
Algoritmi di clustering per l.analisi di dati...
34
Algoritmi per l’analisi di dati di espressione genica in serie temporale. Presentazione per l’esame del corso di “Algoritmi per la bioinformatica” di Stefano Colombo Giambattista Tiepolo, “Il tempo scopre la verità” (dettaglio).
Transcript of Algoritmi di clustering per l.analisi di dati...

Algoritmi per l’analisi di dati di espressione genica in serie
temporale.
Presentazione per l’esame del corso di “Algoritmi per la bioinformatica” di Stefano Colombo
Giambattista Tiepolo, “Il tempo scopre la verità” (dettaglio).
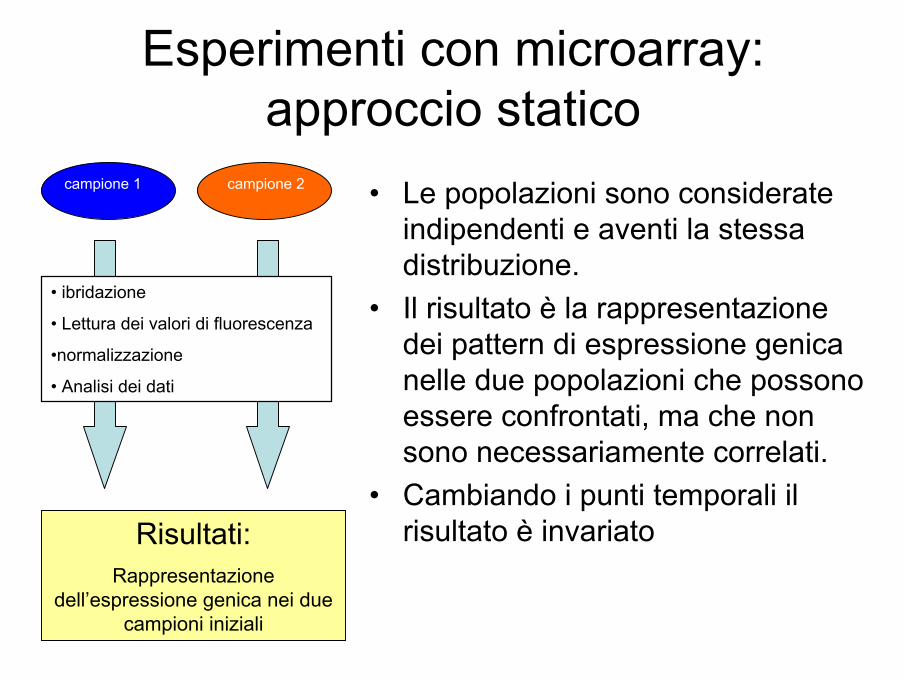
Esperimenti con microarray: approccio statico
• Le popolazioni sono considerate indipendenti e aventi la stessa distribuzione.
• Il risultato è la rappresentazione dei pattern di espressione genica nelle due popolazioni che possono essere confrontati, ma che non sono necessariamente correlati.
• Cambiando i punti temporali il risultato è invariato
campione 1 campione 2
• ibridazione
• Lettura dei valori di fluorescenza
•normalizzazione
• Analisi dei dati
Risultati:Rappresentazione
dell’espressione genica nei due campioni iniziali

Esperimenti con microarray: approccio “Time Series”
• I dati successivi di analisi “Time Series” mostrano una forte auto-correlazione.
• Non ho informazioni solo sullo stato della popolazione iniziale e finale, ma anche dati riguardanti gli stati intermedi.
• Risultato: Misura di un processo temporale.
• Il risultato dipende dalla sequenza di punti temporali adottata
campione 1
campione 5
campione 4
campione 3
campione 2
t0 =0
t2= t0 + ∆t
t3= t2 + ∆t
t4= t3 + ∆t
t5= t4 + ∆t
Risultati:misurazione dell’espressione genica in una serie temporale
Esempi di processi naturali in serie temporali
• Ritmi Circadiani
• Infezioni Virali
• Ciclo Cellulare
•Sviluppo Embrionale

Problemi degli studi di espressione “Time Series”
• Tradizionalmente gli algoritmi di normalizzazione, analisi e clustering dei dati sono implementati per esperimenti statici e non tengono conto della correlazione temporale dei dati e altre caratteristiche delle serie temporali.
• Bisogna scegliere l’intervallo di tempo e la durata dell’esperimento spesso in modo arbitrario.
• È necessaria una sicronizzazionedei dati.

Le serie temporalicampione 1
campione 5
campione 4
campione 3
campione 2
Una serie temporale (time serie) è una serie di valori o dati presi in periodi di tempo successivi.
I punti temporali (time points, tp) sono gli istanti nel tempo in cui vengono prese le misurazioni.
L’intervallo di campionamento (sampling interval) è la distanza tra i punti temporali; questa può essere costante o variabile.
I dati ottenuti nel contesto di una serie temporale possono avere una struttura interna che non si deve ignorare.
t0 =0
t2= t0 + ∆t
t3= t2 + ∆t
t4= t3 + ∆t
t5= t4 + ∆t

Analisi statistica di dati in serie temporale
Obbiettivo: identificare le natura del fenomeno rappresentato dalla serie osservata e predire i futuri valori delle variabili della serie temporale.
Le proprietà statistiche più importanti delle serie temporali sono:
Autocorrelazione: cioè la correlazione della serie temporale con i suoi stessi valori passati e futuri.
Trend: cioè la componente lineare o non lineare che cambia nel tempo senza ripetersi.
Seasonality (stagionalità): fluttuazioni periodiche mostrate da più serie temporali.
Stazionarietà: è un’assunzione comune di molti metodi di analisi, conferisce alla serie temporale la proprietà di non cambiare la media, la varianza e la struttura dell’autocorrelazione nel tempo.
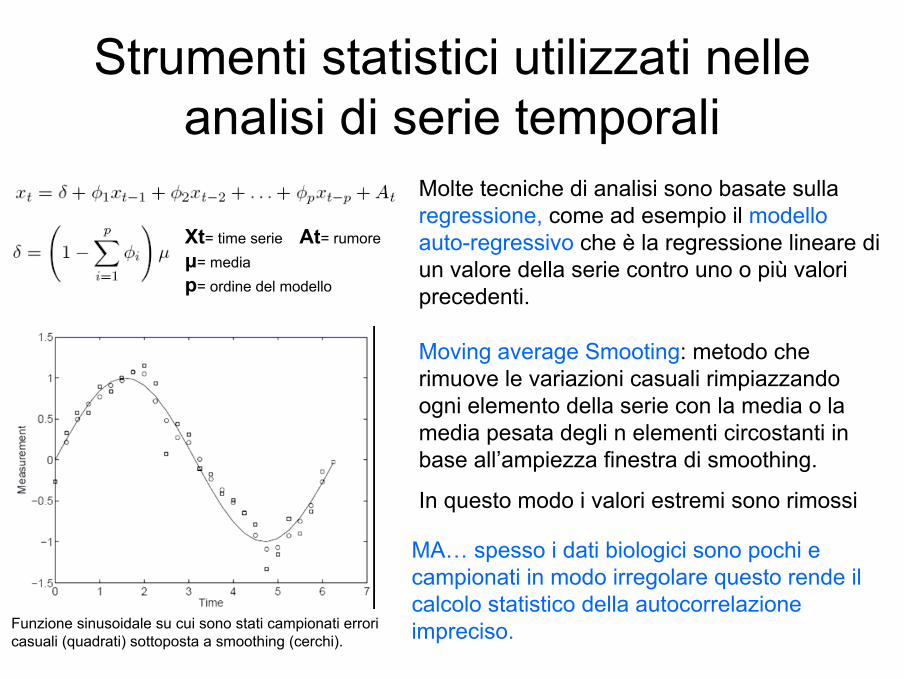
Strumenti statistici utilizzati nelle analisi di serie temporali
Funzione sinusoidale su cui sono stati campionati errori casuali (quadrati) sottoposta a smoothing (cerchi).
Molte tecniche di analisi sono basate sulla regressione, come ad esempio il modello auto-regressivo che è la regressione lineare di un valore della serie contro uno o più valori precedenti.
Xt= time serie At= rumore µ= media p= ordine del modello
Moving average Smooting: metodo che rimuove le variazioni casuali rimpiazzando ogni elemento della serie con la media o la media pesata degli n elementi circostanti in base all’ampiezza finestra di smoothing.
In questo modo i valori estremi sono rimossi
MA… spesso i dati biologici sono pochi e campionati in modo irregolare questo rende il calcolo statistico della autocorrelazioneimpreciso.

Il ClusteringIl clustering è un tecnica che permette il “data mining”, cioè un estrazione non traviante o implicita di informazioni sconosciute o potenzialmente utili dai dati, sviluppano sottoinsiemi significativi di individui o oggetti.
Nel contesto dell’analisi dell’espressione genica viene usato per identificare sottoinsiemi di geni che si comportano in modo simile al passare del tempo in condizioni simili.
Il clustering di serie temporali si sviluppa in 4 step fondamentali:
• Rappresentazione e Modellamenento della serie temporale.
• Definizione della Misura di Similarità tra serie temporali con cui dare un senso al modello/rappresentazione scelto.
• Definizione dell’Algoritmo di Clustering con il quale raggruppare i dati in base alla definizione di similarità scelta.
• Validazione e Scoring dei risultati del clustering.

Rappresentazione e Modellamenento
Il modello adottato incide in modo sostanziale sui successivi steps infatti da questo dipende la misura di similarità da adottare e ha un notevole impatto sulla scelta dell’algoritmo di clustering.
I tipi di rappresentazione di sequenze temporali possono essere divisi in 4 gruppi:
• Time-domain continuous representation:La serie è rappresentata usando gli elementi originali ordinati in base al tempo di occorrenza senza alcun processamento.
• Transformation based: Si basa sulla trasformazione della sequenza iniziale in un altro dominio e l’uso dei punti del nuovo dominio per rappresentare la serie originale.
• Discretisation based: converte la serie temporale iniziale di elementi con valori reali in una serie discretizzata.
• Generative models: si basa sull’idea di ottenere il modello che ha generato la serie osservata.

SimilaritàLa misura di similarità è necessaria per il confronto delle serie temporali e dovendo trattare elementi quali punti “outlying”, rumore, scaling-traslazioni e altre distorsioni dell’asse temporale è correlata al modello che li ha prodotti.
Tipi di similarità principali:
• Distanza Minkowski : ogni serie di lunghezza k è vista come un punto nello spazio k-dimensionale viene quindi calcolata la distanza tra i punti.
• Pearson: misura statistica della relazione lineare tra due variabili
• Hamming Distance: si basa sul confronto diretto dei simboli delle serie misura le coordinate per cui questi differiscono.
• Likelihood: la similarità tra sequenze è ottenuta in base a quanto strettamente i dati di queste si adattano al modello scelto.

ClusteringI principali algoritmi di clustering possono essere divisi in 3 gruppi:
• Hierarchical: procedono in modo iterativo raggruppando clusters piccoli in clusters più grandi (agglomerativi) oppure dividendo clusters grandi in clusters più piccoli (divisivi).
• Partitioning-Optimisation: ammette la rilocazione degli elementi, utilizza dei centroidi rappresentativi del cluster per raccogliere gli elementi.
• Based on Maximum Likelihood: i dati vengono uniti per clustering gerarchico o rilocazione basandosi sul criterio di Massima Verosimiglianza. Ogni cluster è rappresentato da un particolare modello adottato. Usato per metodi structure-based.

Similarità di serie temporali di espressione genica.
Obbiettivo del clustering dell’espressione genica: identificare i geni co-espressi cioè geni che hanno simili pattern di espressione. Pattern di espressione sono considerati simili se aumentano o diminuiscono nel tempo in modo coordinato.
Trattando dati di espressione genica la misurare di similarità deve trattare con problemi quali:
• Scaling e Shifting
• Punti di campionamento distribuiti in modo irregolare
• Forma (struttura interna) dei dati
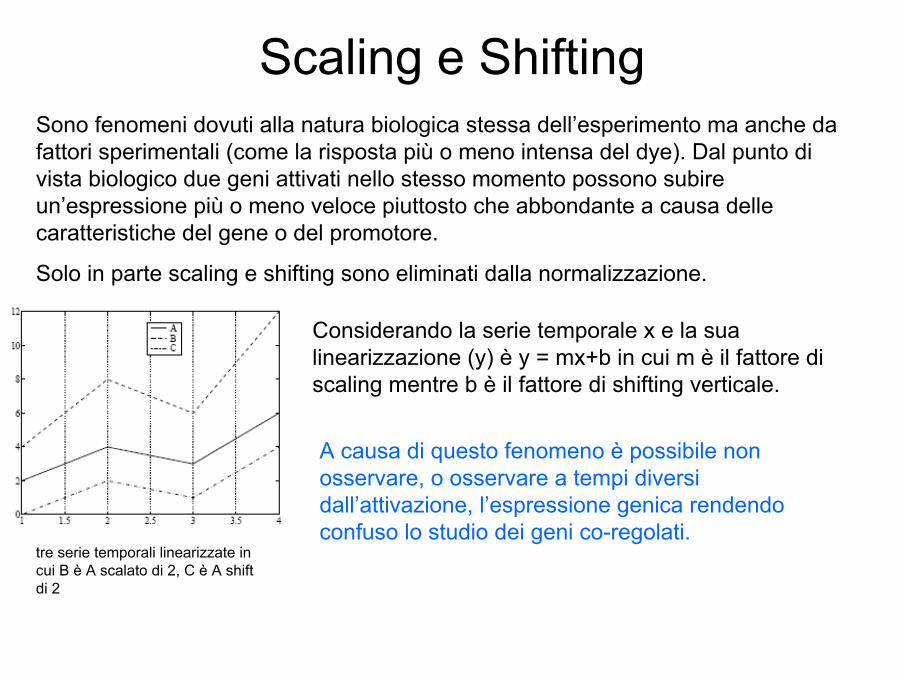
Scaling e Shifting
tre serie temporali linearizzate in cui B è A scalato di 2, C è A shiftdi 2
Sono fenomeni dovuti alla natura biologica stessa dell’esperimento ma anche da fattori sperimentali (come la risposta più o meno intensa del dye). Dal punto di vista biologico due geni attivati nello stesso momento possono subire un’espressione più o meno veloce piuttosto che abbondante a causa delle caratteristiche del gene o del promotore.
Solo in parte scaling e shifting sono eliminati dalla normalizzazione.
Considerando la serie temporale x e la sua linearizzazione (y) è y = mx+b in cui m è il fattore di scaling mentre b è il fattore di shifting verticale.
A causa di questo fenomeno è possibile non osservare, o osservare a tempi diversi dall’attivazione, l’espressione genica rendendo confuso lo studio dei geni co-regolati.
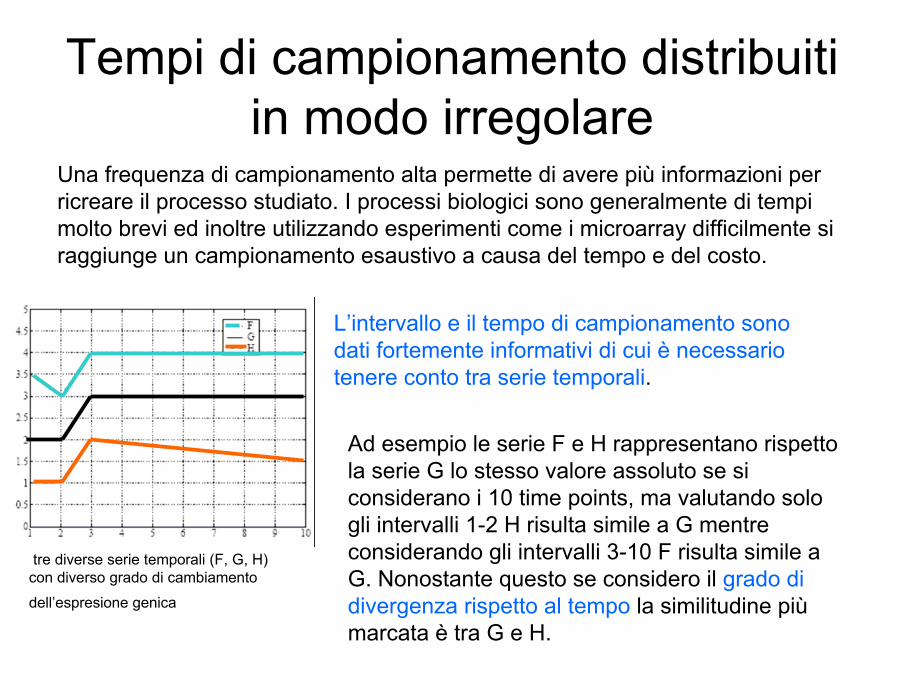
Tempi di campionamento distribuiti in modo irregolare
tre diverse serie temporali (F, G, H) con diverso grado di cambiamento
dell’espresione genica
Una frequenza di campionamento alta permette di avere più informazioni per ricreare il processo studiato. I processi biologici sono generalmente di tempi molto brevi ed inoltre utilizzando esperimenti come i microarray difficilmente si raggiunge un campionamento esaustivo a causa del tempo e del costo.
L’intervallo e il tempo di campionamento sono dati fortemente informativi di cui è necessario tenere conto tra serie temporali.
Ad esempio le serie F e H rappresentano rispetto la serie G lo stesso valore assoluto se si considerano i 10 time points, ma valutando solo gli intervalli 1-2 H risulta simile a G mentre considerando gli intervalli 3-10 F risulta simile a G. Nonostante questo se considero il grado di divergenza rispetto al tempo la similitudine più marcata è tra G e H.

Struttura Interna
Tre serie temporali (J, K, L)
La struttura interna è la principale differenza tra una serie temporale e un set di misurazioni ed è il motivo per cui una serie temporale non può essere trattata come una un set di dati indipendenti con la stessa distribuzione della serie.
Generalmente la struttura interna è legata alla forma della serie.
In un esperimento di microarray non è rilevante l’intensità dell’espressione genica è invece informativa la variazione relativa di intensità che è caratterizzata dalla forma dell’espressione genica.
Se considero l’intensità dell’espressione J e K sono i più simili ma se considero il cambiamento relativo di intensità J è simile a L.
La condizione necessaria per l’esistenza della struttura interna è la misurazione in ordine temporale.

Metodi di similarità di serie temporali
Sono stati identificati in letteratura principalmente i seguenti approcci per comparare serie temporali:
• Linear Transformation (trasformation based)
• Temporal Structure based
• Shape Based

Linear Transformation(trasformation based) (Aach et al, 2001)
Una trasformazione T: Rn Rm è lineare se T(u+v)= T(u)+T(v) e T(cu)= cT(u)
Similarità basata sul grado con il quale una serie temporale può essere espressa come la trasformazione lineare y=mx+b di un’altra.
Tratteggio: differenti trasformazioni lineari della stessa funzione. Linea:dopo la normalizzazione
La distanza Euclidea può essere usata per studiare la forza di correlazione tra due serie temporali, più questa è piccola più la relazione lineare tra le funzioni è forte.
Temporal Database Field: due serie temporali sono simili se esiste una funzione lineare f che permetta di mappare una sequenza X su una sequenza Y.
Dynamic Time Warping: si basa sull’allineamento di due sequenze contro un pattern di riferimento che non può essere fornito in un metodo non supervisionato. Senza un pattern di riferimento i può calcolare la distanza Euclidea ma questo implica una standardizzazione delle funzioni.
La similarità basata sulla trasformazione lineare non considera la struttura interna dell’intervallo di sampling

Temporal structure basedSi tratta di modelli basati sull’assunzione di un modello statistico; considerano la serie temporale caratterizzabile come un processo parametrico casuale i cui parametri stocastici possono essere stimati in modo preciso. Il clustering dei dati ottenuti con questi metodi avviene attraverso la likelihood.
I principali modelli utilizzati sono:
• Normal Mixture
• Autoregressive
• Hidden Markov
• Splines

Normal Mixture model-based(MacLachlan et al, 2002)
Approccio che considera ogni gene derivante da un’insieme di densità normali multivariate. Una “mistura” di distribuzioni è usata per descrivere con formule matematiche il modello statistico di fenomeni casuali.
non considera la struttura temporale dei dati ne’ l’intervallo di campionamento.
x1… xn : n osservazioni p-dimensionali
g :numero di densità normali multivariate iniziali
π1… πn : proporzioni di g ignote
Ф(x:µi,∑i): funzione di probabilità della densitànormale p-variata; µi media, ∑i matrice di covarianza.
Ψ: vettore dei parametri ignoti
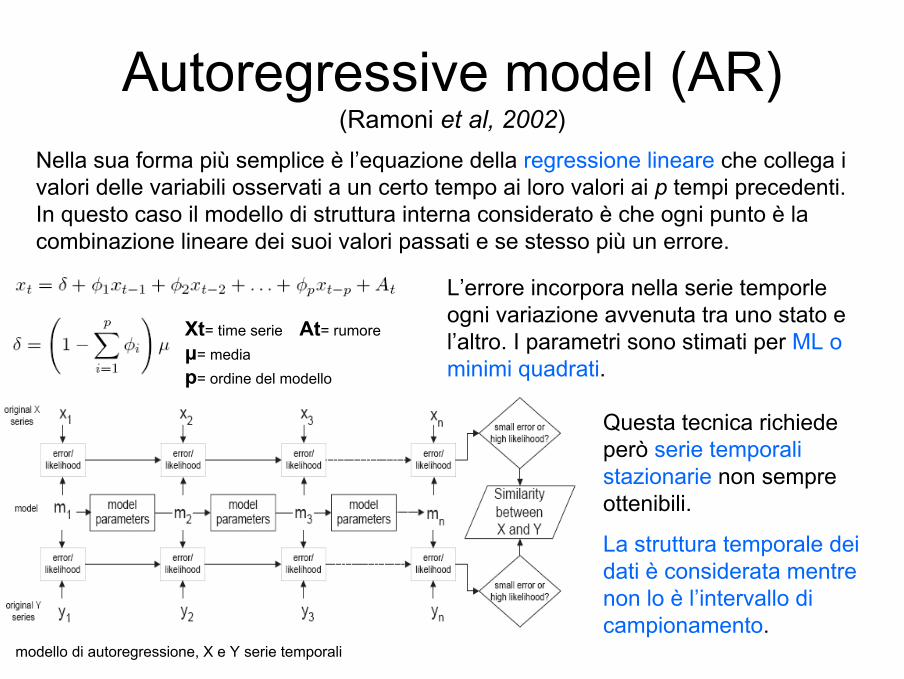
Autoregressive model (AR) (Ramoni et al, 2002)
Xt= time serie At= rumore µ= media p= ordine del modello
Nella sua forma più semplice è l’equazione della regressione lineare che collega i valori delle variabili osservati a un certo tempo ai loro valori ai p tempi precedenti. In questo caso il modello di struttura interna considerato è che ogni punto è la combinazione lineare dei suoi valori passati e se stesso più un errore.
modello di autoregressione, X e Y serie temporali
L’errore incorpora nella serie temporleogni variazione avvenuta tra uno stato e l’altro. I parametri sono stimati per ML o minimi quadrati.
Questa tecnica richiede però serie temporali stazionarie non sempre ottenibili.
La struttura temporale dei dati è considerata mentre non lo è l’intervallo di campionamento.

Modello Spline (Ziv Bar-Joseph et al, 2002)
Le serie temporali vengono rappresentate da un modello che definisce una curva nel tempo. Gli splines sono funzioni polinomiali connesse tra loro in modo omogeneo.
I principali elementi del modello B-spline sono:
• Il numero n di splines connessi.
•Il grado k degli n-splines (cioè il grado dei polinomiali)
• il numero p di punti di connessione detti nodi (knots)
Ogni segmento è descritto da k+1 parametri ma, a causa della costrizione dovuta all’omogeneità che impone continuità agli splines e alla loro derivata secondo l’ordine dei nodi, ogni segmento ha un solo grado di libertà. Q-splines è un metodo che sfrutta polinomiali cubici la cui derivata prima e seconda è continua nei nodi, determinando statisticamente gli spline è possibile definire il profilo di espressione della serie temporale.Se i nodi sono definiti in modo corretto questa funzione considera il profilo e la lunghezza dell’intervallo di campionamento. Non sempre è possibile definire correttamente i nodi ed inoltre questo metodo un’alto grado di campionamento.
pensate che questa spiegazione sia sommaria? chiedete a loro
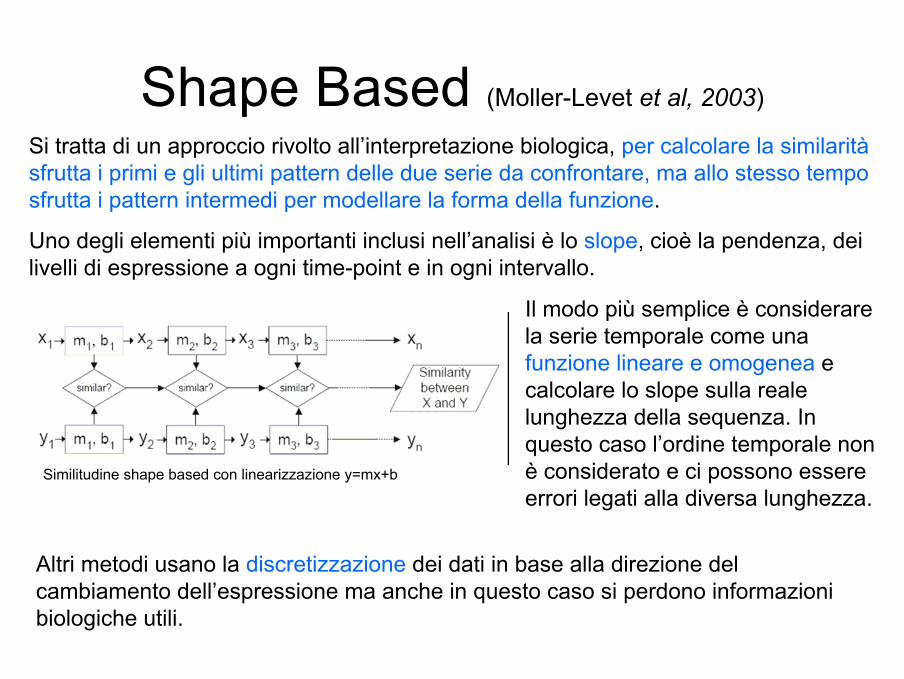
Shape Based (Moller-Levet et al, 2003)
Si tratta di un approccio rivolto all’interpretazione biologica, per calcolare la similarità sfrutta i primi e gli ultimi pattern delle due serie da confrontare, ma allo stesso tempo sfrutta i pattern intermedi per modellare la forma della funzione.
Uno degli elementi più importanti inclusi nell’analisi è lo slope, cioè la pendenza, dei livelli di espressione a ogni time-point e in ogni intervallo.
Il modo più semplice è considerare la serie temporale come una funzione lineare e omogenea e calcolare lo slope sulla reale lunghezza della sequenza. In questo caso l’ordine temporale non è considerato e ci possono essere errori legati alla diversa lunghezza.
Altri metodi usano la discretizzazione dei dati in base alla direzione del cambiamento dell’espressione ma anche in questo caso si perdono informazioni biologiche utili.
Similitudine shape based con linearizzazione y=mx+b
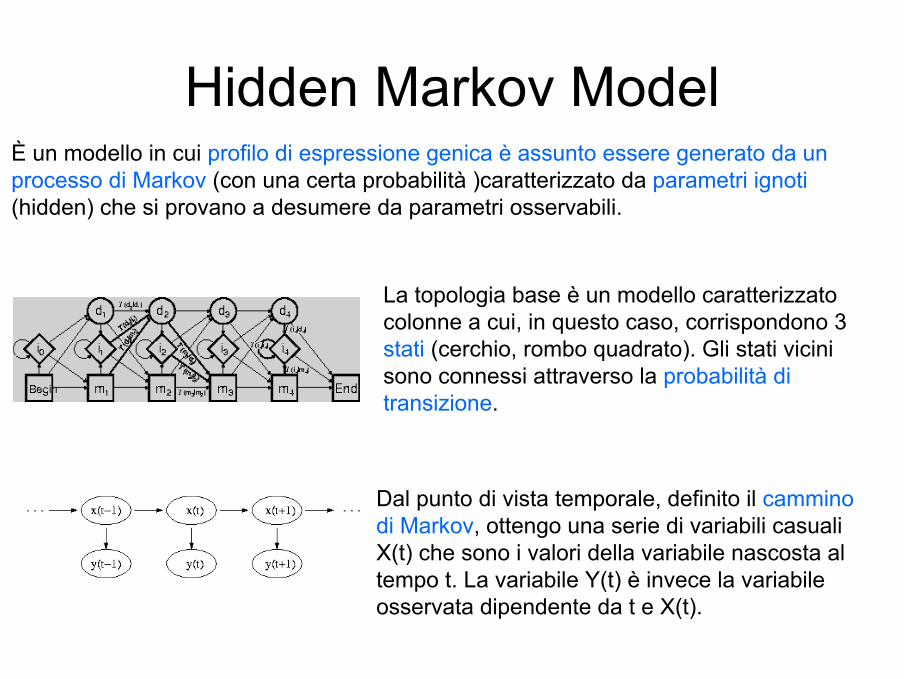
Hidden Markov ModelÈ un modello in cui profilo di espressione genica è assunto essere generato da un processo di Markov (con una certa probabilità )caratterizzato da parametri ignoti(hidden) che si provano a desumere da parametri osservabili.
La topologia base è un modello caratterizzato colonne a cui, in questo caso, corrispondono 3 stati (cerchio, rombo quadrato). Gli stati vicini sono connessi attraverso la probabilità di transizione.
Dal punto di vista temporale, definito il cammino di Markov, ottengo una serie di variabili casuali X(t) che sono i valori della variabile nascosta al tempo t. La variabile Y(t) è invece la variabile osservata dipendente da t e X(t).

Procedimento HMM
Trasformazione della serie temporale
Probabilità di transizione di stato A=aij
Osservazione probabilità di simboli
1. Trasformo la serie temporale secondo un modello alfabetico che associa a ogni stato un simbolo (es: up-regolato=v2, down-regolato=v1, invariato=v3) in base alla relazione (<,>,=) di ogni stato col precedente.
2. Chiamo gli stati S e la probabilità di transizione a.
3. Definisco la probabilità b di osservare un certo simbolo.
4. Fornisco il set di parametri N (numero di stati), M(numero di simboli osservati differenti), A (probabilità di transizione), B (probabilità di osservare un simbolo) e π(distribuzione dello stato iniziale).
5. Continuo a stimare iterativamente parametri fino a trovare il modello che meglio si adatta ai miei dati.
La struttura temporale definita con questo metodo è una funzione probabilistica. Come nell’autoregressione la lunghezza del campionamento non è incorporata nella similarità.

Catene Markov e parametri nascosti
Algoritmo che descrive una catena di markov i parametri ek e ak sono noti
Se non conosco i parametri ek e ak (HMM) posso provare a calcolarli con l’algoritmo Baum-Welch:
I valori Akl e Ekl sono stimati per ogni transizione e utilizzati per costruire un nuovo modello caratterizzato da ak e ek. La re-stima dei parametri è iterata ciclicamente fino al raggiungimento della massima likelihood definita come:

Clustering: Approccio 1 (Schliep et al,2003)
• L’algoritmo rappresenta in ogni cluster rappresenta un HMM.
• I parametri sono aggiustati fino alla massima likelihood con il procedimento Baum-Welch.
• l’apprendimento usato è Parzialmente Supervisionato, alcuni profili biologici noti sono forniti durante l’inizializzazione e i loro parametri possono essere usati nei cicli di re-stima dei parametri senza che il modello iniziale venga fortemente variato.
• Il numero di cluster è deciso automaticamente eliminando i cluster senza il numero minimo di profili e ri-assegnandoli agli altri cluster.
Algoritmo di addestramento Baum-Welch. Questo algoritmo viene ripetuto più volte (50-100) per poter evitare di cadere in un minimo locale.

Clustering: Approccio 2 (Ji et al,2003)
• Le sequenze di espressione sono trasformate in sequenze di fluttuazioneusate come training set per generare gli HMM che descrivono i cluster. Le sequenze di fluttuazione sono descritte dai valori 0,1, 2 definiti da:
L = lenght a = valore soglia
• Per w cluster sono inizializzate w HMMognuno dei quali addestrato separatamente con l’algoritmo Baum-Welch pesato sull’intero set di sequenze di fluttuazione. Il fattore peso differisce per ogni cluster e determina una stima diversa dei parametri.
• Viene calcolata la probabilità per ogni sequenza per ogni HMM generato.
• I risultati ottenuti vengono clusterizzati in base alla likelihood rispetto al modello con cui sono ottenuti.
• Il metodo è non supervisionato.
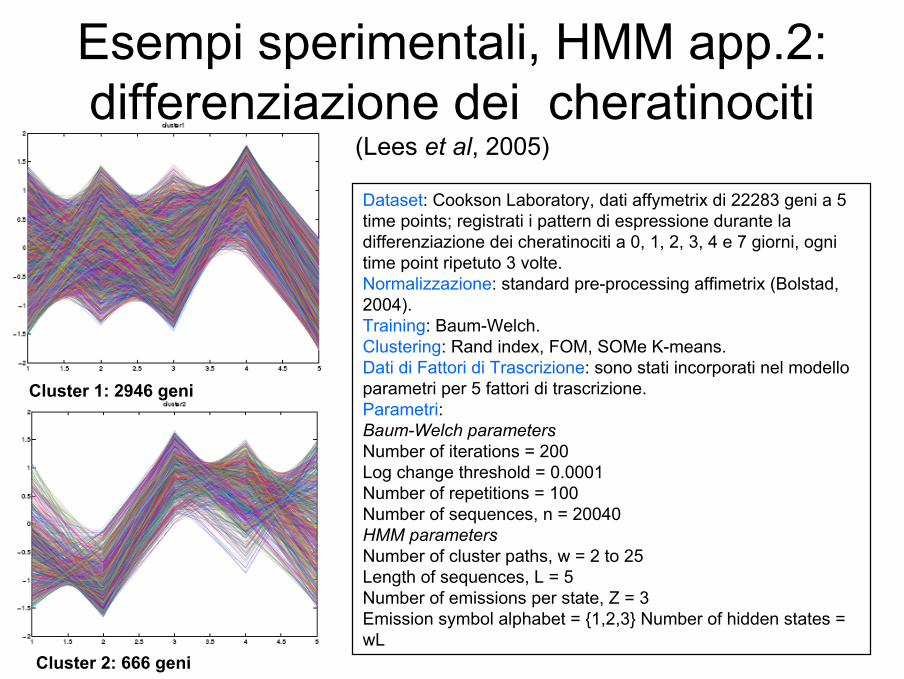
Esempi sperimentali, HMM app.2:differenziazione dei cheratinociti
(Lees et al, 2005)
Dataset: Cookson Laboratory, dati affymetrix di 22283 geni a 5 time points; registrati i pattern di espressione durante la differenziazione dei cheratinociti a 0, 1, 2, 3, 4 e 7 giorni, ogni time point ripetuto 3 volte. Normalizzazione: standard pre-processing affimetrix (Bolstad, 2004). Training: Baum-Welch. Clustering: Rand index, FOM, SOMe K-means. Dati di Fattori di Trascrizione: sono stati incorporati nel modello parametri per 5 fattori di trascrizione.Parametri:Baum-Welch parametersNumber of iterations = 200Log change threshold = 0.0001Number of repetitions = 100Number of sequences, n = 20040HMM parametersNumber of cluster paths, w = 2 to 25Length of sequences, L = 5Number of emissions per state, Z = 3Emission symbol alphabet = {1,2,3} Number of hidden states = wL
Cluster 1: 2946 geni
Cluster 2: 666 geni
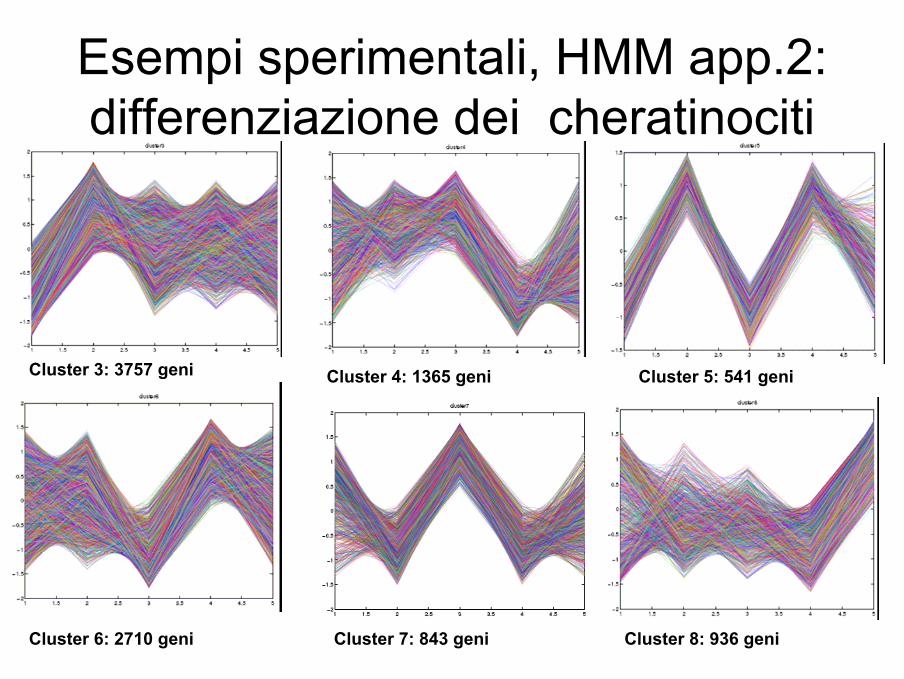
Esempi sperimentali, HMM app.2:differenziazione dei cheratinociti
Cluster 3: 3757 geni Cluster 4: 1365 geni Cluster 5: 541 geni
Cluster 6: 2710 geni Cluster 7: 843 geni Cluster 8: 936 geni
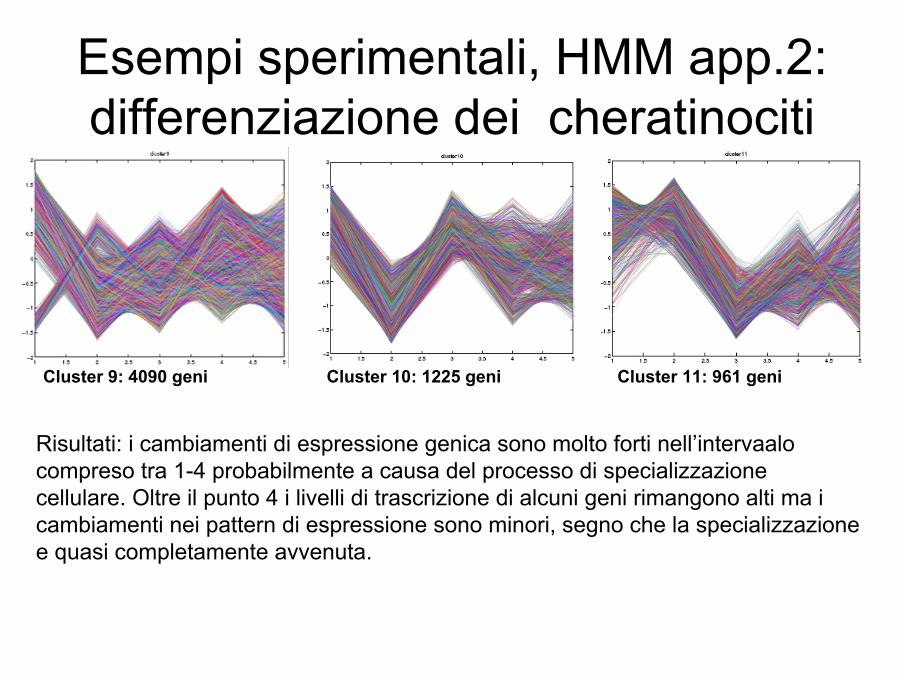
Esempi sperimentali, HMM app.2:differenziazione dei cheratinociti
Cluster 9: 4090 geni Cluster 10: 1225 geni Cluster 11: 961 geni
Risultati: i cambiamenti di espressione genica sono molto forti nell’intervaalocompreso tra 1-4 probabilmente a causa del processo di specializzazione cellulare. Oltre il punto 4 i livelli di trascrizione di alcuni geni rimangono alti ma i cambiamenti nei pattern di espressione sono minori, segno che la specializzazione e quasi completamente avvenuta.

Esempi sperimentali, HMMa1 Vs AR:ciclo cellulare di S.cerevisiae
(Schliep et al,2003)
Dataset: da cellule di lievito sincronizzate con α-factor sono stati misurati i livelli di espressione di 6178 geni ogni 7 minuti per 140 minuti, valutati un totale di 18 time pointsnormalizzati (Spellman et al, 1998). Rimossi i geni che non mostravano particolare over- o under- espressione è stato valutato un set di 1044 geni.
Modelli: utilizzata una collezione di 9 modelli aventi ognuno 9 stati.
Training: Baum-Welch, con parametri di rumori esclusi (de-noise)
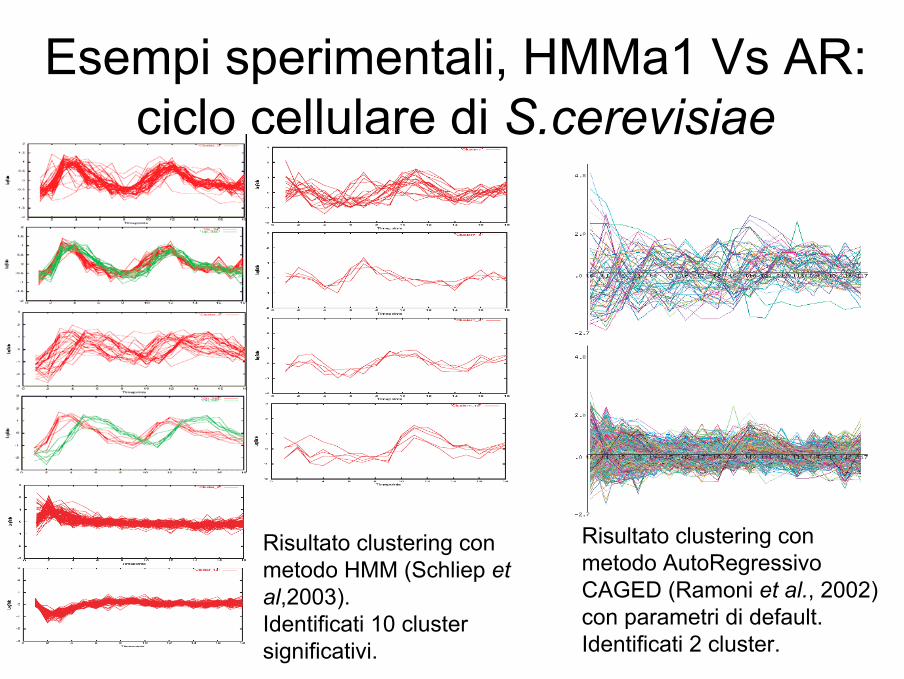
Esempi sperimentali, HMMa1 Vs AR:ciclo cellulare di S.cerevisiae
Risultato clustering con metodo AutoRegressivoCAGED (Ramoni et al., 2002) con parametri di default.Identificati 2 cluster.
Risultato clustering con metodo HMM (Schliep etal,2003).Identificati 10 cluster significativi.

bibliografia• Aach, J. (2001) Aligning gene expression time series with time warping algorithms, Bioinformatics 17(6),
459-508.• Bar-Joseph,Z., Gerber,G., Gifford,D.K. and Jaakkola,T.S. (2002) A new approach to analyzing gene
expression time series data. 6th Annual International Conference on Research in Computational MolecularBiology.
• Bar-Joseph, Z., Gerber, G., Gifford, D. K., Jaakkola, T. S. and Simon, I. (2002) A new approach to analyzing gene expression time series data, Proceedings of RECOMB, Washington DC, USA, pp. 39–48.
• Bolstad,B.M., Irizarry,R.A., A° strand,M., Speed,T.P. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias, Bioinformatics, vol.19(2), 185-193.
• Ji, X., Li-Ling, J. and Sun, Z. (2003) Mining gene expression data using a novel approach based on hiddenMarkov models, FEBS 542, 125–131.
• Lees, K., Taylor, J., Luntr, G., Hein, J. (2005) Identifyng gene clusters and regulatory themes using time course expression data, hidden markov models and trascription factor information. Oxford University
• MacLachlan, G.J., Bean, RW, Peel, D., (2001) A mixture model based approach to the clustering microarrays expression data, Bioinformatics, vol.18(3), 413-422.
• Moller-Levet, C. S., Cho, K.-H. and Wolkenhauer, O. (2003) Microarray data clustering based on temporal variation: FCV with TSD preclustering, Applied Bioinformatics 2(1), 35–45.
• Ramoni,M.F., Sebastiani,P. and Kohane,I.S. (2002) Cluster analysis of gene expression dynamics. Proc. Natl Acad. Sci. USA, 99, 9121–9126.
• Schleip, A., Schonhuth, A. and Steinhoff, C. (2003) Using hidden markov models analyze gene expression time course data, Bioinformatics 19(1), i255–i263.
• Spellman,P.T., Sherlock,G., Zhang,M.Q., Iyer,V.R., Anders,K., Eisen,M.B., Brown,P.O., Botstein,D. and Futcher,B. (1998) Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell, 9, 3273–3797.
Tutti gli articoli (anche non citati) utilizzati sono raccolti nella cartella “biblioteca” del cd allegato. Ringrazio chi ha seguito chi ha seguito la presentazione per la pazienza avuta oggi venerdì 27 luglio.

![ATTI del II Congresso Internazionale Colombiano Famiglia Colombo.pdf · pp. 64-65, doc. LVI] 1296, ottobre 12. Cuccaro Il dominus Ferrario Columbus riceve quietanza per beni acquistati](https://static.fdocumenti.com/doc/165x107/605fe0194cb61d55796abdbf/atti-del-ii-congresso-internazionale-famiglia-colombopdf-pp-64-65-doc-lvi.jpg)














![',02675$=,21( FRQWLQXDelio/DIDATTICA/aa0607/AlgELab/... · ',02675$=,21( frqwlqxd ,o qxphur gl qrgl Ù txlqgl gdwr gdooh uhod]lrql gl ulfruuhq]dulfruuhq]d q qqq q qqq q qqqnnnn qnnnn-](https://static.fdocumenti.com/doc/165x107/5fa206d227a0493a9e62fe7f/0267521-eliodidatticaaa0607algelab-0267521-frqwlqxd-o-qxphur.jpg)

