Corso di Chimica Fisica A - theochem.unito.it · della pressione di un gas in serie di potenze...
28
Universit` a di Torino Corso di Studi in Chimica - Laurea Triennale Anno Accademico 2006-2007 Corso di Chimica Fisica A Appunti delle lezioni Roberto Dovesi Loredana Valenzano (19 febbraio 2007)
Transcript of Corso di Chimica Fisica A - theochem.unito.it · della pressione di un gas in serie di potenze...

Universita di Torino
Corso di Studi in Chimica - Laurea Triennale
Anno Accademico 2006-2007
Corso di Chimica Fisica A
Appunti delle lezioni
Roberto Dovesi
Loredana Valenzano
(19 febbraio 2007)


Indice
1 Le proprieta dei gas 1
1.1 L’equazione di stato dei gas ideali e le unita di misura . . . . . . . . . . . . . . . . . . . . . . . . 11.2 L’equazione di van der Waals . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.3 Le equazioni di Redlich-Kwong (RK) e di Peng-Robinson (PR) . . . . . . . . . . . . . . . . . . . 71.4 L’uso di un’equazione di stato cubica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.5 La legge degli stati corrispondenti . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111.6 Il secondo coefficiente del viriale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131.7 Il potenziale di Lennard-Jones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161.8 Le forze di dispersione di London . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181.9 Il secondo coefficiente del viriale: alcuni casi particolari . . . . . . . . . . . . . . . . . . . . . . . 201.10 Esercizi svolti . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
1.10.1 Esercizio 1.1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 231.10.2 Esercizio 1.2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 231.10.3 Esercizio 1.3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 241.10.4 Esercizio 1.4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 241.10.5 Esercizio 1.5 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
i

Capitolo 1
Le proprieta dei gas
In questo capitolo, verra discussa prima l’equazione del gas ideale; in seguito vedremo alcune estensioni, comel’equazione di van der Waals, che descrive, sebbene in modo approssimato, le deviazioni di un gas dal comporta-mento ideale. Un approccio piu accurato passa attraverso lo sviluppo del viriale, che consiste in un’espansionedella pressione di un gas in serie di potenze della densita. Verranno poi messi in relazione i coefficienti di questaespansione con l’energia di interazione tra le molecole del gas. Questo ci portera a discutere le interazioniintermolecolari, allontanandoci dal caso ideale.
1.1 L’equazione di stato dei gas ideali e le unita di misura
Un gas sufficientemente diluito obbedisce all’equazione di stato
PV = nRT (1.1)
Diluito significa che le molecole che lo compongono sono cosı lontane che la loro mutua interazione puo esseretrascurata. Dividendo entrambi i membri di questa equazione per il numero di moli n si ottiene
P V = RT (1.2)
dove V = V/n e il volume molare. D’ora in poi, la notazione X indichera che X e una grandezza molare. Leequazioni (1.1) e (1.2) sono dette equazioni di stato dei gas ideali: esse esprimono la relazione tra pressione,volume e temperatura per una data quantita di gas ideale.E importante capire la differenza tra V e V che esprimono un’importante caratteristica delle grandezze e dellevariabili utilizzate per descrivere i sistemi macroscopici. Si fa distinzione tra grandezze (o variabili) di tipoestensivo e intensivo: le prime sono direttamente proporzionali alla dimensione del sistema (ne sono esempi: ilvolume, la massa, l’energia); le seconde non dipendono dall’estensione del sistema (ne sono esempi: la pressione,la temperatura e la densita). Dividendo una grandezza estensiva per il numero di moli (o il volume, o la massa)presenti nel sistema, si ottiene una grandezza intensiva. Ad esempio, V (dm3) e una grandezza estensiva, mentreV (dm3· mol−1) e una grandezza intensiva.Nelle equazioni (1.1) e (1.2), non c’e alcuna informazione sulle caratteristiche del gas (forma, grandezza dellemolecole e loro mutua interazione). Sperimentalmente, a 1 atm e 0 ◦C molti gas soddisfano la (1.1) e la (1.2)entro l’1 %.Tali equazioni rendono necessaria una discussione sul sistema di unita (SI) adottato dalla IUPAC (InternationalUnion of Pure and Applied Chemistry).Il volume nel sistema SI e’ espresso in m3 (metro cubo) ma il litro (L) definito esattamente come 1 dm3
(decimetro cubo) e un’unita di volume accettata anche dalla IUPAC. L’unita SI della pressione e il pascal (Pa);si ha che: 1Pa = 1N · m−2 = 1kg · m−1· s−2 dove il newton (N) e l’unita SI della forza. La pressione e quindiuna forza su un’unita di superficie. La pressione puo essere misurata sperimentalmente osservando l’altezzadi una colonna di liquido sostenuto dal gas (vedi figura 1.1). Se m e la massa del liquido e g e la costante diaccelerazione gravitazionale, la pressione e data da:
P =F
A=
mg
A=
ρhAg
A= ρhg (1.3)
1

Tabella 1.1: Unita di misura della pressione e fattori di conversione.
1 pascal = 1 N · m−2 = 1 kg · m−1· s−2
1 atmosfera = 1.01325 × 105 Pa= 1.01325 bar= 101.325 kPa= 1013.25 mbar= 760 torr
1 bar = 105 Pa = 0.1 MPa
dove A e l’area della base della colonna, ρ e la densita del fluido e h e l’altezza della colonna. Sebbene il pascalsia l’unita SI della pressione, l’atmosfera continua ad essere largamente usata. Una atmosfera viene definitacome la pressione che sorregge una colonna di mercurio di altezza pari a 76.0 cm (vedi figura 1.1). Con ilpassaggio alle unita SI, lo standard di pressione e il bar pari a 0.986 atm. Un’altra unita comunemente usata eil torr che e la pressione che sorregge una colonna di mercurio alta 1.00 mm, quindi 1 torr = (1/760) atm.
Figura 1.1: Una atmosfera viene definita come la pressione che sorregge una colonna di mercurio di altezza paria 76.0 cm.
In tabella 1.1 sono riportate varie unita di misura per la pressione.
La scala fondamentale delle temperature si basa sulla legge dei gas ideali. Dal momento che tutti i gas sicomportano in modo ideale nel limite P → 0, definiamo T come
T = limP→0
P V
R(1.4)
La temperatura si misura in kelvin (K). Dal momento che P e V non possono assumere valori negativi, il minimovalore possibile della temperatura e 0 K (zero assoluto) che corrisponde alla temperatura di una sostanza chenon possiede energia termica. Per stabilire l’unita del kelvin, al punto triplo dell’acqua e stata assegnata latemperatura di 273.15 K (il punto triplo di una sostanza corrisponde ad un sistema in equilibrio che contienegas, liquido e solido). Per quanto detto fino ad ora sulla temperatura, siamo in possesso di una definizionedi 0 K e di 273.15 K che generano una scala lineare delle temperature. Un kelvin viene definito quindi come1/273.15 della temperatura del punto triplo dell’acqua.In figura 1.2, e riportato l’andamento del valore sperimentale di V in funzione di T per Ar(g) a diverse pressioni.Come atteso dalla definizione che abbiamo dato delle scala delle temperature, l’estrapolazione di questi dati
2
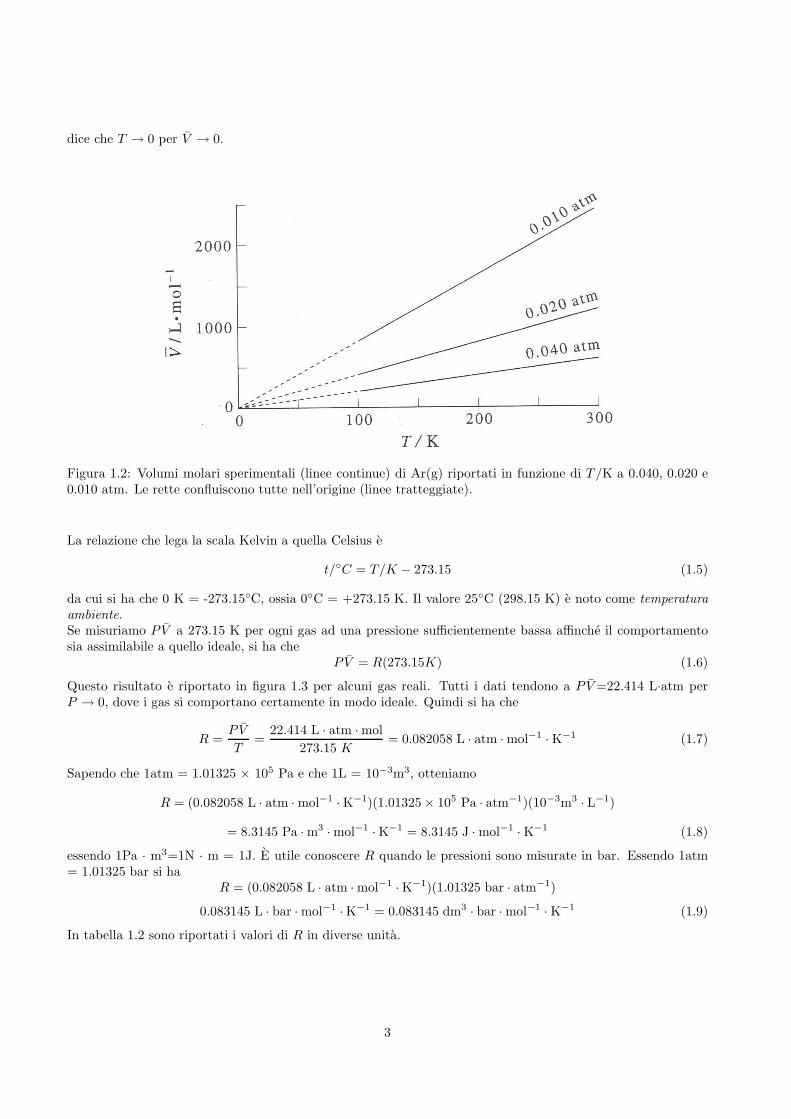
dice che T → 0 per V → 0.
Figura 1.2: Volumi molari sperimentali (linee continue) di Ar(g) riportati in funzione di T /K a 0.040, 0.020 e0.010 atm. Le rette confluiscono tutte nell’origine (linee tratteggiate).
La relazione che lega la scala Kelvin a quella Celsius e
t/◦C = T/K − 273.15 (1.5)
da cui si ha che 0 K = -273.15◦C, ossia 0◦C = +273.15 K. Il valore 25◦C (298.15 K) e noto come temperatura
ambiente.Se misuriamo P V a 273.15 K per ogni gas ad una pressione sufficientemente bassa affinche il comportamentosia assimilabile a quello ideale, si ha che
P V = R(273.15K) (1.6)
Questo risultato e riportato in figura 1.3 per alcuni gas reali. Tutti i dati tendono a P V =22.414 L·atm perP → 0, dove i gas si comportano certamente in modo ideale. Quindi si ha che
R =P V
T=
22.414 L · atm · mol
273.15 K= 0.082058 L · atm · mol−1 · K−1 (1.7)
Sapendo che 1atm = 1.01325 × 105 Pa e che 1L = 10−3m3, otteniamo
R = (0.082058 L · atm · mol−1 · K−1)(1.01325 × 105 Pa · atm−1)(10−3m3 · L−1)
= 8.3145 Pa · m3 · mol−1 · K−1 = 8.3145 J · mol−1 · K−1 (1.8)
essendo 1Pa · m3=1N · m = 1J. E utile conoscere R quando le pressioni sono misurate in bar. Essendo 1atm= 1.01325 bar si ha
R = (0.082058 L · atm · mol−1 · K−1)(1.01325 bar · atm−1)
0.083145 L · bar · mol−1 · K−1 = 0.083145 dm3 · bar · mol−1 · K−1 (1.9)
In tabella 1.2 sono riportati i valori di R in diverse unita.
3
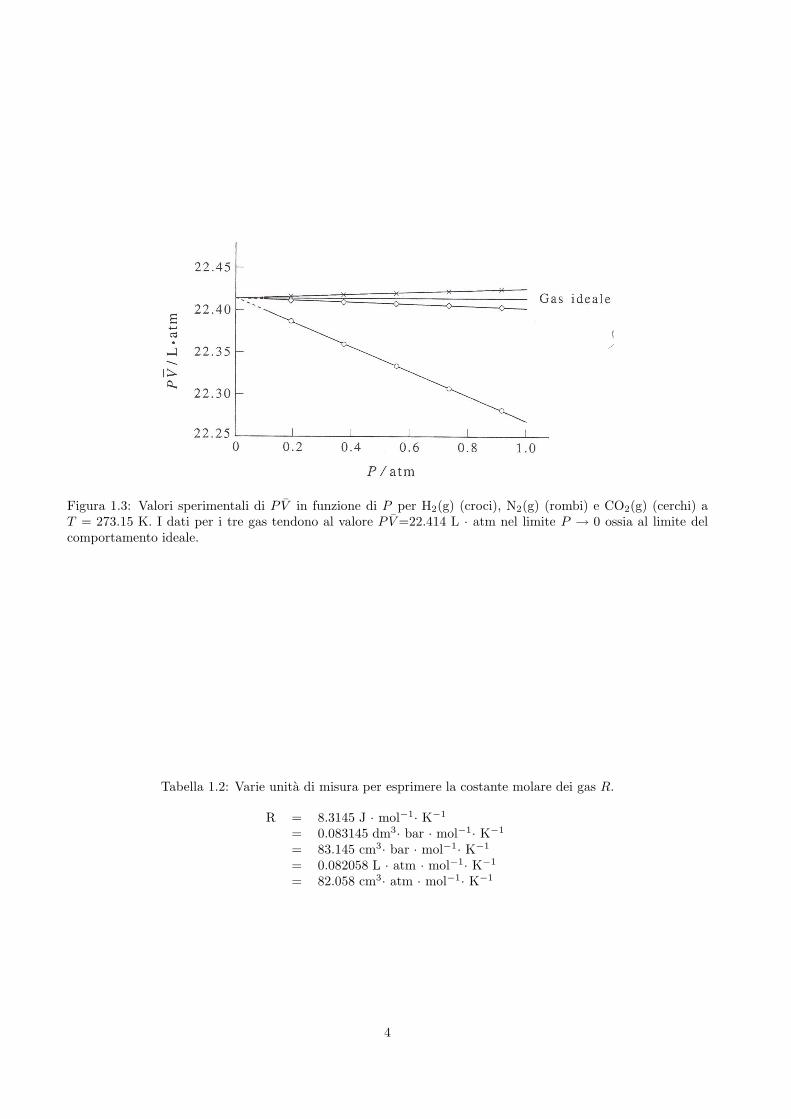
Figura 1.3: Valori sperimentali di P V in funzione di P per H2(g) (croci), N2(g) (rombi) e CO2(g) (cerchi) aT = 273.15 K. I dati per i tre gas tendono al valore P V =22.414 L · atm nel limite P → 0 ossia al limite delcomportamento ideale.
Tabella 1.2: Varie unita di misura per esprimere la costante molare dei gas R.
R = 8.3145 J · mol−1· K−1
= 0.083145 dm3· bar · mol−1· K−1
= 83.145 cm3· bar · mol−1· K−1
= 0.082058 L · atm · mol−1· K−1
= 82.058 cm3· atm · mol−1· K−1
4
1.2 L’equazione di van der Waals
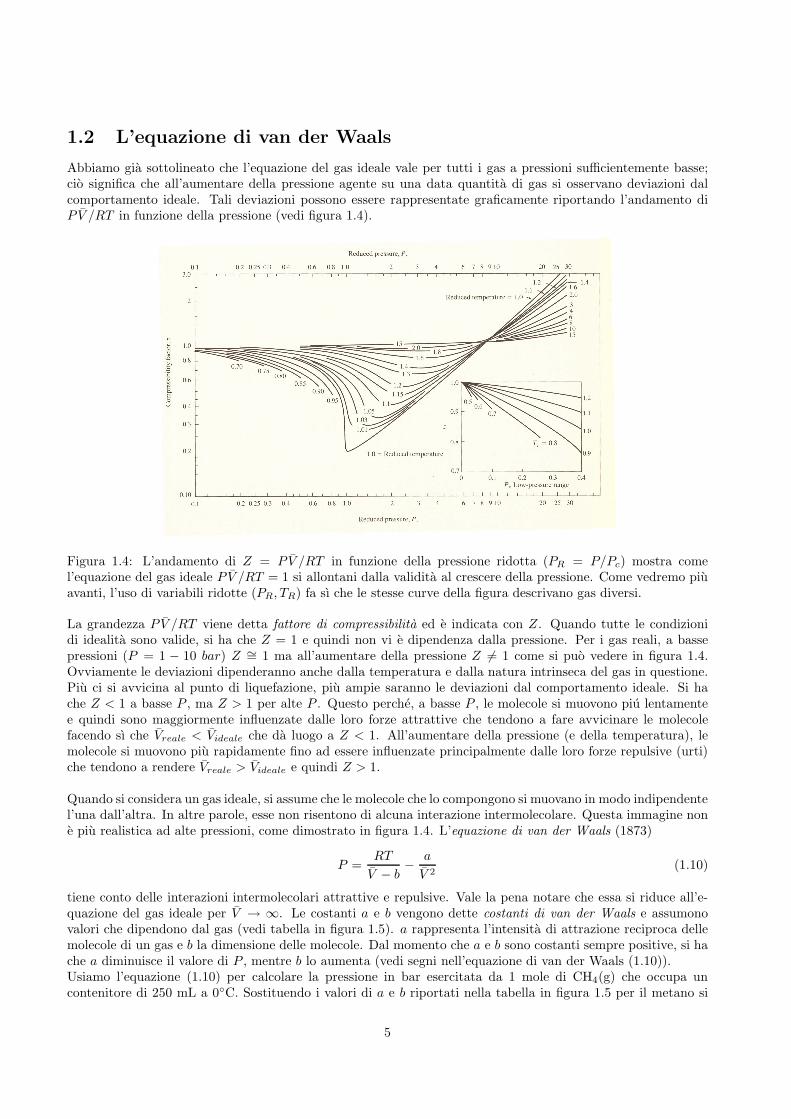
Abbiamo gia sottolineato che l’equazione del gas ideale vale per tutti i gas a pressioni sufficientemente basse;cio significa che all’aumentare della pressione agente su una data quantita di gas si osservano deviazioni dalcomportamento ideale. Tali deviazioni possono essere rappresentate graficamente riportando l’andamento diP V /RT in funzione della pressione (vedi figura 1.4).
Figura 1.4: L’andamento di Z = P V /RT in funzione della pressione ridotta (PR = P/Pc) mostra comel’equazione del gas ideale P V /RT = 1 si allontani dalla validita al crescere della pressione. Come vedremo piuavanti, l’uso di variabili ridotte (PR, TR) fa sı che le stesse curve della figura descrivano gas diversi.
La grandezza P V /RT viene detta fattore di compressibilita ed e indicata con Z. Quando tutte le condizionidi idealita sono valide, si ha che Z = 1 e quindi non vi e dipendenza dalla pressione. Per i gas reali, a bassepressioni (P = 1 − 10 bar) Z ∼= 1 ma all’aumentare della pressione Z 6= 1 come si puo vedere in figura 1.4.Ovviamente le deviazioni dipenderanno anche dalla temperatura e dalla natura intrinseca del gas in questione.Piu ci si avvicina al punto di liquefazione, piu ampie saranno le deviazioni dal comportamento ideale. Si hache Z < 1 a basse P , ma Z > 1 per alte P . Questo perche, a basse P , le molecole si muovono piu lentamentee quindi sono maggiormente influenzate dalle loro forze attrattive che tendono a fare avvicinare le molecolefacendo sı che Vreale < Videale che da luogo a Z < 1. All’aumentare della pressione (e della temperatura), lemolecole si muovono piu rapidamente fino ad essere influenzate principalmente dalle loro forze repulsive (urti)che tendono a rendere Vreale > Videale e quindi Z > 1.
Quando si considera un gas ideale, si assume che le molecole che lo compongono si muovano in modo indipendentel’una dall’altra. In altre parole, esse non risentono di alcuna interazione intermolecolare. Questa immagine none piu realistica ad alte pressioni, come dimostrato in figura 1.4. L’equazione di van der Waals (1873)
P =RT
V − b−
a
V 2(1.10)
tiene conto delle interazioni intermolecolari attrattive e repulsive. Vale la pena notare che essa si riduce all’e-quazione del gas ideale per V → ∞. Le costanti a e b vengono dette costanti di van der Waals e assumonovalori che dipendono dal gas (vedi tabella in figura 1.5). a rappresenta l’intensita di attrazione reciproca dellemolecole di un gas e b la dimensione delle molecole. Dal momento che a e b sono costanti sempre positive, si hache a diminuisce il valore di P , mentre b lo aumenta (vedi segni nell’equazione di van der Waals (1.10)).Usiamo l’equazione (1.10) per calcolare la pressione in bar esercitata da 1 mole di CH4(g) che occupa uncontenitore di 250 mL a 0◦C. Sostituendo i valori di a e b riportati nella tabella in figura 1.5 per il metano si
5
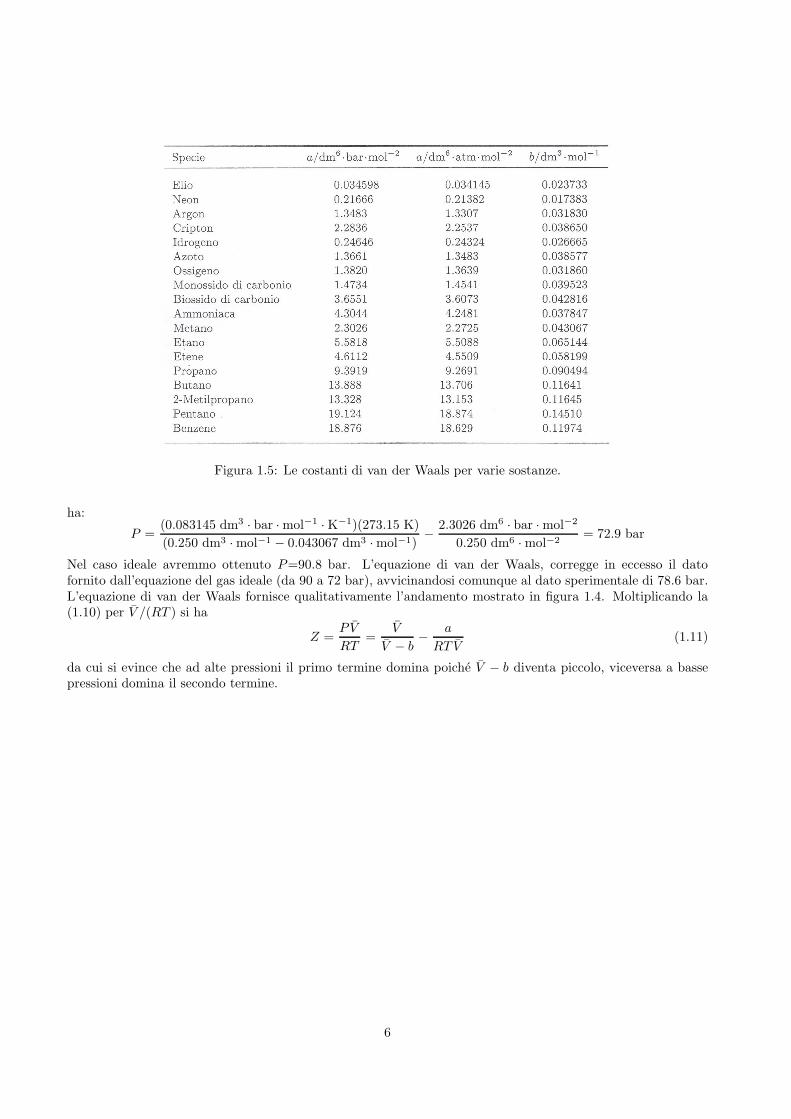
Figura 1.5: Le costanti di van der Waals per varie sostanze.
ha:
P =(0.083145 dm3 · bar · mol−1 · K−1)(273.15 K)
(0.250 dm3 · mol−1 − 0.043067 dm3 · mol−1)−
2.3026 dm6 · bar · mol−2
0.250 dm6 · mol−2= 72.9 bar
Nel caso ideale avremmo ottenuto P=90.8 bar. L’equazione di van der Waals, corregge in eccesso il datofornito dall’equazione del gas ideale (da 90 a 72 bar), avvicinandosi comunque al dato sperimentale di 78.6 bar.L’equazione di van der Waals fornisce qualitativamente l’andamento mostrato in figura 1.4. Moltiplicando la(1.10) per V /(RT ) si ha
Z =P V
RT=
V
V − b−
a
RT V(1.11)
da cui si evince che ad alte pressioni il primo termine domina poiche V − b diventa piccolo, viceversa a bassepressioni domina il secondo termine.
6

1.3 Le equazioni di Redlich-Kwong (RK) e di Peng-Robinson (PR)
Altre due equazioni di stato relativamente semplici, ma piu accurate e quindi piu utili dell’equazione di van derWaals sono l’equazione di Redlich-Kwong (1949)
P =RT
V − B−
A
T 1/2V (V + B)(1.12)
e l’equazione di Peng-Robinson (1976)
P =RT
V − β−
α
V (V + β) + β(V − β)(1.13)
dove A, B, α e β sono parametri che dipendono dal gas e che possono essere tabulati come fatto per i parametria e b che compaiono nell’equazione di van der Waals.
Riprendiamo l’esempio del metano considerato per l’equazione di van der Waals nel paragrafo precedente e cal-coliamo il valore della pressione con l’equazione di Redlich-Kwong. I dati sono gli stessi per temperatura (273.15K) e volume (0.250 dm3·mol−1). Per le costanti A e B, per il metano si ha che: A=32.205 dm6·bar·mol−2·K1/2
e B=0.029850 dm3·mol−1. Sostituendo questi valori nella (1.12) in maniera analoga a come si era fatto nelparagrafo precedente per l’equazione di van der Waals, si ha che P =75.3 bar, piu vicino al dato sperimentaledi 78.6 bar.
Mentre le equazioni di Redlich-Kwong e di Peng-Robinson sono pressoche quantitative, l’equazione di van derWaals cessa di dare previsioni corrette a pressioni maggiori di 200 bar. Inoltre, le equazioni di Redlich-Kwong e diPeng-Robinson continuano ad essere quantitativamente corrette anche nelle regioni in cui il gas diventa liquido.L’equazione di Peng-Robinson si comporta meglio nella regione liquido-vapore, mentre quella di Redlich-Kwongrappresenta meglio il comportamento del gas ad alte pressioni.Per concludere, notiamo come i primi addendi delle tre equazioni considerate (che descrivono la parte attrattiva),hanno la stessa forma funzionale, mentre i secondi sono molto diversi tra loro.
7
1.4 L’uso di un’equazione di stato cubica
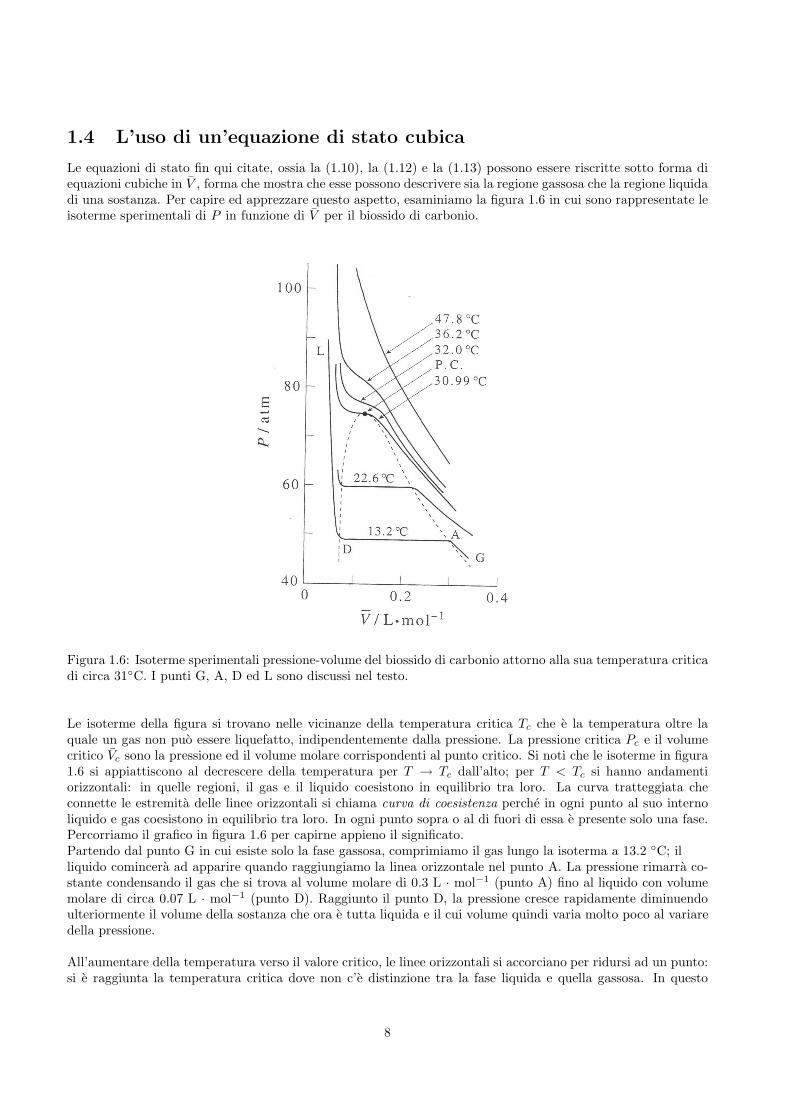
Le equazioni di stato fin qui citate, ossia la (1.10), la (1.12) e la (1.13) possono essere riscritte sotto forma diequazioni cubiche in V , forma che mostra che esse possono descrivere sia la regione gassosa che la regione liquidadi una sostanza. Per capire ed apprezzare questo aspetto, esaminiamo la figura 1.6 in cui sono rappresentate leisoterme sperimentali di P in funzione di V per il biossido di carbonio.
Figura 1.6: Isoterme sperimentali pressione-volume del biossido di carbonio attorno alla sua temperatura criticadi circa 31◦C. I punti G, A, D ed L sono discussi nel testo.
Le isoterme della figura si trovano nelle vicinanze della temperatura critica Tc che e la temperatura oltre laquale un gas non puo essere liquefatto, indipendentemente dalla pressione. La pressione critica Pc e il volumecritico Vc sono la pressione ed il volume molare corrispondenti al punto critico. Si noti che le isoterme in figura1.6 si appiattiscono al decrescere della temperatura per T → Tc dall’alto; per T < Tc si hanno andamentiorizzontali: in quelle regioni, il gas e il liquido coesistono in equilibrio tra loro. La curva tratteggiata checonnette le estremita delle linee orizzontali si chiama curva di coesistenza perche in ogni punto al suo internoliquido e gas coesistono in equilibrio tra loro. In ogni punto sopra o al di fuori di essa e presente solo una fase.Percorriamo il grafico in figura 1.6 per capirne appieno il significato.Partendo dal punto G in cui esiste solo la fase gassosa, comprimiamo il gas lungo la isoterma a 13.2 ◦C; illiquido comincera ad apparire quando raggiungiamo la linea orizzontale nel punto A. La pressione rimarra co-stante condensando il gas che si trova al volume molare di 0.3 L · mol−1 (punto A) fino al liquido con volumemolare di circa 0.07 L · mol−1 (punto D). Raggiunto il punto D, la pressione cresce rapidamente diminuendoulteriormente il volume della sostanza che ora e tutta liquida e il cui volume quindi varia molto poco al variaredella pressione.
All’aumentare della temperatura verso il valore critico, le linee orizzontali si accorciano per ridursi ad un punto:si e raggiunta la temperatura critica dove non c’e distinzione tra la fase liquida e quella gassosa. In questo
8
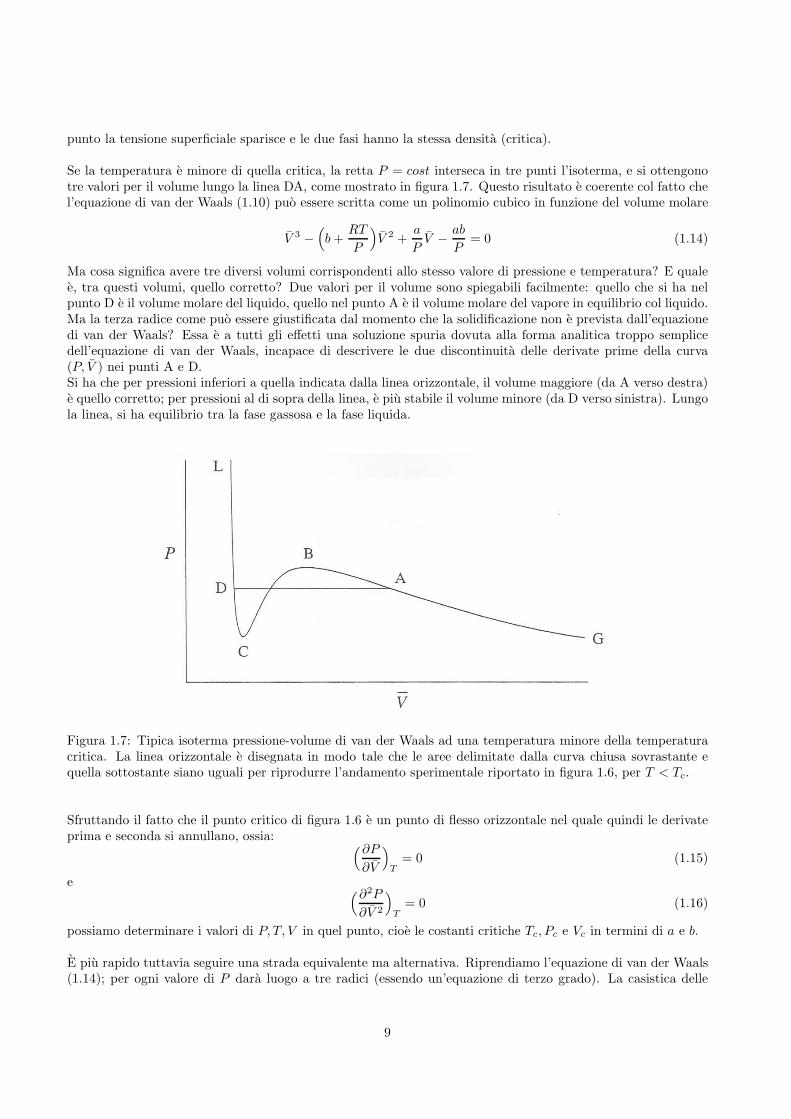
punto la tensione superficiale sparisce e le due fasi hanno la stessa densita (critica).
Se la temperatura e minore di quella critica, la retta P = cost interseca in tre punti l’isoterma, e si ottengonotre valori per il volume lungo la linea DA, come mostrato in figura 1.7. Questo risultato e coerente col fatto chel’equazione di van der Waals (1.10) puo essere scritta come un polinomio cubico in funzione del volume molare
V 3 −(
b +RT
P
)
V 2 +a
PV −
ab
P= 0 (1.14)
Ma cosa significa avere tre diversi volumi corrispondenti allo stesso valore di pressione e temperatura? E qualee, tra questi volumi, quello corretto? Due valori per il volume sono spiegabili facilmente: quello che si ha nelpunto D e il volume molare del liquido, quello nel punto A e il volume molare del vapore in equilibrio col liquido.Ma la terza radice come puo essere giustificata dal momento che la solidificazione non e prevista dall’equazionedi van der Waals? Essa e a tutti gli effetti una soluzione spuria dovuta alla forma analitica troppo semplicedell’equazione di van der Waals, incapace di descrivere le due discontinuita delle derivate prime della curva(P, V ) nei punti A e D.Si ha che per pressioni inferiori a quella indicata dalla linea orizzontale, il volume maggiore (da A verso destra)e quello corretto; per pressioni al di sopra della linea, e piu stabile il volume minore (da D verso sinistra). Lungola linea, si ha equilibrio tra la fase gassosa e la fase liquida.
Figura 1.7: Tipica isoterma pressione-volume di van der Waals ad una temperatura minore della temperaturacritica. La linea orizzontale e disegnata in modo tale che le aree delimitate dalla curva chiusa sovrastante equella sottostante siano uguali per riprodurre l’andamento sperimentale riportato in figura 1.6, per T < Tc.
Sfruttando il fatto che il punto critico di figura 1.6 e un punto di flesso orizzontale nel quale quindi le derivateprima e seconda si annullano, ossia:
(∂P
∂V
)
T= 0 (1.15)
e(∂2P
∂V 2
)
T= 0 (1.16)
possiamo determinare i valori di P, T, V in quel punto, cioe le costanti critiche Tc, Pc e Vc in termini di a e b.
E piu rapido tuttavia seguire una strada equivalente ma alternativa. Riprendiamo l’equazione di van der Waals(1.14); per ogni valore di P dara luogo a tre radici (essendo un’equazione di terzo grado). La casistica delle
9

soluzioni possibili e la seguente:
1) per T > Tc, si hanno una radice reale e due complesse;
2) per T < Tc e P ≈ Pc, tutte le radici sono reali (vedi figura 1.7 e sua discussione);
3) per T = Tc, si ha una sola radice triplamente degenere.
Il caso 3) ci permette di scrivere l’equazione (1.14) nella forma (V − Vc)3 = 0, ossia:
V 3 − 3VcV2 + 3V 2
c V − V 3
c = 0 (1.17)
Confrontando questa equazione con la (1.14) al punto critico, si ha:
3Vc = b +RTc
Pc, (1.18)
3V 2
c =a
Pc, (1.19)
V 3
c =ab
Pc. (1.20)
Eliminando Pc tra le equazioni (1.19) e (1.20) si ottiene
Vc = 3b (1.21)
che sostituito nella (1.20) da l’espressione di Pc
Pc =a
27b2. (1.22)
Infine, sostituendo la (1.21) e la (1.22) nella (1.18) si ha
Tc =8a
27bR. (1.23)
Abbiamo cosı ottenuto Vc, Pc, Tc in termini di a e b.
I valori delle costanti critiche dei parametri A e B dell’equazione di Redlich-Kwong possono essere determinatiin modo analogo.Se a questo punto calcolassimo il rapporto PcVc/RTc a partire dalle espressioni ottenute per le variabili ter-modinamiche critiche secondo van der Waals, Redlich-Kwong e Peng-Robinson otterremmo dei valori pressochecostanti. Questo e un esempio della legge degli stati corrispondenti per cui le proprieta di tutti i gas sono ugualise confrontate nel punto critico. Questo argomento verra ripreso e approfondito nel paragrafo seguente.
10

1.5 La legge degli stati corrispondenti
Riscriviamo l’equazione di van der Waals (1.10) nella forma(
P +a
V 2
)(
V − b)
= RT (1.24)
e in essa sostituiamo l’espessione di a che si ottiene dalla (1.19) e l’espressione di b che si ottiene dalla (1.21):
(
P +3PcV
2c
V 2
)(
V −1
3Vc
)
= RT (1.25)
Dividiamo ambo i membri per PcVc, nella forma
[( 1
Pc
)(
P +3PcV
2c
V 2
)][( 1
Vc
)(
V −1
3Vc
)]
=( 1
PcVc
)
RT (1.26)
e otteniamo( P
Pc+
3PcV2c
V 2Pc
)( V
Vc−
1
3
Vc
Vc
)
=RT
PcVc(1.27)
Semplificando si ha che( P
Pc+
3V 2c
V 2
)( V
Vc−
1
3
)
=RT
PcVc(1.28)
A secondo membro cerchiamoPcVc
RTc=
1
R
( a
27b2
)
(3b)(27bR
8a
)
=3
8(1.29)
che ci permette di scrivere la (1.28) come
( P
Pc+
3V 2c
V 2
)( V
Vc−
1
3
)
=8
3
T
Tc(1.30)
Introduciamo ora le seguenti quantita ridotte (adimensionali): PR = P/Pc, VR = V /Vc e TR = T/Tc che cipermettono di scrivere la (1.30) nella forma
(
PR +3
V 2
R
)(
V 2
R −1
3
)
=8
3TR (1.31)
L’ultima equazione e particolarmente importante perche vale per tutti i gas: essa infatti non contiene alcunaquantita caratteristica di un gas in particolare. Essa afferma che, per esempio, il valore di PR e lo stesso pertutti i gas che si trovano agli stessi valori di TR e di VR. L’equazione (1.31) e la legge degli stati corrispondenti
ottenuta a partire dall’equazione di van der Waals: secondo questa legge (cioe usando variabili ridotte), tutti igas messi a confronto nelle condizioni corrispondenti, si comportano allo stesso modo perche ubbidiscono alla(1.31). In altre parole, la (1.31) e una legge universale che vale per tutti i gas.
Come esempio, consideriamo CO2(g) e N2(g) per VR=20 e TR=1.5. Sostituendo questi valori nella (1.31), si hache PR=0.196. Usando i valori sperimentali delle costanti critiche (Tc=304.1 K, Pc=73.8 bar, Vc=0.094 L·mol−1
e Tc=126.2 K, Pc=34.0 bar, Vc=0.090 L·mol−1) per CO2(g) e N2(g) rispettivamente, si ha che PCO2=14.5 bar,VCO2=1.9 L·mol−1 e TCO2=456 K e che PN2=6.66 bar, VN2=1.8 L·mol−1 e TN2=189 K. Si dice quindi che questidue gas si trovano in stati corrispondenti (hanno cioe gli stessi valori di PR se si fissano gli stessi valori di VR e TR).
Il fattore di compressibilita Z associato con l’equazione di van der Waals obbedisce anch’esso alla legge deglistati corrispondenti. Per dimostrare che cio e vero, consideriamo la (1.11) e sostituiamo la (1.19) per a e la(1.21) per b per ottenere:
Z =P V
RT=
V
V − 1
3Vc
−3PcV
2c
RT V(1.32)
Dalla (1.29) si ha che
PcVc =3
8RTc (1.33)
11
quindi
Z =V
V − 1
3Vc
−3
8
3RTcVc
RT V(1.34)
che semplificata e dopo avere introdotto le variabili ridotte da luogo a
Z =VR
VR − 1
3
−9
8VRTR(1.35)
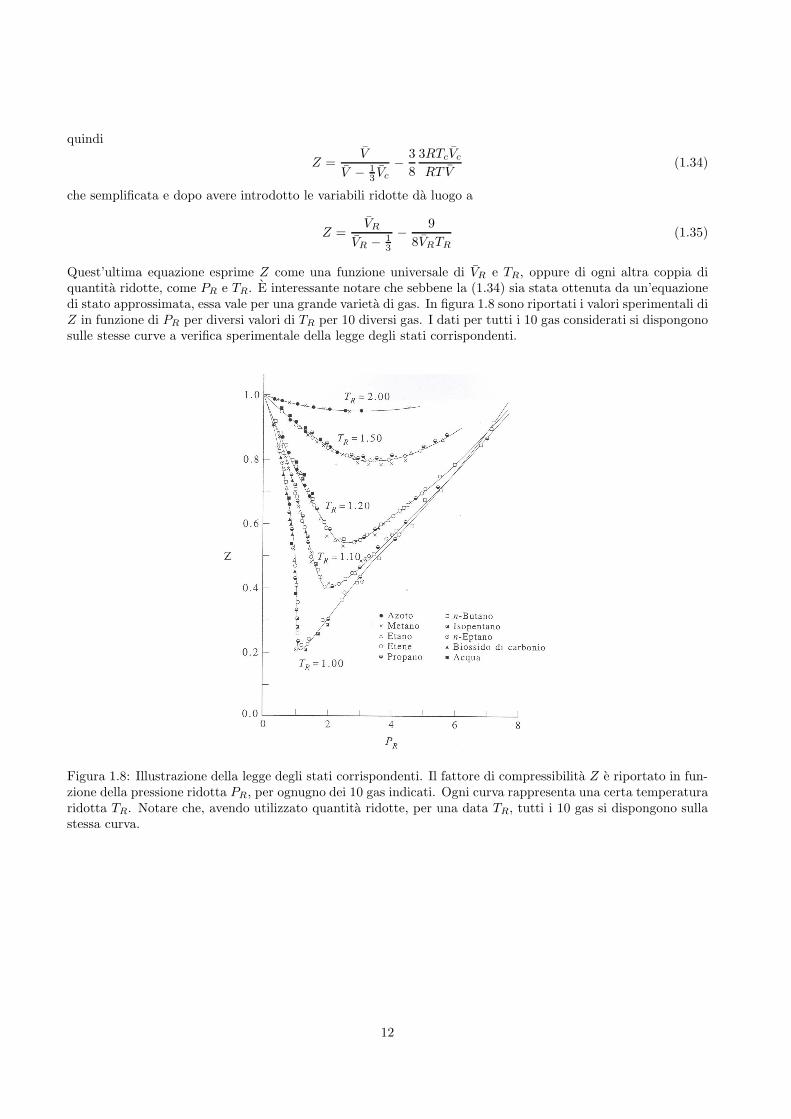
Quest’ultima equazione esprime Z come una funzione universale di VR e TR, oppure di ogni altra coppia diquantita ridotte, come PR e TR. E interessante notare che sebbene la (1.34) sia stata ottenuta da un’equazionedi stato approssimata, essa vale per una grande varieta di gas. In figura 1.8 sono riportati i valori sperimentali diZ in funzione di PR per diversi valori di TR per 10 diversi gas. I dati per tutti i 10 gas considerati si dispongonosulle stesse curve a verifica sperimentale della legge degli stati corrispondenti.
Figura 1.8: Illustrazione della legge degli stati corrispondenti. Il fattore di compressibilita Z e riportato in fun-zione della pressione ridotta PR, per ognugno dei 10 gas indicati. Ogni curva rappresenta una certa temperaturaridotta TR. Notare che, avendo utilizzato quantita ridotte, per una data TR, tutti i 10 gas si dispongono sullastessa curva.
12
1.6 Il secondo coefficiente del viriale
L’equazione di stato che possiede il fondamento teorico piu corretto e l’equazione di stato del viriale che esprimeil fattore di compressibilita come una serie (che si puo dimostrare essere convergente) in V :
Z =P V
RT= 1 +
B2V (T )
V+
B3V (T )
V 2+ ... (1.36)
I coefficienti di questa espressione sono funzioni della temperatura e vengono chiamati coefficienti del viriale; inparticolare: B2V (T ) e detto secondo coefficiente del viriale, B3V (T ) terzo coefficiente del viriale, ecc.
Il fattore di compressibilita puo essere anche espresso come una serie di potenze in P :
Z =P V
RT= 1 + B2P (T )P + B3P (T )P 2 + ... (1.37)
Notiamo che nelle due equazioni precendenti, Z → 1 per V → ∞ o per P → 0, rispettivamente. I coefficientidel viriale B2V (T ) e B2P (T ) sono legati dalla relazione:
B2V (T ) = RTB2P (T ) (1.38)
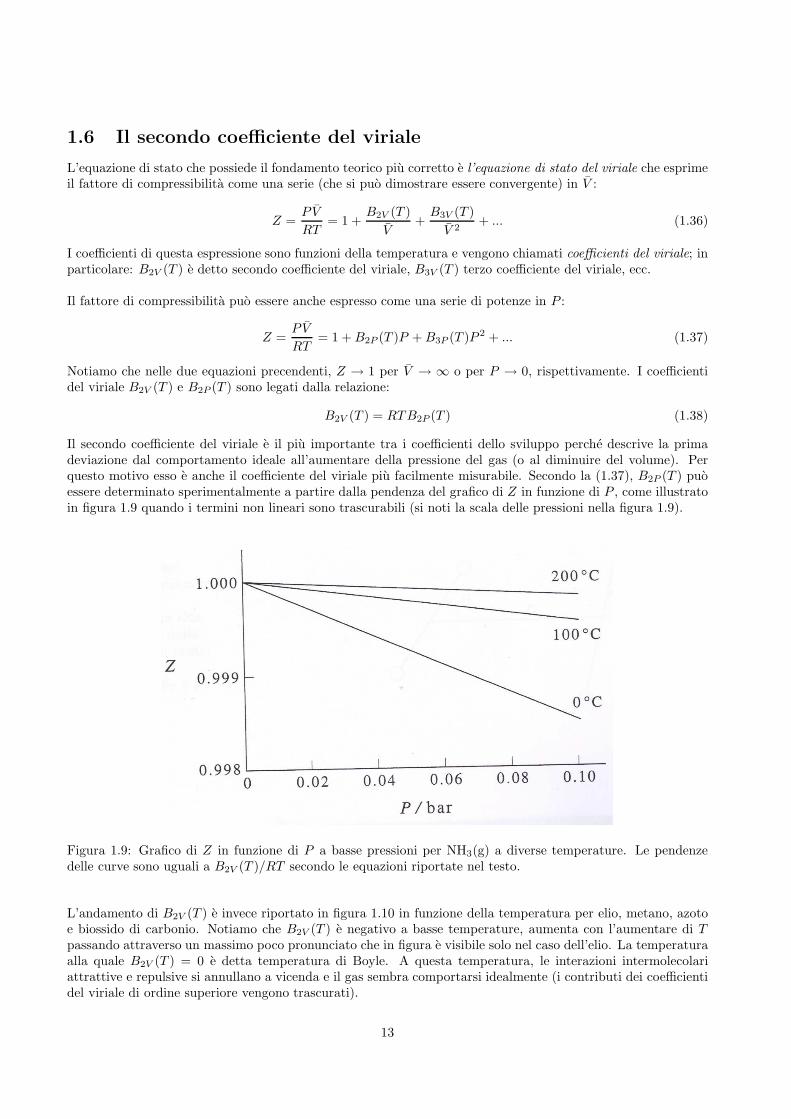
Il secondo coefficiente del viriale e il piu importante tra i coefficienti dello sviluppo perche descrive la primadeviazione dal comportamento ideale all’aumentare della pressione del gas (o al diminuire del volume). Perquesto motivo esso e anche il coefficiente del viriale piu facilmente misurabile. Secondo la (1.37), B2P (T ) puoessere determinato sperimentalmente a partire dalla pendenza del grafico di Z in funzione di P , come illustratoin figura 1.9 quando i termini non lineari sono trascurabili (si noti la scala delle pressioni nella figura 1.9).
Figura 1.9: Grafico di Z in funzione di P a basse pressioni per NH3(g) a diverse temperature. Le pendenzedelle curve sono uguali a B2V (T )/RT secondo le equazioni riportate nel testo.
L’andamento di B2V (T ) e invece riportato in figura 1.10 in funzione della temperatura per elio, metano, azotoe biossido di carbonio. Notiamo che B2V (T ) e negativo a basse temperature, aumenta con l’aumentare di Tpassando attraverso un massimo poco pronunciato che in figura e visibile solo nel caso dell’elio. La temperaturaalla quale B2V (T ) = 0 e detta temperatura di Boyle. A questa temperatura, le interazioni intermolecolariattrattive e repulsive si annullano a vicenda e il gas sembra comportarsi idealmente (i contributi dei coefficientidel viriale di ordine superiore vengono trascurati).
13
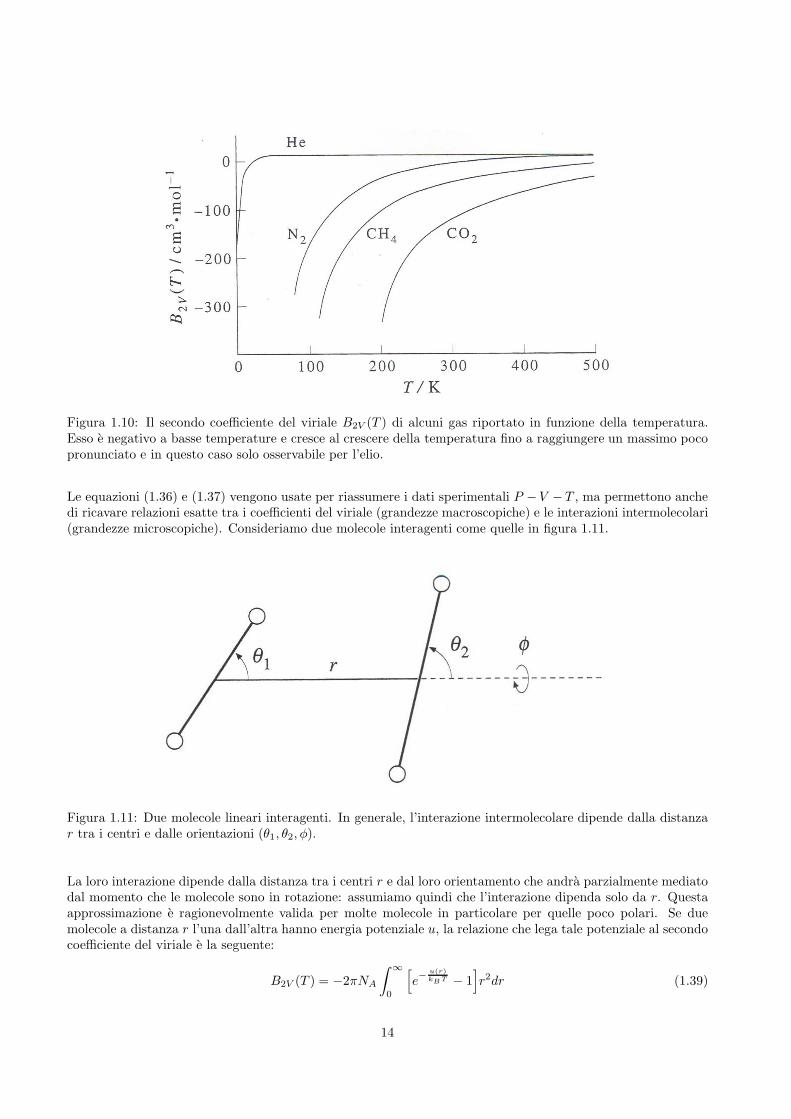
Figura 1.10: Il secondo coefficiente del viriale B2V (T ) di alcuni gas riportato in funzione della temperatura.Esso e negativo a basse temperature e cresce al crescere della temperatura fino a raggiungere un massimo pocopronunciato e in questo caso solo osservabile per l’elio.
Le equazioni (1.36) e (1.37) vengono usate per riassumere i dati sperimentali P −V −T , ma permettono anchedi ricavare relazioni esatte tra i coefficienti del viriale (grandezze macroscopiche) e le interazioni intermolecolari(grandezze microscopiche). Consideriamo due molecole interagenti come quelle in figura 1.11.
Figura 1.11: Due molecole lineari interagenti. In generale, l’interazione intermolecolare dipende dalla distanzar tra i centri e dalle orientazioni (θ1, θ2, φ).
La loro interazione dipende dalla distanza tra i centri r e dal loro orientamento che andra parzialmente mediatodal momento che le molecole sono in rotazione: assumiamo quindi che l’interazione dipenda solo da r. Questaapprossimazione e ragionevolmente valida per molte molecole in particolare per quelle poco polari. Se duemolecole a distanza r l’una dall’altra hanno energia potenziale u, la relazione che lega tale potenziale al secondocoefficiente del viriale e la seguente:
B2V (T ) = −2πNA
∫
∞
0
[
e−
u(r)kB T − 1
]
r2dr (1.39)
14
dove NA e la costante di Avogadro e kB e la costante di Boltzman (kB = R/NA). Notiamo che B2V (T ) = 0quando u(r) = 0, ossia se non ci sono interazioni molecolari, il gas si comporta in modo ideale.
Dalla (1.39) si ha che dato il potenziale u(r), e possibile calcolare facilmente B2V (T ) come funzione dellatemperatura o viceversa. Grazie alla teoria delle perturbazioni, e possibile dimostrare che
u(r) → −c6
r6(1.40)
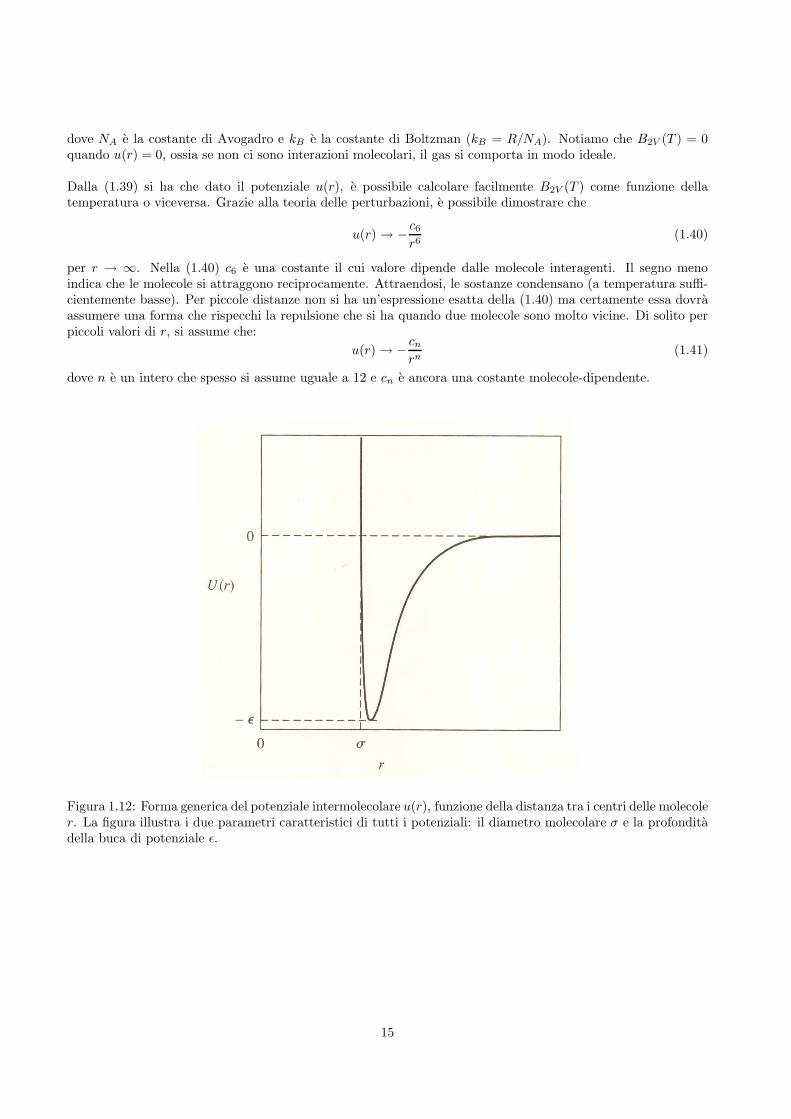
per r → ∞. Nella (1.40) c6 e una costante il cui valore dipende dalle molecole interagenti. Il segno menoindica che le molecole si attraggono reciprocamente. Attraendosi, le sostanze condensano (a temperatura suffi-cientemente basse). Per piccole distanze non si ha un’espressione esatta della (1.40) ma certamente essa dovraassumere una forma che rispecchi la repulsione che si ha quando due molecole sono molto vicine. Di solito perpiccoli valori di r, si assume che:
u(r) → −cn
rn(1.41)
dove n e un intero che spesso si assume uguale a 12 e cn e ancora una costante molecole-dipendente.
Figura 1.12: Forma generica del potenziale intermolecolare u(r), funzione della distanza tra i centri delle molecoler. La figura illustra i due parametri caratteristici di tutti i potenziali: il diametro molecolare σ e la profonditadella buca di potenziale ǫ.
15
1.7 Il potenziale di Lennard-Jones
Un potenziale intermolecolare che comprenda l’andamento a lungo raggio (attrattivo) dell’equazione (1.40) el’andamento a corto raggio (repulsivo) dell’equazione (1.41) e semplicemente la somma delle due. Se conside-riamo n = 12, si ha
u(r) = 4ǫ[(σ
r
)12
−(σ
r
)6]
(1.42)
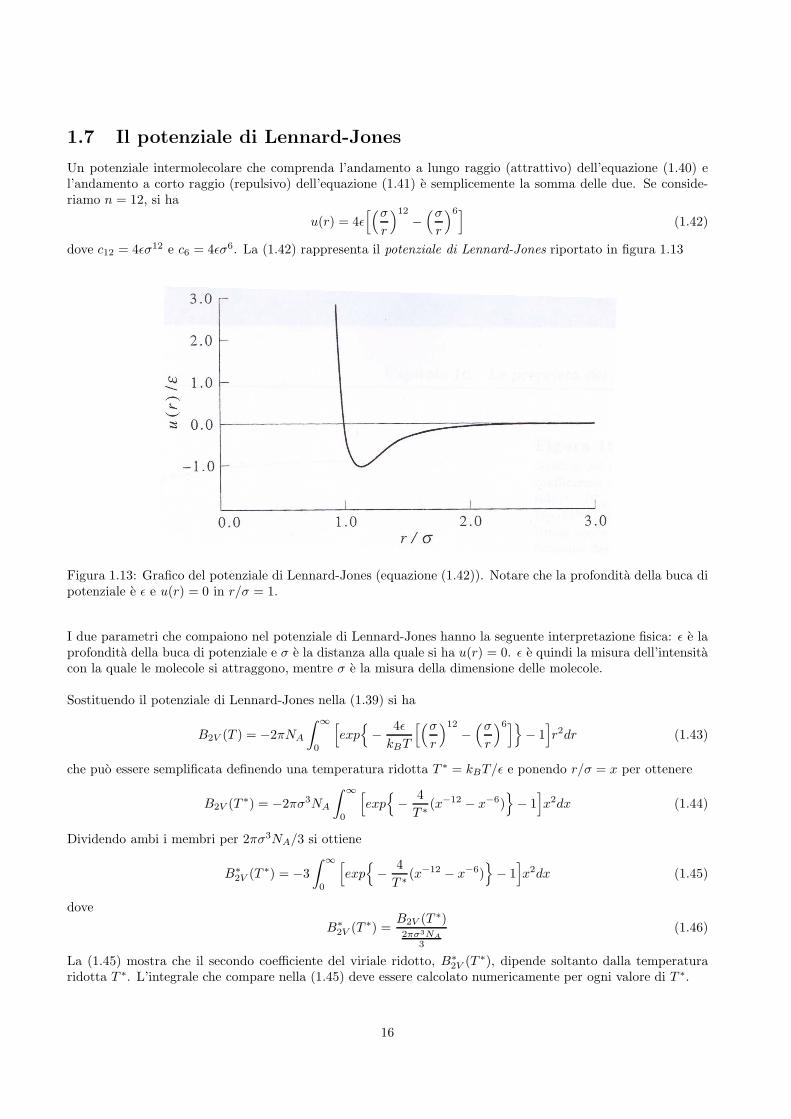
dove c12 = 4ǫσ12 e c6 = 4ǫσ6. La (1.42) rappresenta il potenziale di Lennard-Jones riportato in figura 1.13
Figura 1.13: Grafico del potenziale di Lennard-Jones (equazione (1.42)). Notare che la profondita della buca dipotenziale e ǫ e u(r) = 0 in r/σ = 1.
I due parametri che compaiono nel potenziale di Lennard-Jones hanno la seguente interpretazione fisica: ǫ e laprofondita della buca di potenziale e σ e la distanza alla quale si ha u(r) = 0. ǫ e quindi la misura dell’intensitacon la quale le molecole si attraggono, mentre σ e la misura della dimensione delle molecole.
Sostituendo il potenziale di Lennard-Jones nella (1.39) si ha
B2V (T ) = −2πNA
∫
∞
0
[
exp{
−4ǫ
kBT
[(σ
r
)12
−(σ
r
)6]}
− 1]
r2dr (1.43)
che puo essere semplificata definendo una temperatura ridotta T ∗ = kBT/ǫ e ponendo r/σ = x per ottenere
B2V (T ∗) = −2πσ3NA
∫
∞
0
[
exp{
−4
T ∗(x−12 − x−6)
}
− 1]
x2dx (1.44)
Dividendo ambi i membri per 2πσ3NA/3 si ottiene
B∗
2V (T ∗) = −3
∫
∞
0
[
exp{
−4
T ∗(x−12 − x−6)
}
− 1]
x2dx (1.45)
dove
B∗
2V (T ∗) =B2V (T ∗)2πσ3NA
3
(1.46)
La (1.45) mostra che il secondo coefficiente del viriale ridotto, B∗
2V (T ∗), dipende soltanto dalla temperaturaridotta T ∗. L’integrale che compare nella (1.45) deve essere calcolato numericamente per ogni valore di T ∗.
16
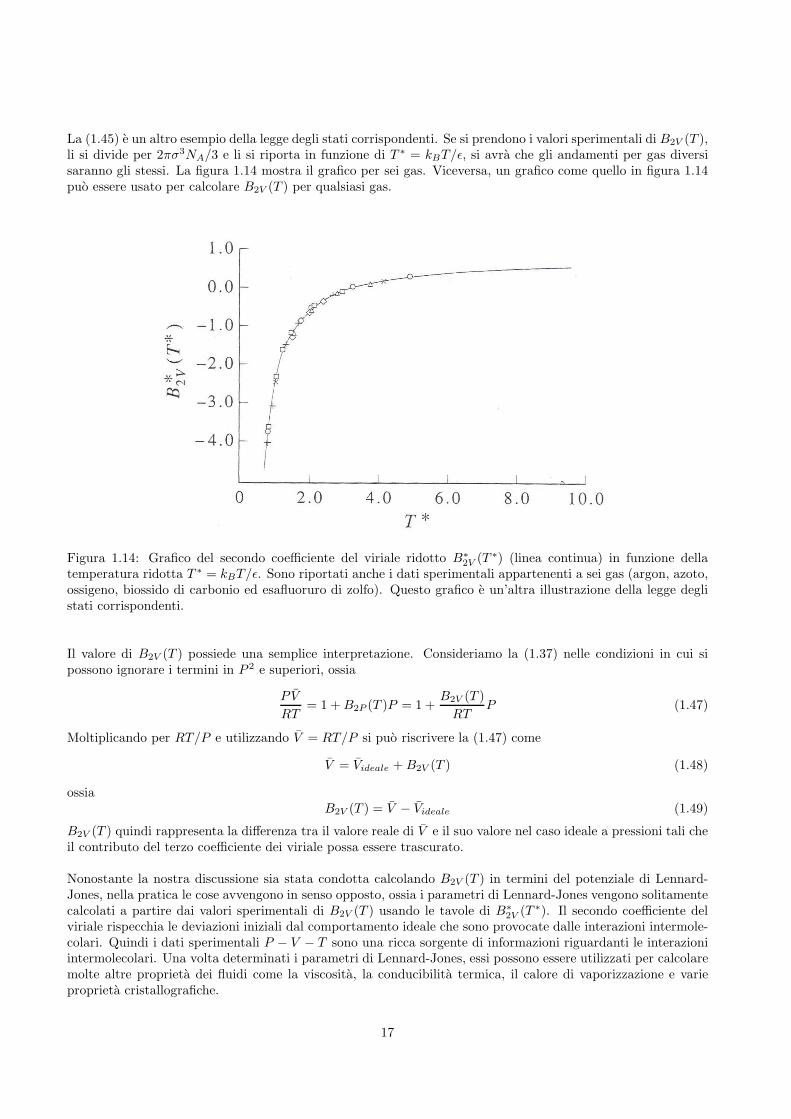
La (1.45) e un altro esempio della legge degli stati corrispondenti. Se si prendono i valori sperimentali di B2V (T ),li si divide per 2πσ3NA/3 e li si riporta in funzione di T ∗ = kBT/ǫ, si avra che gli andamenti per gas diversisaranno gli stessi. La figura 1.14 mostra il grafico per sei gas. Viceversa, un grafico come quello in figura 1.14puo essere usato per calcolare B2V (T ) per qualsiasi gas.
Figura 1.14: Grafico del secondo coefficiente del viriale ridotto B∗
2V (T ∗) (linea continua) in funzione dellatemperatura ridotta T ∗ = kBT/ǫ. Sono riportati anche i dati sperimentali appartenenti a sei gas (argon, azoto,ossigeno, biossido di carbonio ed esafluoruro di zolfo). Questo grafico e un’altra illustrazione della legge deglistati corrispondenti.
Il valore di B2V (T ) possiede una semplice interpretazione. Consideriamo la (1.37) nelle condizioni in cui sipossono ignorare i termini in P 2 e superiori, ossia
P V
RT= 1 + B2P (T )P = 1 +
B2V (T )
RTP (1.47)
Moltiplicando per RT/P e utilizzando V = RT/P si puo riscrivere la (1.47) come
V = Videale + B2V (T ) (1.48)
ossiaB2V (T ) = V − Videale (1.49)
B2V (T ) quindi rappresenta la differenza tra il valore reale di V e il suo valore nel caso ideale a pressioni tali cheil contributo del terzo coefficiente dei viriale possa essere trascurato.
Nonostante la nostra discussione sia stata condotta calcolando B2V (T ) in termini del potenziale di Lennard-Jones, nella pratica le cose avvengono in senso opposto, ossia i parametri di Lennard-Jones vengono solitamentecalcolati a partire dai valori sperimentali di B2V (T ) usando le tavole di B∗
2V (T ∗). Il secondo coefficiente delviriale rispecchia le deviazioni iniziali dal comportamento ideale che sono provocate dalle interazioni intermole-colari. Quindi i dati sperimentali P − V − T sono una ricca sorgente di informazioni riguardanti le interazioniintermolecolari. Una volta determinati i parametri di Lennard-Jones, essi possono essere utilizzati per calcolaremolte altre proprieta dei fluidi come la viscosita, la conducibilita termica, il calore di vaporizzazione e varieproprieta cristallografiche.
17

1.8 Le forze di dispersione di London
Nel paragrafo precedente, si e usato il potenziale di Lennard-Jones (equazione (1.42)) per rappresentare il po-tenziale intermolecolare che agisce tra le molecole. Il termine r−12 tiene conto della repulsione a corta distanzamentre il termine r−6 tiene conto dell’attrazione a grande distanza. La vera forma del termine repulsivo none ben determinata; lo e invece la dipendenza r−6 del termine attrattivo. In questo paragrafo verranno discussitre contributi al termine di attrazione r−6 e verranno confrontate le loro importanze relative.
L’interazione tra due molecole dipolari con momenti di dipolo µ1 e µ2 dipendera da come questi dipoli sonoorientati l’uno rispetto all’altro. L’energia variera da repulsiva, quando sono orientati “testa a testa” comemostrato in figura 1.15a ed attrattiva, quando sono orientati “testa a coda” come in figura 1.15b.
Figura 1.15: Due dipoli permanenti orientati (a) “testa a testa” e (b) “testa a coda”. L’orientazione “testa acoda” e energeticamente favorita. (c) Una molecola con un momento di dipolo permanente indurra un momentodi dipolo di una molecola vicina. (d) La correlazione istantanea dipolo-dipolo qui illustrata e cio che porta aduna attrazione di London tra tutti gli atomi e le molecole.
In fase gassosa, entrambe le molecole ruotano, e facendo la media di entrambi i dipoli casualmente orientati, leinterazioni dipolo-dipolo risulterebbero nulle. Tuttavia, dal momento che le diverse orientazioni hanno energiediverse, esse non sono equiprobabili. L’orientazione di bassa energia “testa a coda” e ovviamente quella favoritarispetto all’orientazione repulsiva “testa a testa”. Tenendo conto delle diverse configurazioni, si ha che la mediacomplessiva dell’interazione tra le due molecole da luogo ad un termine attrattivo r−6 della forma
ud−d(r) = −2µ2
1µ22
(4πǫ0)2(3kBT )
1
r6(1.50)
Questo quando entrambe le molecole hanno un momento di dipolo permanente.Nel caso in cui una molecola non possegga un momento di dipolo permanente, essa avra un momento di dipoloindotto dall’altra. Cio e possibile perche tutti gli atomi e le molecole sono polarizzabili. Infatti, quando unatomo o una molecola interagisce con il campo elettrico creato da un’altra molecola, gli elettroni (negativi)vengono spostati in una direzione e i nuclei (positivi) si spostano nella direzione opposta (vedi figura 1.15c).Questa separazione di carica, che genera un momento di dipolo, e proporzionale all’intensita del campo elettrico.Se indichiamo con µindotto il momento di dipolo indotto e con E il campo elettrico, si ha che µindotto ∝ E. Lacostante di proporzionalita, che denominiamo α, e detta polarizzabilita. Si ha quindi che
µindotto = αE (1.51)
Le unita del campo elettrico E sono V·m−1 quindi le unita di α nella (1.51) sono
C · m
V · m−1= C · m2 · V−1
18

dove C=coulomb e V=volt. Si ha quindi che il momento di dipolo e misurato in C·m. Sapendo che
energia =(carica)2
4πǫ0(distanza)
in unita SI si ha
joule ∼C2
(4πǫ0)m=
C2m−1
4πǫ0
Allo stesso modo, dall’elettrostatica si ha che
joule = coulomb× volt = C · V
Uguagliando queste due espressioni e semplificando rispetto ai joule si ottiene
C · V =C2m−1
4πǫ0→ C · V−1 = (4πǫ0)m
che sostituita nelle unita di α da:α ∼ (4πǫ0)m
3
da cui si ha che la quantita α/(4πǫ0), a cui ci si riferisce come al volume di polarizzabilita, ha le unita di una(distanza)3. Piu facilmente il campo elettrico riesce a deformare la distribuzione atomica o molecolare, maggioree la polarizzabilita. La polarizzabilita di un atomo o di una molecola e proporzionale alle sue dimensioni e alnumero dei suoi elettroni.
Torniamo ora all’interazione di momento di dipolo indotto illustrata in figura 1.15c. Visto che il momentodi dipolo indotto si trova sempre nell’orientazione “testa a coda” rispetto al momento di dipolo permanente,l’interazione e sempre attrattiva ed e data da
µindotto(r) = −µ2
1α2
(4πǫ0)2r6−
µ22α1
(4πǫ0)2r6(1.52)
dove il primo termine rappresenta un momento di dipolo permanente nella molecola 1 ed un momento di dipoloindotto nella molecola 2, mentre il secondo rappresenta la situazione opposta.
La (1.50) e la (1.52) sono nulle quando nessuna delle molecole possiede un momento di dipolo permanente.
Il terzo contributo al termine r−6 del potenziale di Lennard-Jones (1.42) e non nullo anche se entrambe lemolecole non sono polari. Questo contributo fu calcolato per la prima volta nel 1930 dallo scienziato tedescoFritz London e viene detto attrazione di dispersione di London. Nonostante questa attrazione sia un fenomenoquantomeccanico, essa puo essere interpretata in modo classico come segue. Consideriamo due atomi che sitrovano a distanza r (vedi figura 1.15d). Gli elettroni appartenenti ad un atomo non schermano completamentela forte carica positiva del nucleo dagli elettroni dell’altro atomo. Dal momento che la molecola e polarizzabile,la funzione d’onda elettronica si puo modificare per abbassare ulteriormente l’energia di interazione. Facendola media quantistica di questa attrazione elettronica, si ottiene un termine attrattivo che si comporta come r−6.Una forma approssimata del risultato finale e
µdisp(r) = −3
2
( I1I2
I1 + I2
) α1α2
(4πǫ0)21
r6(1.53)
dove Ij e l’energia di ionizzazione dell’atomo o della molecola j. Notiamo che la (1.53) non contiene un momentodi dipolo permanente e che l’energia di interazione e proporzionale al prodotto dei volumi di polarizzabilita.Quindi, il peso di µdisp(r) aumenta con la dimensione del sistema e per questo e spesso il contributo dominanteal termine r−6 del potenziale di Lennard-Jones (1.42).
Per concludere, il contributo totale al termine r−6 del potenziale di Lennard-Jones (1.42) e dato dalle equazioni(1.50), (1.52) e (1.53), quindi:
c6 =2µ4
3(4πǫ0)2kBT+
2αµ2
(4πǫ0)2+
3
4
Iα2
(4πǫ0)2(1.54)
per atomi o molecole identici.
19

1.9 Il secondo coefficiente del viriale: alcuni casi particolari
Nonostante il potenziale di Lennard-Jones sia abbastanza realistico, esso e difficile da utilizzare. Infatti, ilsecondo coefficiente del viriale deve essere calcolato numericamente. Per questo motivo, per stimare le proprietadei gas si usano spesso potenziali intermolecolari che possono essere calcolati analiticamente. Il piu semplice diquesti potenziali e il cosiddetto potenziale di sfera rigida (vedi figura 1.16a) che ha forma matematica
u(r) =
{
∞ per r < σ0 per r > σ
(1.55)
Figura 1.16: (a) Illustrazione schematica di un potenziale di sfera rigida e (b) di un potenziale di buca quadrata.Il parametro σ e il diametro delle molecole, ǫ e la profondita della buca attrattiva e (λ−1)σ e la larghezza dellabuca.
La (1.55) rappresenta sfere rigide di diametro σ e descrive la regione repulsiva con un andamento infinitamenteripido piuttosto che come r−12. Notiamo anche come questo potenziale tenga conto della dimensione finita dellemolecole, caratteristica fondamentale nella determinazione della struttura dei liquidi e dei solidi ma manchidi un termine attrattivo. Ad alte temperature (rispetto al termine ǫ/kB), le molecole si muovono con energiasufficiente affinche il potenziale attrattivo sia trascurabile.
Nel caso del potenziale a sfera rigida, e facile calcolare il secondo coefficiente del viriale. Sostituendo la (1.55)nella (1.39) si ha
B2V (T ) = −2πNA
∫
∞
0
[
e−
u(r)kB T − 1
]
r2dr
= −2πNA
∫ σ
0
[0 − 1]r2dr − 2πNA
∫
∞
σ
[e0 − 1]r2dr =2πσ3NA
3(1.56)
che e uguale a quattro volte il volume di NA sfere dal momento che σ e il diametro delle sfere. Si ha quindi cheil secondo coefficiente del viriale calcolato per il potenziale a sfera rigida non dipende dalla temperatura.
Un altro semplice potenziale spesso utilizzato e il potenziale di buca quadrata (vedi figura 1.16b):
u(r) =
∞ per r < σ−ǫ per σ < r < λσ0 per r > λσ
(1.57)
Il parametro ǫ e la profondita della buca e (λ−1)σ e la sua larghezza. Questa potenziale da luogo ad una regioneattrattiva, sebbene grossolana. Nel caso del potenziale a buca quadrata, il secondo coefficiente del viriale puoessere calcolato analiticamente:
B2V (T ) = −2πNA
∫ σ
0
[0 − 1]r2dr − 2πNA
∫ λσ
σ
[eǫ/kBT − 1]r2dr − 2πNA
∫
∞
λσ
[e0 − 1]r2dr
20
=2πσ3NA
3−
2πσ3NA
3(λ3 − 1)(eǫ/kBT − 1) =
2πσ3NA
3[1 − (λ3 − 1)(eǫ/kBT − 1)] (1.58)
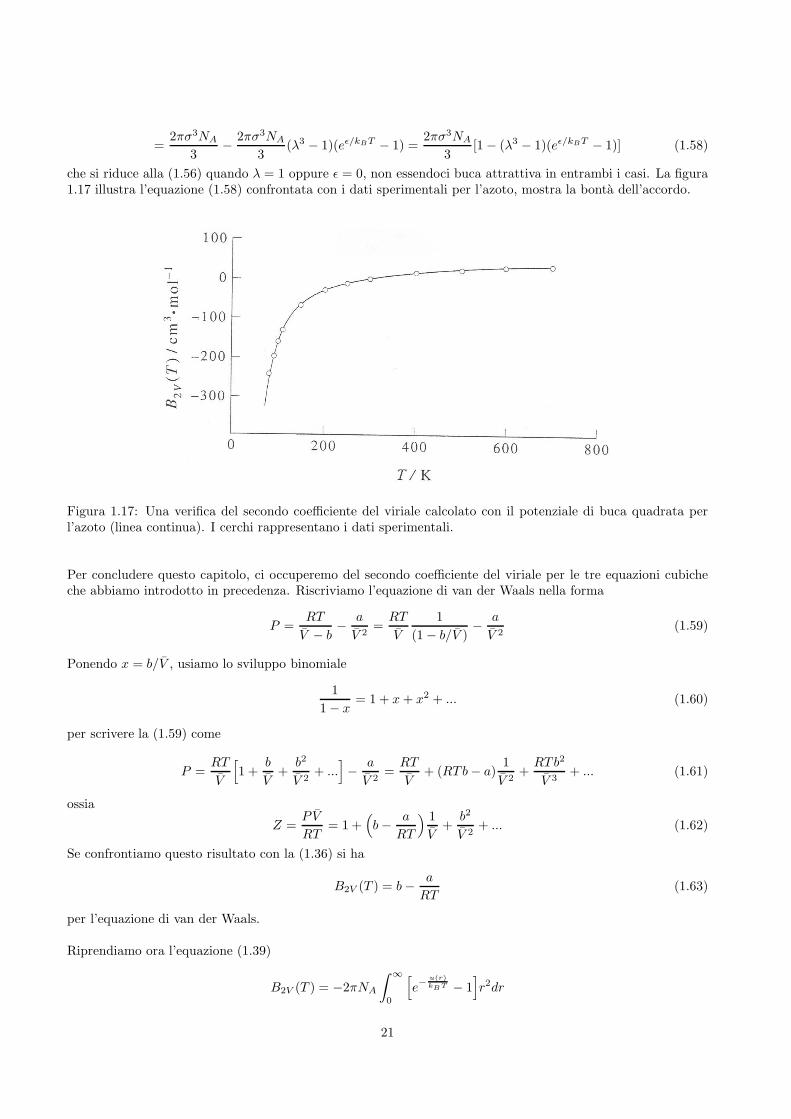
che si riduce alla (1.56) quando λ = 1 oppure ǫ = 0, non essendoci buca attrattiva in entrambi i casi. La figura1.17 illustra l’equazione (1.58) confrontata con i dati sperimentali per l’azoto, mostra la bonta dell’accordo.
Figura 1.17: Una verifica del secondo coefficiente del viriale calcolato con il potenziale di buca quadrata perl’azoto (linea continua). I cerchi rappresentano i dati sperimentali.
Per concludere questo capitolo, ci occuperemo del secondo coefficiente del viriale per le tre equazioni cubicheche abbiamo introdotto in precedenza. Riscriviamo l’equazione di van der Waals nella forma
P =RT
V − b−
a
V 2=
RT
V
1
(1 − b/V )−
a
V 2(1.59)
Ponendo x = b/V , usiamo lo sviluppo binomiale
1
1 − x= 1 + x + x2 + ... (1.60)
per scrivere la (1.59) come
P =RT
V
[
1 +b
V+
b2
V 2+ ...
]
−a
V 2=
RT
V+ (RTb − a)
1
V 2+
RTb2
V 3+ ... (1.61)
ossia
Z =P V
RT= 1 +
(
b −a
RT
) 1
V+
b2
V 2+ ... (1.62)
Se confrontiamo questo risultato con la (1.36) si ha
B2V (T ) = b −a
RT(1.63)
per l’equazione di van der Waals.
Riprendiamo ora l’equazione (1.39)
B2V (T ) = −2πNA
∫
∞
0
[
e−
u(r)kB T − 1
]
r2dr
21

e da essa ricaviamo un risultato simile alla (1.63). I parametri a e b verranno interpretati in termini dei parametrimolecolari. Sostituiamo il seguente potenziale intermolecolare
u(r) =
{
∞ per r < σ− c6
r6 per r > σ(1.64)
che e un ibrido tra il potenziale a sfera rigida e il potenziale di Lennard-Jones, nella (1.39) e otteniamo
B2V (T ) = −2πNA
∫ σ
0
(−1)r2dr − 2πNA
∫
∞
σ
[
ec6
kB T r6 − 1]
r2dr (1.65)
Nel secondo integrale, assumiamo che c6/kBTr6 ≪ 1 ed usiamo lo sviluppo per ex
ex = 1 + x +x2
2!+ ... (1.66)
fermandoci al prim’ordine per ottenere
B2V (T ) =2πσ3NA
3−
2πNAc6
kBT
∫
∞
σ
r2dr
r6=
2πσ3NA
3−
2πNAc6
3kBTσ3(1.67)
Confrontando questa equazione con la (1.63) e ricordando che kB = R/NA si ha che
a =2πN2
Ac6
3σ3
b =2πσ3NA
3
Si ha quindi che a e direttamente proporzionale a c6 che e il coefficiente di r−6 nel potenziale intermolecolareattrattivo, e che b e uguale al quadruplo del volume delle molecole. Da un punto di vista molecolare, l’equazionedi van der Waals si basa su un potenziale intermolecolare che a piccole distanze si comporta come un potenzialedi sfera rigida e a grandi distanze ha le caratteristiche di un potenziale debolmente attrattivo, ossia tale chec6/kBTr6 ≪ 1.
In maniera analoga si possono ottenere il secondo coefficiente del viriale per l’equazione di Redlich-Kwong
B2V (T ) = B −A
RT 3/2(1.68)
e per l’equazione di Peng-Robinson
B2V (T ) = β −α
RT(1.69)
Quest’ultimo ha la stessa forma funzionale del secondo coefficiente del viriale ottenuto per l’equazione di van derWaals, ma ha ovviamente valori numerici diversi perche i valori delle costanti sono diversi. Inoltre, il parametroα e una funzione della temperatura.
22

1.10 Esercizi svolti
1.10.1 Esercizio 1.1
Un gas ideale subisce una compressione a T costante che ne riduce il volume di 2.20 dm3. La pressione e ilvolume finali del gas sono rispettivamente 3.78·103 Torr e 4.65 dm3.Calcolate la pressione iniziale del gas (a) in Torr, (b) in atm.
Soluzione
a) Applichiamo l’equazione di stato dei gas ideali PV = nRT .Dato che n e T sono costanti e R e una costante allora PV = cost. (Legge di Boyle).Quindi vale PiVi = PfVf da cui:
Pi (Torr) =PfVf
Vi
=3.78 · 103 Torr · 4.65 dm3
6.85 dm3= 2.56 · 103 Torr
b) Poiche 760 Torr equivalgono ad un’atmosfera, si puo convertire la pressione da Torr ad atm:
Pi (atm) =1 atm · 2560 Torr
760 Torr= 3.37 atm
1.10.2 Esercizio 1.2
Un recipiente del volume di 1.0 L contiene 131 g di Xe alla temperatura di 25◦C.Calcolare la pressione del gas nell’ipotesi che si comporti (a) come un gas perfetto (b) come un gas di van derWaals. Si assuma per lo xenon: a=4.194 dm6·atm·mol−2 e b=5.105·10−2 dm3·mol−1.
Soluzione
a) Convertiamo per prima cosa i grammi in moli. Si ha che
nXe =131 g
131 g · mol−1= 1.0 mol
Per la legge dei gas ideali, vale PV = nRT . Quindi
P (atm) =1.0 mol · 0.082058 dm3 · atm · mol−1 · K−1 · 298 K
1.0 dm3= 24 atm
b) L’equazion di van der Waals e:
P =nRT
V − nb−
an2
V 2
Quindi:
P =1.0 mol · 0.082058 dm3 · atm · mol−1 · K−1 · 298 K
1.0 dm3 − (1.0 mol · 5.105 · 10−2dm3 · mol−1(1.70)
−4.194 dm6 · atm · mol−2 · (1.0)2 mol2
(1.0)2 dm6(1.71)
= 22 atm (1.72)
23

1.10.3 Esercizio 1.3
Dimostrare che il minimo del potenziale di Lennard-Jones si trova in rmin = 21/6σ = 1.12σ. Calcolare u(r) inrmin.
Soluzione
Per trovare rmin, deriviamo l’equazione (1.42):
du
dr= 4ǫ
[
−12σ12
r13+
6σ6
r7
]
= 0
che da r6min = 2σ6, ossia rmin = 21/6σ. Quindi,
u(rmin) = 4ǫ[( σ
21/6σ
)12
−( σ
21/6σ
)6]
= 4ǫ(1
4−
1
2
)
= −ǫ
ǫ e la profondita della buca di potenziale, relativamente al valore che si ha ad una distanza infinita.
1.10.4 Esercizio 1.4
A 300 K e a 20 atm di pressione il fattore di compressibilita di un certo gas risulta di 0.86. Calcolare (a) il vo-lume di 8.2 mmol del gas ai suddetti valori di T e P, (b) il valore approssimato del secondo coefficiente del viriale.
Soluzione
Z si puo esprimere, usando l’equazione di stato del viriale, come
Z =PVm
RT= 1 +
B2V (T )
Vm+
B3V (T )
V 2m
+ ... (1.73)
Quindi, trascurando il termine in B3V (T )
Vm =ZRT
P=
0.86 · 0.082058 dm3 · atm · mol−1 · K−1 · 300 K
20atm= 1.059 dm3 · mol−1
a)
V = nVm = 8.2 · 10−3 mol · 1.059 dm3 · mol−1 = 8.7 · 10−3 dm3
b) Troncando l’espansione del viriale al secondo termine si puo riscrivere la (1.73) come
B2V = Vm
(
PVm
RT− 1
)
= Vm(Z − 1) (1.74)
= 1.059 dm3 · mol−1 · (0.86 − 1) = −0.15 dm3 · mol−1 (1.75)
24

1.10.5 Esercizio 1.5
Un campione di argon di volume molare 17.2 L·mol−1 viene mantenuto a 10.0 atm e 280 K. A che volumemolare, pressione e temperatura si troverebbe in uno stato corrispondente un campione di azoto?Si usino i dati seguenti:
Pc (atm) Vc (cm3·mol−1) Tc (K)Ar 48.0 75.3 150.7N2 33.5 90.1 126.3
Soluzione
Ricordando che Pr = P/Pc, Vr = Vm/Vc e Tr = T/Tc e usando i dati sulle costanti critiche dell’argon calcoliamole sue variabili ridotte
Vr =17.2 dm3 · mol−1
0.0753 dm3 · mol−1= 228.4
Pr =10.0 atm
48.0 atm= 0.2083
Tr =280 K
150.7 K= 1.858
Poi si usano le costanti critiche dell’azoto per trovare i valori di volume, pressione e temperatura che corrispondoalle variabili ridotte dell’argon
Vm = 228.4 · 0.0901 dm3 · mol−1 = 20.6dm3 · mol−1
P = 0.2083 · 33.5 atm = 6.98atm
T = 1.858 · 126.3 K = 235 K
25