
Riepilogo 1^ lezione - IL SITO UFFICIALE DEGLI STUDENTI DI ... Spina BIOCHIMICA 1 2011.pdf · La...
96
Riepilogo 1^ lezione
Transcript of Riepilogo 1^ lezione - IL SITO UFFICIALE DEGLI STUDENTI DI ... Spina BIOCHIMICA 1 2011.pdf · La...

Riepilogo 1^ lezione

• I 20 amminoacidi che si trovano comunemente nelle proteine sono uniti l’uno all’altro da legami peptidici.
• La sequenza lineare degli amminoacidi legati contiene l’informazione necessaria a generare una proteina con una forma tridimensionale esclusiva.
• La struttura di una proteina è complessa: organizzazione in 4 livelli gerarchici(struttura primaria, secondaria, terziaria, quaternaria).
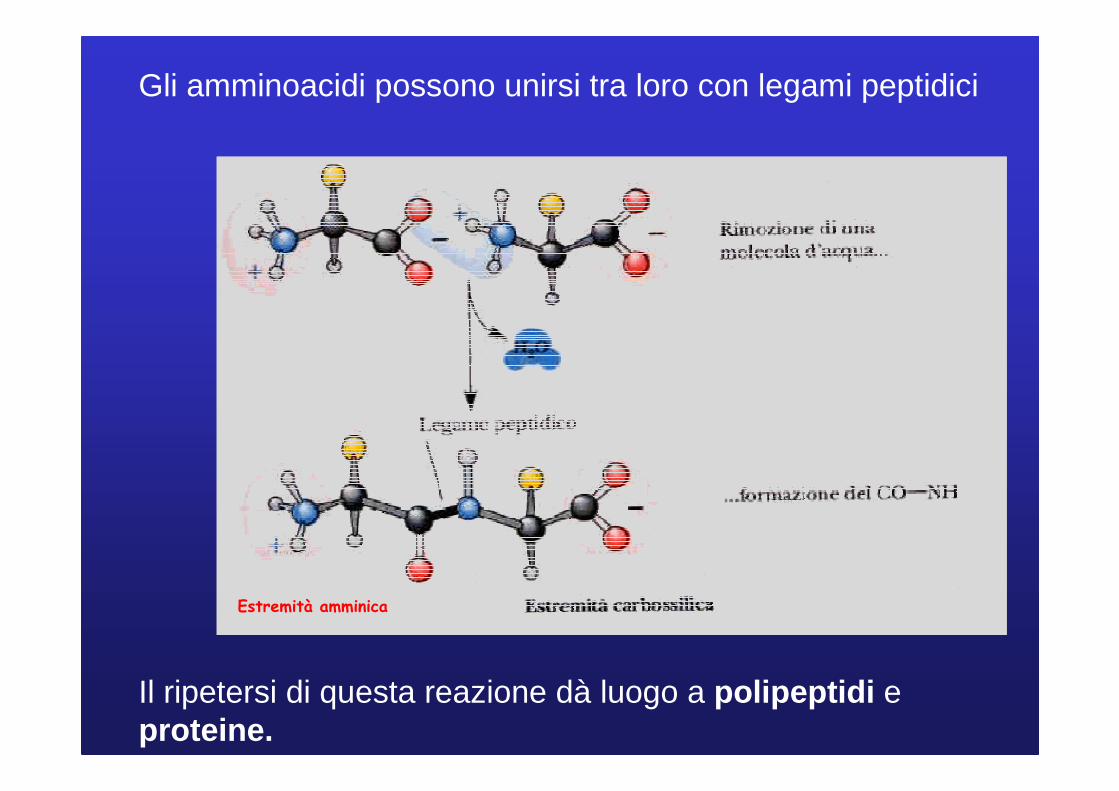
Gli amminoacidi possono unirsi tra loro con legami peptidici
Il ripetersi di questa reazione dà luogo a polipeptidi e proteine.
Estremità amminica

Proprieta’ del legame peptidico:Planare, ha una forza intermedia tra il legame semplice ed il
legame doppio.
C CNH
H
H
O
OH
R
C CNH
H
H
O
OH
R
+
CC
N
H
HH
O
RN
C C
H
O
OH
R
H

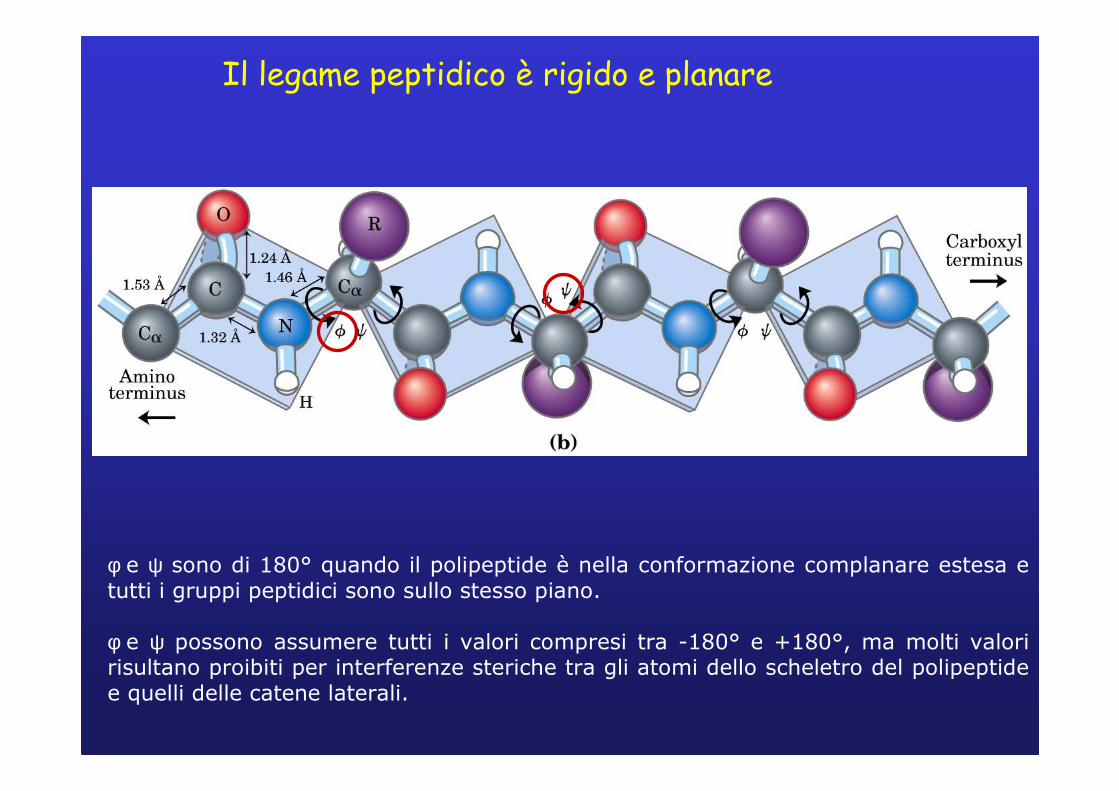
Il legame peptidico è rigido e planare
Gli atomi di Cα di amminoacidi adiacenti sono separati da tre legami covalenti: O H
Cα – C – N – Cα
PROPRIETA’ DEL LEGAME PEPTIDICO� I 6 atomi del gruppo peptidico giacciono sullo stesso piano →l’ossigeno legato al carbonio del gruppo carbonilico e l’atomo di idrogeno legato all’azoto amminico, si trovano in trans.� L’ossigeno carbonilico ha una parziale carica negativa e l’azoto amminico ha una parziale carica positiva → ciò genera un parziale dipolo elettrico.� I legami ammidici C-N hanno un parziale carattere di doppio legame per effetto della risonanza→ non possono ruotare liberamente.� La rotazione è permessa solo attorno ai legami N-Cα e Cα-C.
Il legame peptidico è rigido e planare
φ e ψ sono di 180° quando il polipeptide è nella conformazione complanare estesa e tutti i gruppi peptidici sono sullo stesso piano.
φ e ψ possono assumere tutti i valori compresi tra -180° e +180°, ma molti valori risultano proibiti per interferenze steriche tra gli atomi dello scheletro del polipeptide e quelli delle catene laterali.

Caratteristiche del legame peptidico• Ha il carattere di un doppio legame parziale (è più corto
di un legame singolo).• E’ rigido e planare (non è possibile la rotazione attorno
al legame tra il carbonio carbonilico e l’azoto del legame peptidico).
• In genere è un legame di tipo trans, a causa di interferenze steriche tra i gruppi -R (i legami tra un Cαe un gruppo α-amminico o α-carbossilico possono ruotare!)
• I gruppi -C=O ed -NH del legame peptidico non hanno una carica elettrica (a differenza del gruppo α-amminico all’estremità N-terminale ed α-carbossilico al C-terminale) ma sono polari e partecipano alla formazione di legami a idrogeno.

• I singoli amminoacidi in una catena peptidica sono chiamati residui amminoacidici.
• In genere le proteine sono composte da 50-2000 residui amminoacidi.
• La struttura primaria di una proteina èdefinita dalla sequenza lineare dei residui amminoacidici.

La peculiare sequenza amminoacidica di una catena polipeptidica rappresenta la struttura primaria
Lisozima

Struttura secondaria
• Si riferisce alla conformazione locale della catena polipeptidica.
• E’ determinata da interazioni di tipo legame a idrogeno fra l’ossigeno di un gruppo carbonilico del legame peptidico e l’idrogenodel gruppo ammidico di un altro legame peptidico.
• Esistono due tipi di strutture secondarie: l’ α-elica ed il foglietto β.

proteine: struttura secondariastrutture dovute ad interazioni “locali” di tipo ponte-H
α-elica• ponte-H ogni 3,6 aminoacidi•Il legame H siinstaura tra l’Hdell’azoto amidicoe l’O del gruppocarbonilico• residui esternialla spirale
β-foglietto• legami idrogeno fra aminoacidi di catene diverse• foglietto piegato

Struttura secondaria (αααα-elica)• E’ una struttura in cui la catena polipeptidica è
avvolta a spirale .• Le catene laterali degli amminoacidi (-R) si
protendono verso l’esterno rispetto all’asse della spirale.
• L’α-elica è stabilizzata da legami idrogenointracatena che si formano tra l’ossigeno carbonilico di un legame peptidico e l’idrogeno ammidico di un legame peptidico situato a 4 residui di distanza sulla catena.
• La prolina interrompe l’α-elica!!!• Gli amminoacidi con catene laterali (-R ) voluminose
o cariche possono interferire con la formazione dell’α-elica.

Struttura secondaria: alfa elica
Le proprietà idrofobiche o idrofiliche di una alfa-elicadipendono dalle catenelaterali degli aa
Legame idrogeno

Champe et al., Le basi della biochimica, Ed. Zanichelli
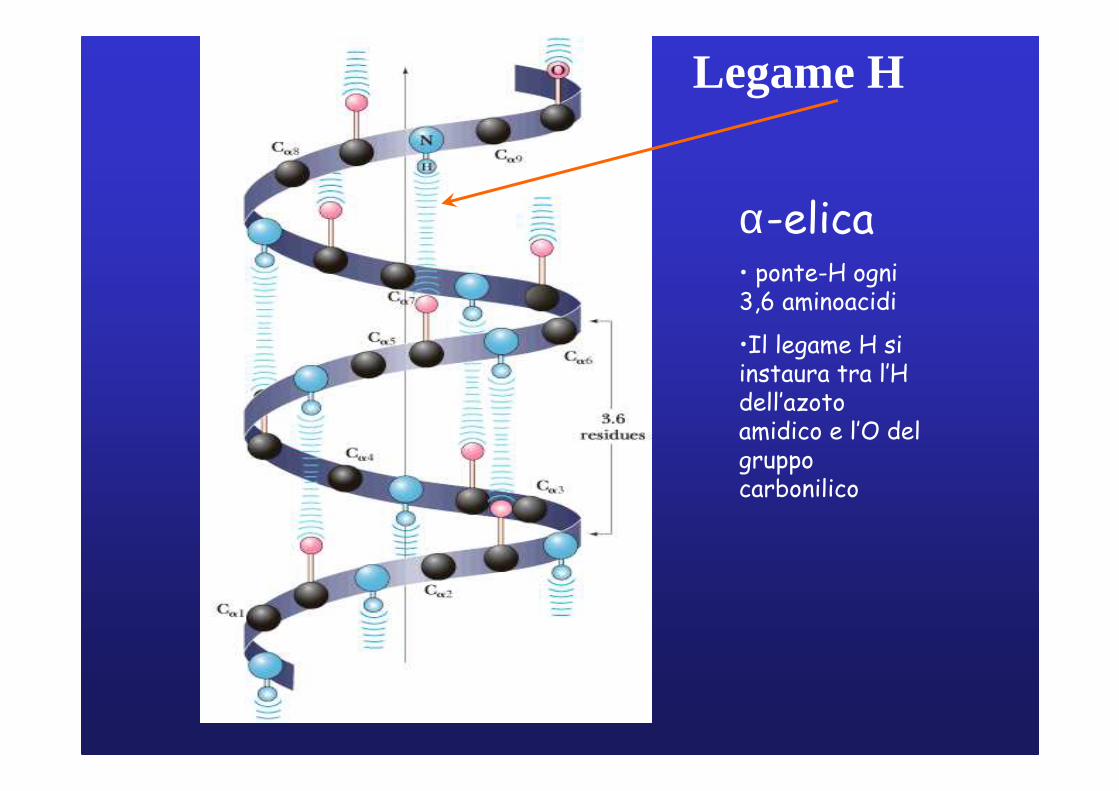
Legame H
α-elica• ponte-H ogni3,6 aminoacidi
•Il legame H siinstaura tra l’Hdell’azotoamidico e l’O del gruppocarbonilico

Esempio di proteina composta da alfa eliche

Struttura secondaria (foglietto ββββ)
• E’ una struttura ripiegata, formata da 2 o piùcatene polipeptidiche (filamenti) quasi completamente distese.
• I legami a idrogeno sono intercatena e perpendicolari allo scheletro del peptide.
• Tutti i componenti di un legame peptidico partecipano alla formazione di legami a idrogeno.
• Tali legami si realizzano tra l’ossigeno di un gruppo carbonilico di un legame peptidico e l’idrogeno del gruppo ammidico di un altro legame peptidico appartenente ad un filamento diverso.

Struttura secondaria: foglietto beta

Nei foglietti pieghettati ci sono ancora dei legami ad idrogeno, ma stavolta sono tra fogli adiacenti (sheet)

Struttura secondaria (foglietto β)
• I polipeptidi che formano un foglietto β possono disporsi in modo parallelo o anti-parallelo.
• Un foglietto β può essere formato anche da una singola catena polipeptidica ripiegata su se stessa: in tal caso i legami a H sono legami intracatena.
• La superficie dei foglietti β è “pieghettata”.

ββββ SheetStabilizzata da legami H intercatenatra N-H & C=O
2 OrientationsParallel
Not optimum H-bonds; less stable
Anti-parallel
Optimum H-bonds; more stable

Struttura secondaria(sequenze non ripetitive)
• Queste strutture non ripetitive non sono “casuali”.
• Hanno una forma meno regolare rispetto all’α-elica ed al foglietto β.
• La catena polipeptidica assume una conformazione ad anse ed avvolgimenti.

• Circa un terzo dei residui che costituiscono le proteine globulari sono coinvolti in ripiegamenti "a gomito" che invertono la direzione della catena polipeptidica alla superficie della molecola e rendono possibile la struttura globulare. Data la loro frequenza, questi ripiegamenti vengono classificati come terzo tipo di struttura secondaria oltre alle α-eliche e ai foglietti β.
• α-elica• Foglietto-β• β-turns
Esistono diversi tipi di ripiegamento che coinvolgono diversi residui. I più frequenti sono i β-turns (tradotti in italiano come curve β, ripiegamenti β, etc.), che spesso uniscono due filamenti β antiparalleli a formare un'ansa a forcina. I β-turns sono definiti da 4 residui che occupano le posizioni designate da i a i+3
• i• i+1• i+2• i+3• Nell'ambito dei β-turns si possono identificare diversi tipi, ma i più frequenti sono
quelli cosiddetti di Tipo I e Tipo II, anche se il Tipo I è 2-3 volte più frequente del Tipo II.

I ripiegamenti β sono frequenti nelle proteine . Le proteine globulari presentano circa 1/3 dei residui aa sotto forma di ripiegamenti o anse, dove la catena proteica inverte la sua direzione, i ripiegamenti β sono i più diffusi,collegano due estremità di un foglietto β antiparallelo, 4 residui con un angolazione di 180° e formazione di un ponte di H tra il Carbonile del I a. e l’H dell’N del IV amminoacido. Gly e Pro sono frequenti in questa conformazione.
Pro perché l’imminoacido promuove la forma cis degli a a. coinvolti nel legameGly perché piccola e flessibile

Super-secondary Structureβ-turns in una proteina permettono che eliche e
foglietti si allineino
βαββαββαββαβ αααααααα ββββ meander


Qui sopra sono riprodotte le 2 conformazioni descritte per la stessa proteina, la proteina prionica, la cui alterazione conformazionale provoca la cosiddetta malattia della Mucca Pazza(v.”Connessioni Biochimiche” , pag. 105 del Campbell-Farrell). A sinistra è illustrata la struttura a) della proteina prionica “buona”, che èpresente in moltissimi organismi, uomo compreso. A destra (b) è riportata la struttura della proteina prionica“cattiva”. Come si può constatare nella forma a) esistono 2 tratti ad α-elica, 1 a sinistra (verde), ed 1 a destra (rosso). Nel passaggio alla forma b) il tratto “verde” cambia ripiegamento e genera 2 tratti β antiparalleli. Pure il tratto “rosso” fa altrettanto e genera i 2 tratti “rossi” β antiparalleli nella forma b),producendo , in definitiva, un unico foglietto β formato da 4 tratti β contigui.


Le strutture primarie sono mantenute da legami peptidici.Le strutture secondarie sono mantenute da legami idrogeno tra atomi di residui aminoacidici.

• Primaria: la sequenza lineare degli amino acidi tenuti insieme da legami peptidici.
• Secondaria: l’organizzazione di parti di una catena polipeptidica (es.: l’α elica o il foglietto β), tenute da legami a ponte di H.
• Terziaria: la struttura tridimensionale completa di una catena polipeptidica, con molti tipi di legami e interazioni di cui solo uno covalente il ponte dS.
• Quaternaria: l’associazione di due o più polipeptidi in una struttura complessa multi-subunità
Quattro livelli di struttura determinano la forma di una proteina
Beta
alfa

collagene
mioglobina
Per funzionare una proteina deve assumere una struttura tridimensionale precisa

proteine: struttura terziariaDetermina la struttura 3DStabilizzata da
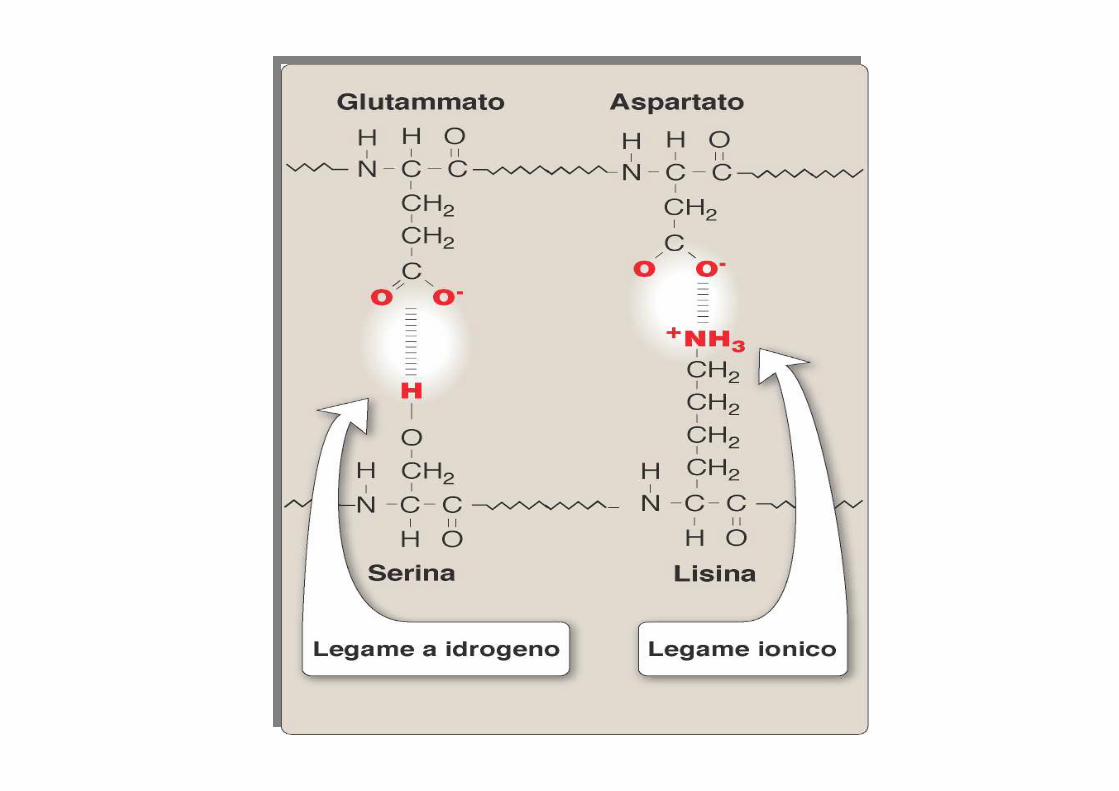
• ponti S-S• interazioni idrofobiche• interazioni elettrostatiche (legami ionici)• legami di Wan der Waals
Suscettibile di denaturazione-rinaturazione
• R apolari verso l’interno (eccetto in proteine integrali di membrana)
• R polari verso l’esterno (solvatati da H2O)

ponti disolfuro

Il Ponte Di-S si forma tra gruppi sulfidrilici adiacenti dicysteine (-S-H).
La formazione avviene mediante ossidazione, il taglio in residui disulfidici mediante riduzione.
Denatured inactive ribonuclease

Struttura terziaria : relazione a lungo raggio tra amminoacidi anche molto distanti tra di loro, I protagonisti sono i gruppi R che possono appartenere anche a filamenti con diverse strutture secondarie.



proteine: struttura terziariaeffetto dell’interazione idrofobica
Non polareCatena laterale
Legame a Hsi può formare sulla catenalaterale sullato esternodella molecola
Polipeptide senza struttura terziaria
Conformazione con struttura terziariain ambiente acquoso
Core idrofobico contenente catene laterali non polari
struttura 3D
ORIGINE
DELLA
STRUTTURA
TERZIARIA

L'effetto idrofobico è la forza motrice
del ripiegamento di una proteina.
La struttura terziaria si genera grazie alle interazioni tra i gruppi R, i quali si posizioneranno in risposta a attrazione o repulsione, generando
la struttura finale .

Le strutture terziarie sono sempre Le strutture terziarie sono sempre compattecompatte
La superficie delle proteine La superficie delle proteine èè polare polare mentre l'interno mentre l'interno èè prevalentemente prevalentemente apolare eccezione per le proteine di apolare eccezione per le proteine di membrana, dove membrana, dove èè ll ’’opposto.opposto.

L'avvolgimento della catena deve L'avvolgimento della catena deve
essere tale da esporre sempre al essere tale da esporre sempre al
solvente acquoso le catene laterali solvente acquoso le catene laterali
idrofile. Catene laterali cariche idrofile. Catene laterali cariche
possono trovarsi all'interno di una possono trovarsi all'interno di una
proteina solo se la loro carica netta proteina solo se la loro carica netta
viene neutralizzata.viene neutralizzata.

Legami responsabili della struttura Legami responsabili della struttura terziariaterziaria
Forze di Wan der Waals 1-2 Kcal/moleLegami a H 3-7 Kcal/moleLegami ionici 5 Kcal/moleLegami S-S 50 Kcal/mole



Le proteine con peso molecolare superiore a 50.000 sono OLIGOMERICHEOLIGOMERICHE
Sono costituite cioè da più
catene polipeptidiche
PROTOMERI O SUBUNITPROTOMERI O SUBUNITÀÀ

Le proteine oligomeriche presentano un ulteriore livello di organizzazione srutturale la struttura quaternaria
LA STRUTTURA QUATERNARIA descrive il modo in cui le singole catene polipeptidiche sono disposte l'una rispetto all'altra.

Biofisica
Classificazione generale delle struttureterziarie
Proteine con predominanza
di α elica
Proteine con predominanza di β sheets
Proteine miste

Per la sintesi di una catena polipeptidicadi 4000 residui aminoacidici è necessario un gene che contenga almeno 4OOO x 3 = 12.000 basi azotate.
Per la sintesi di 2O copie di una stessa catena polipeptidica di 200 residui è sufficiente un gene che contenga solo 200 x 3 = 600 basi

EmoglobinacollagenoEmoglobina e
Collageno

La struttura terziaria è generata dal ripiegamento e dalla conformazione della catena polipeptidica.
La struttura quaternaria è l’organizzazione di polipeptidi in un’unica unità funzionale che consiste di più di unasubunità polipeptidica.

Rappresentazioni grafiche differenti della stessa proteina



In alcune catene In alcune catene polipeptidichepolipeptidiche
particolarmente lunghe (piparticolarmente lunghe (pi ùù di 200 di 200
residui) si ritrovano 2 o piresidui) si ritrovano 2 o pi ùù zone zone
distinte (30distinte (30 --150 residui) a struttura 150 residui) a struttura
globulare e compatta, congiunte da globulare e compatta, congiunte da
segmenti di catena segmenti di catena polipeptidicapolipeptidica
relativamente flessibili.relativamente flessibili.
Queste strutture si definisconoQueste strutture si definiscono
DOMINI

LA STRUTTURA QUATERNARIA DI UNA PROTEINA PUÒ SUBIRE MODIFICHE CONFORMA ZIONALI REVERSIBILI AD OPERA DI LIGANDI, DEFINITI EFFETTORI ALLOSTERICI.
LE MODIFICHE CONFORMAZIONALI POSSONO ALTERARE LA FUNZIONE DI UNA PROTEINA, REALIZZANDO IN TAL MODO UN IMPORTANTE MECCANISMO DI CONTROLLO DELLA SUA ATTIVITÀBIOLOGICA.

Riepilogo 2^lezione

Le proteine oligomeriche presentano un ulteriore livello di organizzazione srutturale la struttura quaternaria
LA STRUTTURA QUATERNARIA descrive il modo in cui le singole catene polipeptidiche sono disposte l'una rispetto all'altra.


Biofisica
La struttura quaternaria riguarda proteine costituite da 2 o piùcatene polipeptidiche o da più domini strutturali (es. proteine regolatrici). E’ possibile classificare le proteine in due gruppi:Proteine fibrose con catene disposte in lunghi fasci o foglietti e Proteine globulari con catene polipeptidiche ripiegate a formare forme globulari o sferiche
Esempio: la emoglobina
La struttura quaternaria delle proteine
Le interazioni tra le subunità consentono grandi variazioni nell’attività catalitica

Biofisica
Per la sintesi di una catena polipeptidicadi 4000 residui aminoacidici è necessario un gene che contenga almeno 4OOO x 3 = 12.000 basi azotate.
Per la sintesi di 2O copie di una stessa catena polipeptidica di 200 residui èsufficiente un gene che contenga solo 200 x 3 = 600 basi

Biofisica
In alcune catene In alcune catene polipeptidichepolipeptidiche
particolarmente lunghe (piparticolarmente lunghe (pi ùù di di
200 residui) si ritrovano 2 o pi200 residui) si ritrovano 2 o pi ùù
zone distinte (30zone distinte (30 --150 residui) a 150 residui) a
struttura globulare e compatta, struttura globulare e compatta,
congiunte da segmenti di catena congiunte da segmenti di catena
polipeptidicapolipeptidica relativamente relativamente
flessibili.flessibili.
Queste strutture si definisconoQueste strutture si definiscono
DOMINI

Struttura terziaria (i domini)• Le catene polipeptidiche formate da più di 200 amminoacidi in
genere comprendono 2 o più piccole unità compatte: i domini.• I domini sono le unità strutturali e funzionali di una proteina.• Ciascun dominio è una regione globulare, compatta, che si
forma per la combinazione di più elementi strutturali secondari(α-eliche, foglietti β, sequenze non ripetitive).
• Strutturalmente, ciascun dominio è indipendente da altri domini della stessa catena polipeptidica.
• La struttura terziaria riguarda sia il ripiegamento di ciascun dominio sia la disposizione reciproca finale dei domini di un polipeptide.

Le proteine con peso molecolare superiore a 50.000 sono OLIGOMERICHEOLIGOMERICHE
Sono costituite cioè da più
catene polipeptidiche
PROTOMERI O SUBUNITPROTOMERI O SUBUNITÀÀ

Vantaggi della struttura quaternaria
Risparmio di DNA
Minimizzazione degli errori casuali durante la biosintesi proteica
Presenza di interazioni allosteriche

La Ferritina ha un peso molecolare di circa 480.000 daltons. Non è costituita da una sola catena polipeptidica di 400 a.a. ma di 20 catene identiche di circa 200 residui ciascuna.

LA GLICERALDEIDE-3-FOSFATO DEIDROGENASI È COSTITUITA DA 4 SUBUNITÀ IDENTICHE DI 330 RESIDUI (330 X 3 = 990 NUCLEOTIDI)
SE LA PROTEINA CONSISTESSE IN 1 CATENA DI (330 X 4) 1320 RESIDUI OCCORREREBBE UN GENOMA DI 3 X 1320=3960 NUCLEOTIDI.



Generalmente nella formazione delle proteine oligomeriche le subunitàdifettose sono scartate .

Nelle cellule le proteine si sintetizzano ad una velocità molto elevata.
Le cellule di E.Coli producono una molecola proteica biologicamente attiva contenente 100 residui aminoacidici in 5 sec a 37°.

COME FANNO LE PROTEINE AD COME FANNO LE PROTEINE AD
AVVOLGERSI NEL TEMPO DI POCHI AVVOLGERSI NEL TEMPO DI POCHI
SECONDI?SECONDI?

�Supponiamo che ciascuno dei 2 angoli di torsione, φ ψ,φ ψ,φ ψ,φ ψ, di una proteina con n residui possa assumere 3 conformazioni stabili, le conformazioni possibili per questa proteina saranno 3 2n circa 100 n
�Se la proteina può esplorare una conformazione ogni 10-13 secondi
Il tempo in sec necessario per esplorare tutte le conformazioni possibili sarà
t = 10 n / 1013
Per n= 100 t = 10 87 ( 20 miliardi di anni!)

Le proteineLe proteine
non ricercano casualmente la
conformazione nativa fra le molte
possibili
si ripiegano seguendo vie direttesi ripiegano seguendo vie dirette

PICCOLI TRATTI DI STRUTTURA SECONDARIA SERVONO DA MEDIATORI DEL PROCESSO DI AVVOLGIMENTO.

LA FORMAZIONE DI CORTI SEGMENTI DI STRUTTURA SECONDARIA È MOLTO VELOCE.

Questi piccoli tratti (circa 15 residui) si stabilizzano formando dei complessi (es 2αααα, 2β, β, β, β, αβαβαβαβ) che si chiamano
unitunit àà di avvolgimento.di avvolgimento.
Intorno a questi centri si stabilizzano poi altri tratti di struttura secondaria
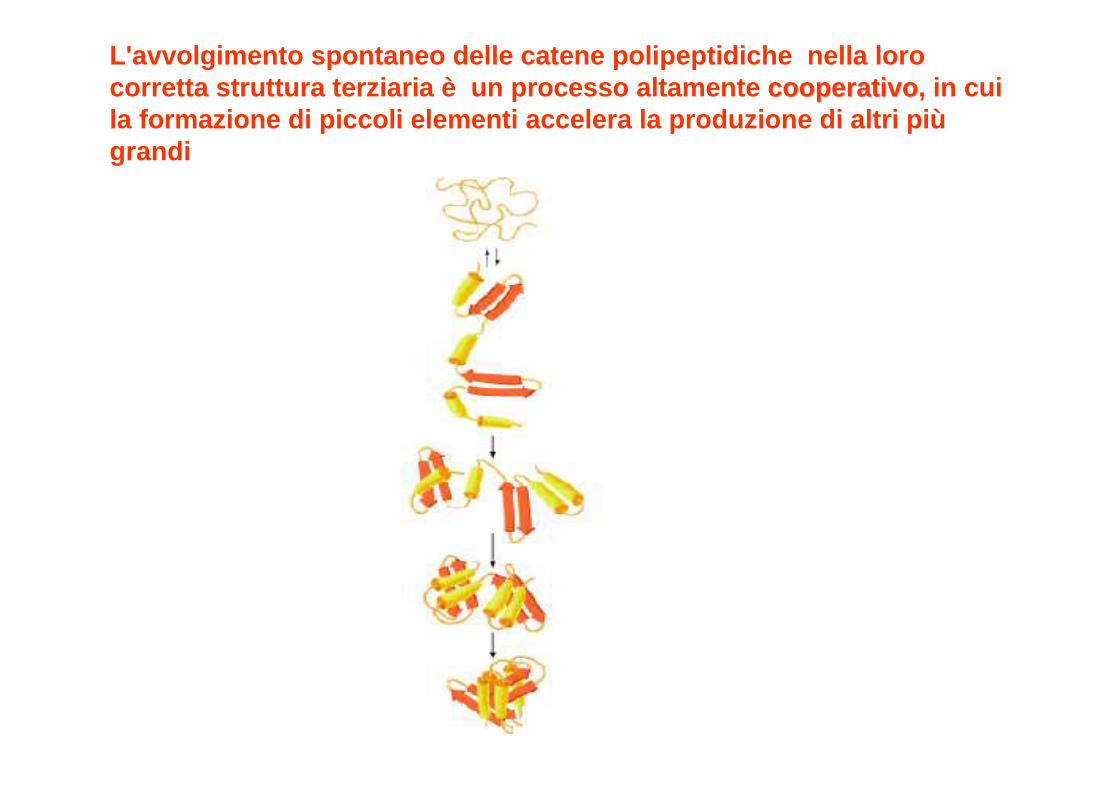
L'avvolgimento spontaneo delle catene polipeptidich e nella loro corretta struttura terziaria è un processo altamente cooperativocooperativo , in cui la formazione di piccoli elementi accelera la produzio ne di altri piùgrandi

Il processo di ripiegamento di una proteina procede da uno stato ad alta energia ed alta entropia ad uno a bassa energia e bassa entropia

IL processo di avvolgimento delle proteine può essere accelerato dall’enzima “ proteina disolfuro isomerasi”



Per alcune proteine a tale processo partecipano gli chaperonichaperoni molecolarimolecolariche :
contribuiscono al corretto avvolgimento di una proteina nascente
consentono alle proteine ripiegate in modo non corretto di raggiungere la conformazione nativa

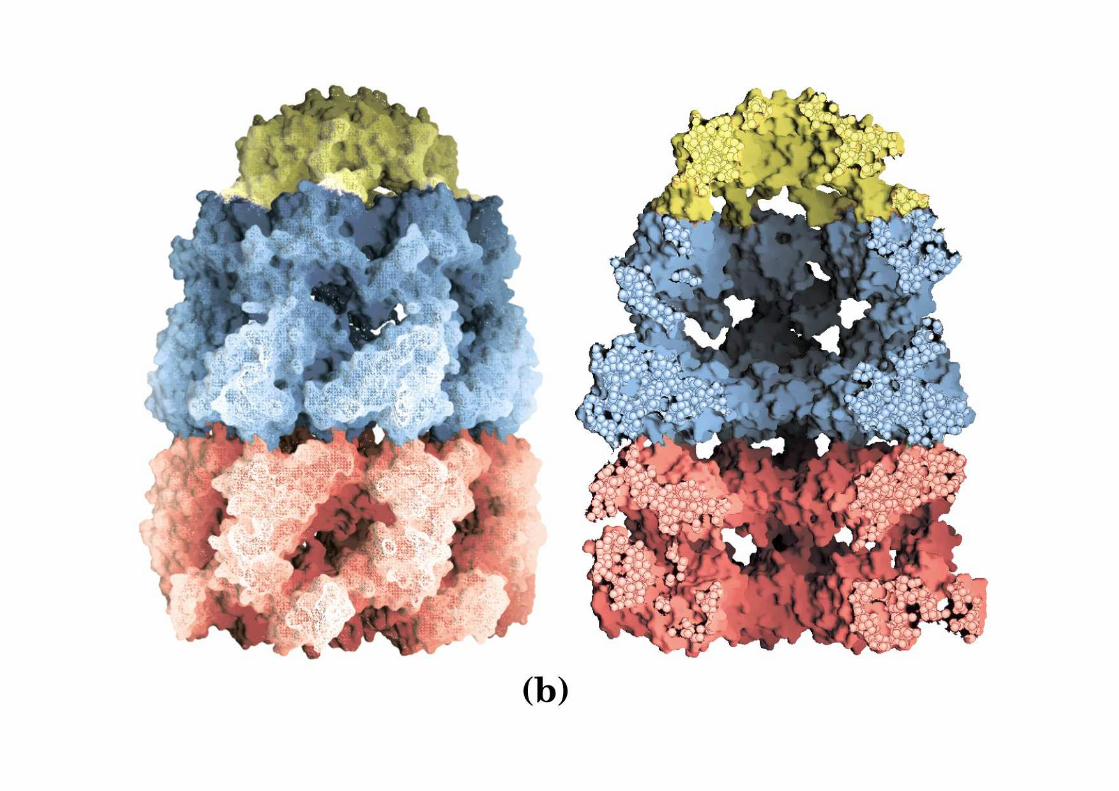
Esistono due classi di chaperonichaperoni molecolarimolecolari : la famiglia Hsp70Hsp70 e le chaperoninechaperonine (Hps60 o GroEL ed o Hsp10 GroES )

Le chaperonine sono costituite da due tipi di proteine HP60 (GroEL) e HP10 (GroES)
GroEL 14 subunità identiche (549 aa) disposte in due anelli sovrapposti (7+7)
GroES 7 subunità identiche (97aa) formano un anello eptamerico

Conformazione, modificazione e degradazione delle proteine
• Una catena polipeptidica appena sintetizzata deve conformarsi e spesso subire modificazioni chimiche per generare la proteina finale
• Tutti i polipeptidi con la stessa sequenza amminoacidicaassumono, in condizioni standard, la stessa conformazione (lo stato nativo), che è la più stabile conformazione che la molecola può assumere.

L’ informazione per il “folding” della proteina è contenuta nella sequenza

Proteine denaturare al calore, con acidi, o chimici perdono lastruttura terziaria e secondaria e la funzione biologica.
Il processo è reversibile

Le chaperonine assistono le proteine nella fase di folding, prevenendo il legame con ligandi inappropriati.


Molte malattie sono dovute al difettoso ripiegamento di una proteina
Alcune patologie derivano da proteine che non sono in grado di raggiungere la loro struttura funzionale e che tendono a formaregrossi aggregati (fibrille o forme amiloidi): Alzheimer, Parkinson, encefalopatia spongiforme, diabete di tipo II.
In altri casi mutazioni puntiformi generano proteine che non raggiungono la loro locazione finale o che non sono più in grado di svolgere la loro funzione perché incapaci di legare i loro substrati.
La fibrosi cistica è un difetto nella proteina transmembrana che agisce come un canale degli ioni cloro nelle cellule epiteliali (CFTR: 1480 amminoacidi). La mutazione più comune è la delezione di un amminoacido (Phe 508) e la proteina mutata non si avvolge correttamente.
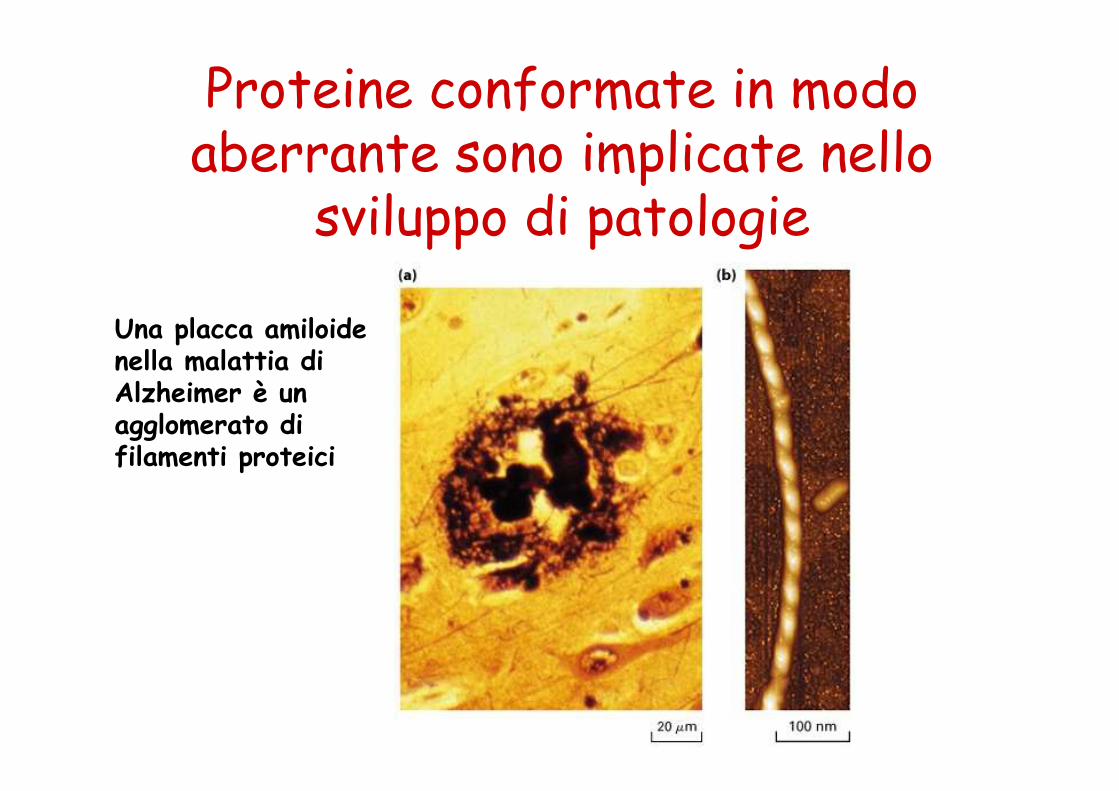
Proteine conformate in modo aberrante sono implicate nello
sviluppo di patologie
Una placca amiloide nella malattia diAlzheimer è un agglomerato di filamenti proteici

Hum αglobina: mvlspadktn vkaawgkvga hageygaeal Bovis: mvlsaadkgn vkaawgkvgg haaeygaeal Pig: vlsaadkan vkaawgkvgg qagahgaeal
ermflsfptt ktyfphfdls hgsaqvkghg kkvadaltna vahvddmpna ermflsfptt ktyfphfdls hgsaqvkghg akvaaaltka vehlddlpga ermflgfptt ktyfphfnls hgsdqvkahg kvadaltka vghlddlpga
lsalsdlhah klrvdpvnfk llshcllvtl aahlpaeftp avhasldkfl lselsdlhah klrvdpvnfk llshsllvtl ashlpsdftp avhasldkfllsalsdlhah klrvdpvnfk llshcllvtl aahhpddfnp svhasldkfl
asvstvltsk yranvstvltsk yranvstvltsk yr
Comparazione sequenze

L’omologia delle sequenze suggerisce relazioni funzionali ed evolutive tra le
proteine


![sinapsi 1 [modalità compatibilità] - sunhope.it · Biofisica e Fisiologia Corso di Laurea Magistrale in “Medicina e Chirurgia” Sinapsi Il neurone Caratteristica peculiare delle](https://static.fdocumenti.com/doc/165x107/5c6a621509d3f27a7e8c9aed/sinapsi-1-modalita-compatibilita-biofisica-e-fisiologia-corso-di-laurea.jpg)















