
Corso di laurea magistrale in Ingegneria Elettronica...
53
Nanoelettronica 1 Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis Corso di laurea magistrale in Ingegneria Elettronica
Transcript of Corso di laurea magistrale in Ingegneria Elettronica...

Nanoelettronica
1
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Introduzione alle simulazioni atomistiche di nano-materiali
2
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
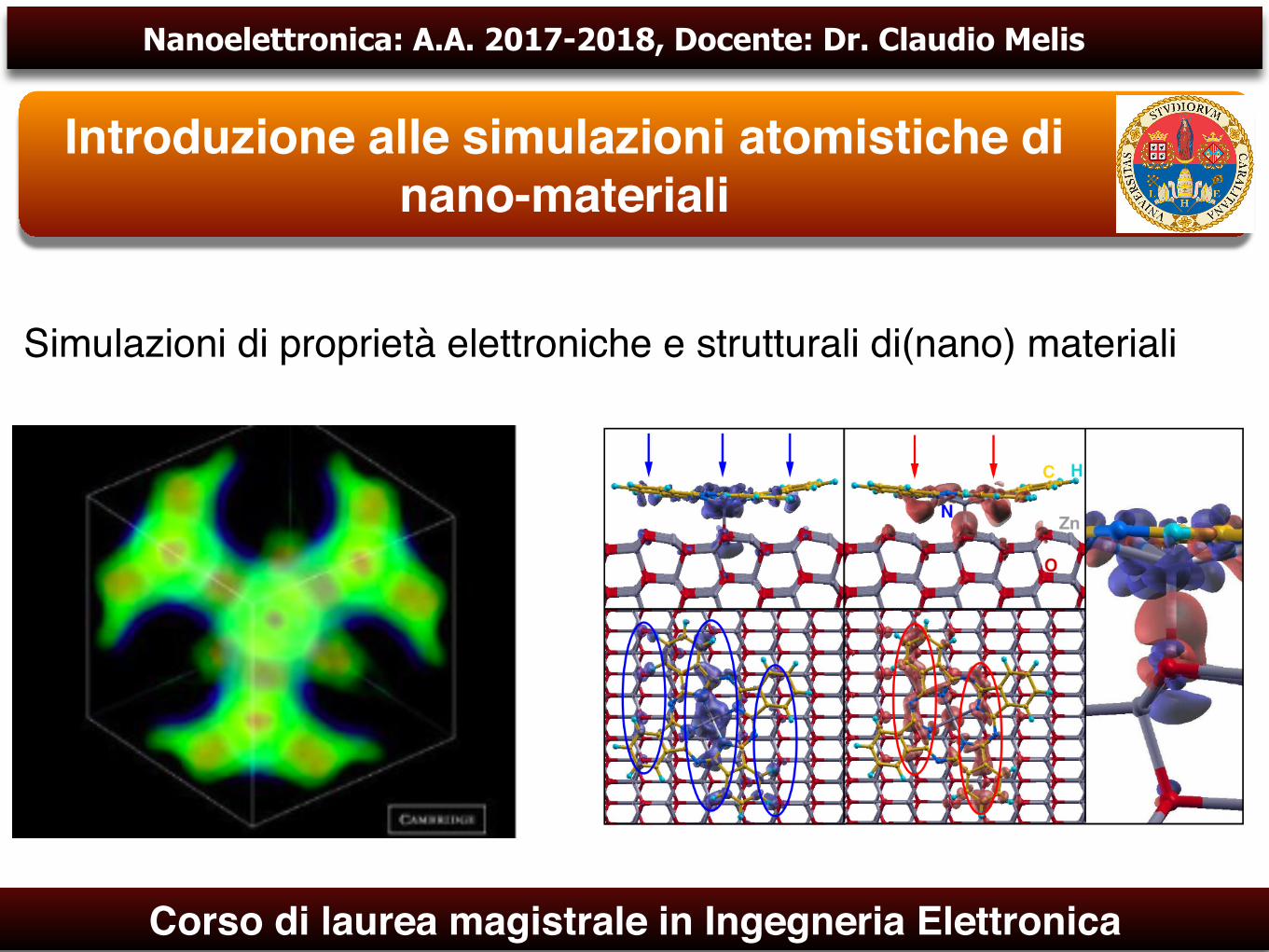
Simulazioni di proprietà elettroniche e strutturali di(nano) materiali

Introduzione alle simulazioni atomistiche di nano-materiali
3
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Architettura: 10 BGQ Frame Modello: IBM-BG/QProcessore : IBM PowerA2, 1.6 GHzNumero di Core: 163840 Numero di nodi: 10240 Performance di picco: 2PFlop/s
APPARATO SPERIMENTALE: CALCOLATORI AD ALTE PRESTAZIONI

Introduzione alle simulazioni atomistiche di nano-materiali
4
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Approssimazione diBorn-Oppenheimer

Introduzione alle simulazioni atomistiche di nano-materiali
5
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
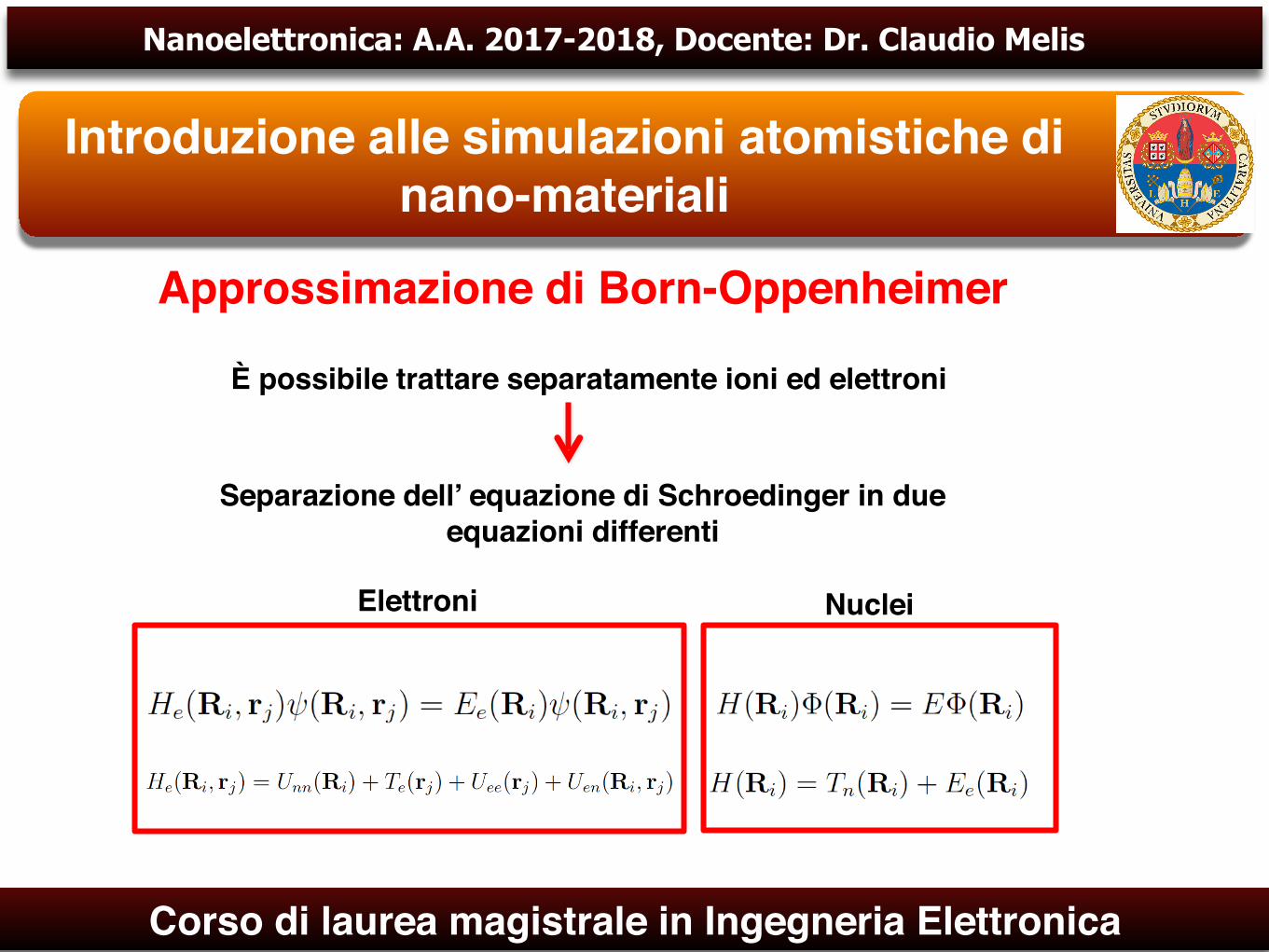
Approssimazione di Born-OppenheimerSistema a molti-corpi
Non si puo’ risolvere esattamente per sistemi contenenti più’ di due elettrone
Sono necessarie diverse approssimazioni
Equazione di Schrodinger dipendente dal tempo
Introduzione alle simulazioni atomistiche di nano-materiali
6
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
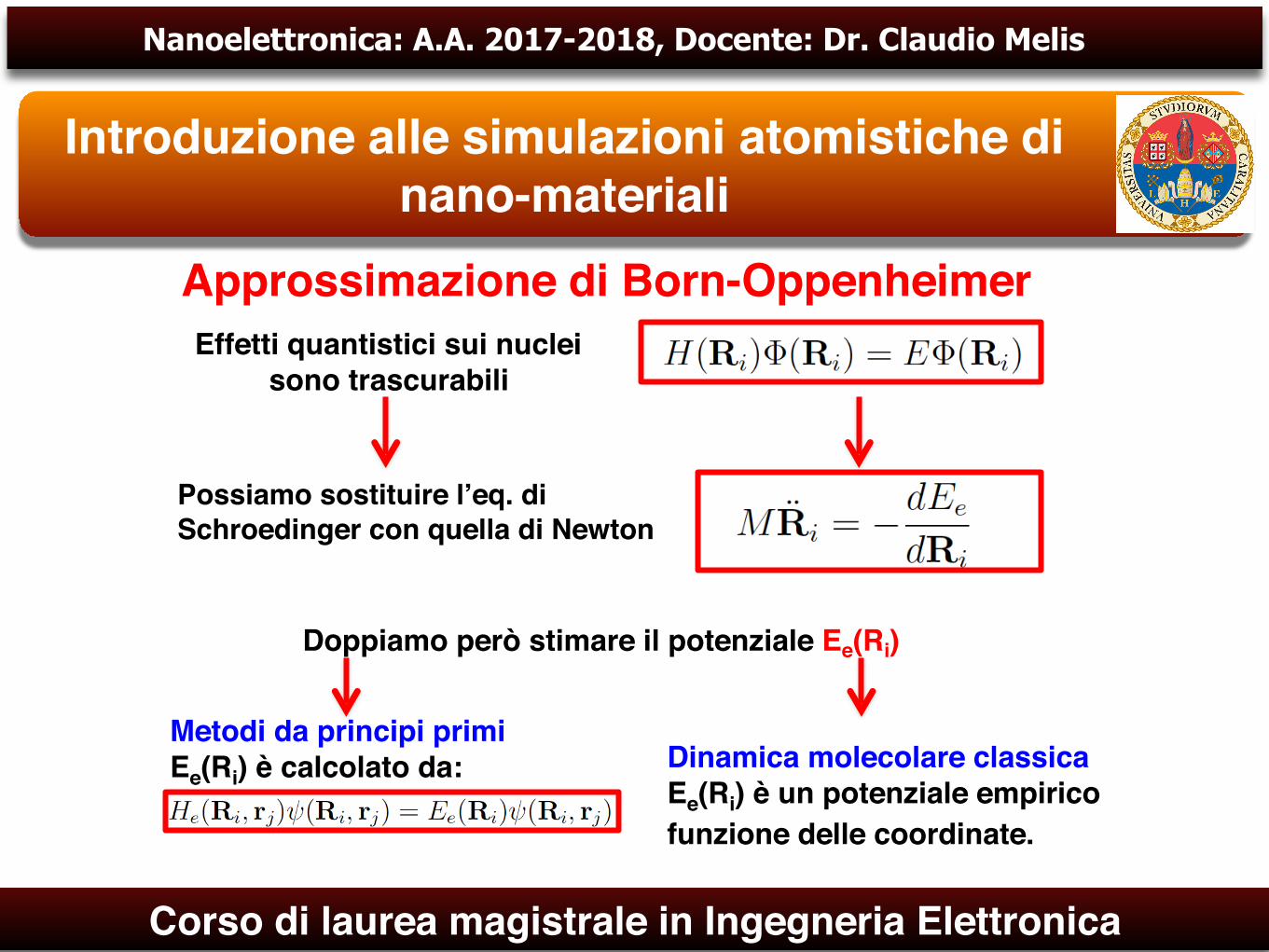
Approssimazione di Born-Oppenheimer
È possibile trattare separatamente ioni ed elettroni
Elettroni
Separazione dell’ equazione di Schroedinger in due equazioni differenti
Nuclei
Introduzione alle simulazioni atomistiche di nano-materiali
7
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Approssimazione di Born-OppenheimerEffetti quantistici sui nuclei
sono trascurabili
Possiamo sostituire l’eq. di Schroedinger con quella di Newton
Doppiamo però stimare il potenziale Ee(Ri)
Metodi da principi primiEe(Ri) è calcolato da: Dinamica molecolare classica
Ee(Ri) è un potenziale empirico funzione delle coordinate.

Introduzione alle simulazioni atomistiche di nano-materiali
8
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Configurazione nucleare al tempo t

Introduzione alle simulazioni atomistiche di nano-materiali
9
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Calcolo della funzione d’onda elettronica per lo stato fondamentale → V(R)
Configurazione nucleare al tempo t

Introduzione alle simulazioni atomistiche di nano-materiali
10
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Soluzione dell’equazione di Newton per il moto degli ioni ….
Configurazione nucleare al tempo t
Introduzione alle simulazioni atomistiche di nano-materiali
11
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
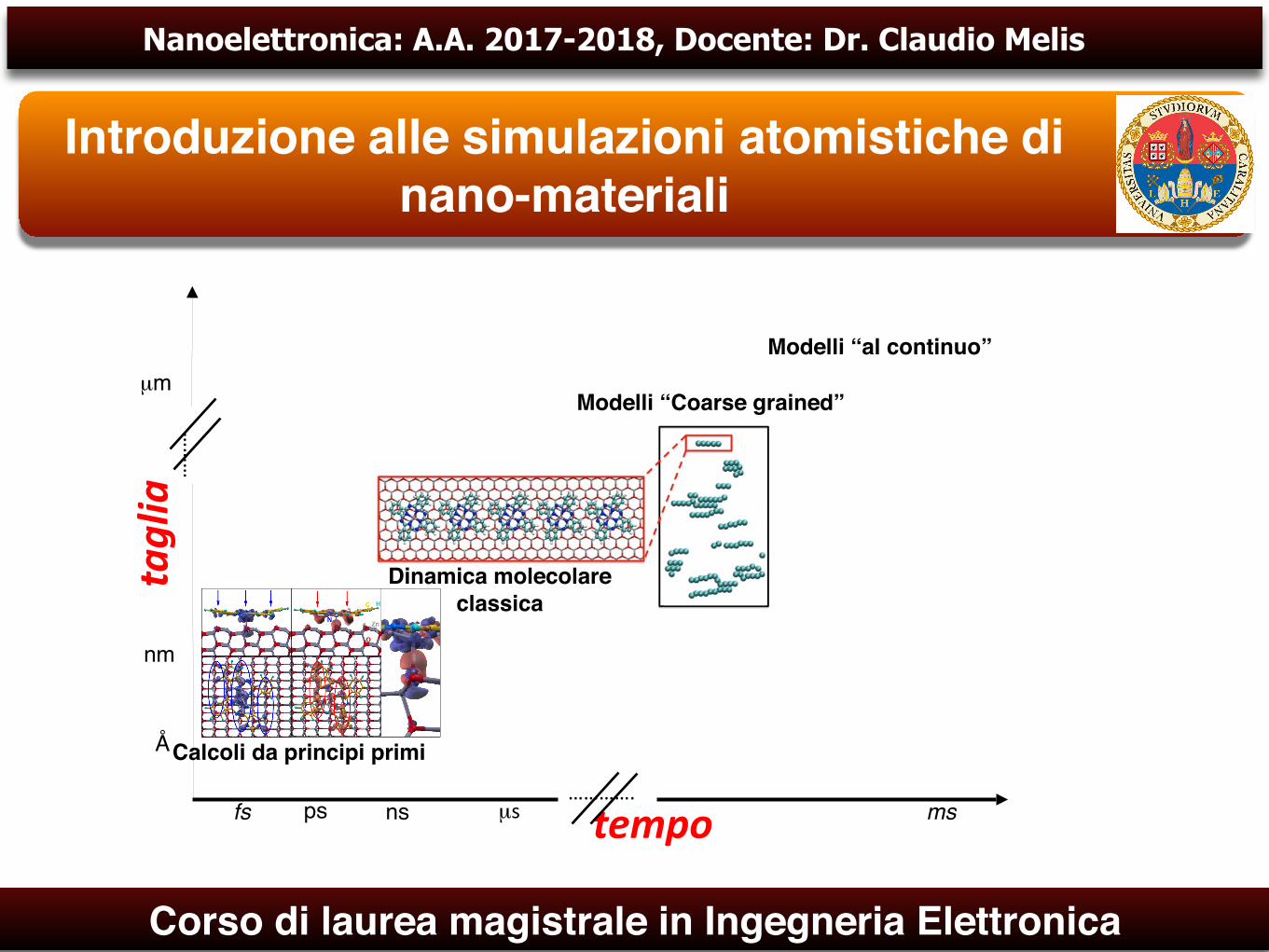
taglia
nm
µm
fs mstempo
Dinamica molecolare classica
Modelli “Coarse grained”
Modelli “al continuo”
ns………….
µs
………
ps
Calcoli da principi primiÅ

Introduzione alle simulazioni atomistiche di nano-materiali
Nanoelettronica -Dipartimento di Ingegneria Elettrica ed Elettronica
TEORIA DEL FUNZIONALE
DENSITA’

Introduzione alle simulazioni atomistiche di nano-materiali
13
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Calcoli da principi primi
Funzione d’onda Densità elettr.
Hartree-Fock DFT
TD-DFTMP2-CI
DFT è computazionalmente piu’ leggera di Hartree-Fock and MP2-CI
Introduzione alle simulazioni atomistiche di nano-materiali
14
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
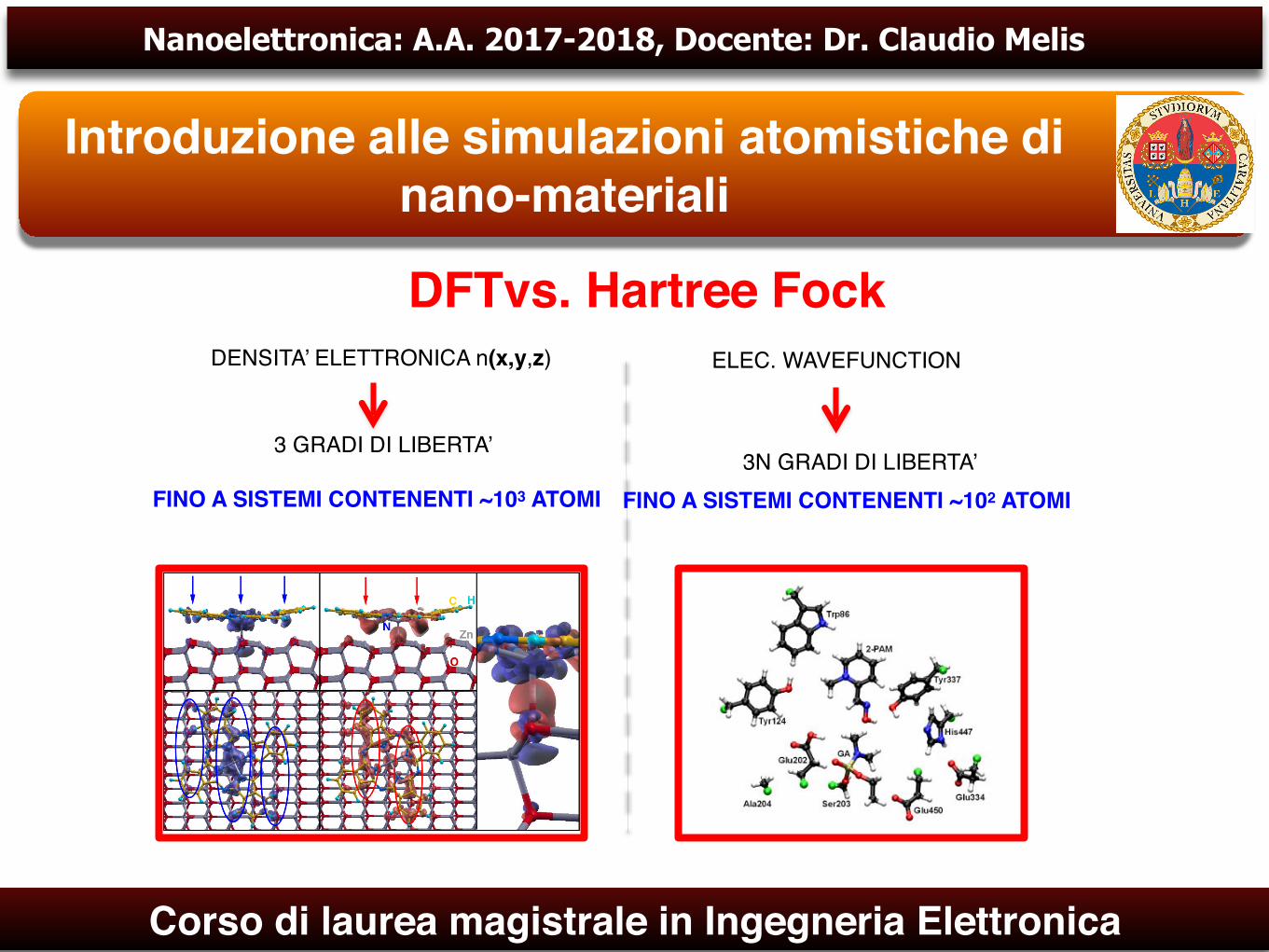
DFTvs. Hartree FockDENSITA’ ELETTRONICA n(x,y,z) ELEC. WAVEFUNCTION
FINO A SISTEMI CONTENENTI ~103 ATOMI
3 GRADI DI LIBERTA’
FINO A SISTEMI CONTENENTI ~102 ATOMI 3N GRADI DI LIBERTA’

Introduzione alle simulazioni atomistiche di nano-materiali
15
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Introduzione alle simulazioni atomistiche di nano-materiali
16
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
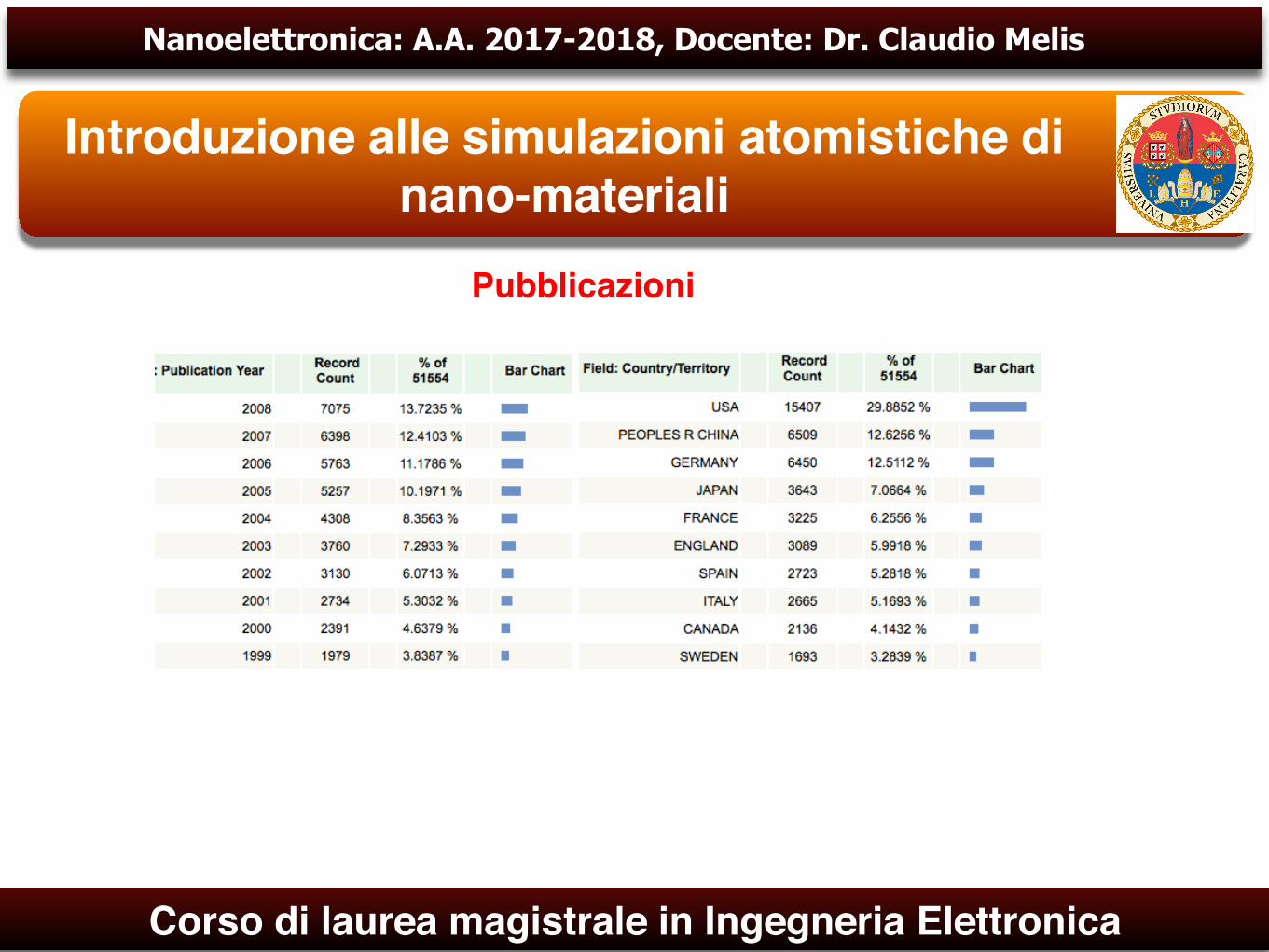
Pubblicazioni
Introduzione alle simulazioni atomistiche di nano-materiali
17
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
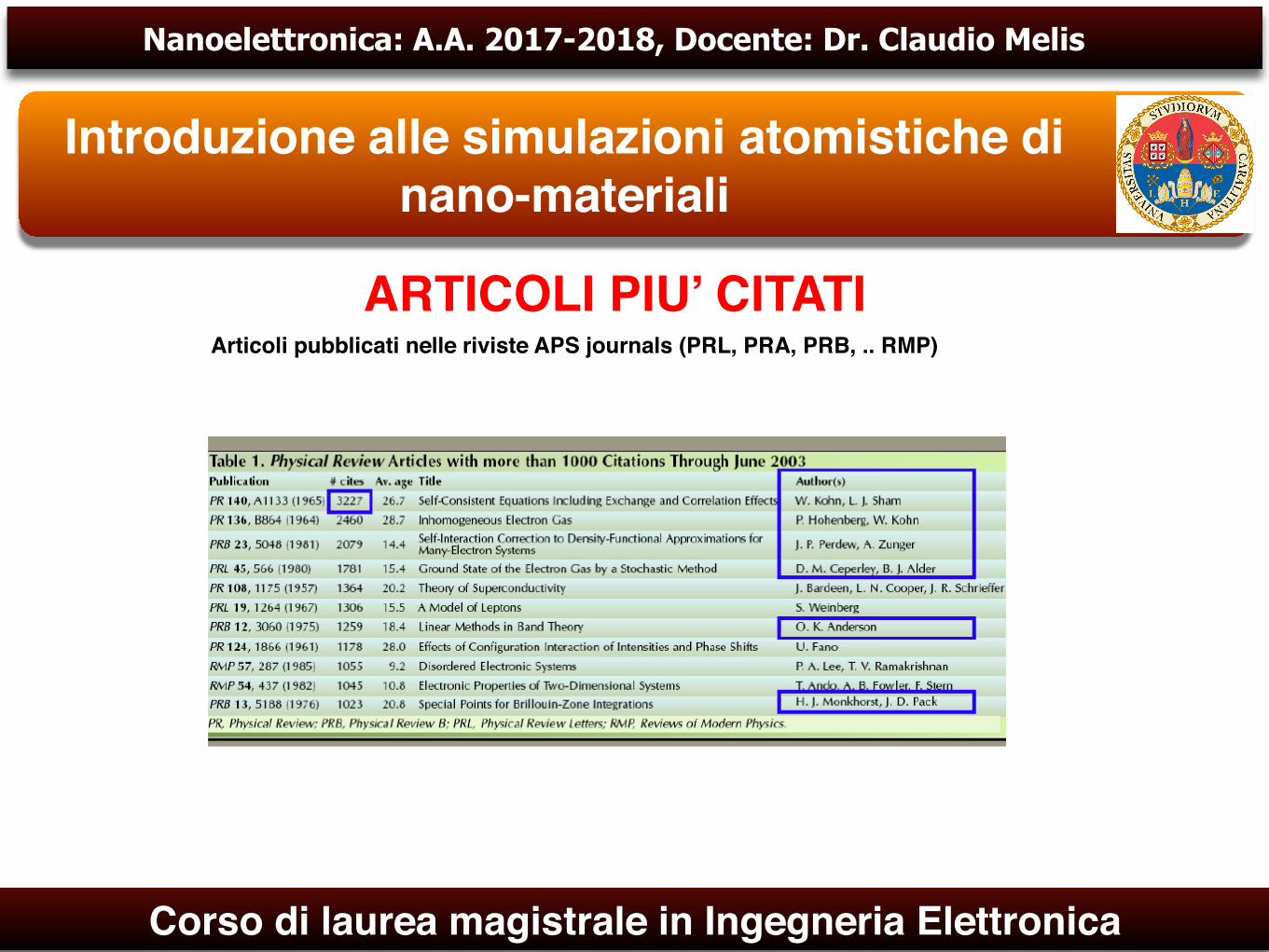
ARTICOLI PIU’ CITATIArticoli pubblicati nelle riviste APS journals (PRL, PRA, PRB, .. RMP)

Introduzione alle simulazioni atomistiche di nano-materiali
18
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
REFERENZA ALLA DFT
Richard M. Martin
Cambridge University Press, 2004
Electronic Structure : Basic Theory and Practical Methods
(ISBN: 0521782856)
http : //www.cambridge.org/uk/catalogue/catalogue.asp?isbn=0521782856

Introduzione alle simulazioni atomistiche di nano-materiali
19
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
DFT◆ La DFT risolve l’eq. di Sch. di un sistema multi-corpi riducendo il problema ad un
sistema di elettroni non-interagenti
◆ Cio’ è fatto utilizzando come variablile fondamentale la densità di carica elettroni invece della funzione d’onda.
◆ La base teorica della DFT è il teorema di Hohnberg-Kohn in cui si studia un sistema multi-elettronico soggetto ad un potenziale esterno

Introduzione alle simulazioni atomistiche di nano-materiali
20
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Hohenberg e Kohn (I)
“The ground state density of a many-electron system determines univocally the external potential, modulo a constant.”
Considerimo un insieme di elettroni aventi stessa energia cinetica stessa interazione elettrone-elettrone ma diverso potenziale esterno
Il potenziale esterno è un funzionale della densità
Lo stato fondamentale avrà una diversa densità
Anche l’energia totale è un funzionale della densità n, EV [n]

Introduzione alle simulazioni atomistiche di nano-materiali
21
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Hohenberg e Kohn (II) Lo stato fondamentale è il minimo del funzionale EV [n] rispetto alla densità
Il minimo è ottenuto quando n è nello stato fondamentale
L’energia dello stato fondamentale è:
F[n] è una funzione universale della densità, definita dalla energia cinetica Te e dall’interazione elettrone-elettrone Uee
Il problema è che non esiste una forma analitica per F

Introduzione alle simulazioni atomistiche di nano-materiali
22
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Equazioni di Kohn and Sham Kohn and Sham proposero una espressione approssimata per
F[n] Considerando un problema equivalente di elettroni non interagenti
“for every system of interacting electrons, a corresponding system of non-interacting particles, described by single particle orbitals , exists subject to an external potential with the
same ground state density as the interacting system”
Exc[n] è l’energia di scambio e correlazione

Introduzione alle simulazioni atomistiche di nano-materiali
23
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
F[n] è variazionale rispetto alla densità di carica n, sotto l condizione che il numero di elettroni sia conservato
λ¸ è un moltiplicatore di lagrange che vincola la densità ad essere normalizzata al numero totale di elettroni.
La soluzione dell’equazione variazione fornisce le seguenti eq:

Introduzione alle simulazioni atomistiche di nano-materiali
24
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Equazioni di Kohn and Sham
Tali equazioni devono essere risolte in modo autoconsistente dal momento che il potenziale Vks dipende dalla densita’ di carica
Lo stato fondamentale è ottenuto risolvendo le seguenti equazioni di singola particella:
Le autofunzioni vengono dette gli orbitali di kohn e sham

Introduzione alle simulazioni atomistiche di nano-materiali
25
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Equazioni di Kohn and Sham
Tali equazioni devono essere risolte in modo autoconsistente dal momento che il potenziale Vks dipende dalla densita’ di carica
Lo stato fondamentale è ottenuto risolvendo le seguenti equazioni di singola particella:
Le autofunzioni vengono dette gli orbitali di kohn e sham

Introduzione alle simulazioni atomistiche di nano-materiali
26
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
◆ Il compito più’ difficile di una simulazione DFT è gestire il potenziale di scambio e correlazione Exc
◆ L’energia di scambio deriva dal principio di esclusione di Pauli che impone l’antisimmetria della funzione d’onda multielettronica producendo una separazione spaziale fra gli eletroni aventi lo stesso spin sistema. Tale riduzione si è detta energia di scambio
◆ Il metodo Hartree-Fock descrive esattamente l’energia di scambio. La differenza tra l’enrgia calcolata con Hartree-Fock e l’energia calcolada DFT è detta energia di correlazione
Energia di scambio e correlazione

Introduzione alle simulazioni atomistiche di nano-materiali
27
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
◆ Il calcolo dell’energia di corelazione è molto complicato. L’unico conto esatto è stato fatto utilizzando il metodo quantum montecarlo.
◆ Le approssimazioni piu’ popolari per l’energia di scambio e correlazione è ka Local Density Approximation (LDA) e la Generalised Gradient Approximation (GGA)
Energia di scambio e correlazione
◆ L’energia di scambio e correlazione e generalmente espressa come l’integrale della energia di scambio e correlazione locale

Introduzione alle simulazioni atomistiche di nano-materiali
28
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
◆ LDA è il metodo piu’ semplice per descrivere l’energia di scambio e correlazione. Si assume che l’energia di scambio e correlazione locale εxc[n] sia uguale all’energia di scambio e correlazione locale di un gas omogeneo di elettroni avente la stessa densità
◆ LDA assume che l’energia di scambio e correlazione sia un funzionale puramente locale ignorando le correzioni dovute alle interazioni all’energia di scambio e correlazione nel punto r dovute alle inomogeneità della densità di carica
Energia di scambio e correlazione-LDA

Introduzione alle simulazioni atomistiche di nano-materiali
29
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
APPLICAZIONI DELLA DFT

Introduzione alle simulazioni atomistiche di nano-materiali
30
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
J.Phys.Chem.C2012;116:15439 J.Phys.Chem.B2006;110:26313
Appliedphysicsletters200485:4902
Introduzione alle simulazioni atomistiche di nano-materiali
31
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
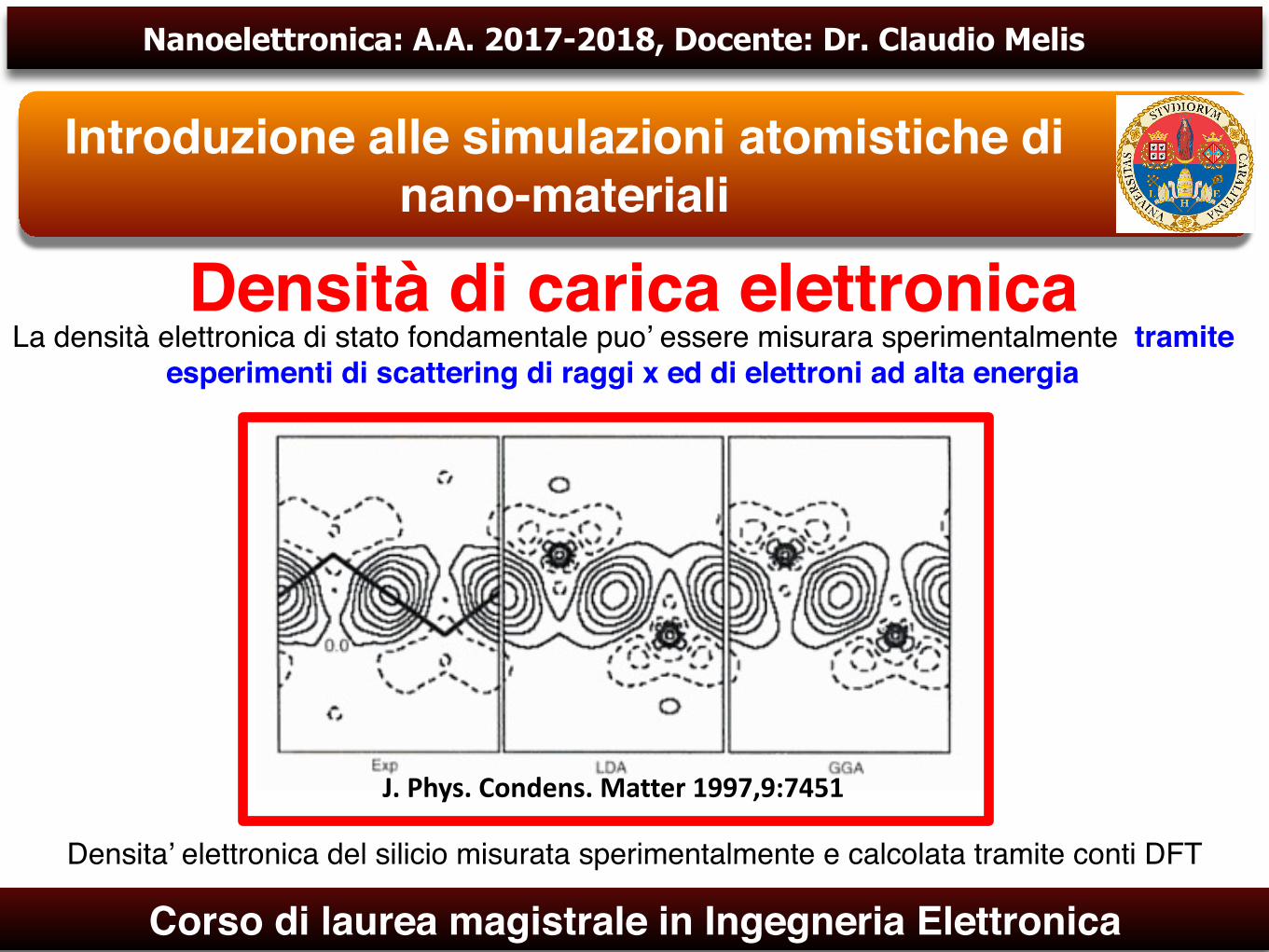
Densità di carica elettronicaLa densità elettronica di stato fondamentale puo’ essere misurara sperimentalmente tramite
esperimenti di scattering di raggi x ed di elettroni ad alta energia
Densita’ elettronica del silicio misurata sperimentalmente e calcolata tramite conti DFT
J.Phys.Condens.Matter1997,9:7451

Introduzione alle simulazioni atomistiche di nano-materiali
32
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Volume pressione e moduli elastici
La descrizione della struttura di equilibrio di un sistema determina varie proprietà meccaniche.
In particolare le quantità che possono essere paragonate con gli esperimenti sono sono
l’energia E, la pressione P e il modulo di bulks B
E = E(V)
P= − dEdV
B= −V dPdV
=V d2EdV 2
Introduzione alle simulazioni atomistiche di nano-materiali
33
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
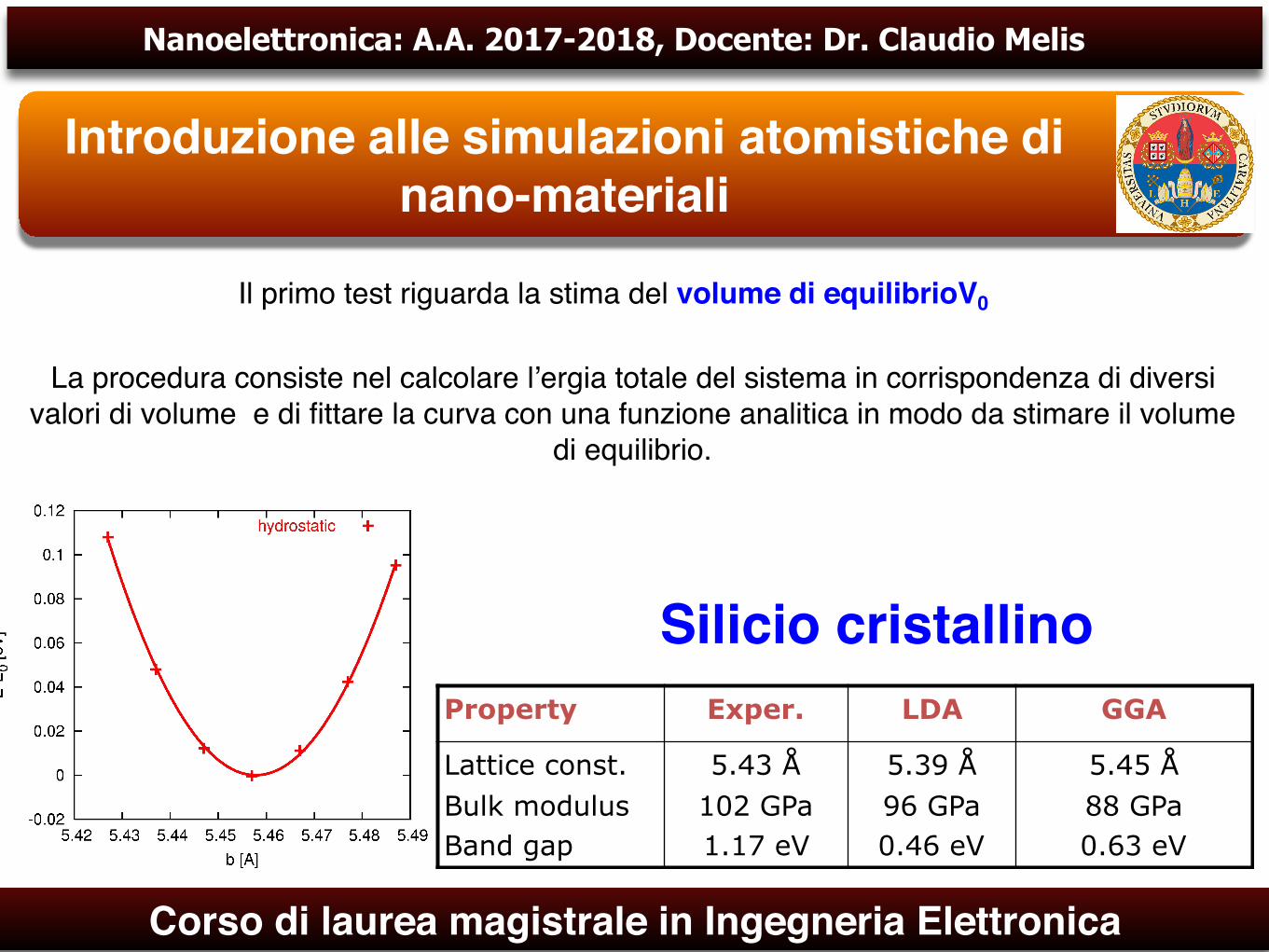
Il primo test riguarda la stima del volume di equilibrioV0
La procedura consiste nel calcolare l’ergia totale del sistema in corrispondenza di diversi valori di volume e di fittare la curva con una funzione analitica in modo da stimare il volume
di equilibrio.
Property Exper. LDA GGA
Lattice const. Bulk modulus Band gap
5.43 Å 102 GPa 1.17 eV
5.39 Å 96 GPa 0.46 eV
5.45 Å 88 GPa 0.63 eV
Silicio cristallino
Introduzione alle simulazioni atomistiche di nano-materiali
34
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
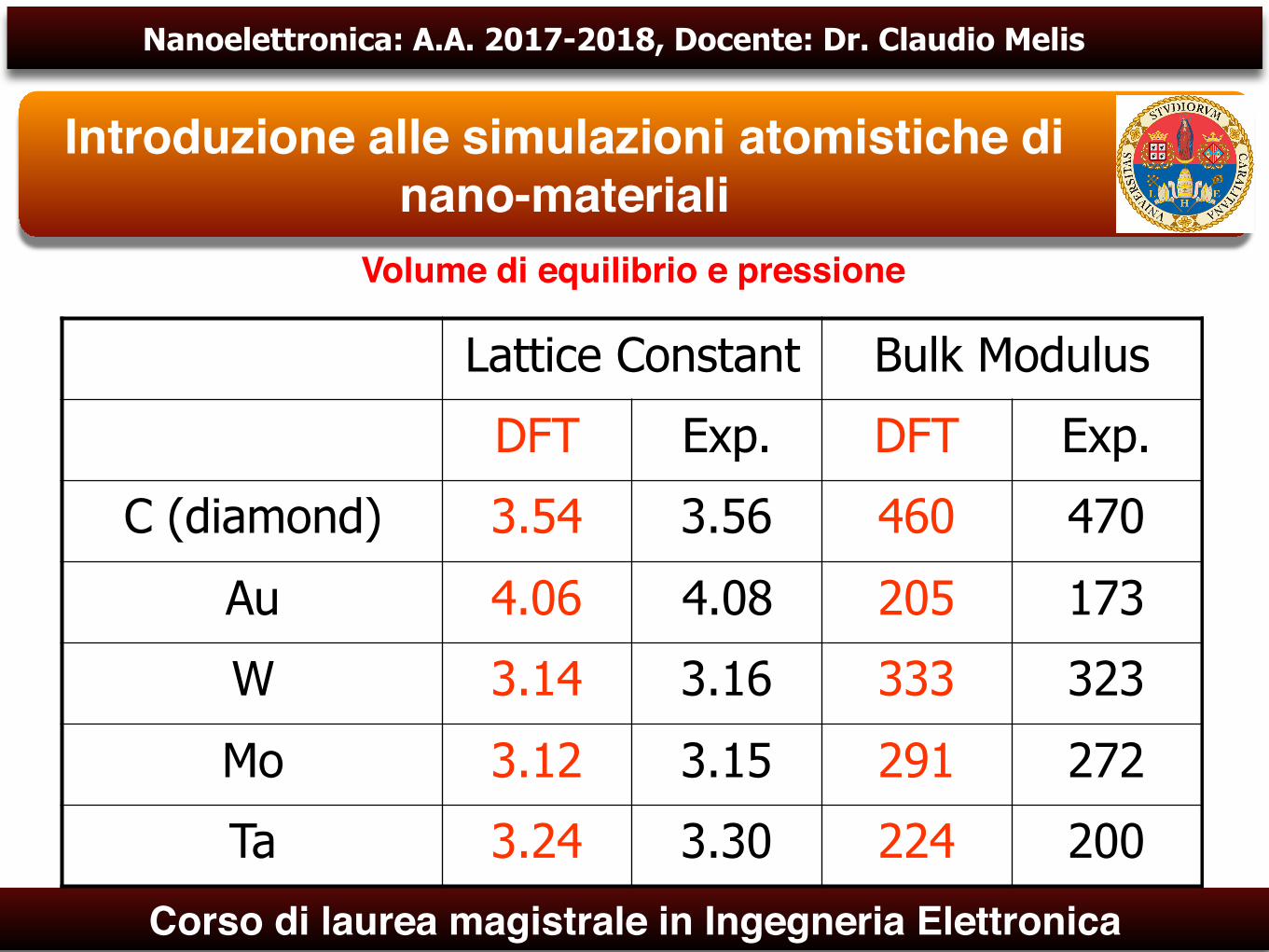
Volume di equilibrio e pressione
Lattice Constant Bulk Modulus
DFT Exp. DFT Exp.
C (diamond) 3.54 3.56 460 470
Au 4.06 4.08 205 173
W 3.14 3.16 333 323
Mo 3.12 3.15 291 272
Ta 3.24 3.30 224 200
Introduzione alle simulazioni atomistiche di nano-materiali
35
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
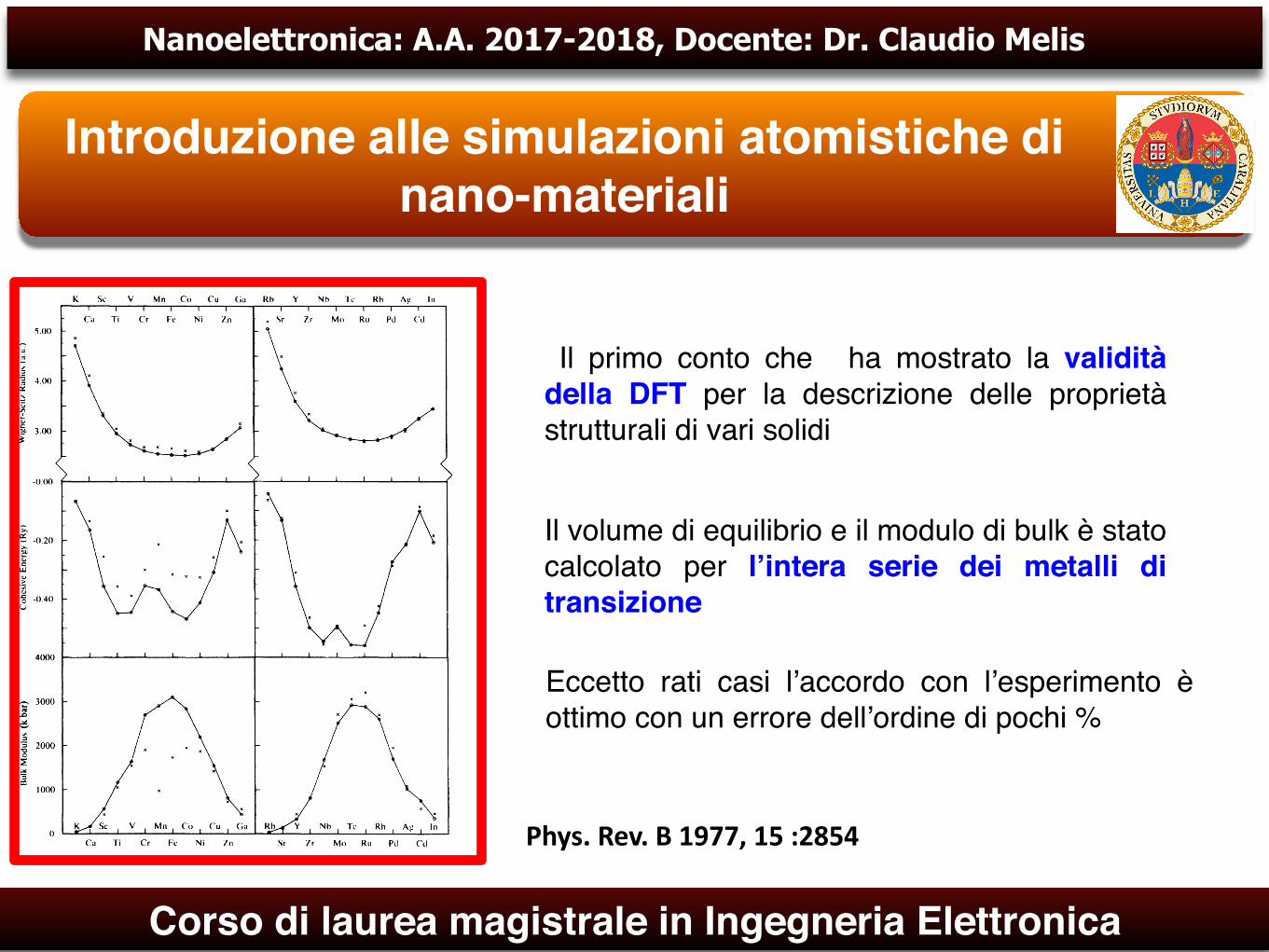
Il primo conto che ha mostrato la validità della DFT per la descrizione delle proprietà strutturali di vari solidi
Il volume di equilibrio e il modulo di bulk è stato calcolato per l’intera serie dei metalli di transizione
Eccetto rati casi l’accordo con l’esperimento è ottimo con un errore dell’ordine di pochi %
Phys.Rev.B1977,15:2854
Introduzione alle simulazioni atomistiche di nano-materiali
36
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
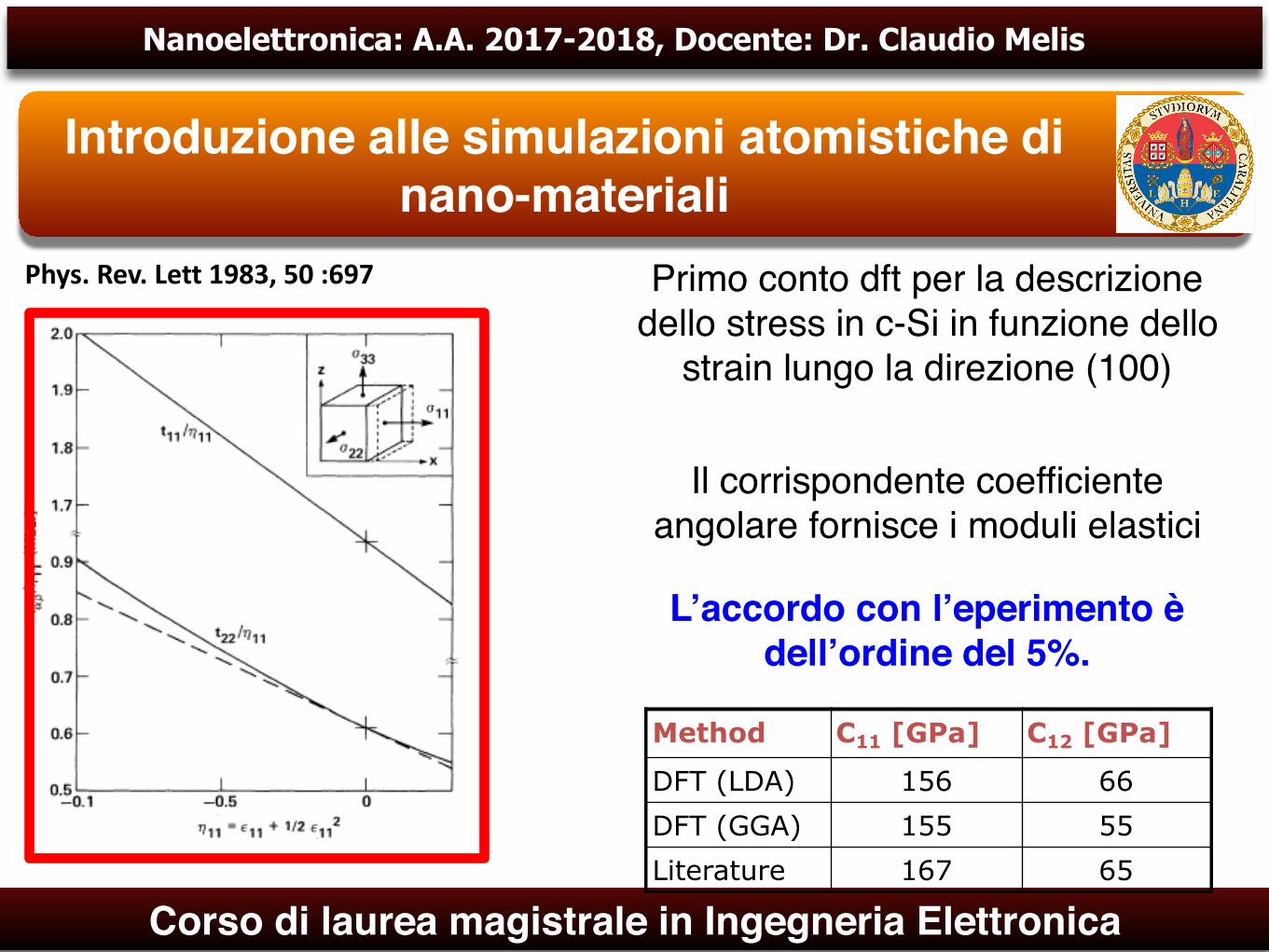
Primo conto dft per la descrizione dello stress in c-Si in funzione dello
strain lungo la direzione (100)
Il corrispondente coefficiente angolare fornisce i moduli elastici
L’accordo con l’eperimento è dell’ordine del 5%.
Method C11 [GPa] C12 [GPa]
DFT (LDA) 156 66DFT (GGA) 155 55Literature 167 65
Phys.Rev.Lett1983,50:697

Introduzione alle simulazioni atomistiche di nano-materiali
37
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
DINAMICA MOLECOLARE CLASSICA

Introduzione alle simulazioni atomistiche di nano-materiali
38
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
● 1955 – Fermi-Pasta-Ulam. Integrazione numerica delle equazioni del moto di una catena dioscillatori armonici monodimensionali con lo scopo di verificare l'ergodicit. e l'equipartizioneenergetica del sistema.
● 1957 – Alder, Wainwright. Primo calcolo di dinamica molecolare in fase condensata di unsistema di sfere rigide.
● 1964 – Rahman, 1967 – Verlet. Prima dinamica molecolare di un sistema realistico di 864 atomi di Argon descritto con una funzione potenziale continua.
● 1968 – Harp, Berne. Prima dinamica molecolare di liquidi diatomici.
● 1971 – Stillinger, Rahman. Prima dinamica molecolare di acqua liquida.
● 1976 – McCammon, Karplus. Prima dinamica molecolare di una proteina in solvente esplicito.…● 1985 – Car, Parrinello. Dinamica molecolare ab initio, dove il potenziale agente sui nuclei .calcolato “on the fly” dalla struttura elettronica.
● 2011 – Shaw, 2011 – De Fabritiis. Primi eventi di binding tra proteina/farmaco osservati indinamica molecolare.

Introduzione alle simulazioni atomistiche di nano-materiali
39
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
1) Scelta di una forma appropriata per V
2) Soluzione numerica delle equazioni di Newton per il moto degli atomi
MD classica è più facile e veloce e permette di studiare sistemi più grandi, ma
l’affidabilità dei risultati dipende totalmente da V
Dinamica Molecolare classica (MD)

Introduzione alle simulazioni atomistiche di nano-materiali
40
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Prima approssimazione: interazioni a coppie
( )∑≠
−≈ji
jiN RRRRV!!!
…!
v21),,( 1

Introduzione alle simulazioni atomistiche di nano-materiali
41
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
CAMPO DI FORZA
per MD

Introduzione alle simulazioni atomistiche di nano-materiali
42
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Potenziali di interazioni utilizzati in MD
CLASS I (proteins, DNA)Amber (Assisted Model Building
with Energy Refinement)Charmm,Gromos,OPLS…
CLASS II (polymers)COMPASS (Condensed-phase
Optimized Molecular Potentials for Atomistic SimulationStudies,
CVFF,CVFF91
Stillinger Weber: C, Si
Tersoff :C, Si
Embedded atom model: Metals
Sitemi OrganicI Sitemi InorganicI
Buckingham form:Ionic systems (ZnO,TiO2)

Introduzione alle simulazioni atomistiche di nano-materiali
43
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica

Introduzione alle simulazioni atomistiche di nano-materiali
44
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Dinamica Molecolare classica
• Soluzione delle equazioni di Newton per un sistema molecolare:
iii amF !!=

Introduzione alle simulazioni atomistiche di nano-materiali
45
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Integrazione delle equazioni di Newton
Metodi alle differenze finite: il tempo è discretizzato.
Passo temporale Δt (in generale dell’ordine del femtosecondo 10-15 s)
…+Δ+Δ+Δ+=Δ+ 3...
2...
)(61)(
21)()()( ttxttxttxtxttx
I vari algoritmi cercano di ridurre l’errore di troncamento.

Introduzione alle simulazioni atomistiche di nano-materiali
46
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Integratore: Algoritmo di Verlet
)()()()(2)( 42 tOtatttrtrttr Δ+Δ+Δ−−=Δ+
)()(21)(v)()( 32 tOtattttrttr Δ−Δ+Δ−=Δ−
)()(21)(v)()( 32 tOtattttrttr Δ+Δ+Δ+=Δ+
Posizione iniziale {r(t), v(t)}, integriamo sino a {r(t+Δt), v(t+Δt)}:
{r(t), v(t)}
{r(t+Δt),v(t+Δt)}Lanuovaposizioneat+Δt:
Analogamente, la vecchia posizione a t-Δt:
Sommando:
Sottraendo: )())()((21)()(v 2tOttrttrt
trt Δ+Δ−−Δ+Δ
== !

Introduzione alle simulazioni atomistiche di nano-materiali
47
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Gli integratori devono soddisfare tutta una serie di requisiti. Tra questi:Proprietà degli integratori1. Efficienza: numero di integrazioni eseguite nell'unità di tempo. Non è molto importante: la frazione di tempo spesa per integrare le equazioni del moto è irrilevante rispetto al tempo speso per il calcolo delle interazioni (calcolo delle forze).Gli integratori di dinamica molecolare dovrebbero contemplare idealmente una sola valutazione di forze per ciclo.2. Accuratezza: divergenza della traiettoria numerica da quella analitica (instabilità di Lyapunov). Algoritmi di ordine superiore sono più accurati, ma richiedono più memoria specialmente se il calcolo coinvolge un elevato numero di atomi.In genere per calcoli di dinamica molecolare vengono impiegati integratori efficienti di basso ordine.3. Stabilità: preservazione delle leggi di conservazione. La stabilità degli integratori è una stabilità condizionale in relazione al tempo d'integrazione. Stabilità per timestep aggressivi è definita robustezza.La stabilità a lungo termine è estremamente importante per il sampling corretto dello spazio delle fasi. Algoritmi stabili non sono necessariamente accurati.4. Reversibilità temporale: è strettamente legata alla stabilità. Si dimostra che la reversibilità temporale è il requisito minimo per ottenere stabilità a lungo termine.

Introduzione alle simulazioni atomistiche di nano-materiali
48
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica

Introduzione alle simulazioni atomistiche di nano-materiali
49
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
● In una simulazione di dinamica molecolare si identificano idealmente 3 fasi:
1. Inizializzazione: l'integratore . in grado di propagare le equazioni del moto a partire da dellecondizioni iniziali note. Tali condizioni iniziali sono definite specificando coordinate e velocità.per ogni atomo del sistema.
• Le coordinate vengono ricavate da informazioni sperimentali (principalmente spettroscopia araggi X) o da modelli teorici.• Le velocità. vengono assegnate in modo casuale in modo da riprodurre la distribuzione diMaxwell-Boltzmann per la temperatura di interesse:
2. Equilibrazione: rilassamento dalla configurazione arbitraria iniziale per raggiungere unacondizione di equilibrio (perdita di memoria della condizione iniziale). Durante la fase diequilibrazione le medie d'insieme non sono stabili per cui vengono scartate (la fase diequilibrazione viene protratta finch. tali medie non sono stabili).
3. Produzione: accumulazione di statistica.
Fasi della dinamica molecolare

Introduzione alle simulazioni atomistiche di nano-materiali
50
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
● Raggio di cutoff. Per ridurre il peso del calcolo delle interazioni viene introdotto un raggio di cutoff (rc). Per ciascun atomo vengono calcolate solo le interazioni con gli atomi rientranti all'interno del raggio di cutoff (tipicamente compreso tra 8 e 15 Å).
● Condizioni periodiche al contorno. Per evitare effetti di discontinuità. ai bordi, ciascun atomo della cella di simulazione interagisce:
• con gli atomi della cella di simulazione reale (cerchi grigi);
• con gli atomi di copie periodiche della cella di simulazione reale (cerchi bianchi).
● Convenzione dell'immagine minima. Per evitare che gli atomi appartenenti alla cella reale interagiscano con le loro copie periodiche (ossia per evitare che interagiscano con se stessi) il raggio di cutoff deve essere impostato in modo da soddisfare:
Cella di simulazione e confini

Introduzione alle simulazioni atomistiche di nano-materiali
51
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
Termostati● Termostati: Algoritmi che alterano le equazioni del moto in modo che la distribuzione di microstati sia compatibile con la funzione densità. di probabilità. dell'insieme canonico (ρNVT).
L'uso di un termostato richiede la definizione di una temperatura istantanea:
tale temperatura T (istantanea, microscopica) verrà. comparata con la temperatura di referenza T0 (termodinamica, macroscopica) del bagno termico (virtuale) a cui il sistema . accoppiato.Attenzione: un termostato (algoritmo) . molto diverso da un termostato “reale”. Il trasferimento di calore (che consiste in una progressiva diffusione di calore dalla superficie verso il centro o vice versa implicando una disomogeneità. spaziale di temperatura nel campione) non vienee splicitamente simulato (occorrerebbe un sistema di dimensioni macroscopiche).
● Poichè. la temperatura istantanea . direttamente in relazione con le velocità., mantenere la temperatura costante in MD significa in genere introdurre qualche modalità. di controllo sulle velocità

Introduzione alle simulazioni atomistiche di nano-materiali
52
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
CODICE DI DINAMICA MOLECOLARE: LAMMPS

Introduzione alle simulazioni atomistiche di nano-materiali
53
Nanoelettronica: A.A. 2017-2018, Docente: Dr. Claudio Melis
Corso di laurea magistrale in Ingegneria Elettronica
CODICE DI DINAMICA MOLECOLARE: LAMMPSLarge-scaleAtomic/MolecularMassivelyParallelSimulatorhttp://lammps.sandia.gov
CodicedidinamicamolecolareclassicaOpensource,portableC++Solidiinorganici,organici,ibridi
Particlesimulatoratvaryinglengthandtimescaleselectrons)atomistic)coarse-grained)continuumSpatial-decompositionofsimulationdomainforparallelismMD,non-equilibriumMD,energyminimizationGPUandOpenMPenhancedCanbecoupledtootherscales:QM,kMC,FE,CFD,...


















