
SezIone VIII - Doctor33 · parte dei disordini autoimmuni sia di origine multifatto- ... la febbre,...
47
31. Malattie reumatologiche, 603 Reumatologia VIII SEZIONE
Transcript of SezIone VIII - Doctor33 · parte dei disordini autoimmuni sia di origine multifatto- ... la febbre,...

31. Malattie reumatologiche, 603
Reumatologia
VIIIS e z I o n e


C a p I t o l o
603
Malattie reumatologicheG. Bartolozzi
31
Generalità
La reumatologia è una specialità della pediatria, svilup-patasi da una trentina di anni: essa è per la maggior parte una specialità essenzialmente descrittiva, perché la fisiopa-tologia delle singole malattie incluse in questa disciplina non è ancora completamente conosciuta.
Le malattie reumatologiche dell’infanzia (chiamate anche malattie infiammatorie del tessuto connettivo o malattie del collageno) sono caratterizzate da modificazioni flogistiche del tessuto connettivo nei vari distretti dell’organismo, al di sotto delle quali si ipotizza o è già stata dimostrata la presenza di autoanticorpi di vario tipo.
In ognuna delle numerose malattie reumatologiche l’infiammazione cronica, spesso intermittente, contribuisce con il tempo alla distruzione dell’organo bersaglio, che è spesso sede di deposizione di complessi immuni. I risultati ottenuti dalla genomica confermano l’importanza della genetica nella comparsa di questo gruppo di malattie: i nuovi studi sottilineano che l’interazione dei componenti ambientali con i fattori genetici dell’ospite è alla base del fenomeno dell’autoimmunità, il cui riconoscimento può essere utile per la prevenzione e per il trattamento.
Più di 200 loci genetici sono risultati associati con una o più malattie autoimmuni.
Come si sa, il ruolo del sistema immunitario è quello di mantere l’equilibro fra le varie componenti del nostro organismo. Ciò comporta la capacità di riconoscere agenti patogeni esterni o tumorali, per instaurare una difesa immune appropriata sia umorale, sia cellulare. Nello stesso tempo il sistema immunitario riconosce gli antigeni self, cioè appartenenti allo stesso organismo, verso i quali le cellule dell’organismo non reagiscono, per cui si instaura uno stato di tolleranza. Un’alterazione di queste due condizioni provoca rispettivamente uno stato di immunodeficienza o uno stato di autoimmunità: in quest’ultima condizione vengono prodotti anticorpi e/o cellule che attaccano i normali componenti dell’organismo. Da qui il nome di autoimmunità, cioè di un’immunità rivolta verso se stessi, per la perdita della tolleranza. Nonostante la maggior parte dei disordini autoimmuni sia di origine multifatto-
riale, lo studio dei disordini monogenici ha dimostrato spesso la presenza contemporanea di immunodeficienza e di autoimmunità.
Le malattie reumatologiche sono infatti mediate dalla risposta immune, che passa attraverso l’attivazione dei monociti, dei linfociti, dei polimorfonucleati, del comple-mento e la presenza di veri e propri anticorpi. Le citochine, i fosfolipidi, le chinine e altri mediatori dell’infiammazione sono liberati in grande quantità a livello dei tessuti colpiti, insieme a enzimi idrolitici (come proteasi e collagenasi), ai quali è dovuto il danno locale a carico dell’articolazio-ne e dell’osso. La flogosi della sinovia, che ne determina l’ipertrofia (panno sinoviale), finisce per interessare lo strato cartilagineo e gli strati ossei sottostanti: gli enzimi idrolitici concorrono alla distruzione, demineralizzazione e sensibilizzazione all’azione delle forze della postura e degli insulti meccanici che agiscono sull’articolazione.
La patogenesi riconosce classicamente, accanto a eventi ambientali scatenanti (infezioni, traumatismi), una predi-sposizione genetica, dimostrata dal fatto che molte delle di-verse fome cliniche hanno uno specifico assetto HLA, come se di fronte a un evento esogeno, per quanto specifico, la risposta clinica dipendesse da un assetto genetico, diverso per ogni sottogruppo. Riferendosi all’artrite idiopatica giovanile, mentre per il sottogruppo a inizio sistemico e per il sottogruppo poliarticolare, fattore reumatoide negativo, non è stato identificato alcun assetto HLA specifico, per gli altri tre sottogruppi l’associazione è ormai sufficientemente dimostrata (Figura 31.1). I legami fra spondiloartrite e malattie collegate, da un lato, e HLA B27, dall’altro, sono così stretti da rappresentare ormai una certezza.
Anche se i singoli quadri patologici verranno descritti separatamente, esiste nella realtà una sovrapposizione evidente di sintomi e di segni fra un quadro e l’altro, che talvolta può rendere difficile la diagnosi clinica.
Le manifestazioni cliniche delle malattie reumatiche si riferiscono a diverse localizzazioni:• sinovite e artrite, per infiammazione della sinovia e delle
strutture osteocartilaginee dell’articolazione;• entesite o entesopatia, per infiammazione di un lega-
mento alla sua inserzione sull’osso (come avviene nella

CapItolo 31 Malattie reumatologiche604
tendinite del tendine di Achille, nella fasciite plantare, nell’artrite dell’articolazione sacroiliaca);
• sierosite, per infiammazione delle sierose;• miosite, per infiammazione di un muscolo;• vasculite, per interessamento di vasi di vario calibro.
L’infiammazione può essere localizzata a un’articolazione o a un gruppo di tendini, o può interessare poche o la maggioranza delle articolazioni.
I sintomi locali sono quelli classici dell’infiammazione: do-lore, tumefazione, rossore, calore e alterazione della funzione; i sintomi generali dell’infiammazione sono: la febbre, lo stato di malessere, l’anoressia, la perdita di peso e le mialgie.
Di fronte a un bambino che si lamenta o che sembra lamentarsi di dolori articolari, il pediatra deve cercare di rispondere inizialmente a due domande:• il bambino con i dolori è in ottima salute e sembra star
bene, oppure è ammalato e ha la febbre?• è colpita una sola articolazione o sono interessate anche
altre sedi?
Se il bambino appare complessivamente come compro-messo e c’è una netta tumefazione di un’articolazione, il primo pensiero deve essere rivolto all’artrite settica (vedi Capitolo 52), eventualmente associata a osteomielite.
Una tumefazione articolare in un bambino che sta bene difficilmente è un’artrite settica; più spesso si tratta di un’artrite reattiva, successiva a una malattia infettiva, o un interessamento articolare da trauma o più di rado, quando la situazione si protragga da qualche settimana, un’artrite cronica giovanile all’inizio. Altre cause sono riportate nella Tabella 31.1.
Spesso nella raccolta dell’anamnesi risulta che i geni-tori del bambino presentano o hanno presentato dolori articolari di vario genere.
Quando il bambino, a un attento esame, risulti colpito anche a carico di altre articolazioni e appaia cronicamente
ammalato, la diagnosi si sposta verso l’artrite idiopatica gio-vanile, sia pauciarticolare (fino a 5 articolazioni interessate) sia poliarticolare. Ma vanno prese in considerazione an-che situazioni più gravi, come la leucemia, i linfomi e il
Il bambino con sofferenza articolare
Interessamento di una sola articolazione
Il bambino appare malato Il bambino apparentemente sta bene
Artrite setticaOsteomieliteAscesso dello psoas
Artrite reattiva, postinfettivaSofferenza post-traumaticaArtrite idiopatica giovanile
pauciarticolareEmofiliaFratturaNecrosi asettica dell’ossoTumori
Sarcoma osteoideSarcoma di EwingSarcoma osteogenico
Interessamento di più articolazioni
Il bambino appare malato Il bambino apparentemente sta bene
Artrite idiopatica giovanile poliarticolare
Artrite idiopatica giovanile pauciarticolare
Malattia reumaticaPorpora di Schönlein-HenochMalattia da sieroLeucemia linfoblastica acutaNeuroblastomaMalattia metabolicaSarcoidosi
Artrite reattiva, postinfettivaSoggetto HLA B27 positivoEmofiliaDermatomiositeSclerodermia
tabella 31.1
FIgura 31.1 - Patogenesi dell’artrite idiopatica. (Da: Klareskog L, Catrina AI, Paget S: Rheumatoid arthritis, Lancet 373(9664):659-672, 2009; modificata.)

605CapItolo 31 Malattie reumatologiche
neuroblastoma. Altre possibilità diagnostiche sono riportate nella Tabella 31.2.
Si può concludere che i dolori articolari e l’interes-samento articolare possano essere originati da una miriade di situazioni patologiche pediatriche, che vanno molto al di là del campo specifico delle malattie reumatologiche.
La prevalenza viene giudicata di 3,7 casi su 1.000 al di sotto dei 12 anni.
Vi sono alcuni sintomi e segni che, sebbene non siano specifici, suggeriscono la presenza di una malattia reuma-tologica (Tabella 31.3).
Il coinvolgimento del sistema nervoso centrale è relativa-mente frequente nelle malattie reumatologiche in generale: esso può andare da manifestazioni lievi di difficile rilievo a quadri clinici gravi, che possono mettere in gioco la vita stes-sa del paziente. Fra le malattie reumatologiche, quelle che più spesso si associano a interessamento cerebrale sono:• il lupus eritematoso sistemico;• le vasculiti sistemiche;• l’artrite idiopatica giovanile;• la CINCA (sindrome cronica, infantile, neurologica,
cutanea e articolare);• la sclerodermia;• la dermatomiosite giovanile.
Metodi di studio
Pur nella loro assoluta aspecificità, esistono numerose pro-ve utili al pediatra per confermare la presenza di una ma-lattia di interesse reumatologico o meglio per individuare
quale, fra queste, essa sia. Non esistono infatti prove per la diagnosi sicura della malattia reumatologica.
proteine della fase acuta
La velocità di sedimentazione degli eritrociti (VES), pur nella sua estrema aspecificità e nella sua vetustà, è la pro-va di laboratorio più usata per determinare l’attività di un’affezione reumatologica. Buoni indicatori di questa attività sono anche la determinazione della proteina C reattiva, che viene sintetizzata dal fegato, la ricerca della procalcitonina, la conta delle piastrine (il cui numero aumenta nelle flogosi croniche), la determinazione del livello di complemento totale (CH50), l’elevazione della curva delle a2-globuline all’elettroforesi delle proteine, l’aumento del fibrinogeno, l’aumento delle mucoproteine, l’abbassamento della sideremia e altri esami. Ognuno di questi può essere utile nel porre la diagnosi iniziale e nel seguire la malattia del paziente.
Fattore reumatoide
I fattori reumatoidi sono anticorpi diretti contro la por-zione Fc delle immunoglobuline G (IgG); classicamente i fattori reumatoidi appartengono alla classe delle IgM e sono dimostrabili con metodiche di agglutinazione con il latex. I fattori reumatoidi appartenenti alle classi IgG, IgA e IgE sono misurabili con altre tecniche di laboratorio. Questi anticorpi non sono specifici per le Ig dell’ospite e possono essere diretti verso Ig di altri individui o di altre
Cause di artrite nel bambino
Comuni Meno comuni RareArtrite idiopatica giovanileLupus eritematoso sistemicoArtrite reattiva, postinfettivaPorpora di Schönlein-HenochMalattia di KawasakiMalattia di LymeInfezione da piogeni (artrite settica)
Malattia reumaticaLeucemiaSindrome di ReiterMalattia infiammatoria cronicaDermatomiosite
Artrite psoriasicaGranulomatosi di WegenerSarcoidosiSclerodermiaDiabete mellito
tabella 31.2
Sintomi che fanno pensare a una malattia reumatologica
Sintomo Malattia reumatologica altre affezioni non reumatiche che causano sintomi simili
Febbre Artrite idiopatica giovanile sistemica Tumori maligni, infezioni, malattia infiammatoria intestinaleArtralgia Artrite idiopatica giovanile, lupus eritematoso
sistemico, dermatomiosite, sclerodermiaTrauma, artrite reattiva, infezioni osteoarticolari
Stanchezza Dermatomiosite Distrofie muscolariEsantema al volto Lupus eritematoso sistemico, dermatomiosite Dermatite da fotosensibilitàDolore al torace Artrite idiopatica giovanile, lupus eritematoso
sistemico (con pericardite o osteocondrite)Costocondrite, frattura di costa, spondilolisi, spondilolistesi
Dolore lombare Artrite idiopatica giovanile, spondiloartropatia Microfratture vertebrali, disciite, tumori endospinali
(Da: Nelson textbook of pediatrics, ed 18, Philadelphia, 2007, WB Saunders; p 996; modificata.)
tabella 31.3
CapItolo 31 Malattie reumatologiche606
specie. Va subito sottolineato che la positività della ricerca del fattore reumatoide non è sicuramente diagnostica di artrite idiopatica giovanile (nell’adulto solo un terzo dei soggetti positivi per il fattore reumatoide ha clinicamen-te un’artrite cronica), perché in soggetti con epatopatia (epatite B, soprattutto) o con altre malattie autoimmuni di tipo reumatologico (lupus eritematoso sistemico – LES –, porpora di Schönlein-Henoch) o con malattie infettive (endocardite batterica subacuta, tubercolosi, infezioni con-genite da toxoplasmosi, rosolia, citomegalovirus ed herpes simplex) la prova può risultare ugualmente positiva.
Nell’artrite idiopatica giovanile il fattore reumatoide si ritrova in un piccolo sottogruppo (artrite poliarticolare) che è più simile all’artrite dell’adulto, come quadro clinico e come decorso. D’altra parte è ormai chiaro che il fattore reumatoide non è causa di malattia articolare, anche se può avere un ruolo nel mantenimento della flogosi sinoviale.
anticorpi antinucleo e anticitoplasma
In corso di malattie reumatologiche sono stati riscon-trati autoanticorpi diretti verso componenti nucleari e
citoplasmatici. Questi anticorpi si ritrovano non solo nelle malattie del tessuto connettivo (LES, sindrome di Sjögren, artrite reumatoide, sclerodermia), ma anche in sogget-ti sani (bambini sani, adulti e anziani nei quali le false positività possono raggiungere il 35-75%) e in soggetti con situazioni patologiche non reumatologiche, come la mononucleosi, l’endocardite, l’epatite acuta e l’epatite cronica attiva, la cirrosi epatica e biliare, la leucemia, la miastenia gravis, l’insufficienza renale cronica e la malaria cronica. È più facile riscontrare una falsa positività se la ricerca viene fatta sulle cellule Hep2 e non sul fegato di ratto. Gli anticorpi antinucleo (ANA) si determinano mediante le tecniche di immunofluorescenza indiretta, im-munodiffusione, ELISA (Enzyme-Linked ImmunoSorbent Assay) o Western blot. Anche se i valori superiori rispetto al normale differiscono da un laboratorio a un altro, un titolo di 1:40 è la diluizione più bassa da considerarsi positiva.
Gli anticorpi antinucleo specifici (Tabella 31.4) si ri-trovano tipicamente nelle corrispondenti malattie; tuttavia essi possono essere riscontrati in soggetti senza le manife-stazioni tipiche di queste patologie.
Specificità anticorpale e associazione con malattie
anticorpo Malattia prevalenza (%) Specificità
Antinucleo (ANA) Lupus, artrite idiopatica giovanile, dermatomiosite, sclerodermia, artrite psoriasica, malattia mista del tessuto connettivo
– Associati con aumentato rischio di uveite nell’artrite idiopatica giovanile e nell’artrite psoriasica. Oltre il 30% dei bambini senza malattie reumatologiche risultano positivi alla prova
DNA a doppia elica Lupus eritematoso sistemico 60-70 Alta specificità per il lupus, associato a nefrite lupica
Smith (Sm) Lupus eritematoso sistemico 20-30 Alta specificità per il lupus, associato a nefrite lupicaMuscolo liscio (Sm) Epatite autoimmune – –Pm-Scl (polimiosite,scleroderma)
Sclerodermatomiosite – –
SSA (Ro), associato all’RNA citoplasmatico
Lupus, sindrome di Sjögren 25-30 Associato a lupus neonatale, lupus cutaneo subacuto e trombocitopenia
SSB (La), associato alla RNA III polimerasi
Lupus, sindrome di Sjögren 25-30 Usualmente coesiste con l’anticorpo anti-SSA
Proteina ribonucleasi (RNP) Malattia mista del tessuto connettivo, lupus
30-40 Suggestiva per malattia mista del tessuto connettivo, a meno che non ci siano i criteri per il lupus
Istone Lupus indotto dai farmaci, lupus – –Centromero Sclerosi sistemica cutanea limitata 70 Non specifico della sclerosi sistemicaTopoisomerasi I (Scl-70) Sclerosi sistemica – Rara nel bambinoAnticorpi anticitoplasma dei
neutrofili (ANCA)Vasculite – –
Anticitoplasma (c-ANCA) – I c-ANCA si associano alla granulomatosi di Wegener e alla fibrosi cistica
Perinucleare (p-ANCA) – I p-ANCA si associano a poliangite microscopica, poliarterite nodosa, lupus, malattia infiammatoria intestinale, fibrosi cistica, colangite sclerosante primaria, porpora di Schönlein-Henoch, malattia di Kawasaki
(Da: Rabinovich CE: Rheumatic diseases in children. In: Nelson textbook of pediatrics, ed 19, New York, 2011, Elsevier, p 829; modificata.)
tabella 31.4

607CapItolo 31 Malattie reumatologiche
Gli anticorpi che reagiscono con i granuli dei neutrofili so-no chiamati anticorpi anticitoplasma dei neutrofili (ANCA). Questi anticorpi, dimostrabili con l’immunofluorescenza, vennero per la prima volta trovati nel siero di pazienti con granulomatosi di Wegener. Con la colorazione si differenziano due aspetti: uno che riguarda una diffusa colorazione dei granuli (c-ANCA) e uno a localizzazione perinucleare (p-ANCA). Con l’esperienza è risultato che i c-ANCA non sono specifici della granulomatosi di We-gener, ma si ritrovano anche in altre forme di vasculite, compresa la malattia di Kawasaki. I p-ANCA si ritrovano invece in un gran numero di condizioni patologiche, fra le quali la glomerulonefrite necrotizzante o a semilune, la colite ulcerosa, la colangite, l’epatite cronica attiva e la malattia di Schönlein-Henoch. I due tipi si ritrovano anche nei soggetti con infezione da HIV. Essi sono utili anche per seguire l’evoluzione della malattia (Tabella 31.5).
Più di recente sono stati identificati altri anticorpi, que-sta volta diretti verso molti antigeni fosfolipidici: sono gli anticorpi antifosfolipidi. Questi anticorpi comprendono i lupus-anticoagulanti, gli anticorpi anticardiolipina, e gli anticorpi che reagiscono con il substrato usato per l’esecuzione della prova VRDL della sifilide. Gli anticorpi
antifosfolipidi si ritrovano in circa un terzo dei casi di lupus: la loro presenza si accompagna a tendenza alla trombosi, alla trombocitopenia, all’anemia emolitica, alla corea e all’infarto.
Complemento e immunoglobuline
Nelle malattie reumatologiche i vari componenti del com-plemento risultano tutti aumentati. Nel LES, soprattutto nella glomerulonefrite in corso di LES, i livelli di comple-mento possono risultare abbassati. In molti pazienti con malattie del connettivo si riscontra un aumento assoluto delle immunoglobuline, con conseguente inversione del rapporto albumina-globulina.
Sistema Hla
L’associazione di particolari aplotipi di HLA (Human Leucocyte Antigen) con forme cliniche diverse di malattie reumatologiche e in particolare con sottotipi di artrite idiopatica giovanile è stata spesso riscontrata. Gli antigeni HLA sono localizzati sulla superficie di numerose cellule umane: di questi si riconoscono molti loci, conosciuti
Valutazione di laboratorio a seconda del sospetto diagnostico
Malattia sospetta Valutazione iniziale Ulteriore valutazione Valutazione particolare
Lupus eritematoso sistemico, malattia mista del tessuto connettivo
Emocromo, VES, ANA, ALT, AST, CK, creatinina, albumina, analisi delle urine, pressione arteriosa, profilo tiroideo
Se gli ANA sono positivi: anti-SSA (Ro), anti-SSB (La), anti-Smith, anti-RNP Abs, anti-dsDNA Ab, C3, C4, Coombs, urine, rapporto proteine/creatinina, Rx torace
Antifosfolipidi Abs, lupus- anticoagulante, anti-b2-glicoproteine, ECG, considerare la biopsia renale, prove respiratorie, broncoscopia con lavaggio, Rx polmone ad alta risoluzione, considerare la biopsia polmonare
Dermatomiosite giovanile Emocromo, CK, ALT, AST, LDH, aldolasi, SNA, cercare il riflesso faringeo
Risonanza magnetica del muscolo
Considerare l’elettromiografia e la biopsia muscolare, prove respiratorie, studio della deglutizione, neopterina sierica
Artrite idiopatica giovanile Emocromo, VES, creatinina, ALT, AST, considerare il TAS, anti-DNAasi B, anticorpi EBV, anticorpi Lyme, parvovirus B19, Rx delle articolazioni
Considerare la ricerca degli anticorpi verso i comuni agenti infettivi, PPD, fattore reumatoide, ANA, HLA B27
Risonanza magnetica
Granulomatosi di Wegener Emocromo, ANCA, AST, ALT, albumina, creatinina, VES, urine, Rx torace, pressione arteriosa
Rapporto urinario proteine/creatinina, anticorpi antimieloperossidasi e antiproteinasi-3, prove respiratorie
Broncoscopia con lavaggio, Rx del polmone ad alta risoluzione, considerare la biopsia renale e polmonare
Sarcoidosi Emocromo, elettroliti, AST, ALT, albumina, creatinina, calcio, fosforo, enzima angiotensina-convertente, pressione arteriosa
Rx del polmone, prove respiratorie
Rx del polmone ad alta risoluzione, considerare la biopsia renale e polmonare
Scleroderma localizzato Biopsia cutanea, emocromo, VES Immunoglobuline G sieriche, ANA, fattore reumatoide, anticorpi anti-DNA a unica elica
Scleroderma generalizzato ANA, emocromo, VES, pressione arteriosa, CK, creatinina, Rx torace
Anti-Sci70, prove respiratorie Prove respiratorie, ECG, Rx gastrointestinale alta
(Da: Rabinovich CE: Rheumatic diseases in children. In: Nelson textbook of pediatrics, ed 19, New York, 2011, Elsevier, p 829; modificata.)
tabella 31.5

CapItolo 31 Malattie reumatologiche608
come A, B, C, D, DR e DQ, ognuno dei quali con nume-rosi alleli. A parte la loro importanza nel determinare la compatibilità tissutale, il loro preciso ruolo biologico non è ancora ben conosciuto. Poiché gli antigeni HLA sono geneticamente determinati e possono essere identificati con esattezza, essi sono in grado di fornire informazioni sull’associazione con alcune malattie. Nella Tabella 31.6 sono riportate le associazioni più frequenti fra malattie reumatologiche e HLA.
Il liquido sinoviale nell’artrite idiopatica giovanile è di aspetto torbido ed è ricco di proteine. Le cellule, rap-presentate essenzialmente da neutrofili, sono aumentate di numero e variano da qualche migliaio a molte decine di migliaia. I livelli di glucosio sono bassi e quelli del com-plemento possono essere normali o diminuiti.
Diagnostica per immagini
L’indagine radiologica tradizionale si dimostra di grande utilità sia per la conferma diagnostica, sia per il decorso di molte malattie reumatologiche che interessino i capi articolari e i tessuti circostanti. Attraverso la radiografia è possibile dimostrare la tumefazione del tessuto molle, l’osteoporosi, le eventuali fratture, le alterazioni dello spazio e dei capi articolari, la periostite, gli eventuali di-sturbi dell’accrescimento.
Negli ultimi anni l’indagine ecografica ha acquisito un ruolo sempre più importante, come indagine di prima istanza, nello studio dei tessuti molli articolari e periartico-lari, e per l’individuazione di versamenti (artrite reattiva). La non invasività, il basso costo, la rapida esecuzione e quindi la possibilità di indagini ripetute nel tempo sono tutte ragioni che giustificano il suo sempre più frequente impiego.
La tomografia computerizzata (TC) e la risonanza magne-tica (RM) hanno acquisito, anch’esse di recente, un importante
ruolo nella reumatologia pediatrica (Figura 31.2). La TC permette di rilevare le più piccole e precoci lesioni distruttive a carico dell’osso, mentre con la RM è possibile avere una migliore risoluzione delle parti molli articolari, dei muscoli, dei tendini, della cartilagine e in parte anche dell’osso. Per l’artrite idiopatica giovanile la RM, consentendo di rilevare lo spessore della sinoviale, permette di differenziarla da un eventuale versamento.
Anche la scintigrafia ha dimostrato la sua importanza nella localizzazione del danno scheletrico iniziale, spes-so prima che la radiografia tradizionale sia in grado di metterlo in evidenza. Tuttavia, essa poco o nulla è in grado di dire sull’eziologia della lesione.
Principi di trattamento
Il principio più importante nel trattamento con farmaci delle malattie reumatologiche consiste nell’indurre un controllo, il più precoce possibile, della malattia, con il minor danno di tessuto o di organo, in attesa del mi-glioramento della prognosi, a breve e a lungo periodo. La cura deve allontanare l’invalidità, deve incentivare al massimo il movimento e la qualità della vita dei bambini
Associazioni più frequenti fra HLA e malattie reumatologiche
Malattia Hla più spesso incontrato
LES B8, DR2, DR3Dermatomiosite infantile B8, DR3Spondilite anchilosante B27Sindrome di Reiter B27Artrite in corso di malattie infiammatorie croniche B27Artrite reattiva, successiva a infezioni batteriche
intestinaliB27
Artrite pauciarticolare ANA positiva, con iridociclite DR8, DR6, DR5, DPw2, DQ
Artrite pauciarticolare dei bambini di età più avanzata
B27
Artrite poliarticolare, fattore reumatoide positiva DR4, DR1Artrite psoriasica B27
tabella 31.6
FIgura 31.2 - risonanza magnetica con gadolinio di un bambino di 10 anni con artrite idiopatica giovanile. Il segnale bianco nella sinovia, vicino all’estremo distale del femore, della parte prossimale della tibia e della patella, riflette l’infiammazione. (Da: Kliegman RM, et al, editors: Pediatria di Nelson, ed 19, Milano, 2012, Elsevier.)

609CapItolo 31 Malattie reumatologiche
colpiti, deve far scomparire il dolore, prevenire e ridurre il danno di organo e deve evitare o limitare la tossicità dei farmaci. I medicamenti vanno scelti fra le diverse classi terapeutiche sulla base della diagnosi, della gravità della malattia e della prognosi a lungo termine, nella dose indicata in rapporto alle misure antropometriche del paziente. I farmaci usati per il trattamento delle malattie reumatologiche del bambino hanno diversi meccanismi di azione, ma hanno in comune la soppres-sione dell’infiammazione. Sia i farmaci antireumatici biologici, sia quelli non biologici, agiscono direttamente sul sistema immune.
La terapia non farmacologica è un’aggiunta importan-te al trattamento medico delle malattie reumatologiche. Un gruppo multidisciplinare di reumatologia pediatrica offre servizi coordinati per i bambini e le loro famiglie. È fondamentale il coordinamento delle sottospecialità e dei servizi di riabilitazione, compresa la scuola; il gruppo di lavoro deve prevedere:• il reumatologo pediatra;• il pediatra;• le infermiere;• le assistenti sociali;• gli addetti alla riabilitazione fisica e alla occupational
therapy;• i consulenti: oftalmologo, nefrologo, ortopedico, der-
matologo, gastroenterologo;• i nutrizionisti;• le insegnanti della scuola.
I vaccini vivi attenuati sono controindicati nei pazienti trattati con dosi elevate di corticosteroidi, mentre i vaccini inattivati non sono controindicati. Anzi, la vaccinazione contro l’influenza viene consigliata. Una prova alla tu-bercolina dovrebbe esere eseguita prima di cominciare il trattamento con i farmaci biologici.
antinfiammatori non steroidei
La maggior parte dei bambini risponde agli antinfiam-matori non steroidei (FANS) per bocca, come l’ibu-profene, il naproxene o il meloxicam (l’Aspirina è stata esclusa dal trattamento nei bambini al di sotto dei 9-12 anni). L’effetto antinfiammatorio nell’artrite idiopatica giovanile è visibile dopo 4-6 settimane di trattamento: questi farmaci agiscono principlamente inibendo l’en-zima ciclo-ossigenasi (COX), che è fondamentale per la sintesi delle prostaglandine, una famiglia di sostanze che promuove l’infiammazione. L’entrata in commer-cio, qualche anno fa, di farmaci antinfiammatori non steroidei ad attività inibitoria prevalente sulla ciclo-os-sigenasi 2 (anti-COX-2: celecoxib, rofecoxib), e quindi accompagnati meno di frequente a sofferenza gastrica, ha portato alla comparsa di sofferenze miocardiche e di insufficienza cardiaca congestizia, tanto che l’uso del celecoxib (Celebrex) è sconsigliato nel bambino
(Informatore Farmaceutico, 2011). I più frequenti ef-fetti collaterali legati all’assunzione dei FANS sono la nausea, la perdita di appetito e i dolori addominali. Le gastriti e le ulcere gastroduodenali sono eccezionali nel bambino.
Farmaci non biologici, modificanti la malattia
Il metotrexato, un antimetabolita, è la pietra miliare del trattamento, nella reumatologia pediatrica, per la sua elevata efficacia e per la sua bassa tossicità, anche per lunghi periodi di tempo. Il metotrexato inibisce gli enzimi folato-dipendenti, importanti per la sintesi de novo delle purine, per cui la sua assunzione porta a un blocco della loro sintesi, fino all’immunosoppressione. La dose è quel-la di 10 mg/m2 una volta alla settimana (Methotrexate, compresse da 2,5 mg). È usato nell’artrite idiopatica giovanile e nella dermatomiosite giovanile, anche come agente risparmiatore di steroidi. Gli effetti collaterali sono i sintomi gastrointestinali, la stomatite, l’aumento delle aminotransferasi, la cefalea, la leucopenia. Può es-sere usato l’acido folico per ridurre gli effetti collaterali. La continuazione del trattamento con metotrexato dopo l’induzione della remissione non riduce l’incidenza delle recidive.
L’idrossiclorochina 6 (Plaquenil, compresse da 200 mg, 3-6 mg/kg/die) è un antimalarico, importante per il trat-tamento del LES e della dermatomiosite. Non ha alcun effetto nell’artrite idiopatica giovanile; il maggiore effetto collaterale è la tossicità retinica: un esame oftalmologico completo deve essere eseguito ogni 6-12 mesi di tratta-mento.
La leflunomide (Arava, compresse da 10, 20 e 100 mg, 10 mg per bambini da 10 a 20 kg, 15 mg per bambini da 20 a 40 kg, 20 mg per bambini di peso >40 kg per os, una volta al giorno) è usata per il trattamento dell’artrite idiopatica govanile. Fra gli effetti collaterali sono presenti parestesie e neuropatia periferica, intolleranza gastrointe-stinale, aumento delle aminotransferasi e insufficienza epatica.
La sulfasalazina (Salazopyrin EN, compresse da 500 mg, 50 mg/kg/die; dose massima per l’adulto 3.000 mg/die) è usata nell’artrite idiopatica giovanile, nelle forme pauciar-ticolari e nelle spondiloartropatie periferiche. Si associa a gravi reazioni di ipersensibilità, inclusa la sindrome di Stevens-Johnson.
Il micofenolato mofenil (non in commercio in Italia) è un farmaco immunosoppressivo usato nel LES, nella uveite e nelle manifestazioni cutanee autoimmuni.
I corticosteroidi, usati per via orale, endovenosa, oculare, topica e intrarticolare, fanno parte del tratta-mento delle malattie reumatologiche: dal lupus, nelle forme moderate e in quelle lievi, alla dermatomiosi-te e alla maggior parte delle malattie vasculitiche. Il loro impiego prolungato si associa a complicazioni dose-dipendenti, come la soppressione della crescita

CapItolo 31 Malattie reumatologiche610
in altezza, l’aspetto cushingoide, l’osteoporosi, la ne-crosi avascolare, l’ipertensione, l’alterata tolleranza al glucosio, i disturbi dell’umore e l’aumento delle in-fezioni cutanee. Fra i corticosteroidi, il più utilizzato è il metilprednisolone (Urbason 25, 40, 250 mg/mL, 10-30 mg/kg/dose, fino a un massimo di 1.000 mg, in una somministrazione al giorno, ev, per 1-5 giorni). Per le infiltrazioni intrarticolari viene usato il triamcino-lone esacetonide. Gli steroidi oculari vanno prescritti dagli oftalmologi, in gocce o per iniezione nei tessuti molli, intorno al bulbo oculare in caso di uveite attiva. Gli steroidi intrarticolari (triamcinolone acetonide = Kenacort sospensione 40 mg/mL) sono somministrati di frequente per la terapia inziale di bambini con artrite idiopatica giovanile pauciarticolare e quando manchi una risposta al trattamento.
Farmaci biologici
I farmaci biologici, geneticamente preparati, sono diretti verso siti specifici della cascata infimmatoria, come le citochine, le molecole sulla superficie cellulare (recettori delle citochine) e le molecole di adesione. Negli ultimi anni sono state preparate numerose sostanze che interferiscono con le citochine, i mediatori solubili dell’infiammazione, che si legano sulla superficie delle cellule, su recettori specifici. Esse possono essere divise in proinfiammatorie, come il Tumor Necrosis Factor a (TNF-a) e l’interleuchina 6 (IL-6), e antinfiammatorie come l’interleuchina 10 e
l’interleuchina 1, antagonisti dei recettori (IL-1ra). La maggior parte dei farmaci biologici si lega alle citochine solubili o previene il loro legame con i recettori specifici.
Questi farmaci sono in generale delle proteine di fusio-ne, formate da una parte costituita da un recettore della superficie cellulare, fuso con la regione C di una IgG1 allo scopo di creare un recettore solubile che si leghi alle citochine circolanti (Tabella 31.7).
I farmaci biologici sono gravati da specifici effetti collaterali, legati strettamente alla loro stessa modalità di azione, e da specifiche controindicazioni (Tabelle 31.8 e 31.9). Nei pazienti trattati con natalizumab, un farmaco biologico in commercio in Italia per il trattamento della sclerosi multipla e non usato nel trattamento dell’artrite idiopatica giovanile, è stata riscontrata di frequente la riattivazione subclinica di un virus polioma JC, in qualche raro caso con il quadro della leucoencefalopatia multifo-cale progressiva.
Le applicazioni di questi farmaci in pediatria sono in continuo aumento, soprattutto per quei bambini che non rispondono ai vecchi farmaci. Con l’aumento delle cono-scenze, il tipo di farmaco biologico da usare sarà ritagliato su misura sul profilo genetico citochinico del paziente e sul tipo e sulla gravità della malattia.
Citotossici
La ciclofosfamide (Endoxan, compresse da 50 e 500 mg e fiale da 1 g per im o ev) viene metabolizzata dal fegato
Farmaci biologici, già in uso o in via di sviluppo, per il trattamento dell’artrite idiopatica giovanile
Farmaco Categoria mab obiettivo Dose terapeutica
Natalizumab (Tysabri)* Anticorpo monoclonale umanizzatoEtanercept (Enbrel)* Proteina di fusione TNF-a 0,8 mg/kg/dose, 1 volta alla settimana, massimo 50 mg/dose.
Iniezione sottocutaneaInfliximab (remicade)* Chimera TNF-a 6-10 mg/kg/dose, 1 volta alle settimane 0, 2, 6 e poi ogni
4-8 settimaneAdalimumab (Humira)* Totalmente umano TNF-a 24 mg/m2 ogni 2 settimane, massimo 40 mg/dose. Iniezione
sottocutaneaAnakinra (Kineret)* Antagonista recettoriale totalmente
umanoRecettore IL-1 1-2 mg/kg/die, massimo 100 mg/dose
Rilonacept (arkalyst) Proteina di fusione IL-1 2,2-4,4 mg/kg 1 volta alla settimana. Iniezione sottocutanea
Abatacept (Orencia)* Proteina di fusione Antigene 4 dei linfociti T citotossici
10 mg/kg alle settimane 0, 2, 4 e poi ogni 4 settimane, massimo 1.000 mg/dose
Rituximab (Mabthera) Chimera CD20 750 mg/m2 dose ogni 2 settimane o 375 mg/m2; 4 dosi settimanalmente per 4 volte, massimo 1.000 mg/dose. Per via venosa
Tocilizumab (roactemra)* Umanizzato IL-6 8-12 mg/kg ogni 2 settimane. Per via venosaCanakinumab (Ilaris)* Totalmente umano IL-1b 2 mg/kg dose ogni 8 settimane. Iniezione venosa o sottocute
*In commercio in Italia.(Da: Breda L, Del Torto M, De Sanctis S, Chiarelli F: Biologics in children’s autoimmune disorders: efficacy and safety, Eur J Pediatr 170:157-167, 2011; modificata.)
tabella 31.7
611CapItolo 31 Malattie reumatologiche
nella sua forma attiva, che porta all’immunosoppressione per l’inibizione della mitosi in fase S2. Si associa a una diminuzione dei linfociti T e B. L’infusione di ciclofo-sfamide (500-1.000 mg/m2, una volta al mese per 6 mesi) viene usata nel lupus e nella vasculite generalizzata; gli effetti collaterali sono frequenti: nausea, vomito, anores-sia, alopecia, mucosite, cistite emorragica, soppressione midollare. Fra le complicazioni a distanza vanno consi-derate la sterilità e la comparsa di cancro, specialmente leucemia, linfoma e cancro della vescica. Le bambine e le adolescenti corrono un rischio più basso: la soppressione
ovarica con un inibitore dell’ormone liberante le gonado-tropine preserva la fertilità.
altri farmaci e rimedi
In bambini che non avevano risposto ai farmaci sopra riportati è stata impiegata con evidenti miglioramenti la talidomide.
La fisioterapia, l’applicazione di docce e il movimento concorrono a raggiungere una stabilizzazione e spesso un miglioramento.
Effetti collaterali e relative controindicazioni per l’uso del TNF-a
principali effetti collaterali Controindicazione per l’uso
Reazioni nella sede dell’iniezione AssolutaReazioni all’infusione endovenosa Infezioni attiveInfezioni Storia di infezioni ricorrenti o cronicheRiattivazione di un’infezione latente Tubercolosi precedente, non trattataMalattia demielinizzante Sclerosi multipla o neurite otticaEffetti collaterali neuropsichici, come stanchezza, cefalea, vertigini,
depressione, sindrome da amplificazione del dolore, ansietàTrattamento combinato con anakinra
Insufficienza cardiaca Storia attiva e recente di cancro (negli ultimi 10 anni), esclusi i tumori della pelle
Cancri RelativaImmunogenicità Gravidanza, allattamento, infezioni da HIV, HBV e HCV
(Da: Breda L, Del Torto M, De Sanctis S, Chiarelli F: Biologics in children’s autoimmune disorders: efficacy and safety, Eur J Pediatr 170:157-167, 2011; modificata.)
tabella 31.8
Indicazioni comparative sui farmaci approvate dall’FDA e dall’EMA*
Farmaco approvato dall’FDa approvato dall’eMa
Etanercept Artrite reumatoide, artrite psoriasica, spondilite anchilosante, psoriasi a placche, artrite idiopatica giovanile poliarticolare
Artrite reumatoide, artrite psoriasica, spondilite anchilosante, psoriasi a placche, sclerosi multipla, psoriasi pediatrica a placche (>8 anni)
Infliximab Artrite reumatoide, malattia di Crohn, artrite psoriasica, spondilite anchilosante, sclerosi multipla, colite ulcerosa, malattia di Crohn (>6 anni)
Artrite reumatoide, malattia di Crohn, artrite psoriasica, spondilite anchilosante, psoriasi a placche, colite ulcerosa, malattia di Crohn pediatrica (>6 anni)
Adalimumab Artrite reumatoide, artrite psoriasica, spondilite anchilosante, malattia di Crohn, psoriasi a placche, artrite idiopatica giovanile poliarticolare (>4 anni)
Artrite reumatoide, artrite psoriasica, spondilite anchilosante, malattia di Crohn, psoriasi a placche, artrite idiopatica giovanile poliarticolare, artrite idiopatica giovanile poliarticolare (13-17 anni)
Anakinra Artrite reumatoide Artrite reumatoideRilonacept Sindrome periodica criopirina-associata (>12 anni) Sindrome periodica criopirina-associata (>12 anni)Abatacept Artrite reumatoide, artrite idiopatica giovanile poliarticolare
(>6 anni)Artrite reumatoide, artrite idiopatica giovanile poliarticolare
(>6 anni)Rituximab Artrite reumatoide, linfoma non-Hodgkin, leucemia linfocitica
cronicaArtrite reumatoide, linfoma non-Hodgkin, leucemia linfocitica
cronicaTocilizumab Artrite reumatoide Artrite reumatoideCanakinumab Sindrome periodica criopirina-associata (>4 anni) Sindrome periodica criopirina-associata (>4 anni)
*In corsivo le malattie di interesse pediatrico.(Da: Breda L, Del Torto M, De Sanctis S, Chiarelli F: Biologics in children’s autoimmune disorders: efficacy and safety, Europ J Pediatr 170:157-167, 2011; modificata.)
tabella 31.9

CapItolo 31 Malattie reumatologiche612
Artrite idiopatica giovanile
Si definisce artrite idiopatica giovanile un’artrite di una o più articolazioni, che risponda ai seguenti requisiti:• insorganeiprimi16annidietà;• duripiùdi6settimane;• siadacausasconosciuta.Il sottotipo della malattia e il suo decorso sono definiti dopo 6 mesi dall’inizio del quadro clinico.
La denominazione artrite idiopatica giovanile ha sostituito in ambito europeo la denominazione artrite reumatoide giovanile, largamente usata nella letteratura angloamericana: l’intento iniziale dei reumatologi pediatri europei è stato quello di comprendere sotto un unico termine anche le forme di spondiloartrite, HLA B27- collegate. Tuttavia oggi le due denominazioni possono essere considerate equivalenti.
In seno all’artrite idiopatica giovanile vengono individua-te tre forme distinte, sulla base delle manifestazioni cliniche iniziali e della loro evoluzione nell’arco di 3-6 mesi:• forma sistemica: rappresenta il 10-20% del totale.
Colpisce in prevalenza bambini della seconda infanzia, con elevata temperatura iniziale (>39-39,5 °C) per 2 settimane, spesso accompagnata al momento dell’acme da un esantema aspecifico; quasi sempre manca l’inte-ressamento articolare, che può comparire nel decorso della malattia;
• forma poliarticolare: rappresenta circa il 20-25% del totale. Anche in questa forma la febbre è bassa o può mancare, mentre l’interessamento articolare riguarda 5 o più articolazioni, sempre alla fine di un periodo iniziale di osservazione di almeno 6 mesi; si distinguono una forma fattore reumatoide positiva e una fattore reumatoide negativa;
• forma pauciarticolare: rappresenta circa il 50-60% del totale. Si tratta di pazienti senza febbre e con febbre
molto bassa, con interessamento di 4 articolazioni o meno, alla fine dei 6 mesi iniziali di osservazione. Se ne conoscono un tipo I e un tipo II.
Come vedremo la forma poliarticolare viene ulteriormente suddivisa (Tabella 31.10). Accanto a queste tre forme clas-siche esistono altre forme, più rare, quali l’artrite associata a entesite e l’artrite psoriasica. Circa il 10% di tutti i casi sfugge ai criteri sopra esposti: a questi casi viene dato il nome di artrite indifferenziata.
L’incidenza e la prevalenza della malattia in Europa sono state variamente calcolate; si tratta comunque di una malattia non rara, come si credeva una volta, con un’inci-denza per anno compresa fra 0,8 e 22,6 casi su 100.000 soggetti di età al di sotto dei 16 anni e con una prevalenza di 7-401 casi su 100.000 soggetti in età inferiore ai 16 anni; in Italia i soggetti con artrite, in età inferiore ai 15 anni, sarebbero fra 5.400 e 10.800. I costi diretti e indiretti per l’artrite e le altre malattie reumatiche negli USA, nel 1997, è stato di 86,2 miliardi di dollari (di cui 51,1 per costi diretti e 35,1 per costi indiretti), circa l’1% del prodotto interno lordo di allora nell’America del Nord.
A tutt’oggi la causa della malattia non è ben cono-sciuta. Negli ultimi anni, ricerche in campo virologico e batteriologico hanno rafforzato l’ipotesi che l’evento scatenante sia di tipo infettivo (vedi Borrelia burgdorferi nella malattia di Lyme, parvovirus B19, micoplasmi), ma che la cronicizzazione avvenga soltanto in determinati soggetti anche in rapporto al loro corredo HLA. Recenti ricerche su gemelli, tuttavia, hanno stabilito che i geni hanno un’importanza minore dei fattori ambientali nello sviluppo dell’artrite reumatoide.
Vi sono alcune caratteristiche comuni alle diverse for-me, come di seguito descritto.• Un certo grado di rigidezza e di dolore al mattino è
caratteristico dell’artrite idiopatica giovanile come del-l’artrite reumatoide dell’adulto. Il ricorso a un bagno caldo al mattino riduce questa difficoltà mattutina dei
Gruppi e sottogruppi dell’artrite idiopatica giovanile
Item a inizio sistemico
poliarticolareFR*-negativa
poliarticolareFR*-positiva
pauciarticolare tipo I pauciarticolare tipo II
% sul totale 10-20 25-35 4-5 40-50 10-15Sesso 60% maschi 90% femmine 80% femmine 80% femmine 90% maschiEtà all’inizio 1a-2a infanzia Tutte le età 3a infanzia-adolescenza 1a-2a infanzia 3a infanzia-adolescenzaArticolazioni Tutte Tutte Tutte Poche grandi articolazioni Poche grandi articolazioniSacroileite No No Rara No ComuneIridociclite No Rara No 30% iridociclite cronica 10-20% iridociclite acutaFattore reumatoide Negativo Negativo 100% Negativo Negativo/positivoAnticorpi antinucleo Negativi 25% 75% 90% NegativiHLA ? ? HLA DR4 HLA DR5, DR6 HLA B27Assetto finale Grave artrite
nel 25%Grave artrite
nel 10-15%Grave artrite
nel 50%Danno oculare 10%Poliartrite 20%
Spondiloartropatia successiva (?)
*FR: fattore reumatoide.
tabella 31.10
613CapItolo 31 Malattie reumatologiche
movimenti. Dopo un periodo di immobilità, dovuto anche alla malattia, il grado di irrigidimento aumenta: ne consegue che, per quanto è possibile, i bambini con artrite debbono essere lasciati liberi di muoversi duran-te tutto il giorno, anche mentre sono a scuola.
• Il dolore è un sintomo variabile; spesso i bambini che avvertono meno dolore sono proprio quelli che nelle età più avanzate hanno le più gravi conseguenze dell’ar-trite, perché uno dei sintomi principali non è stato utile come campanello di allarme. Vi sono d’altra parte altri bambini che avvertono dolori di alta intensità, tanto da rendere loro difficili i movimenti. È evidente che un dolore muscoloscheletrico isolato, in assenza di altri segni e sintomi, non ha alcun valore predittivo di una malattia reumatologica del bambino.
• Nelle forme sistemiche e nella grave malattia poliarti-colare è spesso evidente un ritardo generalizzato di cre-scita, spesso accentuato dall’uso dei corticosteroidi. Se la malattia si attenua, la crescita staturale può ripren-dere, ma quando, come avviene nella maggioranza dei bambini, la malattia si prolunghi nel tempo, l’altezza finale può rimanere gravemente deficitaria.
• In qualche caso l’alterazione della crescita è localiz-zata, sia nel senso di una sottocrescita, sia di una cre-scita esuberante. La prima conseguenza è un’evidente asimmetria, soprattutto quando questa localizzazione sia a carico di un arto inferiore, come avviene di fre-quente nell’artrite pauciarticolare tipo I. Nella maggior parte dei casi si assiste a una crescita eccessiva, per accelerazione della maturazione epifisaria dei segmenti
ossei vicini all’articolazione interessata. A carico della mandibola, quando vi sia un’artrite dell’articolazione temporo-mandibolare, si può manifestare una crescita ridotta, con marcata micrognatia e grave deformità facciale e ortodontica.
Forma a inizio sistemico
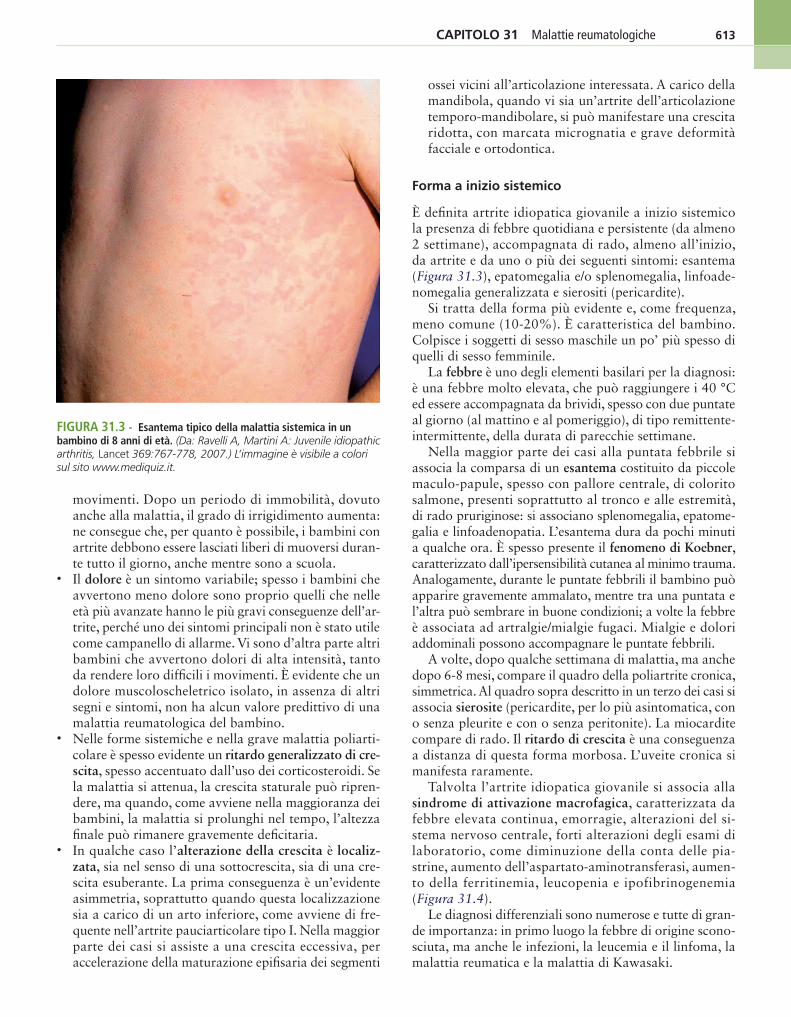
È definita artrite idiopatica giovanile a inizio sistemico la presenza di febbre quotidiana e persistente (da almeno 2 settimane), accompagnata di rado, almeno all’inizio, da artrite e da uno o più dei seguenti sintomi: esantema (Figura 31.3), epatomegalia e/o splenomegalia, linfoade-nomegalia generalizzata e sierositi (pericardite).
Si tratta della forma più evidente e, come frequenza, meno comune (10-20%). È caratteristica del bambino. Colpisce i soggetti di sesso maschile un po’ più spesso di quelli di sesso femminile.
La febbre è uno degli elementi basilari per la diagnosi: è una febbre molto elevata, che può raggiungere i 40 °C ed essere accompagnata da brividi, spesso con due puntate al giorno (al mattino e al pomeriggio), di tipo remittente-intermittente, della durata di parecchie settimane.
Nella maggior parte dei casi alla puntata febbrile si associa la comparsa di un esantema costituito da piccole maculo-papule, spesso con pallore centrale, di colorito salmone, presenti soprattutto al tronco e alle estremità, di rado pruriginose: si associano splenomegalia, epatome-galia e linfoadenopatia. L’esantema dura da pochi minuti a qualche ora. È spesso presente il fenomeno di Koebner, caratterizzato dall’ipersensibilità cutanea al minimo trauma. Analogamente, durante le puntate febbrili il bambino può apparire gravemente ammalato, mentre tra una puntata e l’altra può sembrare in buone condizioni; a volte la febbre è associata ad artralgie/mialgie fugaci. Mialgie e dolori addominali possono accompagnare le puntate febbrili.
A volte, dopo qualche settimana di malattia, ma anche dopo 6-8 mesi, compare il quadro della poliartrite cronica, simmetrica. Al quadro sopra descritto in un terzo dei casi si associa sierosite (pericardite, per lo più asintomatica, con o senza pleurite e con o senza peritonite). La miocardite compare di rado. Il ritardo di crescita è una conseguenza a distanza di questa forma morbosa. L’uveite cronica si manifesta raramente.
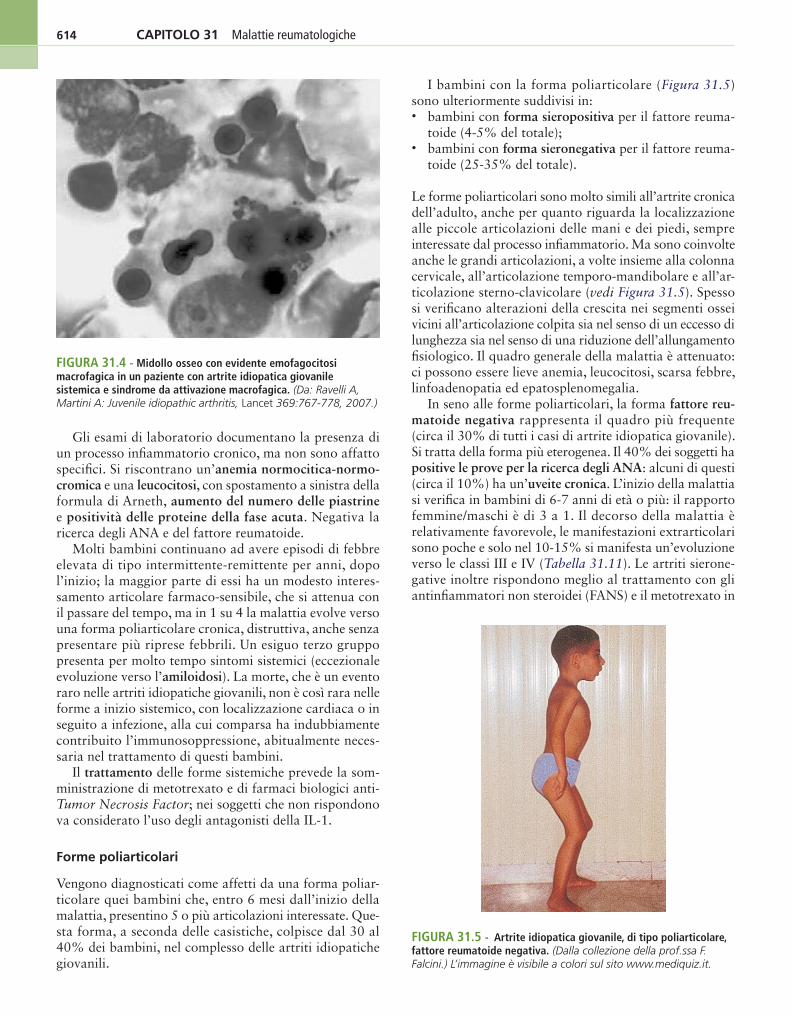
Talvolta l’artrite idiopatica giovanile si associa alla sindrome di attivazione macrofagica, caratterizzata da febbre elevata continua, emorragie, alterazioni del si-stema nervoso centrale, forti alterazioni degli esami di laboratorio, come diminuzione della conta delle pia-strine, aumento dell’aspartato-aminotransferasi, aumen-to della ferritinemia, leucopenia e ipofibrinogenemia (Figura 31.4).
Le diagnosi differenziali sono numerose e tutte di gran-de importanza: in primo luogo la febbre di origine scono-sciuta, ma anche le infezioni, la leucemia e il linfoma, la malattia reumatica e la malattia di Kawasaki.
FIgura 31.3 - Esantema tipico della malattia sistemica in un bambino di 8 anni di età. (Da: Ravelli A, Martini A: Juvenile idiopathic arthritis, Lancet 369:767-778, 2007.) L’immagine è visibile a colori sul sito www.mediquiz.it.
CapItolo 31 Malattie reumatologiche614
Gli esami di laboratorio documentano la presenza di un processo infiammatorio cronico, ma non sono affatto specifici. Si riscontrano un’anemia normocitica-normo-cromica e una leucocitosi, con spostamento a sinistra della formula di Arneth, aumento del numero delle piastrine e positività delle proteine della fase acuta. Negativa la ricerca degli ANA e del fattore reumatoide.
Molti bambini continuano ad avere episodi di febbre elevata di tipo intermittente-remittente per anni, dopo l’inizio; la maggior parte di essi ha un modesto interes-samento articolare farmaco-sensibile, che si attenua con il passare del tempo, ma in 1 su 4 la malattia evolve verso una forma poliarticolare cronica, distruttiva, anche senza presentare più riprese febbrili. Un esiguo terzo gruppo presenta per molto tempo sintomi sistemici (eccezionale evoluzione verso l’amiloidosi). La morte, che è un evento raro nelle artriti idiopatiche giovanili, non è così rara nelle forme a inizio sistemico, con localizzazione cardiaca o in seguito a infezione, alla cui comparsa ha indubbiamente contribuito l’immunosoppressione, abitualmente neces-saria nel trattamento di questi bambini.
Il trattamento delle forme sistemiche prevede la som-ministrazione di metotrexato e di farmaci biologici anti-Tumor Necrosis Factor; nei soggetti che non rispondono va considerato l’uso degli antagonisti della IL-1.
Forme poliarticolari
Vengono diagnosticati come affetti da una forma poliar-ticolare quei bambini che, entro 6 mesi dall’inizio della malattia, presentino 5 o più articolazioni interessate. Que-sta forma, a seconda delle casistiche, colpisce dal 30 al 40% dei bambini, nel complesso delle artriti idiopatiche giovanili.
I bambini con la forma poliarticolare (Figura 31.5) sono ulteriormente suddivisi in:• bambini con forma sieropositiva per il fattore reuma-
toide (4-5% del totale);• bambini con forma sieronegativa per il fattore reuma-
toide (25-35% del totale).
Le forme poliarticolari sono molto simili all’artrite cronica dell’adulto, anche per quanto riguarda la localizzazione alle piccole articolazioni delle mani e dei piedi, sempre interessate dal processo infiammatorio. Ma sono coinvolte anche le grandi articolazioni, a volte insieme alla colonna cervicale, all’articolazione temporo-mandibolare e all’ar-ticolazione sterno-clavicolare (vedi Figura 31.5). Spesso si verificano alterazioni della crescita nei segmenti ossei vicini all’articolazione colpita sia nel senso di un eccesso di lunghezza sia nel senso di una riduzione dell’allungamento fisiologico. Il quadro generale della malattia è attenuato: ci possono essere lieve anemia, leucocitosi, scarsa febbre, linfoadenopatia ed epatosplenomegalia.
In seno alle forme poliarticolari, la forma fattore reu-matoide negativa rappresenta il quadro più frequente (circa il 30% di tutti i casi di artrite idiopatica giovanile). Si tratta della forma più eterogenea. Il 40% dei soggetti ha positive le prove per la ricerca degli ANA: alcuni di questi (circa il 10%) ha un’uveite cronica. L’inizio della malattia si verifica in bambini di 6-7 anni di età o più: il rapporto femmine/maschi è di 3 a 1. Il decorso della malattia è relativamente favorevole, le manifestazioni extrarticolari sono poche e solo nel 10-15% si manifesta un’evoluzione verso le classi III e IV (Tabella 31.11). Le artriti sierone-gative inoltre rispondono meglio al trattamento con gli antinfiammatori non steroidei (FANS) e il metotrexato in
FIgura 31.4 - Midollo osseo con evidente emofagocitosi macrofagica in un paziente con artrite idiopatica giovanile sistemica e sindrome da attivazione macrofagica. (Da: Ravelli A, Martini A: Juvenile idiopathic arthritis, Lancet 369:767-778, 2007.)
FIgura 31.5 - artrite idiopatica giovanile, di tipo poliarticolare, fattore reumatoide negativa. (Dalla collezione della prof.ssa F. Falcini.) L’immagine è visibile a colori sul sito www.mediquiz.it.

615CapItolo 31 Malattie reumatologiche
confronto alle artriti sieropositive. Nei pazienti che non rispondono vanno usati i farmaci biologici.
La diagnosi differenziale corre con la malattia di Lyme, con la malattia reumatica e con il LES.
I soggetti con forma sieropositiva (meno del 10% di tutte le forme di artrite idiopatica giovanile) hanno al-l’inizio un’età superiore ai 10 anni, con forte prevalenza nel sesso femminile (9:1): la maggior parte ha una forma grave, caratterizzata da un’artrite erosiva e distruttiva, già dopo un anno di malattia. La presenza di noduli (al gomito, alle ginocchia, al cuoio capelluto) è più frequente nella forma sieropositiva. Si riscontrano spesso lesioni di tipo vasculitico. Circa la metà dei pazienti è colpita da una malattia grave, senza remissioni, con una prognosi funzio-nale delle classi III e IV. La malattia si prolunga nell’età adulta, con una storia naturale simile a quella dell’artrite reumatoide iniziata nell’età adulta. La metà dei casi ha la ricerca degli ANA positiva; l’associazione più frequente è con l’HLA DR4 e DR1. Le remissioni spontanee sono rare. La terapia deve essere aggressiva.
Forme pauciarticolari
Fra il 50 e il 60% di tutti i bambini con artrite idiopatica gio-vanile ha una forma che colpisce 4 articolazioni o meno.
Le manifestazioni cliniche dell’artrite pauciarticolare sono caratteristiche del bambino nei primi 6 anni di vita: esse sono rappresentate dall’interessamento asimmetrico di una grande articolazione dell’arto inferiore (ginocchio soprattutto e poi caviglia), seguito da quelli degli arti superiori (polso, gomito, spalla) con assenza di segni e sintomi generali. Le anche non sono quasi mai interes-sate; alle mani e ai piedi il bambino può mostrare una tumefazione transitoria. Nei soggetti di sesso femminile la
frequenza è maggiore (4:1). Fra le diverse forme di artrite idiopatica giovanile, la forma pauciarticolare è quella nella quale i fattori genetici hanno un ruolo predisponente maggiore: tuttavia la possibilità che nella stessa famiglia vi siano due figli colpiti è molto bassa.
Il dolore è lieve, ma può anche mancare, per cui in gran parte dei casi la diagnosi viene posta dopo mesi di un rigonfiamento asintomatico, già quando si sono sviluppate contratture, in un bambino peraltro in ottime condizioni generali. Le lesioni articolari di rado sono di tipo distrutti-vo. È possibile riscontrare un’alterazione dell’accrescimento dei segmenti ossei corrispondenti all’articolazione colpita: il fenomeno è particolarmente grave quando l’articolazione coinvolta sia il ginocchio, perché in questo caso l’eccessivo allungamento dell’arto interessato si ripercuote sulla deam-bulazione, sull’atteggiamento della gamba e in ultima ana-lisi sulla statica del bambino (Figura 31.6). Le conseguenze cliniche dell’uveite (uni- o bilaterale) sono limitate (quando siano presenti, si hanno perdita dell’acutezza visiva, dolore, fotofobia e arrossamento dell’occhio): la diagnosi in questi casi viene posta, con la lampada a fessura, solo sulla spinta della lesione articolare. Purtroppo, quando la diagnosi di uveite cronica venga troppo ritardata, l’evoluzione verso la cecità è frequente.
La maggior parte degli esami di laboratorio è normale, come il livello di emoglobina, la velocità di sedimentazione e la conta dei globuli bianchi. Anche la ricerca del fattore reumatoide è negativa, ma oltre la metà di questi pazienti ha una prova della ricerca degli ANA positiva. La presenza di ANA in un soggetto di sesso femminile, nei primi anni di vita, con un’artrite pauciarticolare, si associa frequente-mente allo sviluppo di un’iridociclite cronica, per cui, ogni volta che ci si trovi di fronte a una bambina di questo tipo, la consulenza oculistica deve essere sempre richiesta: la mancata ricerca dell’uveite rappresenta un grave errore, per le possibilità evolutive della malattia oculare. La malattia si associa in particolare con HLA DRB1*08.
Il trattamento richiede l’impiego dei FANS e dei corti-costeroidi per via articolare. A volte è richiesto l’uso del
Criteri di classificazione dello stato funzionale nell’artrite idiopatica giovanile
Classe Criteri di giudizio*
Classe I Completamente capace di svolgere le attività normali della vita quotidiana
Classe II Capace di accudire se stesso e svolgere le attività necessarie per la vita, ma con capacità limitate per i passatempi
Classe III Capace di accudire se stesso, ma limitato nello svolgere le attività necessarie per la vita e quelle dedicate ai passatempi
Classe IV Limitate capacità di accudire se stesso e svolgere le attività necessarie per la vita e quelle dedicate ai passatempi
*Le attività normali comprendono il vestirsi, il mangiare, il lavarsi, il mettersi in ordine e l’uso del gabinetto. Le attività necessarie per la vita sono la scuola, il lavoro e il lavoro domestico. Le attività di passatempo sono quelle dedicate al tempo libero, comprese quelle ricreative.
tabella 31.11
FIgura 31.6 - artrite idiopatica giovanile, pauciarticolare, di tipo I. (Dalla collezione della prof.ssa G. Bernini.) L’immagine è visibile a colori sul sito www.mediquiz.it.

CapItolo 31 Malattie reumatologiche616
metotrexato. Nel 10% dei soggetti l’artrite ha un’evolu-zione sfavorevole. La prognosi è buona anche nelle forme persistenti, nelle quali la malattia si attenua con il tempo senza lasciare, quando trattata correttamente, alcun segno articolare importante.
In alcuni pazienti l’artrite pauciarticolare rimane limi-tata a poche articolazioni, lungo tutto il decorso della ma-lattia (oligoartrite persistente), in altri (meno della metà) si estende dopo i primi 6 mesi di malattia fino a colpire 5 o più articolazioni (oligoartrite estesa), prevedibile per una VES elevata e per l’interessamento anche del polso: fino a qualche anno fa si pensava che si trattasse di due forme distinte, mentre è molto probabile che si tratti di una stessa malattia con differente gravità e con interes-samento articolare più o meno esteso.
Uveite nell’artrite idiopatica giovanileL’uveite è un processo flogistico, inizialmente vasculitico, localiz-zato all’uvea (suddivisa in senso antero-posteriore in iride, corpo ciliare, pars plana e coroide), che può estendersi posteriormente, coinvolgendo il vitreo e la retina.Nell’80% dei casi l’uveite anteriore è associata all’artrite idio-patica giovanile: nelle forme a esordio sistemico il rischio è bassissimo, nelle forme poliarticolari è basso, mentre è alto nelle forme pauciarticolari. Se consideriamo l’artrite idiopatica giovanile nel suo complesso, l’incidenza è del 15%, ma se consideriamo le forme pauciarticolari essa sale al 40%. Nel 6% dei casi l’uveite precede la comparsa dell’artrite, ma il periodo nel quale più spesso si riscontra è nei 4 anni successivi alla diagnosi di artrite, con un picco entro i 2 anni.La presenza di uveite è spesso asintomatica; di frequente è bilaterale. Il modo migliore per diagnosticarla è quello di ricer-carla sistematicamente in tutti i bambini con artrite idiopatica giovanile, soprattuto nei primi 2 anni dalla diagnosi di artrite. Oltre il 20% dei soggetti con alterazioni della visione prima dei 18 anni ha alla base una patologia reumatologica.Il trattamento dell’uveite con gocce di steroidi locali e agenti dilatanti la pupilla è, nella maggior parte dei casi, seguito da successo.
artrite con entesite
È probabile che l’artrite con entesite rappresenti per il bambino una forma di spondiloartropatia, come sembra essere dimostrato dalla frequentissima positività dell’HLA B27. La presenza di entesite è il sintomo specifico, dal quale scaturisce la diagnosi. Come abbiamo visto le en-tesiti sono processi infiammatori a carico delle entesi, cioè nei punti di inserzione dei tendini, dei legamenti e della capsula articolare sulla superficie dell’osso: le se-de preferenziale è la superficie posteriore o inferiore del calcagno (inserzione del tendine di Achille e della fascia plantare, alle teste dei metatarsi, alla tuberosità tibiale o alla superficie inferiore della rotula). A volte si manifesta
una calcificazione dell’entesi interessata. L’artrite è limitata essenzialmente agli arti inferiori e, a differenza delle altre forme di artrite, interessa l’articolazione dell’anca (coxite). A livello delle mani e dei piedi si osserva una tumefazione a salsicciotto (dattilite), limitata spesso a un singolo dito, per la presenza di un’artrite e di una tenosinovite.
L’artrite assiale (colonna vertebrale e articolazioni sacroiliache, usualmente bilaterale) è poco frequente e tardiva nel bambino (Figura 31.7). Colpisce i bambini dai 9 ai 12 anni, con un rapporto femmine/maschi di 1 a 7. Rappresenta il 10% di tutti i casi di artrite idiopatica giovanile.
Nella maggior parte dei casi, il bambino ha una storia di una lieve diminuzione della forza nello svolgere eser-cizi, presenta stato di malessere, senso di dolenzia fino al vero e proprio dolore. Spesso si associa uno stato lieve di depressione emotiva. A volte si manifestano sintomi sistemici, accompagnati da perdita di peso, febbre, anores-sia e mialgie.
Anche in questo tipo di artrite si manifesta uveite, ma si tratta in questo caso di un’uveite acuta anteriore, di tipo non distruttivo; i sintomi acuti (eritema, fotofobia, dolore intenso, riduzione dell’acutezza visiva) sono tali da indurre la famiglia a sottoporre il bambino a una visita oculistica. A volte c’è associazione con le malattie infiammatorie dell’intestino.
L’80% dei casi è HLA B27 positivo.La cura si basa sulla somministrazione di FANS e di
corticosteroidi intrarticolari: da prendere in considerazio-ne la sulfasalazina e il metotrexato.
artrite psoriasica
Secondo la classificazione ILAR, l’artrite psoriasica vie-ne definita dalla presenza contemporanea di artrite e di psoriasi (circa il 50% dei casi). In assenza di psoriasi, la
FIgura 31.7 - grave interessamento delle articolazioni sacroiliache in un ragazzo di 13 anni con artrite idiopatica giovanile a inizio sistemico. La radiografia mostra la distruzione della testa del femore e dell’acetabolo, il restringimento dello spazio articolare e la sublussazione dell’anca. Il paziente ha ricevuto corticosteroidi sistematicamente per 9 anni. (Da: Kliegman RM, et al, editors: Pediatria di Nelson, ed 19, Milano, 2012, Elsevier.)
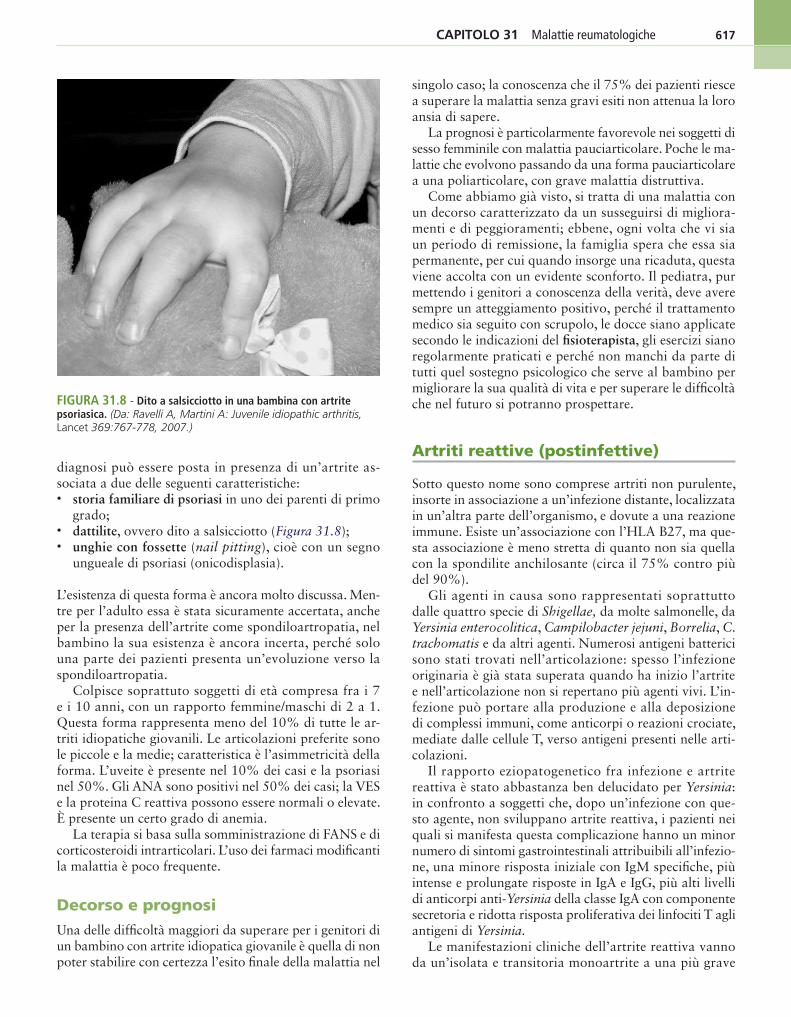
617CapItolo 31 Malattie reumatologiche
diagnosi può essere posta in presenza di un’artrite as-sociata a due delle seguenti caratteristiche:• storia familiare di psoriasi in uno dei parenti di primo
grado;• dattilite, ovvero dito a salsicciotto (Figura 31.8);• unghie con fossette (nail pitting), cioè con un segno
ungueale di psoriasi (onicodisplasia).
L’esistenza di questa forma è ancora molto discussa. Men-tre per l’adulto essa è stata sicuramente accertata, anche per la presenza dell’artrite come spondiloartropatia, nel bambino la sua esistenza è ancora incerta, perché solo una parte dei pazienti presenta un’evoluzione verso la spondiloartropatia.
Colpisce soprattuto soggetti di età compresa fra i 7 e i 10 anni, con un rapporto femmine/maschi di 2 a 1. Questa forma rappresenta meno del 10% di tutte le ar-triti idiopatiche giovanili. Le articolazioni preferite sono le piccole e la medie; caratteristica è l’asimmetricità della forma. L’uveite è presente nel 10% dei casi e la psoriasi nel 50%. Gli ANA sono positivi nel 50% dei casi; la VES e la proteina C reattiva possono essere normali o elevate. È presente un certo grado di anemia.
La terapia si basa sulla somministrazione di FANS e di corticosteroidi intrarticolari. L’uso dei farmaci modificanti la malattia è poco frequente.
Decorso e prognosiUna delle difficoltà maggiori da superare per i genitori di un bambino con artrite idiopatica giovanile è quella di non poter stabilire con certezza l’esito finale della malattia nel
singolo caso; la conoscenza che il 75% dei pazienti riesce a superare la malattia senza gravi esiti non attenua la loro ansia di sapere.
La prognosi è particolarmente favorevole nei soggetti di sesso femminile con malattia pauciarticolare. Poche le ma-lattie che evolvono passando da una forma pauciarticolare a una poliarticolare, con grave malattia distruttiva.
Come abbiamo già visto, si tratta di una malattia con un decorso caratterizzato da un susseguirsi di migliora-menti e di peggioramenti; ebbene, ogni volta che vi sia un periodo di remissione, la famiglia spera che essa sia permanente, per cui quando insorge una ricaduta, questa viene accolta con un evidente sconforto. Il pediatra, pur mettendo i genitori a conoscenza della verità, deve avere sempre un atteggiamento positivo, perché il trattamento medico sia seguito con scrupolo, le docce siano applicate secondo le indicazioni del fisioterapista, gli esercizi siano regolarmente praticati e perché non manchi da parte di tutti quel sostegno psicologico che serve al bambino per migliorare la sua qualità di vita e per superare le difficoltà che nel futuro si potranno prospettare.
Artriti reattive (postinfettive)
Sotto questo nome sono comprese artriti non purulente, insorte in associazione a un’infezione distante, localizzata in un’altra parte dell’organismo, e dovute a una reazione immune. Esiste un’associazione con l’HLA B27, ma que-sta associazione è meno stretta di quanto non sia quella con la spondilite anchilosante (circa il 75% contro più del 90%).
Gli agenti in causa sono rappresentati soprattutto dalle quattro specie di Shigellae, da molte salmonelle, da Yersinia enterocolitica, Campilobacter jejuni, Borrelia, C. trachomatis e da altri agenti. Numerosi antigeni batterici sono stati trovati nell’articolazione: spesso l’infezione originaria è già stata superata quando ha inizio l’artrite e nell’articolazione non si repertano più agenti vivi. L’in-fezione può portare alla produzione e alla deposizione di complessi immuni, come anticorpi o reazioni crociate, mediate dalle cellule T, verso antigeni presenti nelle arti-colazioni.
Il rapporto eziopatogenetico fra infezione e artrite reattiva è stato abbastanza ben delucidato per Yersinia: in confronto a soggetti che, dopo un’infezione con que-sto agente, non sviluppano artrite reattiva, i pazienti nei quali si manifesta questa complicazione hanno un minor numero di sintomi gastrointestinali attribuibili all’infezio-ne, una minore risposta iniziale con IgM specifiche, più intense e prolungate risposte in IgA e IgG, più alti livelli di anticorpi anti-Yersinia della classe IgA con componente secretoria e ridotta risposta proliferativa dei linfociti T agli antigeni di Yersinia.
Le manifestazioni cliniche dell’artrite reattiva vanno da un’isolata e transitoria monoartrite a una più grave
FIgura 31.8 - Dito a salsicciotto in una bambina con artrite psoriasica. (Da: Ravelli A, Martini A: Juvenile idiopathic arthritis, Lancet 369:767-778, 2007.)

CapItolo 31 Malattie reumatologiche618
malattia multisistemica. In molti casi dall’anamnesi risulta un’antecedente infezione, 1-4 settimane prima dell’inizio dei sintomi. I sintomi generali sono rappresentati da stan-chezza, stato di malessere, febbre e, alla lunga, perdita di peso. L’artrite è acuta, asimmetrica, con la partecipazione di nuove articolazioni nell’arco di qualche settimana. La diagnosi viene posta solo sui reperti clinici, perché non esiste alcun accertamento diagnostico, né di laboratorio né strumentale, che la possa suffragare. Si possono riscon-trare quadri di entesite (dita a salsicciotto).
I pazienti con artrite reattiva HLA B27 positivi hanno una frequenza aumentata di uveite.
Il trattamento si basa sulla somministrazione di FANS.La sindrome di Reiter (artrite, uretrite e congiuntivite) è
rara nel bambino: essa si sviluppa da 1 a 3 settimane dopo la diarrea e l’uretrite. È generalmente asimmetrica e interessa le grandi articolazioni, specialmente degli arti inferiori.
Numerosi virus sono associati con l’artrite postinfet-tiva; sono soprattuto il virus della rosolia (sia malattia, sia vaccinazione) e quello dell’epatite B che colpiscono le piccole articolazioni, mentre il virus del morbillo e quello della varicella interessano principalmente le grandi arti-colazioni, specialmente il ginocchio. La sindrome epatite B-dermatite è caratterizzata da esantema orticarioide e da una poliartrite generalizzata migratoria.
Anche il parvovirus B19 può causare artralgia, tumefa-zione articolare simmetrica e rigidità al risveglio.
Di recente è stata individuata un’artrite reattiva post-streptococcica, che fa seguito a un’infezione da streptococ-co gruppo A o gruppo G (Tabella 31.12). Poiché sono state dimostrate in qualche caso lesioni valvolari all’ecocardio-grafia, alcuni reumatologi pediatri considerano questa forma un quadro incompleto di una malattia reumatica. Alcuni HLA (DRB1*01) possono predisporre i bambini a presentare un’artrite reattiva poststreptococcica, mentre in una malattia reumatica vera e propria predomina l’-HLA DRB1*16. Il tipo di artrite è oligoarticolare: può
colpire le articolazioni degli arti inferiori, sia le grandi sia le piccole; può persistere per mesi, a differenza del-l’andamento dell’interessamento articolare della malattia reumatica, che è migrante e di breve durata. I sintomi sono lievi e tendono a ridursi progressivamente con il tempo. La risposta all’Aspirina in fase acuta è scarsa. Anche sul tipo di trattamento preventivo non c’è accordo: alcuni ritengono necessario trattare questa situazione come una vera e propria malattia reumatica, cioè con benzatina, ogni 25 giorni, almeno per 5 anni, mentre altri consigliano di seguire attentamente il bambino, senza iniziare alcun tipo di trattamento preventivo.
La sinovite transitoria dell’anca, la più frequente delle artriti reattive del bambino, rientra in questo gruppo di affezioni: essa insorge dopo un’infezione delle vie aeree superiori. È quasi sempre monolaterale: la sede più spesso interessata è la destra. Sono in generale colpiti bambini fra i 3 e i 10 anni di età, con prevalenza del sesso maschile: la VES e il numero dei globuli bianchi sono in genere normali. Un esame ECO dimostra un ingrandimento dello spazio articolare, per presenza di liquido al suo interno, che persiste al massimo per 2 settimane: una durata su-periore rende necessaria una rivalutazione completa del paziente. A volte è necessario procedere all’aspirazione del liquido articolare nel sospetto di un’artrite settica. Nel 4-17% dei casi la sinovite transitoria dell’anca può ripresentarsi entro 6-12 mesi dalla prima comparsa.
Un’artrite non purulenta è stata riscontrata, soprattutto negli adolescenti, in occassione di un’acne grave del dorso.
Spondiloartrite anchilosante e altre spondiloartriti
Si tratta di una malattia dell’adolescente e dell’adulto, che interessa soprattutto il sesso maschile ed è carat-terizzata inizialmente dalla rigidità e dal dolore in sede
Caratteristiche cliniche dell’artrite reattiva poststreptococcica (ARPS), della malattia reumatica (MR) e dell’artrite idiopatica giovanile (AIG)
Caratteristiche anamnestiche e cliniche aRpS MR aIG
Infezione antecedente da streptococco gruppo A Sì Sì NoPeriodo di latenza fra l’infezione e l’inizio dell’artrite <2 settimane 3 settimane –Artrite:• simmetrica• migrante• interessantelepiccolearticolazioni• interessantelacolonnavertebrale
SìNoSìSì
NoSìRaramenteNo
Sì/NoNoSìSì
Risposta all’aspirina o ad altri FANS Scarsa e lenta Pronta LentaDurata della sintomatologia Protratta Breve ProtrattaInteressamento cardiaco:• pericardite• miocardio/valvulite
Rara6%
Rara50%
SìNo
(Da: Ahamed S, Ayoub EM: Post streptococcal reactive arthritis, Pediatr Infect Dis J 20:1081-1082, 2001; modificata.)
tabella 31.12

619CapItolo 31 Malattie reumatologiche
dorso-lombare, per l’interessamento delle articolazioni sacroiliache. La malattia può successivamente progredire fino al coinvolgimento di tutta la colonna, compresa quel-la cervicale. Circa la metà dei pazienti ha interessamento anche delle articolazioni periferiche e spesso vi sono i segni dell’entesopatia.
C’è una fortissima associazione con l’HLA B27.
Malattia reumatica
Fino agli anni Sessanta, la malattia reumatica (termine da preferire al vecchio nome, reumatismo articolare acuto) ha rappresentato, soprattutto per le sue conseguenze a distanza, uno dei più importanti problemi pediatrici. Ma dal 1970 a qualche anno fa, sia in Europa sia negli USA, l’incidenza della malattia è improvvisamente diminuita a livelli più che 10 volte inferiori a quelli precedenti (0,3- 2 casi/100.000/anno). Negli ultimi anni, anche in alcune zone d’Italia, la malattia reumatica si è fatta più frequente (vedi Capitolo 44).
Lupus eritematoso sistemico giovanile
Il lupus eritematoso sistemico (LES) giovanile è una malattia autoimmune che insorge entro il 16° anno di vita ed è caratterizzata da un processo infiammatorio a carico di numerosi organi e apparati: cute, rene, sistema nervoso centrale, midollo, articolazioni, apparato gastrointestinale e altri.
Il LES entra in diagnosi differenziale con numerosi quadri patologici dell’età evolutiva. L’inizio può essere acuto, con l’interessamento di più organi e apparati, o può essere insidioso, con manifestazioni intermittenti, come febbre, stato di malessere, esantemi, artrite e pleurite. Il sospetto di LES deve sorgere di fronte a un bambino che abbia la ricerca degli ANA positiva: tuttavia, anche se è vero che tutti i LES hanno questa prova positiva, non è vero che la sola positività di questa prova sia sufficiente per stabilire una diagnosi di LES.
Nei criteri di classificazione del LES sono state incluse numerose manifestazioni di malattia (Tabella 31.13). An-che se lo scopo di questa classificazione non è quello di facilitare la diagnosi di LES, tuttavia essa si è dimostrata utile nell’iter diagnostico, sia nel LES dei bambini sia in quello degli adulti.
epidemiologia
La malattia non è rara nel bambino: i bambini e i giova-ni al di sotto dei 20 anni rappresentano circa il 12% di tutti i casi. La prevalenza nei bambini e negli adolescenti è di 1-6 casi/100.000 e negli adulti di 20-70/100.000.
L’incidenza annuale viene calcolata in 0,6 casi/100.000 soggetti. I soggetti di sesso femminile sono colpiti 5 volte più spesso dei soggetti di sesso maschile: prima della pu-bertà questo rapporto è un po’ più basso, cioè di 3 a 1. Il LES in età evolutiva è più frequente in età adolescenziale ed è visto raramente prima dei 5 anni.
eziopatogenesi
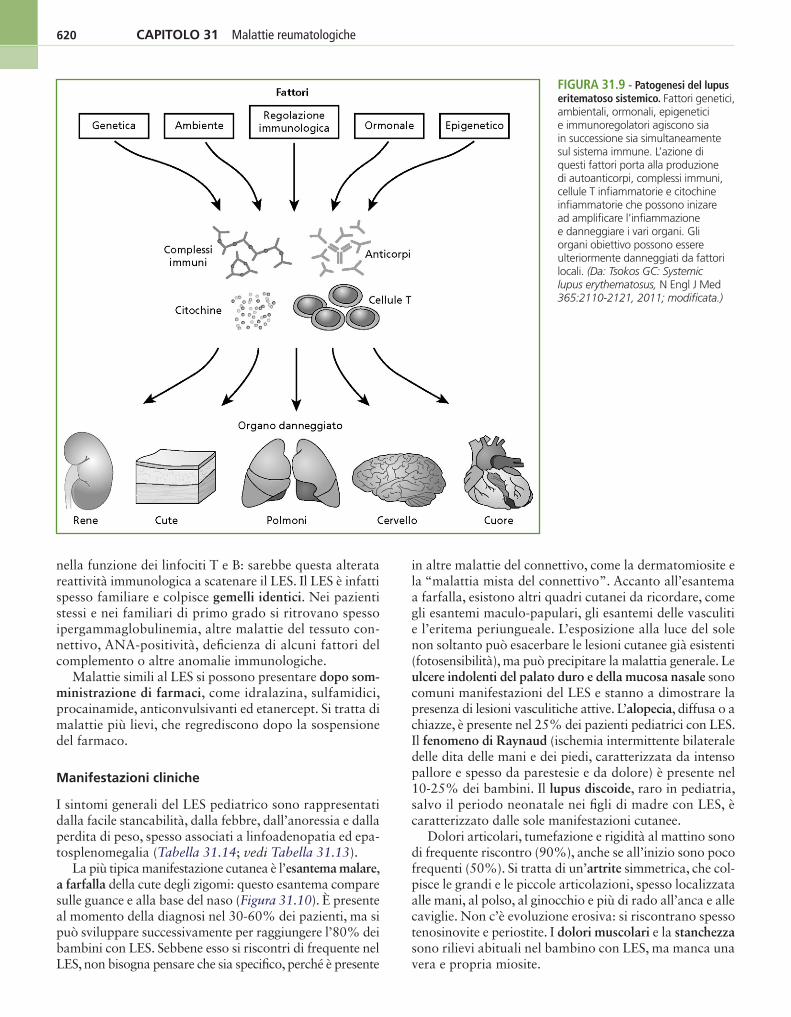
Con grande probabilità l’eziologia del LES è multifattoriale: la genetica, l’ambiente e la risposta immunologica interagi-scono fra loro. La produzione di autoanticorpi è l’elemento principe della malattia; l’attivazione del complemento, mediata dai complessi immuni, è essenziale per produrre il danno renale e quello vascolare (Figura 31.9).
L’inizio del quadro è spesso associato a infezioni inter-correnti, che possono essere facilitate da un’aumentata suscettibilità di questi bambini, che presentano alterazioni
Segni e sintomi di presentazione del LES
Manifestazioni generali Stanchezza, anoressia, perdita di peso, febbre di lunga durata, linfoadenopatia
Manifestazioni muscoloscheletriche
Artrite, miosite, tendinite, artralgie, mialgie, necrosi avascolare, osteoporosi
Cute Esantema malare (a farfalla degli zigomi, con tendenza a diffondere alle pieghe naso-labiali), esantema discoide, esantema come reazione insolita alla luce del sole, vasculite cutanea, livedo reticularis, alterazioni dei capillari subungueali, fenomeno di Raynaud, alopecia, ulcere orali e nasali
rene Ipertensione, proteinuria, ematuria, edema, sindrome nefrosica, insufficienza renale
apparato cardiovascolare
Pericardite, miocardite, alterazioni della conduzione, endocardite di Libman-Sacks
Encefalo Convulsioni, psicosi, cerebrite, stroke, mielite trasversa, depressione, alterazioni cognitive, cefalea, pseudotumor, neuropatia periferica, corea, neurite ottica, paralisi dei nervi cranici
apparato polmonare Pleurite, malattia interstiziale, emorragie polmonari, ipertensione polmonare, embolia polmonare
apparato emopoietico
Citopenia immunomediata (anemia emolitica, trombocitopenia, leucopenia), anemia dell’infiammazione cronica, ipercoagulabilità, microangiopatia trombotica trombocitopenica
apparato gastrointestinale
Epatosplenomegalia, pancreatite, vasculite intestinale, enteropatia proteino-disperdente
tabella 31.13
CapItolo 31 Malattie reumatologiche620
nella funzione dei linfociti T e B: sarebbe questa alterata reattività immunologica a scatenare il LES. Il LES è infatti spesso familiare e colpisce gemelli identici. Nei pazienti stessi e nei familiari di primo grado si ritrovano spesso ipergammaglobulinemia, altre malattie del tessuto con-nettivo, ANA-positività, deficienza di alcuni fattori del complemento o altre anomalie immunologiche.
Malattie simili al LES si possono presentare dopo som-ministrazione di farmaci, come idralazina, sulfamidici, procainamide, anticonvulsivanti ed etanercept. Si tratta di malattie più lievi, che regrediscono dopo la sospensione del farmaco.
Manifestazioni cliniche
I sintomi generali del LES pediatrico sono rappresentati dalla facile stancabilità, dalla febbre, dall’anoressia e dalla perdita di peso, spesso associati a linfoadenopatia ed epa-tosplenomegalia (Tabella 31.14; vedi Tabella 31.13).
La più tipica manifestazione cutanea è l’esantema malare, a farfalla della cute degli zigomi: questo esantema compare sulle guance e alla base del naso (Figura 31.10). È presente al momento della diagnosi nel 30-60% dei pazienti, ma si può sviluppare successivamente per raggiungere l’80% dei bambini con LES. Sebbene esso si riscontri di frequente nel LES, non bisogna pensare che sia specifico, perché è presente
in altre malattie del connettivo, come la dermatomiosite e la “malattia mista del connettivo”. Accanto all’esantema a farfalla, esistono altri quadri cutanei da ricordare, come gli esantemi maculo-papulari, gli esantemi delle vasculiti e l’eritema periungueale. L’esposizione alla luce del sole non soltanto può esacerbare le lesioni cutanee già esistenti (fotosensibilità), ma può precipitare la malattia generale. Le ulcere indolenti del palato duro e della mucosa nasale sono comuni manifestazioni del LES e stanno a dimostrare la presenza di lesioni vasculitiche attive. L’alopecia, diffusa o a chiazze, è presente nel 25% dei pazienti pediatrici con LES. Il fenomeno di Raynaud (ischemia intermittente bilaterale delle dita delle mani e dei piedi, caratterizzata da intenso pallore e spesso da parestesie e da dolore) è presente nel 10-25% dei bambini. Il lupus discoide, raro in pediatria, salvo il periodo neonatale nei figli di madre con LES, è caratterizzato dalle sole manifestazioni cutanee.
Dolori articolari, tumefazione e rigidità al mattino sono di frequente riscontro (90%), anche se all’inizio sono poco frequenti (50%). Si tratta di un’artrite simmetrica, che col-pisce le grandi e le piccole articolazioni, spesso localizzata alle mani, al polso, al ginocchio e più di rado all’anca e alle caviglie. Non c’è evoluzione erosiva: si riscontrano spesso tenosinovite e periostite. I dolori muscolari e la stanchezza sono rilievi abituali nel bambino con LES, ma manca una vera e propria miosite.
FIgura 31.9 - Patogenesi del lupus eritematoso sistemico. Fattori genetici, ambientali, ormonali, epigenetici e immunoregolatori agiscono sia in successione sia simultaneamente sul sistema immune. L’azione di questi fattori porta alla produzione di autoanticorpi, complessi immuni, cellule T infiammatorie e citochine infiammatorie che possono inizare ad amplificare l’infiammazione e danneggiare i vari organi. Gli organi obiettivo possono essere ulteriormente danneggiati da fattori locali. (Da: Tsokos GC: Systemic lupus erythematosus, N Engl J Med 365:2110-2121, 2011; modificata.)

621CapItolo 31 Malattie reumatologiche
In circa il 15% dei bambini con LES si manifesta una necrosi asettica, avascolare, a carico della testa del femore, della spalla, del gomito o del polso, soprattutto in bam-bini che abbiano seguito un trattamento prolungato con corticosteroidi.
A carico del sistema nervoso centrale, interessato nel 15-45% dei bambini con LES e secondo, come localizzazio-ne, soltanto al rene, si possono manifestare sintomi e segni già prima che ne compaiano altri, di tipo sia neurologico sia neuropsichiatrico. Le più comuni manifestazioni compren-dono le convulsioni (10-20% dei casi), la cefalea, le psicosi funzionali, la meningite asettica, la corea, la paralisi dei nervi cranici e la neuropatia periferica (vedi Tabella 31.14).
L’interessamento renale è molto frequente: si può pre-sentare con proteinuria o con ematuria o con ambedue; vi
possono essere ipertensione e, sindrome nefrosica, fino all’in-sufficienza renale cronica. La classificazione internazionale prevede la suddivisione delle lesioni anatomopatologiche renali in 5 classi (Tabella 31.15). La biopsia renale è sempre indicata in tutti i bambini con LES affetti da patologia renale, perché essa permette un’esatta valutazione della situazione renale e guida il tipo e l’intensità del trattamento.
I reperti ematologici vanno dall’anemia alla trom-bocitopenia, alla leucopenia fino alle alterazioni della coagulazione. In un numero ridotto di pazienti si può sviluppare un’anemia emolitica, Coombs positiva. A ca-rico del polmone, l’evento più comune è la pleurite, ma si possono presentare infezioni con tendenza alla cronicità, emorragie, embolie e trombosi.
La più frequente delle manifestazioni cardiache è la pericardite e, più di rado, la miocardite e l’arteriosclerosi prematura.
esami di laboratorio
La prova più utile per la diagnosi di LES è la ricerca degli ANA, anche se la positività di questa prova non è diagnosti-ca per il LES, perché può risultare positiva anche nel 2-4% della popolazione normale. Per l’importanza della ricerca di altri anticorpi vedi Tabella 31.16. Gli autoanticorpi sono tipicamente presenti alcuni anni prima della diagnosi di LES. Il livello del C3 e del C4 è diminuito quando sia in atto un interessamento renale. La VES è elevata; spesso vi è un aumento delle gammaglobuline. È sempre utile, una volta stabilita la diagnosi di LES, esplorare l’attività dei vari organi, per scoprire un eventuale loro interessamento.
In corso di LES, è possibile riscontrare positività per gli anticorpi antifosfolipidi.
Decorso e prognosi
Il decorso del LES, come quello della maggioranza delle malattie legate alla presenza di autoanticorpi, è caratteriz-zato da un susseguirsi di miglioramenti e di peggioramenti. In linea di massima nuovi organi non sono interessati prima di 1-2 anni di malattia, con eccezione del sistema nervoso centrale. La malattia, fatale nel passato, ora permette una sopravvivenza del 95% a 5 anni e dell’85% a 10 anni. Mentre molti anni fa l’insufficienza renale era la causa più frequente di morte, oggi a esserlo sono le infezioni.
Manifestazioni cliniche del LES in età pediatrica
Sintomi e segni percentuale sul totale dei pazienti
Malessere, perdita di peso, ritardo di crescita
96
Alterazioni cutanee 96Alterazioni ematologiche 91Febbre 84Nefrite 84Alterazioni muscoloscheletriche 82Affezioni pleuropolmonari 67Epatosplenomegalia, linfoadenopatia 58Malattia neurologica 49Alterazioni cardiache 38Ipertensione 33Alterazioni oculari 31Sintomi gastrointestinali 27Fenomeno di Raynaud 13
(Da: Lehman TJ, et al: Serum complement abnormalities in the antinuclear antibody-positive relatives of children with systemic lupus erythematosus, Arthritis Rheum 22:954-958, 1979; modificata.)
tabella 31.14
FIgura 31.10 - Esantema malare, a farfalla degli zigomi. (Dalla collezione della prof.ssa F. Falcini.) L’immagine è visibile a colori sul sito www.mediquiz.it.
Classificazione delle lesioni patologiche renali (OMS)
Classi tipo di lesione
Classe I Reperti normaliClasse II Nefrite mesangialeClasse III Nefrite proliferativa focaleClasse IV Nefrite proliferativa diffusaClasse V Nefrite membranosa
tabella 31.15

CapItolo 31 Malattie reumatologiche622
trattamento
Il trattamento di ogni bambino con LES è dettato dal-l’estensione e dalla gravità della sua malattia, soprattutto per quanto riguarda l’interessamento renale.
Se è presente solo il coinvolgimento della cute e delle articolazioni, sono sufficienti i FANS (farmaci antinfiam-matori non steroidei, come tolmetina o naproxene) da soli o in associazione all’idrossiclorochina (5-7 mg/kg/die). Per le lesioni cutanee possono essere utili gli steroidi per applicazione locale, insieme a fattori di protezione dalla luce solare.
Se si associa una lesione di un organo principale, l’impie-go dei corticosteroidi diviene essenziale: la dose piena inizia-le di 2 mg/kg/die di prednisone per 3-4 settimane è seguita da una graduale, lenta diminuzione. L’impiego di farmaci risparmiatori di steroidi nella cura del LES è ormai diffuso: metotrexato, azatioprina. Nelle forme più gravi viene usata anche la ciclofosfamide per bocca e per via venosa. Per la terapia di mantenimento del LES, il micofenolato mofetile si è dimostrato superiore all’azatioprina nel mantenere una risposta renale al trattamento e nel prevenire le ricadute in pazienti che avevano risposto alla terapia induttiva.
A questi farmaci di base vanno associati farmaci spe-cifici, per risolvere alcune situazioni come le convulsioni, l’ipertensione, le psicosi, il fenomeno di Raynaud o le trombosi. Il paziente e la famiglia vanno coinvolti nel decidere la cura e nell’attuarla.
In una larga esperienza, randomizzata e placebo-control-lata, il belimumab, un anticorpo monoclonale G1-l immu-noglobulina, completamente umana, si è dimostrato efficace e sicuro nel trattamento di pazienti con LES attivo.
Un trattamento con rituximab, un anticorpo mono-clonale anti-CD20 ad azione anti-cellule B, si è dimostrato poco efficace, come coterapia: il suo uso, d’altra parte, si accompagna a una frequenza elevata di eventi avversi, che possono raggiungere anche il 45% dei trattati. Sono in studio altri agenti biologici.
Molto indicata è anche la somministrazione di calcio e vitamina D per limitare l’osteoporosi.
LES neonataleLa somministrazione di contracettivi per bocca non au-menta il rischio di peggioramento in donne con LES, nelle quali la malattia sia stabilizzata.
Si tratta di una sindrome che insorge nei figli di madri che abbiano anticorpi anti-Ro (SSA) e anti-La (SSB): è caratterizzata da lesioni cutanee fotosensibili e da blocco cardiaco congenito. L’origine del blocco cardiaco risie-de nel fatto che gli antigeni Ro e La sono presenti sulla superficie delle cellule cardiache, in prossimità del nodo atrioventricolare, in modo tale da essere accessibili agli autoanticorpi materni. I legame antigene-anticorpo induce una risposta immune infiammatoria con fibrosi del sistema di conduzione.
Le madri possono avere un LES completo o un’altra malattia del connettivo, anche se il 50-60% di esse è com-pletamente asintomatico. In seguito al passaggio degli anticorpi attraverso la placenta, i neonati hanno la ricerca degli ANA, degli anti-Ro e degli anti-La positive. Questi anticorpi persistono per 3-6 mesi dalla nascita.
L’esantema tipico del LES neonatale è di tipo anulare, eritematoso, fotosensibile; esso inizia ore o giorni dal parto. A volte la comparsa dell’esantema ritarda fino alla prima esposizione alla luce del sole. Nel 50% dei casi di LES neonatale è presente un blocco congenito completo di cuore, di tipo permanente. Il blocco può già dare segni di sé in utero o può manifestarsi alla nascita con insufficienza cardiaca e idrope fetale o neonatale. Il blocco cardiaco può accompagnarsi a un 10-15% di letalità precoce e può richiedere precocemente l’inserzione di un pacemaker. Altre manifestazioni possono essere la trombocitopenia, la leucopenia, l’anemia emolitica e più di rado l’epatite neonatale.
Le donne che in gravidanza hanno anticorpi anti-Ro e anti-La devono ricevere immunoglobuline e corticosteroidi per prevenire o limitare le alterazioni della conduzione cardiaca fetale. Nei bambini con blocco di cuore è indicato alla nascita l’uso del pacemaker e, nei casi gravi, addirittura il trapianto di cuore.
Autoanticorpi, comunemente associati con il LES
anticorpo associazione clinica
Anti-DNA a doppia elica Correlato con l’attività della malattia, specialmente nefrite, in alcuni casi di LESAnti-Smith Specifico per la diagnosi di LESAntiribonucleoproteina Aumenta il rischio di fenomeno di Raynaud e di ipertensione polmonare
Alti titoli possono suggerire la diagnosi di disordine misto del tessuto connettivoAnti-Ro (anticorpo anti-SSA)Anti-La (anticorpo anti-SSB)
Associato con la sindrome sicca; può suggerire la diagnosi di malattia di SjögrenAumenta il rischio di LES neonatale (blocco congenito del cuore)Può essere associato con manifestazioni cutanee e polmonari del LESPuò essere associato a LES discoide isolato
Antifosfolipidi (inclusi anticorpi anticardiolipina) Aumenta il rischio di episodi trombotici arteriosiAnti-istone Presente nella maggioranza dei pazienti con lupus indotto dai farmaci
Può essere presente nel LES
tabella 31.16

623CapItolo 31 Malattie reumatologiche
Poiché gli anticorpi cominciano a passare attraverso la placenta alla 16a settimana di gestazione, il feto va monito-rato dal pediatra cardiologo con l’elettrocardiografia fino al momento del parto. Se si riscontra una bradicardia in utero, vanno ricercati gli anticorpi anti-Ro e anti-La.
Una madre con LES che ha avuto un primo figlio con blocco cardiaco ha il 15% di probabilità che un blocco si possa verificare anche al secondo.
Sindrome antifosfolipidi
La sindrome antifosfolipidi è una situazione di trombo-filia acquisita, mediata da anticorpi, e caratterizzata da trombosi ricorrenti, venose e arteriose; gli anticorpi sono diretti contro le proteine circolanti, fra i cui componenti vi siano anche i fosfolipidi. Gli anticorpi sono diretti princi-palmente contro un’apolipoproteina, conosciuta come b2- glicoproteina I, e contro la protrombina. Un altro gruppo di anticorpi, chiamato lupus anticoagulante, prolunga il tempo di coagulazione in vitro.
Alla fine del 2007 erano stati registrati nel mondo 121 casi (65 femmine e 56 maschi) di sindrome antifosfolipidi in età inferiore ai 18 anni: circa il 50% di essi aveva una malattia autoimmune sottostante.
La sindrome antifosfolipidi può essere primitiva o secondaria ad altre malattie autoimmuni, come il LES, nel quale un terzo dei pazienti possiede questi anticorpi, mentre nelle altre malattie autoimmuni la loro presenza va dal 6 al 15%. Questi anticorpi possono essere associati con una recente infezione virale e non rappresentano un fattore di rischio per la trombosi.
Anticorpi antifosfolipidi, leganti le proteine del pla-sma, si ritrovano nell’1-5% della popolazione; la loro prevalenza aumenta con l’età. Circa un terzo dei pazienti con anticorpi antifosfolipidi manifesta effetti trombotici o complicazioni della gravidanza.
Il quadro clinico è la conseguenza, diretta o indiretta, della trombosi venosa o arteriosa: trombosi delle vene cerebrali, della retina, del polmone; la livedo reticularis insorge per trombosi dei capillari e delle venule della cute. La sindrome antifosfolipidi va sospettata quando si presenti una manifestazione vascolare cerebrale prima dei 55 anni di età o in gravidanza, in presenza di livedo reticularis o di trombocitopenia.
I pazienti con questa sindrome vanno sottoposti a un trattamento continuativo con warfarin (dicumarolo).
Dermatomiosite giovanile
La dermatomiosite è una malattia multisistemica che inte-ressa prevalentemente la muscolatura striata (stanchezza progressiva dei muscoli prossimali) e la cute (eritema elio-tropo, di colorito violaceo, a carico delle palpebre superiori, della radice del naso e dei solchi fra naso e guance).
La dermatomiosite giovanile colpisce i soggetti in età compresa fra i 4 e i 10 anni; essa si differenzia dalla forma dell’adulto, che si associa a tumori, soprattutto a carico dell’esofago, e che colpisce soggetti in età fra 45 e 64 anni. Nei bambini l’associazione con i neoplasmi è as-solutamente sconosciuta. La malattia si differenzia inoltre dalla polimiosite sotto molti aspetti, per la mancanza in quest’ultima dell’esantema caratteristico e per la frequenza nettamente superiore (un rapporto di 10-20 volte supe-riore); il rapporto femmine/maschi è di 2 a 1. L’incidenza è di 3 casi su 1.000.000 di soggetti.
eziologia e patogenesi
L’eziologia è sconosciuta; tuttavia la dermatomiosite viene fatta rientrare fra le malattie autoimmuni. I linfociti dei pazienti con questa malattia liberano infatti linfotossine che ledono le cellule muscolari in vitro; nelle biopsie dei soggetti affetti, inoltre, si ritrovano immunoglobuline e depositi di complemento a carico dei vasi del muscolo. Molti virus sono stati oggetto di discussione da parte di diversi studiosi, come agenti “grilletto” per la malattia: viene spesso riportata una storia di infezioni nei 3 mesi precedenti. La malattia è associata all’HLA B8/DR3; ne-gli USA si associa a HLA DRB1*0301 e DQA1*0501. È più rara dell’artrite idiopatica giovanile, del LES e della porpora di Schönlein-Henoch.
L’interferon tipo 1 sembra sia importante nella pato-genesi della dermatomiosite: esso attiva i geni dell’immu-noregolazione e soprattutto la maturazione delle cellule natural killer e delle cellule dendritiche.
Manifestazioni cliniche
L’inizio, nella maggioranza dei casi, è insidioso ed è carat-terizzato da lesioni cutanee, dalla comparsa di una pro-gressiva stanchezza, da irritabilità e da perdita di peso.
Le lesioni cutanee precedono o seguono le manifestazio-ni a carico dei muscoli. Nel 50% dei casi costituiscono il primo sintomo; solo nel 25% dei casi si associano inizial-mente a sofferenza muscolare. Si notano eritema ed edema alle palpebre, unito da una parte all’altra attraverso la radice del naso ed estendentesi al solco fra naso e guance; l’eritema è di colorito violaceo, favorito dall’esposizione al sole (Figura 31.11). È evidente una somiglianza con l’esan-tema a farfalla del LES (vedi Figura 31.10). L’eritema può essere esteso alla superficie estensoria degli arti superiori e inferiori e alle dita. In corrispondenza della cute che copre i metacarpi e le articolazioni interfalangee prossimali si possono notare delle papule ipertrofiche di color rosso pallido (papule di Gottron). È possibile ritrovare zone di cute sottile e atrofica in corrispondenza delle articolazioni. Manifestazioni evidenti di vasculite (teleangectasie, piccoli infarti dell’epitelio della bocca, ulcerazioni delle estremità delle dita) si ritrovano nelle forme più gravi. È ben visibile una chiara fotosensibilità. La comparsa di calcificazioni

CapItolo 31 Malattie reumatologiche624
in seno al tessuto sottocutaneo e ai muscoli è anch’essa caratteristica delle forme più gravi e di più lunga durata: si riscontra nel 20-30% dei casi. Il fenomeno di Raynaud è presente nel 10-20% dei pazienti.
La presenza dell’infiammazione dei piccoli vasi è spesso visibile alle unghie e alle gengive: le anse capillari sono ispessite, tortuose o assenti.
L’altro gruppo di sintomi e di segni deriva dall’interes-samento della muscolatura striata della parte prossimale degli arti. La sofferenza muscolare è documentata dalla stan-chezza nel salire le scale, nell’alzarsi da una sedia o dall’uso degli arti superiori per mantenere la stazione eretta o per avanzare; il segno di Gower è positivo (uso delle mani sulle gambe per alzarsi). A volte la pressione sui corpi muscolari determina dolore; il bambino tende a tenere le articolazioni in posizione di semiflessione (posizione acamatica), fino a manifestare contratture in flessione degli arti. Alla conse-guente immobilità fa seguito la comparsa di osteoporosi.
Alla sofferenza cutanea e muscolare si associano spes-so sintomi a carico dell’apparato gastrointestinale. Un certo grado di difficoltà alla deglutizione e la presenza di reflussi gastroesofagei possono derivare dalla diminuita motilità esofagea. I fenomeni vasculitici possono insorgere anche a carico dell’intestino, fino all’ulcerazione e alla perforazione.
Si riscontra spesso un interessamento del sistema car-diorespiratorio, manifestato da alterazioni aspecifiche del-l’ECG, come alterazioni asintomatiche della conduzione, e da diminuizione della capacità respiratoria, evidenziata in oltre i tre quarti dei pazienti.
esami di laboratorio
In corso di flogosi del muscolo e quindi anche nella der-matomiosite, gli enzimi muscolari presentano nel siero un’aumentata attività (creatin-chinasi, aminotransferasi, aldolasi e lattico-deidrogenasi) (Tabella 31.17); tuttavia
questa associazione non è sempre presente. Si riscontra anemia per la presenza di una malattia cronica. Gli anti-corpi antinucleo si ritrovano positivi in più dell’80% dei casi: inizialmente sono ad alto titolo. L’elettromiografia mostra spesso tracciati patologici, con alterazioni di tipo miopatico e neuropatico.
La RM può localizzare nel muscolo le sedi della malattia attiva per l’esecuzione di un’eventuale biopsia muscolare.
Diagnosi
Nelle forme tipiche, la diagnosi non presenta per il pedia-tra alcuna difficoltà. Nelle forme che si allontanino dal quadro classico, la diagnosi può presentare qualche pro-blema per la differenziazione con altre forme di malattia reumatologica o con altre malattie muscolari, fra le quali principalmente la polimiosite (Tabella 31.18). Esistono alcune forme amiopatiche, nelle quali mancano i segni muscolari, come la stanchezza e la dolenzia.
trattamento e prognosi
Gli steroidi sono il trattamento di scelta (Deltacortene, compresse da 5 e da 25 mg, alla dose di 1-2 mg/kg/die per 2 settimane; successivamente, scendere alla dose minima per ottenere il controllo clinico della malattia). Per i pazienti che non rispondono agli steroidi sono stati usati il metotrexato (0,5-1 mg/kg o 15-20 mg/m2), la ciclosporina, l’azatioprina o l’idrossichinolina. Qualche successo è stato ottenuto anche con le immunoglobuline per via endovenosa. Occupational therapy e fisioterapia
FIgura 31.11 - Dermatomiosite: eritema alle palpebre di colore violaceo. (Dalla collezione della prof.ssa F. Falcini.) L’immagine è visibile a colori sul sito www.mediquiz.it.
Reperti di laboratorio nella dermatomiosite giovanile
tipo di esame Risultato in corso di dermatomiosite giovanile
Anticorpi antinucleo Positivo di tipo punteggiato• anti-DNAadoppiaelica Negativo• anti-Sm Negativo• antiribonucleoproteine NegativoAnticorpi antimiosite (MSAs) Raramente positivoAnticorpi anti-Jo-1 e anti-Mi-2 PositivoAnti-SSA-Ro e anti-SSA-La NegativoAntigene relativo al fattore
di von Willebrand, formato dalle cellule endoteliali
Elevato
Creatin-chinasi ElevatoAspartato-aminotransferasi ElevatoLattico-deidrogenasi ElevatoAldolasi ElevatoVES Normale o elevatoConta dei GB Normale o elevatoEmoglobina Normale o diminuitoProva con il D-xilosio Alterato
tabella 31.17

625CapItolo 31 Malattie reumatologiche
sono essenziali per il mantenimento di una sufficiente funzione muscolare.
Va evitata l’esposizione al sole. Utile l’uso della vita-mina D e del calcio.
Con un trattamento precoce e ben condotto la prognosi nei bambini viene considerata buona. Una volta moriva circa un terzo dei bambini con dermatomiosite e un altro terzo aveva degli esiti invalidanti: oggi la letalità è intorno all’1%.
Sclerodermia
La sclerodermia è una malattia caratterizzata da un’altera-zione fibrotica cronica del tessuto connettivo, che interessa principalmente la cute, ma anche l’apparato gastrointesti-nale, il cuore, i polmoni, i reni e la sinovia. Si tratta di una malattia rara nel bambino. Se ne conoscono due forme:• sclerosi sistemica (scleroderma): di tipo diffuso o limita-
to, spesso associata ad altre malattie reumatologiche;• scleroderma localizzato: morfea e scleroderma lineare
(colpo di sciabola) con interessamento fibroso limitato alla cute (Figura 31.12).
Il fenomeno di Raynaud si associa spesso (70%) alla scle-rodermia, qualche mese dopo la comparsa delle prime manifestazioni.
Fra le manifestazioni viscerali della sclerodermia siste-mica vanno ricordate l’ipertensione dell’arteria polmonare e la dismotilità intestinale (presente nel 25% dei bambini), caratterizzata da disfagia, reflusso e dispepsia.
I pazienti con sclerosi sistemica sono positivi per au-toanticorpi antinucleo (ANA) autoanticorpi SSc, antifi-brillarina e anti-RNA polimerasi I e III. Nella morfea sono utilizzati i corticosteroidi topici e la terapia ultravioletta. Per le lesioni generalizzate sono utili le associazioni di corticosteroidi e di metotrexato.
Mentre la prognosi della forma localizzata è favorevole, quella sistemica ha una prognosi variabile, con sopravvivenze a 5, 10 e 15 anni, rispettivamente, dell’89, 80-87 e 74-87%.
Fenomeno di RaynaudIl fenomeno di Raynaud indica un cambiamento di colore della parte distale delle estremità, in risposta al freddo o allo stress. Il fenomeno può essere primitivo o può essere secondario a una malattia sottostante come la sclerodermia e il LES. È rela-tivamente raro nei bambini: colpisce più il sesso femminile ed è prevalente nella sua forma primitiva. Nelle forme secondarie si ritrova positiva la ricerca degli anticorpi antinucleo, mentre i capillari del letto unguale si presentano anormali. Gli anticorpi antifosfolipidi sono spesso positivi.
Malattia di Behçet
La malattia di Behçet è un’affezione autoimunitaria che interessa molti sistemi, descritta originariamente come costituita da ulcere aftose ricorrenti della bocca, da ulcere genitali, da uveite e da lesioni cutanee ricorrenti. Frequente
Criteri diagnostici nella dermatomiosite giovanile*
Esantema classico Esantema eliotropo delle palpebrePapule di Grotton
più tre dei seguenti segniStanchezza Simmetrica
ProssimaleElevazione degli enzimi
muscolari (≥1)Creatin-chinasiAspartato-aminotransferasiLattico-deidrogenasiAldolasi
Modificazioni elettromiografiche
MiopatiaDenervazione
Biopsia muscolare NecrosiInfiammazione
*Per la diagnosi sono necessari la presenza dell’esantema caratteristico e tre degli altri quattro sintomi e segni.
tabella 31.18
FIgura 31.12 - Scleroderma lineare a colpo di sciabola alla fronte: area di indurimento, ipopigmentazione e depressione. (Da: Vallongo C, Martoini G, Zulian F: Quando la pelle si fa dura: la sclerodermia localizzata, Area Pediatr 9:6-12, gennaio 2008.) L’immagine è visibile a colori sul sito www.mediquiz.it.

CapItolo 31 Malattie reumatologiche626
l’associazione ad artrite cronica, segni neurologici, va-scolari e gastrointestinali. Tutte le manifestazioni sono transitorie, salvo le lesioni oculari che, recidivando, pos-sono portare alla cecità.
La malattia è relativamente rara: in Europa ha una prevalenza da 0,6 a 6,4/100.000. La malattia è ancora più rara nei bambini, che rappresentano il 5% del totale dei casi. L’incidenza della malattia è elevata nei Paesi orientali e in Turchia (80-370 casi/100.000). L’età media d’inizio è a 7 anni e mezzo. Il rapporto femmine/maschi va da 1,2:1 a 1,4:1. La suscettibilità alla malattia è legata all’HLA B51.
Il quadro clinico è dominato dalla presenza saltuaria di piccole ulcere, dolorose, superficiali, di 2-10 mm di diame-tro, localizzate sulla mucosa buccale, sulla gengiva, sulle labbra e sulla lingua. Esse possono perdurare da pochi giorni a settimane; appaiono singolarmente o a gruppi. Le lesioni oculari sono rappresentate da uveite anteriore o posteriore. L’artrite è comune, ricorrente, asimmetrica, di tipo poliarticolare, a carico soprattutto delle grandi articolazioni.
L’impiego della colchicina è molto spesso efficace nei bambini. Molto utili si sono dimostrati anche la talido-mide e i farmaci biologici (infliximab).
Sindrome di Sjögren
Si tratta di una sindrome infiammatoria, cronica, ca-ratterizzata da infiltrazione linfocitica progressiva delle ghiandole salivari e lacrimali.
È eccezionale nei bambini: predilige le donne in età compresa tra 35 e 45 anni. L’età pediatrica più colpita è quella che va dai 9 ai 10 anni, con il 75% di soggetti di sesso femminile. A volte è primitiva e a volte è secondaria all’associazione con altre malattie reumatologiche (LES, sclerodermia o malattia mista del tessuto connettivo), che la seguono di qualche anno.
Sindromi periodiche febbrili ereditarie
Le sindromi della febbre periodica ereditaria sono un raro gruppo di malattie autoinfiammatorie causate da un errore congenito nel sistema immune innato. Esse sono caratterizzate da episodi ricorrenti di febbre che si auto-limitano e avvengono in assenza di infezioni o di reazioni autoimmuni, sia legate agli anticorpi, sia alle cellule. Il sistema immune innato fornisce la prima linea di difesa contro la maggior parte dei microbi e dei virus, usando recettori per il riconoscimento dell’assetto (PRR), come i recettori toll-like, che riconoscono un limitato numero di strutture molecolari virali o batteriche, conosciute come aspetti molecolari associati ai patogeni (PAMP).
I recettori delle molecole di riconoscimento stimolano l’infiammazione attivando proteine intracellulari che mediano la regolazione del fattore nucleare kB (NF-kB),
l’apoptosi cellulare e l’interleuchina 1b (IL-1b) attraverso le comuni vie di segnalazione. Mutazioni in queste pro-teine intracellulari portano a un’aumentata produzione e secrezione di IL-1b, responsabile dei sintomi clinici.
Viene comunemente indicata con il nome di febbre periodica una febbre che ha 3 o più episodi nell’arco di 6 mesi, non accompagnata da una ben definita malattia medica che spieghi la febbre, e con un intervallo di alme-no 7 giorni fra un episodio e l’altro. Questa definizione differenzia le febbri periodiche dalle febbri persistenti, che, durando più di 2 settimane, rientrano nelle febbri di origine sconosciuta. Poiché le eziologie delle febbri periodiche sono completamente differenti da quelle delle febbri persistenti, il preciso riconoscimento del tipo di febbre rappresenta un importante fattore differenziale.
Criopirina e inflammasomaLo studio delle malattie periodiche ha messo in evidenza l’esi-stenza di febbri, abbastanza rare, dovute a un’alterazione dei meccanismi di controllo della risposta infiammatoria. Tutto è nato nel 2001, quando fu osservato che la mutazione del gene CIAS1, che codifica per la criopirina, causa tre sindromi ereditarie periodiche (Tabella 31.19).Si sapeva che i macrofagi e i neutrofili formano l’inflamma-soma, un complesso di proteine che hanno ruoli distinti nel sistema innato di difesa. È stato visto inoltre che la criopirina contribuisce alla sintesi dell’interleuchina 1b, una citochina proinfiammatoria. Circa la metà delle mutazioni che causa-no le sindromi delle febbri periodiche ereditarie deriva una criopirina iperattiva, che porta a un’aumentata secrezione di interleuchina 1b.Prove cliniche mostrano che gli antagonisti del recettore dell’in-terleuchina 1b (anakinra) migliorano effettivamente i sintomi clinici delle tre sindromi, legate al gene CIAS1. Si può concludere che nel futuro, modificando l’attività della criopirina, potremo proteggerci da diversi microrganismi, aumentare l’efficacia dei vaccini e facilitare le caratteristiche infiammatorie della gotta, ma anche trattare le forme di febbre periodica legate a mutazione del gene CIAS1.
Pur all’oscuro della causa ultima della periodicità, è ri-sultato evidente che un singolo attacco può essere causato dagli stimoli più diversi:• traumi, anche minimi;• piccoli interventi;• vaccinazioni;• infezioni virali delle vie aeree superiori;• stress, anche psicologici.
Calcolando insieme le forme ereditarie e le forme non ereditarie, si raggiunge solo il 50% di tutte le febbri pe-riodiche (Tabella 31.20; vedi Tabella 31.19). Nella sua pratica professionale, per la loro frequenza, il pediatra può incontrare qualche caso di PFAFA (Periodic Fever, Aphtous stomatitis, Pharyngitis, cervical Adenitis) e un caso di
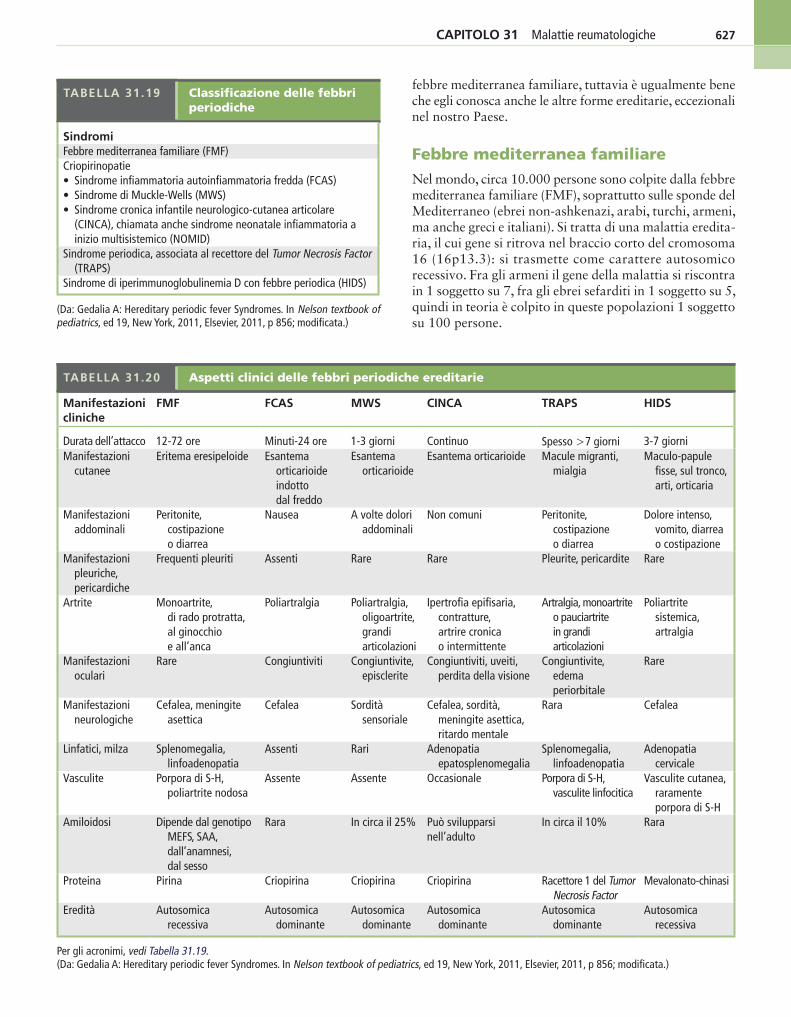
627CapItolo 31 Malattie reumatologiche
febbre mediterranea familiare, tuttavia è ugualmente bene che egli conosca anche le altre forme ereditarie, eccezionali nel nostro Paese.
Febbre mediterranea familiareNel mondo, circa 10.000 persone sono colpite dalla febbre mediterranea familiare (FMF), soprattutto sulle sponde del Mediterraneo (ebrei non-ashkenazi, arabi, turchi, armeni, ma anche greci e italiani). Si tratta di una malattia eredita-ria, il cui gene si ritrova nel braccio corto del cromosoma 16 (16p13.3): si trasmette come carattere autosomico recessivo. Fra gli armeni il gene della malattia si riscontra in 1 soggetto su 7, fra gli ebrei sefarditi in 1 soggetto su 5, quindi in teoria è colpito in queste popolazioni 1 soggetto su 100 persone.
Classificazione delle febbri periodiche
SindromiFebbre mediterranea familiare (FMF)Criopirinopatie• Sindromeinfiammatoriaautoinfiammatoriafredda(FCAS)• SindromediMuckle-Wells(MWS)• Sindromecronicainfantileneurologico-cutaneaarticolare
(CINCA), chiamata anche sindrome neonatale infiammatoria a inizio multisistemico (NOMID)
Sindrome periodica, associata al recettore del Tumor Necrosis Factor (TRAPS)
Sindrome di iperimmunoglobulinemia D con febbre periodica (HIDS)
(Da: Gedalia A: Hereditary periodic fever Syndromes. In Nelson textbook of pediatrics, ed 19, New York, 2011, Elsevier, 2011, p 856; modificata.)
tabella 31.19
Aspetti clinici delle febbri periodiche ereditarie
Manifestazioni cliniche
FMF FCaS MWS CInCa tRapS HIDS
Durata dell’attacco 12-72 ore Minuti-24 ore 1-3 giorni Continuo Spesso >7 giorni 3-7 giorniManifestazioni
cutaneeEritema eresipeloide Esantema
orticarioide indotto dal freddo
Esantema orticarioide
Esantema orticarioide Macule migranti, mialgia
Maculo-papule fisse, sul tronco, arti, orticaria
Manifestazioni addominali
Peritonite, costipazione o diarrea
Nausea A volte dolori addominali
Non comuni Peritonite, costipazione o diarrea
Dolore intenso, vomito, diarrea o costipazione
Manifestazioni pleuriche, pericardiche
Frequenti pleuriti Assenti Rare Rare Pleurite, pericardite Rare
Artrite Monoartrite, di rado protratta, al ginocchio e all’anca
Poliartralgia Poliartralgia, oligoartrite, grandi articolazioni
Ipertrofia epifisaria, contratture, artrire cronica o intermittente
Artralgia, monoartrite o pauciartrite in grandi articolazioni
Poliartrite sistemica, artralgia
Manifestazioni oculari
Rare Congiuntiviti Congiuntivite, episclerite
Congiuntiviti, uveiti, perdita della visione
Congiuntivite, edema periorbitale
Rare
Manifestazioni neurologiche
Cefalea, meningite asettica
Cefalea Sordità sensoriale
Cefalea, sordità, meningite asettica, ritardo mentale
Rara Cefalea
Linfatici, milza Splenomegalia, linfoadenopatia
Assenti Rari Adenopatia epatosplenomegalia
Splenomegalia, linfoadenopatia
Adenopatia cervicale
Vasculite Porpora di S-H, poliartrite nodosa
Assente Assente Occasionale Porpora di S-H, vasculite linfocitica
Vasculite cutanea, raramente porpora di S-H
Amiloidosi Dipende dal genotipo MEFS, SAA, dall’anamnesi, dal sesso
Rara In circa il 25% Può svilupparsinell’adulto
In circa il 10% Rara
Proteina Pirina Criopirina Criopirina Criopirina Racettore 1 del Tumor Necrosis Factor
Mevalonato-chinasi
Eredità Autosomica recessiva
Autosomica dominante
Autosomica dominante
Autosomica dominante
Autosomica dominante
Autosomica recessiva
Per gli acronimi, vedi Tabella 31.19.(Da: Gedalia A: Hereditary periodic fever Syndromes. In Nelson textbook of pediatrics, ed 19, New York, 2011, Elsevier, 2011, p 856; modificata.)
tabella 31.20

CapItolo 31 Malattie reumatologiche628
Su 496 pazienti con FMF, 254 pazienti (31,2%) ebbero il primo attacco in età ≤2 anni, con una media di 1,1 ± 0,8 anni; 242 ebbero la prima manifestazione fra 2 e 16 anni: la maggior parte di essi era omozigote per la mutazione M694V ed erano di estrazione nord-africana (ebrei sefarditi).
Il gene MEFV codifica per una proteina, chiamata pi-rina o marenostrina, a carico della quale sono state ritro-vate oltre 70 diverse mutazioni, principalmente missense, concentrate nell’esone 10 e sull’esone 2. Questa proteina è stata codificata: essa è costituita da 781 aminoacidi e ha un peso molecolare di 86.000 Da. Si ritiene che queste mutazioni siano avvenute in un antenato comune, vissuto in Medio Oriente circa 2.500 anni fa. Non si conosce ancora quale sia il fattore di selezione.
Circa il 70% dei pazienti con mutazioni cliniche sono eterozigoti e posseggono una o due mutazioni. La più comune (M694V, per sostituzione della metionina con la valina al codone 694), è presente nel 20-67% dei pazienti. L’omozigosi M694V si riscontra nelle forme più gravi, che più spesso evolvono verso l’amiloidosi.
La pirina agisce come un fattore antinfiammatorio, inibendo la trasformazione della citochina pro-IL-1b nella forma attiva.
Si pensa che una deficienza dell’inibitore del C5 (fat-tore 5 del complemento, potente anafilotossina e potente agente chemiotattico), conseguenza della disfunzione della pirina/marenostrina, induca un accumulo di C5, che porta all’attacco acuto.
Nel 65% dei casi le manifestazioni cliniche iniziano prima dei 5 anni e nel 90% prima dei 20 anni.
Il quadro clinico (Figura 31.13) è caratterizzato da:• febbre periodica, della durata di 1-4 giorni;• dolori addominali ricorrenti per flogosi del peritoneo
(80-90%): i pazienti vengono spesso operati per ap-pendicite acuta, per colecistite, per peritonite o per laparoscopia esplorativa;
• dolore toracico per flogosi della pleura (41%);• dolore articolare per flogosi della sinovia (41%);• mialgie (12%);• sierosite della vaginale del testicolo (4%);• esantema maculo-papulare durante la febbre.
Fra le complicazioni va ricordata l’amiloidosi renale.La diagnosi si basa sull’anamnesi, con riferimento al-
l’etnia e alla familiarità, all’esame obiettivo, all’emocromo, al dosaggio delle classi di immunoglobuline e infine alla ricerca del gene della FMF (Tabella 31.21), che diviene molto importante nelle aree nelle quali la malattia è più rara. Per lo screening genetico, in generale vengono usate la PCR (reazione polimerasica a catena) e l’analisi di re-strizione. Poiché molti laboratori ricercano soltanto da 10 a 15 delle numerose mutazioni, il sospetto clinico diviene essenziale per la diagnosi.
Fra gli esami di laboratorio sono da ricordare l’aumento di numero dei neutrofili e della loro chemiotassi, l’aumentato numero delle piastrine, l’aumento della VES e della PCR.
Il trattamento preventivo consiste essenzialmente nella somministrazione di colchicina alla dose di 0,02-0,03 mg/kg/die (dose massima 2 mg) in 1 o 2 dosi. In generale la dose iniziale è di 0,5 mg nei bambini in età inferiore a 5 anni, di 1 mg/die nei bambini da 5 a 10 anni e di 1,5 mg nei ragazzi in età superiore ai 10 anni. Circa il 65% dei pazienti va in remissione, il 20-30% migliora e il 5-10% non risponde. La colchicina ha un effetto favorevole anche sull’amiloide. Fra gli effetti collaterali, i più importanti sono la diarrea e i dolori addominali. La colchicina non ha alcun effetto quando venga data durante l’attacco. L’uso prolungato della colchicina non ha conseguenze sulla ferti-lità, sulla gravidanza o sullo sviluppo del feto. Nei casi che non rispondono alla colchicina, viene usata l’anakinra.
Il 30-35% dei soggetti non trattati e il 75% degli adulti con FMF sviluppa un’amiloidosi renale.
Sindrome da iper-IgD (aciduria mevalonica; malattia olandese e francese)Le IgD sono una delle cinque classi di immunoglobuline, accanto alle IgG, IgA, IgM e IgE. Non si ritrovano nel neonato. Nelle età successive sono presenti in quantità variabile: sono dosate in mg/dL o in UI/mL. Sono
Criteri diagnostici della febbre mediterranea familiare
Criteri Sintomi e segni
Criteri maggiori. Attacchi ricorrenti di:
1. Febbre (1-3 giorni)2. Peritonite (1-3 giorni)3. Pleurite unilaterale, pericardite (1-3 giorni)4. Monoartrite (anca, ginocchio, caviglia) (1-30 giorni)
Criteri minori. Attacchi incompleti a carico di:
1. Addome2. Torace3. Articolazioni (non in una sede tipica)4. Dolore da sforzo alle gambe5. Risposta favorevole alla colchicina
Criteri di appoggio
1. Storia familiare di febbre mediterranea familiare
2. Origine etnica appropriata3. Età inferiore ai 20 anni, all’inizio della malattia
4-6. Aspetti dell’attacco (grave, risoluzione spontanea, intervallo libero da sintomi)
7. Risposte infiammatorie della fase acuta transitorie
8. Proteinuria ed ematuria episodiche9. Laparotomia esplorativa infruttuosa
10. Consanguineità dei genitori
La diagnosi di febbre mediterranea familiare richiede 1 o più criteri maggiori, o 2 o più criteri minori, o un criterio minore e 5 o più criteri di appoggio, o 1 criterio minore più 4 o più dei primi 5 criteri di appoggio.(Da: Samuels J, et al: Familial Mediterranean fever at the millennium: Clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health, Medicine 77:268-97, 1998; modificata.)
tabella 31.21
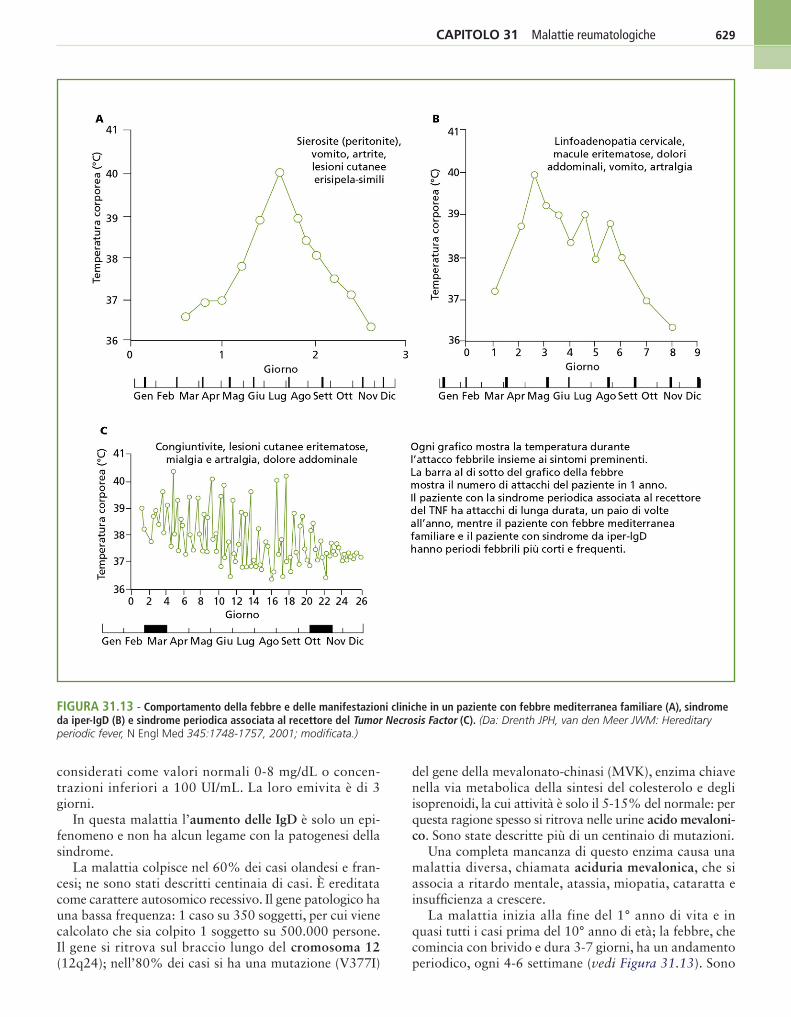
629CapItolo 31 Malattie reumatologiche
considerati come valori normali 0-8 mg/dL o concen-trazioni inferiori a 100 UI/mL. La loro emivita è di 3 giorni.
In questa malattia l’aumento delle IgD è solo un epi-fenomeno e non ha alcun legame con la patogenesi della sindrome.
La malattia colpisce nel 60% dei casi olandesi e fran-cesi; ne sono stati descritti centinaia di casi. È ereditata come carattere autosomico recessivo. Il gene patologico ha una bassa frequenza: 1 caso su 350 soggetti, per cui viene calcolato che sia colpito 1 soggetto su 500.000 persone. Il gene si ritrova sul braccio lungo del cromosoma 12 (12q24); nell’80% dei casi si ha una mutazione (V377I)
del gene della mevalonato-chinasi (MVK), enzima chiave nella via metabolica della sintesi del colesterolo e degli isoprenoidi, la cui attività è solo il 5-15% del normale: per questa ragione spesso si ritrova nelle urine acido mevaloni-co. Sono state descritte più di un centinaio di mutazioni.
Una completa mancanza di questo enzima causa una malattia diversa, chiamata aciduria mevalonica, che si associa a ritardo mentale, atassia, miopatia, cataratta e insufficienza a crescere.
La malattia inizia alla fine del 1° anno di vita e in quasi tutti i casi prima del 10° anno di età; la febbre, che comincia con brivido e dura 3-7 giorni, ha un andamento periodico, ogni 4-6 settimane (vedi Figura 31.13). Sono
FIgura 31.13 - Comportamento della febbre e delle manifestazioni cliniche in un paziente con febbre mediterranea familiare (a), sindrome da iper-IgD (B) e sindrome periodica associata al recettore del Tumor Necrosis Factor (C). (Da: Drenth JPH, van den Meer JWM: Hereditary periodic fever, N Engl Med 345:1748-1757, 2001; modificata.)

CapItolo 31 Malattie reumatologiche630
presenti linfoadenopatia, specialmente latero-cervicale, artralgia, dolori addominali con vomito e diarrea, esan-tema maculo-papulare, a volte petecchie e manifestazioni orticarioidi, epatosplenomegalia, ulcere aftose del cavo orale e della vagina. Il paziente sta bene fra gli attacchi. Un interessamento articolare, sotto forma di artralgie o di artrite oligoarticolare, asimmetrica, è abbastanza comune. L’amiloidosi può costituire una possibile complicazione a lungo termine della malattia.
Le IgD superano gli 80 mg/dL o >100 UI/mL nell’80% dei casi, ma possono essere normali in bambini al di sotto dei 3 anni o in qualche paziente di oltre 3 anni di età; nell’80% dei casi è elevato anche il livello di IgA (500 mg/dL e più). D’altra parte va ricordato che gli elevati livelli di IgD circolanti non sono patognomonici, perché un aumento delle IgD può essere ritrovato, specialmente sotto i 2 anni di vita, in pazienti con altre forme di feb-bre ricorrente. Durante l’attacco sono elevate la VES, la PCR e l’amiloide A; si riscontrano leucocitosi e at-tivazione delle citochine. Durante gli episodi febbrili i soggetti presentano, come abbiamo visto, un aumento dell’escrezione dell’acido mevalonico. Il reperto è più sensibile e più specifico di quanto non sia il dosaggio delle immunoglobuline D, tanto che alcuni autori preferi-scono indicare questa sindrome come febbre periodica con deficit incompleto di MVK.
La diagnosi si basa sull’anamnesi, sul dosaggio delle IgD e delle IgA e sulla ricerca dell’acido mevalonico nelle urine. Non c’è nessuna cura: la colchicina non ha alcun effetto. In qualche paziente gli attacchi mancano per anni.
L’anakinra e l’etanercept danno buoni risultati, come anche l’impiego della simvastatina.
Febbre periodica associata al recettore del Tumor Necrosis Factor (malattia irlandese o iberniana)La sindrome è frequente fra gli irlandesi e gli scozzesi. È trasmessa come carattere autosomico dominante: il gene si ritrova sul braccio corto del cromosoma 12 (12p13), che codifica per il recettore del TNF (TNFRSF1A); si cono-scono almeno 50 mutazioni; la mancanza è soprattutto a carico della forma solubile del recettore. La mancata attività del recettore aumenta la presenza in circolo di Tumor Necrosis Factor, che, come si sa, induce la sintesi di molte citochine, attiva i leucociti, determina febbre e tendenza alla cachessia. La sua unione con il recettore ne neutralizza l’attività e quindi agisce come un suo inibitore. La mancanza del recettore induce una forte infiammazione non controllata dai meccanismi inibitori.
A carico del gene sono state trovate numerose muta-zioni, delle quali la più frequente e la più grave è quella legata alla sostituzione di un residuo di cisteina negli esoni codificanti la porzione extracellulare della proteina; altre mutazioni (R92Q e P46L) hanno bassa penetranza e sono associate a un quadro clinico meno importante.
La malattia è caratterizzata da febbre periodica (con crisi 2-4 volte per anno), con attacchi di durata variabile (da 4 a 6 giorni; fino a 3 settimane), intervallati fra loro anche di molti mesi (vedi Figura 31.13), da forti dolori addominali, nausea e vomito; più di rado sono presenti eruzione cutanea, eritema doloroso al tronco e agli arti, mialgie localizzate o migranti, artrite o artromialgia ri-corrente, linfoadenomegalia, congiuntivite dolorosa (che-ratite?) ed edema palpebrale. In alcuni casi si osservano lesioni eritematose fisse, calde e dolenti, con infiltrato sottocutaneo, tali da assumere l’aspetto di una panniculite. Anche in questi casi l’amiloidosi rappresenta una possibile complicazione a lungo termine.
La diagnosi si basa sull’anamnesi, sui bassi livelli del recettore solubile del TNF (grande variabilità), sull’analisi molecolare, sulla neutrofilia, sull’aumento della PCR, sull’attivazione del complemento, sull’aumento delle IgA, a volte delle IgD, ma sempre a livelli inferiori a quelli della sindrome da iper-IgD.
La malattia non risponde alla colchicina, mentre ri-sponde ai cortisonici e alla somministrazione di etaner-cept, un recettore del TNF-a umano, da usare alla dose di 25 mg 2 volte alla settimana, sottocute. L’etanercept si lega al TNF-a umano e ne neutralizza l’azione. Non c’è ancora una casistica sull’uso dell’infliximab, un anticorpo monoclonale, antagonista diretto del TNF-a umano.
Febbre periodica associata a mutazioni del gene CIAS1 (criopirina)Le sindromi associate a circa 50 mutazioni del gene CIAS1 (situato nel cromosoma 1q44) sono rappresentate da un gruppo di affezioni monogeniche, autosomico-dominanti, secondarie a mutazioni differenti a carico dello stesso gene, che codifica per una proteina, chiamata criopirina, coinvolta nella regolazione della secrezione e dell’attiva-zione dell’interleuchina 1b (IL-1b). Le mutazioni della criopirina portano a un’aumentata produzione di interleu-china 1b, causa di una delle tre sindromi sotto riportate. Quadri simili, ma molto più rari, riconoscono come causa mutazioni del gene ILIRN, che codifica un antagonista del recettore dell’interleuchina 1b.
Le forme più comuni sono tre, delle quali la forma più lieve è costituita dalla sindrome familiare infiammatoria da freddo, tipica dell’adulto e caratterizzata da accessi febbrili e lesioni orticarioidi, scatenate dall’esposizione al freddo, talvolta associata ad artralgia, dolori addominali e congiuntivite.
Una seconda forma (la sindrome di Muckle-Wells) è caratterizzata da elementi simil-orticarioidi, non sempre pruriginosi, a inizio nei primi anni di vita, di natura non vasculitica. Con gli anni possono comparire una poliar-trite non erosiva, un’amilodosi renale e una sordità neu-rosensoriale.
La forma più grave è rappresentata dalla sindrome CINCA (Chronic Infantile Neurological Cutaneous

631CapItolo 31 Malattie reumatologiche
Articular), una malattia infiammatoria febbrile, sistemica, caratterizzata da febbricola ad andamento cronico e da lesioni cutanee di tipo orticarioide, a inizio neonatale; gli indici di flogosi sono molto elevati ed è presente anemia ipocromica. Possono associarsi un grave interessamento articolare (artrite e displasie ossee), sordità neurosensoria-le, deficit intellettivo di grado variabile, cefalea subcronica, legata a un quadro di meningite asettica, e coinvolgimento oculare (iridociclite, vasculite retinica).
I portatori di mutazioni del gene CIAS1 presentano un’aumentata secrezione di interleuchina 1b da parte dei monociti circolanti. In terapia, l’impiego dell’anakirna (un antagonista recettoriale della IL-1b) ha portato a risultati eccellenti, alla dose di 1-2 mg/kg di peso per giorno, per via sottocutanea, per molti mesi. La sospensione del far-maco si accompagna a una ripresa della sintomatologia.
Buoni risultati sono stati ottenuti anche con la terapia biologica (canakinumab, un anticorpo umano monoclonale anti-interleuchina 1b = Ilaris 150 mg, per iniezione sotto-cutanea, 2 mg/kg fra 15 e 40 kg, ogni 8 settimane; ovvero rilonacept = Arcalyst 220 mg, per iniezione sottocutanea, da 12 a 17 anni, 4,4 mg/kg, dose massima 320 mg, 1 volta alla settimana – non in commercio in Italia).
Sindromi PAPA e di BlauLa PAPA (Pyogenic Arthritis, Pyoderma gangrenoso, Acne) è una sindrome autosomica dominante con mutazione del gene che codifica la proteina adattatore della 1a proteina serina-prolina-treonina, fosfatasi interagente, localizzato sul cromosoma 15 (15q24). Pioderma gangrenoso e grave acne cistica si associano a ulcerazioni cutanee delle estremi-tà, determinate da un trauma. È presente artrite sterile.
La sindrome di Blau è una rara malattia autosomica dominante, caratterizzata da artrite granulomatosa, uveite, esantema, contratture delle dita; è dovuta alle mutazioni del gene codificante CARD15 (Caspase Recruitment Do-main 15 Protein), conosciuto anche come NOD2 (Nu-cleotide-binding Oligomerization Domain 2 protein), localizzato sul cromosoma 16 (16q12).
Febbre periodica con stomatite aftosa, faringite e adenopatieLa PFAFA (Periodic Fever, Aphtous stomatitis, Pharyngitis, cervical Adenitis), detta anche sindrome di Marshall, ha un’incidenza di 0,4 casi/1.000 bambini/anno, senza pre-dilezione etnica. Essa inizia in soggetti con età inferiore ai 5 anni (80% dei casi); la febbre è superiore ai 38 °C, con periodicità di 28 giorni (da 21 a 28 giorni), con circa 8-12 episodi per anno.
Sono stati descritti casi in gemelli monocoriali e in fratelli.
Essa è caratterizzata da:• febbre per 4-6 giorni, spesso con brivido;• dolori addominali;
• faringo-tonsillite;• nausea e vomito;• stomatite aftosa, con afte piccole;• adenopatia latero-cervicale;• aumento degli indici di flogosi durante il periodo
di acuzie;• indici peraltro normali negli intervalli liberi;• assenza di un difetto genetico, ereditario.
Fra gli esami di laboratorio è importante l’esecuzione di un emocromo (per escludere una neutropenia ciclica) e di un dosaggio delle classi di immunoglobuline (per escludere una sindrome da iper-IgD); talvolta il tasso di IgD può essere un po’ più alto del normale, ma non raggiunge mai i livelli di 100 UI/mL, che si ritrovano nella sindrome da IgD. Durante l’attacco febbrile aumentano i monociti e diminuiscono gli eosinofili. I livelli di proteina C reattiva aumentano durante la crisi.
In una vasta casistica italiana di 383 bambini con feb-bre periodica, 210 soddisfacevano i criteri per la PFAFA e 43 erano portatori di una mutazione, di cui 33 con mevalonato-chinasi deficienza, 7 avevano la FMF e 3 la sindrome periodica associata al recettore del Tumor Ne-crosis Factor; 37 avevano mutazioni con bassa penetranza o genotipi incompleti e 130 avevano le prove negative.
La malattia risolve bruscamente, entro 24 ore, dopo una singola dose di corticosteroidi (prednisone 1-2 mg/kg); in qualche caso come profilassi è stata usata la cimeti-dina (Cimetidina Teva, compresse da 400 mg, da 20 a 40 mg/kg/die in 3-4 dosi). L’impiego dei comuni anti-piretici ha uno scarso effetto. Dopo ripetuti episodi, la tonsillectomia risolve definitivamente la malattia, ma non in tutti i bambini.
La prognosi è favorevole nell’arco di 4-8 anni.
Neutropenia ciclicaÈ un’affezione che si tramette con modalità autosomico- dominante: si associa a mutazione del gene dell’elastasi neutrofila (ELA2), che porta a un’apoptosi accelerata per la presenza di una proteina anormale.
Si tratta di una malattia più rara della PFAFA: essa si presenta con una periodicità di 21 giorni (in media da 14 a 45 giorni). Anch’essa inizia nei bambini al di sotto dei 5 anni e si associa a stomatite, gengivite, linfoadenopatia cervicale e febbre; spesso viene ereditata. I bambini con neutropenia ciclica possono avere anche ripetute infezioni batteriche, ma la febbre tipica dei bambini con neutrope-nia ciclica si verifica in maniera caratteristica in assenza di infezione.
Va ricordato che la neutropenia può essere assente al momento della febbre.
Per dimostrare la presenza di una neutropenia ciclica, la conta dei globuli bianchi con la formula va eseguita 2 o 3 volte alla settimana per un periodo di 6 settimane, durante il quale il bambino presenterà almeno un episodio febbrile.

CapItolo 31 Malattie reumatologiche632
La diagnosi si basa sulla conta assoluta dei neutrofili, che deve essere inferiore ai 500 per mm3 e che, senza alcun tipo di trattamento, torna al normale. Nei casi in cui è stato eseguito un puntato midollare è stato dimostrato un arresto della linea mieloide allo stadio dei mielociti. Per i bambini che abbiano questa malattia da molto tempo, è possibile ottenere buoni risultati con la somministrazione del fattore stimolante le colonie di granulociti, che riduce la durata della neutropenia e quindi diminuisce in modo evidente il rischio di infezione.
La malattia si fa meno frequente con l’aumentare dell’età.
Amiloidosi
L’amiloidosi comprende un gruppo di malattie caratteriz-zate dalla deposizione extracellulare di proteine insolubili, fibrose amiloidi in vari tessuti dell’organismo.
L’analisi biochimica dell’amiloide ha messo in evidenza, negli USA, che essa è costituita da frammenti monoclonali della catena leggera delle immunoglobuline. Si riscontra nel corso di numerose malattie periodiche febbrili ereditarie.
Sarcoidosi
La sarcoidosi è una malattia granulomatosa di origine sconosciuta, che colpisce organi diversi: si pensa che essa risulti dall’esposizione di soggetti geneticamente suscetti-bili a uno o più antigeni non identificati. L’incidenza, di 0,22-0,24/100.000 bambini, aumenta con l’età, con un picco a 20-39 anni (11 casi/100.000).
Le lesioni granulomatose della sarcoidosi, non soggette alla caseificazione, possono avvenire in ogni parte del cor-po. Nella maggior parte dei casi queste lesioni guariscono, ma in circa il 20% segue un tessuto cicatriziale. È caratteri-stico che i macrofagi, presenti nel granuloma, producano e secernano 1,25-diidrossi-colecalciferolo, la forma attiva della vitamina D, di regola formata dal parenchima renale. Ne possono conseguire ipercalcemia e ipercalciuria, con quadri di intossicazione da vitamina D.
Nei bambini la malattia inizia con perdita di peso, tosse, stanchezza, dolori ossei e articolari e anemia.
Il polmone è l’organo più spesso colpito: si ritrovano infiltrati parenchimali, noduli miliari e linfoadenopatia ilare e paratracheale. Si riscontrano di frequente uveite e irite, lesioni cutanee e sofferenza epatica e artrite, con le caratteristiche dell’artrite idiopatica giovanile. I bambini in età inferiore ai 4 anni possono presentare un esantema maculo-papulare, senza segni di sofferenza polmonare.
Anemia, leucopenia ed eosinofilia sono spesso riscon-trate. Altri reperti sono l’ipergammaglobulinemia, l’au-mento delle proteine della fase acuta, la VES e la PCR. L’ipercalcemia e l’ipercalciuria si ritrovano solo in una piccola percentuale di bambini. Le cellule epitelioidi del
granuloma formano anche l’enzima angiotensina-con-vertente, per cui il suo dosaggio dimostra un’elevazione. La prova di Kveim-Siltzbach si basa sull’iniezione intra-dermica di materiale da una lesione della sarcoidosi: la positività della prova è documentata dalla comparsa di un granuloma molte settimane più tardi.
Il trattamento è sintomatico. Si può avere una guari-gione spontanea dopo mesi o anni, o la situazione può cronicizzarsi con una progressiva sofferenza polmonare. I corticosteroidi possono migliorare le situazioni acute e il metotrexato può costituire un trattamento di fondo. I farmaci biologici hanno presentato uno scarso effetto.
Malattia di Kawasaki
La malattia di Kawasaki è un’affezione acuta febbrile, di origine sconosciuta, che colpisce soggetti in età pediatrica, dovuta a una vasculite delle coronarie e a un vasto insieme di sintomi e segni generali.
Dopo la descrizione iniziale di Kawasaki in Giappone, nel 1967, essa è risultata presente in ogni parte del mondo: il suo aspetto clinico è risultato molto vicino a quello di un’altra vasculite, la poliarterite infantile. È la più comune causa di malattia acquisita di cuore nei bambini dei Paesi occidentali.
eziologia e patogenesi
Gli aspetti epidemiologici e clinici propendono per un’ori-gine infettiva. Diversi agenti infettivi, batterici e virali, sono stati isolati da pazienti con malattia di Kawasaki: l’ultimo di questi, il coronavirus New Haven, sembrava proprio che avesse le caratteristiche per essere l’agente scatenante, ma studi successivi hanno fatto cadere defini-tivamente questa ipotesi.
Recentemente si è supposto che la malattia sia dovuta a tossine batteriche, simili alla tossina stafilococcica della sindrome da shock tossico, che agiscono nella patogenesi della malattia di Kawasaki come un superantigene. Il re-perto di un aumentato numero di una sottopopolazione di cellule T (cellule TVb2-positive) è in accordo con l’ipotesi che sia in gioco una tossina che agisce come superan-tigene. Non vi sono segni che l’autoimmunità abbia un ruolo determinante, tuttavia è ammessa una suscettibilità immunologica.
I geni associati con l’attivazione delle piastrine e dei neutrofili sono espressi ai più alti livelli nei pazienti con malattia di Kawasaki rispetto ai pazienti con infezioni da adenovirus o con reazioni avverse ai farmaci, ma sono pres-soché uguali a quelli che si riscontrano nella scarlattina.
Durante la fase acuta della malattia i livelli di immuno-globuline sono elevati, come se fosse presente una vigorosa risposta anticorpale.

633CapItolo 31 Malattie reumatologiche
A sostenere l’ipotesi di una forte componente genetica, vi è la prevalenza della malattia nella popolazione asiatica e nei discendenti di queste popolazioni, anche quando emi-grati nei Paesi occidentali, la comparsa in figli di genitori precedentemente affetti, la maggiore incidenza nei fratelli e soprattutto nei gemelli.
epidemiologia
Non ci sono prove di contagiosità della malattia, tuttavia studi sulla distribuzione geografica e temporale di centina-ia di pazienti hanno dimostrato che è un agente infettivo a determinare la malattia di Kawasaki.
La maggior parte dei pazienti ha un’età compresa fra pochi mesi e 6-8 anni, con un massimo a 2-3 anni (l’80% dei pazienti ha meno di 4 anni). L’incidenza cade bru-scamente oltre gli 8 anni. Nei pazienti con età inferiore ai 6 mesi, è più frequente una presentazione clinica incompleta. Nei soggetti al di sopra dei 6 anni di età le forme incom-plete hanno un’incidenza simile a quella riscontrata nei soggetti più giovani, mentre il rischio di sequele cardiova-scolari è risultato più elevato. Vi è una predominanza di maschi, con un rapporto 1,5-1,8:1. Non vi sono chiare differenze geografiche o urbano-rurali nell’incidenza della malattia nello stesso Giappone; la malattia sembra più frequente nei mesi invernali e primaverili.
Poiché essa è nettamente più frequente nei bambini in Giappone e fra i giapponesi delle isole Hawaii (so-no stati complessivamente riportati in Giappone più di 140.000 casi), è stata sospettata la presenza di fattori genetici che ne aumentino la suscettibilità: l’antigene di istocompatibilità HLA Bw22, sottotipo 12, si è dimostrato fra gli ammalati con una frequenza doppia di quella della popolazione generale.
Negli USA l’incidenza annuale è stata calcolata fra 4 e 15 casi/100.000 in bambini al di sotto dei 5 anni. Negli ultimi anni la malattia risulta in lieve in aumento, ma è probabile che che esso sia dovuto a un aumento del riconoscimento delle dilatazioni delle arterie coronarie.
Fra i predittori di un decorso sfavorevole sono stati riconosciuti: il sesso, l’età minore, la gravità delle alte-razioni degli esami di laboratorio, l’etnia asiatica e delle isole del Pacifico. Anche la durata della febbre elevata è un buon predittore.
Manifestazioni cliniche
Accanto a forme classiche (Tabella 31.22), si manifestano con una certa frequenza casi atipici (o incompleti), con pochi segni clinici iniziali, che evolvono successivamente nelle tipiche alterazioni coronariche: spesso si tratta di soggetti nel primo anno di vita, nei quali viene posta una diagnosi generica di gastroenterite o di malattia delle vie aeree superiori o addirittura di sepsi. In questi la letalità è elevata, perché non è stato preso alcun provvedimento preventivo delle lesioni coronariche. Da ricordare che
l’irritabilità rappresenta un segno importante, presente in quasi tutti i casi, anche se essa non è stata inclusa nei criteri diagnostici: il quadro è probabilmente legato alla meningite asettica.
Nelle forme classiche, la malattia è caratterizzata da un decorso trifasico, con una fase acuta febbrile, una fase subacuta e una fase di convalescenza.
La febbre è il primo segno (oltre 39,5 °C): essa dura oltre 5 giorni e non risponde al trattamento antibiotico e antifebbrile (Figura 31.14); alla febbre si associano irritabilità o letargia, discreta iniezione della congiuntiva bulbare, senza secrezione (Figura 31.15) e manifestazioni orofaringee (faringe arrossato, labbra rosse, secche, fis-surate, lingua a fragola, uguale a quella che siamo soliti vedere nelle infezioni streptococciche). Compare insieme un esantema eritemato-maculare (di tipo morbilliforme o scarlattiniforme, o orticarioide o polimorfo; eccezional-mente vescicolare, bolloso o petecchiale) non specifico, diffuso al tronco, al volto e alle estremità. Dopo qualche giorno dalla comparsa della febbre, le mani e i piedi divengono tumefatti, edematosi con edema duro, dolenti; le dita assumono un aspetto fusiforme; la pelle è secca e lucida. Il palmo delle mani e la pianta dei piedi mostrano un colore rosso diffuso nella fase acuta. All’esantema si associa una linfoadenopatia latero-cervicale, presente per tutto il decorso della malattia; la linfoadenopatia è di dimensioni notevoli, non tende alla suppurazione e non è fortemente dolorosa. In Oriente la linfoadenopatia è presente solo nel 24% dei casi ed è correlata direttamente all’età: la sua assenza può ritardare la diagnosi. Non ha un’associazione complessiva con le lesioni coronariche.
Criteri diagnostici principali della malattia di Kawasaki
Febbre, che duri almeno 5 giorni, inizialmente superiore ai 39 °CCongiuntivite: bulbare, bilaterale senza essudato purulento
e interessamento cornealeAlterazioni delle labbra e della mucosa orale:• eritema,fissurazioneecrostesullelabbra• diffusoarrossamentofaringeo• linguaafragolaAlterazioni a carico degli arti:• eritema,indurimentoededemadel palmo delle mani
e della pianta dei piedi, nella fase acuta febbrile• desquamazionedelleditadellemaniedeipiedi,2settimane
dopo l’inizio della febbre, estendentesi alle palme e alle piante• solchitrasversalisulleunghie2o3mesidopol’iniziodellamalattiaEsantema polimorfo, eritematoso, di tipo morbilliforme
o scarlattiniforme, senza vescicole o crosteLinfonodi ingranditi, di oltre 1,5 cm di diametro, usualmente
da un solo lato nella regione laterale del colloEsclusione di altre malattie con reperti simili
Per la diagnosi al 4° giorno di malattia, sono necessari ≥4 dei segni; la febbre è un elemento indispensabile.
tabella 31.22
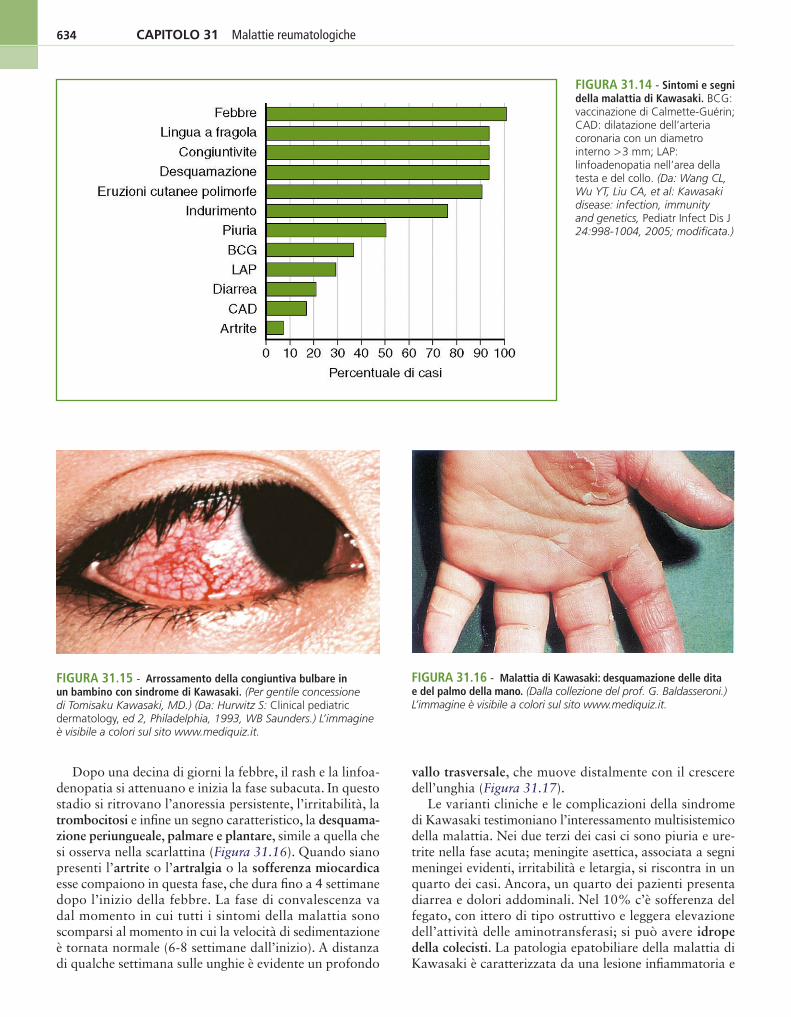
CapItolo 31 Malattie reumatologiche634
Dopo una decina di giorni la febbre, il rash e la linfoa-denopatia si attenuano e inizia la fase subacuta. In questo stadio si ritrovano l’anoressia persistente, l’irritabilità, la trombocitosi e infine un segno caratteristico, la desquama-zione periungueale, palmare e plantare, simile a quella che si osserva nella scarlattina (Figura 31.16). Quando siano presenti l’artrite o l’artralgia o la sofferenza miocardica esse compaiono in questa fase, che dura fino a 4 settimane dopo l’inizio della febbre. La fase di convalescenza va dal momento in cui tutti i sintomi della malattia sono scomparsi al momento in cui la velocità di sedimentazione è tornata normale (6-8 settimane dall’inizio). A distanza di qualche settimana sulle unghie è evidente un profondo
vallo trasversale, che muove distalmente con il crescere dell’unghia (Figura 31.17).
Le varianti cliniche e le complicazioni della sindrome di Kawasaki testimoniano l’interessamento multisistemico della malattia. Nei due terzi dei casi ci sono piuria e ure-trite nella fase acuta; meningite asettica, associata a segni meningei evidenti, irritabilità e letargia, si riscontra in un quarto dei casi. Ancora, un quarto dei pazienti presenta diarrea e dolori addominali. Nel 10% c’è sofferenza del fegato, con ittero di tipo ostruttivo e leggera elevazione dell’attività delle aminotransferasi; si può avere idrope della colecisti. La patologia epatobiliare della malattia di Kawasaki è caratterizzata da una lesione infiammatoria e
FIgura 31.14 - Sintomi e segni della malattia di Kawasaki. BCG: vaccinazione di Calmette-Guérin; CAD: dilatazione dell’arteria coronaria con un diametro interno >3 mm; LAP: linfoadenopatia nell’area della testa e del collo. (Da: Wang CL, Wu YT, Liu CA, et al: Kawasaki disease: infection, immunity and genetics, Pediatr Infect Dis J 24:998-1004, 2005; modificata.)
FIgura 31.15 - arrossamento della congiuntiva bulbare in un bambino con sindrome di Kawasaki. (Per gentile concessione di Tomisaku Kawasaki, MD.) (Da: Hurwitz S: Clinical pediatric dermatology, ed 2, Philadelphia, 1993, WB Saunders.) L’immagine è visibile a colori sul sito www.mediquiz.it.
FIgura 31.16 - Malattia di Kawasaki: desquamazione delle dita e del palmo della mano. (Dalla collezione del prof. G. Baldasseroni.) L’immagine è visibile a colori sul sito www.mediquiz.it.

635CapItolo 31 Malattie reumatologiche
distruttiva dei dotti interlobulari, che può evolvere verso la loro scomparsa.
La più temibile espressione della malattia è quella car-diaca; nella fase subacuta una parte dei pazienti presenta alterazioni cardiache cliniche, come insufficienza acuta di cuore, versamento pericardico, insufficienza acuta della mitrale o infarto acuto del miocardio. Dal 10 al 40% dei bambini colpiti presenta una vasculite delle arterie corona-rie entro le prime settimane dall’inizio della malattia, con dilatazione e formazione di aneurismi (visibili con ecografia bidimensionale). Questo esame è talmente indicativo della malattia che tutti i bambini che presentino una malattia di Kawasaki sicura o solo sospettata vanno sottoposti all’esa-me, che d’altra parte non è invasivo, nelle prime 2 settimane di malattia. La posta in gioco è talmente importante che è necessario usare tutti i mezzi a disposizione per non lasciarsi sfuggire un caso di interessamento coronarico. Sotto questo aspetto, la malattia di Kawasaki non può più infatti essere considerata come una malattia febbrile benigna, perché lo 0,5-1% dei pazienti muore improvvisamente durante la fase subacuta o in quella di convalescenza, per una malattia delle arterie coronarie (dilatazione, aneurismi e trombosi).
Reperti di laboratorio
Le modificazioni degli esami di laboratorio sono poco specifiche e quindi poco diagnostiche. Si trova una leuco-citosi (globuli bianchi oltre i 20.000/mm3 con aumento delle forme immature), associata a trombocitosi (piastrine oltre le 500.000/mm3) nella 2ª-3ª settimana di malattia. La proteina C reattiva è spesso più elevata della VES nella fa-se acuta della malattia: esse tornano al normale dopo 6-l0 settimane. Le immunoglobuline sieriche sono aumentate, specialmente le IgE. La ricerca degli ANA e del fattore reumatoide è negativa (Tabella 31.23).
Gli enzimi epatici (aminotransferasi) e il complemento possono risultare aumentati nel siero. Per valutare l’in-
tensità della colangiolisi può essere utile il dosaggio delle g-glutamiltranspeptidasi (g-GT) sieriche. L’attività della creatin-chinasi e della lattico-deidrogenasi è elevata nei pazienti con miocardite o infarto del miocardio.
L’ECG può essere patologico per aritmie, diminuzione del voltaggio e segni di ipertrofia ventricolare sinistra.
La valutazione della situazione cardiaca deve rappre-sentare l’obiettivo principale per il pediatra: è necessario eseguire una Rx del torace, un ECG, un ecocardiogramma bidimensionale, un 2D-eco-Doppler o meglio un eco-color-Doppler, per dimostrare una dilatazione o un aneurisma delle coronarie. Questo tipo di ricerche va eseguito appena sorga il sospetto, e in seguito dopo 2-3 settimane e dopo 6-8 settimane dall’inizio della malattia, per valutarne il decorso. Il bambino con aneurismi coronarici va seguito per almeno 5 anni.
Diagnosi
Di recente l’attenzione dei pediatri si è rivolta alla diagnosi precoce di casi che non rispondono in pieno ai criteri clas-sici, riportati nella Tabella 31.22, perché è stato visto che es-si richiedono lo stesso trattamento dei casi tipici, in quanto esposti allo stesso rischio di coinvolgimento coronarico. La difficoltà risiede nel valutare, caso per caso, i limiti di allontamento dai criteri classici per la diagnosi: la comparsa di un esantema aspecifico con febbre elevata, che duri più di 4-5 giorni, deve costituire un sospetto diagnostico.
Per la diagnosi di “forma completa” sono necessari una febbre per 5 o più giorni e quattro dei rimanenti sintomi, riportati nella Tabella 31.22, oppure la presenza di febbre e di aneurisma coronarico, insieme a tre degli altri criteri.
Nel giugno 2000, un gruppo di pediatri del Children Hospital di Los Angeles ha proposto una modifica dei criteri per la diagnosi della malattia di Kawasaki “atipica” (Figura 31.18):• febbre da 5 o più giorni;• + due dei criteri maggiori;
FIgura 31.17 - Malattia di Kawasaki: solchi ungueali dopo 8 settimane dall’inizio dei sintomi. (Dalla collezione del prof. G. Baldasseroni.) L’immagine è visibile a colori sul sito www.mediquiz.it.
Reperti di laboratorio nella fase acuta della malattia di Kawasaki
• Leucocitosiconneutrofiliaepresenzadiformeimmature• Velocitàdisedimentazionedelleemazieelevata• ProteinaCreattivaelevata• Anemia• Alterazionideilipidiplasmatici• Ipoalbuminemia• Trombocitosidopo1settimana• Piuriasterile• Elevazionedelletransaminasisieriche• Pleiocitosidelliquidocerebrospinale• Leucocitosidelliquidosinoviale
(Da: Son MBF, Newburger JW. In Nelson textbook of pediatrics, ed 19, New York, 2011, Elsevier, pp 862-867; modificata.)
tabella 31.23
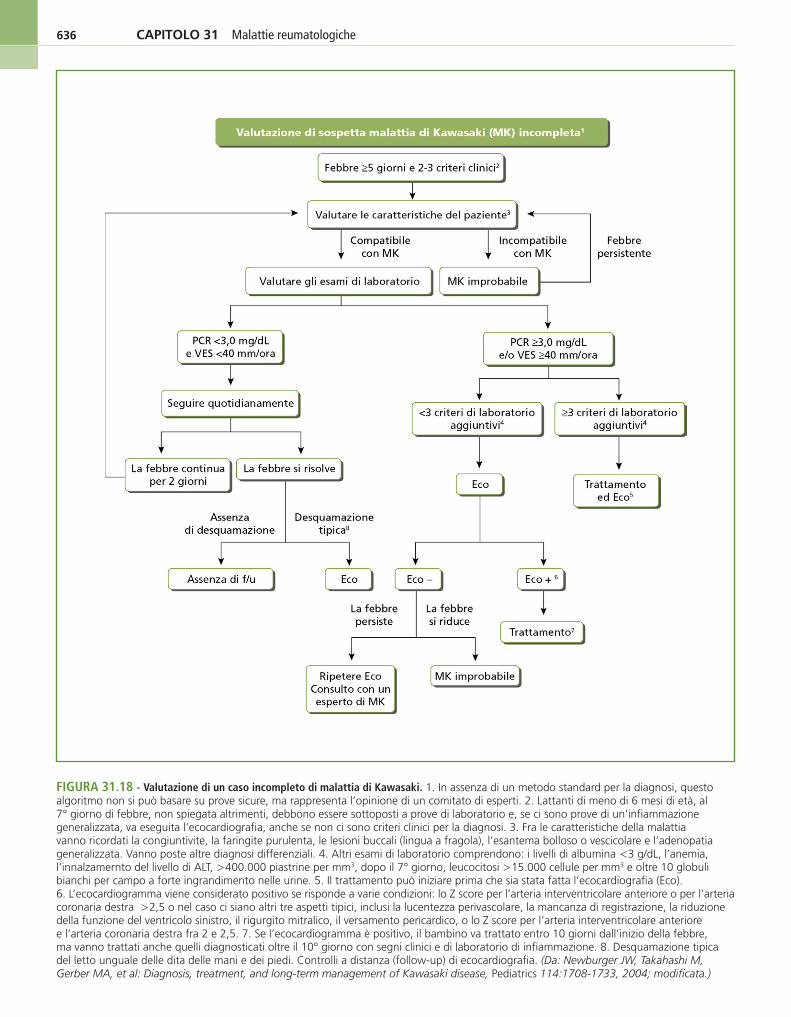
CapItolo 31 Malattie reumatologiche636
FIgura 31.18 - Valutazione di un caso incompleto di malattia di Kawasaki. 1. In assenza di un metodo standard per la diagnosi, questo algoritmo non si può basare su prove sicure, ma rappresenta l’opinione di un comitato di esperti. 2. Lattanti di meno di 6 mesi di età, al 7° giorno di febbre, non spiegata altrimenti, debbono essere sottoposti a prove di laboratorio e, se ci sono prove di un’infiammazione generalizzata, va eseguita l’ecocardiografia, anche se non ci sono criteri clinici per la diagnosi. 3. Fra le caratteristiche della malattia vanno ricordati la congiuntivite, la faringite purulenta, le lesioni buccali (lingua a fragola), l’esantema bolloso o vescicolare e l’adenopatia generalizzata. Vanno poste altre diagnosi differenziali. 4. Altri esami di laboratorio comprendono: i livelli di albumina <3 g/dL, l’anemia, l’innalzamernto del livello di ALT, >400.000 piastrine per mm3, dopo il 7° giorno, leucocitosi >15.000 cellule per mm3 e oltre 10 globuli bianchi per campo a forte ingrandimento nelle urine. 5. Il trattamento può iniziare prima che sia stata fatta l’ecocardiografia (Eco). 6. L’ecocardiogramma viene considerato positivo se risponde a varie condizioni: lo Z score per l’arteria interventricolare anteriore o per l’arteria coronaria destra >2,5 o nel caso ci siano altri tre aspetti tipici, inclusi la lucentezza perivascolare, la mancanza di registrazione, la riduzione della funzione del ventricolo sinistro, il rigurgito mitralico, il versamento pericardico, o lo Z score per l’arteria interventricolare anteriore e l’arteria coronaria destra fra 2 e 2,5. 7. Se l’ecocardiogramma è positivo, il bambino va trattato entro 10 giorni dall’inizio della febbre, ma vanno trattati anche quelli diagnosticati oltre il 10° giorno con segni clinici e di laboratorio di infiammazione. 8. Desquamazione tipica del letto unguale delle dita delle mani e dei piedi. Controlli a distanza (follow-up) di ecocardiografia. (Da: Newburger JW, Takahashi M, Gerber MA, et al: Diagnosis, treatment, and long-term management of Kawasaki disease, Pediatrics 114:1708-1733, 2004; modificata.)

637CapItolo 31 Malattie reumatologiche
• + due dei seguenti criteri (detti minori): ipoalbumi-nemia, riduzione dell’ematocrito, elevazione di ALT, elevazione di VES e/o PCR.
La diagnosi, in mancanza di un esame specifico, si basa sui reperti clinici e sull’esclusione di altre possibili cause; tuttavia la ricerca dell’interessamento delle arterie coro-narie all’ecocardiografia rappresenta un elemento indi-spensabile, perché il rilievo di questo aspetto patologico è caratteristico della malattia.
Il quadro ecografico della linfoadenopatia cervicale è tipico della malattia.
La diagnosi differenziale include malattie batteriche o da spirochete (scarlattina, sindromi esfoliative stafilococ-ciche, leptospirosi), gli esantemi virali (morbillo e altri), le rickettsiosi, la toxoplasmosi, la sindrome di Stevens- Johnson e l’artrite idiopatica giovanile.
Se la desquamazione delle mani e dei piedi e la trombo-citosi non compaiono entro 12-20 giorni dall’inizio della febbre, la diagnosi di malattia di Kawasaki può essere messa in dubbio.
prognosi
La guarigione è completa nei pazienti che non abbiano una va-sculite coronarica dimostrabile all’esame ecocardiografico.
La letalità viene calcolata fra lo 0,5 e l’1% ed è sempre in relazione con la complicazione cardiaca. Sfortunata-mente le morti non sono sempre predicibili: il 50% di esse avviene nel primo mese dall’inizio della malattia, il 75% entro 2 mesi e il 95% entro 6 mesi. Ma la morte può giungere anche dopo 10 anni e può essere ugualmente im-provvisa e inaspettata. La letalità per malattia di Kawasaki in Giappone è inferiore allo 0,14%.
Secondo studi giapponesi dal 10 al 20% dei pazienti svi-luppa aneurismi coronarici, diagnosticabili all’ecocardiogra-fia, che tuttavia possono regredire in buona parte nell’anno successivo all’insorgenza. Per tutto questo e per riconoscere le lesioni comparse tardivamente si richiede, anche nei casi che inizialmente non mostrano un reperto coronarico patologico, un controllo cardiologico 3 mesi dopo la dimissione e poi ogni 6 mesi-1 anno per 3-5 anni. Le linee guida internazionali consigliano tuttavia di ridurre i controlli nei bambini che risultino indenni alla fine della malattia.
Circa il 50% delle lesioni coronariche regredisce en-tro 5 anni. Gli aneurismi giganti (>8 mm) è difficile che regrediscano.
Non ci sono prove che la malattia di Kawasaki aumenti l’incidenza dell’arteriosclerosi nelle età successive, tuttavia sono state riscontrate significative differenze nei livelli di colesterolo e di apolipoproteina B dopo aver superato la malattia.
trattamento
L’introduzione del trattamento con immunoglobuline (Ig) per via endovenosa ha rappresentato una svolta decisiva
nella cura della malattia: la risposta clinica al loro uso nel periodo acuto febbrile della malattia è completa e decisiva. La febbre e gran parte della rimanente sintomatologia si attenuano e scompaiono anche entro 24 ore con l’uso delle immunoglobuline. Il loro impiego inoltre previene la com-parsa dell’interessamento coronarico. La dose è di 2 g/kg in una singola dose: per avere il massimo del successo sulla complicazione cardiaca è necessario che esse siano usate nei primi 10 giorni dall’inizio della malattia. La sommini-strazione nei primi 5 giorni di malattia avrebbe qualche vantaggio. Non tutti i pazienti rispondono a una prima dose per via venosa: per quelli che non rispondono (si tratta per lo più di soggetti trattati molto precocemente) si richiede una seconda dose (circa nel 23% dei casi).
Nei soggetti refrattari sia alla prima sia alla seconda dose di immunoglobuline, a dose piena, viene consigliato l’uso dei corticosteroidi, della ciclosporina A e più di recente dell’infliximab, un anticorpo monoclonale contro il Tumor Necrosis Factor a, che si è dimostrato molto efficace.
Anche l’Aspirina trova un utile impiego, per la sua azione antinfiammatoria e antiaggregante piastrinica. La dose varia da 20 a 30 mg/kg/die in una o più sommini-strazioni nelle prime fasi della malattia, per passare a dosi più basse (5 mg/kg/die) per 6-8 settimane, almeno finché la velocità di sedimentazione rimanga elevata o siano ancora presenti lesioni coronariche (vedi Figura 31.18).
L’associazione, una singola dose elevata di Ig endo-vena e Aspirina, offre le migliori possibilità di impedire lo sviluppo di aneurismi a carico delle arterie coronarie, riducendo la frequenza allo 0-0,05%.
Di recente è stata tentata con successo la terapia con abciximab, un inibitore del recettore della glicoproteina IIb/IIIa delle piastrine: i bambini trattati con questa so-stanza hanno dimostrato una regressione del diametro degli aneurismi maggiore di quella che si verificava nei soggetti trattati con la terapia standard.
Un controllo ECG ed ecografico bidimensionale è necessario per alcuni mesi e meglio per qualche anno, a intervalli di 6 mesi.
L’ospedalizzazione è utile nella fase acuta.Per l’esecuzione delle vaccinazioni, sia quelle con virus
vivi attenuati sia le altre, viene consigliato di attendere 3 mesi dall’inizio della malattia.
Sindromi vasculitiche
La vasculite è un processo clinico-patologico caratterizzato dall’infiammazione e dal danno dei vasi sanguigni. Il processo interessa essenzialmentre il lume del vaso, per cui la lesione si associa a ischemia dei tessuti che ricevono sangue.
Sotto la denominazione di vasculiti sono comprese numerose malattie che hanno un comune denominatore: l’infiammazione dei vasi sanguigni.

CapItolo 31 Malattie reumatologiche638
La distribuzione delle lesioni vascolari comprende i piccoli vasi (capillari, arteriole e venule postcapillari), i vasi di medio calibro (arterie renali, mesenteriche e co-ronarie) e i grandi vasi (aorta e sue branche prossimali) (Tabella 31.24).
I sintomi legati all’alterazione di un vaso dipendono in primo luogo dalla sua grandezza e dalla sua sede.
Quando sono colpiti i piccoli vasi, non dotati di tonaca muscolare, il quadro più comune è rappresentato dalla porpora anafilattoide di Schönlein-Henoch. L’interes-samento delle arterie di media grandezza, dotate di to-naca muscolare, è caratteristico della poliarterite nodosa (malattia relativamente rara nel bambino) e della malattia di Kawasaki, relativamente frequente. Nell’arterite di Takayasu (detta anche malattia senza polsi) sono colpiti l’aorta e i grossi vasi: si tratta di un’affezione eccezionale nei bambini italiani.
L’interessamento flogistico dei vasi sanguigni avviene anche nel corso di numerose malattie reumatologiche, come il LES, la dermatomiosite e la sclerodermia.
Le cause di queste affezioni sono sconosciute: la ma-lattia di Schönlein-Henoch e la poliarterite nodosa pos-sono seguire un’infezione (per esempio epatite B), l’uso di farmaci o l’incontro con allergeni di vario tipo. Nella malattia da siero, le manifestazioni sono dovute alla de-posizione nella cute di complessi immuni.
Porpora di Schönlein-Henoch
Si tratta di una vasculite che interessa tutto l’organismo del bambino: la cute, con manifestazioni emorragico-ponfoidi nelle parti declivi, le articolazioni, con tumefazioni e dolore, l’intestino, con dolori ed emorragie, e il rene, di frequente con minimi segni di sofferenza e di rado con un’evoluzione nettamente sfavorevole.
L’incidenza va da 14 a 20 casi/100.000 bambini per anno.
In circa il 90% di tutti i casi, la porpora di Schönlein-Henoch (PSH) compare in bambini da 3 a 11 anni. Col-pisce prevalentemente il sesso maschile con un rapporto maschi/femmine di 1,8:1,2.
L’eziologia non è ancora chiara. Il 75% dei pazienti ha una storia di infezione delle vie aeree superiori risa-lente a giorni prima dell’inizio della sindrome. Il ruolo di Bartolonella henselae nell’eziologia della porpora sembra essere elevato (67%) in confronto alle altre cause infettive. Anche la faringite streptococcica sem-bra avere un ruolo nella genesi della malattia, ma la ricerca degli anticorpi specifici fra i soggetti che hanno sofferto della malattia non è diversa da quella del resto della popolazione. In qualche caso la malattia ha fatto seguito a una vaccinazione o alla somministrazione di alcuni farmaci (penicillina, ampicillina, eritromicina, chinidina, chinino), all’uso di alcuni cibi, all’esposizione al freddo e alla puntura di insetti. La malattia è rara nei mesi estivi.
La patogenesi sembra dovuta a una vasculite IgA-me-diata dei piccoli vasi negli organi interessati (cute, rene, articolazioni, intestino). Si ritiene che una stimolazione antigenica sconosciuta determini un aumento del livello di IgA, specialmente IgA1, attivando le diverse vie pato-genetiche che portano alla vasculite necrotizzante. Lin-fociti, neutrofili, complessi immuni circolanti, IgA, C3 e fibrina infiltrano tutti i vasi dell’organismo, in quantità più o meno intensa. La lesione renale non si distingue istopatologicamente dalla nefropatia da IgA (malattia di Berger): probabilmente nell’ospite immaturo i complessi immuni circolanti possono causare il quadro diffuso a tutto l’organismo, mentre nell’adulto essi possono dar luogo soltanto alle manifestazioni renali della malattia. Alleli HLA-B34 e HLA-DRB1*01 sono legati alla nefrite in corso di PSH.
Delle manifestazioni cliniche, quella che costituisce l’aspetto tipico è la comparsa di un esantema emorragico-ponfoide (non dovuto a piastrinopenia), associato a do-lori addominali, artrite e nefrite. La malattia può seguire decorsi diversi (Tabella 31.25).
Poiché non esiste una prova che sia specifica del-la malattia, la diagnosi viene posta soltanto quando compare il tipico esantema, ma probabilmente vi sono forme che si presentano anche senza di esso; comunque al momento attuale la malattia è costantemente carat-terizzata dall’esantema emorragico-ponfoide (100% dei casi), che nel 50% dei casi costituisce il segno di esordio (Tabella 31.26).
Le lesioni sono estese agli arti inferiori, alle natiche e di rado agli arti superiori. Classicamente sono costituite da ponfi orticarioidi, da maculo-papule eritematose, da vere e proprie petecchie palpabili e lesioni emorragiche più grandi, che complessivamente, per il loro numero,
Dimensione dei vasi e vasculiti
Calibro del vaso Malattie
Prevalentemente dei piccoli vasi
granulomatose, aNCa-positiveGranulomatosi di WegenerSindrome di Churg-StraussNon granulomatosePoliangite microscopicaPorpora di Schönlein-HenochVasculite leucocitoclastica isolata cutaneaVasculite orticariosa ipocomplementemica
Vasi di medio calibro Malattia di KawasakiPoliarterite nodosa dell’infanziaPoliarterite nodosa cutanea
Grossi vasi Arterite di Takayasu
tabella 31.24

639CapItolo 31 Malattie reumatologiche
meritano la denominazione di porpora (Figura 31.19). Le lesioni sono simmetriche. Le zone emorragiche cambiano di colore (dal rosso al verde, al giallo) con il passare dei giorni, fino a svanire in 3-10 giorni. La comparsa di nuove lesioni avviene giorno dopo giorno, per cui si ritrovano manifestazioni in fase diversa di evoluzione. L’esantema può essere transitorio o persistere, con nuove gittate, per settimane. A volte recidiva anche a distanza di 4 mesi. La posizione eretta ne aumenta l’intensità e la durata, almeno nella fase iniziale della malattia.
A questo tipo di lesioni si associa un angioedema nel 20-40% dei pazienti, che non lascia il segno della fovea e può interessare anche le labbra, le palpebre, il dorso delle mani e dei piedi, la parte bassa della schiena, il perineo e lo scroto.
A carico delle articolazioni nel 68-75% dei casi si può avere artralgia o vera e propria artrite (nelle sue 5 clas-siche componenti: tumefazione, rossore, calore, dolore e alterazione della funzione), che talvolta (un quarto dei casi) precede la comparsa dell’esantema. L’interes-samento è soprattutto periarticolare e non si verificano mai, all’interno dell’articolazione, né un versamento sieroso, né uno emorragico. Le anche e le caviglie sono le articolazioni più spesso colpite; l’artralgia o l’artrite sono assolutamente transitorie, ma possono ricomparire quando la malattia ricada; esse preferenzialmente sono localizzate in vicinanza dell’esantema. Sono spesso sim-metriche.
Accanto all’esantema e ai dolori articolari, compongono il quadro clinico i dolori addominali, che si manifestano in circa l’80% dei casi e si accompagnano a diarrea, ileo paralitico, melena. Di rado le manifestazioni addominali precedono l’esantema (15% dei casi). Più spesso i dolori addominali assumono il carattere della colica, che si può associare a vomito. Nella metà dei casi le feci mostrano la presenza di sangue visibile a occhio nudo o occulto.
Sintomi e segni della porpora di Schönlein-Henoch
organo o apparato interessato
Sintomi e segni
Cuore Infarto del miocardio (vasculite coronarica)
Sistema nervoso centrale ApatiaSonnolenzaCefaleaIperattivitàIrritabilità
Apparato gastrointestinale VomitoDolori addominali tipo colicaAppendicite acutaOcclusione intestinale per invaginazione
ileo-ilealeEmorragie gastrointestinali (ematemesi,
melena, presenza di sangue nelle feci)Epatomegalia, idrope della cistifelleaPancreatite
Apparato genitourinario Ematuria e proteinuriaEmaturia microscopicaSegni di nefrite e di nefrosiInteressamento edematoso dello scroto
Apparato muscoloscheletrico
Artralgia/artriteEmorragie all’interno del muscolo
Segni generali FebbreInfezione precedente
Apparato polmonare Emorragia polmonareVersamento pleurico sieroematico
Cute Maculo-papule eritematosePetecchie, porporaEdema sottocutaneoOrticaria
tabella 31.25
Criteri classificativi della porpora di Schönlein-Henoch
Criteri classificativi della European League against Rheumatism/Rheumatology European Society
Porpora palpabile (in assenza di coagulopatia o di piastrinopenia) e uno o più dei seguenti segni:
• doloreaddominalediffuso• artriteoartralgia• biopsiadimostranteladeposizioneprevalentediAgA
Criteri classificativi dell’American College of RheumatologyDevono essere presenti due dei seguenti criteri:• porporapalpabile• età≤20 anni• interessamentoaddominale:doloreaddominalepostprandiale,
diarrea ematica• biopsiadimostrantegranulocitiintramuralinellearteriole
e/o nelle venule
tabella 31.26
FIgura 31.19 - Sindrome di Schönlein-Henoch: porpora e tumefazione dei piedi. (Dalla collezione del prof. G. Baldasseroni.) L’immagine è visibile a colori sul sito www.mediquiz.it.

CapItolo 31 Malattie reumatologiche640
Si può avere ematemesi. Nella maggioranza dei casi, i dolori addominali sono dovuti alla fuoriuscita dai vasi di liquidi e sangue all’interno della parete intestinale, che può ulcerarsi e versare sangue nel lume: tutto questo è dovuto alla presenza di vasculite a carico dei rami dell’arteria mesenterica. La presenza di un ispessimento della parete dell’ileo, in seguito al versamento di liquido emorragico, interrompe il flusso delle normali onde peristaltiche, che, forzando il passaggio, possono portare a un’invaginazione, per lo più a sede ileo-ileale, come avviene nel 2-3% dei pazienti; in questi casi si ha una brusca intensificazione delle crisi di dolore addominale, che deve far sospettare un’occlusione e che richiede l’intervento del chirurgo pe-diatra. L’esame ecografico è diagnostico. La perforazione intestinale è rara.
L’interessamento renale iniziale è molto frequente (dal 40 al 50% dei casi), ma nella maggioranza dei casi è limitato a una lieve proteinuria e a ematuria micro-scopica: esso è assolutamente transitorio. Solo in meno dell’1% dei casi, la nefropatia diviene persistente e può portare all’insufficienza renale terminale. I pazienti che hanno un’evoluzione renale sfavorevole (macroe-maturia, proteinuria, segni di insufficienza renale), lo dimostrano nella maggioranza dei casi entro 1 mese dall’inizio della malattia: un danno renale permanen-te non si sviluppa mai nei casi con esame delle urine normale inizialmente, anche dopo molti anni. Nella maggior parte dei casi non c’è rapporto fra la gravità del quadro extrarenale e l’evoluzione renale sfavorevo-le. Alla biopsia renale, i casi a evoluzione sfavorevole dimostrano una grave glomerulonefrite con semilune, ipercellularità, sclerosi segmentale, fibrosi e presenza di depositi glomerulari diffusi, formati da IgA, C3, fibrina, IgG, properdina e IgM.
La partecipazione dello scroto alle lesioni cutanee avviene nel 2-35% dei casi a seconda delle statisti-che: per lo più si tratta di un ispessimento della parete scrotale per la presenza di liquido e di sangue. Tutto passa senza lasciare segni. In diagnosi differenziale bisogna pensare a una torsione del testicolo o delle sue appendici.
Nel corso della malattia non solo non si ritrova una piastrinopenia, ma spesso si ritrova addirittura una pia-strinosi.
Il sistema nervoso centrale può soffrire per la presenza di lesioni di tipo vasculitico: il suo interessamento è rela-tivamente raro.
La diagnosi dipende essenzialmente dall’anamnesi e dal quadro clinico. Gli esami di laboratorio possono essere utili, ma non sono diagnostici. L’esame delle urine è la prova più importante, da ripetere anche a distanza dalla guarigione della malattia (per almeno 1 anno), perché documenta un eventuale tardivo interessamento renale. La ricerca del sangue occulto nelle feci dimostra la partecipa-zione della parete intestinale alla malattia. Le IgA sieriche possono presentare un aumento dei valori.
Non esiste un trattamento specifico. Ogni farmaco che venisse preso dal bambino al momento della malattia, va, se è possibile, sospeso. I FANS si dimostrano utili nel ridurre il dolore articolare e quello addominale. I corticosteroidi hanno effetto nelle forme con manife-stazioni cutanee e addominali più gravi, ma non riducono la durata complessiva della malattia, né hanno alcuna influenza sull’evoluzione renale sfavorevole. L’associa-zione prednisone e azatioprina, iniziata precocemente nelle forme di nefrite grave, offre qualche possibilità di arrestare l’evoluzione delle lesioni renali. Nelle forme con complicazione renale, è richiesto comunque l’intervento del pediatra nefrologo.
In assenza di una malattia renale tardiva e di un interes-samento precoce del sistema nervoso centrale, la malattia ha sempre un’evoluzione favorevole, dopo una durata di 4-6 settimane.
Le donne che hanno avuto una storia di PSH durante l’infanzia sono ad aumentato rischio di complicazioni (proteinuria e ipertensione) durante la gravidanza, per cui devono essere monitorate in maniera più accurata.
Arterite di TakayasuFra le altre forme di vasculite, va ricordata la malattia di Takayasu, detta malattia senza polsi, una vasculite dei grandi vasi a eziologia sconosciuta, che interessa l’aorta e i suoi rami principali. È una malattia rara che colpisce soggetti da 10 a 40 anni. Interessa soprattutto il sesso femminile con un rapporto di 2-4 a 1.
L’eziologia è sconosciuta.Esiste una fase precedente la scomparsa del polso,
caratterizzata da febbre, stato di malessere, perdita di peso, cefalea, ipertensione, mialgie e artralgie. In qualche caso non è riconoscibile questa fase. Sono stati propo-sti dei criteri diagnostici precisi, riportati nella Tabel-la 31.27.
Il trattamento comprende i corticosteroidi, la ciclofo-sfamide, il metotrexato e la terapia biologica.
Nella maggioranza dei casi la malattia evolve con ricadute e miglioramenti. La sopravvivenza a 5 anni è del 93%, quella a 10 anni dell’87%.
Criteri per la diagnosi di malattia di Takayasu
Alterazioni angiografiche dell’aorta o dei suoi rami principali (convenzionali, TC, angiografia, RM) e almeno uno dei seguenti criteri:
• diminuitipolsiperifericiarteriosie/oclaudicatio intermittens• differenzedipressionedellearterieperiferichefrabraccia
e gambe >10 mmHg• soffidell’aortaodellesuebrancheprincipali• ipertensione
tabella 31.27

641CapItolo 31 Malattie reumatologiche
Vasculiti anticorpi anticitoplasma dei neutrofili-associateCome abbiamo visto, a questo gruppo corrispondono due malattie: la granulomatosi di Wegener e la malattia di Churg-Strauss, ambedue rare nel bambino, caratterizzate dalla presenza di anticorpi anticitoplasma dei neutrofili (ANCA) e dalla deposizione di complessi nei tessuti colpiti.
Si deve pensare alla granulomatosi di Wegener di fron-te a un bambino con sinusiti resistenti al trattamento, infiltrati polmonari e nefrite. Il quadro della malattia di Churg-Strauss è simile al precedente: ne differisce per la maggiore rarità e per l’assenza della componente sinusale.
Malattia mista del tessuto connettivo
Si tratta di una sindrome che accomuna i caratteri del LES con quelli dell’artrite idiopatica giovanile, della derma-tomiosite e della sclerodermia, a testimonianza che tutte queste malattie hanno una patogenesi comune.
Colpisce soprattutto soggetti di sesso femminile, in età superiore ai 6 anni. È una malattia molto rara.
Eritema nodoso
La malattia colpisce soprattutto soggetti in età superiore ai 6 anni, è caratterizzata dalla presenza di noduli ovalari (con l’asse più lungo parallelo a quello dell’arto), dolenti, di consistenza dura, coperti da cute lucida e tesa, ros-sa, calda al termotatto, rilevati sulla cute circostante, di grandezza da 1 a 6 cm di diametro. La sede più comune è la faccia anteriore della gamba; i noduli sono quasi sempre simmetrici e possono sorgere anche posterioremente, ai polpacci, alle cosce, alle natiche e perfino agli arti supe-riori. Spesso alla comparsa dei noduli si associano febbre, stato generale di sofferenza e artralgia.
Il nodulo ha un’evoluzione caratteristica: dopo 1 settimana il colore rosso-violaceo si attenua e il nodulo diminuisce di grandezza; successivamente il colore vira al marrone e al giallastro. La lesione scompare nell’arco di 3-4 settimane, ma nel frattempo ne possono insorgere altre in fase diversa di evoluzione (Figura 31.20).
La malattia, nel passato, era strettamente connessa con l’infezione tubercolare; oggi è più spesso legata a un’infezione streptococcica o a situazioni diverse, come un’infezione da virus di Epstein-Barr, la tularemia, la psittacosi, la leptospirosi, la malattia da graffio di gatto o, abbastanza spesso, all’uso di farmaci, sulfamidici a lenta eliminazione per primi. Può associarsi a malattie sistemiche reumatologiche.
La VES è elevata. La somministrazione di Aspirina in bambini di età superiore ai 9 anni, è sufficiente, come sintomatico.
Dolori di crescita
Circa il 10-20% dei bambini, soprattutto di 4-8 anni, soffre dei cosiddetti dolori di crescita, notturni, simme-trici, agli arti inferiori: si tratta di dolori muscoloschele-trici di tipo intermittente, che interessano soprattutto la parte anteriore delle cosce e i polpacci, risparmiando le articolazioni. I bambini li descrivono comunemente come dolori crampiformi, profondi, che insorgono dopo il tramonto o nella notte. Il dolore spesso sveglia il bambino, ma si risolve precocemente con il massag-gio e le carezze, per cui non è più presente al mattino. L’esame obiettivo è normale e la deambulazione non è alterata.
Essi sono da considerare come una situazione benigna, limitata nel tempo. Rappresentano una parte della sindro-me di amplificazione del dolore, di cui faranno parte pochi anni dopo i dolori addominali e più tardi la cefalea. Spesso uno dei due genitori, o ambedue, sono cefalalgici e hanno sofferto da bambini di dolori alle gambe o all’addome.
Nessun trattamento farmacologico è indicato: valgono solo la rassicurazione, l’informazione e genericamente l’igiene del sonno.
Fibromialgia
La fibromialgia primitiva giovanile è una comune sin-drome dolorosa muscoloscheletrica che interessa dal 10 al 20% dei bambini e degli adolescenti. Sono coinvolte almeno tre sedi (Figura 31.21); il dolore deve continuare per almeno 3 mesi, in assenza di malattie sottostanti. Gli esami di laboratorio sono normali e l’esame obiettivo
FIgura 31.20 - Eritema nodoso. (Dalla collezione della prof.ssa G. Bernini.) L’immagine è visibile a colori sul sito www.mediquiz.it.
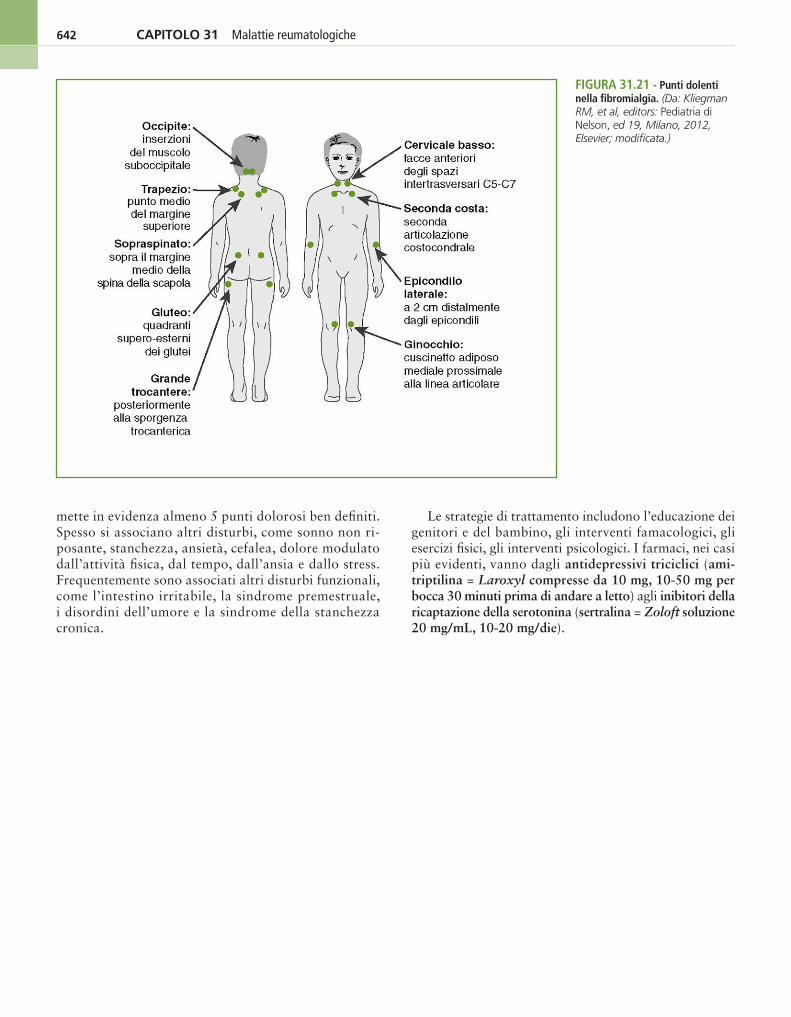
CapItolo 31 Malattie reumatologiche642
mette in evidenza almeno 5 punti dolorosi ben definiti. Spesso si associano altri disturbi, come sonno non ri-posante, stanchezza, ansietà, cefalea, dolore modulato dall’attività fisica, dal tempo, dall’ansia e dallo stress. Frequentemente sono associati altri disturbi funzionali, come l’intestino irritabile, la sindrome premestruale, i disordini dell’umore e la sindrome della stanchezza cronica.
Le strategie di trattamento includono l’educazione dei genitori e del bambino, gli interventi famacologici, gli esercizi fisici, gli interventi psicologici. I farmaci, nei casi più evidenti, vanno dagli antidepressivi triciclici (ami-triptilina = Laroxyl compresse da 10 mg, 10-50 mg per bocca 30 minuti prima di andare a letto) agli inibitori della ricaptazione della serotonina (sertralina = Zoloft soluzione 20 mg/mL, 10-20 mg/die).
FIgura 31.21 - Punti dolenti nella fibromialgia. (Da: Kliegman RM, et al, editors: Pediatria di Nelson, ed 19, Milano, 2012, Elsevier; modificata.)

C a p i t o l o
e31-1
Malattie reumatologiche
31
Metodi di studioBosch X, Guilabert A, Font J: Antineutrophil cytoplasmic
antibodies, Lancet 368:404–418, 2006. Corradi A, Tramontana F, Gattinara M, Lomater C: Artrite
reumatoide giovanile: diagnostica per immagini, Riv Ital Pediatr 20(Suppl):68–80, 1994.
Fischer A, Barone A, Musumeci S: Artrite reumatoide giovanile: le indagini di laboratorio, Riv Ital Pediatr 20(Suppl):59–67, 1994.
Hahn BH: Antibodies to DNA, N Engl J Med 338:1359–1366, 1998.
Principi di trattamentoBardare M, Corona F, Cimaz R, Cohen E: La terapia dell’artrite
cronica giovanile, Riv Ital Pediatr 20(suppl):81–91, 1994. Bathon JM, Martin RW, Fleischmann RM, et al: A comparison of
etanercept and methotrexate in patients with early rheumatoid arthritis, N Engl J Med 343:1586–1593, 2000.
Bongartz T, Sutton AJ, Sweeting MJ, et al: Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies, JAMA 295:2275–2285, 2006.
Breda L, Del Torto M, De Sanctis S, Chiarelli F: Biologics in children’s autoimmune disorders: efficacy and safety, Europ J Pediatr 170:157–167, 2011.
Cash JM, Klippel JH: Second-line drug therapy for rheumatoid arthritis, N Engl J Med 330:1368–1375, 1994.
Cassidy JT: Medical management of children with juvenile rheumatoid arthritis, Drugs 58:831–850, 1999.
Choi HK, Hernàn MA, Seeger JD, et al: Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study, Lancet 359:1173–1177, 2002.
Deeks JJ, Smith LA, Bradley MD: Efficacy, tolerability, and upper gastrointestinal safety of celecoxib for treatment of osteoarthritis and rheumatoid arthritis: sistematic review of randomised controlled trials, BMJ 325:619, 2002.
Deng G-M, Zheng L, Chan FK-M, Lenardo M: Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors, Nat Med 11:1066–1072, 2005.
Drigo I, Saccari A, Bacchin C, et al: Resistenza e ipersensibilità ai corticosteroidi Dalla clinica alle basi molecolari: andata e ritorno, Medico & Bambino 25:241–247, 2006.
Edwards JCW, Szczepanski L, Szechinski J, et al: Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis, N Engl J Med 350:2572–2581, 2004.
FitzGerald GA, Patrono C: The coxibs, selective inhibitors of cyclooxygenase-2, N Engl J Med 345:433–442, 2001.
Foel D, Wulffraat N, Wedderburn LR, et al: Methotrexate withdrawal at 6 vs 12 months in juvenile idiopathic arthritis in remission A randomized clinical trial, JAMA 303:1266–1273, 2010.
Franks ME, Macpherson GR, Figg MD: Thalidomide, Lancet 363:1802–1811, 2004.
Gedalia A, Shetty AK: Chronic steroid and immunosuppressant therapy in children, Pediatr Rev 25:425–434, 2004.
Giannini EH, Brewer EJ, Kuzmina N, et al: Methotrexate in resistant juvenile rheumatoid arthritis, N Engl J Med 326:1043–1049, 1992.
Gorman JD, Sack KE, Davis J: Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor a, N Engl J Med 346:1349–1356, 2002.
Graham DJ, Campen D, Hui R, et al: Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study, Lancet 365:475–481, 2005.
Hashkes PJ, Laxer RM: Medical treatment of juvenile idiopathic arthritis, JAMA 294:1671–1684, 2005.
Hawkey CJ: COX-2 inhibitors, Lancet 353:307–314, 1999.
Huppertz H-I, Tschammler A, Horwitz AE, Schwab KO: Intra-articular corticosteroids for chronic arthritis in children: efficacy and effects on cartilagine and growth, J Pediatr 127:317–321, 1995.
Jackson LM, Hawkey CJ: COX-2 selective nonsteroidal anti-inflammatory drugs Do they really offer any advantages? Drugs 59:1207–1216, 2000.
Jarvis B, Faulds D: Etanercept. A review of its use in rheumatoid arthritis, Drugs 57:945–966, 1999.
Kremer JM, Westhivens R, Leon M, et al: Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig, N Engl J Med 249:1907–1915, 2003.
Kugathasan S, Newman AJ, Dahms BB, Boyle JT: Liver biopsy findings in patients with juvenile rheumatoid arthritis receiving long-term, weekly methotrexate therapy, J Pediatr 128:149–151, 1996.
Ilowite NT: Current treatment of juvenile rheumatoid arthritis, Pediatrics 109:109–115, 2002.
Lehman TJA, Striegel KH, Onel KB: Thalidomide therapy for recalcitrant systemic onset juvenile rheumatoid arthritis, J Pediatr 140:125–127, 2002.
Lipsky PE, van der Heijde DM, St Clair EW, et al: Infliximab and methotrexate in the treatment of rheumatoid arthritis Anti-Tumor Necrosis Factor trial in rheumatoid arthritis with concomitant therapy Study Group, N Engl J Med 343:1594–1602, 2000.
Lovell DJ, Giannini EH, Reiff A, et al: Etanercept in children with polyarticular juvenile rheumatoid arthritis, N Engl J Med 342:763–769, 2000.

Malattie reuMatologiChee31-2
Maini R, St Clair EW, Breedveld F, et al: Infliximab (chimeric anti-tumor necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial ATTRACT Study Group, Lancet 354:1932–1939, 1999.
Mayer ML, Sandborg CI: Mellins ED: Role of pediatric and internist rheumatologists in treating children with rheumatic diseases, Pediatrics 113:e173–e181, 2004.
Marchetti F, Lenhardt A, Lazzerini M, et al: La talidomide, Medico & Bambino 22:517–523, 2003.
Markham A, Lamb HM: Infliximab. A review of its use in the management of rheumatoid arthritis, Drugs 50:1341–1349, 2000.
Oberholzer-Gee F, Inamdar SN: Merck’s recall of rofecoxib A strategic perspective, N Engl J Med 351:2147–2149, 2004.
O’Dell JR, Haire CE, Erikson N, et al: Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine and hydroxychloroquine, or a combination of all three medications, N Engl J Med 334:1287–1291, 1996.
Ramanan AV, Whitworth P, Baildam EM: Use of methotrexate in juvenile idiopathic arthritis, Arch Dis Child 88:197–200, 2003.
Ravelli A, De Benedetti F, Viola S, Martini A: Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine, J Pediatr 128:275–278, 1996.
Rhen T, Cidlowski JA: Antiinflammatory action of glucocorticoids – New mechanism for old drugs, N Engl J Med 353:1711–1723, 2005.
Scott DL, Kingsley GH: Tumor necrosis factor inhibitors for rheumatoid arthritis, N Engl J Med 355:704–712, 2006.
Silverman E, Mouy R, Spiegel L, et al: Leflunomide or methotrexate for juvenile rheumatoid arthritis, N Engl J Med 352:1655–1656, 2005.
Simon LE, Weaver AL, Graham DY, et al: Anti-inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis: a randomised controlled trial, JAMA 282:1921–1928, 1999.
Smolen JS, Kalden JR, Scott DL, et al: Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: a double-blind, randomised, multicentre trial, Lancet 353:259–266, 1999.
Traversa G, Bianchi C, Pisetzky F, Rossi M: Nuovi antinfiammatori in medicina generale, BEN 15(giugno), 2002.
Vanags D, Williams B, Johnson B, et al: Therapeutic efficacy and safety of chaperonin 10 in patients with rheumatoid arthritis: a double-blind randomised trial, Lancet 368:855–863, 2006.
Weinblatt ME, Kremer JM, Bankhurst AD, et al: A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate, N Engl J Med 340:253–259, 1999.
Artrite idiopatica giovanileBuoncompagni A, Loy A, Picco P, et al: L’artrite idiopatica
giovanile, Area Pediatr 9(febbario:):I–XX, 2008. CDC: Direct and indirect costs of arthritis and other rheumatic
conditions United States 1997, MMWR 53:388–389, 2004. Cho JH, Gregersen PK: Genomics and the multifactorial nature of
human autoimmune disease, N Engl J Med 265:1612–1623, 2011.
Choy EHS, Panavi GS: Cytokine pathways and joint inflammation in rheumatoid arthritis, N. Engl J Med 344:907–916, 2001.
Falcini F, Alessio M, Cortis E, et al: Diagnosi differenziale delle monoartriti nel bambino e adolescente, Quaderni Pediatr 2:163–169, 2003.
Fantini F: Recenti acquisizioni sulla immunogenetica dell’artrite reumatoide giovanile, Riv Ital Pediatr 20(suppl):5–16, 1994.
Fautrel B, Bourgeois P: Affections rhumatismales Généralités, Drugs 59(Special issue 1):1–9, 2000.
Gerloni V: L’interessamento oculare nell’artrite reumatoide giovanile, Riv Ital Pediatr 20(suppl):41–51, 1994.
Jacobsson LTN, Jaconsson ME, Askling J, Knowler WC: Perinatal characteristics and risk of rheumatoid arthritis, BMJ 326:1068–1069, 2003.
Martini A: Polimorfismo clinico, Riv Ital Pediatr 20(suppl): 26–35, 1994.
Martini A: L’artrite idiopatica giovanile, Prospet Pediatr 38:52–58, 2008.
McGhee JL, Burks FN, Sheckels JL, Jarvis JN: Identifying children with chronic arthritis based on chief complaints: absence of predictive values for muscoloskeletal pain as an indicator of rheumatic disease in children, Pediatrics 110:354–359, 2002.
McInnes IB, Schett G: The pathogenesis of rheumatoid arthritis, N Engl J Med 365:2205–2219, 2011.
Prakken, Albani S, Martini A: Juvenile idiopathic arthritis, Lancet 2138–2149, 2011.
Prince FHM, Otten MH, van Suijlekom-Smit LWA: Diagnosis and management of juvenile idiopathic arthritis, BMJ 342:95–102, 2011.
Rabinovich CE: Rheumatic diseases in children. Nelson texbook of pediatrics, ed 19, USA, 2011, Elsevier, pp 829.
Ravelli A, Magni Manzoni S, Moretti C, Martini A: Interessamento cerebrale in corso di connettiviti, Riv Ital Pediatr 27:684–687, 2001.
Ravelli A, Magni-Manzoni S, Pistorio A, et al: Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis, J Pediatr 146:598–604, 2005.
Ravelli A, Martini A: Juvenile idiopathic arthritis, Lancet 369:767–778, 2007.
Schanberg LE, Anthony KK, Gil KM, et al: Family pain history predict child health status in children with chronic rheumatic disease, Pediatrics 108:p.e47, 2001.
Scholz S, Albert ED: Immunogenetics aspects of juvenile chronic arthritis, Clin Exper Rheumat 11(suppl 9):37–41, 1993.
Sewell KL, Trentham DE: Pathogenesis of rheumatoid arthritis, Lancet 341:283–286, 1993.
Schneider R, Lang BA, Reilly BJ, et al: Prognostic indicators of joint destruction in systemic-onset juvenile rheumatoid arthritis, J Pediatr 120:200–205, 1992.
Sullivan KE: Inflammation in juvenile idiopathic arthritis, Pediatr Clin North Am 53:335–358, 2005.
Svendsen AJ, Holm NV, Kyvik K, et al: Relative importance of genetic effects in rheumatoid arthritis: historical cohort study of Danish nationwide twin population, BMJ 324:264–266, 2002.
Wu EJ, Van Mater HA, Rabinovich CE: Juvenile idiopathic arthritis. Nelson textbook of pediatrics, ed 19, USA, 2011, Elsevier, pp 829–839.
Zannin ME, Martini G, Pinello L, et al: L’uveite nell’artrite idiopatica giovanile Un esempio di collaborazione tra pediatra e oculista, Medico & Bambino 23:383–388, 2004.
Artrite reattivaAhmed S, Ayoub EM: Poststreptococcal reactive arthritis, Pediatr
Infect Dis J 20:1081–1082, 2001. Ayoug EM, Majeed HA: Poststreptococcal reactive arthritis,
Current Opin Rheumatol 12:306–310, 2000. Barash J, Mashiach E, Navon-Elkan P, et al: Differentiation of
post-streptococcal reactive arthritis from acute rheumatic fever, J Pediatr 153:696–699, 2008.
Calabrese LH, Naides SJ: Virtal arthritis, Infect Dis North Am 19:963–980, 2005.
Giani T, Falcini F: L’artrite transitoria dell’anca, Area Pediatr 7, 2006, aprile.
Petersel DL, Sigal LH: Reactive arthritis, Infect Dis North Am 19:863–883, 2005.

e31-3Malattie reuMatologiChe
Townes JM: Reactive arthritis after enteric infections in the United States. The problem of definition, Clin Infect Dis 50:247–254, 2010.
Spondiloartrite anchilosanteBurgos-Vargas R: Juvenile onset spondyloarthropathies:
therapeutic aspects, Arch Dis Child 88:212–218, 2003. Dougados M, Baeten D: Spondyloarthritis, Lancet 377:2127–
2137, 2011. Lenhardt A, Marchetti F, Cont G, et al: Le spondiloartropatie in
età pediatrica, Medico e Bambino 25:429–437, 2006.
Lupus eritematoso sistemicoArbuckle MR, McClain MT, Rubertone MV, et al: Development
of autoantibodies before the clinical onset of systemic lupus erythematosus, N Engl J Med 349:1526–1533, 2003.
Asanuma Y, Oeser A, Shintani AK, et al: Premature coronary-artery atherosclerosis in systemic lupus erythematosus, N Engl J Med 349:2407–2415, 2003.
Buyon JP: Neonatal lupus: bedside to bench and back, Scand J Rheumatol 25:271–276, 1996.
Cameron JS: Lupus nephritis in childhood and adolescence, Pediatr Nephrol 8:230–XXX, 1994.
D’Cruz DP, Khamashta MA, Hughes GRV: Systemic lupus erythematosus, Lancet 369:587–596, 2007.
Dooley MA, Jayne D, Ginzel EM, et al: Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis, N Engl J Med 365:1886–1895, 2011.
Gottlieb BS, Howite NT: Systemic lupus erythematosus in children and adolescent, Pediatr Rev 27:323–329, 2006.
Iqbal S, Sher MR, Good RA, Cawkwell GD: Diversità in presenting manifestations of systemic lupus erythematosus in children, J Pediatr 135:500–505, 1999.
Lang BA, Silverman EP: A clinical overview of systemic lupus erythematosus in childhood, Pediatr Rev 14:194–201, 1993.
Massengill SE, Richard GA, Donnelly WH: Infantile systemic lupus erythematosus with onset simulating congenital nephrotic syndrome, J Pediatr 124:27–XXX, 1994.
CDC: Trends in deaths from lupus erythematosus – United States, 1979-1998, MMWR 51:371–374, 2002.
Meyer O: Neonatal cutaneous lupus and congenital heart block: it’s not all antibodies, Lancet 362:1596–1597, 2003.
Mills JA: Systemic lupus erythematosus, N Engl J Med 330:1871–1879, 1994.
Navarra SV, Guzmán RM, Gallacher AE, et al: Efficacy and safety of belimumab in patients with active systemic lupus erythematosus a randomised, placebo-controlled, phase 3 trial, Lancet 377:721–731, 2011.
Petri M, Kim MY, Kalunian KC, et al: Combined oral contraceptive in women with systemic lupus erythematosus, N Engl J Med 353:2550–2558, 2005.
Pisetsky DS, Gilkeson G, Clair EW: Systemic lupus erythematosus Diagnosis and treatment, Med Clin North Am 81:113–128, 1997.
Ravelli A, Magni Manzoni S, Moretti C, Martini A: Interessamento cerebrale in corso di connettiviti, Riv Ital Pediatr 27:684–687, 2001.
Roman MJ, Shanker B-A, Davis A, et al: Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus, N Engl J Med 349:2399–2406, 2003.
Ruiz-Irastorza G, Khamashta MA, Castellino G, Hughes GRV: Systemic lupus erythematosus, Lancet 357:1027–1032, 2001.
Shakoor N, Michalska M, Harris CA, Block JA: Drug-induced systemic lupus erythematosus associated with etanercept therapy, Lancet 359:579–580, 2002.
Seaman DE, Londino AV, Kwoh CK, et al: Antiphospholipids antibodies in pediatric systemic lupus erythematosus, Pediatrics 96:1040–1045, 1995.
Traynor AE, Schroeder J, Rosa RM, et al: Treatment of severe systemic lupus erythematosus with high-dose chemotherapy and haemopoietic stem-cell transplantation: a phase I study, Lancet 356:701–707, 2000.
Tsokos GC: Systemic lupus erythematosus, N Engl J Med 365:2110–2121, 2011.
Willems M, Haddad E, Niaudet P, et al: Rituximab therapy for childhood-onset systemic lupus erythematosus, J Pediatr 148:623–627, 2006.
Sindrome antifosfolipidiAvcˇin T, Cimaz R, Silverman BD, et al: Pediatric antiphospholipid
syndrome: clinical and immunologic features of 121 patients in an international registry, Pediatrics 122:e1100–e1107, 2008.
Dermatomiosite giovanileBlock JA, Sequeira W: Raynaud’s phenomenon, Lancet 357:2042–
2048, 2001. Callen JP: Dermatomyositis, Lancet 355:53–57, 2000. Dalakas MC, Illa I, Dambrosia SA, et al: A controlled trial of
high-dose intravenous immune-globulin infusions as treatment for dermatomyositis, N Engl J Med 329:1993–2000, 1993.
Dalakas MC, Hohlfeld R: Polymyositis and dermatomyositis, Lancet 362:971–982, 2003.
Levy DM, Bingham CA, Kahn PJ, et al: Favorable outcome of juvenile dermatomyositis treated without systemic corticosteroids, J Pediatr 156:302–307, 2010.
SclerodermiaBlock JA, Sequeira W: Raynaud’s phenomenon, Lancet 357:2042–
2048, 2001. Charles C, Clements P, Furst DE: Systemic sclerosis:
hypothesis-driven treatment strategies, Lancet 367:1683–1691, 2006.
Gabrielli A, Avvedimento EV, Krieg T: Scleroderma, N Engl J Med 360:1989–2001, 2009.
Leighton C: Drug treatment of sclerodermia, Drtugs 61:419–427, 2001.
Nigrovic PA, Fuhlbrigge RC, Sundel RP: Raynaud’s phenomenon in children: a retrospective review of 123 patients, Pediatrics 111:715–721, 2003.
Svegliati Baroni S, Santillo M, Bevilacqua F, et al: Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis, N Engl J Med 354:2667–2676, 2006.
Tan FK: Autoantibodies against PDGF receptor in scleroderma, N Engl J Med 354:2709–2711, 2006.
Vallongo C, Martoini G, Zulian F: Quando la pelle si fa dura: la sclerodermia localizzata, Area Pediatr 9(gennaio):6–12, 2008.
Malattia di BehçetOzen S, Bilginer Y, Besbas N, et al: Behçet disease: treatment of
vascular involvement in children, Eur J Pediatr 427–430, 2010. Sakane T, Takeno M, Suzuki N, Inaba G: Behçet disease, N Engl J
Med 341:1284–1291, 1999.
Sindrome di SjögrenFox RI: Sjögren’s syndrome, Lancet 366:321–331, 2005. Ng WF, Browman SJ, Griffiths B: UKPSSR study group: United
Kingdon primary Sjögren’s syndrome registry-a united effort to tackle an orphan rheumatic disease, Rheumatology 50:32–39, 2011.
Sindromi periodiche febbrili ereditarieAdachi M, Watanabe A, Nishiyama A, et al: Familiar cases of
periodic fever with aphthous stomatitis, pharyngitis, and cervical adenitis syndrome, J Pediatr 158:155–159, 2011.
Aksentijevich I, Torosyan Y, et al: Familial mediterranean fever at the millennium: clinical spectrum, ancient mutations, and a survey of 100 american referrals to the National Institutes of Health, Medicine 77:268–297, 1998.

Malattie reuMatologiChee31-4
Besoli G: Gruppo di Studio sulla PFAFA del Friuli-Venezia Giulia: Incidenza e storia naturale della PFAFA in Friuli-Venezia Giulia: una ricerca collaborativa dei PdF, Medico e Bambino 20:234–238, 2001.
Brick R, Shinavi M, Kepten I, et al: Familial mediterranean fever: clinical and genetic characterization in a mixed pediatric population of jewish and arab patients, Pediatrics 103:p.e70, 1999.
Chandy CJ, Gilsdorf JR: Recurrent fever in children, Pediatr Infect Dis J 21:1071–1080, 2002.
Cimaz R, Casadei E, Meregalli P, Careddu P: Febbri periodiche e febbri ricorrenti a confronto: prospettive terapeutiche, Riv Ital Pediatr 27:622–625, 2001.
D’Agaro P, Panizon F, Ventura A, Zucconi E: La tonsillite ricorrente e la tonsillite focale: una rivisitazione, Medico & Bambino 20:231–234, 2001.
Drenth JPH, van den Meer JWM: Hereditary periodic fever, N Engl J Med 345:1748–1757, 2001.
D’Osualdo A, Ferlito F, Prigione I, et al: Neutrophils from patients with TNFRSF1A mutations display resistance to Tumor Necrosis Factor-induced apoptosis: pathogenetic and clinical implications, Arthritis Rheum 54:998–1008, 2006.
Drenth JP, van den Meer JW: Hereditary periodic fever, N Engl J Med 345:1748–1757, 2001.
Drenth JP, van der Meer JW: The inflammasome – A linebacker of innate defense, N Engl J Med 355:730–732, 2006.
Garavello W, Romagnoli M, Gaini RM: Effectiveness of adenotonsillectomy in PFAFA syndrome: a randomized study, J Pediatr 155:250–253, 2009.
Gattorno M, Martini A: Inherited autoinflammatory syndromes: an expanding new group of chronic inflammatory diseases, Clin Exp Rheumatol 23:133–136, 2005.
Gattorno M, Caorsi R, Meini A, et al: Differentiating PFAFA syndrome from monogenic periodic fever, Pediatrics 124:ed721–728, 2009.
Goldbach-Mansky R, Dailey NJ, Canna SW, et al: Neonatal-onset multisystem inflammatory disease responsive to interleukin-1b inhibiton, N Engl J Med 355:581–592, 2006.
Kallinich T, Haffner D, Niehues T, et al: Colchicine use in children and adolescent with familial mediterranean fever: literature review and consensus statement, Pediatrics 119:e474–e483, 2007.
Kastner D: Pathogenesis and diagnostic tools in the hereditary periodic fever, Riv Ital Pediatr 27:618–621, 2001.
Long SS: Syndrome of periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAFA) – What it isn’t What is it? J Pediatr 135:1–5, 1999.
Meini A, Cattalini M, Lougaris V, et al: Le febbri periodiche in età pediatrica: approccio diagnostico, Area Pediatr 8(settembre):I–XVII, 2007.
Mimouni A, Magal N, Stoffman N, et al: Familial mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis, Pediatrics 105: e70, 2000.
Obici L, Manno C, Muda AO, et al: First report of systemic reactive (AA) amyloidosis in a patient with the hyperimmunoglobulinemia D with periodic fever syndrome, Arthritis Rheum 50:2966–2969, 2004.
Ozen S: A new insight into periodic and recurrent fever Clinical aspects and differential diagnosis of periodic fever syndromes, Riv Ital Pediatr 27:616–617, 2001.
Ozen S: Familial mediterranean fever: revisiting an ancient disease, Eur J Pediatr 162:449–454, 2003.
Padeh S, Brezniak N, Zemer D, et al: Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome: clinical characteristics and outcome, J Pediatr 135:98–101, 1999.
Padeh S, Livneh A, Pras E, et al: Familial mediterranean fever in the first two years of life: a unique phenotype of disease in evolution, J Pediatr 156:985–989, 2010.
Picco P, Ceccherini I, Martini A: The inherited periodic fever syndromes, Ital J Pediatr 28:285–291, 2002.
Pietrogrande MC, Dellepiane RM, Panisi C, Bardare M: Ipergammaglobulinemia D e febbre periodica: una sindrome da conoscere, Riv Immunol Allergol Pediatr 13:105–110, 1999.
Reddy S, Jia S, Geoffrey R, et al: An autoinflammatory disease due to homozygous deletion of the IL1RN locus, N Engl J Med 360:2438–2444, 2009.
Salisbury FT: Hyperimmunoglobulinemia D and periodic fever syndrome (HIDS) in a child with normal serum IgD, but increased serum IgA concentration, J Pediatr 143:127–129, 2003.
Thomas KT, Feder HM, Lawton AR, Edwards KM: Periodic fever syndrome in children, J Pediatr 135:15–21, 1999.
Weyhreter H, Schwartz M, Kristensen D, et al: A new mutation causing autosomal dominant periodic fever syndrome in a danish family, J Pediatr 142:191–193, 2003.
SarcoidosisBaughman RP, Lower EE, du Bois RM: Sarcoidosis, Lancet
361:1111–1118, 2003. Ho L-P, Urban BC, Thickett DR, et al: Deficienct of a subset
of T-cells with immunoregulatory properties in sarcoidosis, Lancet 365:1062–1072, 2005.
Iannuzzi MCV, Rybicki BA, Teirestein AS: Sarcoidosis, N Engl J Med 357:2153–2165, 2007.
Iannuzzi MC, Fontana JR: Sarcoidosis. Clinical presentation, immunopathogenesis, and therapeutics, JAMA 305:391–399, 2011.
Wang C-L, Wu Y-T, Liu C-A, et al: Kawasaki disease Infection, immunity and genetics, Pediatr Infect Dis J 24:998–1004, 2005.
Malattia di KawasakiBaughman RP, Lower EE, du Bois RM: Sarcoidosis, Lancet
361:1111–1118, 2003. Belay ED, Maddox RA, Holman RC, et al: Kawasaki syndrome
and risk factors for coronary artery abnormalities United States, 1994-2003, Pediatr Infect Dis J 25:245–249, 2006.
Brogan PA, Bose A, Burgner D, et al: Kawasaki disease: an evidence based approach to diagnosis, and proposal for future research, Arch Dis Child 86:286–290, 2002.
Burns JC, Glodé MP: Kawasaki syndrome, Lancet 364:533–544, 2004.
Burns JC, Mason WH, Hauger SB, et al: Infliximab treatment for refractory Kawasaki syndrome, J Pediatr 146:662–667, 2005.
Chang R-KR: Hospitalizations for Kawasaki disease among children in the United States, 1988-1997, Pediatrics 109:p.e87, 2002.
Chang F-Y, Hwang B, Chen S-J, et al: Characteristics of Kawasaki disease in infants younger than six months of age, Pediatr Infect Dis J 25:241–244, 2006.
Dergun M, Kao A, Hauger SB, et al: Familial occurrence of Kawasaki syndrome in North America, Arch Pediatr Adolesc Med 159:876–881, 2005.
de Zorzi A, Colan SD, Gaivreau K, et al: Coronary artery dimensions may be misclassified as normal in Kawasaki disease, J Pediatr 133:254–258, 1998.
Fimbres AM, Shulman ST: Kawasaki disease, Pediatr Rev 29:308–315, 2008.
Gupta-Malhotra M, Gruber D, Abraham SS, et al: Atherosclerosis in survivors of Kawasaki disease, J Pediatr 155:572–577, 2009.
Harnden A, Alves B, Sheikh A: Rising incidence of Kawasaki disease in England: analysis of hospital admission data, BMJ 324:1424–1425, 2002.
Hirata S, Nakamura Y, Matsumoto K, Yanagawa H: Long-term consequences of Kawasaki disease among first-year junior high school students, Arch Pediatr Adolesc Med 156:77–80, 2002.

e31-5Malattie reuMatologiChe
Kao AS, Getis A, Brodine S, Burns JC: Spatial and temporal clustering of Kawasaki syndrome cases, Pediatr Infect Dis J 27:981–985, 2008.
Leung DYM, Meissner HC, Shulman ST, et al: Prevalence of superantigen-secreting bacteria in patients with Kawasaki disease, J Pediatr 140:742–746, 2002.
Marchesi A, Gonfiantini M, de Zorzi A, et al: La malattia di Kawasaki: diagnosi, stratificazione di rischio e strategie terapeutiche, Area Pediatr 7(maggio), 2006.
Marchesi A, Pongiglione G, Rimini A, et al: Malattia di Kawasaki: linee guida italiane, Prosp Pediatr 38:266–283, 2008.
Muta H, Ishii M, Sakaue T, et al: Older age is a risk factor for the development of cardiovascular sequelae in Kawasaki disease, Pediatrics 114:751–754, 2004.
Newburger JW, Takahashi M, Gerber MA, et al: Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professional from the Committee on rheumatic fever, endocarditis, and Kawasaki disease, Council on cardiovascular disease in the young, American Heart Association, Circulation 110:2747–2771, 2004.
Nomura Y, Yoshiunaga M, Masuda K, et al: Maternal antibody against toxic shock syndrome toxin-1 may protect infants younger than 6 months of age from developing Kawasaki syndrome, J Infect Dis 185:1677–1680, 2002.
Pellegrini MC, Taddio A, Ventura A, Lepore L: La malattia di Hawasaki: ancora una sfida per il pediatra, Medico e Bambino 30:236–241, 2011.
Popper SJ, Watson VE, Shimizu C, et al: Gene transcript abundance profiles distinguish Kawasaki disease from adenovirus infection, J Infect Dis 200:657–666, 2009.
Rowling AH: Incomplete (atypical) Kawasaki disease, Pediatr Infect Dis J 21:563–565, 2002.
Son MBF, Newburger JW: Kawasaki Disease. In Kliegman RM et al: Nelson Textbook of Pediatrics, ed 19, Philadelphia, USA, 2011, Elsevier Saunders, pp 862–867.
Son MB, Gauveau K, Burn JC, et al: Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study, J Pediatr 158:644–649, 2011.
Sung RYT, Ng Y-M, Choi K-C, et al: Lack of association of cervical lymphadenopathy and coronary artery complications in Kawasaki disease, Pediatr Infect Dis J 25:521–525, 2006.
Tashiro N, Matsubara T, Uccida M, et al: Ultrasonographic evaluation of cervical lymph nodes in Kawasaki disease, Pediatrics 109:e77, 2002.
Tse SML, Silverman ED, McCrindle BW, Yeung RSM: Early treatment with immunoglobulin in patients with Kawasaki disease, J Pediatr 140:450–455, 2002.
Wang C-L, Wu Y-T, Liu C-A, et al: Kawasaki disease, Infection, immunity and genetics, Pediatr Infect Dis J 24:998–1004, 2005.
Weyand CM, Goronzy JJ: Medium- and large-vessel vasculitis, N Engl J Med 349:160–169, 2003.
Wilder MS, Palinkas LA, Kao AS, et al: Delayed diagnosis by physicians contributes to the development of coronary artery aneurysms in children with Kawasaki syndrome, Pediatr Infect Dis J 26:256–260, 2007.
Yellen ES, Gauvrenau K, Takahaschi M, et al: Performance of 2004 American Heart Association recommendations for treatment of Kawasaki disease, Pediatrics 125:e234–241, 2010.
VasculitiPavan M, Marchetti F, Lepore L: Le vasculiti “maggiori” in età
pediatrica, Medico e Bambino 30:223–228, 2011.
Porpora di Schönlein-HenochAyoub EM, McBride J, Schmiederer M, Anderson B: Role of
Bartonella henselae in etiology of Henoch-Schönlein purpura, Pediatr Infect Dis 21:28–31, 2002.
Chartapisak W, Opastiraku S, Willis INS, et al: Prevention and treatment of renal disease in Henoch-Schönlein purpura: a systematic review, Arch Dis Child 94:132–137, 2009.
Davin J-C, Weening JJ: Henoch-Schönlein purpura nephritis: an update, Eur J Pediatr 160:689–695, 2001.
Foster BJ, Bernard C, Drummond KN, Sharma AK: Effective therapy for severe Henoch-Schönlein purpura nephritis with prednisone and azathioprine: a clinical and histopathologic study, J Pediatr 136:370–375, 2000.
Jauhola O, Ronkainen J, Koskimies O, et al: Clinical course of extrarenal symptoms in Henoch-Schönlein purpura; 6-month prospective study, Arch Dis Child 95:81–86, 2010.
Jauhola O, Ronkainen J, Koskimies O, et al: Renal manifestations of Henoch-Schönlein purpura in a 6-month prospective study of 223 children, Arch Dis Child 95:877–882, 2010.
Kato S, Ebina K, Naganuma H, et al: Intestinal IgA deposition in Henoch-Schöenlein purpura with severe gastro-intestinal manifestations, Eur J Pediatr 155:91–95, 1996.
McCarthy HJ, Tizard EJ: Diagnosis and management of Henoch-Schönlein purpura, Eur J Pediatr 169:643–650, 2010.
Narchi H: Risk of long term impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review, Arch Dis Child 90:916–920, 2005.
Orfila C, Lepert JC, Modesto A, et al: Henoch-Schöenlein purpura and membranoproliferative-like glomerulonephritis, Nephron 74:209–213, 1996.
Rizzoni G, El Hachem M, Diomedi Camassei F: La porpora di Schönlein-Henoch, OPGB. Form Cont Pediatr 1:17–28, 2006.
Ronkainen J, Nuutinen M, Koskimies O: The adult kidney 24 years after childhood Henoch-Schönlein purpura: a retrospective cohort study, Lancet 360:666–670, 2002.
Szer IS: Henoch-Schönlein purpura: when to treat and how, Riv Ital Pediatr 27:564–568, 2001.
Malattia di TakayasuOzen A, Duzova A, Bakkaloglu A, et al: Takayasu arteritis in
children: preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate, J Pediatr 150:72–76, 2007.
Vasculiti ANCA-associatePagnoux C, Mahr A, Hamidou MA, et al: Azathioprine or
methotrexate maintenance for ANCA-associated vasculitis, N Engl J Med 359:2790–2803, 2008.
FibromialgiaAnthony KK, Schanberg LE: Fibromyalgia. Textbook of
Pediatrics, ed. 19, USA, 2011, Elsevier, pp 878–879.


















