EREDITARIE: Alterazioni della membrana Deficit enzimatici ... · flessibilità della membrana...
29
ANEMIE EMOLITICHE O DA AUMENTO DELLA PERDITA ANEMIE EMOLITICHE Anomalie intrinseche (intracorpuscolari): EREDITARIE: Alterazioni della membrana SFEROCITOSI ELLISSOCITOSI Difetti della sintesi lipidica Deficit enzimatici Deficit di enzimi glicolitici (piruvato chinasi, esochinasi) Deficit degli enzimi dello shunt dei pentoso-fosfati (G6PDH, glutatione sintetasi) Deficit della sintesi dell’emoglobina Sindromi talassemiche Anemia drepanocitica Emoglobine instabili ACQUISITE: Alterazioni della membrana emoglobinuria parossistica notturna
Transcript of EREDITARIE: Alterazioni della membrana Deficit enzimatici ... · flessibilità della membrana...

ANEMIE EMOLITICHE O DA AUMENTO DELLA PERDITA
ANEMIE EMOLITICHE
Anomalie intrinseche (intracorpuscolari):EREDITARIE:
Alterazioni della membrana SFEROCITOSIELLISSOCITOSIDifetti della sintesi lipidica
Deficit enzimaticiDeficit di enzimi glicolitici (piruvato chinasi, esochinasi)
Deficit degli enzimi dello shunt dei pentoso-fosfati (G6PDH, glutatione sintetasi)
Deficit della sintesi dell’emoglobinaSindromi talassemicheAnemia drepanociticaEmoglobine instabili
ACQUISITE:
Alterazioni della membrana
emoglobinuria parossistica notturna

Anomalie estrinseche (extracorpuscolari):MEDIATE DA ANTICORPI:
ISOEMOAGGLUTININE (ALLOANTICORPI): eritroblastosi fetale, reazioni trasfusionali
AUTOANTICORPI: da Ab caldi, da Ab freddi, da Ab bitermici
DA FARMACI
DA TRAUMA MECCANICO E DA AGENTI FISICI:Emoglobinuria da marciaAnemia emolitica cardiaca traumaticaAnemia emolitica microangiopaticaUstioni
IPERSPLENISMODA AGENTI CHIMICI: farmaci, xenobiotici
DA INFEZIONI:
BATTERI: clostridi spp, Diplococcus pneumoniae, Piogenus aureus
VIRUS: Mycoplasma pneumoniae, HBV, HBV e HCV, citomegalovirus, herpes virus
PROTOZOI: Plasmodium falciparum
AUMENTO DELLA PERDITA
EMORRAGIA CRONICAEMORRAGIA ACUTA

CONSIDERAZIONI GENERALI
Le anemie emolitiche sono caratterizzate da:
1. Accorciamento della t½ dei GR
2. Accumulo dei prodotti del catabolismo dell’Hb (bilirubinemia, urobilinogeno)
3. Iperplasia compensativa del midollo osseo
• L’emolisi può essere intravasale: emoglobinemia, emoglobinuria e metaemoglobinuria,metaemealbuminuria, ittero, emosidernuria, diminuzione dei livelli sierici di aptoglobina
• L’emolisi può essere extravasale: in sede splenica, si verica quando le emazie sono danneggiate o rese “estranee” all’organismo o perdono la loro plasticità. Sequestro splenico ⇒ splenomegalia. Non compaiono emoglobinemia ed emoglobinuria, tuttavia possono essere presenti ittero e diminuzione di aptoglobina per aumento del catabolismodell’Hb.
LINEE GUIDA PER LA DIAGNOSI:
Il paziente è pallido, subitterico o itterico, può presentare splenomegalia. Frequentemente si ha emissione di urine scure o francamente rossastre. Anamnesi familiare.
• incremento della conta reticolocitaria
• aumento della bilirubinemia indiretta
• Ipersideremia
• diminuzione dell’aptoglobina se l’emolisi è intravasale

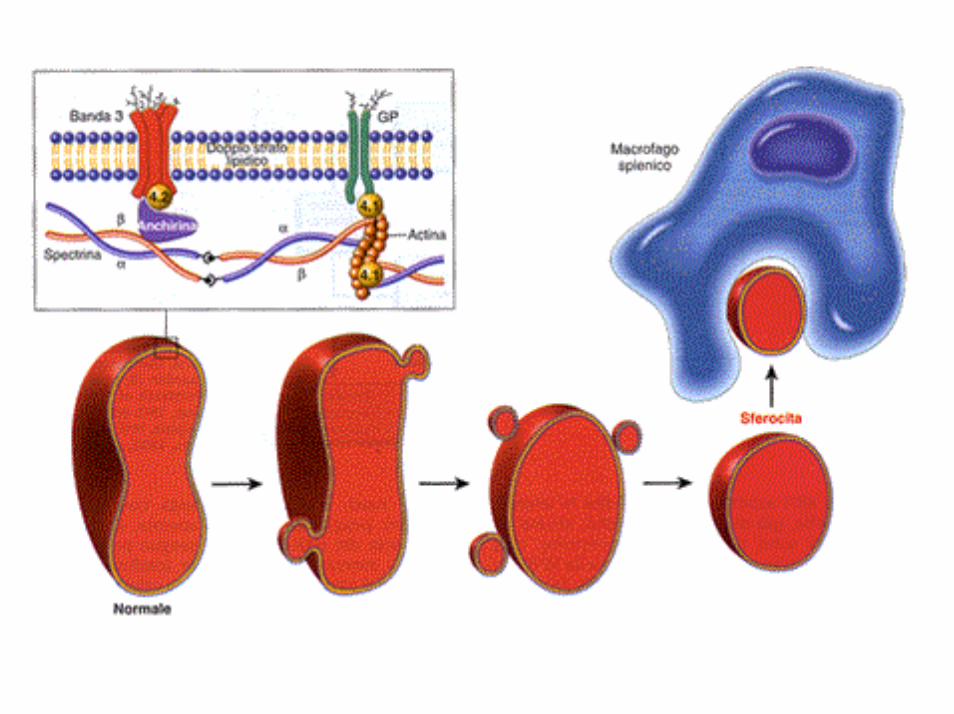
SFEROCITOSI EREDITARIA o malattia di Minkowsky-Chauffard
EZIOLOGIA: difetto intrinseco del citoscheletro della membrana degli eritrociti ereditato come carattere autosomico dominante.
La SE è stata riscontrata in molte razze. Negli USA ed in Europa ha un’incidenza di 1:5000 ed è diagnosticata ad ogni età. Negli USA è stata riscontrata raramente nei neri ed in altri gruppi razziali. In Europa potrebbe essere sottostimata: si è notata una prevalenza del 1% di aumentata fragilità osmotica nei donatori di sangue apparentemente sani. La SE ha nel 75% dei casi carattere autosomico dominante. Nel restante 25%, non avendo riscontrato familiari malati, si è ipotizzata o una trasmissione di tipo recessivo, o l’insorgenza di nuove mutazioni. Il gene della SE dovrebbe trovarsi sul cromosoma 6 vicino al locus dell’HLA o sul cromosoma 12.

PATOGENESI
La forma sferica dell’eritrocita appare il risultato di un difetto fondamentale dello scheletro di membrana, il difetto primario non è noto con certezza. Le proteine chiamate in causa nella patogenesi della SE sono quattro; si può avere, infatti, una carenza di: spectrina, anchirina(con deficit secondario di spectrina), banda 3, proteina 4.1.
• la spectrina è la principale proteina delcitoscheletro di membrana e consta di 2 catenepolipeptidiche α e β a formare un dimero elicoidale. I singoli dimeri rappresentano gli elementi di un’estesa rete di collegamento con altre proteine del citoscheletro.
• l’anchirina costituisce un ponte tra le molecole di spectrina e la proteina banda 3 che funge da trasportatore di ioni Cl- e HCO3
-
• la proteina 4.1 collega la spectrina allaglicoforina
L’insieme di questi complessi sovramolecolariconferisce la forma, l’elasticità e la flessibilità della membrana eritrocitaria
Il difetto di spectrina è l’anomalia più comune in tutte le forme di SE

• Il contenuto di spectrina in questi pazienti varia dal 60 al 90% ed è correlato con la gravità della patologia.
• Tutte le mutazioni che si osservano nella sferocitosi ereditaria comportano un difetto nella struttura del citoscheletro ed, in particolar modo, inficiano le interazioni verticali con ilbilayer di membrana.
• sono state identificate mutazioni anche nel gene dell’anchirina e nel gene della banda 3 (20% dei casi).
Il deficit di spectrina è accompagnato da ridotta stabilità di membrana e da perdita spontanea di frammenti della membrana cellulare eritrocitaria quando le cellule sono sottoposte alle sollecitazioni che incontrano in circolo.
L’assunzione della forma sferica modifica le proprietà della membrana eritrocitaria: se normalmente il globulo rosso riesce a deformarsi aumentando anche del 230% la propria lunghezza (ciò gli è indispensabile quando deve passare attraverso i cordoni di Billroth ed entrare nei sinusoidi splenici che hanno dimensioni inferiori di 2-3 µm), gli sferociti se da un lato non riescono a deformare la propria membrana, dall’altro sono più sensibili agli stress osmotici.
Sequestro splenico degli sferociti

DECORSO CLINICO E DIAGNOSI:
Gli aspetti clinici caratteristici sono: anemia, splenomegalia ed ittero. La gravità della malattia varia da un paziente all’altro. Molti pazienti sviluppano calcoli biliari bilirubinici in seguito all’iperemolisi cronica.
• in una minoranza di pazienti la patologia può comparire sin dalla nascita con ittero intenso ⇒ trasfusione
• nel 20-30% dei casi la malattia è asintomatica, l’emolisi è modesta e compensata da aumento dell’eritropoiesi.
• nella maggior parte dei casi anemia emolitica cronica non compensata ⇒ anemia moderata o lieve. Talvolta si manifestano crisi aplastiche (parvovirus) in cui possono essere necessarie trasfusioni.
La DIAGNOSI si basa sull’anamnesi familiare, sui reperti ematologici e sul test di fragilitàosmotica.
La maggior parte dei pazienti trae beneficio dalla splenectomia.

CARENZA DI G6PDH o FAVISMO
EZIOLOGIA: deficit ereditario della glucoso-6-fosfato deidrogenasi. L’eredità è legata al cromosoma X (locus Xq28), nei maschi il deficit enzimatico è decisamente marcato, le donne possono essere portatrici sane o affette.
La carenza di G6PDH è una malattia rara, ne sono affette 400 milioni di persone al mondo, in Italia circa 400000, circa lo 0,8% della popolazione. E’ diffusa soprattuttto in Africa, in Asia meridionale, e nel bacino del Mediterraneo. In Italia l’incidenza più alta si ha in Sardegna, nella zona del delta del Po ed in Veneto. Anche per la diffusione della carenza di G6PDH ha avuto un ruolo importante la malaria.

PATOGENESI
L’enzima G6PDH catalizza la reazione di deidrogenazione del G6P a F6P con produzione di NADPH(H+)
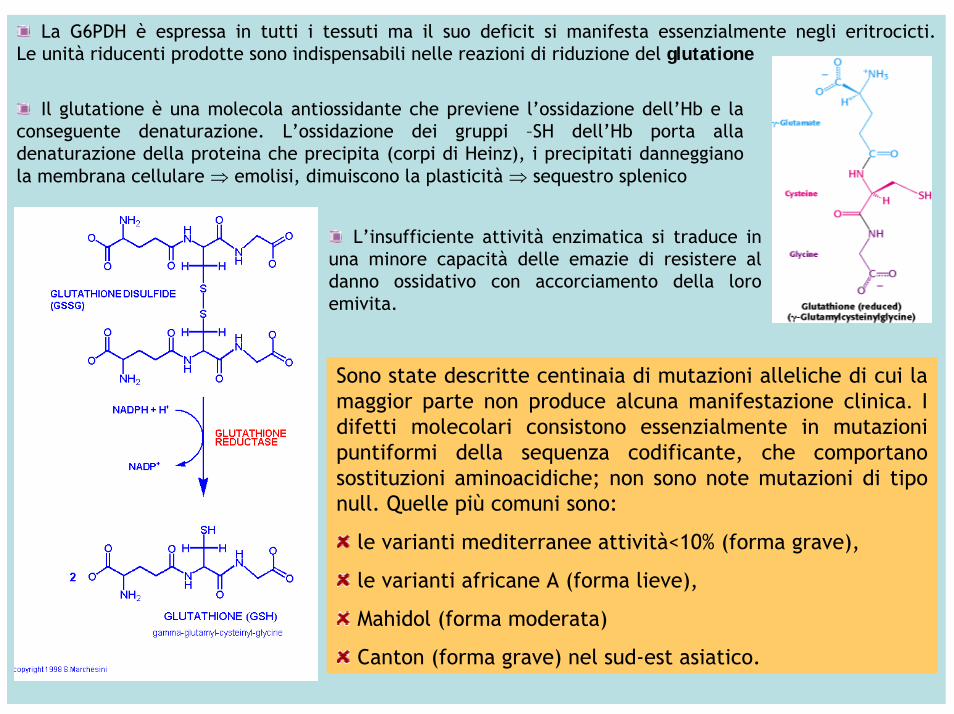
La G6PDH è espressa in tutti i tessuti ma il suo deficit si manifesta essenzialmente negli eritrocicti. Le unità riducenti prodotte sono indispensabili nelle reazioni di riduzione del glutatione
Il glutatione è una molecola antiossidante che previene l’ossidazione dell’Hb e la conseguente denaturazione. L’ossidazione dei gruppi –SH dell’Hb porta alladenaturazione della proteina che precipita (corpi di Heinz), i precipitati danneggiano la membrana cellulare ⇒ emolisi, dimuiscono la plasticità ⇒ sequestro splenico
L’insufficiente attività enzimatica si traduce in una minore capacità delle emazie di resistere al danno ossidativo con accorciamento della loroemivita.
Sono state descritte centinaia di mutazioni alleliche di cui la maggior parte non produce alcuna manifestazione clinica. I difetti molecolari consistono essenzialmente in mutazioni puntiformi della sequenza codificante, che comportano sostituzioni aminoacidiche; non sono note mutazioni di tiponull. Quelle più comuni sono:
le varianti mediterranee attività<10% (forma grave),
le varianti africane A (forma lieve),
Mahidol (forma moderata)
Canton (forma grave) nel sud-est asiatico.

DECORSO CLINICO E DIAGNOSI:
La sintomatologia clinica dipende dal tipo di mutazione. La maggior parte delle persone affette è asintomatica, anche se alcuni pazienti presentano ittero neonatale e anemia emolitica acuta, dopo infezione o ingestione di farmaci ossidanti (compresi alcuni farmaci antimalarici) o di fave (favismo) ⇒ emolisi acuta autolimitantesi in seguito a stressossidativo.
INFEZIONI: epatiti virali, polmonite, febbre tifoide.
La crisi emolitica si manifesta improvvisamente a 12-48 ore dall’ingestione dei farmaci o dei cibi indicati, possono insorgere tachicardia, astenia, respiro difficoltoso, pallore abnorme, ittero, febbre, urine di colore scuro, dolori muscolari, perdita di conoscenza. Terapia: riposo assoluto, idratazione, ospedalizzazione ed eventuale emotrasfusione.


DIAGNOSI: al di fuori delle crisi emolitiche questi individui sono ematologicamentenormali. L’emolisi è sia intra- che extravascolare
emazie con corpi di Heinz, reticolocitosi spiccatissima
dosaggio dell’attività enzimatica della G6PDH rispetto ad altri enzimi (piruvato chinasi o esochinasi)
iperbilirubinemia indiretta

EMOGLOBINURIA PAROSSISTICA NOTTURNA
EZIOLOGIA: mutazione del gene per il glicosilfosfatidilinositolo glicano A (PIGA) essenziale per il corretto ancoraggio di alcune proteine alla membrana cellulare mediante glicosilfosfatidilinositolo. Non è ereditaria.
PATOGENESI
Il glicosilfosfaditilinositolo è un fosfolipide che ancora alcune proteine alla membrana cellulare
La mutazione somatica del gene PIGA colpisce le cellule staminali pluripotenti: la progenie clonale (eritrociti, piastrine, leucociti) presenta deficit di tutte le proteine che sono ancorate medianteGPI, tra queste alcune inattivano il complemento ⇒ sensibilità alla lisi mediata dal complemento.

DECORSO CLINICO E DIAGNOSI:
Nel 25% dei casi si ha emolisi intravascolare che si presenta in forma parossistica e notturna. La maggior parte dei casi presenta emolisi cronica senza che si manifesti una drammatica emoglobinuria. Sono manifestazioni cliniche le frequenti trombosi venose delle vene epatiche, portali e cerebrali che sono fatali nel 50% dei casi. La sopravvivenza media è di 10 anni. Talora evolve in altre alterazioni delle cellulestaminali: anemia aplastica, leucemia acuta.
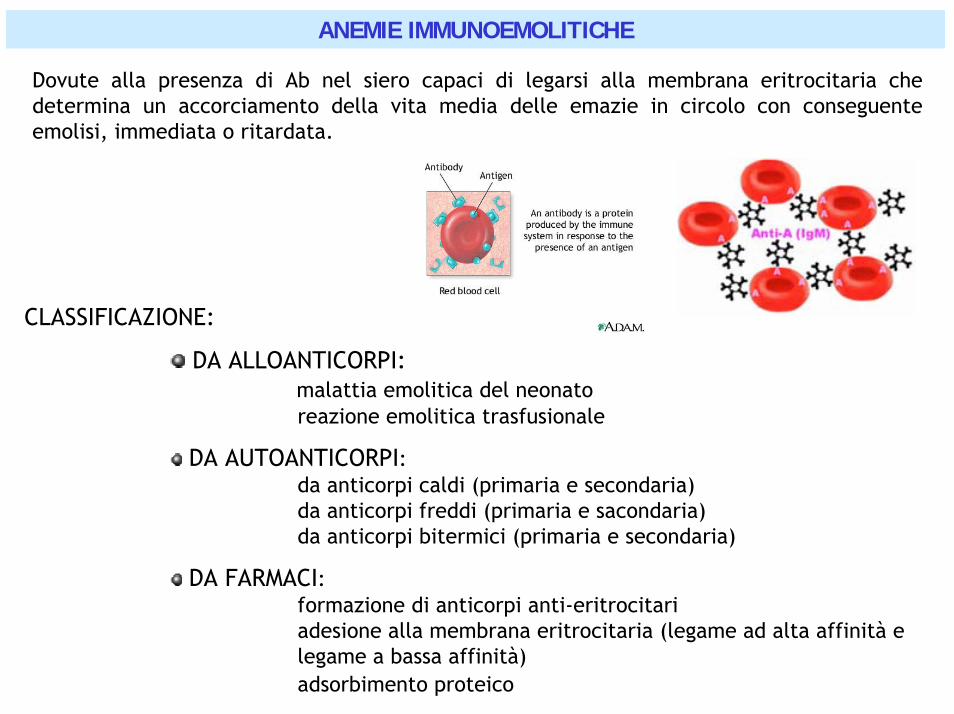
ANEMIE IMMUNOEMOLITICHE
Dovute alla presenza di Ab nel siero capaci di legarsi alla membrana eritrocitaria che determina un accorciamento della vita media delle emazie in circolo con conseguente emolisi, immediata o ritardata.
CLASSIFICAZIONE:
DA ALLOANTICORPI: malattia emolitica del neonatoreazione emolitica trasfusionale
DA AUTOANTICORPI: da anticorpi caldi (primaria e secondaria)da anticorpi freddi (primaria e sacondaria)da anticorpi bitermici (primaria e secondaria)
DA FARMACI:formazione di anticorpi anti-eritrocitariadesione alla membrana eritrocitaria (legame ad alta affinità e legame a bassa affinità)adsorbimento proteico

I GRUPPI SANGUIGNI
Nell’uomo sono stati identificati 26 gruppi sanguigni che comprendono 228 antigeni. Esistono, inoltre, anche altri tipi di antigeni che non sono stati attribuiti a nessun gruppo. Gli antigeni eritrocitari possono essere:
PROTEINE
GLICOPROTEINE
GLICOLIPIDI
La maggior parte degli antigeni sono sintetizzati dai GR stessi, mentre altri vengono adsorbiti a livello di membrana dal plasma. Alcuni sono GR specifici, altri sono espressi da tutte le cellule dell’organismo.
Nel 1980 la International Society of Blood Transfusion Terminology ha fornito le linee guida per una comune classificazione e nomenclatura dei gruppi sanguigni. Questi vengono classificati in sistemi, collezioni e serie. Aggiornamento del 2001: 26 sistemi, 5 collezioni, e 2 serie.
SISTEMA: gruppo di antigeni geneticamente discreti codificati da un locus o da pochi loci strettamente correlati (sistema ABO, sistema Rh, Sistema Kell, Sistema MS, …..)
COLLEZIONE: raggruppamenti di antigeni strettamente correlati tra loro (geneticamente, biochimicamente, sierologicamente) ma che non sono geneticamente distinti da tutti gli altri raggrupamenti
SERIE: serie 700 che comprende gli antigeni a bassa frequenza (< 1% della popolazione), serie 901 che comprende gli antigeni ad alta frequenza (> 90%)

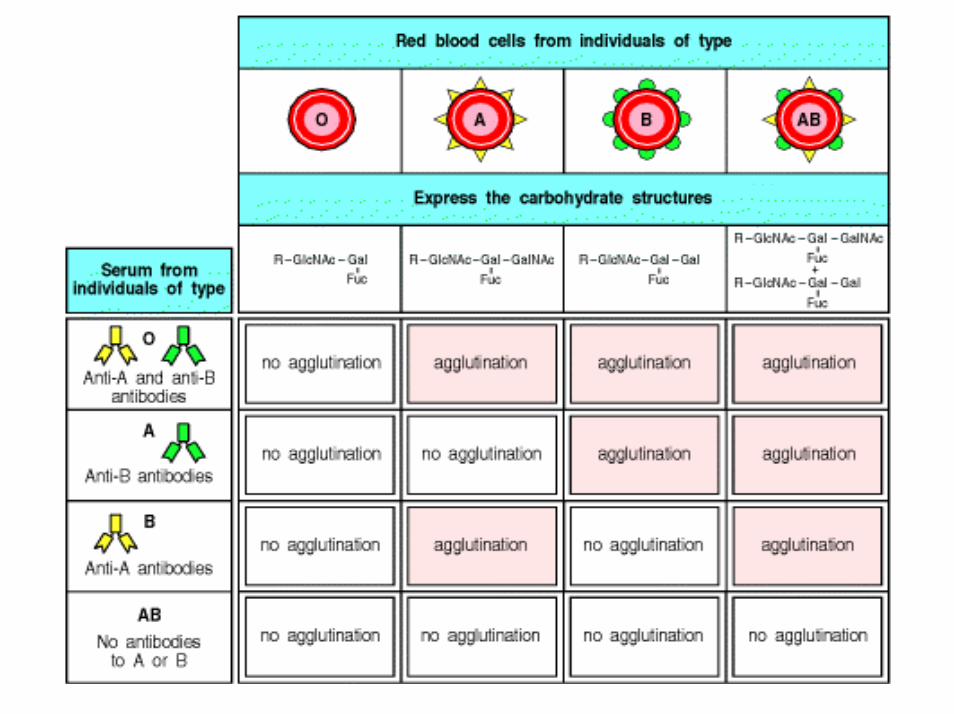
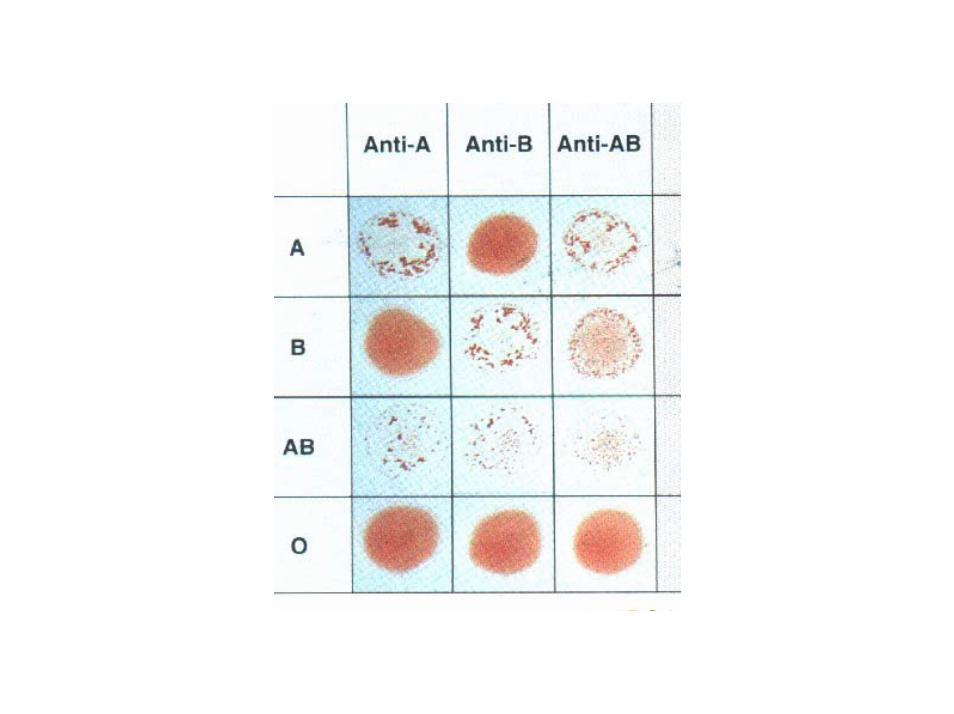
IL SISTEMA AB0• identificato nel 1900 da Landsteiner: i GR di alcuni individui posi a contatto con il siero di altri agglutinavano.
• da allora è rimasto il sistema più importante.
• sulla base della presenza o assenza di questi Ag sui GR si distinguono 4 gruppi: A, B, AB e 0. Successivamente lo stesso Landsteiner identificò un altro AG del gruppo A dotato di minor potere antigenico e lo identificò come A2.. Quindi, i gruppi sono 6.
• I gruppi più frequenti sono il gruppo 0 ed il gruppo A: 39% e 43% della popolazione rispettivamente. Il gruppo B costituisce il 12%, l’AB è il meno frequente, 3%.
• nel siero degli individui di gruppo A sono presenti agglutinine antiB, nel siero degli individui di gruppo B sono presenti agglutinine antiA, nel siero degli individui AB non sono presenti agglutinine, nel siero dei pazienti di gruppo 0 sono presenti entrambe le agglutinine.
• gli Ag sono presenti nei GR fin dalla nascita mentre le corrispondenti agglutinine sono assenti al momento della nascita; la loro produzione inizia al momento della nascita ed al 6°mese raggiungono livelli pari a quelli dell’adulto.
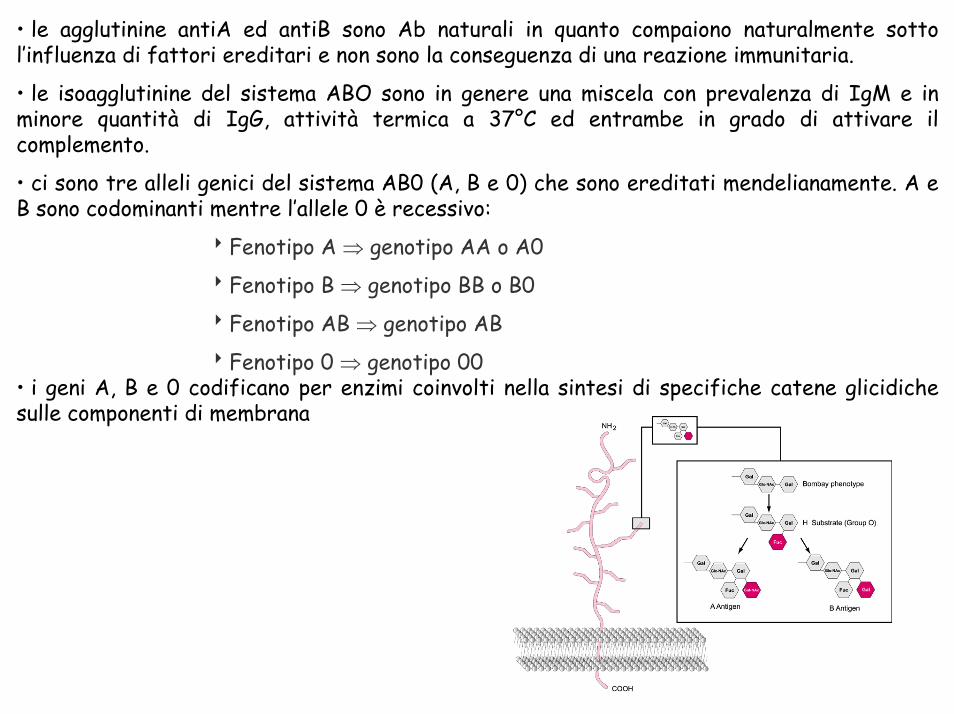
• le agglutinine antiA ed antiB sono Ab naturali in quanto compaiono naturalmente sotto l’influenza di fattori ereditari e non sono la conseguenza di una reazione immunitaria.
• le isoagglutinine del sistema ABO sono in genere una miscela con prevalenza di IgM e in minore quantità di IgG, attività termica a 37°C ed entrambe in grado di attivare il complemento.
• ci sono tre alleli genici del sistema AB0 (A, B e 0) che sono ereditati mendelianamente. A e B sono codominanti mentre l’allele 0 è recessivo:
8Fenotipo A ⇒ genotipo AA o A0
8Fenotipo B ⇒ genotipo BB o B0
8Fenotipo AB ⇒ genotipo AB
8Fenotipo 0 ⇒ genotipo 00• i geni A, B e 0 codificano per enzimi coinvolti nella sintesi di specifiche catene glicidiche sulle componenti di membrana

IL SISTEMA Rh• identificato nel 1940 da Landsteiner e Weiner: iniezione di GR di Macacus rhesus in conigli ottenedo la produzione di un Ab inizialmente indentificato con Ab Rh, oggi anti-D.
• il siero di coniglio posto successivamente a contatto con sangue umano era in grado di agglutinare i GR nell’85% dei casi
• in base a questa osservazione Landsteiner e Weiner stabilirono che l’85% degli individui possedeva Ag uguale a quello del Macacus ⇒ Rh+, il restante 15% ⇒ Rh-.
• 3 tipi di nomenclatura sono stati utilizzati per identificare gli Ag e gli alleli del sistema Rh.
• gli antigeni del sistema sono 45 e sono codificati da 2 geni: RHD e RHCE . I due geni codificano 2 polipeptidi simili (417 aa), sulla membrana prendono contatto con una glicoproteina RhAG.
• Gli Ag principali sono D, C, E, c ed e, gli individui Rh+ esprimono l’Ag D.
• l’antigene D è fortemente immunogenico: causa la formazione di Ab anti D nel 70% degli individui Rh- esposti a sangue Rh+.

MALATTIA IMMUNOEMOLITICA DEL NEONATO (ERITROBLASTOSI FETALE)
EZIOLOGIA: si definisce malattia emolitica del neonato la sindrome da iperdistruzione eritrocitaria fetale conseguente all’incompatibilità antigenica materno-fetale.
PATOGENESI
• si verifica nei casi in cui il feto ha fenotipo Rh+ e la madre fenotipo Rh-
• la MEN si sviluppa solitamente attraverso una catena di fenomeni patogenetici incentrati sulla sintesi da parte della madre Rh- di Ab contro l’antigene D del sistema Rh presente sulle emazie del feto
• la produzione di Ab avviene soprattutto nelle fasi terminali della gravidanza e durante il parto ⇒ quantità sufficienti di emazie fetali passano la barriera placentare risposta anticorpale primaria, lenta IgM
• la presenza di di una incompatibilità AB0 esercita effetto protettivo
• alla seconda gravidanza anche piccole quantità di emazie Rh+ evocano una risposta immunitaria secondaria IgG, capaci di passare la barriera placentare.

DECORSO CLINICO E DIAGNOSI:
A seconda della gravità del processo emolitico si può avere morte fetale tra la 25-35 settimana. Se non interviene morte si possono avere quadri clinici differenti:
• idrope fetale universale e pre-idrope: neonati prematuri, pallidi, versamenti pleurico ed addomianle, ipervolemia, deficit ventilatorio, scompeso cardio-circolatorio ⇒ morte entro poche ore dalla nascita. Pre-idrope, meno grave, sopravvivenza nel 50% dei casi.
• ittero grave del neonato a 12-24 ore dalla nascita se non trattati ⇒ kernicterus
DIAGNOSI: test di Coombs diretto nel neonato, indiretto nella madre.

ANEMIE IMMUNOEMOLITICHE DA AUTOANTICORPI
EZIOLOGIA: è ignota nel 50% dei casi definiti idiopatici (primaria), nella maggior parte dei casi si associa ad una malattia sistemica che coinvolge direttamente od indirettamente il sistema immunitario (secondaria). Quest’ultima frequentemente associata a malattie linfoproliferative (linfomi, leucemie), connettiviti (LES, artrite reumatoide, sclerodermia), meno requentemente a malattie infettive (polmonite, mononucleosi, parotite, epatiti) e malattie autoimmuni (morbo di Crohn).
PATOGENESI
Non sono completamente noti i meccanismi attraverso i quali cloni linfocitari sfuggono ai normali controlli timici che reprimono linfociti riconoscenti Ag self.
Sulla base delle caratteristiche chimico-fisiche e sierologiche degli autoanticorpi si distinguono 3 tipi di anemie emolitiche autoimmuni:
1. AUTOANTICORPI CALDI: forma più comune. Questi Ab hanno un optimum termico a 37°C appartengono prevalentemente alla classe IgG. Le IgG sono Abincompleti non in grado di agglutinare (incompleti), nel 70-80% dei casi reagiscono contro Ag del sistema Rh (e). I GR rivestiti di IgG interagiscono con macrofagi che tentano di fagocitarli ⇒ sferociti ⇒ sequestro splenico ⇒ emolisi extravasale.

2. ANTICORPI FREDDI: sono Ab di tipo IgM presenti nel siero a titolo elevato. Hanno potere agglutinante elevato con range termico piuttosto ampio 0°-32°C con optimum a 0°-4°C. Tali Ab si manifestano in modo acuto durante la fase di convalescenza di alcune malattie infettive (polmonite, mononucleosi). L’emolisi è solitamente autolimitantesi. La sintomatologia clincia consegue all’agglutinazione degli eritrociti nelle estremità del corpo dove è più facilel’ipotermia.
3. ANTICORPI BITERMICI: Ab di tipo IgG capaci di legare le emazie a temperature comprese tra 0° e 20°C e di attivare il complemento ma a 37°C (emoglobinuria parossistica da freddo). Consegue ad alcuni tipi di malattie infettive (polmonite, morbillo, parotite, sindromi influenzali).

ANEMIE IMMUNOEMOLITICHE DA FARMACI
EZIOLOGIA: approssimativamente il 10% delle anemie emolitiche su base immunologica può essere messa in relazione alla precedente assunzione di un farmaco.
PATOGENESI
Principali meccanismi:
1. Induzione della formazione di un Ab diretto contro le strutture della membrana eritrocitaria: in genere, IgG contro Ag Rh. Alfa-metil dopa, levodopa, acido mefenamico
2. Adesione del farmaco alla membrana eritrocitaria con legame ad alta affinità: penicillina, cefalosporine, tetracicline. Legame diretto del farmaco alla membrana eritrocitaria dose-dipendente ⇒ lisi extravascolare
3. Adesione del farmaco alla membrana eritrocitaria con legame a bassa affinità: chinidina. Formazione di un complesso ternario: farmaco + Ag di membrana + Ab
4. Asorbimento di proteine su base non immunologica: dosi elevate e croniche di cefalosporine. Queste danneggiano la membrana e facilitano l’adsorbimento di pt plasmatiche.