
STUDIO CLINICO PROSPETTICO: Induzione e consolidamento ... · Induzione e consolidamento della...
41
1 STUDIO CLINICO PROSPETTICO : Induzione e consolidamento della remissione completa nella leucemia mieloide acuta dell’adulto con idarubicina, citosina arabinoside ad alta intensità di dose, G-CSF, e “mini”-autotrapianto di cellule emopoietiche staminali circolanti: fattibilità, tossicità ed efficacia per classe di rischio (con valutazione comparativa verso allotrapianto di midollo osseo nei casi selezionati per la procedura) Divisione di Ematologia, Ospedali Riuniti, Bergamo (Prof. T. Barbui) Opsedali Riuniti, Largo Barozzi 1, 24100 Bergamo tel 035 269 493 fax 035 266 157 e-mail: [email protected] Preparato il: 14 Gennaio 2001 Coordinamento: Dott. R. Bassan INDICE
Transcript of STUDIO CLINICO PROSPETTICO: Induzione e consolidamento ... · Induzione e consolidamento della...

1
STUDIO CLINICO PROSPETTICO:
Induzione e consolidamento della remissione completa nella leucemia mieloide
acuta dell’adulto con idarubicina, citosina arabinoside ad alta intensità di
dose, G-CSF, e “mini”-autotrapianto di cellule emopoietiche staminali
circolanti: fattibilità, tossicità ed efficacia per classe di rischio (con valutazione
comparativa verso allotrapianto di midollo osseo nei casi selezionati per la
procedura)
Divisione di Ematologia, Ospedali Riuniti, Bergamo (Prof. T. Barbui)
Opsedali Riuniti, Largo Barozzi 1, 24100 Bergamo
tel 035 269 493
fax 035 266 157
e-mail: [email protected]
Preparato il: 14 Gennaio 2001
Coordinamento: Dott. R. Bassan
INDICE

2
TITOLO/Sezione pag. cfr.
1. PREMESSE
1.1 Consolidamento della remissione nella leucemia mieloide acuta 5
dell’adulto (LMA-a): opzioni e fattori di rischio.
1.1.1 Opzioni 5
1.1.2 Fattori di rischio 5
1.2 Esperienza del gruppo L-B-V (London-Bergamo-Vicenza) 6
1.2.1 Studi clinici 6 Tab. 1
1.2.2 Autotrapianto vs ara-C ad alte dosi 6 Fig. 1
1.3 Autotrapianto con cellule staminali periferiche (studio BXIV) 7
1.3.1 Risultati 7
1.4 Tossicità da trattamento 7
1.4.1 Mortalità da complicanze 7 Tab. 2
1.4.2 Mortalità in remissione da ara-C ad alte dosi 8
Tab. 3
1.5 Sintesi delle attuali possibilità terapeutiche 9
1.5.1 Prognosi 9
1.5.2 Variabili correlate al trattamento 9
1.5.3. Scelta di modalità e intensità terapeutica 9
1.5.4. Ruolo di ara-C ad alte dosi 9
1.5.5. Ruolo dei fattori di crescita granulocitaria 10
1.5.6. Ruolo delle cellule staminali circolanti 10
2. NUOVO STUDIO CLINICO: ASPETTI TEORICI
2.1 Razionale 11

3
2.1.1 Razionale 1 11
2.1.2 Razionale 2 11 Tab. 4
2.2 Scopo dello studio 12
2.2.1 Obiettivi 12
2.3 Criteri di eleggibilità 12
2.3.1 Diagnosi 12
2.3.2 Età 13
2.3.4 Consensi informati 13
2.4 Definizioni 13
2.4.1 Sopravvivenza e EFS 13
2.5 Considerazioni statistiche 13
2.5.1 Calcolo del campione 13
2.5.2 End-points statistici 13 Tab. 5
3. DISEGNO E APPLICAZIONE
3.1 Disegno 15
3.1.1 Disegno 15
3.2 INDUZIONE RC 16
3.2.1 Induzione di RC con schema ‘ICE’ : 16
3.2.2 Risposta al trattamento: definizioni 16
3.2.3 Secondo ciclo “IC” per casi in RC 16
3.2.4 Schema di salvataggio “SPLIT” 16
3.3 Trattamento post-RC: fase comune “A8” e cellule staminali circolanti 17
3.3.1 Secondo consolidamento/mobilizzazione 17

4
3.3.2 Raccolta e impiego cellule staminali CD34+ 18
3.4 Trattamento post-RC: attribuzione e somministrazione 18
3.4.1 Criteri di diversificazione del trattamento 18
3.4.2 Definizione di classe di rischio 19
Tab. 6
3.4.3 Trattamento postremissionale finale per classe di rischio 21
3.4.4 Intervalli di trattamento 21
3.4.5. Osservazione 22
4 PROGRAMMA RECIDIVE
4.1 Razionale 23
4.1.1 Razionale 1 23
4.1.2 Razionale 2 23
4.2 Studio recidive 23
4.2.1 Reinduzione RC per recidiva 23 Fig. 3
4.2.2 Trattamento dopo RC 24
5 OBIETTIVI, ARRUOLAMENTO, STATISTICA,
CENTRI PARTECIPANTI, ORGANIZZAZIONE
5.1 Obiettivi 25
5.2 Arruolamento 25
5.3 Svolgimento dello studio, monitoraggio 25
5.4 Statistica 25
5.5 Raccolta dati e software per analisi 26
5.6 Centri partecipanti 26
5.7 Coordinamento 27
6. ASPETTI ETICI

5
6.1 Aspetti etici 28
6.2 Consenso informato 28
6.3 Aderenza al protocollo 28
6.4 Comitato etico 28
7. Referenze bibliografiche 29-33
8. Appendici 1-3
9. Schema riassuntivo
1. PREMESSE
1.1 Consolidamento della remissione nella leucemia mieloide acuta dell’adulto
(LMA-a): opzioni e fattori di rischio.
1.1.1 Opzioni Il trattamento primario con polichemioterapia permettte di ottenere una
remissione completa (RC) nel 70-80% dei pazienti con LMA-a (età 15-60 anni). Caratteristicamente,
in questa fascia di età si situa la gran parte dei casi guaribili con chemioterapia, intendendosi con
questo termine uno stato di RC perdurante a 5 anni e oltre. Per consolidare la remissione della LMA-a
e quindi ottenere un possibile risultato di cura sono disponibili tre diverse opzioni terapeutiche:
trattamento con farmaci a dosi convenzionali (in via di abbandono e generalmente riservato ai pazienti
più anziani), con alte dosi (comprendente citosina arabinoside/ara-C), oppure con schemi mieloablativi
che necessitano di un supporto trapiantologico (allogenico o autologo) con cellule emopoietiche
staminali. A parte numerosi studi di fase II, sono noti i risultati di alcuni studi randomizzati dai quali si
evidenzia una sostanziale parità di “event-free survival/EFS” tra i trattamenti comprendenti ara-C ad
alte dosi, allotrapianto, ed autotrapianto, in genere con una ridotta incidenza di recidiva ma aumento
della mortalità dopo allotrapianto1-5.
1.1.2 Fattori di rischio I risultati di EFS variano a seconda del profilo di rischio: escludendo la
LMA promielocitica, attualmente trattata in modo autonomo con programmi a base di acido retinoico,

6
per rischio intermedio-alto si intendono i casi con cariotipo apparentemente normale o con alterazioni
cromosomiche diverse da t(8;21) e inv(16)6-10, e ciò vale sia per programmi chemioterapici che
trapiantologici11,12. Anche i casi con fenotipo funzionale MDR-1+ (e forse MRP+ e LRP+), RC
tardiva (dopo il primo ciclo) con LMA secondaria/post-mielodisplasia (MDS) sembrano appartenere
prevalentemente alla categoria alto rischio, come pure i casi con blasti elevati (livelli variabili) ed età
avanzata (livelli variabili ma generalmente >50 anni) dei pazienti. Nella nostra esperienza, è stato
confermato un ruolo prognostico sfavorevole per cariotipo, epatosplenomegalia, LAM-MDS e
leucocitosi >50.000/mmc (per l’ottenimento della RC in quest’ultimo caso)13-17. Poichè con i
protocolli a base di idarubicina (ICE e similari)18 e, nel caso di iniziale resistenza, con schema
SPLIT19 vengono indotti in RC più casi con blasti >50.000/mmc, è molto probabile che l’impatto
prognostico negativo di una blastosi elevata riguardi successivamente anche la durata di RC. In vista
delle buone correlazioni cliniche, della relativa facilità di esecuzione e della disponibilità di sonde
molecolari per t(8;21) e inv(16), che permettono di discernere con sicurezza almeno i casi a profilo di
rischio favorevole, l’indice di rischio citogenetico appare un ottimo indicatore prognostico generale
per un nuovo studio clinico, affiancato da altri criteri clinici (blastosi, epatosplenomegalia, e MDS) nei
casi a profilo di rischio citogenetico intermedio.
1.2 Esperienza del gruppo L-B-V (London-Bergamo-Vicenza)
1.2.1 Studi clinici A partire dal 1984 sono stati attivati dal gruppo L-B-V 3 studi clinici
collaborativi osservazionali prospettici (fase II) nei quali è stato esplorato il ruolo terapeutico di un
consolidamento convenzionale a breve termine (STT, 1984), e successivamente dell’ autotrapianto di
midollo osseo (BXIII, 1988) e di cellule emopoietiche circolanti (BXIV, 1994). I risultati di questi
studi13-17, confrontando i risultati di BXIII/BXIV vs STT sono sintetizzati di seguito (Tabella 1).
Tabella 1. RC e EFS degli strudi STT, BXIII, BXIV (“intention-to-treat”).
________________________________________________________________________________
Programma N. RC (%) EFS (5 anni) p Fattori
________________________________________________________________________________
STT* 396 243 (61) 30% - MDS, età,
blasti (RC) M3,

7
BXIII** 144 106 (73) 52% 0.002
citogenetica (CR),
epatosplenomegalia (EFS)
BXIV*** 141 108 (77) 40% (4 anni) 0.008
citogenetica
________________________________________________________________________________
________________
*69 con FAB M3; **età max. 50 anni; no M3; ***età max. 60 anni, no M3
1.2.2 Autotrapianto vs ara-C ad alte dosi L’analisi di un sottogruppo di pazienti consolidati con
ara-C per esigenze di protocollo (BXIII: età >50 anni) o altri problemi (ara-C: 1-2 cicli ad 1
g/m2/dose, totale 12-24 g/m2), ma senza autotrapianto, risulta particolarmente importante. Infatti,
come evidenziato nella Figura 1, il risultato di EFS per i gruppi di trattamento effettivo con ara-C o
autotrapianto è risultato essere sovrapponibile, confermando anche in questa esperienza la parità
terapeutica tra i due diversi tipi di intensificazione.
Figura 1. EFS BXIII-BXIV: ara-C alte dosi vs autotrapianto.

8
1.3 Autotrapianto con cellule staminali periferiche (studio BXIV)
1.3.1 Risultati Con questo programma17 sono stati trattati (cioè realmente autotrapiantati) 47
pazienti. L’impiego di consolidamento mieloablativo con supporto autotrapiantologico di tipo
periferico non ha modificato sostanzialmente la curva di EFS rispetto al programma BXIII
(autotrapianto midollare); inoltre non è stato notato alcun effetto “favorente” la recidiva nei pazienti
autotrapiantati con cellule staminali periferiche, sia in generale che in rapporto al numero di cellule
nucleate totali, CD34+ e CFU-GM reinfuse.
1.4 Tossicità da trattamento
1.4.1 Mortalità da complicanze Un aspetto importante correlato all’autotrapianto con cellule
circolanti è quello della rapida ripresa emopoietica e del breve periodo di pancitopenia. Ogni altro
consolidamento ad alte dosi comporta una lunga fase di mielosoppressione, con elevato rischio di
complicanze infettive e/o emorragiche letali. Nel nostro caso, ad esempio, una quota significativa di
pazienti autotrapiantati con cellule midollari o consolidati con ara-C sviluppava complicanze
pancitopeniche letali, contro nessuno dei casi autotrapiantati con periferico (Tabella 2).
Tabella 2. Dati comparativi di mortalità in remissione. _________________________________________________________________________________ Programma (trattamento) N. decessi in RC / n. casi trattati (%) _________________________________________________________________________________ BXIII (autotrapianto MIDOLLO) 4/42 (9.5%) BXIII/XIV (Ara-C alte dosi) 3/38 (8%) BXIV (autotrapianto PERIFERICO) 0/16
Questa differenza era dovuta alla breve durata di neutropenia (e piastrinopenia) nell’ultimo gruppo di
pazienti, che ha notevolmente ridotto la gravità delle complicanze infettive. L’esperienza di altri
gruppi con autotrapianto di periferico è analoga. Le complicanze letali da mielosoppressione
prolungata sono occasionali e la durata della citopenia è breve. Un recupero ematologico (neutrofili
>500-1000/mmc, piastrine >20000/mmc) sufficientemente rapido sembra ottenersi anche con la
reinfusione di una quota relativamente modesta di cellule CD34+, intorno a 1x106/kg.
9
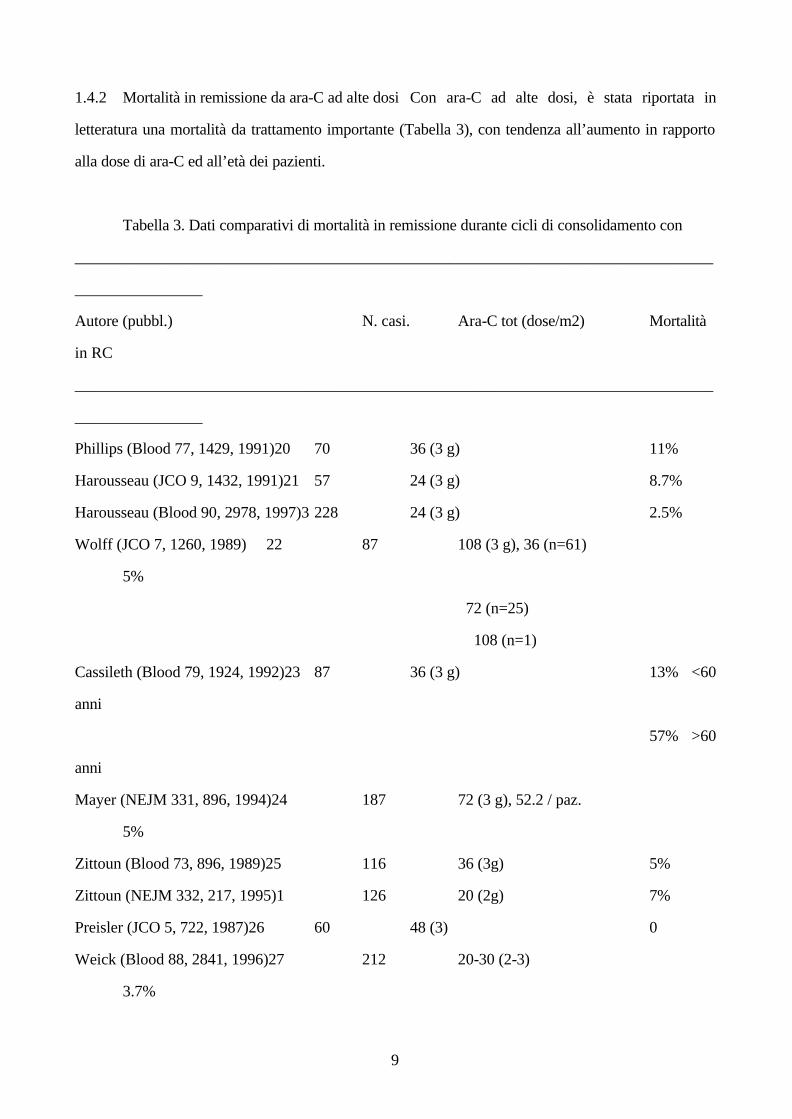
1.4.2 Mortalità in remissione da ara-C ad alte dosi Con ara-C ad alte dosi, è stata riportata in
letteratura una mortalità da trattamento importante (Tabella 3), con tendenza all’aumento in rapporto
alla dose di ara-C ed all’età dei pazienti.
Tabella 3. Dati comparativi di mortalità in remissione durante cicli di consolidamento con
________________________________________________________________________________
________________
Autore (pubbl.) N. casi. Ara-C tot (dose/m2) Mortalità
in RC
________________________________________________________________________________
________________
Phillips (Blood 77, 1429, 1991)20 70 36 (3 g) 11%
Harousseau (JCO 9, 1432, 1991)21 57 24 (3 g) 8.7%
Harousseau (Blood 90, 2978, 1997)3 228 24 (3 g) 2.5%
Wolff (JCO 7, 1260, 1989) 22 87 108 (3 g), 36 (n=61)
5%
72 (n=25)
108 (n=1)
Cassileth (Blood 79, 1924, 1992)23 87 36 (3 g) 13% <60
anni
57% >60
anni
Mayer (NEJM 331, 896, 1994)24 187 72 (3 g), 52.2 / paz.
5%
Zittoun (Blood 73, 896, 1989)25 116 36 (3g) 5%
Zittoun (NEJM 332, 217, 1995)1 126 20 (2g) 7%
Preisler (JCO 5, 722, 1987)26 60 48 (3) 0
Weick (Blood 88, 2841, 1996)27 212 20-30 (2-3)
3.7%

10
________________________________________________________________________________
________________
Inolte, la tossicità non letale ha spesso causato omissione/riduzione forzata della terapia. Il dosaggio
massimo proponibile è circa 72 g/m2. Ciò si raggiunge con difficoltà. Nello studio di Wolff la
maggioranza dei pazienti ha ricevuto 36 g contro un teorico di 108 (1 solo paziente!), e solo 25
pazienti sono stati in grado di ricevere 72 g totali. Peraltro, la durata di RC è stata migliore in
quest'ultimo gruppo. Nello studio di Mayer la dose media è stata di 52.2 g per paziente contro un
teorico di 72 g. Ancora, un dosaggio cumulativo elevato di ara-C ha migliorato notevolmente i
risultati nelle categorie con alterazioni tipo CBF (“core binding factor”) e con cariotipo normale28,29.
1.5 Sintesi delle attuali possibilità terapeutiche
1.5.1 Prognosi La prognosi della malattia è principalmente dettata dalle caratteristiche di
rischio, ma il tipo di trattamento prescelto (alte dosi ara-C, trapianto) condiziona fortemente il risultato
finale.
1.5.2 Variabili correlate al trattamento L’intensità del trattamento postremissionale può
parzialmente influenzare l’indice di rischio. E’ possibile, e vi sono indicazioni in tal senso, che
l’intensificazione con ara-C migliori i risultati in tutte le classi di rischio, peraltro con effetti
progressivamente decrescenti nel rischio alto ed altissimo. Ad esempio, sono noti i risultati largamente
positivi, con EFS a lungo termine di 80% e oltre, raggiunti dal gruppo CALGB nei casi
citogeneticamente favorevoli come LMA con inv(16) o t(8;21) trattati con i dosaggi cumulativi più alti
di ara-C28,29.
1.5.3. Scelta di modalità e intensità terapeutica Mentre non vi sono alternative ai farmaci
disponibili (antracicline, ara-C alte dosi) ed alle modalità di trattamento ablativo, permangono
incertezze, alimentate dai fenomeni di morbidità e mortalità, sul dosaggio ottimale di ara-C, sul
numero dei cicli con ara-C, e sulla tossicità a breve, medio e lungo termine sia da ara-C che da terapia
per trapianto (che può comprendere agenti alchilanti, irradiazione corporea totale,
immunosoppressione), e quindi sulla opportunità di quale trattamento scegliere per quali pazienti. Per

11
il trapianto allogenico, gravato dalla maggiore tossicità a lungo termine ma anche dalla maggiore
efficacia antileucemica, potrebbe delinearsi una situazione dicotomica: impiego in prima linea per i
casi a rischio medio-alto (incluso trapianto da donatore volontario non correlato - o MUD) e impiego
in seconda linea, dopo recidiva, nei soggetti a rischio medio-basso. Questo schema potrebbe
aumentare la sopravvivenza globale ed al tempo stesso escludere dalla procedura più tossica i casi a
maggior “rischio di cura” con metodi cosiddetti tradizionali.
1.5.4. Ruolo di ara-C ad alte dosi Per quanto riguarda il dosaggio di ara-C nei cicli ad alte dosi, vi
è notevole incertezza su cosa possa essere ritenuto ottimale. Le dosi unitarie più alte (3g/m2/dose)
comportano tossicità notevole, particolarmente di tipo ematologico e neurologico nei pazienti con età
>50-60 anni, ma tale dosaggio consente anche la maggiore attività antileucemica, come dimostrato
dalla ricordata proporzionalità dose-risultato e da studi condotti su casi con malattia refrattaria.
Probabilmente, il dosaggio cumulativo di ara-C è molto importante. Questo è chiaramente suggerito
dal citato studio CALGB e da altri , ma solo una parte dei pazienti in RC sembra in grado di ricevere
il dosaggio cumulativo più elevato. Ovviamente, cicli ripetuti di ara-C ad alte dosi comportano un
proporzionale aumento del rischio di tossicità ematologica subletale e letale.
1.5.5. Ruolo dei fattori di crescita granulocitaria In nessuno degli studi di consolidamento con
ara-C alte dosi riportati nella Tabella 3 veniva impiegato un fattore di crescita granulocitario (G-
CSF/GM-CSF). Uno studio randomizzato recente con G-CSF documenta un possibile effetto
favorente una riduzione del periodo di neutropenia febbrile dopo ara-C alte dosi durante terapia di
induzione30. Nella nostra esperienza (Ematologia Bergamo), l’impiego estensivo precoce di G-CSF
dopo ara-C alte dosi (35 cicli, 19 pazienti) ha comportato una lieve riduzione di neutropenia severa
(10 vs 13 giorni) rispetto ad un precdente gruppo trattato senza G-CSF on con G-CSF tardivo (24
pazienti, 24 cicli), ma non ha comunque escluso una alta incidenza di neutropenia severa prolungata
(>15 giorni: 43%) con mortalità correlata al trattamento (10% vs 8% senza G-CSF). Pertanto, non
sembrerebbe che l’impiego isolato di un fattore di crescita granulocitario possa abrogare l’incidenza
della neutropenia severa prolungata ed il connesso rischio infettivo dopo ara-C ad alte dosi.

12
1.5.6. Ruolo delle cellule staminali circolanti La reinfusione di cellule staminali circolanti di
derivazione autologa può ridurre significativamente la tossicità ematologica (e la mortalità ad essa
collegata) di trattamenti mieloablativi e sub-ablativi (come ara-C ad alte dosi). Questo effetto è
correlato al rapido recupero granulocitario garantito dalla reinfusione di progenitori circolanti periferici
(più eventuale G-CSF). Una ridotta morbidità e mortalità da infezioni/stato pancitopenico correla
infatti con il contenimento della neutropenia severa <500/mmc. entro un periodo di 10-15 giorni.
Nello studio BXIV con autotrapianto di cellule staminali periferiche, questo periodo è stato
mediamente di 13 giorni, contro 25 dopo autotrapianto midollare. Per le piastrine, il recupero ad oltre
20.000/mmc è evvenuto dopo 16 giorni, contro 42 dopo autotrapianto midollare. Queste differenze
sono significative ed hanno contribuito a produrre i diversi dati di mortalità da trattamento illustrati
nella Tabella 2.
2. NUOVO STUDIO CLINICO: ASPETTI TEORICI
2.1 Razionale
2.1.1 Razionale 1 Le informazioni principali della nostra ed altrui esperienza sono riassunte nella
Figura 1 e nelle Tabelle 1-3: la probabilità di EFS è favorevolmente influenzata da un consolidamento
con ara-C ad alte dosi oppure da un programma autotrapiantologico con supporto midollare o
periferico ma, eccetto che con l’ autotrapianto da periferico, questi ultimi trattamenti sono gravati da
importante morbidità ematologica e da mortalità correlata.
2.1.2 Razionale 2 L’ EFS dei due principali gruppi di trattamento intensivo (Figura 1), con
autotrapianto oppure ara-C alte dosi, è risultato sovrapponibile. Nonostante la presenza di svariati
fattori di bias statistico-interpretativo quali la natura degli studi (fase II), il diverso numero di casi, gli
schemi di induzione e consolidamento precoce, il variabile dosaggio di ara-C ad alte dosi, l’assenza di
antraciclina nei cicli ara-C ad alte dosi, le caratteristiche demografiche, il profilo di rischio, e la diversa
fonte di supporto autotrapiantologico, questi risultati depongono globalmente a favore del trattamento
con ara-C (Tabella 4).

13
Tabella 4. Valutazione comparata di tossicità ed efficacia del consolidamento con ara-C alte dosi oppure terapia mieloablativa con autotrapianto da midollo o per
_________________________________________________________________________
Parametro autotrapianto
(incidenza) ara-C alte dosi midollo periferico
EFS = = =
mielotossicità media alta bassa
tossicità mucosa media alta medio-alta
infezioni medio-alta alta medio-
bassa
mortalità in RC media media bassa-
assente
sterilità bassa molto alta molto alta
trapianto se recidiva possibile difficile difficile
manipolazione cellule no si si
2.2 Scopo dello studio
2.2.1 Obiettivi Il nuovo studio intende valuare prospettivamente la fattibilità e l’efficacia di
una strategia postremissionale flessibile nella LMA-a. Tale strategia è funzionale al profilo di rischio
omogeneamente definito dal pattern citogenetico (v. oltre) integrato da informazioni cliniche, secondo
il seguente schema:
PIANO DI TRATTAMENTO SECONDO CLASSE DI RISCHIO (opzioni)
-rischio standard (1) “ara-C alte dosi” con mini-autotrapianto
-rischio alto: (2) allotrapianto midollo osseo da consanguineo**
*(3) “ara-C intermedia” se (1) non possibile
**(1) se (2) non possibile, (3) se (2) non possibile

14
Il piano di trattamento considera tutte le possibili opzioni per la terapia di prima linea (ovviamente le
opzioni obbligatorie per le due categorie di rischio sono la numero (1) e la numero (2) mentre la
numero (3) si riferisce solo ai casi nei quali le prime due non siano praticabili. In particolare,
confermate le indicazioni per allotrapianto da consanguineo od eventualmente MUD31, lo studio si
propone di verificare se la reinfusione di una quota minima prefissata (mini-autotrapianto: cellule
CD34+ 1-2x106/kg) di cellule staminali circolanti autologhe, più G-CSF, consente di somministrare
senza ritardi ed omissioni e con bassa tossicità ematologica e clinica una elevata dose totale di ara-C
(60 g/m2 in tre cicli consecutivi, più 8 g/m2 nel precedente ciclo di mobilizzazione), secondo un
concetto di trattamento recentemente impiegato in tumori solidi e nei linfomi32-36. L’effetto
combinato dell’aumento di intensità di dose (atteso incremento di efficacia) e della ridotta
mielotossicità (attesa ridotta mortalità ed aumentata compliance al trattamento) potrebbe tradursi in un
significativo aumento di EFS rispetto al dato osservato (1) in un gruppo di controllo storico e (2) nei
pazienti trattati con “ara-C intermedia” (20 g/m2 in due cicli consecutivi, più 8 g/m2 ciclo precedente).
La somministrazione di una dose cumulativa elevata di ara-C senza il supporto del mini-autotrapianto
non è ritenuta eticamente proponibile dato il rischio già noto di grave tossicità ematologica.
2.3 Criteri di eleggibilità
2.3.1 Diagnosi Prima diagnosi di LMA (esclusa LMA promielocitica e promielocitica variante;
inclusa LMA post mielodisplasia (MDS) o secondaria a chemio-radioterapia per altra neoplasia già
guarita oppure in remissione clinica e/o con aspettativa di vita >12 mesi).
2.3.2 Età Età 15-65 anni.
2.3.3 Condizioni cliniche Assenza di grave patologia collaterale epatica, renale, polmonare,
cardiaca, metabolica, allergica, infettiva (HIV, tubercolosi, epatite cronica attiva), neuropsichiatrica
etc., che controindichi la somministrazione del previsto piano di trattamento, a giudizio del
responsabile locale dello studio.
2.3.4 Consensi informati Consenso informato scritto a terapia trasfusionale con emoderivati.
Consenso informato scritto allo studio (consenso dei genitori o del tutore per i minori).

15
2.4 Definizioni
2.4.1 Sopravvivenza e EFS Sopravvivenza: intervallo tra data diagnosi e data morte da ogni causa.
EFS: intervallo tra data RC e data evento avverso (recidiva in ogni sede o morte in RC da ogni causa).
2.5 Considerazioni statistiche
2.5.1 Calcolo del campione L'analisi degli studi recenti di fase III con ara-C ad alte dosi (Tabella 3)
e di quelli di fase II del gruppo L-B-V (Tabella 1) mostra un numero medio di pazienti trattati per
braccio >50 e generalmente compreso, con rare eccezioni, tra 100-200. Calcolando un rateo di RC di
circa 80%, per ottenere un gruppo omogeneo ottimale minimo-massimo di 150-200 pazienti valutabili
in RC sarebbe necessario un arruolamento totale di circa 200-250 casi. Perchè lo studio possa essere
concluso in 5 anni (nel qual caso, con un arruolamento costante la metà dei pazienti valutabili-cioé
circa 100 casi-avrebbero un follow-up di almeno 2.5 anni (che permetterebbe una buona analisi ad
interim) dovrebbero essere arruolati circa 40 pazienti/anno, il che rientra nelle possibilità teoriche di
arruolamento del gruppo cooperativo.
2.5.2 End-points statistici L’analisi dei risultati sarà condotta secondo il principio di intenzione al
trattamento. La valutazione comprenderà tutti i punti esposti nella Tabella 5, più l’analisi prognostica
classica secondo i fattori prognostici convenzionali. I potenziali end-points statistici includono:
a. verifica della riduzione di mortalità da trattamento con ara-C alte dosi più mini-autotrapianto e G-
CSF. Se stimata intorno al 8% (v. introduzione), una riduzione di mortalità significativa con
probabilità di errore non superiore a 0.05 (con potenza 80%) dovrebbe prevedere l’arruolamento di
circa 600 pazienti. Ovviamente, poiché questo numero non potrà essere raggiunto, si raccoglieranno
indicazioni generali in tal senso e dati utilizzabili per promuovere un eventuale studio comparativo
prospettico.
b. per la verifica di efficacia in termini di EFS, essendo il valore ottenuto nel gruppo di controllo
storico circa 40% (BXIV) e 50% (BXIII), un significativo aumento di efficacia nel braccio
sperimentale potrà essere verificato con probabilità di errore non superiore a 0.05 (con potenza 80%)
includendo nel nuovo studio circa 65 pazienti (RRR 50%), 100 pazienti (RRR 40%), e 200 pazienti

16
valutabili (RRR 30%). Questo tipo di arruolamento appare possibile ed è conforme a quanto
prospettato al punto a.
Tabella 5. Analisi comparativa dei risultati di trattamento.
__________________________________________________________________________
_
Parametri Verifica Gruppi
__________________________________________________________________________
_
FATTIBILITA’ N. casi trattati/N. casi valutabili “A20”, “A10”
Tempo a fine trattamento (intensità di dose) storico
TOSSICITA’ Ematologica (durata citopenia, trasfusioni) idem
Infettiva idem
(allotrapianto)
Extraematologica idem
EFFICACIA EFS globale e per gruppo di rischio idem
Sopravvivenza idem
___________________________________________________________________________
17
3. DISEGNO E APPLICAZIONE
3.1 Disegno
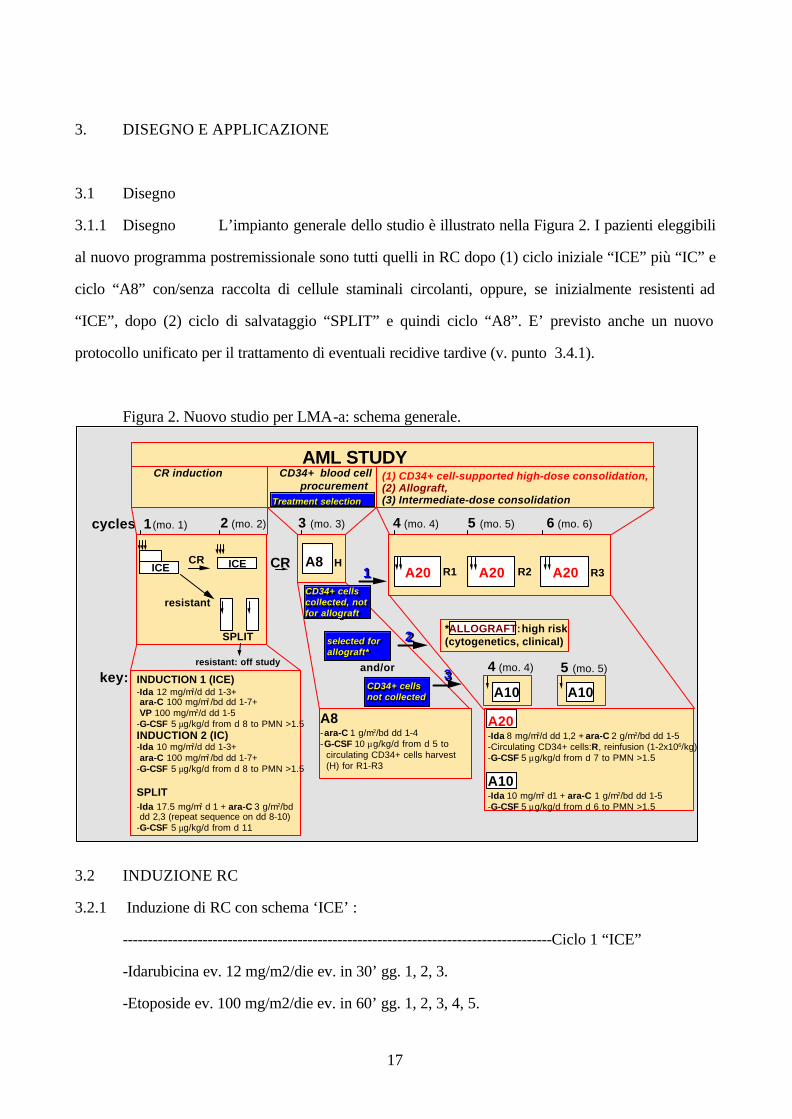
3.1.1 Disegno L’impianto generale dello studio è illustrato nella Figura 2. I pazienti eleggibili
al nuovo programma postremissionale sono tutti quelli in RC dopo (1) ciclo iniziale “ICE” più “IC” e
ciclo “A8” con/senza raccolta di cellule staminali circolanti, oppure, se inizialmente resistenti ad
“ICE”, dopo (2) ciclo di salvataggio “SPLIT” e quindi ciclo “A8”. E’ previsto anche un nuovo
protocollo unificato per il trattamento di eventuali recidive tardive (v. punto 3.4.1).
Figura 2. Nuovo studio per LMA-a: schema generale.
AML STUDY
cycles 1 (mo. 1) 2 (mo. 2) 3 (mo. 3) 4 (mo. 4) 5 (mo. 5)
A8 -ara-C 1 g/m2/bd dd 1-4 -G-CSF 10 µg/kg/d from d 5 to circulating CD34+ cells harvest (H) for R1-R3
A20 -Ida 8 mg/m2/d dd 1,2 + ara-C 2 g/m2/bd dd 1-5 -Circulating CD34+ cells: R, reinfusion (1-2x106/kg) -G-CSF 5 µg/kg/d from d 7 to PMN >1.5 A10 -Ida 10 mg/m2 d1 + ara-C 1 g/m2/bd dd 1-5 -G-CSF 5 µg/kg/d from d 6 to PMN >1.5
CR
CR induction CD34+ blood cell procurement
(1) CD34+ cell-supported high-dose consolidation, (2) Allograft, (3) Intermediate-dose consolidation
A20 A20 R2R1
SPLIT
resistant
resistant: off study
CR
INDUCTION 1 (ICE) -Ida 12 mg/m2/d dd 1-3+ ara-C 100 mg/m2 /bd dd 1-7+ VP 100 mg/m2/d dd 1-5 -G-CSF 5 µg/kg/d from d 8 to PMN >1.5 INDUCTION 2 (IC) -Ida 10 mg/m2/d dd 1-3+ ara-C 100 mg/m2 /bd dd 1-7+ -G-CSF 5 µg/kg/d from d 8 to PMN >1.5 SPLIT -Ida 17.5 mg/m2 d 1 + ara-C 3 g/m2/bd dd 2,3 (repeat sequence on dd 8-10) -G-CSF 5 µg/kg/d from d 11
key:
A8A20 R3
selected for allograft* selected for allograft*
CD34+ cells not collected CD34+ cells not collected
H
6 (mo. 6)
*ALLOGRAFT: high risk (cytogenetics, clinical)
ICE ICE
A10
4 (mo. 4) 5 (mo. 5)
CD34+ cells collected, not for allograft
CD34+ cells collected, not for allograft
and/or
11
22
33
Treatment selection Treatment selection
A10
3.2 INDUZIONE RC
3.2.1 Induzione di RC con schema ‘ICE’ :
--------------------------------------------------------------------------------------Ciclo 1 “ICE”
-Idarubicina ev. 12 mg/m2/die ev. in 30’ gg. 1, 2, 3.
-Etoposide ev. 100 mg/m2/die ev. in 60’ gg. 1, 2, 3, 4, 5.

18
-ara-C ev. 100 mg/m2 x2/die ev. in 30’ gg. 1, 2, 3, 4, 5, 6, 7.
A fine ciclo (dal g. 8 e fino a conteggio neutrofili >1.500/mmc): G-CSF 5 mcg/kg/die s.c.
Associare profilassi antibatterica/antifungina (secondo schema istituzionale).
-----------------------------------------------------------------------------------------------------------
3.2.2 Risposta al trattamento: definizioni Lo stato di RC, valutato dopo il primo ciclo al g. 28 o
diversamente secondo altra indicazione clinica prevede un midollo normocellulare o moderatamente
ipocellulare in rigenerazione, con chiara evidenza di attiva eritropoiesi, granulopoiesi e
megacariocitopoiesi, e quota di cellule immature/blastiche <5%; e inoltre: emoglobina >10 g/dl,
leucociti >2000/mmc., piastrine >50000/mmc. La non risposta (NR), o refrattarietà, viene
documentata da residuo midollare di cellule blastiche >5% e qualifica per trattamento alternativo con
programma “SPLIT”. I decessi durante il primo ciclo (o secondo ciclo) vengono conteggiati come
morti precoci (ED, “early death”).
3.2.3 Secondo ciclo “IC” per casi in RC I pazienti in RC dopo “ICE” ricevono a partire dal g. 28
un secondo ciclo di induzione “IC”:
-----------------------------------------------------------------------------------------Ciclo 2 “IC”
-Idarubicina ev. 10 mg/m2/die ev. in 30’ gg. 1, 2, 3.
-ara-C ev. 100 mg/m2 in 30’ x2/die ev. gg. 1, 2, 3, 4, 5, 6, 7.
A fine ciclo (dal g. 8 e fino a conteggio neutrofili >1.500/mmc): G-CSF 5 mcg/kg/die s.c.
Associare profilassi antibatterica/antifungina (secondo schema istituzionale).
-----------------------------------------------------------------------------------------------------------
3.2.4 Schema di salvataggio “SPLIT” La percentuale di risposta nei pazienti refrattari al primo
ciclo di chemioterapia si prevede compresa tra il 30-40% sia ripetendo un identico ciclo che
impiegando schemi alternativi tipo MEC oppure HAM o HAM/HAI sequenziale. Recentemente,
dopo uno studio preliminare in vitro, è stato introdotto il programma denominato ‘SPLIT’, derivato
dal regime HAI sequenziale, nel quale è stata impiegata idarubicina a dosaggio scalare (studio di fase
I) più ciclosporina A come modulatore negativo di farmacoresistenza MDR-1. Con ‘SPLIT’ sono stati
sinora trattati a Bergamo 9 pazienti refrattari al primo ciclo ‘ICE’, ottenendosi 8 remissioni complete

19
ed una risposta parziale, e 18 pazienti totali con LMA refrattaria (no risposta induzione, recidiva
precoce, seconda o ulteriore recidiva) ottenendo 11 remissioni su 12 pazienti con profilo di
performance ECOG 0-1 (92%) contro nessuna risposta in 6 casi con ECOG >1. Il programma
“SPLIT”, supportato da G-CSF, è tollerabile fino ad un livello di idarubicina di 17.5 mg/m2/dose (per
2 dosi). Il programma “SPLIT” viene quindi proposto come terapia di salvataggio precoce (ciclo 2)
per i casi resistenti al primo ciclo ‘ICE’. In questi casi, infatti, è prevedibile un miglior profilo di
performance rispetto ad una eventuale refrattarietà dopo un secondo ciclo convenzionale.
----------------------------------------------------------------------------------------Ciclo 2 “SPLIT”
-Idarubicina ev. 17.5 mg/m2/die ev. in 30’ gg. 1, 8
-ara-C ev. 3000* mg/m2 in 3 ore x2/die gg. 2, 3, 9, 10.
*2000 se età >55 anni
A fine ciclo (dal g. 11 e fino a conteggio neutrofili >1.500/mmc): G-CSF 5 mcg/kg/die s.c.
Associare profilassi antibatterica/antifungina (secondo schema istituzionale).
Somministrare nutrizione parenterale totale da g. 11.
Mieloaspirato g. 11 e g. 28.
---------------------------------------------------------------------------------------------------------------
NB. Uno studio collaborativo (F. Gieseler, Kiel, Germania) di farmacologia cellulare riguarda la
somministrazione di idarubicina in 30’ oppure in 180’ (partecipazione facoltativa).
3.3 Trattamento post-RC: fase comune “A8” e cellule staminali circolanti
3.3.1 Secondo consolidamento/mobilizzazione Come mostrato in Figura 2 tutti i pazienti,
indipendentemente dalla classe di rischio, vengono sottoposti al ciclo “A8”, con ara-C a dose
intermedia e G-CSF incrementato, che ha lo scopo di consolidare mobilizzando progenitori
emopoietici circolanti CD34+.
---------------------------------------------------------------------------------------------Ciclo 3 “A8”
-ara-C ev. 1000 mg/m2 ev. in 3 ore x2/die gg. 1, 2, 3, 4.
A fine ciclo (dal g. 8 e fino a raccolta cellule circolanti CD34+): G-CSF 10 mcg/kg/die s.c.
Associare profilassi antibatterica/antifungina (secondo schema istituzionale).

20
---------------------------------------------------------------------------------------------------------------
I dettagli riguardanti le modalità di raccolta e criopreservazione delle cellule staminali autologhe
CD34+ vengono forniti di seguito.
3.3.2 Raccolta e impiego cellule staminali CD34+ Tutti i pazienti in RC vengono sottoposti a
raccolta di cellule staminali circolanti autologhe CD34+ dopo ciclo “A8”. Nei pazienti che
prevedibilmente non saranno sottoposti a mini-autotrapianti/cicli “A20” (per qualsiasi ragione), il
prodotto aferetico con almeno >1x106/kg cellule CD34+ verrà criocongelato e conservato per 5 anni
per un eventuale uso tardivo con il programma di salvataggio per recidiva (v. punto 4.2.1). Si prevede
di effettuare, invece, nei casi destinati ai cicli “A20”, una reinfusione di cellule staminali (mini-
autotrapianto) dopo ogni ciclo. Inoltre, la raccolta di cellule staminali dopo “A8” dovrà puntare a
ottenere un quantitativo frazionato di cellule CD34+ sufficiente a supportare almeno 2 dei 3 cicli
previsti di ara-C (numero minimo per attivazione dei cicli “A20”, in caso contrario il paziente viene
trattato con cicli “A10” senza reinfusione), secondo il seguente schema riassuntivo:
RACCOLTA CELLULE CIRCOLANTI CD34+:
(1) Quando: dopo ciclo 3 denominato “A8”
(2) Come: con G-CSF 10 mcg/kg fino a raccolta
(3) Quanto: 1-2 x106/kg per 2-3 cicli “A20” (minimo 2 cicli), più una quota >1x106/kg per
eventuale ciclo alla recidiva.
Pertanto il quantitativo minimo necessario per entrare nella fase “A20” sara di 2x106/kg (1x106/kg x2
= 2 cicli “A20”) mentre quello massimo richiesto sarà di 6x106/kg (2x106/kg x3 = 3 cicli “A20”) più
una quota >1x106/kg per l’eventuale ciclo per recidiva. La definizione della dose minima di cellule
CD34+ circolanti per questo studio (che impiega trattamento non mieloablativo) deriva da dati della
letteratura37, mentre il posizionamento della raccolta (dopo il terzo ciclo, o secondo di
consolidamento), è volto a minimizzare il rischio di reinfusione di malattia come suggerito da una
recente analisi retrospettiva EBMT38.
3.4 Trattamento post-RC: attribuzione e somministrazione

21
3.4.1 Criteri di diversificazione del trattamento Come anticipato il criterio guida per
determinare la fase finale del trattamento è costituito dalla classe di rischio. Poichè negli studi
precedenti del gruppo L-B-V l’analisi citogenetica ha permesso di individuare differenze prognostiche
significative anche in gruppi di pazienti sottoposti a trattamenti ad alte dosi, ciò in linea con numerose
esperienze internazionali, lo studio corrente si propone di variare l’intensità della parte finale della
terapia e di valutarne la risposta prevalentemente in base alle caratteristiche di rischio citogenetico di
ogni singolo paziente (v. punto 2.2.1). L’allotrapianto di midollo osseo è riservato ai casi ad rischio
alto/intermedio-alto (per i quali è anche possibile in casi particolari l’attivazione della ricerca di un
donatore non correlato, o MUD), mentre nei restanti casi (rischio standard) si attua un consolidamento
con “ara-C ad alte dosi” (dosaggio cumulativo 60+8 g/m2), con elevata intenstà di dose e ridotta
tossicità ematologica grazie all’impiego combinato di G-CSF e mini-autotrapianto con cellule
staminali periferiche. L’opzione secondaria, applicabile a pazienti di entrambe le categorie, consiste in
un consolidamento con ara-C “dose alta o intermedia”, secondo possibilità, similmente a quanto
impiegato in alcuni gruppi di pazienti dei programmi BXIII e BXIV.
3.4.2 Definizione di classe di rischio La classe di rischio (ALTO, STANDARD) viene definita come
segue (Tabella 6). Inoltre appartiene di diritto alla categoria ALTO rischio ogni paziente che abbia
conseguito RC dopo ciclo “SPLIT” e chi, pur con criteri citogenetici intermedi, si presenti con fattori
clinico-diagnostici precedentemente identificati come sfavorevoli (LMA secondaria/postMDS,
epatosplemenomegalia, iperleucocitosi >50.000, FAB M0,M6,M7).
Tabella 6. Categorie di rischio.
__________________________________________________________________________
RISCHIO STANDARD
(1) CITOGENETICA: -anomalie RISCHIO BASSO senza altre anomalie
rischio ALTO e
indipendentemente dal profilo clinico
t(8;21), inv(16), t(16;16), del(16q)
(2) CITOGENETICA + CLINICA:

22
-anomalie RISCHIO INTERMEDIO senza altre ALTO
normale (diploide), +8, +6, +21, +22, del(12p), +22, -Y,
altre anomalie numeriche/strutturali non ALTO rischio
-citogenetica non nota
-clinica non ALTO rischio (v. sotto)
-------------------------------------------------------------------------------------------------------------
--- continua
ALTO RISCHIO
(1) CITOGENETICA: -anomalie ALTO RISCHIO
-5/del(5q), -7, t(11q23), t(9;22), abn 3q, 9q, 11q, 20q,
21q, 17p, iso(17q), t(3;5), t(6;9), t(9;22),
complessa*
*clone con almeno 3 anomalie cromosomiche non
correlate
(2) RC TARDIVA -dopo “SPLIT”
(3) CITOGENETICA -anomalie RISCHIO INTERMEDIO (v. sopra)
+ CLINICA -ALTO rischio clinico
secondaria/post MDS, epatosplenomegalia, GB
>50.000, FAB M0,6,7
Questa classificazione di rischio prevalentemente citogenetico è basata sull’analisi degli studi
CALGB, MRC e SWOG/ECOG recentemente pubblicati9,10,28,29, nonchè su altri dati similari6-12
e, prevalentemente per la parte clinica, sull’esperienza personale15-17. Nello studio MRC che ha
impiegato un criterio di rischio esclusivamente citogenetico, nella categoria ALTO rischio la
sopravvivenza a 5 anni è stata del 14%, 41% nella categoria INTERMEDIO e 65% nella categoria

23
BASSO. Mentre per i due poli estremi appare relativamente facile una scelta terapeutica molto
diversificata (allotrapianto verso ara-C alte dosi, sebbene l’impatto negativo nel gruppo ALTO rischio
permanga dopo allotrapianto, con sopravvivenza solo del 13%), nella categoria rischio
INTERMEDIO questa scelta appare più difficile senza ricorrere a criteri clinici aggiuntivi, ma è
proprio in questa categoria che si situano la gran parte dei pazienti (approssimativamente: almeno i
due terzi). Per quanto concerne la parte clinica, i fattori di rischio vengono specificati di seguito.
RISCHIO CLINICO ELEVATO SE PRESENTE ALMENO UNO DEI SEGUENTI FATTORI:
(1) LMA insorta dopo trattamento comprendente chemioterapici e/o radioterapia per altra
(2) LMA insorta dopo MDS (RA, RAS, REAB, RAEB-T) oppure preceduta da anomalie
(anemia <11 g%, leucopenia <3000/mmc, piastrinopenia <120.000/mmc isolatamente o in
(3) Epatosplenomegalia: fegato >2 cm e/o milza >1 cm oltre l’arco costale.
(4) RC non raggiunta dopo ciclo 1, raggiunta dopo ciclo SPLIT.
(5) Iperleucocitosi >50.000/mmc.
3.4.3 Trattamento postremissionale finale per classe di rischio (1) I casi a rischio STANDARD
(citogenetica a basso rischio oppure rischio intermedio senza criteri clinici sfavorevoli), nei quali sia
stato possibile ottenere dopo ciclo “A8” un numero sufficiente di cellule staminali CD34+ vengono
avviati al trattamento con ara-C alte dosi + G-CSF + mini-autotrapianto di cellule staminali periferiche
(cicli “A20”), secondo quanto indicato in Figura 2 e specificato di seguito. (2) I casi ALTO rischio
con disponibilità di donatore famigliare istocompatibile devono essere avviati a procedura di trapianto
allogenico di cellule staminali emopoietiche. Nei pazienti eleggibili per età etc. va ricercata la
possibilità di ricerca ed esecuzione di allotrapianto da MUD, se ritenuto opportuno. La terapia pre-
trapianto ad interim consiste in uno/due cicli “A10”, a seconda della organizzazione temporale del
trapianto, che va comunque eseguito entro 6 mesi dalla RC. La modalità di allotrapianto
(condizionamento, fonte di cellule staminali) è libera per ogni centro di trattamento.
--------------------------------------------------------------------------------------Cicli 4,5,6 “A20”
-Idarubicina ev. 8 mg/m2 ev. in 30’ gg. 1, 2.
-ara-C ev. 2000* mg/m2 x2/die ev. in 3 ore gg. 1, 2, 3, 4, 5.

24
*se tossicità neurologica insorta/documentata > grado 1 CTC, ridurre successivamente a 1500
mg/m2/dose (età <55 anni), 1200 mg/m2/dose (età >55 anni)
Autotrapianto cellule CD34+ 1-2x106/kg g. 6.
A fine ciclo (dal g. 6 e fino a neutrofili >1500/mmc.: G-CSF 5 mcg/kg/die s.c.
Associare profilassi antibatterica/antifungina (secondo schema istituzionale).
---------------------------------------------------------------------------------------------------------------
(3) Tutti i casi nei quali non sia stato possibile per qualsiasi ragione procedere a (1) o (2) vengono
trattati con due cicli di ara-C a dose intermedia denominati “A10” (Figura 2)
-----------------------------------------------------------------------------------------Cicli 4,5 “A10”
-Idarubicina ev. 10 mg/m2 ev. in 30’ gg. 1.
-ara-C ev. 1000 mg/m2 x2/die ev. in 3 ore gg. 1, 2, 3, 4, 5.
A fine ciclo (dal g. 6 e fino a neutrofili >1500/mmc.: G-CSF 5 mcg/kg/die s.c.
Associare profilassi antibatterica/antifungina (secondo schema istituzionale).
---------------------------------------------------------------------------------------------------------------
3.4.4 Intervalli di trattamento Per uniformare il trattamento si adottano i segg. criteri: intervallo
teorico minimo tra un ciclo qualsiasi ed il successivo 1 mese; per la ripresa della terapia è necessario
un conteggio minimo di leucociti di 3000/mmc. e di piastrine 100000/mmc.; nonchè la risoluzione di
eventuali complicanze, soprattutto infettive, una condizione di apiressia (temperatura <37.7 °C), e
livelli di GOT/GPT <Nx2 e bilirubina <2 mg%.
3.4.5. Osservazione A fine trattamento è prevista osservazione ambulatoriale secondo lo schema:
controllo clinico + emocromo + SMAC + ETF proteica ogni 6 settimane per il primo anno, ogni 10
settimane fino al secondo anno di RC, quindi ogni 3-6 mesi dal terzo al quinto anno, in seguito
annualmente. Eseguire terapia sostitutiva con immunoglobuline umane se riscontro di livelli Ig <0.5
g/dl. Profilassi secondaria con acyclovir per 6-12 mesi dopo episodio infettivo HSV/VZV. Non sono
previsti mieloaspirati di sorveglianza, l’esame del midollo osseo verrà eseguito nel sospetto clinico di
recidiva.

25
4 PROGRAMMA RECIDIVE
4.1 Razionale
4.1.1 Razionale 1 La risposta terapeutica dopo recidiva di LMA-a è condizionata dai precedenti
trattamenti e dalla durata della remissione. I pazienti che recidivano precocemente, particolarmente
dopo terapie ad alte dosi (ara-C, trapianto) hanno un probabilità di seconda remissione del 30-40%.
Poichè dopo programmi di questo tipo le recidive sono relativamente precoci (entro 1-2 anni), i
programmi convenzionali hanno un ruolo decisamente limitato. La causa di ciò risiede, in pari grado,

26
in fenomeni di chemioresistenza e nelle complicanze da mielosoppresione. In questi pazienti, un
incremento di dose rispetto ai noti programmi MEC/s-HAM/s-HAI (tipo SPLIT)/FLAG e similari
appare proibitivo. Inoltre non sono al momento disponibili per impiego su larga scala altri nuovi
farmaci o combinazioni di farmaci di maggiore efficacia.
4.1.2 Razionale 2 Recentemente, sono stati pubblicati dati riguardanti trattamenti ad alte dosi con
autotrapianto in pazienti in recidiva39, usando midollo criopreservato ottenuto durante precedente RC.
I risultati sono stati apprezzabili, sia in termini di risposta (76%) che di ridotta mielotossicità. Va anche
ricordato che talora pazienti con donatore istocompatibile sono ri-allotrapiantati direttamente alla
recidiva, ancora con un discreti risultati (compatibilmente con l'incidenza e la gravità delle
complicanze da GVHD, ovviamente assenti nell'ambito dell'autotrapianto).
4.2 Studio recidive
4.2.1 Reinduzione RC per recidiva Dato queste premesse, il trattamento della recidiva potrebbe
comprendere uno schema a dosi medio-alte (sub-ablativo) associato a reinfusione di cellule periferiche
autologhe CD34+, raccolte e criopreservate nella fase della prima RC (ciclo “A8”, Figura 2), e
conservate per almeno 5 anni a partire dalla data di prima RC. In tal modo si prevede di sostenere
efficaciemente la mielotossicità del programma, con un possibile incremento del rateo di risposta. Il
programma di ritrattamento proposto è esposto in dettaglio nella Figura 3.
N.B. Prima del trattamento eseguire valutazione della funzionalità cardiaca: ridurre mitoxantrone a 50
mg/m2 se frazione eiezione ventricolo sx <45% (associando eventualmente razoxane).
Figura 3. Programma per trattamento recidive.

27
Giorno 1 2 3 4
Ara-C 800 mg/m2/die infusione continua (de-bulking)
6
Etoposide 2 g/m2
(in 12 ore)
8
Mitoxantrone 65 mg/m 2
(in 3 ore)
11
CD34 >1x10e6/kg
REINFUSIONE + G-CSF
4.2.2 Trattamento dopo RC Per i pazienti in seconda RC dopo ritrattamento e con donatore
famigliare o MUD compatibile (HLA, DR) disponibile, si prevede allotrapianto di midollo osseo. In
caso di indisponibilità di donatore entro 3 mesi dalla data d’inizio della ricerca, si propone espianto di
midollo osseo (“purging” in vitro con mafosfamide/altro metodo) per autotrapianto con programma
comprendente irradiazione corporea totale.
5 OBIETTIVI, ARRUOLAMENTO, ANALISI STATISTICA, CENTRI
5.1 Obiettivi

28
Obiettivo primario dello studio è il miglioramento della prognosi dei pazienti adulti con LAM rispetto
ai risultati degli studi precedenti. In particolare, rimanendo prevedibilmente costante il tasso di RC
iniziale (circa 80%), l’obiettivo principale è il miglioramento del risultato di DFS (“disease-free
survival” ovvero sopravvivenza libera da malattia in prima RC), per sottogruppo di malattia (classe di
rischio), a 3-5 anni rispetto ai programmi B-XIII/B-XIV ed a quanto generalmente riportato nella
letteratura corrente citata in precedenza. Gli obiettivi secondari correlati riguardano l’analisi di
sopravvivenza globale, dei risultati conseguiti in fase di recidiva/resistenza, e della tossicità acuta e
cronica del trattamento.
5.2 Arruolamento
Gli studi precedenti hanno fornito informazioni prognostiche e terapeutiche ben precise dopo un
arruolamento di circa 100-150 pazienti totali nell’arco di 3-5 anni. Questi numeri e la lunghezza media
del follow-up permettono una valutazione degli eventi avversi a breve-medio termine (entro i primi 3
anni dalla data di RC), che qualificano la gran parte dei diversi sottogruppi prognostici e permettono
con una buona approssimazione di prevedere il risultato di DFS a 5 anni. Data l’attuale strutturazione
del gruppo collaborativo LMA-a (v. di seguito la lista dei centri partecipanti) e l’ulteriore
differenziazione terapeutico-prognostica del piano di trattamento, è prevedibile che informazioni di
questo tipo possano essere raccolte nell’arco di 5 anni in una popolazione iniziale di circa 200 pazienti
(40 pazienti/anno), dei quali circa 160 otterranno la RC iniziale.
5.3 Svolgimento dello studio, monitoraggio
Analogamente agli studi precedenti, lo studio prevede per il primo anno uno stretto monitoraggio
trimestrale/semestrale dei risultati e della tossicità, al fine di correggere rapidamente eventuali
imperfezioni del piano di trattamento. Successivamente, a fine arruolamento è previsto un
aggiornamento/incontro con analisi semestrale/annuale dei dati fino alla conclusione dello studio.
5.4 Analisi statistica
L’obiettivo primario dello studio è di consentire, tramite la somministrazione di un trattamento
postremissionale variabile per classe di rischio e comprendente citarabina ad alta intensità di dose
(STUDIO DI FATTIBILITA’), un miglioramento statisticamente significativo (test log-rank con

29
p<0.05) del DFS a 3-5 anni rispetto al protocollo immediatamente precedente (B-XIII-B-XIV), in una
popolazione non selezionata e consecutiva di pazienti in RC stratificati per classe di rischio
citogenetico-clinico.
Si definiscono infine i seguenti end-points clinici e statistici:
-Durata sopravvivenza globale (OS) = intervallo data diagnosi a data morte (da qualsiasi
causa);
-DFS = intervallo data RC a data recidiva di LAL (in qualsiasi sede) o data morte in RC (da
-“Censoring” = i pazienti deceduti per cause non attinenti alla terapia della LAL ed alle sue
previsto per gli altri casi.
5.5 Raccolta dati e software per analisi
Viene approntata una scheda paziente per raccolta dati (appendice 1), da compilarsi accuratamente in
stampatello da ogni centro partecipante e da inoltrare al completamento di ognuna delle seguenti fasi
al centro coordinatore (Bergamo):
-Arruolamento e diagnosi iniziale (registrazione)
-Fine ciclo 2 (valutazione iniziale)
-Semestralmente per 5 anni (valutazione)
-Ad ogni evento avverso: recidiva/morte
-Quando richiesto dal centro coordinatore
La scheda prevede l’analisi di tossicità da trattamento mediante impiego dei Common Toxicity
Criteria (CTC, appendice 2) I dati verranno controllati ed immagazzinati in un apposito archivio
elettronico File-Maker Pro per schedatura e analisi dati in modalità univariata e multivariata mediante
software/hardware IBM compatibile.
L’aggiornamento della scheda dati va inviato al coordinatore dello studio, via fax (allo 035 266147,
Dr R. Bassan, Div. Ematologia Ospedali Riuniti, Bergamo).
5.6 Centri partecipanti
Hanno dato adesione al nuovo studio collaborativo LAM-a i seguenti centri:

30
Dott. T. Chisesi, M. Vespignani: Ematologia Osp. VENEZIA
Prof. P. Coser, Dott. P. Fabris: Ematologia Osp. BOLZANO
Dott. M. Musso: Osp. Onc. La Maddalena PALERMO
Prof. E. Pogliani, G. Corneo: Ematologia Osp. MONZA
Dott. A. Porcellini: Onc. Med. Ematol. Osp. NOALE (VE)
Dott. G. Rossi, T. Izzi: Ematologia Osp. BRESCIA
Prof. R. Mozzana, Medicina Osp. GALLARATE (MI)
5.7 Coordinamento
Il centro coordinatore-organizzatore è costituito presso la Div. di Ematologia, Ospedali Riuniti,
BERGAMO:
Responsabile Prof. T. Barbui,
Coordinatore protocollo Dott. R. Bassan,
Gestione dati e analisi statistica Dott. E. Oldani.

31
6. ASPETTI ETICI
6.1 Aspetti etici
Lo studio dovrà essere condotto secondo le specifiche del protocollo, approvate dagli aderenti, e in
accordo con le regole per la buona pratica clinica (ICH Harmonized Tripartite Guidelines for Good
Clinical Practice 1996; Direttiva 91/507/EEC, The Rules Governing Medicinal Products in the
European Community; Dichiarazione di Helsinki riguardante la ricerca clinica sull’uomo,
Recommendatiuons Guiding Physicians in biomedsical research Involving Human Subjects, Helsinki
1964, emendamenti Tokyo 1975-Venice 1983-Hong Kong 1989-Somerset west 1996).
6.2 Consenso informato
Data la struttura innovativa orientata al rischio citogenetico-clinico, viene richiesto al paziente un
consenso informato scritto (appendice 3). Al paziente andrà spiegata la formulazione generale del
programma nell’ambito della propria classe di rischio, come pure le ragioni per la scelta del tipo
consolidamento finale. Ovviamente, in caso di mancata accettazione, il paziente verrà trattato con il
programma tipo cicli “A10”, corrispondente al concetto di consolidamento semi-intensivo secondo i
canoni di trattamento tradizionali, oppure con allotrapianto (dopo discussione).
6.3 Aderenza al protocollo
Scelte terapeutiche diverse da quanto previsto dal piano di definizione prognostica del protocollo non
sono eticamente accettabili in questo ambito, ed eventuali casi di violazione delle linee guida
prognostico-terapeutiche verranno esclusi da ogni valutazione.
6.4 Comitato etico

32
Prima dell’inizio dello studio il protocollo completo di tutte le sue parti sarà presentato per
approvazione al Comitato Etico degli Ospedali Riuniti di Bergamo. 7. Referenze bibliografiche
1. Zittoun RA, Mandelli F, Willemze R et al. Autologous or allogeneic bone marrow transplantation
compared with intensive chemotherapy in acute myelogeneous leukemia. N Engl J Med 1995, 332,
217.
2. Reiffers J, Stoppa AM, Attal M et al. Allogeneic vs autologous stem cell transplantation vs
chemotherapy in patients with acute myeloid leukemia in first remission: the BGMT 87 study.
Leukemia 1996, 10: 1874.
3. Harousseau J-L, Cahn J-Y, Pignon B et al. Comparison of autologous bone marrow transplantation
and intensive chemotherapy as postremission therapy in adult acute myeloid leukemia. Blood 1997,
90, 2978.
4. Cassileth PA, Harrington DP, Appelbaum FR et al. Chemotherapy compared with autologous or
allogeneic bone marrow transplantation in the mAnagement of acute myeloid leukemia in first
remission. N Engl J Med 1998, 339: 1649.
5. Burnett AK, Goldstone AH, Stevens RMF et al. Randomised comparison of addition of autologous
bone marrow transplantation to intensive chemotherapy for acute myeloid leukaemia in first remission:
results of MRC AML 10 trial. The lancet 1998, 351: 700.

33
6. Keating MJ, Cork A, Broach Y et al. Toward a clinically relevant cytogenetic classification of
acute myelogenous leukemia. Leuk Res 1987, 11: 119.
7. Keating MJ, Smith TL, Kantarjian H et al. Cytogenetic pattern in acute myelogenous leukemia: a
major reproducible determinant of outcome. Leukemia 1988, 2: 403.
8. Dastugue N, Payen C, Lafage-Pochitaloff M et al. Prognostic significance of karyotype in de novo
adult acute myeloid leukemia. Leukemia 1995, 9: 1491.
9. Grimwade D, Walker H, Oliver F et al. The importance of diagnostic cytognetics on outcome in
AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood 1998, 92: 2322.
10. Slovak ML, Kopecky KJ, Cassileth PA et al. Karyotypic analysis predicts outcome of
preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology
Group/Eastern cooperative Oncology group study. Blood 2000, 96: 4075.
11. Gale RP, Horowitz MM, Weiner Rs et al. Impact of cytogenetic abnormalities on outcome of
bone marrow transplants in acute myelogenous leukemia in first remission. Bone Marrow Transplant
1995, 16: 203.
12. Ferrant A, Labopin M, Frassoni F et al. karyotype in cute myeloblastic leukemia: prognostic
significance for bone marrow ytransplantation in first remission: a European Group for Blood and
Marrow Transplantation study. Blood 1997, 90: 2931.
13. Battista R, Bassan R, D’Emilio a et al. Short-term remission induction and consolidation therapy
for adult acute myelogenous leukemia. Hematol Oncol 1991, 9: 43.
14. Rohatiner AZS, Battista R, Bassan R et al. Short term therapy (STT) for acute myelogenous
leukaemia (AML). Leukemia 1992, 6 (suppl 2): 85.

34
15. Bassan R, Raimondi R, Lerede T et al. Outcome assessment of age group-specific (+50 years)
post-remission consolidation with high-dose cytarabine or bone marrow autograft for adult acute
myelogenous leukemia. Haematologica 1998, 83: 627.
16. Rohatiner AZS, Bassan R, Raimondi R et al. High-dose treatment with autologous bone marrow
support as consolidation of first remission in younger patients with acute myelogenous leukemia. Ann
Oncol 2000, 11: 1007.
17. Rohatiner AZS, Bassan R, Bjorkholm M et al. High-dose treatment (HDT) with peripheral blood
progenitor cell (PBPC) support as consolidation of first remission in younger patients (pts) with acute
myelogenous leukaemia (AML). Blood 1998, 92 (suppl 1): 293a.
18. Bassan R, Barbui T. Remission induction therapy for adults with acute myelogenous leukemia:
towards the ICE age? Haematologica 1995, 80: 82.
19. Bassan R, Lerede T, Barbui T. Escalating doses of idarubicin (Ida) with multidrug blockade by
short course cyclosporin a (CSA) and sequential high-dose ara-C for refractory acute leukemia: phase
I/II clinical trial. Blood 2000, 96 (suppl 1): 323a.
20. Phillips GL, Reece DE, Shepherd JD et al. High-dose cytarabine and daunorubicin induction and
postremission chemotherapy for the treatment of acute myelogenous leukemia in adults. Blood 1991,
77: 1429.
21. Harousseau JL, Milpied N, Briere J et al. Double intensive chemotherapy in adult acute myeloid
leukemia. J Clin Oncol 1991, 9: 1432.
22. Wolff SN, Herzig RH, Fay JW et al. High-dose cytarabine and daunorubicin as consolidation
therapy for acute myeloid leukemia in firts remission: long-term follow-up and results. J Clin Oncol
1989, 7: 1260.

35
23. Cassileth PA, Lynch E, Hines JD et al. Varying intensity of postremission therapy in acute
myeloid leukemia. Blood 1992, 79: 1924.
24. Mayer RJ, Davis RB, Schiffer CA et al. Intensive postremission chemotherapy in adults with
acute myeloid leukemia. N Engl J Med 1994, 331: 896.
25. Zittoun R, Jehn U, Fière D et al. Alternating v repeated postremission treatment in adult acute
myelogenous leukemia: a randomized phase III study (AML6) of the EORTC leukemia cooperative
group. Blood 1989, 73: 896.
26. Preisler HD, Raza A, Early A et al. Intesnsive remission consolidation therapy in the treatment of
acute nonlymphocytic leukemia. J Clin Oncol 1987, 5: 722.
27. Weick JK, Kopecky KJ, Appelbaum FR et al. A randomized investigation of high-dose versus
standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute
myeloid leukemia: a Southwest Oncology Group study. Blood 1996, 88: 2841.
28. Bloomfield CD for the CALGB. Adult acute myeloid leukemia (AML) patients (pts) with
t(8;21)(q22;q22) have a superior outcome when repetitive cycles of high-dose cytarabine (HDAC) are
administered. Ann Hematol 1999, 78 (suppl 2): S13.
29. Bloomfield CD, Lawrence D, Byrd JC et al. Frequency of prolonged remission duration after
high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer
Res 1998, 58: 4173.
30. Bradstock KF, Matthews JP, Young GAR et al. Effects of lenograstim (glycosylated huG-GSF)
after high-dose cytarabine-based induction chemotherapy for adult acute myeloid leukemia (AML).
Blood 1999, 10 (suppl 1): 299a.
31. Sierra J, Storer B, Hansen JA et al. Unrelated donor marrow transplantation for acute myeloid
leukemia: an update of the Seattle experience. Bone Marrow Transplant 2000, 26: 397.

36
32. Stoppa AM, Bouabdallah R, Chabannon C et al. Intensive sequential chemotherapy with repeated
blood stem-cell support for untreated poor-prognosis non-Hodgkin’s lymphoma. J Clin Oncol 1997,
15: 1722.
33. Shea T, Mason JR, Storniolo AM et al. Sequential cycles of high-dose carboplatin administered
with recombinant human granulocyte-macrophage colony-stimulating factor and repeated infusions of
autologous peripheral-blood progenitor cells: a novel and effective method for delivering multiple
courses of dose-intensive therapy. J Clin Oncol 1992, 10: 464.
34. Schilder RJ, Johnson S, Gallo J et al. Phase I trial of multiple cycles of high-dose chemotherapy
supported by autologous peripheral-blood stem cells. J Clin Oncol 1999, 17: 2198.
35. Wandt H, Birkman J, Denzel T et al. Sequential cycles of high-dose chemotherapy with dose
escalation of carboplatin with or without paclitaxel supported by G-CSF mobilized peripheral blood
progenitor cells: a phase I/II study in advanced ovarian cancer. Bone Marrow Transplant 1999, 23:
763.
36. Pettengell R, Woll PJ, Thatcher N et al. Multicyclic, dose-intensive chemotherapy supported by
sequential reinfusion of hematopoietic progenitors in whole blood. J Clin Oncol 1995, 13: 148.
37. Weaver CH, Potz J, Redmond J et al. Engraftment and outcomes of patients receiving
myeloablative therapy followed by autologous peripheral blood stem cells with a low CD34+ cell
content. Bone Marrow Transplant 1997, 19: 1103.
38. Reiffers J, Labopin M, Sanz M et al. Autologous blood cell vs marrow transplantation for acute
myeloid leukemia in complete remission: an EBMT retrospective analysis. Bone Marrow Transplant
2000, 25: 1115.

37
39. de la Rubia J, Sanz GF, Martin G et al. Autologous bone marrow transplantation for patients with
acute myeloblastic leukemia in relapse after autologous blood stem cell transplantation. Bone Marrow
Transplant 1996, 18: 1167.

38
Consenso informato allo studio clinico LMA-a (preparato il 15/1/2001) Luogo e data: Egregio Sig./Sig.a, 1. La Sua malattia del sangue rientra nel gruppo delle malattie neoplastiche potenzialmente curabili con chemioterapia. Fortunatamente, nel Suo come in molti altri casi, la terapia iniziale ha prodotto una remissione completa, ma la chemioterapia deve proseguire per ridurre il rischio di una ricaduta. 2. In questa malattia le possibilità di cura offerte dalla chemioterapia somministrata dopo la remissione dipendono, oltrechè dal tipo e dalla intensità della chemioterapia stessa, anche della presenza o meno di determinati fattori prognostici. 3. Nel Suo caso, la prognosi dipende in modo particolare dal risultato dell’analisi cromosomica (citogenetica), che può identificare i casi ad alto rischio oppure a basso rischio. Criteri clinici aggiuntivi possono essere necessari in alcuni casi secondo nostro giudizio. 4. Per questo motivo riteniamo giustificato proporLe un programma finale di trattamento che tiene conto della classe di rischio del Suo caso specifico, secondo uno schema che identifica queste possibilità di trattamento: 4.1 Rischio BASSO Terapia senza trapianto di midollo allogenico (da donatore) o autologo. Il trattamento previsto consiste in tre cicli consecutivi che impiegano ara-C ad alto dosaggio. Questo farmaco è ritenuto particolarmente efficace in questa categoria di rischio. Per ridurre la tossicità del trattamento, che può essere rilevante, useremo dopo ogni ciclo un piccolo quantitativo di cellule emopoietiche staminali autologhe raccolte nel suo stesso sangue periferico, utili per garantire una ripresa dei valori dell’emocromo in tempi rapidi. 4.2 Rischio ALTO Il trapianto di midollo osseo da famigliare compatibile appare la scelta migliore, e questo sarà il trattamento proposto se le sue condizioni generali e cliniche saranno giuducate adeguate. Per casi particolari senza donatore, attiveremo la ricerca di un donatore di midollo estraneo alla famiglia, da ricercare quindi nell’ambito dei registri nazionali e internazionali dei donatori volontari. L’opzione precedente 4.1 resta valida in caso di impossibilità di procedere al trattamento trapiantologico in questa categoria di rischio. 4.3 Nel caso non sia indicato o possibile, per qualsiasi motivo (incluso il Suo rifiuto a partecipare a questo studio), seguire una delle due opzioni terapeutiche principali, Le verrà somministrato un programma tradizionale consistente in due cicli con ara-C a dose intermedia. 5. La somministrazione di questa terapia orientata al rischio richiede il Suo consenso. I medici responsabili del Suo caso possono su richiesta fornirLe ogni altra informazione riguardante la Sua malattia e la terapia. Dichiarazione di consenso informato: Il/la sottoscritto/a Sig./Sig.a_____________________________________________ avendo letto, discusso e compreso i sovrastanti punti 1-5,

39
ACCONSENTE/NON ACCONSENTE ad essere sottoposto/a, nell’ambito del presente studio clinico ad un trattamento finale ad intensità variabile secondo classe di rischio. Firma del paziente/genitore/tutore_______________________________ Firma/timbro del medico ____________________________________

40

41


















