
IMMUNODEFICIENZE PRIMARIE O EREDITARIE · ancora chiarite (difetto intrinseco dei linfociti B o...
77
IMMUNODEFICIENZE PRIMARIE O EREDITARIE
Transcript of IMMUNODEFICIENZE PRIMARIE O EREDITARIE · ancora chiarite (difetto intrinseco dei linfociti B o...

IMMUNODEFICIENZE
PRIMARIE O EREDITARIEPRIMARIE O EREDITARIE

Granulociti
Neutrofili
Eosinofili
Basofili
Leucociti Monociti
Linfociti B
Natural Killer
T
T helper
T citotossici

Valutazione delle componenti umorali
del sistema immunitario

IMMUNODEFICIENZA
• quadro clinico e anatomo-patologico eterogeneo
• aumentata suscettibilità alle infezioni
• aumentata incidenza di neoplasie
• aumentata incidenza di manifestazioni autoimmuni
IMMUNODEFICENZA PRIMITIVA
O CONGENITA
(dovuti ad un difetto ereditario)
IMMUNODEFICIENZA SECONDARIA
O ACQUISITA
(causati da una noxa esterna)

IMMUNODEFICIENZA PRIMITIVAIMMUNODEFICIENZA PRIMITIVA
IMMUNITA’ NATURALE
• deficit dei fagociti
• deficit del complemento
IMMUNITA’ SPECIFICA
(dovuti ad un anomalo sviluppo, funzione
effettrice o immunoregolatoria)
• deficit dell ’’’’ immunità umorale• deficit dell ’’’’ immunità umorale
(infezioni ricorrenti da piogeni, aumentata
suscettibilità ad alcune infezioni virali e ad
alcuni parassiti intestinali)
• deficit dell ’’’’ immunità cellulo-mediata
(aumentata suscettibilità alle infezioni da
virus, miceti, batteri intra-cellulari e
protozoi; infezioni opportunistiche)
• deficit combinati

Le infezioni ricorrenti suggeriscono
una diagnosi di immunodeficienza
• Storia di infezioni ricorrenti
• Da batteri piogeni legata prevalentemente a
carenze anticorpali del complemento o carenze anticorpali del complemento o
fagocitarie
• Da virus legata prevalentemente a difetti
della risposta mediata dai linfociti T
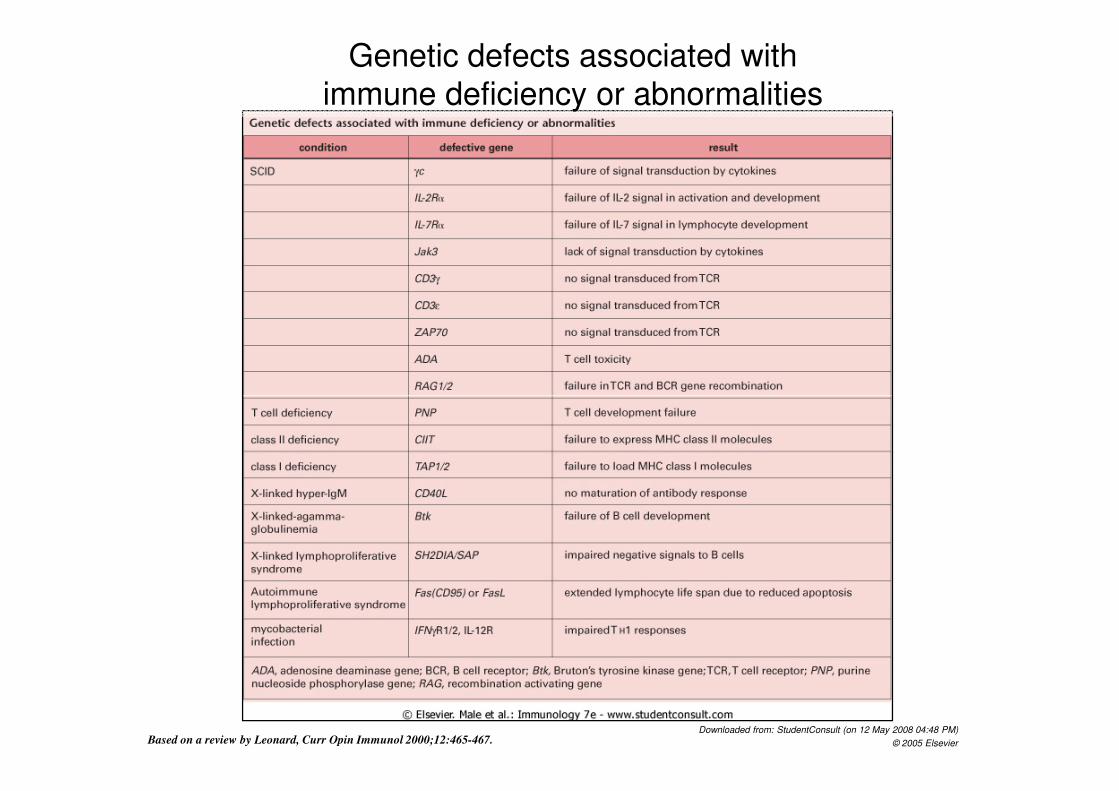
Genetic defects associated with
immune deficiency or abnormalities
Downloaded from: StudentConsult (on 12 May 2008 04:48 PM)
© 2005 Elsevier Based on a review by Leonard, Curr Opin Immunol 2000;12:465-467.

Sindromi da immunodeficienza

Sindromi da immunodeficienza
9

Sindromi da immunodeficienza
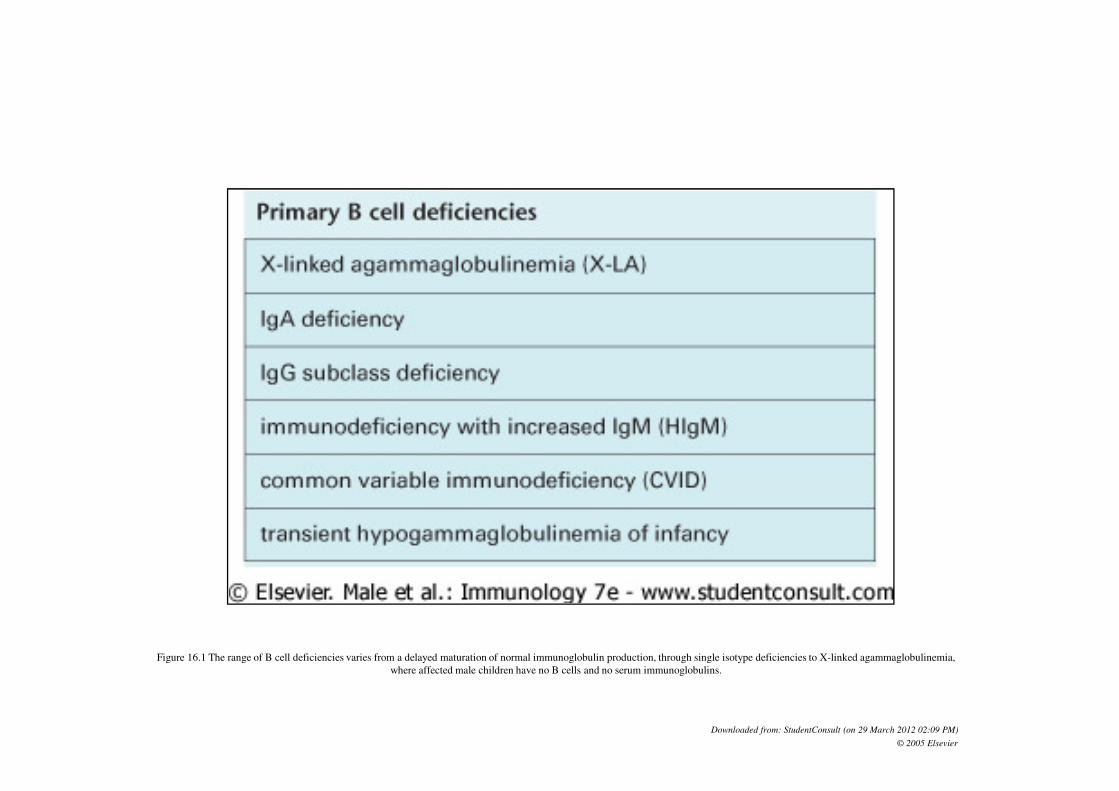
Figure 16.1 The range of B cell deficiencies varies from a delayed maturation of normal immunoglobulin production, through single isotype deficiencies to X-linked agammaglobulinemia,
where affected male children have no B cells and no serum immunoglobulins.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

Figure 16.2 The genes for many immunodeficiency diseases are located on the X chromosome. The genetic defects have been identified for all these diseases. (Adapted from Schwaber J,
Rosen FS. X chromosome linked immunodeficiency. Immunodefic Rev 1990;2:233-251)
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier
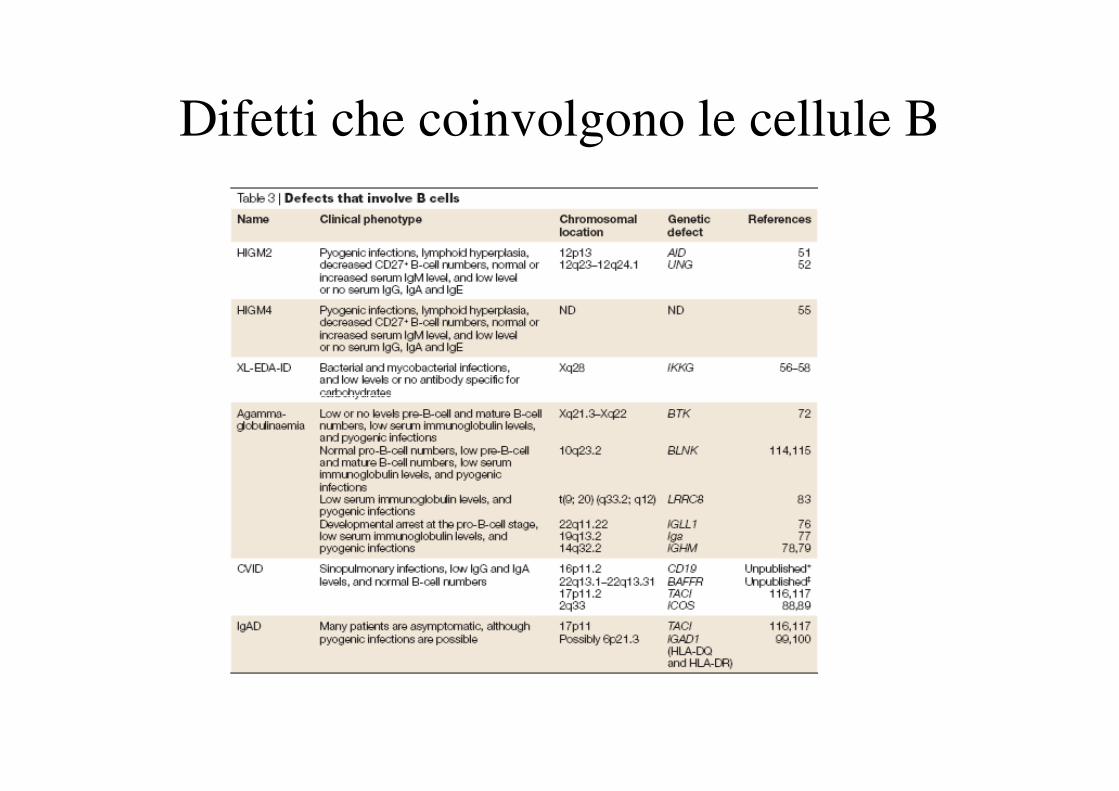
Difetti che coinvolgono le cellule B

Figure 16.3 In X-LA, affected male infants have no B cells and no serum immunoglobulins, except for small amounts of maternal IgG. In IgA deficiency, IgA-bearing B cells, and in some
cases IgG2- and IgG4-bearing B cells, are unable to differentiate into plasma cells. People with HIgM lack IgG and IgA. In CVID, B cells of most isotypes are unable to differentiate into
plasma cells. Black bars denote points of inhibition.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

B cells differentiate from lymphoid stem cells into naive B cells and may then be driven by antigen to become memory cells or plasma
cells. Upon antigen stimulation, the B cell proliferates and develops into a plasma cell or a memory cell after a phase of proliferation,
activation, and blast transformation. During early B cell development, the heavy chain gene (IgH) undergoes a DJ recombination,
followed by a VDJ recombination. The recombined heavy chain is initially expressed with a surrogate light chain and the Igα and
Igβ chains (CD79) to produce the pre-B cell receptor. Later, light chain genes undergo a VJ recombination and surface IgM is
produced as the B cell receptor (BCR). Pre-B cells express cytoplasmic μ chains only. The immature B cell has surface IgM, and the
mature B cell other immunoglobulin isotypes. The diagram also shows the time of expression of a number of B cell markers.
Downloaded from: StudentConsult (on 12 May 2008 04:48 PM)
© 2005 Elsevier
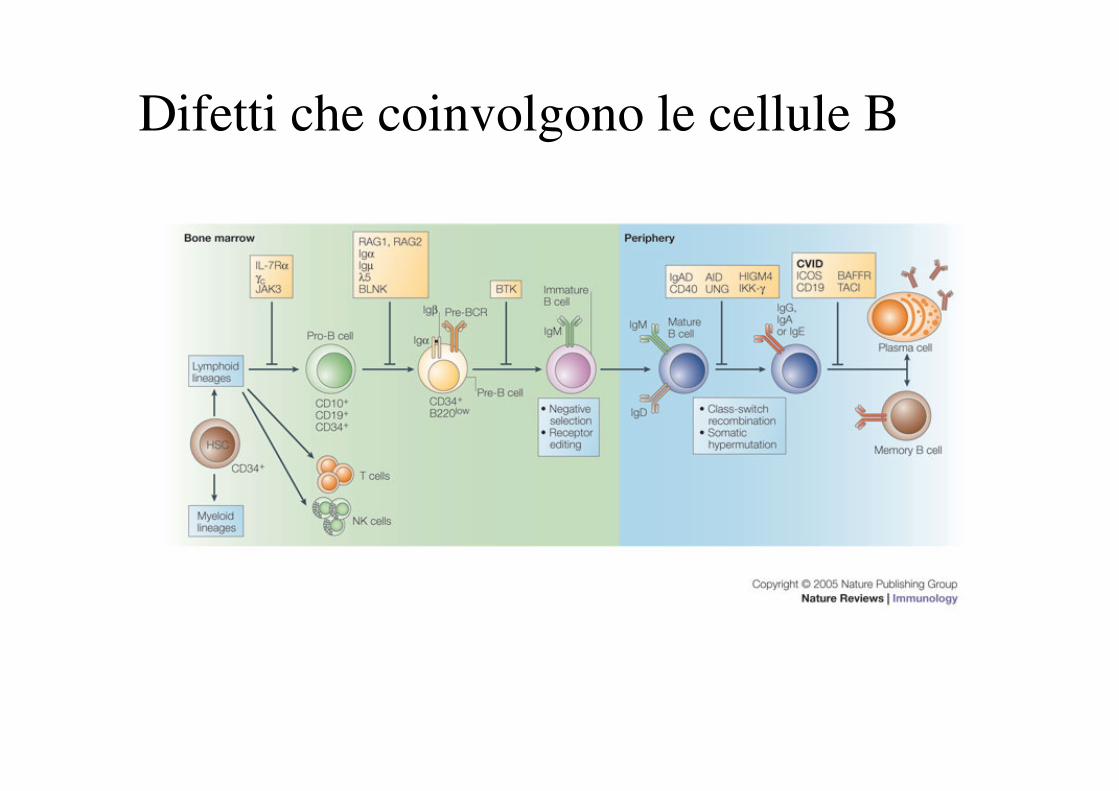
Difetti che coinvolgono le cellule B

Per l’attivazione dei linfociti B
è richiesto un secondo segnale
17Sindrome da iper-IgM

Le cellule T adiuvanti stimolano la proliferazione e
poi il differenziamento (switch isotipico) delle cellule B
X18
X X

PROTEIN AND GENE DEFECTS IN B-CELL DEVELOPMENT AND FUNCTION
Da: C Cunningham-Rundles, PP Ponda. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol. 2005, 5:
880.

AGAMMAGLOBULINEMIA DI BRUTON
Malattia caratterizzata dalldall’’’’’’’’assenzaassenza didi gammaglobulinegammaglobuline nelnel sierosiero; è causata dalla mutazione
di un gene situato sul braccio lungo del cromosoma X che codifica per una tirosin-chinasi (btk)
specificamente espressa dai linfociti durante le fasi di differenziazione dei linfociti B. Gli
individui che presentano tale deficit hanno cellulecellule prepre--BB concon catenecatene µµµµµµµµ normalinormali in cui nonnon sisi
verificaverifica ilil successivosuccessivo riarrangiamentoriarrangiamento delledelle catenecatene leggereleggere, a causa della mancata
produzione di una btk funzionale. Quindi le cellule pre-B sono presenti in numero normale nelproduzione di una btk funzionale. Quindi le cellule pre-B sono presenti in numero normale nel
midollo osseo, mentre i linfociti B sono assenti o fortemente ridotti in circolo e nei tessuti
linfoidi.
Sono colpiti solo gli individui di sesso maschilemaschile, mentre le donne, che sono portatrici sane,
presentano un fenotipo normale.
I neonati sono asintomatici, per la presenza di immunoglobuline di origine materna.

IMMUNODEFICIENZA COMUNE VARIABILE
Questo gruppo di immunodeficit è caratterizzato da un difetto nella produzione anticorpale, ed
è ereditato come tratto autosomico.
E’ solitamente diagnosticato nella tarda infanzia o nell’adolescenza.
I livelli sierici di IgGIgG eded IgAIgA sono bassi, mentre circa la metà dei pazienti presenta livelli
sierici di IgM normali. Il numero di linfociti B circolanti è normale o ridotto, e queste cellule
possono proliferare in risposta alla stimolazione antigenica, ma nonnon riesconoriescono aa differenziarsidifferenziarsi
inin plasmacelluleplasmacellule..Diversamente dai pazienti con agammaglobulinemia legata al sesso, la proliferazione dei
linfociti T in risposta ai mitogeni è diminuita nel 40% dei pazienti con immunodeficienza
comune variabile. Questi pazienti presentano un’alta incidenza di tumori linfoidi e a carico delcomune variabile. Questi pazienti presentano un’alta incidenza di tumori linfoidi e a carico del
tratto gastrointestinale, nonché di malattie autoimmuni.
Questa immunodeficienza è stata attribuita a molteplici alterazioni, quali un difetto intrinseco
dei linfociti B, o una deficitariadeficitaria cooperazionecooperazione dada parteparte delledelle cellulecellule TT. Le cause molecolari
sono note solo per un ristretto gruppo di pazienti. Ad esempio, in alcuni di questi sono state
riscontrate delezionidelezioni deldel genegene codificantecodificante ICOSICOS ((inducibleinducible TT--cellcell costimulatorcostimulator),), unauna
proteinaproteina transmembranatransmembrana espressaespressa selettivamenteselettivamente sullasulla superficiesuperficie deidei linfocitilinfociti TT attivatiattivati. Il
legame di ICOS al suo ligando induce un aumento nella proliferazione cellulare T e nella
produzione di citochine, specialmente di IL-10, che è stata implicata nel differenziamento dei
linfociti B in plasmacellule.

DEFICIT SELETTIVI DI ISOTIPI IG
• deficit selettivo di IgA: forma più comune di deficit selettivo di isotipi di Ig (incidenza di
1:800); deficit primario (modalità di trasmissione autosomica dominante o recessiva) o
acquisito (in seguito all’azione di farmaci o ad infezioni contratte durante la vita intra-
uterina); caratteristiche cliniche variabili, che vanno dalla normalità all’aumentata incidenza
di infezioni dell ’ apparato respiratorio e gastrointestinale, associate ad una maggiore
suscettibilità a malattie autoimmunitarie; patogenesi dovuta ad un blocco della
differenziazione terminale dei linfociti B verso plasmacellule secernenti IgA, per ragioni non
ancora chiarite (difetto intrinseco dei linfociti B o alterata funzione dei linfociti T helper).
• deficit selettivo delle sottoclassi di IgG: normali livelli totali di IgG totali, ma• deficit selettivo delle sottoclassi di IgG: normali livelli totali di IgG totali, ma
concentrazioni di una o più sottoclassi ridotte.
• deficit di IgG ed IgA con aumento delle IgM (sindrome da iper-IgM): notevole carenza
di IgG, IgA, e IgE sieriche, associata ad un marcato aumento di IgM, per mancatomancato switchswitch
isotipicoisotipico delladella catenacatena pesantepesante; linfociti B in circolo, esprimenti IgM di membrana, presenti
in numero normale o aumentato; sono stati identificati diversi difetti molecolari responsabili
di questa patologia, che nei due terzi dei casi è dovuta a unauna mutazionemutazione aa caricocarico deldel dominiodominio
extracellulareextracellulare deldel CDCD4040LL, (espresso sulla superficie dei linfociti T attivati); in altri pazienti è
stata individuata la mancata espressione di CD40 sulla superficie dei linfociti B, macrofagi e
cellule dendritiche (in entrambi i casi, questi individui presentano anche una maggiore
sensibilità verso infezioni opportunistiche).

Figure 16.6 There is a wide range of causes for T cell deficiencies, ranging from absence of lymphocytes, to enzyme deficiency, through to MHC deficiency. All affect the ability of T cells
to function, which leads to combined T and B cell deficiency.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

Difetti di sviluppo dei linfociti T

PROTEIN AND GENE DEFECTS IN T-CELL DEVELOPMENT AND FUNCTION
Da: C Cunningham-Rundles, PP Ponda. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol. 2005, 5: 880.

Difetti di sviluppo e attivazione dei linfociti T.
Es.: sindrome di Di George (aplasia insufficienza timica, CD3)

SINDROME DI DI GEORGESINDROME DI DI GEORGE
Patologia, denominata anche ipoplasiaipoplasia timicatimica, caratterizzata dall’assenza del timo e delle
paratiroidi; si manifesta subito dopo la nascita ed è causato da un anomalo sviluppo della terza
e quarta tasca faringea. E’ associata ad altre malformazioni. La sua comparsa si associa:
• ad abuso di alcool da parte della madre,
• a traslocazioni che coinvolgono il cromosoma 22,
• ereditarietà di tipo autosomico dominante.
Come conseguenza dell’ipoplasia timica, i linfocitilinfociti TT nonnon maturanomaturano, e nonnon sonosono presentipresenti ininCome conseguenza dell’ipoplasia timica, i linfocitilinfociti TT nonnon maturanomaturano, e nonnon sonosono presentipresenti inin
periferiaperiferia o lo sono in numero molto ridotto, e, in questo ultimo caso nonnon proliferanoproliferano inin
rispostarisposta agliagli attivatoriattivatori policlonalipoliclonali; oltre ad una disfunzione delle reazioni immuni di tipo
cellulo-mediato può anche essere presente una alterazione della sintesi di immunoglobuline.
Con l’età la funzione T tende a migliorare, probabilmente grazie sia alla presenza di residui
timici che al processo di maturazione dei linfociti T in tessuti extra-timici.
Recentemente, è stato identificato un difetto genetico in un membro della famiglia dei fattori di
trascrizione T-box, TBX1, come causa dei difetti presenti in questa sindrome.

TOPO SCIDTOPO SCID
Modello animale di immunodeficienza di tipo SCID. Originatosi da una mutazione spontanea
in un ceppo inbred; sono assenti sia i linfociti T che B per un blocco precoce nella
maturazione dai precursori midollari. Il difetto consiste in un’alterazione di DNA-PKCS,
(DNA-dependent protein kinase), una proteina coinvolta nel processo di riparazione delle
rotture del doppio filamento di DNA, tappa finale del processo di riarrangiamento V(D)J
necessario per la generazione di recettori specifici per l’antigene dei linfociti T e B. il
recettore per l’antigene non viene espresso ed i linfociti in via di maturazione vengono
eliminati in vivo.
In una piccola percentuale di animali è presente un piccolo numero di linfociti T e B maturi;In una piccola percentuale di animali è presente un piccolo numero di linfociti T e B maturi;
questo fenotipo, definito “leaky”, è caratterizzato da un repertorio linfocitario limitato.
Un altro tipo di modello animale che presenta un fenotipo simile a quello dei topi SCID, è
rappresentato dai topi knockout per il gene RAG-1 o RAG-2 (Recombination-Activating
Gene), geni i cui prodotti proteici sono anche essi coinvolti nel processo di riarrangiamento
delle sequenze V(D)J.

TOPO NUDOTOPO NUDO
Modello animale di immunodeficienza. Un difetto ereditario localizzato in un gene recessivo
localizzato sul cromosoma 11, che interessa le cellule epiteliali cutanee e il pavimento della
terza e quarta tasca faringea. Nei topi omozigoti si manifesta il fenotipo caratterizzato
dall’assenza del pelo e dall’ ipoplasia timica. Come per l’uomo, non si verifica la
maturazione dei linfociti T che sono assenti o quasi nei tessuti linfoidi periferici. Non sono
quindi presenti risposte immunitarie cellulo-mediate, quali il rigetto di allotrapianti, la DTH e
la risposta anticorpale verso antigeni proteici timo-dipendenti. I topi nudi sono suscettibili a
molte infezioni ma riescono ad eliminare alcuni batteri-intracellulari, grazie alla presenza di
un numero normale di linfociti natural killer (NK), che, tramite la produzione di IFN-γ, sono
in grado di attivare le capacità microbicide dei macrofagi.

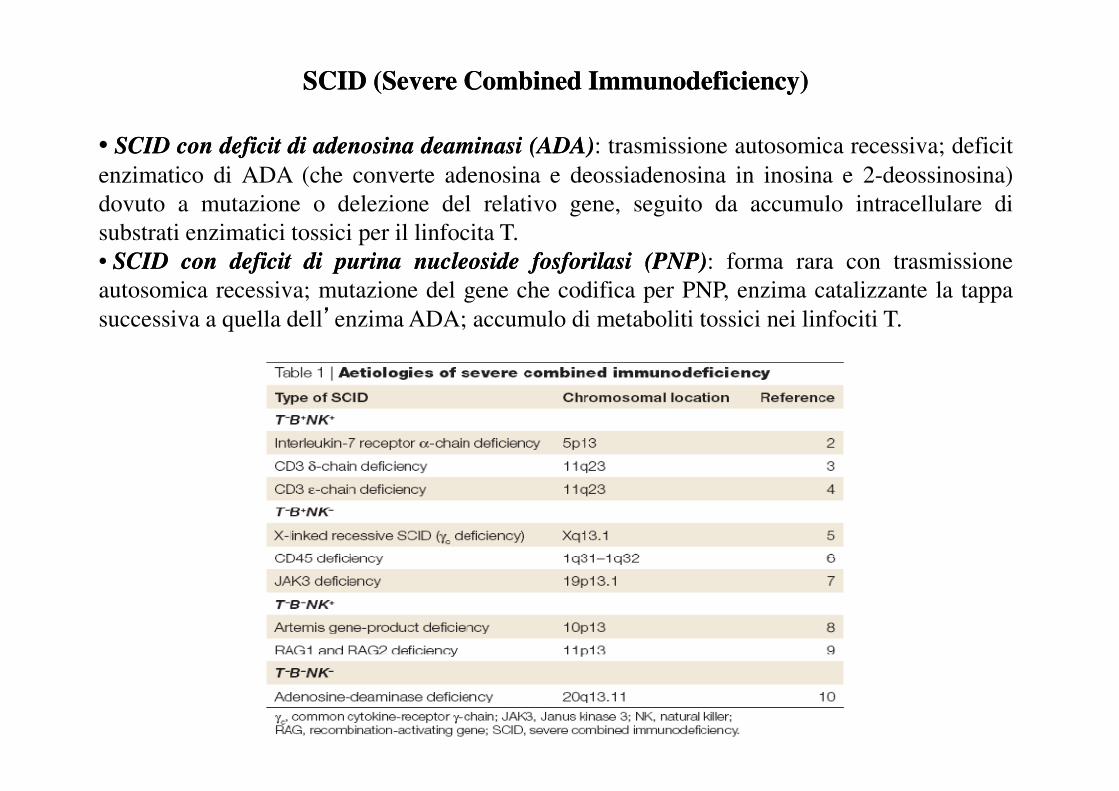
SCID (Severe Combined Immunodeficiency) SCID (Severe Combined Immunodeficiency)
Questa immunopatologia comprende un gruppo eterogeneo di sindromi cliniche, caratterizzate
dalla forte diminuzione o assenza di linfociti T funzionali, associata in vario modo a deficit di
altre popolazioni linfocitarie e leucocitarie. E’ la conseguenza di mutazioni che avvengono in
geni diversi, ereditate in modo autosomico recessivo o recessivo legato al cromosoma X. Tutti
i tipi di microrganismi determinano infezioni nei pazienti, con prevalenza delle infezioni
opportunistiche. L’immunodeficienza, in mancanza di un opportuno intervento, porta a morte
i pazienti entro il primo anno di vita.
• SCIDSCID concon ipoplasiaipoplasia emopoieticaemopoietica generalizzatageneralizzata: forma più grave di SCID, con deficit della
serie cellulare linfoide e mieloide per un difetto differenziativo della cellula staminaleserie cellulare linfoide e mieloide per un difetto differenziativo della cellula staminale
totipotente (assenza di immunità specifica e naturale).
• AgammaglobulinemiaAgammaglobulinemia didi tipotipo svizzerosvizzero: trasmissione ereditaria autosomica recessiva;
mancato sviluppo della cellula staminale linfoide.
• SindromeSindrome deldel linfocitalinfocita nudonudo: tramissione autosomica recessiva; assenza delle molecole
MHC II, dovuta a mutazioni che interessano i geni regolatori dell’espressione delle molecole
MHC; incapacità delle APC di presentare l’antigene ai linfociti T CD4, con conseguente
deficit delle risposte DTH e delle risposte anticorpali T-dipendenti.
• SCIDSCID concon trasmissionetrasmissione recessivarecessiva legatalegata alal cromosomacromosoma XX: mutazione del gene codificante
per la catena γ comune utilizzata da diversi recettori delle citochine, quali IL-2, IL-4, IL-7, IL-
9, IL-15 e IL-21.

Figure 11.25 (1) A defect in the common chain (γc) of the cytokine receptors IL-2, IL-15, IL-4, IL-7, and IL-9
leads to a SCID, with loss of both T and NK cells. A similar deficiency results from mutation in the janus kinase (Jak3),
which transduces signals from the γc chain. Note that IL-2 and IL-15 have three chains in their high-affinity
receptor, whereas IL-4, IL-7, and IL-9 have only two chains. (2) Absence of the specific IL-7R chain also produces a
severe immunodeficiency, but this primarily affects T cell development.
Downloaded from: StudentConsult (on 12 May 2008 04:48 PM)
© 2005 Elsevier
SCID (Severe Combined Immunodeficiency) SCID (Severe Combined Immunodeficiency)
• SCIDSCID concon deficitdeficit didi adenosinaadenosina deaminasideaminasi (ADA)(ADA): trasmissione autosomica recessiva; deficit
enzimatico di ADA (che converte adenosina e deossiadenosina in inosina e 2-deossinosina)
dovuto a mutazione o delezione del relativo gene, seguito da accumulo intracellulare di
substrati enzimatici tossici per il linfocita T.
• SCIDSCID concon deficitdeficit didi purinapurina nucleosidenucleoside fosforilasifosforilasi (PNP)(PNP): forma rara con trasmissione
autosomica recessiva; mutazione del gene che codifica per PNP, enzima catalizzante la tappa
successiva a quella dell’enzima ADA; accumulo di metaboliti tossici nei linfociti T.
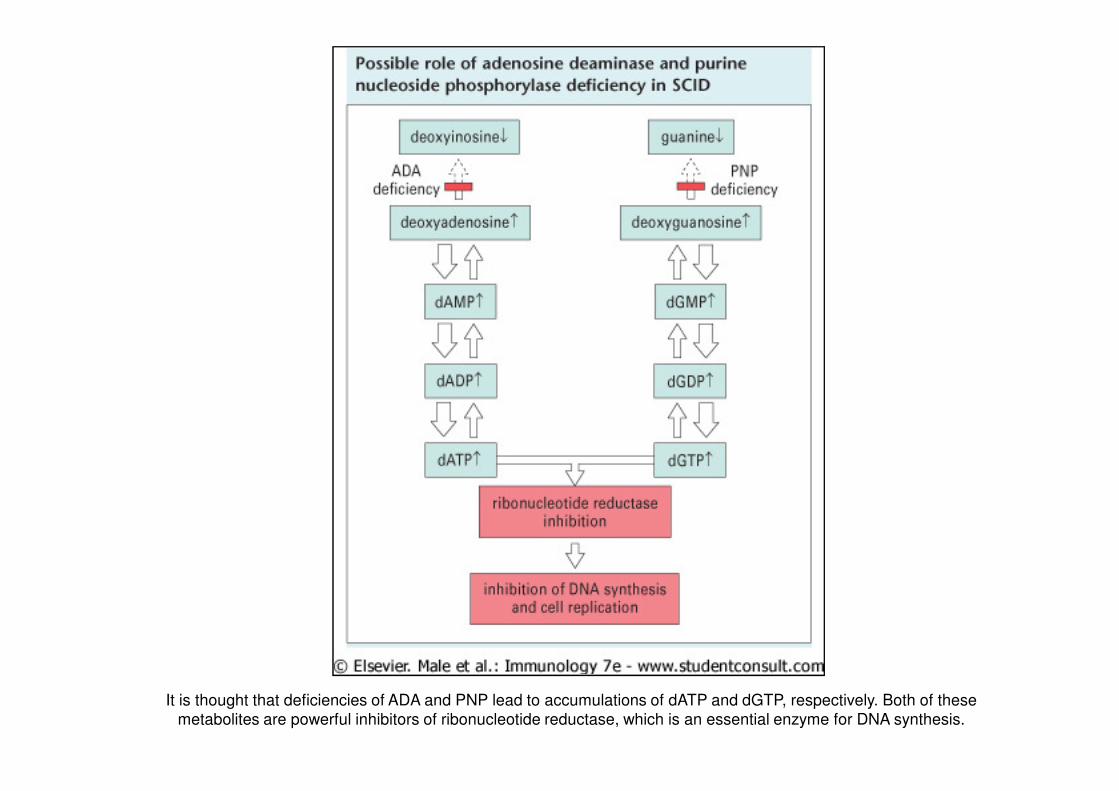
It is thought that deficiencies of ADA and PNP lead to accumulations of dATP and dGTP, respectively. Both of these
metabolites are powerful inhibitors of ribonucleotide reductase, which is an essential enzyme for DNA synthesis.
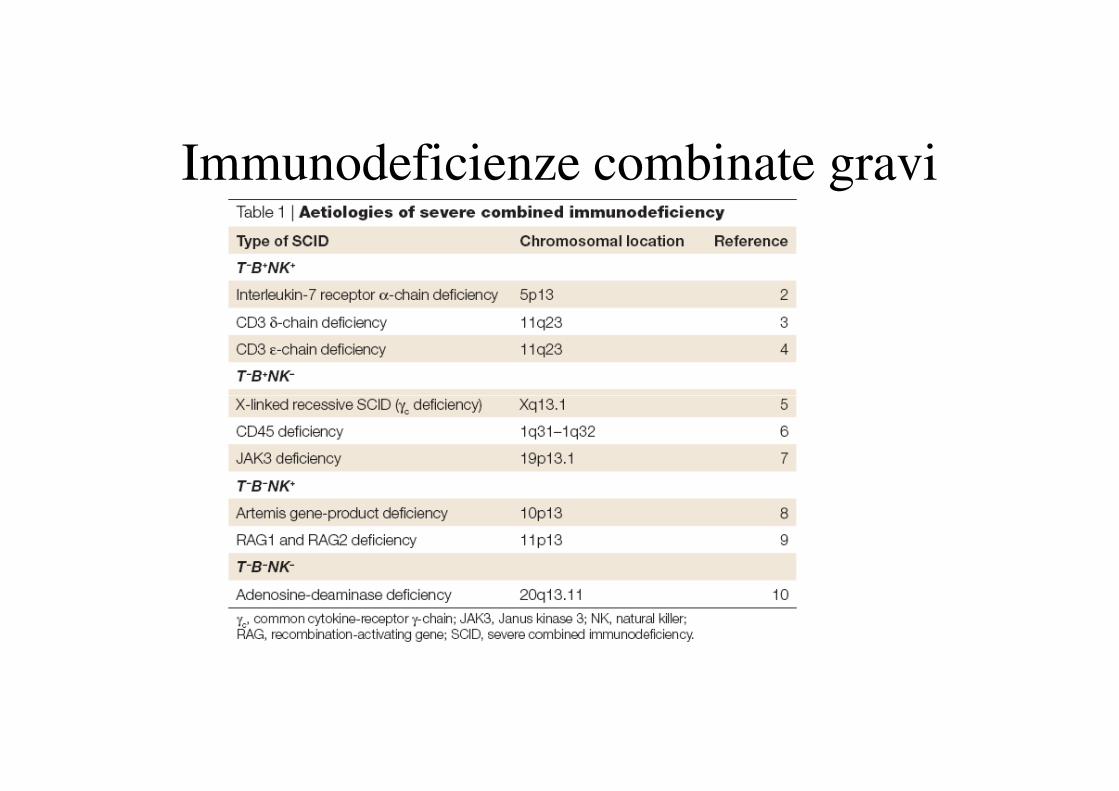
Immunodeficienze combinate gravi
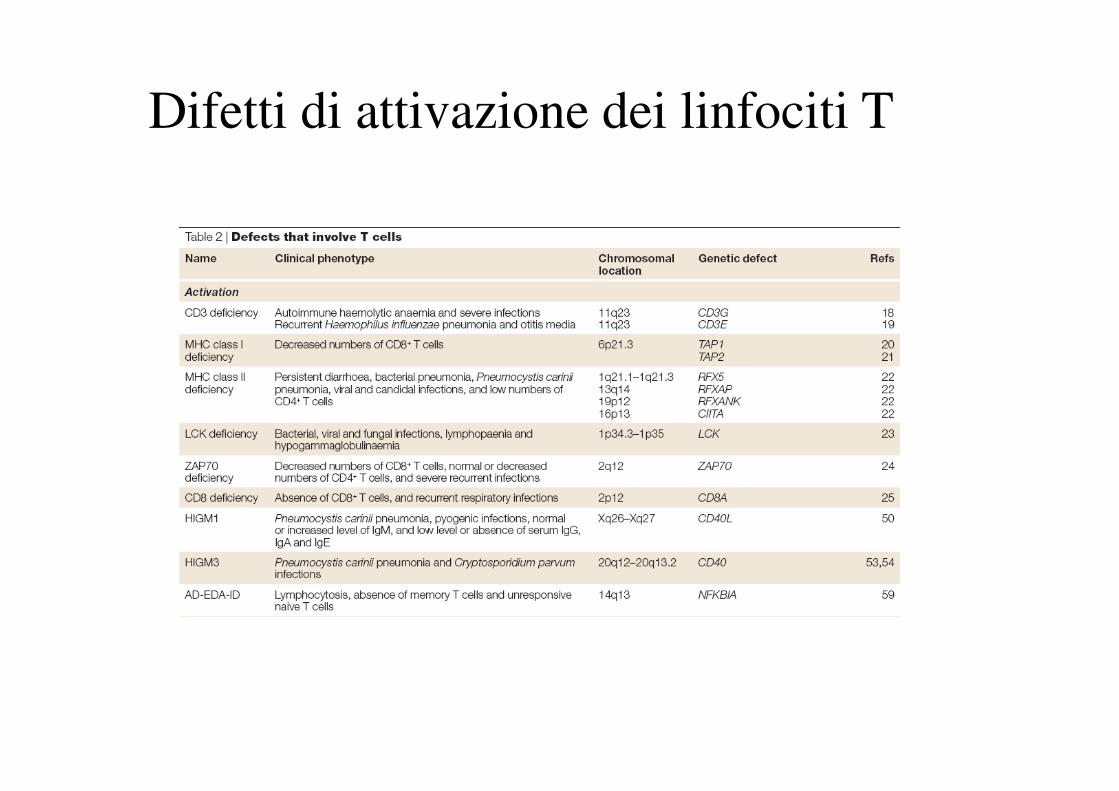
Difetti di attivazione dei linfociti T

Difetti di sviluppo e attivazione dei linfociti T.
Es.: deficienza di molecole di classe II


L’assenza di molecole di classe II nel timo non
permette la selezione positiva dei linfociti T CD4+
Cellule epitelialidella corticale timica
MHC classe II +
Cellule epitelialidella corticale timica
MHC classe II -
IN PIU’’’’
Assenza di presentazione antigenica
38
Linfociti T CD4+ Linfociti T CD8+

Difetti funzionali e regolatori
dei linfociti T

Esempio di patologia in cui un difetto immunitario
caratterizzato da una risposta proliferativa abnorme dei
linfociti T causa immunodeficienza. In tali pazienti è
stata individuata una mutazione del gene SH2D1A,
codificante per SAP (SLAM-associated protein), una
proteina citoplasmatica in grado di legare SLAM
(signalling lymphocytic activation molecule). SLAM è
SINDROME LINFOPROLIFERATIVA LEGATA AL CROMOSOMA X (XLP)SINDROME LINFOPROLIFERATIVA LEGATA AL CROMOSOMA X (XLP)
SH2D1A gene: SLAM-Associated Protein (SAP),
Signalling Lymphocytic Activation Molecule (SLAM)
(signalling lymphocytic activation molecule). SLAM è
una proteina transmembrana la cui espressione sulla
superficie dei linfociti aumenta dopo attivazione
cellulare; il legame tra SAP e SLAM media un segnale
inibitorio.
Per ragioni non ancora chiare, mutazioni a carico di
SAP determinano una proliferazione abnorme dei
linfociti T di individui infettati dal virus di Epstein-
Barr, una eliminazione virale inefficace, la comparsa di
linfoma o ipogammaglobulinemia.

Difetti funzionali e regolatori
dei linfociti T

ALPS:
autoimmune lymphoproliferative syndrome
dysregulation of apoptosis mediated by CD95

Difetti funzionali e regolatori
dei linfociti T

SINDROME IPEX SINDROME IPEX
(IMMUNODYSREGULATION, POLYENDOCRINOPATHY AND (IMMUNODYSREGULATION, POLYENDOCRINOPATHY AND
ENTEROPATHY, XENTEROPATHY, X--LINKED)LINKED)
Esempio di immunodeficienza determinata da alterata
immunoregolazione. Patologia della prima infanzia, con
esito spesso fatale, caratterizzata a livello clinico dalla
presenza di diabete, enteropatia e dermatite. Questa
Linfociti T effettori self reattivi non sono
inibiti da Treg
Mutazioni di FOXP3 comporta perdita di Treg
sindrome è associata ad autoimmunità e ad infezioni
ricorrenti con Enterococcus e Staphylococcus. Il difetto
molecolare è stato individuato nella mutazione del gene
FOXP3, che codifica per un fattore di trascrizione.
FOXP3 è principalmente espresso e rappresenta un
marker dei linfociti T regolatori CD4+ CD25hi. Tale
mutazione determina l’assenza di linfociti T regolatori e
quindi la mancata inibizione dei linfociti T effettori
autoreattivi.

Difetti delle cellule fagocitarie(persistenza di infezioni batteriche)
45

DEFICIT DEI FAGOCITIDEFICIT DEI FAGOCITI
• DeficitDeficit didi adesioneadesione deidei leucocitileucociti didi tipotipo 11 (LAD(LAD--11)): patologia autosomica recessiva
caratterizzata da infezioni batteriche e fungine ricorrenti, con incapacità a produrre essudato
purulento e una normale cicatrizzazione. Patogenesi dovuta a diminuita o assente espressione
delle integrine β2 (famiglia di glicoproteine CD18/CD11) causata da una deficitaria sintesi
della catena β (CD18). In questi pazienti sono alterate la maggior parte delle funzioni
leucocitarie mediate da queste molecole di adesione, come:
• l’aderenza agli endoteli
• l’aggregazione e la chemiotassi dei neutrofili
• la fagocitosi• la fagocitosi
• l’attività cititossica mediata da neutrofili, NK e CTL.
• MalattiaMalattia granulomatosagranulomatosa cronicacronica (MGC)(MGC): malattia rara, a trasmissione ereditaria
(autosomica recessiva o legata al cromosoma X), caratterizzata da gravi e ricorrenti infezioni
di batteri e funghi. Il meccanismo patogenetico della MGC risiede nell’assenza del burst
respiratorio causata da un difetto di produzione dell’anione superossido, per alterata
funzionalità della NADPH ossidasi. Si osserva la formazione di granulomi in seguito al
rilascio di microbi vivi da parte dei fagociti e al conseguente richiamo di nuovi fagociti.

Figure 4.1 Complement has a central role in inflammation causing chemotaxis of phagocytes, activation of mast cells and phagocytes, opsonization and lysis of pathogens, and clearance of
immune complexes.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

Each of the activation pathways generates a C3 convertase, which converts C3 to C3b, the central event of the complement pathway. C3b in turn
activates the terminal lytic membrane attack pathway. The first stage in the classical pathway is the binding of antigen to antibody. The alternative
pathway does not require antibody and is initiated by the covalent binding of C3b to hydroxyl and amine groups on the surface of various
microorganisms. The lectin pathway is also triggered by microorganisms in the absence of antibody, with sugar residues on the pathogen surface
providing the binding sites. The alternative and lectin pathways provide non-specific 'innate' immunity, whereas the classical pathway represents a
more recently evolved link to the adaptive immune system.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

A summary of the clinical consequences of the various complement deficiencies.
Black arrows denote pathway, red arrows show strong effects, and blue arrows indicate weak effects.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

Difetti nei componenti del complemento(suscettibilità a infezioni e accumulo di immunocomplessi)
50
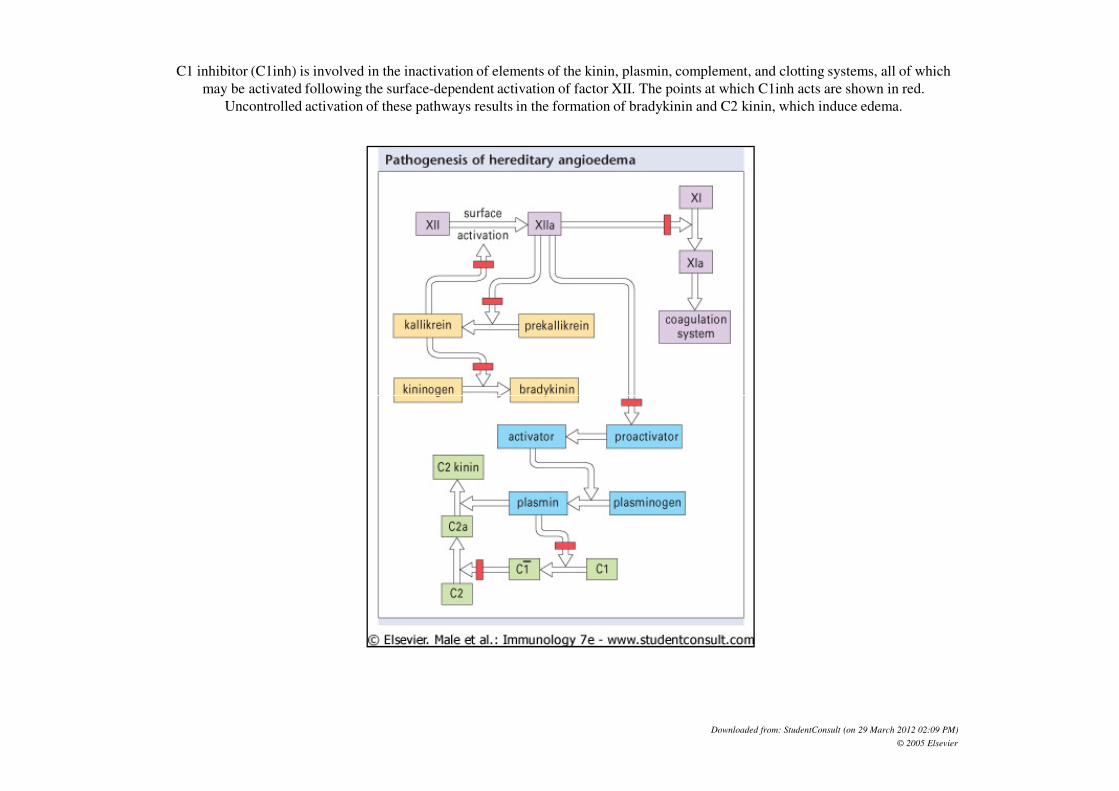
C1 inhibitor (C1inh) is involved in the inactivation of elements of the kinin, plasmin, complement, and clotting systems, all of which
may be activated following the surface-dependent activation of factor XII. The points at which C1inh acts are shown in red.
Uncontrolled activation of these pathways results in the formation of bradykinin and C2 kinin, which induce edema.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

Figure 4.18 The clinical appearance of hereditary angioedema, showing the local transient swelling that affects mucous membranes.
Downloaded from: StudentConsult (on 29 March 2012 02:09 PM)
© 2005 Elsevier

Gli strumenti per il monitoraggio

Valutazione delle componenti cellulari
del sistema immunitario
54

Serum protein electrophoresis.

Agammaglobulinemia legata all’X
56

Ricerca anticorpi antigene specifici:
Enzyme-linked immunoassorbent assay
5757
Conta con emocitometro
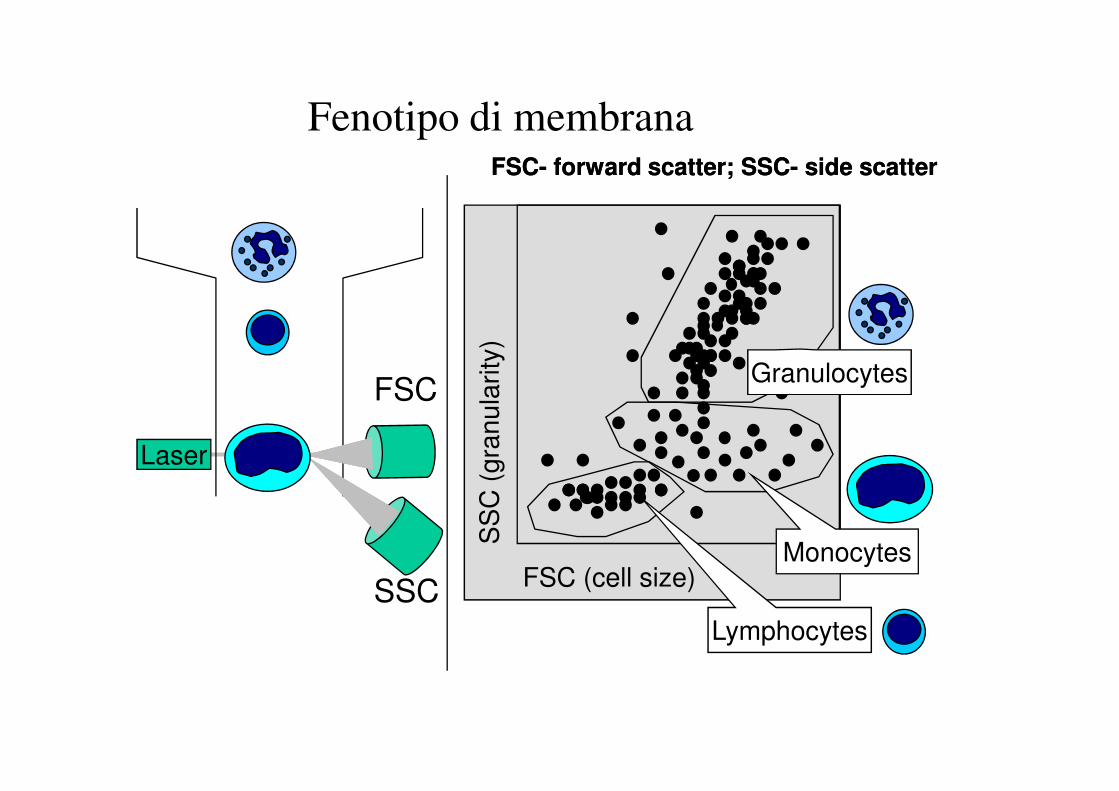
Fenotipo di membrana
SS
C (
gra
nu
lari
ty)
GranulocytesFSC
FSCFSC-- forward scatter; SSCforward scatter; SSC-- side scatterside scatter
Laser
FSC (cell size)
SS
C (
gra
nu
lari
ty)
Monocytes
Lymphocytes
FSC
SSC

GRANULOCYTESGRANULOCYTES
MONOCYTES
LYMPHOCYTES

Antigene A
Anticorpo anti-A
Antigene B
Anticorpo anti-B
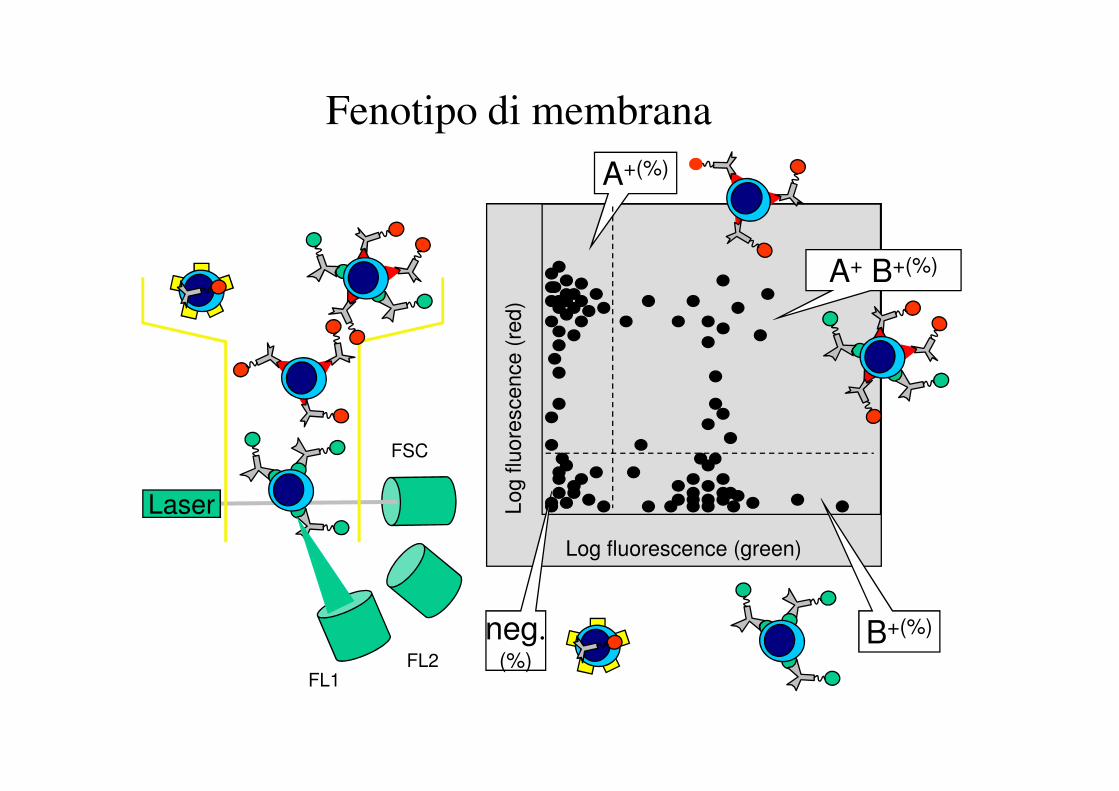
Fenotipo di membrana
Lo
g flu
ore
sce
nce
(re
d)
A+(%)
A+ B+(%)
Laser
Log fluorescence (green)
Lo
g flu
ore
sce
nce
(re
d)
B+(%)neg.(%)
FSC
FL1FL2

CD4
Fenotipo di membrana
fluorescenzafluorescenza
CD4
CD8

La conta assoluta dei linfociti T CD4

Lymphocyte subsets by Flow Cytometry

Test funzionali in vitro
66

IMMUNODEFICIENZE SECONDARIEIMMUNODEFICIENZE SECONDARIE
• Immunodeficit secondari ad un processo morboso: malnutrizione, neoplasie ed infezioni.
• Immunodeficit causati da terapie mirate a combattere altre malattie (immunodeficienze
iatrogene): farmaci ad attività immunosoppressiva (corticosteroidi, ciclosporina), farmaci
anti-neoplastici.

Downloaded from: StudentConsult (on 12 May 2008 04:39 PM)




Measles virus (MV) infection induces a profound immunosuppression responsible for a high rate of mortality in
malnourished children. MV can encounter human dendritic cells (DCs) in the respiratory mucosa or in the secondary
lymphoid organs. The purpose of this study was to investigate the consequences of DC infection by MV, particularly
concerning their maturation and their ability to generate CD8 T cell proliferation.
•MV-infected Langerhans cells or monocyte-derived DCs undergo a maturation process
similarly to the one induced by TNF-a or LPS, respectively.
•CD40 ligand (CD40L) expressed on activated T cells is shown to induce terminal
differentiation of DCs into mature effector DCs.differentiation of DCs into mature effector DCs.
•In contrast, the CD40L-dependent maturation of DCs is inhibited by MV infection, as
demonstrated by CD25, CD69, CD71, CD40, CD80, CD86, and CD83 expression down-
regulation.
•Moreover, the CD40L-induced cytokine pattern in DCs is modified by MV infection with
inhibition of IL-12 and IL-1a/b and induction of IL-10 mRNAs synthesis.
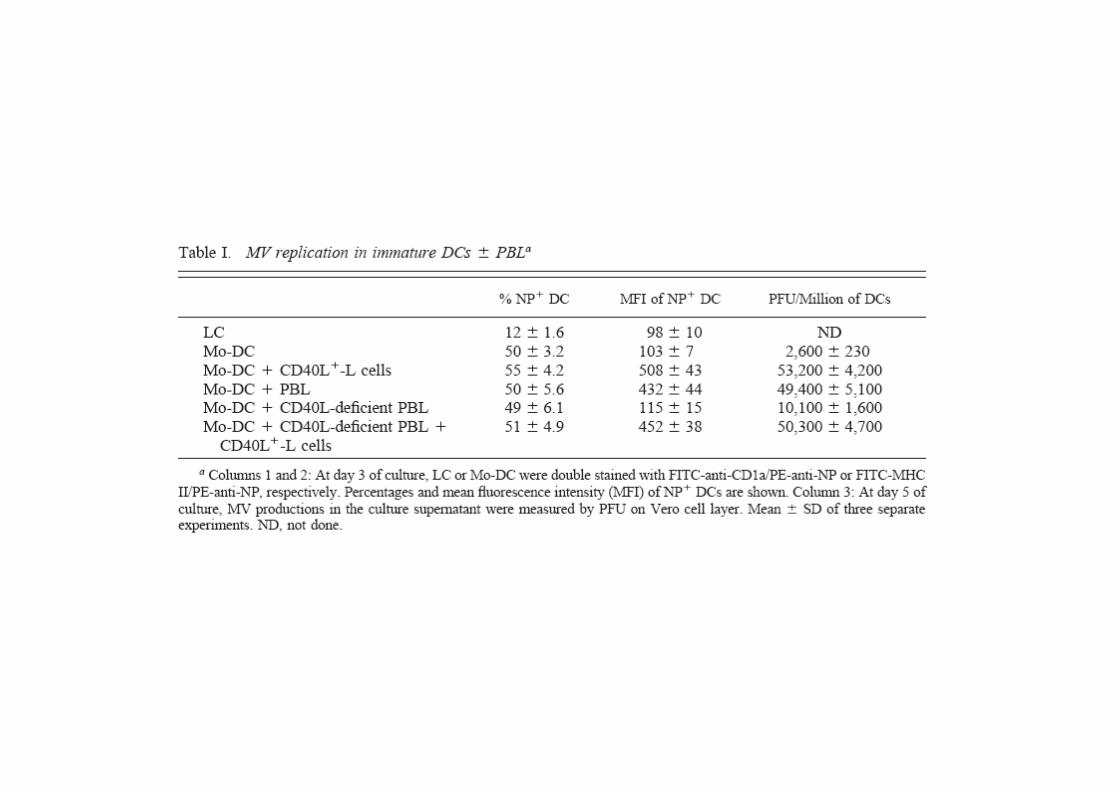



Using peripheral blood lymphocytes from CD40L-deficient patients, wedemonstrate that MV infection of DCs prevents the CD40L-dependentCD8 T cell proliferation. In such DC-PBL cocultures, inhibition of CD80and CD86 expression on DCs was shown to require both MV replicationand CD40 triggering. Finally, for the first time, MV was shown to inhibittyrosine-phosphorylation level induced by CD40 activation in DCs. Ourdata demonstrate that MV replication modifies CD40 signaling in DCs,data demonstrate that MV replication modifies CD40 signaling in DCs,thus leading to impaired maturation. This phenomenon could play apivotal role in MV-induced immunosuppression.














![I linfociti T [modalità compatibilità] · 2008. 6. 26. · I linfociti T sono le cellule dell’immunità adattativa responsabili della protezione verso le infezioni ad opera dei](https://static.fdocumenti.com/doc/165x107/60f946bca41c975adb05678c/i-linfociti-t-modalit-compatibilit-2008-6-26-i-linfociti-t-sono-le-cellule.jpg)


