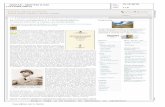
Alois Alzheimer (1864 - 1915) · 2019. 6. 5. · 1906 – Tubinga: Congresso della Società degli...
39
1906 – Tubinga: Congresso della Società degli Psichiatri Tedeschi del sud-ovest “Una caratteristica malattia della corteccia cerebrale” Alois Alzheimer (1864 - 1915) Descrisse il caso di una donna di 51 anni con progressivo declino cognitivo e deliri. All’esame autoptico: • atrofia cerebrale • “presenza di neurofibrille aggrovigliate” • “su tutta la corteccia di un gran numero di foci di deposito di una peculiare sostanza”
Transcript of Alois Alzheimer (1864 - 1915) · 2019. 6. 5. · 1906 – Tubinga: Congresso della Società degli...
-
1906 – Tubinga: Congresso della Società degliPsichiatri Tedeschi del sud-ovest
“Una caratteristica malattia della cortecciacerebrale”
Alois Alzheimer (1864 - 1915)
Descrisse il caso di una donna di 51 anni conprogressivo declino cognitivo e deliri.
All’esame autoptico:
• atrofia cerebrale
• “presenza di neurofibrille aggrovigliate”
• “su tutta la corteccia di un gran numero di foci di deposito di una peculiare sostanza”
-
Auguste D. età 51 anni e mezzo … non era alcolizzata e non c’era nella sua famiglia alcuno affetto da malattia mentale...
Il caso Auguste D. - 1907
da malattia mentale... Una donna pulita … gentile.All’improvviso ... divenne gelosa. Cominciò a soffrire di amnesia e divenne incapace di cucinare. A novembre entrò in ospedale. Mentre mangiava della carne, alla domanda cosa stesse mangiando, rispose patate ...
-
1906 – a Monaco gli venne
affidato lo studio del caso
Auguste D.
Gaetano Perusini (1879 -1915)
Descrisse quattro casi clinici� degenerazione neurofibrillare� placche
1910 – Osservazioni istologiche
e cliniche in alcune malattie
psichiatriche degli anziani.
-
Oltre 100 cause di Demenza
A . Degenerative• Malattia di Alzheimer
• Malattia di Pick
• Malattia di Huntington
• Atrofia Frontotemporale senza corpi di Pick
B. Vascolari• Multi-infartuale• Malattia di Binswanger• Vasculiti • Ematoma Subdurale• Infarto Strategico • Ipoperfusione
• Malattia a corpi di Lewy
• Malattia di Parkinson
• Malattia di Wilson
• Paralisi Sopranucleare Progressiva
• Degenerazione Spinocerebellare
• Degenerazione Corticobasale
• Afasia Progressivea
• Demenza Semantica
• Atrofia corticale posteriore
• Ipoperfusione• Postemorragica
C. Miste vascolari e degenerative
D. Miscellanea (malattie respiratorie ostruttive; sleep apnea; radiationi; dialisi;privazione di sonno; ipossia)
-
• E. Metaboliche
• Malattie della Tiroide
• Malattie delle Paratiroidi
• Disfunzioni Epatiche
• Malattia di Cushing
• Ipopituitarismo
Oltre 100 cause di Demenza (segue)
F. Tossiche• Farmaci (anticolinergici, antiistaminici, tranquillanti antiipertensivi, cimetidina, digossina)• Politerapie in range terapeutico• Alcool• Metalli pesanti (As,Pb,Hg)
• Ipopituitarismo
• Carenza di Estrogeni
• Uremia
• Porfiria
• Carenza di B12
• Carenza di Folati
• Carenza di Tiamina
• Alterazioni di Electroliti
•
G. Infettive• Sifilide• TBC• Meningite micotica• Malattia di Lyme • AIDS dementia complex• Encefalite erpetica• Meningiti batteriche
H. Cause psichiatriche(mania, depressione, late onset schizofrenia, delusional disorders)
-
• G. Infiammatorie Demielinizzanti
• Sclerosi Multipla
• Sarcoidosi
• Lupus
Oltre 100 cause di Demenza (segue)
I. TraumaticheTrauma cranicoInsulto post anossicoConcussione
J. Idrocefalo• Encefalite Limbica
H. Neoplasie- Tumori primari del cervello
a. glioma lobo frontaleb. glioma corpo calloso
- Metastasi cerebrali - Carcinomatosi meningea
J. IdrocefaloOstruttivoNon-ostruttivoNormoteso
K. Prion DiseaseCreutzfeldt-Jakob
-
Deterioramento cognitivo lieve (MCI)
� Deficit soggettivo di memoria, convalidato da una persona informata (familiare, amico).
Funzioni cognitive generali conservate.� Funzioni cognitive generali conservate.
� Capacità funzionali quotidiane intatte.
� Deficit di memoria, corretto per livello di istruzione ed età, oggettivo (tests).
(Petersen et al. Int Psychoger 1997;9:37-43)
-
Deterioramento cognitivo lieve (MCI)
2. Quadri clinici
� MCI amnestico MCI con deficit limitato alla memoria.
� MCI senza demenza MCI con deficit misurabile/i di:
– Orientamento
– Linguaggio
– Funzioni psicomotorie e motorie
– Attività di vita quotidiana
� Le alterazioni cognitive sono indipendenti dal deficit di memoria, che può essere presente o meno.
� I deficit presenti, misurabili, non inducono Demenza (ClinicalDementia Rating CDR = 0.5).
-
Deterioramento Cognitivo Lieve (MCI)
3.Sintomi non cognitivi e comportamentali.
� Nelle persone con MCI:
– Sintomi depressivi: 11 - 20%
– Sintomi da ansia:10%– Sintomi da ansia:10%
– Sintomi psicotici: 3 - 5%
� Sintomi rilevanti per una evoluzione sfavorevole in Demenza:
– Preoccupazioni inutili (ansia e tratti ossessivi compulsivi)
– Ritiro sociale
– Sospettosità
-
Quale modello?
La transizione tra normalità e demenza è un continuum che passa modello? che passa attraverso il MCI
-
Le funzioni cognitive da indagare sono molteplici e includono:
Attenzione e concentrazione
Abilità prassiche
Memoria
Linguaggio
Critica e Giudizio
Abilità prassiche
Intelligenza
Abilità visuospaziale
Ragionamento astratto
Calcolo
-
Malattia di Alzheimer
“Patologia degenerativa con decorso caratterizzato da un
prevalente ed iniziale deficit di memoria che si
accompagna ad impoverimento delle funzioni cognitiveaccompagna ad impoverimento delle funzioni cognitive
quali: linguaggio, orientamento, abilità visuo-spaziali,
capacità di astrazione e problem solving, prassia.“
DSM IV
-
DEMENZE: EPIDEMIOLOGIA
o Crescente aumento a causa del trend demografico
> 5% nei soggetti dai 65 anni
o Prevalenza
> del 40% dopo gli 85 anni
In Italia i malati di Alzheimer sono tra i 430.000 e i 450.000.
-
Malattia di Alzheimer
Esordio:
- AD INSORGENZA PRECOCE (
-
Alterazioni morfologiche quantitative:
- riduzione di peso e di volume dell’organo
- dilatazione cavità ventricolari
Aspetti Neuropatologici
- dilatazione cavità ventricolari
- ampliamento dei solchi e delle
scissure a livello della corteccia
- maggiore atrofia
-
PLACCHE DI AMILOIDE :
depositi amorfi extracellulari di proteina β-amiloide (Aβ) sotto forma di placche
Patologia della malattia di Alzheimer
GROVIGLI NEUROFIBRILLARI :
intreccio intracellulare
di neurofibrille formate da proteina TAU anomala
-
Degenerazione neurofibrillare
Demenza di Alzheimer: neuropatologia
Marcata e diffusa atrofia corticale a partenza temporo-frontale
Placche senili
Amiloidosi vascolarecerebrale
-
Formazione di placche amiloidi
Per placca amiloide si intende generalmente un ammassoextracellulare di elementi dendridici, assonali e gliali aggregati.
Caratteristica è la presenza della proteina ß-amiloide che tende adautoaggregarsi in maniera complessa.
Il precursore dell'amiloide (APP) è una proteina più grande codificata dalcromosoma 21. Il rischio di ammalarsi di AD è maggiore nei pazientiaffetti dalla trisomia 21.
Placche amiloidi si trovano anche nelle persone anziane non affette da AD(placche senili), ma in maniera molto ridotta.
L'ippocampo e le strutture limbiche sono le prime strutture interessate daidepositi di amiloide, e sono anche le più gravemente compromesse. Ciòspiegherebbe in parte i deficit a carico delle funzioni di memoria.
-
� CLASSICHE : sono accumuli extracellulari , costituiti da un nucleocentrale di β-amiloide, circondato da un anello di neuroni distrofici,microglia e astrociti
� DIFFUSE: sono una forma preliminare rispetto alle classiche elocalizzate in aree celebrali non sintomatiche dell’AD
Placche di amiloide
-
Sono costituite da una forma insolubile del peptide che deri va dalla proteinaprecursore del ββββ-amiloide (APP) e queste placche possono avere un nucleodenso centrale.Una porzione variabile di tali placche contiene anche altre m olecole proteiche enon, e possono essere associate a cellule non neuronali e a pr ocessi neuriticianormali.
Placche di amiloide
Colorazione rosso congoColorazione rosso congo
-
PLACCHE DI AMILOIDE :
depositi amorfi extracellulari di proteina β-amiloide (Aβ) sotto forma di placche
Patologia della malattia di Alzheimer
GROVIGLI NEUROFIBRILLARI :
intreccio intracellulare
di neurofibrille formate da proteina TAU anomala
-
I grovigli neurofibrillari sono formati da matasse di filamenti elicoidaliaccumulate nel corpo cellulare .
Nelle placche si osservano elementi dendridici e assonali e anche depositiamiloidi. Ogni filamento consiste in due fibrille disposte ad elica. Questeformazioni non assomigliano alle proteine normali del citoscheletro del
Patologia neurofibrillare
formazioni non assomigliano alle proteine normali del citoscheletro delneurone.
I grovigli si formano in neuroni di grandi dimensioni nell'ippocampo, nellacorteccia olfattiva, nell’amigdala, nei nuclei del proencefalo basale e inparecchi nuclei del tronco cerebrale.
I grovigli neurofibrillari non sono esclusivi della AD, in quanto si osservanoanche in altre forme neurodegenerative, quali la sindrome di Down e lademenza dei pugili.
-
Colorazione con Sali d’argento
Grovigli neurofibrillari
-
PROTEINA TAU
GROVIGLI
Grovigli neurofibrillari
I grovigli neufibrillari sono dovuti a fasci di filamenti insolubili che derivano daalterata fosforilazione delle proteine TAU , associate al citoscheletro deineuroni, che si accumulano nel corpo neuronale
L’iperfosforilazione riduce l’affinità delle TAU per i microtubuli causando unaperdita di stabilità nel neurone e può portare alla modifica zione delmetabolismo dell’APP
-
Demenza di Alzheimer: neuropatologia
o Placche senili: β amiloide APP
o Ammassi neurofibrillari
Proteina Precursore
dell’Amiloide
(o grovigli neurofibrillari)accumuli di neuriti distrofici
ricchi di proteina τ iperfosforilata
o Amiloidosi vascolare cerebrale
(o grovigli neurofibrillari) ricchi di proteina τ iperfosforilata
angiopatia da infiltrazionedelle pareti dei piccoli emedi vasi cerebrali
-
Atrofia Cerebrale e Riduzione dei Neuroni Colinergici
Lobo frontale
Lobo parietale
Marcata perdita di neuroni nel nucleo basale di Meynert
Amigdala
Ippocampo
PROGRESSIONE MALATTIA
-
Atrofia cerebrale
- atrofia corticale con assottigliamento della sostanzagrigia da perdita neuronale .
- Più colpiti lobo frontale, lobo temporale e loboparietale. L'ippocampo può essere severamenteparietale. L'ippocampo può essere severamenteatrofico.
- generalmente interessate anche il locus coerulus e learee limbiche (probabile causa dei disturbidell'umore) ed il nucleo basale di Meynert.
-
Il deficit colinergico dell’AD determina la sintomatologia clinica
� Deficit colinergico
– Perdita progressivadei neuroni colinergici
N.basale del Meynert
– Riduzione progressivadell’ACh disponibile
– Compromissione di ADL,comportamento e funzioni cognitive
IppocampoCorteccia
N.basale del Meynert
Bartus et al., 1982; Cummings and Back, 1998, Perry et al., 1978
-
Degenerazione dei neuroni colinergici del nucleo basale di Meynert
Il nucleo basale di Meynertè un'importante struttura implicatanel circuito della memoria per le sue connessioni e proiezionicolinergiche che raggiungono la corteccia, l'ippocampo,l'amigdala, il bulbo olfattivo, il talamo e il troncodell'encefalo.dell'encefalo.
Riceve afferenze dall'ipotalamo, dall'amigdala e dalmesencefalo.
La degenerazione del nucleo di Meynert può spiegare la gravediminuzione della concentrazione di acetilcolina nellacorteccia (60-70% dell'attività corticale).
Su questa osservazione si basa il trattamento con inibitori diacetilcolin-transferasi.
-
Genetica della Malattia di Alzheimer
� 4 geni identificati con un possibile ruolo :
– APP – Amyloid precursor protein (cromosoma 21) rararara
– PS1 – Presenilin 1 la forma più comune di AD familiare ad eseordio precoce
– PS2 – Presenilin 2
– ApoE – Apolipoprotein E (allele 4) codifica proteina che trasporta il colesterolo e si lega all'amiloide
-
Complesso: non c’è nessun singolo meccanismo di trasmissione chegiustifica la sua ereditarietà;
Eterogeneo : le mutazioni e i polimorfismi nei geni multipli sono coinvoltiinsieme a fattori non genetici;
Genetica della Malattia di Alzheimer
Dicotomico: le mutazioni dell’AD familiare ad insorgenza precoce sono:- rare,- altamente penetranti,- a trasmissione autosomica-dominante,
- Mutazioni in:presenilin 1, amyloid precursor protein, presenilin 2
���� early-onset familial AD
- Polimorfismi in Apolipoproteina E���� associati alla forma idiopatica
-
Individui non dementi, con depositi di β-amiloide nella neocorteccia possono avere deficit cognitivi
quindi
Stadio precoce di AD
Forse fase preclinica, prevista dalla cascata patologica dell’ADdell’AD
Lo sviluppo delle placche nelle aree neocorticali precede la patologia
neurofibrillare e la demenza clinica
-
Storia naturale della DA
17
25Sintomi cognitivi
Disturbi del comportamentoDecadi
Pre-DA Lieve-Moderata Intermedia Grave
MM
SE
Adattata da Gauthier S. ed. Clinical Diagnosis and Management of Alzheimer’s Disease. 1996.
0
5
10
0 2 4 6 8 10
Anni
Perdita dell’autosufficienza
Ricovero in strutture sanitarie
Morte
MM
SE
-
Criteri per la diagnosi di probabile demenza di Alzheimer
a) Criteri che devono essere presenti contemporaneamente
− demenza stabilita dall’esame clinico e documentata da test oggettivi (ad es. MMS);
− deficit in due o più funzioni
b) Criteri a supporto della diagnosi:
− progressivo deterioramento di specifiche funzioni cognitive quali linguaggio (afasia), capacità motoria (aprassia) e percezione (agnosia);
− deficit in due o più funzioni cognitive;
− progressivo deterioramento della memoria e di almeno un’altra funzione cognitiva;
− nessun disturbo della coscienza;
− comparsa tra i 40 e i 90 anni;
− assenza di altre patologie del SNC o malattie sistemiche che possano causare demenza.
(agnosia);
− riduzione della indipendenza nello svolgimento delle attività quotidiane;
− storia familiare di disturbi simili;
− eventuale quadro di neuroimaging (ad es. atrofia cerebrale).
-
AD : quadro clinico
� Stadio 1: anamnestico
Stadio 2: demenza conclamata� Stadio 2: demenza conclamata
� Stadio 3: vegetativo
-
AD : quadro clinico
� Stadio1
a) dominato dai disturbi della memoria
b) frequentemente precoci disturbi del linguaggio b) frequentemente precoci disturbi del linguaggio
c) pazienti ripetitivi, perdono oggetti, faticano a trovare le parole, tendono a smarrirsi, possono presentare squilibri emotivi talora con grave instabilità
� Durata media: 2-4 anni
-
AD : quadro clinico
� Stadio 2
a) perdita grave delle facoltà cognitive con progressiva perdita dell’autonomia;
b) frequentemente perdita d’interesse con atteggiamenti disturbanti, deliri od allucinazioni
c) necessità di assistenza a tempo pieno.
� Durata media: 2-10 anni
-
AD : quadro clinico
� Stadio 3
a) perdita completa dell’autonomia (pazienti che non si alimentano, non comunicano, non badano all’igiene personale) all’igiene personale) b) necessaria assistenza continua di tipo contenitivo c) morte per insorgenza di malattie intercorrenti.
� Durata media: 1-3 anni