
Come a riera ioa unttiii a aronano le roematie irriote ... · storia naturale delle malattie,...
13
25 Gennaio-Marzo 2016 • Vol. 46 • N. 181 • Pp. 25-37 Prospettive in Pediatria Malattie metaboliche ereditarie Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie Giancarlo Parenti Nicola Brunetti-Pierri Dipartimento di Scienze Mediche Traslazionali, Sezione di Pediatria, Università Federico II, Napoli, Telethon Institute of Genetics and Medicine, Pozzuoli (NA) Over the past few decades, major advances have been made in the diagnosis and treat- ment of inherited metabolic diseases. Following such important successes, it is time to draw the first conclusions on the diagnostic strategies and efficacy of these therapies. While for some diseases the results of treatments are indisputable, for others the therapeu- tic efficacy is limited and significant unmet medical needs still exist. Future research will have to address these problems. Thanks to significant progress in biomedical research, it is possible to envisage the strategies that may address the challenges posed by inherited metabolic diseases. This article examines a few examples of these new strategies. The diagnostic approach will likely take advantage of new large-scale genome sequencing technologies. New tools for improving bioavailability and production of therapeutic agents (e.g. recombinant enzymes), or approaches based on gene therapy and genome editing, will likely improve therapeutic efficacy and prognosis. Summary Nel corso degli ultimi decenni sono stati fatti importanti progressi nella diagnosi e terapia delle malattie metaboliche ereditarie. Ora, dopo anni di esperienze, è possibile fare i primi bilanci sugli approcci diagnostici e sull’efficacia delle terapie. Mentre per alcune malattie metaboliche i risultati dei trattamenti sono stati brillanti e indiscutibili, per altre l’efficacia terapeutica è stata parziale e sono emerse problematiche ancora insolute. La ricerca degli anni a venire dovrà affrontare e risolvere queste problematiche. Alla luce dei progressi delle metodologie in campo biomedico, tuttavia, già si cominciano a intravedere le strade da per- correre per affrontare le sfide poste dalle malattie metaboliche. In questo articolo vengono presi in esame alcuni esempi di nuove strategie per migliorare e rendere più efficiente la diagnosi e il trattamento delle malattie metaboliche ereditarie. L’approccio alla diagnosi potrà avvalersi delle nuove tecniche di sequenziamento su larga scala del genoma. Nuo- vi strumenti per migliorare la biodisponibilità e la produzione degli agenti terapeutici (ad esempio enzimi ricombinanti), o approcci basati sulla terapia genica e genome editing, probabilmente contribuiranno a migliore l’efficacia delle terapie e la prognosi dei pazienti. Riassunto Metodologia della ricerca bibliografica Gli autori hanno selezionato dalla letteratura più re- cente i contributi scientifici che a loro giudizio erano più rilevanti sulla diagnosi e terapia delle malattie me- taboliche ereditarie. La ricerca degli articoli è stata ef- fettuata su banca bibliografica (Medline), utilizzando come motore di ricerca PubMed e le seguenti parole chiave per i diversi argomenti: “inborn errors of me-
Transcript of Come a riera ioa unttiii a aronano le roematie irriote ... · storia naturale delle malattie,...

25
Gennaio-Marzo 2016 • Vol. 46 • N. 181 • Pp. 25-37 Prospettive in Pediatria
Malattie metaboliche ereditarie
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
Giancarlo Parenti Nicola Brunetti-Pierri
Dipartimento di Scienze Mediche Traslazionali, Sezione di Pediatria,
Università Federico II, Napoli, Telethon Institute of Genetics
and Medicine, Pozzuoli (NA)
Over the past few decades, major advances have been made in the diagnosis and treat-ment of inherited metabolic diseases. Following such important successes, it is time to draw the first conclusions on the diagnostic strategies and efficacy of these therapies. While for some diseases the results of treatments are indisputable, for others the therapeu-tic efficacy is limited and significant unmet medical needs still exist. Future research will have to address these problems. Thanks to significant progress in biomedical research, it is possible to envisage the strategies that may address the challenges posed by inherited metabolic diseases. This article examines a few examples of these new strategies. The diagnostic approach will likely take advantage of new large-scale genome sequencing technologies. New tools for improving bioavailability and production of therapeutic agents (e.g. recombinant enzymes), or approaches based on gene therapy and genome editing, will likely improve therapeutic efficacy and prognosis.
Summary
Nel corso degli ultimi decenni sono stati fatti importanti progressi nella diagnosi e terapia delle malattie metaboliche ereditarie. Ora, dopo anni di esperienze, è possibile fare i primi bilanci sugli approcci diagnostici e sull’efficacia delle terapie. Mentre per alcune malattie metaboliche i risultati dei trattamenti sono stati brillanti e indiscutibili, per altre l’efficacia terapeutica è stata parziale e sono emerse problematiche ancora insolute. La ricerca degli anni a venire dovrà affrontare e risolvere queste problematiche. Alla luce dei progressi delle metodologie in campo biomedico, tuttavia, già si cominciano a intravedere le strade da per-correre per affrontare le sfide poste dalle malattie metaboliche. In questo articolo vengono presi in esame alcuni esempi di nuove strategie per migliorare e rendere più efficiente la diagnosi e il trattamento delle malattie metaboliche ereditarie. L’approccio alla diagnosi potrà avvalersi delle nuove tecniche di sequenziamento su larga scala del genoma. Nuo-vi strumenti per migliorare la biodisponibilità e la produzione degli agenti terapeutici (ad esempio enzimi ricombinanti), o approcci basati sulla terapia genica e genome editing, probabilmente contribuiranno a migliore l’efficacia delle terapie e la prognosi dei pazienti.
Riassunto
Metodologia della ricerca bibliograficaGli autori hanno selezionato dalla letteratura più re-cente i contributi scientifici che a loro giudizio erano
più rilevanti sulla diagnosi e terapia delle malattie me-taboliche ereditarie. La ricerca degli articoli è stata ef-fettuata su banca bibliografica (Medline), utilizzando come motore di ricerca PubMed e le seguenti parole chiave per i diversi argomenti: “inborn errors of me-

26
G. Parenti, N. Brunetti-Pierri
tabolism”, “lysosomal storage disorders”, “next gene-ration sequencing”, “gene therapy”, “enzyme replace-ment therapy”, ”drug repositioning”, “drug repurposing”, “chaperone therapy”.
IntroduzioneFino a non moltissimi anni fa le malattie metaboliche ereditarie (MME) erano viste come patologie dalla gestione estremamente complessa, sia per le difficol-tà nell’approccio diagnostico, sia perché considerate quasi invariabilmente associate a una elevata morta-lità o, nel migliore dei casi, gravate da una pesante morbidità. Non era raro che medici o pediatri genera-listi considerassero l’iter per arrivare a una definizio-ne diagnostica in un soggetto con sospetta MME uno sforzo poco gratificante o addirittura frustrante, viste le scarse prospettive terapeutiche per questi pazienti. Chi negli ultimi due decenni ha avuto modo di segui-re attivamente pazienti con queste malattie ha invece assistito a una rapida evoluzione nelle conoscenze che ha portato a ribaltare le attitudini dei medici nei confronti della prognosi e della terapia delle MME. Nel corso degli ultimi anni è infatti intervenuta una favo-revole e fortunata combinazione di diversi fattori che hanno progressivamente cambiato il panorama e le prospettive in questo ambito.Innanzitutto, lo sviluppo delle tecnologie, come quelle basate sul DNA ricombinante, ha dato un’accelera-zione decisiva alla ricerca di nuove terapie. Queste tecnologie a loro volta hanno facilitato lo sviluppo di modelli animali per molte, se tutte queste malattie. La disponibilità di modelli animali ha reso possibili studi in vivo, fondamentali per la comprensione dei meccanismi patogenetici e per valutazioni precliniche dell’efficacia di nuove terapie. Lo sviluppo delle cono-scenze sulla fisiopatologia delle MME ha consentito di mettere a punto migliori strategie di intervento tera-peutico, oltre che identificare nuovi target terapeutici. Molti ricercatori si sono avvicinati allo studio di queste malattie, perché rappresentavano un ottimo modello per lo studio di vie metaboliche e della fisiologia cel-lulare. Infine, l’accresciuto interesse da parte dell’in-dustria farmaceutica, promosso in gran parte dalla le-gislazione sui farmaci orfani, ha reso queste malattie, una volta neglette, un target di interesse commerciale. L’introduzione di nuove terapie, spesso con il suppor-to dell’industria farmaceutica, ha a sua volta favorito, innescando un circolo virtuoso, studi clinici multicen-trici (ad esempio registri di malattie, collaborazioni in-ternazionali), con un’ampia condivisione dei dati sulla storia naturale delle malattie, sull’efficacia dei nuovi approcci terapeutici e un’accresciuta esperienza sulle terapie disponibili. Grazie a tutti questi fattori non è sbagliato considerare le MME un eccezionale e, per ora, abbastanza fortunato modello e una “palestra” per lo sviluppo di strategie potenzialmente applicabili anche ad altri campi della medicina.
Dopo diversi anni di esperienze, con il coinvolgimento di gruppi sia clinici che di ricerca di base, è giunto il momento di iniziare a fare i primi bilanci. Da una parte l’ottimismo rispetto alla prognosi dei pazienti è note-volmente aumentato. D’altra parte per alcuni aspetti le MME hanno rappresentato un “bersaglio mobile”. È vero che le nuove terapie hanno risolto alcuni impor-tanti problemi, basti pensare alla sopravvivenza dei pazienti che si è allungata o normalizzata. Tuttavia, proprio per questo motivo e per la complessità delle MME, sono emerse una serie di problematiche nuove (i cosiddetti unmet medical needs), a volte del tutto inattese, che pongono nuove sfide alla ricerca in que-sto gruppo di malattie.Lo scopo di questa review è di analizzare, con le co-noscenze a oggi disponibili, quali potrebbero esse-re le risposte a queste sfide. Non sarà, ovviamente, possibile trattare in maniera esaustiva tutte le proble-matiche aperte e relative ai singoli gruppi di malattie. Pertanto ci limiteremo a considerare alcuni esempi significativi ed esemplificativo in grado di illustrare i potenziali futuri sviluppi nella diagnosi e terapia delle MME.
La diagnosi delle MME: le nuove tecnologie e i challenge futuri Lo screening neonatale ha introdotto importanti cam-biamenti nel campo delle MME e più in generale sta ampliando il concetto di medicina preventiva. Alle me-todiche tradizionali, ormai di interesse più che altro storico, negli ultimi anni si è sostituito lo screening neonatale, basato sulla spettrometria di massa (o screening neonatale esteso) che con un’unica analisi su spot di sangue raccolto alla nascita su cartoncino consente, mediante l’uso di sofisticate apparecchia-ture (la tandem mass spectrometry – TMS), di iden-tificare circa 40 diverse malattie del metabolismo in-termedio. Questo approccio ha portato a un aumento esponenziale delle diagnosi di MME, che però non è stato privo di problemi e insidie legate soprattutto ai falsi positivi e all’incompleta conoscenza della storia naturale di molte MME. In alcune malattie, come la fenilchetonuria, le anoma-lie del profilo dei metaboliti sono molto caratteristiche e non generano dubbi diagnostici. Tuttavia, vi sono alterazioni riscontrate allo screening neonatale che possono essere di più difficile interpretazione e, no-nostante la positività allo screening, alcuni bambini non ricevono una diagnosi di MME per diversi motivi. Ad esempio, poiché i primi giorni dopo la nascita sono un periodo di catabolismo nei difetti di ossidazione degli acidi grassi, il profilo delle acilcarnitine può risul-tare anormale nel campione di sangue raccolto per lo screening, per poi normalizzarsi (o diventare dubbio) in campioni raccolti successivamente. Inoltre, per altri difetti metabolici possono esserci so-vrapposizioni tra profili metabolici che possono cau-

27
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
sare ambiguità o ritardi nella diagnosi. Un esempio particolarmente significativo in questo senso è quello del deficit della deidrogenasi degli acidi grassi a cate-na molto lunga (VLCAD), un disturbo dell’ossidazione degli acidi grassi relativamente frequente tra quel-li identificati dallo screening neonatale allargato. Lo screening identifica sia il fenotipo severo (con cardio-miopatia, miopatia, malattia epatica e morte improv-visa), che quello lieve (asintomatico) e probabilmente benigno. Quei neonati con deficit VLCAD identificati con lo screening che sono rimasti asintomatici du-rante l’infanzia sono probabilmente affetti dalla forma lieve ma è difficile, allo stato attuale delle conoscenze, distinguere un neonato con fenotipo potenzialmente severo da quello che rimarrà asintomatico durante vita (Schiff et al., 2013). I programmi di screening mirano, infatti, a individua-re quanti più possibili bambini affetti (vale a dire, evi-tando di perdere gli affetti, ossia i falsi negativi) allo stesso tempo tollerando un numero accettabile di falsi positivi. L’obiettivo è di individuare non solo i neona-ti gravemente affetti, ma anche quelli che presenta-no fenotipi più lievi e che pertanto hanno anormalità biochimiche che spesso si sovrappongono a quelli di neonati non affetti. Tuttavia le difficoltà diagnostiche possono avere un impatto negativo sui genitori e le famiglie (Hewlett e Waisbren, 2006).Lo screening neonatale esteso, quindi, nonostante rappresenti un successo della medicina preventiva, pone nuovi problemi che derivano dalla possibile am-biguità dei risultati (stesso metabolita alterato in più malattie), impatto psicologico negativo sui genitori e la conseguente necessità di fornire indicazioni preci-se nel più breve tempo possibile. A questi problemi si aggiungono la lunghezza, indaginosità e invasività dell’iter necessario per la conferma della diagnosi. Una possibile soluzione offerta dalle tecnologie e dal-la ricerca più recenti potrebbe venire dalla sempre più diffusa disponibilità di metodiche di analisi molecola-re basate sul cosiddetto next generation sequencing (NGS). Questa modalità di sequenziamento del DNA, che si avvale di apparecchiature e tecnologie minia-turizzate ed estremamente avanzate, consente l’ana-lisi simultanea di un numero elevatissimo di geni (e quindi l’identificazione di mutazioni), fino al sequen-ziamento dell’intero esoma (i.e. intera porzione codi-ficante del genoma) o dell’intero genoma. Una review su questa metodica e sulle sue potenzialità cliniche è stata pubblicata di recente su Prospettive in Pediatria (Nigro, 2015). Frequenti applicazioni della NGS sono quelle basate su panel di geni, come potrebbero es-sere, ad esempio, i geni che sono mutati nelle MME sottoposte a screening neonatale esteso. Già si parla della possibilità di avvalersi del sequenziamento di un panel specifico ai fini della conferma diagnostica dei risultati dello screening, in quelle situazioni in cui la conferma basata su approcci biochimici tradizionali sia particolarmente indaginosa o dubbia.
In aggiunta alle possibili applicazioni relative alla conferma dello screening neonatale, la NGS appare particolarmente promettente come strumento diagno-stico per gruppi di MME. Panel specifici sono già stati sviluppati, ad esempio, per patologie lisosomiali (Di Fruscio et al., 2015), malattie mitocondriali, malattie perossisomiali, difetti congeniti di glicosilazione, e così via. In alcuni di questi casi il vantaggio è legato alla possibilità di passare rapidamente dal sospetto clinico alla conferma diagnostica, evitando procedu-re diagnostiche intermedie invasive (ad esempio, la biopsia muscolare per patologie mitocondriali), costo-se (ad esempio, esami neuroradiologici) o difficilmen-te accessibili (ad esempio, dosaggi enzimatici). È verosimile che nel prossimo futuro sarà anche pos-sibile utilizzare questo approccio per l’analisi non in-vasiva dell’intero genoma fetale nel sangue materno fin dalla quinta settimana di gestazione. Questi meto-di high-throughput probabilmente forniranno un’ulte-riore nuova risorsa per lo screening prenatale e neo-natale non invasivo delle malattie genetiche, tra cui le MME. Va tuttavia segnalato, a tale proposito, che per l’applicazione di tali procedure restano da risolvere di-verse questioni di natura tecnica, politica sanitaria e soprattutto etica (Scala et al., 2012).La NGS potrebbe quindi aumentare in maniera signi-ficativa il numero di malattie identificate, oltre a identi-ficare le varianti genetiche che aumentano il rischio di suscettibilità alle malattie del neonato e per estensio-ne dei suoi familiari. Ancora una volta, nonostante queste enormi poten-zialità, la NGS nello screening neonatale solleva mol-te domande e serie preoccupazioni. Alcune di queste preoccupazioni erano già evidenti agli albori dello screening neonatale tradizionale (Wilcken, 2013). L’eccesso di diagnosi e di trattamento per la fenilche-tonuria è stato, ad esempio, un problema (Brosco e Paul, 2013). Inizialmente si riteneva che ciascun bam-bino con fenilalanina elevata avesse la fenilchetonuria e dovesse essere trattato con terapia dietetica. Nel giro di pochi anni, tuttavia, si è dimostrato che alcuni bambini identificati mediante screening avevano una variante di fenilchetonuria con iperfenilalaninemia che non richiedeva terapia. Con lo sviluppo dello scree-ning neonatale allargato, il problema dell’eccesso di diagnosi e di trattamento è notevolmente aumentato e se prima questo problema riguardava uno o due disturbi, adesso interessa molte condizioni (Wilcken, 2013). Molto probabilmente ciascuna MME ha una variante benigna che si manifesta con anomalie bio-chimiche, ma senza problemi clinici. Queste anoma-lie identificate allo screening metabolico comportano ‘medicalizzazione’ e consequenti alti costi sanitari e psicologici per la famiglia.Oltre alle varianti benigne, vi sono poi disturbi iden-tificati dallo screening allargato che sono probabil-mente benigni come deficit di acil-CoA deidrogenasi degli acidi grassi a corta catena (SCADD), il deficit di

28
G. Parenti, N. Brunetti-Pierri
metionina adenosiltrasferasi (MATI/III), il deficit d iso-butirril-CoA deidrogenasi e della 3-metilcrotonil-CoA carbossilasi (3-MCC). Per alcune di queste anoma-lie biochimiche, i pazienti sono riferiti a un centro di malattie metaboliche per eseguire i test di conferma spesso seguiti da visite mediche e altri test di labo-ratorio nei mesi e talvolta negli anni successivi allo screening neonatale.Queste problematiche nel loro complesso non sono sufficienti per influire sul giudizio complessivo sullo screening metabolico esteso che rimane molto posi-tivo. Ciononostante rappresentano dei segnali di cui tenere conto con attenzione, soprattutto nel conside-rare ampliamenti del numero di malattie sottoposte a screening neonatale, e impongono un’attenta e intelli-gente programmazione per evitare conseguenze dan-nose per molte famiglie. Sebbene lo screening gene-tico possa diventare uno strumento molto potente di medicina preventiva, permettendo la diagnosi pre-sin-tomatica di molte malattie genetiche e possibilmente terapie più efficaci, esso solleva anche molte proble-matiche etiche di non facile risoluzione. Per esempio, è probabile che ogni neonato sottoposto a questo tipo di indagine risulti portatore di diverse malattie o affetto da malattie con esordio nell’età adulta. Come e chi trasmetterà tutte queste informazioni ai genitori per essere incorporate nella cura del bambino? Attual-mente, i minori asintomatici non vengono valutati con test genetici per malattie con esordio nell’età adulta e uno screening genomico neonatale potrebbe dare informazioni non desiderate e che riguardano epoche della vita lontane a venire.
Che cosa può complicare l’interpretazione di dati biochimici?Il dogma per le malattie mendeliane è stato che le mutazioni che le causano sono molto penetranti e quasi mai influenzate dall’ambiente. Tuttavia progres-sivamente è emerso il concetto che le MME hanno un livello di complessità non inferiore a quello delle più comuni malattie multifattoriali. Secondo questo punto di vista, le MME non solo formano uno spettro nell’ambito di una specifica malattia metabolica, ma le MME e le malattie comuni multifattoriali fanno entram-be parte di uno spettro di difetti metabolici. In questa visione le malattie genetiche sono un continuum che va da difetti primari di un singolo gene influenzato da pochi geni modificatori, a difetti più lievi di un gene sotto l’influenza di più geni (Lanpher et al., 2006). La variazione biochimica esiste in ogni individuo indi-pendentemente da una diagnosi di MME ed in un re-cente studio di associazione genetica (GWAS) in una popolazione sana è stata riscontrata un’associazione tra i livelli plasmatici di C8-carnitina, il metabolita usa-to per la diagnosi di deficit deidrogenasi degli acidi grassi a catena media (MCAD), e polimorfismi (SNP) al locus ACADM che codifica per l’enzima responsa-
bile della MCAD (Shin et al., 2014). Lo stesso studio ha evidenziato anche un’associazione tra i livelli ema-tici di fenilalanina e un locus adiacente al gene PAH codificante per fenilalanina idrossilasi responsabile della fenilchetonuria (Shin et al., 2014). Nel comples-so, queste osservazioni evidenziano un continuum di fenotipi biochimici con varianti comuni in geni respon-sabili di MME che possono dar luogo a fenotipi più lievi ad un’estremità dello spettro e varianti estreme più severe responsabili di MME all’altra.
Il monitoraggio dei pazienti con MME: l’importanza di nuovi biomarcatoriUna delle problematiche emergenti nel campo delle MME è quello della disponibilità di biomarker quan-titativi e affidabili. Questi marcatori possono essere definiti come “analiti che indicano la presenza di un processo biologico legato alle manifestazioni cliniche e all’evoluzione di una malattia” (Aerts et al., 2011) e sono sicuramente di importanza fondamentale, non solo ai fini diagnostici, ma ancora di più per il monitoraggio nel tempo dei pazienti e per la valuta-zione dell’efficacia di un trattamento. È evidente che per valutare l’efficacia di nuovi approcci terapeutici è fondamentale da una parte conoscere bene la storia naturale delle malattie (e quindi avere gli strumenti per seguirla) e d’altra parte avere una misura obietti-va e possibilmente quantitativa di quanto una terapia incide sulla storia naturale della malattia.Mentre per alcune patologie (ad esempio, le ami-noacidopatie come la fenilchetonuria, la tirosinemia o la malattia delle urine a sciroppo d’acero) i livelli plasmatici del metabolita in questione rappresenta-no il marcatore ideale per monitorare l’efficacia della terapia, per altre patologie la situazione non è così semplice e immediata.Per alcune malattie lisosomiali, ad esempio, gli indi-ci di efficacia di una terapia sono legati a valutazioni cliniche (il 6-minute-walk test, test di funzionalità re-spiratoria, valutazioni mediante questionari o patient reported outcomes), che hanno il difetto di essere molto dipendenti dall’esaminatore e dalla collabora-zione del paziente, o estremamente soggettivi.Fortunatamente, anche in questo campo, grazie a nuo-ve tecnologie quali quelle basate sulla metabolomica (o probabilmente in futuro la proteomica) ed i microRNA si stanno facendo importanti progressi ed è possibile ipo-tizzare che il futuro riserverà ancora nuove acquisizioni.Per restare nel campo delle malattie lisosomiali, in particolare quelle con la maggiore prevalenza, sono stati identificati marcatori biochimici, quali metaboliti (il GB3 e il liso-GB3 nella malattia di Fabry) (Aerts et al., 2008; Sueoka et al., 2015), enzimi (la chitotriosi-dasi nella malattia di Gaucher) (Hollak et al., 1994), alcune citochine (PARC/CCL18, ancora nella malattia di Gaucher) (Boot et al., 2004).

29
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
Più recentemente, proprio grazie allo sviluppo del-le tecnologie analitiche, sono stati identificati diversi nuovi metaboliti, come la glucosfingosina (Mirzaian et al., 2015) nella malattia Gaucher, alcuni ossiste-roli (prodotti di ossidazione di colesterolo) (Porter et al., 2010; Jiang et al., 2011) e la lisosfingomielina-509 (Giese et al., 2015) per le malattie di Niemann-Pick tipo C. È interessante notare che alcuni di questi mar-ker sembrano correlare con i risultati di interventi te-rapeutici e in questo senso potrebbero essere molto utili.
Terapia delle MMEAnche nel campo della terapia delle MME, come si è già detto, i progressi sono stati entusiasmanti. Lo sviluppo dei primi approcci terapeutici e il tentativo di manipolare le vie metaboliche interessate, risale alla metà del ventesimo secolo, con lo sviluppo della dieta per la fenilchetonuria. Più di sessanta anni fa, sulla scorta delle allora scarse conoscenze sulla biochimi-ca della malattia, gli studi pionieristici di un medico te-desco, Horst Bickel (1918-2000), consentirono di met-tere a punto una dieta a ridotto apporto di fenilalanina (Bickel et al., 1953). Questo approccio consentiva di mantenere i livelli plasmatici dell’aminoacido entro li-miti accettabili e non dannosi nonostante l’esistenza di un blocco enzimatico (deficit di fenilalanina idrossi-lasi, l’enzima che trasforma la fenilalanina in tirosina). A distanza di vari decenni il principio della dietotera-pia per la fenilchetonuria è rimasto invariato ed anzi è stato esteso a diverse altre malattie del metaboli-smo degli aminoacidi, acidemie organiche, difetti del ciclo dell’urea. Ormai l’efficacia di questo approccio è ampiamente documentata e validata (Poustie e Wildgoose, 2010; Camp et al., 2014) ed è possibile affermare senza molti dubbi che la dietoterapia per la fenilchetonuria rappresenti uno dei più formidabili successi della medicina.Nonostante i brillantissimi risultati di questa terapia tuttavia esistono alcune problematiche che ancora devono essere affrontate. Gli alimenti speciali per le diete a ridotto apporto di aminoacidi sono in genera-le poco gradevoli sul piano organolettico, in partico-lare le miscele di aminoacidi. È esperienza comune di chi gestisce questi pazienti che in particolari epo-che della vita (ad esempio, nell’adolescenza) le diete sono mal tollerate e la compliance diminuisce dra-sticamente. Un importante obiettivo dell’industria far-maceutica al momento è proprio quello di sviluppare prodotti alimentari e integratori dietetici che abbiano un minore impatto sulle caratteristiche organolettiche della dieta e sulla qualità di vita dei pazienti. Allo stes-so tempo sono stati messi a punto approcci alterna-tivi o complementari alla dieta (Blau e Longo, 2015), ognuno basato su un razionale diverso, quali la tetrai-drobiopterina (il cofattore della fenilalanina idrossilasi) (Muntau e Gersting, 2010; Longo et al., 2015; Thiele et
al., 2015), nuove miscele di aminoacidi neutri (large neutral amino acids – LNAA) (Matalon et al., 2006; Matalon et al., 2007) e l’uso della fenilalanina ammo-nia liasi (Sarkissian et al., 1999; Longo et al., 2014; Rossi et al., 2014). Alcuni di questi tipi di approcci sono stati discussi in maggior dettaglio nell’articolo di Cerone et al (Cerone et al., 2012).È possibile ipotizzare che nel futuro anche lo sviluppo di alimenti geneticamente modificati o batteri sintetici in grado di degradare aminoacidi nel tubo digerente potrà contribuire a rendere le diete per la fenilcheto-nuria e per altre malattie del metabolismo intermedio più appetibili e meno impegnative per i pazienti. Un altro approccio che potrà portare in tempi relativa-mente rapidi nuove terapie per MME è il drug repo-sitioning (conosciuto anche come drug repurposing). Lo sviluppo di farmaci è un processo che richiede tempi molto lunghi e costi molto alti. Si stima che in media sono necessari 10 anni e almeno 1 miliardo di dollari per portare un farmaco sul mercato (Drug Re-purposing and Repositioning: Workshop Summary, 2014). Considerati questi tempi e costi per lo sviluppo de novo dei farmaci, le aziende farmaceutiche sono sempre più interessate a trovare nuove applicazioni per i farmaci già esistenti sul mercato. Questo proces-so è denominato il drug repositioning. Finora questo processo è stato in gran parte non-intenzionale o for-tuito, spesso scaturito dall’osservazione di effetti non desiderati di un farmaco. Uno degli esempi più noti del drug repositioning è quello del sildenafil, commer-cializzato come Viagra o con altri nomi commerciali. Originariamente sviluppato come un anti-ipertensivo, il sildenafil è stato riproposto per il trattamento della disfunzione erettile e dell’ipertensione arteriosa pol-monare. Essendo la farmacodinamica e farmacodi-stribuzione ben caratterizzata per farmaci già in uso nell’uomo, con il drug repositioning i farmaci possono essere rapidamente sviluppati per una nuova indica-zione clinica saltando tutti gli studi pre-clinici e clinici di tossicità, guadagnando in maniera considerevole tempo e riducendo in maniera drammatica i costi.Nell’ambito delle MME, due studi recenti hanno per-messo il drug repositioning del fenilbutirrato, un far-maco già in uso in pazienti con difetti congeniti del ciclo dell’urea, per la malattia delle urine a sciroppo d’acero e il deficit di piruvato deidrogenasi (Brunetti-Pierri et al., 2011; Ferriero et al., 2013). Per queste malattie il meccanismo di azione del fenilbutirrato è legato all’aumento dell’attività residua della deidro-genasi degli aminoacidi a catena ramificata e della piruvato deidrogenasi responsabili della malattia delle urine a sciroppo d’acero e del deficit di piruvato dei-drogenasi, rispettivamente. Per queste applicazioni il ruolo del fenilbutirrato sui due enzimi è stato ipotizza-to e poi dimostrato partendo dall’osservazione clinica che i pazienti con malattie del ciclo dell’urea in tera-pia con il fenilbutirrato sviluppano una riduzione della concentrazione sierica degli aminoacidi a catena ra-

30
G. Parenti, N. Brunetti-Pierri
mificata, che suggeriva un’attivazione della deidroge-nasi degli amminoacidi a catena ramificata. L’effetto terapeutico del fenilbutirrato in effetti è legato a un au-mento dell’attività enzimatica residua. Per la malattia delle urine a sciroppo d’acero uno studio clinico è in corso (NCT01529060), mentre è in fase di sviluppo per il deficit di piruvato deidrogenasi.
L’esempio delle malattie lisosomiali e della terapia enzimatica sostitutiva. I limiti della terapia (biodisponibilità, costi) e le strategie per migliorarne l’efficacia La ricerca di nuove terapie per la cura delle malattie lisosomiali è stata nel corso degli ultimi venticinque anni l’ambito caratterizzato dal più vivace progresso, con diversi approcci innovativi, alcuni ormai approva-ti, altri tuttora in fase di sviluppo clinico. La terapia enzimatica sostitutiva (enzyme replace-ment therapy – ERT) ha rappresentato probabilmente il più rilevante e importante avanzamento in questo campo. Lo scopo della ERT è quello di correggere il deficit enzimatico responsabile della malattia fornen-do lo specifico enzima, prodotto con tecnologia del DNA ricombinante, mediante infusioni endovenose periodiche. Questo tipo di approccio appare partico-larmente vantaggioso nelle malattie lisosomiali, per-ché gli enzimi lisosomiali sono normalmente equipag-giati con residui di mannosio-6-fosfato. Il mannosio-6-fosfato, infatti, viene riconosciuto da uno specifico recettore (cation-independent mannose-6-phosphate receptor – CI-MPR) che consente all’enzima ricombi-nante di raggiungere le cellule e gli organelli (i liso-somi) dove la sua attività è richiesta per correggere il deficit enzimatico e rimuovere i substrati accumulati. Questo tipo di terapia, dopo un esordio agli inizi degli anni Novanta (quindi non recentissimo), caratterizza-to da notevole successo nella cura della malattia di Gaucher, è stato esteso o è tuttora in corso di studio, per diverse altre malattie lisosomiali.Alla prima fase di sviluppo della ERT sta ora suben-trando una fase di attenta valutazione dei suoi suc-cessi e dei suoi limiti (Parini, questo volume). Per alcune malattie trattabili con ERT, infatti, sono ormai disponibili dati clinici relativi all’outcome di centina-ia (o migliaia, come nel caso della malattia di Gau-cher) di pazienti per periodi di osservazione molto lunghi. Per questi motivi la ERT è oggi probabilmente il miglior esempio di una terapia innovativa su cui è possibile fare un bilancio critico della efficacia e delle problematiche che restano ancora da affrontare e da risolvere.Nella maggior parte delle malattie lisosomiali tratta-bili con ERT la terapia si è dimostrata efficace nel migliorare la performance generale dei pazienti, nel ridurre l’accumulo e le conseguenze anatomopato-
logiche dell’accumulo nei visceri (fegato, milza), nel ridurre l’escrezione urinaria di metaboliti. Per alcuni casi, come la malattia di Pompe infantile classica, caratterizzata da una severa cardiomiopatia, l’ERT è risultata efficace nel ridurre l’ipertrofia cardiaca e nel prolungare la sopravvivenza dei pazienti. Le malattie lisosomiali, tuttavia, sono tipicamente multisistemiche. Gli enzimi ricombinanti devono per-ciò essere in grado di raggiungere livelli terapeutici (correttivi) in tutte le cellule, tessuti e organi coinvolti dalla malattia. In questo l’ERT ha mostrato i maggiori limiti. Lo scarso effetto a livello scheletrico in diver-se di queste malattie, la scarsa risposta delle mani-festazioni oculari e di quelle cardiache nelle muco-polisaccaridosi, la limitata correzione della patologia muscolare scheletrica nella malattia di Pompe, sono tipici esempi di queste problematiche (Parini, questo volume; Wraith 2009).Alla base dell’insufficiente biodisponibilità degli enzi-mi ricombinanti somministrati ci sono diversi fattori. Uno di questi è il fatto che gli enzimi ricombinanti uti-lizzati come agenti terapeutici sono grosse macromo-lecole che non diffondono liberamente attraverso le membrane cellulari e non sono in grado di raggiun-gere concentrazioni terapeutiche in alcuni dei tessuti bersaglio. Questo è particolarmente vero per il siste-ma nervoso centrale (SNC), dove gli enzimi ricom-binanti non raggiungono livelli terapeutici, in quanto non sono in grado di attraversare la barriera emato-encefalica (blood-brain barrier – BBB). Considerato che in due terzi delle malattie lisosomiali è possibile un coinvolgimento del SNC, con neurodegenerazione progressiva, spesso responsabile di gravi deficit neu-rologici, è evidente che la correzione della patologia neurologica rappresenta una delle sfide più importan-ti da affrontare con la ricerca degli anni futuri.Già oggi esistono diversi approcci che sono stati spe-rimentati per migliorare la distribuzione di enzimi liso-somiali ricombinanti al SNC. La strategia più diretta e scontata è quella basata sull’iniezione distrettuale dell’enzima ricombinante. L’obiettivo principale di que-sto approccio è quello di alleviare la compressione del midollo spinale e, forse, di migliorare il decorso neurologico o cognitivo dei pazienti. La somministra-zione, studiata in modelli animali di vari tipi di mu-copolisaccaridosi, è stata fatta per via intratecale, a livello lombare o nella cisterna magna. La sommini-strazione intratecale dell’ERT è stata tradotta in tera-pia umana per le mucopolisaccaridosi di tipo I, II e VI (Giugliani et al., 2014; Dickson et al., 2015; Muenzer et al., 2016). La somministrazione richiede tuttavia procedure invasive e sono allo studio dispositivi spe-cifici per minimizzare l’impatto sui pazienti. Un altro approccio proposto è basato su modificazio-ni chimiche della componente oligosaccaridica degli enzimi, in modo da modificarne l’affinità per il CI-MPR e/o l’emivita plasmatica. Ad esempio, la beta-glucu-ronidasi, l’enzima deficitario nella mucopolisaccarido-
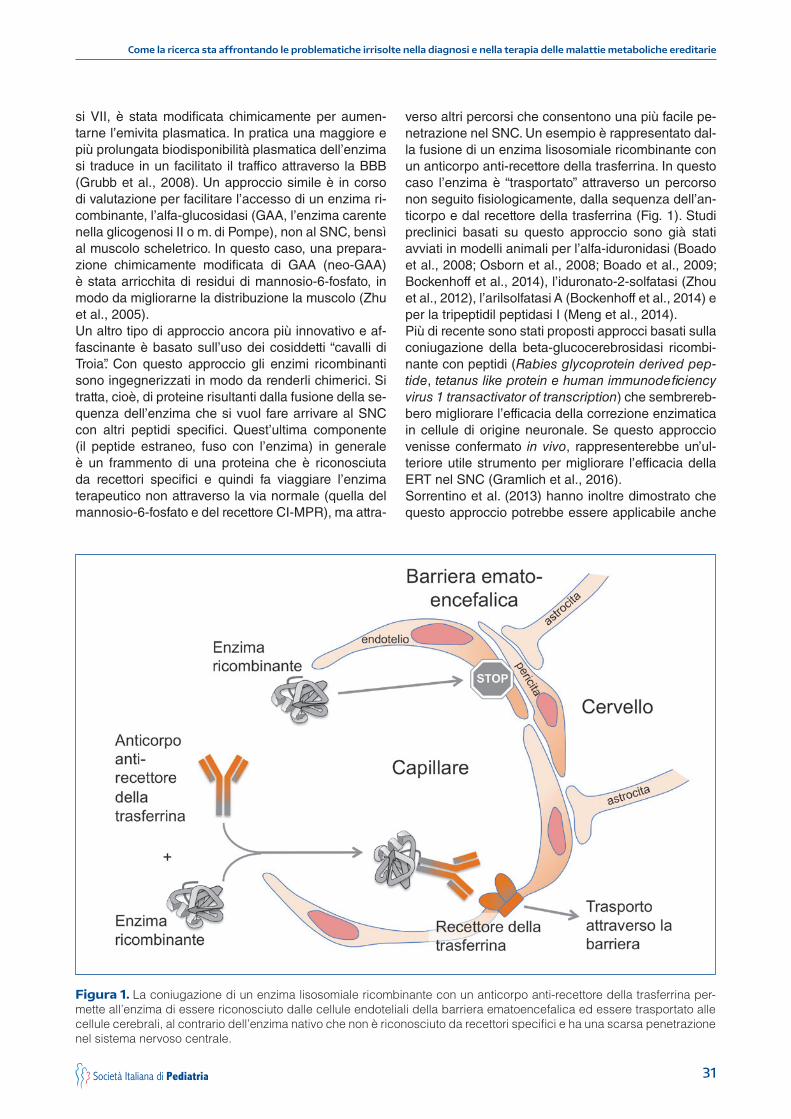
31
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
si VII, è stata modificata chimicamente per aumen-tarne l’emivita plasmatica. In pratica una maggiore e più prolungata biodisponibilità plasmatica dell’enzima si traduce in un facilitato il traffico attraverso la BBB (Grubb et al., 2008). Un approccio simile è in corso di valutazione per facilitare l’accesso di un enzima ri-combinante, l’alfa-glucosidasi (GAA, l’enzima carente nella glicogenosi II o m. di Pompe), non al SNC, bensì al muscolo scheletrico. In questo caso, una prepara-zione chimicamente modificata di GAA (neo-GAA) è stata arricchita di residui di mannosio-6-fosfato, in modo da migliorarne la distribuzione la muscolo (Zhu et al., 2005).Un altro tipo di approccio ancora più innovativo e af-fascinante è basato sull’uso dei cosiddetti “cavalli di Troia”. Con questo approccio gli enzimi ricombinanti sono ingegnerizzati in modo da renderli chimerici. Si tratta, cioè, di proteine risultanti dalla fusione della se-quenza dell’enzima che si vuol fare arrivare al SNC con altri peptidi specifici. Quest’ultima componente (il peptide estraneo, fuso con l’enzima) in generale è un frammento di una proteina che è riconosciuta da recettori specifici e quindi fa viaggiare l’enzima terapeutico non attraverso la via normale (quella del mannosio-6-fosfato e del recettore CI-MPR), ma attra-
verso altri percorsi che consentono una più facile pe-netrazione nel SNC. Un esempio è rappresentato dal-la fusione di un enzima lisosomiale ricombinante con un anticorpo anti-recettore della trasferrina. In questo caso l’enzima è “trasportato” attraverso un percorso non seguito fisiologicamente, dalla sequenza dell’an-ticorpo e dal recettore della trasferrina (Fig. 1). Studi preclinici basati su questo approccio sono già stati avviati in modelli animali per l’alfa-iduronidasi (Boado et al., 2008; Osborn et al., 2008; Boado et al., 2009; Bockenhoff et al., 2014), l’iduronato-2-solfatasi (Zhou et al., 2012), l’arilsolfatasi A (Bockenhoff et al., 2014) e per la tripeptidil peptidasi I (Meng et al., 2014). Più di recente sono stati proposti approcci basati sulla coniugazione della beta-glucocerebrosidasi ricombi-nante con peptidi (Rabies glycoprotein derived pep-tide, tetanus like protein e human immunodeficiency virus 1 transactivator of transcription) che sembrereb-bero migliorare l’efficacia della correzione enzimatica in cellule di origine neuronale. Se questo approccio venisse confermato in vivo, rappresenterebbe un’ul-teriore utile strumento per migliorare l’efficacia della ERT nel SNC (Gramlich et al., 2016).Sorrentino et al. (2013) hanno inoltre dimostrato che questo approccio potrebbe essere applicabile anche
Figura 1. La coniugazione di un enzima lisosomiale ricombinante con un anticorpo anti-recettore della trasferrina per-mette all’enzima di essere riconosciuto dalle cellule endoteliali della barriera ematoencefalica ed essere trasportato alle cellule cerebrali, al contrario dell’enzima nativo che non è riconosciuto da recettori specifici e ha una scarsa penetrazione nel sistema nervoso centrale.

32
G. Parenti, N. Brunetti-Pierri
per migliorare l’efficacia a livello del SNC della terapia genica diretta al fegato. Questo gruppo ha messo a punto un costrutto virale basato su un vettore adeno-associato (AAV) che codifica per una eparan sulfami-dasi (l’enzima carente nella mucopolisaccaridosi IIIA, o m. di Sanfilippo) fusa con il signal peptide di un altro enzima lisosomiale altamente secreto, l’idurona-to-2-solfatasi, e con una sequenza dell’apolipoprotei-na B (ApoB). Dopo iniezione per via intravenosa nel modello murino di mucopolisaccaridosi IIIA, il gene codificante per l’enzima modificato è stato trasferito nelle cellule del fegato (che in questo modo rappre-sentano una fonte costante di enzima), prodotto e secreto nel plasma. Attraverso la circolazione san-guigna l’enzima viene distribuito, grazie alla compo-nente ApoB, a tutti i tessuti dotati di questo recettore, compreso il SNC. Questo si è tradotto in un’efficiente correzione del deficit enzimatico a livello cerebrale ed una migliore clearance del substrato accumulato nel SNC.Oltre alla limitata biodisponibilità degli enzimi lisoso-miali, la ERT si associa ad altre problematiche. Una di queste, molto sentita nei paesi che dispongono di mi-nori risorse economiche (paesi del terzo mondo, pae-si in fase di revisione del bilancio finanziario), è quella degli elevati costi di queste terapie. Il trattamento di un singolo paziente affetto da malattia lisosomiale può costare fino a diverse centinaia di migliaia di euro all’anno. I costi di produzione e soprattutto gli inve-stimenti in ricerca e sperimentazione contribuiscono al costo elevato degli enzimi lisosomiali ricombinanti. Un’attenta analisi di questi aspetti è stata fatta nel Re-gno Unito alcuni anni fa (Wyatt et al., 2012). Come è possibile affrontare questo problema? In realtà nuove tecnologie per la produzione di pro-teine ricombinanti sono già in avanzato stato di spe-rimentazione. In particolare la produzione di proteine umane in piante geneticamente modificate risulta mol-to interessante e promette di abbassare notevolmente i costi degli enzimi ricombinanti. Una beta-glucoce-rebrosidasi ricombinante è stata prodotta in cellule di carota (Shaaltiel et al., 2007) ed è già approvata dal 2012 per l’uso clinico nella malattia di Gaucher. Altri processi di produzione, ad esempio in semi di riso (oryza sativa), sono stati segnalati (Patti et al., 2012).
Altri approcci terapeutici per la cura delle malattie lisosomialiNonostante alcuni buoni risultati conseguiti dalla ERT in alcune malattie lisosomiali, con rare eccezioni le terapie oggi disponibili non hanno risolto interamente le problematiche cliniche associate a queste malattie. Le malattie lisosomiali restano perciò tuttora respon-sabili di unmet medical needs. In realtà anche in que-sto senso la ricerca attuale è estremamente attiva e molto si sta facendo per sviluppare approcci alternati-vi o complementari alla ERT.
Uno di questi approcci, il primo a comparire sulla scena dopo la ERT, è quello basato sulla cosiddet-ta “riduzione del substrato” (substrate reduction the-rapy – SRT). Le manifestazioni anatomopatologiche e cliniche delle malattie lisosomiali, come è noto, sono dovute allo sbilanciamento dell’equilibrio tra sintesi di un substrato (in genere molecole complesse come mucopolisaccaridi, sfingolipidi, oligosaccaridi) e la loro degradazione a opera degli enzimi lisosomiali. Se la ERT ha come scopo quello di rimpiazzare li-velli sufficienti di enzima con la somministrazione di enzimi ricombinanti, la SRT ha invece lo scopo di ri-durre il carico di substrato, inibendone (parzialmente) la sintesi. Questo compito è generalmente realizzato con piccole molecole inibitrici di enzimi coinvolti nella biosintesi dei diversi substrati.Questo approccio è già in uso clinico in alcune ma-lattie. Il miglustat, è stato approvato per il trattamento della malattia di Gaucher tipo 1 (Cox et al., 2000; El-stein et al., 2007) e di Niemann-Pick di tipo C (Pat-terson et al., 2007; Fecarotta et al., 2015; Patterson et al., 2015). Un altro inibitore della sintesi di substrato, l’eliglustat tartrato, è stato introdotto di recente e valu-tato in uno studio clinico di fase II (Lukina et al., 2014; Cox et al., 2015), ancora per il trattamento della ma-lattia di Gaucher. La genisteina flavonoide è stata pro-posta come trattamento per alcune mucopolisacca-ridosi (Piotrowska et al., 2011). Uno studio clinico di fase III con alte dosi di genisteina orale aglicone è tut-tora in corso (www.mahsc.ac.uk/projects/clinical-trial-genistein-novel-treatment-sanfilippo-diseases/). Altri farmaci che inibiscono gli enzimi EXTL2 and EXTL3 (implicati nella biosintesi dei mucopolisaccaridi) sono in corso di studio per possibili applicazioni nella cura delle mucopolisaccaridosi (Canals et al., 2015).Un altro approccio terapeutico promettente è quello basato sulla terapia con chaperones farmacologici (pharmacological chaperone therapy – PCT). La PCT si basa sul concetto che spesso malattie con perdita di funzione enzimatica, come le malattie lisosomiali, sono causate da mutazioni missenso che, piuttosto che rendere gli enzimi inattivi, alterano la loro con-formazione tridimensionale o struttura terziaria e ne causano il misfolding. Questi enzimi alterati nella loro conformazione possono essere riconosciuti dai siste-mi di controllo della qualità del reticolo endoplasmati-co (ER) e degradati, possono essere impropriamente glicosilati, o possono non raggiungere la destinazione corretta nelle cellule (in questo caso i lisosomi) (Ger-main e Fan, 2009; Parenti, 2009; Parenti et al., 2015). Pertanto, in queste malattie la perdita della funzione non è dovuta alla perdita di attività catalitica, ma piut-tosto è il risultato della degradazione o della localiz-zazione aberrante della proteina enzimatica (Fig. 2). Piccole molecole e chaperones farmacologici pos-sono interagire con le proteine mutanti, favorirne la conformazione nativa, migliorarne la loro stabilità e consentirne il traffico corretto ai lisosomi. Come risul-
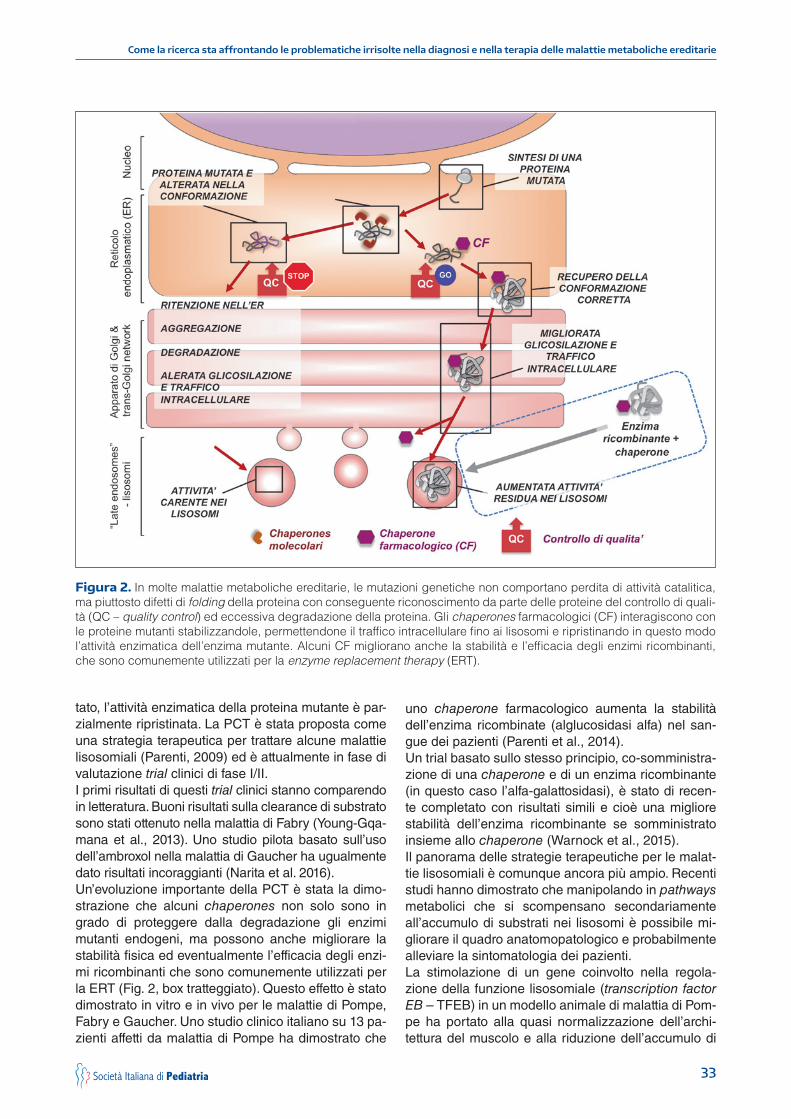
33
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
tato, l’attività enzimatica della proteina mutante è par-zialmente ripristinata. La PCT è stata proposta come una strategia terapeutica per trattare alcune malattie lisosomiali (Parenti, 2009) ed è attualmente in fase di valutazione trial clinici di fase I/II.I primi risultati di questi trial clinici stanno comparendo in letteratura. Buoni risultati sulla clearance di substrato sono stati ottenuto nella malattia di Fabry (Young-Gqa-mana et al., 2013). Uno studio pilota basato sull’uso dell’ambroxol nella malattia di Gaucher ha ugualmente dato risultati incoraggianti (Narita et al. 2016).Un’evoluzione importante della PCT è stata la dimo-strazione che alcuni chaperones non solo sono in grado di proteggere dalla degradazione gli enzimi mutanti endogeni, ma possono anche migliorare la stabilità fisica ed eventualmente l’efficacia degli enzi-mi ricombinanti che sono comunemente utilizzati per la ERT (Fig. 2, box tratteggiato). Questo effetto è stato dimostrato in vitro e in vivo per le malattie di Pompe, Fabry e Gaucher. Uno studio clinico italiano su 13 pa-zienti affetti da malattia di Pompe ha dimostrato che
uno chaperone farmacologico aumenta la stabilità dell’enzima ricombinate (alglucosidasi alfa) nel san-gue dei pazienti (Parenti et al., 2014).Un trial basato sullo stesso principio, co-somministra-zione di una chaperone e di un enzima ricombinante (in questo caso l’alfa-galattosidasi), è stato di recen-te completato con risultati simili e cioè una migliore stabilità dell’enzima ricombinante se somministrato insieme allo chaperone (Warnock et al., 2015).Il panorama delle strategie terapeutiche per le malat-tie lisosomiali è comunque ancora più ampio. Recenti studi hanno dimostrato che manipolando in pathways metabolici che si scompensano secondariamente all’accumulo di substrati nei lisosomi è possibile mi-gliorare il quadro anatomopatologico e probabilmente alleviare la sintomatologia dei pazienti. La stimolazione di un gene coinvolto nella regola-zione della funzione lisosomiale (transcription factor EB – TFEB) in un modello animale di malattia di Pom-pe ha portato alla quasi normalizzazione dell’archi-tettura del muscolo e alla riduzione dell’accumulo di
Figura 2. In molte malattie metaboliche ereditarie, le mutazioni genetiche non comportano perdita di attività catalitica, ma piuttosto difetti di folding della proteina con conseguente riconoscimento da parte delle proteine del controllo di quali-tà (QC – quality control) ed eccessiva degradazione della proteina. Gli chaperones farmacologici (CF) interagiscono con le proteine mutanti stabilizzandole, permettendone il traffico intracellulare fino ai lisosomi e ripristinando in questo modo l’attività enzimatica dell’enzima mutante. Alcuni CF migliorano anche la stabilità e l’efficacia degli enzimi ricombinanti, che sono comunemente utilizzati per la enzyme replacement therapy (ERT).

34
G. Parenti, N. Brunetti-Pierri
glicogeno nei muscoli scheletrici (Spampanato et al., 2013). Risultati simili sono stati ottenuti anche in altre malattie lisosomiali e non-lisosomiali (Medina et al., 2011; Ballabio, 2016). Un gene simile (TFE3) è stato di recente identificato come altro potenziale target tera-peutico nella malattia di Pompe (Martina et al., 2014).La stimolazione farmacologica della produzione di HSP70 (una heat shock protein che aiuta le protei-ne native a raggiungere la corretta conformazione e le protegge dalla degradazione) porta alla stabilizza-zione dei lisosomi in modelli di malattia di Niemann-Pick tipo C e al miglioramento delle anomalie cellulari (Kirkegaard et al., 2010). Un trial clinico con arimoclo-molo (il farmaco che appunto stimola HSP70) è ora in corso.Infine, studi sul ruolo dell’infiammazione nelle ma-lattie lisosomiali, particolarmente sulle manifesta-zioni scheletriche (Simonaro, 2010; Kollmann et al., 2013; Clarke e Hollak, 2015) hanno suggerito ulteriori strategie terapeutiche. Un trial clinico basato sull’u-so di pentosan solfato (un farmaco modulatore dei pathway dell’infiammazione) è attualmente già in cor-so (Schuchman et al., 2013).
La terapia genica: prospettive future La terapia genica ha il potenziale di poter curare in maniera definitiva molte MME agendo sulla causa pri-maria delle malattie, ossia la mutazione genetica. La terapia genica per le malattie genetiche ha avuto un considerevole sviluppo a livello preclinico negli ultimi anni ed in un numero crescente di casi è stata intra-presa la sperimentazione nell’uomo. Gli approcci per il trasferimento genico sono stati molteplici e ciascuno di essi ha specifici vantaggi e svantaggi. Queste ma-lattie sono ottimi candidati, perché per molte di esse
anche piccoli aumenti di attività enzimatica (spesso anche del 10%) possano essere sufficienti per ottene-re un beneficio clinico.Nonostante i problemi di tossicità, come ad esem-pio l’immunogenicità del vettore e/o il prodotto del transgene, la prevalenza di immunità preesistente contro il vettore, la cancerogenicità inserzionale di alcuni vettori e la perdita di espressione a causa del-la proliferazione tissutale per vettori non-integranti, i risultati finora ottenuti lasciano sperare che questi problemi possano essere superati. Nuove tecnolo-gie (zinc finger, TALEN, e CRISPR/Cas9) sono state recentemente sviluppate per ottenere la correzione genomica della mutazione (cosiddetto gene editing) e rappresentano la prossima generazione di farmaci per la terapia genica. Anche se questi approcci sono ancora lontani dall’essere utilizzati in applicazioni cli-niche a causa di problemi rilevanti legati alla sicurez-za (alterazioni genomiche off-target), essi potrebbero superare molti degli ostacoli dei vettori attualmente disponibili per la terapia genica, quali la perdita di espressione del transgene secondaria alla prolifera-zione cellulare e potrebbero consentire l’espressione del gene nel suo fisiologico contesto genomico.In conclusione, la terapia di sostituzione genica ha conseguito importanti successi in clinica e, sulla base del costante progresso fino a oggi, ci aspettiamo che diverse MME saranno indagate nei pazienti nell’im-mediato futuro. Maggiori dettagli sugli approcci di terapia genica sono stati precedentemente discussi in articoli di Prospettive in Pediatria (Brunetti-Pierri, 2008; Mussolino, 2012) e si rimanda il lettore a queste revisioni e altri articoli disponibili in letteratura (Picco-lo e Brunetti-Pierri, 2015; Ginocchio e Brunetti-Pierri, 2016).
Box di orientamento
• Cosa si sapeva primaNel corso degli ultimi decenni sono stati fatti enormi progressi nella diagnosi e terapia delle malattie me-taboliche ereditarie. Per molte malattie sono state sviluppate terapie altamente efficaci.
• Cosa sappiamo adessoSappiamo tuttavia che per molte malattie l’efficacia terapeutica è stata parziale e sono emerse proble-matiche ancora insolute. La ricerca futura dovrà affrontare e confrontarsi con le sfide poste da queste malattie metaboliche e dalle problematiche irrisolte ad esse associate. Nuove strategie diagnostiche (ad esempio, basate su next generation sequencing) e terapeutiche (ad esempio, tecniche per migliorare la biodisponibilità dei farmaci, terapie geniche e genome editing) sono già in corso di sviluppo.
• Quali ricadute sulla pratica clinicaLe nuove metodologie entreranno sempre di più nella pratica clinica e contribuiranno a migliorare e ot-timizzare l’approccio diagnostico, l’efficacia delle terapie e la prognosi dei pazienti. I pediatri, non solo quelli impegnati nella cura di pazienti con malattie metaboliche ereditarie, dovranno acquisire familiarità con queste metodiche.

35
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
Bibliografia
Roundtable on Translating Genomic-Based Research for Health; Board on Health Sciences Policy; Institute of Medi-cine. Drug Repurposing and Repositioning: Workshop Summary. Washington (DC): National Academies Press (US); 2014 Aug.
** Un interessante punto della situazio-ne sulle prospettive e potenziali sviluppi del “riposizionamento” dei farmaci.
Aerts JM, Groener JE, Kuiper S, et al. El-evated globotriaosylsphingosine is a hall-mark of Fabry disease. Proc Natl Acad Sci U S A 2008;105:2812-7.
Aerts JM, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and in-hibodies. J Inherit Metab Dis 2011;34:605-19.
Ballabio A. The awesome lysosome. EMBO Mol Med 2016;8:73-6.
Bickel H, Gerrard J, Hickmans EM. Influ-ence of phenylalanine intake on phenylke-tonuria. Lancet 1953;265:812-3.
Blau N, Longo N. Alternative therapies to address the unmet medical needs of patients with phenylketonuria. Expert Opin Pharmacother 2015;16:791-800.
Boado RJ, Zhang Y, Zhang Y, et al. Ge-netic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol Bioeng 2008;99:475-84.
Boado RJ, Zhang Y, Wang Y, et al. Engi-neering and expression of a chimeric trans-ferrin receptor monoclonal antibody for blood-brain barrier delivery in the mouse. Biotechnol Bioeng 2009;102:1251-8.
* Un esempio di approccio innovativo per il superamento della barriera emato-encefalica.
Bockenhoff A, Cramer S, Wolte P, et al. Comparison of five peptide vectors for improved brain delivery of the lyso-somal enzyme arylsulfatase A. J Neurosci 2014;34:3122-9.
Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surro-gate marker for assessing therapeutic in-tervention. Blood 2004;103:33-9.
Brosco JP, Paul DB. The political history of PKU: reflections on 50 years of newborn screening. Pediatrics 2013;132:987-9.
Brunetti-Pierri N. Terapia genica per le malattie metaboliche ereditarie. Prospetti-ve in Pediatria 2008;152:241-8.
** Una interessante review sulle pro-spettive della terapia genica per le malattie metaboliche.
Brunetti-Pierri N, Lanpher B, Erez A, et al. Phenylbutyrate therapy for ma-ple syrup urine disease. Hum Mol Genet 2011;20:631-40.
Camp KM, Parisi MA, Acosta PB, et al. Phenylketonuria Scientific Review Conference: state of the science and fu-ture research needs. Mol Genet Metab 2014;112:87-122.
* Uno studio sullo stato dell’arte nella terapia della fenilchetonuria e sui futuri sviluppi del management dei pazienti.
Canals I, Beneto N, Cozar M, et al. EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction ther-apy for Sanfilippo C syndrome. Sci Rep 2015;5:13654.
Cerone R, Schiaffino MC, Fantasia AR, et al. Aggiornamento sulle malattie meta-boliche ereditarie. Prospettive in Pediatria 2012;45:195-201.
Clarke LA, Hollak CE. The clinical spec-trum and pathophysiology of skeletal com-plications in lysosomal storage disorders. Best Pract Res Clin Endocrinol Metab 2015;29:219-35.
Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000;355:1481-5.
Cox TM, Drelichman G, Cravo R, et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet 2015;385:2355-62.
Di Fruscio G, Schulz A, De Cegli R, et al. Lysoplex: an efficient toolkit to de-tect DNA sequence variations in the au-tophagy-lysosomal pathway. Autophagy 2015;11:928-38.
Dickson PI, Kaitila I, Harmatz P, et al.; Mucopolysaccharidosis I.I.R.C. Safety of laronidase delivered into the spinal canal for treatment of cervical stenosis in mu-copolysaccharidosis I. Mol Genet Metab 2015;116:69-74.
Elstein D, Dweck A, Attias D, et al. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme re-placement. Blood 2007;110:2296-301.
Fecarotta S, Romano A, Della Casa R, et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet J Rare Dis 2015;10:22.
Ferriero R, Manco G, Lamantea E, et al. Phenylbutyrate therapy for pyruva-te dehydrogenase complex deficiency and lactic acidosis. Sci Transl Med 2013;5:175ra31.
Germain DP, Fan JQ. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: in vitro and preclinical studies. Int J Clin Pharmacol Ther 2009;47(Suppl 1):S111-7.
Giese AK, Mascher H, Grittner U, et al. A novel, highly sensitive and specific bio-marker for Niemann-Pick type C1 disease. Orphanet J Rare Dis 2015;10:78.
Ginocchio VM, Brunetti-Pierri N. Prog-ress toward improved therapies for inborn errors of metabolism. Hum Mol Genet 2016;25:R27-35.
Giugliani R, Herber S, Lapagesse L, et al. Therapy for mucopolysaccharidosis VI: (Maroteaux-Lamy syndrome) present sta-tus and prospects. Pediatr Endocrinol Rev 2014;12(Suppl 1):152-8.
Gramlich PA, Westbroek W, Feldman RA, et al. A peptide-linked recombinant gluco-cerebrosidase for targeted neuronal deliv-ery: design, production, and assessment. J Biotechnol 2016;221:1-12.
Grubb JH, Vogler C, Levy B, et al. Chemi-cally modified beta-glucuronidase crosses blood-brain barrier and clears neuronal stor-age in murine mucopolysaccharidosis VII. Proc Natl Acad Sci U S A 2008;105:2616-21.
Hewlett J, Waisbren SE. A review of the psychosocial effects of false-positive results on parents and current communi-cation practices in newborn screening. J Inherit Metab Dis 2006;29:677-82.
Hollak CE, van Weely S, van Oers MH. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher dis-ease. J Clin Invest 1994;93:1288-92.
Jiang X, Sidhu R, Porter FD, et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J Lipid Res 2011;52:1435-45.
Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lyso-somal pathology. Nature 2010;463:549-53.
Kollmann K, Pestka JM, Kuhn SC, et al. Decreased bone formation and in-creased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol Med 2013;5:1871-86.
Lanpher B, Brunetti-Pierri N, Lee B. In-born errors of metabolism: the flux from Mendelian to complex diseases. Nat Rev Genet 2006;7:449-60.
Longo N, Arnold GL, Pridjian G, et al. Long-term safety and efficacy of saprop-terin: the PKUDOS registry experience. Mol Genet Metab 2015;114:557-63.
Longo N, Harding CO, Burton BK, et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, mul-ticentre, phase 1 dose-escalation trial. Lan-cet 2014;384:37-44.
* La prima sperimentazione clinica di un approccio basato su una terapia enzimatica sostitutiva per la cura della fenilchetonuria

36
G. Parenti, N. Brunetti-Pierri
Lukina E, Watman N, Dragosky M, et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: phase 2 trial results after 4 years of treatment. Blood Cells Mol Dis 2014;53:274-6.
Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal bio-genesis, and clearance of cellular debris. Sci Signal 2014;7:ra9.
Matalon R, Michals-Matalon K, Bhatia G, et al. Large neutral amino acids in the treatment of phenylketonuria (PKU). J In-herit Metab Dis 2006;29:732-8.
Matalon R, Michals-Matalon K, Bhatia G, et al. Double blind placebo control trial of large neutral amino acids in treatment of PKU: effect on blood phenylalanine. J In-herit Metab Dis 2007;30:153-8.
Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 2011;21:421-30.
Meng Y, Sohar I, Sleat DE, et al. Effective intravenous therapy for neurodegenerative disease with a therapeutic enzyme and a peptide that mediates delivery to the brain. Mol Ther 2014;22:547-53.
Mirzaian M, Wisse P, Ferraz MJ, et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol Dis 2015;54:307-14.
Muenzer J, Hendriksz CJ, Fan Z, et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysac-charidosis II. Genet Med 2016;18:73-81.
Muntau AC, Gersting SW. Phenylke-tonuria as a model for protein misfolding diseases and for the development of next generation orphan drugs for patients with inborn errors of metabolism. J Inherit Me-tab Dis 2010;33:649-58.
* Una revisione delle conoscenze sul ruolo del misfolding proteico nella pato-genesi di malattie metaboliche ereditarie e sulle prospettive per lo sviuluppo di nuovi approcci terapeutici basati su tali cono-scenze.
Mussolino C. Nuove frontiere per la tera-pia genica: enzimi artificiali per correggere le mutazioni genetiche. Prospettive in Pe-diatria 2012;42:236-42.
Narita A, Shirai K, Itamura S, et al. Am-broxol chaperone therapy for neurono-pathic Gaucher disease: a pilot study. Ann Clin Transl Neurol 2016;3:200-15.
Nigro V. La next generation sequencing è entrata nella pratica pediatrica? Prospettive in Pediatria 2015;45:137-42.
Osborn MJ, McElmurry RT, Peacock B, et al. Targeting of the CNS in MPS-IH using a nonviral transferrin-alpha-L-idu-
ronidase fusion gene product. Mol Ther 2008;16:1459-66.
Parenti G. Treating lysosomal storage diseases with pharmacological chaper-ones: from concept to clinics. EMBO Mol Med 2009;1:268-79.
Parenti G, Fecarotta S, la Marca G, et al. A chaperone enhances blood alpha-gluco-sidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther 2014;22:2004-12.
* La prima sperimentazione clinica di una terapia basata sulla combinazione di una farmaco”caheprone” e la terapia enzi-matica sostitutiva per la cura di una malat-tia lisosomiale.
Parenti G, Andria G, Valenzano KJ. Phar-macological chaperone therapy: preclinical development, clinical translation, and pros-pects for the treatment of lysosomal stor-age disorders. Mol Ther 2015;23:1138-48.
Patterson MC, Vecchio D, Prady H, et al. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol 2007;6:765-72.
Patterson MC, Mengel E, Vanier MT, et al., investigators N.P.C.R. Stable or im-proved neurological manifestations during miglustat therapy in patients from the inter-national disease registry for Niemann-Pick disease type C: an observational cohort study. Orphanet J Rare Dis 2015;10:65.
Patti T, Bembi B, Cristin P, et al. Endo-sperm-specific expression of human acid beta-glucosidase in a waxy rice. Rice (N Y) 2012;5:34.
Piccolo P, Brunetti-Pierri N. Gene thera-py for inherited diseases of liver metabo-lism. Hum Gene Ther 2015;26:186-92.
Piotrowska E, Jakobkiewicz-Banecka J, Maryniak A. Two-year follow-up of Sanfilippo Disease patients treated with a genistein-rich isoflavone extract: assessment of effects on cognitive functions and general status of pa-tients. Med Sci Monit 2011;17:CR196-202.
Porter FD, Scherrer DE, Lanier MH, et al. Cholesterol oxidation products are sensi-tive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med 2010;2:56ra81.
Poustie VJ, Wildgoose J. Dietary inter-ventions for phenylketonuria. Cochrane Database Syst Rev 2010:D001304.
Rossi L, Pierige F, Carducci C, et al. Erythrocyte-mediated delivery of phenyl-alanine ammonia lyase for the treatment of phenylketonuria in BTBR-Pah(enu2) mice. J Control Release 2014;194:37-44.
* Uno studio che propone un approccio originale ed innovativo per la terapia della fenilchetonuria.
Sarkissian CN, Shao Z, Blain F, et al. A different approach to treatment of phenyl-ketonuria: phenylalanine degradation with
recombinant phenylalanine ammonia lyase. Proc Natl Acad Sci U S A 1999;96:2339-44.
Scala I, Parenti G, Andria G. Universal screening for inherited metabolic diseases in the neonate (and the fetus). J Matern Fe-tal Neonatal Med 2012;25:4-6.
Schiff M, Mohsen AW, Karunanidhi A, et al. Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 2013;109:1-7.
Schuchman EH, Ge Y, Lai A, et al. Pen-tosan polysulfate: a novel therapy for the mucopolysaccharidoses. PLoS One 2013;8:e54459.
Shaaltiel Y, Bartfeld D, Hashmueli S, et al. Production of glucocerebrosidase with termi-nal mannose glycans for enzyme replacement therapy of Gaucher’s disease using a plant cell system. Plant Biotechnol J 2007;5:579-90.
Shin SY, Fauman EB, Petersen AK, et al. An atlas of genetic influences on human blood metabolites. Nat Genet 2014;46:543-50.
** Una estesa analisi di loci e geni che influenzano i livelli di più di 400 metaboliti ematici.
Simonaro CM. Cartilage and chondro-cyte pathology in the mucopolysaccharido-ses: The role of glycosaminoglycan-medi-ated inflammation. J Pediatr Rehabil Med 2010;3:85-8.
Sorrentino NC, D’Orsi L, Sambri I, et al. A highly secreted sulphamidase engi-neered to cross the blood-brain barrier corrects brain lesions of mice with mu-copolysaccharidoses type IIIA. EMBO Mol Med 2013;5:675-90.
* Uno studio su un approccio innovativo per superare la barriera emato-encefalica.
Spampanato C, Feeney E, Li L, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 2013;5:691-706.
Sueoka H, Ichihara J, Tsukimura T, et al. Nano-LC-MS/MS for quantification of lyso-Gb3 and its analogues reveals a use-ful biomarker for Fabry disease. PLoS One 2015;10:e0127048.
Thiele AG, Rohde C, Mutze U, et al. The challenge of long-term tetrahydrobiopterin (BH4) therapy in phenylketonuria: effects on metabolic control, nutritional habits and nutrient supply. Mol Genet Metab Rep 2015;4:62-7.
Warnock DG, Bichet DG, Holida M, et al. Oral migalastat HCl leads to greater systemic exposure and tissue lev-els of active alpha-galactosidase A in Fabry patients when co-administered with infused agalsidase. PLoS One 2015;10:e0134341.
Wilcken B. Medicine. Newborn screen-ing: gaps in the evidence. Science 2013;342:197-8.

37
Come la ricerca sta affrontando le problematiche irrisolte nella diagnosi e nella terapia delle malattie metaboliche ereditarie
** Una interessante revisione critica dell’efficacia degli screening neonatali.
Wraith JE. Enzyme replacement therapy for the management of the mucopolysac-charidoses. Int J Clin Pharmacol Ther 2009;47(Suppl 1):S63-5.
Wyatt K, Henley W, Anderson L, et al. The effectiveness and cost-effective-ness of enzyme and substrate replace-ment therapies: a longitudinal cohort study of people with lysosomal stor-
age disorders. Health Technol Assess 2012;16:1-543.
** Uno studio ampio ed estremamente ac-curato sui risultati e sui costi delle terapie per malattie lisosomiali su tutti i pazienti affetti da queste patologie nel Regno Unito.
Young-Gqamana B, Brignol N, Chang HH, et al. Migalastat HCl reduces globo-triaosylsphingosine (lyso-Gb3) in Fabry transgenic mice and in the plasma of Fab-ry patients. PLoS One 2013;8:e57631.
Zhou QH, Boado RJ, Lu JZ, et al. Brain-penetrating IgG-iduronate 2-sulfatase fu-sion protein for the mouse. Drug Metab Dispos 2012;40:329-35.
Zhu Y, Li X, McVie-Wylie A, et al. Car-bohydrate-remodelled acid alpha-glu-cosidase with higher affinity for the cat-ion-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem J 2005;389:619-28.
Corrispondenza
Giancarlo ParentiDipartimento di Scienze Mediche Traslazionali, Sezione di Pediatria, Università Federico II, via Pansini 5, 80131 Napoli - E-mail: [email protected]


















