che codificano la sequenza del secondo loop intracellulare γ2 o
le sequenze del COOH-terminale del recettore D5 hanno bloccato la modulazione di GABAA del
D5 hanno indotto l'accumulazione di cAMP. L'uso delle chimere D1/D5 hanno dato l’ulteriore

54
prova per l'implicazione cruciale del complesso COOH-terminale del recettore D5 dell’agonista-
indotto/γ2 nella regolazione dell’agganciamento alla proteina G del recettore D5. Un altro
significato funzionale di questo dinamico complesso eteromerico potrebbe essere un ruolo nel
traffico del recettore [147]. Così, esistono lì delle indicazioni che l'attivazione del l’un l’altro
recettore agonista GABAA o il D5 fa l'endocytosis possibile di entrambi i recettori e il così
cotrafficking [147]. Dovrebbe essere anche considerato che la subunità γ2 potrebbe essere
essenziale per il gruppo dei recettori del GABAA postsinaptici. Un ruolo potente di questo
complesso recettore GABAA/recettore D5 nella patofisiologia della schizofrenia è stato anche
postulato nella veduta dei fatti che le alterazioni nel D1/D5 e nel GABAA γ2 contengono i
recettori e le loro funzioni potrebbero esistere nel cervello schizofrenico [148-151]. Molto
recentemente Liu ha ottenuto la prova che i recettori D1 possono interagire direttamente
tramite interazione proteine/proteine con i recettori NMDA. Così, l'eteromerizzazione del
NMDA/D1 potrebbe avere un ruolo nella regolazione della trasmissione del glutammato. La
dimostrazione del GABAA/D5 e forse dei complessi del recettore NMDA/D1 stimola molto,
poiché aprono una nuova maniera per capire come i segnali della trasmissione di volume (VT) e
la trasmissione di cablaggio (WT) possono diventare integrati [20,152,153]. Così, i recettori
GABAA e NMDA sono dei classici recettori, sinaptici veloci che operano tramite la regolazione
dei loro canali ionici, poiché il GPCR è lento e principalmente localizzato estrasinapticamente e
raggiunto dai segnali VT nello spazio extracellulare. L'eteromerizzazione dei recettori di canale
ionico e GPCR offre un nuovo meccanismo per l'integrazione di WT e VT e su come controllare la
forza sinaptica di importanza cruciale per apprendere e memorizzare [154]. Potrebbe essere
menzionato che diversi anni fa abbiano ottenuto delle indicazioni che gli agonisti del recettore
GABAA potrebbero modulare le caratteristiche obbligatorie dei recettori D2-like nelle
preparazioni di membrana dello striatal [155]. Così, il muscimol agonista del GABAA ha ridotto
l'affinità dell'agonista del recettore D2 di alta affinità situato mentre ha mostrato competizione
negli esperimenti con [H]raclopride contro la dopamina. Così, i complessi di recettore
eteromerici di GABAA/D2 potrebbero anche esistere, poiché tali interazioni al livello di
riconoscimento sono state considerate [20] come gli indicatori biochimici dell'esistenza di un

55
complesso eteromerico, in questo caso tra i recettori GABAA e D2. È anche di interesse che
potremmo riferire la capacità dei recettori agonista-attivato α2 adrenergico e D1
sostanzialmente per modulare [H]nicotine che lega nelle preparazioni di membrana dalle regioni
encefaliche [156,157]. Quanto visto sopra, sembra pertinente per testare l'esistenza dei
complessi eteromerici tra recettori nicotinico e α2 adrenergico e tra i recettori nicotinico ed i D1
coinvolti nel controllo dei meccanismi allosterici ai recettori di acetilcolina nicotinici [158]. È
anche di interesse sostanziale che la cross-inibizione tra certi canali di catione trasmettitore-
recintati è stata mostrata esistere sulla loro coattivazione [159] riflettendo probabilmente
l'eteromerizzazione tra questi due recettori di canale ionico quando coattivati.

56
15. COMPLESSI OLIGOMERICI E RECETTORI TIROSIN-CHINASICI
Recentemente, è stato riportato che i recettori del fattore di crescita epidermico (EGF) possono
diventare frequenti con i recettori dell'ormone della crescita (GH) e con i recettori β2 agonisti
sulla loro stimolazione rispettivamente dai recettori GH e β2 agonisti [160,161]. La prova è
stata fornita dalla dimostrazione che la loro associazione fisica è risultata in una trans
attivazione del recettore EGF. Gli esempi, si concentreranno su come il recettore β2 adrenergico
può produrre la dimerizzazione del recettore EGF, l'autofosforilazione della tirosina e
l'internalizzazione del recettore EGF conducendo all'attivazione delle MAPK. Un presupposto
per tale trans attivazione del RTK appare essere la formazione di β2 agonista-indotto di un
complesso multiproteico che contiene non solo il recettore β2 e il recettore EGF ma anche la β-
arrestina e il c-Src, un recettore tirosin chinasico. Gli esperimenti della coimmunoprecipitazione
dimostrata dei complessi eteromerici dei recettori β2 adrenergici/EGF che hanno formato
rapidamente un picco dopo il trattamento correlato di β2 agonista con l’attivazione di MAPK β2
agonista indotto. Questo complesso potrebbe anche essere rilevato sotto le condizioni basali e
probabilmente ha connesso alla costitutiva attività del recettore β adrenergico, poiché questo
complesso del recettore β2 adrenergico/EGF è stato segnatamente ridotto da un recettore β2
adrenergico agonista inverso. È stato mostrato che gli inibitori della Src kinase hanno bloccato la
formazione del complesso recettoriale β2 adrenergico/EGF e attivazione delle MAPK, indicando
un ruolo critico dell’attività catalitica Src. Inoltre, la β-arrestina recluta la proteina c-Src al
recettore β2 adrenergico [162] dopo che la β-arrestina è venuta ha collegarsi al recettore β2
adrenergico attraverso la sua fosforilazione di agonista indotto tramite il recettore della
proteina-G chinasica. È stato notato che la trans attivazione dell'EGFR dal recettore β2
adrenergico è stato bloccato dall'inibizione endocytosis mediata di clathrin. Potrebbe essere
quindi che la clathrin ha ricoperto dei buchi che possono rappresentare i micro domini per
ottimizzare il segnalare fra le proteine montate, conducendo all'attivazione di RAS-dipendente
del Raf dopo che è accaduta la trans attivazione del recettore EGF. Maudsley [161] notò che
questi risultati aprono un meccanismo importante per come i GPCRs ed anche i recettori delle

57
citochine [163,164] potrebbero controllare il segnalare tropico, cioè attraverso i complessi
eteromerici di agonista-indotti con i recettori EGF che conducono alla regolazione della trans
attivazione seguita dall'attivazione delle MAPK. Questi studi sono degli ottimi esempi di come i
complessi delle multiproteine formano un ruolo cruciale anche nel segnale tropico.

58
16. COMPLESSI CALCITONINA/RAMP1-3
I peptidi della famiglia della calcitonina, come calcitonina, peptidi gene connessi con calcitonina
(CGRP), adrenomedullin, e amylin agiscono tramite GPCR. Fra gli altri, il recettore calcitonin-
recettore-like (CRL) è stato clonato ma non potrebbe legare il peptide CGRP. Come tentativo è
stato quindi fatto clonare il gene per il recettore CGRP dall’espressione di clonazione [165]. In
questo studio, un singolo cDNA è stato finalmente mostrato segnatamente per migliorare le
risposte a un recettore CGRP endogeno nell'oocytes Xenopus. Il cDNA è stato mostrato per
codificare una proteina con un singolo dominio TM e uno extracellulare NH2 terminale (RAMP1).
L’espressione di questa proteina ha migliorato selettivamente le azioni di CGRP-indotto
nell'oocytes. Questo era la maniera in cui è stato scoperto il RAMP1 [165]. Gli esperimenti
successivi sulle linee cellulari (HEK-293 T ) hanno dimostrato che la coespressione del RAMP1 era
necessaria per la legatura del ligando del recettore CRL e la funzione in termini di accumulazione
di cAMP. La domanda era come RAMP1 abbia causato lo sviluppo di un recettore funzionale CGRP
basato sulla coespressione del recettore CRL e sul RAMP1. Per mezzo del recettore CRL epitopo-
etichettato e per mezzo del RAMP1 nella combinazione con la classificazione cellulare di
fluorescenza-attivata, è stato mostrato che la loro coespressione ha fatto il possibile per
l'espressione sulla superficie cellulare di entrambi i recettori. Era di interesse sostanzioso che la
sds-page potrebbe mostrare cross-linking dei I-CGRP1 a due proteine dalla superficie delle cellule
HEK-293 T. Le due bande sono sembrate corrispondere al recettore nativo CGRP ed a RAMP1,
rispettivamente, che potrebbe formare un complesso eteromerico facilmente rotto secondo
queste scoperte. Gli ulteriori esperimenti hanno dimostrato che la coespressione del RAMP1
dell’epitopo-etichettato e il recettore CRL hanno condotto alla scomparsa di una banda di 58 kDa
trovata con l'espressione del recettore CRL solo con l'apparenza di una banda che diffonde a 66
kDa che mette in correlazione nella misura con la banda cross-linked a I-CGRP1. Quest'aumento
addizionale della misura da 8 kDa potrebbe non essere dovuto a un'associazione con il RAMP1 del
epitopo-etichettato che ha una misura di M 14 kDa. Invece, gli esperimenti con l'endoglycosidases
F e H hanno indicato che è stato collegato a una glicosilazione terminale del recettore CRL, non

59
hanno trovato quando è stato solo espresso e soggetto solo al centro della glicosilazione. Il
recettore CRL della glicosilazione terminale trovato in presenza del RAMP1 indica che il recettore
CRL può essere ora espresso sulla superficie cellulare come una glicoproteina matura, capace di
essere un recettore CGRP. Tuttavia, sembra probabile che anche il RAMP1 coespresso con il
recettore CRL terminalmente glicosilato sulla superficie cellulare e capace di diventare cross-
linked con I-CGRP1 può contribuire alla regolazione della selettività e alla funzione dei ligandi del
recettore CRL dalle interazioni fisiche e/o indirette. Recentemente è stato anche osservato che i
recettori multipli di amylin possono essere formati dalle interazioni di RAMP con il prodotto
genetico del recettore della calcitonina [166]. Due altre proteine di RAMP sono chiamate RAMP2 e
RAMP3 che sono state anche scoperte [165], ed è stato trovato che RAMP2 ed il recettore CRL
potrebbero generare un recettore per l'adrenomedullin (ADM). È stato trovato che RAMP2 ha
consentito il trasporto all’espressione di un recettore CRL glicosilato al centro sulla superficie
cellulare. Questo è stato mostrato per non essere una regolazione del recettore CGRP. È stato
dimostrato in modo elegante che oocytes esprime i recettori RAMP2 e i CRL rispondono
sostanzialmente alle concentrazioni basse di frammenti di ADM ma solo debolmente a CGRP.
Nella coespressione delle cellule HEK-293 T queste proteine specifiche di legame con ADM e
aumenti di ADM-mediato dell’accumulazione di cAMP potrebbero essere dimostrati. Così, il
recettore CRL trasportato RAMP2 diventa recettore ADM sulla superficie della cellula. Tutti questi
processi potrebbero condurre alla diversità di sviluppo dei recettori con le specificità dei differenti
ligandi e CGRP, ADM, ed i sottotipi del recettore amylin [167,168] può essere formato basato sulle
interazioni del recettore CRL e sulla calcitonina con i diversi tipi di RAMP. Così, il recettore di CRL e
forse altro GPCR possono alterare segnatamente la sua tasca obbligatoria dalle interazioni con le
sole proteine di dominio TM. Il ruolo della glicosilazione centrale e terminaIe contro le interazioni
di proteina del recettore/RAMP che risultano nei presunti complessi eteromerici rimane essere
determinato. Quando leggendo sui RAMP uno si rende conto della possibilità che molti recettori
non potrebbero funzionare nella loro propria ragione ma principalmente come i partner nei
complessi eteromerici e nell'associazione con le altre proteine di membrana-associate.

60
17. COMPLESSO D1/CALCIONE
In un tentativo di capire come i recettori D1 possono agganciare alle proteine-G multiple,
Goldman, Rakic e Bergson hanno iniziato a cercare l’interazione delle proteine al recettore D1
[169]. Hanno trovato una singola proteina transmembrana di 24 kDa, chiamata calcyon, che
potrebbe interagire con il recettore D1 e produrre miglioramenti del segnale recettore
D1indotto Ca++. Il calcyon potrebbe essere quindi considerato come un RAMP dove
l'interazione è concentrata sull'agganciamento alla proteina-G e non sulla selettività
obbligatoria della tasca poiché descritto sopra per RAMP1-2. Il calcyon appare per avere un
NH2-terminale localizzato nella porzione extracellulare e un COOH-terminale localizzato nella
porzione intracellulare il dominio-like del RAMP1-3 contiene anche l'oligosaccaride del N-linked.
L’immunocitochimica hanno dimostrato che il recettore D1 e calcyon sono localizzati nella
stessa popolazione cellulare a piramide della corteccia cerebrale ed in uno strato contenenti i
recettori D1 dello striatal. È di interesse che sia i recettori calcyon e sia i recettori D1 sono stati
localizzati nelle spine dorsali perifericamente a una posizione postsinaptica. Esperimenti sulla
coimmunoprecipitazione hanno indicato che il recettore D1 ed il calcyon hanno formato un
complesso eteromerico nelle cellule HEK-293. Inoltre, usando una proteina di fusione GST con la
sequenza di esca del recettore D1 (GSTD1) ed una proteina di fusione batterica con la parte di
COOH-terminale del calcyon (S-calcyon), potrebbe essere mostrato che GSTD1 venga diretta alla
S-calcyon. Così, un'interazione diretta COOH-terminale potrebbe essere coinvolta nella
formazione di questo complesso eteromerico calcyon/recettore D1, dove le sequenze 421-435
del recettore D1 COOH-terminale appaiono cruciali poiché questo peptide ha evitato la legatura.
La formazione del complesso eteromerico calcyon/recettore D1, è risultato in un cambiamento
marcato nel segnale del recettore D1 nelle cellule HEK-293. Dopo la carica attivazione dei
recettori ATP P2Y, i recettori D1 agonisti SKF 81297 hanno prodotto un aumento rapido nel
segnale dipendente Ca++ sulla liberazione dalla scorta intracellulare di Ca++ fornito che il
calcyon era stato eseguito. Questa risposta Ca++ era simile nella misura a quel prodotto dal
recettore P2Y collegato a Gq/11. Questi risultati possono essere spiegati presumendo che

61
l’attivazione p2Y può aumentare l'agganciamento del recettore D1 alla proteina Gq/11, un
aumento che potrebbe essere rafforzato dalla formazione del complesso recettore D1/calcyon.
In contrasto, il recettore D1 che segnala sopra il Gs, all’adenilato ciclasico conduce agli aumenti
di accumulazione di cAMP era inalterato dalla formazione di questo complesso. Così, il doppio
segnale del recettore D1 con l'implicazione anche del segnale del Ca2 + tramite Gq/11 diventa
più pronunciato da questo complesso eteromerico con il calcyon. La formazione di questo
complesso è apparsa essere inibita dall'espressione nelle cellule del D1, che ha ridotto le
risposte di Ca++ del recettore D1 stimolato probabilmente gareggiando con il recettore D1 per il
calcyon. La formazione del complesso eteromerico è aumentata dall'attivazione del recettore
D1 e ha ridotto da un inibitore del PKC. Infatti, la parte del COOH-terminale del calcyon può
diventare fosforilata da PKC e potrebbe legare fosfoinositol-4,5- bifosfato. Queste osservazioni
danno un’aumentata comprensione all'importanza dell'interazione diretta tra le parti di COOH-
terminale del recettore D1 e il calcyon per l'aumento dell’agganciamento della proteina Gq/11
del recettore D1 con l'attivazione della fosfolipase C (PLC) e la formazione di inositol 1,4,5-
trifosfato (lP3). È di interesse sostanzioso che la stimolazione dei recettori endogeni muscarinici
M1 agganciati Gq/11 come attivazione ATP P2Y, prima dell'attivazione del recettore D1 ha fatto
anche un possibile forte aumento del recettore-indotto nel segnale Ca2 + dal complesso
eteromerico recettore D1/calcyon. Sembra possibile che il meccanismo basilare delle azioni
primarie dell’attivazione recettoriale di ATP ed M, può essere severalfold con il risultato finale
che è un aumento dell’agganciamento Gq/11 del recettore D1. Così, l'indicazione delle
interazioni del recettore multiplo con la formazione di alti complessi eteromerici di ordine che
contiene ATP recettori D1e calcyon o recettori di M1 e D1 ed il calcyon potrebbero essere
considerati condurre all'aumento dell’agganciamento del recettore D1 alla proteina Gq/11.
L'attivazione di cascate della fosforilazione intracellulare che coinvolge, per esempio, PKC può
essere anche coinvolta con la fosforilazione del calcyon COOH-terminale, soprattutto poiché
l'azione prima è stata solo osservata fino ad ora con GPCR agganciato al Gq/11. Il lavoro
pionieristico di Goldman-Rakic ha dimostrato il ruolo prominente del recettore D1 nella
corteccia cerebrale nella modulazione del recettore di glutammato che segnala durante le

62
operazione di memoria. Questo lavoro avrà quindi la pertinenza per capire la plasticità corticale.
Ispira anche che i recettori M1, come i recettori D1, sono localizzati sulle cellule nervose
piramidali delle spine dorsali [170]. Così, l'interazione recettore/recettore M1/D1 tramite i
possibili complessi eteromerici potrebbe esistere quindi nelle spine dorsali dendritiche ed ha
una pertinenza funzionale [171].

63
18. COMPLESSO A1/ADENOSINA DEAMMINASE
Franco e collaboratori [172-174] hanno ottenuto la prova che l’adenosina deaminase (ADA) può
formare un complesso eteromerico con i recettori di adenosina A1. ADA è un enzima capace di
convertire l’adenosina nell'inosina. ADA è anche una proteina multifunzionale che appare sulla
superficie cellulare ancorata alle diverse proteine [175-177]. Può agire quindi enzimaticamente
ma anche extra enzimaticamente [172,178] come nel caso della formazione di complessi del
recettore ADA/A1. La formazione di complessi del recettore ADA/A1 è stata dimostrata negli
esperimenti che coinvolge la microscopia di laser, la coimmunoprecipitazione e la cromatografia
di affinità. Così, i recettori coimmunoprecipitati di ADA e A1 ed i recettori A1 sono stati
trattenuti in una matrice di ADA-Sepharose. Finalmente i recettori A1 sono localizzati con ADA
sulle membrane della cellula, compreso la superficie cellulare dei neuroni corticali [179]. La
legatura di ADA ai recettori A1 appare per essere essenziale per la legatura di agonista di alta-
affinità dei recettori A1, dando un ruolo funzionale di quest'interazione fisica nella trasmissione
recettore-mediato di A1 [180-182]. Così, ADA ha un ruolo non solo come un ectoenzyme
degradante ma anche come un recettore A1 di proteina attività-modulante. Esso quindi viene di
interesse nel studiare un ruolo possibile di ADA nel complesso del recettore eteromerico A1/D1.
In uno studio recente [183] è stato usato lo stesso recettore A1/D1 delle linee cellulari dei
fibroblasti come descritto sopra, la prova di ciò è stata ottenuta nelle cellule non permiabilizzate
del recettore A1/D1, ma non nelle cellule con il recettore D1, ADA esiste sulla plasma
membrana. Questi risultati hanno indicato che ADA può essere mirato alla membrana dai
recettori A1 ma non dai recettori D1. Inoltre l'agonista del recettore A1 risulta preincubato nella
coaggregazione di entrambi i recettori A1 e ADA e recettori D1 e ADA nelle cellule dei fibroblasti
del recettore A1/D1. Questi risultati hanno suggerito che dopo il trattamento di agonista del
recettore A1 con la coaggregata eteromerizzazione del A1/D1 sono formati che contengono
l'alto-ordine di strutture molecolari [183] coinvolgendo i complessi eteromerici ADA/recettore
A1/recettore D1 e altri che interagiscono delle proteine che hanno un ruolo speciale funzionale,
soprattutto nel traffico del recettore. Comunque, ADA non sembra essere direttamente

64
collegato ai recettori D1. In linea con questa veduta, questi aggregati del recettore ADA/D1 non
sono più presenti dopo il pre trattamento di agonista del recettore D1 rompendo
l'eteromerizzazione del recettore A1/D1 che conduce al solo recettore D1 aggregato, mentre
l'immunoreattività del recettore ADA/A1 rimane diffusamente localizzata [183]. L’impatto del
complesso del recettore ADA/A1 sul segnale del recettore D1 è stato dimostrato dal blocco
agonista-indotto del recettore A1, dal blocco irreversibile del recettore D1 della funzione ADA
che usa il deoxycoformycin. Questa controazione non era correlata all'alba di livelli di adenosina
endogeni [183]. Così, ADA è la parte del complesso del recettore eteromerico A1/D1
direttamente collegato solo al recettore A1, dove rende possibile lo stato di alta-affinità del
recettore A1 per gli agonisti, consentendo che lui modula l'operazione del recettore D1.

65
19. COMPLESSO A1/HSC 73
Sarriò e collaboratori [184] hanno dimostrato che i recettori adenosina A1 interagiscono con
una proteina della famiglia delle proteine della scossa di calore. Dalla cromatografia di affinità la
scossa di calore proteina analoga hsc73 è stata identificata come un componente del cytosolic
capace di interagire con il recettore del terzo loop intracellulare. Come dimostrato dalla
risonanza di superficie del plasmon, i recettori A1 purificati interagiscono specificatamente con
hsc73 con una costante di dissociazione nella gamma nanomolar (0,5 ± 0,1 nM). L’interazione
recettore hsc73/A1 conduce a una riduzione marcata nell'affinità dei ligandi di agonista del
recettore A1, una riduzione di legatura di antagonista del recettore A1 ed evita l'attivazione
delle proteine G, come dedotto da [S]GTPyS i test di legatura. In modo interessante,
quest'effetto sulla legatura di agonista del recettore A1 era più forte di quello esercitato
dall’analogo nucleotide della guanina, che separa dei recettori dalle proteine G ed è stato
completamente evitato da ADA, che interagisce con i domini extracellulari dei recettori A1.
Come valutato dall'immunoprecipitation, un'alta percentuale di recettori A1 nelle cellule lysates
sono agganciati a hsc73. I membri della famiglia dell'hsp70 interagisce con un numero di
proteine cellulari. Dovuto alla funzione della compagnia molecolare delle proteine hsp70,
appaiono capaci del riconoscimento delle forme delle proteine non-native. Questo non è il caso
per i recettori A1 per alcune ragioni. Uno è dovuto al fatto che i recettori A1 solubilizzati sono
molto sensibili alla composizione del mezzo, che colpisce la legatura dei ligandi alla molecola
solubile. Così, una combinazione precisa di detergente ed i sali è richiesta a conseguire un’alto
recupero dei siti di legame nelle preparazioni solubilizzate dei recettori A1. L'effetto forte della
concentrazione nanomolare del hsc73 sulla legatura di ligandi ai recettori A1 purificati solubili è
la prova per un'interazione specifica tra hsc73 ed i recettori A1 funzionali. La specificità
dell'interazione è stata dimostrata anche nelle culture primarie dei neuroni dove altri GPCR non
sono colocalizzati con hsc 73. D'altra parte, la colocalization tra i recettori A1 e hsc73 non è
limitata a una zona specifica della cellula, anche nelle cellule esprimendo naturalmente le
proteine, e questo è la prova che l'interazione accade con i recettori completamente piegati

66
funzionali. A prescindere dal ruolo regolatore dell'interazione nella legatura di ligandi, ci sono
dei dati che sostengono l'idea che hsc73 è pertinente per il traffico dei recettori A1. In realtà, la
localizzazione tra hsc73 ed i recettori A1 è stato rilevato nelle regioni specifiche del cervelletto
del topo e nei corpi cellulari nervosi dei neuroni corticali ma non nelle dendriti o nelle sinapsi.
Inoltre, sembra che l’internalizzazione del recettore di agonista-indotto conduce all'endocytosis
dei recettori A1 da due tipi di vescicola qualitativamente diversi, uno in cui è localizzato nei
recettori A1 e hsc73 ed un altro in cui hsc73 è assente. Questi risultati aprono la possibilità
interessante che il segnale ed il traffico di GPCR potrebbe essere regolato dalle proteine di
scossa di calore. Le nuove scoperte presentate da Sarriò [184] suggeriscono un ruolo specifico
per le proteine hsp70 nel regolare l'attivazione e l'operazione di recettori A1. Sebbene un ruolo
pertinente per la compagnia nel segnalare dai recettori di ormone steroide è stato già
dimostrato, i nostri risultati sono la prima prova che suggerisce un controllo da hsc73 del
segnale tramite GPCR di plasma membrana. Dovrebbe essere anche notato che questo membro
del GPCR può essere diversamente regolato da una proteina che interagisce con i domini
extracellulari del recettore e da una proteina del cytosolic che interagisce con il terzo loop
intracellulare del recettore.

67
20. CONCLUSIONI
Questa nuova strada nel studiare la formazione dei complessi eterodimerici apre la possibilità di
scoprire i meccanismi molecolari, fisiologici e patologici finora sconosciuti ed allo stesso tempo
apre anche un campo nello sviluppo di farmaci che dovrebbero essere specificatamente mirati a
influenzare la formazione di complessi proteina/proteina o a modulare la funzione di questi
complessi proteici. Bisogna tener conto che le interazioni proteina/proteina molto spesso
includono parti della sequenza proteica. Così è difficile sviluppare molecole di basso peso
molecolare capaci di alterare l’interazione della proteina G. Comunque, anche piccole proteine
sono troppo grandi ad essere usate come farmaci. Così, diviene importante l'uso di piccoli
peptidi o meglio molecole peptidiche mimetiche come farmaci. Un approccio importante nello
sviluppo di peptidi che ostacolano le interazioni proteina/proteina è quello di sostituire qualche
amminoacido naturale con amminoacido non naturali.
Come possibili bersagli per l'azione dei farmaci sui complessi eteromerici, il recettore a proteina
G o l'adenilato ciclasi sono i migliori bersagli.
Oltre al classico approccio mirato all'attivazione o all'inibizione del recettore dovuto
all'occupazione del farmaco nella tasca di legame per il ligando naturale, è possibile concepire
altri interventi farmacologici utilizzando sequenze del recettore utilizzabili per la localizzazione
del farmaco.
a) Il farmaco è sviluppato per un recettore centrale per modulare un altro recettore centrale a
livello del sito di riconoscimento. Un esempio è di avere un antagonista A2A che agisce sul
recettore centrale A2A nei complessi dei recettori eterodimerici antagonisti A2A/D2 a produrre
il miglioramento del segnale del recettore D2. Tale farmaco potrebbe diventare un nuovo
farmaco antiparkinsoniano con meno effetti collaterali. Il migliore approccio sarà di bloccare
selettivamente la tasca di legame A2A nel complesso eterodimerico e non quei recettori A2A
non collegati ai recettori D2.
b) Il farmaco è sviluppato per un recettore centrale per indirizzare l'agganciamento della
proteina G e la selettività della proteina G di un altro recettore centrale o l'attività di un

68
recettore a canale ionico esistente nello stesso complesso eterodimerico. Un esempio è il
complesso eterodimerico del recettore D5/GABA A dove l’attivazione del recettore D5 può
ridurre la forza sinaptica del recettore GABA A. Tali farmaci potrebbero essere utilizzati nel
trattamento dei disordini neuropsichiatrici.
c) Il farmaco è sviluppato per uno o entrambi i recettori centrali al controllo del traffico del
recettore del complesso eterodimerico. Un esempio è il complesso eterodimerico A2A/D2 dove
il trattamento prolungato combinato con gli agonisti dei recettori centrali per l’A2A e il D2 ha
prodotto una forte internalizzazione e desensibilizzazione coinvolgendo anche il recettore D2.
Questo farmaco darà una nuova conoscenza nel sviluppare bersagli per il trattamento di
malattie neuropsichiatriche.

69
BIBLIOGRAFIA
1 Ferre, S. et al. (2009) Building a new conceptual framework for reeeptor heteromers. Nat.
Chem. Biol. 5, 131-134 2 AbdAlla, S. et al. (2000) AT1-receptor heterodimers show enhanced G protein activation and altered receptor sequestration. Nature 407, 94- 98 3 Gomes, I. et al. (2000) Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 20, RC110 4 Jordan, B.A. e Devi, L.A. (1999) G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399, 697-700 5 Rocheville, M. et al. (2000) Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science 288, 154-157 6 George, S.R. et al. (2000) Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J. Biol. Chem. 275, 26128-26135 7 Pfeiffer, M. et al. (2001) Homo- and heterodimerization of somatostatin receptor subtypes. Inactivation of sst(3) receptor function by heterodirnerization with sst(2A). J. Biol. Chem. 276, 14027-14036 8 Zhu, W.Z. et al. (2005) Heterodimerization of beta1- and beta2- adrenergic receptor subtypes optimizes beta-adrenergic modulation of cardiac contractility. Circ. Res. 97,244-251 9 Rozenfeld, R. et al. (2006) Heterodimers of G protein-coupled receptors as novel and distinct drug targets. Drug Disov. Today: Ther. Strat. 3, 437-443 10 Jordan, B.A. et al. (2003) Functional interactions between mu opioid and alpha 2A-adrenergic receptors. Mol. Pharmacol. 64, 1317-1324 11 Vilardaga, J.P. et al. (2008) Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat. Chem. Biol. 4, 126-131 12 Breit, A. et al. (2006) Simultaneous activation of the delta opioid receptor (deltaOR)/sensory neuron-specific receptor-4 (SNSR-4) hetero-oligomer by the mixed bivalent agonist bovine adrenal medulla peptide 22 activates SNSR-4 but inhibits deItaOR signaling. Mol. Pharmacol. 70, 686-696

70
13 Rashid, A.J. et al. (2007) D1-D2 dopamine receptor hetero-oligorners with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci.
USA 104, 654-659 14 So, C.H.et al. (2007) Desensitization of the dopamine D1 and D2 receptor hetero-oligomer mediated calcium signal by agonist occupancy of either receptor. Mol. Pharmacol.72, 450-462 15 Fan, T. et al. (2005) A role for the distaI carboxyl tails in generating the noveI pharmacology and G protein activation profìle of mu and delta opioid receptor hetero-oligomers. J. Biol. Chem.
280,38478—38488 16 Small, KM. et al. (2006) Alpha2A and alpha2C adrenergic receptors form homo and heterodimers: the heterodimeric state impairs agonist-promoted GRK phosphorylation and beta-arrestin recruitmenl. Biochemistry 45, 4760-4767 17 Rozenfeld, R. e Devi, L.A. (2007) Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 21, 2455-2465 18 Agnati LF, Fuxe K, Zini I, Lenzi P, and Hokfelt T (1980) Aspects on receptor regulation and isoreceptor identification. Med Biol (Helsinki) 58:182-187
19 Agnati LF, Celani MF, and Fuxe K (1983b) Cholecystokinin peptides in vitro modulate the characteristics of striatal [3H] N-propylnorapomorphine binding sites. Acta Physiol Scand
118:79-81.
20 Zoli M, Agnati LF, Hedlund PB, Li XM, Ferré S, and Fuxe K (1993) Receptor/receptor interactions as an integrative mechanism in nerve cells. Mol Neurobiol 7:293-334.
21 Hollenberg MD (]991l Structure-activity relationship for transmembrane signalling: the receptor's turn FASEB J 5:178-186.
22 Schlessinger J (1988) Signal transduction by allosteric reeeptor oligomerization. Trends
Biochem Sci 13:443-447.
23 Schlessinger J (2000) Celi signaling by receptor tyrosine kinases. Cell 103:211-225. 24 Helldin CH (1995) Dimerization of cell surface receptors in signal transduction. Cell 80:213-223. 25 Margeta-Mitrovic M, Jan YN, and Jan LY (2000) A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27:97-106.

71
26 Agnati LF, Fuxe K, Zoli M, Rondanini C, and Ogren SO (1982) New vistas on synaptic pasticity: mosaic hypothesis on the engram. Med Biol 60:183-190.
27 Zoli M, Agnati LF, Hedlund PB, Li XM, Ferré S, and Fuxe K (1993) Receptor/receptor interactions as an integrative mechanism in nerve cells. Mol Neurobiol 7:293-334.
28 Agnati LF and Fuxe K (2000) Volume transrnission as a key fcature of information handling in the central nervous system. Possible new interpretative value of the Turing's B-type machine. Prog Brain Res 125:3-19.
29 Kaupmann K, Malitschek B, Schuler V, Heid J, Froesti W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, et al. (1998) GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature (Lond) 396:683-687.
30 Jones KA. Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, et al. (1998) GABAB receptors function as a heterorneric assembly of the subunits GABABR1 and GABABR2. Nature (Lond) 396:674-682.
31 White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, and Marshall FH (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature (Lond) 396:679-682.
32 Kuner R, Kohr G, GrUnewald S, Eisenhardt G, Bach A, and Kornau HC (1999) Role of heteromer formation in GABAB receptor function. Science (Wash DC) 283:74-77.
33 Ng GY, Clark J, Coulombe N, Ethier N, Hebert TE, Sullivan R, Kargman S, Chateauneuf A, Tsukamoto N, McDonald T, et al. (1999) Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J Biol Chem 274:7607-7610.
34 Gordon YK, Clark J, Coulombe N, Ethier N, Herbert TE, Sullivan R, Kargman S, Chateauneuf A, Tsukamoto N, McDonald T, et al. (1999) Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J Biol Chem 274:7607-7610.
35 Mitrovic MM, Nung Jan Y, and Yeh Jan L (2000) A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27:97-106.
36 Alger BE and Nicoll RA (1979) GABA mediated biphasic inhibitory responses in hippocampus. Nature (Lond) 281:315-317.
37 Bettler B, Kaupmann K, and Bowery NG (1998) GABAB receptors: drugs meet clones. Curr
Opin Neurobiol 8:345-350.

72
38 Kammerer RA, Frank S, Schulthess T, Landwehr R, Lustig A, and Engel J (1999)
Heteromerization of a functional GABAB receptor is mediated by parallel coiled-coil α helices. Biochemistry 38:13263-13269.
39 Fuxe K and Agnati LF (1985) Receptor/receptor interactions in the centraI nervous system. A new integrative mechanism in synapses. Med Res Reu 5:441-482.
40 Fuxe K and Agnati L (1987) Reeeptor / receptor Interactions. A New Intramembrane
Integrative Mechanism, Macmillan, London.
41 Ng GY, Bertrand S, Sullivan R, Ethier N, Wang J, Yergey J, Belley M, Trimble L, Bateman K, Alder L, et al. (2001) Gamma-aminobutyric acid type B receptors with specific heterodimer composition and postsynaptic actions in hippocampal neurons are targets of anticonvulsant gabapentin action. Mol Pharmacol 59:144-152.
42 Gamier M, Zaratin PF, Ficalora G, Valente M, Fontanella l, Rhee MH, Blumer KJ, Scheideler MA (2003) Up-regulation of regulator of G protein signaling 4 expression in a model of neuropathic pain and insensitivity to morphine. J Pharmacol Exp Ther 304(3):1299-1306.
43 Gaveriaux-Ruff C, Kieffer BL (2002) Opioid receptor genes inactivated in mice: The highlights. Neuropeptides 36(2-3):62-71.
44 Cahill CM, Morinville A, Hoffert C, O'Oonneli D, Beaudet A: Upregulation and trafficking of δ opioid receptor in a model of chronic inflammation (2003) Implications for pain control. Pain
101(1-2):199-208. 45 Bao l, Jin SX, Zhang C, Wang lH, Xu ZZ, Zhang FX, Wang lC, Ning FS, Cai HJ, Guan JS, Xiao HS et
al (2003) Activation of δ opioid receptors induces receptor insertion and neuropeptide secretion. Neuron 37(1):121-133. 46 Kieffer BL (1999) Opioids: first lessons from knockout mice, TIPS 20:19-26.
47 Levac BA, O'Dowd BF, and George SR (2002) Oligomerization of opioid receptors: generation of novel signaling units. Curr Opin. Pharmacol 2:76-8l.
48 Howlett AC (2002) The cannabinoid receptors. Prostaglandins Other Lipid Mediat 68-69:619-631. 49 Malan TP Jr, Ibrahim MM, lai J, Vanderah TW, Makriyannis A, Porreca F (2003) CB2 cannabinoid receptor agonists: Pain relief without psychoactive effects? Curr Opin Pharmacol
3(1):62-67.

73
50 Zhang J, Hoffert C, Vu H-K, Groblewski T, Ahmad S, O'Oonneli D (2003) Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci 17(12):2750-2754. 51 Lembo PMC, Grazzini E, Groblewski T, O'Oonneli D, Roy M-O, Zhang J, Hoffert C, Cao J, Schmidt R, Pelletier M, labarre M (2002) Proenkephalin A gene products activate a new family of sensory neuron-specific GPCRs. Nat Neurosci 5(3):201-209. 52 Dong X, Han S, Zylka MJ, Simon MI, Anderson D (2001) A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106(5):619-632. 53 Han S-K, Oong X, Hwang J-I, Zylka MJ, Anderson DJ, Simon MI (2002) Orphan G protein-coupled receptors MrgA1 and MrgC11 are distinctively activated by RF-amide-related peptides through the Gmq/11 pathway. Proc Natl Acad Sci USA 99(23):14740-14745. 54 Bender E, Buist A, Jurzak M, langlois X, Baggerman G, Verhasselt P, Erken M, Guo HO, Wintmolders C, Van den Wyngaert l, Van Oers I et al (2002) Characterization of an orphan G protein-coupled receptor localized in the dorsal root ganglia reveals adenine as a signaling molecule. Proc Natl Acad Sci USA 99(13):8573-8578. 55 Yu XH, Cao CO, Mennicken F, Puma C, Oray A, O'Oonneli D, Ahmad S, Perkins M (2003) Pro-nociceptive effects of neuromedin U in rat, Neuroscience 120(2):467-474. 56 Xu XJ, Hokfelt T, Bartfai T, Wiesenfeld-Hallin Z (2000) Galanin and spinal nociceptive mechanisms: Recent advances and therapeutic implications. Neuropeplides 34(3-4):137-147. 57 Ji RR, Zhang O, Bedecs K, Arvidsson J, Zhang X, Xu XJ, Wiesenfeld- Hallin Z, Bartfai T, Hokfelt T (1994) Galanin antisense oligonucleotides reduce galanin levels in dorsal root ganglia and induce autotomy in rats after axotomy. Proc Natl Acad Sci USA 91(26):12540- 12543. 58 Blakeman KH, Hao JX, Xu XJ, Jacoby AS, Shine J, Crawley JN, lismaa T, Wiesenfeld-Hallin Z (2003) Hyperalgesia and increased neuropathic pain-like response in mice lacking galanin receptor 1 receptors. Neuroscience 117(1):221-227. 59 Usdin TB, Oobolyi A, Ueda H, Palkovits M (2003) Emerging functions for tuberoinfundibular peptide of 39 residues. Trends Endocrinol Melab 14(1):14-19. 60 Dobolyi A, Ueda H, Uchida H, Palkovits M, Usdin TB (2002) Anatomical and physiological evidence for involvement of tuberoinfundibular peptide of 39 residues in nociception. Proc Natl
Acad Sci USA 99(3):1651-1656.

74
61 Ferré S, von Euler G, Johansson B, Fredholm BB, and Fuxe K (1991d) Stimulation of high-affinity adenosine A-2 receptors decreases the affinity of dopamine D-2 receptors in rat striatal membranes. Proc Natl Acad Sci USA 88:7238-7241.
62 Ferré S and Fuxe K (1992) Dopamine denervation leads to an increase in the membrane interaction between adenosine A2 and doparnine D2 receptors in the neostriatum. Brain Res
594:124-130.
63 Ferré S, Fredholm BB, Morelli M, Popoli P, and Fuxe K (1997) Adenosine-dopamine receptor/receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci
20:482-487.
64 Fuxe K, Ferré S, Snaprud P, von Euler G, Johansson B, and Fredholm B (1993) Antagonistic A2A/D2 receptor interactions in the striatum as a basis for adenosinel dopamine interactions in the central nervous system. Drug Dev Res 28:374-380.
65 Fuxe K, Ferré S, Zoli M, and Agnati LF (1998) Integrated events in central dopamine transmission as analyzed at multiple levels. Evidence for intramernbrane adenosine A2A/dopamine D2 and adenosine A1/dopamine D1 receptor interactions in the basaI ganglia. Brain Res Reu 26:258-273.
66 Lepiku M, Rinken A, Jarv J, and Fuxe K (1997) Modulation of [3H]quinpirole binding to dopaminergic receptors by adenosine A2A receptors. Neurosci Lett 239:61-64. 67 Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, et al. (2002) Coaggregation, cointernalization and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem 277:18091-18097.
68 Salim H, Ferré S, Dalal A, Peterfreund RA, Fuxe K, Vincent JD, and Lledo PM (2000) Activation of adenosine Al and A2A receptors modulates dopamine D2 receptorinduced responses in stably transfected human neuroblasloma cells. J Neurochem 74:432-439. 69 Kull B, Fer-ra S, Arslan G, Svenningsson P, Fuxe K, Owman C, and Fredholm BB (1999) Reciprocal interactions between adenosine A2A and dopamine D2 receptors in Chinese harnster ovary cclls co-transfectcd with the two receptors, Biochem: Pharmacol 158:1035-1045.
70 Kozell LB, Machida CA, Neve RL, and Neve KA (]994) Chimeric D1/D2 dopamine receptors. Distinct determinants of selective efficacy, potency and signal transducticn. J Biol Chem.
269:30299-30306.
71 Kozell LB and Neve KA (1997) Constitutive activity of a chimeric D2/D1 dopamine receptor. Mol Pharmacol 52:1137-1149

75
72 Zeng EV, Pearce RK, MacKenzie GM, and Jenner P (2000) Alterations in preproenkephalin and adenosine-2a receptor mRNA, but not preprotachykinin mRNA correlate with occurence of dyskinesia in normal monkeys chronically treated with L-DOPA. Eur J Neurosci 12:1096-1104
73 Torvinen M, Liu Y, Kozell LB, Neve KA, Ferré S, Ibanez C, and Fuxe K (2001) Dopamine D2 receptor domains involved in the intramembrane interaction between adenosine A2A and dopamine D2 receptors. Soc Neurosci Abstr 27:379.3
74 Garrett BE and Holtzman SG (1994) Caffeine cross-tolerance to selective dopamine D1 and D2 receptor agonists but not to their synergistic interaction. Eur J Pharmacol 262:65-75.
75 Fenu S, Cauli O, and Morelli M (2000) Cross-sensitization between the motor activating effects of bromocriptine and caffeine: role of adenosine A2A receptors. Behav Brain Res 114:97-
105.
76 Hillion, L, Canals, M., Torvinen, M., Casa do, V., Scott, R., Terasmaa, A., Hansson, A., Watson, S., Olah, M. E., Mallol, J., Canela, E. I., Zoli, M., Agnati, L. F., Ibanez.C. F., Lluis, C, Franco, R., Ferre, S., and Fuxe, K. (2002) J. Biol. Chem. 277, 18091-18097 77 Ferre, S., von Euler, G., Johansson, B., Fredholm, B. B., and Fuxe, K. (1991) Proc. Natl. Acad.
Sci. U.5.A. 88,7238-7241 78 Ferré, S., Fredholm, B. B., Morelli, M., Popoli, P., and Fuxe, K. (1997) Trends Neurosci. 20, 482-487
79 Ferré, S., Ciruela, F., Quiroz, C, Lujàn, R., Popoli, P., Cunha, R.A., Agnati, L. F., Fuxe, K., Woods, A. S., Lluis, C, and Franco, R. (2007) Scientific WorldJournal 7, 74- 85 80 Ferré, S., Quiroz, c., Woods, A. S., Cunha, R., Popoli, P., Ciruela, F., Lluis, c., Franco, R., Azdad, K., and Schiffmann, S. N. (2008) Curr. Pharm. Des. 14,1468-1474 81 Canals, M., Marcellino, D., Fanelli, F., Ciruela, F., de Benedetti, P., Goldberg, S. R., Neve, K., Fuxe, K., Agnati, L. F., Woods, A. S., Ferré, S., Lluis, C, Bouvier, M., and Franco, R. (2003) J. Biol.
Chem. 278, 46741-46749 82 Altamura, A. C, Bassetti, R., Cattaneo, E., and Vismara, S. (2005) World J. Biol. Psychiatry 6, Suppl. 2, 23-30 83 Ciruela, F., Burgueno, L, Casadò, V., Canals, M., Marcellino, D., Goldberg, S. R., Bader, M., Fuxe, K., Agnati, L. F., Lluis, c, Franco, R., Ferré, S., and Woods, A. S. (2004) Anal. Chem. 76, 5354-5363

76
84 Woods, A. S., and Ferré, S. (2005) J. Proteome Res. 4, 1397-1402 85 Woods, A. S., Ciruela, F., Fuxe, K., Agnati, L. F., Lluis, c., Franco, R., and Ferré, S. (2005) J Mol.
Neurosci. 26, 125-132 86 Bofill-Cardona, E., Kudlacek, O., Yang, Q., Ahorn, H., Frcissmuth, M., and Nanoff, C. (2000) J. Biol. Chem. 275, 32672-32680 87 Liu, Y., Buck, D. C, Macey, T. A., Lan, H., and Neve, K. A. (2007) J. Recept. Signal. Transduct.
Res. 27,47-65 88 Woods, A. S., Marcellino, D., Iackson, S. N., Franco, R., Ferré, S., Agnati, L. F., and Fuxe, K. (2008) J. Proteome Res. 7,3428-3434 89 Woods, A. S. (2004) J. Proteome Res. 3,478-484 90 Woods, A. S., and Huestis, M. A. (2001) J. Am. Soc. Mass Spectrom. 12, 88-96 91 Nimchinsky EA, Hof PR, Janssen WC, Morrison JH, and Schmauss C (1997) Expression of dopamine D3 receptor dimers and tetramers in brain and in transfected cells. J Biol Chem
272:29229-29237.
92 Lee SP, Xie Z, Varghese G, Nguyen T, O'Dowd BF, and George SR (2000) Oligomerization of dopamine and serotonin receptors. Neuropsychopharmacology 23:S32-840. 93 Scarselli M, Novi F, Schallmach E, Lin R, Baragli A, Colzi A, Griffon N, Corsini GU, Sokoloff P, Levenson R, et al. (2001) D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J Biol Chem 276:30308-30314.
94 Gouldson PR, Snell CR, Bywater RP, Higgs C, and Reynolds CA (1998) Domain swapping in G-protein coupled receptor dimers, Protein Eng 11:1181-1193.
95 Gouldson PR, Higgs C, Smith RE, Dean MK, Gkoutos GV, and Reynolds CA (2000) Dimerization and domain swapping in G-protein-coupled receptors: a computational study. Neuropsychopbarmacology 23:60-77.
96 Maggio R, Vogel Z, and Wess J (1993) Coexpression studies with mutant muscarinid adrenergic receptors provide evidence for intermolecular 'cross-talk' between Gprotein- linked receptors. Proc Natl Acad Sci USA 90:3103-3107.

77
97 Le Moine C and Bloch B (1996) Expression of the D3 dopamine receptor in peptideric neurons of the nucleus accumbens: comparison with the D1 and D2 dopamine receptors. Neuroscience 73:131-143.
98 Gurevich EV and Joyce JN (999) Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropeychophormacology 20:60-80.
99 Rocheville M, Lange DC, Kumar U, Sasi R, Patel RC, and Patel YC (2000b) Subtypes of the somatostatin receptor assemble as functional home- and heterodimers. J Biol Chem 275:7862-
7869.
100 Cohn ML and Cohn M (1975) "Barrel rotation" induced by somatostatin in the nonlesioned rat. Brain Res 96:138
101 Havlicek V, Rezek M, and Friesen H (1976) Somatostatin and thyrotropin releasing hormone: centraI effect on sleep and motor system. Pharmacol Biochem Behav 4:455-459. 102 Kastin AJ, Coy DH, Jacquet Y, Schally AV, and PlotnikoffNP (1978) CNS effects of somatostatin. Metabolism 27:1247-1252.
103 Chneiweiss J, Glowinski J, and Premont J (1985) Modulation by monoamines of somatostatin-sensitive adenylate cyclase on neuronal and glial cells from the mouse brain in primary cultures. Neurochemistry 44:1825.
104 Ginés S, Hillion J, Torvinen M, LeCrom S, Casado V, Canela E, Rondin S, Lew J, Watson S, Zoli M, et al. (2000) Dopamine D1 and adenosine A1 receptors assemble into functionally interacting heteromeric complexes. Proc Natl Acad Sci USA 97:8606-8611.
105 Martin-Iverson MT, Radke JM, and Vincent SR (1986) The effects of cysteamine on dopamine-mediated behaviors: evidence for dopamine-somatostatin interactions in the striatum. Pharmacol Biochem Behau 24:1707-1714.
106 Leblanc R, Gauthier S, Gauvin M, Qu.irion R, Palmour R, and Masson H (1988) Neurobehavioral effects of intrathecal somatostatinergic treatment in subhuman primates. Neurology 38:1887-1890.
107 Izquierdo-Claros RM, Boyano-Adanez MC, Larsson C, Gustavsson L, and Arilia E (1997) Acute effects of D1 and D2 receptor agonist and antagonist drugs on somatostatin binding, inhibition of adenylyl cyclase activity and accumulation of inositol 1,4,5- trisphosphate in the rat striatum. Brain Res Mol Brain Res 47:99-107.

78
108 Rodriguez-Sanchez MN, Puebla L, Lopee-Samudo S, Rodriguez-Martin E, Martin- Espinosa A, Rodriguez-Pena MS, Juarranz MG, and Arilla E (997) Dopamine enhances sornatostatin receptor-mediated inhibition of adenylate cyclase in rat striatum and hippocampus, J Neurosci
Res. 47:238-248.
109 Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, and Patel YC (2000a) Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science (Wash DC) 288:154-157.
110 Hukovic N, Panetta R, Kumar U, Rocheville M, and Patel YC (1998) The cytoplasmic tail of the human somatostatin receptor type 5 is crucial for interaction with adenylate cyclase and in mediating desensitization and internalization. J Biol Chem 273:21416-21422.
111 Ferré S, Popoli P, Giménez-Llort L, Finnman U-B, Martinez E, Scotti de Carolis A, and Fuxe K (1994b) Postsynaptic antagonistic interaction between adenosine A1 and dopamine D1 receptors. Neuroreport 6:73-76.
112 Ferré S, Torvinen M, Antoniou K, Irenius E, Civellì O, Arenas E, Fredholm BB, and Fuxe K (1998) Adenosine A1 receptor-mediated modulation of dopamine D1 receptors in stably cotransfected fibroblast cells. J Biol Chem 273:4718-4724.
113 Fuxe K, Ferré S, Torvinen M, Hillion J, Stromberg I, Franzén O, Ibànez C, Zoli M, Lluis C, Agnati LF, et al. (2002) Heteromerization of adenosine and dopamine receptor subtypes. Relevance for neuronal integration in normal and pathological states, in Catecholamine
Research: From Molecular Insights to Clinical Medicine, 9 th Int Catecholamine Symposium,
Kyoto, Japan; 2001; in press; Kluwer Academid Plenum Publishers, New York. 114 Franco R, Ferré S, Torvinen M, Ginés S, Hillion J, Ciruela F, Canela EI, Mallol J, Casadé V, Lluis C, et al. (2001) Adenosioe/dopamine receptor/receptor interactions in the central nervous system. Drug Dev Res 52:296-302.
115 Lefkowitz RJ (2000)The superfamily of heptahelical receptors. Nat Cell Biol 2:EI33- E136.
116 McDonald PH and Lefkowitz RJ (2001) βArrestins: new roles in regulating heptahelical receptors' functions. Cell Signal 13:683-689.
117 Ciruela F, Casad6 V, Mallol J, Canela EI, Lluis C, and Franco R (1995) Immunological identification of A1 adenosine receptors in brain cortex. J Neurosci Res 42:818-828. 118 Ferré S, Popoli P, Rimondini R, Reggio R, Kehr J, and Fuxe K (1999a) Adenosine A2A and group I metabotropic glutamate receptors synergistically modulate the binding characteristics or dopamine D2 receptors in the rat striatum. Neuropharmacology 39:129-140.

79
119 Ciruela F, Escriche M, Burgueno J, Angulo E, Casado V, Soloviev MM, Canela EI, Mallol J, Chan WY, Lluis C, et al. (2001a) Metabotropic glutamate 1alpha and adenosine A1 receptors assemble into functionally interacting complexes. J Biol Chem 276:18345-18351.
120 Ciruela F, Eseriche M, Soloviev MM, Canela EI, Burgeno J, Mallol J, Chan WY, Lluis C, Mcllhinney RAJ, and Franco R (2001b) Adenosine-glutamate receptor/receptor interaetions in the centraI nervous system. Drug Dev Res 52:316-322.
121 Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamarnoto M, Kumasaka T, Nakanishi S, Jingami H, and Morikawa K (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature (Lond) 407:971-977.
122 Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escoribuela RM, Fernandez-Teruel A, Wiesenf'eld-Hallin Z, Xu XJ, et al. (2001) Hyperalgesia, anxiety and decreased hypoxic neuroprotection in mice lacking the adenosine A1 reccptor. Proc
Natl Acad Sci USA 98:9407-9412.
123 Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu WK, Lanahan AA, Sheng M, et al. (1999) Coupling of mGluR/Homer and PSD-95 complexes by the shank family of postsynaptic density proteins. Neuron 23:583-592. 124 Xiao B, Tu JC, and Worley PF (2000) Homer: a Iink between neural activity and glutamate receptor function. Curr Opin Neurobiol 10:370-374.
125 Ciruela F, Soloviev MM, Chan WY, and McCbinney RA (2000) Homer-1c/Vesl-1L modulates the cell surface targeting of metabotropic glutamate receptor type 1a: evidence for an anchoring function. Mol Cell Neurosci 15:36-50.
126 Yoshioka K, Saitoh O, and Nakata H (2001) Heterorueric association creates a P2Y-like adenosine receptor. Proc Natl Acad Sci USA 98:7617-7622.
127 Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, and Williams M (1994) Nomenclature and classification of purinoceptors. Pharmacol Rev 46:143-
156.
128 Mendoza-Femandez V, Andrew RD, and Barajas-Lépez C (2000) ATP inhibits glutamate synaptic release by acting at P2Y receptors in pyramidal neurons of bippocampus slices. J
Pharmacol Exp Ther 293:172-179.
129 Fredholm BB (1995a) Purinoceptors in the nervous system. Pharmacol Toxicol 76:228-239.

80
130 Ferré Sand Fuxe K (2000) Adenosine as a volume transmiss ion signal. A feedback detector of neuronal activation, in Progress in Brain. Research (Agnati LF, Fuxe K, Nicholson C, and Sykovà E eds) vol 125, pp 353-361, Eisevier Science B.V., Amsterdam. 131 Fredholm BB, Uzerman AP, Jacobson KA, Klotz KN, and Linden J (2001) International union of pharmacology. XXV. Nomenclature and classification of adenosine receptor. Pharmacol Rev
53:527-552.
132 Ferré S, Karcz-Kubicha M, Hope BT, Popoli P, Burgueno J, Gutierrez MA, Casado V, Fuxe K, Goldberg SR, Lluis C, et al. (2002) Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc Natl Acad Sci USA 99:11940-
11945.
133 Popoli P, Pezzola A, Torvinen M, Reggio R, Pintor A, Scarchilli L, Fuxe K, and Ferré S (2001) The selective mGlu(5) receptor agonist CHPG inhibits quinpirole-induced turning in 6-hydroxydopamine-lesioned rats and modulates the binding characteristics of dopamine D(2) receptors in the rat striatum: interactions with adenosine A(2a) receptors. Neurapsyehopharmaealagy 25:505-513.
134 Ferré S, Popoli P, Rimondini R, Reggio R, Kehr J, and Fuxe K (1999a) Adenosine A2A and group I metabotropic glutamate receptors synergistically modulate the binding characteristics or dopamine D2 receptors in the rat striatum. Neuropharmacology 39:129-140.
135 Rimondini R, Fuxe K, and Ferré S (999) Multiple intramembrane receptor/receptor intcractions in the regulation of striatal dopamine D2 receptors, Neuroreport 10:2051-2054. 136 Milligan G and White JH (2001) Protein-protein interactions at G-protein-coupled receptors. Trends Pharmacol Sci 22:513-518.
137 Sheng M and Kim E (2000) The Shank family of scaffold proteins. J Cell Sci 113:1851-1856. 138 Cho K and Bashir Z (2002) Cooperation between mglu receptors: a depressing mechanism? Trends Neurosci 25:405-411.
139 Dale LB, Babwah AV, and Ferguson SSG (2002) Mechanisms of metabotropic glutamate receptor desensitization: role in the patterning or effector enzyme activation. Neurachem Tnt
41:319-326.
140 Breysse N, Baunez C, Spooren W, Gasparini F, and Amalric M (2002) Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic delicits n a rat model of Parkinsonism. J Neurosci 22:5669-5678.

81
141 Fior DR, Hedluod PB, and Fuxe K (1993) Autoradiographic evidence for a radykinin/angiotensin II receptor/receptor interaction in the rat br-ain. Neurosci Lett 163:58-62. 142 AbdAlla S, Lother H, and Quietterer U (2000) AT1 receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature (Lond) 407:94-98. 143 AbdAlla S, Lother H, el Massiery A, and Quitterer U (2001) Increased AT1 receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med
7:1003-1009.
144 Liu F, Wan Q, Pristupa ZB, Yu XM, Wang YT, and Niznik HB (2000) Direct protein-protein coupling enables cross-talk between dopamine D5 and -y-aminobutyric acid A receptors. Nature
(Lond) 403:274-280.
145 Bergson C, Mazljak L, Sniley JF, Pappy M, Levenson R, and Goldrnan-Rakic PS (1995) Regional cellular and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci 15:7821-7836.
146 Nusser Z, Roberts JD, Baude A, Richards JG, and Somogyi P (1995) Immunocytochemical
localization of α 1 and β2/3 subunits of the GABAA receptor in relation to specific GABAergic synapses in the dentate gyrus. Eur J Neurosci 7:630-636.
147 Wang YT (2002) Dopamine D5/GABA-A heteromerization a mechanism to regulate synaptic weight. Collegium Intemationale Neuro- Psychopharmacologicum 2002, June 23-27: Abstract, refnr: 901, Anirnula Publishing House, Budapest, Hungary. 148 Goldman-Rakic PS and Selemon LD (997) Functional and anatomical aspects of prefrontal pathology in schizopbrenia. Schizophr Bull 23:437-458.
149 Okubo Y, Suhara T, Suzuki K, Kobayasbi K, Inoue O, Terasaki O, Somcya Y, Sassa T, Sudo Y, Matsushima E, et al. (1997) Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature (Lond) 385:634- 636 150 Huntsman M:M, Tran BV, Potkin SG, Bunney WR Jr, and Jones EG (1998) Altered ratios of altematively spliced long and short -y2 subunit mRNAs of the GABAA receptor in prefrontal cortex of schizophrenia. Proc Natl Acad Sci USA 95:15066-15071.
151 Keverne EB (1999) GABA-ergic neurons and the neurobiclogy of sehizophrenia and other psychosis. Brain Res Bull 48:467-473.

82
152 Agnati LF, Fuxe K, Merlo-Pich E, Zoli M, Zini I, Benfenati F, Harfstrand A, and Goldstein M (1987) Aspects on the integrative capabilities of the central nervous system: evidence for "volume transmission" and its possible relevance far receptor/ receptor interactions in Receptor
I receptor Interactions (Fuxe, K and Agnati LF eds) pp 236-249, Macmillan, London. 153 Agnati LF and Fuxe K (2000) Volume transmission as a key feature of information handling in the central nervous system. Possible new interpretative value of the Turing's B-type machine. Prog Brain Res 125:3-19.
154 Abel T and Kandel E (1998) Positive and negative regulatory mechanims that mediate long-term memory storage. Brain Res Rev 26:360-378.
155 Pérez de la Mora M, Ferré S, and Fuxe K (1997) GABA-dopamine receptor/receptor interactions in neostriatal membranes of the rat. Neurochem Res 22:1051-1054.
156 Fuxe K, von Euler G, Agnati LF, and Ògren SO (1988c) Galanin selectively modulates 5-hydroxytryptamine 1A receptors in the rat ventral limbic cortex. Neurosci Lett 85:163-167.
157 Fuxe K, Agnati LF, von Euler G, Benfenati F, Zoli M, Harfstrand A, and Fredholm B (1989) Receptor/receptor interactions and development of psychoactive drugs, in Neurachemieal
Pharmacology (Costa E ed) pp 211-227, Raven Press Ltd., New York 158 Changeux J and Edelstein SJ (2001) Allosteric mechanisms in normal and pathological nicotinic acetylcholine receptors. Curr Opin Neurobial 11:369-377.
159 Khakh BS, Zhou X, Sydes J, Galligan JJ, and Lester HA (2000) State-dependent cross-inhibition between transmitter-gated cation channels. Nature (Lond) 406: 405-410. 160 Yamauchi T, Ueki K, Tobe K, Tamemoto H, Sekine N, Wada M, Honjo M, Takahashi M, Takahashi T, and Hirai T (1997) Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature (Lond) 390:91-96
161 Maudsley S, Pierce KL, Zamah M, Miller WE, Abn S, Daaka Y, Lefkowitz RJ, and Luttrell LM
(2000) The β2-adrenergic receptor mediates extracellular signalregulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem
275:9572-9580. 162 Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu
H, Owada K, Luttrell DK, et al. (1999) β-arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science (Wash DC) 283:655-661.

83
163 Phonphok Y and Rosenthal KS (1991) Stabilization of clathrin vesicles by amantadine, tromantadine and other hydrophobic amines. FEBS Lett 281:188-190.
164 Quijano VJ Jr and Sheflield LG (1998) Prolactin decreases epidermal growth factor receptor kinase activity via a phosphorylalion-dependent mechanism. J Biol Chem 273:1200-1207.
165 McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, and Foord SM (1998) Ramps regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature (Lond) 393:333-339.
166 Christopoulos G, Perry KJ, Morlis M, Tilakaratne N, Gao Y, Fraser NJ, Main MJ, Foord SM, and Sexton PM (1999) Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol 56:235-242.
167 Chen WJ, Armour S, Way J, Chen G, Watson C, Irveng P, Cobb J, Kadwell L, Beaumont K, Rimele T, et al. (1997) Expression cloning and receptor pharmacology of human calcitonin receptors from MCF-7 cells and their relationship to amylin receptors. Mol Pharmacol 52:1164-
1175.
168 Perry KJ, Quiza M, Myers DE, Morlis M, Christopoulos G, and Sexton PM (1997) Characterization of amylin and calcitonin receptor binding in the mouse alphathyroid- stimulating hormone thyrotroph cell line. Endocrinology 138:3486-3496.
169 Lezcano N, Mrzljak L, Eubanks S, Levenson R, Goldman-Rakic P, and Bergson C (2000) Dual signaling regulated by Calcyon, a D1 dopamine receptor interacting protein. Science (Wash DC)
287:1660-1664.
170 Mrzljak L, Levey Al, and Goldman-Rakic PS (993) Association of m1 and m2 muscarinic receptor proteins with asymmetric synapses in the primate cerebral cortex: morphological evidence for cholinergic modulation of excitatory neurotransmission. Proc Natl Acad Sci USA
90:5194-5198.
171 Wang JQ and McGinty JF (1997) The full D1 dopamine receptor agonist SKF-82958 induces neuropeptide mRNA in the normosensitive striatum of rats: regulation of D1/D2 interactions by muscarinic receptors. J Pharmacol Exp Ther 281:972-982.
172 Franco R, Casad6 V, Ciruela F, Saura C, Mallol J, Canela EI, and Lluis C (1997) Cell surface adenosine deaminase: much more than an ectoenzyme. Prog Neurobiol (Oxf) 52:283-294.
173 Franco R, Mallol J, Casad6 V, Llui C, Canela EI, Saura C, BIanco J, and Ciruela F (1999) Ecto-adenosine deaminase: an ectoenzyme and a costimulatory protein acting on a variety of cell surface receptors. Drug Dev Res 45:261-268.

84
174 Franco R, Ferré S, Agnati L, Torvinen M, Gines S, Hillion J, Casado V, Lledo PM, Zoli M, Lluis C, et al. (2000) Evidence far adenosine/dopamine receptor interactions: indications for heteromerization. Neuropsychopharmacology 23:850--859.
175 Lluis C, Cordero O, and Franco R (1998) Ecto-adenosine deaminase may play a relevant role in the development of the immune syste. Immunol Today 19:533-534
176 Mirabet M, Herrera C, Cordero OJ, Mallol J, Lluis C, and Franco R (1999) Expression of A2B adenosine receptors in human Iymphocytes: their role in T cell activation. J Cell Sci 112:491-502.
177 Herrera C, Casad6 V, Ciruela F, Schoficld P, Mallol J, L1ufs C, and Franco R (2001) Adenosine A2B receptors behave as an alternative anchoring protein for cell surface adenosine dearninase in Iymphocytcs and cultured cells. Mol Pharmacol 59:127-134. 178 Franco R, Valenzuela A, Lluts C, and BIanco R (1998) Enzymatic and extraenzymatic role of ecto-adenosine deaminase in lymphocytes. Immunol Rev 161:27-42.
179 Ruiz MA, Escriche M, Lluis C, Franco R, Martin M, Andrea A, and Ros M (2000) Adenosine A1 receptor in cultured neurons from reat cerebral cortex: colocalization with adenosine deaminase. J Neurochem 75:656-664.
180 Ciruela F, Saura C, Canela EI, Mallol J, Lluis C, and Franco R (1996) Adenosine deaminase affects ligand-induced signalling by interacting with cell surface adenosine receptors. FEBS Lett
380:219-223.
181 Saura C, Ciruela F_ Casad6 V, Canela EI, Mallol J, Lluis C, and Franco R (1996) Adenosine deaminase interacts with A1 adenosine receptors in pig brain cortical membranes. J Neurochem
66:1675-1682.
182 Saura C, Mallol J, Canela EI, Llufs C, and Franco R (1998) Adenosine deaminase and A, adenosine receptors internalize together following agonist-induced receptor desensitization. J
Biol Chem 27:17610-17617.
183 Torvinen M, Ginés S, Hillion J, Latini S, Canals M, Ciruela F, Bordoni F, Staines W, Pedata F, Agnati LF, et al. (2002) Interactions among adenosine deaminase, adenosine A1 receptors and dopamine DI receptors in stably cotransfected fibreblast cells and neurons. Neuroscience
113:709-719.
184 Sarriò S, Casadò V, Escriche M, Ciruela F, Mallol J, Canela EI, Lluis C, and Franco R (2000) The heat shock cognate protein hsc73 assembles with A1 adenosine receptors to form functional modules in the cell membrane. Mol Cell BioI 20:5164- 5174.IUINDI


















