
ISSN: 2281-4884 dcth · C. Delfino, V. Grandi, A. Pileri, S. Gunnella, L. Rigacci, R. Alterini, N....
82
DCTH Drugs Cell Therapies Hematology and Editor in Chief Alberto Bosi in OPEN ACCESS www.dcth.org Vol. 1 • N. 4 • 2013 ISSN: 2281-4884
Transcript of ISSN: 2281-4884 dcth · C. Delfino, V. Grandi, A. Pileri, S. Gunnella, L. Rigacci, R. Alterini, N....

dcthDrugs Cell Therapies Hematology
and
Editor in Chief
Alberto Bosi
inopen access
www.dcth.org
Vol. 1 • N. 4 • 2013
ISSN: 2281-4884

Levact ® i.v. (bendamustina HCI)
RIASSUNTODELLE CARATTERISTICHEDEL PRODOTTO
01 RCP Medico Levact.indd 1 03/08/12 12.48
LA STORIA PIÙ BELLAÈ QUELLA CHE DEVIANCORA VIVERE.Cancidas è il primo farmaco e il più usato nella classe delle echinocandine.1 Grazie alla sua efficacia e al suo elevato profilo di tollerabilità,2 in 10 anni ha raggiunto il traguardo di quasi 2 milioni di pazienti trattati in tutto il mondo. La sua storia è la storia di tutti i medici e di tutti i pazienti, che insieme hanno combattuto un’infezione fungina per riprendersi quello che spettava loro di diritto. Il futuro.
®
www.msd-italia.it www.contattamsd.it [email protected] www.univadis.it
Esemplare fuori commercio. Omaggio per i Sigg. Medici AINF-1074266-0000-CAN-J-02/2015 Materiale depositato presso l’ AIFA il 04.03.2013
Prima della prescrizione, consultare il riassunto delle caratteristiche del prodotto accluso.
Proteggiamo il futuro
1. Dati IMS - YTD Dicembre 2012
2. Walsh TJ, Teppler H, Donowitz GR, et al. Caspofungin versus liposomal amphotericin B for empirical antifungal therapy in patients with persistent fever and neutropenia. N Engl J Med. 2004;351(14):1391–1402.

Vol. 1 ⋅ N. 4 ⋅ 2013
Publisher
Editor-in-Chief
Alberto Bosi
Endorsed by
Gruppo Italiano per il Trapianto di Midollo Osseo
(GITMO)
Società Italiana di Emaferesi e Manipolazione Cellulare
(SIdEM)
Edizioni Internazionali srl
Divisione EDimEsEdizioni medico-scientifiche - Pavia
Via Riviera 39 - 27100 PaviaTel. +39 0382 526253 r.a. - Fax +39 0382 423120
E-mail: [email protected]
Director-in-ChiefPaolo E. Zoncada
Autorizzazione Tribunale di Milanon. 423 del 12 Novembre 2012
Associate Editors
Luca Pierelli, SIdEM - PresidentAlessandro Rambaldi, GITMO - PresidentValeria Santini Alessandro M. Vannucchi
Managing EditorsFrancesca Buchi
Section Editorsn Acute lymphoblAstic leukemiA Renato Bassan, italy
n Acute myeloid leukemiA Sergio Amadori, italy Claude Gorin, France
n AnemiAs Lucio Luzzato, italy Joan-Lluis Vives Corrons, spain
n bone mArrow cells And environment Roberto Lemoli, italy Caroline Le Bousse, France
n bone mArrow trAnsplAntAtion Alessandro Rambaldi, italy Dietger Niederwieser, Germany
n chimerism And engrAftment Andrea Bacigalupo, italy Aron Nagler, israel
n chronic myeloid leukemiA Michele Baccarani, italy
n hemApheresis Luca Pierelli, italy Miguel Lozano, spain
n hemostAsis Francesco Rodeghiero, italy
n infections Gian Maria Rossolini, italy Murat Akova, Turkey
n lymphoproliferAtive neoplAsiAs Gianluca Gaidano, italy Anna Sureda, UK
n myelodysplAsiAs Valeria Santini, italy Pierre Fenaux, France
n myeloproliferAtive neoplAsiAs Alessandro M. Vannucchi, italy Ayalew Tefferi, UsA
n pediAtric hemAtology Giorgio Dini, italy Christina Peters, Austria
n preclinicAl And clinicAl studies Roberto Marchioli, italy
n phArmAcogenomics/phArmAcogenetics Romano Danesi, italy Renato V. La Rocca, UsA
n plAsmAcells disorders Giampaolo Merlini, italy Robert A. Kyle, UsA
n stem cell reseArch And regenerAtive medicine Paolo Rebulla, italy Zbigniew M. Szczepiorkowski, Lebanon
Gruppo Italiano InfermieristicoMobilizzazione ed Aferesi

© Copyright 2014 by
Edizioni Internazionali srlDivisione EDIMES - Edizioni Medico-Scientifiche - PaviaVia Riviera, 39 - 27100 PaviaTel. 0382/526253 r.a. - Fax 0382/423120E-mail: [email protected]
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any formby any means electronic, mechanical, photocopying, recording, or otherwise, without prior written consent of the publisher.

REVIEW◗◗◗ Targeting the minimal residual disease in acute
myeloid leukemia: the role of alloreactive natural killer cells .................. 245 S. Parisi, R.M. Lemoli, A. Curti
◗◗◗ Ultrasound and contrast enhanced ultrasound sonography evaluation of intestinal acute graft-vs-host disease ............................................... 253
E. Benedetti
◗◗◗ Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation; usefulness of the pretransplant scoring systems ............................................................................................................................................................................ 261
R. Raimondi, A. Tosetto, C. Borghero, F. Rodeghiero
FORUM◗◗◗ CD30 expression in lymphoid neoplasms: from diagnostic
marker to target of therapy ................................................................................................................................. 279 L. Leoncini, M.R. Ambrosio, S. Lazzi, B.J. Rocca, P. Tosi
◗◗◗ Brentuximab vedotin in CD30-expressing cutaneous T-cell lymphoma ....................................................................................................................................................................... 301
C. Delfino, V. Grandi, A. Pileri, S. Gunnella, L. Rigacci, R. Alterini, N. Pimpinelli
Contents

IL PAZIENTEAL CENTRODEL NOSTRO MONDO
www.mundipharma.it
mundipharma 200x240:Layout 1 05/10/12 13.51 Pagina 1
IL PAZIENTEAL CENTRODEL NOSTRO MONDO
www.mundipharma.it
mundipharma 200x240:Layout 1 05/10/12 13.51 Pagina 1
IL PAZIENTEAL CENTRODEL NOSTRO MONDO
www.mundipharma.it
mundipharma 200x240:Layout 1 05/10/12 13.51 Pagina 1

DCTH - 4•2013 - 245-252
Key words: acute myeloid leukemia, nat-ural killer cells, minimal residual disease.
Correspondence:Antonio Curti, MDDepartment of ExperimentalDiagnostic and Specialty Medicine, Institute of Hematology “ L. and A. Seràgnoli”, S. Orsola-Malpighi HospitalUniversity of BolognaVia Massarenti, 9 - 40138 Bologna, ItalyE-mail: [email protected]
SUMMARYThe clinical results of AML patients, especially if elderly, are particularly dismal, although the achievement of CR with MRD after combined chemotherapy appears as possible in a good fraction of patients. Indeed, the persistence of MRD leads to progression and patients ultimately die. For these reasons, alternative approaches for the preven-tion of relapse in CR patients are necessary and are currently under active investiga-tion. Allogeneic stem cell transplantation (SCT), which combines the cytotoxic effect of conditioning regimen with adoptive immunotherapy, has been shown to offer a clear advantage in terms of relapse prevention, thus providing the proof-of-principle of the capacity of immune cells of eradicating MRD. However, such approach has several and important limitations and is not applicable to all the patients, especially if elderly. In this scenario, the role of immunological therapies in the post-remission management of adult AML patients, such as NK therapy, beside the SCT setting, have been recently exploited with promising results in terms of immunological and clinical responses.
Targeting the minimal residual disease in acute myeloid leukemia: the role of alloreactive natural killer cellsS. Parisi1, R.M. Lemoli2, A. Curti11Department of Specialistic, Diagnostic an Experimental Medicine, Institute of Hematology “L. and A. Seràgnoli”, University of Bologna, Bologna, Italy; 2Chair of Hematology, Department of Internal Medicine (DIMI), University of Genoa, Genoa, Italy
◗◗◗ INTRODUCTION
Acute myeloid leukemia (AML) treat-ment in adult patients is based on intensive chemotherapy regimens
REVIEW
containing multiple cycles of anthra-cyclines and cytosine arabinoside, of-ten followed by allogeneic stem cell transplantation (SCT). Complete remission (CR) rate after chemotherapy in adult AML patients range from 60 to 85% in patients young-er than 60 years, but 5 year-Overall Sur-vival (OS) is 40% mainly due to a high rate of subsequent relapse. These results are even worse in elderly patients, whose OS falls down to about 10% due to a more frequent incidence of poor prognostic features at diagno-sis (secondary AML, complex cytoge-netics), resulting both in reduced CR rate and, whenever CR is achieved, in the inability to complete intensive con-

246 S. Parisi, et al.
solidation program due to co-morbidi-ties. In the last years many efforts have been made to improve OS and long-term disease free survival in AML and a broad spectrum of chemotherapeutic regimens and targeted therapies have been proposed. Nevertheless, the clinical results demonstrate that a significant num-ber of patients who achieve CR after induction therapy still harbor a minimal residual disease (MRD) often resistant to further chemotherapeutic treat-ments which eventually leads to re-lapse and disease progression. These data demonstrate the impor-tance of preventing relapse by ad-dressing MRD with novel strategies oth-er than chemotherapy. Immunological therapies, which act in a different way than cytotoxic drugs, may significant-ly impact on the eradication of MRD, thus resulting in improved clinical out-come. In this scenario, allogeneic SCT, which combines the cytotoxic effect of con-ditioning regimen with adoptive im-munotherapy, has been shown to offer a clear advantage in terms of relapse prevention, thus providing the proof-of-principle of the capacity of immune cells of eradicating MRD. However, such approach has several and important limitations and is not applicable to all the patients.
◗◗◗ NK CELLS: BIOLOGICAL PILLS
Within allogeneic SCT, donor lympho-cytes are able to recognize and de-stroy recipient’s residual leukemic cells. The demonstration that such process, known as graft versus leukemia (GvL) effect, plays a major role in the ther-
apeutic effect of SCT has provided the background for investigating the mechanisms underlying such effect and to promote the development of novel strategies of adoptive immuno-therapy before and after SCT. In particular, donor lymphocyte in-fusion (DLI) is widely used to prevent relapse in myeloid malignancies after allogeneic stem cell transplantation. This procedure was primarily investigat-ed in chronic myeloid leukemia (CML) patients who relapsed after allogene-ic bone marrow transplantation; in this cohort of patients DLI demonstrated to be more effective than chemotherapy in obtaining and maintaining a second complete remission. In the last years, DLI has become a standard practice to treat patients who present mixed chimerism or initial relapse after allogeneic SCT. In partic-ular, several studies report about the therapeutical effect of DLI in high-risk AML patients, relapsing after allogene-ic SCT (1). Although most of the data are referred to the effect of allogeneic T cells in mediating GvL, it is known that other subsets of circulating lymphocytes, such as natural killer (NK) cells, may sig-nificantly act as effector cells against leukemia in the post-transplantation setting. NK cells are a subset of peripheral lym-phocytes defined by the expression of CD56 and CD16 and by the absence of CD3 and are involved in the innate immune response. NK cells play a critical role in cancer im-munosurveillance, being able to con-trol tumor development and growth; they recognize and kill transformed cell lines in an MHC-unrestricted fash-ion (2). NK cells activity depends on

247Targeting the minimal residual disease in acute myeloid leukemia
the expression on their surface of sev-eral activating and inhibitory receptors that recognize MHC class I molecules (Figure 1); the inhibitory receptors are named killer cell immunoglobulin-like receptors (KIRs) and they recognize al-lotypic determinants shared by certain groups of HLA class I alleles. NK cells that express a KIR whose li-gand is a HLA class I which is absent on allogeneic targets sense the missing expression of the self class I KIR ligand and mediate alloreactions. NK cell re-ceptors that recognize antigens at the HLA-A, -B, or -C loci are members of the immunoglobulin super family and are termed killer immunoglobulin re-ceptors or KIRs. Engagement of these NK cell recep-tors results in stimulation or inhibition of NK cell effector function, which ul-
timately depends on the net effect of activating and inhibitory receptors. Recently, other inhibitory receptors on NK cells have been recognized such as CD94/NKG2A receptors that rec-ognize a non-classical MHC-I (HLA-E). CD94/NKG2A continuously recycle from the cell surface through endoso-mal compartments, thus facilitating its inhibitory capacity (3). Moreover, activating receptors ex-pressed on NK cells include Fcgam-maRIIIA, activating forms of KIRs, NK-G2D. These receptors are able to trigger an-tibody-dependent cellular cytotoxicity (ADCC) on opsonized target cells and on tumor cells. Integrins also play a central role in me-diating adhesion to target cells and degranulation (4).
FigURe 1 • Receptors and ligand involved in NK cell-mediated cytotoxicity.
NCRs
KIRs
CD 94, NKG2A
2 B4, NTBA
NKG2D
???
HLA
HLA-E
CD 48 ???
MIC A, MIC B,ULBPs
+
-
-
-+
+

248 S. Parisi, et al.
◗◗◗ THE ROLE OF NK CELLS IN THE SETTING OF ALLOGENEIC SCT
Haploidentical SCTSeveral preclinical and clinical investi-gations demonstrated that haploiden-tical KIR-mismatched NK cells play the main role as anti-leukemia effector cells and they exert their cytotoxic activity within 4-5 days after transplant (Table 1). In addition, AML patients with KIR ligand mismatch are significantly pro-tected against leukemia relapse (5). Preliminary studies at the pre-clinical level demonstrated that alloreactive NK cells, infused into human AML-en-grafted NOD/SCID mice, were capa-ble of clearing leukemia and improving survival. Based on these premises, the first sem-inal study by Ruggeri et al. evaluated the impact of donor-versus-recipient NK cell alloreactivity on the outcome of 112 high-risk acute leukemia pa-tients, undergoing haploidentical he-matopoietic SCT. Patients were divided in two subgroups, accordingly to KIR li-gand incompatibility in the graft versus host direction. The 5-year event free survival was 5% in the group with KIR-L incompatibility and 60% in the group without KIR-L in-compatibility, this demonstrating that KIR ligand incompatibility was the only independent predictive factor of sur-vival in AML patients (5). High-risk AML patients with a KIR-ligand mismatch in the GVH direction had a relapse
rate of 0% compared to KIR-ligand matched patients who had a relapse rate of 75%. Furthermore, alloreactive mismatched NK cells facilitate hema-topoietic engraftment after infusion of haploidentical stem cells, and inhibit the onset of GVHD by targeting host antigen-presenting cells. A recent work by Stern et al. showed the results of a phase II multicenter study in which purified NK cells were administered pre-emptively in recipi-ents of T-cell depleted haploidentical SCT. Sixteen young patients diagnosed with high-risk leukemia or highly malig-nant solid tumors were included in this protocol and received NK-DLI on day 40 and on day 100 after transplanta-tion. This study demonstrated the feasi-bility of the procedure. However, the trial showed a high inci-dence of acute GVHD (perhaps due to contaminating T cells in NK DLI cell preparation in the context of T-cell de-pleted haploidentical SCT) whereas the antileukemic activity appeared to be very limited following these late in-fusions (1). Unrelated SCTDifferently from haploidentical SCT, the role of NK cells alloreactivity in the field of unrelated SCT is controversial, even though several studies have already investigated this setting (Table 2). Giebel et al. conducted a study involv-ing 130 patients with hematological malignancies who underwent alloge-
TAbLe 1 • Most relevant papers reporting the impact of KIR-L mismatch in haploidentical SCT.Authors Survival TRM Relapse gVHDRuggeri et al. (2007) ↑ ↓ ↓ ↓a
Stern et al. (2013) not assessed not assessed → →a
Symons et al. (2011) → ↓ → ↑a
TRM, transplant-related mortality; ATG, anti-lymphocyte globulin.

249Targeting the minimal residual disease in acute myeloid leukemia
neic SCT and receiving Cyclosporine, ATG and short-term methotrexate as GHVD prophylaxis. With a median fol-low up of 4.5 years overall survival was 87% in patients with a KIR mismatch in the donor direction versus 48% in non KIR-mismatched patients; disease-free survival was 87% in the first group com-pared with 39% in the second one. Transplant-related mortality was 6% in the KIR-mismatched patients and 40% in non-mismatched patients (6). These results were not confirmed by the study published by Davies et al., which in-cluded 175 recipients of unrelated donor SCT; in this cohort no survival benefit was observed among KIR mis-matched patients (OS 38% in KIR non mismatched patients versus 13% in mis-matched patients) (7). Bornhauser et al. reported an in-creased rate of relapse in patients receiving grafts from KIR-ligand mis-matched donors, with no significant differences in survival (8). Schaffer et al. performed a retrospective analysis about the role of KIR-ligand mismatch in 190 unrelated transplantations. In this study KIR-ligand mismatch was associated with significantly inferior sur-vival attribuible to higher transplant-re-lated mortality mostly due to infections. This controversial data demonstrate that the role of NK cells have to be bet-ter clarified in the setting of unrelated stem cell transplantation. Several factors, such as post-transplan-
tation immunosuppressive therapies, different stem cell sources and stem cell doses, T-cell depletion, play a sig-nificant role in this setting of patients (9).
◗◗◗ NK CELLS AS ADOPTIVE IMMUNOTHERAPY OUTSIDE SCT
Infusion of NK cells has been already used in vivo as a means of adoptive immunotherapy outside the SCT-set-ting. Partially purified haploidentical NK cells have been infused (10, 11) af-ter labelling with 111In to track, in vivo, their kinetics and organ distribution, in patients with renal cancer (11). These studies demonstrated the clearance of NK cells from the peripheral blood within 7 days, a distribution to the whole body, with preference for liv-er, spleen and BM, after a short initial uptake in the lungs. The half-life in all body tissues remained almost constant over 6 days suggesting the extended survival of haploidentical cells in the host organism (11). Clinical scale selection of NK cells for cellular immunotherapy has been re-cently developed (12).A two-step procedure for purification of CD56+CD3- NK cells from leukapheresis is based on immunomagnetic tech-nique. The procedure is performed in a closed, sterile and endotoxin-free tubing set. Tubing sets and cell selec-
TAbLe 2 • Most relevant papers reporting the impact of KIR-L mismatch in unrelated SCT.Authors Survival TRM Relapse gVHD ATgDavies et al. (2002) ↓ not assessed → ↑a, b NoGiebel et al. (2003) ↑ ↓ ↓a ↓a, c YesBornhäuser et al. (2004) → → ↑ → YesSchaffer et al. (2005) ↓ ↑ → → Yes
aTrend = P-value between 0.05 and 0.09; bGVHD grade II–IV; cGVHD grade III–IV.

250 S. Parisi, et al.
tion reagents are manufactured ac-cording to medical device regulation and can be applied to clinical trials. Immunomagnetic selection of NK cells results in the depletion of 3.5-4 Log of T cells from a final product containing >90% CD56+CD3- NK cells (yield rang-ing from 30 to 60%) (12). Miller et al. published in 2005 the re-sults of a seminal study in which up to 1.5x107/haploidentical NK cells/Kg were safely infused in AML and cancer patients following Fludarabine/Cyclo-phosphamide (Flu/Cy) immunosup-pressive chemotherapy; in this study some clinical responses without GVHD had been observed. Circulating haploidentical NK cells were found up to 28 days after infusion especially when exogenous IL-2 was given for 9 doses. In vivo expansion of NK cells was correlated with a high IL-
15 serum concentration. In particular, 19 poor risk AML patients, together with 10 metastatic melanoma patients and 13 metastatic renal cell carcinoma patients received a cell population containing a median of 8.5±0.5x106 and 1.75±0.3x105 NK and T cells, re-spectively. Five out of 19 AML patients achieved CR and NK cells adoptive im-munotherapy was well tolerated and no hematological toxicity was regis-tered. The maximum tolerated dose of NK cells was not achieved and GVHD was not observed despite the relative-ly high number of haploidentical T cells infused.However, it should be noted that NK cells were only partially purified after a single round of depletion of CD3+ cells which resulted in less than 2 logs reduction of T cells (13). More recently, Rubnitz et al. reported
FigURe 2 • Percentage of long-term CR patients after NK cell infusion.Thirteen AML patients, 5 with active disease, 2 in molecular relapse and 6 in morphological complete remission (CR) were treated with alloreactive NK cells, after fludarabine/cyclophos-phamide immunosuppressive chemotherapy. Only 1 of the 5 patients with active disease achieved transient CR, whereas the other 4 patients had no clinical benefit. On the contrary, 5/8 patients showed response, which in some cases was long-lasting CR.
70
60
50
40
30
20
10
0Relapsed patients CR patients
Resp
onse
rate
to N
K (%
)

251Targeting the minimal residual disease in acute myeloid leukemia
their experience with haploidentical KIR-HLA mismatched NK cell transplan-tation in a cohort of ten pediatric AML patients. In this cohort of patients, who underwent NK therapy after an im-munosuppressive regimen, the 2-year event-free survival was 100%. Notably, all the enrolled patients were consid-ered at low-risk of relapse, with a signif-icant fraction harboring good-progno-sis cytogenetics as intermediate and high risk patients were candidates for allogeneic stem cell transplantation. Furthermore, as children weigh less than adults, the median number of in-fused NK cells was significantly higher than in adult trial and the separation procedure consisted in highly purified NK cells (14). Our group has recently published the results of a study reporting about adoptive immunotherapy with Natu-ral Killer Cells in 13 AML patients, 5 with active disease, 2 in molecular relapse and 6 in morphological complete re-mission (CR) with a median age of 62 years (range 53-73). These patients re-ceived highly purified CD56+CD3- NK cells from haploidentical KIR-ligand mismatched donors after fludarabine/cyclophosphamide immunosuppres-sive chemotherapy, followed by IL-2 administration. The median number of infused NK cells was 2.74 x 106/kg. T cells were under 105/kg. No NK cell related tox-icity, including GVHD, was observed. One of the 5 patients with active dis-ease achieved transient CR, whereas the other 4 patients had no clinical benefit. Both patients in molecular re-lapse achieved CR lasting for 9 and 4 months, respectively.Three patients in CR were disease-free after 34, 32 and 18 months of follow
up (Figure 2). After infusion, donor NK cells were found in the peripheral blood of all evaluable patients with a peak value on day 10. Donor-ver-sus-recipient alloreactive NK cells were demonstrated in vivo by the detection of donor-derived NK clones that killed recipient’s targets. Adoptively transferred NK cells were alloreactive against recipient’s cells, including leukemia (15).
◗◗◗ REFERENCES1. Stern M, Passweg JR, Meyer-Monard S,
et al. Pre-emptive immunotherapy with purified natural killer cells after hap-loidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant 2013; 48: 433-8.
2. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene 2008; 27: 5932-43.
3. Borrego F, Masilamani M, Kabat J, et al. The cell biology of the human natural killer cell CD94/NKG2A inhibitory recep-tor. Mol Immunol. 2005; 42:485-8.
4. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future di-rections. J Allergy Clin Immunol. 2013; 132: 536-44.
5. Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hemato-poietic transplants. Science 2002; 295: 2097-100.
6. Giebel S, Locatelli F, Lamparelli T, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated do-nors. Blood 2003; 102: 814-9.
7. Davies SM, Ruggieri L, DeFor T, et al. Evaluation of KIR ligand incompatibili-ty in mismatched unrelated donor he-matopoietic transplants. Killer immuno-globulin-like receptor. Blood 2002; 100: 3825-7.
8. Bornhäuser M, Schwerdtfeger R, Martin H, et al. Role of KIR ligand incompatibili-

252 S. Parisi, et al.
ty in hematopoietic stem cell transplan-tation using unrelated donors. Blood. 2004; 103(7): 2860-1
9. Malmberg KJ, Schaffer M, Ringdén O, Remberger M, Ljunggren HG. KIR-li-gand mismatch in allogeneic hemato-poietic stem cell transplantation. Mol Immunol. 2005; 42: 531-4.
10. Hsu KC, Gooley T, Malkki M, et al. KIR ligands and prediction of relapse after unrelated donor hematopoietic cell transplantation for hematologic malig-nancy. Biol Blood Marrow Transplant. 2006; 12: 828-36.
11. Meller B, Frohn C, Brand JM, et al. Mon-itoring of a new approach of immu-notherapy with allogenic (111)In-la-belled NK cells in patients with renal cell carcinoma. Eur J Nucl Med Mol Imag-ing. 2004;31:403-7.
12. Passweg JR, Tichelli A, Meyer-Monard S,
et al. Purified donor NK-lymphocyte in-fusion to consolidate engraftment after haploidentical stem cell transplanta-tion. Leukemia. 2004; 18: 1835-8.
13. Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human hap-loidentical NK cells in patients with can-cer. Blood. 2005; 105: 3051-7.
14. Rubnitz JE, Inaba H, Ribeiro RC, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in child-hood acute myeloid leukemia. J Clin Oncol. 2010; 28: 955-9.
15. Curti A, Ruggeri L, D’Addio A, et al. Successful transfer of alloreactive hap-loidentical KIR ligand-mismatched nat-ural killer cells after infusion in elderly high risk acute myeloid leukemia pa-tients. Blood. 2011; 118: 3273-9.

DCTH - 4•2013 - 253-260
Key words: intestinal GVHD; CEUS; ultra-sound sonography.
Correspondence:Edoardo Benedetti, Via Roma, 67 - 56100 Pisa, Italy E-mail: [email protected]
SUMMARYIntestinal acute graft-vs-host disease (I-GVHD) is a life-threatening complication after al-lografting. Non-invasive bedside procedures to evaluate extension and treatment re-sponse are still lacking. Standard ultrasound sonography (US) detects bowel wall thicken-ing (BWT) in I-GVHD and helps to identify its extension (single or multiple sites at once). Color doppler US detects blood flow at arterioles level. Contrast-enhanced ultrasound sonography (CEUS) can detect microcirculation changes (MVC) of the bowel wall at capillary level, real time, and even bedside. CEUS allows evaluating, with dedicated software, quantitatively wash-in and washout curves of blood flowing through the thick-ened bowel wall, giving time intensity curves, which help to monitor treatment response as for Chron’s disease. Patients with I-GVHD with clinical relevant improvement may still have quiescent active disease. CEUS allows identifying, qualitatively and quantitatively, patients with I-GVHD with clinical improvement but with still active disease.
Ultrasound and contrast enhanced ultrasound sonography evaluation of intestinal acute graft-vs-host disease E. BenedettiDivision of Hematology at the S. Chiara Hospital, University of Pisa, Italy; SIUMB Basic Course & Emergency School of Ultrasonography (SIUMB - Italian Society of Ultrasound in Medicine and Biology)
◗◗◗ INTRODUCTION
Intestinal acute GVHD (I-GVHD), is a major cause of non-relapse mortality following allogeneic transplants (1).Diarrhea volume is generally used to determine the severity of the intestinal involvement, but its clinical reliability is highly limited (2, 3). Diagnosis remains problematic for some patients with this pathology in the
REVIEW
midgut. Overall, non-invasive specific and sensitive techniques to diagnose intestinal GVHD, to evaluate both the extention of intestinal involvement and treatment response and to guide the duration of immunosuppression are still lacking. Standard transabdominal ul-trasonography (US) has already been used at diagnosis and during clinical follow up (4-6) and, more widely, in a variety of other intestinal diseases (7-9) including inflammatory bowel diseases (10-13). Recent studies have highlight-ed neovascularization in early stages of GVHD (14).We, and other groups have report-ed on Ultrasound, Colordoppler ultra-sound (4, 5, 6), and CEUS in intestinal aGVHD (15, 16).

254 E. Benedetti
◗◗◗ STANDARD US TECHNIQUE
Standard (B-mode) US can be per-formed bed-side with a portable so-nographer (16) without any prepara-tion, at the onset of I-GVHD symptoms. The entire gastrointestinal tract is sub-mitted to a gray-scale ultrasound ex-amination (B-mode US). The colon can be examined from the cecum to the sigmoid colon. The en-tire small bowel is examined with par-ticular attention to the last portion of the terminal ileum, most commonly in-volved site of GVHD (4-6).The following parameters can be as-sessed:1) bowel wall thickness (BWT) defined
as abnormal if <3 mm in the large bowel and <2 mm in the duodenum and small bowel (17);
2) bowel wall layers: the superficial mucosal interface, the deep muco-sa, the submucosa, the muscolaris propria and the serosa (18, 19);
3) degree of dilation (20);4) motility (17);5) bowel content defined as gas, food
stuff of feces, mixtures of the two, or fluid-filled (21);
6) presence of haustral or dehaustra-tion (17, 22);
7) presence/absence of free abdom-inal fluid in all four quadrants and/or upper abdominal organ patholo-gies other than GVHD.
◗◗◗ STANDARD US RESULTS
In patients with acute I-GVHD stan-dard US reveals increased BWT mostly related to mucosal edema (16). The intestinal segments involved may vary from patient to patient. In a previous
prospective study 9/14 patients had more than one site involved at the on-set of symptoms (16), in accordance with others (4, 5). The BW layers could be identified in 11 patients whereas in the others bound-aries were poorly defined, indicating a more inflamed bowel wall. Moreover, standard US showed normal intesti-nal features in patients with localized stomach a GVHD (16).
◗◗◗ CONTRAST ENHANCED ULTRASOUND SONOGRAPHY (CEUS)
Real-time microvascular imaging has recently been made possible by novel echo-contrast enhancing agents and low mechanical-index harmonic so-nography. CEUS has been extensively used in active Chron’s disease in which the neovascularization of the small bowel walls has been described (23-29). Importantly, recent studies have highlighted neovascularization in early stages of GVHD (24). Table 1 summa-rizes the main experiences with CEUS in aGVHD and other inflammatory bowel diseases.GVHD pathophysiology is character-ized by neovascularization, mainly driv-en by vasculogenesis, during its early inflammatory phase. At a later stage, the vasculature itself becomes a tar-get of allo-reactive donor T cells lead-
TABlE 1 • Main experiences with CEUS in aG-VHD and other inflammatory bowel diseases.Main experiences ReferencesCEUS in intestinal GVHD
15, 16
CEUS in inflammatory bowel diseases
23 and references therein, 24-29

255GVHD evaluation by CEUS
ing to fibrosis and rarefaction of blood vessels (14). Overall, the increased arterial microvascular enhancement evidentiated by CEUS may correlate with an early neovascularization of the bowel walls (14). TechniqueAfter standard US, the ultrasound con-trast agent (UCA) is administered i.v. and CEUS performed on diseased in-testine (23) with the same sonogra-pher equipped with contrast specific real-time imaging technology defined as contrast tuned imaging (CnTI) (16). A second generation echo-contrast agent, SonoVue® (Bracco, Milan, Ita-ly), is injected as i.v. bolus into an ante-cubital vein. SonoVue® is a non-neph-rotoxic contrast agent which consists of 2.5 mm-diameter microbubbles sta-bilized with phospholipids and filled with sulphur hexafluoride which flow through the pulmonary microcircula-tion and remain within the vascular space (25). SonoVue® is approved in Europe for clinical use and has a wide range of clinical applications (26, 27).CnTI exploits the resonance proper-ty of the microbubbles and prevent them from bursting during insonation. This allows for real-time imaging of the microcirculation, without gray-scale echoes, and provides continuous per-fusion data on viscera (28).After injection the contrast agent reaches the intestinal wall in about 10-15 seconds and its peak concentra-tion after approximately 30 seconds. In the intestine this arterial phase is fol-lowed by the venous phase in which the contrast agent, after distributing to the whole intestinal capillary bed, is exhaled through the lungs (23). Contin-
uous imaging is usually recorded from injection throughout the entire arterial and venous phases as previously de-scribed (29). Distinct digital cine-clips for basic US and for CEUS scans are stored for com-puted analysis. Echo-signal intensity of the vascularity of the bowel segments selected with CEUS, defined by the operator as re-gions of interest (ROI) may be quan-titatively analyzed with dedicated softwares (e.g. Q-ontrast;e-AMID-Ad-vanced Medical Imaging Develop-ment, Italy distributed from Bracco, Milan (16, 30-29). Q-ontrast generates chromatic maps of the ROI perfusion patterns, and automatically compen-sate for motion artifacts during data acquisition (30). The Q-ontrast analysis of ROI gener-ates curves representing echo-signal intensity vs time (time intensity curves, TIC). For all patients TIC parameters, in-cluding the slope of the first ascending tract of the curve, the curve shape, time to peak enhancement, the area under the curve (AUC), regional blood flow and mean transit time (MTT) may be recorded for a subsequent quanti-tative analysis (29).CEUS can be safely performed in pa-tients with renal insufficiency (23).
◗◗◗ CEUS RESULTS
CEUS was previously reported as a di-agnostic tool in I-GVHD (15), showing passage of microbubbles from the BW into the intestinal lumen and consid-ering this phenomenon as diagnostic of I-GVHD. We were no able to repro-duce their results in our prospective study (16).

256 E. Benedetti
Possible explanations are:1) in our prospective study we found
passage of microbubbles from the damaged mucosa into the lumen also in patients with neutropenic enterocolitis (which was one of our control group), thus indicating that the contrast media, which is blood pool, can pass from the bowel wall into the intestinal lumen when there is a damaged mucosal barrier, inde-pendent from the cause. This impli-cates that in patients who received an allogeneic transplant and with onset of intestinal symptoms (such as diarrhea, and abdominal pain), histology is mandatory to diagnose GVHD, and NEC has to be exclud-ed having both the same standard ultrasound features (BWT) and pos-sibly, depending on the degree of damaged mucosal barrier, passage of microbubbles from the bowel wall into the lumen.
2) It has to be taken into account that to see a passage of contrast me-dia from the bowel wall into the lumen, the lumen per se has to be fluid-filled and distended, in order to be able to visualize the micro-bubbles floating in the lumen. If the I-GVHD involves for example the colon causing BWT without dilation and without a fluid filled lumen, this process will not be seen, and it is going to lose its hypothesizes diag-nostic power.
CEUS can instead be used to monitor I-GVHD after biopsy-proven diagno-sis. CEUS allows assessing response to immunosuppressive treatment (with a quantitative and qualitative analysis of microvascular blood in-flow and wash out phase, of the bowel wall) (16, 29) as for Chron’s disease (29).
CEUS and US findings are superimpos-able at diagnosis and in patients in complete remission of symptoms (16), but CEUS is significantly more sensitive and specific than standard US to iden-tify sub-clinical GVHD activity, predic-tive of clinical flare, in patients without complete resolution of symptoms (clin-ical relevant improvement) (1, 16).
◗◗◗ CONCLUSIONS AND PERSPECTIVES
Acute GVHD and its complications are major causes of non-relapse mortality following an allograft. Reliable non in-vasive procedures to evaluate its ex-tension and treatment response are still lacking (3). Acute I-GVHD response to treatments correlates with longer survival (1). Acute I-GVHD shows an increased mi-crovessel network circulation (31) and graft-vs-host reactions are associated with increased neovascularization (14). Extention of intestinal involvement by aGVHD, which might be patchy, is seen with standard US (4, 16), with pos-itron emission tomography (PET) (32), and computed tomography (CT) (33), but there are limitations using PET and CT due to:1) difficult to repeat frequently to as-
sess response to immunosuppressive treatment;
2) the patients must leave the isolation room;
3) concerns due to radiation expo-sure. On the contrary US and CEUS can be both easily repeated bed-side.
Up to now intestinal biopsy remains mandatory for diagnosis. CEUS pro-posed as a diagnostic tool (15) has

257GVHD evaluation by CEUS
some important limitations and is yet to be validated prospectively.Standard US:1) reveals extention of I-GVHD;2) it reveals the extent of BWT;3) it reveals sites involved and reveals,
if present, a patchy pattern of intes-tinal involvement;
4) patients with localized stomach aGVHD have normal intestinal US features, suggesting that in this subset of patients, there is the pos-sibility to utilize less immunosuppres-sive treatment as compared with I-GVHD (16);
5) easily repeatable during the pa-tient’s follow up (weekly in patients with stable symptoms, even more frequently if there is a worsening of symptoms and every two to three weeks in patients with clinical rele-vant improvement of symptoms);
6) it allows to screen both intestine and for abdominal organs patholo-gy and free abdominal fluid;
7) if used bed-side, the patients do not leave the isolation room (helpful es-pecially if neutropenic and clinically infirm);
8) competence ensured by adequate training is a prerequisite to achieve correct diagnoses when using ultra-sonography and especially CEUS. EFSUMB has defined three levels of training for a physician in its minimal training requirements (23). Table 2 summarizes main characteristics and advantages of CEUS.
Patients with clinical relevant improve-ment without complete resolution of symptoms are a challenging subgroup at high risk of flare. Close follow up is mandatory. Standard US only allows monitoring BWT. By contrast CEUS allows detecting mi-crovascular changes at a capillary lev-el, underlying persistent inflammatory activity, also in patients with normal BWT (16).
TABlE 2 • Synoptic table of main characteristics and advantages of CEUS.CEUS Characteristics CEUS Advantages1. CEUS is a real-time microvascular diag-
nostic imaging technique at capillary level.
2. The cost of this technique: in Hospitals and Health care institutions is approxi-mately 65 Euros/vial. Usually from ½ to 1 vial is used per examination.
1. Applicable Bed side with sonographers equipped for this technique, thus pa-tients do not have leave the Hematolo-gy/Transplant ward.
2. Non radiation-based technique.3. Low side effects (Life threatening ana-
phylactoid reactions have been report-ed with a rate of less than 0.002%) (23).
4. Ultrasound Contrast Agents are not nephrotoxic and do not interact with the thyroid gland and it is therefore not nec-essary to perform laboratory tests before their administration (23).
5. Previous allergic/anayphylactoid reac-tion to X-ray iodinated contrast agents does not necessitate the prophylactic use of steroids or antihistamines prior to UCA injection since the two types of agent are completely different (23).

258 E. Benedetti
CEUS can thus be used:1) to monitor I-GVHD after diagnosis is
made (biopsy-proven), to assess re-sponse to immunosuppressive treat-ment (using quantitative assess-ment of microvascular blood flow in the involved bowel wall) (16);
2) it reveals quiescent I-GVHD (16) as for other inflammatory bowel dis-eases (10, 27, 29);
3) CEUS resulted to be more sensitive and specific in respect to standard US to identify patients with clinical relevant improvement (16) but with still quiescent active disease;
4) it can be performed in patients with renal impaired function (23), where CT or MRI with contrast media are contraindicated (34);
5) the reported incidence of severe hypersensitivity or allergic events to CEUS contrast agents is extremely low (0.001%) (23);
6) it is a cheap, radiation-free, bed-side technique (4-6, 16).
In perspective CEUS shows microcircu-lation changes of the bowel wall which correlates with clinical symptoms of biopsy-proven intestinal GVHD and its treatment response both at diagnosis, at follow up and at flare (16). In conclusion, though not diagnostic, CEUS findings might result a useful tool to manage immunosuppressive treat-ment especially in the challenging subgroup of patients with clinical rel-evant improvement of symptoms, but with still active aGVHD.
◗◗◗ REFERENCES
1. McDonald GB, Shulman HM, Sullivan KM, Spencer GD. Intestinal and hepatic complications of human bone marrow transplantation. Part I. Gastroenterolo-gy 1986; 90: 460-77.
2. Sale GE, Shulman HM, McDonald GB, Thomas ED. Gastrointestinal graft-ver-sus-host disease in man. A clinicopath-ologic study of the rectal biopsy. Am J Surg Pathol 1979; 3: 291-9.
3. Fisk JD, Shulman HM, Greening RR, et al. Gastrointestinal radiographic features of human graft-vs.-host disease. Am J Roentgenol 1981; 136: 329-36.
4. Klein SA, Martin H, Schreiber-Dietrich D, et al. A new approach to evaluating in-testinal acute graft-versus-host disease by transabdominal sonography and colour Doppler imaging. Br J Haematol 2001; 115: 929-34.
5. Görg C, Wollenberg B, Beyer J, et al. High-resolution ultrasonography in gas-trointestinal graft-versus-host disease. Ann Hematol 2005; 84:33-9.
6. Haber HP, Schlegel PG, Dette S, Ruck P, Klingebiel T, Niethammer D. Intestinal acute graft-versus-host disease: findings on sonography. Am J Roentgenol 2000; 174: 118-20.
7. Cammarota T, Sarno A, Robotti D, et al. US evaluation of patients affected by IBD: how to do it, methods and findings. Eur J Radiol 2009; 69: 429-37.
8. Robotti D, Cammarota T, Debani P, et al. Activity of Crohn disease: value of Color-Power-Doppler and contrast-en-hanced ultrasonography. Abdom Im-aging 2004; 29: 648-52.
9. Gritzmann N, Hollerweger A, Macheiner P, Rettenbacher T. Transabdominal so-nography of the gastrointestinal tract. Eur Radiol 2002; 12: 1748-61.
10. Maconi G, Imbesi V, Bianchi Porro G. Doppler ultrasound measurement of in-testinal blood flow in inflammatory bow-el disease. Scand J Gastroenterol 1996; 31: 590-3.
11. Giovagnorio F, Diacinti D, Vernia P.

259GVHD evaluation by CEUS
Doppler sonography of the superior mesenteric artery in Crohn’s disease. Am J Roentgenol 1998; 170: 123-6.
12. Ludwig D, Wiener S, Brüning A, et al. Mes-enteric blood flow is related to disease activity and risk of relapse in Crohn’s disease: a prospective follow-up study. Am J Gastroenterol 1999; 94: 2942-50.
13. Mayer D, Reinshagen M, Mason RA, et al. Sonographic measurement of thick-ened bowel wall segments as a quan-titative parameter for activity in inflam-matory bowel disease. Z Gastroenterol 2000; 38: 295-300.
14. Penack O, Socié G, Van den Brink MRM. The importance of neovascularization and its inhibition for allogeneic hemato-poietic stem cell transplantation. Blood 2011; 117: 4181-9.
15. Schreyer AG, Landfried K, Zorger N, et al. Transmural penetration of intrave-nously applied microbubbles during contrast-enhanced ultrasound as a new diagnostic feature in patients with GVHD of the bowel. Bone Marrow Trans-plant. 2011; 46: 1006-11.
16. Benedetti E, Bruno B, McDonald GB, et al. Prospective qualitative and quanti-tative non-invasive evaluation of intesti-nal acute GVHD by contrast-enhanced ultrasound sonography. Bone Marrow Transplant 2013; 48: 1421-8.
17. Kuzmich S, Howlett DC, Andi A, et al. Transabdominal sonography in assess-ment of the bowel in adults. AJR Am J Roentgenol 2009; 192: 197-212.
18. Bolondi L, Casanova P, Santi V, et al. The sonographic appearance of the normal gastric wall: an in vitro study. Ul-trasound Med Biol 1986; 12: 991-8.
19. Lim JH, Jeong YM. Sonography of the stomach: an in vitro study to determine the anatomic cause of inner hypere-choic and hypoechoic layers of the gastric wall. AJR Am J Roentgenol 1994; 162: 335-8.
20. Hollerweger A. Colonic diseases: the value of US examination. Eur J Radiol 2007; 64: 239-49.
21. Cartoni C, Dragoni F, Micozzi A, et al.
Neutropenic Enterocolitis in Patients With Acute Leukemia: Prognostic Sig-nificance of Bowel Wall Thickening De-tected by Ultrasonography Journal of Clinical Oncology 2001; 19: 756-61.
22. Hagiu C, Badea R. Applicability of ab-dominal ultrasonography in inflamma-tory bowel disease. J Gastrintestin Liver Dis 2007; 16: 205-9.
23. Piscaglia F, Nolsøe C, Dietrich CF, et al. The EFSUMB Guidelines and Recom-mendations on the Clinical Practice of Contrast Enhanced Ultrasound (CEUS): Update 2011 on non-hepatic applica-tions. Ultraschall Med 2012; 1: 33-59.
24. Penack O, Socié G, Van den Brink MRM. The importance of neovascularization and its inhibition for allogeneic hemato-poietic stem cell transplantation. Blood 2011; 117: 4181-9.
25. Morel DR, Schwieger I, Hohn L, et al. Human pharmacokinetics and safety evaluation of SonoVue, a new contrast agent for ultrasound imaging. Invest Ra-diol 2000; 35: 80-5.
26. Claudon M, Cosgrove D, Albrecht T, et al. Guidelines and good clinical prac-tice recommendations for contrast enhanced ultrasound (CEUS) - update 2008. Ultraschall Med 2008; 29: 28-44.
27. Quaia E, Calliada F, Bertolotto M, et al. Characterization of focal liver lesions with contrast-specific US modes and a sulfur hexafluoride-filled microbub-ble contrast agent: diagnostic perfor-mance and confidence. Radiology 2004; 232: 420-30.
28. Migaleddu V, Scanu AM, Quaia E, et al. Contrast-enhanced ultrasonograph-ic evaluation of inflammatory activity in Crohn’s disease. Gastroenterology 2009; 137: 43-52.
29. Serra C, Menozzi G, Labate AM, et al. Ultrasound assessment of vasculariza-tion of the thickened terminal ileum wall in Crohn’s disease patients using a low-mechanical index real-time scan-ning technique with a second gener-ation ultrasound contrast agent. Eur J Radiol 2007; 62: 114-21.

260 E. Benedetti
30. Quaia E, Migaleddu V, Baratella E, et al. The diagnostic value of small bowel wall vascularity after sulfur hexafluoride-filled microbubble injection in patients with Crohn’s disease. Correlation with the therapeutic effectiveness of specific anti-inflammatory treatment. Eur J Radi-ol 2009; 69: 438-44.
31. Shidham VB, Chang CC, Shidham G, et al. Colon biopsies for evaluation of acute graft-versus-host disease (A-GVHD) in allogeneic bone marrow transplant pa-tients. BMC Gastroenterol 2003; 3: 5.
32. Stelljes M, Hermann S, Albring J, et al.
Clinical molecular imaging in intestinal graft-versus-host disease: mapping of disease activity, prediction, and mon-itoring of treatment efficiency by posi-tron emission tomography. Blood 2008; 111: 2909-18.
33. Shimoni A, Rimon U, Hertz M, et al. CT in the clinical and prognostic evaluation of acute graft-vs-host disease of the gastrointestinal tract. Br J Radiol 2012; 85: e416-23.
34. Katzberg RW, Haller C. Contrast-induced nephrotoxicity: clinical landscape. Kid-ney Int Suppl. 2006; S3-7.

DCTH - 4•2013 - 261-277
Key words: comorbidity, allogeneic trans-plantation, risk score.
Correspondence:Roberto RaimondiU.O. Ematologia, Ospedale San BortoloViale Rodolfi, 3736100 Vicenza, ItalyE-mail: [email protected]
SUMMARYIn the recent years some scoring systems have been proposed with the aim to improve the ability to predict the non-ralapse mortality risk of the allogeneic hematopoietic stem cell transplantation. They have been applied both in wide cohorts of patients with different haematological diseases, than in specific-disease groups. Although these systems are based on different parameters and the validation studies have given conflicting results, they represent at the moment effective tools for estimate the NRM risk before transplant and stratify the patients into risk categories.
Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation; usefulness of the pretransplant scoring systemsR. Raimondi, A. Tosetto, C. Borghero, F. RodeghieroDipartimento di Terapie Cellulari ed Ematologia, Unità Operativa di Ematologia, Ospedale San Bortolo, Vicenza, Italy
◗◗◗ INTRODUCTION
Although in the recent years the out-come of allogeneic hematopoietic stem cell transplantation (HSCT) im-proved and the overall mortality re-duced, non-relapse mortality (NRM) still remains a major problem and its re-duction a challenge for the next future (1, 2). NRM risk is influenced by several fac-
REVIEW
tors like type of transplant procedure, donor and stem cell source, and the patient’s risk profile which includes age, performance status and comor-bidities. The broadening indications for HSCT, the increased number of patients eli-gible for transplant, the trend to trans-plant early in the course of the disease, the possibility to modulate the proce-dure (i.e. the conditioning regimen) and the donor increased availability (sibling, unrelated, aploidentical, cord blood) make more relevant nowadays a careful assessment of the risk/benefit ratio before transplantation. The traditional parameters used to esti-mate the risk of the transplant are age, performance status and tests evaluat-ing for single organ function (e.g. left ventricular ejection fraction for the
262 R. Raimondi, et al.
heart, spirometry and carbon oxide diffusion capacity for lung, creatinine clearance for kidney and bilirubin and transaminases for liver function). Each of these single parameters is not able in itself to predict the risk of NRM and studies evaluating their impact on NRM have given conflicting results.For this reason in the last years at-tempts have been made to put to-gether some of these parameters to create a scoring system with the aim to improve the ability to predict the risk of NRM.
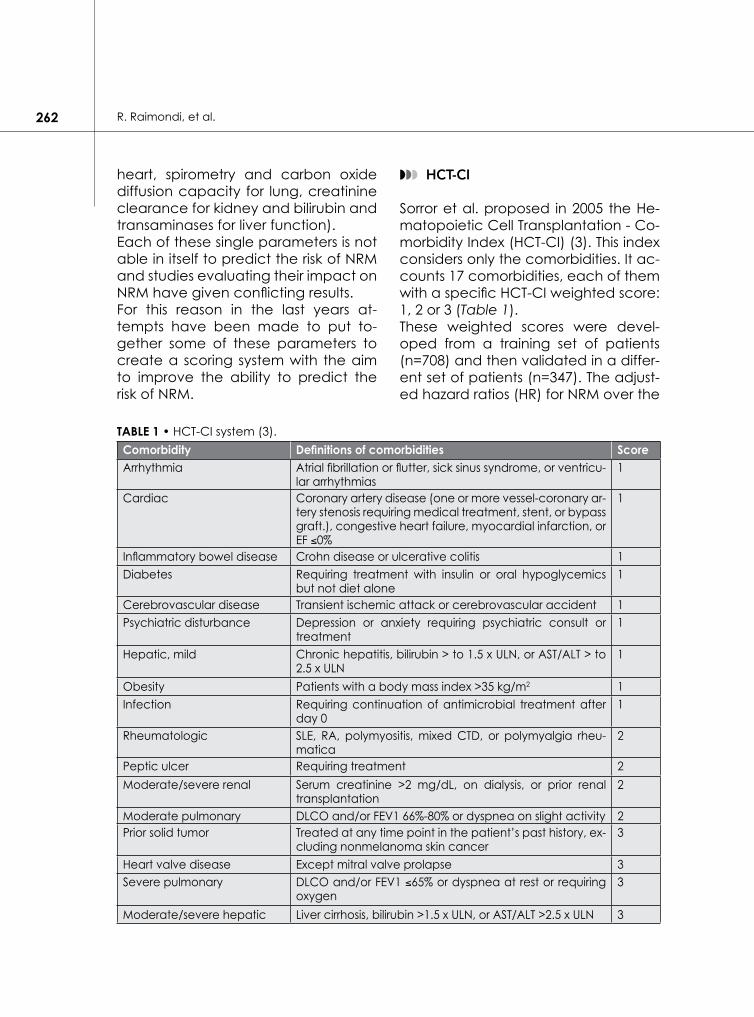
◗◗◗ HCT-CI
Sorror et al. proposed in 2005 the He-matopoietic Cell Transplantation - Co-morbidity Index (HCT-CI) (3). This index considers only the comorbidities. It ac-counts 17 comorbidities, each of them with a specific HCT-CI weighted score: 1, 2 or 3 (Table 1).These weighted scores were devel-oped from a training set of patients (n=708) and then validated in a differ-ent set of patients (n=347). The adjust-ed hazard ratios (HR) for NRM over the
TABle 1 • HCT-CI system (3).Comorbidity Definitions of comorbidities ScoreArrhythmia Atrial fibrillation or flutter, sick sinus syndrome, or ventricu-
lar arrhythmias1
Cardiac Coronary artery disease (one or more vessel-coronary ar-tery stenosis requiring medical treatment, stent, or bypass graft.), congestive heart failure, myocardial infarction, or EF ≤0%
1
Inflammatory bowel disease Crohn disease or ulcerative colitis 1Diabetes Requiring treatment with insulin or oral hypoglycemics
but not diet alone1
Cerebrovascular disease Transient ischemic attack or cerebrovascular accident 1Psychiatric disturbance Depression or anxiety requiring psychiatric consult or
treatment1
Hepatic, mild Chronic hepatitis, bilirubin > to 1.5 x ULN, or AST/ALT > to 2.5 x ULN
1
Obesity Patients with a body mass index >35 kg/m2 1Infection Requiring continuation of antimicrobial treatment after
day 01
Rheumatologic SLE, RA, polymyositis, mixed CTD, or polymyalgia rheu-matica
2
Peptic ulcer Requiring treatment 2Moderate/severe renal Serum creatinine >2 mg/dL, on dialysis, or prior renal
transplantation2
Moderate pulmonary DLCO and/or FEV1 66%-80% or dyspnea on slight activity 2Prior solid tumor Treated at any time point in the patient’s past history, ex-
cluding nonmelanoma skin cancer3
Heart valve disease Except mitral valve prolapse 3Severe pulmonary DLCO and/or FEV1 ≤65% or dyspnea at rest or requiring
oxygen3
Moderate/severe hepatic Liver cirrhosis, bilirubin >1.5 x ULN, or AST/ALT >2.5 x ULN 3

263Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
first 2 years after transplantation were then converted to integer weights. Original comorbidities with HR ≤1.2 were excluded, comorbidities with HR of 1.3 to 2 were assigned a weight of 1 and comorbidities with HR of 2.1 to 3 or ≥3.1 were assigned a weight of 2 and 3 respectively; the HCT-CI score is the sum of each comorbidity weight.This system allowed Sorror and coll. to stratify patients into 3 risk groups: HCT-CI =0 (low risk), HCT-CI =1-2 (intermedi-ate risk), HCT-CI ≥3 (high risk).In the validation set the 2-year NRM was 14, 21 and 41% respectively for low, intermediate and high risk groups. Also the overall survival (OS) resulted different in the 3 groups (71, 60 and 34% respectively). The HCT-CI proved to be more effec-tive in the setting of HSCT to capture pretransplant comorbidities and to assess NRM than the previously used Charlson Comorbidity Index.Several studies tested HCT-CI. Some Authors found HCT-CI to be predic-tive for NRM and overall survival (4-17), while others did not (18-25). Other Authors found HCT-CI to be predictive only if modified (26, 27). Taking in account the largest studies, the HCT-CI has confirmed its predictive value in acute myeloid leukemia (AML) (15), acute lymphoblastic leukemia (ALL) (19), myelodisplastic syndrome (MDS) (28), chronic myelomonocytic leukemia (CMML) (29), non-Hodgkin lymphomas (NHL) and multiple myelo-ma (MM) (12) and in chronic lympho-cytic leukemia (CLL) (10). Michelis et al. (30) confirmed the prog-nostic value of HCT-CI in AML patients transplanted in 2nd complete remis-sion, Quan Le et al. (31) and Ratan et al. (32) found that HCT-CI maintains its
predictive ability also in ex vivo T-cell depleted HSCT, whereas Paun et al. (33) didn’t find a correlation between HCT-CI and NRM in a cohort of pa-tients undergoing umbilical cord blood transplantation. Recent reports analyzing a large co-hort of patients confirmed that HCT-CI could be an useful tool to predict overall survival also in hematologic non-malignant diseases (34) and to predict NRM in autologous hemato-poietic stem cell transplantation (35). Its value in predicting survival, but not NRM, after autologous hematopoiet-ic stem cell transplantation in patients with multiple myeloma has also been described (36).Almost all these studies were retro-spective, often with a small number of patients from a single Centre and in some cases considering transplants performed even 10 or 15 years ago.Recently the HCT-CI has been validat-ed in a prospective multicentre study from GITMO (Gruppo Italiano Trapi-anto di Midollo Osseo) (37). A large co-hort of 1937 Italian adult patients trans-planted from January 2008 to February 2011 for malignant and non-malignant haematological diseases have been analysed. HCT-CI was strongly correlat-ed with both 2-year NRM (14.7%, 21.3%, and 27.3% in patients having an HCT-CI score of 0, 1-2, and ≥3, respective-ly) and overall survival (56.4%, 54.5%, and 41.3%, respectively). There was an excellent calibration between the predicted and observed 2-year NRM in patients having an HCT-CI score of 0 and 1-2, whereas in the group with score ≥3 predicted NRM from the orig-inal study of Sorror overestimated the observed NRM (41% vs. 27.3%). HCT-CI alone was the strongest predictor of

264 R. Raimondi, et al.
NRM in patients with lymphoma, my-elodisplastic syndrome and acute my-eloid leukemia in first remission.Nevertheless some problems still re-main. The discriminative capacity (c-statistics) for NRM is 0.60-0.65 and should be improved. Moreover some comorbidities could be incorrectly calculated. Examples of difficulties in defining and scoring the comorbidities in the clinical setting are arrhythmia, heart valve disease and pulmonary comorbidities.Arrhythmia has to be defined and scored only for any type of arrhythmia that has necessitated the delivery of a specific antiarrhythmic treatment at any time in the patient’s past med-ical history, even if the patient was in normal sinus rhythm at the time of pretransplant evaluation. No score is assigned to transient arrhythmias that never required treatment. Heart valve disease has to be defined and scored only in the presence of 1 or more of the following 3 clinical presentations:1) at least a moderate or severe de-
gree of valve stenosis or insufficien-cy, as determined by echocardio-gram, whether that valve was mitral, aortic, tricuspid, or pulmonary;
2) prosthetic mitral or aortic valve;3) symptomatic mitral valve prolapse. Pulmonary comorbidity needs to be defined and scored by the results of pulmonary function tests, in particular corrected diffusion capacity of car-bon monoxide (DLCO) and forced expiratory volume in 1 second (FEV1) percentages. Measured DLCO needs to be corrected for the concurrent haemoglobin value using the Dinakara equation. For the definition and scor-ing of moderate or severe pulmonary comorbidity the shortness of breath
and/or the need for oxygen supple-mentation have been also added.In a recent report Coffey et al. (38) have compared two methods for ad-justing DLCO for haemoglobin level, i.e. the Dinakara and the Cotes equations. They showed that the method used may significantly affect the interpreta-tion of the HCT-CI. They found that the Cotes method may overestimate NRM predicted by the HCT-CI in as many as a third of patients and recommend that for the purpose of determining a patient’s HCT-CI score the Dinakara method be used for adjusting DLCO for haemoglobin level.Recently Sorror (39) has drawn the at-tention on the reliability of the comor-bidities assessment, showing that only a fair inter-observer agreement rate could be detected when comorbidi-ty scoring was tested across different evaluators. A training program and guidelines for comorbidity coding have been pro-posed and a Webbased application and calculator, available at http://www.hctci.org has been implement-ed. The HCT-CI score is also useful to cal-culate the comorbidity weight on the results of clinical trials and to compare the different trials; actually it has been incorporated among the parameters and the inclusion/exclusion criteria of most of the new clinical trials.Recently the Centre for International Blood and Marrow Transplantation Re-search (CIBMTR) has incorporated the HCT-CI in routine data collection from transplant Centres.Another road that the HCT-CI has opened is the hope to find a correla-tion between a specific comorbidity and a specific cause of mortality (or

265Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
morbidity) with the aim to be able to implement effective specific strategies to prevent that risk. At the moment however no study has addressed this topic with a real clinical impact. A modified HCT-CI has been proposed by DeFor et al. (27). They hypothesized that the discriminating and predictive power of the HCT-CI for mortality could be improved by eliminating the assign-ment of categorical weights to comor-bidities and replacing them with haz-ard ratios (HR) from a Fine and Gray adjusted regression model. They found that certain comorbidities in the HCT-CI could be uninformative either for a low prevalence or for a truly low predictive power on NRM. Instead of converting the adjusted HR to categorical weights they used a pure multiplicative model of the ex-act parameter estimates from the HR, obtaining the final score directly by exponentiation the sum of all parame-ter estimates. Using the original HCT-CI in their cohort the 2-year NRM for the low, intermediate and high-risk groups were 18%, 23% and 27% respectively, whereas using the Modified HCT-CI, the 2-year NRM for the low, interme-diate and high-risk new groups were 15%, 23% and 34%. However in this study the patient numbers did not provide enough power to create a separate and independent validation cohort, and a major disadvantage is that the equation for the final score is quite complex in comparison with the original HCT-CI that is very easy to be calculated. So the Authors suggest to use the ‘exp’ function in a software spreadsheet such as Microsoft Excel or alternatively an application online such as the one at http://bmt.ahc.umn.edu: 8082/hct/.
◗◗◗ PAM
In 2006 Parimon et al. (40) proposed a risk score to calculate the overall mortality risk of HSCT. This system has been called Pretransplantation Assess-ment of Mortality (PAM) and has been constructed on 8 pretransplantation clinical variables (patient age, donor type, disease risk, conditioning regi-men, FEV1, carbon monoxide diffu-sion capacity, serum creatinine level, and serum alanine aminotransferase concentration). It derives from a retro-spective cohort of 2802 patients treat-ed with HSCT between 1990 and 2002.In the PAM system at each of these parameters is assigned a score (Table 2) and the final score is the sum of the single scores. The Authors stratified patients into 4 categories according to the final scores: category 1 (final score ranged from 9 to 16) with probability of death <25%, category 2 (final score ranged from 17 to 23) with probability of death 25% to 50%, category 3 (final score ranged from 24 to 30) with probability of death 50% to 75%, category 4 (final score ranged from 31 to 44) with prob-ability of death >75%.In the original paper the results are pre-sented for the 3 most common disease categories: chronic myelogenous leu-kemia, acute myelogenous leukemia and the myelodisplastic syndromes.It must be emphasized that the PAM has been tested and is only useful for predicting the risk for death for any cause within the first 2 years after he-matopoietic cell transplantation, not the NMR risk; it was not designed to predict NRM.Very few studies have validated the PAM system in different cohorts.

266 R. Raimondi, et al.
TABle 2 • PAM system (40).Variable Definitions ScoreAge <20 years 1
20-30 years 130-40 years 140-50 years 150-60 years 3>60 years 5
Donor type Related, matched 1Unrelated 3Related, mismatched 4
Disease risk Low 1Intermediate 8High 12
Conditioning regimen Nonmyeloablative 1Non-total-body irradiation 4Total-body irradiation with ≤12 Gy 8Total-body irradiation with >12 Gy 9
FEV1 >80% 170%-80% 3<70% 6
Carbon monoxide diffusing capacity >80% 170%-80% 1<70% 4
Serum alanine aminotransferase level ≤49 U/L 1>49 U/L 2
Serum creatinine level ≤106 mol/L (1.2 mg/dL) 1>106 mol/L (1.2 mg/dL) 8
Barba et al. (26) have found that a “flexible” HCT-CI, using a different risk group stratification in comparison with the original HCT-CI, i.e. low risk group for score 0-3, intermediate risk group for score 4-5 and high risk group for score ≥6, was associated with an high-est predictive capacity for NRM and overall survival, whereas the original HCT-CI and the PAM score were not associated with predictive value in their cohort of HSCT with reduced-in-tensity conditioning regimen.Xhaard et al. (22) compared a re-duced version of the HCT-CI (without pulmonary function tests that were
not available in their population) with an adjusted version of the PAM and found that only the adjusted version of the PAM excluding FEV1 and DLCO al-lowed to discriminate 3 risk groups with distinct 2-year overall survival.
◗◗◗ EBMT RISK SCORE
In 2009 Gratwohl et al. (41) published the results of a large retrospective study on 56.505 HSCTs performed in Europe from 1990 to 2005 and included in the EBMT (European Group for Blood and Marrow Transplantation) data base.

267Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
In different haematological malig-nant diseases, the Authors tested five parameters (age of patient, disease stage, time interval from diagnosis to transplant, donor type and donor-re-cipient sex combination) that in a pre-vious study published in 1998 demon-strated to be predictive of NMR and OS after HSCT in the setting of chronic myeloid leukemia. A score point 0 or 1 or 2 was assigned for each factor and a final score from 0 to 7 could be cal-culated (Table 3).These five parameters confirmed their significant influence on survival and NRM. Five-year survival rates decreased from 71% for patients with final score 0 to 24% for patients with final score 6 or 7, and 5-year NRM rates increased from 15% in patients with final score 0 to 47% for patients with risk score 6 or 7. This study showed a continuous gra-dient towards an increase of NRM and a decrease of overall survival with the increasing of the score, and that pre-transplant risk elements act additively for an individual patient.The EBMT risk score system has been tested in several studies and is now-
adays widely used. Terwey et al. (19) have found that in ALL patients a modified version of the EBMT risk score, where the definition of disease stage was adapted for ALL (“disease stage”, scores of 0, 1 or 2 were assigned to patients transplanted in CR1, in CR >1 and with active disease) and where the parameter “time from diagnosis to transplantation” was omitted due to multiple sources of bias, was prognos-tic for NRM and OS. Higher mEBMT was associated with inferior OS (HR =1.50), higher NRM (HR =1.36) and higher re-lapse mortality (HR =1.68).Hemmati et al. (42) confirmed the prognostic value of the mEBMT risk score for OS in a cohort of 214 patients transplanted for AML. Rezvani et al. (43) analysing patients who underwent a second transplanta-tion using an allogeneic donor after ei-ther a first autologous or first allogeneic transplant, have found that the EBMT risk score can identify patients most likely to benefit from a second trans-plantation. The 5-year OS was 51% for low risk scores 0-3, 29% for intermediate risk score 4, and 10% for risk scores 5-7.
TABle 3 • EBMT risk score (41).Risk factor Definitions ScoreAge of the patient <20 years 0
20–40 years 1>40 years 2
Disease stage1 Early 0Intermediate 1Late 2
Time interval from diagnosis to transplant2 <12 months 0>12 months 1
Donor type HLA-identical sibling donor 0Unrelated donor, other 1
Donor recipient sex combination All other 0Female donor, male recipient 1
1Disease stage does not apply for aplastic anemia (score 0).2Does not apply for patients transplanted in first CR (score 0).

268 R. Raimondi, et al.
Lodewyck et al. (44) evaluated the prognostic impact of the EBMT risk score combined with the HLA match-ing in a cohort of 327 patients with poor-risk AML or MDS who received a T-cell depleted unrelated donor HSCT. Patients with EBMT risk scores of 1-2, 3, 4 and 5-7 had 5-year OS estimates of 53, 43, 30 and 20% respectively. The five-year OS was better with an 8/8 donor and an EBMT risk score low. An overall survival of 74% for fully matched pa-tients with a low-risk EBMT score com-pared with an overall survival of 39% for EBMT low-risk patients with ≤7/8 do-nors suggests that incorporating both the EBMT risk score and the degree of HLA-matching could be useful in the risk assessment prior to unrelated do-nor T-cell depleted HSCT.Michelis et al. (45) showed that the mEBMT risk score is an independent prognostic factor for both NRM and OS in patients with AML undergoing HSCT and is superior to the HCT-CI.
◗◗◗ COMPARISIONS AND COMBINATIONS
Some studies compared the three scor-ing systems (HCT-CI, PAM and EBMT risk score). Castagna et al. (20) in a cohort of elderly patients transplanted with a reduced-intensity conditioning found that in this specific cohort neither the HCT-CI nor the EBMT risk score, nor the PAM score were predictive for NRM and OS. Steckel et al. (46) found that only the PAM risk score maintained a prognostic value in a cohort of patients transplanted with refractory AML.Barba et al. (47) tried to determine whether the integration of the HCT-CI and the EBMT risk score would improve
individual capacity for stratification of high-risk HSCT candidates. They built a new model with 6 risk groups: groups 1 and 2 with HCT-CI 0 and EBMT risk score 0-3 or 4-7, groups 3 and 4 with HCT-CI 1-2 and EBMT risk score 0-3 or 4-7, and groups 5 and 6 with HCT-CI ≥3 and EBMT risk score 0-3 or 4-7. The groups 1 and 2 had similar risk of NRM (20%) independently of their EBMT score and also the groups 3 and 4 had similar risk of NRM (28%), whereas group 5 had lower NRM risk compared with group 6 (25% and 40% respectively). So the Au-thors conclude that the combination of the HCT-CI and the EBMT risk score might contribute to a better identifica-tion of high-risk patients. Versluis et al. (48) integrated the pre-dominant parameters of each score (HCT-CI and EBMT risk score) into a novel score in patients with AML in 1st complete remission transplanted with a reduced-intensity conditioning regi-men. From HCT-CI they took 9 specific comorbidities (infection, peptic ulcer, cerebrovascular disease, arrhythmia, severe liver function abnormalities, obesity, rheumatologic disease, heart valve disease and renal disease) and from the EBMT risk score they took age, donor-type, interval from diagnosis to transplant and CMV-serology. Each significant parameter was attribut-ed 1, 2 or 3 points, as determined by its quantified HR. Combining all signif-icant parameters into an integrated score, NRM at 2 years was 9% for low-risk patients with scores 0-2, 15% for in-termediate-risk patients with score 3, and 28% for high-risk patients with a score ≥4. NRM increased to 11%, 24% and 36% at 5 years from transplant in the low-, intermediate- and high-risk groups respectively.

269Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
◗◗◗ DISEASE SPECIFIC SCORE SYSTEMS
There are also specific risk score sys-tems for specific diseases, for example for Myelofibrosis. Bacigalupo et al. (49) proposed a system based on 3 vari-ables (number of transfusions before HSCT, spleen size and type of donor) that permits to stratify the patients in 2 groups, low (score 0-1) and high risk (score 2-3) with NRM 8% and 41% re-spectively and overall survival 77% and 8% respectively.Alchalby et al. (50) proposed anoth-er scoring system based on 3 different variables (JAK2 status, age and consti-tutional symptoms). Depending on the presence of one, two or all of these factors, hazard ratio of death was 3.08, 4.70 and 16.61 respectively. Scott et al. (51) showed that the Dy-namic International Prognostic Scoring System for Myelofibrosis (DIPSS) calcu-lated by age, constitutional symptoms, haemoglobin level, leukocyte count and circulating blasts, is useful also to predict the HSCT outcome. For NRM the hazard ratio was 1, 1.41, 3.19 and 3.41 for DIPSS low risk, intermediate-1,
intermediate-2 and high risk patients. In Table 4 are summarized these three systems. For acute leukemia and MDS Armand et al. (52) have proposed a specific risk score based on 5 variables: age, disease, stage at transplantation, cy-togenetics, and pre-transplantation ferritin associated with conditioning regimen (Table 5). The defined 3 risk categories, low-risk (score ≤2), interme-diate risk (score =3) and high risk (score ≥4) had a 5-year overall survival of 56%, 22% and 5% respectively, and a 5-year NRM of 24%, 25% and 36% respectively.The CIBMTR has developed a predic-tive scoring system for patients with acute leukemia not in complete remis-sion at the moment of the transplant, based on 4-5 pretransplantation vari-ables different for acute myeloid leu-kemia (AML) and acute lymphoblastic leukemia (ALL) (53). For AML patients these pretransplantation variables are: disease group, cytogenetics, HLA match, circulating blasts and Karnof-sky or Lansky score. For ALL patients the variables are: disease group, donor CMV, bone marrow blasts and age. (Table 6).
TABle 4 • Pretransplantation scoring systems for Myelofibrosis.Scoring system Variables ScoreBacigalupo (49) Number of transfusions be-
fore transplant>20 1
Spleen size >22 cm 1Type of donor other than HLA identical sibling 1
Alchalby (50) JAK2 V617F status wild-type 1Age ≥57 years 1Constitutional symptoms yes 1
DIPSS (51) Age >65 years 1Constitutional symptoms yes 1Hemoglobin level <10 g/dL 2Leukocyte count >25 x 10^9/L 1Circulating blasts ≥1% 1

270 R. Raimondi, et al.
TABle 5 • Pretransplantation scoring systems for Acute Leukemia and MDS (52).Variable Definitions ScoreAge <40 years 0
≥40 years 1Disease Low-risk MDS (RA, RARS, or RCMD), AML, ALL 0
High-risk MDS (RAEB)/transformed MDS 1Cytogenetics* Favourable 0
Intermediate 1Adverse 2
Stage CR1 or Untreated MDS 0CR >1 or Induction failure 1AML/ALL untreated or active relapse 2
Ferritin and condi-tioning regimen
Reduced-intensity conditioning regimen OR ferritin <2500 ng/dL 0Myelo-ablative conditioning regimen AND ferritin ≥2500 ng/dL 1
*For ALL, t(9;22) and t(4;11) are considered adverse, and all others intermediate. AML cytogenetics are grouped according to the MRC classification scheme, and MDS cytogenetics according to the IPSS scheme.
TABle 6 • Pretransplantation CIBMTR scoring systems for Acute Leukemia not in complete re-mission (53).
Disease Variables ScoreAML Disease group Primary induction failure or duration first
CR >6 months0
Duration of first CR <6 months 1Cytogenetics Good or intermediate 0
Poor 1HLA match group HLA identical sibling or well matched or
partially matched unrelated0
Mismatched unrelated 1Related other than HLA identical sibling 2
Circulating blasts Absent 0Present 1
Performance status (Karnofsky or Lansky score)
90-100 0<90 1
ALL Disease group Primary induction failure or first untreat-ed relapse
0
First refractory relapse 1Second and additional relapse 2
Donor CMV Negative 0Positive 1
Bone marrow blasts <25% 0>25% 1
Age 1-9 years 010-39 years 1>40 years 2

271Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
AML patients with a score of 0 had a 42% 3-year OS whereas patients with a score ≥3 had only 6% 3-year OS. ALL patients with a score of 0 or 1 had 46% survival at 3 years whereas patients with a score ≥3 had only 10% 3-year OS. The Authors conclude that, in gen-eral, patients with a score ≥3 have a dismal outcome and alternative ther-apy should be considered, while for patients with a risk score ≤2 they sug-gest strong consideration of HSCT be-cause the predicted 3-year survival is between 15% and 46%.In a recent study on behalf of GITMO, Todisco et al. (54) have validated this CIBMTR prognostic score in a relative-ly large patient population with active AML. They confirmed that the CIBMTR score is an effective and reproducible approach for predicting survival of this group of AML patients, also for trans-plants performed with reduced-intensi-ty conditioning regimen, condition not evaluated in the original CIBMTR study that analyzed only transplants with my-eloablative conditioning regimens.
◗◗◗ INCORPORATION OF RISK SCORE SYSTEMS IN A DYNAMIC APPROACH
One of the most interesting approach-es is that suggested by Cornelissen et al. (55).For AML patients in first complete re-mission they propose to integrate both disease-related and transplant-re-lated factors for the decision to pro-ceed either to allogeneic HSCT or to a non-transplant strategy. Disease-related factors are cytoge-netic, molecular prognostic markers,
time to complete remission, number of blasts appearing early after induction therapy and quantified minimal resid-ual disease after induction or consol-idation. The transplant-related factors are obtained from HCT-CI and EBMT risk score. For patients with AML in first complete remission who are receiving myeloab-lative HSCT a low EBMT risk score (be-tween 0 and 1) predicts a NRM rate of less than 15%; an EBMT risk score of 2–3 predicts a NRM of 20–25% whereas an elevated EBMT risk score (>4) shows en-hanced NRM of approximately 35%.A HCT-CI of 0, 1 or 2 points resulted in a 2-year nonrelapse mortality rate of approximately 10%, 15-20% and 25%, respectively. A higher HCT-CI score of 3 or ≥4 resulted in nonrelapse mortality rates of 35-40%.The disease-related factors permit to calculate the risk of relapse and mor-tality following a consolidation ap-proach only, without transplant, and the transplant-related factors permit to calculate the risk of NRM after trans-plantation. Combining all these data, in a dynamic manner at different phases of the clinical course, would permit to evaluate whether the HSCT could be “advantageous” as defined by the Authors, i.e. a transplant that is expected to improve the overall sur-vival at least 10% compared with a non-transplant strategy. This approach has been applied only in acute myeloid leukemia in first com-plete remission, one of the settings in which often the choice towards the transplant can be difficult. But the same approach could potentially be applied to other haematological ma-lignant diseases. Armand et al. (56) described a Disease Risk Index (DRI)

272 R. Raimondi, et al.
for all the haematological malignant diseases. This DRI could be compared with the transplant risk scores; indeed in the same cohort of that study the HCT-CI maintained its prognostic value independently from the DRI.
◗◗◗ A DIFFERENT APPROACH
From a different point of view a new approach has recently been suggest-ed. It uses two techniques, Machine Learning, a field in artificial intelligence stemming from computer science, and the Data Mining, a multidisciplinary field based on statistics, mathematics, computer science and artificial intelli-gence to create algorithms for analys-ing large and complex data sets. These tools could be able to process multi-plicity of variables, describe complex non-linear interactions such as those in the setting of HSCT and create accu-rate prediction models for outcomes. A review on the potential application of this approach for clinical predictive modelling in HSCT has been written by Shouval et al. (57). A first application of these methods has been presented by Shouval et al. (58) at the last ASH 2013 meeting. Us-ing the Acute Leukemia Working Party registry of the EBMT the Authors devel-oped a decision tree algorithm taking in account twenty two variables in-cluding diagnosis, disease status, Karn-ofsky performance status, conditioning regimen, graft type, GVHD prophy-laxis regimens, age, donor-recipient HLA-matching, number of transplants in each Centre per year, donor-pa-tient CMV serostatus match, donor-re-cipient sex match, year of transplant, etc. For both overall mortality and NRM
at day +100 after HSCT this system as-signed scores and the final results were a continuous proportional increase of poorer outcome with a correspond-ing increase of the score, from 4.9% to 52.8% for overall mortality and from 4.1% to 32.2% for NRM at day +100.
◗◗◗ CONCLUSION
It stands to reason that the pretrans-plant risk evaluation systems above mentioned are based on different pa-rameters: HCT-CI considers exclusively the comorbidities that are only partly included in the PAM and are absent in the EBMT risk score. On the other hand in the HCT-CI are not included some characteristics like age, performance status, type of donor and status of the disease that are present, although in different manner, in the other two systems. That is to say that these ap-proaches look at different aspects.It is to be hoped that in the near future it will be possible to achieve a unique system that incorporates the most significant parameters to predict the transplant risk. In short, we can say that the search of a pretransplant prognostic index is still at the beginning, but in the same time we can say that HCT-CI and EBMT risk score represent at the moment the most effective tools for estimate the NRM risk before HSCT and stratify the patients into risk categories. The challenge for the future is to im-prove the ability of the pretransplant scoring systems to predict NRM, to make these tools more and more reli-able, if possible integrating them with biological markers, with the aim to ob-tain individualized, tailored risk estima-

273Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
tion for a specific patient with a specif-ic transplant procedure.
Conflict of interests: the authors de-clare no conflict of interests.
◗◗◗ REFERENCES
1. Gooley TA, Chien JW, Pergam SA, Hin-gorani S, Sorror ML, Boeckh M, et al. Re-duced mortality after allogeneic hema-topoietic-cell transplantation. N Engl J Med 2010; 363: 2091-101.
2. Horan JT, Logan BR, Agovi-Johnson MA, Lazarus HM, Bacigalupo AA, Ballen KK, et al. Reducing the risk for transplan-tation-related mortality after allogene-ic hematopoietic cell transplantation: how much progress has been made? J Clin Oncol 2011; 29: 805-13.
3. Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogene-ic HCT. Blood 2005; 106: 2912-9.
4. Sorror ML, Sandmaier BM, Storer BE, Fran-ke GN, Laport GG, Chauncey TR, et al. Long-term outcomes among older pa-tients following nonmyeloablative con-ditioning and allogeneic hematopoi-etic cell transplantation for advanced hematologic malignancies. JAMA 2011; 306: 1874-83.
5. Pavlů J, Kew AK, Taylor-Roberts B, Auner HW, Marin D, Olavarria E, et al. Optimiz-ing patient selection for myeloablative allogeneic hematopoietic cell trans-plantation in chronic myeloid leukemia in chronic phase. Blood 2010; 115: 4018-20.
6. Bokhari SW, Watson L, Nagra S, Cook M, Byrne JL, Craddock C, et al. Role of HCT-comorbidity index, age and dis-ease status at transplantation in pre-dicting survival and non-relapse mortal-ity in patients with myelodysplasia and leukemia undergoing reduced-intensi-ty-conditioning hemopoeitic progen-
itor cell transplantation. Bone Marrow Transplant 2012; 47: 528-34.
7. Smith AR, Majhail NS, MacMillan ML, DeFor TE, Jodele S, Lehmann LE, et al. Hematopoietic cell transplantation co-morbidity index predicts transplantation outcomes in pediatric patients. Blood 2011; 117: 2728-34.
8. Martino R, Valcárcel D, Brunet S, Sure-da A, Sierra J. Comparable non-relapse mortality and survival after HLA-identical sibling blood stem cell transplantation with reduced or conventional-intensity preparative regimens for high-risk myel-odysplasia or acute myeloid leukemia in first remission. Bone Marrow Trans-plant 2008; 41: 33-8.
9. Lim ZY, Ingram W, Brand R, Ho A, Ken-yon M, Devereux S, et al. Impact of pretransplant comorbidities on alemtu-zumab-based reduced-intensity condi-tioning allogeneic hematopoietic SCT for patients with high-risk myelodysplas-tic syndrome and AML. Bone Marrow Transplant 2010;45:633-9.
10. Sorror ML, Storer BE, Sandmaier BM, Maris M, Shizuru J, Maziarz R, et al. Five-year follow-up of patients with ad-vanced chronic lymphocytic leukemia treated with allogeneic hematopoietic cell transplantation after nonmyeloab-lative conditioning. J Clin Oncol 2008; 26: 4912-20.
11. Sorror ML, Storer BE, Maloney DG, Sand-maier BM, Martin PJ, Storb R. Outcomes after allogeneic hematopoietic cell transplantation with nonmyeloablative or myeloablative conditioning regimens for treatment of lymphoma and chronic lymphocytic leukemia. Blood 2008; 111: 446-52.
12. Farina L, Bruno B, Patriarca F, Spina F, Sorasio R, Morelli M, et al. The hema-topoietic cell transplantation comor-bidity index (HCT-CI) predicts clinical outcomes in lymphoma and myelo-ma patients after reduced-intensity or non-myeloablative allogeneic stem cell transplantation. Leukemia 2009; 23: 1131-8.

274 R. Raimondi, et al.
13. Kataoka K, Nannya Y, Ueda K, Kumano K, Takahashi T, Kurokawa M. Differential prognostic impact of pretransplant co-morbidity on transplant outcomes by disease status and time from transplant: a single Japanese transplant centre study. Bone Marrow Transplant 2010; 45: 513-20.
14. Sorror M, Storer B, Sandmaier BM, Ma-loney DG, Chauncey TR, Langston A, et al. Hematopoietic cell transplanta-tion-comorbidity index and Karnofsky performance status are independent predictors of morbidity and mortality after allogeneic nonmyeloablative he-matopoietic cell transplantation. Can-cer 2008; 112: 1992-2001.
15. Sorror ML, Giralt S, Sandmaier BM, De Lima M, Shahjahan M, Maloney DG, et al. Hematopoietic cell transplantation specific comorbidity index as an out-come predictor for patients with acute myeloid leukemia in first remission: com-bined FHCRC and MDACC experienc-es. Blood 2007; 110: 4606-13.
16. El Kourashy S, Williamson T, Chaudhry MA, Savoie ML, Turner AR, Larratt L et al. Influence of comorbidities on transplant outcomes in patients aged 50 years or more after myeloablative conditioning incorporating fludarabine, BU and ATG. Bone Marrow Transplant 2011; 46: 1077-83.
17. Sorror ML, Sandmaier BM, Storer BE, Maris MB, Baron F, Maloney DG, et al. Co-morbidity and disease status based risk stratification of outcomes among pa-tients with acute myeloid leukemia or myelodysplasia receiving allogeneic hematopoietic cell transplantation. J Clin Oncol 2007; 25: 4246-54.
18. Birninger N, Bornhäuser M, Schaich M, Eh-ninger G, Schetelig J. The hematopoietic cell transplantation-specific comorbidity index fails to predict outcomes in high-risk AML patients undergoing allogeneic transplantation--investigation of poten-tial limitations of the index. Biol Blood Marrow Transplant 2011; 17: 1822-32.
19. Terwey TH, Hemmati PG, Martus P, Di-
etz E, Vuong LG, Massenkeil G, et al. A modified EBMT risk score and the hema-topoietic cell transplantation-specific comorbidity index for pre-transplant risk assessment in adult acute lymphoblas-tic leukemia. Haematologica 2010; 95: 810-8.
20. Castagna L, Fürst S, Marchetti N, El Cheikh J, Faucher C, Mohty M, et al. Retrospective analysis of common scoring systems and outcome in pa-tients older than 60 years treated with reduced-intensity conditioning regimen and alloSCT. Bone Marrow Transplant 2011; 46: 1000-5.
21. Guilfoyle R, Demers A, Bredeson C, Rich-ardson E, Rubinger M, Szwajcer D, et al. Performance status, but not the hema-topoietic cell transplantation comor-bidity index (HCT-CI), predicts mortality at a Canadian transplant center. Bone Marrow Transplant 2009; 43: 133-9.
22. Xhaard A, Porcher R, Chien JW, de La-tour RP, Robin M, Ribaud P, et al. Impact of comorbidity indexes on non-relapse mortality. Leukemia 2008; 22: 2062-9.
23. Popat U, de Lima MJ, Saliba RM, An-derlini P, Andersson BS, Alousi AM, et al. Long-term outcome of reduced-inten-sity allogeneic hematopoietic SCT in patients with AML in CR. Bone Marrow Transplant 2012; 47: 212-6.
24. Krauter J, Wagner K, Stadler M, Dam-mann E, Zucknick M, Eder M, et al. Prognostic factors in allo-SCT of elderly patients with AML. Bone Marrow Trans-plant 2011; 46: 545-51.
25. Slack JL, Dueck AC, Fauble VD, Sproat LO, Reeder CB, Noel P, et al. Reduced toxicity conditioning and allogeneic stem cell transplantation in adults using fludarabine, carmustine, melphalan, and antithymocyte globulin: outcomes depend on disease risk index but not age, comorbidity score, donor type, or human leukocyte antigen mismatch. Biol Blood Marrow Transplant 2013; 19: 1167-74.
26. Barba P, Piñana JL, Martino R, Valcár-cel D, Amorós A, Sureda A, et al. Com-

275Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
parison of two pretransplant predictive models and a flexible HCT-CI using dif-ferent cut off points to determine low-, intermediate- and high-risk groups: the flexible HCT-CI Is the best predictor of NRM and OS in a population of patients undergoing allo-RIC. Biol Blood Marrow Transplant 2010; 16: 413-20.
27. DeFor TE, Majhail NS, Weisdorf DJ, Brun-stein CG, McAvoy S, Arora M, et al. A modified comorbidity index for hema-topoietic cell transplantation. Bone Marrow Transplant 2010; 45: 933-8.
28. Boehm A, Sperr WR, Leitner G, Worel N, Oehler L, Jaeger E, et al. Comorbidity predicts survival in myelodysplastic syn-dromes or secondary acute myeloid leukaemia after allogeneic stem cell transplantation. Eur J Clin Invest 2008; 38: 945-52.
29. Eissa H, Gooley TA, Sorror ML, Nguyen F, Scott BL, Doney K, et al. Allogene-ic hematopoietic cell transplantation for chronic myelomonocytic leukemia: relapse-free survival is determined by karyotype and comorbidities. Biol Blood Marrow Transplant 2011; 17: 908-15.
30. Michelis FV, Atenafu EG, Gupta V, Kim DD, Kuruvilla J, Lambie A, et al. Dura-tion of first remission, hematopoietic cell transplantation-specific comorbidity in-dex and patient age predict survival of patients with AML transplanted in sec-ond CR. Bone Marrow Transplant 2013; 48: 1450-5.
31. Quan Le R, Jain NA, Tian X et al. Co-morbidity Measures In Ex Vivo T Cell De-pleted Allogeneic Hematopoietic Stem Cell Transplantation (HCT). Blood (ASH Annual Meeting) 2013; 122. Abstract 2124.
32. Ratan R, Ceberio I, Hilden P et al. The Hematopoietic Cell Transplant-Co-Mor-bidity Index (HCT-CI) Predicts Outcomes After T Cell Depleted (TCD) Allogeneic HCT For AML and MDS. Blood (ASH An-nual Meeting) 2013 122. Abstract 2045.
33. Paun OV, Kindwall-Keller TL, Lazarus HM et al. Charlson Comorbidity Index (CCI) and Hematopoietic Cell Transplanta-
tion Specific-Comorbidity Index (HCT-CI) Do Not Predict Transplant Related Mortality (TRM) and Post-Transplant Outcomes In Young Patients Under-going Reduced Intensity Conditioning (RIC) Umbilical Cord Blood (UCB) Trans-plantation. Blood (ASH Annual Meeting) 2013; 122. Abstract 4582.
34. Thakar MS, Logan BR, Pasquini MC et al. The Hematopoietic Cell Transplantation Comorbidity Index (HCT-CI) Can Pro-spectively Discriminate Risks Affecting Overall Survival in Pediatric and Adult Patients with Non-Malignant Diseases. Blood (ASH Annual Meeting) 2012; 120. Abstract 737.
35. Pasquini MC, Logan BR, Ho VT et al. Co-morbidity Index (CI) in Autologous He-matopoietic Cell Transplantation (HCT) for Malignant Diseases: Validation of the HCT-CI. Blood (ASH Annual Meet-ing) 2012; 120. Abstract 814.
36. Saad A, Mahindra A, Zhang MJ, Zhong X, Costa LJ, Dispenzieri A, et al. Hema-topoietic Cell Transplant Comorbidity Index Is Predictive of Survival after Au-tologous Hematopoietic Cell Transplan-tation in Multiple Myeloma. Biol Blood Marrow Transplant. 2013 Dec 14.
37. Raimondi R, Tosetto A, Oneto R, Cavazzina R, Rodeghiero F, Bacigalupo A, Fanin R, et al. Validation of the He-matopoietic Cell Transplantation-Spe-cific Comorbidity Index: a prospective, multicenter GITMO study. Blood 2012; 120: 1327-33.
38. Coffey DG, Pollyea DA, Myint H Smith C, Gutman JA. Adjusting DLCO for Hb and its effects on the Hematopoietic Cell Transplantation-specific Comorbid-ity Index. Bone Marrow Transplant 2013; 48: 1253-6.
39. Sorror ML. How I assess comorbidities before hematopoietic cell transplanta-tion. Blood 2013; 121: 2854-63.
40. Parimon T, Au DH, Martin PJ, Chien JW. A risk score for mortality after allogene-ic hematopoietic cell transplantation. Ann Intern Med 2006; 144: 407-14.
41. Gratwohl A, Stern M, Brand R, Apper-

276 R. Raimondi, et al.
ley J, Baldomero H, de Witte T, et al. Risk score for outcome after allogeneic hematopoietic stem cell transplanta-tion: a retrospective analysis. Cancer 2009;115:4715-26.
42. Hemmati PG, Terwey TH, le Coutre P, Vuong LG, Massenkeil G, Dörken B, et al. A modified EBMT risk score predicts the outcome of patients with acute myeloid leukemia receiving allogeneic stem cell transplants. Eur J Haematol 2011; 86: 305-16.
43. Rezvani K, Kanfer EJ, Marin D, Gabriel I, Rahemtulla A, Taylor A,bet al. EBMT risk score predicts outcome of alloge-neic hematopoietic stem cell trans-plantation in patients who have failed a previous transplantation procedure. Biol Blood Marrow Transplant 2012; 18: 235-40.
44. Lodewyck T, Oudshoorn M, van der Holt B, Petersen E, Spierings E, von dem Borne PA, et al. Predictive impact of al-lele-matching and EBMT risk score for outcome after T-cell depleted unrelat-ed donor transplantation in poor-risk acute leukemia and myelodysplasia. Leukemia 2011; 25: 1548-54.
45. Michelis FV, Messner HA, Uhm J et al. The Modified Pre-Transplant EBMT Risk Score Is Superior To The HCT-CI Score In Predicting Overall Survival and Non-Re-lapse Mortality After Allogeneic Hema-topoietic Cell Transplantation In Pa-tients With Acute Myeloid Leukemia. Blood (ASH Annual Meeting) 2013; 122. Abstract 2153.
46. Steckel NK, Ditschkowski M, Groth C et al. Comparison Of Different Pretrans-plant Predictive Scores In Patients With Refractory Acute Myeloid Leukemia After Allogeneic Stem Cell Transplanta-tion Including Highdose Melphalan: Re-sults Of a Double-Center Observation-al Study. Blood (ASH Annual Meeting) 2013; 122. Abstract 4525.
47. Barba P, Martino R, Pérez-Simón JA, Fernández-Avilés F, Castillo N, Piñana JL, et al. Combination of the hematopoiet-ic cell transplantation comorbidity index
and the European group for blood and marrow transplantation score allows a better stratification of high-risk patients undergoing reduced-toxicity allogene-ic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014; 20: 66-72.
48. Versluis J, Labopin M, Niederwieser D et al. Prediction of Non-Relapse Mortality in Recipients of Reduced Intensity Con-ditioning Allo-HSCT with Acute Myeloid Leukemia in First Complete Remission: Integrating the Seattle Comorbidity In-dex (HCT-CI) and EBMT Scoring Systems. Blood (ASH Annual Meeting) 2012; 120. Abstract 734.
49. Bacigalupo A, Soraru M, Dominietto A, Pozzi S, Geroldi S, Van Lint MT, et al. Al-logeneic hemopoietic SCT for patients with primary myelofibrosis: a predictive transplant score based on transfusion requirement, spleen size and donor type. Bone Marrow Transplant 2010; 45: 458-63.
50. Alchalby H, Yunus DR, Zabelina T, Kob-be G, Holler E, Bornhäuser M, et al. Risk models predicting survival after re-duced-intensity transplantation for my-elofibrosis. Br J Haematol 2012; 157: 75-85.
51. Scott BL, Gooley TA, Sorror ML, Rezvani AR, Linenberger ML, Grim J, et al. The Dynamic International Prognostic Scor-ing System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood 2012; 119: 2657-64.
52. Armand P, Kim HT, Cutler CS, Ho VT, Ko-reth J, Ritz J, et al. A prognostic score for patients with acute leukemia or my-elodysplastic syndromes undergoing al-logeneic stem cell transplantation. Biol Blood Marrow Transplant 2008; 14: 28-35.
53. Duval M, Klein JP, He W, Cahn JY, Cai-ro M, Camitta BM, et al. Hematopoietic stem-cell transplantation for acute leu-kemia in relapse or primary induction failure. J Clin Oncol 2010; 28: 3730-8.
54. Todisco E, Ciceri F, Oldani E, Boschini

277Evaluation of non-relapse mortality risk in hematopoietic stem cell transplantation
C, Micò C, Vanlint MT, et al. The CIBMTR score predicts survival of AML patients undergoing allogeneic transplantation with active disease after a myeloabla-tive or reduced intensity conditioning: a retrospective analysis of the Gruppo Italiano Trapianto Di Midollo Osseo. Leu-kemia 2013; 27: 2086-91.
55. Cornelissen JJ, Gratwohl A, Schlenk RF Sierra J, Bornhäuser M, Juliusson G, et al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nat Rev Clin Oncol 2012; 9: 579-90.
56. Armand P, Gibson CJ, Cutler C, Ho VT, Koreth J, Alyea EP, et al. A disease
risk index for patients undergoing allo-geneic stem cell transplantation. Blood 2012; 120: 905-13.
57. Shouval R, Bondi O, Mishan H, Shimo-ni A, Unger R, Nagler A. Application of machine learning algorithms for clinical predictive modeling: a data-mining ap-proach in SCT. Bone Marrow Transplant 2013 Oct 7.
58. Shouval R, Labopin M, Bondi O et al. Prediction Of Allogeneic Hematopoiet-ic Stem Cell Transplantation (allo-HSCT) Related Mortality in Acute Leukemia: Generation Of a Machine Learn-ing-Based Model Using The Data Set of The Acute Leukemia Working Party (ALWP) Of The EBMT. Blood (ASH Annual Meeting) 2013; 122. Abstract 409.

Sales & Info:http://worldwide.grifols.com
Fully automated instrument with a high processing capacity for pre-transfusion compatibility tests using the unique DG Gel® 8-column cards
Erytra®
Upright designIncreases the workload without taking up extra space in the laboratory
Highest throughput100 cards/h ---- 800 tests/h
Continuous loadingMulti-access for reagents, samples and cards
Nonstop running
AutonomyIndependent operation for long periods of time
Excellent capacity• 96 samples• 54 reagents• 400 DG Gel® cards• 16L waste solution• 16L washing solutions
www.theerytradimension.com
4723 Anunci Erytra iatlia12.indd 1 05/10/12 10:00

DCTH - 4•2013 - 279-300
Key words: CD30, Ki-1, monoclonal anti-body, Hodgkin’s lymphoma, anaplastic large cell lymphoma, target therapy, brentuximab vedotin.
Correspondence:Lorenzo LeonciniSection of Pathology, Department of Medical BiotechnologyUniversity of SienaVia delle Scotte, 6 - Siena, ItalyE-mail: [email protected]
SUMMARYSince its discovery in 1982, CD30 continued to fascinate the scientific community and there is a growing body of evidence on a changing landscape in the treatment of CD30-positive lymphomas. Initially termed Ki-1, this antigen was later clustered as CD30, showing a very strong expression on Hodgkin and Reed-Sternberg cells in Hodgkin’s lymphoma. Shortly thereafter, CD30 was also found on the malignant cells of anaplastic large cell lymphoma and other lymphoid and non-lymphoid malignant neoplasms. The expression of CD30 by all these tumors have suggested this antigen as a potential target for thera-py, especially in relapsed and refractory Hodgkin’s lymphoma and in anaplastic large cell lymphoma. A number of murine monoclonal antibodies against CD30, both in na-tive form or linked to a variety of different toxins, radioisotopes or cytostatic drugs, were evaluated for their therapeutic effects. However, most of these drugs gave disappoint-ing clinical results. The scenery changed dramatically with the advent of Brentuximab vedotin whose mechanism of action determines a high specific potency. Nonetheless, a number of key questions still remain unanswered and we need additional trials to fully evaluate the role of target therapy in CD30-positive lymphoproliferative disorders.
CD30 expression in lymphoid neoplasms: from diagnostic marker to target of therapyL. Leoncini, M.R. Ambrosio, S. Lazzi, B.J. Rocca, P. TosiSection of Pathology, Department of Medical Biotechnologies, University of Siena, Italy
◗◗◗ INTRODUCTION
Historical backgroundThe CD30 antigen was originally iden-tified in 1982 by Schwab and cowork-ers thank to the establishment of a permanent Hodgkin cell line, the L428
FORUM
(1). Briefly, the authors searched for tu-mor-specific antigens on Hodgkin cells (HC) and Reed-Sternberg (RS) cells by using a rabbit antiserum raised against this cell line (1). After absorption with various normal human cells, the an-ti-L428 antiserum proved to be selec-tively reactive with L428 cells and HL and RS cells (1). Then they produced hybridoma antibodies directed against HC and RS cells. Among the 57 mono-clonal antibodies tested on frozen sec-tions of tissue with Hodgkin’s lymphoma (HL), the clone Ki-1 was the only one to react with HC and RS cells and not with other cells (1). Therefore, the Ki-1 anti-gen was considered a specificfeature of HC and RS cells, and subsequently its characterization assumed an import-ant role in the differential diagnosis of

280 L. Leoncini, et al.
HL (2). Since its initial description, there has been an overwhelming flood of information concerning the molecule and it was shown that Ki-1 antigen was not restricted to HL (3-9). In 1985 exten-sive immunohistochemical studies with the Ki-1 monoclonal antibody led Stein and coworkers to identify a group of large-cell neoplasms characterized by subtotal effacement of lymph node architecture, paracortical growth pat-tern, spread to sinuses, polymorphic appearance and expression of Ki-1 antigen by virtually all neoplastic cells (3). These neoplasms, frequently misdi-agnosed, proved to constitute a mor-phologically distinct type of lymphoid large-cell neoplasms. Because of their specific reactivity with the monoclonal antibody anti-Ki-1, they were designed as Ki-1 lymphomas, or Ki-1 positive large cell lymphomas or Ki-1 positive large cell anaplastic lymphomas. All these terms have generated confusion in the medical literature and the sta-tus of Ki-1 lymphoma as a single entity has long been controversial. The sub-sequent generation of additional anti-bodies (e.g. Ber-H2, Ber-H4, HRS-3, HRS-4) detecting different epitopes of the Ki-1 molecule on formalin-fixed paraf-fin-embedded tissues, led to the iden-tification of the so-called CD30 cluster and introduce the term CD30-positive anaplastic lymphoma (10). Howev-er, all these antibodies were positive in both HL and large cell lymphoma cells and, like anti-Ki-1, were not able to discriminate between the two dis-eases (11-14). In 1990, Leoncini et al. examined the grey zone between HL and large cell lymphoma, aiming at focusing on the differences and the similarities between these two entities (15). Only ten years later, anaplastic
lymphoma has been included into the World Health Organization (WHO) clas-sification as a separate entity, named anaplastic large cell lymphoma (ALCL) due to the demonstration of transloca-tion t(2;5) in a subset and of the T-cell origin (16).In the last WHO classification of tumour of hematopoietic and lymphoid tissue, the grey zone remainsand includes the B-cell lymphoma unclassifiable, with features intermediate between diffuse large B-cell lymphoma (DLBCL) and classical HL (17).
◗◗◗ STRUCTURe aND expReSSION OF CD30 ClUSTeR
CD30 receptorThe CD30 gene is localized at chro-mosome 1p36, closely linked to other members of the Transcription necro-sis factor (TNF) receptor super-family, such as the human TNFR2 and OX40 genes. It encodes for a precursor form of 84kDa which is processed during its passage through the Golgi complex to a mature form of 120 kDa. The ma-ture form of CD30 antigen, represents a type I transmembrane protein which shares a variable degree of homology. The protein analysis of human CD30 demonstrates that this molecule has a 18-residue leader peptide, followed by a 365 amino-acids (aa) extracytoplas-mic domain, a 24 aa transmembrane region, and a cytoplasmic domain of 188 aa. The human CD30 extra-cyto-plasmic domain can be divided into six cysteine-rich motifs of approximate-ly 40 amino acids that contain six cys-teines with the exception of motif1B and 3B, which are truncated. A hinge region ofapproximately 50 amino ac-

281CD30 expression in lymphoid neoplasms
ids, which contains multiple serine, threonine, and proline residues and is O-glycosylated, connects the two cysteine-rich domains. There are two sites for N-linked glycosylation. The in-tracellular domain of human CD30 is long andits sequence diverges signifi-cantly from the intracellular sequences of all the other TNF-R family members. The cytoplasmic tail of CD30 contains several TNF receptorassociated factor (TRAF)-binding sequences that can mediate activation of nuclear factor kappa-B (NF-kB) and extracellular sig-nal-regulated kinase signaling path-ways. Within the intracellular domain a conserved motif for ATP-binding (Gx-GxxG) found in many protein kinases is also present. Finally, potential phos-phorylation sites for tyrosine kinase and serine/threonine kinases are located in the extracellular and intracellular do-mains, respectively (18, 19).
Soluble form of CD30 The extracellular part of the mem-brane-bound CD30 can be cleaved by the action of a zinc-metallopro-tease. This produces a soluble form of CD30 (sCD30) with a molecular mass of 85-90 kDa. Shedding of CD30 occurs as an active process of via-ble CD30 positive cells. Soluble CD30 can interfere with signaling by mem-brane-bound CD30 through binding to CD30L and blocking its interaction with CD30 on the cell membrane. To be effective, this type of interference requires that the soluble form ofthe re-ceptor binds to the ligand with high af-finity. Alternatively, soluble CD30 could interfere with transmembrane signaling by associating with membrane-bound CD30 to form complexes thatcause dominant-negative interference. It has
been suggested that sCD30 provides a mechanism by which CD30-positive tu-mors can escape immunosurveillance and the apoptosis-induced activity of CD30L. Increased serum sCD30 levels have been noted in various conditions: neoplasms (HL, ALCL, embryonal car-cinoma of the testis, nasopharyngeal carcinoma), infectious diseases (infec-tious mononucleosis, B and C hepati-tis, human immunodeficiency virus in-fection) and inflammatory conditions (systemic lupus erythematosus, prima-ry biliary cirrhosis, inflammatory bowel disease, rheumatoid arthritis, Hashimo-to’s thyroiditis, systemic sclerosis) char-acterized by strong B-cell or T-cell ac-tivation. In patients with HL and ALCL sCD30 appears to be a reliable tumor burden marker. Several reports have identified correlations between serum sCD30 levels and poor prognosis in CD30-positive lymphomas (20-23).
CD30 ligandThe human CD30 ligand (CD30L) gene has been mapped to chromosome 9q33 and encodes for a 40 kDa type II membrane glycoprotein. Human CD30L protein has an extracellular do-main comprising carboxy-terminal 172 amino acids, and cytoplasmic domain of N-terminal 40 residues. The extracel-lular domain shows significant homol-ogy to TNF-a, TNF-b and the CD40L. CD30L exhibits classic pleiotropic cyto-kine activities (18).
CD30 expression in normal cellsThe exact function of CD30 and CD30L in healthy individuals remains poor-ly understood, as no human diseases have been linked with defects of the CD30 receptor and CD30L genes. CD30 receptor expression is preferen-

282 L. Leoncini, et al.
tially restricted to activated B, T or null lymphoid cells. In normal peripheral lymphoid organs, CD30-positive cells are seen within the parafollicular areas and in the rim of the follicular centers. These interfollicular B and T cells have abundant cytoplasm and evident nucleoli, are proliferating, and often co-express Bcl-6. In addition, CD30-pos-itive centroblasts are found within ger-minal centers. CD30-positive cells are also identified in the medulla of the thymus, frequently around the Hassal’s corpuscles. A variable percentage (3–31%) of human peripheral blood lym-phocytes have been demonstrated to express surface CD30. Many of these cells belong to the CD8subset and pro-duce IFN-g and IL-4. In addition toB and T lymphocytes, lung macrophages, activatedNK cells, endothelial cells and decidual cells also express CD30 to variable extent. CD30L is expressed in both resting and activated B cells, activated T cells and natural killer (NK) cells, eosinophils, granulocytes, mono-cytes and mast cells (11, 18, 19).
CD30 signalingThe molecular details of how the inter-action between CD30 and CD30L re-sults in the trigger of the downstream signaling pathways are just beginning to be addressed. Emerging evidenc-es have demonstrated that the acti-vation of CD30 signaling may, alter-natively, provide positive or negative signals depending on cell type, stage of differentiation, and the presence of other stimuli (e.g. cytokines). Following engagement with its ligand, CD30 may induce different cellular responses as cell proliferation and survival, but also a block of cell division and induction of cell death (24).
Like other TNF superfamily proteins, CD30L is a homotrimer. The interaction between CD30 receptor and CD30L is largely mediated by the cysteine-pseu-dorepeats of the receptor which fold tightly together, forming intimate lon-gitudinal contacts with the neighbors. A soluble recombinant trimeric form of CD30L, however, does not trigger effective signaling via CD30 unless it is cross-linked by an antibody, suggest-ing that the minimal form of CD30L that is capable of effective signaling is a hexamer. Hexameric CD30L allows the formation of stable complexes be-tween the receptor and two groups of signal transducers, the TNFR-associ-ated proteins (TRAF) andproteins with a death domain (FADD and TRADD). The former interact with the cytoplas-mic tails of the surface receptors and serve asadapter proteins to recruit downstream signal transducers such as NF-k B-inducing kinase (NIK), re-sponsible for activation of NF-k B. NIK, which has structural homology to the MAPKinase (MAPKs) family, activates I-kB kinase (IKK)-a and IKK-b,which in turn phosphorylate I-kB, and induces degradation leading to nuclear trans-location of NFkB. All the members of the TRAF family contain a conserved C-terminal TRAF domain that serves for homo- or hetero-oligomerization among the TRAF family, and for inter-actions with the cytoplasmicregions of the TNFR superfamily. In additionto the TRAF domain, most TRAF proteins con-tain an N-terminal RING finger and sev-eral zinc finger structuresthat appear critical for effector functions. CD30 has been shown to interact with TRAF1, 2, 3 and 5, and the binding regions have been characterized. TRAF1, TRAF2 and TRAF3 bind the C-terminal 42 ami-

283CD30 expression in lymphoid neoplasms
no-acids, that can be subdivided into two elements, 2A and 2B; both regions bind TRAF1 and TRAF2, while only 2A can bind TRAF3. TRAF5 also interacts with CD30 and mediates NF-kB acti-vating signals. In this case, the C-ter-minal 100-amino acid region is the sig-nal transducing domain, composed of three independent subdomains of approximately 30 amino-acid residues, D1, D2 and D3. TRAF2 and TRAF5 bind to D2 and D3, respectively. The D1 sub-domain can independently mediate signals to activate NF-kB when overex-pressed and without interacting with any known TRAF proteins. It was report-ed that binding of TRAF2 to the cyto-plasmic domain of CD30 results in rapid depletion of TRAF2 and the associated protein TRAF1 byproteolysis. These find-ings suggest a model in which CD30 limits its own ability to transduce cell survival signals through signal-coupled depletion of TRAF2. Depletion of intra-cellular TRAF2 and its associated pro-teins also increases the sensitivity of the cell to undergo apoptosis during activation of death-inducing recep-tors such as TNFR1. CD30 also activates another MAPKs by TRAF2, MEKK1 which selectively activates IKKb, resulting in NF-kB activation. MEKK1 activation by TRAF2 also results in activation of the stress-activated kinase (SAPK) c-jun N-terminal kinase (JNK) through activa-tion of MEK4. Thus, signals from the TNFR family diverge downstream of TRAF2, leadingto activation of two important transcription factors, NF-kB and c-jun, one through interaction withNIK, the other via MEKK1 and SAPKJNK. In addi-tion to TRAF2, NIK associates with other members of the TRAF family, including TRAF1, TRAF3, TRAF5 and TRAF6.NF-kB activation by TRAF5 and TRAF6 is inhib-
ited by catalytically inactive NIK mu-tants, suggesting that these TRAF pro-teins activate NF-kB by interacting with NIK. LikeTRAF2, overexpression of TRAF5 and TRAF6 activated SAPKJNK. Anoth-er stress-activated kinase, p38 MAP, is induced bycross-linking of CD30 as well as the extracellular-regulated ki-nase (ERK). The mechanism of acti-vation of ERK by CD30 has not been clearly elucidated, although studies on the related TNFR superfamily member CD40 have demonstrated a role for Src family kinases and the MAP3K Tpl2/Cot in ERK activation (25, 26). The signaling pathways activated by CD30 are illustrated in Figure 1.
Biologic effects on B cellsIt has been shown that the interaction between CD30 and CD30L represents an important signal for B-lymphocyte proliferation, dependent on IL-4 and IL-5. CD30L determines enhancement of antigen-specific immunoglobulin
FigURe 1 • Signal transduction pathways ac-tivated by CD30.

284 L. Leoncini, et al.
production (dependent on IL-2 and IL-5 or IL-4 and IL-5) and activates class-switch DNA recombination. Fur-thermore CD30 signals inhibit germ line transcription of the e gene (27).
Biologic effects on T cellsIt has been demonstrated that the activation of the CD30 receptor plays a critical role in the T cell-dependent immune response, a characteristic shared with some other members of the TNFR superfamily (28). The biologic effects elicited by CD30 are multiple:• mitogenic co-stimulation in the pres-
ence of T-cell receptor activation;• promotion of human T helper type2-
like T cells;• production of cytokines exerting in-
flammatory effects (IL-2, IL-5, IL-17A, TNF, IFN-g), involved in TH1 response
against infections and in TH17 differ-entiation;
• induction of cell surface molecules (CD54, CD80, CD86);
• negative selection in the thymus to avoid auto-reactive reaction.
Biologic effects on other hematopoietic cell population CD30/CD30L down-regulates the ex-pression of cytotoxic effector mole-cules, Fas ligand, perforin, granzyme B, and abrogates cytotoxicity of NK cells through B7/CD28 interaction (18).
CD30 expression in neoplastic cellsFollowing the initial discovery of CD30 expression on RS cells of HL, CD30 was also observed in numerous lymphoid malignancies of B-, T-, and NK-cell or-igin. Therefore, CD30-positive lympho-
TABLe 1 • Expression of CD30 in tumors.HeMATOPOieTiC MALigNANCieSB-cell lymphomas Diffuse large B-cell lymphoma, not otherwise specified Diffuse large B-cell lymphoma of the elderly Diffuse large B-cell lymphoma associated to chronic inflammation Lymphomatoid granulomatosis Primary effusion lymphoma Primary mediastinal B-cell lymphoma Lymphoma unclassifiable with features intermediate between Hodgkin lymphoma and
diffuse large B-cell lymphomaT-cell lymphomas Primary cutaneous T-cell lymphoma Peripheral T-cell lymphoma not otherwise specified Angioimmunoblastic T-cell lymphoma Anaplastic large cell lymphoma (ALK-positive, ALK-negative) Adult T-cell leukemia/lymphomaHodgkin’s lymphomaPost-transplant lymphoproliferative disordersHiV-related lymphomaNON HeMATOPOieTiC MALigNANCieSGerm cell tumorsNasopharingeal carcinomaMesenchymal tumors (leiomyoma, rhabdomyosarcoma, synovial sarcoma, malignant fibrous histiocytoma, osteosarcoma)

285CD30 expression in lymphoid neoplasms
ma cells may represent malignant transformation of activated lymphoid T-cells or, less commonly, B-cells. CD30 is also expressed on non-lymphoid germ cells tumors, and can be seen occasionally in nasopharyngeal carci-noma and mesenchymal tumors (Ta-ble 1) (29, 30).In Hodgkin’s lymphoma, CD30L en-hances cytokine production, such as IL-6 and TNFa by activating NF-kB pathway, as well as intercellular adhe-sion molecule-1 (ICAM-1) expression and shedding in RS cells. Activation of NF-kB signaling induces expression of anti-apoptotic genes, such as cFLIP, XIAP and BclXl, in RS and HL cells, determining their prolifer-ation. In addition, activation of CD30/CD30L signaling promotes CD54 sur-face expression and induces CD80 and CD86expression. In classical HL, neutrophils and eosin-ophils express high levels of surface CD30L andof IL-5, IL-3, and GM-CSF produced by RS cells, which are able to enhance CD30L. In addition, T cells within the HL lesions also express CD30L. These may suggest a possible role for CD30/CD30L in the interaction between malignant cells and other cells within the microenvironment in HL (32-34).In anaplastic large cell lymphoma, over-expression of CD30 results in self-activation in the absence of a li-gand and this determines the induc-tion of two different downstream sig-naling pathways depending on the presence of the chimeric oncoprotein nucleophosmin (NPM)- anaplastic lymphoma kinase (ALK). In ALK-nega-tive and cutaneous anaplastic large cell lymphoma, activation of CD30 receptor initiates the TRAF-IKK-IkB-NF-
kB pathway as in HL and this results in induction of homotypic aggregation, expression of cell surface molecules CD54, CD80, CD86 and enhance-ment of cytokine secretion. This path-way is disrupted in NPM-ALK-positive systemic ALCL due to the association of NPM-ALKwith TRAF proteins. NPM usually functions as a nucleolar shut-tle, whilst ALK is amonomeric receptor tyrosine kinase belonging to the insu-lin receptor subfamily that is normally expressed exclusively on neural tissues. NPM-ALK is created by a chromosom-al translocation t(2;5)(p23;q35) that fuses the NPM sequenceslocated on chromosome 5 to ALK sequences on chromosome 2. This effectively replaces the ALK pro-moter, usually silent in cells of lymphoid origin, with the strong NPM promoter. The resultant chimera contains the N-terminalportion of NPM and the C-terminal catalytic domainof ALK. In-teraction between the NPM domain of monomers leads to formation of an NPM-ALK hexamer. This oligomerisation is required for the constitutive kinase activity characteristic of the NPM-ALK oncoprotein. Activation of ALK tyrosine kinase promotes the growth of ALCL cells (32, 35, 36).
◗◗◗ CD30- pOSITIVe lYMpHOMaS
According to the last WHO classifica-tion of tumors of haematopoietic and lymphoid tissues, CD30 expression is detectable in numerous B- and T-cell lymphomas (17). Table 2 summarizes the clinical, mor-phological, immunophenotypical and genetic features of the lymphoid neo-plasms expressing this antigen (37).

286 L. Leoncini, et al.
TABLe 2 • Features of CD30-positivelymphomas.Clinic Morphology iHC Molecular
BiologyDLBCL, NOS adult and elderly
with enlarging mass in nodal and extranodal (gastrointestinal) sites; stage IV in 1/3 of cases
diffuse growth pattern with centroblastic, im-munoblastic, anaplastic or mixed morphology
pan-B cell antigens positive with expression of germinal center markers in a subset of cases; proliferative index (Ki-67): 30-40%
t(14;18); translocation involving 3q27; two groups identified by gene expression profile, the ABC and the CG type
eBV-positive DLBCL of the elderly
patients >50 year old without history of immunodeficiency or lymphoma, presenting with extranodal mass + lymphadenopathy; association with EBV
diffuse growth pattern with a monomorphous or polymorphous infiltrate of large cells, sometimes resembling HL and RS cells; sheets of necrosis, often with geographic pattern
CD20+, CD79a+, PAX-5+, MUM-1+, CD30+/-; high Ki-67;EBV type III latency pattern (LMP-1+, EBNA2+, EBER+)
Monoclonal IgH gene rearrangement
DLBCL associated with chronic inflammation
adult, M, with a longstanding history of chronic inflammation; 90% of cases are PAL
diffuse growth pat-tern of large cells with centroblastic or im-munoblastic cytology; massive angiodestruc-tive necrosis
non-GC B-cell immunophenotype, high Ki-67, EBNA-2+, LMP-1+/-, CD30+/-
Ig gene rearrangement, p53 mutation
Primary mediastinal large B-cell lymphoma
M 20-35 years; enlarging mass, often bulky, in anterior-superior mediastinum
diffuse to vaguely nodular pattern with sclerosis and compar-timentalization of small groups of intermediate to large lymphoid cells, sometimes resembling HL and RS cells; reac-tive infiltrate of T-cells
CD20+, CD79a+, PAX-5+, IRF-4/MUM-1+, CD23+, OCT-2+, BOB-1+, CD45/LCA+, CD30+/-, CD15-, EBV-
gain in 9p24, 2p15, 12q31, chromosome X, loss of 1p, 3p, 13q, 15q, 17p; translocations involving CCND1, BCL2, BCL6, MYC; high level of expression of IL-13 receptor and JAK-2 and STAT-1
PeL pleural, peritoneal and pericardial effusion without distinct extracavitary tumor masses and organomegaly; association with HHV8
small number of neoplastic cells with im-munoblastic, plasmab-lastic and/or anaplastic cytology adherent to mesothelial surface
plasma cell-associated markers (CD38, CD138, IRF-4/MUM-1) +; pan B-cell antigens -; CD30+; HHV8 + is essential for the diagnosis
complex karyotype with several chromosome gains and losses; somatic hypermutation of IgH variable region

287CD30 expression in lymphoid neoplasms
DLBCL/HL M 20-40 years with anterior mediastinal mass and advanced clinical stage
mixture of areas with large pleomorphic large cells resembling DLBCL and areas with scattered HL and RS cells; fibrosis and focal necrosis
co-expression of HL and DLBCL markers, MAL+, ALK-, EBV-
LYg young adult M; lung most frequent site of involvement; associationwith EBV
angiocentric and angiodestructivelym-phohistiocytic infiltrate with variable extent of necrosis and reactive T-cell background; three grades based on number of EBV+ B-cells and extent of necrosis
pan B-cell markers +; CD30+/-, EBV/LMP-1+/-
Monoclonal IgH rearrangement in grade 2 and 3
Adult T-cell leukemia/lymphoma
M, 20-80 years; higher incidence in Japan, sub-Saharan Africa and South America; associationwith HTLV-1
paracortical involve-ment by small, medium, large and/or anaplastic cells
pan-T-cell antigens+, CD4+, CD8-, FOXP-3+, CD30+/-, CD15+/-; high Ki-67
TCR rearrangement
enteropathy-associated T-cell lymphoma
median age: 60 years; clinical history of celiac disease; sites of involvement: jejeunum and ileum
two types: classical vari-ant and monomorphic variant
pan-T-cell antigens+, CD4-, CD8-
deletion 16q12.1
Primary cutaneous CD30-positive T-cell lympho-proliferative disorders
Primary cutaneous anaplastic large cell lymphomaAdult, M; nodules or papules + ulcerations
diffuse infiltrate of large neoplastic cells with a reactive background of T-cells, histiocytes, eosinophils, neutrophils
CD30+, CD4+, cytotoxic markers+, CD56+/-, CD15-, ALK-1-
gains in 7q, 17q, 21
Lymphomatoidpapulosis45 years, M; popular, papulonodular and nodular skin lesion in trunk and extremities, frequent spontaneous regression
4 histological types (A-B-C-D) based on cytological features and extent of epi-dermotropism
A and C: CD30+, ALK-1-, CD2+, CD3+, CD5+, cytotoxic markers+B: CD3+, CD4+, CD30-D: CD30+, CD3+, CD8+, CD4-
PTCL, NOS middle-aged M paracortical infil-trate, high endothe-lial venules, reactive background (eo-sinophils, epitheli-oidhistiocytes)
mature T-cell im-munophenotype, CD30+ and expres-sion of cytotoxic markers in a subset
TCRrearrange-ment; t(5;9) and t(14;19)

288 L. Leoncini, et al.
B-cell lymphomas
Diffuse large B-cell lymphoma, not other-wise specifiedDiffuse large B-cell lymphoma (DLB-CL) is a neoplasm of large B lymphoid cells with nuclear size equal to or ex-ceeding normal macrophage nuclei or more than twice the size of a normal lymphocyte. It has a diffuse growth pattern. CD30 may be especially ex-pressed in the anaplastic variant (17) (Figure 2).
EBV-positive diffuse large B-cell lympho-ma of the elderlyEBV-positive diffuse large B-cell lym-phoma of the elderly is an EBV-posi-tive clonal B-cell lymphoid proliferation that occurs in patients >50 years and
without any known immunodeficiency or prior lymphoma. The cells are vari-ably CD30 positive (17).
Diffuse large B-cell lymphoma associated with chronic inflammationDLBCL associated with chronic inflam-mation is a lymphoid neoplasm oc-curring in the context of long-standing chronic inflammation and showing association with EBV. Most cases in-volve body cavities or narrow spaces (e.g. pyothorax-associated lympho-ma-PAL). CD30 can be expressed by neoplastic cells (17).
Lymphomatoid granulomatosisLymphomatoid granulomatosis is an angiocentric and angiodestructive lymphoproliferative disease involv-
ALK+ ALCL children and young adult, M; advanced clinical stage and B symptoms
hallmark cells CD30+, ALK-1+, cytotoxic markers+, CD20-, Pax-5, CD15-, EBV-
t(2;5)
ALK- ALCL No age and no sex preference
Hallmark cells, extensive sinus involvement
CD30+, ALK-1-, CD15-, EBV-, cytotoxic markers+, B-cell antigens-
HL middle aged person with lymph node involvement and B-symptoms in a variable percentage of cases
HC and RS cells; four variants (nodular sclerosis, mixed cellularity, lymphocytes-rich, lymphocyte-depleted)
CD30+, CD15+, LMP-1+, CD20-, PAX-5+, EBV+
Immunodefi-ciency-associated lympho-proliferative disorders
patients with primary or secondary immunodeficency
depending on histological type
depending on histological type
Abbreviations: DLBCL, diffuse large B-celllymphoma; NOS, nototherwisespecified; ABC, activated B-cells; GC, germinal center; IHC, immunohistochemistry; M, male; EBV, Epstein-Barr virus; HC, Hodgkincells; RS, Reed-Sternberg; PAL, pyo-thoraxassociatedlymphoma; LYG, lymphomatoidgranulomatosis; PEL, pleuraleffusionlymphoma; DLBCL/HL, B-celllym-phoma, unclassifiable, with features intermediate between diffuse large B-celllymphoma and classicalHodgkinlym-phoma ;PTCL, peripheral T-celllymphoma; ALCL, anaplastic large lymphoma; HL, Hodgkin’slymphoma;

289CD30 expression in lymphoid neoplasms
FigURe 2 • Histo-logical aspects of DLBCL, NOS (A) ex-pressing CD30 (B). A, Haematoxylin and Eosin (H&E); B, CD30 stain. Original Mag-nification (OM): A-B, x200.
FigURe 3 • Histolog-ical aspects of PM-LBCL (A) expressing CD30 (B) A, H&E; B, CD30 stain. OM: A-B, x200.
ing extranodal sites, composed of EBV-positive B-cells admixed with re-active T-cells, which usually predomi-nate. In grade 3 lesions, the EBV-posi-tive B-cells may be variably positive for CD30 (17).
Primary mediastinal (thymic) large B-cell lymphomaPrimary mediastinal (thymic) large B-cell lymphoma (PMLBCL) is a DLBCL
arising in the mediastinum and with distinctive clinical, immunophenotypic and genotypic feature. CD30 is pres-ent in more than 80% of the neoplastic cells (17) (Figure 3).
Primary effusion lymphomaPrimary effusion lymphoma (PEL) is a large B-cell neoplasm usually present-ing as serous effusion without detect-able tumour masses. It is universally

290 L. Leoncini, et al.
associated with human herpes virus 8 (HHV8). CD30 is often positive (17).
B-cell lymphoma, unclassifiable, with fea-tures intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical HL (DLBCL/HL) is defined as a B-lineage lymphoma that demon-strates overlapping clinical, morpho-logical and/or immunophenotypic features between DLBCL, especially primary mediastinal large B-cell lym-phoma,and classical HL. CD30 is pos-itive (17) (Figure 4).
T-cell lymphomas
Adult T-cell leukemia/lymphomaAdult T-cell leukemia/lymphoma is a peripheral T-cell neoplasm most often composed of highly pleomorphic lym-phoid cells, caused by a human retro-virus, the human T-cell leukemia virus type 1 (HTLV-1). The large transformed cells may be positive for CD30 (17).
Enteropathy-associated T-cell lymphomaEnteropathy-associated T-cell lympho-ma is an intestinal tumour of intra-epi-thelial T lymphocytes, showing varying degrees of transformation but usual-ly presenting as a tumour composed of large lymphoid cells, often with an inflammatory background. The adja-cent small intestinal mucosa shows vil-lous atrophy with crypt hyperplasia. In almost all cases, a varying proportion of the tumour cells express CD30 (17).
Primary cutaneous CD30-positive T-cell lymphoproliferative disordersPrimary cutaneous CD30-positive T-cell lymphoproliferative disorders are the second most common group of the cutaneous T-cell lymphomas, account-ing for approximately 30% of the cases (17). This group includes:• Primary cutaneous anaplastic large
cell lymphoma is composed of large cells with an anaplastic, pleo-morphic or immunoblastic cytomor-phology; CD30 antigen is expressed by the majority (>75%) of the tumour cells.
FigURe 4 • Histolog-ical aspects of DLB-CL/HL (A) expressing CD30 (B). A, H&E; B, CD30 stain. OM: A-B, x200.
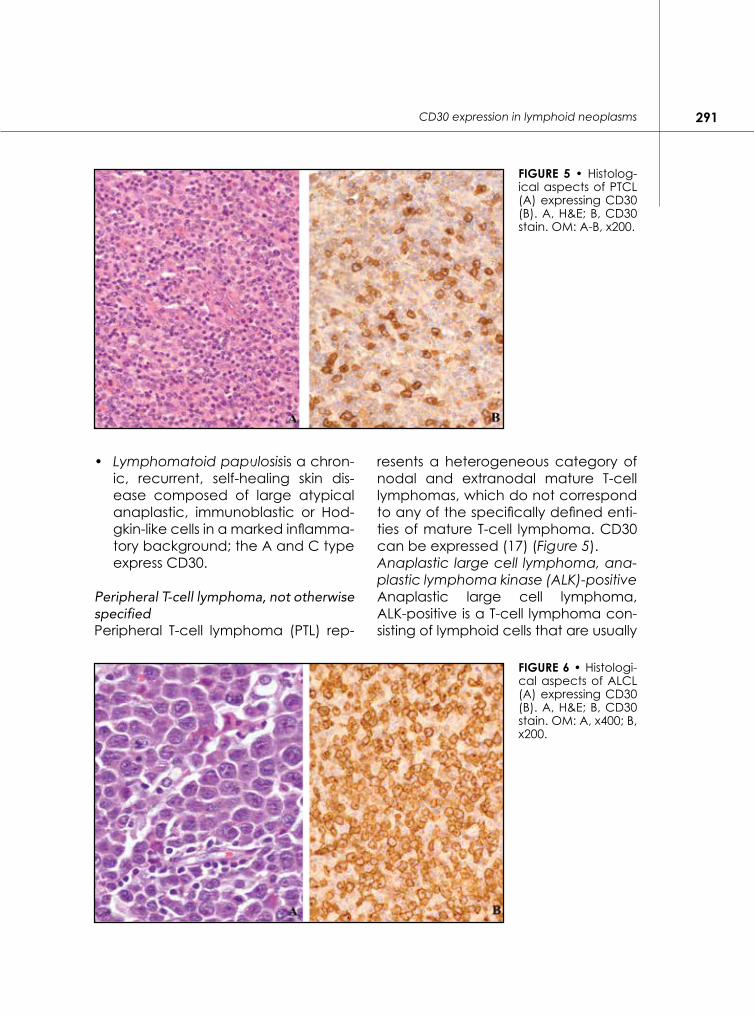
291CD30 expression in lymphoid neoplasms
• Lymphomatoid papulosisis a chron-ic, recurrent, self-healing skin dis-ease composed of large atypical anaplastic, immunoblastic or Hod-gkin-like cells in a marked inflamma-tory background; the A and C type express CD30.
Peripheral T-cell lymphoma, not otherwise specifiedPeripheral T-cell lymphoma (PTL) rep-
resents a heterogeneous category of nodal and extranodal mature T-cell lymphomas, which do not correspond to any of the specifically defined enti-ties of mature T-cell lymphoma. CD30 can be expressed (17) (Figure 5).Anaplastic large cell lymphoma, ana-plastic lymphoma kinase (ALK)-positiveAnaplastic large cell lymphoma, ALK-positive is a T-cell lymphoma con-sisting of lymphoid cells that are usually
FigURe 5 • Histolog-ical aspects of PTCL (A) expressing CD30 (B). A, H&E; B, CD30 stain. OM: A-B, x200.
FigURe 6 • Histologi-cal aspects of ALCL (A) expressing CD30 (B). A, H&E; B, CD30 stain. OM: A, x400; B, x200.

292 L. Leoncini, et al.
large with abundant cytoplasm and pleomorphic, often horseshoe-shaped nuclei; there is a translocation involving the ALK gene; ALK protein is expressed, as well as CD30 (17) (Figure 6).
Anaplastic large cell lymphoma, anaplas-tic lymphoma kinase (ALK)-negativeAnaplastic large cell lymphoma, ALK-negative is defined as a CD30-pos-itive T-cell neoplasm that is not distin-guishable on morphological grounds from ALK-positive ALCL, but lacks ALK protein (17).
Classical Hodgkin’s lymphomaClassical Hodgkin lymphoma is a monoclonal lymphoid neoplasm (in most instances derived from B-cells) composed of mononuclear Hodgkin cells and multinucleated Reed-Stern-berg cells residing in an infiltrate con-taining a variable mixture of non-neo-plastic small lymphocytes, eosinophils, neutrophils, histiocytes, plasma cells, fibroblasts and collagen fibers. CD30 is over-expressed in all the four variants (17) (Figure 7).
Immunodeficiency-associated lymphop-roliferative disordersLymphoproliferative diseases associat-ed with primary immune disordersLymphoproliferative diseases associ-ated (LPD) with primary immune disor-ders (PID) are lymphoid proliferations (non-neoplastic and neoplastic) that arise as a result of immune deficiency due to primary immunodeficiency or immunoregulatory disorders. Lympho-mas occurring in patients with PID do not generally differ in their morphology from those occurring in immunocom-petent hosts and are mainly represent-ed by lymphomatoid granulomatosis, DLBCL, HL and peripheral T-cell lym-phoma. All these histotypes are char-acterized by EBV-positive CD30-posi-tive neoplastic cells (17).
Post-transplant lymphoproliferative disor-dersPost-transplant lymphoproliferative disorders (PTLD) are lymphoid or plas-macytic proliferations that develop as a consequence of immunosuppres-sion in a recipient of solid organ, bone
FigURe 7 • Histologi-cal aspects of ALCL (A) expressing CD30 (B). A, H&E; B, CD30 stain. OM: A-B, x200.

293CD30 expression in lymphoid neoplasms
marrow or stem cell allograft. PTLD comprise a spectrum ranging from in-fectious mononucleosis to B- or T-cell lymphoma and HL. In the latter diseas-es (so called monomorphic PTLDs) the neoplastic population is EBV-infected and express CD30 (17).
Lymphomas associated with HIV infectionAmong the lymphomas that develop in HIV setting, DLBCL, PEL and HL ex-press CD30 (17).
◗◗◗ CD30-TaRGeTeD THeRapY
In addition to diagnostic value of CD30, its highly restrictedexpression pattern made it ideal for target therapy using monoclonal antibodies (38, 39). After the first attempt to target CD30 in pa-tients with relapsed HL in 1992, several investigatorshave evaluated different strategies to bind CD30 by monoclonal antibodies (40-44). These strategies included a variety of naked and conjugated antibodies. However, most of the first- and sec-ond-generation anti-CD30 immuno-conjugates (including rich A-chain, radioisotopes or cytostatic drugs) were either too immunogenic or not effec-tive enough for further clinical devel-opment (41, 42). In addition, human or humanized monoclonal antibodies against CD30 gave disappointing clini-cal results (43, 44). Thirty years after Ki-1 discovery and numerous unsuccess-ful attempts, CD30 is back on center stage as a major therapeutic targetin the management of poor-prognosis HL, and, alone or in combination, in the setting up-front and/or salvage therapy for HL, ALCL, and possibly oth-er CD30-positive malignancies (lym-
phomatoid papulosis, subtypes of DL-BCL and PTCL expressing CD30, PEL and PTLD) (45-49). The landscape has changed dramatically with the ad-vent of brentuximab vedotin. Brentuximab vedotin (ADCETRIS, SGN-35) is an antibody–drug conjugate (ADC) consisting of three components: the monoclonal antibody cAC10, the cytotoxic agent monomethylauristatin E (MMAE) and a protease-cleavable covalent linker that attaches cAC10 to MMAE. cAC10 (SGN-30) is a chi-meric anti-CD30 specific monoclonal antibody derived from the fusion of the variable heavy and light region of the murine anti-CD30 antibody AC10, with the constant gamma1-heavy and kappa-light region of the human im-munoglobulin (50). In an attempt to enhance the antitu-mor properties of SGN-30, MMAE was conjugated to the monoclonal anti-body generating the cAC10-vcMMAE ADC (SGN-35, brentuximab vedotin). MMAE is a synthetic derivative of do-lastatin 10, a natural cytostatic pseu-dopeptide originally isolated from the marine shell-less mollusk Dorabella au-ricularia. MMAE exerts its potent cyto-static effect by inhibiting microtubule assembly, tubulin-dependent GTP hy-drolysis and polymerization. It has been demonstrated that both dolastin 10 and MMAE have signifi-cant activity against various cell lines of hematopoietic tumours in vitro. The points of MMAE attachment are -SH groups of cysteine residues produced by mild reduction of the inter-chain disulfide bonds. The linker consists of a thiol-reactive maleimidocaproyl spac-er, the dipeptide valine–citrulline linker, and a self-immolative p-aminobenzyl-carbamate spacer. Upon binding to

294 L. Leoncini, et al.
CD30-expressing neoplastic cells, the ADC is internalized by endocytosis. Ly-sosomal degradation causes selective cleavage of the linker, allowing release of the MMAE in the tumour microenvi-ronment. MMAE binds to tubulin, dis-rupting the microtubule network with resultant cell cycle arrest and apopto-sis of bystander cells (51). Based on the encouraging results of in vitro studies, clinical trials were carried out.
phase I studiesTwo phase I studies investigating two different regimens of brentuximab ve-dotin were undertaken (51). In the first study, the ADC was admin-istered intravenously every 3 weeks at doses of 0.1 to 3.6 mg/kg according to a traditional dose-escalation design followed by a cohort expansion phase. The primary objective was to deter-mine the maximum tolerated dose (MTD), while the secondary objectives included assessment of antitumor ac-tivity. The study enrolled a total of 45 patients: 42 with HL, 2 with ALK-positive ALCL and one with angioimmunoblas-tic T-cell lymphoma. All participants had relapsed or refrac-tory disease to multiple prior regimens; the mean number of prior chemother-apies was 3 (range: 1-7) while 73% had failed autologous stem cell transplan-tation (SCT). The MTD and the dose for further clinical investigations were determined at 1.8 mg/kg every three weeks. The drug was well tolerated with mild to moderate adverse events. The most common adverse events includ-ed fatigue, pyrexia, diarrhea, nausea, neutropenia and cumulative dose-re-lated peripheral neuropathy. The lat-ter occurred in 36% of patients after a median of three cycles. The overall ob-
jective response rate was 38.6% (25% complete and 13.6% partial response rate), whereas 81% of patients with disease-related symptoms at baseline experienced resolution regardless of response status. At the MTD, the over-all response rate was 50%. The mean duration of response was at least 9.7 months and the progression-free sur-vival 5.9 months (52). In the second phase I study, the MTD was determined at 1.2 mg/kg weekly every three weeks of a 4-week cycle. The primary objective was the deter-mination of MTD and safety profile of a weekly regimen, while secondary objective included assessment of an-titumor efficacy. The study enrolled 44 patients with relapsed (55%) or refrac-tory (45%) CD30-positive hematologic malignancies: 38 patients with HL, 5 with ALCL (1 ALK-positive, 4 ALK-neg-ative), and 1 patient with peripheral T-cell lymphoma not otherwise speci-fied. The patients characteristics were similar to the previous phase I study: the mean number of prior systemic therapies was 3 (range: 1-8) and 68% of patients had failed prior autologous SCT. Hyperglycemia and diarrhea con-stituted the dose-limiting toxicities; no dose-limiting toxicities were seen at the MTD. The most common adverse events were fatigue, nausea, arthral-gia, diarrhea, upper respiratory tract infection, infusion reactions, and pri-marily mild to moderate peripheral neuropathy (sensory and motor). The latter occurs in 73% of patients; grade III peripheral neuropathy developed in 14% of the patients. The incidence of the adverse effects was higher in respect to the phase I previous study. The overall response rate was 59% with complete remission achieved in 34%.

295CD30 expression in lymphoid neoplasms
At the MTD, the overall and complete response rates were 58% and 25%, re-spectively. With a median follow up of 11 months, the mean progression-free survival was approximately 7 months, whereas the mean overall survival has not been reached (53). Both studies have indicated a dose-re-sponse therapeutic effect, but efficacy was not their primary objective. Week-ly infusion of brentuximab vedotin seems to be more advantageous in in-ducing rapid responses in patients with bulky or symptomatic disease respect to a every 3-week regimen, but cu-mulative peripheral neuropathy may limit prolonged administrations. Both phase I studies allowed retreatment with brentuximab vedotin in patients who achieved reductions in tumor volume to the investigational therapy but relapsed. The regimen consisted of 1.8 mg/kg every 3 weeks or 1 mg/kg weekly, depending on the prior study. Preliminary data on seven patients (six with HL and one with ALCL) demon-strated remarkable responses, two and four out of eight patients achieved-complete and partial remissions. Only one patient needed autologous SCT. Adverse events were similar to those reported in the phase I studies includ-ing peripheral neuropathy and upper respiratory tract infections (51).
phase II studiesThe encouraging results of the phase I studies prompted to carry out two singlearm multicenter phase II clinical trials with brentuximab vedotin at 1.8 mg/kg every three weeks up to 16 cy-cles in relapsed or refractory HL and ALCL (51). In both studies, the primary aim was the objective response rate.The phase II study in HL enrolled 98 pa-
tients having failed prior autologous SCT. The mean age of the patients was 31 years (range: 15-77) and the mean number of prior treatments was 3.5 (range: 1-13) in addition to high-dose chemotherapy followed by autolo-gous SCT. The objective response rate was 75% with complete remission rate of 34% and a partial response in 40% of patients. The 95% of the patients achieved reductions in tumor size and 83% of subjects with B symptoms at baseline experienced resolution of these symptoms. The follow-up last 9 months and the reported estimated 12-month overall survival was 88%. The most common adverse events were peripheral neuropathy, fatigue, nau-sea, diarrhea, and neutropenia, and cytopenias; 18% of patients discontin-ued treatment due to adverse events (54).The phase II study in ALCL enrolled a total of 58 patients (28% ALK-positive and 72% ALK-negative) with relapsed or refractory disease (55). The mean age of the participants was 52 years (range: 14-76) and the mean number of prior treatments was 2 (range: 1-6) %). The objective response rate was 86% with complete remission rate of 53% and partial response in 33%. More than 90% of patients with secondary cutaneous involvement experienced resolution of the lesions after a median of 4.9 weeks. The mean duration of response ranged from 0.3 to 45.3 weeks. Interesting-ly, the response pattern was similar in ALK-positive and ALK-negative dis-ease. The treatment-related adverse events were similar in nature and inci-dence to those reported in the phase II study in HL, cytopenia and periph-eral neuropathy constituting the most

296 L. Leoncini, et al.
common grade III toxicities. In both studies brentuximab vedotin showed remarkable antitumor activity and a manageable toxicity (51).
phase III studiesA randomized double-blind, place-bo-controlled phase III study (AETHERA clinical trial) is currently ongoing in pa-tients at high risk for residual HL after SCT and two distinct clinical trials are evaluating the potential role of bren-tuximab vedotin (1.8 mg/kg every 21 days) in combination with currently used chemotherapy protocols in HL (50). The study enrolled 329 patients, the primary aim was the measure of the progression-free survival; secondary aims were the measure of incidence of adverse events and laboratory ab-normalities.
IndicationsGiven the unprecedented antitumor efficacy in a patient population with few other treatment options, in 2011 the Food and Drug Administration (FDA) granted accelerated approval of brentuximab vedotin in relapsed or refractory HL and ALCL (56, 57). In HL, the indication includes failure of autol-ogous SCT or at least two prior multi-agent chemotherapy regimens in pa-tients ineligible for SCT (56). Given the paucity of effective sec-ond-line regimens in ALCL (especially ALK-negative ALCL), the indication in ALCL includes failure of at least one prior multiagent chemotherapy. Owing to cumulative toxicities, the treatment should not exceed 16 cycle, although effective retreatment with brentuximab vedotin should be con-sidered investigational (57).
◗◗◗ a lOOK FORWaRD
We now have a better understanding as to the cell populations that express CD30 and the mechanisms by which this expression is controlled. Analysis of CD30 expression in malignant cells provides a useful tool for diagnosis and classification of lymphomas, and also the basis forimmunotherapy targeted to the CD30 antigen. Brentuximab ve-dotin has already changed treatment approaches in relapsed and refractory HL and ALCL. Our understanding of the function of CD30 in HL and ALCL is continuous-ly improving, and the basis for CD30 overexpression in these tumours is be-coming clear. Still, there are a num-ber of new and possibly even more relevant questions to be addressed, including the use of the drug in earli-er stages (58). The next challenge will be to understand the precise nature of theevents that trigger signal transduc-tion by CD30. Intensive work will be required to fur-ther characterize intracellularsignaling molecules and to interpret the cross-talkbetween major signaling pathways (59). Another challenge will be to map the cellular pathways responsible for the induction of cell death by CD30, which should allow us to better devise CD30-targeted strategies for the treat-ment of HL and ALCL. The effectiveness of brentuximab ve-dotin in other CD30-positive tumours is an area of study that will soon be in the spotlight and is likely to become the next focus of clinical research with this and other drugs (i.e. panobistonat, a histone deacetylase inhibitor) (59). There are a number of currently ongo-ing studies that are investigating the

297CD30 expression in lymphoid neoplasms
use of these new drugs in CD30-positive lymphoid neoplasms (i.e. CD30-pos-itive DLBCL, PEL, T-lymphoma) even early in the disease course (45, 59, 60). Treatment of CD30-positive lymphoid neoplasms with brentuximab vedotin (and perhaps with other drugs) may improve patient outcome. This is the reason why CD30 could be converted from a diagnostic into a therapeutic tool.
◗◗◗ ReFeReNCeS1. Schwab U, Stein H, Gerdes J, et al. Pro-
duction of a monoclonal antibody spe-cific foer Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 1982; 299: 65-7.
2. Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease as-sociated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocyt-ic malignancies are derived from ac-tivated lymphoid cells. Blood 1985; 66: 848-58.
3. Andreesen R, Osterholz J, Lohr G, et al. Hodgkin cell-specific antigen is ex-pressed on a subset of auto and alloac-tivated T (helper) lymphoblasts. Blood 1984; 63: 1299-302.
4. Gerdes J, Schwarting R, Stein H. High proliferative activity of Reed Sternberg associated antigen Ki-1 positive cells in normal and lymphoid tissue. J Clin Pathol 1986; 39: 993-7.
5. Ralfkiaer E, Bosq J, Gatter KC, et al. Ex-pression of a Hodgkin and Reed-Ster-nberg cell associated antigen (Ki-1) in cutaneous lymphoid infiltrate. Arch Der-matol Res 1987; 279: 285-92.
6. Froese P, Lemke H, Gerdes J, et al. Bio-chemical characterization and byosin-thesis of the Ki-1 antigen in Hodgkin-de-rived and virus transformed human B and T lymphoid cell lines. J Immunol 1987; 139: 2081-7.
7. O’ Connor NT, Stein H, Gatter KC, et al. Genotypic analysis of large cell lym-phoma which express the Ki-1 antigen. Histopathology 1987; 11: 733-40.
8. Andreesen R, Brugger W, Lohr GW, et al. Human macrophages can express the Hodgkin’s cell-associated antigen Ki-1 (CD30). Am J Pathol 1989; 134: 187-92.
9. Cambiaggi A, Cantoni C, Marciano S, et al. Cultured human NK cells express the Ki-1/CD30 antigen. Br J Haematol 1993; 85: 270-6.
10. Falini B, Flenghi L, Fedeli L, et al. In vivo targeting of Hodgkin and Reed-Ster-nberg cells of Hodgkin’s disease with monoclonal antibody Ber-H2 (CD30): immunohistological evidence. Br J Hae-matol 1992; 82: 38-45.
11. Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995; 85: 1-14.
12. Kaudewitz P, Stein H, Dallenbach F. Pri-mary and secondary cutaneous Ki-1+ (CD30+) anaplastic large cell lympho-ma. Morphologic, immunohistologic, and clinical characteristics. Am J of Pathol 1989; 135: 359-67.
13. Stein H, Herbst H, Anagnostopoulos I, et al. The nature of Hodgkin and Reed-Ster-nberg cells, their association with EBV, and their relationship to anaplastic large-cell lymphoma. Ann Oncol 1991; 2: 33-8.
14. Herbst H, Dallenbach F, Hummel M, et al. Epstein-Barr virus DNA and latent gene products in Ki-1 (CD30)-positive anaplastic large cell lymphomas. Blood 1991; 78: 2666-73.
15. Leoncini L, Del Vecchio MT, Kraft R, et al. Hodgkin’s disease and CD30-posi-tive anaplastic large cell lymphomas--a continuous spectrum of malignant disor-ders. A quantitative morphometric and immunohistologic study. Am J Pathol. 1990; 137: 1047-57.
16. Chan JK. The new World Health Organi-zation classification of lymphomas: the

298 L. Leoncini, et al.
past, the present and the future. Hema-tol Oncol. 2001; 19: 129-50.
17. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Hae-matopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC Press; 2008.
18. Horie R, Watanabe T. CD30: expression and function in health and disease. Im-munology 1998; 10: 457-70.
19. Chiarle R, Podda A, Prolla G, et al. Mol-ecule of the month. CD30 in normal and neoplastic cells. Clinical Immunol-ogy 1999; 90: 157-64.
20. Josimovic-Alasevic O, Durkop H, Schwarting R, et al. Ki-1 (CD30) antigen is released by Ki-1-positive tumor cells in vitro and in vivo. Partial characterization of soluble Ki-1 antigen and detection of the antigen in cell culture supernatants and in serum by an enzyme-linked im-munosorbent assay. Eur J Immunol 1989; 19: 157-62.
21. Nadali G, Vinante F, Stein H, et al. Serum levels of soluble form of CD30 molecule as a tumor marker in CD30+ anaplastic large-cell lymphoma. J Clin Oncol 1995; 13: 1355-60.
22. Pizzolo G, Vinante F, Chilosi M, et al. Serum levels of soluble CD30 molecule (Ki-1 antigen) in Hodgkin’s disease: re-lationship with disease activity and clin-ical stage. Br J Hematol 1990; 75: 282-4.
23. Nadali G, Vinante F, Stein H, et al. Serum levels of soluble form of CD30 molecule as a tumor marker in CD30 anaplastic large-cell lymphoma. J Clin Oncol 1995; 13: 1355-60.
24. McDonald PP, Cassatella MA, Bald A, et al. CD30 ligation indices nuclear fac-tor-kappa B activation in human T cell lines. Eur J Immunol 1995; 25: 2870-6.
25. Lee SY, Lee SY, Kandala G, et al. TNF receptor-associated factor interaction: NF-kappa B activation and binding specificity. Proc Natl Acad Sci 1996; 93: 9699-703.
26. Boucher L-M, Marengère LE, Lu Y, et al. Binding sites of cytoplasmic effec-tors TRAF1, 2 and 3 on CD30 and other members of the TNF receptor superfam-
ily. Biochem Biophys Comm 1997; 233: 592-600.
27. Jumper MD, Nishioka Y, Davis LS, et al. Regulation of human B cell proliferation by recombinantCD40 ligand and oth-er TNF-related ligands. J Immunol 1995; 155: 2369-78.
28. Hamann D, Hilkens CM, Grogan JL, et al. CD30expression does not discrimi-nate between human Th1- andTh2-type T cells. J. Immunol. 1996; 156: 1387-91.
29. Durkop H, Foss HD, Eitelbach F, et al. Ex-pression of the CD30 antigen in non-lym-phoid tissues and cells. JOP 2000; 190: 613-8.
30. Latza U, Foss HD, Durkop H, et al. CD30 antigen in embryonal carcinoma and embryogenesis and release of the solu-ble molecule. Am J of Pathol 1995; 146: 463-71.
31. Al-Shamkhani A. The role of CD30 in the pathogenesis of haematopoietic ma-lignancies. Current opinion in pharma-cology 2004; 4: 355-9.
32. Ho DS, Rea AJ, Abraham LJ. Functional aspects of the CD30 gene in Hodgkin’s lymphoma and anaplastic large cell lymphoma. Oncol Rev 2009; 3: 89-101.
33. Durkop H, Oberbamscheidt M, Latza U, et al. The restricted expression pattern of the Hodgkin’s lymphoma-associated cytokine receptor CD30 is regulated by a minimal promoter. JOP 2000; 192: 182-93.
34. Durkop H, Oberbamscheidt M, Latza U, et al. Structure of the Hodgkin’s lympho-ma-associated human CD30 gene and the influence of a microsatellite region on its expression in CD30 (+) cell lines. Biochim Biophys Acta 2001; 1519: 185-91.
35. Durkop H, Burkhard H, Hahn C, et al. Dif-ferential expression and function of A20 and TRAF1 in Hodgkin lymphoma and anaplastic large cell lymphoma and their induction by CD30 stimulation. JOP 2003; 200: 229-39.
36. Hirsch B, Hummel M, Bentink S, et al. CD30-induced signaling is absent in Hodgkin’s cells but present in anaplas-

299CD30 expression in lymphoid neoplasms
tic large cell lymphoma cells. Am J of Pathol 2008; 172: 510-20.
37. Jaffe ES, Harris NL, Vardiman JW, et al. Hematopathology. 1st ed. Saunders El-sevier, 2011.
38. Younes A. CD30-targeted antibody ther-apy. Curr Opin Oncol 2011; 23: 587-93.
39. Deutsch YE, Tadmor T, Podack ER, et al. CD30: an important new target in he-matologic malignancies. Leukemia and Lymphoma 2011; 52: 1641-54.
40. Falini B, Bolognesi A, Flenghi L, et al. Re-sponse to refractory Hodgkin’s disease to monoclonal anti-CD30 immunotoxin. Lancet 1992; 339: 1195-6.
41. Tazzari PL, Bolognesi A, de Totero D, et al. Ber-H2 (anti-CD30)-saporinimmuno-toxin: a new tool for the treatment of Hodgkin’s disease and CD30+ lympho-ma: in vitro evaluation. Br J Hematol 1992; 81: 203-11.
42. Engert A, Burrows F, Jung W, et al. Eval-uation of ricin A chain-containing im-munotoxins directed against the CD30 antigen as potential reagents for the treatment of Hodgkin’s disease. Cancer Res 1990; 50: 84-8.
43. Sforzini S, Bolognesi A, Meazza R, et al. Targeting of type 1 ribosome-inactivat-ing proteins to CD30+ or CD25+ hema-tologic neoplasia by bispecific antibod-ies. J Hematother 1995; 4: 429-32.
44. Terenzi A, Bolognesi A, Pasqualucci L, et al. Anti-CD30 (BER-H2) immunotox-ins containing the type-1 ribosome-in-activating proteins momordin and PAP-S (pokeweed antiviral protein from seeds) display powerful antitumor ac-tivity against CD30+ tumour cells in vitro and in SCID mice. Br J Haematol 1996; 92: 872-9.
45. Steinhoff M, Hummel M, Anagnost-opoulos I, et al. Single-cell analysis of CD30+ cells in lumphomatoid papulosis demonstrates a common clonal T-cell origin. Blood 2000; 100: 578-84.
46. Hu S, Xu-Monette ZY, Balasubramanyam A, et al. CD30 expression defines a nov-el subgroup of diffuse large B-cell lym-phoma with favorable prognosis and
distinct gene expression signature: a re-port from the International DLBCL Ritux-imab-CHOP Consortium Program Study. Blood 2013; 121: 2715-24.
47. Sabattini E, Pizzi M, Tabanelli V, et al. CD30 expression in peripheral T-cell lym-phoma. Haematologica 2013; 98: e81.
48. Bhatt S, Ashlock BM, Natkunam Y. CD30 targeting with brentuximab vedotin: a novel therapeutic approach to prima-ry effusion lymphoma. Blood 2013; 122: 1233-42.
49. Theurich S, Wennhold K, Wedemeyer I, et al. CD30-targeted therapy with bren-tuximab vedotin and DLI in a patient with T-cell posttransplantion lymphoma: induction of clinical remission and cel-lular immunity. Transplantation 2013; 96: e16-8.
50. Terriou L, Bonnet S, Debarri H, et al. Bren-tuximab vedotin: new treatment for CD30+ lymphomas. Bull Cancer 2013; 100: 775-9.
51. Vaklavas C, Forero-Torres A. Safety and efficacy of brentuximab vedotin in pa-tients with Hodgkin lymphoma or sys-temic anaplastic large cell lymphoma. Ther Adv Hematol 2012; 3: 209-25.
52. Younes A. Novel treatment strategies for patients with relapsed classical Hod-gkin lymphoma. Hematology Am Soc Hematol Educ Program. 2009, 507-19.
53. Fanale MA, Forero-Torres A, Rosenblatt JD, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed /refractory CD30-positive he-matologic malignancies. Clin Cancer Res 2012; 18: 248-55.
54. Chen RW, Gopal AK, Smith SE, et al. Results from a pivotal phase II study of brentuximab vedotin (SGN-35) in pa-tients with relapsed or refractory Hod-gkin lymphoma (HL). J Clin Oncol 2011; 29: 8031.
55. Pro B, Advani R, Brice P, et al. Brentux-imab vedotin (SGN-35) in patients with relapsed or refractory systemic ana-plastic large-cell lymphoma: results of a phase II study. J Clin Oncol 2012; 30: 2190-6.

300 L. Leoncini, et al.
56. Rothe A, Sasse S, Goergen H, et al. Bren-tuximab vedotin for relapsed or refrac-tory CD30+ hematologic malignancies: the German Hodgkin Study Group ex-perience. Blood 2012; 120: 1470-2.
57. Zinzani PL, Viviani S, Anastasia A. Bren-tuximab vedotinin relapsed/refractory Hodgkin lymphoma: the Italian experi-ence and results of its use in daily clini-cal practice outside clinical trials. Hae-matologica 2013; 98(8): 1232-6.
58. Engert A. CD30-positive malignant lymphoma: time for a change of man-
agement? Haematologica 2013; 98: 1165-8.
59. Canellos GP. Brentuximab vedotin and Panobinostat: new drugs for Hodgkin’s lymphoma – can they make one of medical oncology’s chemotherapy suc-cess stories more successful? J Clin On-col 2012; 30: 2171-2.
60. Assaf C, Hirsch B, Wagner F, et al. Dif-ferential expression of TRAF1 aids in the distinction of cutaneous CD30-positive lymphoproliferations. J Investigative Der-matol 2007; 127: 1898-904.

DCTH - 4•2013 - 301-309
Key words: CD30, brentuximab vedotin, CTCL, skin, lymphoma.
Correspondence:Nicola Pimpinelli, MD, PhDDept. Surgery and Translational MedicineDivision DermatologyUniversity of Florence Medical SchoolViale Michelangiolo, 4150125 Florence, ItalyE mail: [email protected]
SUMMARYBrentuximab vedotin (BV) is an anti-CD30 monoclonal antibody conjugated with the cytotoxic drug monomethylyauristatin E (MMAE), a potent microtubule inhibitor. In re-lapsed/refractory Hodgkin lymphoma and systemic anaplastic large cell lymphoma, BV achieved durable response with low and transitory toxicity. Accordingly, it was approved by FDA in 2011 for patients affected by these malignancies. In some case reports, BV obtained rapid response in CD30-expressing cutaneous T-cell lymphoma, as transformed mycosis fungoides and relapsed/refractory primary cutaneous ACL lymphoma. Currently, two phase 2 trials are ongoing to assess the potential use of brentuximab in patients af-fected by refractory/relapsed CD30+ T-cell lymphoproliferative disorders and in patients affected by mycosis fungoides/Sézary syndrome, respectively. Finally, one phase 3 trial is comparing brentuximab to methotrexate or bexarotene in CD30-expressing cutaneous T-cell lymphomas. Among the several new drugs tested in the last 5 years, BV showed the most promising and challenging results to be translated in the clinical practice.
Brentuximab vedotin in CD30-expressing cutaneous T-cell lymphomaC. Delfino1, V. Grandi1, A. Pileri2, S. Gunnella1, L. Rigacci3, R. Alterini3, N. Pimpinelli11Dept. Surgery and Translational Medicine, Division Dermatology; University of Florence, Italy; 2Dept. Internal Medicine and Medical Specialties, Division Dermatology; University of Bologna, Italy; 3Dept. Clinical and Experimental Medicine, Division Hematology; University of Florence, Italy
◗◗◗ INTRODUCTION
Brentuximab vedotin (SGN-35, BV) is an antibody-drug conjugate (ADC) com-posed by the anti-CD30 monoclonal antibody cAC10 conjugated with the cytotoxic drug monomethylyauristatin E (MMAE), a potent microtubule inhib-itor. CD30 is a transmembrane protein
FORUM
belonging to the tumor necrosis fac-tor receptor (TNFR) superfamily, and is physiologically expressed in activated T cells, B cells and NK cells. Moreover, it is currently well known that CD30 is ex-pressed in Hodgkin lymphoma (HL), an-aplastic large cell lymphoma (ALCL), specific subtypes of B-cell non-Hod-gkin lymphomas (NHL), mature T cell lymphomas and germ-line malignan-cies (1). In August 2011, the Food and Drug Ad-ministration (FDA) approved the use of BV for patients affected by HL after fail-ure of autologous stem cell transplan-tation (autoSCT) or failure of at least two prior multiagent chemotherapy (polyCT) regimens in autoSCT-ineligible candidates, and for the treatment of systemic ALCL after failure of at least

302 C. Delfino, et al.
one polyCT regimen. An increasing in-terest concerns the use of BV in other CD30+ malignancies (2). Particularly, some case reports describe fast re-mission and high tolerance in CD30+ cutaneous T-cell lymphomas (CTCL) treated with BV (3-6). Finally, its use in CD30+ cutaneous T-cell lymphomas (CTCL) and lymphoprolipherative disor-ders (LPD) is currently under investiga-tion in a phase 2 study (NCT01352520) (Duvic M, et al. Results of a phase II trial of brentuximab vedotin (SGN-35) for CD30+ cutaneous T-cell lymphomas and lymphoproliferative disorders. ASH 2012 Annual Meeting Abstract; 120: Abstract 3688).
◗◗◗ BReNTUxIMaB veDOTIN: MOleCUlaR aspeCTs aND MeChaNIsMs OF aCTION
Brentuximab Vedotin is an ADC of nov-el generation. Part of the molecule is a mouse-human chimeric IgG1 An-ti-CD30 mAb (cAC10; SGN-30) that binds a different epitope than other anti-CD30 antibodies (7); the mAb is conjugated with monomethylaurista-tin E (MMAE), a synthetic analog of do-lastatin, and each mAb molecule car-ries an average of 4 MMAE groups (8). BV binds to CD30 receptor, and it is in-ternalized by endocytosis. After being exposed to proteolytic lisosomal en-zymes, MMAE molecules are liberated in cytoplasm (9), where they bind to tu-bulin The binding causes the disruption of microtubule network and the induc-tion of G2/M-phase cell cycle arrest and apoptosis (10). The high stability of the molecule in the plasma, associated with the rapid intracellular enzymatic cleavage only in antigen-positive cells,
allows a lower in vivo toxicity and high-er efficacy when compared to other conjugates (8). Moreover, BV induces tumor cell death thanks to mecha-nisms also led by SGN-30: antigen-de-pendent cellular phagocytosis (ADCP) and direct effects on tumor cell signal-ing. In contrast, another important ef-fect of this drug is a result of the free diffusion of a variable percentage of bystander MMAE molecules in the mi-croenvironment outside CD30+ cells, where MMAE kill the surrounding cells that are able to support tumor prolif-eration (9). Some authors hypothesize that this effect could explain the high response rates observed in a group of patients who have a low occupation ratio (<3%) of CD30-binding sites by BV molecules (11).
◗◗◗ appROveD INDICaTIONs
In August 2011, FDA approved the use of BV in patients with HL who have failed auto-SCT or who were not suit-able for auto-SCT after at least two polyCT regimens; other approved indi-cation is in patients affected by sALCL after failure of at least one polyCT reg-imen (12). Classic HL is an uncommon lymphoid neoplasm (13) characterized by the presence of CD30+ Hodgkin/Reed Ster-nberg cells in a background of reac-tive inflammatory and accessory cells. After the introduction and improving of polyCT and radiotherapy regimens, early stage classic HL has become a highly curable disease. However, up to 30% of treated patients progresses or relapses to more advanced stages of disease, and up to 50% of these can still be cured by high-dose chemotherapy

303Brentuximab vedotin in CTCL
and auto-SCT. Nevertheless, median overall survival (OS) after auto-SCT fail-ure is approximately less than 2 years, and a considerable percentage of patients are not eligible for auto-SCT. Moreover, survival is lower in elderly patients. Accordingly, these subsets of patients are an important therapeutic challenge (14-17).Systemic ALCL (sALCL) is a rare CD30+ T cell neoplasm, representing about 2-8% of all adult NHL (18); it is histolog-ically characterized by the presence of large, pleomorphic, neoplastic cells which strongly express CD30. Two dis-tinct subtypes of disease differ for ge-netic aspects and clinical course, and may be recognizable by the expression of Anaplastic Lymphoma Kinase (ALK) (19). Usually, sALCL has an aggressive behavior, especially ALK- negative cases (20, 21). Recommended first-line treatment is a multiagent, anthracy-cline-containing regimen, although the use of anthracycline in ALK-nega-tive cases is still debated (22). To date, the treatment of relapsed/refractory cases is unsatisfactory, with overall re-sponses of about 35% with new drugs as Pralatrexate (23). Responses to al-logeneic SCT are also disappointing, especially in patients with refractory and progressive disease at the time of transplantation (24).Two single-arm multicenter phase II clinical trials tested safety and effica-cy of BV at 1.8 mg/kg q3 weeks up to 16 cycles in relapsed or refractory HL (25) and sALCL (26). Both studies had objective response rate as primary ob-jective. In the trial by Younes et al. (25), 102 patients affected by HL refractory or relapsing after auto-SCT failure were enrolled. The objective response rate was 75%, with a median duration of re-
sponse of 6.7 months, and 34% of these patients achieved complete remission (CR). Median duration of CR was 20.5 months. Although the median follow up was only 9 months, they estimated that 12 months OS could be about 88% and that the overall disease control rate (CR + PR + stable disease) should be 96%. The median time to objective response was 5.7 weeks, and the medi-an time to CR was 12 weeks. Concerning safety data, after a medi-an number of 9 cycles, the most com-mon (>10%) treatment-related adverse events (AE) were peripheral sensory neuropathy (42%), nausea (35%), fa-tigue (34%), neutropenia (19%), diar-rhea (18%), pyrexia (14%), vomiting (13%), arthralgia (12%), pruritus (12%), myalgia (11%), peripheral motor neu-ropathy (11%), and alopecia (10%). Fifty five % patients experienced AE of grade 3 or higher. Except for peripheral sensory neu-ropathy, the major severe adverse events (SAE) were laboratory abnor-malities, such as neutropenia, anemia and thrombocytopenia, and no drug AE-related death occurred. Grade 1 and 2 peripheral sensory neuropathy cases were characterized by numb-ness and tingling of fingers and toes, with a median time of onset of about 12 weeks. Otherwise, in more severe cases of peripheral sensory neuropa-thy the median time of onset was 38 weeks. Complete resolution has been observed in 50% patients, with a medi-an time of improvement or resolution of about 13 weeks.In the study published by Pro et al. (26), 58 patients affected by sALCL were treated (42 ALK- positive, 16 ALK- neg-ative). Objective clinical response was obtained in the 86% of patients, with

304 C. Delfino, et al.
a CR rate of 57%. The median dura-tion of response has been of about 12.6 months, and the median progres-sion- free survival was 13.3 months. Al-though the follow up was too short, the estimated 12 months survival rate has been calculated around 70%. About safety, the most common AE were peripheral sensory neuropathy (41%), nausea, fatigue, followed by pyrexia, diarrhea, cutaneous rash, constipation and neutropenia. 60% of patients ex-perienced grade 3 or more SAE, mainly neutropenia, thrombocytopenia, pe-ripheral sensory neuropath and ane-mia. No adverse event was fatal. 24% of patients discontinued the treatment due to SAE, mainly peripheral sensory neuropathy. Again, peripheral sensory neuropathy and neutropenia were the commonest cause of BV dose delay, and so for dose reduction from 1.8 mg/kg to 1.2 mg/kg in 7 patients. No base-line factors were identified to be asso-ciated with an increased incidence of peripheral sensory neuropathy.
◗◗◗ CD30+ CUTaNeOUs T-Cell lyMphOMas
CTCL are a heterogeneous group of extranodal NH lymphomas primarily presenting in the skin without extra cu-taneous involvement at diagnosis. Sev-eral different entities are recognized on the basis of clinical, histological and immunophenotypic aspects (27, 28). CD30 is expressed in transformed my-cosis fungoides (MF) and, by definition, in primary CD30+ cutaneous lymphop-roliferative disorders (LPD).MF neoplastic cells are small-medium sized lymphoid cells with a mature CD3+, CD4+, CD45RO+ and CD8-
memory T-cell phenotype; CD30 may be heavily expressed when transforma-tion into diffuse large cell lymphoma occurs (27). Usually, the transformation to large T-cell lymphoma suggests an aggressive behavior (29), which is not related to CD30 expression.The group of CD30+ LPD includes a spectrum of clinically indolent entities, which are characterized by the fin-ing of large atypical CD30+ lymphoid cells at presentations, indolent clinical course, and relatively frequent spon-taneous regression (27, 28, 30-33). Pri-mary cutaneous anaplastic large cell lymphoma (pcALCL) and lymphoma-toid papulosis (LyP) are at the extrem-ities of the spectrum, which comprises all cases not clearly recognized as pcALCL or as LyP (“borderline” cases) (27, 28, 30).According to the recommendations of the International Society for Cutaneous Lymphomas (ISCL), aadvanced stage MF (IIB-IV) require radiotherapy (con-ventional and/or total skin electron beam irradiation, TSEBI) and/or system-ic therapy, including single agent che-motherapy (monoCT) or polyCT and biologic response modifiers (BRM), as alpha-interferon (IFN-α) and oral bex-arotene. The main indication for Stage III MF is extracorporeal photochemo-terapy (ECP) (34-38). When patients affected by advanced stage MF became refractory or relapse after 2 cycles of systemic treatment, the therapeutic choices are scarce. The possible, advisable recommenda-tion to address patients to transplan-tation procedures in indeed not suit-able for elderly patients, who are the majority. Moreover, polyCT causes side effects that can be problematic in pa-tients with often severe comorbidities.

305Brentuximab vedotin in CTCL
Finally, monoCT can induce remark-able (up to 88%), yet not durable re-sponse rates (38). CD30+ LPD usually have an indolent clinical behaviour, and aggressive treatment is not necessary. According to the EORTC, ISCL, and USCLC con-sensus recommendation, monoCT or polyCT should be administered only in case of pcALCL with extracutane-ous spread (39). Nevertheless, relapses are frequently observed, even after polyCT. Due to its high specificity against CD30, BV is an ideal targeted therapy against CD30+ malignancies. Very good re-sponse rates have been obtained in heavily pretreated relapsed/refracto-
ry LH and sALCL (40). Accordingly, it seems rational to use BV in CTCL that express CD30. In the literature, only a few case reports (3-5) concern BV administration in el-derly patients affected by transformed MF and pcALCL. All patients had expe-rienced many lines of treatment (Table I), because of the refractory/relapsing behavior of the disease or because of SAE onset. The results achieved were very prom-ising: all patients had a rapid response after the first cycle, and CR was usual-ly achieved after 2 cycles of BV. As in patients affected by HL and by sALCL, the main side effect was peripheral neuropathy; nevertheless, the grade
TAbLe 1 • Schematic overview of the published reports about brentuximab vedotin and CTCL (3-6).Studio Pts Disease Prev tp bV dose better
responseAe
Desai A, 2012 (5)
Male, 74-y-o
pcALCL Topical carmustineEBRTTomotherapyMethotrexate
1.8 mg/kg every 3 weeks
CR after 1 cycle
Diarrhea (G1)
Moby K, 2013 (3)
Male, 75-y-o
Male, 87-y-o
Transformed MF
pcALCL
GemcitabineCHOP
Peg-doxonbUVB
1.8 mg/kg every 3 weeks, → 1.2 mg/kg every 3 weeks
1.2 mg/kg every 3 weeks
CR after 2 cycles
CR after 2 cycles
Dysgeusia
-
Broccoli A, 2013 (4)
Fe-male, 73-y-o
Relapsing pcALCL
Carboplatin and paclitaxelRadiotpVincristine
1.8 mg/kg every 4 weeks
CR FatigueIncontinentiaMuscolar weaknessDistal hypoesthesia
Kaffenberg BH,2013 (6)
Male, 60-y-o
pcALCL MTX 1.8 mg/kg every 3 weeks
CR after 2 cycles
Infusion reaction

306 C. Delfino, et al.
of toxicity was low and AE were transi-tory, with a complete recovery. Currently, some ongoing trials are evaluating the use of BV in CTCL (41). A phase 2 study (NCT01352520) is en-rolling patients affected by pcALCL, transformed MF and extensive LyP in order to assess efficacy and toxicity of BV in these cutaneous malignancies. MF and pcALCL patients must have progressed or relapsed after local ra-diation therapy, phototherapy, topical therapy, or have failed systemic ther-apy after at least one single agent or one multi-agent chemotherapy. LyP patients must have scarring or ac-tive lesions (at least 10 new lesion per month), or any number of active lesion on face, hands and feet. BV is adminis-tered at the dose of 1.8 mg/kg every 3 weeks. Estimated primary completion date is June 2015. Preliminary results show activity in MF apparently not re-lated to CD30 expression (results pre-sented at the meetings: “EORTC-CLTF”, Vienna sept 7-9 2012 and “2009-2012. T-cell lymphomas: work in progress” Bologna sept 17-19 2012). Another phase 2 study (NCT01396070) is evaluating the efficacy of BV 1.8 mg/kg every 3 weeks in patients affect-ed by MF/SS, stage IB-IVB, who failed at least one standard systemic ther-apy. Advanced MF and SS respond to systemic therapy but the duration of response is usually short; since BV achieves good response regardless from the percentage of occupancy of CD30-binding sites (11), it would be in-teresting to value BV efficacy in these two malignancies even if neoplastic cells variably express CD30. Enrolled patients were grouped in three sub-groups by CD30 expression levels mea-sured by tissue immunohistochemistry.
Finally, a randomized, open-Label, Phase 3 Trial compares BV to another treatment chosen by physician (meth-otrexate or bexarotene) in patients with CD30+ CTCL (NCT01578499). Pa-tients with MF must have received at least one prior systemic therapy, while patients with pcALCL must have re-ceived prior radiation therapy or at least one prior systemic therapy.
◗◗◗ CONClUsIONs
MF with its variants and CD30+ LPD are the two more frequent groups of CTCL. Their clinical behavior is usually indolent, even if transformed MF has a more aggressive course. Nevertheless, CD30+ LPD commonly recur even after systemic polyCT and sometimes have an extracutaneous spread. The need to find new targeted therapy able to achieved a durable response with low toxicity is urgent, mostly in el-derly patients affected by high comor-bidity.Due to its selectivity, BV is an intrigu-ing therapeutic proposal in selected patients affected by CD30-expressing cutaneous T-cell lymphomas. Case re-ports in the literature describe response already after the first cycle, and CR was usually observed after the second cycle. Finally, it is well tolerated even in elder-ly patients, and its common side ef-fects (as peripheral sensory neuropa-thy, neutropenia, fatigue, nausea) are temporary. It is noteworthy to empha-size that, among the several new drugs tested in the last 5 years, BV showed the most promising and challenging results (42, 43) to be translated in the clinical practice.

307Brentuximab vedotin in CTCL
◗◗◗ ReFeReNCes
1. van de Donk NWCJ, Dhimolea E. Bren-tuximab vedotin. MAbs 2012; 4: 458-65.
2. Ranjana A, Oki Y, Shustov AR, et al. Brentuximab vedotin for relapsed or re-fractory non-Hodgkin lymphoma: pre-liminary results from a phase II study. J Clin Oncol 2012: (Suppl. abstr 8070).
3. Mody K, Wallace JS, Stearns DM, Bowers G, Lacy SR, Levy NB, et al. CD30 posi-tive cutaneous T-cell lymphoma and re-sponse to brentuximab vedotin: 2 illus-trative cases. Clin Lymphoma Myeloma Leuk 2013; 13: 319-23.
4. Broccoli A, Derenzini E, Pellegrini C, Narducci R, Stefani G, Casadei B, et al. Complete response of relapsed system-ic and cutaneous anaplastic large cell lymphoma using brentuximab vedotin: 2 case reports. Clin Lymphoma Myelo-ma Leuk 2013; 13: 493-5.
5. Desai A, Telang GH, Oliszewski AJ. Re-mission of primary cutaneous anaplas-tic large cell lymphoma after a brief course of brentuximab vedotin. Ann Hematol 2013; 92: 567-8.
6. Kaffenberger BH, Winardi FK, Freder-ickson J, Porcu P, Wong HK. Periocular cutaneous anaplastic large cell lym-phoma clearance with brentuximab vedotin. J Clin Aesthet Dermatol 2013; 6: 29-31.
7. Borchmann P, Treml JF, Hansen H, Gott-stein C, Schnell R, Staak O, et al. The hu-man anti-CD30 antibody 5F11 shows in vitro and in vivo activity against malig-nant lymphoma. Blood 2003; 102: 3737-42.
8. Doronina SO, Toki BE, Torgov MY, Men-delsohn BA, Cerveny CG, Chace DF, et al. Development of potent monoclonal antibody auristatin conjugates for can-cer therapy. Nat Biotechnol 2003; 21: 778-84;
9. Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conju-gate. Clin Cancer Res 2010; 16: 888-97.
10 Francisco JA, Cerveny CG, Mey-
er DL, Mixan BJ, Klussman K, Chace DF, et al. cAC10-vcMMAE, an an-ti-CD30-monomethyl auristatin E conju-gate with potent and selective antitu-mor activity. Blood 2003; 102: 1458-65.
11. Fromm JR, McEarchern JA, Kennedy D, Thomas A, Shustov AR, Gopal AK. Clini-cal binding properties, internalization ki-netics, and clinic-pathologic activity of brentuximab vedotin: an antibody-drug conjugate for CD30-positive lymphoid neoplasms. Clin Lymphoma Myeloma Leuk 2012: 12; 280-3.
12. de Claro RA, McGinn K, Kwitkowski V, Bullock J, Khandelwal A, Habtemariam B, et al. U.S. Food and Drug Adminis-tration approval summary: brentux-imab vedotin for the treatment of re-lapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lympho-ma. Clin Cancer Res 2012; 18: 5845-9.
13. Siegel R, Naishadham D, Jemal A. Can-cer statistics, 2012. CA Cancer J Clin 2012; 62: 10-29.
14. Majhail NS, Weisdorf DJ, Defor TE, Mill-er JS, McGlave PB, Slungaard A, et al. Long-term results of autologous stem cell transplantation for primary refracto-ry or relapsed Hodgkin’s lymphoma. Biol Blood Marrow Transplant 2006; 12: 1065-72.
15. Josting A, Müller H, Borchmann P, Baars JW, Metzner B, Döhner H et al. Dose intensity of chemotherapy in patients with relapsed Hodgkin’s lymphoma. J Clin Oncol 2010; 28: 5074-80.
16. Moskowitz AJ, Perales MA, Kewalra-mani T, Yahalom J, Castro-Malaspina H, Zhang Z, et al. Outcomes for patients who fail high dose chemo-radiotherapy and autologous stem cell rescue for re-lapsed and primary refractory Hodgkin lymphoma. Br J Haematol 2009; 146: 158-63.
17. Kuruvilla J, Keating A, Crump M. How I treat relapsed and refractory Hodgkin lymphoma. Blood 2011; 117: 4208-17.
18. Merkel O, Hamacher F, Sifft E, Kenner L, Greil R. European Research Initiative on Anaplastic Large Cell Lymphoma.

308 C. Delfino, et al.
Novel therapeutic options in anaplastic large cell lymphoma: molecular targets and immunological tools. Mol Cancer Ther 2011; 10: 1127-36.
19. Amin HM, Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood 2007; 110: 2259-67.
20. Corradini P, Tarella C, Zallio F, Dodero A, Zanni M, Valagussa P, et al. Longterm follow-up of patients with peripheral T-cell lymphomas treated up-front with high-dose chemotherapy followed by autologous stem cell transplantation. Leukemia 2006; 20: 1533-8.
21. Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, et al. ALK- ana-plastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise spec-ified: report from the International Pe-ripheral T-Cell Lymphoma Project. Blood 2008; 111: 5496-504.
22. Vose J, Armitage J, Weisenburger D. In-ternational peripheral T-cell and natural killer/Tcell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 2008; 26: 4124-30.
23. O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 2011; 29: 1182–9.
24. Le Gouill S, Milpied N, Buzyn A, De Latour RP, Vernant JP, Mohty M, et al. Graft versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Societe Francaise de Greffe de Mo-elle et de Therapie Cellulaire. J Clin On-col 2008; 26: 2264-71.
25. Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lym-phoma. J Clin Oncol 2012; 30: 2183-9.
26. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Bren-tuximab vedotin (SGN-35) in patients
with relapsed or refractory systemic an-aplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 2012; 30: 2190-6.
27. Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EO-RTC classification for cutaneous lym-phomas. Blood 2005; 17: 3768-85.
28. Swerdlow SH, Campo E, Harris NL, et al. (Eds). World Health Organization Classi-fication of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon 2008.
29. Diamandidou E, Colome-Grimmer M, Fayad L, Duvic M, Kurzrock R. Transfor-mation of mycosis fungoides/Sézary syndrome: clinical characteristics and prognosis. Blood 1998; 92: 1150-9.
30. Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30+ lymphoprolif-erative disorders. A proposal for classifi-cation and guidelines for management and treatment. J Am Acad Dermatol 1993; 28: 973-80.
31. Paulli M, Berti E, Rosso R, Boveri E, Kindl S, Klersy C, et al. CD30/Ki-1 positive lymphoproliferative disorders of the skin--clinicopathologic correlation and statistical analysis of 86 cases: a multi-centric study of the European Organi-zation of Research and Treatment of Cancer. Cutaneous Lymphoma Project Group. J Clin Oncol 1995; 13: 1343-54.
32. Bekkenk M, Geelen FA, van Voorst Vad-er PC, Heule F, Geerts ML, van Vloten WA, et al. Primary and secondary cuta-neous CD30-positive lymphoprolifera-tive disorders: long term follow-up data of 219 patients and guidelines for diag-nosis and treatment. A report from the Dutch Cutaneous Lymphoma Group. Blood 2000; 95: 3653-61.
33. Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. CD30+ cutaneous lymphoproliferative disorders: The Stan-ford experience in lymphomatoid pap-ulosis and primary cutaneous anaplas-tic large cell lymphoma. J Am Acad Dermatol 2003; 49: 1049-58.
34. Olsen E, Vonderheid E, Pimpinelli N, Wil-

309Brentuximab vedotin in CTCL
lemze R, Kim Y, Knobler R, et al. Revisions to the staging and classification of my-cosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007; 110(6): 1713-22.
35. Trautinger F, Knobler R, Willemze R, Peris K, Stadler R, Laroche L, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary Syndrome. Eur J Cancer 2006; 42: 1014-30.
36. Olsen EA, Rook AH, Zic J, Kim Y, Porcu P, Querfeld C, et al. Sézary syndrome: immunopathogenesis, literature review of therapeutic options, and recommen-dations for therapy by the United States Cutaneous Lymphoma Consortium (US-CLC). J Am Acad Dermatol 2011; 64: 352-404.
37. Wong HK, Mishra A, Hake T, Porcu P. Evolving insights in the pathogenesis and therapy of cutaneous T-cell lym-
phoma (Mycosis Fungoides and Sézary Syndrome). Br J Haematol 2011; 155: 150-66.
38. Lasingan F, Foss F. Current and emerg-ing treatment strategies for cutaneous T-cell lymphoma. Drugs 2010; 70: 273-86.
39. Kempf W, Pfaltz K, Vermeer MH, Cozzio A, Ortiz-Romero PL, Bagot M, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lympho-matoid papulosis and primary cutane-ous anaplastic large-cell lymphoma. Blood 2011; 118: 4024-35.
40. Deng C, Pan B, O’Connor OA. Brentux-imab vedotin. Clin Cancer Res 2013; 19: 22-7.
41. www.clinicaltrials.gov42. Zinzani PL. Advanced/aggressive CTCL:
improving the efficacy of treatment. G Ital Dermatol Venereol 2012; 147: 563-72.
43. Jain S, Zain J, O’Connor O. Novel thera-peutic agents for cutaneous T-cell lym-phoma. J Haematol Oncol 2012; 5: 24.

Your Clean-Room Solution for Advanced Therapy
More than 100 rooms Misterium have been validated and are operational in both the industrial and hospital settings.More than 20.000 m2 of Clean-Rooms constructed.Meets the classification of rooms (B, C or D) as well as the UNI-EN12128 levels of laboratory containment of microbiological.
� Quick and easy installation� Adaptable according to the measurement required� Conforms to the Ph. Eur. and to G.M.P.
Some References:
Stem Cells Laboratory� Centro Riferimento Oncologico, Aviano� Ospedale San Maurizio, Bolzano� Università degli Studi di Napoli, Federico II� Università degli Studi di Torino, Centro di Criopreservazione
Tissue Bank� Banca dei Tessuti Regione Veneto, Treviso
Cell Factory (AIFA approval on going)
� Istituto Giannina Gaslini, Genova

Riassunto delle caratteristiche del prodotto1. DENOMINAZIONE DEL MEDICINALE: CANCIDAS 50 mg polvere per concentrato per soluzioneper infusione. 2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA: Ciascun flaconcino con-tiene caspofungin 50 mg (come acetato). Eccipienti con effetti noti: ciascun flaconcino da 50 mg con-tiene 35,7 mg di saccarosio. Per l’elenco completo degli eccipienti, vedere paragrafo 6.1. 3. FORMAFARMACEUTICA: Polvere per concentrato per soluzione per infusione endovenosa. Prima della ri-costituzione, la polvere è una polvere compatta di colore bianco-biancastro. 4. INFORMAZIONICLINICHE. 4.1 Indicazioni terapeutiche: • Trattamento della candidiasi invasiva, in pazientiadulti o pediatrici. • Trattamento della aspergillosi invasiva in pazienti adulti o pediatrici refrattari ointolleranti alla terapia con amfotericina B, formulazioni lipidiche di amfotericina B e/o itracona-zolo. Vengono definiti refrattari alla terapia i pazienti con infezioni che progrediscono o non miglio-rano dopo un periodo minimo di 7 giorni di trattamento con dosaggi terapeutici di terapia antifunginaefficace. • Terapia empirica di infezioni fungine presunte (come Candida o Aspergillus) in pazientiadulti o pediatrici neutropenici con febbre. 4.2 Posologia e modo di somministrazione: La te-rapia con caspofungin deve essere iniziata da medici esperti nella gestione delle infezioni fungine in-vasive. Posologia: Pazienti adulti: Un dosaggio singolo da carico di 70 mg deve essere somministratonel primo giorno di trattamento seguito quindi da 50 mg al giorno. In pazienti di peso corporeo su-periore a 80 kg, dopo il dosaggio da carico iniziale di 70 mg, è raccomandato un dosaggio di70 mg/die di caspofungin (vedere paragrafo 5.2). Non è necessario alcun aggiustamento del do-saggio in base a sesso o razza (vedere paragrafo 5.2). Pazienti pediatrici (da 12 mesi a 17 anni):In pazienti pediatrici (da 12 mesi a 17 anni di età) il dosaggio deve essere basato sull’area di su-perficie corporea del paziente (vedere Istruzioni per l’Uso in pazienti Pediatrici, formula di Mostel-ler1). Per tutte le indicazioni, un dosaggio singolo da carico di 70 mg/m2 (non si deve superare undosaggio effettivo di 70 mg) deve essere somministrato nel primo giorno di trattamento, seguitoquindi da 50 mg/m2 al giorno (non si deve superare un dosaggio effettivo di 70 mg al giorno). Seil dosaggio giornaliero di 50 mg/m2 è ben tollerato ma non fornisce un’adeguata risposta clinica, ildosaggio giornaliero può essere aumentato a 70 mg/m2 al giorno (non si deve superare un dosag-gio effettivo giornaliero di 70 mg). La sicurezza e l'efficacia di caspofungin non sono state suffi-cientemente studiate in studi clinici su neonati e lattanti di età inferiore a 12 mesi. Si raccomandacautela quando si trattano pazienti in questa fascia di età. Dati limitati suggeriscono che si può pren-dere in considerazione la terapia con caspofungin al dosaggio di 25 mg/m2 al giorno in neonati elattanti (di età inferiore ai 3 mesi) e al dosaggio di 50 mg/m2 al giorno in giovani bambini (da 3 a11 mesi di età) (vedere paragrafo 5.2). Durata del trattamento: La durata della terapia empirica deveessere basata sulla risposta clinica del paziente. Si deve proseguire con la terapia fino ad un massimodi 72 ore dopo la risoluzione della neutropenia (ANC≥500). I pazienti ai quali viene diagnosticatauna infezione fungina devono essere trattati per un minimo di 14 giorni e il trattamento deve con-tinuare per almeno 7 giorni dopo la risoluzione sia della neutropenia che dei sintomi clinici. La du-rata del trattamento della candidiasi invasiva deve essere basata sulle condizioni della risposta clinicae microbiologica del paziente. A seguito del miglioramento dei segni e sintomi della candidiasi inva-siva e dopo esito negativo delle colture, si può prendere in considerazione un passaggio alla terapiaantifungina orale. In generale, la terapia antifungina deve proseguire per almeno 14 giorni dopo l’ul-tima coltura positiva. La durata del trattamento dell’aspergillosi invasiva va valutata caso per casoe deve essere basata sulla gravità della patologia di base del paziente, sull’entità del miglioramentoclinico dell’immunosoppressione e sulla risposta clinica. In generale, il trattamento deve continuareper almeno 7 giorni dopo la risoluzione dei sintomi. Le informazioni sulla sicurezza per trattamentidi durata superiore a 4 settimane sono limitate. Tuttavia, i dati disponibili suggeriscono che caspo-fungin continua ad essere ben tollerato con cicli di terapia più lunghi (fino a 162 giorni in pazientiadulti e fino a 87 giorni in pazienti pediatrici). Popolazioni speciali: Pazienti anziani: Nei pazientianziani (65 anni di età e oltre), l’area sotto la curva (AUC) è aumentata di circa il 30 %. Non si ri-chiede tuttavia un aggiustamento sistematico del dosaggio. L’esperienza con il trattamento in pa-zienti di età uguale o superiore ai 65 anni è limitata (vedere paragrafo 5.2). Compromissione renale:Non è necessario alcun aggiustamento del dosaggio in base alla compromissione renale (vedere pa-ragrafo 5.2). Compromissione epatica: Per pazienti adulti con compromissione epatica di grado lieve(punteggio di Child-Pugh da 5 a 6), non è necessario alcun aggiustamento del dosaggio. Per i pazientiadulti con compromissione epatica moderata (punteggio di Child-Pugh da 7 a 9), in base a dati di far-macocinetica si raccomanda di somministrare 35 mg/die di caspofungin. Si deve somministrare undosaggio da carico di 70 mg al Giorno 1. Non sono disponibili dati clinici in pazienti adulti con com-promissione epatica grave (punteggio di Child-Pugh maggiore di 9) e in pazienti pediatrici con qual-siasi grado di compromissione epatica (vedere paragrafo 4.4). Co-somministrazione con induttoridegli enzimi metabolici: Dati limitati suggeriscono un aumento del dosaggio giornaliero di caspo-fungin fino a 70 mg, dopo il dosaggio da carico di 70 mg, quando caspofungin viene somministratoin pazienti adulti in concomitanza ad alcuni induttori degli enzimi metabolici (vedere paragrafo 4.5).Quando caspofungin è somministrato a pazienti pediatrici (da 12 mesi a 17 anni di età) in concomi-tanza con gli stessi induttori degli enzimi metabolici (vedere paragrafo 4.5), si deve prendere in con-siderazione un dosaggio di caspofungin di 70 mg/m2 al giorno (non si deve superare un dosaggio
effettivo di 70 mg al giorno). Modo di somministrazione: Dopo ricostituzione e diluizione, la soluzionedeve essere somministrata per infusione endovenosa lenta in circa 1 ora. Per istruzioni sulla ricosti-tuzione vedere paragrafo 6.6. Sono disponibili entrambi i flaconcini da 50 mg e da 70 mg. Caspo-fungin deve essere somministrato come infusione endovenosa singola giornaliera. 4.3Controindicazioni: Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti riportati nelparagrafo 6.1. 4.4 Avvertenze speciali e precauzioni di impiego: Dati limitati suggerisconoche i lieviti non-Candida e le muffe non-Aspergillus meno comuni non sono coperti da caspofungin.L’efficacia di caspofungin nei confronti di questi funghi patogeni non è stata accertata. L’uso conco-mitante di caspofungin con ciclosporina è stato valutato in volontari sani adulti ed in pazienti adulti.Alcuni volontari sani adulti che hanno ricevuto due dosaggi di ciclosporina da 3 mg/kg con caspo-fungin hanno mostrato aumenti transitori della alanina transaminasi (ALT) e aspartato transaminasi(AST) inferiori o uguali a 3 volte il limite superiore della norma (LSN), che si sono risolti con la so-spensione del trattamento. In uno studio retrospettivo su 40 pazienti trattati da 1 a 290 giorni (me-diana 17,5 giorni) con caspofungin e ciclosporina dopo l’immissione in commercio del prodotto, nonsono stati osservati reazioni avverse serie a livello epatico. Questi dati suggeriscono che caspofunginpuò essere utilizzato in pazienti trattati con ciclosporina quando i benefici potenziali sono superioriai rischi potenziali. In caso di somministrazione concomitante di caspofungin e ciclosporina si deveoptare per un attento monitoraggio degli enzimi epatici. In pazienti adulti con compromissione epa-tica lieve e moderata, l’AUC è aumentata di circa il 20 % ed il 75 %, rispettivamente. Nella compro-missione epatica moderata, si raccomanda per gli adulti una riduzione a 35 mg del dosaggiogiornaliero. Non vi sono dati clinici negli adulti con compromissione epatica grave o in pazienti pe-diatrici con qualsiasi grado di compromissione epatica. E’ prevedibile una esposizione maggiore ri-spetto a quella della compromissione epatica moderata e caspofungin deve essere usato con cautelain questi pazienti (vedere paragrafi 4.2 e 5.2). Questo medicinale contiene saccarosio. I pazienti conrari problemi ereditari di intolleranza al fruttosio, o insufficienza di saccarasi-isomaltasi non devonoassumere questo medicinale (vedere paragrafo 2). 4.5 Interazioni con altri medicinali ed altreforme d’interazione: Gli studi in vitro mostrano che caspofungin non è un inibitore di alcun en-zima del sistema del citocromo P450 (CYP). Negli studi clinici, caspofungin non ha indotto il meta-bolismo di altri medicinali mediato dal CYP3A4. Caspofungin non è un substrato della glicoproteinaP ed ha una debole azione di substrato per gli enzimi del citocromo P450. E’ stato tuttavia dimostratoin studi clinici e farmacologici che caspofungin interagisce con altri medicinali (vedere più avanti). Indue studi clinici condotti in soggetti sani adulti, la ciclosporina A (1 dosaggio da 4 mg/kg o 2 dosaggida 3 mg/kg a distanza di 12 ore) ha aumentato la AUC del caspofungin di circa il 35 %. Questi au-menti della AUC sono probabilmente dovuti alla ridotta captazione di caspofungin da parte del fe-gato. Caspofungin non ha aumentato i livelli plasmatici di ciclosporina. Quando caspofungin è statosomministrato insieme alla ciclosporina, sono stati osservati incrementi transitori delle ALT e AST epa-tiche minori o uguali a 3 volte i limiti superiori della norma (LSN), che si sono risolti con l’interru-zione della terapia. In uno studio retrospettivo su 40 pazienti trattati da 1 a 290 giorni (mediana17,5 giorni) con caspofungin e ciclosporina dopo l’immissione in commercio del prodotto, non sonostate osservate reazioni avverse serie a livello epatico (vedere paragrafo 4.4). In caso di sommini-strazione concomitante dei due medicinali si deve optare per un attento monitoraggio degli enzimiepatici. Caspofungin ha ridotto del 26 % la concentrazione minima di tacrolimus in volontari saniadulti. Per i pazienti che ricevono entrambe le terapie si raccomanda il monitoraggio standard delleconcentrazioni ematiche di tacrolimus e gli aggiustamenti appropriati di dosaggio di tacrolimus. Studiclinici effettuati su volontari sani hanno mostrato come la farmacocinetica di caspofungin non vengamodificata a livelli significativi dal punto di vista clinico da itraconazolo, amfotericina B, micofeno-lato, nelfinavir o tacrolimus. Caspofungin non ha influenzato la farmacocinetica di amfotericina B,itraconazolo, rifampicina o micofenolato mofetile. Sebbene i dati di sicurezza siano limitati, non sem-bra siano necessarie precauzioni particolari quando amfotericina B, itraconazolo, nelfinavir o mico-fenolato mofetile vengono somministrati insieme a caspofungin. Rifampicina ha causato un aumentodel 60 % nell’AUC ed un aumento del 170 % nella concentrazione minima di caspofungin al primogiorno di somministrazione concomitante quando la terapia con i due medicinali è stata iniziata con-temporaneamente in volontari sani adulti. I livelli minimi di caspofungin sono diminuiti gradual-mente dopo somministrazione ripetuta. Rifampicina ha avuto un effetto limitato sull’AUC dopodue settimane di somministrazione ma i livelli minimi sono risultati minori del 30 % rispetto ai sog-getti adulti ai quali è stato somministrato caspofungin da solo. Il meccanismo alla base dell’intera-zione potrebbe in qualche modo essere dovuto ad una inibizione iniziale ed alla susseguenteinduzione di proteine di trasporto. Si può prevedere un effetto simile per altri medicinali che indu-cono enzimi metabolici. Dati limitati di farmacocinetica di popolazione indicano che l’uso concomi-tante di caspofungin con gli induttori efavirenz, nevirapina, rifampicina, dasametasone, fenitoina ocarbamazepina, può risultare in una diminuzione dell’AUC di caspofungin. Nel caso di somministra-zione concomitante di induttori degli enzimi metabolici si deve prendere in considerazione in pa-zienti adulti un aumento del dosaggio giornaliero di caspofungin fino a 70 mg, a seguito del dosaggioda carico di 70 mg (vedere paragrafo 4.2). Tutti gli studi di interazione farmacologica sopra descritti,
1 Mosteller RD: Simplified Calculation of Body Surface Area. N Engl J Med 1987 Oct 22;317(17):1098 (letter).

condotti negli adulti, sono stati effettuati con dosaggi giornalieri di caspofungin di 50 o 70 mg. L’in-terazione di dosaggi più alti di caspofungin con altri medicinali non è stata formalmente studiata.In pazienti pediatrici, i risultati derivanti dalle analisi di regressione di dati di farmacocinetica sug-geriscono che la somministrazione concomitante di desametasone con caspofungin può causare ri-duzioni clinicamente significative delle concentrazioni di valle del caspofungin. Questa constatazionepuò indicare che i pazienti pediatrici avranno con gli induttori riduzioni simili a quelle viste negliadulti. Quando caspofungin viene somministrato in pazienti pediatrici (da 12 mesi a 17 anni di età)in concomitanza a induttori della clearance del medicinale, quali rifampicina, efavirenz, nevirapina,fenitoina, desametasone, o carbamazepina, si deve prendere in considerazione un dosaggio di ca-spofungin di 70 mg/m2 al giorno (non si deve superare un dosaggio effettivo di 70 mg al giorno).4.6 Fertilità, gravidanza e allattamento: Gravidanza: I dati sull’uso di caspofungin nelle donnein gravidanza non sono disponibili o sono limitati. Caspofungin non deve essere usato durante lagravidanza a meno che non sia chiaramente necessario. Studi sull'animale hanno dimostrato sviluppodi tossicità (vedere paragrafo 5.3). Negli studi sull’animale è stato dimostrato che caspofungin at-traversa la barriera placentare. Allattamento: Non è noto se caspofungin sia escreto nel latte umano.I dati farmacodinamici/tossicologici disponibili negli animali hanno dimostrato che caspofungin èescreto nel latte. Le donne che assumono caspofungin non devono allattare. Fertilità: Per caspofun-gin, non ci sono stati effetti sulla fertilità in studi condotti in ratti maschio e femmina (vedere para-grafo 5.3). Non ci sono dati clinici per caspofungin per valutare il suo impatto sulla fertilità. 4.7Effetti sulla capacità di guidare veicoli e sull’uso di macchinari: Non sono stati effettuati studisulla capacità di guidare veicoli e sull’uso di macchinari. 4.8 Effetti indesiderati: Sono stati riportatipossibili sintomi mediati dal rilascio di istamina incluse segnalazioni di rash, gonfiore del viso, an-gioedema, prurito, sensazione di calore o broncospasmo. E’ stata segnalata anafilassi durante lasomministrazione di caspofungin. In pazienti con aspergillosi invasiva sono stati riportati inoltreedema polmonare, sindrome da distress respiratorio dell’adulto (ARDS) ed infiltrati radiografici. Pa-zienti adulti: Negli studi clinici, 1.865 persone adulte sono state trattate con dosaggi singoli o multi-pli di caspofungin: 564 pazienti neutropenici con febbre (studio sulla terapia empirica), 382 pazienticon candidiasi invasiva, 228 pazienti con aspergillosi invasiva, 297 pazienti con infezioni localizzateda Candida,e 394 persone arruolate negli studi clinici di fase I. Nello studio sulla terapia empirica i pa-zienti erano stati trattati con chemioterapia per neoplasia maligna o erano stati sottoposti a trapiantocon cellule ematopoietiche staminali (inclusi 39 trapianti allogenici). Negli studi con i pazienti con in-fezioni documentate da Candida, la maggior parte dei pazienti con infezioni invasive da Candidaavevano serie condizioni mediche di base (ad es.: ematopatie maligne od altre condizioni oncologi-che, recenti importanti interventi chirurgici, HIV), tali da richiedere la somministrazione concomi-tante di diversi medicinali. I pazienti nello studio non comparativo sull’Aspergillus avevano spessogravi patologie di base predisponenti (ad es.: trapianto di midollo o di cellule staminali periferiche,ematopatie maligne, tumori solidi o trapianti d’organo), tali da richiedere la somministrazione con-comitante di diversi medicinali. La flebite è stata una reazione avversa frequentemente riportata alsito di iniezione in tutte le popolazioni di pazienti. Altre reazioni localizzate sono state eritema, do-lore/dolorabilità, prurito, secrezione e sensazione di bruciore. Le anormalità cliniche e di laborato-rio riportate nel totale degli adulti trattati con caspofungin (in tutto 1.780 pazienti) sono statetipicamente lievi ed hanno raramente condotto alla interruzione della terapia. Sono state segnalatele seguenti reazioni avverse: [Molto comune (≥1/10), Comune (≥1/100, <1/10), Non comune(≥1/1.000, <1/100)]. Patologie del sistema emolinfopoietico: Comune: diminuzione dellaemoglobinemia, diminuzione dell’ematocrito, diminuzione della conta dei leucociti. Non comune:anemia, trombocitopenia, coagulopatia, leucopenia, aumento della conta degli eosinofili, diminu-zione della conta delle piastrine, aumento della conta delle piastrine, diminuzione della conta dei lin-fociti, aumento della conta dei leucociti, diminuzione della conta dei neutrofili. Disturbi delmetabolismo e della nutrizione: Comune: ipokalemia. Non comune: sovraccarico di fluidi, ipo-magnesemia, anoressia, squilibrio elettrolitico, iperglicemia, ipocalcemia, acidosi metabolica. Di-sturbi psichiatrici: Non comune: ansia, disorientamento, insonnia. Patologie del sistemanervoso: Comune: cefalea. Non comune: capogiro, disgeusia, parestesia, sonnolenza, tremore, ipoe-stesia. Patologie dell’occhio: Non comune: ittero oculare, visione offuscata, edema della palpe-bra, aumento della lacrimazione. Patologie cardiache: Non comune: palpitazioni, tachicardia,aritmia, fibrillazione atriale, insufficienza cardiaca congestizia. Patologie vascolari: Comune: fle-bite. Non comune: tromboflebite, arrossamento, vampata, ipertensione, ipotensione. Patologie re-spiratorie, toraciche e mediastiniche: Comune: dispnea. Non comune: congestione nasale, dolorefaringolaringeale, tachipnea, broncospasmo, tosse, dispnea parossistica notturna, ipossia, rantoli, si-bili. Patologie gastrointestinali: Comune: nausea, diarrea, vomito. Non comune: dolore addo-minale, dolore nel tratto superiore dell’addome, secchezza delle fauci, dispepsia, disturbo allostomaco, distensione addominale, ascite, costipazione, disfagia, flatulenza. Patologie epatobi-liari: Comune: incremento degli indicatori di funzionalità epatica (alanina aminotrasferasi, aspartatoaminotrasferasi, fosfatasi alcalina, bilirubina coniugata, bilirubina ematica). Non comune: colestasi,epatomegalia, iperbilirubinemia, ittero, alterata funzione epatica, epatotossicità, disturbi del fegato.Patologie della cute e del tessuto sottocutaneo: Comune: rash, prurito, eritema, iperidrosi.Non comune: eritema multiforme, rash maculare, rash maculo-papulare, rash pruriginoso, orticaria,dermatite allergica, prurito generalizzato, rash eritematoso, rash generalizzato, rash morbilliforme,lesione cutanea. Patologie del sistema muscolo scheletrico e del tessuto connettivo: Co-mune: artralgia. Non comune: mal di schiena, dolore alle estremità, dolore osseo, debolezza mu-scolare, mialgia. Patologie renali e urinarie: Non comune: insufficienza renale, insufficienzarenale acuta. Patologie sistemiche e condizioni relative alla sede di somministrazione: Co-mune: piressia, brividi, prurito nella sede di infusione. Non comune: dolore, dolore nella sede del ca-tetere, stanchezza, sensazione di freddo, sensazione di caldo, eritema nella sede di infusione,indurimento nella sede di infusione, dolore nella sede di infusione, gonfiore nella sede di infusione,flebite nella sede di iniezione, edema periferico, iperestesia, disturbo toracico, dolore toracico, edemadel volto, sensazione di variazione della temperatura corporea, indurimento, stravaso nella sede diinfusione, irritazione nella sede di infusione, flebite nella sede di infusione, rash nella sede di infu-sione, orticaria nella sede di infusione, eritema nella sede di iniezione, edema nella sede di inie-zione, dolore nella sede di iniezione, gonfiore nella sede di iniezione, malessere, edema. Esamidiagnostici: Comune: diminuzione della kaliemia, diminuzione dell’albuminemia. Non comune: au-mento della creatininemia, eritrociti nelle urine, diminuzione delle proteine totali, proteine nelle
urine, tempo di protrombina prolungato, tempo di protrombina ridotto, diminuzione della sodiemia,aumento della sodiemia, diminuzione della calcemia, aumento della calcemia, diminuzione della clo-remia, aumento della glicemia, riduzione della magnesemia, riduzione della fosforemia, aumentodella fosforemia, aumento della uremia, aumento della gamma-glutamiltrasferasi, tempo di trom-boplastina parziale attivata prolungato, diminuzione dei bicarbonati ematici, aumento della cloremia,aumento della kaliemia, aumento della pressione sanguigna, diminuzione dell’acido urico ematico,ematuria, rumori respiratori anormali, diminuzione dell’anidride carbonica, aumenti dei livelli dei me-dicinali immunosoppressori, aumento della INR, cilindri urinari, leucociti nelle urine, aumento del pHurinario. Caspofungin è stato valutato al dosaggio di 150 mg al giorno (fino a 51 giorni) in 100 pa-zienti adulti (vedere paragrafo 5.1). Lo studio ha confrontato caspofungin al dosaggio di 50 mg algiorno (dopo un dosaggio da carico da 70 mg al giorno 1) versus 150 mg al giorno nel trattamentodella candidiasi invasiva. In questo gruppo di pazienti il profilo di sicurezza di caspofungin a questodosaggio più alto è risultato generalmente simile a quello dei pazienti che ricevevano caspofunginal dosaggio di 50 mg al giorno. La proporzione di pazienti con una reazione avversa seria farmaco-correlata o con una reazione avversa farmaco-correlata che ha portato all’interruzione della terapiacon caspofungin è stata comparabile nei 2 gruppi di trattamento. Pazienti pediatrici: I dati derivantida 5 studi clinici completati in 171 pazienti pediatrici suggeriscono che l’incidenza globale di espe-rienze avverse cliniche (26,3%; 95% IC -19,9, 33,6) non è peggiore rispetto a quella riportata negliadulti trattati con caspofungin (43,1%; 95% IC -40,0, 46,2). Tuttavia, i pazienti pediatrici probabil-mente hanno un profilo di eventi avversi differente rispetto a quello dei pazienti adulti. Le espe-rienze avverse cliniche più comuni correlate con il medicinale riportate nei pazienti pediatrici trattaticon caspofungin sono state piressia (11,7%), eruzione cutanea (4,7%) e cefalea (2,9%). Sono statesegnalate le seguenti reazioni avverse: [Molto comune (≥1/10), Comune (≥1/100, <1/10)]. Pa-tologie del sistema emolinfopoietico: Comune: eosinofilia. Patologie del sistema nervoso:Comune: cefalea. Patologie cardiache: Comune: tachicardia. Patologie vascolari: Comune: ar-rossamento, ipotensione. Patologie epatobiliari: Comune: incremento degli enzimi di funziona-lità epatica (AST, ALT). Patologie della cute e del tessuto sottocutaneo: Comune: rash, prurito.Patologie sistemiche e condizioni relative alla sede di somministrazione: Molto comune:febbre. Comune: brividi, dolore nel sito di inserzione del catetere. Esami diagnostici: Comune: di-minuzione della kaliemia, ipomagniesiemia, aumento della glicemia, riduzione della fosforemia, edaumento della fosforemia. Come nei pazienti adulti anche nei pazienti pediatrici sono stati riportatisintomi mediati dal rilascio di istamina. Esperienza post-marketing: Sono state segnalate le seguentireazioni avverse post-marketing: Patologie epatobiliari: Disfunzione epatica. Patologie siste-miche e condizioni relative alla sede di somministrazione: Gonfiore ed edema periferico.Esami diagnostici: Ipercalcemia. 4.9 Sovradosaggio: E’ stata segnalata la somministrazione ac-cidentale di caspofungin fino a 400 mg in un giorno. Tali evenienze non hanno dato luogo a reazioniavverse clinicamente significative. Caspofungin non è dializzabile. 5. PROPRIETÀ FARMACO-LOGICHE. 5.1 Proprietà farmacodinamiche: Categoria farmacoterapeutica: antimicotici peruso sistemico, codice ATC: J 02 AX 04. Caspofungin acetato è un lipopeptide semisintetico (echino-candina) sintetizzato da un prodotto di fermentazione di Glarea lozoyensis. Caspofungin acetato ini-bisce la sintesi del beta (1,3) - D - glucano, un componente essenziale della parete cellulare di moltifunghi filamentosi e lieviti. Il beta (1,3) - D - glucano non è presente nelle cellule dei mammiferi. L’at-tività fungicida di caspofungin è stata dimostrata contro i lieviti del genere Candida. studi in vitro edin vivo dimostrano che l’esposizione di Aspergillus a caspofungin dà luogo a lisi e morte delle estre-mità delle ife apicali e dei punti di ramificazione dove hanno luogo crescita e divisione cellulare. Ca-spofungin possiede attività in vitro nei confronti delle specie di Aspergillus (Aspergillus fumigatus[N = 75], Aspergillus flavus [N = 111], Aspergillus niger [N = 31], Aspergillus nidulans [N = 8] eAspergillus terreus [N = 52]) e Aspergillus Candidus [N=3]. Caspofungin possiede inoltre attivitàin vitro nei confronti delle specie di Candida (Candida albicans [N = 1032], Candida dubliniensis[N = 100], Candida glabrata [N = 151], Candida guilliermondii [N = 67], Candida kefyr [N = 62],Candida krusei [N = 147], Candida lipolytica [N = 20], Candida lusitaniae [N = 80], Candida pa-rapsilosis [N = 215]), Candida rugosa [N = 1] e Candida tropicalis [N = 258], inclusi gli isolati conmutazioni di trasporto con resistenza multipla e quelli con resistenza acquisita o intrinseca a fluco-nazolo, amfotericina B e 5-flucitosina. I test di sensibilità sono stati eseguiti in base a modifiche adentrambi i metodi M38-A2 (per le specie di Aspergillus) e M27-A3 (per le specie di Candida) del Cli-nical and Laboratory Standards Institute (CLSI, precedentemente conosciuto come National Commit-tee for Clinical Laboratory Standards [(NCCLS]). Tecniche standardizzate per testare la sensibilitàsono state stabilite per i lieviti dall’EUCAST. Tuttavia, non sono stati approvati dall’EUCAST break-point interpretativi per caspofungin. Isolati di Candida con ridotta sensibilità al caspofungin sono statiidentificati in un piccolo numero di pazienti durante il trattamento (MIC per caspofungin >2 mg/l (au-menti della MIC da 4 a 30 volte) sono state riportate usando tecniche di prova standardizzate per laMIC approvate dal CLSI). Il meccanismo della resistenza identificato consiste in mutazioni nel geneFKS1/FKS2. Questi casi sono stati associati con esiti clinici scarsi. È stato identificato lo sviluppo dellaresistenza in vitro a caspofungin da parte di Aspergillus sp.. Nel corso di limitate esperienze cliniche,è stata osservata resistenza a caspofungin in pazienti con aspergillosi invasiva. Il meccanismo dellaresistenza non è stato determinato. L’incidenza della resistenza a caspofungin da parte di vari isolaticlinici di Candida ed Aspergillus è rara. Candidiasi invasiva in pazienti Adulti: sono stati arruolati due-centotrentanove pazienti in uno studio iniziale volto a confrontare caspofungin ed amfotericina B neltrattamento della candidiasi invasiva. Ventiquattro pazienti erano affetti da neutropenia. Le diagnosipiù frequenti sono state di infezioni del circolo ematico (candidemia) (77%, n=186) e di peritoniteda Candida (8%, n=19); i pazienti con endocardite, osteomielite o meningite da Candida sono statiesclusi dallo studio. Caspofungin è stato somministrato al dosaggio di 50 mg in monosomministra-zione giornaliera dopo un dosaggio da carico di 70 mg, mentre amfotericina B è stata somministrataal dosaggio di 0,6 -– 0,7 mg/kg/die in pazienti non neutropenici o al dosaggio di 0,7-1,0 mg/kg/diein pazienti neutropenici. La durata media della terapia endovenosa è stata di 11,9 giorni, con unavariabilità da 1 a 28 giorni. Per considerare una risposta come favorevole sono stati richiesti sia larisoluzione dei sintomi che la scomparsa dell’infezione da Candida dal punto di vista microbiologico.Duecentoventiquattro pazienti sono stati inclusi nell’analisi primaria sull’efficacia (analisi MITT) dellarisposta alla fine della terapia endovenosa; i tassi di risposta favorevole per il trattamento della can-didiasi invasiva fra caspofungin (73 % [80/109]) e amfotericina B (62 % [71/115]) [differenza per-centuale 12,7 (95,6 % IC -0,7, 26,0)] sono risultati paragonabili. Fra i pazienti con candidemia, i tassi

di risposta favorevole alla fine della terapia endovenosa in studio sono stati paragonabili fra caspo-fungin(72 % [66/92]) e amfotericina B (63 % [59/94]) nell’analisi primaria di efficacia (analisiMITT) [differenza percentuale 10,0 (95,0 % IC –4,5, 24,5)]. I dati provenienti da pazienti con sitodi infezione non ematologico sono stati più limitati. I tassi di risposta faverevole nei pazienti neu-tropenici sono stati 7/14 (50 %) nel gruppo caspofungin e 4/10 (40%) nel gruppo amfotericina B.Tali dati limitati sono suffragati dall’esito dello studio sulla terapia empirica. In un secondo stu-dio, pazienti con candidiasi invasiva hanno ricevuto dosaggi di 50 mg di caspofungin in monosom-ministrazione giornaliera (dopo un dosaggio da carico di 70 mg al Giorno 1) o dosaggi di 150 mgdi caspofungin in monosomministrazione giornaliera (vedere paragrafo 4.8). In questo studio, il do-saggio di caspofungin è stato somministrato in 2 ore (invece della abituale somministrazione in 1 ora).Da questo studio sono stati esclusi pazienti con endocardite, meningite o osteomielite da Candida. Poi-ché questo era uno studio di terapia primaria, sono stati esclusi anche i pazienti che erano refrattariad una precedente terapia con medicinali antifungini. È stato anche limitato (8,0%) il numero di pa-zienti neutropenici arruolati in questo studio. L’efficacia era un endpoint secondario in questo studio.Nell’analisi dell’efficacia sono stati inclusi pazienti che avevano soddisfatto i criteri di inclusione e cheavevano ricevuto uno o più dosaggi di caspofungin. Alla fine della terapia con caspofungin i tassi dirisposta favorevole globale sono stati simili nei 2 gruppi di trattamento: 72 % (73/102) e 78 %(74/95) rispettivamente per i gruppi di trattamento caspofungin 50-mg e 150-mg (differenza 6,3 %[95 % IC-5,9, 18,4]). Aspergillosi invasiva in pazienti Adulti: sono stati arruolati in uno studio noncomparativo in aperto 69 pazienti adulti (età compresa tra 18 e 80 anni) con aspergillosi invasivaper valutare la sicurezza, la tollerabilità e l’efficacia di caspofungin. I pazienti arruolati erano o re-frattari (malattia in evoluzione o mancato miglioramento con altre terapie antifungine sommini-strate per almeno 7 giorni) (84 % dei pazienti arruolati) o intolleranti (16 % dei pazienti arruolati)ad altre terapie antifungine standard. La maggior parte dei pazienti presentava patologie di base(emopatie maligne [N = 24], trapianto allogenico del midollo o trapianto di cellule staminali[N = 18], trapianto d’organo [N = 8], tumore solido [N = 3] o altre patologie [N = 10]). Per la dia-gnosi di aspergillosi invasiva e per la risposta alla terapia (per una risposta favorevole era necessa-rio un miglioramento clinicamente significativo sia nelle immagini radiografiche che nei segni e neisintomi) sono state usate definizioni rigorose, formulate seguendo le indicazioni del Mycoses StudyGroup Criteria. La durata media della terapia è stata di 33,7 giorni, con una variabilità fra 1 e162 giorni. Un comitato indipendente di specialisti ha valutato che il 41 % (26/63) dei pazienti aiquali era stato somministrato almeno un dosaggio di caspofungin ha risposto in modo favorevole.Tra i pazienti ai quali era stato somministrato caspofungin per più di 7 giorni, il 50 % (26/52) haavuto una risposta favorevole. I tassi di risposta favorevole per i pazienti refrattari od intolleranti alleterapie precedenti sono stati del 36 % (19/53) e del 70 % (7/10), rispettivamente. Sebbene in 5 pa-zienti arruolati come refrattari i dosaggi delle terapie antifungine precedenti fossero inferiori a quellispesso somministrati per il trattamento dell’aspergillosi invasiva, il tasso di risposte favorevoli durantela terapia con caspofungin in questi pazienti è risultato simile a quello osservato negli altri pazientirefrattari (2/5 vs 17/48, rispettivamente). I tassi di risposta favorevole fra i pazienti con malattiapolmonare ed extrapolmonare sono stati del 47 % (21/45) e del 28 % (5/18), rispettivamente. Frai pazienti con malattia extrapolmonare, hanno avuto una risposta favorevole anche 2 su 8 pazienticon coinvolgimento del SNC certo, probabile o possibile. Terapia empirica in pazienti adulti neutro-penici con febbre: un totale di 1.111 pazienti con febbre persistente e neutropenia sono stati arruo-lati in uno studio clinico e trattati o con caspofungin 50 mg in monosomministrazione giornaliera dopoun dosaggio da carico di 70 mg o con amfotericina B liposomiale 3,0 mg/kg/die. I pazienti eleggi-bili erano stati trattati con chemioterapia per neoplasie maligne o erano stati sottoposti a trapiantocon cellule staminali ematopoietiche, presentavano neutropenia (<500 cellule/mm3 per 96 ore) efebbre (>38,0°C) che non aveva risposto a ≥96 ore di trattamento antibatterico parenterale. I pa-zienti dovevano essere trattati fino ad un massimo di 72 ore dopo la risoluzione della neutropenia,per una durata massima di 28 giorni. I pazienti con infezione fungina documentata potevano tutta-via essere trattati più a lungo. In caso di buona tolleranza al medicinale ma di persistenza della feb-bre e di deterioramento delle condizioni cliniche dopo 5 giorni di terapia, il dosaggio del medicinalein studio poteva essere aumentato a 70 mg/die di caspofungin (13,3% dei pazienti trattati) o a5,0 mg/kg/die di amfotericina B liposomiale (14,3% dei pazienti trattati). Nell’analisi di efficacia pri-maria modificata per intenzione di trattamento (MITT) sulla risposta favorevole globale erano inclusi1095 pazienti; caspofungin (33,9 %) è risultato efficace quanto amfotericina B liposomiale (33,7 %)[% differenza 0,2 (95,2 % IC –5,6, 6,0)].Per una risposta globale favorevole veniva richiesto disoddisfare i 5 seguenti criteri: (1) trattamento soddisfacente di qualsiasi infezione fungina al basale(caspofungin 51,9 % [14/27], amfotericina B liposomiale 25,9 % [7/27]), (2) assenza di nuove in-fezioni fungine nel corso della somministrazione del medicinale in studio o entro 7 giorni dal com-pletamento della terapia (caspofungin 94,8 % [527/556], amfotericina B liposomiale 95,5 %[515/539]), (3) sopravvivenza per 7 giorni dopo il completamento della terapia in studio (caspo-fungin 92,6 % [515/556], amfotericina B liposomiale 89,2 % [481/539]), (4) assenza di interru-zioni dalla terapia in studio a causa di tossicità correlata al medicinale o di mancanza di efficacia(caspofungin 89,7 % [499/556], amfotericina B liposomiale 85,5 % [461/539]), e (5) risoluzionedella febbre durante il periodo di neutropenia (caspofungin 41,2% [229/556], amfotericina B lipo-somiale 41,4 % [223/539]). I tassi di risposta a caspofungin e amfotericina B liposomiale per le in-fezioni al basale causate da Aspergillus sp. sono state, rispettivamente, 41,7% (5/12) e 8,3% (1/12),e per Candida sp. sono state 66,7 % (8/12) e 41,7 % (5/12). Nei pazienti nel gruppo caspofunginsi sono verificate nuove infezioni fungine dovute ai seguenti lieviti e muffe non comuni: Trichospo-ron sp. (1), Fusarium sp. (1), Mucor sp. (1), e Rhizopus sp. (1). Pazienti pediatrici: La sicurezzae l’efficacia di caspofungin sono state valutate in pazienti pediatrici di età compresa tra 3 mesi e17 anni in due studi clinici prospettici, multicentrici. Il disegno dello studio, i criteri diagnostici, ed icriteri per la valutazione dell’efficacia erano simili a quelli dei corrispondenti studi effettuati nei pa-zienti adulti (vedere paragrafo 5.1). Il primo studio, in cui sono stati arruolati 82 pazienti di etàcompresa tra i 2 e i 17 anni, era uno studio randomizzato, doppio cieco volto a confrontare caspo-fungin [50 mg/m2 EV al giorno susseguente ad un dosaggio da carico di 70 mg/m2 al Giorno 1 (nonera consentito superare i 70 mg al giorno)] e amfotericina B liposomale (3 mg/kg EV al giorno) inuno schema di trattamento 2:1 (56 pazienti trattati con caspofungin e 26 con amfotericina B lipo-somale) come terapia empirica in pazienti pediatrici con persistente febbre e neutropenia. I tassi glo-bali di successo terapeutico in base ai risultati della analisi MITT, aggiustata per strati di rischio, sono
stati i seguenti: 46,6 % (26/56) per caspofungin e 32,2% (8/25) per amfotericina B liposomale. Ilsecondo studio era prospettico, in aperto, non comparativo atto a valutare la sicurezza e l’efficaciadi caspofungin in pazienti pediatrici (di età compresa tra 3 mesi e 17 anni) con candidiasi invasiva,candidiasi esofagea e aspergillosi invasiva (come terapia di salvataggio). Sono stati arruolati qua-rantanove pazienti che sono stati trattati con caspofungin 50 mg/m2 EV una volta al giorno dopo undosaggio da carico di 70 mg/m2 al Giorno 1 (non era consentito superare i 70 mg al giorno), di que-sti 48 sono stati inclusi nell’analisi MITT. Di questi pazienti 37 avevano candidiasi invasiva, 10 ave-vano aspergillosi invasiva e 1 paziente aveva candidiasi esofagea. Il tasso di risposta favorevole,per indicazione, alla fine della terapia con caspofungin è stato nell’analisi MITT il seguente: 81 %(30/37) nella candidiasi invasiva, 50 % (5/10) nella aspergillosi invasiva e 100 % (1/1) nella can-didiasi esofagea. 5.2 Proprietà farmacocinetiche: Distribuzione: Caspofungin si lega ampia-mente all’albumina. La frazione plasmatica non legata di caspofungin varia dal 3,5% nei volontarisani al 7,6% in pazienti con candidiasi invasiva. La distribuzione gioca un ruolo prominente nellafarmacocinetica plasmatica di caspofungin ed è la fase critica di passaggio in entrambe le fasi di di-sposizione alfa e beta. La distribuzione tissutale ha raggiunto il picco da 1,5 a 2 giorni dopo il do-saggio quando il 92 % del dosaggio era distribuito all’interno dei tessuti. E’ probabile che solo unapiccola parte del caspofungin assorbito dai tessuti torni successivamente nel plasma come compostoimmodificato. Di conseguenza, l’eliminazione avviene in assenza di un equilibrio di distribuzione eduna stima reale del volume di distribuzione di caspofungin è attualmente impossibile da ottenere. Bio-trasformazione: Caspofungin va incontro ad un processo spontaneo di degradazione in un compostoad anello aperto. Il metabolismo successivo comprende idrolisi peptidica ed N-acetilazione. Due pro-dotti intermedi, formati durante la degradazione di caspofungin a tale composto ad anello aperto,formano addotti covalenti con le proteine plasmatiche determinando un legame di basso livello, ir-reversibile, con le proteine plasmatiche. Studi in vitro mostrano che caspofungin non è un inibitoredegli enzimi 1A2, 2A6, 2C9, 2C19, 2D6 o 3A4 del citocromo P450. Negli studi clinici il caspofunginnon ha indotto o inibito il metabolismo di altri medicinali mediato dal citocromo CYP3A4. Caspofun-gin non è un substrato della glicoproteina P e ha scarsa attività di substrato per gli enzimi del cito-cromo P450. Eliminazione ed escrezione: L’eliminazione di caspofungin dal plasma è lenta con unaclearance di 10-12 ml/min. Le concentrazioni plasmatiche di caspofungin diminuiscono secondo unandamento polifasico a seguito di infusioni endovenose singole della durata di 1 ora. Una breve fasealfa si verifica immediatamente dopo l’infusione endovenosa, seguita da una fase beta con emivitadalle 9 alle 11 ore. Si verifica anche una fase gamma addizionale con una emivita di 45 ore. Il mec-canismo dominante sulla clearance plasmatica è rappresentato dalla distribuzione piuttosto che dallaescrezione o dalla biotrasformazione. E’ stato recuperato all’incirca il 75 % del dosaggio radioattivonell’arco di 27 giorni: 41 % nelle urine e 34 % nelle feci. Vi è una bassa escrezione o biotrasforma-zione di caspofungin durante le prime 30 ore dopo la somministrazione. L’escrezione è lenta e l’emi-vita terminale della radioattività è stata da 12 a 15 giorni. Una piccola quantità di caspofungin èescreta immodificata nelle urine (circa 1,4 % del dosaggio). Caspofungin mostra una farmacocine-tica moderata non lineare con incremento dell’accumulo all’aumentare del dosaggio e una dose-di-pendenza nel tempo fino al raggiungimento dello stato di equilibrio con somministrazione a dosaggiomultiplo. Popolazioni speciali: E’ stata osservata una maggiore esposizione a caspofungin in pazientiadulti con compromissione renale e lieve compromissione epatica, nelle donne, e negli anziani. Ge-neralmente l’aumento è stato limitato e non ampio abbastanza da giustificare un aggiustamento deldosaggio. In pazienti adulti con moderata compromissione epatica o in pazienti di maggiore peso cor-poreo, può essere necessario un aggiustamento del dosaggio (vedere sotto). Peso: è stato riscon-trato che il peso influenza la farmacocinetica di caspofungin nell’analisi della farmacocinetica dipopolazione nei pazienti adulti affetti da candidiasi. Le concentrazioni plasmatiche diminuiscono al-l’aumentare del peso. Si prevede che l’esposizione media in un paziente adulto di 80 kg di peso siaminore del 23 % circa rispetto a quella di un paziente adulto di 60 kg (vedere paragrafo 4.2). Com-promissione epatica: in pazienti adulti con compromissione epatica lieve e moderata, l’AUC è au-mentata rispettivamente del 20 e del 75 %. Non vi sono dati clinici in pazienti adulti concompromissione epatica grave e in pazienti pediatrici con qualsiasi grado di compromissione epatica.In uno studio a dosaggio multiplo, è stato dimostrato che una riduzione del dosaggio giornaliero a35 mg in pazienti adulti con compromissione epatica moderata determina una AUC simile a quellaottenuta in soggetti adulti con funzione epatica normale ai quali viene somministrato un regime stan-dard (vedere paragrafo 4.2). Compromissione renale: in uno studio clinico con singoli dosaggi da70 mg, la farmacocinetica di caspofungin è risultata simile nei volontari adulti con compromissionerenale lieve (clearance della creatinina 50-80 ml/min) e nei controlli. La compromissione renalemoderata (clearance della creatinina da 31 a 49 ml/min), avanzata (clearance della creatinina da5 a 30 ml/min) e allo stadio terminale (clearance della creatinina <10ml/min e dialisi-dipendenza)ha aumentato moderatamente le concentrazioni plasmatiche di caspofungin a seguito di sommini-strazione di dosaggio singolo (AUC da 30 a 49 %). Tuttavia, per i pazienti adulti con candidiasi in-vasiva, candidiasi esofagea o aspergillosi invasiva ai quali sono stati somministrati dosaggi giornalierimultipli di caspofungin 50 mg, la compromissione da lieve ad avanzata della funzione renale nonha avuto un effetto significativo sulle concentrazioni di caspofungin. Non è necessario aggiustamentodel dosaggio nei pazienti con compromissione renale. Caspofungin non è dializzabile, non è pertantorichiesto un dosaggio supplementare dopo emodialisi. Sesso: le concentrazioni plasmatiche di caspo-fungin sono state in media più alte del 17-38 % nelle donne rispetto agli uomini. Anziani: un mode-sto aumento dell’AUC (28 %) e del C24h (32 %) è stato osservato negli anziani di sesso maschilerispetto agli uomini giovani. Nei pazienti trattati con terapia empirica o affetti da candidiasi invasiva,è stato osservato un simile effetto modesto dell’età negli anziani rispetto ai giovani. Razza: i dati difarmacocinetica dei pazienti indicano che non sono state osservate differenze clinicamente signifi-cative nella farmacocinetica di caspofungin fra caucasici, neri, ispanici e meticci. Pazienti pediatrici:In adolescenti (età compresa tra 12 e 17 anni) trattati con caspofungin a 50 mg/m2 al giorno (al mas-simo 70 mg al giorno), la AUC0-24h plasmatica di caspofungin è stata generalmente confrontabilecon quella riscontrata in adulti trattati con caspofungin a 50 mg al giorno.Tutti gli adolescenti hannoricevuto dosaggi > 50 mg al giorno, e, infatti, 6 su 8 hanno ricevuto il massimo dosaggio di70 mg/die. Le concentrazioni plasmatiche di caspofungin in questi adolescenti erano ridotte rispettoa quelle di adulti trattati con 70 mg al giorno, il dosaggio somministrato con maggiore frequenza agliadolescenti. In bambini (età compresa tra 2 e 11 anni) trattati con caspofungin 50 mg/m2 al giorno(al massimo 70 mg al giorno), la AUC0-24h plasmatica di caspofungin dopo dosaggi multipli è stata

confrontabile con quella riscontrata in adulti trattati con caspofungin a 50 mg al giorno. In bambinipiccoli ed in bambini che compiono i primi passi (età compresa tra 12 e 23 mesi) trattati con caspo-fungin 50 mg/m2 al giorno (al massimo 70 mg al giorno), la AUC0-24h plasmatica di caspofungindopo dosaggi multipli è stata confrontabile con quella riscontrata in adulti trattati con caspofungina 50 mg al giorno e con quella riscontrata in bambini più grandi (da 2 a 11 anni di età) trattati conil dosaggio da 50 mg/m2 al giorno. Complessivamente, i dati disponibili di farmacocinetica, effica-cia e sicurezza sono limitati nei pazienti da 3 a 10 mesi di età. I dati di farmacocinetica relativi adun bambino di 10 mesi trattato con il dosaggio da 50 mg/m2 al giorno indicano valori di AUC0-24hcompresi in un range simile a quello osservato in bambini più grandi e in adulti trattati rispettiva-mente con dosaggi da 50 mg/m2 e 50 mg, mentre in un bambino di 6 mesi trattato con il dosaggioda 50 mg/m2, la AUC0-24h è stata un poco più alta. In neonati e lattanti (< 3 mesi) trattati con ca-spofungin 25 mg/m2 al giorno (corrispondenti ad un dosaggio giornaliero medio di 2,1 mg/kg), laconcentrazione di picco di caspofungin (C1h) e la concentrazione di valle di caspofungin (C24h) dopodosaggi multipli erano comparabili con quanto riscontrato in adulti trattati con caspofungin 50 mgal giorno. In questi neonati e lattanti rispetto agli adulti al Giorno 1, la C1h era comparabile e la C24hera modestamente elevata (36 %). Comunque è stata riscontrata variabilità sia nella C1h (la mediageometrica al Giorno 4 era di 11,73 mg/ml, range da 2,63 a 22,05 mg/ml) che nella C24h (lamedia geometrica al Giorno 4 era di 3,55 μg/ml, range da 0,13 a 7,17 mg/ml). In questo studionon sono state fatte misurazioni della AUC0-24h a causa della scarsità dei campioni plasma. Da no-tare, che l’efficacia e la sicurezza di caspofungin non sono stati adeguatamente studiati in studi cli-nici prospettici che abbiano coinvolto neonati e lattanti di età inferiore ai 3 mesi. 5.3 Dati preclinicidi sicurezza: Studi di tossicità a dosaggi ripetuti in ratti e scimmie con dosaggi fino a 7-8 mg/kgendovena hanno mostrato reazioni nel sito di iniezione in ratti e scimmie, segni di rilascio di istaminain ratti ed evidenza di effetti avversi a livello del fegato nelle scimmie. Studi di tossicità sull’accre-scimento nei ratti hanno mostrato che caspofungin ha causato diminuzioni del peso corporeo fetaleed incrementi nell’incidenza di calcificazione incompleta a livello delle vertebre, sternebre ed ossa delcranio a dosaggi di 5 mg/kg insieme a reazioni avverse nelle madri quali segni di rilascio di istaminanei ratti in gravidanza. E’ stato anche osservato un aumento nell’incidenza di coste cervicali. Caspo-fungin è risultato negativo in una serie di saggi in vitro per genotossicità potenziale e nel test cro-mosomico in vivo su midollo osseo di topo. Non sono stati condotti studi a lungo termine in animaliper valutare il potenziale cancerogeno. Per caspofungin, non ci sono stati effetti sulla fertilità in studicondotti in ratti maschio e femmina fino a 5 mg/kg/die. 6. INFORMAZIONI FARMACEUTI-CHE. 6.1 Elenco degli eccipienti: Saccarosio. Mannitolo. Acido acetico glaciale. Sodio idrossido (perl’aggiustamento del pH). 6.2 Incompatibilità: Non mescolare con diluenti contenenti glucosio, poi-ché CANCIDAS non è stabile in diluenti contenenti glucosio. In assenza di studi sulla compatibilità, que-sto medicinale non deve essere mescolato con altri medicinali. 6.3 Periodo di validità: 2 anni.Concentrato ricostituito: deve essere usato immediatamente. Dati di stabilità hanno mostrato che ilconcentrato per soluzione per infusione endovenosa può essere conservato fino a 24 ore se il fla-concino è conservato a temperatura uguale o inferiore a 25°C e ricostituito con acqua per prepara-zione iniettabile. Soluzione di infusione endovenosa diluita per il paziente: deve essere usataimmediatamente. Dati di stabilità hanno mostrato che il prodotto può essere usato entro 24 ore seconservato a temperatura uguale o inferiore a 25°C, o entro 48 ore quando la sacca (flacone) perl’infusione endovenosa è conservata in ambiente refrigerato (da 2 a 8°C) e diluita con una soluzioneper infusione endovenosa di sodio cloruro 9 mg/ml (0,9 %), 4,5 mg/ml (0,45 %), o 2,25 mg/ml(0,225 %), o con una soluzione di Ringer lattato. CANCIDAS non contiene conservanti. Da un puntodi vista microbiologico, il prodotto deve essere usato immediatamente. Se non viene usato imme-diatamente, i tempi di conservazione durante l’uso e le condizioni di conservazione prima dell’usosono responsibilità dell’operatore e non devono normalmente superare le 24 ore a 2 – 8°C, a menoche la ricostituzione e la diluizione non abbiano avuto luogo in condizioni asettiche controllate e va-lidate. 6.4 Precauzioni speciali per la conservazione: Flaconcini intatti: conservare in frigori-fero (da 2°C a 8°C). Per le condizioni di conservazione dopo ricostituzione e diluizione del medicinale,vedere paragrafo 6.3. 6.5 Natura e contenuto del contenitore: Flaconcino in vetro Tipo I da10 ml con tappo in butile grigio e capsula di chiusura di plastica con fascetta di alluminio rossa. For-nito in confezioni da 1 flaconcino. 6.6 Precauzioni particolari per lo smaltimento e la ma-nipolazione: Ricostituzione di CANCIDAS: NON USARE DILUENTI CONTENENTI GLUCOSIO, poichéCANCIDAS non è stabile in diluenti contenenti glucosio. NON MESCOLARE O SOMMINISTRARE NELLASTESSA VIA ENDOVENOSA CANCIDAS CON QUALSIASI ALTRO MEDICINALE, poiché non sono disponi-bili dati sulla compatibilità di CANCIDAS con altre sostanze, additivi o medicinali per uso endovenoso.Controllare visivamente la soluzione per infusione endovenosa per verificare la presenza di particelleo alterazioni di colore. ISTRUZIONI PER L’USO IN PAZIENTI ADULTI: Fase 1 Ricostitu-zione di flaconcini convenzionali: Per ricostituire la polvere, portare il flaconcino a temperaturaambiente ed aggiungere asetticamente 10,5 ml di acqua per preparazione iniettabile. La concen-trazione del flaconcino ricostituito risulterà di 5,2 mg/ml. La polvere liofilizzata compatta bianco-bian-castra deve essere completamente dissolta. Mescolare leggermente fino ad ottenere una soluzionelimpida. Le soluzioni ricostituite devono essere controllate visivamente per verificare la presenza diparticelle o alterazioni di colore. Questa soluzione ricostituita può essere conservata fino a 24 ore atemperature uguali o inferiori a 25°C. Fase 2 Aggiunta di CANCIDAS ricostituito alla solu-zione di infusione endovenosa per il paziente: I diluenti per la soluzione di infusione endo-venosa finale sono: soluzione per preparazione iniettabile di sodio cloruro, o soluzione di Ringerlattato. La soluzione per infusione endovenosa viene preparata aggiungendo asetticamente la quan-tità adeguata del concentrato ricostituito (come mostrato nella tabella che segue) a una sacca o a unflacone da infusione da 250 ml. Se necessario dal punto di vista medico possono essere usati volumidi infusione ridotti a 100 ml per i dosaggi quotidiani da 50 mg o 35 mg. Non usare se la soluzionepresenta opacità o precipitati.
PREPARAZIONE DELLA SOLUZIONE PER L’INFUSIONE IN ADULTIDOSAGGIO* Volume di CANCIDAS Preparazione standard Volume di infusione
ricostituito per il (CANCIDAS ricostituito ridotto (CANCIDAStrasferimento alla aggiunto a 250 ml) ricostituito aggiuntosacca o al flacone per concentrazione finale a 100 ml) soluzione endovenosa concentrazione finale
50 mg 10 ml 0,20 mg/ml -50 mg avolume ridotto 10 ml - 0,47 mg/ml35 mg per lacompromissione epatica moderata (da 1 flaconcino da 50 mg) 7 ml 0,14 mg/ml -35 mg per lacompressione epaticamoderata (da 1 flaconcino da 50 mg) a volume ridotto 7 ml - 0,34 mg/ml* devono essere usati 10,5 ml per ricostituire tutti i flaconciniISTRUZIONI PER L’USO IN PAZIENTI PEDIATRICI: Calcolo dell’Area della Superficie Corporea(BSA) per il dosaggio pediatrico: Prima della preparazione dell’infusione, calcolate l’area di superfi-cie corporea (BSA) del paziente usando la seguente formula: (Formula di Mosteller).
Preparazione dell’infusione da 70 mg/m2 per pazienti pediatrici di età > 3 mesi (utliz-zando un flaconcino da 50 mg): Stabilire il dosaggio da carico appropriato da usare nei pazientipediatrici utilizzando la BSA del paziente (come sopra calcolato) e la seguente equazione: BSA (m2)X 70 mg/m2 = Dosaggio da Carico. Il massimo dosaggio da carico al Giorno 1 non deve superare i70 mg indipendentemente dalla dosaggio calcolato del paziente. Portate il flaconcino di CANCIDAS re-frigerato a temperatura ambiente. Aggiungete asetticamente 10,5 ml di acqua per preparazione iniet-tabile.a Questa soluzione ricostituita può essere conservata fino a 24 ore a temperatura uguale oinferiore a 25°C.b Questa fornirà una concentrazione finale di caspofungin nel flaconcino di 5,2 mg/ml.Prendere dal flaconcino il volume del medicinale corrispondente al dosaggio da carico calcolato (fase1). Trasferire asetticamente questo volume (ml)c di CANCIDAS ricostituito in una sacca da infusione EV(o bottiglia) contenente 250 ml di sodio cloruro per preparazione iniettabile 0,9 %, 0,45 % o 0,225 %,oppure Ringer lattato per preparazione iniettabile. In alternativa, il volume (ml) c di CANCIDAS rico-stituito può essere aggiunto ad un volume ridotto di sodio cloruro per preparazione iniettabile 0,9 %,0,45 % o 0,225 %, oppure Ringer lattato per preparazione iniettabile, senza superare una concen-trazione finale di 0,5 mg/ml. Questa soluzione per infusione deve essere usata entro 24 ore se con-servata a temperatura uguale o inferiore a 25°C o entro 48 ore se conservata in ambiente refrigeratotra 2 e 8°C. Preparazione dell’infusione da 50 mg/m2 per pazienti pediatrici di età >3 mesi (utlizzando un flaconcino da 50 mg): Stabilire il dosaggio giornaliero di mantenimentoappropriato da usare nel paziente pediatrico utilizzando la BSA del paziente (come sopra calcolato)e la seguente equazione: BSA (m2) X 50 mg/m2 = Dosaggio di Mantenimento giornaliero. Il dosag-gio di mantenimento giornaliero non deve superare i 70 mg indipendentemente dal dosaggio calco-lato del paziente. Portate il flaconcino di CANCIDAS refrigerato a temperatura ambiente. Aggiungeteasetticamente 10,5 ml di acqua per preparazione iniettabile.a Questa soluzione ricostituita può essereconservata fino 24 ore a temperatura uguale o inferiore a 25°C.b Questa fornirà una concentrazionefinale di caspofungin nel flaconcino di 5,2 mg/ml. Prendere dal flaconcino il volume del medicinalecorrispondente al dosaggio giornaliero di mantenimento calcolato (fase 1). Trasferire asetticamentequesto volume (ml) c di CANCIDAS ricostituito in una sacca da infusione EV (o bottiglia) contenente 250ml di sodio cloruro per preparazione iniettabile 0,9 %, 0,45 % o 0,225 %, oppure Ringer lattato perpreparazione iniettabile. In alternativa, il volume (ml)c di CANCIDAS ricostituito può essere aggiuntoad un volume ridotto di sodio cloruro per preparazione iniettabile 0,9 %, 0,45 % o 0,225 %, oppureRinger lattato per preparazione iniettabile, senza superare una concentrazione finale di 0,5 mg/ml.Questa soluzione per infusione deve essere usata entro 24 ore se conservata a temperatura ugualeo inferiore a 25°C o entro 48 ore se conservata in ambiente refrigerato tra 2 e 8°C.Note per la preparazione: a. Il composto da bianco a bianco-biancastro si scioglierà completamente. Mescolare delicatamentefino a quando la soluzione diventa limpida.b. Ispezionate visivamente la soluzione ricostituita per verificare la presenza di particelle o altera-zioni di colore durante la ricostituzuione e prima dell’infusione. Non usare se la soluzione non è lim-pida o contiene precipitati.c. CANCIDAS è formulato per fornire il pieno dosaggio indicato sull’etichetta (50 mg) quando ven-gono estratti dal flaconcino 10 ml.7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO: Merck Sharp& Dohme Ltd. Hertford Road, Hoddeson. Hertforshire EN11 9BU. Regno Unito. 8. NUMERO(I)DELL’AUTORIZZAZIONE (DELLE AUTORIZZAZIONI) ALL’IMMISSIONE IN COMMER-CIO: EU/1/01/196/001. 9. DATA DELLA PRIMA AUTORIZZAZIONE/ RINNOVO DEL-L’AUTORIZZAZIONE: Data della prima autorizzazione: 24 ottobre 2001. Data dell’ultimorinnovo: 07 settembre 2011. 10. DATA DI REVISIONE DEL TESTO: Luglio 2012.
www.msd-italia.it - www.contattamsd.it [email protected] - www.univadis.it
Medicinale soggetto a prescrizione medica limitativa, utilizzabile esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile OSPClasse H - Prezzo al pubblico: 50 mg polvere € 662,14Tali prezzi potrebbero essere soggetti a variazioni determinate da provvedimenti legislativi.
Informazioni più dettagliate su questo medicinale sono disponibili sul sito web della Agenzia Europea dei Medicinali http://www.ema.europa.eu

Riassunto delle Caratteristiche del Prodotto
1. DENOMINAZIONE DEL MEDICINALELevact 2,5 mg/ml polvere per concentrato per soluzione per infusione.
2. COMPOSIZIONE QUALITATIVA E QUANTITATIVAUn flaconcino contiene 25 mg di bendamustina cloridrato. Un flaconcino contiene 100 mg di bendamustina cloridrato. 1 ml di concentrato contiene 2,5 mg di bendamustina cloridrato quando ricostituito come indicato al paragrafo 6.6. Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
3. FORMA FARMACEUTICAPolvere per concentrato per soluzione per infusione. Polvere bianca, microcristallina.
4. INFORMAZIONI CLINICHE
4.1 Indicazioni terapeuticheTrattamento di prima linea della leucemia linfatica cronica (stadio Binet B o C) in quei pazienti per i quali non è appropriata una chemioterapia contenente fludarabina. Linfoma non-Hodgkin indolente come monoterapia in pazienti che hanno avuto una progressione malattia durante o entro 6 mesi dal trattamento con rituximab o con un regime terapeutico contenente rituximab. Trattamento di prima linea del mieloma multiplo (stadio Durie – Salmon II con progressione o stadio III) in associazione con prednisone in pazienti oltre i 65 anni di età che non sono eleggibili a trapianto autologo di cellule staminali e che presentano neuropatia clinica al momento della diagnosi che precluda l’uso di un trattamento contenente talidomide o bortezomib.
4.2 Posologia e modo di somministrazione Per infusione endovenosa della durata di 30 – 60 minuti (vedere paragrafo 6.6). L’infusione deve essere somministrata sotto la supervisione di un medico qualificato ed esperto nell’uso di agenti chemioterapici. La riduzione della funzione midollare è correlata all’aumento della tossicità ematologica indotta dai chemioterapici. Il trattamento non deve essere iniziato se i valori dei leucociti e/o delle piastrine scendono rispettivamente a < 3.000/µl o < 75.000/µl (vedere paragrafo 4.3). Monoterapia per leucemia linfatica cronica. 100 mg/m2 di superficie corporea di bendamustina cloridrato nei giorni 1 e 2; ogni 4 settimane Monoterapia per Linfoma non-Hodgkin indolente refrattario al rituximab. 120 mg/m² di superficie corporea di bendamustina cloridrato nei giorni 1 e 2; ogni 3 settimane Mieloma multiplo. 120 - 150 mg/m² di superficie corporea di bendamustina cloridrato nei giorni 1 e 2 e 60 mg/m² per superficie corporea di prednisone i.v o per os nei giorni da 1 a 4; ogni 4 settimane. Il trattamento deve essere interrotto o ritardato se i valori dei leucociti e/o delle piastrine scendono rispettivamente a < 3.000/µl o < 75.000/µl. Il trattamento può essere continuato dopo che i valori dei leucociti sono aumentati a > 4.000/µl e quelli delle piastrine a > 100.000/ µl. Il valore minimo (nadir) di leucociti e piastrine è raggiunto dopo 14-20 giorni, con rigenerazione dopo 3-5 settimane. Si raccomanda un rigoroso monitoraggio della conta ematica durante gli intervalli liberi dalla terapia (vedere paragrafo 4.4). In caso di tossicità non ematologica le riduzioni di dose devono essere basate sul grado peggiore secondo i Criteri Comuni di Tossicità (CTC) osservato nel ciclo precedente. Una riduzione del 50% della dose è raccomandata in caso di CTC di grado 3. Si raccomanda l’interruzione del trattamento in caso di CTC di grado 4. Se il paziente richiede una variazione della dose, la dose ridotta calcolata individualmente deve essere somministrata nei giorni 1 e 2 del rispettivo ciclo di trattamento. Per istruzioni di preparazione e somministrazione vedere il paragrafo 6.6. Insufficienza epatica Sulla base dei dati di farmacocinetica, non è necessario un aggiustamento della dose nei pazienti con insufficienza epatica lieve (bilirubina sierica < 1,2 mg/dl). Si raccomanda una riduzione della dose del 30% nei pazienti con insufficienza epatica moderata (bilirubina sierica 1,2 - 3,0 mg/dl). Non sono disponibili dati in pazienti con insufficienza epatica severa (bilirubina sierica > 3,0 mg/dl) (vedere paragrafo 4.3). Insufficienza renale Sulla base dei dati di farmacocinetica, non è necessario un aggiustamento della dose nei pazienti con clearance della creatinina > 10 ml/min. L’esperienza relativa a pazienti con insufficienza renale severa è limitata. Pazienti pediatrici Non ci sono esperienze su bambini e adolescenti trattati con Levact. Pazienti anziani Non ci sono evidenze che aggiustamenti di dose siano necessari nei pazienti anziani (vedere paragrafo 5.2).
4.3 Controindicazioni Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti (vedere paragrafo 6.1). Durante l’allattamento al seno. Insufficienza epatica severa (bilirubina sierica > 3,0 mg/dl). Ittero. Severa soppressione midollare e severe alterazioni della conta ematica (valori di leucociti e piastrine scesi rispettivamente a < 3.000/ µl o < 75.000/ µl). Interventi di chirurgia maggiore entro 30 giorni dall’inizio del trattamento. Infezioni, soprattutto quando comportano leucopenia. Vaccinazione contro la febbre gialla.
4.4 Speciali avvertenze e precauzioni d’uso. Mielosoppressione I pazienti trattati con bendamustina cloridrato possono manifestare mielosoppressione. In caso di mielosoppressione dovuta al trattamento, leucociti, piastrine, emoglobina e neutrofili devono essere controllati almeno settimanalmente. Prima di iniziare il ciclo di terapia, sono raccomandati i seguenti parametri: valori di leucociti e/o di piastrine rispettivamente > 4.000/ µl o > 100.000 µl. Infezioni Sono state segnalate infezioni, incluse polmoniti e sepsi. In rari casi l’infezione si è associata ad ospedalizzazione, shock settico e morte. Pazienti con neutropenia e/o linfopenia dopo trattamento con bendamustina cloridrato sono più suscettibili alle infezioni. I pazienti con mielosoppressione dopo trattamento con bendamustina cloridrato devono essere avvisati di contattare un medico in caso abbiano sintomi o segni di infezione, inclusi febbre o sintomi respiratori. Reazioni cutanee È stato riportato un certo numero di reazioni cutanee. Questi eventi hanno incluso rash, reazioni tossiche cutanee ed esantema bolloso. Alcuni eventi si sono manifestati quando bendamustina cloridrato è stata somministrata in associazione con altri agenti anticancerogeni, e per questo motivo è incerta la precisa correlazione. Quando insorgono reazioni cutanee, queste possono progredire e aumentare di gravità
01 RCP Medico Levact.indd 3 03/08/12 12.48

con ulteriori trattamenti. Se le reazioni cutanee peggiorano, la somministrazione di Levact va interrotta o sospesa. Per reazioni cutanee gravi dove si sospetta una relazione con bendamustina cloridrato, il trattamento deve essere sospeso. Pazienti con disturbi cardiaci Durante il trattamento con Bendamustina cloridrato, deve essere strettamente monitorata la concentrazione di potassio nel sangue, devono essere somministrati supplementi di potassio in presenza di valori di K+
< 3,5 mEq/l (o < 3,5 mmol/l), e vanno eseguite registrazioni ECG. Nausea, vomito Può essere somministrato un antiemetico per il trattamento sintomatico di nausea e vomito. Sindrome da lisi tumorale Nel corso delle sperimentazioni cliniche è stata riportata sindrome da lisi tumorale associata al trattamento con Levact. La sindrome da lisi tumorale insorge generalmente entro le 48 ore dalla prima dose di Levact e, senza interventi, può condurre ad insufficienza renale acuta e morte. Misure preventive includono un adeguato stato volemico, lo stretto monitoraggio degli esami ematochimici, in particolare potassiemia ed uricemia. L’uso di allopurinolo durante le prime due settimane di trattamento con Levact può essere preso in considerazione, ma non è obbligatorio. Tuttavia, quando bendamustina e allopurinolo sono stati somministrati in concomitanza, sono stati segnalati sporadici casi di sindrome di Stevens – Johnson e di Necrolisi Epidermica Tossica. Anafilassi Negli studi clinici si sono verificate comunemente reazioni infusionali alla bendamustina cloridrato. I sintomi sono generalmente lievi e includono febbre, brividi, prurito e rash. Severe reazioni anafilattiche e anafilattoidi si sono verificate in rari casi. I pazienti devono essere interrogati in merito a sintomi indicativi di reazioni infusionali dopo il primo ciclo di terapia. Si devono prendere in considerazione misure per prevenire reazioni severe, inclusi gli antistaminici, gli antipiretici e i corticosteroidi nei cicli successivi nei pazienti che hanno in precedenza manifestato reazioni infusionali. Pazienti che hanno manifestato reazioni di tipo allergico di grado 3 o maggiore, tipicamente non sono stati ritrattati. Contraccezione Bendamustina cloridrato è teratogena e mutagena. Durante il trattamento le donne non dovrebbero iniziare una gravidanza. I pazienti maschi non devono concepire un figlio durante e nei 6 mesi successivi al trattamento. Devono essere informati in merito alla conservazione dello sperma prima del trattamento con bendamustina cloridrato, a causa di possibile infertilità irreversibile. Stravaso infusionale Una iniezione extravasale deve essere interrotta immediatamente. L’ago deve essere rimosso dopo una breve aspirazione. Dopo ciò, l’area tissutale coinvolta dallo stravaso deve essere raffreddata e il braccio sollevato. Trattamenti supplementari, come l’uso di corticosteroidi, non sono di chiaro beneficio.
4.5 Interazioni con altri prodotti medicinali e altre forme di interazione Non sono stati condotti studi d’interazione in vivo. Quando Levact è somministrato in associazione con agenti mielosoppressivi, l’effetto sul midollo osseo di Levact e/o dei co-medicamenti può venire potenziato. Qualunque trattamento che riduca lo stato funzionale del paziente o che peggiori la funzione midollare può aumentare la tossicità di Levact. La associazione di Levact con ciclosporina o tacrolimus può produrre una eccessiva immunosoppressione, con rischio di linfoproliferazione. I citostatici possono ridurre la formazione di anticorpi dopo vaccinazione con virus vivo, ed aumentare il rischio di infezioni ad esito fatale. Il rischio è aumentato nei soggetti che sono già immunodepressi a causa della loro malattia di base. Il metabolismo di bendamustina coinvolge il citocromo P450(CYP) isoenzima 1A2 (vedere paragrafo 5.2). Pertanto esiste la possibilità di interazione con gli inibitori CYP 1A2, quali fluvoxamina, ciprofloxacina, aciclovir, cimetidina.
4.6 Gravidanza e allattamento Gravidanza I dati sull’uso di Levact nelle donne gravide sono insufficienti. In studi non clinici, bendamustina cloridrato è risultata embrio- e feto-letale, teratogenica e genotossica (vedere paragrafo 5.3). Durante la gravidanza Levact non deve essere usato, se non strettamente necessario. La madre deve essere informata sui rischi per il feto. Se il trattamento con Levact è assolutamente necessario durante la gravidanza o se la gravidanza si verifica durante il trattamento,la paziente deve essere informata sui rischi per il nascituro, e deve essere monitorata attentamente. Si deve considerare la possibilità di una consulenza genetica. Donne potenzialmente fertili/contraccezione. Donne in età potenzialmente fertile devono usare metodi di contraccezione, sia prima che durante la terapia con Levact. Agli uomini in trattamento con Levact si raccomanda di non concepire figli durante e sino a sei mesi dopo la cessazione del trattamento. Devono essere informati in merito alla conservazione dello sperma prima del trattamento, a causa della possibilità di infertilità irreversibile dovuta alla terapia con Levact. Allattamento Non è noto se bendamustina sia escreta nel latte materno, perciò Levact è controindicato durante il periodo di allattamento (vedere paragrafo 4.3). L’allattamento deve essere interrotto durante il trattamento con Levact.
4.7 Effetti sulle capacità di guida ed utilizzo di macchinari Non sono stati condotti studi sugli effetti sulla capacità di guidare e sull’uso dei macchinari. Tuttavia, sono state segnalate atassia, neuropatia periferica e sonnolenza durante il trattamento con Levact (vedere paragrafo 4.8). I pazienti devono essere avvisati che, nel caso in cui manifestino tali sintomi, devono evitare attività potenzialmente pericolose come guidare ed utilizzare macchinari.
4.8 Effetti indesiderati Le più comuni reazioni avverse con bendamustina cloridrato sono reazioni avverse ematologiche (leucopenia, piastrinopenia), tossicità dermatologica (reazioni allergiche), sintomi costituzionali (febbre), sintomi gastrointestinali (nausea, vomito).La tabella seguente riflette i dati ottenuti negli studi clinici con bendamustina cloridrato. Sono stati osservati alcuni casi di sindrome di Stevens-Johnson e di Necrolisi Epidermica Tossica in pazienti che hanno usato bendamustina in associazione con allopurinolo, o con allopurinolo e rituximab. Il rapporto CD4/CD8 può essere ridotto. È stata osservata una riduzione della conta dei linfociti. Nei pazienti immuno-soppressi potrebbe aumentare il rischio di infezione (es. herpes zoster). Ci sono stati casi isolati di necrosi in seguito a somministrazione accidentale extra – vascolare, necrosi epidermica tossica, sindrome da lisi tumorale, e anafilassi. Ci sono segnalazioni di tumori secondari, inclusi sindrome mielodisplastica, malattie mieloproliferative, leucemia mieloide acuta e carcinoma bronchiale. Non è stata accertata la correlazione con la terapia con Levact.
01 RCP Medico Levact.indd 4 03/08/12 12.48

ClassificazionesistemicaorganicaMedDra
Infezionie infestazioni
Tumori benigni,maligni
Patologiedel sistemaemolinfopoietico
Disturbi del sistemaimmunitario
Patologiedel sistemanervoso
Patologiecardiache
Patologievascolari
Patologie respiratorie,toraciche emediastiniche
Patologiegastrointestinali
Patologie della cute e del tessutosottocutaneo
Patologiedell’apparatoriproduttivo o dellamammella
Patologiesistemichee condizionirelative alla sede di sommistrazione
Esami diagnostici
Molto comune≥ 1/10
Infezione NOS*
Leucopenia NOS*,Piastrinopenia
Nausea, vomito
Infiammazionidelle mucose, affaticamento,piressia
Calo di emoglobina,aumento di creatinina,aumento di urea
Comune≥ 1/100 e < 1/10
Sindrome da lisi tumorale
Emorragia, anemia, neutropenia
Ipersensibilità NOS*
Insonnia
Disfunzione cardiaca, come palpitazioni, anginapectoris, aritmia
Ipotensione,ipertensione
Disfunzione polmonare
Diarrea, stipsi, stomatite
Alopecia, patologie della cute NOS*
Amenorrea
Dolore, brividi,disidratazione, anoressia
Aumento di AST, aumento di ALT, aumento difosfatasi alcalina, aumento di bilirubina,ipokaliemia
Non comune≥ 1/1.000e < 1/100
Versamentopericardico
Raro≥ 1/10.000 e < 1/1.000
Sepsi
Reazioneanafilattica,reazioneanafilattoide
Sonnolenza,Afonia
Insufficienzacircolatoria acuta
Eritema, dermatiti,prurito, rashiperidrosiiperidrosi
Molto raro < 1/10.000
Polmoniteprimaria atipica
Emolisi
Shock anafilattico
Disgeusia,parestesia,neuropatia sensorialeperiferica, sindromeanticolinergica, patologieneurologiche, atassia,encefalite
Tachicardia,infarto del miocardio,insufficienzacardiaca
Flebite
Fibrosi polmonare
Esofagite emorragica,emorragiagastrointestinale
Infertilità
Insufficienzamulti-organo
Non nota (non può essere definita sulla basedei dati disponibili)
* NOS = non altrimenti specificato
01 RCP Medico Levact.indd 5 03/08/12 12.48

4.9 SovradosaggioDopo una somministrazione di 30 minuti di infusione di Levact una volta ogni 3 settimane, la dose massima tollerata (MTD) è stata 280 mg/m². Sono insorti eventi cardiaci CTC grado 2 compatibili con modificazioni ischemiche dell’ECG, e che sono stati considerati “dose limitanti”. In uno studio successivo con infusione di Levact di 30 minuti nei giorni 1 e 2 ogni 3 settimane, la MTD è stata 180 mg/m². La tossicità “dose limitante” è stata rappresentata da piastrinopenia di grado 4. Con questo schema, la tossicità cardiaca non è stata di tipo “dose-limitante”. Contromisure Non esiste un antidoto specifico. Come contromisure efficaci nel controllo degli effetti collaterali ematologici, possono essere effettuati il trapianto di midollo osseo e trasfusioni (piastrine, eritrociti concentrati), oppure possono essere somministrati fattori di crescita ematologici. Bendamustina cloridrato e i suoi metaboliti sono dializzabili in misura ridotta.
5. PROPRIETÀ FARMACOLOGICHE
5.1 Proprietà farmacodinamiche Categoria farmacoterapeutica: Agenti Antineoplastici, agenti alchilanti, codice ATC: L01AA09. Bendamustina cloridrato è un agente alchilante antitumorale con peculiare attività. L’azione antineoplastica e citocida della bendamustina cloridrato si basa fondamentalmente sul cross-linking per alchilazione sul DNA a singola e doppia elica. Di conseguenza, le funzioni di matrice, sintesi e riparazione del DNA sono compromesse. L’azione anti-tumorale della bendamustina cloridrato è stata dimostrata in numerosi studi in vitro condotti su differenti linee cellulari di tumori umani (tumore della mammella, tumore polmonare a piccole cellule e non a piccole cellule, carcinoma dell’ovaio e differenti leucemie) e studi in vivo in differenti modelli sperimentali di tumore con tumori del topo, ratto e di origine umana (melanoma, tumore della mammella, sarcoma, linfoma, leucemia e tumore polmonare a piccole cellule). Bendamustina cloridrato ha mostrato un profilo d’attività in linee di cellule tumorali umane differente da quello di altri agenti alchilanti. Il principio attivo ha rivelato una bassa o nulla resistenza crociata nelle linee cellulari di tumori umani con differenti meccanismi di resistenza, almeno in parte legati ad una interazione comparativamente persistente sul DNA. È stato inoltre dimostrato in studi clinici che non sussiste una completa resistenza crociata della bendamustina con antracicline, agenti alchilanti o rituximab. Tuttavia il numero di pazienti valutati è esiguo. Leucemia linfatica cronica L’indicazione per l’impiego nella leucemia linfatica cronica è supportata da un solo studio in aperto, che ha confrontato bendamustina con clorambucil. In questo studio prospettico, multicentrico, randomizzato sono stati inclusi 319 pazienti non precedentemente trattati con leucemia linfatica cronica stadio Binet B o C Il trattamento di prima linea con bendamustina cloridrato 100 mg/m² i.v. nei giorni 1 e 2 (BEN) è stato confrontato con clorambucil 0,8 mg/kg nei giorni 1 e 15 (CLB), per 6 cicli in entrambi i gruppi di trattamento. Ai pazienti è stato somministrato allopurinolo al fine di prevenire la sindrome da lisi tumorale. I pazienti con BEN hanno mostrato una sopravvivenza mediana libera da progressione significativamente più lunga rispetto a pazienti trattati con CLB (21,5 mesi versus 8,3 mesi, p < 0,0001 nell’ultimo follow-up). La sopravvivenza complessiva non ha mostrato una differenza statisticamente significativa (mediana non raggiunta). La durata mediana della remissione è stata 19 mesi con BEN, e 6 mesi con CLB (p < 0,0001). La valutazione della sicurezza in entrambi i gruppi trattati non ha mostrato effetti indesiderati inaspettati per tipologia e frequenza. Il dosaggio di BEN è stato ridotto nel 34% dei pazienti. Il trattamento con BEN è stato interrotto nel 3,9% dei pazienti a seguito di reazioni allergiche. Linfoma non-Hodgkin indolente L’indicazione nel linfoma non-Hodgkin indolente si basa su due studi non controllati di fase II. Nel principale studio prospettico multicentrico in aperto, 100 pazienti con linfoma non-Hodgkin indolente a cellule B refrattario a rituximab in monoterapia o in associazione, sono stati trattati con BEN in monoterapia. I pazienti avevano ricevuto mediamente 3 cicli preliminari di chemioterapia o terapia biologica. Il numero mediano di cicli precedenti contenenti rituximab era 2. I pazienti non avevano mostrato risposta o erano progrediti entro 6 mesi dal trattamento con rituximab. Il dosaggio di BEN è stato 120 mg/m² i.v. nei giorni 1 e 2, pianificato per almeno 6 cicli. La durata del trattamento era legata alla risposta (6 cicli pianificati). Il tasso complessivo della risposta è stata del 75% e include il 17% di risposte complete (CR e CRu) ed il 58% di risposte parziali, come valutato da un comitato revisore indipendente. La durata mediana della remissione è stata 40 settimane. BEN è stata generalmente ben tollerata quando somministrata secondo questi dosaggi e schemi di terapia. L’indicazione è inoltre supportata da un altro studio prospettico multicentrico in aperto su 77 pazienti. La popolazione dei pazienti era più eterogenea e comprendeva linfomi non-Hodgkin indolenti o trasformati a cellule B refrattari a rituximab in monoterapia o in associazione. I pazienti non avevano risposta o erano progrediti entro i 6 mesi dal trattamento, o avevano avuto una reazione sfavorevole con il precedente trattamento con rituximab. I pazienti avevano ricevuto mediamente 3 precedenti cicli di chemioterapia o di terapia biologica. Il numero mediano dei precedenti cicli di terapia con rituximab era 2. Il tasso complessivo di risposta è stato del 76%, con una durata mediana di risposta di 5 mesi (29 settimane [95% CI 22,1- 43,1]). Mieloma multiplo In uno studio clinico prospettico multicentrico randomizzato in aperto, sono stati inclusi 131 pazienti con mieloma multiplo in fase avanzata (Durie-Salmon stadio II in progressione, o stadio III). La terapia di prima linea con bendamustina cloridrato associata a prednisone (BP) è stata confrontata ad un trattamento con melphalan e prednisone (MP). Né l’eleggibilità a trapianto, né la presenza di specifiche co-morbilità hanno influenzato l’inclusione dei pazienti nello studio. Il dosaggio usato è stato bendamustina cloridrato 150 mg/m² i.v. nei giorni 1 e 2, oppure melphalan 15 mg/m² i.v. nel giorno 1, ciascuno in associazione con prednisone. La durata del trattamento era dipendente dalla risposta, ed è stato mediamente di 6,8 cicli nel gruppo BP e 8,7 nel gruppo MP. I pazienti trattati con BP hanno avuto una sopravvivenza mediana libera da progressione più lunga rispetto ai pazienti trattati con MP (15 mesi [95% CI 12-21] versus 12 mesi [95% CI 10-14]) (p = 0,0566). Il tempo mediano al fallimento del trattamento è stato 14 mesi nel gruppo trattato con BP e 9 mesi con MP. La durata della remissione è stata 18 mesi con il trattamento BP e 12 mesi con MP. La sopravvivenza globale non è stata significativamente differente (35 mesi con BP versus 33 mesi con MP). La tollerabilità in entrambi i gruppi di trattamento è stata in linea con il profilo di sicurezza conosciuto dei rispettivi medicinali, con una riduzione della dose significativamente maggiore nel gruppo trattato con BP.
01 RCP Medico Levact.indd 6 03/08/12 12.48

5.2 Proprietà farmacocinetiche Distribuzione L’emivita di eliminazione t1/2ß dopo 30 minuti di infusione i.v. di 120 mg/m² per superficie corporea in 12 soggetti è stata pari a 28,2 minuti. Dopo 30 minuti di infusione i.v. il volume centrale di distribuzione è stato 19,3 l. In condizioni stabili a seguito di un’iniezione in bolo i.v., il volume di distribuzione è stato 15,8 – 20,5 l. Oltre il 95% della sostanza è legata alle proteine plasmatiche (principalmente albumina). Metabolismo La principale via d’eliminazione di bendamustina è l’idrolisi a monoidrossi- e diidrossi-bendamustina. La formazione di N- desmetil-bendamustina e gamma-idrossi-bendamustina da parte del metabolismo epatico è ad opera del citocromo P450 (CYP) isoenzima 1A2. Un’altra importante via metabolica della bendamustina è la coniugazione con il glutatione. Negli studi in vitro, bendamustina non inibisce CYP 1A4, CYP 2C9/10, CYP 2D6, CYP2E1 e CYP 3A4. Eliminazione La clearance media totale dopo 30 minuti di infusione di 120 mg/ m² di superficie corporea in 12 soggetti è stata 639,4 ml/min. Circa il 20% della dose somministrata è stata ritrovata nelle urine entro le 24 ore. Le quantità escrete nelle urine sono state nell’ordine: monoidrossi-bendamustina> bendamustina> diidrossi-bendamustina > metabolita ossidato >N-desmetil-bendamustina. Nella bile sono primariamente eliminati i metaboliti polari. Insufficienza epatica In pazienti con il 30 – 70% di coinvolgimento tumorale del fegato ed insufficienza epatica lieve (bilirubina sierica < 1,2 mg/dl), il comportamento farmacocinetico non è cambiato. Non ci sono state significative differenze rispetto ai pazienti con normale funzione epatica e renale per quanto riguarda Cmax, tmax, AUC, t1/2ß volume di distribuzione e clearance. AUC e clearance corporea totale di bendamustina correlano inversamente con la bilirubina sierica. Insufficienza renale Nei pazienti con clearance della creatinina > 10 ml/min, compresi i pazienti dipendenti da dialisi, non sono state osservate significative differenze rispetto a pazienti con normale funzione epatica e renale per quanto riguarda Cmax, tmax AUC, t1/2ß, volume di distribuzione e clearance. Pazienti anziani Soggetti di età fino a 84 anni sono stati inclusi negli studi di farmacocinetica. L’età più elevata non influenza la farmacocinetica di bendamustina.
5.3 Dati preclinici di sicurezza Le reazioni avverse non osservate durante gli studi clinici, ma osservate in studi animali per livelli di esposizione simili all’esposizione clinica e con possibile rilevanza per uso clinico, sono state le seguenti: Indagini istologiche nei cani hanno mostrato iperemia macroscopica della mucosa e emorragia nel tratto gastrointestinale. Indagini microscopiche hanno mostrato estese modifiche del tessuto linfatico, indicanti immuno-soppressione e modifiche tubulari dei reni e testicoli, così come modifiche atrofiche necrotiche dell’epitelio della prostata. Studi negli animali hanno mostrato che bendamustina è embriotossica e teratogena. Bendamustina induce aberrazione dei cromosomi ed è mutagena in vivo come in vitro. Bendamustina è risultata cancerogena in studi di lungo termine nei topi femmina.
6. INFORMAZIONI FARMACEUTICHE
6.1 Elenco degli eccipienti Mannitolo
6.2 Incompatibilità Questo prodotto medicinale non deve essere miscelato con altri prodotti, ad eccezione di quelli menzionati nel paragrafo 6.6.
6.3 Periodo di validità 3 anni. La polvere deve essere ricostituita immediatamente dopo l’apertura del flaconcino. Il concentrato ricostituito deve essere immediatamente diluito con soluzione di cloruro di sodio 0,9%. Soluzione per infusione Dopo la ricostituzione e la diluizione, la stabilità chimica e fisica è stata dimostrata per 3,5 ore a 25C° / 60% RH e per 2 giorni da 2°a 8°C in sacche di polietilene. Da un punto di vista microbiologico, la soluzione deve essere immediatamente usata. Se non usata immediatamente, i tempi di conservazione durante l’uso e le condizioni prima dell’uso sono sotto la responsabilità dell’utilizzatore.
6.4 Precauzioni particolari per la conservazione Conservare il flaconcino nel cartone esterno al fine di proteggerla dalla luce. Per le condizioni di conservazione del medicinale ricostituito o diluito, vedere paragrafo 6.3
6.5 Natura e contenuto del contenitore Flaconcini di vetro marrone tipo I di 26 ml o 60 ml con tappo di gomma e cappuccio di protezione in alluminio. Il flaconcino da 26 ml contiene 25 mg di bendamustina cloridrato ed è disponibile in confezioni da 5, 10 e 20 flaconcini. Il flaconcino da 60 ml contiene 100 mg di bendamustina cloridrato ed è disponibile in confezioni da 5 flaconcini. È possibile che non tutte le confezioni siano commercializzate.
6.6 Precauzioni particolari per lo smaltimento e la manipolazione Quando si maneggia Levact deve essere evitata l’inalazione, il contatto con la pelle o il contatto con le mucose (indossare guanti ed abiti di protezione!). Parti del corpo contaminate devono essere accuratamente risciacquate con acqua e sapone, gli occhi devono essere risciacquati con soluzione fisiologica salina. Se possibile, si raccomanda di lavorare su speciali banchi da lavoro di sicurezza (flusso laminare) utilizzando foglio assorbente usa-e-getta impermeabile a liquidi. Il personale in gravidanza deve essere escluso dal maneggiare citostatici. La polvere per concentrato per soluzione per infusione deve essere ricostituita con acqua per iniezione, diluita con soluzione per iniezione di sodio cloruro 9 mg/ml (0,9%) e poi somministrata tramite infusione intravenosa. Deve essere usata una tecnica asettica. 1. Ricostituzione Ricostituire ogni flaconcino di Levact contenente 25 mg di bendamustina cloridrato in 10 ml di acqua per iniezione
01 RCP Medico Levact.indd 7 03/08/12 12.48

agitando la soluzione. Ricostituire ogni flaconcino di Levact contenente 100 mg di bendamustina cloridrato in 40 ml di acqua per iniezione agitando la soluzione. Il concentrato ricostituito contiene 2,5 mg di bendamustina cloridrato per ml ed appare come una soluzione chiara priva di colore. 2. Diluizione Appena si ottiene una soluzione chiara (in genere dopo 5-10 minuti) diluire immediatamente la dose totale raccomandata di Levact con soluzione di NaCL 0,9% per produrre un volume finale di circa 500 ml. Levact deve essere diluito con soluzione di NaCL 0,9% e con nessun’altra soluzione iniettabile. 3. Somministrazione La soluzione è somministrata tramite infusione intravenosa della durata di 30 – 60 min. I flaconcini sono solo per uso singolo. Il medicinale non utilizzato ed i rifiuti derivanti da tale medicinale devono essere smaltiti in conformità alla normativa locale vigente.
7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIOAstellas Pharma GmbH, Postfach 50 01 66, D-80971 München.
8. NUMERO DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIOAIC n. 040175010/M - “2,5 mg/ml polvere per concentrato per soluzione per infusione” 5 flaconcini in vetro da 25 mg AIC n. 040175022/M - “2,5 mg/ml polvere per concentrato per soluzione per infusione” 10 flaconcini in vetro da 25 mg AIC n. 040175034/M - “2,5 mg/ml polvere per concentrato per soluzione per infusione” 20 flaconcini in vetro da 25 mg AIC n. 040175046/M - “2,5 mg/ml polvere per concentrato per soluzione per infusione” 5 flaconcini in vetro da 100 mg
9. DATA DELLA PRIMA AUTORIZZAZIONE/RINNOVO DELL’AUTORIZZAZIONEOttobre 2011
10. DATA DI REVISIONE DEL TESTOOttobre 2011
Prezzi*:Levact 5 flaconcini da 25mg Euro 292,42Levact 20 flaconcini da 25mg Euro 1.169,67Levact 5 flaconcini da 100mg Euro 1.169,67 * Prezzo netto di cessione alle strutture del SSN Regime di rimborsabilità:Classe H
Depo
sita
to p
ress
o AI
FA in
dat
a 01
/08/
2012
Co
d LE
V 02
1
01 RCP Medico Levact.indd 8 03/08/12 12.48


















