Algoritmi bioinformatici per la classificazione sintattica delle lingue
48
1 Università degli Studi di Trieste Dipartimento di Ingegneria e Architettura Laurea Triennale in Ingegneria dell’Informazione Curriculum Informatica Algoritmi bioinformatici per la classificazione sintattica delle lingue Relatore: Prof. Andrea Sgarro Correlatore: Prof. Alberto Casagrande Candidato: Devis Meka
-
Upload
devis111 -
Category
Technology
-
view
282 -
download
1
Transcript of Algoritmi bioinformatici per la classificazione sintattica delle lingue

1
Università degli Studi di Trieste
Dipartimento di Ingegneria e Architettura
Laurea Triennale in Ingegneria dell’Informazione
Curriculum
Informatica
Algoritmi bioinformatici per la classificazione
sintattica delle lingue
Relatore: Prof. Andrea Sgarro
Correlatore: Prof. Alberto Casagrande
Candidato: Devis Meka

2

3
Indice:
Introduzione 4
1.Algoritmi bioinformatici 6
1.1 Concetti generali 6
1.2 Metodi basati sui caratteri 8
1.3 Metodi basati sulle distanze 11
1.4 Metodi di consenso 13
2.Algoritmi bioinformatici nell’ambito della linguistica 14
2.1 Il problema della classificazione delle lingue 14
2.2 La Massima Parsimonia e i parametri linguistici 15
2.3 Il Metodo di Massima Verosimiglianza 20
2.4 Analisi dei risultati 23
3.Due tipi di distanze e una filosofia distance-based 26
3.1 Applicazioni di UPGMA e NJ sulle distanze di Hamming 26
3.2 Le distanze fuzzy di Hamming 32
3.3 Perche l’algoritmo NJ non funziona bene 38
4.Conclusioni 44

4
1.Introduzione
Il concetto d’algoritmo è indispensabile quando si vuole trovare una soluzione ad un
problema. L’algoritmo si può definire come una sequenza d’istruzioni che si devono seguire
in modo da risolvere un problema (Jones N, Pevzner P : 2004). In altre parole, un algoritmo è
il metodo in cui un dato input si trasforma nell’output. L’input può essere per esempio una
serie di numeri interi e l’output può essere il massimo tra questi numeri. Si può dimostrare
che esistono infiniti algoritmi che risolvono lo stesso problema, ognuno dei quali avrà
bisogno di un certa quantità di tempo per dare una risposta. La complessità di un problema
è, intuitivamente, il minor tempo necessario alla soluzione di una delle sue istanze. In molti
casi, dato un problema, è possibile stabilire la sua complessità e identificare un insieme di
algoritmi che risolvono tutte le sue istanze nel minor tempo possibile. In altri casi, la
complessità del problema è ignota e, nel caso in cui algoritmi che lo risolvano siano noti, è
disponibile una sola stima per eccesso.
La Bioinformatica è una disciplina recente che ha cercato di beneficiare dei mezzi informatici
per risolvere i problemi della Biologia. Così problemi come l’analisi delle somiglianze e le
differenze tra le varie sequenze dei geni o delle proteine vengono risolti grazie agli algoritmi
bioinformatici. Alcuni di questi algoritmi prendono in input un insieme di sequenze oppure
qualche misura di distanza tra ogni coppia di sequenze e restituiscono un albero che
rappresenta delle ipotetiche relazioni di parentela tra le varie specie. Tale albero è detto
albero filogenetico. Le foglie di un albero filogenetico rappresentano le specie considerate,
mentre i nodi interni sono associati a delle ipotetiche specie progenitrici. In particolare, ogni
nodo è progenitore delle foglie del sottoalbero radicato in esso. Se due specie vengono
rappresentate da nodi che giacciono su uno stesso ramo dell’albero filogenetico, la loro
distanza genetica è il numero di nodi che li separano.
Gli obbiettivi di questa tesi saranno: verificare il fatto che i metodi basati sulle distanze
funzionano meglio dei metodi basati sui caratteri in termini di classificazione e fare un
analisi dettagliata sui pregi e difetti delle distanze di Hamming e sulle distanze fuzzy di
Hamming come strumenti di misura delle divergenze tra le lingue.

5
Nel capitolo 1 viene fatto una rappresentazione generale dei metodi basati sui caratteri e dei
metodi basati sulle distanze, nonché dei metodi di consenso.
Nel capitolo2 vengono applicati i metodi basati sui caratteri su una griglia di parametri
sintattici e si prova a creare una classificazione delle lingue, ricostruendo diversi alberi
filogenetici.
Nel capitolo 3 vengono applicati gli algoritmi NJ e UPGMA prima sulle distanze di Hamming,
poi sulle distanze fuzzy di Hamming. In questo capitolo viene analizzato dettagliatamente il
funzionamento di NJ e vengono evidenziate le imprecisioni causate da questo metodo.

6
Desidero ricordare tutti coloro che mi hanno aiutato nella stesura della tesi con suggerimenti, critiche ed osservazioni: a loro va la mia gratitudine, anche se a me spetta la responsabilità per ogni errore contenuto in questa tesi.
Ringrazio anzitutto il professor Andrea Sgarro, Relatore, ed il professor Alberto Casagrande, Correlatore: senza il loro supporto e la loro guida sapiente questa tesi non esisterebbe.
Vorrei infine ringraziare le persone a me più care: i miei amici e la mia famiglia.

7
1.Algoritmi bioinformatici
1.1 Concetti generali
I metodi che vengono utilizzati per ricostruire l’albero filogenetico sono molteplici. In base al
formato dei dati in input possiamo distinguere due sottogruppi1:
a) Metodi basati sui caratteri
b) Metodi basati sulle distanze
I primi prendono in input un insieme di sequenze e valutano passo dopo passo ogni carattere
dell’alfabeto nucleotidico o amminoacidico, confrontando i caratteri che si trovano nella
stessa posizione in ogni sequenza. Mentre nei secondi si considera come input una matrice
di distanze, le cui componenti matriciali rappresentano le divergenze tra le differenti
sequenze.
L’input di questi metodi sarà un insieme di lingue e non un insieme di specie. Gli algoritmi
valuteranno sequenze di caratteri ‘+’, ‘-‘, ‘0’, che rappresentano rispettivamente la presenza,
l’assenza e la non applicabilità di alcune strutture sintattiche in una determinata lingua.
Queste sequenze sono state riassunte nella Figura 1. Nelle righe sono rappresentati i
parametri sintattici mentre nelle colonne le lingue da classificare.
1 Wing-Kin Sung, Algorithms in Bioinformatics, A Practical Introduction.

8
Figura 1. Tabella dei parametri, Longobardi ed altri, 2013

9
1.2 Metodi basati sui caratteri
Nei metodi basati sui caratteri l’input è costituito da un insieme di sequenze di caratteri.
Questi metodi ricostruiscono l’albero filogenetico, confrontando i caratteri corrispondenti
delle sequenze. L’input è una matrice M il cui elemento nella n-esima riga e m-esima colonna
corrisponde allo stato del carattere che si trova nella posizione m della sequenza n. In base al
criterio di ottimizzazione utilizzato per ricostruire questi alberi filogenetici si possono
distinguere 3 metodi distinti:
1) Massima Parsimonia
2) Compatibilità
3) Massima Verosimiglianza
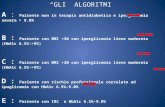
1.2.1 Massima Parsimonia
Il metodo di Massima Parsimonia probabilmente è tra i più diffusi e utilizzati tra quelli sovra
elencati. Il criterio della Massima Parsimonia afferma che tra tutti gli alberi che descrivano le
relazioni filogenetiche tra le sequenze, si deve scegliere l’albero che richiede il minor numero
di cambiamenti per spiegare la variabilità tra le sequenze. L’output di questo metodo può
essere un albero, o più di uno, con altezza pari al numero totale di cambiamenti nei rami
dell’albero. L’obbiettivo di questo metodo è proprio la minimizzazione di questa altezza.
Il processo attraverso il quale è possibile andare a determinare l’albero/gli alberi più
parsimoniosi può essere suddiviso in due problemi:
- Il piccolo problema parsimonioso (The Small parsimony problem). Presa in esame
una topologia d’albero, si vuole trovare l’altezza dell’albero e la distribuzione dei nodi
interni, che spiega l’evoluzione dell’ entità, in modo che ci sia il minimo numero di
cambiamenti. Per questo problema esistono vari algoritmi che danno una risposta in tempo
polinomiale.

10
- Il grande problema parsimonioso (The large parsimony problem). Presa in esame
una matrice di caratteri M, si vuole trovare l’albero più parsimonioso che spiega questa
matrice. Questo è un problema NP-hard, 2cioè esistono solo algoritmi che nei migliore dei
casi possono dare una risposta in tempo esponenziale. Comunque esistono diversi algoritmi
proposti per risolvere il problema, tra cui l’algoritmo di Fitch, il metodo di Hendy e Penny e
l’algoritmo approssimato.
1) Algoritmo di Fitch.
Questo algoritmo è stato proposto da Fitch e indipendentemente da Hartigan. L’algoritmo di
Fitch risolve il Piccolo Problema Parsimonioso in tempo polinomiale. Esso può essere usato
anche per risolvere il Grande Problema Parsimonioso. L’algoritmo di Fitch deve confrontare
(2*n-5)!! alberi filogenetici senza radice, dove n è il numero di sequenze di caratteri, per
trovare l’albero più parsimonioso. Questo algoritmo è basato sulla programmazione
dinamica.
In questa ricerca è stato utilizzato l’algoritmo “Pars” del software “PHYLIP 3.695”. Questo
algoritmo implementa la strategia di Wagner. Secondo Wagner, i cambiamenti tra i caratteri
sono equiprobabili in tutte le direzioni. In questo caso, ciò sta a significare che la
trasformazione +→- ha la stessa probabilità della trasformazione -→+. Questo algoritmo ci
dà la possibilità di inserire anche il simbolo ‘?’ per rappresentare quei casi dove manca
l’informazione oppure c’ è una dipendenza tra i parametri. L’albero filogenetico riprodotto
sarà senza radice. Questo problema può essere risolto usando un’entità qualsiasi come
radice. “Pars” ci offre la possibilità di mischiare anche l’ordine delle entità in input e di
decidere il numero massimo di alberi che vogliamo salvare.
2) Il metodo di Hendy e Penny
Questo metodo è stato proposto da Hendy e Penny nel 1982. Esso procede inizialmente
creando un albero costituito solo da 2 entità /specie, nel caso di un albero con radice oppure
costituito da 3 sequenze/specie nel caso di un albero senza radice. Di seguito aggiunge
un'altra entità all’ albero corrente e siccome un albero senza radice con n entità ha (2*n-3)
rami, allora l’algoritmo può inserire l’entità in qualsiasi delle (2*n-3) posizioni. In questa
maniera si riescono ad ottenere tutti gli alberi possibili che sono in numero (2*n-5)!!. L’idea
2 Graham and Foulds, 1982; Day, 1983.

11
di Hendy e Penny fu di interrompere il procedimento nel caso in cui l’albero considerato
superasse l’altezza di un albero già noto. Perciò appena ciò avviene viene interrotta
l’espansione di questo albero e si torna indietro rimuovendo l’entità appena aggiunta.
Questo metodo sfrutta la strategia della ricerca in profondità. E’ un algoritmo abbastanza
lento e chiede alcuni informazioni in anticipo, per esempio le lunghezze di un certo numero
di alberi già completati e non assicura sempre la soluzione .
Nel lavoro a seguire verrà utilizzato il metodo “Penny”, un algoritmo del software “PHYLIP
3.695”, al fine di ricostruire alberi basandoci sul metodo sovra descritto. “Penny”
implementa il criterio di Wagner, non permette di mischiare l’ordine delle entità in input,
accetta il carattere ‘?’ nei casi dove non ci sia informazione ma è molto lento. Infatti per
trovare l’albero più parsimonioso con questo algoritmo si sono dovute impiegare 12 ore.
3)Algoritmo approssimato
Questo algoritmo non trova una soluzione esatta ma una soluzione approssimata. Nei
migliori dei casi si ottiene una soluzione di grado 1.55. Questo valore rappresenta il rapporto
tra il risultato dell’algoritmo e i suoi costi ottimali, nel presente studio questo algoritmo
troverà un albero che avrà altezza al massimo 1.55 volte più grande dell‘ albero più
parsimonioso. Questo algoritmo riduce il problema della ricostruzione di un albero
filogenetico con il criterio di massima parsimonia nel problema del minimo albero di
connessione, cioè di quell‘albero connesso che minimizza i costi degli archi il quale è
risolvibile in tempo polinomiale.
1.2.2 Compatibilità
Il metodo di compatibilità assume che la maggior parte dei caratteri/stati che costituiscono
le entità/specie alterino il loro valore raramente e al massimo una volta nella loro storia.
Questo metodo ricostruisce l’albero filogenetico nel quale il numero dei caratteri che
alterino il loro valore una volta è massimo. La compatibilità assume che i valori che possono
prendere i caratteri in input siano binari, cioè 1 o 0.

12
1.2.3 Massima Verosimiglianza
Il metodo di Massima Verosimiglianza, diversamente dal metodo di Massima Parsimonia, usa
un modello esplicito di riferimento. Questo metodo è stato proposto da Felsenstein,
A.Edwards e E.Thompson. La matrice dell’input in questo caso si ipotizza sia il risultato di
alcuni modelli di evoluzione. L’albero che ricostruirà questo metodo sarà l’albero che meglio
spiega la matrice di input basato su questo modello di evoluzione.
In questo elaborato è stato impiegato il programma “Dnaml” del medesimo software
precedentemente indicato “PHYLIp 3.695”. Il modulo che implementa questo programma e’
il Modello Nascosto di Markov3. Quest’ultimo permette di impostare diversi tassi di
evoluzione per le entità. In più si possono impostare anticipatamente le probabilità di
evoluzione. Nel caso specifico la scelta è ricaduta su un tasso di evoluzione costante per
tutte le lingue.
1.3 Metodi basati sulle distanze
Questi metodi, diversamente da quelli precedenti, prendono in input una matrice M
simmetrica, dove un valore Mij al i-esima riga e j-colonna corrisponde alla distanza che
l’entità i-esima ha dall’entità j-esima. Questi metodi, a partire dalla matrice M,
ricostruiscono l’albero filogenetico e siccome vengono scelte 2 entità alla volta, l’albero
ricostruito sarà binario. Esistono vari metodi basati sulle distanze che differiscono tra loro
dal modo in cui vengono scelte le entità da fondere insieme.
A seguire verranno elencati alcuni dei metodi distance-based più importanti:
1.3.1 Il metodo Unweighted Pair Group with Aritmetic Means (UPGMA)
Questo metodo si basa sul fatto che entità simili devono trovarsi vicine nell’ albero
filogenetico e sull’ approccio bottom-up, cioè risolve il problema risalendo dal basso verso
l’alto. UPGMA è un metodo ultrametrico, ossia l’albero ricostruito avrà tutti i rami di ugual
lunghezza. Il metodo usa un algoritmo iterativo, il quale in n-1 iterazioni, dove n è il numero
delle specie/entità, raggruppa ad ogni passo le 2 entità che più si assomigliano (più
3 Trattato da Felsenstein e Churchill in una pubblicazione del 1996.

13
precisamente le 2 entità che hanno la distanza media più piccola). In questo modo il metodo
ricostruisce l’albero filogenetico in un tempo O(n^2)4. L’algoritmo UPGMA è robusto ma non
sempre riesce a trovare l’albero più parsimonioso e questo accade quando l’evoluzione tra
l’entità non è uniforme.
Per implementare questo metodo è stato scelto il programma “UPGMA ” del software
“PHYLIP 3.695”. UPGMA ha la caratteristica di produrre un albero con radice senza aiuto
esterno.5 Questo programma accetta le matrici di distanze sia in forma triangolare superiore
che triangolare inferiore.
1.3.2 Il metodo di Neighbor-joining(NJ)
Questa metodica è stata proposta nel 1987 ed è simile alla precedente. NJ parte da un
albero senza radice e con una topologia a stella, dove le 3 entità scelte all’inizio non hanno
nessuna relazione tra di loro. Inseguito, proseguendo nell’iterazione si scelgono le 2 entità
che hanno distanza minore e si collegano ad un padre comune. Da questo punto le 2 entità
vanno trattate come una singola entità. NJ si basa sulla proprietà additiva, ossia la distanza
tra una coppia di foglie dell’albero deve essere uguale alla somma delle distanze tra i nodi
che costituiscono il percorso tra le foglie. La complessità di NJ è uguale alla complessità di
UPGMA, O(n^2). L’albero ricostruito dall’algoritmo NJ può avere rami di lunghezza diversa e
il fatto che le entità possano avere una evoluzione non uniforme, non è una restrizione per
questo algoritmo.
1.4 Metodi di consenso
I metodi di consenso si usano per trovare un albero di consenso partendo da un insieme di
alberi. Questi metodi sono particolarmente interessanti quando si cerchi di ottenere un
albero di bootstrapping6 da un insieme relativamente grande di alberi di partenza. Ci sono
4 Questo notazione sta ad indicare che un raddoppio della dimensione dell’input, nei casi peggiori, quadruplica
il tempo di risposta dell’algoritmo. 5 Il fatto che UPGMA consideri che tutte le lingue abbiano un tasso di evoluzione costante implica che deve
esserci un antenato comune e le lunghezze dei rami debbano essere uguali. 6 Il concetto di bootstrapping viene trattato in seguito.

14
vari metodi di consenso proposti che differiscono tra di loro dal modo di scegliere i rami
dell’albero filogenetico finale. A seguire ne verranno descritti in breve:
a) La regola della maggioranza.
Questa regola è stata introdotta da Margush e McMorris nel 1981. L’albero di consenso
ricostruito basandosi su questo metodo avrà esattamente quei rami che compaiono almeno
nella metà degli alberi di partenza e si ottiene sempre un albero di consenso unico.
b) La regola stretta
Questa regola è stata introdotta da R.R.Sokol e F.J.Rohlf. L’albero di consenso che questa
regola ricostruisce contiene esattamente i rami che compaiono in tutti gli alberi di partenza.
Come nel metodo della maggioranza, l’albero ricostruito esiste sempre ed è unico.
c) La regola della maggioranza estesa
Questa regola si basa sullo stesso principio della regola della maggioranza, ma per
completare l’albero, dopo aver scelto i rami che compaiono in almeno la metà degli alberi di
partenza, usa anche gli altri rami nell’ordine di comparizione. In questo lavoro di tesi si è
scelto proprio questo metodo di consenso per trovare gli alberi di bootstrapping. Il
programma usato è stato Consense del software PHYLIP 3.695.
d) La regola della mediana
Questa regola considera le distanze tra tutte le coppie degli alberi di partenza. Per distanza
tra gli alberi si intende il numero di rami che compaiono in un albero ma non nell’altro.
L’albero di consenso proposto da questo metodo minimizza la somma delle distanze tra
tutte le coppie degli alberi. Questo albero esiste sempre ma non è unico.

15
2.Algoritmi bioinformatici nell’ambito della linguistica
2.1 Il problema della classificazione delle lingue
Dal diciannovesimo secolo l’evoluzione biologica e la storia delle lingue hanno seguito strade
parallele ma con la stessa meta. L’obbiettivo comune era classificare la popolazione umana e
le lingue in famiglie genealogiche, evidenziare le relazione di parentela e trovare l’origine
comune. Questo obbiettivo ha fatto in modo che queste due discipline apparentemente
lontane, possano condividere gli stessi meccanismi e procedure di comparazione e
ricostruzione.
Per classificare le lingue ed evidenziare le relazioni di parentela, all’inizio si è investigato sugli
elementi più facilmente accessibili come gli elementi lessicali: l’insieme delle parole e i
morfemi.7 Dall’altra parte la classificazione delle lingue raramente è stata basata sulle regole
sintattiche, le quali non sono cosi immediatamente accessibili. La classificazione basata
sull’entità lessicali è stata, fondamentalmente, rappresentata dal Metodo Classico di
Comparazione e dal Metodo Multilaterale. Il primo metodo indubbiamente è stato uno dei
successi più grandi delle scienze umane e riesce a risolvere parzialmente il problema delle
classificazione delle lingue senza usare metodi sofisticati. Questo metodo è stato introdotto
da Bopp F. e Rask R. e sfrutta le relazioni lessicali e fonetiche per classificare le lingue.
Presenta però una forte limitazione in quanto la velocità di evoluzione di queste entità
lessicali fa si che non si possano recuperare relazioni in un periodo di tempo più grande di
10,000 anni (Sgarro ed altri, 2011:1).
Il Metodo Multilaterale, conosciuto anche come metodo di Greenberg (1987, 2000), si basa
nuovamente sulle entità lessicali. Ma Greenberg pensava che bisognasse fare un confronto
dell’insieme delle parole non solo su una coppia di lingue, ma su un insieme più grande di
lingue nello stesso tempo. Questo metodo in teoria può essere applicato ad ogni lingua,
indipendentemente quanto remota possa essere. Anche questa strategia, però, fallisce nel
trovare misure precise nelle somiglianze di suoni o significati.8
Per aggirare questi ostacoli, in particolare la limitazione di applicabilità e l’impossibilità di
misurare distanze esatte, si è iniziato a cercare altri strumenti e metodi. Cosi inevitabilmente
7 Longobardi C.(2009:1680).
8 Longobardi C.( 2009:1679).

16
l’attenzione si è concentrata sulla sintassi ed in particolare sul Metodo Comparativo dei
Parametri(PCM). La pietra miliare in questa direzione fu la pubblicazione di
“Principles&Parameters”.9 In questa pubblicazione, i parametri vengono concepiti come
parte di una Grammatica Universale(UG) e come delle scelte aperte tra 2 valori binari.
Questo approccio verso la sintattica ha dato una svolta al problema della classificazione delle
lingue tenendo conto anche del fatto che le entità sintattiche cambiano più lentamente nel
tempo rispetto alle entità lessicali.10 Infatti i parametri sono immuni dalla selezione naturale,
al contrario delle entità lessicali che possono cambiare con il cambiamento dell’oggetto che
descrivano. I parametri sembrano essere immuni anche dal libero cambiamento individuale,
cioè dalla trasformazione, anche se piccola, che ognuno può dare alle entità lessicali come le
parole o i suoni. Nel PCM non c’è spazio per ambiguità siccome si conoscono perfettamente
le entità che vanno confrontate, diversamente dal caso delle entità lessicali.
2.2 Massima parsimonia e i parametri linguistici
In questo paragrafo si cercherà di implementare il metodo di massima parsimonia sulla
griglia dei parametri aventi a disposizione. Il metodo di MP è il metodo character-based più
facile per ricostruire alberi filogenetici ed è stato proposto per la prima volta in Edwards e
Calvalli-Sforza nel 1963. Il criterio della Massima Parsimonia si basa sul trovare l’albero che
richiede il minimo numero di cambiamenti.11 Ossia la Massima Parsimonia MP valuta diversi
alberi e fornisce l’albero (o gli alberi) che minimizza il numero dei cambiamenti di stati nei
suoi rami. Nel trovare l’albero oppure gli alberi che minimizzano il numero di cambiamenti,
questo algoritmo non solo prova ad aggiungere nuove foglie nei rami esistenti dell’albero,
ma cerca anche di aggiungerle alla fine dei nuovi rami.11 Dall’ altra parte questo metodo non
usa tutte le sequenze che vengono date in input, poiché alcune le considera senza
informazione.
Perché i parametri? I parametri sono universali e anche binari (al massimo ternari, nei casi
dove manca l’informazione o c’è una relazione di dipendenza). In questo modo, avendo una
lista di parametri universali che possono prendere solo due valori, si ha la possibilità di fare
misurazioni precise tra le lingue, usufruendo degli strumenti matematici e degli algoritmi
9 Chomsky, 1981.
10 Longobardi&Guardiano, 2009:1694.
11 J. Felsenstein, 2008.

17
bioinformatici. Questi parametri sono infatti applicabili a qualsiasi lingua,
indipendentemente se è una lingua recente o remota.
MP e i parametri sembrano fatti l’uno per l’altro, ma non risulta essere così. MP assume
infatti che i parametri siano indipendenti tra di loro in modo che possano essere considerati
informativi12. Dall’ altra parte i parametri linguistici hanno un grado di correlazione tra di
loro e questo problema si rifletterà nell’albero filogenetico ricostruito.
Nel caso trattato in questo studio, i parametri prendono i valori da un alfabeto ternario. Il
caso ‘0’ verrà escluso dalla computazione siccome è un caso dove non è presente
informazione, oppure il valore è prevedibile. Questo vuol dire che le coppie dove compare
almeno un simbolo ‘0’ non influenzeranno sulla parsimonia dell’albero. Ai fini della
computazione, anche le coppie dove appare almeno un ‘?’ non avranno effetto sulla
parsimonia dell’albero.
Facendo riferimento alla Tabella 7, dove nelle righe compaiono le lingue e nelle colonne
compaiono i vari parametri, ed utilizzando il programma “Seeqboot” del software Phylip
3.695, si sono ricostruite 2999 matrici di bootstrapping. Il bootstrapping è una tecnica
statistica di ricampionamento con riemissione che si usa per approssimare la distribuzione
campionaria di una statistica. Gli alberi ricostruiti dai metodi basati sui caratteri oppure sulle
distanze possono essere instabili, infatti se si rimuovono alcuni caratteri o se ne aggiungono
altri, è possibile avvenga un cambiamento della loro struttura. Il bootstapping viene
utilizzato appunto per fare diventare stabili questi alberi.
Una matrice di bootstrapping è una matrice che ha lo stesso numero di colonne della
matrice originaria. Per costruire questa matrice si scelgono casualmente diverse colonne
dalla matrice originaria, una o più volte. Queste matrici serviranno come input per il
programma “Pars” dello stesso software. In questo modo sono stati ricostruiti almeno 30000
alberi usando il metodo di Massima Parsimonia. Il fatto che ci siano almeno 30000 alberi,
partendo da 2999 matrici di dati, si spiega con la proprietà di questo metodo, cioè che
spesso non torna un albero unico ma un insieme di alberi parsimoniosi.
12
MP considera non informativi i parametri che hanno per esempio solo un carattere ‘+’ in confronto con un grande numero di ‘-‘ e ‘0’.

18
La figura raffigurante tutti gli alberi trovati non è stata riportata in questa tesi per mancanza
di spazio, ma partendo da questi alberi e usando il programma “Consense ” dello stesso
software è stato possibile ottenere un albero di consenso costruito basandosi sulla strategia
della “Regola della maggioranza estesa” (Figura 2). Questa strategia sceglie, inizialmente,
quei rami dall’insieme degli alberi che compaiono almeno nella metà delle volte e
successivamente, in base alla frequenza, sceglie quei rami che completa l’albero. I numeri
presenti sui rami indicano quante volte sono comparse su un insieme di 2999 alberi. Come si
può osservare la coppia Polacco-Russo la troviamo in 1369 alberi mentre la coppia formata
dalle lingue Hindi e Marathi compare in 2679 alberi. Questo albero in generale è coerente
con quello che si sarebbe dovuto ottenere. In questo modo i sottogruppi delle lingue slave
viene correttamente evidenziato con il Polacco, raggruppato a sua volta al Russo. Il Rumeno
viene rappresentato come esterno alle lingue Latine ed anche questo risulta essere corretto.
Ma dall’altra parte MP comporta alcuni imprecisioni. Ad esempio, come si può osservare
nella Figura 2 le lingue Germaniche vengono unite con le lingue Celtiche ed inoltre i dialetti
italiani (Siciliano, Salentino e Calabrese) vengono fusi insieme con i dialetti Greci (Bovese e
Grico) ed il Greco.

19
Figura 2. Albero di consenso basato sulla Massima Parsimonia.

20
Per avere una panoramica più chiara su come applicare la filosofia character-based in
generale ed il metodo della massima parsimonia in particolare sui parametri linguistici, si è
cercato di ricostruire anche un albero di consenso usando l’algoritmo di Hendy e Penny,
partendo da un insieme iniziale di 1000 alberi. L’albero ricostruito viene rappresentato nella
Figura 3. Di nuovo i numeri sui rami mostrano quante volte gli stessi rami compaiono su un
insieme di 1000 alberi di partenza. Questo metodo fa uso dell’algoritmo “branch and bound”
che non garantisce di trovare l’albero più parsimonioso (J.Felsenstein, PHYLIP) e risulta
essere abbastanza lento. Solo per un insieme di 10 entità, se il procedimento viene eseguito
fino in fondo, si riescono a ricostruire 34,459,425 alberi. Il particolare software, che è stato
utilizzato, è dipendente dall’ordine in cui le lingue compaiono in input. Questo algoritmo
inizialmente sceglie due lingue casualmente, e le unisce in un unico ramo. Dopo di che prova
ad aggiungere le altre lingue in ordine dell’input, nella posizione migliore che possa
trovare13. Per esimere tale dipendenza, prima della computazione è stato eseguito un
mescolamento nell’ordine dell’input per 1000 volte. In questo caso come radice esterna è
stata utilizzata la lingua Basca.14
L’albero ricostruito in generale, si comporta meglio della MP. Infatti, i dialetti italiani in
questo caso vengono correttamente raggruppati con le lingue romane. Dall’altra parte
questo algoritmo distingue bene le lingue Germaniche da quelle Celtiche. Ma anche in
questo caso sono presenti alcune imperfezioni. Ad esempio il Rumeno, anche se nella
posizione più esterna, viene raggruppato con il Greco, ed i suoi dialetti con le lingue Latine.
Una leggera imprecisione compare anche nel sottogruppo dei dialetti italiani dove il Siciliano
viene raggruppato con l’Italiano, ed il Calabrese compare nella posizione più esterna.
I risultati ottenuti finora, basandosi sulla Massima Parsimonia sono abbastanza soddisfacenti
e coerenti con ciò che concerne la classificazione delle lingue Indo-Europee, ma rimangono,
comunque, alcune imprecisioni.
13
Felsenstein, 2008. 14
Il Basco è stato selezionato come radice esterna perché è una lingua isolata che si differenzia da tutte le lingue indoeuropee. Il Basco, inoltre, viene considerato come una lingua senza legami con le altre lingue.(Trask, R.L :1997).

21
2.3 Il metodo di Massima Verosimiglianza
Per verificare se le imprecisioni siano frutto della Massima Parsimonia in particolare oppure
dei metodi basati sui caratteri in generale, è stato eseguito un terzo tentativo: un albero di
consenso basato sul metodo di Massima Verosimiglianza(Figura 3). All’inizio sono state
ricostruite 2999 matrici di bootstrapping, le quali sono servite come input al software DNAml
dell’PHYLIP, al fine di poter costruire 2999 alberi di Massima Verosimiglianza. Questo
algoritmo si basa sul Modello Nascosto di Markov (Hidden Markov Model). A seguito è stato
determinato un albero di consenso con la Regola della Maggioranza estesa, dove i numeri sui
rami mostrano quante volte quest’ultimi compaiano su un totale di 2999 alberi. I vantaggi
della Massima Verosimiglianza sono individuabili in questo metodo in quanto risulta ben
fondato statisticamente e considera come informativi tutte le sequenze dell’input
(F.Opperdoes:1997). Dall’altra parte questo metodo è relativamente lento e dipende dal
modello scelto come riferimento.

22
Figura 3. Albero di consenso basato sul metodo di Hendy-Penny.

23
Figura 4. Albero di consenso basato sul metodo di Massima Verosimiglianza

24
2.4 Analisi dei risultati
Come si osserva dai tre alberi ricostruiti con i metodi character-based (Figura 2, Figura 3,
Figura 4), le differenze tra di loro sono minime. Il primo albero di consenso costruito con il
metodo di massima parsimonia, utilizzando il software “Phylip 3.695” e partendo da più di
30000 alberi iniziali, riporta in modo corretto la posizione della lingua Bulgara nel
sottogruppo delle lingue slave. Risulta essere corretta anche la posizione della lingua
Rumena. Le imprecisioni presenti sono il mescolamento dei sottogruppi delle lingue
Germaniche e Celtiche ed il collasso in un unico sottogruppo dei dialetti greci e di quelli
italiani.
Il mescolamento tra le lingue Germaniche e Celtiche può essere stato causato da un certo
numero di parametri che i due sottogruppi condividono. Per esempio se prendiamo l’Inglese
ed il Gallese come rappresentanti dei due sottogruppi, solo i seguenti parametri non
vengono condivisi:
Le rimanenti coppie dei parametri sono uguali oppure presentano almeno un ‘0’. Questo
numero elevato di parametri uguali può essere conseguenza di uno sviluppo parallelo di
questi sottogruppi15. Anche il mescolamento tra i dialetti Greci, il Greco ed i dialetti italiani
può essere stata conseguenza di un’affinità tra questi due sottogruppi. Infatti dalla griglia è
possibile osservare che il numero dei parametri non condivisi risulta essere molto piccolo16.
In conclusione possiamo affermare che il numero ridotto dei parametri, nonché l’effetto
dello sviluppo parallelo tra alcuni sottogruppi, rendono difficile la distinzione tra tali
sottogruppi.
Il secondo albero, ricostruito col metodo di Hendy e Penny, classifica erroneamente il
Rumeno con i dialetti greci. Questa imprecisione può essere frutto del fatto che il Rumeno è
15
Questi sottogruppi sono stati sempre in contatto uno con l’altro, come ad esempio durante l’invasione Scandinava. Ad oggi il contatto e i conflitti tra l’Inglese e le lingue Celtiche è un dibattito che risulta essere ancora aperto. 16
Prendendo ad esempio il Siciliano ed il Greco come rappresentanti di ciascun sottogruppo, il numero dei parametri dove divergono i due e’ 4.

25
la lingua più esterna del sottogruppo Latino cosi come può essere dovuto anche all’usuale
problema del numero ridotto dei parametri in input.
Anche l’albero ricostruito con il metodo della Massima Verosimiglianza rappresenta alcuni
imprecisioni abbastanza evidenti. Infatti siamo in presenza di un mescolamento tra le lingue
Germaniche e quelle Celtiche, come nel caso dell’albero di Massima Parsimonia. Inoltre il
Farsi viene raggruppato dentro questo gruppo mescolato in 893 alberi su 2999, andando ad
aumentare l’imprecisione di questo metodo. Per il resto l’albero rappresenta abbastanza
bene i sottogruppi e colloca correttamente il Rumeno con le lingue Latine, come l’elemento
più esterno. La stessa cosa vale anche per il Bulgaro presente nel sottogruppo delle lingue
slave.
La collocazione della lingua Farsi nel sottogruppo delle lingue Celtiche può essere
conseguenza di un fenomeno definito come “Long branch attraction”17. Questo fenomeno
viene descritto come la tendenza di 2 sequenze che evolvono rapidamente e diversamente
dalle altre sequenze, di collassare in un unico nodo. In questo caso le 2 sequenze che
vengono unite non hanno un’affinità tra loro, ma sono presenti nello stesso ramo solo a
causa di questa attrazione tra i rami lunghi. Nel caso specifico, il Farsi è la lingua più esterna
all’insieme delle lingue Indo-Europee. Anche il sottogruppo delle lingue Celtiche si trova
all’estremità di questa classificazione. Queste lingue collassano in un unico gruppo 893 volte
su un totale di 2999 alberi.
Gli errori presenti anche nell’ultimo albero, come si può osservare dalla Figura 4, non sono
dovuti solo alla Massima Parsimonia in particolare ma ai metodi basati sui caratteri in
generale. Ma quali possono essere i fattori che generano questi comportamenti?
La risposta a questa domanda deve essere cercata nell’insieme dei parametri che sono stati
utilizzati per ricostruire questi alberi. Essi rispettano alcuni criteri e sono scelti con cura
(Longobardi&Guardiano:2009, Sgarro e collaboratori: 2011). Cosi i parametri linguistici
possono avere delle dipendenze parziale ma che siano note. Inoltre i parametri devono
essere il frutto di una quantità grande di dati intersecati. Ma cosa si aspetta in input un
metodo basato sui caratteri? Secondo J. Felsenstein (1986-2008), nell’ ipotesi della Massima
Parsimonia del software PHYLIP, deve essere garantita l’indipendenza evolutiva tra i
17
L’attrazione tra i rami lunghi, trattato da Felsenstein nel 1978, Gribaldo e Philippe nel 2002.

26
parametri ed inoltre il numero dei parametri deve essere relativamente grande in modo che
si possa ricostruire un albero affidabile.
I parametri di questo lavoro rappresentano legami di dipendenza, infatti i simboli ‘0’ e ‘?’
rappresentano quei casi dove non ci sia informazione, oppure quel parametro per una
precisa lingua dipende dal valore di un altro parametro. Nella Tabella1 vengono
rappresentati 4 parametri e 3 lingue.
Tabella 1. La dipendenza tra i parametri
Il valore del parametro 11 dipende direttamente dai parametri 5,6 e 8. Come si può
osservare anche in Tabella1, nel caso del Sloveno, se il parametro 5 non è ‘-‘, oppure il
parametro 6 non è ‘-‘ o il parametro 8 non è ‘+’, allora il parametro 11 non potrà essere
valutato e verrà posto a ‘0’. Perciò un unico cambiamento dà luogo ad una reazione a catena
che tende a cambiare la parsimonia dell’albero.
Usando la notazioni standard delle proposizioni logiche, una regola per il parametro 11
potrebbe essere18:
18
Sgarro ed altri, 2011:11.
Parametri Norwegian Serbo-Croat Slovenian
5.+/- feature spreadN + + +
6.+/-numb.on N + + +
8.grammar.def + 0 0
11.grammar.dist.art - 0 0

27
3. Due tipi di distanze e una filosofia distance-based
In questo capitolo si cercherà d’implementare i metodi basati sulle distanze nell’ ambito
della linguistica al fine di riuscire a classificare le lingue Indo-Europee. Poiché questi metodi
chiedono in input una matrice simmetrica costituita dalle distanze tra le entità (lingue nel
nostro caso), a partire dalla Tabella 7 si andranno a misurare 2 tipi diversi di distanze:
distanza di Hamming normalizzata e una distanza fuzzy di Hamming. In base a queste
distanze, usufruendo degli algoritmi distance-based, verranno ricostruiti vari alberi
filogenetici e infine verranno analizzati i risultati.
3.1 Applicazioni di NJ e UPGMA sulle distanze di Hamming
La distanza di Hamming tra 2 stringhe binarie della stessa lunghezza è il numero delle
posizioni nelle quali le 2 stringhe differiscono tra di loro. La distanza di Hamming è una
distanza metrica e come tale soddisfa anche la disuguaglianza triangolare:
dove a,b,c sono delle stringhe binarie.
Nel nostro caso l’alfabeto dei caratteri non è un alfabeto binario ma è un alfabeto ternario
{+,-,0}. Basandosi sulla Tabella 1, un carattere ‘+’ nella i-esima riga e j-esima colonna vuole
significare che il parametro alla posizione i-esima e condiviso dalla lingua nella posizione j-
esima, un carattere ‘-‘ vuole dire che la lingua j-esima non rispetta il parametro i-esima
mentre un carattere ‘0’ significa che è impossibile assegnare uno tra i valori ‘+’ o ’-’ oppure il
valore è prevedibile. Le coppie dei caratteri dove appare almeno uno ‘0’, indipendentemente
dal carattere con il quale viene associato, verranno esclusi dal calcolo della distanza di
Hamming tra le coppie delle lingue.
Nella seguente griglia sono state prese 2 lingue come esempio: l’Italiano e il Francese.
Indicando con ‘d’ il numero delle posizioni dove le 2 lingue differiscono, con ’n’ il numero
totale dei parametri e ‘z’ le posizioni dove compare almeno un ‘0’, la distanza di Hamming
normalizzata risulterà essere:

28
In questo caso d = 2, z = 14 e n = 56. La distanza di Hamming normalizzata sulle posizioni
sani sarà 0,0476. La matrice completa con le distanze tra tutte le coppie è presentata nelle
Figura 5, Figura 6 e Figura 7. Questa matrice è simmetrica e la diagonale principale
rappresenta le distanze identità.
Tabella 2. Griglia dei parametri per l’Italiano ed il Francese
Figura 5. La matrice delle distanze di Hamming tra le coppie delle lingue.
Ita + + + - + + + + + - - - 0 - - + - + 0 - + + + 0 + 0 + -
Fra + + + - + - + + + 0 - - 0 - - + - 0 0 - + + + 0 + 0 + -
Ita + - + - - - + 0 - 0 - - - 0 + + - - - + 0 0 0 0 0 - - +
Fra + - + - - + + 0 - 0 - - - 0 + + - - - + 0 0 0 0 0 - - +

29
Figura 6. Matrice delle distanze di Hamming tra le coppie delle lingue
Figura 7. Matrice delle distanze di Hamming tra le coppie delle lingue

30
Nella Figura 8 viene rappresentato un albero filogenetico ricostruito basandosi sul metodo
UPGMA. Questo metodo è un metodo tipico di clustering19 e unisce in un unico nodo quelle
entità che minimizzano la distanza media tra di loro costruendo un albero con radice e
binario. Yu-Rong Ch.(2012:1) . Dall’albero si vede che questo metodo dà i risultati più
soddisfacenti fin adesso. Tutti i sottogruppi sono ben distinti, con il Rumeno nella posizione
più esterna delle lingue romane, il Bulgaro nella posizione più esterna delle lingue slave del
sud, il Sloveno che giustamente viene unito con il Serbo-Croato(709 volte su 1000 alberi), le
lingue germaniche sono distinte dalle lingue Celtiche e i dialetti Greci sono correttamente
rappresentati. Anche i dialetti italiani sono ben collocati, con l’Italiano esterno al
sottogruppo. I numeri sui rami come sempre indicano il numero di volte che gli stessi rami
compaiono in un insieme di 1000 alberi. Per arrivare a questo albero ho usato la Regola della
Maggioranza estesa.
19
Il metodo di clustering procede accoppiando ad ogni iterazione due diverse specie che hanno la minima distanza tra di loro.

31
Figura 8. Albero di consenso basato sul metodo UPGMA

32
Figura 9. Albero di consenso basato sul metodo NJ

33
Nella Figura8 viene rappresentato un altro albero di consenso, questa volta ricostruito con il
metodo di Neighbor-Joining, il quale non assume che ci sia un tasso di evoluzione uguale per
tutte le lingue, e di conseguenza l’albero ricostruito non sarà ultrametrico. Questo metodo è
piu veloce dell’ UPGMA ed accetta anche valori di distanze negativi. NJ funziona bene
quando il tasso di evoluzione da un’entità ad un’altra e molto diverso. ( Felsenstein.J,
2008:PHYLIP). Di nuovo questo albero è un albero di consenso ricostruito con la Regola della
Maggioranza estesa e partendo da 2999 alberi.
Come si vede dall’albero,NJ funziona relativamente bene ma rispetto UPGMA presenta
alcune imprecisioni.
3.2 Le distanze fuzzy di Hamming
La distanza fuzzy di Hamming20, si può considerare come una estensione della distanza di
Hamming ‘classica’. Date 2 stringhe della stessa lunghezza con caratteri da un alfabeto
ternario {+,-,b}, dove b rappresenta il caso della mancanza dell’informazione oppure il
risultato è prevedibile, per trovare la distanza fuzzy di Hamming, diversamente da come
abbiamo fatto prima, non possiamo più ignorare le posizioni dove compare almeno una ‘b’.
A questi posizioni gli daremmo un peso specifico ‘w’ preso dall’intervallo continuo [0,1]. Il
caso particolare quando w=1/2 rappresenta quella che si chiama la distanza fuzzy di
Hamming. In altre parole questo vuol dire che, ogni volta che in una coppia di caratteri che
confrontiamo,compare almeno un zero (b) si aumenta il numero di differenze con 0,5.
La scelta particolare w=0,5 e in generale w>=0,5 dipende dal fatto che al di sotto di questo
valore, la proprietà triangolare non vale ed l’uso dei algoritmi come UPGMA e NJ può dare
risultati errati(Bortolussi, L., Sgarro, A., 2007:11).
La distanza fuzzy di Hamming rispetta la disuguaglianza triangolare (Sgarro, 2007:2 ) ed e ‘
stata usata in precedenza dal linguista Muljačić per classificare le lingue romane nel 1967.
In questo paragrafo io proverò a fare la stessa cosa con le lingue Indo-Europee, ricostruendo
alberi filogenetici basandomi sugli algoritmi distance-based e usando proprio le distanze
20
Sgarro Distance, come nel Dictionary of Distances, Deza E., Deza M. M;

34
fuzzy di Hamming. Riprenderò le 2 lingue usate in precedenza, l’Italiano e il Francese e
misurerò la distanza fuzzy di Hamming.
Ita + + + - + + + + + - - - 0 - - + - + 0 - + + + 0 + 0 + -
Fr
a
+ + + - + - + + + 0 - - 0 - - + - 0 0 - + + + 0 + 0 + -
Ita + - + - - - + 0 - 0 - - - 0 + + - - - + 0 0 0 0 0 - - +
Fr
a
+ - + - - + + 0 - 0 - - - 0 + + - - - + 0 0 0 0 0 - - +
Tabella 3. La griglia dei parametri per l’Italiano ed il Francese
Come prima, sia ‘d’ il numero dei caratteri dove differiscono le 2 lingue escludendo le
coppie dove appare almeno un 0 , ’b’ il numero delle coppie dove appare almeno un
carattere 0. Nel nostro caso d=2, b=14. Siccome i caratteri ‘0’ devono essere pesati con
w=0,5 allora il valore totale del loro contributo sarà 7. La distanza fuzzy di Hamming sarà:
La matrice completa con tutte le coppie delle distanze tra le lingue è rappresentata nella
Tabella3. In questo caso le distanze identità che si trovano nella diagonale principale, cioè
d(x,x) dove ‘x’ è una qualsiasi lingua, sono forzate a 0. Infatti la distanza fuzzy di Hamming
viola uno dei criteri per essere una pseudo-distanza metrica : si può avere una distanza
identità strettamente maggiore di zero. (Sgarro, 2007:8).
Ma questo fatto è irrilevante ai fini del nostro scopo. La matrice è stata rappresentata nella
forma triangolare superiore .
La distanza fuzzy di Hamming è più recente ed è stata introdotta per correggere un
inconvenienza della distanza di Hamming normale. Quest’ultima fa delle misurazioni
assolute senza tener conto della vicinanza o del quasi uguale. Mentre la distanza fuzzy di
Hamming considera anche la vicinanza tra due stringhe, con distanze minori alle stringhe più
vicine. Per questo motivo Muljačić ha scelto questa distanza per fare la sua classificazione,
dando peso ai casi “parziali”. Ma diversamente dal caso di Muljačić dove si fa la

35
comparazione basandosi sulle entità lessicale21 e il fatto di somiglianze parziali ha
significato, nel nostro caso i caratteri ‘0’ stanno a significare una mancanza d’informazione
oppure una dipendenza dei parametri.
Tabella 4. Le distanze fuzzy di Hamming tra tutte le coppie.
Nella Figura10 viene rappresentato l’albero di consenso per il metodo Neighbor-Joining
basato sulle distanze fuzzy di Hamming. Questo metodo non fa una ricostruzione abbastanza
accurata come UPGMA ma ricompensa questa imprecisione con la sua velocità di esecuzione
(Felsenstein, 2008 : PHYLIP). Inizialmente, partendo dalla matrice originaria, vengono
ricostruite 100 matrice di bootstrapping. In seguito vengono ricostruite 100 alberi
filogenetici basandosi sul metodo di Neighbor-Joining e infine viene trovato l’albero di
consenso in base alla Regola di Maggioranza estesa. Viene usato il Basco come radice
dell’albero.
21
Sgarro ed altri, 2011.

36
Come si vede nella Figura 10 ci sono alcune imprecisioni in confronto allo stesso metodo
basato sulle distanze di Hamming classica. NJ sulle distanze fuzzy classifica in modo errato il
Rumeno unendolo con il Greco ed i suoi dialetti(38 volte su 100 alberi), cosi come classifica
in modo errato anche il Bulgaro come l’elemento più esterno di questo maxi-gruppo di
lingue Latine e Greche(25 volte su 100 alberi). Quest’ultimo errore può essere spiegato con il
fatto che il Bulgaro è la lingua più esterna del sottogruppo delle lingue slave e dall’altra parte
il Bulgaro è attratto dal Rumeno. Ma queste imprecisioni sono dovute al metodo specifico
oppure alle distanze fuzzy che non rappresentano correttamente le distanze tra le lingue?
Per rispondere a questa domanda viene applicato sulle stesse distanze fuzzy il metodo
UPGMA(Figura 11). Questo metodo ha funzionato molto bene con le distanze classiche ma
con le distanze fuzzy le cose non vanno cosi bene. L’albero di consenso con il metodo
UPGMA partendo da 100 alberi iniziali classifica in modo errato il Bulgaro raggruppandolo
addirittura con le lingue germaniche (45 volte su 100 alberi), mentre vengono confermate le
coppie Serbo-Croato con lo Sloveno(74/100), il Rumeno come elemento esterno delle lingue
romane ed il Farsi come l’elemento più esterno della classificazione(90/100 alberi). Questo
albero riesce anche a distinguere le lingue nord-germaniche dalle lingue germaniche
dell’ovest,con il Norvegese che viene raggruppato con il Danese e l’Islandese,e l’Inglese con il
Tedesco come lingue esterne a queste sottogruppo. Ma come ho sottolineato l’errore più
grave resta la classificazione del Bulgaro con quest’ultimo sottogruppo.

37
Figura 10. Albero di consenso NJ sulle distanza fuzzy

38
Figura 11. Albero di consenso UPGMA sulle distanze fuzzy

39
3.3 Perché l’algoritmo NJ non funziona bene?
Se vengono confrontati gli alberi ricostruiti dai metodi NJ e UPGMA sulle distanze di
Hamming e quelle fuzzy, si vedono delle piccole differenze. Cosi partendo con l’algoritmo
Unweighted Pair Group Method with Arthimetic Mean , l’albero sulle distanze di Hamming si
comporta relativamente meglio di quello basato sulle distanze fuzzy. In particolare la
posizione del Bulgaro è sbagliata. Il Bulgaro viene raggruppato con le lingue Germaniche,
invece che con le lingue Slave come succede con le distanze di Hamming “classiche” .
Nella Tabella 4 ho provato a confrontare il Bulgaro con altre lingue Slave e Germaniche, in
termini di distanze. Le distanze sono calcolate come la media della distanze del Bulgaro da
ogni lingua del rispettivo sottogruppo.
Tabella 5. Confronto tra 2 tipi di distanze.
Come si vede da questa tabella, la situazione cambia con le distanze fuzzy. Cosi nel caso delle
distanze di Hamming “classica”, il Bulgaro è più vicino alle lingue Slave rispetto alle lingue
Germaniche. Mentre nel caso delle distanze fuzzy, il Bulgaro è più vicino alle lingue
Germaniche che quelle Slave. Il motivo di questo ribaltamento lo possiamo trovare nella
Tabella1 dei parametri. Cosi i parametri 7, 8, 11, 12, 13 e 14 per quanto riguarda le lingue
Slave prendono il valore ‘0’. Questi parametri fanno si che il Bulgaro sia più distante dalle
lingue Slave piuttosto che dalle lingue Germaniche. Questo è il motivo della classificazione
errata del Bulgaro nell’albero UPGMA delle distanze fuzzy.
Se facciamo un confronto tra i due algoritmo basati sulle distanze, indipendentemente dalle
distanze usate, si vede che UPGMA funziona leggermente meglio di NJ. Cosi il Neighbor-
Joining sulle distanze di Hamming “classiche” classifica erroneamente il Bulgaro con le lingue
Indo-Iraniane invece che con le lingue Slave. Mentre il Neighbor-Joining sulle distanze fuzzy
fa ancora peggio, poiché classifica erroneamente sia il Bulgaro che il Rumeno con i dialetti
greci. Ma in questo caso, diversamente da prima, la causa principale di questi imprecisioni e
Lingua Bulgara Lingue Slave Lingue Germaniche
Distanza di Hamming 0,1102 0,1532
Distanza fuzzy di Hamming 0,2633 0,2321

40
l’algoritmo stesso. Per avere una spiegazione di questo comportamento viene preso un
insieme di lingue piccolo e viene implementato sia il metodo di NJ, sia il metodo UPGMA
basandosi sulle distanze fuzzy di Hamming. Cosi vengono scelto le lingue latine, i dialetti
greci, il Rumeno ed il Bulgaro come input. La matrice delle distanze è rappresentato nella
Tabella In questo caso le distanze sono calcolate come la media delle distanze tra il Rumeno,
il Bulgaro e le singole lingue dei sottogruppi.
Lingua Rumeno Lingue latine Dialetti grechi Bulgaro
Rumeno 0.0 0.249 0.2827 0.2946
Lingue latine 0.2493 0.0 0.2864 0.2640
Dialetti grechi 0.2827 0.2864 0.0 0.3243
Bulgaro 0.2946 0.2640 0.3243 0.0
Tabella 6. Le distanze fuzzy iniziali.
ll Rumeno è l’unica lingua del blocco dell’Est che è parte del sottogruppo delle lingue latine
ed per secoli è stata isolata dallo sviluppo del mondo Neolatino. Nel corso della storia, il
Rumeno è stato influenzato da un certo numero di lingue e di culture. Cosi le lingue slave, la
lingua Turca, l’Albanese, la lingua Greca, l’Ungherese ma soprattutto le lingue latine hanno
dato il loro contributo nella trasformazione del Rumeno, una trasformazione che non è
ancora finita(Niculescu A.,1981). Questa diversità si traduce in difficoltà di classificazione
negli alberi filogenetici.
Neighbor-Joining usa un algoritmo iterativo di clustering per ricostruire l’albero. Questo
metodo parte da un albero senza radice a topologia a stella ed unisce ad un padre comune
quelle lingue che hanno la distanza minima. Ma siccome questo algoritmo considera anche
entità che hanno un tasso di evoluzione molto diversa, può non bastare cercare tra le foglie
più vicine in questa matrice. Può succedere che si abbiano due foglie vicine ma la distanza

41
tra una delle due entità e un'altra foglia sia più piccola22. Infatti NJ non usa questa matrice
direttamente ma ne ricostruisce un’altra. La nuova matrice ricostruita è rappresentata nella
Figura 6. Per arrivare a questa matrice, l’algoritmo NJ prima calcola la divergenza per ogni
lingua, cioè la somma delle distanze per ogni riga . La matrice delle divergenze è
rappresentata nella Tabella6 mentre la formula per ricavare questo divergenze è:
Dove N è il numero delle lingue.
Tabella 7. Le divergenze delle lingue.
In un passo successivo viene calcolato la matrice finale. Per calcolare, per esempio, la
distanza finale tra la lingua Rumena e i dialetti greci occorre trovare la differenza tra la
distanza iniziale e la somma delle divergenze. In questo modo si trovano le distanze tra tutte
le coppie.
Tabella 8. Le distanze fuzzy finali
Da questa matrice si possono scegliere le coppie che hanno la distanza più piccola le quali
andranno unite in un unico nodo. Come si vede le coppie con la distanza più piccola sono il
22
Questo può succedere quando le lunghezze dei rami sono molto diverse.
Rumeno Lingue latine Dialetti grechi Bulgaro
0.4133 0.3998 0.4467 0.4414
Lingua Rumeno Lingue latine Dialetti grechi Bulgaro
Rumeno 0.0
Lingue latine -0.5638 0.0
Dialetti grechi -0.5773 -0.5601 0.0
Bulgaro -0.5601 -0.5772 -0.5638 0.0

42
Rumeno e i Dialetti Grechi, e l’algoritmo le classifica insieme . Da questo punto in poi, queste
due coppie saranno un’unica unita. Quindi il procedimento viene ripetuto di nuovo, fino a
trovare due elementi. L’albero ricostruito viene rappresentato nella Figura 12. La lingua
Basca viene usata come radice dell’albero. I numeri sui rami indicano in quale passaggio
dell’algoritmo vengono uniti i seguenti rami.
Figura12. Albero NJ sulle distanze fuzzy
Come si vede il Rumeno viene classificato in modo errato con i dialetti Grechi, cosi come il
Bulgaro. Ma cosa succede con l’algoritmo UPGMA?
Il metodo UPGMA si basa sull’ipotesi che il tasso di evoluzione sia quasi uguale tra tutte le
entità. Per ricostruire l’albero filogenetico, questo metodo non ricostruisce un’altra matrice.
UPGMA trova direttamente le coppie che hanno la distanza minima nella matrice di
partenza. Cosi nella matrice delle distanze, l’UPGMA classifica insieme il Rumeno con le
lingue Latine, visto che hanno la distanza più piccola tra tutte le coppie delle lingue. L’albero
ricostruito è rappresentato nella Figura 13. Di nuovo i numeri sui rami rappresentano il passo
dove i stessi rami sono riuniti. Come si vede dall’albero, per prima si riunisce il Rumeno con
le lingue Latine. Quindi si aggiunge il Bulgaro.

43
Figura13. Albero UPGMA sulle distanze fuzzy
Come si vede(Figura 12, Figura 13), l’UPGMA funziona meglio di NJ. Questa differenza può
essere conseguenza di alcuni fattori. Cosi per esempio NJ non fa una ricostruzione molto
accurata quando le specie hanno un tasso di evoluzione quasi uguale 23 ed è molto sensibile
al modello di evoluzione scelto. Dall’altra parte NJ funziona bene con un insieme grande di
dati, ed è molto utile per fare operazioni di consenso o di bootstrapping24. Il metodo UPGMA
ipotizza che il tasso di evoluzione tra tutte le lingue sia quasi uguale e ciò ci permette una
ricostruzione corretta.
Figura 14. Albero NJ sulle distanze fuzzy
23
Kuhner e Felsenstein hanno dimostrato nel 1994 con simulazioni nel computer che NJ fa una valutazione meno accurata della UPGMA . 24
Felsenstein:PHYLIP

44
Nella Figura14 viene rappresentato lo stesso insieme di lingue, meno il Bulgaro. Il Bulgaro
viene lasciato fuori dalla computazione per vedere il comportamento del Rumeno rispetto
alle lingue Latine e i dialetti Greci. Come si vede, in questo caso, il Rumeno viene collocato
correttamente con le lingue Latine. Questo esperimento sostiene il fatto che NJ comporta
delle imprecisioni quando le entità da classificare sono lingue contemporanee con un tasso
di evoluzione più o meno uguale.

45
4.Conclusioni
In questa ricerca si confrontano due diverse filosofie e due diverse distanze. La filosofia
basata sui caratteri viene confrontata con quella basata sulle distanze. Dall’altra parte viene
fatto un confronto anche tra le distanze di Hamming e le distanze fuzzy di Hamming.
Dall’analisi fatta, si conclude che i metodi basati sui caratteri in generale funzionano bene
con i parametri sintattici e gli alberi ricostruiti sono coerenti con quello che ci aspettiamo.
Comunque ci sono delle imprecisioni dovuti non tanto ai metodi stessi ma al fatto che il
numero dei parametri in grado di identificare in modo chiaro i sottogruppi delle lingue è
piccolo. Dall’altra parte, anche la dipendenza tra i parametri usati fa si che ci siano delle
imprecisioni negli alberi filogenetici ricostruiti.
Per quanto riguarda i metodi basati sulle distanze,l’UPGMA funziona meglio di NJ. UPGMA
applicato sulle distanze di Hamming è quello che ricostruisce la migliore classificazione delle
lingue Indo-Europee. Mentre NJ riporta alcuni imprecisioni sopratutto per quanto riguarda il
mescolamento tra le lingue Germaniche e quelle Celtiche, e il posizionamento del Bulgaro.
Le distanze fuzzy in generale danno vita a classificazioni meno accurate e con tanti
imprecisioni, sia applicando UPGMA che NJ. Probabilmente questa mancanza di accuratezza
può essere spiegato con il fatto che queste distanze non rappresentano un vero strumento
di misura della diversità tra le lingue.
In futuro, usando i metodi basati sui caratteri su un numero maggiore di dati da valutare, si
potranno ottenere risultati migliori.

46
Bibliografia
[1] Bortolussi L., Sgarro A., 2007, Hamming-like Distances for Ill-Defined Strings in Lingusitic
Classification, 2007, RIMUT.
[2] Bortolussi L., Sgarro A., Liviu, P., D., Measures of fuzzy disarray in linguistic typology, 2009,
RIMUT.
[3] Chomsky N., Lasnik H., “Principles and Parameters Theory”, in Syntax: An International Handbook
of Contemporary Research, 1993.
[4] Bortolussi, L., Longobardi, G., Guardiano, C, Sgarro, A., 2011, How many languages are there?
Biology, Computation and Linguistics: New interdisciplinary Paradigms.
[5] Deza, E., Deza, M., M., Dictionary of Distances, 2006, Elsevier.
[6] Dinu, L., P., Sgarro, A., A low complexity distance for DNA strings, 2006, Fundamenta
Informaticae.
[7] Felsenstein, J., Inferring Phylogenis, 2004, Sinauer Associates.
[8] Felsenstein, J., PHYLIP, version 3.695.
[9] Jones, N., C., Pevzner, P., A., An introduction to bioinformatics algorithms, 2004, Bredford Books.
[10] Longobardi, G., Guardiano, C., Evidence for syntax as a signal of historical relatedness, 2009,
Lingua 119.
[11] Minett W. J., Wang Williams S.-Y., On detecting borrowing: distance-based and character-based
approaches, Hong-Kong, P.R.C.
[12] Nakhleh L., Warnow T., Ringe D., Evans N., S., 2005, Transactions of the Philological Society, Rice
University.

47
[13] Saitou N., Masatoshi N., The Neighbor-joining Method: A new Method for Riconstruing
Phylogenetic Trees, 1987, University of Tokyo, Tokyo.
[14] www.evolution.genetics.washington.edu/phylip.html.
[15] www.icp.ucl.ac.be/~opperd/private
[16] www.online.liebertpub.com/doi/abs/10.1089/106652702761034136.

48