Biochimica del selenio e della selenocisteina e loro ruolo ... · PDF fileparticolare fegato e...
78
UNIVERSITA’ DEGLI STUDI DI PISA DIPARTIMENTO DI FARMACIA Corso di Laurea in Farmacia Tesi di laurea BIOCHIMICA DEL SELENIO E DELLA SELENOCISTEINA E LORO RUOLO NELLA FISIOLOGIA E FISIOPATOLOGIA UMANA Candidato: Veronica Colangelo Relatore: Prof.Antonio Lucacchini Correlatore: Prof.Gino Giannaccini Anno Accademico: 2014-2015
Transcript of Biochimica del selenio e della selenocisteina e loro ruolo ... · PDF fileparticolare fegato e...

UNIVERSITA’ DEGLI STUDI DI PISA
DIPARTIMENTO DI FARMACIA
Corso di Laurea in Farmacia
Tesi di laurea
BIOCHIMICA DEL SELENIO E DELLA SELENOCISTEINA E LORO
RUOLO NELLA FISIOLOGIA E FISIOPATOLOGIA UMANA
Candidato:
Veronica Colangelo
Relatore:
Prof.Antonio Lucacchini
Correlatore:
Prof.Gino Giannaccini
Anno Accademico: 2014-2015

Introduzione ...................................................................................................................................................... 2
Capitolo1 ........................................................................................................................................................... 3
Selenio e selenocisteina .................................................................................................................................... 3
Identificazione: cenni storici .............................................................................................................................. 3
Aspetti nutrizionali generali .............................................................................................................................. 4
Vie metaboliche del selenio nella specie umana ............................................................................................... 5
Assorbimento e metabolismo: .......................................................................................................................... 7
Distribuzione sistemica: .................................................................................................................................... 7
Escrezione: ........................................................................................................................................................ 7
Capitolo2 ........................................................................................................................................................... 8
Le selenoproteine ............................................................................................................................................. 8
Cenni generali ................................................................................................................................................... 8
Selenoproteine specifiche: .............................................................................................................................. 11
Glutatione perossidasi .................................................................................................................................... 11
Tioredossina reduttasi .................................................................................................................................... 13
Iodotironina iodasi .......................................................................................................................................... 15
Selenoproteina 15 e selenoproteina M .......................................................................................................... 15
Selenoproteina S e K ....................................................................................................................................... 16
Selenoproteina W ........................................................................................................................................... 17
Selenoproteina H ............................................................................................................................................ 17
Selenoproteina T ............................................................................................................................................. 17
Selenoproteina V ............................................................................................................................................. 18
Selenoproteina P ............................................................................................................................................. 18
Selenofosfato sintetasi .................................................................................................................................... 19
Selenoproteina R ............................................................................................................................................. 20
Selenoproteina N ............................................................................................................................................ 21
Selenoproteina I .............................................................................................................................................. 21
Selenoproteina O ............................................................................................................................................ 21
Capitolo3 ......................................................................................................................................................... 22
Selenio e patologie nell’uomo ........................................................................................................................ 22
Selenodeficienza e selenotossicità ................................................................................................................. 22
Il selenio come medicinale: ............................................................................................................................. 34
Conclusioni ...................................................................................................................................................... 49
Bibliografia………………...………………………………..……………………………………………………………………………………………50
Ringraziamenti……………………………………………………………………………………………………………………………..75

3
Capitolo1
Selenio e selenocisteina
Identificazione: cenni storici
A oggi, il selenio è stato identificato come un semimetallo essenziale, correlato a molte
importanti funzioni biologiche e fisiopatologiche nell’uomo e in altri mammiferi, nei quali
è considerato un elemento fondamentale per molti processi biochimici (Hatfield ET al.,
2012). Tuttavia, esso fu inizialmente considerato una tossina, ritenuta responsabile di una
malattia da cui erano affetti vari capi di bestiame allevati nelle pianure del Nebraska e del
Dakota (USA), intorno alla metà del 1800. Questa malattia fu descritta, infatti, per la prima
volta nel 1856 dal chirurgo dell’esercito C.C. Madison (Madison, A., 1860), che riportò
che, i cavalli, che pascolavano attorno a Fort Randall (Nebraska) manifestavano necrosi
agli zoccoli e diradamento progressivo di coda e criniera; solo nel 1930 tali manifestazioni
furono correlate all’ingestione, da parte di questi animali, di piante selenifere che avevano
causato un eccessivo accumulo di selenio nell’organismo. Il selenio continuò a essere visto
come una tossina, addirittura cancerogena, fino al 1957, anno in cui i medici Schwartz e
Foltz scoprirono che esso preveniva la necrosi epatica nei topi (Schwartz, K. e Foltz, C. M,
1958). Essi finirono che tal elemento era dunque tossico a elevate concentrazioni, ma a
livelli bassi era un micronutriente essenziale nella dieta. Da allora, gli effetti benefici del
selenio furono rapidamente riconosciuti nell’allevamento di bestiame. Nei casi di
selenodeficienza si manifestavano, infatti, disordini di varia natura: patologie riguardanti il
muscolo striato; miopatie generalizzate (ovini e bovini); insufficienza pancreatica e diatesi
essudativa (specie avicole); riduzione della fertilità e patologie epatiche (suini, ovini,
bovini). Si scoprì, dunque, che integrando la dieta del bestiame con selenio si potevano
ridurre milioni di dollari di perdite (Re Illy, C, 1996). A oggi, è attribuita una grandissima
importanza al selenio anche per quanto riguarda la salute dell’uomo, soprattutto per il suo
ruolo nella prevenzione del cancro; espletato attraverso l’incorporazione di tal elemento
nelle proteine. Con meccanismo co-traslazionale, il selenio diviene, infatti, parte
dell’amminoacido selenocisteina, ossia il 21° utilizzato dall’organismo nella sintesi
proteica. Le selenoproteine così formatesi hanno a loro volta un ruolo fondamentale come
antiossidanti, come regolatori delle reazioni di ossido-riduzione e come regolatori del
metabolismo e della spermatogenesi. Fino ad ora ne sono state identificate venticinque, ma

4
solo di alcune sono chiari i processi metabolici che le coinvolgono. Tuttavia, negli ultimi
anni sono stati fatti studi che dimostrano che livelli insufficienti di selenio nell’organismo
sono associabili a condizioni patologiche umane quali :cancro, diabete, disordini
immunologici e sistemici, cardiomiopatie e osteocondropatia cronica (Rayman, C.P;
Combs, G.F, 1996)
Aspetti nutrizionali generali
Il modo principale di fornire un corretto apporto di selenio all’organismo è la dieta, mentre
l’acqua e l’aria non sono considerabili come fonti. La quantità totale di selenio nella dieta
varia in base al tipo di cibo e alla sua composizione, e gli alimenti che contribuiscono
maggiormente all’assunzione della corretta quantità di questo minerale sono pane e cereali,
seguiti da carne, pesce, uova e prodotti caseari (Rayman, M.P., 2008; US department of
agricolture, 2013; Alarcon, M. N, 2008). I livelli di selenio nelle colture dipendono da
quelli presenti nel suolo, e la biodisponibilità è regolata dalle condizioni fisico-chimiche
del terreno come il pH, la salinità e i materiali organici in esso presenti. Le piante
generalmente hanno poco bisogno di selenio per la loro crescita, tuttavia, esse
rappresentano la fonte maggiore di questo metallo, perché largamente consumate da tutta
la popolazione mondiale. In particolare, le piante più ricche di selenio sono di cereali (10-
550 microgrammi) e della famiglia Allium (es. cipolle, aglio), seguite da broccoli, funghi e
noci brasiliane (68/83/96 microgrammi). Per quanto riguarda invece i prodotti di origine
animale, si può introdurre una buona quantità di selenio assumendo pesce, uova, carne e in
particolare fegato e reni (da quarantanove a 500 microgrammi) (Re Illy, C., 2006). I
nutrienti contenenti selenio nelle verdure sono rappresentati soprattutto da
selenometionina, selenati e seleniti, e in misura minore da selenometilselenocisteina e
gammaglutamilseleniometilselenocisteina. Tali nutrienti variano considerevolmente in
base al tipo di terreno di coltivazione, alle piante di cui si cibano gli animali, in base
all’ambiente e anche alla singola specie animale o vegetale considerata. Ogni specie è,
infatti, caratterizzato da specifici modi di assorbire/assimilare il selenio, che può essere
circa efficace, perciò analisi sistematiche condotte in questo senso possono essere la chiave
per ritrarre al meglio il rapporto tra assunzione di selenio e stato di salute, in particolar
modo nei casi in cui ci sia una selenodeficienza e sia necessario integrare questo minerale

5
(Burke, Levander, 2005). Infatti, il selenio è un elemento essenziale che presenta un
confine di dosaggio molto labile tra insufficienza nell’apporto di giusta quantità e dose
tossica. La quantità ottimale che dovrebbe essere assunta è ancora in discussione. Fino a
pochi anni fa, la maggior parte degli studi riguardanti la valutazione della quantità di
selenio da assumere ricercava solo l’ammontare totale di questo semimetallo nei fluidi o
nei tessuti corporei. La sua concentrazione nel plasma, nel siero e nell’urina secreta era
considerata un utile biomarcatore dell’assorbimento e dell’apporto tramite la dieta presto,
mentre per esami a lungo termine si ricercava la concentrazione del selenio negli eritrociti,
nei capelli o nelle unghie. Tuttavia, di recente è stato scoperto (Thompson, 2004) che la
concentrazione di selenio totale non è rappresentativa dell’attività funzionale del selenio,
perché questo elemento è incorporato in una vastissima quantità di proteine con diverse
funzioni biologiche, e la distribuzione del selenio tra le selenoproteine segue una rigida
“gerarchia”, che va dall’assunzione giornaliera media, allo stato di salute del singolo,
includendo attività fisica, età, stile di vita, fumo e polimorfismi genetici delle
selenoproteine. Di conseguenza, la misurazione dell’attività delle singole selenoproteine è
un biomarcatore molto più accurato riguardo allo stato della funzionalità del selenio, e per
questo motivo il parametro usato più frequentemente oggigiorno è l’attività della
selenoproteina plasmatica Glutatione perossidasi (Horst ET al 2005; Burk ET al 2006).
Comunque, in generale si afferma che la RDA (recommended dietary allowance, o dose
giornaliera raccomandata) di selenio è all’incirca di venti microgrammi il giorno, in
conformità a studi epidemiologici che accertano che tale dose (Levander, Wagner, 1996)
sia quella minima per evitare complicanze in senso patologico (es. Morbo di Keshan). Se
consideriamo una prevenzione più in generale di sintomi clinicamente rilevanti derivanti
da selenodeficienza, l’Organizzazione Mondiale della Sanità ha corretto questa cifra a
sedici microgrammi /di e per le donne e diciannove per gli uomini, tenendo in
considerazione anche il peso corporeo. Tali dosi evitano, infatti, con sicurezza che sia
raggiunta una dose tossica di questo elemento, visto il labile confine tra giusto apporto e
tossicità da iperselenemia, es: cambiamenti a livello delle unghie. (Sonde, 2000).
Vie metaboliche del selenio nella specie umana
La via di assorbimento principale del selenio introdotto con la dieta (IP, 1998) consiste
nella riduzione delle differenti specie di nutrienti contenenti selenio in ione idroselenuro

6
(HSe-); questa specie gioca un ruolo fondamentale sia per l’utilizzo sia per l’escrezione del
selenio. La detossificazione in caso di eccesso di selenio avviene attraverso un
meccanismo di metilazione sequenziale che porta alla formazione del dimetil-selenuro
escreto attraverso l’espirazione polmonare, e di selenozuccheri e trimetilselenuro escreti
nelle urine. L’assorbimento delle selenospecie avviene soprattutto nell’intestino tenue con
vari meccanismi e percorsi, molte volte condivisi con i loro analoghi solforici.
Normalmente tutte le forme del selenio, sia organiche sia inorganiche, sono prontamente
assorbite con un’efficienza media che va dal 70 al 90% in normali condizioni fisiologiche.
Le seleniti sono un’eccezione perché il loro assorbimento diretto non supera il 60% di
efficienza, ma alla presenza di glutatione in forma ridotta, come avviene nei fluidi
intestinali, l’assorbimento di seleniti aumenta consistentemente (Gammelgaard ET al.,
2012). In queste condizioni, le seleniti reagiscono in maniera non enzimatica con i gruppi
tionici del GSH per formare il selenodiglutatione, poi scomposto a selenuro dalla
glutatione reduttasi. La frazione di seleniti assorbita per via diretta subisce la stessa
riduzione negli eritrociti. In definitiva tutto la quantità di selenio introdotto è convertito in
selenuro. In alternativa, il selenito può fare da substrato al sistema della tioredoxina, in
modo da essere ridotto direttamente a selenuro con una cascata di reazioni simile a quella
della glutatione reduttasi. Il diglutatione non fa da substrato per la tioredoxina reduttasi,
tuttavia l’inserimento di un atomo di selenio lo rende adatto come substrato di questo
enzima e capace di fare il ciclo di ossidoriduzioni alla presenza di ossigeno (Gabel-Jensen
ET al., 2006). Il selenato è assorbito per via paracellulare con meccanismo di diffusione
passiva a elevata efficienza, è ridotto a selenito dopo l’assorbimento tramite una molecola
non ancora ben identificata, definita come selenio-isologo della 3 fosfoadenosina 5
fosfosolfato (Nickel ET al., 2009). Gli amminoacidi contenenti selenio (selenocisteina e
selenometionina) sono assorbiti con meccanismi trans cellulari mediati da co-carrier che
trasportano anche i loro analoghi solforati; la selenometionina è assorbita tramite un
processo sodio-dipendente, e può essere incorporata in proteine non specifiche come
emoglobina o albumina serica, oppure trasformata in selenocisteina e poi in selenuro con
meccanismo di trans-selenazione, analogo a quello di trans-solforazione (Suzuki ET al.,
2006).

7
Il selenuro è trasformato tramite ATP e selenofosfato-sintetasi in selenofosfato, una specie
donatrice di selenio che ne permette l’utilizzo. Il selenofosfato insieme alla serina fa si che
si crei un fosfoseril-RNA transfert e dona un atomo di selenio alla cisteina RNA in questo
modo legge l’amminoacido selenilcisteina e lo integra nella sequenza amminoacidi che
porta alla formazione di selenoproteine, in maniera ribosoma-mediata.Il catabolismo delle
selenoproteine porta alla liberazione di selenocisteina che è ciclicamente riconvertita in
selenuro (TURANOV, 2011).
Assorbimento e metabolismo:
Distribuzione sistemica:
A livello grossolano,le selenospecie assorbite nel tratto gastrointestinale sono portate
innanzitutto al fegato. La selenilmetionina è trasportata in forma di selenoabumina mentre i
selenati e le altre specie organiche sono trasportati generalmente intatti o con meccanismi
non ancora del tutto chiari. Il fegato è dunque l’organo principale per il metabolismo del
selenio, perché sintetizza la maggior parte delle selenoproteine ed espelle i metaboliti. Le
proteine sintetizzate nel fegato sono poi rilasciate nella circolazione sistemica e distribuite
agli organi in cui sono poi prodotti altri tipi di selenoproteine (Elson ET al 2007/2008).
L’uptake locale di selenio dal plasma avviene mediante endocitosi mediata da recettori per
l’apolipoproteina. In questo modo il fegato regola la distribuzione in tutto l’organismo del
selenio tramite la sintesi di selenoproteine e il processo dei metaboliti che saranno escreti,
in modo che la percentuale di selenio che non può essere usata per la sintesi proteica entra
direttamente nel percorso eliminatorio (Burk, 2009).
Escrezione:
L’escrezione del selenio in eccesso segue due possibili vie che conducono entrambe a
specie metilate. Se il selenio presente rasenta la dose tossica, è prodotto prevalentemente
Trimetilselenio tramite una metiltransferasi, che produce sia, trimetilselenio che
dimetilselenio, entrambi escreti rapidamente, l’uno per via renale e l’altro per via
polmonare. Quando l’eccesso di selenio è molto di sotto la dose tossica, il selenuro è
convertito in un selenozucchero, poi metilato e trasformato in seleniometilen-
acetilgalattosamide, escreto solo nelle urine; la seconda via è leggermente più lenta, perché
non vi è nessuna necesità di espulsione rapida del selenio nel sangue(OHT, Suzuki, 2008).

8
Le selenoproteine
Cenni generali
Le selenoproteine sono una classe di peptidi contenenti uno o più atomi di selenio in stretta
congiunzione tra loro, e furono identificate inizialmente grazie alla loro capacità di essere
marcate con selenio radioattivo Se75. Il selenio è, infatti, l’elemento chiave del sito attivo
di molte selenoproteine aventi funzionalità biologiche essenziali; ne sono state identificate
venticinque nel proteoma umano (Papp ET al., 2007), e 24 in quello di topo e di ratto
(Barnes ET al., 2009).La maggior parte di esse contiene selenio nella sua forma di
amminoacido Selenocisteina. Il quale è codificato dal corrispondente mRNA come codone
UGA, e viene in seguito inserito nelle proteine tramite una SECIS, ossia una sequenza
d’inserzione di selenocisteina.La parte fondamentale della biosintesi delle selenoprotreine
è l’incorporazione della selenocisteina, (21’amminoacido), nel polipeptide in crescita, e di
questo processo multistadio fanno parte due diversi percorsi (figura).Uno: generalmente il
codone UGA funziona da tripletta di stop nella traslazione della proteina. Stranamente
l’inserzione della selenocisteina è condotta da un codone UGA “in frame” seguito da un
elemento d’inserzione per la selenocisteina, ossia la sequenza SECIS. Due: dopo l'affinità
della proteina di legame per il SECIS (SBP2) a questa sequenza, per fornire selenocisteina
è coinvolto il tRNA; infatti, solo in questo caso la sintesi della selenocisteina dai metaboliti
del selenio è effettuata esclusivamente a questo tRNA, che acquista il nome specifico di
Sec-tRNA[ser]sec. La selenocisteina è un amminoacido molto reattivo con un valore di
pKa eccezionalmente basso (5,2) e quindi non si ha un pool libero di selenocisteina (Lu ET
al., 2009; Donovan ET al., 2010; Hatfield ET al., 2006).
Nonostante questi step richiedano un alto quantitativo di energia, il fatto che le
selenoproteine si siano preservate nell’evoluzione della specie è attribuibile al fatto che la
selenocisteina contenuta in questi enzimi ha una nucleofilicità molto elevata. Conferisce
una maggiore resistenza all’inattivazione indotta da specie ossidanti rispetto alla normale
cisteina, cosa che permette ai selenoenzimi di avere un’attività da dieci a cento volte
maggiore rispetto alle non selenoproteine. Si lega a diversi substrati in maniera migliore

9
(Arnèr, 2010; Honda 2011; Snider ET al., 2013; Koishi ET al., 2000; Bar-Noy ET al.,
2001; Schomburg ET al., 2004).
Tra le varie selenoproteine trovate negli esseri umani, la maggior parte dimostra attività
antiossidante. Altri processi più specifici si sono dimostrati essere ricollegabili a queste
proteine, ad esempio la biosintesi del deossiribonucleoside trifosfato (dNTPs) nel DNA, la
riduzione di membrane e proteine ossidate, la regolazione dei processi di ossidoriduzione
nei fattori di trascrizione, la regolazione dell’apoptosi, l’immunomodulazione, la
sistemazione degli ormoni tiroidei, il trasporto e l’immagazzinamento del selenio,
l’assemblaggio delle proteine nel reticolo endoplasmatico e la loro demolizione. Tutte le
selenoproteine tranne la selenoproteina P contengono un solo residuo di selenocisteina che
ha un ruolo fondamentale nel definire la loro attività biochimica; secondo dove è
localizzato tale residuo, si possono, infatti, suddividere le selenoproteine in due gruppi: nel
primo la selenocisteina è collocata nella regione C-terminale e di esso fanno parte le
tioredossina-reduttasi, ossia SelK, SelS, SelR, SelO e SelI; il secondo include tutte le altre
selenoproteine con il residuo di selenocisteina nella regione N-terminale. Di seguito sono
spiegate tutte le caratteristiche principali delle selenoproteine scoperte di recente, con
particolare attenzione alla loro biochimica.
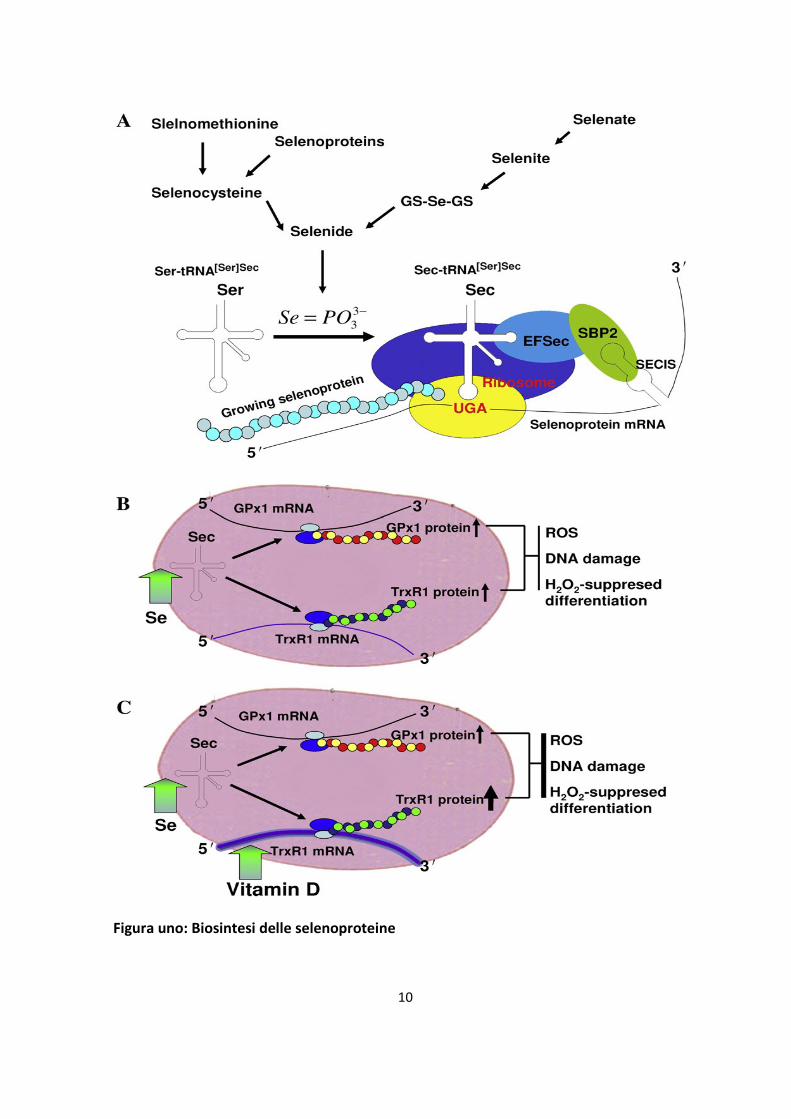
10
Figura uno: Biosintesi delle selenoproteine

11
Selenoproteine specifiche:
Glutatione perossidasi
Le Glutatione perossidasi sono una famiglia di enzimi con attività antiossidante, ne fanno
parte otto isoforme, ma solo cinque di esse contengono un residuo di selenocisteina e
catalizzano la riduzione del perossido d’idrogeno e degli idroperossidi lipidici usando GSH
come cofattore (Gromer ET al., 2005). Questo gruppo comprende il GPx citosolico
ubiquitario, il GPx gastrointestinale, quello plasmatico, il GPx idroperossido fosfolipidico
e il GPx dell’epitelio olfattivo. Il residuo di selenocisteina è ossidato dal perossido con la
formazione di acido selenico, che è poi ridotta una seconda volta a selenolato dai tioli (vedi
FIG. quattro). Il gruppo selenolico del sito attivo del GPx fa parte di una triade catalitica di
residui selenocisteinici, tiamina pirofosfato e glutammina, ed è sia stabilizzato sia attivato
dall’instaurarsi di legami a ponte idrogeno (Roy ET al., 2005).
Ogni GPx è caratterizzato dalla quantità di selenio incorporato, che si pensa sia
rappresentativa della loro importanza a livello biologico: GPx2>GPx4>GPx3=GPx1.
Glutatione perossidasi-1
La Glutatione perossidasi-1 è una proteina omotetramerica ubiquitaria localizzata
nel citosol e nei mitocondri. Questo enzima utilizza esclusivamente GSH come
substrato per la riduzione del perossido d’idrogeno e di una piccola quantità di altri
idroperossidi organici. Le reazioni mediate dal GPx1 dimostrano che tal enzima è
implicato in processi cellulari mediati dagli idroperossidi, tra cui l’apoptosi e i
segnali delle citochine. Nella famiglia delle glutatione-perossidasi, questo enzima è
uno dei più sensibili alla variazione di concentrazione del selenio nell’organismo e
alle condizioni di stress ossidativo, ma poiché la sintesi proteica totale è ridotta in
condizioni di stress in modo da preservare le risorse della cellula, sembra che il
GPx1 si riprenda più rapidamente in termini di funzionalità rispetto ad altre
selenoproteine (Sunde ET al., 2009).
Glutatione perossidasi-2
La Glutatione perossidasi-2 è un enzima omotetramerico secreto, espresso
soprattutto a livello della mucosa gastrointestinale, dell’epitelio squamoso
dell’esofago e, anche se in minor quantità, nel fegato. La sua espressione

12
nell’intestino non è uniforme, ma è maggiore nel fondo delle cripte e diminuisce
gradualmente nell’arrivare alla superficie del lume, cosa che suggerisce un suo
ruolo nella proliferazione cellulare (Florian ET al., 2001).
La funzione principale del GPx2 è di proteggere l’epitelio intestinale dallo stress
ossidativo e di garantire l’omeostasi della mucosa. Questo enzima mostra una
specificità di substrato simile al GPx1 (perossido d’idrogeno,
terzialbutilidroperossido, idroperossido dell’acido linoleico), ma l’espressione del
GPx2 è molto più resistente rispetto al primo quando si ha una situazione di
selenodeficienza globale. Infatti, la sua resistenza e anche la sua posizione
suggeriscono che questa selenoproteina sia una difesa di prima linea dallo stress
ossidativo indotto da ingestione di molecole pro-ossidanti (Brigelius-Flohé, 2009).
Glutatione perossidasi-3
La Glutatione perossidasi -3 è l’unico enzima extracellulare della famiglia. E’ una
proteina omotetramerica glicosilata prodotta nelle cellule dell’epitelio tubulare
prossimale e nelle cellule della capsula di Bowman a livello renale. Parte del GPx3
è poi secreta nel plasma, il quale va a costituire circa il 15/20% del selenio totale,
ma la quantità maggiore di questo enzima resta legata alla membrana basale del
rene. Questa sua capacità di legarsi alle membrane cellulari è stata riscontrata anche
a livello gastrointestinale, polmonare, dell’apparato riproduttivo maschile, e, anche
se in minor misura, nel cuore e nella tiroide. Si pensa abbia un ruolo come
antiossidante localizzato in tali distretti.
Diversamente dal GPx1, il GPx3 ha una specificità di substrato per i perossidi
molto ristretta. Nonostante possa ridurre il perossido d’idrogeno e altri
idroperossidi, la sua attività è fino a dieci volte minore rispetto al GPx1
(Malinouski ET al 2012; Burk ET al., 2011).
La Glutatione perossidasi-4 è un enzima monomerico intracellulare che presenta
tre isoforme: citosolico, mitocondriale e nucleare. L’espressione e l’attività di
questa proteina sono state riscontrate in vari tessuti, in particolare a livello
endocrino e nei mitocondri degli spermatozoi, ed è regolata dagli ormoni.
Diversamente dagli altri GPx, questo può utilizzare direttamente l’idroperossido
fosfolipidico come substrato e riduce il perossido d’idrogeno, gli idroperossidi del
colesterolo, degli esteri e della timina usando elettroni dai tioli delle proteine come

13
dal GSH. Il GPx4 gioca un ruolo fondamentale come antiossidante cellulare
durante la differenziazione nello sviluppo embrionale ed è coinvolto anche nella
condensazione della cromatina durante la spermatogenesi. E’ inoltre una proteina
strutturale degli spermatozoi: l’isoforma nucleare contribuisce alla condensazione
della cromatina post-testicolare mentre quella mitocondriale partecipa
all’organizzazione strutturale del tratto intermedio dello spermatocita. Inoltre, uno
studio recente ha confermato che il GPx4 ha un importante ruolo protettivo dei
fotorecettori dallo stress ossidativo (Imai-Nakagawa, 2003; Conrad ET al., 2007;
Chabory ET al., 2010).
Tioredossina reduttasi
Le Tioredossina-reduttasi sono enzimi omodimerici appartenenti alla famiglia del flavo
proteine, che tra le altre includono lipoamide-idrogenasi, glutatione-reduttasi e ione
mercurico reduttasi. Nei mammiferi sono state identificate tre isoforme: citosolica (TrxR1),
mitocondriale (TrxR2) e tioredossina glutatione reduttasi (TrxR3 o TGR). Come nel caso
delle altre proteine flavonoiche, ogni monomero delle TrxR contiene un gruppo prostetico
di FAD, un sito di legame per NADPH e un sito attivo costituito da un disolfuro che agisce
sulle ossidoriduzioni. Le due subunità partecipano all’attività dell’enzima in maniera
coordinata: gli elettroni sono trasferiti dal NADPH tramite il FAD al sito attivo disolforico
dell’enzima, che poi riduce il substrato come rappresentato in FIG 5. Le TrxR riducono in
modo specifico le tioredossine ossidate, un gruppo di piccoli peptidi ubiquitari che possono
formare ponti disolfuro a livello del DNA, causando alterazioni nella trascrizione dei geni,
e possono avere azione inibitrice dell’apoptosi, causando eccessiva proliferazione
cellulare. Oltre a questi oligopeptidi, sono stati identificati altri substrati endogeni per tal
enzima, tra cui acido lipoico, idroperossido lipidico, il peptide citotossico NK-lisina, la
proteina oncosoppressore p-53, l’acido deidroascorbico e il radicale libero ascorbile. Il
fatto che tal enzima sia in grado di ridurre quest’ultima specie suggerisce che esso possa
avere un ruolo secondario nel riciclo dell’acido ascorbico: gli esseri umani, infatti, non
sono capaci di sintesi.

14
Tissutale: la forma uno è maggiormente espressa nel fegato, nel rene, nella tiroide e nella
ghiandola pituitaria, la forma due nella tiroide, nel sistema nervoso centrale, nel muscolo
scheletrico e nella ghiandola pituitaria. La forma tre presenta un modello di espressione più
specifico ed è presente soprattutto in tessuti embrionali e neonatali, cosa che la fa
considerare come un enzima fetale, perché la sua eliminazione causa uno sviluppo
anormale. E’ assodato che la DIO1 è responsabile del controllo dei livelli circolanti
dell’ormone T3, mentre la DIO2 e tre sono coinvolti in processi locali di regolazione della
de iodurazione, ma, come per le altre famiglie di selenoproteine, non sono ancora
perfettamente chiare le differenze funzionali tra le varie isoforme. Per funzionare, tali
proteine assorbono una gran quantità del selenio, un importante antiossidante che protegge
tutti i tipi di cellule dallo stress ossidativo, quindi il suo riciclo che avviene tramite questo
enzima, e il suo apporto con la dieta sono essenziali per mantenere un giusto livello di
ascorbato nell’organismo. La correlazione tra ciclo dell’acido ascorbico e attività della
Tioredossina reduttasi è stata dimostrata dal fatto che topi sottoposti a dieta priva di selenio
(selenodeficiente) presentavano diminuzione della quantità di ascorbato, TrxR e GPx nel
fegato (Mustacich ET al., 2000). Curiosamente, anche altre specie contenenti selenio
fungono da substrati per questo enzima (seleniti, selenocisteina e altri), cosa che indica che
questi selenoenzimi sono coinvolti anche nella formazione delle altre selenoproteine
perché sono capaci di generare s Oggi, in particolare, si sa che le isoforme uno e due della
tioredossina reduttasi sono essenziali per l’embriogenesi, in particolare l’isoforma due
protegge la cellula embrionale dallo stress ossidativo mitocondrio-mediato e dall’apoptosi.
L’isoforma tre è ancora poca nota, ma si sa che è maggiormente espressa nelle cellule
germinali maschili e si pensa che abbia un ruolo nella maturazione degli spermatozoi
formando ponti disolfuro tra le proteine strutturali (Arner, 2009).Oggi, in particolare, si sa
che le isoforme uno e due della tioredossina reduttasi sono essenziali per l’embriogenesi, in
particolare l’isoforma due protegge la cellula embrionale dallo stress ossidativo
mitocondrio-mediato e dall’apoptosi. L’isoforma tre è ancora poca nota, ma si sa che è
maggiormente espressa nelle cellule germinali maschili e si pensa che abbia un ruolo nella
maturazione degli spermatozoi formando ponti disolfuro tra le proteine strutturali .
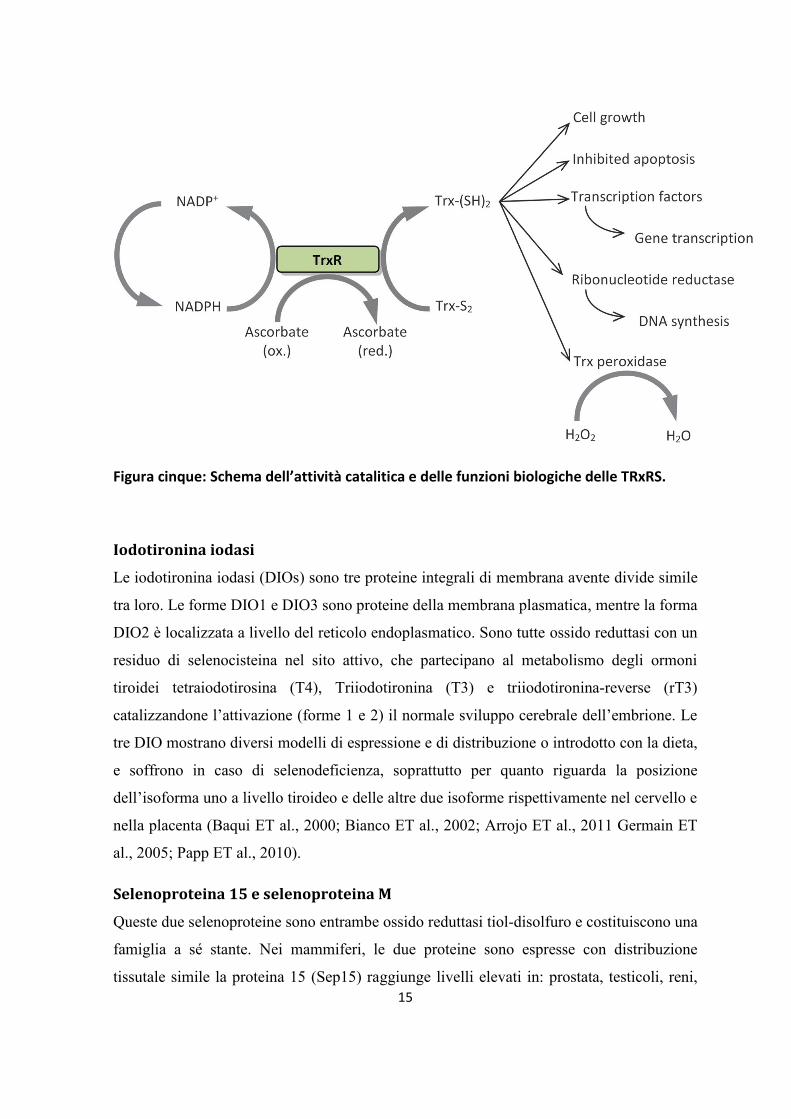
15
Figura cinque: Schema dell’attività catalitica e delle funzioni biologiche delle TRxRS.
Iodotironina iodasi
Le iodotironina iodasi (DIOs) sono tre proteine integrali di membrana avente divide simile
tra loro. Le forme DIO1 e DIO3 sono proteine della membrana plasmatica, mentre la forma
DIO2 è localizzata a livello del reticolo endoplasmatico. Sono tutte ossido reduttasi con un
residuo di selenocisteina nel sito attivo, che partecipano al metabolismo degli ormoni
tiroidei tetraiodotirosina (T4), Triiodotironina (T3) e triiodotironina-reverse (rT3)
catalizzandone l’attivazione (forme 1 e 2) il normale sviluppo cerebrale dell’embrione. Le
tre DIO mostrano diversi modelli di espressione e di distribuzione o introdotto con la dieta,
e soffrono in caso di selenodeficienza, soprattutto per quanto riguarda la posizione
dell’isoforma uno a livello tiroideo e delle altre due isoforme rispettivamente nel cervello e
nella placenta (Baqui ET al., 2000; Bianco ET al., 2002; Arrojo ET al., 2011 Germain ET
al., 2005; Papp ET al., 2010).
Selenoproteina 15 e selenoproteina M
Queste due selenoproteine sono entrambe ossido reduttasi tiol-disolfuro e costituiscono una
famiglia a sé stante. Nei mammiferi, le due proteine sono espresse con distribuzione
tissutale simile la proteina 15 (Sep15) raggiunge livelli elevati in: prostata, testicoli, reni,

16
fegato e cervello, entrano la proteina M (SeM) sono espressi maggiormente nel cervello.
Entrambe sono localizzate nel reticolo endoplasmatico ed entrambe codificano per un N-
peptide che è scisso una volta traslocato nel reticolo endoplasmatico. Inoltre, la Sep15
nativa ha mostrato proprietà migratorie; essa contiene un dominio Cistein-ricco nella
porzione N-terminale della proteina, tramite il quale l'amminoacido forma un complesso
con UDP-glucosio, cioè la glicoproteina Glucosiltranferasi (UGGT). Il complesso formato
dalla proteina Sep15 e UGGT è responsabile di trattenere nel reticolo endoplasmatico la
proteina stessa e potrebbe anche essere implicata nell’assemblaggio e nella secrezione di
glicoproteine. La Sep15 presenta un dominio tiodedossina-simile con una superficie dotata
di motiv ossido riduttivo, in cui la selenocisteina e la cisteina formano un ponte seleno-
solfuro reversibile. Sul suo potenziale ossido riduttivo, è possibile che la Sep15 abbia un
ruolo secondario di catalizzatore la riduzione dei ponti disolfuro, e nella regolazione
dell’apoptosi. La proteina M ha 31% della sequenza amminoacidica in comune con la
Sep15, ma ha un diverso motiv ossido riduttivo quindi il suo ruolo specifico non è ancor
stato stabilito con certezza (KorotKov ET al., 2002; Kumaraswamy ET al., 2000; Zhou ET
al., 2001; Hatfield ET al., 2007; Labunskyy ET al., 2005; Apostolou ET al., 2004;
Novoselov ET al., 2006).
Selenoproteina S e K
Queste due selenoproteine condividono una sequenza N-terminale corta nel lume del
reticolo endoplasmatico, un’elica trans membrana a singolo filamento N-terminale, e un
sito attivo C-terminale contenente selenocisteina. Le selenoproteine K e S hanno simili
caratteristiche strutturali, siti di legame e reazioni tipiche, perché coinvolte nella
regolazione dello stress ossidativo del reticolo endoplasmatico con l’incarico di proteine
trans membrana su di esso localizzate.La K è una proteina ubiquitaria, espressa per la
maggior percentuale nella milza, nel cervello, nel cuore e nelle cellule del sistema
immunitario. Il suo sito catalitico ha un motiv in cui la selenocisteina non è accoppiata con
cisteina, serina o tirosina, ciò significa che un possibile formatore di legame a idrogeno si
avvicini solo in base alla struttura della proteina. Fanno pensare che sia la proteina S sia la
K adatta, le loro funzioni di reduttasi in conformità a diversi substrati. Le loro attività
tuttavia non sono ancora del tutto note (Lu ET al; 2006; Zhang ET al; 2008; Liu ET al;
2012).

17
Selenoproteina W
La selenoproteina W è una piccola proteina con il residuo di selenocisteina localizzato
nella porzione terminale del motiv ossido riduttivo; è espressa principalmente nel cervello,
in cui è immagazzinata in caso di futura selenodeficienza (cosa che fa pensare che abbia
uno specifico ruolo qui localizzato), ma è ubiquitaria. La sua elevata affinità per il GSH, la
sua iperespressione a livello del tessuto muscolare e la sua stessa struttura suggeriscono
che essa abbia un’attività antiossidante. I processi molecolari non sono ancora chiari e la
sua funzione specifica è sconosciuta. Si ipotizza tuttavia che tale proteina possa avere un
ruolo nell’interrompere la mitosi cellulare con un meccanismo regolato da un ossido
riduzione e abbia dunque capacità di prevenire la formazione di cellule cancerosa (Dikiy
ET al., 2007; Jeong ET al., 2002; Fomenko ET al., 2007; Bellingham ET al., 2003;
Amantana ET al., 2004; Kim ET al., 2005).
Selenoproteina H
La selenoproteina H è una proteina nucleare tioredossina-simile con proprietà che le
permettono di legarsi al DNA. E’ espressa moderatamente in vari tessuti marini, mentre
nell’uomo è espressa maggiormente nel cervello durante lo sviluppo embrionale e in vari
tumori, ad esempio quelli al fegato, alla tiroide, al polmone o allo stomaco. Questi dati
suggeriscono dunque un possibile ruolo della SelH nella proliferazione cellulare durante lo
sviluppo o la crescita tumorale. Questa proteina è, infatti, coinvolto anche nell’up-
regulation dei livelli di GSH e della glutatione perossidasi e ha effetti protettivi contro i
superossidi e i raggi ultravioletti di tipo B, probabilmente tramite attivazione di un segnale
di biogenesi mitocondriale (Panee ET al., 2007; Novoselov ET al., 2007; Jilani ET al.,
2007).
Selenoproteina T
La Selenoproteina T è una proteina trans membrana glicosilata e fa parte della famiglia
delle proteine tioredossina-simili. Nel topo è localizzata quasi esclusivamente nell’apparato
di Golgi, nella membrana plasmatica e nel reticolo endoplasmatico mentre nell’uomo è
ubiquitaria ed è soprattutto presente nei testicoli, nella tiroide e nella ghiandola pituitaria.
Si trova in maggior concentrazione durante la fase embrionale che poi va diminuendo con
la crescita; la sua espressione è regolata dal neuro-polipeptide trofico pituitario attivante
l’adenilato-ciclasi PACAP (pituitary adenylate cyclase-activating polypeptide). E'stata

18
riscontrata un’alta percentuale di selenoproteina T nel fegato di topi che avevano subito
parziale epatectomia, e nel cervello di ratti in cui era stata indotta artificialmente una
condizione d’ipossia. Si ritiene che essa abbia un importante ruolo nell’ontogenesi, nella
rigenerazione e maturazione dei tessuti e nel metabolismo cellulare dello SNC e del
sistema endocrino. La selenoproteina T presenta inoltre varie analogie strutturali con la W,
cosa che potrebbe indicare un’interrelazione funzionale tra la due, cosa dimostrata che una
diminuzione dei livelli di SelT in fibroblasti di topo è compensata da un aumento di SelW
(Moustafa ET al., 2012; Grumolato ET al., 2008; Tanguy ET al., 2011; Ikematsu ET al.,
2007; Sengupta ET al., 2009).
Selenoproteina V
La selenoproteina V fa parte della famiglia tioredossina-simile ed è espressa solo negli
spermatociti; ha un dominio N-terminale prolin-ricco e un residuo di selenocisteina
collocata in una porzione idrofoba. Non sono ancora note le sue funzionalità, ma poiché
sembra accoppiarsi con un’acetilglucosamina-transferasi, potrebbe essere un soppressore
del segnale tra le citochine (Varlamova ET al., 2012).
Selenoproteina P
La selenoproteina P è l’unica selenoproteina che contiene dieci residui di selenocisteina sia
nei ratti sia nei topi sia negli umani. E’ una proteina glicosilata che presenta tre siti di N-
glicosilazione occupati e un altro occupato da O-glicosilazione, ha diversi ponti disolfuro e
selenildisolfuro, che avrebbero funzionalità strutturali e di protezione dei gruppi selenolici.
Uno studio recente ha riscontrato la possibilità di identificare e dividere tre isoforme
distinte di SelP nel plasma umano rispettivamente da quarantacinque, quarantanove e
cinquantasette kDa, di cui la prima è un’isoforma tronca che manca di alcune unità di
selenio. La SelP è prodotta principalmente dal fegato e poi rilasciata nel plasma, dove
incorpora la maggior parte del selenio presente nell’organismo, ma è espressa e
probabilmente secreta anche in altri tessuti, come per esempio: cuore e cervello. Ci sono
prove evidenti che dimostrano che questa proteina regola il trasporto e l’omeostasi di tutto
il selenio all’interno dell’organismo. E’ stato riscontrato che in topi privati artificialmente
di tale proteina andavano incontro a grave selenodeficienza. In caso di dieta povera di
selenio, questa proteina mantiene l’elemento a concentrazioni adeguate nel cervello e nei
testicoli piuttosto che nel fegato, dove decresce molto rapidamente. Infine, la

19
localizzazione di SelP in prossimità delle cellule endoteliali dei vasi sanguigni sembra
essere in accordo con un suo eventuale ruolo di antiossidante a livello delle membrane (Ma
ET al., 2003; Ballihaut ET al., 2012; Hill ET al., 2002; Kasaikina ET al., 2012; Schomburg
ET al., 2003; Burk ET al., 2003).
Selenofosfato sintetasi
Il ruolo di questo enzima è di trasferire un gruppo gammafosforile dell’ATP sul selenuro,
diventando quindi donatrice chiave di selenio per la biosintesi delle selenoproteine,
reazione da esso catalizzata. Ne esiste tuttavia un’altra isoforma il cui unico substrato è la
selenocisteina, e il cui ruolo non è ancora chiaro, ma si pensa che sia coinvolta nel riciclo
di quest’amminoacido (Tamurra ET al., 2004; Xu ET al., 2007; Small-Howard ET al.,
2007).

20
Figura uno: Via di sintesi delle selenoproteine.
Selenoproteina R
La selenoproteina R appartiene alla famiglia della metionin-solfossido reduttasi, che sono
responsabili della riconversione dei residui di metionina alla loro forma ossidata metionina
solfossido, che potrebbe influire negativamente su molte funzioni biologiche, formando
insieme ai radicali liberi dell’ossigeno due forme diastereoisomere, alla loro struttura
ridotta (azione cellulo-protettiva).Questa proteina è anche uno zinco-enzima, e si ritiene
che il metallo vada a legarsi a quattro residui cisteinici, avendo così funzionalità strutturale
(Kim ET al., 2007, 2004-2005; Kryukov, 2002; Olry ET al., 2005).

21
Selenoproteina N
La selenoproteina N è una glicoproteina ubiquitaria molto espressa nei tessuti fetali,
soprattutto nel muscolo, nel cervello e nei polmoni. Il sito catalitico ha dei moti di tipo
serina cisteina selenocisteina glicina, quindi è possibile che abbia funzione di reduttasi,
tuttavia l’assenza di domini di legame per NADPH e FAD fa pensare che ci sia una
maggiore specificità di substrato, per molecole non ancora identificate con certezza.
Questa proteina si colloca sulla membrana del reticolo endoplasmatico con la regione N-
terminale rivolta verso il citoplasma e la coda proteica e il sito attivo restano dentro il lume
del reticolo. Ha un ruolo confermato nei processi ossido riduttivi associati all’assorbimento
e al rilascio di calcio in circolo nel tessuto muscolare in formazione nella fase embrionale,
e una sua mutazione causa una miopatia specifica.
Selenoproteina I
La selenoproteina I, detta anche etanolaminafosfotransferasi uno, è stata scoperta di
recente, è presente in maniera ubiquitaria ma è maggiormente concentrata a livello del
cervelletto e si pensa che essa partecipi alla biosintesi della fosfatidiletanolamina, che è
collocata nella parte più vicina al citosol della membrana plasmatica, dove costituisce circa
il 25% del pool di fosfolipidi facenti, parte delle cellule dei mammiferi. Questo fosfolipide
è inoltre un precursore di una proteina di ancoraggio per un neurotrasmettitore cerebrale, la
n-acetiletanolamina, ed è quindi coinvolta nell’assemblaggio delle proteine di membrana e
nella fusione di quelle neuronali durante la sinapsi (Horibata et.al., 2007; Vance ET al.,
2013).
Selenoproteina O
La selenoproteina O è stata identificata perché selenoproteina più grande presente nei
mammiferi, ed è una della più misteriosa. Di recente le è stato attribuito una doppia
funzione di molecola segnale di chinasi e promotrice di ossidoriduzioni, ma non sono
possibili altre caratterizzazioni funzionali (Dudkiewicz ET al., 2012).

22
Capitolo3
Selenio e patologie nell’uomo
E’ stato comprovato negli ultimi due decenni che il selenio può essere collegato circa
direttamente a una gran quantità di problemi legati alla salute umana. La maggior parte di
queste associazioni è legate al ruolo degli enzimi Glutatione per ossidasi (GPx) e
Tioredossina reduttasi (TrxR) le quali svolgono un ruolo nell’ambito della protezione
cellulare dallo stress ossidativo: processo che è stato identificato come principale
responsabile dello sviluppo e della progressione di molte patologie. Altre selenoproteine
sono coinvolte anche in processi più specifici come i segnali trasmessi dagli ioni calcio

23
(quindi legati alla costituzione ossea), funzionalità cerebrale e spermatogenesi.
Alterazioni genetiche o insufficiente espressione di tali enzimi dovuta a selenodeficienza
sono state identificate come possibili cause di patologie corrispondenti. Tuttavia non siamo
ancora giunti alla completa comprensione dei meccanismi che fanno sì che alla mancata
azione delle selenoproteine corrispondano malattie specifiche. Emergono, infatti, dei dati
apparentemente conflittuali dagli innumerevoli studi epidemiologici condotti ed esaminati,
in cui la concentrazione totale di selenio tra dieta e integratori alimentari, sangue e unghie
sono stati posti in relazione con l’avvio della progressione di uno stato patologico. Trovare
la connessione causale tra la cellula, l’individuo e la popolazione sono una vera e propria
sfida. E’ stato riscontrato che la maggior parte delle scleroproteine svolge un’azione
benefica su molte condizioni patologiche umane, mentre un’insufficienza di
concentrazione delle suddette proteine può corrispondere alla manifestazione o al
peggioramento di tali condizioni.
Selenodeficienza e selenotossicità
Una condizione di grave selenodeficienza è direttamente associata a due malattie
endemiche diffuse principalmente nelle zone con terreni poveri di selenio della Cina e
della Russia: il morbo di Kashin-Beck e quello di Keshan. La malattia di Kashin-Beck è
un’osteoartrite caratterizzata da atrofia, degenerazione e necrosi del tessuto cartilagineo,
che si manifesta prevalentemente in bambini tra i cinque e i tredici anni. La patologia ha
come risultato l’allargamento delle articolazioni, l’accorciamento delle dita di mani e piedi
e in casi estremi, anche nanismo (Lì ET al., 2007). La malattia di Keshan è una
cardiomiopatia endemica giovanile che si manifesta in bambini tra i due e i dieci anni,
caratterizzata da ingrossamento cardiaco, anomalie nell’elettrocardiogramma, shock
cardiotonico e insufficienza cardiaca congestizia, con necrosi miocardica multifocale
(Chen ET al., 2012).
Nonostante il selenio sia principalmente noto come antiossidante e quindi come protettore
della funzionalità dell’organismo a causa della sua incorporazione nella selenocisteina e di
conseguenza nelle selenoproteine, può essere anche tossico in caso d’iperselenemia. Gli
effetti tossici del selenio, tutti strettamente concentrazione e specie-dipendenti, possono
avvenire in seguito ad inalazione diretta oppure dopo un’eccessiva assunzione per via orale

24
con la dieta.La tossicità acuta da selenio da inalazione causa mal di stomaco, mal di testa,
un gran numero di sintomi respiratori, come edema polmonare, spasmi bronchiali, asfissia,
bronchite persistente, o ancora, elevata frequenza cardiaca, diminuzione della pressione
arteriosa, vomito, nausea e irritabilità (US Dipartente of Health and Human Service, 2003).
Se l’inalazione è cronica, si può avere irritazione e malfunzionamento dei seni nasali,
spasmi bronchiali, tosse e infiammazione generalizzata di tutte le vie respiratorie.
Un’esposizione acuta a selenio avvenuta per via orale provoca invece nausea, vomito,
diarrea, tachicardia, e se la sovra-assunzione è cronica si può avere un vero e proprio
avvelenamento, che sfocia in una patologia tipica definita “selenosi, ”, la quale è
caratterizzata da perdita di capelli, deformazione e caduta delle unghie e dei denti, alito
agliato, disturbi gastrointestinali, rash cutanei e alterazioni nella funzionalità del sistema
nervoso, quali paralisi, emiplegia occasionale e intorpidimenti (Yang ET al., 1983). Gli
effetti tossici a essa correlati sono dovuti all’alterazione delle funzioni endocrine, della
sintesi degli ormoni (tiroidei e della crescita) e del metabolismo dei fattori di crescita
insulino-simili; in particolare, alti livelli di selenio nella dieta sono associabili con
sicurezza anche a epatotossicità e deplezione delle cellule natural killer del Sistema
Immunitario.
Veniamo adesso alla descrizione della localizzazione e delle specifiche tipologie di danni
causati da eccessiva o insufficiente concentrazione di selenio nell’organismo umano.
a) Disordini Muscolari
Oltre al Morbo di Keshan, descritto in precedenza, anche alcune forme di distrofia
muscolare (miopatia del multiminicore, distrofia muscolare della spina rigida e miopatie
legate ai corpi di Mallory) sono state collegate a problemi riguardanti le selenoproteine, in
particolare a mutazioni del gene codificante per la selenoproteina N, il SEPN1. Tutte
queste patologie condividono, infatti, un quadro clinico simile ed è riferibile al gruppo
delle miopatie concernenti il SEPN1, tuttavia il ruolo della selenoproteina N nelle distrofie
muscolari non è ancora stato delucidato, perché la funzione biologica di questa proteina
non è ancora del tutto chiara (Zorzato ET al., 2007).

25
b) Epatopatie
Le epatopatie sono una classe di malattie che sono state collegate ad alti livelli di stress
ossidativo, in cui gli enzimi ad attività antiossidante potrebbe avere un’azione benefica
(Pemberton ET al., 2005). La malattia del fegato provocata dall’alcolismo è caratterizzata
dall’infiltrazione dei leucociti e dalla formazione di collagene negli epatociti. Questo
processo è causato e guidato dall’aumentata produzione dei radicali liberi, che portano alla
perossidazione dei lipidi delle membrane cellulari. L’azione del citocromo P450 3E1 e
dell’aldeide-ossidasi. Rispettivamente sull’etanolo e sull’acetaldeide, genera, infatti,
superossidi, che producono a loro volta radicali idrossilici, i quali reagiscono con l’etanolo
creando radicali idrossietilici. In questa situazione, è stata proposta la teoria che enzimi
antiossidanti come la glutatione perossidasi, potrebbero giocare un ruolo fondamentale
nell’opporsi all’aumento sproporzionato di radicali liberi, che si formano a causa
dell’elevata assunzione di alcool. Considerando i dati emersi da studi condotti in pazienti
con cirrosi epatica posti a confronto con un gruppo di controllo, molti studiosi
suggeriscono che una somministrazione integrativa di selenio possa migliorare il
funzionamento epatico nel trattamento di questa patologia, e l’effetto benefico è stato
confermato anche in trials successivi (Kolachi ET al., 2012; Stewart ET al., 2007). In
genere i pazienti affetti da cirrosi epatica seguono una dieta povera di selenio perché
l’apporto eccessivo di alcool è accompagnato da una diminuzione nel consumo di cibo di
altra natura, cosa che fornisce un altro supporto al potenziale beneficio che può apportare
un supplemento di selenio nella dieta. In questa patologia il selenio può essere utile anche
come terapia antiossidante. (Gonzalez-Reimers ET al., 2008; Petrovski ET al., 2012).
c) Insufficienza renale
Si è notato che l’attività dei glutatione perossidasi tre e i livelli di selenio negli eritrociti,
nel plasma e nel sangue intero diminuiscono in maniera espressiva nei pazienti affetti da
insufficienza renale cronica o sottoposti a emodialisi rispetto ai gruppi di controllo sani, e
ciò è stato spesso associato anche alla progressione della malattia (Zachara ET al., 2001)
Per spiegare quest’associazione sono state proposte varie teorie, tra cui una diminuzione
dell’apporto di selenio con la dieta, aumento della perdita del minerale con l’escrezione
urinaria o con la dialisi, diminuzione dell’assorbimento a livello intestinale, e anomalie nei

26
legami del selenio alle proteine di trasporto, ma i risultati conclusivi non sono
soddisfacenti. Tuttavia, qualsiasi sia il motivo di base, si pensa che un’integrazione di
selenio in questi pazienti possa essere d’aiuto con la sua azione antiossidante, anche se ciò
non è valido per tutti i casi. Per questo motivo saranno necessari altri studi per rivelare il
ruolo di fattori specifici che condizionano l’assorbimento del selenio nell’organismo.
d) Difesa immunitaria e problemi di natura infiammatoria
Il sistema immunitario consta di vari processi che avvengono in modo tale da proteggere
l’organismo dai patogeni, tra cui la generazione di radicali liberi dell’ossigeno (ROS), la
regolazione coordinata di molecole di adesione e l’espressione di mediatori solubili e dei
loro recettori (es. Citokine, icosanoidi). Il selenio influenza l’immunità umana tramite vari
meccanismi, che sono stati analizzati di recente. Facendo parte del sistema antiossidante, la
glutatione perossidasi e la Tioredossina-reduttasi contribuiscono a controllare la
produzione di radicali liberi, distruggendoli quando essi sono prodotti in concentrazioni
eccessive durante una reazione immunitaria. Le selenoproteine stesse partecipano a un
complesso equilibrio di reazioni di ossidoriduzione che media la trasmissione del segnale
tra le cellule immunitarie (Huang ET al., 2012). La glutatione perossidasi ha la funzione di
messaggero secondario nell’attivazione leucocitaria, poiché media l’azione del perossido
d’idrogeno (Fomenko ET al., 2011). Il quale, secondo le teorie tradizionali, agisce
direttamente come molecola segnale per l’ossidazione dei residui adiacenti di cisteina e per
la formazione di ponti disolfuro in proteine con residui cisteinici, cambiando il loro stato di
attivazione. In questa situazione, la diminuzione della concentrazione del perossido
d’idrogeno causata dalla glutatione perossidasi può interrompere tutti i processi di
passaggio di segnali che avvengono nel Sistema Immunitario e inoltre, la Tioredossina-
reduttasi media, a livello dei linfociti T, la riduzione dei ponti disolfuro. Tramite la
tioredossina, che una volta ridotta libera tioli e stimola il rilascio di molecole segnale
indotte dai recettori delle cellule T, tra cui lo ione calcio Ca++ e i fattori nucleari delle
cellule T attivate, che sono i processi coinvolti nella genesi di sostanze ossidanti e nella
regolazione delle citochine. Perciò, l’equilibrio che s’instaura tra Glutatione perossidasi e
Tioredossina Reduttasi è un fattore chiave nella modulazione della risposta immunitaria:

27
studi condotti utilizzando cellule T prive di glutatione perossidasi hanno evidenziato che,
rispetto a normali cellule T, causavano un’espressione molto maggiore del recettore per
l’interleukina due e una produzione eccessiva d’interferone gamma (che aumenta le
reazioni di ossidoriduzione nell’organismo), in accordo con gli effetti causati da un’azione
incontrollata della tioredossina reduttasi. Una selenodeficienza globale portava all’effetto
opposto, dovuto alla riduzione totale dell’attività e della concentrazione delle
selenoproteine (Hoffman ET al., 2010; Won ET al., 2010). Ciò suggerisce, dunque, che
nelle cellule T la tioredossina reduttasi stimola la propagazione del segnale infiammatorio
dei recettori delle cellule T, mentre la glutatione perossidasi limita l’estendersi della
risposta infiammatoria dopo che tale segnale si è già propagato.
Un’altra selenoproteina coinvolta specificamente nella risposta immunitaria è la
selenoproteina S: la sua espressione a livello degli epatociti è, infatti, regolata dalle
citokine infiammatorie e dalla concentrazione di glucosio extracellulare (Gao ET al.,
2006). La selenoproteina S ha un ruolo antiapoptotico e riduce lo stress ossidativo a livello
del reticolo endoplasmatico nei macrofagi. Un particolare polimorfismo del gene che
codifica per la SelS sembra essere la causa dell’aumento dei livelli di citokine
infiammatorie nel sangue. Ciò potrebbe aumentare il rischio di andare incontro a varie
patologie di tipo infiammatorio (es. morbo di chron, colite ulcerosa), anche se la causalità
diretta non è stata ancora comprovata. (Bellinger ET al., 2009; Kime t al., 2007; Curran ET
al., 2005).
Nel complesso, il selenio partecipa alla risposta immunitaria tramite vari processi: regola
gli equilibri delle vie di sintesi degli icosanoidi, portando allo schema di favore di
leucotrieni e prostacicline piuttosto che quella di trombossano e prostaglandine, e riduce
l’espressione delle citokine e delle molecole di adesione (Hoffman, 2007). Inoltre, causa
l’up-regulation dei recettori per l’interleukina due e aumenta l’attività sia dei linfociti T sia
B e delle cellule natural killer. In uno studio (Shrimali ET al., 2008), sui topi su cui era
stata fatta una delezione specifica a livello del Trna hanno manifestato la perdita totale
delle selenoproteine a livello delle cellule T e ciò ha causato la diminuzione della loro
funzionalità, ridotta produzione d’immunoglobuline antigene-specifiche in vivo e atrofia di
timo, milza e linfonodi. Gli animali hanno inoltre manifestato aumentata suscettibilità a
malattie di origine virale dovuta a un’esagerata risposta immunitaria in senso pro-

28
infiammatorio.La delezione del selenio a livello della glutatione perossidasi ha permesso ai
virus di mutare più velocemente aumentando la loro virulenza (Rivera ET al., 2002; Beck
ET al., 2001; Nelson ET al., 2001).
HIV
Le implicazioni del selenio nel Sistema Immunitario sono incuriosite gli studiosi riguardo
al ruolo che questo semimetallo potrebbe avere nei soggetti affetti da AIDS, e riguardo ai
modi in cui esso potrebbe contrastare l’HIV. Negli stadi precoci e avanzati della malattia,
infatti, è stato riportato un quadro di stress ossidativo cronico, collegabile all’apoptosi delle
cellule T indotta dal virus, e al conseguente sviluppo progressivo dell’AIDS, al sarcoma di
Kaposi e a danni neuronali (Bogden-Oleske, 2007; Tapiero ET al., 2003). Alcuni studi
riguardanti la quantità di selenio nell’organismo e la progressione della malattia ha
osservato che è possibile che ci sia un’associazione diretta tra la bassa concentrazione di
selenio nel plasma/siero, la bassa attività della glutatione perossidasi eritrocitaria (GP1) e
la ridotta conta degli anticorpi CD4+, la progressione rapida del virus e l’aumento della
mortalità (Kupka ET al., 2004). Tuttavia, altri studi non hanno riscontrato livelli
preoccupantemente bassi di selenio in persone affette, e ciò farebbe pensare che gli unici
pazienti che manifestano HIV accompagnato da selenodeficienza sono quelli che seguono
una dieta povera di questo minerale, come chi abusa di droghe in endovena o chi vive in
condizioni di estrema povertà. Perciò, mantenere una concentrazione di selenio ottimale
nell’organismo potrebbe aiutare ad aumentare la difesa enzimatica, migliorando lo stato di
salute generale e riducendo il rischio di ospedalizzazione dei pazienti per infezioni
opportunistiche e per complicazioni (Abuye ET al., 2005; Stephensen ET al., 2007;
Ximena Burbano ET al., 2002).
f) Infertilità maschile
Una lieve seleno-deficienza causa una diminuzione della motilità e dell’alterazione nella
morfologia del tratto intermedio degli spermatozoi, che porta spesso a disconnessione tra
coda e testa, mentre in caso di selenodeficienza severa si ha la completa perdita della
spermatogenesi (Flohe, 2007). L’importante azione strutturale e antiossidante esercitata dai
glutatione perossidasi quattro negli spermatozoi umani rendono questa molecola la
maggiore sospettata di essere la causa di tali effetti. Questa selenoproteina è stata

29
riconosciuta, infatti, come una delle possibili cause di oligoastenozoospermia, una forma
d’infertilità caratterizzata dalla riduzione sia nel numero sia nella motilità degli
spermatozoi (Imai ET al., 2001). Inoltre, una diminuzione dell’espressione dei glutatione
perossidasi quattro negli spermatozoi causa un errore nell’incorporazione della rodamina
123, con perdita del potenziale di membrana mitocondriale che ne modifica la morfologia.
Perché enzima antiossidante, il glutatione perossidasi quattro riduce l’idroperossido
fosfolipidico e il perossido d’idrogeno, che sono entrambi importanti messaggeri secondari
nel processo di spermatogenesi. Infatti, queste due specie, da un lato favoriscono
l’importante processo di condensazione del DNA spermatico, ma dall’altro aumentano lo
stress ossidativo e possono causare effetti drammatici sull’integrità e sulla motilità degli
spermatozoi, perciò è necessaria una modulazione molto delicata di questi due messaggeri,
e la glutatione perossidasi gioca un ruolo chiave proprio nel mantenimento di questi
equilibri. Tuttavia, anche se elevati livelli di stress ossidativo mediati da perossido
d’idrogeno sono associabili a una diminuita fertilità maschile, e a una diminuzione nella
concentrazione o nell’attività della glutatione perossidasi, che però non sembra avere con
essa un rapporto di causalità verificato negli esseri umani (Noblanc ET al., 2011).
g) Disturbi del sistema endocrino
Condizioni patologiche causate da deficienza dell’ormone iodotironina deiodasi negli
umani non sono ancora state documentate, tuttavia molti disordini che coinvolgono il
metabolismo degli ormoni tiroidei sono caratterizzati dalla regolazione anomala di
queste selenoproteine. Molti di questi disordini hanno origine genetica. Un recente
studio ha identificato una mutazione omozigote missenso del gene SBP2 come
responsabile di alcune anomalie nella funzionalità della tiroide dovute alla diminuita
attività degli iodotironina deiodasi due e alla mancata espressione delle isoforme uno e
tre dello stesso enzima, un difetto che non può essere corretto dall’integrazione di
selenio attraverso la dieta (Dumitrescu ET al., 2005; Schomburg ET al., 2009).
In altri disordini endocrini, alterazioni nei livelli della iodotironina deiodasi possono
essere correlati all’assunzione di selenio attraverso la dieta: la combinazione di un
insufficiente apporto di selenio e iodio sembra essere la causa del cretinismo
mixedematoso endemico (Duntas, 2010).

30
Alcuni studi hanno collegato una moderata selenodeficienza a tiroidite di origine
autoimmune, dimostrando che fornendo un supplemento di selenometionina o di seleniti
causa la riduzione degli anticorpi anti perossidasi tiroidea nella maggior parte dei casi,
con effetti positivi sul decorso della malattia.
L’ipertiroidismo di Graves è un altro esempio di patologia tiroidea di origine
autoimmune, ed è causata dalla produzione di autoanticorpi anti tsh (tireotropina), un
recettore che stimola l’attività della iodotironina deiodasi uno per la produzione di
ormoni T3 e T4. In caso di moderata selenodeficienza, sembra che gli integratori
alimentari di selenio favoriscano la normalizzazione del metabolismo degli ormoni
tiroidei, quindi una dieta povera di selenio potrebbe essere un fattore di rischio per varie
forme di tiroidite di origine autoimmune, in particolare in quei soggetti geneticamente
predisposti (Vrca ET al., 2004).
h) Diabete
L’associazione tra selenio e diabete di tipo due coinvolge vari meccanismi (figura). Nello
specifico, il diabete di tipo due è caratterizzato da anomalie nella secrezione e nell’azione
dell’insulina, causate dall’incapacità delle cellule dell’organismo di rispondere alla
presenza d’insulina (fenomeno definito come “insulino-resistenza”). Normalmente, il
legame dell’insulina al proprio recettore dà inizio a una serie di segnali a cascata, che
inducono una serie di ossidoriduzioni abbastanza blande, in cui il perossido d’idrogeno
agisce come messaggero secondario. Questa molecola, infatti, ossida i residui di cisteina,
causando la disattivazione della tirosina fosfatasi 1B (che inattiva poi il substrato dei
recettori per l’insulina), e della proteina omologa della fosfatasi e della tensina (che in un
secondo momento inibisce la fosfatidilinositolokinasi3), causando la stimolazione globale
delle vie di segnalazione per l’uptake del glucosio. La glutatione perossidasi riduce il
perossido d’idrogeno, quindi si pensa che eserciti un’azione di tipo inibitorio sui segnali a
cascata, e tale ipotesi è stata confermata da prove sperimentali ottenute in uno studio in cui
dei topi transgenici con iperespressione della glutatione perossidasi manifestavano
insulino-resistenza, mentre altri, in cui era stato eliminato completamente questo enzima,
erano più sensibili alle variazioni di concentrazione dell’insulina (Wang ET al., 2008; Loh
ET al., 2009). La conferma che ciò avverrebbe anche negli esseri umani è arrivata da uno

31
studio che ha dimostrato che l’aumento dell’attività della glutatione perossidasi
eritrocitaria è associato a una lieve insulino-resistenza nelle donne incinte, mentre una
deficienza di selenoproteine sistemica causata da mutazione del gene SBP2 aumenta la
sensibilità all’insulina (Hawkes, 2004; Schoenmakers ET al., 2010).
Tuttavia, anche altre selenoproteine partecipano al metabolismo del glucosio, rendendo gli
effetti globali del selenio più complessi. Ad esempio, si pensa che la selenoproteina P
inibisca i segnali dell’insulina inattivando la protein-kinasi attivata da adenosina
monofosfato (AMPK), un regolatore in senso positivo della sintesi insulinica a livello delle
cellule beta pancreatiche (Misu et al., 2010). Studi in vitro hanno dimostrato anche che
l’espressione della selenoproteina P negli adipociti sottocutanei umani è aumentata
dall’insulina (Olsson ET al., 2011).
La tioredossina reduttasi invece potrebbe influenzare positivamente la propagazione dei
segnali insulinici, fornendo specie riducenti che riduce l’ossido nitrico endoteliale prodotto
soprattutto in corrispondenza di strutture muscolari, molecola che normalmente causa una
certa insulino resistenza in questi distretti (Perreault, 2001; Carvalho-Filho, 2005;
Sengupta, 2012).
Studi osservazionali riguardanti la somministrazione di dosi supplementari di selenio,
hanno mostrato anche che questo elemento potrebbe avere proprietà insulino-mimetiche,
poiché effettivamente stimola l’uptake del glucosio sia in vitro sia in vivo, e regola la
glicolisi, la gluconeogenesi, la sintesi degli acidi grassi e la via dei pentosi (Erbayraktar ET
al., 2007). In particolare, si pensa che il selenato influenzi due importanti meccanismi
coinvolti nel fenomeno dell’insulino-resistenza: innanzitutto riduce l’attività delle tirosina-
fosfatasi citosoliche a livello epatico perché regolatori in senso inibitorio delle vie di
segnalazione dell’insulina, e in secondo luogo aumenta l’espressione del PPARgamma,
cioè il recettore gamma attivato dai perossisomi. Questi due meccanismi sono responsabili
dei cambiamenti del metabolismo intermedio, in particolare della gluconeogenesi e del
metabolismo lipidico (Mueller-Pallauf, 2006).
Sono stati fatti molti studi osservazionali di tipo caso-controllo per denotare un’effettiva
correlazione di tipo causa-effetto tra la concentrazione di selenio nell’organismo e
l’incidenza di diabete mellito due ma molti risultati sembrano contraddittori: alcuni studi

32
hanno dimostrato che le concentrazioni medie di selenio plasmatiche, sieriche o nel sangue
intero e l’attività della glutatione perossidasi erano minori nei pazienti affetti rispetto ai
controlli sani (Roman ET al., 2010; Whiting ET al., 2008; Kljai-runje, 2001; Kornhauser
ET al., 2008). Quantità inferiori di selenio nel plasma e nel siero sono state riscontrate
anche in donne in gravidanza affette da diabete gestazionale rispetto a normali gravidanze
(Molnar ET al., 2008; Kilinc ET al., 2008). Altri studi hanno trovato livelli di selenio più
elevati in pazienti affetti da diabete, o non hanno riscontrato differenze espressive (Serdar
ET al., 2009; Bleys ET al., 2007). Infine, in alcuni casi la differenza nella concentrazione
sistemica di selenio era limitata ad alcuni sottogruppi di pazienti, ad esempio maschi o
malati da meno di due anni (Akbaraly ET al., 2007). I risultati più recenti da studi
randomizzati conferma questi dati divergenti, mostrando che un aumento di selenio nella
dieta potrebbe sia aumentare sia non il rischio di diabete di tipo due. Nel complesso,
l’associazione causa-effetto diabete2-selenio sembra seguire graficamente una forma a U,
ciò potrebbe significare che gli effetti variano in base al livello base di assunzione di
selenio della popolazione presa in esame (Stranges ET al., 2007; Lippman ET al., 2009).
i)Fisiologia e fisiopatologia dell’osso
Un inadeguato apporto di selenio che compromette la biosintesi delle selenoproteine è
ritenuto da molto tempo dannoso per la salute, in particolare per quella delle ossa. D’altro
canto, se il selenio è presente in concentrazione eccessiva, anche superiore a quella
richiesta per formare le selenoproteine, esso può provocare tramite vari meccanismi alcune
alterazioni patologiche, inibendo reazioni di tipo infiammatorio, attivando la riparazione
Figura otto: Schema del potenziale ruolo della selenoproteina nella regolazione del segnale a cascata dell’insulina.

33
del DNA, inducendo gli enzimi chemio protettivi e promuovendo l’apoptosi con blocco
della proliferazione cellulare. I potenziali effetti farmacologici del selenio introdotto a
livelli sovra nutrizionali sono mediati principalmente da metaboliti del selenio a basso peso
molecolare come il selenodiglutatione e il metilselenolo, che hanno una natura molto
reattiva perché il selenio di questi composti ha elevato, nucleofilicità. (Kasaikina ET al.,
2012; Brown ET al., 2001; zen 2013; Zhang ET al., 2002; Zamamiri-Davis ET al., 2002;
Allan ET al., 1999; Xiao, Parkin, 2006; Irons ET al 2006; Ganther ET al 1999)
Un rimodellamento scheletrico a norma è mantenuto da un equilibrio sottile tra le attività
degli osteoblasti mesenchimali derivati da staminali e degli osteoclasti originati
dall’ematopoiesi, sulla superficie dell’osso. Gli osteoblasti che depositano la matrice
organica extracellulare in una sequenza ordinata sono responsabili della formazione del
nuovo materiale osseo mentre gli osteoclasti multinucleati facilitano la degradazione del
vecchio materiale osseo secernendo acido ed enzimi litici (Boyle ET al 2003). Quando
questo equilibrio si altera e avviene il disaccoppiamento delle funzionalità di questi due
tipi cellulari senza possibilità di riparazione nel tempo, tendono a manifestarsi vari tipi di
patologie ossee, come osteolisi, osteopetrosi, osteoartrite e osteoporosi (Suda ET al 1999).
Lo stress ossidativo presente in questo sistema a causa dell’aumento della concentrazione
di specie reattive dell’ossigeno (ROS) è deleterio per la normale fisiologia ossea perché tali
specie sopprimono la differenziazione degli osteoblasti, mentre promuovono quella
dell’osteoclasto genesi (Bai ET al 2005; Manolagas 2010).
Si è visto, infatti, che rimuovendo artificialmente il gene che codifica per il tRNA
coinvolto nel processo d’incorporazione della selenocisteina nelle selenoproteine a livello
delle cellule progenitrici del materiale osseo, diminuisce lo sviluppo scheletrico, e
mutazioni del gene SBP2, essenziale perché questo procedimento avvenga, causano ritardo
nella crescita e nella maturazione delle ossa soprattutto nei bambini, probabilmente a causa
della perturbazione dell’equilibrio tra gli ormoni tiroidei causata dalla diminuzione
dell’attività enzimatica della DIO. L’eliminazione artificiale della selenoproteina P,
responsabile del trasporto e dell’immagazzinamento nei tessuti, causa perdita di materiale
osseo. Tutti questi risultati insieme dimostrano quindi che è essenziale preservare il buon
funzionamento delle selenoproteine nell’osso per ottenere e mantenere una buona
funzionalità scheletrica.

34
Veniamo adesso alla descrizione di problematiche ossee connesse alla selenodeficienza:
Sviluppo Scheletrico
Uno studio condotto da Cao ET al. nel 2012 ha dimostrato che in topi in cui era stato
ridotto a circa il 10 % il selenio a livello epatico, l’enzima glutatione perossidasi
aumentava l’attività degli osteoclasti e il riassorbimento del materiale osseo, mentre
Moreno-Reyes ET al. (2001) ha dimostrato che in topi nei quali la concentrazione epatica
di selenio si aggirava intorno all’1/2% di quella normale, presentava ritardo della crescita e
diminuzione dello sviluppo scheletrico, con bassa densità ossea e calcio escreto nelle urine.
Osteoporosi
L’osteoporosi è un fenomeno che normalmente si presenta in età abbastanza avanzata, e in
particolare nelle donne in menopausa. E’caratterizzata da perdita del materiale osseo, che
diventa molto poroso e fragile e maggiormente soggetto a rischio di fratture. La perdita
della struttura e l’impoverimento del materiale osseo derivano da alterazioni che occorrono
nell’equilibrio che c’è tra la formazione degli osteoblasti e il riassorbimento degli
osteoclasti, infatti, è stato documentato che l’eccessiva presenza di specie reattive
dell’ossigeno dovuta all’età, insieme alla riduzione della quantità di ormoni estrogeni in
circolo, aumenta il riassorbimento osseo e la formazione di osteoclasti (Manolagas ET al.,
2010). Di conseguenza, una situazione di selenodeficienza in cui è compromessa la
biosintesi di un gran numero di antiossidanti, quali sono appunto i selenoenzimi, potrebbe
scatenare o peggiorare l’osteoporosi. (Raisz, 2005; Ebert, Jakob, 2007).
L’artrite reumatoide
L’artrite reumatoide è una patologia cronica autoimmune che colpisce i tessuti sinoviali a
livello di gran parte delle articolazioni, il cui marcatore è il fattore nucleare NF kB, un
elemento di trascrizione che L’artrite reumatoide è una patologia cronica autoimmune
che colpisce i tessuti sinoviali a livello di gran parte delle articolazioni, il cui marcatore è
il fattore nucleare NF kB, un elemento di trascrizione che aumenta la produzione di
molecole mediatrici dell’infiammazione. L’inattivazione di tale fattore è alle ossa.

35
Un’eccessiva percentuale di radicali liberi dell’ossigeno promuove l’attivazione del fattore
NFkB nel citoplasma, mentre i residui di cisteina a livello di questo fattore nella porzione
in cui si trova il suo sito di legame con il DNA sono essenziali per la sua attivazione nel
nucleo. Nel citoplasma, la glutatione perossidasi e la tioredossina reduttasi ne bloccano
l’attivazione neutralizzando i radicali liberi e aumentando la produzione di molecole
derivanti dal metabolismo dell’acido arachidonico. Tuttavia, nel nucleo, la tioredossina
reduttasi legandosi alla tioredossina aumenta la capacità del DNA di legarsi al fattore
nucleare; ciò la rende, di fatto, una molecola pro-ossidante che attiva il fattore NFkB (Tak
et al., 2001; Miagkov ET al 1998; Han ET al., 1998; Tsao ET al., 1997; Tomita ET al.,
1999; zen 2007). Per questo motivo, i livelli di sselenio presenti nell’organismo di un
paziente affetto da artrite reumatoide non dovrebbero essere né troppo bassi, né troppo alti
(sovra nutrizionali), per evitare di incorrere in involontari peggioramenti del danno
cartilagineo/osseo.
Il selenio come medicinale:
a) Malattie Cardiovascolari
Lo stress ossidativo danneggia l’endotelio vascolare ed esacerba malattie cardiache come
l’aterosclerosi, l’ipertensione e l’insufficienza cardiaca congestizia (Schnabel-
Blankenberg, 2007). Poiché molte delle sieroproteine sopra descritte sono espresse a
livello cardiaco e sono coinvolte nella difesa cellulare ad azione antiossidante, è stato
ipotizzato che l’assunzione di adeguati livelli di selenio possa avere un effetto preventivo
sui disordini cardiovascolari non infettivi. Questa ipotesi è stata esaminata a lungo, con
diversi studi epidemiologici basati sia su fattori clinici che osservazionali, ma non ha
ancora avuto una risposta certa. Non si è riscontrata una chiara connessione di tipo causa-
effetto, soprattutto a causa di altri antiossidanti già presenti nella dieta dei pazienti, che
avrebbero azione antiossidante supplementare a quella apportata dal selenio (Rees ET al.,
2013). Tuttavia, a parte un effetto preventivo, le selenoproteine in generale hanno un ruolo
molto più evidente per quanto riguarda la difesa dai danni cellulari causati dalle specie

36
reattive dell’ossigeno (ROS), durante o dopo lo sviluppo dei problemi cardiovascolari. E’
stata osservata, infatti, una correlazione tra i livelli di glutatione perossidasi tre,
Tioredossina-reduttasi uno e selenoproteina R e la diminuzione della produzione di radicali
liberi dell’ossigeno in casi d’ipertrofia cardiaca (Hoffman ET al., 2011). Infatti, il
funzionamento della Tioredossina-reduttasi è modulato direttamente dalla Tioredossina,
che in questi casi regola la risposta alle modificazioni enzimatiche e al rimodellamento
anatomico dei ventricoli dopo l’infarto miocardico. Di conseguenza, si è visto che un
aumento in concentrazione del selenio, porta a un aumento dell'attività della glutatione
perossidasi e della Tioredossina-reduttasi, che si è dimostrata efficace nella riduzione dei
danni ossidativi dopo riperfusione post ischemia cardiaca (Ago-Sadoshima, 2006; Tanguy
ET al., 2011).
Un’altra proteina associata a patologie cardiovascolari tramite un più specifico
meccanismo d’azione è la iodotironina deiodasi (DIO1). Questa proteina controlla la
trasformazione dell’ormone tiroideo T4 in T3, che è la versione attiva vera e propria di
quest’ormone e controlla il normale metabolismo lipidico. L’ipotiroidismo causa
cambiamenti qualitativi delle lipoproteine circolanti, aumentandone il potenziale
aterogenico, quindi un’inadeguata attività della iodotironina deiodasi associata a
ipercolesterolemia. E’particolarmente importante per preservare l’omeostasi del
metabolismo lipidico mediato da una concentrazione sufficiente di ormone T3 (Dhingra-
Bansal, 2006).
Anche la selenoproteina S è stata associata a patologie cardiovascolari, perché dotata di
effetti protettivi per gli astrociti in caso d’ischemia, ma la sua azione non è ancora del tutto
chiara (Fradejas ET al., 2008).
c) Omeostasi del selenio e delle selenoproteine antiossidanti nel cervello:
implicazioni nelle malattie neurodegenerative del sistema nervoso centrale
Come abbiamo detto in precedenza il selenio è incorporato in forma di selenocisteina in
proteine antiossidanti come glutatione per ossidasi, tioredossina reduttasi e selenoproteina

37
P. Il cervello nonostante richiedi quantità relativamente inferiori di selenio rispetto ad altri
distretti dell’organismo, ha la priorità sul trasporto e sulla ritenzione di questo elemento
quando si ha una condizione di selenodeficienza. La proteina di trasporto Sepp1
epatoderivata è la maggior fonte di selenio per il cervello ed è definita come un fattore di
sopravvivenza per i neuroni, infatti, l’eliminazione artificiale di tale proteina porta a
un’ablazione neurone specifica della biosintesi delle selenoproteine causando disfunzioni
neurologiche nei topi e si è visto che sono gli astrociti, generalmente meno vulnerabili dei
neuroni allo stress ossidativo, ad aumentare l’espressione dei selenoproteine antiossidanti
in caso di danno cerebrale di questo tipo. Infatti, sono stati riportati disordini neurologici in
pazienti con livelli di selenio inadeguati nell’organismo o con una mutazione del gene
codificante per la selenocisteina sintetasi, uno degli enzimi principali coinvolti nella
biosintesi delle selenoproteine. In vari test una bassa concentrazione di selenio è stata
associata a scarsa coordinazione motoria e a un declino più rapido delle funzioni cognitive.
Si pensa che il cervello sia particolarmente vulnerabile allo stress ossidativo a causa
dell’elevato consumo di ossigeno, della presenza di un’alta concentrazione di acidi grassi
insaturi che fungerebbero da substrato per la perossidazione dei lipidi e dell’elevato
contenuto di metalli di transizione attivi sulle ossidoriduzioni come ferro e rame (Shin ET
al 2011; Halliwell, 1992). Di conseguenza i particolari mezzi di difesa del cervello dallo
stress ossidativo sono di grande interesse nel campo della fisiopatologia e, in questi
processi i selenoenzimi hanno un ruolo fondamentale (Sies, 1993).
Il cervello utilizza una concentrazione di selenio relativamente bassa, tuttavia è capace di
trattenere al suo interno tutto il selenio utilizzato, a spese degli altri organi come fegato,
muscolo scheletrico e tessuto adiposo, anche per lunghi periodi in cui la quantità di selenio
fornita all’organismo è eccessivamente bassa. Circa il 20% del selenio totale presente nel
cervello è incorporato nell’enzima glutatione perossidasi e mantiene la sua attività anche in
condizioni alle quali normalmente altri organi soccomberebbero. Ad esempio in un
esperimento effettuato su topi sottoposti a dieta adeguata in termini di apporto di selenio,
l’attività del glutationeperossidasi era molto più alta nel fegato piuttosto che nel cervello
ma una volta sottoposti a una condizione di selenodeficienza grave voluta, l’attività della
glutatione perossidasi epatica era diminuita del 92%, arrivando quindi a perdere quasi
completamente la propria funzionalità. Nel cervello era diminuita solo marginalmente

38
(Buckman ET al 1993). Il selenio era maggiormente presente nelle regioni anatomiche
formate da alte percentuali di materia grigia come cervelletto e corteccia, in particolar
modo ventricoli cerebellari e fluido cerebro spinale (Prohaska ET al 1976; Kuhbacher ET
al 2009).
Il meccanismo che mantiene adeguati livelli di selenio nel cervello a spese degli altri
organi anche in situazioni di selenodeficienza complessiva, è diretto dalla selenoproteina P,
che rappresenta la più importante riserva di selenio per i diversi tipi di tessuti. Il ruolo di
questa proteina è, infatti, quello di fornire selenio alle cellule ed è facilitato dalla presenza
di dieci forme di siti di legame per gli atomi di selenio. Uno è localizzato nella regione N-
terminale più ampia e agisce come un enzima, e gli altri nove si trovano in un dominio C-
terminale ricco in cisteina, e sono provvisti di un meccanismo che permette di trasportare
l’atomo di selenio legandosi a tali residui, quindi in forma di selenocisteina (Ferguson ET
al., 2012). Burk ET al. 2014.Hanno recentemente proposto in uno studio i meccanismi con
cui il selenio è trasportato nel cervello, la selenoproteina P presente nel plasma è endocitata
da un’apolipoproteina dell’APOER2, a livello della barriera ematoencefalica, passa nelle
cellule dei capillari cerebrali e in quelle epiteliali del plesso coroideo, che, con
meccanismo ancora ignoto, facilitano l’immissione di selenio nel cervello in forma chimica
sconosciuta. Il selenio è inoltre incorporato direttamente nelle cellule cerebrali tramite
un’altra selenoproteina P, prodotta negli astrociti a livello dell’endotelio capillare di tali
cellule, che rappresenta dunque, oltre che un altro metodo di trasporto, anche una riserva a
sé stante tal elemento. I neuroni in un secondo momento regolano poi autonomamente i
livelli di selenio tramite un uptake APOER3-mediato della selenoproteina neuronale
(Clatworthy ET al., 1999).
Ci si è reso conto che il selenio ha un ruolo molto sfaccettato a livello del sistema nervoso
centrale. Molti studi dimostrano che la sua attività contribuisce al normale funzionamento
del cervello e che quando l’attività antiossidante di alcune selenoproteine specifiche viene
a mancare, in particolare a causa di una dieta scorretta e insufficiente, il cervello sviluppa
una maggiore suscettibilità ad alcune patologie neurodegenerative (Fang ET al., 2013).
Quest’organo contiene un’elevata percentuale di acidi grassi polinsaturi, ed è quindi più
vulnerabile alle reazioni di perossidazione.

39
Di seguito riportiamo le malattie neurodegenerative in cui le selenoproteine sono
maggiormente coinvolte (Cardoso ET al., 2015).
Morbo di Alzheimer
Il morbo di Alzheimer è una malattia neurodegenerativa caratterizzata dalla
progressiva perdita di memoria e della cognizione del mondo circostante, e
compromette le attività giornaliere del soggetto affetto. Le sue caratteristiche
fisiopatologiche sono morte neuronale, perdita delle connessioni sinaptiche,
formazioni di depositi extracellulari di beta-amiloide, placche formate da questa
proteina e precipitazione intracellulare di amminoacidi iperfosforilati, che portano alla
formazione di reticoli neuro fibrillari (Reddy, 2011; Mao ET al., 2011; Yoshiyama ET
al., 2013). Lo stress ossidativo è dunque un elemento centrale della patogenesi di
questa malattia, perché i radicali liberi dell’ossigeno causano danni alla funzionalità
mitocondriale, alla trasmissione sinaptica, al trasporto, assonale, e stimolano la neuro
infiammazione (Swerdlow ET al., 2014; Picco ET al., 2013; Haider ET al., 2014).
Molti studi condotti sugli esseri umani hanno rivelato una correlazione in senso
negativo tra declino cognitivo e livelli insufficienti di selenio, soprattutto nello stadio
eritrocitario. Pazienti affetti da morbo di Alzheimer (anche allo stadio iniziale) o da
lieve diminuzione cognitiva dimostravano livelli molto minori di selenio nei globuli
rossi rispetto al gruppo di controllo sano (Cardoso ET al., 2010/2014; Gonzalez-
Dominguez ET al., 2014; Olde ET al., 2014; Vural ET al., 2010), e questi dati sono
tuttora in accordo con studi precedenti che correlano una situazione di
selenodeficienza ad aumentato rischio di demenza senile. Si è visto che il ripristino dei
corretti livelli di selenio nell’organismo attraverso la dieta (una noce brasiliana,
contenente circa 288 microgrammi di selenio, il giorno, per sei mesi) ha migliorato le
performance cognitive dei pazienti (Cardoso ET al., 2015), facendo pensare
all’introduzione obbligatoria di queste noci nella dieta di soggetti selenodeficienti
come fonte di tal elemento.
Infatti, una dieta insufficiente in termini di apporto di selenio è stata associata anche
all’aumento della formazione delle placche beta-amiloidi nel cervello. In vari
esperimenti condotti su modelli murini (primo su tutti, quello di Haratake ET al.,

40
condotto nel 2013), è stato visto che il trattamento con selenato di sodio riusciva a
prevenire e a rendere reversibili i deficit motori e di memoria, de fosforilando le
proteine precipitate e antagonizzando l’inibitore delle fosfatasi proteiche. Preveniva la
formazione dei reticoli neuro fibrillari e la degenerazione. Anche un altro composto
seleno-organico, usato a livello dell’ippocampo e delle cellule corticali, è stato
associato a diminuita attività delle beta amiloide secretasi con riduzione della
formazione delle placche beta-amiloidee (Corcoran ET al., 2010; Ishrat ET al., 2009;
Gwon ET al., 2010).
In altri studi è stato visto che anche la selenoproteina P è, come accennato in
precedenza, correlata alla presenza di morbo di Alzheimer. In esami condotti post-
mortem su soggetti malati, è stato, infatti, perché la concentrazione di tale proteina era
particolarmente elevata a livello delle placche senili, dei reticoli neuro fibrillari e del
liquido cerebrospinale (Bellinger, 2008; Rueli ET al., 2015). Ciò fa pensare che i
livelli di selenoproteina P siano aumentati proprio in quei distretti come mezzo di
protezione dallo stress ossidativo, molto elevato proprio in tali zone dello SNC,
quando si manifesta la malattia di Alzheimer. Si pensa, infatti, che tale proteina agisca
sia fornendo selenio ai distretti cerebrali danneggiati, sia con un meccanismo proprio
antiossidante, poiché essa presenta due siti di legame dotati di elevata affinità per i
metalli di transizione (principalmente Rame), e potrebbe potenzialmente bloccare la
deposizione delle placche beta-amiloidee, mediata proprio da questo tipo di metalli,
che generano in un secondo momento anche radicali liberi dell’ossigeno (Du ET al.,
2013/2014). Anche in questo caso, si ritiene che una dieta ricca di selenio possa in
questo modo andare ad agire su queste fasi della progressione della patologia.
Morbo di Parkinson
Il morbo di Parkinson è una malattia neurodegenerativa caratterizzata dalla perdita
dei neuroni dopaminergici pigmentati nella parte compatta della substantia nigra, e
dalla presenza contemporanea d’inclusioni proteiche intraneuronali dette “corpi di
Lewy”. I neuroni dopaminergici sono particolarmente vulnerabili allo stress
ossidativo, principalmente per la loro propensione ad accumulare ioni ferro con
l’avanzare dell’età, per cui lo stress ossidativo ha un ruolo centrale nella

41
patogenesi di questa malattia ed è quindi il target dell’attività delle selenoproteine
antiossidanti (Zecca, 2004; Hare ET al., 2013; Venkateshappa ET al., 2011; Gaki,
2014; Recasens, 2014).
Tuttavia, come riportato nello studio di Shahar ET al., condotto nel 2010, è
difficile definire con certezza un rapporto di causa-effetto tra selenio e
fisiopatologia del morbo di Parkinson, in particolar modo se si considera che il
funzionamento motorio compromesso sia spesso collegato a uno stato di
malnutrizione e quindi a bassi livelli in circolo sia di selenio, che di molti altri
minerali e sostanze che potrebbero avere impatti altrettanto importanti sulla
progressione della patologia.
Ciononostante, alcuni studi hanno suggerito che una dieta selenodeficiente
potrebbe contribuire a una maggior vulnerabilità allo stress ossidativo da parte
delle cellule dei neuroni dopaminergici. In particolare prima all’esposizione a
neurotossine di tipo parkinsoniano (Vizuete ET al., 1994; Kim ET al., 2000; Hare
ET al., 2013). Si è visto che un trattamento preparatorio a base di selenio ha in
qualche modo diminuito la deplezione della dopamina proveniente dalla zona
striata in maniera dose-dipendente, e in particolar modo quando è stata raggiunta la
dose di 3mg/kg in forma d’iposelenito tre (Ellwanger ET al., 2015).
Sclerosi multipla
Sono stati affrontati vari studi riguardanti la possibile associazione tra metabolismo
del selenio, attività delle selenoproteine e disordini neurologici. Ad esempio, la
Sclerosi Multipla è una malattia di tipo infiammatorio cronico e progressivo, che
colpisce il sistema nervoso centrale e che è probabilmente collegata in qualche modo
al selenio. Nonostante l’eziologia di tale disturbo sia ancora poco conosciuta, si pensa
che il suo manifestarsi sia correlato a fattori sia genetici sia ambientali. La patogenesi
di questa malattia è, infatti, caratterizzato da risposta di tipo autoimmune contro
determinati auto-antigeni, cosa che porta a intensa neuro degenerazione con
demielinizzazione neuronale molto marcata, e infiammazione dovuta ad attivazione di
autoleucociti pro infiammatori con distruzione della barriera ematoencefalica, morte
degli oligodendrociti e danneggiamento assonale (Gandhi ET al., 2010; Broux, 2013;

42
Ortiz ET al., 2014). Si è sempre più portati a ritenere che il coinvolgimento dello stress
ossidativo nella patogenesi della sclerosi multipla sia un fattore cruciale che dà inizio e
perpetra i meccanismi che causano la degenerazione neuronale. A supporto di ciò,
alcuni studi hanno dimostrato che la glutatione perossidasi diminuisce sia in
concentrazione sia in funzionalità nei soggetti malati rispetto ai controlli (Tasset ET
al., 2012). Infatti, è stato visto che i livelli complessivi tendono a diminuire (Mehrpour
ET al., 2013; Socha ET al., 2014), in modelli animali il trattamento con difenil-
diselenuro riusciva a ridurre i segni della malattia, a prevenire la caratteristica perdita
di peso che ne accompagna la progressione, a diminuire la risposta autoimmune, e di
conseguenza la gravità dei sintomi (Chanaday ET al., 2011).
Sclerosi Amiotrofica Laterale
La sclerosi amiotrofica laterale è un’altra patologia neurodegenerativa, in questo caso
caratterizzata dalla perdita di neuroni motori a livello del midollo spinale e della corteccia
cerebrale, che causa atrofia muscolare, debolezza e fascicolazioni (Rowland, Shneider,
2001).
Nonostante le cause alla base della malattia siano ancora sconosciute, alcuni studi indicano
che c’è un collegamento tra fattori genetici e ambientali; in particolare questi ultimi sono
legati a un’eccessiva esposizione al selenio, osservata intorno al 1977 in una zona a suolo
molto selenifero, in cui la sclerosi amiotrofica laterale era molto più diffuso rispetto ad
altre zone (Killness ET al., 1977). Infatti, in una regione italiana del nord dove la maggior
parte degli abitanti consumava acqua contenente quantitativo di selenio insolitamente alti,
era stato osservato un aumento del rischio di sviluppare questo tipo di sclerosi, con
meccanismo dose-dipendente. Infatti, i pazienti presentavano a livello ematico
concentrazioni eccessive di ione iposelenito, cosa strana, poiché questo tipo di composto
del selenio normalmente non si trova nel sangue e nel plasma, perché è tossico e viene
quindi rapidamente convertito dal GSH a una forma non riducente. Da ciò, si ha ragione di
ritenere che il selenito in eccesso abbia effetti neurotossici a livello dei neuroni motori
(Vinceti ET al., 1996/2010/2013).

43
Corea di Huntington
La Corea di huntington è una malattia neurodegenerativa autonomia dominante causata
dall’aumentata ripetizione delle triplette CAG nell’esone uno del gene Huntington,
causando la produzione di una proteina alterata, eccessivamente lunga, detta Huntingtina.
Questa mutazione fa sì che la proteina è più soggetta ad aggregarsi, cosa che causa la
compromissione dell’attività mitocondriale aumentando lo stress ossidativo contribuendo
alla degenerazione neuronale (Vonsattel ET al., 1985; Bush ET al., 1994; Ju ET al., 2014;
Ha ET al., 2012; Paulsen, 2001), e portando alla comparsa di sintomi quali movimenti
incontrollati, declino cognitivo e disturbi psichiatrici. Nonostante i livelli di selenio nel
plasma non fossero diversi tra soggetti affetti e controlli sani, in uno studio condotto da Lu
ET al. nel 2014, i pazienti presentavano una diminuzione dei livelli di selenio nella regione
del cervello, soprattutto nel putamen, nella corteccia prefrontale dorso-laterale, in quella
visiva primaria e nel cervelletto. In topi con lo stesso tipo di problema, il trattamento con
selenito ha diminuito la perdita di materiale cerebrale e ha ridotto l’unione dell’Huntingtina
e lo stress ossidativo.
a) COMPOSTI DEL SELENIO ATTIVI SUI PROCESSI OSSIDORIDUTTIVI:
TRATTAMENTI PER IL CANCRO
Il selenio è generalmente noto come un antiossidante per la sua presenza nella
selenocisteina e nelle selenoproteine, ma è anche tossico. Gli effetti tossici del selenio sono
tuttavia strettamente concentrazione-dipendenti e connessi alla particolare specie chimica.
In particolare, una classe dei composti del selenio è un potente inibitore della crescita
cellulare con alta specificità per le cellule tumorali, infatti, ha come attività quella di
ossidare tali cellule agendo come pro farmaco.
I composti del selenio che naturalmente possiedono tale proprietà sono: seleniti, selenati,
selenocisteina e il metabolita metilselenolo della selenometionina e della
selenometilselenocisteina. Oggi sono tuttavia oggetti di studio anche composti di nuova
generazione che potranno manifestare in maniera più efficiente e più diretta la loro attività
pro-ossidante sulle cellule tumorali liberando in situ i metaboliti naturalmente dotati di tali
proprietà sopra citati. Gli effetti citotossici di tutti i selenocomposti sono, infatti,
dipendenti dalla generazione di radicali liberi del selenio e di selenati da parte di enzimi

44
reduttasi o tramite l’ossidazione dei tioli (Bjornstedt ET al., 1992; Kumar ET al 1992;
Wallemberg ET al 2010) e la loro azione benefica per soggetti malati di cancro sta nelle
loro proprietà antiproliferative. Alcuni composti del selenio sono inoltre perfetti candidati
come chemioterapici perché sono in un certo senso tumore-selettivi, sia in termini di sito di
assorbimento sia in termini d’interazioni con il microambiente della cellula tumorale.
Alcune volte i fenotipi di cancro resistenti ai normali farmaci antitumorali rispondono in
maniera migliore a una terapia effettuata con i composti del selenio.
In generale, le reazioni di ossidoriduzione sono essenziali per il mantenimento
dell’omeostasi fisiologica sia negli organismi unicellulari sia pluricellulari e il termine
stress ossidativo è definito come la perturbazione dell’equilibrio fisiologico delle
ossidoriduzioni in senso riduttivo piuttosto che ossidativo (Halliwell, 2007). Antiossidanti
sia enzimatici sia non, possono sovvertire quest’equilibrio quando la cellula non è in grado
di auto ripararsi quando subisce un danno. Per quanto riguarda il selenio, le proprietà
antiossidanti sono esplicate principalmente in seguito alla sua incorporazione nelle
selenoproteine che possono catalizzare la riduzione dei ponti disolfuro nelle proteine e tali
proprietà riduttive sono importanti per la protezione degli elementi cellulari ed
extracellulari dall’ossidazione che senza di esse avverrebbe in modo discriminato tramite
ossidanti intrinseci ed estrinseci (Labunskyy ET al 2014; Moghadasszadeh ET al 2006).
C’è una profonda diversità tra le specie chimiche del selenio dovuta ai diversi stati di
ossidazione di questo elemento (-2,0, +4, +6) e tutti i composti con il loro diverso numero
di ossidazione sono implicati nell’aumento dell’espressione delle selenoproteine sia in
vitro sia in vivo, quindi tutti gli stati di ossidazione sono potenzialmente biodisponibili per
la biosintesi delle selenoproteine.
Questi composti del selenio chimicamente eterogenei sono utilizzati per le funzioni
fisiologiche tramite processi e vie specifiche, unici per ogni composto, con percentuali
variabili di metaboliti intermedi. Ad esempio, mentre il selenuro acido è uno dei maggiori
metaboliti intermedi dei composti inorganici del selenio, quelli metilati seleniferi sono i
maggiori metaboliti intermedi dei composti organici del selenio. In conformità a
un’aumentata capacità di sintesi delle selenoproteine da parte di entrambe queste classi di
composti negli eucarioti, è possibile che essi siano metabolizzati per creare una riserva

45
comune di selenuro acido. Esso è in seguito convertito in selenofosfato dalla selenofosfato
sintetasi, una step chiave per la biosintesi della selenocisteina e delle selenoproteine.
C’è una regolazione fisiologica molto rigida per quanto riguarda la capacità dell’organismo
di sintetizzare le selenoproteine da una riserva di selenio (in qualsiasi forma chimica esso
si trovi). Quando i livelli di selenio sono superiori a quelli che servono per formare il pool
enzimatico e proteico necessario per l’organismo, si ha un aumento nel metabolismo di
questo metallo, ma le selenoproteine non sono più prodotte. Si parla dunque di livelli sovra
nutrizionali di selenio, ed è da questo punto che s’iniziano ad avere dosaggi tossici, che a
causa della loro concentrazione modificano in vivo la loro attività da antiossidante a pro-
ossidante. Infatti, molti prodotti derivanti dal catabolismo del selenio, che aumenta in
condizioni sovra nutrizionali, hanno una spiccata capacità di generare specie reattive
dell’ossigeno (ROS), reagendo con i tioli (Wallenberg ET al., 2014; Weekley ET al., 2013;
Guimaraes ET al., 1996; Forstrom ET al., 1978; Spallholz ET al., 1994).
Sono proprio queste specie chimiche del selenio, identificati come COMPOSTI DEL
SELENIO ATTIVI SULLE OSSIDORIDUZIONI, a causare stress ossidativo,
manifestando la loro funzionalità come ipotetici farmaci antitumorali, e si tratta perlopiù di
ione Selenuro, selenocisteina, acido metilselenico e selenometilseleniocisteina.
E’ dunque ovvio che, affinché il selenio si comporti da specie antitumorale e induca lo
stress ossidativo anziché contrastarlo, sia necessario un supplemento nella dieta che lo
porti a concentrazioni sistemiche sovra nutrizionali. Ciò non sarebbe mai una cosa
vantaggiosa per un organismo in salute, ma una potenziale applicazione di questi composti
nella terapia per il cancro potrebbe essere risolutiva per tutte quelle forme tumorali che
richiedono farmaci attivi sui radicali liberi (Gorrini ET al., 2013; Wondrak ET al., 2009).
Il concetto che fa pensare a un’effettiva utilità di questi composti è che le cellule cancerose
di solito mostrano una quantità basale di specie reattive dell’ossigeno molto maggiore della
norma: ciò porta di conseguenza a un up-regulation dei sistemi di difesa antiossidanti,
perché adattamento patofisiologico, che però favorisce lo sviluppo e la malignità del
tumore (Trachootham ET al., 2009). Tuttavia, le cellule cancerose hanno una minore
tolleranza rispetto alle cellule normali, nei confronti dell’aumento dei radicali liberi non
prodotti da loro stesse, e ciò è considerato un punto di partenza ideale per sostenere la
46
teoria che livelli sovra nutrizionali di selenio pro ossidante possano agire andando a
rallentare la proliferazione tumorale (Kong ET al., 2000).
.
FIGURA uno: Effetti dei livelli elevati di ROS in cellule normali e tumorali.
Come accennato in precedenza, i composti del selenio sono tossici in base alla loro forma
chimica, alla dose e al tempo di esposizione del soggetto. Per far sì che il selenio acquisti
proprietàpro-ossidanti, esso deve reagire con i tioli e generare superossidi dal GSH (figura
2.) (Feigal, West, 1947; Tsen et al., 1958; Rhead et al., 1974; Spallholz et l., 1992; Seko et
al., 1997). Inoltre, nel 1997, Chaudière ET al. dimostrò che partendo dalla selenocistamina
diselenuro si formavano, tramite la riduzione con GSH, due molecole di selenuri ossidanti,

47
che andavano a formare a loro volta un ciclo ossido riduttivo alla presenza di GSH in
eccesso, e generavano superossido usando ossigeno. Da ciò, si evinse che la specie tossica
del selenio che deve essere formata una volta in vivo per avere attività pro-ossidante, e
quindi fungere da antitumorale, è proprio lo ione superossido.
Figura due: Reazioni che portano alla formazione di superossido, responsabile dell’attività pro-ossidante.
A oggi, i composti che sono noti per la loro capacità di generare l’anione superossido sono:
SELENIO METIL SELENOCISTEINA (SMC)
La SMC non è un composto del selenio attivo sulle ossidoriduzioni di per sé, ma agisce
“da pro farmaco”. E’ metabolizzato dalla glutammina transaminasi a metilselenolo, che
invece è la molecola attiva in questo senso. La SMC è molto biodisponibile, come indicato
dall’aumento immediato dei livelli di selenoproteina P e glutatione perossidasi quando è
fornita nella dieta, ma essa è meno tossica per le cellule tumorali rispetto ad altre specie del
selenio (Hoefig ET al., 2011). Tuttavia, da dati derivanti da alcuni studi clinici condotti su
modelli animali, è emersa una proprietà antitumorale piuttosto buona. Ad esempio, una
volta somministrata in topi in cui era stato inserito un modello umano di carcinoma del
colon, essa ha ridotto il volume ematico e la densità dei micro vasi creati dal tumore,
riducendone quindi visibilmente il rischio di metastasi (Bhattacharya ET al., 2011). Si
pensa che tali effetti inibitori dell’angiogenesi tumorali siano mediati dalla down-
regulation di VEGF, COX2, HIF-1alfa e Inos (fattore di crescita vascolare - endoteliale,
ciclo ossigenasi 2, fattore1-alfa ipossia-indotto, sintesi dell’acido nitrico inducibile) [Yin
ET al., 2006]. Inoltre, la SMC non ha solo inibito l’angiogenesi, ma anche aumentato la
maturazione vascolare e ha ridotto la pressione del fluido interstiziale, e tutti questi effetti
sono comparsi selettivamente nelle cellule tumorali, ma non negli organi sani. Tali
osservazioni sembrano dunque incoraggiare l’altro studio di meccanismi di drug-delivery a

48
livello dei tumori, senza temere troppo la comparsa di effetti collaterali su altri organi
vicini. E ultimamente quest’obiettivo è stato raggiunto, è stata, infatti, comprovato
l’attività antitumorale della SMC in associazione con quattro diversi farmaci ad azione
citostatica. Quest’associazione ha permesso la remissione del carcinoma colo rettale di
Ward sia nei topi sia in pazienti umani (Cao et al., 2014). Ancora più importante, la SMC
ha dimostrato di riuscire a proteggere l’organismo dalla severa mielosuppressione indotta
dall’oxaliplatino nei topi. Perciò, il potenziale unico di questo selenocomposto incoraggia
molto altri studi in modo che possa, un giorno essere applicato anche nel trattamento di
vari tipi di cancro negli esseri umani.
SELENOCISTINA
La selenocistina (CysSeSeCys) è uno dei composti del selenio chiave per la sua attività sui
processi ossido riduttivi, con funzionalità doppia. Essa agisce, infatti, sia come
antiossidante sia come pro-ossidante. Il composto in sé non mostra alcun tipo di attività,
finché non è ridotta da tioli a basso peso molecolare e dall’enzima disolfuro reduttasi, a
selenocisteina, che invece è molto reattivo in termini di ossidoriduzioni. Infatti, in
ambiente riducente, la selenocisteina ha un’azione del tutto simile a quella della glutatione
perossidasi.
A livelli nutrizionali normali si pensa che la selenocistina funzioni come uin antiossidante
molto biodisponibile (Hoefig ET al., 2011), ma una volta metabolizzata a selenocisteina,
nonostante la sua potente capacità d’inattivazione elettrofila, essa diventa un potente
agente citotossico se presente in eccesso. Uno dei metaboliti intermedi chiave della
selenocisteina è, infatti, lo ione selenuro acido, generato dall’enzima selenocisteina liasi in
maniera dipendente dal piridossal-fosfato. Poiché questo è anche un metabolita che
normalmente si forma dal selenito di sodio, ci si aspetterebbe un profilo di tossicità simile
tra selenocisteina e selenito, poiché le loro vie di assorbimento sono simili. In alcuni studi
(Chen ET al., 2009; Kumar ET al., 2009) si è notato che il selenito va ad agire in maniera
tossica anche sulle cellule sane, che sono più sensibili a questa selenospecie che alla
selenocisteina. Ciò suggerisce che ci sia un determinato grado di selettività per le cellule
tumorali da parte di quest’amminoacido quando somministrato a livelli sovra nutrizionali,
come dimostrato da uno studio condotto nel 1980 su modelli murini maschi, in cui il

49
selenito si è dimostrato molto più tossico rispetto alla selenocisteina somministrata negli
stessi dosaggi.
I dati farmacocinetici emersi da due studi (Hasegawa, 1994; Lane, 1990) suggeriscono che
per l’essere umano, diversamente dai modelli animale, è più vantaggiosa la
somministrazione per via endovenosa, per far sì che il composto manifesti le sue proprietà
di mantenimento costante dei livelli di selenio all’interno del plasma.
Gli effetti più evidenti della selenocistina negli umani si sono manifestati nel trattamento
della leucemia mieloide cronica e acuta (Weisberger, Suhrland, 1956). Gli scienziati hanno
riportato un maggiore effetto sui leucociti immaturi rispetto a quelli maturi, senza effetti
indesiderati degni di nota sul midollo osseo. Un altro aspetto interessante emerso dallo
stesso studio clinico è stato che si è manifestata una maggiore risposta al trattamento con
selenocistina nei pazienti refrattari ad altri trattamenti con agenti chemioterapici allora
disponibili. Ciò non fa altro che confermare i dati emersi da altri studi, in cui appunto molti
pazienti che non rispondevano a farmaci ad azione citostatica, al trattamento successivo
con selenito (Brodin ET al., 2015). Infatti, tutti i dati a oggi in nostro possesso indicano che
la selenocistina potrebbe essere un ottimo canditato come antitumorale, soprattutto per
quanto riguarda la leucemia.
SELENITO
Come dimostrato di recente (Brodin ET al., 2015), il selenuro non è solo un antiossidante,
ma possiede proprietà pro-ossidanti alla presenza di specifici substrati. Il selenito è, infatti,
capace di ossidare i politioli ai corrispondenti disolfuri, ma non reagisce con i monotioli. E
sono proprio i politioli a essere maggiormente presenti quando ci sono condizioni
riducenti, come accade nei tessuti tumorali ipossici.Questi politioli possono, a turno
iniziare uno scambio disolforico con le proteine del plasma, cui si legano prevalentemente
tramite il fibrinogeno, per formare un polimero fibrino-simile insolubile e resistente alle
proteasi. Ciò porta le cellule tumorali a essere, di fatto, circondate e protette da uno strato
di tale polimero, che maschera gli antigeni specifici del tumore, permettendo quindi alla
massa di cancro di sfuggire al riconoscimento da parte dei linfociti Natural Killer e quindi
di essere distrutta.

50
Il selenito, proprio ossidando i tioli presenti sulle membrane cellulari, può prevenire la
formazione di questo strato protettivo e rendere di conseguenza le cellule tumorali
vulnerabili al sistema immunitario, e quindi distruttibili. Inoltre, tale molecola può attivare
le cellule Natural Killer, e anche l’angiogenesi tumorale, senza alcuna diminuzione
indesiderata nel potenziale ossidativo dell’ambiente cellulare sano.
Questo semplice composto del selenio deve essere somministrato per via endovenosa,
perché se dato per trasmissione orale è una scarsissima percentuale della molecola,
arriverebbe attivo nel plasma. Di recente, è stato completato un trial clinico di fase uno in
cui sono state stabilite sicurezza, proprietà farmacocinetiche e massima dose tollerata (dati
non ancora pubblicati, Brodin ET al., 2015).
Si può quindi affermare in conclusione che questo composto potrà essere utilizzato per
studi sempre più specifici, e si sta già distinguendo come candidato ottimale per divenire
un farmaco antitumorale a tutti gli effetti.
Conclusioni
Negli ultimi decenni sono stati fatti notevoli progressi nelle conoscenze riguardanti i
processi che regolano l’azione biologica del selenio e delle sue specie. Ne è emerso un
quadro complesso, in cui molte selenoproteine collaborano nella regolazione dei
meccanismi di trascrizione, nel combattere lo stress ossidativo e nel complesso equilibrio
delle reazioni di ossido-riduzione. Esperimenti condotti su modelli animali, come ad
esempio topi privati di determinate selenoproteine, o nei quali esse venivano iperespresse,
hanno permesso di indagare più a fondo sull’azione esercitata dalla singola selenoproteina
o specie chimica del selenio, anche se i dati emersi non sono stati sufficientemente
esaustivi per traslatare le scoperte fatte nei modelli murini anche all’uomo.
Purtroppo, il fatto che non ci siano ancora parametri esatti che descrivano quali siano i
livelli di selenio “normali” nel sangue, e di conseguenza anche la mancanza di metodi per
stabilirli, ha fatto sì che questi fattori riguardanti l’elemento e il suo coinvolgimento nella
salute umana non sia ancora stato identificato con esattezza.
La complessità di quest’argomento si basa su vari punti di vista tutti connessi tra di loro:

51
- Le selenospecie bioattive intervengono in varie misure sulle funzioni biologiche
dell’organismo: ad esempio, per quanto riguarda la risposta immunitaria, le selenoproteine
agiscono in modo diverso secondo il substrato sul quale vanno ad agire, e di come
interagiscono tra di loro, quindi i processi specifici sono difficili da indagare per la singola
specie chimica.
Il polimorfismo genetico delle selenoproteine sembra essere una variabile molto
importante da considerare per quanto riguarda l’apporto di selenio nell’organismo e il suo
coinvolgimento nella comparsa, nella progressione e nella prognosi delle patologie.
-E’ necessario fare studi epidemiologici su larga scala per avere un quadro generale più
dettagliato di tutti gli aspetti di questo metallo e delle sue specie bioattive, in particolare
per quanto riguarda la caratterizzazione genetica degli individui coinvolti (vedi
polimorfismi genetici di cui sopra).
-Infine, ci sono ancora molte domande irrisolte cui rispondere, come ad esempio le
dinamiche precise e la regolazione della sintesi delle selenoproteine, come anche la
comprensione esatta delle loro funzionalità biochimiche specifiche (molte non sono ancora
state identificate)
-In conclusione, possiamo affermare tuttavia che esiste una disciplina emergente, la
biologia dei sistemi, che sembra offrire informazioni sempre più dettagliate man mano che
gli scienziati proseguono i loro esperimenti in questo senso. Tali informazioni potranno in
futuro essere utilizzate come base per costruire un prospetto di quali siano i processi e le
dinamiche regolate dalle selenoproteine, per prevedere i cambiamenti che avvengono in
condizioni normali o patologiche e per costituire studi epidemiologici affidabili.

52
BIBLIOGRAFIA:
1) A. Turanov, X.-M.Xu, B.A. Carlson, M.-H.Yoo,
V.N.Gladyshev and D.L.Hatfield, Adv.Nutr;2011, 2,122-
128.
2) A.Amantana, W.R Vorachek, J.A. Butler, W. Ream and
P.D.Whanger,J.Inorg. Biochem., 2004, 98, 1513-1520.
3) A.Bush, W.Pettingell, G.Multhaup, M.D.Paradis,
J.Vonsattel, J.Gusella, K.Beyreuther, C.Masters and
R.Tanzi, Science, 1994, 265, 1464-1467.
4) A.C.Bianco, D. Salvatore, B. Gereben, M.J.Berry and
P.R.Larsen, Endocr.Rev., 2002, 23,38-89.
5) A.D.Ha and V.S.C.Fung, Curr.Opin. Neurol., 2012, 25, 491-
498.
6) A.Dikiy, S. V. Novoselov, D.E. Fomenko, A.Sengupta, B.A.
Carlson, R.L. Cerny, K. Ginalski, N.V.Grishin, D.L.
Hatfield and V.N. Gladishev, Biochemistry, 2007, 46, 6871-
6882.
7) A.E.Clatworthy, W.Stockinger, R.H.Christie,
W.J.Schneider, J.Nimpf, B.T.Hyman and G.W.Rebeck,
Neuroscience, 1999, 90, 903-911.
8) A.Ghose, J.Fleming, K.El-Bayoumy, P.R.Harrison,
Enhanced sensitivity of human oral carcinomas to induction
of apoptosis by selenium compounds: involvement of
mitogen –activated protein kinase and Fas pathways, Cancer
Res. 61 (2001) 7479-7487.
9) A.M.Dumitrescu,X.-H.Liao,M.S.Y.Abdullah, J.Lado-Abeal,
F.A.Majed, L.C.Moeller, G.Boran, L.Schomburg, R.E.Weiss
and S.Refetoff, Nat.Genet., 2005, 37, 1247-1252.
10) A.Nickel,G.Kottra, G.Schmidt,J.Danier, T.Hofmann and
H.Daniel, Chem.-Biol.Interact., 2009, 177, 234-241.
11) A.Noblanc, A.Kocer, E.Chabory, P.Vernet,F.Saez, R.Cadet,
M.Conrad and J.R.Drevet, J.Androl., 2011, 32, 641-650.
12) A.Picco, M.C.Polidori, M.Ferrara, R.Cecchetti, D.Arnaldi,
M.Baglioni, S.Morbelli, P.Bastiani, I.Bossert, G.Fiorucci,
A.Brugnolo, M.E.Dottorini, F.Nobili and P.Mecocci,
Eur.J.Nucl.Med.Mol.Imaging, 2013, 41, 764-775.

53
13) A.-R Gwon, J.-S.Park, J.-H.Park, S.-H.Baik, H.-Y.Jeong, D.-
H.Hyun, K.W.Park and D.-G.Jo, Neurosci.Lett., 2010, 469,
391-395.
14) A.Recasens and B.Dehay, Front.Neuroanat., 2014, 8, a159.
15) A.S.Mueller and J.Pallauf, J.Nutr.Biochem., 2006, 17, 584-
560.
16) A.Sengupta, B.A. Carlson, V.M.Labunskyy, V.N. Gladyshev
and D.L.Hatfield, Biochem. Cell Biol., 2009, 87, 953-961.
17) A.Shahar, K.V.Patel, R.D.Semba, S.Bandinelli, D.R.Shahar,
L.Ferrucci and J.M.Guralnik, Mov. Disord., 2010, 25, 1909-
1915.
18) A.Small-Howard, N.Morozova, Z.Stoytcheva, E.P.Forry,
J.B.Mansell, J.W.Harney, B.A.Carlson, X.M. Xu,
D.L.Hatfield and M.J.Berry, Mol. Cell. Biol., 2006, 26,
2337-2346.
19) A.V.Miagkov, D.V.Kovalenko, C.E.Brown, J.R.Didsbury,
J.P.Cogswell, S.A.Stimpson, A.S.Baldwin, S.S.Makarov,
NF-kappaB activation provides the potential link between
inflammation and hyperplasia in the arthritic joint,
Proc.Natl.Acad.Sci. U.S.A. 95 (1998) 13859-13864.
20) A.W.Kilness and F.H.Hichberg, JAMA,J.Am.Med.Assoc.,
1977, 237, 2843-2844.
21) Ansong, E..,Yang, W..,Diamond,A.M.Molecular cross-talk
between members of distinct families of selenium containing
proteins.Mol.Nutr.Food Res.2014, 58, 117-123.
22) B.A. Zachara, A.Adamowicz, U.Trafikowska,
A.Trafikowska, J.Manitius and E.Nartowicz, J.Trace
Elem.Med.Biol., 2001, 15, 201-208.
23) B.A.Zachara, U.Trafikowska, A. Adamowicz, E.Nartowicz
and J.Manitius, J.Trace Elem.Med.Biol., 2001, 15, 161-166.
24) B.Broux, P.Stinissen and N.Hellings, Crit. Rev. Immunol.,
2013, 33, 283-306.
25) B.E.Petrovski, V.Pataki, T.Jenei, R.Adany and Z.Voko,
J.Trace Elem.Med.Biol., 2012, 26, 31-35.
26) B.Gammelgaard, L.Rasmussen, C.Gabel-Jensen and
B.Steffansen, Biol. Trace Elem. Res., 2012, 145, 248-256.
27) B.Halliwel, J.Neurochem.59(1992) 1609-1623.
28) B.R.Cardoso, D.Apolinario, V.da Silva Bandeira,
A.L.Busse, R.M.Magaldi, W.Jacob-Filho and
S.M.F.Cozzolino, Eur.J.Nutr., 2015 1-10, DOI:10.1007/
s00394-014-0829-2.M.Haratake, S.Yoshida, M.Mandai,

54
T.Fuchigami and M.Nakayama, Metallomics, 2013, 5, 479-
483.
29) B.R.Cardoso, T.P.Ong, W.Jacob-Filho, O.Jaluul,
M.I.D.A.A.Freitas and S.M.F.Cozzolino, Br.j.Nutr., 2010,
103, 803-806.
30) B.R.Cardoso, V.Silva Bandeira, W.Jacob-Filho and
S.M.Franciscato Cozzolino, J.Trace Elem.Med.Biol., 2014,
28, 422-426.
31) Bellinger, F.P..,Raman, A.V..,Reeves, M.A..,Berry,
M.J.Regulation and function of selenoproteins in human
disease.Biochem.J.2009, 422, 11-22-
32) Berry, M.J.., Banu, L..,Chen,
Y..,Mandel,S.J..,Kiefer,J.D..,Harney,J.W..,Larsen,P.R.Recog
nition of UGA as a selenocysteine codon in type
I.deiodinase requires sequences in the 3’untranslated
region.Nature 1991, 353, 273-276.
33) Bhattacharya, A.., Turowski, S.G.., San Martin, I.D..,
Rajiput, A..,Rustum, Y.M..,Hoffman, R.M..,Seshadri,
M.Magnetic resonance and fluorescence-proteim imaging of
the anti-angiogenic and anti-tumor efficacy of selenium in
an orthotopic model of human colon cancer. Anticancer Res.
2011, 31, 387-393.
34) Bhattacharya, A..,Seshadri, M..,Oven, S.D..,Toth,
K..,Vaughan, M.M..,Rustum, Y.M.Tumor vascular
maturation and improved drug delivery induced by
methylselenocysteine leads to therapeutic synergy with
anticancer drugs. Clin.Cancer Res. 2008, 14, 3926-3932.
35) Bhattacharya, A.Methylselenocysteine: A promising
antiangiogenic agent for overcoming drug delivery barriers
in solid malignancies for therapeutic synergy with anticancer
drugs. Expert Opin.Drug Deliv. 2011, 8, 749-763.
36) Bjornstedt, M.., Kumar, S..,Holmgren,
A.Selenodiglutathione is a highly efficient oxidant of
reduced thioredoxin and a substrate for mammalian
thioredoxin reductase. J.Biol.Chem. 1992, 267, 8030-8034.
37) Brodin, O..,Eksborg, S..,Wallenberg, M..,Asker-Hagelberg,
C..,Huusfeldt Larsen, E..,Mohlkert, D..,Lenneby-Helleday,
C..,Jacobsson, H..,Linder, S..,Misra, S.., et
al.Pharmacokinetics and toxicity of sodium selenite in the
treatment of patients with carcinoma in a phase 1 clinical

55
trial: The SECAR study. Nutrients 2015, submitted for
publication.
38) Broncato, J..,Chervona, Y..,Costa, M.Molecular responses to
hypoxia-inducible factor 1 alpha and
beyond.Mol.Pharmacol. 2014, 85, 651-657.
39) C. Ip,J.Nutr., 1998, 128, 1845-1854.
40) C.Abuje, A.Tsegaye, C.E.West, P.Versloot, E.J.Sanders,
D.Wolday, D.Hamman, T.F.R.Wit and A.L.F ontanet,
J.Clin.Immunol., 2005, 25, 127-133.
41) C.B.Allan, G.M.Lacourciere, T.C.Stadtman, Responsiveness
of selenoproteins to dietary selenium, Annu.Rev.Nutr.19
(1999) 1-16.
42) C.B.Stephenson, G.S.Marquis, S.D.Douglas, L.A.Kruzich
and C.M.Wilson, Am.J.Clin.Nutr., 2007, 85, 173-181.
43) C.D.Thomson, Eur.J. Clin. Nutr., 2004, 58, 391-402.
44) C.Di Cosmo, N.McLellan, X.H.Liao, K.K.Khanna,
R.E.Weiss, L.Papp and S.Refetoff,
J.Clin.Endocrinol.Metab., 2009,94, 4003-4009.
45) C.Gabel-Jensen, B. Gammelgaard, L. Bendahl, S. Sturup
and O.Jons, Anal. Bioanal. Chem., 2006, 384, 697-702.
46) C.Ip, H.J.Thompson, Z.Zhu, H.E.Ganther, in vitro and vivo
studies of methylseleninic acid: evidence that a
monomethylated selenium metabolite is critical for cancer
chemoprevention, Cancer Res. 60 (2000) 2882-2886.
47) C.Kornhauser, J.R.Garcia-Ramirez, K.Wrobel, E.-L.Pèrez-
Luque, M.E.Garay-Sevilla and K.Wrobel, Prim. Care
Diabetes, 2008, 2, 81-85.
48) C.Lu, F. Qiu, H Zhou, Y. Peng, W.Hao,J. Xu, J. Yuan, S.
Wang, B. QIANG, C.Xu and X. Peng, FEBS Lett., 2006
,580, 5189-5197.
49) C.Reilly, Selenium in Food and Health, Springer-Verlag,
New York, 2nd edn, 2006.
50) C.Venkateshappa, G.Harish, R.B.Mythri, A.Mahadevan,
M.M.S.Bharath and S.K.Shankar, Neurochem. Res., 2011,
37, 358-369.
51) Cao, S..,Durrani, F.A.., Toth, K..,Rustum, Y.M. Se-
methylselenocysteine offers selective protection against
toxicity and potentiates the antitumor activity of anticancer
drugs in preclinical animal models. Br.J.Cancer 2014, 110,
1733-1743.

56
52) Centres for Diseas Control and Prevention, Morb. Mortal.
Weekly Rep., 1984, 33, 157-158.
53) Chaudiere, J..,Courtin, O..,Leclaire, J.Glutathione oxidase
activity of selenocystamine: A mechanistic study.
Arch.Biochem.Biophys. 1992, 296, 328-336.
54) Chen, T..,Wong, Y.S.Selenocysteine induces reactive
oxygen species-mediated apoptosis in human cancer cells.
Biomed.Pharmacother. 2009, 63, 105-113.
55) Combs, G.F. and Combs, S.B., eds (1986) the role of
selenium in nutrition, academic press
56) D.E.Fomenko, A. Koc, N.Agisheva, M.Jacobsen, A.Kaya,
M.Malinouski, J.C.Rutherford, K.L.Siu, D.Y.Jin, D.R.Winge
and V.N.Gladyshev, Proc.Natl.Acad.Sci. U.S.A., 2011, 108,
2729-2734.
57) D.Hare, S.Ayton, A.Bush and P.Lei, Front.Aging Neurosci.,
2013, 5, 34.
58) D.J.Hare, P.A.Adlard, P.A.Doble and D.I.Finkelstein,
Metallomics, 2013, 5, 91-109.
59) D.L.Hatfield, B.A.Carlson, X.M.Xu, H.Mix,V.N.Gladyshev,
Selenocysteine incorporation machinery and the role of
selenoproteins in development and health, Prog.Nucleic
Acid Res.Mol.Biol.81(2006) 97-142.
60) D.L.St Germain, A. Hernandez, M. J. Schneider and
V.A.Galton, Thyroid, 2005, 15, 905-916.
61) D.M.Gerlag, L.Ransone, P.P.Tak, Z.Han, M.Palanki,
M.S.Barbosa, D.Boyle, A.M.Manning, G.S.Firestein, The
effect of a T cell-specific NF-KappaB inhibitor on in vitro
cytochine production and collagen-induced arthritis,
J.Immunol. 165 (2000) 1652-1658.
62) D.Mustacich and G.Powis, Biochem.J., 2000, 346, 1-8.
63) D.Su, S.V.Novoselov, Q.-A. Sun, M.E. Moustafa, Y. Zhou,
R.O ko, D.L. Hatfield and V.N. Gladischev,J.Biol.Chem,
2005, 280, 26491-26498.
64) D.W.Jeong, M.H.Yoo, T.S.Kim, J.H.Kim,I.Y. Kim,
Protection of mice from allergen-induced asthma by selenite:
prevention of eosinophil infriltation by inhibition of NF-
kappa B activation, J.Biol.Chem. 277 (2002) 17871-17876.
65) D.W.Jeong, T.S Kim, Y.W.Chung, B.J.Lee and I.Y.Kim,
FEBS Lett., 2002, 517, 225-228.
66) D.Wang. E.W.Taylor, Y.Wang. X.Wan, J.Zhang,
Encapsulated nanoepigallocatechin-3-gallate and elemental

57
selenium nanoparticles as paradigms for
nanochemoprevention, Int.j.Nanomedicine 7 (2012) 1711-
1721.
67) Davis,C.D..,Tsuji, P.A..,Milner, J.A.Selenoproteins and
Cancer Prevention.Annu.Rev.Nutr. 2012, 32, 73-95.
68) Driscoll, D.M..,Copeland, P.R.Mechanism and regulation of
selenoprotein synthesis.Annu.Rev.Nutr.2003, 23, 17-40.
69) E. Chabory, C. Damon, A. L enoir, J. Henry- Berger, P.
Vernet, R. Cadet, F. Saez and J.R.Drevet, J. Anim. Sci.,
2010, 88, 1321-1331.
70) E.G.Varlamova and V.I.Novoselov, Mol. Biol., 2012, 46,
735-737.
71) E.G.Varlamova and V.I.Novoselov,Mol. Biol., 2012, 46,
250-257.
72) E.Gonzalez-Reimers, L.Galindo-Martin, F. Santolaria-
Fernandez, M.J.Sanchez-Perez, J.Alvisa-Negrin, E.Garcia-
Valdecasas-Campelo, J.M.Gonzalez-Perez and M.C. Martin-
Gonzalez, Biol. Trace Elem. Res., 2008, 125, 22-29.
73) E.J.Shin, J.H.Jeong, Y.H.Chung, W.K.Kim, K.H.Ko,
J.H.Bach, J.S.Hong, Y.Yoneda, H.C.Kim, Neurochem.Int.59
(2011) 122-137.
74) E.Kumaraswamy, A. Malykh, K.V.Korotkov,
S.Kozyavkin,Y.Hu, S.Y.kwon, M.E.Moustafa, B.A.Carlson
M.J.Berry, B.J.Lee,D.L.Hatfield, A.M.Diamond and
V.N.Gladyshev, J.Biol.Chem., 2001. 276, 15330-15336.
75) E.S.Arner, Selenoproteins-what unique properties can arise
with selenocysteine in place of cysteine? Exp.Cell.Res. 316
(2010) 1296-1303.
76) E.S.J.Arnèr, Biochim. Biophys. Acta, 2009, 1790, 495-526.
77) E.Schoenmakers, M.Agostini, C.Mitchell, N.Schoenmakers,
L.Papp, O.Rajanayagam, R.Padidela, L.Ceron-Gutierrez,
R.Doffinger, C.Prevosto, J.Luan, S.Montano, J.Lu,
M.Castanet, N.Clemons, M.Groeneveld, P.Castets,
M.Karbaschi, S.Aitken, A.Dixon, J.Williams, I.Campi,
M.Blount, H.Burton, F.Muntoni, D.O’Donovan, A.Dean,
A.Warren, C.Brierley, D.Baguley, P.Guicheney,
R.Fitzgerald, A.Coles, H.Gaston, P.Todd, A.Holmgren,
K.K.Khanna, M.Cooke ,R.Semple, D.Halsall, N.Wareham,
J.Schwabe, L.Grasso, P.Beck-Peccoz, A.Ogunko, M.Dattani,
M.Gurnell and K.Chatterjee, J.Clin.Invest., 2010, 120, 4220-
4235.

58
78) Eggler, A.L..,Gay, K.A..,Mesecar, A.D.Molecular
mechanisms of natural products in
chemoprevention:Induction of cytoprotective enzymes by
Nrf2.Mol.Nutr.Food Res. 2008, 52 (Suppl.1),S84-S94.
79) F.L. Aachmann, D.E. Fomenko, A.Soragni, V.N. Gladyshev
and A.Dikiy, JBiol.Chem., 2007, 282, 37036-37044.
80) F.P.Bellinger, A.V.Raman, M.A.Reeves and M.J.Berry,
Biochem.J., 2009, 422, 11-22.
81) F.P.Bellinger, Q.P.He, M.T.Bellinger and Y.Lin,
J.Alzheimer’s Dis., 2008, 15, 465, 472.
82) F.W.Hoffmann, A.C.Hashimoto, L.A.Shafer, S.Dow,
M.J.Berry and P.R.Hoffmann, J.Nutr., 2010, 140, 1155-
1161.
83) F.W.Hoffmann, A.S.Hashimoto, B.C.Lee, A.H.Rose,
R.V.Shoet and P.R.Hoffmann, Arch.Biochem.Biophys.,
2011, 512, 38-44.
84) F.Wauquier, L.Leotoing, V.Coxam, J.Guicheux, Y.Wittrant,
Oxidative stress in bone remodeling and disease, Trends
Mol.Med. 15 (2009) 468-477.
85) F.Zamamiri-Davis, Y.Lu, J.T.Thompson, K.S.Prabhu,
P.V.Reddy, L.M.Sordillo, C.C.Reddy, Nuclear factor-kappa
B mediates over expression of cyclooxygenase-2 during
activation of RAW 264.7 macrophages in selenium
deficiency.Free Radic.Biol.Med. 32 (2002) 890-897.
86) F.Zhang, W.Yu, J.L.Hargrove, P.Greenspan,
R.G.Dean,E.W.Taylor,D.K.Hartle, inhibition of TNF-alpha
induced ICAM-1 VCAM-1 and E-selectin expression by
selenium, Atherosclerosis 161 (2002) 381-386.
87) F.Zorzato, H. Jungbluth, H. Zhou, F.Muntoni and
S.Treves,IUMB life, 2007,59, 14-20.
88) Feigl, F..,West, P.W.Test for selenium based on a catalyc
effect. Anal.Chem. 1947, 19, 351-353.
89) Flohe, L.The labour pains of biochemical selenology:The
history of selenoprotein biosynthesis.Biochim.Biophys.Acta
2009,1790, 1389-1403.
90) Forstrom, J.W..,Zakowki, J.J..,Tappel, A.L.Identification of
the catalytic site of rat liver glutathione peroxidase as
selenocysteine.Biochemistry 1978, 17, 2639-2644.
91) Franke, K.W. (1934) A new toxicant occurring naturally in
certain samples of plant foodstuffs.I.Results obtained in
preliminary feeding trials.J.nutr. 8,597-608

59
92) G.Ballihaut, L.E.Kilpatrick, E.L. Kilparick and
W.C.Davis,Metallomics, 2012, 4, 533-538.
93) G.E. Olson, V. P. Winfrey, S.K.NagDas, K.E.Hill and R.F.
Burk,J.Biol.Chem., 2007, 282, 12290-12297.
94) G.E.Olson, V.P.Winfrey, K.E. Hill and
R.F.Burk,J.Biolo.Chem., 2008, 283, 6854-6860.
95) G.F.Combs Jr., L.C.Clark, B.W.Turnbull, An analysis of
cancer prevention by selenium, Biofactors 14 (2001) 153-
159.
96) G.G.Ortiz, F.P.Pacheco-Moisès, M.A.Macias-Islas,
L.J.Flores-Alvarado, M.A.Mireles-Ramirez, E.D.Gonzalez-
Renovato, V.E.Hernandez-Navarro, A.L.Sanchez-Lopez and
M.A.Alatorre-Jimenez, Arch.Med.Res., 2014, 45, 687-697.
97) G.Roy, B.K.Sarma, P.P. Phadnis and G.Mugesh ,
J.Chem.Sci., 2005, 117, 287-303.
98) G.S.Gaki and A.G.Papavassiliou, NeuroMol.Med., 2014, 16,
217-230.
99) G.V.Krykov, R.A.Kumar, A.K oc, Z.Sun and
V.N.Gladyshev, Proc.Natl.Acad.Sci. U.S.A., 2002, 99,
4245-4250.
100) G.W.Snider, E.Ruggles, N.Khan, R.J.Hondal,
Selenocysteine confers resistance to inactivation by
oxidation in thioredoxin reductase: comparison of selenium
and sulfur enzymes,Biochemistry 52 (2013) 5472-5481.
101) G.X.Li, H.J.Lee, Z.Wang, H.Hu, J.D.Liao, J.C.Watts,
G.F.Combs, Jr., Lu, Superior in vivo inhibitory efficacy of
methylseleninic acid against human prostate cancer over
selenomethionine or selenite, Carcinogenesis 29
(2008)1005-1012.
102) G.Yang, S.Wang, R.Zhou and S.Sun, Am.j.Clin.Nutr.,
1983, 37, 872-881.
103) Gilmore,T.D.Introduction to NF-kappaB: Players,
pathways,perspectives.Oncogene 2006, 25, 6680-6684.
104) Gorrini, C.,Harris, I.S..,Mak,T.W.Modulation of
oxidative stress as an anticancer strategy.Nat.Rev.Drug
Discov. 2013, 12, 931-947.
105) Guimares, M.J.., Peterson, D..,Vicari, A..,Cocks,
B.G..,Copeland, N.G.., Gilbert, D.J..,Jenkins, N.A..,Ferrick,
D.A..,Kastelein, R.A..,Bazan, J.F.., et al.Identification of a
novel seld homolog from eukaryotes, bacteria, and archaea:
Is there an autoregulatory mechanism is selenocysteine

60
metabolism? Proc.Natl.Acad.Sci. USA 1996, 93, 15086-
15091.
106) H. Imai and Y.Nakagawa, Free Radical Biol. Med.,
2003, 34, 145-169.
107) H.-C.Kim, W.-KJhoo, E.-J.Shin and G.Bing, Brain
Res., 2000, 862, 247-252.
108) H.E.Ganther, Selenium metabolism, selenoproteins
and mechanism of cancer prevention: complexities with
thioredoxin reductase, Carcinogenesis 20 (1999) 1657-1666.
109) H.Imai,K.Suzuki, K.Ishizaka, S.Ichinose, H.Oshima,
I.Okayasu, K.Emoto, M.Umeda and Y.Nakagawa,
Biol.Reprod., 2001,64, 674-683.
110) H.Misu, T.Takamura, H.Takayama, H.Hayashi,
N.Matsuzawa-Nagata, S.Kurita, K.Ishikura, H. Ando,
Y.Takeshita, T.Ota, M.Sakurai, T.Yamashita, E.Mizukoshi,
M.Honda, K.Miyamoto, T.Kubota, N.Kubota, T.Kadowaki,
H.J.Kim, I.K.Lee, Y.Minokoshi, Y.Saito, K.Takahashi,
Y.Yamada, N.Takakura and S.Kaneko, Cell.Metab., 2010,
12, 483-495.
111) H.Sies, Eur.J.Biochem.215(1993) 213-219.
112) H.Tapiero, D.M.Townsend and K.D.Tew,
Biomed.Pharmacother., 2003, 57, 134-144.
113) H.Ueno, H.Kjihara, H.Nakamura, J.Yodoi,
K.Nakamuro, Contribution of thioredoxin reductase to T-
Cell mitogenesis and NF-kappa B DNA-binding promoted
by selenite, Antioxid.Redox Signal.9 (2007) 115-121.
114) H.Vunta, F.Davis, U.D.Palempalli, D.Bhat, R.J.Arner,
J.T.Thompson, D.G.Peterson, C.C.Reddy, K.S.Prabhu, The
anti-inflammatory effects of selenium are mediated through
15-deoxy-Delta 12, 14- prostaglandin j2 in macrophages,
J.Biol.Chem. 282 (2007) 17964-17973.
115) H.Vural, H.Demirin, Y.Kara, I.Eren and N.Delibas,
J.Trace Elem.Med.Biol., 2010, 24, 169-173.
116) H.Xiao, K.L.Parkin, Induction of phase Il enzyme
activity by various selenium compounds, Nutr.Cancer 55
(2006) 210-223.
117) H.Y.Kim and V.N.Gladyshev, Biochem.J., 2007, 407,
321-329.
118) H.Y.Kim and V.N.Gladyshev, Mol.Biol.Cell, 2004,
15, 1055-1064.

61
119) H.-Y.Kim and V.N.Gladyshev, PLos Biol., 2005, 3,
e375.
120) H.Y.Won, J.H.Sohn, H.J.Min, K.Lee, H.A.Woo,
Y.S.Ho, J.W.Park, S.G.Rhee and E.S.Hwang,
Antioxid.Redox Signaling, 2010, 13, 575-587.
121) H.Zeng, J.J.Cao, G.F.Combs Jr., Selenium in bone
health: roles in antioxidant protection and cell proliferation,
Nutrients 5 (2013) 97-110.
122) Halliwell, B.Biochemistry of oxidative
stress.Biochem.Soc.Trans. 2007, 35, 1147-1150.
123) Hasegawa, T..,Mihara, M..,Nakamuro, K..,Sayato,
Y.Distribution and chemical form of selenium in mice after
administration of selenocysteine.Biol.Pharm Bull. 1994, 17,
1215-1219.
124) Hatfield. D L. et al.,eds (2012)selenium:Its Molecular
Biology and role in human health.Springer science+business
media
125) Hoefig, C.S..,Renko, K.Kohrle, J..,Birringer,
M..,Schomburg, L.Comparison of different
selenocompounds with respect to nutritional value vs.
Toxicity using liver cells in culture. J.Nutr.Biochem. 2011,
22, 945-955.
126) I.Tasset, E.Aguera, F.Sanchez-Lopez, M.Feijoo,
A.I.Giraldo, A.H.Cruz, F.Gascon and I.Tunez,
Clin.Biochem., 2012, 45, 440-444.
127) J. Bellingham, K. Gregory-Evans, M.F.Fox and C.Y.
Gregory Evans, Biochim. Biophys. Acta, 2003, 1627, 140-
146.
128) J. Panee, Z.R. Stoytcheva, W.Liu and M.J.Berry,J.
Biol. Chem., 2007, 282, 23759-23765.
129) J.Bleys, A.Navas-Acien and E.Gallar, Diabetes Care,
2007, 30, 829-834.
130) J.D.Bogden and J.M.Oleske, Nutr. Res., 2007, 27, 69-
77.
131) J.Donovan, P.R.Copeland, Threading the needle :
getting selenocysteine into proteins, Antioxid.Redox Signal.
12 (2010) 881-892.
132) J.E.Curran,J.B.M.Jowett, K.S.Elliott, Y.Gao,
K.Gluschenko, J.Wang, D.M.A.Azim, G.Cai, M.C.Mahaney,
A.G.Comuzzie, T.D.Dyer, K.R.Walder, P.Zimmet,

62
J.W.MacCluer, G.R.Collier, A.H.K issebah and J.Blangero,
Nat.Genet., 2005, 37, 1234-1241.
133) J.E.Vance and G.Tasseva, Biochim. Biophys. Acta,
Mol.Cell. Biol. Lipids, 2013, 1831, 543-554.
134) J.H.Ellwanger, P.Molz, D.R.Dallemole, A.Pereira Dos
Santos, T.E.Muller, L.Cappelletti, M.Goncalves da Silva,
S.I.Rech Franke, D.Pra and J.A.Pegas Henriques, Nutrition,
2015, 31, 359-365.
135) J.J.Cao, B.R.Gregoire, H.Zeng, Selenium deficiency
decreases antioxidative capacity and is detrimental to bone
microarchitecture in mice, J.Nutr. 142 (2012) 1526-1531.
136) J.Lu, A.Holmgren, Selenoproteins, J.Biol.Chem. 284
(2009) 723-727.
137) J.M.Park, A.Kim, J.H.Oh, A.S.Chung,
Methylseleninic acid inhibits PMA-stimulated pro-MMP-2
activation mediated by MT1-MMP expression and further
tumor invasion through suppression of NF-kappaB
activation, Carcinogenesis 28 (2007) 837-847.
138) J.Molnar, Z.Garamvolgyi, M.Herold, N.Adanyi,
A.Somogyi and J.Rigo, Biol.Trace Elem.Res., 2008, 121,
16-22.
139) J.P.Vonsattel, R.H.Myers, T.J.Stevens, R.J.Ferrante,
E.D.Bird and E.P.Richardson jr., J.Neuropathol.Exp.Neurol.,
1985, 44, 559-577.
140) J.Paulsen, R.Ready, J.Hamilton, M.Mega and
J.Cummings, J.Neurol.., Neurosurg. Psychiatry., 2001, 71,
310-314.
141) J.R.Prohaska, H.E.Ganther, J.Neurochem.27 (1976)
1379-1387.
142) J.S.Chen, Asia Pac. J.Clin.Nutr., 2012, 21, 320-326.
143) Jaramillo, M.C..,Zhang, D.D.The emerging role of the
Nrf2-Keap1 signaling pathways in cancer.Genes Dev. 2013,
27, 2179
144) K. Ikematsu, R.Tsuda, S.Tsuruya and I.Nakasono,
Forensic Sci.Int., 2007, 169, 168-172.
145) K.E.Ben Jilani,J.Panee, Q. He, M.J. Berry and P.A.Li,
Int.J.BIOL. Sci., 2007, 3, 198-204.
146) K.E.Hill, J.D.Zhou, W.J.McMahan, A.K.Motley,
J.F.Atkins, R.F.Gesteland and R.F.Burk,J.Biol. Chem.,
2003, 278, 13640-13646.

63
147) K.E.Hill,J.D.Zhou, L.M.Austin, A.K.Motley,
A.J.L.Ham, G.E.Olson, J.F.Atkins, R.F.Gesteland and
R.F.Burk,J.Biol.Chem., 2007, 282, 10972-10980.
148) K.-H.Kim, Y.Gao, K.Walder, G.R.Collier, J.Skelton
and A.H.Kissebah, Biochem.Biophys.Res.Commun., 2007,
354,127-132.
149) K.Kljai and R.Runje, Biol.Trace Elem. Res., 2001, 83,
223-229.
150) K.Loh,H.Deng, A.Fukushima, X.Cai, B.Boivin,
S.Galic, C.Bruce, B.J.Shields, B.Skiba, L.M.Ooms,
N.Stepto, B.Wu,C.A.Mitchell, N.K.Tonks, M.J.Watt,
M.A.Febbraio,P.J.Crack, SAndrikopoulos and T.Tiganis,
Cell.Metab., 2009, 10, 260-272.
151) K.M. Barnes, J.K.Evenson, A.M.Raines and
R.A.Sunde,J.Nutr., 2009, 139, 199-206.
152) K.-M. Fang, F.-C Cheng, Y.-L Huang, S.-Y Chung,
Z.-Y.Jian and M.-C. Lin, Biol. Trace Elem.Res., 2013, 152,
66-74.
153) K.M.Brown, J.R.Arthur, Selenium, selenoproteins and
human health: a review, Public Health Nutr.4 (2001) 593-
599.
154) K.Rees, L.Hartley, C.Day, N.Flowers, A.Clarke and
S.Stranges, Cochrane Database Syst.Rev., 2013,
1,CD009671.
155) K.S.Prabhu, F.Zamamiri-Davis, Y.Lu, J.T.Thompson,
K.S.Prabhu, P.V.Reddy, L.M.Sordillo, C.C.Reddy, Selenium
deficiency increases the expression of inducibile nitric oxide
synthase in RAW 264.7 macrophages: role of nuclear factor-
kappaB in up-regulation, Biochem.j.366 (2002) 203-209.
156) K.Socha, J.Kochanowicz, E.Karpinska,
J.Soroczynska, M.Jakoniuk, Z.Mariak and M.H.Borawska,
Nutr.J., 2014, 13, 62.
157) K.T.Suzuki, C.Doi and N.Suzuki,
Toxicol.Appl.Pharmacol., 2006, 217, 185-195.
158) K.V. Korothov, S.V. Novoselov, D.L. Hatfield and
V.N. Gladyshev, Mol. Cell.Biol., 2002, 22, 1402-1411.
159) Kong, Q..,Beel, J.A.Lillehei,K.O.A threshold concept
for cancer therapy.Med.Hypotheses 2000, 55, 29-35.
160) Kryukov,
G.V..,Castellano,S..,Novoselov,S.V..,Lobanov,
A.V..,Zehtab,O..,Guigo, R..,Gladyshev,

64
V.N.Characterization of mammalian
selenoproteomes.Science 2003, 300, 1439-1443.
161) Kumar, B.S..,Kunwar,., Ahmad, A..,Kumbhare, L.B..,
Jain, V.K.., Priyadarsini, K.I.In vitro radioprotection studies
of organoselenium compounds:Differences between mono-
and diselenides.Radiat.Environ.Biophys. 2009, 48, 379-384.
162) Kumar, S..,Bjornstedt, M..,Holmgren, A.Selenite is a
substrate for calf thymus thioredoxin reductase and
thioredoxin and elicits a large non-stoichiometric oxidation
of NADPH in the presence of oxygen.Eur.J.Biochem.
1992,207, 435-439.
163) L.C.Christensen, N.W.Jensen, A. Vala,
J.Kamarauskaite, L.Johansson, J.R. Winther, K. Hofmann,
K. Teilum and L. Ellegard, J. Biol. Chem., 2012, 287,
26388-26399.
164) L.Flohe, Biol.Chem., 2007,388,987-995.
165) L.G.Raisz, Pathogenesis of osteoporosis: concepts,
conflicts, and prospects, J.Clin.Invest. 115 (2005) 3318-
3325.
166) L.Grumolato, H. Ghzili, M. Montero-Hadjadje,
S.Gasman, J.Lesage, Y. Tanguy, L.Galas, D.Ait-Ali,
J.Leprince, N.C. Guerineau, A.G. Elkahloun, A.Fournier,
D.Vieau, H.Vaudry and Y.Anouar, FASEB J., 2008,
22,1756-1768.
167) L.H.Duntas, Nat.Clin.Pract.Endocrinol.Metab., 2008,
4, 454-460.
168) L.H.Duntas,J.Clin.Endocrinol.Metab., 2010,95, 5180-
5188.
169) L.P.Rowland and N.A.Shneider, N.Engl.J.Med., 2001,
344, 1688-1700.
170) L.R.Ferguson, N.Karunasinghe, S.Zhu and
A.H.Wang, Mutat.Res., 2012, 733, 100-110.
171) L.Schomburg, A.M.Dumitrescu, X.H.Liao, B.Bin-
Abbas, J.Hoeflich, J.Konrle and S.Refetoff, Thyroid, 2009,
19,277-281.
172) L.Schomburg, U.Schweizer, B.Holtmann, L.Flohe,
M.Sendtner and J.Kohrle, Biochem. J., 2003, 370, 397-402.
173) L.Schomburg, U.Schweizer, J.Kohrle, Selenium and
selenoproteins in mammals: extraordinary, essential,
enigmatic, Cell.Mol.Life Sci. 61 (2004) 1988-1995.

65
174) L.V.Papp, A.Holmgren and K.K.Khanna, Antioxid.
Redox Signaling, 2010, 12, 793-795.
175) L.Zecca, A.Stroppolo, A.Gatti, D.Tampellini,
M.Toscani, M.Gallorini, G.Giaveri, P.Arosio,
P.Santambrogio and R.G.Fariello,
Proc.Natl.Acad.Sci.U.S.A., 2004, 101, 9843.
176) Labunskyy, V.M., Hatfield, D.L.Gladyshev,
V.N.Selenoproteins:Molecular pathway and physiological
roles.Physiol.Rev.2014,94, 739-777.
177) Labunskyy, V.M…, Hatfield, D.L.Gladyshev,
V.N.Selenoproteins: Molecular pathways and physiological
roles.Physiol.Rev. 2014, 94, 739-777.
178) Lane, H.W..,Teer, P..,Dukes, J..,Jonson, J..,White,
M.T.The effect of four chemical forms of selenium on
mammary tumor incidence in balb/c female mice treated
with 7-12-dimethylbenz(a)anthracene. Cancer Lett. 1990,
50, 39-44.
179) Lobanov, A.V..,Hatfield, D.L..,Gladyshev,
V.N.Eukaryotic selenoproteins and
selenoproteomes.Biochim.Biophys.Acta 2009, 1790, 1424-
1428.
180) M. Conrad, M. Schneider, A. Seiler and W.
Bornkamm Georg, Biol. Chem., 2007, 338, 1019.
181) M.A.Beck, H.K.Nelson, Q.Shi, P.Van Dael,
E.J.Schiffrin, S.Blum, D.Barclay and O.A.Levander, FASEB
J., 2001, 15,1846-1850.
182) M.A.Carvalho-Filho, M.Ueno, S.M.Hirabara,
A.B.Seabra, J.B.C.Carvalheira, M.G. de Oliveira,
L.A.Velloso, R.Curi and M.J.A.Saad, Diabetes, 2005, 54,
959-967.
183) M.A.Serdar,F.Bakir, A.Hasimi, T.Celik, O.Akin,
L.Kenar, O.Aykut and M.Yildirimkaya, Int.J.Diabetes Dev
Ctries, 2009, 29, 35-40.
184) M.Baqui, D.Botero, B.Gereben, C. Curcio,
J.W.Harney, D.Salvatore, K.Sorimachi, P.R. Larsen and
A.C. Bianco, J.Biol.Chem., 2003, 278, 1206-1211.
185) M.D.Ximena Burbano, P.Maria Jose Miguez-Burbano
Md, D.Kathryn McCollister Ph, M.D.Guoyan Zhang,
M.D.Allan Rodriguez, P.PHILLIP Ruiz Md, B.A.Robert
Lecusay and D.Gail Shor-Posner Ph, HIV Clin.Trials, 2002,
3, 483-491.

66
186) M.Dudkiewicz, T. Szczepinska, M. Grynberg and K.P
awlowski, PLos One, 2012, 7, e32138.
187) M.E. Moustafa and H.A. Antar, Biochem. Genet.,
2012, 50, 736-747.
188) M.G.M.Olde Rikkert, F.R.Verhey, J.W.C.Sijben,
F.H.Bouwman, P.L.J.Dautzenberg, M.Lansik,
W.M.W.Sipers, D.Z.B.van Asselt, A.M.J. van Hees,
M.Stevens, B.Vellas and P.Scheltens, J.Alzheimer’s Dis.,
2014, 41, 261-271.
189) M.J.Christensen, E.T.Nartey, A.L.Hada, R.L.Legg,
B.R.Barzee, High selenium reducesNF-kappaB- regulated
gene expression in uninduced human prostate cancer cells,
Nutr. Cancer 58 (2007) 197-204.
190) M.J.Jurynec, R.Xia, J.J.Mackrill, D.Gunther,
T.Crawford, K. M. Flanigan, J.J.Abramson, M.T.Howard
and D.J.Grunwald, Proc. Natl. Acad. Sci. U.S.A., 2008, 105,
12485-12490.
191) M.Kiline, M.A.Guven,M.Ezer, I.E.Ertas and
A.Coskun, Biol.Trace.Elem.Res., 2008, 123, 35-40.
192) M.Kuhbacher, J.Bartel, B.Hoppe, D.Alber, G.Bukalis,
A.U.Brauer, D.Behne, A.J.Kyriakopoulos, J.Neurochem.110
(2009) 133-142.
193) M.L.Vizuete, V.Steffen, A.Machado and J.Cano,
Eur.J.Pharmacol., 1994, 270, 183-187.
194) M.M.A. Baqui, B. Gereben,J.W.Harney, P.R.Larsen
and A.C.Bianco, Endocrinology, 2000, 141 4309-4312.
195) M.Malinouski, S.Kehr, L.F inney, S. Vogt, B. A.
Carlson, J. Seravalli, R.Jin, D. E. Handy, T.J. Park, J.
Loscalzo, D.L. Hatfield and V.N. Gladishev, Antioxid.
Redox Signaling, 2012, 16, 185-192.
196) M.Mehrpour, A.Kyani, M.Tafazzoli, F.Fathi and
M.T.Joghataie, Magn.Reson. Chem., 2013, 51, 102-109.
197) M.Navarro-Alarcon and C. Cabrera- Vique, Sci. Total
Environ., 2008, 400, 115-141.
198) M.Olsson, B.Olsson,, P.Jacobson, D.S.Thelle,
J.Bjorkegren, A.Walley, P.Froguel, L.M.S.Carlsson and
K.Sjoholm, Metab.,Clin. Exp., 2011, 60, 114-120.
199) M.P.Rayman, Br.J.Nutr., 2008, 100, 254-268
200) M.P.Rayman, Lancet, 2012, 379, 1256-1268
201) M.Perreault and A.Marette, Nat.Med., 2001, 7, 1138-
1143.

67
202) M.Rivera, A.de Souza, A.Moreno, S.Xavier, J.Gomes,
M.Rocha, R.Correa-Oliveira, J.Neve, J.Vanderpas and
T.Araujo-jorge, Am.J.Trop.Med.Hyg., 2002, 66, 706-712.
203) M.Roman, A.Lapolla, P.Jitaru, A.Sechi, C.Cosma,
G.Cozzi, P.Cescon, and C.Barbante, Transi.Res., 2010, 156,
242-250.
204) M.Taccone-Gallucci, A.Noce, P.Bertucci, C.Fabbri,
S.Manca-di-Villahermosa, F.R. Della-Rovere, M.De
Francesco, M.Lonzi, G.Federici, F.Scaccia and M.Dessì,
J.Trace Elem.Med.Biol., 2010, 24, 27-30.
205) M.V.Kasaikina, D.L.Hatfield. V.N.Gladyshev,
Understanding selenoprotein function and regulation through
the use of rodent models, Biochim.Biophys.Acta 1823
(2012) 1633-1642.
206) M.V.Kasaikina,D.L.H atfield and V.N.Gladyshev,
Biochim.Biophys. Acta, Mol.Cell Res., 2012, 1823, 1633-
1642.
207) M.Vinceti, D.Guidetti, M.Pinotti, S.Rovesti,
M.Merlin, L.Vescovi, M.Bergomi and G.Vivoli,
Epidemiology, 1996, 7, 529-532.
208) M.Vinceti, F.Bonvicini, K.J.Rothman, L.Vescovi and
F.Wang, Environ, Health, 2010, 9, 77.
209) M.Vinceti, N.Solovyev, J.Mandrioli, C.M.Crespi,
F.Bonvicini, E.Arcolin, E.Georgoulopoulou and B.Michalke,
Neurotoxicology, 2013, 38, 25-32.
210) Madison, T.C.(1860) sanitary report – Fort Randall.In
Statical Report on the sickness and Mortality in the army of
the united states (Coolidge, R.H., ed).pp., 37- 41, 36 th
congress senate exchange document
211) Milella, M..,Falcone, I..,Conciatori, F..,Incani,
U.C..,del Curatolo, A..,Inzerilli, N..,Nuzzo, C.M..,Vaccaro,
V..,Vari, S..,Cognetti, F..,et al.PTEN:Multiple Functions in
Human Malignant Tumors.Front.Oncol. 2015, 5, 24.
212) Moghadaszadeh, B..,Beggs, A.H.Selenoproteins and
their impact on human health through diverse physiological
pathways.Physiology (Bethesda) 2006, 21, 307-315.
213) N.F Kolachi, T.G.Kazi, H.I.Afridi, N.G.Kazi and
S.Khan, Clin.Nutr., 2012, 31, 967-973.
214) N.Fradejas, M.D.Pastor, S.Mora-Lee, P.Tranque and
S.Calvo,J.Mol.Neurosci., 2008, 35, 259-265.

68
215) N.L. Chanaday, A.F. de Bem and G.A.Roth,
Neurochem.Int., 2011, 59, 1155-1162.
216) N.Li,Z.Gao, D.Luo,X.Tang, D.Chen and Y.Hu, Sci.
Total Environ., 2007, 381, 105-111.
217) N.M.Corcoran, D.Martin, B.Hutter-Paier,
M.Windisch, T.Nguyen, L.Nheu, L.E.Sundstrom,
A.J.Costello andC.M.Hovens,J.Clin.Neurosci., 2010,17,
1025-1033.
218) N.Petit, A.Lescure, M. Rederstorff, A. Krol, B.
Moghadaszadeh, U.M.Wewer and P.Guicheney, Hum. Mol.
Genet., 2003, 12, 1045-1053.
219) N.Y.Alwahaibi, S.B.Budin, J.Mohamed,
A.Alhamdani, Nuclear factor-kappa B as a promising target
for selenium chemoprevention in rat hepatocarcinogenesis,
J.Gastroenterol.Hepatol. 25 (2010) 786-791.
220) O.A. Levander and P.D.Whanger, J. Nutr., 1996, 126,
2427S-2434S.
221) Ost adalovà, I..,Babicky, A.Toxic effect of various
selenium compounds on the rat in the early postnatal period.
Arch. Toxicol. 1980, 45, 207-211.
222) P. Castets, A. Lescure, P. Guicheney and V.
Allamand, J. Mol. Med., 2012, 90, 1095-1107.
223) P.H.Reddy, Brain Res., 2011, 1415, 136-148.
224) P.H.Whiting, A.Kalansooriya, I.Holbrook, F.Haddad
and P.E.Jennings, Br.J.Biomed.Sci., 2008, 65, 71-74.
225) P.Hoffmann, Arch.Immunol.Ther.Exp., 2007, 55,
289-297.
226) P.Mao and P.H.Reddy,
Biochim.Biophys.Acta,Mol.Basis Dis., 2011, 1812, 1359-
1370.
227) P.P.Tak, G.S.Firestein, NF-kappaB: a Key role in
inflammatory diseases, J.Clin.Invest. 107 (2001) 7-11.
228) P.Pemberton, A.Smith and T.Warnes,
Scand.J.Gastroenterol., 2005, 40, 1102-1108.
229) P.W.Tsao, T.Suzuki, R.Totsuka, T.Murata, T.Takagi,
Y.Ohmachi, H.Fujimura, I.Takata, The effect of
dexamethasone on the expression of activated NF-kappaB in
adjuvant arthritis, Clin.Immunol.Immunopathol. 83 (1997)
173-178.

69
230) Perkins, N.D.Integrating cell-signalling pathways with
NF-kappaB and IKK function.Nat.Rev.Mol.Cell Biol. 2007,
8, 49-62.
231) Planchon, S.M..,Waite, K.A..,Eng, C.The nuclear
affairs of PTEN.J.Cell.Sci.2008, 121, (pt 3) 249-253.
232) R. Brigelius-Flohé and A. Kipp, Biochim. Biophys.
Acta, 2009, 1790, 1555-1568
233) R. Hurst, C.N.Armah,J.R.Dainty,D.J.Hart,
B.Teucher,A.J. Goldson, M.R.Broadley, A.K.Motley and
S.J. Fairweather-Tait,Am.J.Clin.Nutr., 2010, 91, 923-931.
234) R.A.Sunde, A.M. Raines, K.M. Barnes and J.K.
Everson, Biosci. Rep., 2009, 29, 329-338.
235) R.Arrojo e Drigo and A.C.Bianco, Int.J.Biochem.Cell
Biol., 2011, 43, 1432-1441.
236) R.Burk and O.Levander, in Modern Nutrition in
Health and Disease, ed.M.E.Shils, M.Shike, A.C.Ross,
B.Caballero and R.J.Cousins, Lippincott Williams &
Wilkins, Philadelphia, US,2005, pp.313-326.
237) R.Ebert, F.Jakob, Selenium deficiency as a putative
risk factor for osteoporosis, Int. Congr.Ser. 1297 (2007) 158-
164.
238) R.F. Burk, G. E. Olson, V. P. Winfrey, K. E. Hill and
D.P. Yin, Am.J. Phisyol.Gastrointest. Liver Physiol., 2011,
301, G32-G38.
239) R.F.Burk and K.E.Hill, Biochim. Biophys. Acta,
2009, 1790, 1441-1447.
240) R.F.Burk, B.K. Norsworthy, K.E. Hill, A.K.Motley
and D.W.Byrne, Cancer Epidemiol.Biomarkers Prev., 2006,
15, 804-810.
241) R.F.Burk, K.E.Hill and A.K. Motley, J.Nutr., 2003,
133, 1517S-1520S.
242) R.F.Burk, K.E.Hill, A.K.Motley, V.P.Winfrey,
S.Kurokawa, S.L.Mitchell and W.Zhang, FASEB J., 2014,
28, 3579-3588.
243) R.Gndhi, A.Laroni and H.L.Weiner,
J.Neuroimmunol., 2010, 221, 7-14.
244) R.Gonzalez-Dominguez, T.Garcia-Barrera and
J.L.Gomez-Ariza, BioMetals, 2014, 27, 539-549.
245) R.H.L.H.Rueli. A.C.Parubrub, A.S.T.Dewing, A.C.
Hashimoto, M.T.Bellinger, E.J.Weeber, J.H.Uyehara-Lock,

70
L.R.White, M.J.Berry and F.P.Bellinger, J. Alzheimer’s
Dis., 2015, 44, 379-383.
246) R.H.Swerdlow, J.M.Burns and S.M.Khan,
Biochom.Biophys.Acta, Mol.Basis Dis, 2014, 1842, 1219-
1231.
247) R.Irons, B.A.Carlson, D.L.Hatfield, C.D.Davis, Both
selenoproteins and low molecular weigh selenocompounds
reduce colon cancer risk in mice with genetically impaired
selenoprotein expression, J.Nutr. 136 (2006) 1311-1317.
248) R.J.Hondal, E.I.Ruggles, Differing views of the role
of selenium in thioredoxin reductase, Amino Acids 41
(2011) 73-89.
249) R.K.Shrimali, R.D.Irons, B.A.Carlson, Y.Sano,
V.N.Gladyshev, J.M.Park and D.L.Hatfield, J.Biol.Chem.,
2008, 283, 20181-20185.
250) R.Koishi, T.Nakamura, T.Takazawa, C.Yoshimura,
N.Seriwava, Production of functional human selenocysteine-
containing KDRF/thioredoxin reductase in E.Coli,
J.Biochem. 127 (2000) 977-983.
251) R.Kupka, G.I.Msamanga, D.Spiegelman, S.Morris,
F.Mugusi, D.L.Hunter and W.W.Fawzi, J.Nutr., 2004, 134,
2556-2560.
252) R.Moreno-Reyes, D.Egrise, J.Nève, J.L.Pasteels,
A.Schoutens, Selenium deficiency-induced growth
retardation is associated with an impaired bone metabolism
and osteopenia, J.Bone Miner. Res. 16 ( 2001) 1556-1563.
253) R.Schnabel and S.Blankenberg, Circulation, 2007,
116, 1338-1340.
254) R.Seetharaman, A.L.Mora, G.Nabozny, M.Boothby,
J.Chen, Essential role of T cell NF-kappaB activation in
collagen-induced arthritis, J.Immunol. 163(1999) 1577-
1583.
255) R.Sengupta and A.Holmgren, Biochim.Biophys. Acta,
2012, 1820, 689-700.
256) R.Sunde, in Biochemical and Physiological Aspects
of Human Nutrition, ed.M. Stipanuk, W.B. Saunders
Company, New York 2000, pp. 782-809.
257) Reilly. C. (ed.)(1996) selenium in Food and Health,
Chapman and Hall

71
258) Rhead, W.J.,Schrauzer,G.N.The selenium catalyzed
reduction of methylene blue by thiols.Bioinorg.Chem. 1974,
3, 225-242.
259) S. Apostolou, J. O. Klein, Y Mitsuuchi,J.N.Shetler,
P.I. Poulikakos, S.C. Jhanwar, W.D. Kruger and J.R. Testa,
Oncogene, 2004, 23, 5032-5040.
260) S.Bar-Noy, S.N.Gorlatov, T.C.Stadman,
Overexpression of wild type and SeCys/Cys mutant of
human thioredoxin reductase in E.Coli: the role of
selenocysteine in the catalytic activity, Free Radic.Biol.Med.
30 (2001) 51-61.
261) S.C.Manolagas, From estrogen-centric to aging and
oxidative stress: a revised perspective of the pathogenesis of
osteoporosis, Endocr. Rev.31 (2010) 266-300.
262) S.Dhingra and M.P.Bansal, Biol.Res., 2006, 39, 307-
319.
263) S.Florian, K. Wingler, K. Schmehl, G. Jacobasch, O.J.
Kreuzer, W. Meyerhof and R. Brigelius-Flohé,Free Radical
Res., 2001, 35, 655-663.
264) S.G.Ma, K.E.Hill, R.F. Burk and R.M.
Caorioli,Biochemistry, 2003, 42, 9703-9711.
265) S.G.Ma, K.E.Hill, R.M.Caprioli and R.F.Burk,J.
Biol.Chem., 2002, 277, 12749-12754.
266) S.Gromer,J.Eubel, B.Lee and J.Jacob, Cell. Mol. Life
Sci., 2005, 62, 2414-2437.
267) S.Haider, S.Saleem, T.Perveen, S.Tabassum,
Z.Batool, S.Sadir, L.Liaquat and S.Mdiha, Age, 2014, 36,
1291-1302.
268) S.Juliger, H.Goenaga-infante,
T.A.Lister,J.Fitzgibbon,S.P.Joel, Chemosensititization of B-
cell lymphomas by methylseleninic acid involves nuclear
factor- kappaB inhibition and the rapid generation of other
selenium species, Cancer Res.67 (2007) 10984-10992.
269) S.Ljubisavljevic, I.Stojanovic, T.Cvetkovic,
S.Vojinovic, D.Stojanov, D.Stojanovic, V.Bojanic,
D.Stokanovic and D.Pavlovic, NeuroImmunoModulation,
2014, 21, 13-20.
270) S.M.Lippmann, E.A.Klein, P.J.Goodman, M.S.Lucia,
I.M.Thompson, L.G.Ford, H.L.Parnes,
L.M.Minasian,J.M.Gaziano,J.A.Hartline, J.K.Parsons,
J.D.Bearden, E.D.Crawford,G.E.Goodmann,J.Claudio,

72
E.Winquist, E.D.Cook, D.D.Karp, P.Walther, M.M.Lieber,
A.R.Kristal, A.K.Darke, K.B.Arnold, P.A.Ganz,
R.M.Santella, D.Albanes, P.R.Taylor, J.L.Probstfield,
T.J.Jagpal, J.J.Crowley, F.L.Meyskens, L.H.Baker and
C.A.Coltman, JAMA,J.Am.Med.Assoc., 2009, 301, 39-51.
271) S.Stewart, M.Prince, M.Bassedine, M.Hudson,
O.James, D.Jones, C.Record and C.P.Day, J. Hepatol., 2007,
47, 277-283.
272) S.Stranges, J.R.Marshall, R.Natarajan, R.P.Donahue,
M.Trevisan, G.F.Combs, F.P.Cappuccio, A.Ceriello
andM.E.Reid, Ann.Intern.Med., 2007, 147, 217-223.
273) S.Tanguy, A.Rakotovao, M.G.Jouan, C.Ghezzi,J. de
Leiris and F.Boucher, Mol.Nutr.Food Res., 2011, 55, 522-
529.
274) S.V.Novoselov, G.V.K ryukov, X.-M.Xu, B.A.
Carlson, D.L. Hatfield and V.N.Gladyshev, J.Biol. Chem.,
2007, 282, 1190-11968.
275) Schwarz, K. and Foltz, C.M.(1958) factor 3 activity of
selenium compound.J.Biol.chem. 233, 245-251
276) Seko, Y..,Imura, N.Active oxygen generation as a
possible mechanism of selenium toxicity,
Biomed.Environ.Sci. 1997, 10, 333-339.
277) Seko, Y..,Saito, Y..,Kitahara, J..,Imura, N.Active
oxygen generation by the reaction of selenite with reduced
glutathione in vitro. In Selenium in Biology and Medicine,
Wendel, A..,Ed..,Springer Berlin Heidelberg:Tubigen,
Germany, 1989.,pp. 70-73.
278) Semenza, G.L.Targeting HIF-1for cancer
therapy.Nat.Rev.Cancer 2003, 3, 721-732.
279) Spallholz, J.E..,Shriver, B.J..,Reid,
T.W.Dimethyldiselenide and methylseleninic acid generate
superoxide in an in vitro chemiluminescence assay in the
presence of glutathione: Implication for the anticarcinogenic
activity of L- selenomethionine and L-
methylselenocysteine.Nutr.Cancer 2001, 40, 34-41.
280) Spallholz, J.E..,Whittam, J.H.Selenium toxicity
interpred from biological, catalytic, chemilurninescent and
scanning electron microscopic data. In Proceeding of the
Fifth International Symposium on Selenium in Biology and
Medicine, Nashville, TN,USA, 20-23 july 1992.

73
281) Spallholz, J.E.On the nature of selenium toxicity and
carcinostatic activity.Free Radic.Biol.Med. 1994, 17, 45-64.
282) T.Ago and J.Sadoshima,J.Mol.Cell.Cardiol., 2006, 41,
762-773.
283) T.-C. Ju, H.-M. Chen, Y.-C. Chen, C.-P. Chang,
C.Chang and Y.Chern, Biochim.Biophys. Acta, Mol.Basis
Dis., 2014, 1842, 1668-1680.
284) T.D.Buckman, M.S.Sutphin, C.D.Eckhert, Biochim.
Biophys. Acta 1163 (1993) 176-184.
285) T.Ishrat, K.Parveen, M.M.Khan, G.Khuwaja,
M.B.Khan, S.Yousuf, A.Ahmad, P.Shrivastav and F.Islam,
Brain Res., 2009, 1281, 117-127.
286) T.N.Akbaraly, J.Arnaud, M.P.Rayman, I.Hininger-
Favier,A.M.Roussel, C.Berr and
A.Fontbonne,J.Nutr.Metab., 2010, 7, 21.
287) T.Suda, N.Takahashi, N.Udagawa, E.Jimi,
M.T.Gillespie, T.J.Martin, Modulation of osteoclast
differentiation and function by the new members of the
tumor necrosis factor receptor and ligand families,
Endocr.Rev.20 (1999) 345-357.
288) T.Tamura, S.Yamamoto, M.Takahata, H.Sakaguchi,
H.Tanaka, T.C.Stadtman and K.Inagaki, Proc.Natl.Acad.Sci.
U.S.A., 2004, 101, 16162-16167.
289) T.Tomita, E.Takeuchi, N.Tomita, R.Morishita,
M.Kaneko, K.Yamamoto, T.Nakase, H.Seki, K.Kato,
Y.Kaneda, T.Ochi, Suppressed severity of collagen-induced
arthritis by in vivo transfection of nuclear factor kappaB
decoy oligodeoxynucleotides as gene therapy, Arthritis
Rheum 42 (1999) 2532-2542.
290) T.Ueta, T.Inoue, T. Furukawa, Y. Tamaki, Y.
Nakagawa, H.Imai and Y. Yanagi,J.Biol. Chem., 2012, 287,
7675-7682.
291) Trachootham, D..,Alexandre, J..,Huang, P.Targeting
cancer cells by ros-mediated mechanism:A radical
therapeutic approach?Nat.Rev.Drug Discov. 2009, 8, 579-
591.
292) Tsen, C.C..,Tappel,A.L.Catalytic oxidation of
glutathione and other sulfhydryl compounds by
selenite.J.Biol.Chem. 1958, 233, 1230-1232.
293) U.G.Gandhi, N.Kaushal, K.C.Ravindra, S.Hegde,
S.M.Nelson, V.Narayan, H.Vunta, R.F.Paulson, K.S Prabhu,

74
Selenoprotein-dependent up-regulation of hematopoietic
prostaglandin D2 synthase in macrophages is mediated
through the activation of peroxisome proliferator-actived
receptor (PPAR) gamma, J.Biol.Chem. 286 (2011) 27471-
27482.
294) U.S.Departement of Agriculture, Agricultural
Research Service, USDA National Nutrient Database for
Standard Reference, Release 25.
http://www.ars.usda.gov/ba/bhnrc/ndl, accessed on
07/06/2013
295) U.S.Department of Health and Human Service,
Toxicological Profile for Selenium, Agency for Toxic
Substances and Disease Registry Division of Toxicology and
Environmental Medicine, Atlanta, U.S.A., 2003.
296) V.B.Vrca, F.Skreb, I.Cepelak, Z.Romic and
L.Mayer,Clin.Chim.Acta, 2004, 341, 55-63.
297) V.M. Labunskyy, A.D. Ferguson, D.E. Fomenko, Y.
Chelliah, D.L. Hatfield and V.N. Gladyshev, J.Biol.Chem.,
2005, 280, 37839-37845.
298) V.M.Labunskyy, D.L. Hatfield and V.N. Gladyshev,
IUBMB Life, 2007, 59, 1-5.
299) W.C.Hawkes, J.Clin.Endocrinol.Metab., 2004, 89,
4772-4773.
300) W.J.Boyle, W.S.Simonet, D.L.Lacery. Osteoclast
differentiation and activation, Nature 423 (2003) 337-342.
301) W.M.EI-Sayed, T.Aboul-Fadl, J.G.Lamb,
J.C.Roberts, M.R.Franklin, Effect of selenium- containing
compounds on hepatic chemoprotective enzymes in mice,
Toxicology 220 (2006)179-188
302) Wallenberg, M.., Misra, S..,Wasik, A.M.., Marzano,
C..,Bjornstedt,M..,Gandin, V..,Fernandes, A.P.Selenium
induces a multi-targeted cell death process in addition to Ros
formation.J.Cell.Mol.Med. 2014, 18, 671-684.
303) Wallenberg, M..,Olm, E..,Hebert, C..,Bjornstedt,
M.Fernandes, A.P.Selenium compounds are substrates for
glutaredoxins: A novel pathway for selenium metabolism
and a potential mechanism for selenium-mediated
cytotoxicity. Biochem.j. 2010, 429, 85-93.
304) Weekley, C.M..,Harris, H.H.Which form is that?The
importance of selenium speciation and metabolism in the

75
prevention and treatment of disease.Chem.Soc.Rev. 2013,
42, 8870-8894.
305) Weisberger, A.S..,Suhrland, L.G.Studies on analogues
of L-cysteine and L-cystine.3.The effect of selenium cystine
on leukemia. Blood 1956, 11, 19-30.
306) Wondrak, G.T.Redox-directed cancer therapeutics:
Molecular mechanism and opportunities. Antioxid.Redox
Signal. 2009, 11, 3013-3069.
307) X.C.Bai, D.Lu,A.L.Liu, Z.M.Zhang, X.M.Li,
Z.P.Zou, W.S.Zeng, B.L.Cheng, S.Q.Luo, Reactive oxygen
species stimulates receptor activator of NF-kappaB ligand
expression in osteoblast, J.Biol.Chem. 280 (2005) 17497-
17506.
308) X.Du, H.Li, Z.Wang, S.Qiu, Q.Liu and J.Ni,
Metallomics, 2013, 5, 861-870.
309) X.Du, Y.Zheng, Z.Wang, Y Chen, R.Zhou, G.Song,
J.Ni and Q.Liu, Inorg. Chem., 2014, 53, 11221-11230.
310) X.Du, Z.Wang, Y.Zheng, H.Li, J.Ni and Q.Liu,
Inorg.Chem., 2014, 53, 1672-1678.
311) X.M.Xu, B.A. Carlson, R.Irons, H.Mix, N.Zhong,
V.N.Gladyshev and D.L.Hatfield, Biochem. J., 2007, 404,
115-120.
312) X.Wang, M.Vatamaniuk, S.Wang, C.Roneker,
R.Simmons and X.Lei, Diabetologia, 2008, 51, 1515-1524.
313) Y. Gao. H. C Feng K. Walder, K. Bolton, T.
Sunderland, N.Bishara, M. Quick,L. Kantham and G.R.
Collier, FEBS Lett., 2004, 563, 185-190.
314) Y. H. Ye, Y. Shibata, C. Yun, D. Ron and T. A.
Rapoport,Nature, 2004, 429, 841-847.
315) Y. Horibata and Y.Hirabayashi, J.Lipid Res., 2007,
48, 503-508.
316) Y.Gao, N.R.F.Hannan, S.Wanyonyi,
N.Konstantopolous, J.Pagnon, H.C.Feng,J.B.M.Jowett, K.-
H.Kim, K.Walder and G.R.Collier, Cytokine, 2006, 33, 246-
251.
317) Y.-J.Kim, Y.-G.Chai and J.-C. Ryu, Biochem.
Biophys. Res. Commun., 2005, 330, 1095-1102.
318) Y.Ohta and K.T. Suzuki, Toxicol. Appl. Pharmacol.,
2008, 226, 169-177.
319) Y.Tanguy, A.Falluel-Morel, S. Arthaud, L. Boukhzar,
D.-L.Manecka, A. Chagraoui, G.Prevost, S.Elias, I.Dorval-

76
Coiffec, J.Lesage, D.Vieau, I.Lihrmann, B.Jègou and Y.
Anouar,Endocrinology, 2011, 152, 4322-4335.
320) Y.Yoshiyama, V.M.Y.Lee and J.Q.Trojanowski,
J.Neurol., Neurosurg.Psychiatry, 2013, 84, 784-795.
321) Y.Zhang, Y. Zhou, U. Schweizer, N.E. Savaskan, D.
Hua, J. Kipnis, D.L.Hatfield and V.N.G ladyshev,
J.Biol.Chem., 2008, 283, 2427-2438.
322) Yin, M.B..,Li, Z.R..,Toth, K..,Cao, S..,Durrani,
F.A..,Hapke, G..,Bhattacharya, A.., Azrak, R.G..,Frank,
C..,Rustum, Y.M.Potentiation of irinotecan sensivity by se-
methylselenocysteine in an in vivo tumor model is
associated with downregulation of cyclooxygenase-2,
inducibile nitric oxide synthase and hypoxia-inducible factor
1 alpha expression, resulting in reduced angiogenesis.
Oncogene 2006, 25, 2509-2519.
323) Z.Erbayraktar, O.Yflmaz, A.E.T.Artmann, R.Cehreli
and C.Coker, Biol.Trace Elem. Res., 2007, 118, 217-226.
324) Z.Han, D.L.Boyle, A.M.Manning, G.S.Firestein, AP-1
and NF-kappaB regulation in rheumatoid arthritis and
murine collagen- induced arthritis, Autoimmunity 28 (1998)
197-208.
325) Z.Huang, A.H.Rose and P.R.Hoffmann,
Antioxid.Redox Signaling, 2012, 16, 705-743.
326) Z.Huang, F.W.Hoffmann, R.L. Norton, A.C.
Hashimoto and P.R. Hoffmann, J.Biol.Chem., 2011, 286,
34830-34838.
327) Z.Lu, E.Marks, J.Chen, J.Moline, L.Barrows,
M.Raisbeck, I.Volitakis, R.A.Cherny, V.Chopra, A.I.Bush,
S.Hersch and J.H.Fox, Neurobiol.Dis., 2014, 71C, 34-42.
328) Z.Wang. C.Jiang, J.Lu, induction of caspase-mediated
apoptosis and cell-cycle G1 arrest by selenium metabolite
methylselenol,, Mol.Carcinog. 34 (2002) 113-120.
329) Review: Alan M.Diamond The Subcellular Location
of Selenoproteins and the impact on Their Function ,
nutrients 2015, 7, 3938-3948; doi
:10.3390/nu7053938.http://www.mdpi.com/journal/nutrients
accessed on 15/05/2015.
330) Review: Barbara Rita Cardoso, Blaine R.Roberts,
Ashley L.Bush and Dominic J.Haret Selenium,
selenoproteins and neurodegenerative diseases. http://
www.rsc.org/metallomics accessed on 18/07/2015 11:27:39.

77
331) Review: Dolph L.Hatfield, Petra A. Tsuji, Bradley A.
Carlson and Vadium N.Gladyshev , Trends in Biochemical
Sciences, march 2014, vol.39, no.3.
332) Review: Holger Steinbrenner, Helmut Sies Selenium
homeostasis and antioxidant selenoproteins in brain:
implication for disorders in the central nervous system
Archives of Biochemisty and Biophysics 536 (2013) 152-
157.
333) Review: Sougat Misra, Mallory Boylan, Arun Selvam,
Julian E. Spallholz and Mikael Bjornstedt Redox- Active
Selenium Compounds-From Toxiciyy and Cell Death to
cancer treatment Nutrients 2015, 7, 3563-3556;
doi:10.3390/nu7053536.http://www.mdpi.com/journal/nutrie
nts accessed on 5/05/2015.
334) Review:Zhichao Zhang, Jinsong Zhang, Jianru Xiao
Selenoproteins and selenium status in bone physiology and
pathology Biochimica et Biophysica Acta 1840(2014) 3246-
3256.http://www.elsevier.com/locate/bbagen accessed on
4/08/2014.

78
Ringraziamento:
Un grazie speciale a tutti quelli che hanno permesso la
realizzazione di questo lavoro, anche se ero un po’scettica, alla
fine ci sono riuscita.
Ringrazio vivamente anche il Professor Lucacchini che mi ha
seguito in questa tesi con massima dedizione e accuratezza.
Grazie ai miei genitori che mi hanno aiutato tanto soprattutto ad
arrivare fino a qui.
Ringraziamenti:
Ai professori, da cui ho appreso lezioni di vita che non
dimenticherò.
A babbo e mamma che mi sostengono e credono in me.
A tutta la mia famiglia che mi è stata sempre accanto.
Ai miei compagni di università con cui ho condiviso gioie e
dolori.
A chi c’è oggi, a chi c’era ieri, comunque sia andata.
Ai miei nonni che mi guardano sicuramente fieri da lassù.
Grazie.