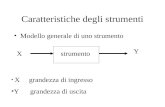
Migliore Punteggio - Lunghezza della sequenza - Grandezza banca dati - Composizione della sequenza.
40
igliore Punteggio - Lunghezza della sequenza - Grandezza banca dati - Composizione della sequenza
-
Upload
lotterio-lisa -
Category
Documents
-
view
226 -
download
1
Transcript of Migliore Punteggio - Lunghezza della sequenza - Grandezza banca dati - Composizione della sequenza.

Migliore Punteggio- Lunghezza della sequenza
- Grandezza banca dati
- Composizione della sequenza

Punteggio sequenze random
PunteggioOPT
100 200 300 400 500 600 700 800 900100
0110
0120
0130
0
Opt = 1070
NumeroSequenzecasuali 70
60
50
40
30
20
10

Significatività statistica
PunteggioOPT
NumeroSequenzecasuali
100 200 300 400 500 600 700 800 900100
0110
0120
0130
0
70
60
50
40
30
20
10
Opt = 1070

Significatività statistica
PunteggioOPT
NumeroSequenzecasuali
100 200 300 400 500 600 700 800 900100
0110
0120
0130
0
70
60
50
40
30
20
10
E = 1.21*10-21
E value =Numero atteso per caso di sequenze con punteggio > opt

Matrici di sostituzione

Sostituzioni osservate

Composizione aminoacidica

Matrice di probabilità A C D E
A 0.14 0.28 0.09 0.09
C 0.14 0.09 0.09
D 0.015
0.03
E 0.01
A C D E
A 0.3 0.15
0.0.. 0.0..
C 0.3 0.0.. 0.0..
D 0.1 0.05
E 0.1
Frequenze osservate
Frequenze attese
Diviso
A C D E
A 2.13
0.53 0 0
C 2.13 0 0
D 6.4 1.6
E 6.4
Matrice di probabilità
=
< 1 Sostituzionesfavorita
> 1 Sostituzionefavorita

Punteggio allineamento
AAADE || | AACEE
f(AA) x f(AA) x f(AC) x f(DE) x f(EE) ---------------------------------------------- a(AA) x a(AA) x a(AC) x a(DE) x a(EE)
= pAA x pAA x pAC x pDE x pEE
= 2.13 x 2.13 x 0.53 x 1.6 x 6.4 = 25
A C D E
A 2.13
0.53 0 0
C 2.13 0 0
D 6.4 1.6
E 6.4
Matrice di probabilità
Scomodo!
Gli Odds non possono essere sommati Per calcolare il puneggio di un allineamentoma debbono essere moltiplicati

Logaritmo delle frequenzeLog ( a x b x c x d ) = log(a) + log(b) + log(c) + log(d)
A C D E
A log(pAA)
log(pCA) log(pAD)
log(pAE)
C log(pAA) log(pCD)
log(pCE)
D log(pDD)
log(pDE)
E log(pEE)
A C D E
A pAA pAC pAD pAE
C pCC pCD pCE
D pDD pDE
E pEE
Prob = pAA x pAA x pAC x pDE x pEE
log( Prob ) = log( pAA x pAA x pAC x pDE x pEE )
Log (Prob) = log(pAA) +log(pAA) +log(pAC) +log(pDE) +log(pEE)

Punteggio di un allineamento
AAADE || | AACEE
A C D E
A 0.3 -0.2 0 0
C 0.3 0 0
D 0.8 0.2
E 0.8
Matrice di punteggio
= 0.3 + 0.3 – 0.2 + 0.2 + 0.8 = 1.4 punteggio di similarità
10 1.4 = 25 = probabilità calcolata precedentemente....
< o Sostituzionesfavorita
> oSostituzionefavorita

Una matrice di punteggio
Identità Sostituzioni avvantaggiateSostituzioni avvantaggiate
Sostituzioni svantaggiate

Distanza delle matrici
A C D E
A 10 -10 -12 -15
C 11 -9 -10
D 13 -8
E 12
A C D E
A 1.0 -1.0 -0.7 -1.5
C 1.5 -1.1 -0.2
D 0.5 -0.9
E 0.8
Da allineamenti di sequenze molto simili
Da allineamenti di sequenze molto divergenti

Matrici PAM Percent Accepted Mutation
PAM 2 = PAM 1 * PAM 1PAM 3 = PAM 2 * PAM 1PAM 4 = PAM 3 * PAM 1 etc..

Matrici BLOSUM
L T A G A R I D E D - - A R I D E D W E D I S L H D W R T E A - - D W L H D W R T D WL T A G A R L D - - - - - - - - E D W E D I S I H E W S T E A - - D W I H E W T T D WL T I G L R I E E - - - - - - D E D A E D I S L H D G R T - - - E D W L H D W R S D WL T A G A R I D E - - - D - - - E D W E D I S L H D W R T E - - - D W L H D W R T D WL T A G A R I D E D W E A R I D E D W E D I S L H D W R T E A I L D W L H D W R T A W
Identità < x %
L T A G A R I DL T A G A R L D
L T A G A R I DL T A G A R I D
L T I G L R I EL T A G A R I D
L T A G A R L DL T I G L R I E

Corrispondenza PAM/BLOSUM
Maggiore divergenza
Minoredivergenza PAM PAM
100100BLOSUM 90BLOSUM 90
PAM PAM 120120
BLOSUM 80BLOSUM 80
PAM PAM 160160
BLOSUM 60BLOSUM 60
PAM 200PAM 200 BLOSUM 52BLOSUM 52
PAM 250PAM 250 BLOSUM 45BLOSUM 45

Alberi Filogenetici
H
GF
ED
B
C
A
Nodi interniUnità tassonomiche sconosciute
Nodi esterni - FoglieUnità tassonomiche operative
Radice
Rami
Tempo
Distanza F-H

Ortologhi e paraloghi
Gene A
Gene A1
Gene A2
Speciazione
Geni Ortologhi
Gene A
Gene A Gene B
Duplicazione
Geni Paraloghi

Filogenesi molecolare
Hb Alpha Gorilla
Filogenesi di geni ortologhi Filogenesi di geni paraloghi
Hb Alpha Uomo
Hb Alpha Ratto
Hb Alpha Topo
Hb Alpha Anatra
Hb EpsilonUomo
Hb GammaUomo
Hb BetaUomo
Hb DeltaUomo
MioglobinaUomo
Hb ZetaUomo
Hb AlphaUomo
Hb ThetaUomo
Emoglobina alfa in specie diverse
Diverse catene di emoglobinanell’ uomo

Similarità e distanza
Singola
Paralleli
Multipli
Convergenti
AGHSVLIWETS
AGHSVLIWETS
Eventi di sostituzione: Avvenuti = 12Osservabili = 3
Sequenza originaria
Duplicazione/Speciazione
Coincidenti
Retro-Sostituzione
AGHSVLIWETS
->I
->T
->E
->A->L
->I
->A
->T
->I->T
->A->E
TGASILLWETTAGESILIWETT

Distanza Genetica
Meglio usare sequenze nucleotidiche:- regioni non codificanti- mutazioni nucleotidiche possono non essere aminoacidiche- modello più facile
Tempo
% Diversità
100%
75%
50%
25%
0%

Distanza Jukes & Cantor
Diversità % Stima distanza genetica
(Sostituzioni per base)
0.10 0.107
0.20 0.232
0.30 0.383
0.40 0.571
0.50 0.823
0.60 1.207
0.70 2.031
sostituzioni per base = - 3/4 * ln( 1 - 4/3 * %Diversità )

Distanza Jukes & Cantor
Tempo
d=DistanzaGenetica
Assunzioni del modello:- Stessa probabilità delle sostituzioni- Stessa probabilità dei siti- Indipendenza dei siti- OROLOGIO MOLECOLARE - Velocità di sostituzione costante- STAZIONARIETA’ - Composizione nucleotidica costante

Matrici di distanze
Scimpanzè Uomo Gorilla Orango Macaco Scim.Ragno
Scimpanzè -
Uomo 0.014 -
Gorilla 0.02 0.015 -
Orango 0.04 0.03 0.04 -
Macaco 0.08 0.07 0.08 0.08 -
Scim. Ragno
0.11 0.10 0.10 0.11 0.12 -
globine

Gerarchico addittivo
1
2
34
5
12
3
45

Allineamenti multipli

Un allineamento multiplo
Riga = sequenza
LT AGARIDED--ARIDEDWEDISLHDWRTEA--DWLHLT AGARLD--------EDWEDISIHEWSTEA--DWIHLT IGLRIEE------DEDAEDISLHDGRT---EDWLHLT AGARIDE---D---EDWEDISLHDWRTE---DWLHLT AGARIDEDWEARIDEDWEDISLHDWRTEAILDWLH
Colonna = posizioneBlocchi conservatiElementi di struttura secondaria ?
Regioni con gaps: Loops?
Famiglia proteine omologhe

Vantaggi
1 PEEKSAVTALW-KVNVDEVGG2 PEEKSAVLALWDKVNEDEVGG
1 PA--TAVKALWGKAGAGEYGA2 AAD-TNVTAAWSKVGAGEYGA3 EHEWQLVLHVW-KVEVAGHGQ
2 sequenze Troppo simili
AllineamentoMultiplo
1 REEKSAVTALN-K--VDEIGG2 K---TA--VIGDKVNIEEV
2 sequenze Troppo divergenti

Colorazione
Cysteine C Negative D, E Positive K, R Alcohol S, TPolar N, QAromatic F, H, W, Y Hydrophobic A, G, I, L, M, P, V

Consensus
100%90%80%70%

Qualità multiallineamento
Punteggio = 1+2+3+4+5+6+7+8+9
= (VG) + (ED) + (DE) + (KK) + …
VEEKSAVTAGEEKAAVLAAADKTNVKALADKTNVKA
123456789
VEDKSAVTAGDEKAAVLA
123456789
Punteggio = 1+2+3+4+5+6+7+8+9
Punteggio = i
P(ci)P(VGAL) = ???

Punteggio di una colonna
xxAxxxxx
xxVxxxxxxxGxxxxx
xxLxxxxx
Punteggio colonna =
i<j Similarità(AiAj)
(VG)+(VA)+(VL)+(GA)+(GL)+(AL)
=A
VG
L

Punteggi alternativi
A
VG L
V
G
L
G
L
LAG
xxAxxxxx
xxVxxxxxxxGxxxxx
xxLxxxxxxxGxxxxxxxLxxxxx
=?
=(LG)+(LG)+(LA)+(LV)+(LL)
=(VL)+(LL)+(LG)+(GG)+(GA)

Programmazione dinamica multi-dimensionale
2 sequenze lunghe 100 = 100*100 = 10.000 quadretti
3 sequenze lunghe 100 = 100*100*100 = 1 milione cubetti4 sequenze lunghe 100 = 100*100*100*100 = 100 milioni di iper-cubetti
TempoO(Ln)

Come NON si costruisce
FEDCBA
VLSAIDWTNVK
VISAGDWTNVRVLTAAE-TNVR
ILSLIDWTQVR
1
2
1) Allineare B con A2) Allineare C con B3) Allineare D con C4) Allineare E con D5) Allineare F con E
3
4
5
VLTLID-SNVR
VLSLAE-TQVK

Costruzione albero guida- 4 6 6 1 9
- 5 5 3 7
- 2 5 5
- 5 4
- 8
-N * (N-1) allineamenti a coppia Matrice di distanze
Albero guida

Allineamento progressivo
FEDCBA
VLSAIDWTNVKVLSLAE-TQVK
VLTLIDSNVRVLTAAETNVR
VISAGDWTNVRVLTLID-SNVRVLTAAE-TNVRILSLIDWTQVR
12
3
4
51) Allineare E con F2) Allineare B con C3) Allineare D con EF4) Allineare BC con DEF5) Allineare A con BCDEF

Allineamento di allineamenti
PEEKSAV--A LW--VNVDEVGGPEE-- GV--A LWDKVNEDEVGGPEEKS GVLGA LWDKVNE---GG
+ PEEKSAVA LW--VNVDEVGGPEE-- GVA LWDKVNEDEVGG
PEEKS GVLGA LWDKVNEGG
=PEEKSAV-AL W--VNVDEVGGPEE-- GVALW DKVNEDEVGG-PEEKS GVLGA LWDKVNEGG
No

LI
KR
DE
..
.
..
.C CL+CI
/2CK+CR/2
CD+CE/2
...
A AL+AI/2
AK+AR/2
AD+AE/2
...
Y YL+YI/2
YK+YR/2
YD+YE/2
...
… ... ... ... ...
LI
KR
DE
...
...CS
CL+CI+SL+SI/4
CK+CR+SK+SR/4
CD+CE+SD+SE/4
...
AA
AL+AI+AL+AI/4
AK+AR+AK+AR/4
AD+AE+AD+AE/4
...
YW
YL+YI+WL+WI/4
YK+YR+WK+WR/4
YD+YE+WD+WE/4
...
..
.... ... ... ...
S&W per allineamenti multipli
1 sequenza con 2 sequenze
2 sequenze con 2 sequenze
LKDKSAIREL- G
CAYKS G