
Presentazione Pavia Copia senza video - Fondazione Mondino · - l’epilessia non scompariva in...
22
1 La SiNDROME di DRAVET OGGI Charlotte Dravet, Rome, Marseille Pavia, 19 Novembre 2016 Prima 2001
Transcript of Presentazione Pavia Copia senza video - Fondazione Mondino · - l’epilessia non scompariva in...

1
La SiNDROME di DRAVET OGGI
Charlotte Dravet,Rome, Marseille
Pavia, 19 Novembre 2016
Prima 2001

2
* inizio prima 1 anno
* crisi convulsive severe
* febbrile poi senza febbre
* mioclonie ++ ,
* assenze atipiche,
* crisi parziali
* rallentamento dello sviluppo psicomotorio
* EEG normale ➜➜➜➜ PO gen. + foc/multifoc
* neuroimagerie normale
Epilessia Mioclonica Severa dell’ Infanzia
Evoluzione :
* crisi farmacoresistente
* stati di male epilettici
* fotosensibilita
* handicap mentale
� Encefalopatia
senza eziologia conosciuta
∆∆∆∆g differenziale : sd di Lennox-Gastaut
���� Epilessia mioclonica severa del lattante

3
FORME BORDERLINE
(SMEB)
� senza mioclonie/assenze atipiche
� « Intractable childhood epilepsies with frequent
GTCS » (Watanabe 1989, Fujiwara, 1992, Kanazawa,2001)
� « Severe idiopathic generalized epilepsy of
infancy with generalized tonic-clonic seizures »
(Doose & al, 1998 )
2001
SMEI diventava la sindrome di Dravet
Perchè?
- alcuni bambini presentavano lo stesso quadro senza crisi miocloniche
- l’epilessia non scompariva in adolescenza e persisteva in età adulta
- “mioclonica” e “infanzia” non erano più appropriate(ILAE
(Classificazione Lega Internazionale , 2001)

4
Dopo 2001
1978-2001SMEI2001 Claes et al
SCN1A mutations
2002 Harkin et al. GABRG2
Dravetsyndrome spectrum
PCDH19 protocadherinin girls Dibbens et al 2008 Depienne et al 2009
CHD2 Suls et al 2013
Other close epilepsy
types

5
� Dravet syndrome is a genetic encephalopathy with epilepsy, cognitive, behavioral and motor impairment
� In most patients it is caused by a mutation in the SCN1A gene.
� However, the diagnosis is based on clinical characteristics
Where are we now ?
Clinical general featuresNo big changes from the first descriptions
�semiological particularities : onset with afebrile seizures or focal seizures, scarcity of febrile seizures, absence of focal seizuresabsence of neurological signs, almost normal psychomotor development in the first 4 years.
�Variety of seizure types
� less patients with photo/pattern-sensitivity but eye-closure as triggering factor of micro-absences
�better knowledge of the motor and orthopedic impairments
�better knowledge of sleep, food, autonomic disorders
�occurrence of SUDEP

6
Variety of seizure types
Video
Clonic seizure
Obtundation status
Are they true atonic seizures in Dravet syndrome ?
Video
1 true atonic seizure and 1 myoclono-atonic seizure in a patient with Epilepsy with myoclonic- atonicseizures (Doose syndrome)
1 atypical absence with atonic component elicited by ILS in a girl with Dravet syndrome (by Dr Cantaloupe courtesy)

7
5 y. generalized myoclonic seizures
5y. Multifocal S and SW
� Photo and pattern sensitivity :
Variable during the time
ILS, patterns, eye closure, environmental light intensity
Very high convulsive susceptibility
Z.A. 3yrs

8
Motor and orthopedic impairment
Video
Genetic background
SCN1A mutations
Sindrome di Dravet 70/80 %GEFS+ 10-20 %
Migrating partial seizures (Carranza et al, 2011, Freilich et al, 2011)
Epilessie focali altre (Shi et al, 2012, McDonald et al, 2016)
Sd di Panayiotopoulos (Grosso et al, 2007, Livingston et al, 2009)
Familial hemiplegic migraine (Thomsen et al, 2007, Vahedi et al, 2009)
Familial autism (Weiss et al, 2003)
Sd di Rasmussen (Ohmori et al, 2008)
...

9
Differential diagnosis
- is not present in all patients with Dravet syndrome
- can be present in other epilepsy types, for example :
Can be difficult knowing that an SCN1A mutation
* Genetic Epilepsy with Febrile Seizures plus (GEFS+ ) syndrome
* focal epilepsies but sharing common traits with Dravetsyndrome
• In the first two years of life
• In childhood
Differential Diagnosis

10
• Dravet syndrome
– Onset before the age of 1 year
– Febrile (<38.5°C) and afebrile seizures
– Long seizures, unilateral or with signs of lateralisation
– Cognitive decline
– For 80%: SCN1A mutation
• Febrile seizures
– Onset rather after the age of 1 year
– Only febrile seizures
– brief seizures
- Normal cognitive outcome
– Possible SCN1A mutation (GEFS+ spectrum)
Be concerned : prescribe rectal diazepam
Hattori et al, 2008. A screening test for prediction of Dravet syndrome before one year
Video
1st clonic seizure after vaccination in a 6 m-old boy
2nd clonic seizure triggered by fever in the same boy
at 10m

11
� Progressive myoclonus epilepsies:
• in the 2nd year when the neurological and behavioural signs appear
• one ceroïd lipofuscinosis can be eliminated throughout :
- interictal myoclonus characteristics
- absence of visual impairment
- absence of fundus abnormalities
- absence of posterior evoked potentials elicited by the
Intermittent Photic Stimulation (IPS) low frequencies
- negative results of biological and genetic investigations
for NCL
4 years Necker Enfants Malades

12
• Dravet syndrome– No perinatal history– Generalised /alternating unilateral
seizures
– Febrile and afebrile seizures
– Other seizure types appear later on:• atypical absences• myoclonic seizures• focal seizures
– EEG: general/multifocal S and SW
– Cognition: normal then global delay
– For 80%: SCN1A mutation
– Aggravated by carbamazepine
– Normal MRI
• Focal epilepsy– With/without perinatal history – Unilateral seizures, always on the
same side
– Febrile and afebrile seizures
– Later on • no atypical absences • no myoclonic seizures• seizures become more focal
- EEG: focal spikes
- Cognition: normal or specific deficits
– Generally no SCN1A mutation
– Normal/abnormal MRI
(Sarisjulis et al; 2OOO)
If you should have any doubts, avoid CBZ and obtain a precise description of the seizures
(home video)
* Other myoclonic epilepsies:
Epilepsy with myoclonic-atonic seizures
(Doose syndrome)
* Other epilepsies
Early Lennox-Gastaut syndrome (not
relevant)
� Relationship with the GEFS+ syndrome
� Relationship with Protocadherin 19 (PCDH19)
In Childhood

13
Video
Doose syndrome: - drop-attack (atonic seizure)
- myoclonic-atonic seizure
LGS: - atonic-tonic atypical absence
- nocturnal axial tonic seizure
� Relationship with GEFS+
(Genetic epilepsy with febrile seizures +)
1/ Family context: several members with FS, FS+, epilepsy
→ GTCS, absences, focal seizures,
rarely myoclonic-astatic epilepsy
* with usually benign outcome, no mental impairment
* SCN1A mutation in 10-20%.
* “There is a possibility of simultaneous involvement of multiple genes for seizure phenotypes.” (Ito et al, 2006)
2/ One patient with Dravet syndrome may be a member
of a GEFS+ family

14
� Relationship with Protocadherin 19 gene
• an almost similar semiology with slight variations
- later onset (mean 7.5 months)
- seizures in clusters
- less frequent
status epilepticus,
myoclonic and atypical absence seizures,
- less severe epilepsy in childhood
• is observed in girls without SCN1A mutation
with PCDH19 mutation on X chromosome
(Dibbens et al;2008 - Marini et al; 2010 - Depienne et al; 2012)
SCN1A-POSITIVE VERSUS PCDH19-POSITIVE
Unpublished data from Leguern et al.
Normal Development before seizure onset
Age at onset ≤ 1 an
Association of febrile /afebrile seizures
Association of generalised / focal seizures
Prolonged seizures or seizures in cluster n=66n=113
SCN1A PCDH19
Mean age at onset: 6 months (1-12)
Highly frequent status epilepticus (82%)
Pharmacoresistance persisting
over time
Highly polymorphic seizures:
Frequent myoclonic jerks (50%)
Frequent atypical absences (85%)
Mean age at onset: 10 months (3-60)
Less frequent status epilepticus (52%)
Intractable seizures at the beginning
Frequency decreases with age
Less polymorphic seizures:
Infrequent myoclonic jerks (20%)
Infrequent atypical absences (20%)

15
The American Journal of Human Genetics 93, 1–9, November 7, 2013
De Novo Loss-of-Function Mutations in CHD2 Cause
a Fever-Sensitive Myoclonic Epileptic Encephalopathy
Sharing Features with Dravet Syndrome
Arvid SULS and the EuroEPINOMICS RES Consortium
Nat Genet. 2013 July ; 45(7): . doi:10.1038/ng.2646.
Targeted resequencing in epileptic encephalopathies
identifies de novo mutations in CHD2 and SYNGAP1
Gemma L. Carvill1, Sinéad B. Heavin2, Simone C. Yendle2, Jacinta M. McMahon2, Brian J.
O’Roak3, Joseph Cook1, Adiba Khan1, Michael O Dorschner4,5, Molly Weaver4,5, Sophie
Calvert6, Stephen Malone6, Geoffrey Wallace6, Thorsten Stanley7, Ann M. E. Bye8, Andrew
Bleasel9, Katherine B. Howell10, Sara Kivity11, Mark T. Mackay10,12,13, Victoria Rodriguez-
Casero14, Richard Webster15, Amos Korczyn16, Zaid Afawi17, Nathanel Zelnick18, Tally
Lerman-Sagie19, Dorit Lev19, Rikke S. Møller20, Deepak Gill15, Danielle M. Andrade21,
Jeremy L. Freeman10,12, Lynette G. Sadleir7, Jay Shendure3, Samuel F. Berkovic2, Ingrid E.
Scheffer2,10,13,22,*, and Heather C. Mefford1,*
CHD2 syndrome
More heterogeneous
- Variable onset age , most often after 1 year (12m-5years)
- Inconstant fever-sensitivity
- Possible delayed development before seizure onset
- Multiple seizure types including myoclonic seizures
- Frequent and strong photosensitivity with autostimulation
- Predominance of generalized anomalies on EEG
This syndrome is clearly different from Dravet syndrome,
even in its incomplete forms
Video
2 brief myoclonic seizures in a boy with CHD2 syndrome,
published by Carvill et al,2013

16
� All the authors have failed to find any significant
correlations genotype / phenotype (Fontana et al, 2004,
Oguni et al, 2005 Akiyama et al, 2010, Brunkhaus et al,
2012...)
Except an earlier age at onset in patients with
truncation, splice site mutation, genomic deletion
(Marini et al, 2010 )
� Researches are ongoing in order to discover other
mutations, variants, modifiers, epigenetic factors
capable to explain the negative cases, and the
variability in the correlation genotype/ phenotype
Genotype/ phenotype correlations
Not consistently found
Clinical outcome
1/ Epilepsy severity
2/ Occurrence of acute encephalopathies
3/ Ocurrence of SUDEP
4/ Different cognitive outcomes
Role of genetics in the different outcomes?

17
Recent clinical studies (Chieffo et al,2011, Nabbout et al, 2013, Villeneuve et al, 2014) are in favour of the effect of mutations rather than epilepsy, supported by experimental studies (Bender et al, 2013, 2016, Tsai et al,2015, Maeda et al, 2016 ) but the mechanisms of these effects are still not well known.
Our findings suggest that SCN1A mutation leads to changes in the dopamine system that may contribute to the behavioral abnormalities in DS.
18
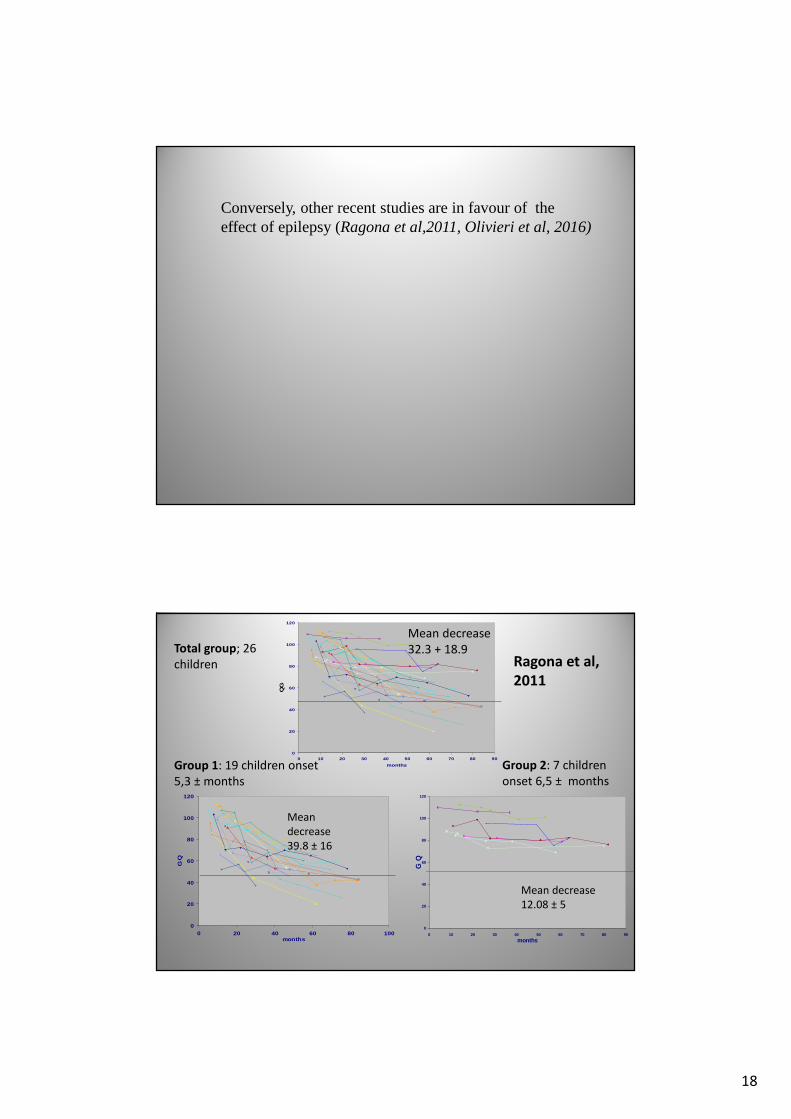
Conversely, other recent studies are in favour of the effect of epilepsy (Ragona et al,2011, Olivieri et al, 2016)
0
20
40
60
80
100
120
0 10 20 30 40 50 60 70 80 90
months
QG
0
20
40
60
80
100
120
0 20 40 60 80 100months
G Q
0
20
40
60
80
100
120
0 10 20 30 40 50 60 70 80 90
months
G Q
Group 1: 19 children onset
5,3 ± months
Group 2: 7 children
onset 6,5 ± months
Mean decrease
12.08 ± 5
Mean
decrease
39.8 ± 16
Mean decrease
32.3 + 18.9Total group; 26
children Ragona et al,
2011

19
Comments of this slide in the article by Ragona et al, 2011
The analysis of individual cognitive profiles showed that
- 19 pts exhibited a steep decrease in GQ (group 1),
- whereas in 7 pts who had been assessed at comparable ages, the
decrease was less steep, and the differential (d)GQ was lower than
20 points (group 2).
On the basis of this arbitrary cut-off of 20 points of dGQ, we
observed that,
although the difference was statistically non significant,
absences and myoclonus during the first 3 years of life
were present in 12 of 19 patients of group 1 (63%)
and in only 1 patient of group 2 (14%).
the higher incidence of complete forms among the cases with more severe neurological and neuropsychological findings,together with more severe epileptic severity in the lastyears (in particular, background EEG abnormalities) would suggest a possible responsibility of some epileptic disorders(early myoclonic seizures and atypical absences, aswell as persistent background slowness) in worsening theneuropsychological outcome, as others already stressed.

20
Cerebellar and extra-striate dorsal stream?
Where are we going to?
???????????
New technologies give us great hope but also great perplexity : next generation sequencing, genome analysis, experimental studies ...

21
New treatment approaches
1/ Fenfluramine
3/ Could we consider the possibility of a true cure through a genic treatment?
2/ Cannabinoids ?Several trials are ongoing and the preliminary results are encouraging
C0NCLUSION
� In 1978, SMEI was only a syndrome
� In 2016, Dravet syndrome is a disease comprising epilepsy, cognitive, behavioral and motor impairment
� due to genetic alterations mainly consisting of mutations in the SCN1A gene, exceptionally in other genes
� The phenotype/genotype relationship remains unclear
� A part of the cases remain without known genetic etiology
� New therapeutic perspectives are now opening

22
My colleagues and friends
In Marseille
J.RogerM.BureauB.Dalla BernardinaC.A. TassinariC.Cassé-PerrotG.Delahaye
In Italy
D.Battaglia R.Guerrini F.Guzzetta D.Chieffo
Château d’If in the Marseille gulf









![03 Caso Clinico.ppt [modalità compatibilità] Gruppi di lavoro/03... · sospensione delle lamivudina, e il flare di transaminasi, peraltro modesto, scompariva spontaneamente. Chemioterapia](https://static.fdocumenti.com/doc/165x107/5c65beab09d3f2916e8d3f16/03-caso-modalita-compatibilita-gruppi-di-lavoro03-sospensione-delle.jpg)








