
12.4 Polimeri stirenici termoplastici...monomeri vinilici, quali acrilonitrile (AN) e altri...
26
12.4.1 Introduzione Già nell’antichità erano note le singolari proprietà di un balsamo aromatico, noto come Styrax liquidus, estratto da Liquidambar orientalis, un albero diffuso nell’Asia Minore. Tale sostanza solidificava in presenza di aria, consentendo così di irrigidire o impermeabilizzare tes- suti e altri materiali. Il nome stirolo si deve al chimico tedesco Eduard Simon, che nel 1839 perfezionò il meto- do di distillazione dello Styrax, ottenendo appunto lo sti- rene monomero (Scheirs Priddy, 2003). Nel corso della sua attività, Simon osservò anche che, in presenza di ossigeno, lo stirolo si trasformava in una sostanza soli- da, che denominò metastirolo. Il carattere macromolecolare del metastirolo fu poi ipotizzato e dimostrato da Hermann Staudinger negli anni Venti del 20° secolo (Staudinger, 1932). A lui si deve anche lo studio strutturale che portò all’individua- zione della relazione tra assenza di stereoregolarità e carattere amorfo del polistirene (PS, polystyrene). Gli studi di Staudinger, e in generale della scuola tedesca, portano a un rapido sviluppo industriale nell’area ger- manica. Già nel 1931, IG Farben inizia la commercia- lizzazione del polistirene cristallino (GPPS, General Purpose PolyStyrene). L’impulso decisivo allo sviluppo dei materiali stirenici coincide però con gli anni della Seconda Guerra Mondiale, quando la difficoltà di impor- tazione della gomma naturale, a causa dei contrapposti blocchi navali, spinse le principali potenze a incremen- tare la produzione di stirene da destinare alla prepara- zione di gomme sintetiche. Nell’immediato dopoguerra, il ripristino dei com- merci con i paesi produttori di lattici naturali causò una notevole sovracapacità di stirene, che diede una spinta irreversibile alla crescita dei materiali polimerici a base stirenica. Grazie anche al volano economico della ri- costruzione edilizia, inizia la rapida diffusione com- merciale del polistirene espandibile (EPS, Expandable PolyStyrene), un altro materiale chiave. Nel 1954 Dow inizia la produzione industriale del polistirene anti- urto (HIPS, High Impact PolyStyrene; Amos et al ., 1954; Amos, 1974), che risolve i tradizionali limiti di resistenza meccanica del GPPS. Il 1959 vede la prima commercializzazione del ter-polimero acrilonitrile- butadiene-stirene (ABS). Negli stessi anni inizia la produzione del copolimero stirene-acrilonitrile (SAN). La tab. 1 riporta la composizione dei principali poli- meri stirenici termoplastici. 837 VOLUME II / RAFFINAZIONE E PETROLCHIMICA 12.4 Polimeri stirenici termoplastici Polimero Stirene PB AN Espandente GPPS (General Purpose Polystyrene) sì – – – SAN (Styrene-Acrylonitrile) sì – sì – EPS (Expandable Polystyrene) sì – – sì HIPS (High Impact Polystyrene) sì sì – – ABS (Acrylonitrile-Butadiene-Styrene) sì sì sì – tab. 1. Definizione e composizione dei principali polimeri stirenici termoplastici PB, gomma polibutadienica; AN, comonomero acrilonitrile
Transcript of 12.4 Polimeri stirenici termoplastici...monomeri vinilici, quali acrilonitrile (AN) e altri...

12.4.1 Introduzione
Già nell’antichità erano note le singolari proprietà di unbalsamo aromatico, noto come Styrax liquidus, estrattoda Liquidambar orientalis, un albero diffuso nell’AsiaMinore. Tale sostanza solidificava in presenza di aria,consentendo così di irrigidire o impermeabilizzare tes-suti e altri materiali. Il nome stirolo si deve al chimicotedesco Eduard Simon, che nel 1839 perfezionò il meto-do di distillazione dello Styrax, ottenendo appunto lo sti-rene monomero (Scheirs Priddy, 2003). Nel corso dellasua attività, Simon osservò anche che, in presenza diossigeno, lo stirolo si trasformava in una sostanza soli-da, che denominò metastirolo.
Il carattere macromolecolare del metastirolo fu poiipotizzato e dimostrato da Hermann Staudinger neglianni Venti del 20° secolo (Staudinger, 1932). A lui sideve anche lo studio strutturale che portò all’individua-zione della relazione tra assenza di stereoregolarità ecarattere amorfo del polistirene (PS, polystyrene). Glistudi di Staudinger, e in generale della scuola tedesca,portano a un rapido sviluppo industriale nell’area ger-manica. Già nel 1931, IG Farben inizia la commercia-lizzazione del polistirene cristallino (GPPS, General
Purpose PolyStyrene). L’impulso decisivo allo sviluppodei materiali stirenici coincide però con gli anni dellaSeconda Guerra Mondiale, quando la difficoltà di impor-tazione della gomma naturale, a causa dei contrappostiblocchi navali, spinse le principali potenze a incremen-tare la produzione di stirene da destinare alla prepara-zione di gomme sintetiche.
Nell’immediato dopoguerra, il ripristino dei com-merci con i paesi produttori di lattici naturali causò unanotevole sovracapacità di stirene, che diede una spintairreversibile alla crescita dei materiali polimerici a basestirenica. Grazie anche al volano economico della ri-costruzione edilizia, inizia la rapida diffusione com-merciale del polistirene espandibile (EPS, ExpandablePolyStyrene), un altro materiale chiave. Nel 1954 Dowinizia la produzione industriale del polistirene anti-urto (HIPS, High Impact PolyStyrene; Amos et al.,1954; Amos, 1974), che risolve i tradizionali limiti diresistenza meccanica del GPPS. Il 1959 vede la primacommercializzazione del ter-polimero acrilonitrile-butadiene-stirene (ABS). Negli stessi anni inizia laproduzione del copolimero stirene-acrilonitrile (SAN).La tab. 1 riporta la composizione dei principali poli-meri stirenici termoplastici.
837VOLUME II / RAFFINAZIONE E PETROLCHIMICA
12.4
Polimeri stirenici termoplastici
Polimero Stirene PB AN Espandente
GPPS (General Purpose Polystyrene) sì – – –
SAN (Styrene-Acrylonitrile) sì – sì –
EPS (Expandable Polystyrene) sì – – sì
HIPS (High Impact Polystyrene) sì sì – –
ABS (Acrylonitrile-Butadiene-Styrene) sì sì sì –
tab. 1. Definizione e composizione dei principali polimeri stirenici termoplastici
PB, gomma polibutadienica; AN, comonomero acrilonitrile

12.4.2 Chimica dellapolimerizzazione
Di seguito sono descritti i meccanismi di sintesi dei mate-riali stirenici termoplastici più diffusi.
Polistirene omopolimeroIl GPPS è prodotto largamente nel mondo per via
radicalica con reazione in fase liquida. Con questo mec-canismo il monomero stirene polimerizza, in assenza diossigeno o di altri inibitori, anche per semplice riscal-damento, poiché la formazione di radicali che inizianole catene polimeriche avviene attraverso un intermediodi reazione di Diels-Alder (addotto Diels-Alder, ossia 1-fenil-1,2,3,8-tetraidronaftalene), instabile, ottenuto percombinazione di due molecole di stirene (Mayo, 1968).In fig. 1 è mostrato il meccanismo complesso della rea-zione di formazione dei radicali di inizio della polime-rizzazione e degli oligomeri ciclici che si formano paral-lelamente al polimero solubile in stirene (ed eventualisolventi aromatici presenti, in qualunque percentuale).
I prodotti dello schema di reazione di fig. 1, nellecondizioni adottate industrialmente, sono principalmentei trimeri ciclici (quattro isomeri, tPhcyH), in quantitàcirca 5-10 volte inferiore rispetto ai difenilciclobuta-ni (due isomeri, dPhcyB). I radicali, che si formano in
bassissima percentuale, iniziano però la reazione di poli-merizzazione che produce catene composte da migliaiadi unità stireniche, per cui il risulato globale che si ottie-ne per riscaldamento dello stirene a 90-180 °C è la for-mazione di polimero per oltre il 99%.
Nella produzione industriale la velocità di polime-rizzazione può essere controllata regolando la tempera-tura e aggiungendo un iniziatore di radicali, comune-mente un perossido. In questo modo si determina la velo-cità con cui iniziano le catene, la cui propagazione haluogo a velocità crescente con la temperatura. La termi-nazione delle catene radicaliche avviene principalmen-te per accoppiamento e per reazione di trasferimento apiccole molecole, che sono il sopra citato dimero inter-medio (Olaj, 1971) della reazione di Diels-Alder ed even-tualmente solventi aromatici o sostanze appositamenteaggiunte per il controllo del peso molecolare quali, peresempio, mercaptani. Lo stadio lento della reazione èquello di formazione dei radicali primi, mentre la pro-pagazione è rapida. Per formare una catena sono suffi-cienti da alcuni centesimi fino a pochi decimi di secon-do dipendentemente dalla temperatura.
Nello schema cinetico semplificato della reazione dipolimerizzazione dello stirene di seguito riportato, checomprende gli stadi di inizio termico e perossidico, dipropagazione, di trasferimento a monomero, a solventee a eventuale regolatore di catena, di terminazione peraccoppiamento e di sproporzione, la reazione comples-sa di inizio termico di fig. 1 è riassunta in un’unica rea-zione e il trasferimento all’addotto Diels Alder è ricon-dotto al trasferimento a monomero ipotizzando che la suaconcentrazione sia proporzionale a quella dello stirene:
Inizio I �� 2 R0�3 M �� 2 R0�R0� � M �� R1�
Propagazione Rn� � M �� R�n�1Trasferimento Rn� � T, M, S �� Pn + T�, M�, S�Terminazione Rn� � Rm� �� Pn + Pm
Rn� � Rm� �� Pn�m
dove I è il perossido, M lo stirene, R� il radicale, T il tra-sferitore di catena, S il solvente e P il polimero.
Le catene di polimero che si ottengono sono a strut-tura lineare, atattica. Per la sintesi di catene a strutturaramificata devono essere impiegati particolari accorgi-menti nella formulazione, come l’aggiunta di comono-meri divinilici o di iniziatori polifunzionali.
A temperature superiori a 100 °C, quelle di maggio-re impiego industriale, il trasferimento di catena all’in-termedio DA-add evita il presentarsi del fenomenodell’autoaccelerazione denominato ‘effetto gel’ o‘Trommsdorf’, che nella polimerizzazione dello stirenea bassa temperatura o per certi copolimeri dello stirenesi manifesta con un aumento incontrollato di velocitàdi reazione e di peso molecolare. Per questo motivo la
838 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
�
�
�
�
�
. .
tPhcyH
dPhcyB
M
DA-add DA-R
SR
Tc
fig. 1. Schema di reazione per la formazione di radicali diinizio di polimerizzazione e degli oligomeri ciclici. M, stirene; tPhcyH, quattro isomeri; dPhcyB, due isomeri;DA-add, addotto Diels-Alder; DA-R, radicale dell’addottoDiels-Alder; SR, 1-fenil-etil-radicale; Tc, trimeri ciclici.
polimerizzazione dello stirene non risulta critica e feno-meni di ‘reazioni fuggitive’ (run-away reactions) o diinstabilità della reazione non avvengono in condizionidi esercizio normali.
A temperature superiori a 200 °C diventano impor-tanti anche le reazioni di depolimerizzazione o degrada-zione delle catene di PS formate o delle catene radicali-che in crescita (Campbell et al., 2003). Per degradazio-ne del PS si formano stirene, difenilbutene, trifenilesene,a-metilstirene e toluene. Lo stirene si produce con mec-canismo inverso alla propagazione, mentre gli altri com-posti sono originati da una migrazione del radicale ter-minale di catena con successivo riarrangiamento.
Copolimeri dello stireneLa copolimerizzazione radicalica dello stirene con
monomeri vinilici, quali acrilonitrile (AN) e altri deriva-ti dell’acido acrilico e metacrilico, è ampiamente utiliz-zata per la produzione di massa di polimeri stirenici ter-moplastici. I comonomeri in miscela con lo stirene poli-merizzano secondo le note leggi della copolimerizzazioneradicalica, che produce polimeri atattici con sequenzecasuali di monomeri in catena (Ito e Yamashita, 1965).La composizione di un copolimero dipende dalla com-posizione della miscela di monomeri e dalla reattivitàrelativa delle catene radicaliche crescenti con i monomeripresenti. Nello schema che segue sono riportate le equa-zioni cinetiche delle reazioni di propagazione per duemonomeri in cui la reattività dipende soltanto dall’ulti-mo monomero legato in catena che supporta il radicale(modello terminale) e non dalla composizione del restodella catena (in questo caso due parametri denominatirapporti di reattività sono sufficienti a determinare la com-posizione media delle catene polimeriche che si forma-no da una miscela monomerica a composizione data):
PA� � A �� PAA� vaa�kaa[PA�][A]PA� � S �� PAS� vas�kas [PA�][S]PS� � S �� PSS� vss�kss [PS�][S]PS� � A �� PSA� vsa�ksa [PS�][A]
dove v è la velocità di reazione e k la costante di velo-cità. I rapporti di reattività sono definiti come ra�vaa/vase rs�vss/vsa.
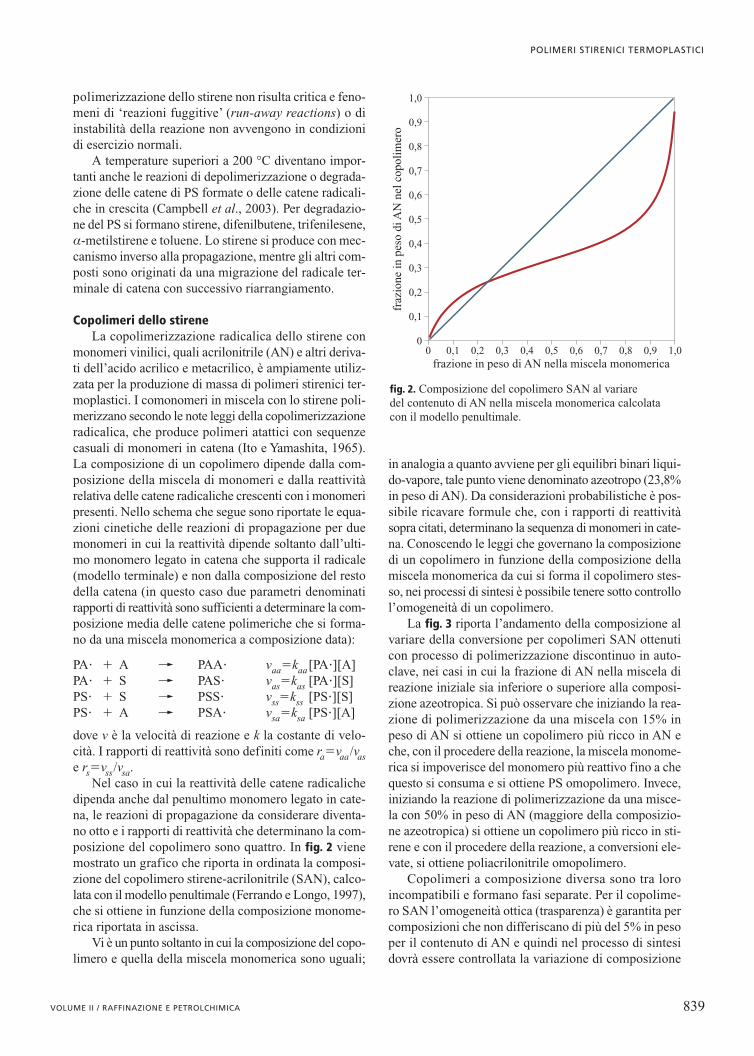
Nel caso in cui la reattività delle catene radicalichedipenda anche dal penultimo monomero legato in cate-na, le reazioni di propagazione da considerare diventa-no otto e i rapporti di reattività che determinano la com-posizione del copolimero sono quattro. In fig. 2 vienemostrato un grafico che riporta in ordinata la composi-zione del copolimero stirene-acrilonitrile (SAN), calco-lata con il modello penultimale (Ferrando e Longo, 1997),che si ottiene in funzione della composizione monome-rica riportata in ascissa.
Vi è un punto soltanto in cui la composizione del copo-limero e quella della miscela monomerica sono uguali;
in analogia a quanto avviene per gli equilibri binari liqui-do-vapore, tale punto viene denominato azeotropo (23,8%in peso di AN). Da considerazioni probabilistiche è pos-sibile ricavare formule che, con i rapporti di reattivitàsopra citati, determinano la sequenza di monomeri in cate-na. Conoscendo le leggi che governano la composizionedi un copolimero in funzione della composizione dellamiscela monomerica da cui si forma il copolimero stes-so, nei processi di sintesi è possibile tenere sotto controllol’omogeneità di un copolimero.
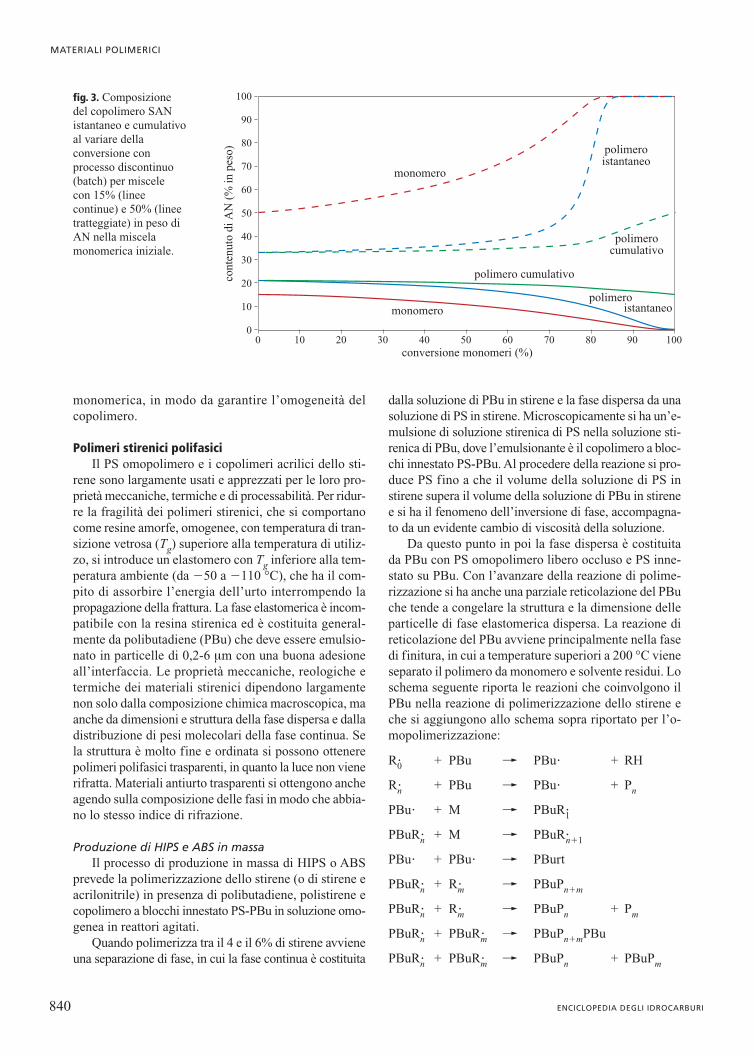
La fig. 3 riporta l’andamento della composizione alvariare della conversione per copolimeri SAN ottenuticon processo di polimerizzazione discontinuo in auto-clave, nei casi in cui la frazione di AN nella miscela direazione iniziale sia inferiore o superiore alla composi-zione azeotropica. Si può osservare che iniziando la rea-zione di polimerizzazione da una miscela con 15% inpeso di AN si ottiene un copolimero più ricco in AN eche, con il procedere della reazione, la miscela monome-rica si impoverisce del monomero più reattivo fino a chequesto si consuma e si ottiene PS omopolimero. Invece,iniziando la reazione di polimerizzazione da una misce-la con 50% in peso di AN (maggiore della composizio-ne azeotropica) si ottiene un copolimero più ricco in sti-rene e con il procedere della reazione, a conversioni ele-vate, si ottiene poliacrilonitrile omopolimero.
Copolimeri a composizione diversa sono tra loroincompatibili e formano fasi separate. Per il copolime-ro SAN l’omogeneità ottica (trasparenza) è garantita percomposizioni che non differiscano di più del 5% in pesoper il contenuto di AN e quindi nel processo di sintesidovrà essere controllata la variazione di composizione
839VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
fraz
ione
in p
eso
di A
N n
el c
opol
imer
o
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
frazione in peso di AN nella miscela monomerica0 0,1 0,2 0,4 0,6 0,80,3 0,5 0,7 0,9 1,0
fig. 2. Composizione del copolimero SAN al variare del contenuto di AN nella miscela monomerica calcolata con il modello penultimale.
monomerica, in modo da garantire l’omogeneità delcopolimero.
Polimeri stirenici polifasiciIl PS omopolimero e i copolimeri acrilici dello sti-
rene sono largamente usati e apprezzati per le loro pro-prietà meccaniche, termiche e di processabilità. Per ridur-re la fragilità dei polimeri stirenici, che si comportanocome resine amorfe, omogenee, con temperatura di tran-sizione vetrosa (Tg) superiore alla temperatura di utiliz-zo, si introduce un elastomero con Tg inferiore alla tem-peratura ambiente (da �50 a �110 °C), che ha il com-pito di assorbire l’energia dell’urto interrompendo lapropagazione della frattura. La fase elastomerica è incom-patibile con la resina stirenica ed è costituita general-mente da polibutadiene (PBu) che deve essere emulsio-nato in particelle di 0,2-6 mm con una buona adesioneall’interfaccia. Le proprietà meccaniche, reologiche etermiche dei materiali stirenici dipendono largamentenon solo dalla composizione chimica macroscopica, maanche da dimensioni e struttura della fase dispersa e dalladistribuzione di pesi molecolari della fase continua. Sela struttura è molto fine e ordinata si possono ottenerepolimeri polifasici trasparenti, in quanto la luce non vienerifratta. Materiali antiurto trasparenti si ottengono ancheagendo sulla composizione delle fasi in modo che abbia-no lo stesso indice di rifrazione.
Produzione di HIPS e ABS in massaIl processo di produzione in massa di HIPS o ABS
prevede la polimerizzazione dello stirene (o di stirene eacrilonitrile) in presenza di polibutadiene, polistirene ecopolimero a blocchi innestato PS-PBu in soluzione omo-genea in reattori agitati.
Quando polimerizza tra il 4 e il 6% di stirene avvieneuna separazione di fase, in cui la fase continua è costituita
dalla soluzione di PBu in stirene e la fase dispersa da unasoluzione di PS in stirene. Microscopicamente si ha un’e-mulsione di soluzione stirenica di PS nella soluzione sti-renica di PBu, dove l’emulsionante è il copolimero a bloc-chi innestato PS-PBu. Al procedere della reazione si pro-duce PS fino a che il volume della soluzione di PS instirene supera il volume della soluzione di PBu in stirenee si ha il fenomeno dell’inversione di fase, accompagna-to da un evidente cambio di viscosità della soluzione.
Da questo punto in poi la fase dispersa è costituitada PBu con PS omopolimero libero occluso e PS inne-stato su PBu. Con l’avanzare della reazione di polime-rizzazione si ha anche una parziale reticolazione del PBuche tende a congelare la struttura e la dimensione delleparticelle di fase elastomerica dispersa. La reazione direticolazione del PBu avviene principalmente nella fasedi finitura, in cui a temperature superiori a 200 °C vieneseparato il polimero da monomero e solvente residui. Loschema seguente riporta le reazioni che coinvolgono ilPBu nella reazione di polimerizzazione dello stirene eche si aggiungono allo schema sopra riportato per l’o-mopolimerizzazione:
R0� + PBu �� PBu� + RH
Rn� + PBu �� PBu� + Pn
PBu� + M �� PBuR1�
PBuRn� + M �� PBuR�n�1
PBu� + PBu� �� PBurt
PBuRn� + Rm� �� PBuPn�m
PBuRn� + Rm� �� PBuPn + Pm
PBuRn� + PBuRm� �� PBuPn�mPBu
PBuRn� + PBuRm� �� PBuPn + PBuPm
840 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60 70 80 90 100
cont
enut
o di
AN
(%
in p
eso)
conversione monomeri (%)
monomero
monomero
polimeroistantaneo
polimero istantaneo
polimerocumulativo
polimero cumulativo
fig. 3. Composizione del copolimero SANistantaneo e cumulativoal variare dellaconversione conprocesso discontinuo(batch) per miscele con 15% (lineecontinue) e 50% (lineetratteggiate) in peso diAN nella miscelamonomerica iniziale.
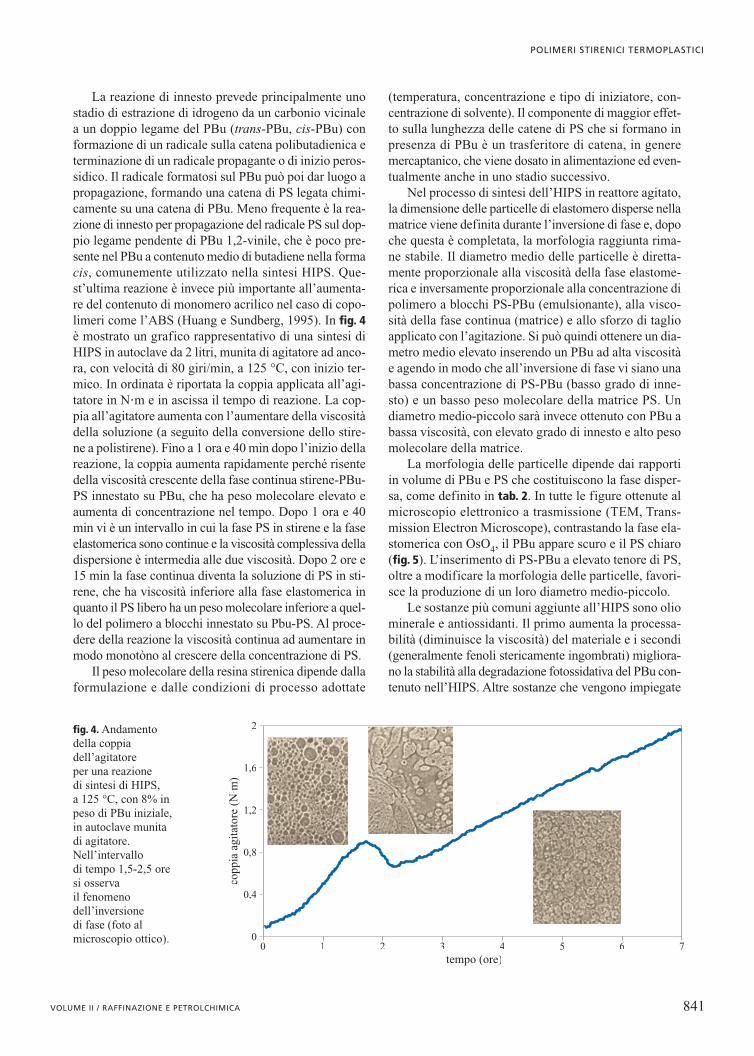
La reazione di innesto prevede principalmente unostadio di estrazione di idrogeno da un carbonio vicinalea un doppio legame del PBu (trans-PBu, cis-PBu) conformazione di un radicale sulla catena polibutadienica eterminazione di un radicale propagante o di inizio peros-sidico. Il radicale formatosi sul PBu può poi dar luogo apropagazione, formando una catena di PS legata chimi-camente su una catena di PBu. Meno frequente è la rea-zione di innesto per propagazione del radicale PS sul dop-pio legame pendente di PBu 1,2-vinile, che è poco pre-sente nel PBu a contenuto medio di butadiene nella formacis, comunemente utilizzato nella sintesi HIPS. Que-st’ultima reazione è invece più importante all’aumenta-re del contenuto di monomero acrilico nel caso di copo-limeri come l’ABS (Huang e Sundberg, 1995). In fig. 4è mostrato un grafico rappresentativo di una sintesi diHIPS in autoclave da 2 litri, munita di agitatore ad anco-ra, con velocità di 80 giri/min, a 125 °C, con inizio ter-mico. In ordinata è riportata la coppia applicata all’agi-tatore in N�m e in ascissa il tempo di reazione. La cop-pia all’agitatore aumenta con l’aumentare della viscositàdella soluzione (a seguito della conversione dello stire-ne a polistirene). Fino a 1 ora e 40 min dopo l’inizio dellareazione, la coppia aumenta rapidamente perché risentedella viscosità crescente della fase continua stirene-PBu-PS innestato su PBu, che ha peso molecolare elevato eaumenta di concentrazione nel tempo. Dopo 1 ora e 40min vi è un intervallo in cui la fase PS in stirene e la faseelastomerica sono continue e la viscosità complessiva delladispersione è intermedia alle due viscosità. Dopo 2 ore e15 min la fase continua diventa la soluzione di PS in sti-rene, che ha viscosità inferiore alla fase elastomerica inquanto il PS libero ha un peso molecolare inferiore a quel-lo del polimero a blocchi innestato su Pbu-PS. Al proce-dere della reazione la viscosità continua ad aumentare inmodo monotòno al crescere della concentrazione di PS.
Il peso molecolare della resina stirenica dipende dallaformulazione e dalle condizioni di processo adottate
(temperatura, concentrazione e tipo di iniziatore, con-centrazione di solvente). Il componente di maggior effet-to sulla lunghezza delle catene di PS che si formano inpresenza di PBu è un trasferitore di catena, in generemercaptanico, che viene dosato in alimentazione ed even-tualmente anche in uno stadio successivo.
Nel processo di sintesi dell’HIPS in reattore agitato,la dimensione delle particelle di elastomero disperse nellamatrice viene definita durante l’inversione di fase e, dopoche questa è completata, la morfologia raggiunta rima-ne stabile. Il diametro medio delle particelle è diretta-mente proporzionale alla viscosità della fase elastome-rica e inversamente proporzionale alla concentrazione dipolimero a blocchi PS-PBu (emulsionante), alla visco-sità della fase continua (matrice) e allo sforzo di taglioapplicato con l’agitazione. Si può quindi ottenere un dia-metro medio elevato inserendo un PBu ad alta viscositàe agendo in modo che all’inversione di fase vi siano unabassa concentrazione di PS-PBu (basso grado di inne-sto) e un basso peso molecolare della matrice PS. Undiametro medio-piccolo sarà invece ottenuto con PBu abassa viscosità, con elevato grado di innesto e alto pesomolecolare della matrice.
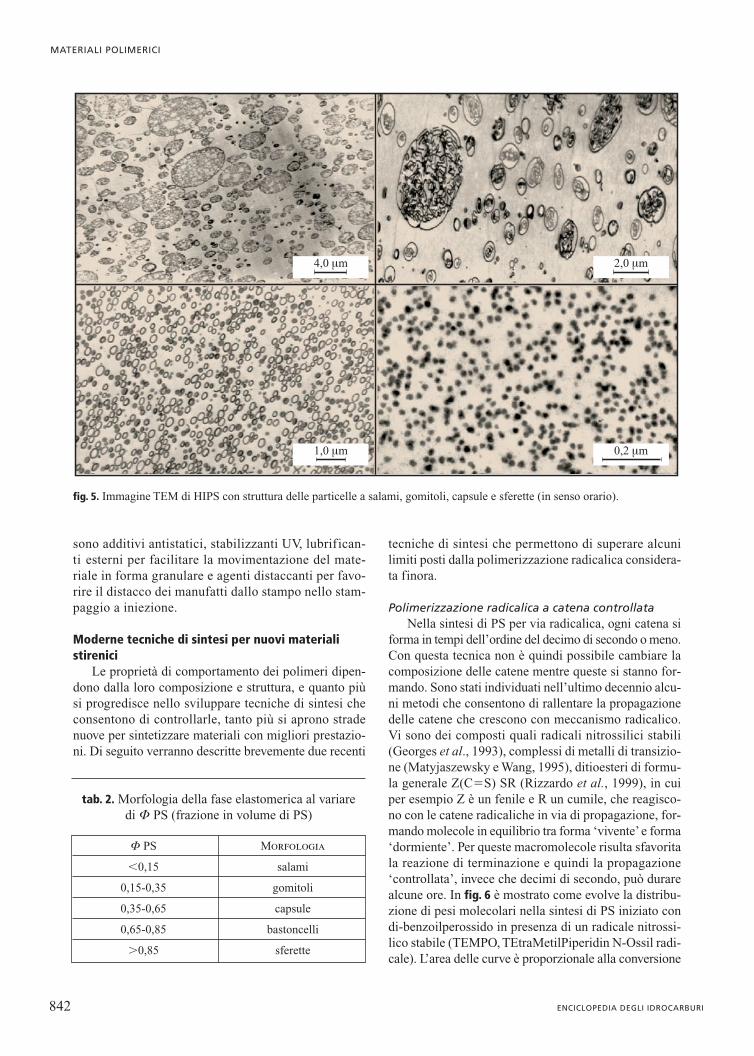
La morfologia delle particelle dipende dai rapportiin volume di PBu e PS che costituiscono la fase disper-sa, come definito in tab. 2. In tutte le figure ottenute almicroscopio elettronico a trasmissione (TEM, Trans-mission Electron Microscope), contrastando la fase ela-stomerica con OsO4, il PBu appare scuro e il PS chiaro(fig. 5). L’inserimento di PS-PBu a elevato tenore di PS,oltre a modificare la morfologia delle particelle, favori-sce la produzione di un loro diametro medio-piccolo.
Le sostanze più comuni aggiunte all’HIPS sono oliominerale e antiossidanti. Il primo aumenta la processa-bilità (diminuisce la viscosità) del materiale e i secondi(generalmente fenoli stericamente ingombrati) migliora-no la stabilità alla degradazione fotossidativa del PBu con-tenuto nell’HIPS. Altre sostanze che vengono impiegate
841VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
copp
ia a
gita
tore
(N
. m)
0
0,4
0,8
1,2
1,6
2
tempo (ore)0 1 32 54 6 7
fig. 4. Andamentodella coppiadell’agitatore per una reazione di sintesi di HIPS, a 125 °C, con 8% inpeso di PBu iniziale,in autoclave munitadi agitatore.Nell’intervallo di tempo 1,5-2,5 oresi osserva il fenomenodell’inversione di fase (foto almicroscopio ottico).
sono additivi antistatici, stabilizzanti UV, lubrifican-ti esterni per facilitare la movimentazione del mate-riale in forma granulare e agenti distaccanti per favo-rire il distacco dei manufatti dallo stampo nello stam-paggio a iniezione.
Moderne tecniche di sintesi per nuovi materialistirenici
Le proprietà di comportamento dei polimeri dipen-dono dalla loro composizione e struttura, e quanto piùsi progredisce nello sviluppare tecniche di sintesi checonsentono di controllarle, tanto più si aprono stradenuove per sintetizzare materiali con migliori prestazio-ni. Di seguito verranno descritte brevemente due recenti
tecniche di sintesi che permettono di superare alcunilimiti posti dalla polimerizzazione radicalica considera-ta finora.
Polimerizzazione radicalica a catena controllataNella sintesi di PS per via radicalica, ogni catena si
forma in tempi dell’ordine del decimo di secondo o meno.Con questa tecnica non è quindi possibile cambiare lacomposizione delle catene mentre queste si stanno for-mando. Sono stati individuati nell’ultimo decennio alcu-ni metodi che consentono di rallentare la propagazionedelle catene che crescono con meccanismo radicalico.Vi sono dei composti quali radicali nitrossilici stabili(Georges et al., 1993), complessi di metalli di transizio-ne (Matyjaszewsky e Wang, 1995), ditioesteri di formu-la generale Z(C�S) SR (Rizzardo et al., 1999), in cuiper esempio Z è un fenile e R un cumile, che reagisco-no con le catene radicaliche in via di propagazione, for-mando molecole in equilibrio tra forma ‘vivente’e forma‘dormiente’. Per queste macromolecole risulta sfavoritala reazione di terminazione e quindi la propagazione‘controllata’, invece che decimi di secondo, può durarealcune ore. In fig. 6 è mostrato come evolve la distribu-zione di pesi molecolari nella sintesi di PS iniziato condi-benzoilperossido in presenza di un radicale nitrossi-lico stabile (TEMPO, TEtraMetilPiperidin N-Ossil radi-cale). L’area delle curve è proporzionale alla conversione
842 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
4,0 mm 2,0 mm
1,0 mm 0,2 mm
F PS Morfologia
�0,15 salami
0,15-0,35 gomitoli
0,35-0,65 capsule
0,65-0,85 bastoncelli
�0,85 sferette
tab. 2. Morfologia della fase elastomerica al variaredi F PS (frazione in volume di PS)
fig. 5. Immagine TEM di HIPS con struttura delle particelle a salami, gomitoli, capsule e sferette (in senso orario).
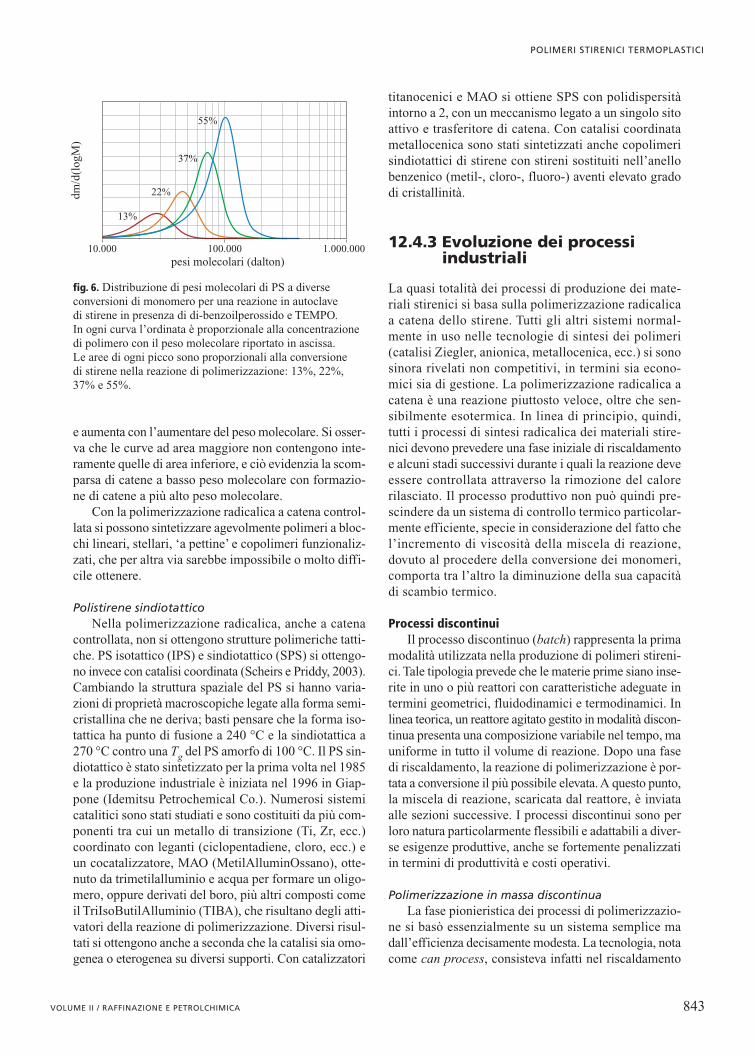
e aumenta con l’aumentare del peso molecolare. Si osser-va che le curve ad area maggiore non contengono inte-ramente quelle di area inferiore, e ciò evidenzia la scom-parsa di catene a basso peso molecolare con formazio-ne di catene a più alto peso molecolare.
Con la polimerizzazione radicalica a catena control-lata si possono sintetizzare agevolmente polimeri a bloc-chi lineari, stellari, ‘a pettine’ e copolimeri funzionaliz-zati, che per altra via sarebbe impossibile o molto diffi-cile ottenere.
Polistirene sindiotatticoNella polimerizzazione radicalica, anche a catena
controllata, non si ottengono strutture polimeriche tatti-che. PS isotattico (IPS) e sindiotattico (SPS) si ottengo-no invece con catalisi coordinata (Scheirs e Priddy, 2003).Cambiando la struttura spaziale del PS si hanno varia-zioni di proprietà macroscopiche legate alla forma semi-cristallina che ne deriva; basti pensare che la forma iso-tattica ha punto di fusione a 240 °C e la sindiotattica a270 °C contro una Tg del PS amorfo di 100 °C. Il PS sin-diotattico è stato sintetizzato per la prima volta nel 1985e la produzione industriale è iniziata nel 1996 in Giap-pone (Idemitsu Petrochemical Co.). Numerosi sistemicatalitici sono stati studiati e sono costituiti da più com-ponenti tra cui un metallo di transizione (Ti, Zr, ecc.)coordinato con leganti (ciclopentadiene, cloro, ecc.) eun cocatalizzatore, MAO (MetilAlluminOssano), otte-nuto da trimetilalluminio e acqua per formare un oligo-mero, oppure derivati del boro, più altri composti comeil TriIsoButilAlluminio (TIBA), che risultano degli atti-vatori della reazione di polimerizzazione. Diversi risul-tati si ottengono anche a seconda che la catalisi sia omo-genea o eterogenea su diversi supporti. Con catalizzatori
titanocenici e MAO si ottiene SPS con polidispersitàintorno a 2, con un meccanismo legato a un singolo sitoattivo e trasferitore di catena. Con catalisi coordinatametallocenica sono stati sintetizzati anche copolimerisindiotattici di stirene con stireni sostituiti nell’anellobenzenico (metil-, cloro-, fluoro-) aventi elevato gradodi cristallinità.
12.4.3 Evoluzione dei processiindustriali
La quasi totalità dei processi di produzione dei mate-riali stirenici si basa sulla polimerizzazione radicalicaa catena dello stirene. Tutti gli altri sistemi normal-mente in uso nelle tecnologie di sintesi dei polimeri(catalisi Ziegler, anionica, metallocenica, ecc.) si sonosinora rivelati non competitivi, in termini sia econo-mici sia di gestione. La polimerizzazione radicalica acatena è una reazione piuttosto veloce, oltre che sen-sibilmente esotermica. In linea di principio, quindi,tutti i processi di sintesi radicalica dei materiali stire-nici devono prevedere una fase iniziale di riscaldamentoe alcuni stadi successivi durante i quali la reazione deveessere controllata attraverso la rimozione del calorerilasciato. Il processo produttivo non può quindi pre-scindere da un sistema di controllo termico particolar-mente efficiente, specie in considerazione del fatto chel’incremento di viscosità della miscela di reazione,dovuto al procedere della conversione dei monomeri,comporta tra l’altro la diminuzione della sua capacitàdi scambio termico.
Processi discontinuiIl processo discontinuo (batch) rappresenta la prima
modalità utilizzata nella produzione di polimeri stireni-ci. Tale tipologia prevede che le materie prime siano inse-rite in uno o più reattori con caratteristiche adeguate intermini geometrici, fluidodinamici e termodinamici. Inlinea teorica, un reattore agitato gestito in modalità discon-tinua presenta una composizione variabile nel tempo, mauniforme in tutto il volume di reazione. Dopo una fasedi riscaldamento, la reazione di polimerizzazione è por-tata a conversione il più possibile elevata. A questo punto,la miscela di reazione, scaricata dal reattore, è inviataalle sezioni successive. I processi discontinui sono perloro natura particolarmente flessibili e adattabili a diver-se esigenze produttive, anche se fortemente penalizzatiin termini di produttività e costi operativi.
Polimerizzazione in massa discontinuaLa fase pionieristica dei processi di polimerizzazio-
ne si basò essenzialmente su un sistema semplice madall’efficienza decisamente modesta. La tecnologia, notacome can process, consisteva infatti nel riscaldamento
843VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
55%
37%
22%
13%
dm/d
(log
M)
pesi molecolari (dalton)10.000 100.000 1.000.000
fig. 6. Distribuzione di pesi molecolari di PS a diverseconversioni di monomero per una reazione in autoclave di stirene in presenza di di-benzoilperossido e TEMPO. In ogni curva l’ordinata è proporzionale alla concentrazionedi polimero con il peso molecolare riportato in ascissa. Le aree di ogni picco sono proporzionali alla conversione di stirene nella reazione di polimerizzazione: 13%, 22%,37% e 55%.

dello stirene in fusti metallici (can); dopo alcuni giorni,il polimero formatosi con una conversione pressochétotale era estratto dai fusti e macinato. Fu però prestocompresa la necessità di introdurre una fase di prepoli-merizzazione (fig. 7) in un reattore discontinuo, condot-ta a bassa temperatura (80 °C) ma per un tempo moltolungo (48 ore). In questo modo era possibile gestire lafase più critica della reazione, cioè quella nella quale ilmonomero ha concentrazioni più elevate, in condizioniblande. Alla conversione di circa il 35%, la miscela erapoi trasferita in un reattore adiabatico composto di varisettori (‘piatti’). Una volta completamente convertito, ilprodotto era estratto e macinato.
Un ulteriore intervento migliorativo fu costituito dal-l’introduzione di una tecnologia semicontinua, che con-sentiva un controllo della reazione più razionale, oltre auna considerevole riduzione dei tempi di produzione. Lamiscela proveniente dai prepolimerizzatori era trasferi-ta in un reattore tubolare (‘a torre’), nel quale la tempe-ratura era portata da 100 °C in ingresso fino a 180 °C inuscita, consentendo alla reazione di procedere fino a unaconversione intorno al 99%. Il reattore tubolare era sca-ricato dal fondo in modalità continua, granulando diret-tamente il polimero senza operazioni di purificazione(devolatilizzazione).
I processi in massa descritti in precedenza, oltre alleproblematiche tipiche delle tecnologie discontinue rela-tivamente a tempi e costi, comportavano problemi diomogeneità e agitazione della massa reagente molto rile-vanti. Un deciso miglioramento in questo senso vennedall’introduzione del processo in sospensione (fig. 8),nel quale il controllo del calore sviluppato dalla reazio-ne è assicurato da un fluido, di caratteristiche tali da noninterferire con la polimerizzazione (generalmente acqua),presente all’interno di un reattore agitato.
La polimerizzazione avviene quindi all’interno diparticelle di monomero sospese in acqua, anche grazieall’utilizzo di opportuni agenti sospendenti (tensioatti-vi), che possono essere aggiunti a diversi stadi di con-versione in modo da modificare la distribuzione dimen-sionale delle perle. È possibile in questa fase utilizzare
iniziatori o trasferitori di catena. A conversione presso-ché completa, il prodotto, in forma di perle, è poi sepa-rato dalla soluzione acquosa per centrifugazione e sot-toposto a essiccamento in aria.
La tecnologia in sospensione ha trovato una certa dif-fusione negli anni Cinquanta, sfruttando le sue già cita-te peculiarità (buon controllo della viscosità e dello svi-luppo termico del sistema). A partire dal decennio suc-cessivo, necessità imprescindibili in termini ambientali,economici e qualitativi porteranno alla sostituzione pres-soché integrale del processo in sospensione con quelloin massa continua nella sintesi dei polimeri stirenici com-patti (v. oltre). Tale tecnologia rimane ancora largamen-te maggioritaria laddove il passaggio alla tecnologia con-tinua non è ancora avvenuto, vale a dire nel campo delpolistirene espandibile (EPS). In questo caso, durante lapolimerizzazione viene incorporato un agente espan-dente organico (tipicamente pentano) che porta nelle fasisuccessive della lavorazione all’espansione della perla equindi alla struttura cellulare, con conseguente diminu-zione della densità del manufatto.
Polimerizzazione in emulsioneParallelamente allo sviluppo del processo in sospen-
sione, si va affermando una tecnologia che trova il campod’applicazione ideale solo nella sintesi di materiali bifa-sici, nella quale, alle già citate problematiche della poli-merizzazione dello stirene, si sommano quelle legate allareazione di graffaggio tra catena macromolecolare e fasegommosa.
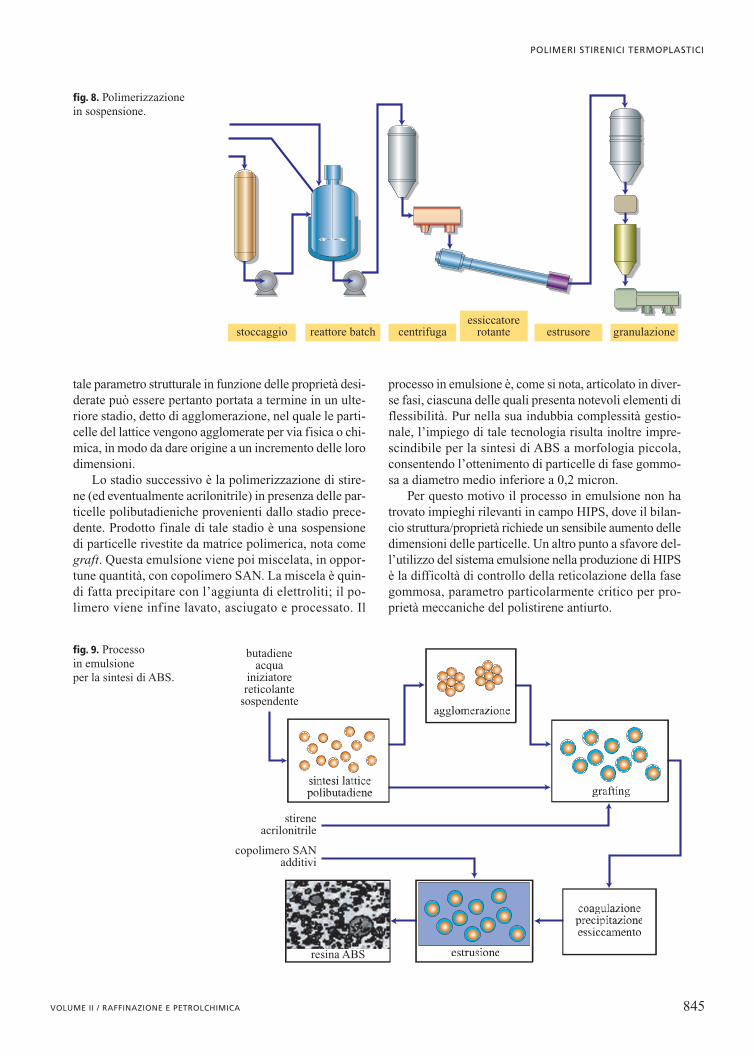
La sintesi via emulsione (fig. 9) è un processo discon-tinuo caratterizzato da diversi stadi.
Il primo stadio prevede la preparazione del lattice digomma: il butadiene è polimerizzato in acqua, in pre-senza di agenti emulsionanti e reticolanti, oltre che di uniniziatore e di un regolatore di peso molecolare. La velo-cità di polimerizzazione del butadiene è relativamentebassa, per cui l’esigenza di ridurre la durata di questafase porta a fermarla a uno stadio di conversione noncompleta, con conseguente penalizzazione della cresci-ta delle dimensioni delle particelle. La regolazione di
844 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
stireneiniziatore
acqua/vapore
acqua/vapore
additivi
prepolimerizzatore reattore a piatti mulino stoccaggio
inerte
fig. 7. Processo in massa discontinuacon reattore a piatti.
tale parametro strutturale in funzione delle proprietà desi-derate può essere pertanto portata a termine in un ulte-riore stadio, detto di agglomerazione, nel quale le parti-celle del lattice vengono agglomerate per via fisica o chi-mica, in modo da dare origine a un incremento delle lorodimensioni.
Lo stadio successivo è la polimerizzazione di stire-ne (ed eventualmente acrilonitrile) in presenza delle par-ticelle polibutadieniche provenienti dallo stadio prece-dente. Prodotto finale di tale stadio è una sospensionedi particelle rivestite da matrice polimerica, nota comegraft. Questa emulsione viene poi miscelata, in oppor-tune quantità, con copolimero SAN. La miscela è quin-di fatta precipitare con l’aggiunta di elettroliti; il po-limero viene infine lavato, asciugato e processato. Il
processo in emulsione è, come si nota, articolato in diver-se fasi, ciascuna delle quali presenta notevoli elementi diflessibilità. Pur nella sua indubbia complessità gestio-nale, l’impiego di tale tecnologia risulta inoltre impre-scindibile per la sintesi di ABS a morfologia piccola,consentendo l’ottenimento di particelle di fase gommo-sa a diametro medio inferiore a 0,2 micron.
Per questo motivo il processo in emulsione non hatrovato impieghi rilevanti in campo HIPS, dove il bilan-cio struttura/proprietà richiede un sensibile aumento delledimensioni delle particelle. Un altro punto a sfavore del-l’utilizzo del sistema emulsione nella produzione di HIPSè la difficoltà di controllo della reticolazione della fasegommosa, parametro particolarmente critico per pro-prietà meccaniche del polistirene antiurto.
845VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
granulazioneestrusoreessiccatore
rotantecentrifugareattore batchstoccaggio
butadieneacqua
iniziatorereticolante
sospendente
stireneacrilonitrile
copolimero SANadditivi
resina ABS
fig. 8. Polimerizzazionein sospensione.
fig. 9. Processo in emulsione per la sintesi di ABS.

Processi continuiAl contrario di quelli sinora trattati, un processo con-
tinuo stazionario si basa sull’alimentazione continua dellematerie prime e sull’uscita continua di prodotti ed even-tuali sottoprodotti o residui di lavorazione. In generale,tale modalità operativa consente di ottimizzare parame-tri chiave quali costi di investimento, costanza qualitati-va dei prodotti e capacità produttiva. Contrariamente aquanto verificatosi per altri polimeri, in primis il polie-tilene, per i quali nel tempo sono stati sviluppati i pro-cessi continui più disparati (alta pressione, soluzione,slurry, fase gassosa, ecc.), lo stato fisico delle materieprime utilizzate per la sintesi dei polimeri stirenici haindirizzato sin dagli albori verso la tecnologia in massa.
Processo in massa continuaCome evidenziato nei paragrafi precedenti, le tec-
nologie in discontinuo, ancorché flessibili e utili per lasintesi di materiali particolari (per esempio ABS lucidi),presentano indubbi inconvenienti dal punto di vista ope-rativo ed economico. Negli ultimi decenni sono poi anda-te evidenziandosi, in modo sempre più imprescindibile,tematiche ambientali legate sia alla qualità del prodottofinito (monomeri e sostanze residue) sia agli aspetti pro-duttivi (quantità e qualità degli effluenti, emissioni nel-l’ambiente di lavoro, ecc.).
Il processo in massa continua non utilizza materieprime o additive estranee alla reazione (quali l’acqua neiprocessi precedenti), permettendo quindi di ridurre laquantità di emissioni in maniera drastica. Questa tecno-logia è poi di norma concepita come ciclo chiuso; tuttociò che non ha reagito al termine della sezione di poli-merizzazione è separato nella sezione di devolatilizza-zione, condensato e alimentato nuovamente all’ingres-so dell’impianto. Solo una minima quota di spurgo, chenei processi più efficienti può essere dell’ordine dell’1,0-1,5%, è resa necessaria per rimuovere dall’impianto icomposti non polimerizzabili che altrimenti tendereb-bero ad accumularsi.
Lo schema classico del processo in massa continua(fig. 10) prevede una sezione di dissoluzione gomma (peri polimeri bifasici), dove il polibutadiene viene solubi-lizzato in stirene, una sezione di reazione, che prevedein genere diversi stadi di reazione in serie, e una sezio-ne di devolatilizzazione (a uno o più stadi), dalla qualeil polimero purificato viene inviato alla granulazione.La polimerizzazione avviene in presenza di un solvente(in genere etilbenzene), che consente di limitare la visco-sità del sistema in caso di reazioni incontrollate, nonchédi contenere la velocità di reazione.
I reattori impiegati nel processo in massa continuaappartengono generalmente a due tipologie: il reattoreCSTR (Continuous Stirred Tank Reactor, reattore con-tinuo agitato) e il reattore PFR (Plug Flow Reactor, reat-tore a flusso a pistone).
Nel reattore CSTR la miscelazione è completa e per-tanto la velocità di reazione, la concentrazione e la tem-peratura hanno lo stesso valore in ogni punto del reatto-re. Ne consegue che le condizioni in uscita sono del tuttocoincidenti con quelle presenti nel reattore stesso. Nellasintesi dei polimeri stirenici la rimozione del calore avvie-ne attraverso l’evaporazione di una parte della misceladi reazione. Tra i principali vantaggi del CSTR si pos-sono annoverare la relativa facilità di pulizia, il buon con-trollo termico e i bassi costi di realizzazione, mentre ilprincipale svantaggio sta nella bassa produttività speci-fica.
Il reattore PFR è assimilabile a un tubo percorso dallamiscela di reazione senza alcuna miscelazione in sensoassiale (assenza di retromiscelazione). Lungo la dire-zione del flusso, ogni sezione presenta caratteristicheuniformi in termini di velocità, composizione e tempe-ratura. Tale sistema è particolarmente premiante nei pro-cessi dove è presente gomma e quindi dove è necessarioun efficace controllo dell’inversione di fase.
Rispetto al CSTR, il PFR risulta più efficiente in ter-mini di produttività specifica, anche se la necessità digarantire il controllo della reazione attraverso l’inseri-mento di elementi di scambio termico interni rende icosti d’investimento in genere più onerosi.
Entrambe le tipologie di reattori utilizzate nel pro-cesso in massa continua prevedono l’uscita dalla sezio-ne di polimerizzazione di una miscela (prepolimero)composta da polimero (65-75%), monomero non reagi-to e composti non polimerizzabili (solvente), a una tem-peratura compresa tra 140 e 170 °C.
Il prepolimero è poi ulteriormente riscaldato in unoscambiatore (pre-heater), dove un sistema a olio diater-mico fornisce il calore necessario sia a raggiungere unatemperatura di norma superiore a 200 °C (calore sensi-bile) sia a mantenere costante la temperatura del poli-mero durante l’evaporazione dei componenti non poli-merici (calore latente di vaporizzazione). Vista la vola-tilità relativamente bassa dei composti in gioco, perlimitare lo stress termico sul prodotto il processo di devo-latilizzazione avviene in condizioni di pressione ridotta.
Particolare importanza riveste inoltre il design delloscambiatore di calore, che deve consentire la distribu-zione del polimero su una superficie di scambio quantopiù possibile elevata, pur ricorrendo ad apparecchiaturedalle dimensioni contenute e senza generare perdite dicarico eccessive. Il processo di devolatilizzazione puòconsistere in uno o due stadi a pressioni diverse. In que-sto secondo caso, la quota maggioritaria di frazioni vola-tili viene separata dal polimero nello stadio a pressionepiù elevata (1,6-2,0 kPa), facilitando così il processo dicondensazione che precede il riciclo in testa al treno direazione. In questa fase la concentrazione di stirene resi-duo si porta a 2.000-3.000 ppm. Nel secondo stadio, cheopera in condizioni di vuoto più spinte (0,3-0,4 kPa), si
846 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI

completa il processo di purificazione, lasciando un resi-duo di monomeri di norma inferiore a 500 ppm.
12.4.4 Struttura e proprietà
Struttura del polistirene omopolimeroLa macromolecola del polistirene, nella forma comu-
nemente ottenuta per polimerizzazione radicalica, haconfigurazione atattica. Allo stato solido (cioè al di sottodella temperatura di transizione vetrosa), il PS è perciòun polimero vetroso amorfo e trasparente.
La temperatura di transizione vetrosa (Tg), misuratamediante calorimetria differenziale a scansione (DSC,Differential Scanning Calorimetry) è di circa 100 °C(Brandrup e Immergut, 1989). Come per tutti i polime-ri termoplastici, il valore di Tg, indipendente dal gradodi polimerizzazione quando quest’ultimo è elevato, dimi-nuisce se la lunghezza di catena scende sotto un valorecritico. Per il PS ciò avviene per valori di peso moleco-lare inferiori a circa 100.000 (grado di polimerizzazio-ne circa uguale a 1.000; Claudy et al., 1983).
La densità del PS allo stato vetroso e a temperatu-ra ambiente è di circa 1,05 g/cm3. La dipendenza delladensità dalla temperatura (coefficiente di espansionetermica di volume) è pari a 2,7�10�4 cm3/g/K al di sot-to di Tg e a 6,0 �10�4 cm3/g/K a temperature superiori aTg (Brandrup e Immergut, 1989; van Krevelen, 1990).
La catena polimerica, a causa dell’ingombrante anel-lo benzenico, è relativamente poco flessibile. Ciò ha comediretta conseguenza che il peso molecolare tra gli entan-glements, Me (il termine entanglements identifica i puntidi intersezione, o di intreccio, tra le catene macromole-colari), assume un valore relativamente elevato (circa19.000) se confrontato con quelli degli altri più comuni
termoplastici (Ferry, 1980; Donald e Kramer, 1982b). Que-sta caratteristica strutturale è importante, come descrittoin seguito, nel determinare le proprietà del materiale.
Nei gradi commerciali di PS è frequente l’aggiuntadi plastificanti, solitamente oli paraffinici, in quantitànon superiori al 5 o 6% in peso, il cui scopo è quello didiminuire la viscosità del polimero alle temperature ditrasformazione. L’effetto strutturale di un plastificantein un polimero è quello di aumentare il volume libero(Ferry, 1980), il che comporta un abbassamento della Tg.Gli oli paraffinici utilizzati nel PS determinano una dimi-nuzione di Tg di circa 4 °C per ogni unità percentuale inpeso additivata.
Proprietà reologiche
ViscositàLa prima conseguenza della struttura molecolare
riguarda la dipendenza della viscosità newtoniana h0 (ilvalore limite della viscosità al tendere a zero del gra-diente di velocità) del polimero fuso con peso moleco-lare M. Come accade in generale per tutti i materialimacromolecolari (Ferry, 1980), al di sotto di un valorecritico Mc di peso molecolare la viscosità cresce linear-mente con M (h0 � M1), mentre per M�Mc la dipendenzaè molto più forte: h0 � M 3,4. Il valore di Mc è diretta-mente correlato a Me (Mc�2Me), e per il PS è di circa35.000 (Ferry, 1980).
I valori di h0 a 220 °C, temperatura tipica di trasfor-mazione del PS, sono di poco superiori a 100 Pa�s quan-do il valore del peso molecolare medio ponderale (Mw)è pari a circa 100.000 e sono invece dell’ordine di 10.000Pa�s per Mw pari a 350.000.
Questi valori, insieme con la dipendenza della Tgdal peso molecolare (v. sopra), determinano il campo
847VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
sistemada vuoto
prodottofinito
condensazione
fornofumi
combustibile
stirene � gomma(dissoluzione)
additivi
stireneprodotti chimici
etilbenzeneperossido
additivi
additivi
trattamento materie prime prepolimerizzazione polimerizzazione devolatilizzazione granulazione
fig. 10. Processo in massa continua per la sintesi di HIPS.
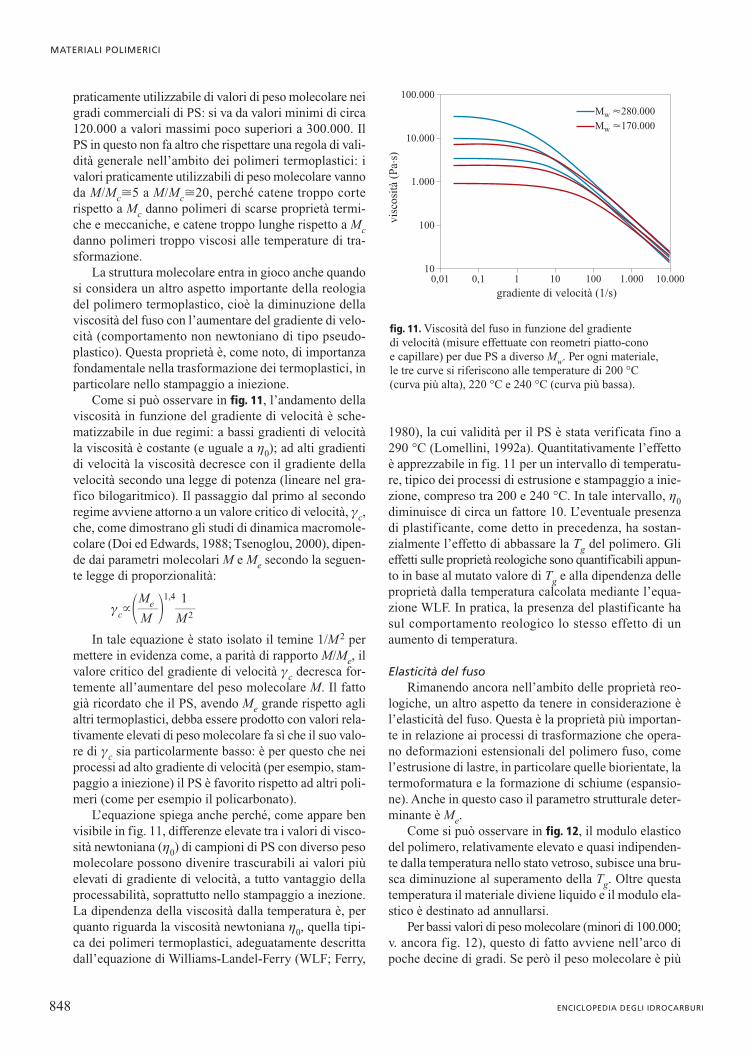
praticamente utilizzabile di valori di peso molecolare neigradi commerciali di PS: si va da valori minimi di circa120.000 a valori massimi poco superiori a 300.000. IlPS in questo non fa altro che rispettare una regola di vali-dità generale nell’ambito dei polimeri termoplastici: ivalori praticamente utilizzabili di peso molecolare vannoda M/Mc�5 a M/Mc�20, perché catene troppo corterispetto a Mc danno polimeri di scarse proprietà termi-che e meccaniche, e catene troppo lunghe rispetto a Mcdanno polimeri troppo viscosi alle temperature di tra-sformazione.
La struttura molecolare entra in gioco anche quandosi considera un altro aspetto importante della reologiadel polimero termoplastico, cioè la diminuzione dellaviscosità del fuso con l’aumentare del gradiente di velo-cità (comportamento non newtoniano di tipo pseudo-plastico). Questa proprietà è, come noto, di importanzafondamentale nella trasformazione dei termoplastici, inparticolare nello stampaggio a iniezione.
Come si può osservare in fig. 11, l’andamento dellaviscosità in funzione del gradiente di velocità è sche-matizzabile in due regimi: a bassi gradienti di velocitàla viscosità è costante (e uguale a h0); ad alti gradientidi velocità la viscosità decresce con il gradiente dellavelocità secondo una legge di potenza (lineare nel gra-fico bilogaritmico). Il passaggio dal primo al secondoregime avviene attorno a un valore critico di velocità, gc,che, come dimostrano gli studi di dinamica macromole-colare (Doi ed Edwards, 1988; Tsenoglou, 2000), dipen-de dai parametri molecolari M e Me secondo la seguen-te legge di proporzionalità:
Me 1gc�12�
1,412
M M2
In tale equazione è stato isolato il temine 1/M2 permettere in evidenza come, a parità di rapporto M/Me, ilvalore critico del gradiente di velocità gc decresca for-temente all’aumentare del peso molecolare M. Il fattogià ricordato che il PS, avendo Me grande rispetto aglialtri termoplastici, debba essere prodotto con valori rela-tivamente elevati di peso molecolare fa sì che il suo valo-re di gc sia particolarmente basso: è per questo che neiprocessi ad alto gradiente di velocità (per esempio, stam-paggio a iniezione) il PS è favorito rispetto ad altri poli-meri (come per esempio il policarbonato).
L’equazione spiega anche perché, come appare benvisibile in fig. 11, differenze elevate tra i valori di visco-sità newtoniana (h0) di campioni di PS con diverso pesomolecolare possono divenire trascurabili ai valori piùelevati di gradiente di velocità, a tutto vantaggio dellaprocessabilità, soprattutto nello stampaggio a inezione.La dipendenza della viscosità dalla temperatura è, perquanto riguarda la viscosità newtoniana h0, quella tipi-ca dei polimeri termoplastici, adeguatamente descrittadall’equazione di Williams-Landel-Ferry (WLF; Ferry,
1980), la cui validità per il PS è stata verificata fino a290 °C (Lomellini, 1992a). Quantitativamente l’effettoè apprezzabile in fig. 11 per un intervallo di temperatu-re, tipico dei processi di estrusione e stampaggio a inie-zione, compreso tra 200 e 240 °C. In tale intervallo, h0diminuisce di circa un fattore 10. L’eventuale presenzadi plastificante, come detto in precedenza, ha sostan-zialmente l’effetto di abbassare la Tg del polimero. Glieffetti sulle proprietà reologiche sono quantificabili appun-to in base al mutato valore di Tg e alla dipendenza delleproprietà dalla temperatura calcolata mediante l’equa-zione WLF. In pratica, la presenza del plastificante hasul comportamento reologico lo stesso effetto di unaumento di temperatura.
Elasticità del fusoRimanendo ancora nell’ambito delle proprietà reo-
logiche, un altro aspetto da tenere in considerazione èl’elasticità del fuso. Questa è la proprietà più importan-te in relazione ai processi di trasformazione che opera-no deformazioni estensionali del polimero fuso, comel’estrusione di lastre, in particolare quelle biorientate, latermoformatura e la formazione di schiume (espansio-ne). Anche in questo caso il parametro strutturale deter-minante è Me.
Come si può osservare in fig. 12, il modulo elasticodel polimero, relativamente elevato e quasi indipenden-te dalla temperatura nello stato vetroso, subisce una bru-sca diminuzione al superamento della Tg. Oltre questatemperatura il materiale diviene liquido e il modulo ela-stico è destinato ad annullarsi.
Per bassi valori di peso molecolare (minori di 100.000;v. ancora fig. 12), questo di fatto avviene nell’arco dipoche decine di gradi. Se però il peso molecolare è più
848 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
visc
osit
à (P
a.s)
10
100
1.000
10.000
100.000
gradiente di velocità (1/s)
Mw �280.000
Mw �170.000
0,01 0,1 1 10 100 1.000 10.000
fig. 11. Viscosità del fuso in funzione del gradiente di velocità (misure effettuate con reometri piatto-cono e capillare) per due PS a diverso Mw. Per ogni materiale, le tre curve si riferiscono alle temperature di 200 °C (curva più alta), 220 °C e 240 °C (curva più bassa).
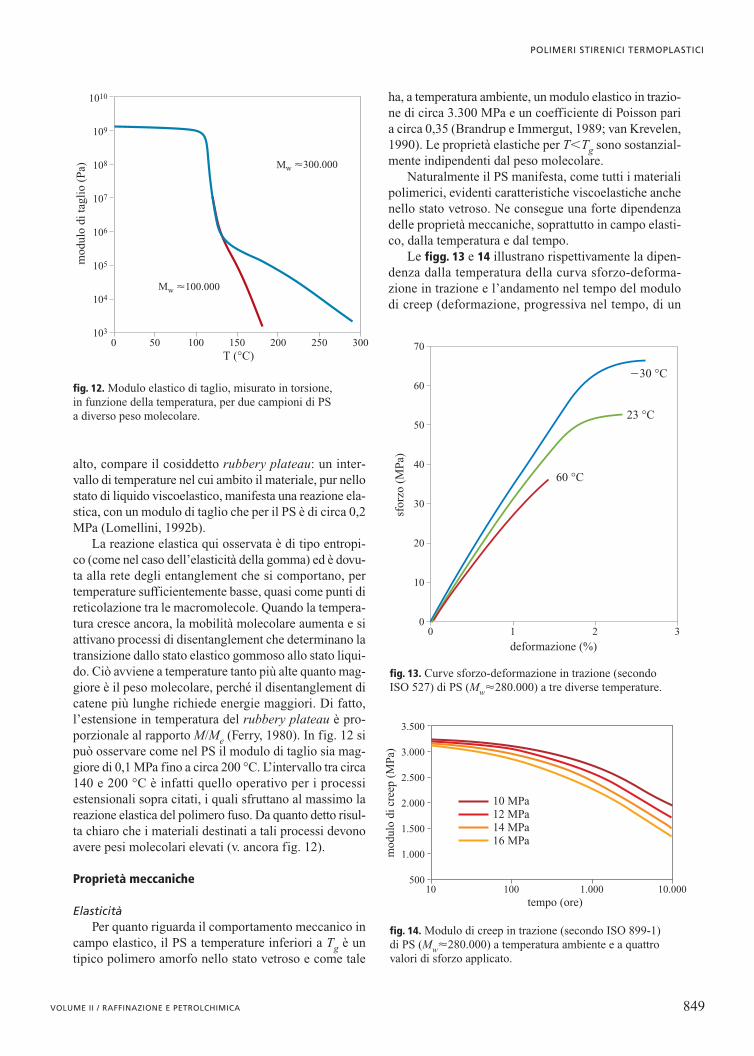
alto, compare il cosiddetto rubbery plateau: un inter-vallo di temperature nel cui ambito il materiale, pur nellostato di liquido viscoelastico, manifesta una reazione ela-stica, con un modulo di taglio che per il PS è di circa 0,2MPa (Lomellini, 1992b).
La reazione elastica qui osservata è di tipo entropi-co (come nel caso dell’elasticità della gomma) ed è dovu-ta alla rete degli entanglement che si comportano, pertemperature sufficientemente basse, quasi come punti direticolazione tra le macromolecole. Quando la tempera-tura cresce ancora, la mobilità molecolare aumenta e siattivano processi di disentanglement che determinano latransizione dallo stato elastico gommoso allo stato liqui-do. Ciò avviene a temperature tanto più alte quanto mag-giore è il peso molecolare, perché il disentanglement dicatene più lunghe richiede energie maggiori. Di fatto,l’estensione in temperatura del rubbery plateau è pro-porzionale al rapporto M/Me (Ferry, 1980). In fig. 12 sipuò osservare come nel PS il modulo di taglio sia mag-giore di 0,1 MPa fino a circa 200 °C. L’intervallo tra circa140 e 200 °C è infatti quello operativo per i processiestensionali sopra citati, i quali sfruttano al massimo lareazione elastica del polimero fuso. Da quanto detto risul-ta chiaro che i materiali destinati a tali processi devonoavere pesi molecolari elevati (v. ancora fig. 12).
Proprietà meccaniche
ElasticitàPer quanto riguarda il comportamento meccanico in
campo elastico, il PS a temperature inferiori a Tg è untipico polimero amorfo nello stato vetroso e come tale
ha, a temperatura ambiente, un modulo elastico in trazio-ne di circa 3.300 MPa e un coefficiente di Poisson paria circa 0,35 (Brandrup e Immergut, 1989; van Krevelen,1990). Le proprietà elastiche per T�Tg sono sostanzial-mente indipendenti dal peso molecolare.
Naturalmente il PS manifesta, come tutti i materialipolimerici, evidenti caratteristiche viscoelastiche anchenello stato vetroso. Ne consegue una forte dipendenzadelle proprietà meccaniche, soprattutto in campo elasti-co, dalla temperatura e dal tempo.
Le figg. 13 e 14 illustrano rispettivamente la dipen-denza dalla temperatura della curva sforzo-deforma-zione in trazione e l’andamento nel tempo del modulodi creep (deformazione, progressiva nel tempo, di un
849VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
mod
ulo
di ta
glio
(P
a)
103
104
105
106
107
108
109
1010
T (°C)0 50 100 150 200 300250
Mw �300.000
Mw �100.000
fig. 12. Modulo elastico di taglio, misurato in torsione, in funzione della temperatura, per due campioni di PS a diverso peso molecolare.
0
10
20
30
40
50
60
70
0 1 2 3
sfor
zo (
MPa
)
deformazione (%)
�30 °C
23 °C
60 °C
fig. 13. Curve sforzo-deformazione in trazione (secondo ISO 527) di PS (Mw�280.000) a tre diverse temperature.
mod
ulo
di c
reep
(M
Pa)
500
1.000
1.500
2.000
2.500
3.000
3.500
tempo (ore)
10 MPa12 MPa14 MPa16 MPa
10 100 1.000 10.000
fig. 14. Modulo di creep in trazione (secondo ISO 899-1) di PS (Mw�280.000) a temperatura ambiente e a quattrovalori di sforzo applicato.

materiale sottoposto a carico costante). In fig. 14 è anchepossibile osservare la non linearità del comportamentoelastico del materiale: per un valore fissato del tempo,il modulo di creep diminuisce al crescere dello sforzoapplicato.
Deformazione plastica e fratturaA differenza delle proprietà elastiche, i valori di resi-
stenza alla deformazione plastica e alla frattura sono for-temente influenzati dal peso molecolare. Ciò è noto dalungo tempo a livello di osservazione sperimentale: è del1959 un lavoro (McCormick et al., 1959) nel quale ven-gono riportati valori di sforzo e deformazione a rotturain trazione e valori di energia assorbita in urto-trazioneper campioni di PS in un ampio campo di pesi moleco-lari. I risultati di tale lavoro mostrano come la resisten-za meccanica cresca fortemente con il peso molecolareper valori di Mw da 40.000 a 120.000 circa, per poi cre-scere più lentamente, tendendo a raggiungere un valorecostante per Mw maggiore di circa 200.000. I dati sonostati sostanzialmente confermati da un lavoro successi-vo (Hauss, 1969).
Per interpretare queste evidenze sperimentali è neces-sario analizzare le modalità di deformazione plastica delmateriale. È noto che nei polimeri termoplastici amorfiallo stato vetroso la deformazione plastica può avveni-re attraverso due meccanismi fondamentali (Haward eYoung, 1997):• lo scorrimento di taglio (shear yielding), dove la
deformazione avviene a volume costante, con la com-parsa di bande di scorrimento e di strizione macro-scopica (necking). Si ha solitamente comportamen-to duttile, con deformazioni relativamente grandiprima della frattura;
• il crazing, in cui si osserva la formazione di difetticaratteristici, detti craze, estesi su piani perpendico-lari allo sforzo applicato. I craze sono simili a frat-ture, ma a differenza di queste possiedono una fittarete di microscopiche fibrille che fanno da ponte trale due superfici (Kramer, 1983). Il crazing è comu-nemente associato a comportamento fragile.Per quanto riguarda la deformazione plastica, il PS
ha un comportamento ben definito: in risposta alla com-ponente tensile dello sforzo applicato il meccanismo cheentra in azione è sempre il crazing, la cui ‘soglia’ d’in-nesco per questo polimero è sempre più bassa (in tra-zione) di quella relativa allo scorrimento di taglio.
Vi è una precisa correlazione tra la struttura mole-colare del PS e la sua netta propensione a deformare percrazing. È stato dimostrato (Donald e Kramer, 1982b;Kramer, 1983) che la caratteristica più importante in que-sto caso è la massima estensibilità (lmax) della strutturamacromolecolare amorfa. Quest’ultima può essere raf-figurata come una ‘rete’ tridimensionale, i cui nodi sonoi punti di entanglement e le cui maglie sono i segmenti
di catena tra due entanglement successivi. Indicando conM0 il peso molecolare dell’unità monomerica, l’estensi-bilità lmax è data dal rapporto tra la lunghezza della maglia,che è proporzionale a Me/M0, e la distanza tra le maglie‘a riposo’, che è proporzionale a (Me/M0)
1/2. Ne risultache, nel confronto tra diversi polimeri, lmax varia pro-porzionalmente a (Me/M0)
1/2. Gli autori citati dimostra-no la correlazione sperimentale tra lmax (e quindi Me/M0)e la prevalenza del meccanismo di crazing su quello discorrimento di taglio: nei polimeri ad alto lmax prevaleil crazing, e viceversa. Come sottolineato precedente-mente, il PS tra i polimeri termoplastici di uso comune,ha uno dei più alti valori di Me, e quindi anche di Me/M0e di lmax. Ciò spiega la decisa predominanza del mec-canismo di crazing in questo materiale.
I lavori di Kramer e collaboratori hanno spiegato intermini di correlazioni struttura-proprietà anche altriimportanti dettagli del meccanismo di crazing nel PS.
Il crazing, come si è detto, è associato a comporta-mento fragile. Sotto sforzo, il craze si estende e le suepareti si separano attraverso il progressivo trasferimen-to di nuovo materiale nelle fibrille, ma ciò permette solopiccole deformazioni macroscopiche prima che un ini-zio di rottura delle fibrille causi la trasformazione cata-strofica del craze in frattura.
Grazie ad analisi TEM di film sottili di PS deforma-ti in trazione, sono stati individuati (Kramer e Berger,1990), su campioni a diverso Mw, valori critici di defor-mazione per la nucleazione (ec) e la frattura (ef) dei craze.Mentre ec risulta indipendente dal peso molecolare, ef èfortemente influenzato dalla lunghezza di catena e cre-sce da valori confrontabili con ec per Mw di circa 40.000(il craze appena nucleato si rompe) a valori dell’ordinedi 10ec per Mw di 200.000, rimanendo poi invariato perMw superiori. I risultati si spiegano in base alla neces-sità del pieno sviluppo della rete di entanglement per lastabilità della struttura fibrillare. Pesi molecolari fino acirca 2Me danno luogo ai valori minimi di resistenza allafrattura; la tenacità cresce poi rapidamente con la lun-ghezza di catena, per raggiungere il massimo quando ilpeso molecolare è dell’ordine di 10Me. Questi risultaticonfermano e spiegano le relazioni empiriche tra Mw ela resistenza meccanica del PS sopra citate (McCormicket al., 1959; Hauss, 1969).
12.4.5 Struttura e proprietà dei copolimeri statisticidello stirene
Una caratteristica generale dei copolimeri statistici con-siste nel fatto che essi hanno proprietà intermedie rispet-to a quelle degli omopolimeri basati sui monomeri con-tenuti nel copolimero stesso. Mediante copolimerizza-zione statistica è perciò a volte possibile ottenere
850 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
‘compromessi’ interessanti, in cui si riesce a sopperire aspecifiche carenze di un omopolimero senza perderetroppo delle sue caratteristiche positive. Nel caso dellostirene, i copolimeri importanti, in quanto industrial-mente realizzati e presenti in campo applicativo, sono ilcopolimero stirene-anidride maleica (SMA), il copoli-mero stirene-metilmetacrilato (SMMA) e il copolimerostirene-acrilonitrile (SAN).
Stirene-anidride maleicaLa possibilità di copolimerizzare stirene e anidride
maleica (MA) è nota da lungo tempo (Scheirs e Priddy,2003). La proprietà più interessante del copolimero è latemperatura di transizione vetrosa, che cresce sensibil-mente con l’aumentare del contenuto di MA.
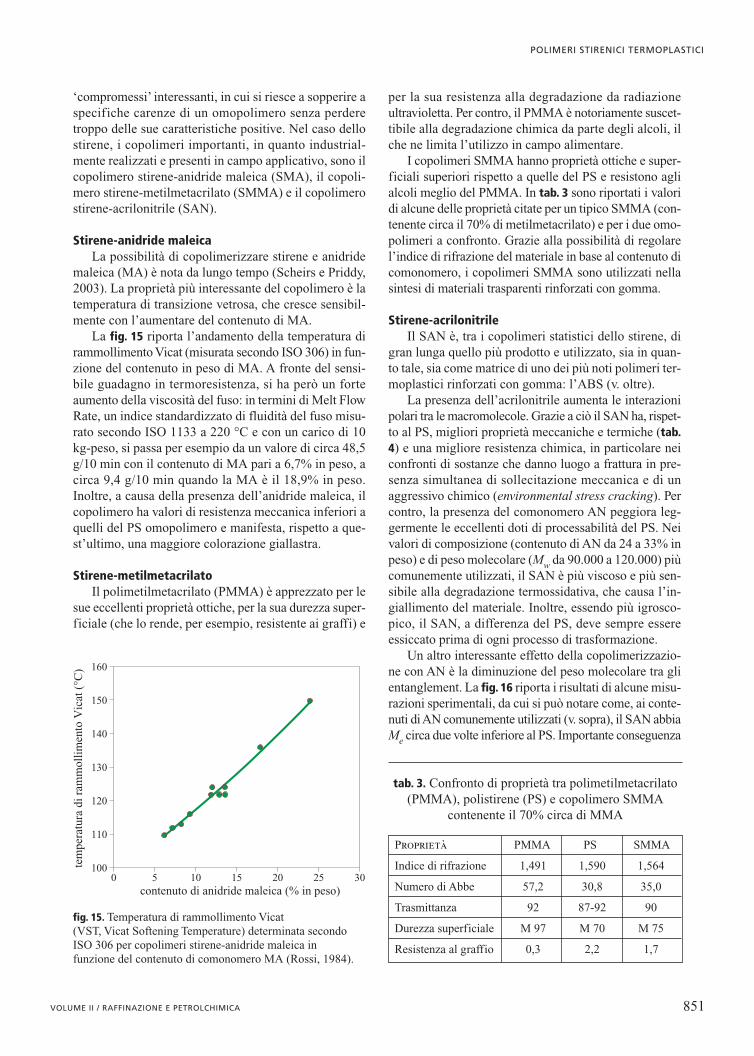
La fig. 15 riporta l’andamento della temperatura dirammollimento Vicat (misurata secondo ISO 306) in fun-zione del contenuto in peso di MA. A fronte del sensi-bile guadagno in termoresistenza, si ha però un forteaumento della viscosità del fuso: in termini di Melt FlowRate, un indice standardizzato di fluidità del fuso misu-rato secondo ISO 1133 a 220 °C e con un carico di 10kg-peso, si passa per esempio da un valore di circa 48,5g/10 min con il contenuto di MA pari a 6,7% in peso, acirca 9,4 g/10 min quando la MA è il 18,9% in peso.Inoltre, a causa della presenza dell’anidride maleica, ilcopolimero ha valori di resistenza meccanica inferiori aquelli del PS omopolimero e manifesta, rispetto a que-st’ultimo, una maggiore colorazione giallastra.
Stirene-metilmetacrilatoIl polimetilmetacrilato (PMMA) è apprezzato per le
sue eccellenti proprietà ottiche, per la sua durezza super-ficiale (che lo rende, per esempio, resistente ai graffi) e
per la sua resistenza alla degradazione da radiazioneultravioletta. Per contro, il PMMA è notoriamente suscet-tibile alla degradazione chimica da parte degli alcoli, ilche ne limita l’utilizzo in campo alimentare.
I copolimeri SMMA hanno proprietà ottiche e super-ficiali superiori rispetto a quelle del PS e resistono aglialcoli meglio del PMMA. In tab. 3 sono riportati i valoridi alcune delle proprietà citate per un tipico SMMA (con-tenente circa il 70% di metilmetacrilato) e per i due omo-polimeri a confronto. Grazie alla possibilità di regolarel’indice di rifrazione del materiale in base al contenuto dicomonomero, i copolimeri SMMA sono utilizzati nellasintesi di materiali trasparenti rinforzati con gomma.
Stirene-acrilonitrileIl SAN è, tra i copolimeri statistici dello stirene, di
gran lunga quello più prodotto e utilizzato, sia in quan-to tale, sia come matrice di uno dei più noti polimeri ter-moplastici rinforzati con gomma: l’ABS (v. oltre).
La presenza dell’acrilonitrile aumenta le interazionipolari tra le macromolecole. Grazie a ciò il SAN ha, rispet-to al PS, migliori proprietà meccaniche e termiche (tab.4) e una migliore resistenza chimica, in particolare neiconfronti di sostanze che danno luogo a frattura in pre-senza simultanea di sollecitazione meccanica e di unaggressivo chimico (environmental stress cracking). Percontro, la presenza del comonomero AN peggiora leg-germente le eccellenti doti di processabilità del PS. Neivalori di composizione (contenuto di AN da 24 a 33% inpeso) e di peso molecolare (Mw da 90.000 a 120.000) piùcomunemente utilizzati, il SAN è più viscoso e più sen-sibile alla degradazione termossidativa, che causa l’in-giallimento del materiale. Inoltre, essendo più igrosco-pico, il SAN, a differenza del PS, deve sempre essereessiccato prima di ogni processo di trasformazione.
Un altro interessante effetto della copolimerizzazio-ne con AN è la diminuzione del peso molecolare tra glientanglement. La fig. 16 riporta i risultati di alcune misu-razioni sperimentali, da cui si può notare come, ai conte-nuti di AN comunemente utilizzati (v. sopra), il SAN abbiaMe circa due volte inferiore al PS. Importante conseguenza
851VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
100
110
120
130
140
150
160
0 5 10 15 20 25 30
tem
pera
tura
di r
amm
olli
men
to V
icat
(°C
)
contenuto di anidride maleica (% in peso)
fig. 15. Temperatura di rammollimento Vicat (VST, Vicat Softening Temperature) determinata secondoISO 306 per copolimeri stirene-anidride maleica in funzione del contenuto di comonomero MA (Rossi, 1984).
Proprietà PMMA PS SMMA
Indice di rifrazione 1,491 1,590 1,564
Numero di Abbe 57,2 30,8 35,0
Trasmittanza 92 87-92 90
Durezza superficiale M 97 M 70 M 75
Resistenza al graffio 0,3 2,2 1,7
tab. 3. Confronto di proprietà tra polimetilmetacrilato(PMMA), polistirene (PS) e copolimero SMMA
contenente il 70% circa di MMA
di ciò è che nella deformazione plastica del SAN il mec-canismo di crazing non è così dominante da escludere loscorrimento di taglio, come accade nel PS (v. sopra); loshear yielding può entrare in azione, incrementando poten-zialmente la duttilità e la tenacità del materiale. In realtànel SAN puro gli sforzi di innesco dei meccanismi di sner-vamento sono elevati e la deformazione plastica è sem-pre fortemente localizzata: il materiale è perciò comun-que fragile. Le potenzialità di tenacità del copolimerovengono invece sfruttate appieno nell’ABS, il cui com-portamento meccanico verrà illustrato più avanti.
12.4.6 Struttura e proprietà dei materiali stirenicirinforzati con gomma
Una delle caratteristiche più limitanti nell’utilizzo pra-tico del PS e dei suoi copolimeri statistici è la fragilità.
Il cedimento meccanico di oggetti realizzati con questimateriali avviene quasi sempre in modo catastrofico, conla propagazione ‘esplosiva’ della frattura e, non di rado,con la formazione di frammenti rigidi e taglienti.
Una tecnica efficace per modificare questo compor-tamento è quella di introdurre nel materiale domini difase dispersa, di dimensioni e quantità opportune (v. anco-ra fig. 5), che abbiano un valore di modulo elastico moltoinferiore a quello della fase continua.
Alla base del comportamento meccanico (Bucknall,1977) dei materiali di questo tipo è il fenomeno dellaconcentrazione di sforzo: quando una forza esterna vieneapplicata al materiale, all’interfaccia tra la fase continuae ogni particella dispersa lo sforzo locale risulta più gran-de di quello applicato esternamente, secondo un fattoreche dipende dalla forma della particella e dal rapportotra i moduli elastici della fase dispersa e di quella con-tinua (Goodier, 1933). Anche la geometria del campo disforzi risulta alterata, con il risultato, per esempio, chesforzi uniassiali applicati al materiale possono dare ori-gine a sforzi triassiali (dilatazionali) nelle immediatevicinanze dei domini di fase dispersa.
I materiali stirenici modificati con fasi disperse rien-trano sostanzialmente in due categorie: quelli in cui lafase continua è polistirene omopolimero, denominatiHIPS, e quelli basati invece sul copolimero stirene-acri-lonitrile, denominati ABS, ASA (acrylonitrile-styrene-acrylic rubber), ecc. Le particelle di fase dispersa sononormalmente realizzate con gomma polibutadiene. Incasi particolari, soprattutto nel caso dei materiali a baseSAN, si possono usare gomme diverse (per esempiogomme acriliche nell’ASA per migliorare la resisten-za alla degradazione fotossidativa). In tutti i casi le par-ticelle di fase gommosa in questi materiali hanno valo-ri di modulo elastico circa 1.000 volte inferiori a quel-li delle rispettive fasi continue: in queste condizioni, econ geometria sferica delle particelle, il valore del fat-tore di concentrazione di sforzo teorico (Goodier, 1933)è circa 2.
HIPSL’effetto della concentrazione di sforzo locale attor-
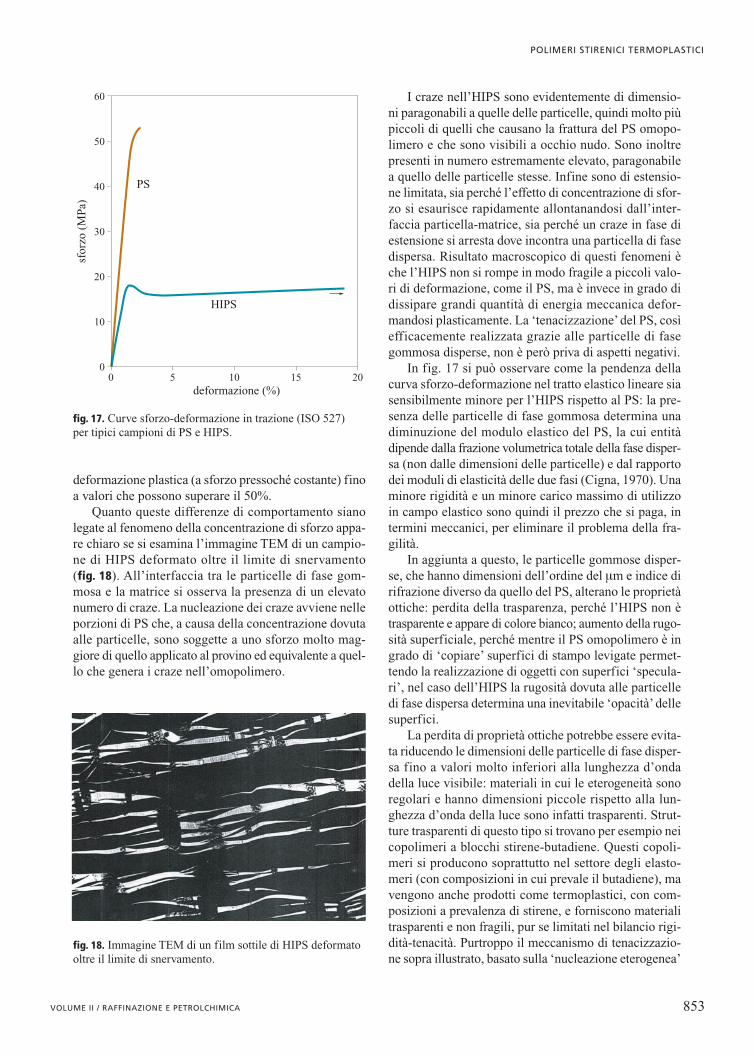
no alle particelle di fase gommosa risulta evidente se siconsiderano le curve sforzo-deformazione in trazione diun PS e di un HIPS, di cui sono riportati tipici esempi infig. 17. Si osserva che il PS ha comportamento elastico(andamento lineare della relazione sforzo-deformazio-ne) fino a un valore limite di sforzo, raggiunto il qualesi ha brusca frattura del provino con quasi totale assen-za di deformazione plastica. La deformazione massimaal momento della rottura non supera di molto il 2%.
Ben diverso è il caso dell’HIPS: il comportamentoè elastico fino a uno sforzo massimo nettamente infe-riore a quello di rottura del PS, raggiunto il quale si haun tipico fenomeno di snervamento con successiva
852 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
PSSAN
PROPRIETÀ(Mw�280.000)
(�24% AN;Mw�115.000)
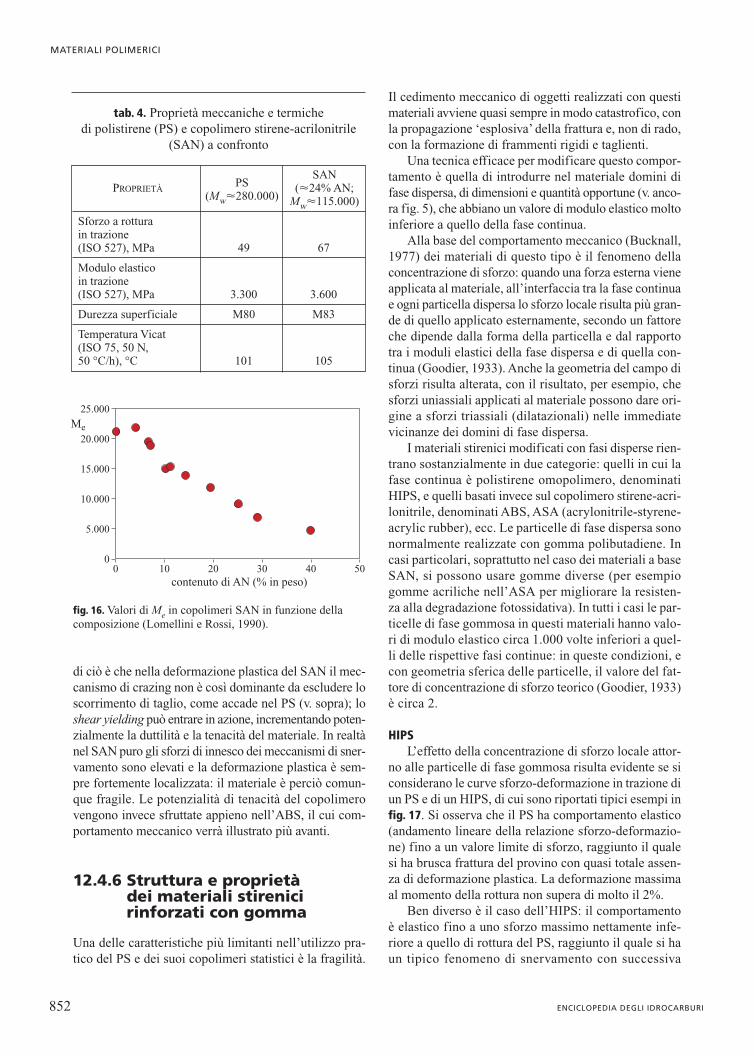
Sforzo a rottura in trazione (ISO 527), MPa 49 67
Modulo elastico in trazione(ISO 527), MPa 3.300 3.600
Durezza superficiale M80 M83
Temperatura Vicat (ISO 75, 50 N,50 °C/h), °C 101 105
tab. 4. Proprietà meccaniche e termiche di polistirene (PS) e copolimero stirene-acrilonitrile
(SAN) a confronto
Me
0
25.000
20.000
15.000
10.000
5.000
contenuto di AN (% in peso)0 10 20 30 40 50
fig. 16. Valori di Me in copolimeri SAN in funzione dellacomposizione (Lomellini e Rossi, 1990).
deformazione plastica (a sforzo pressoché costante) finoa valori che possono superare il 50%.
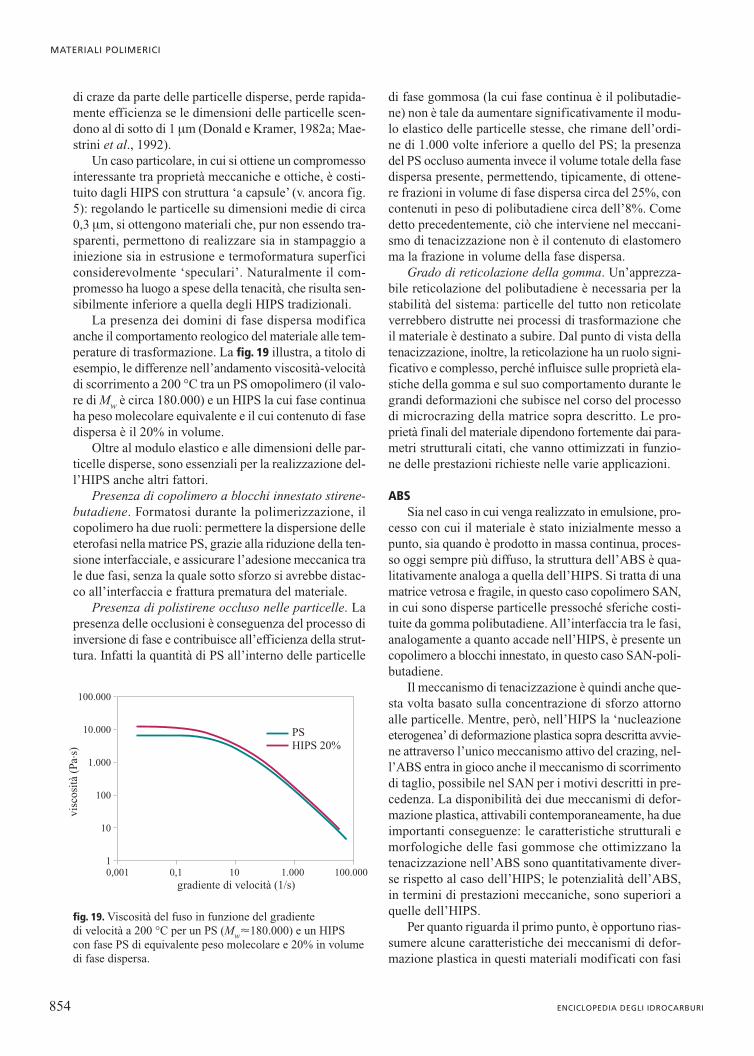
Quanto queste differenze di comportamento sianolegate al fenomeno della concentrazione di sforzo appa-re chiaro se si esamina l’immagine TEM di un campio-ne di HIPS deformato oltre il limite di snervamento(fig. 18). All’interfaccia tra le particelle di fase gom-mosa e la matrice si osserva la presenza di un elevatonumero di craze. La nucleazione dei craze avviene nelleporzioni di PS che, a causa della concentrazione dovutaalle particelle, sono soggette a uno sforzo molto mag-giore di quello applicato al provino ed equivalente a quel-lo che genera i craze nell’omopolimero.
I craze nell’HIPS sono evidentemente di dimensio-ni paragonabili a quelle delle particelle, quindi molto piùpiccoli di quelli che causano la frattura del PS omopo-limero e che sono visibili a occhio nudo. Sono inoltrepresenti in numero estremamente elevato, paragonabilea quello delle particelle stesse. Infine sono di estensio-ne limitata, sia perché l’effetto di concentrazione di sfor-zo si esaurisce rapidamente allontanandosi dall’inter-faccia particella-matrice, sia perché un craze in fase diestensione si arresta dove incontra una particella di fasedispersa. Risultato macroscopico di questi fenomeni èche l’HIPS non si rompe in modo fragile a piccoli valo-ri di deformazione, come il PS, ma è invece in grado didissipare grandi quantità di energia meccanica defor-mandosi plasticamente. La ‘tenacizzazione’del PS, cosìefficacemente realizzata grazie alle particelle di fasegommosa disperse, non è però priva di aspetti negativi.
In fig. 17 si può osservare come la pendenza dellacurva sforzo-deformazione nel tratto elastico lineare siasensibilmente minore per l’HIPS rispetto al PS: la pre-senza delle particelle di fase gommosa determina unadiminuzione del modulo elastico del PS, la cui entitàdipende dalla frazione volumetrica totale della fase disper-sa (non dalle dimensioni delle particelle) e dal rapportodei moduli di elasticità delle due fasi (Cigna, 1970). Unaminore rigidità e un minore carico massimo di utilizzoin campo elastico sono quindi il prezzo che si paga, intermini meccanici, per eliminare il problema della fra-gilità.
In aggiunta a questo, le particelle gommose disper-se, che hanno dimensioni dell’ordine del mm e indice dirifrazione diverso da quello del PS, alterano le proprietàottiche: perdita della trasparenza, perché l’HIPS non ètrasparente e appare di colore bianco; aumento della rugo-sità superficiale, perché mentre il PS omopolimero è ingrado di ‘copiare’ superfici di stampo levigate permet-tendo la realizzazione di oggetti con superfici ‘specula-ri’, nel caso dell’HIPS la rugosità dovuta alle particelledi fase dispersa determina una inevitabile ‘opacità’dellesuperfici.
La perdita di proprietà ottiche potrebbe essere evita-ta riducendo le dimensioni delle particelle di fase disper-sa fino a valori molto inferiori alla lunghezza d’ondadella luce visibile: materiali in cui le eterogeneità sonoregolari e hanno dimensioni piccole rispetto alla lun-ghezza d’onda della luce sono infatti trasparenti. Strut-ture trasparenti di questo tipo si trovano per esempio neicopolimeri a blocchi stirene-butadiene. Questi copoli-meri si producono soprattutto nel settore degli elasto-meri (con composizioni in cui prevale il butadiene), mavengono anche prodotti come termoplastici, con com-posizioni a prevalenza di stirene, e forniscono materialitrasparenti e non fragili, pur se limitati nel bilancio rigi-dità-tenacità. Purtroppo il meccanismo di tenacizzazio-ne sopra illustrato, basato sulla ‘nucleazione eterogenea’
853VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
0
10
20
30
40
50
60
0 5
PS
HIPS
10 15 20
sfor
zo (
MPa
)
deformazione (%)
fig. 17. Curve sforzo-deformazione in trazione (ISO 527) per tipici campioni di PS e HIPS.
fig. 18. Immagine TEM di un film sottile di HIPS deformatooltre il limite di snervamento.
di craze da parte delle particelle disperse, perde rapida-mente efficienza se le dimensioni delle particelle scen-dono al di sotto di 1 mm (Donald e Kramer, 1982a; Mae-strini et al., 1992).
Un caso particolare, in cui si ottiene un compromessointeressante tra proprietà meccaniche e ottiche, è costi-tuito dagli HIPS con struttura ‘a capsule’ (v. ancora fig.5): regolando le particelle su dimensioni medie di circa0,3 mm, si ottengono materiali che, pur non essendo tra-sparenti, permettono di realizzare sia in stampaggio ainiezione sia in estrusione e termoformatura superficiconsiderevolmente ‘speculari’. Naturalmente il com-promesso ha luogo a spese della tenacità, che risulta sen-sibilmente inferiore a quella degli HIPS tradizionali.
La presenza dei domini di fase dispersa modificaanche il comportamento reologico del materiale alle tem-perature di trasformazione. La fig. 19 illustra, a titolo diesempio, le differenze nell’andamento viscosità-velocitàdi scorrimento a 200 °C tra un PS omopolimero (il valo-re di Mw è circa 180.000) e un HIPS la cui fase continuaha peso molecolare equivalente e il cui contenuto di fasedispersa è il 20% in volume.
Oltre al modulo elastico e alle dimensioni delle par-ticelle disperse, sono essenziali per la realizzazione del-l’HIPS anche altri fattori.
Presenza di copolimero a blocchi innestato stirene-butadiene. Formatosi durante la polimerizzazione, ilcopolimero ha due ruoli: permettere la dispersione delleeterofasi nella matrice PS, grazie alla riduzione della ten-sione interfacciale, e assicurare l’adesione meccanica trale due fasi, senza la quale sotto sforzo si avrebbe distac-co all’interfaccia e frattura prematura del materiale.
Presenza di polistirene occluso nelle particelle. Lapresenza delle occlusioni è conseguenza del processo diinversione di fase e contribuisce all’efficienza della strut-tura. Infatti la quantità di PS all’interno delle particelle
di fase gommosa (la cui fase continua è il polibutadie-ne) non è tale da aumentare significativamente il modu-lo elastico delle particelle stesse, che rimane dell’ordi-ne di 1.000 volte inferiore a quello del PS; la presenzadel PS occluso aumenta invece il volume totale della fasedispersa presente, permettendo, tipicamente, di ottene-re frazioni in volume di fase dispersa circa del 25%, concontenuti in peso di polibutadiene circa dell’8%. Comedetto precedentemente, ciò che interviene nel meccani-smo di tenacizzazione non è il contenuto di elastomeroma la frazione in volume della fase dispersa.
Grado di reticolazione della gomma. Un’apprezza-bile reticolazione del polibutadiene è necessaria per lastabilità del sistema: particelle del tutto non reticolateverrebbero distrutte nei processi di trasformazione cheil materiale è destinato a subire. Dal punto di vista dellatenacizzazione, inoltre, la reticolazione ha un ruolo signi-ficativo e complesso, perché influisce sulle proprietà ela-stiche della gomma e sul suo comportamento durante legrandi deformazioni che subisce nel corso del processodi microcrazing della matrice sopra descritto. Le pro-prietà finali del materiale dipendono fortemente dai para-metri strutturali citati, che vanno ottimizzati in funzio-ne delle prestazioni richieste nelle varie applicazioni.
ABSSia nel caso in cui venga realizzato in emulsione, pro-
cesso con cui il materiale è stato inizialmente messo apunto, sia quando è prodotto in massa continua, proces-so oggi sempre più diffuso, la struttura dell’ABS è qua-litativamente analoga a quella dell’HIPS. Si tratta di unamatrice vetrosa e fragile, in questo caso copolimero SAN,in cui sono disperse particelle pressoché sferiche costi-tuite da gomma polibutadiene. All’interfaccia tra le fasi,analogamente a quanto accade nell’HIPS, è presente uncopolimero a blocchi innestato, in questo caso SAN-poli-butadiene.
Il meccanismo di tenacizzazione è quindi anche que-sta volta basato sulla concentrazione di sforzo attornoalle particelle. Mentre, però, nell’HIPS la ‘nucleazioneeterogenea’di deformazione plastica sopra descritta avvie-ne attraverso l’unico meccanismo attivo del crazing, nel-l’ABS entra in gioco anche il meccanismo di scorrimentodi taglio, possibile nel SAN per i motivi descritti in pre-cedenza. La disponibilità dei due meccanismi di defor-mazione plastica, attivabili contemporaneamente, ha dueimportanti conseguenze: le caratteristiche strutturali emorfologiche delle fasi gommose che ottimizzano latenacizzazione nell’ABS sono quantitativamente diver-se rispetto al caso dell’HIPS; le potenzialità dell’ABS,in termini di prestazioni meccaniche, sono superiori aquelle dell’HIPS.
Per quanto riguarda il primo punto, è opportuno rias-sumere alcune caratteristiche dei meccanismi di defor-mazione plastica in questi materiali modificati con fasi
854 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
visc
osit
à (P
a.s)
1
10
100
1.000
10.000
100.000
gradiente di velocità (1/s)
PSHIPS 20%
0,001 0,1 10 1.000 100.000
fig. 19. Viscosità del fuso in funzione del gradiente di velocità a 200 °C per un PS (Mw�180.000) e un HIPScon fase PS di equivalente peso molecolare e 20% in volumedi fase dispersa.

disperse, la cui comprensione è tuttora argomento di atti-va ricerca ed è documentata nella letteratura recente.
Anzitutto, la nucleazione del meccanismo di scorri-mento di taglio da parte delle particelle di fase gommo-sa non sembra condizionata da un valore critico delledimensioni delle particelle, come nel caso del crazing.L’innesco della deformazione plastica per shear yieldingè invece legato alla formazione di cavità nelle parti-celle di gomma (Bucknall et al., 1989; Ramsteiner eHeckmann, 1985): in conseguenza della componentedilatazionale dello sforzo ‘concentrato’ (v. sopra) attor-no alle particelle di fase dispersa, nella gomma presen-te in esse possono nascere e crescere cavità che, alteran-do la distribuzione degli sforzi nella fase continua cir-costante, favoriscono l’innesco dello scorrimento di taglio(Lazzeri e Bucknall, 1995). Questo fenomeno è possibi-le anche per particelle più piccole di quelle in grado digenerare craze e dipende non tanto dalle dimensioni delleparticelle stesse quanto piuttosto da valori opportuni dialtre caratteristiche quali la reticolazione della gomma el’adesione gomma-matrice. Per l’efficiente attivazionedel meccanismo di scorrimento nell’ABS sono adatteparticelle di dimensioni pari a uno o pochi decimi di mm.
Anche il meccanismo di crazing, poi, presenta nellamatrice SAN alcune differenze rispetto al caso del PS.La dimensione critica delle particelle per la nucleazionedei craze è tendenzialmente inferiore nel SAN (Walkere Collyer, 1994); inoltre la funzione di arresto dei crazein propagazione, che nell’HIPS è affidata alle particelledi fase gommosa che anche per questo devono essere suf-ficientemente grandi (Maestrini et al., 1992), nell’ABSè svolta in buona parte dallo sviluppo di zone di scorri-mento di taglio all’apice dei craze (Donald, 1994). Il risul-tato complessivo di queste diversità nei micromeccani-smi è che nell’ABS la tenacizzazione si ottiene con par-ticelle di fase dispersa sensibilmente più piccole di quelletipiche dell’HIPS, in particolare con distribuzioni didimensioni che comprendono sia valori dell’ordine di 0,1mm, adatti all’innesco dello scorrimento di taglio, siavalori di circa 1 mm, utili alla produzione di craze.
In fig. 20 A e B sono illustrati due esempi di mor-fologie della fase dispersa in ABS prodotti rispettiva-mente in emulsione e in massa. Questi esempi sono
solo indicativi: nei prodotti in commercio si possonoosservare differenze notevoli nelle caratteristiche dellefasi disperse, che sono continuo oggetto di ricerca pres-so i vari produttori.
Le differenze di prestazioni meccaniche tra ABS eHIPS si possono adeguatamente descrivere sulla base deirisultati delle prove standard di trazione (per esempiosecondo ISO 527) e di resistenza all’urto (per esempiosecondo ISO 180): tipicamente l’ABS, rispetto all’HIPS,ha maggiore modulo elastico e più alti valori di sforzo edeformazione allo snervamento; consente quindi la rea-lizzazione di oggetti che, a parità di dimensioni, sonopiù rigidi e in grado di sopportare carichi maggiori. Ladeformazione plastica successiva allo snervamento, chenell’HIPS è omogeneamente diffusa su tutto il materia-le, nell’ABS tende a localizzarsi (si osserva strizionenella prova di trazione), dando luogo a valori di defor-mazione nominale a rottura in trazione più piccoli. Taledato è solo apparentemente negativo: in realtà è una diret-ta manifestazione dell’attiva presenza del meccanismodi scorrimento di taglio che, associandosi al crazing,determina nell’ABS una maggiore capacità di dissipareenergia prima della frattura: la resistenza all’urto, misu-rata secondo Izod ISO 180, va da 15 a più di 30 kJ/m2
per gli ABS mentre non supera 10 kJ/m2 per i comunigradi di HIPS.
La minore dimensione delle particelle disperse nel-l’ABS determina anche una minore rugosità superficia-le rispetto all’HIPS e quindi la possibilità di realizzareoggetti dalle superfici ‘speculari’. Per contro, le parti-celle più piccole sono, a parità di frazione in volume difase gommosa totale, più vicine tra loro e questo causaun effetto maggiore (rispetto all’HIPS) della fase disper-sa sulle proprietà reologiche del materiale.
In fig. 21 si osservano le differenze nella curva diflusso (viscosità in funzione del gradiente di velocità) a220 °C per un SAN (Mw circa 90.000, acrilonitrile circail 24% in peso) e un ABS preparato con il medesimoSAN cui è stato aggiunto il 20% in volume di particel-le di polibutadiene con diametro medio pari a 0,13 mm(v., per confronto, fig. 19). Da notare l’incremento diviscosità ai bassi gradienti di velocità, manifestazione diun effetto tissotropico legato alle piccole dimensioni dei
855VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
1 mm 2 mm
a b
fig. 20. Immagine TEMdi un ABS prodotto con processo in emulsione (A) e di un ABS prodotto con processoin massa (B).
A B
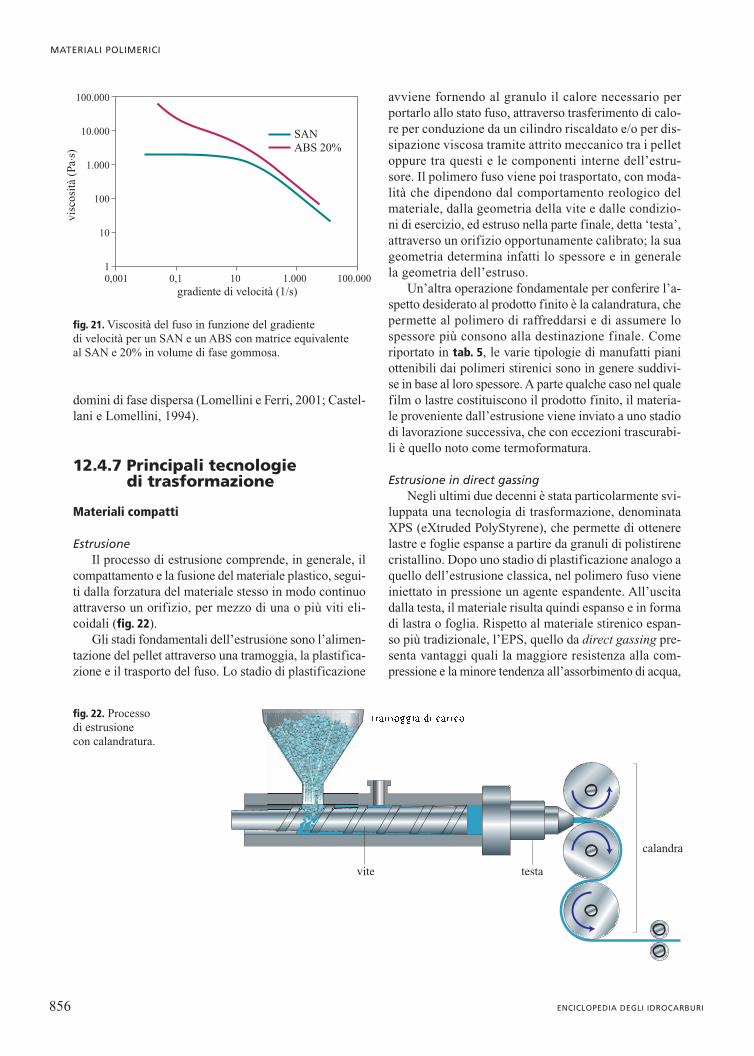
domini di fase dispersa (Lomellini e Ferri, 2001; Castel-lani e Lomellini, 1994).
12.4.7 Principali tecnologie di trasformazione
Materiali compatti
EstrusioneIl processo di estrusione comprende, in generale, il
compattamento e la fusione del materiale plastico, segui-ti dalla forzatura del materiale stesso in modo continuoattraverso un orifizio, per mezzo di una o più viti eli-coidali (fig. 22).
Gli stadi fondamentali dell’estrusione sono l’alimen-tazione del pellet attraverso una tramoggia, la plastifica-zione e il trasporto del fuso. Lo stadio di plastificazione
avviene fornendo al granulo il calore necessario perportarlo allo stato fuso, attraverso trasferimento di calo-re per conduzione da un cilindro riscaldato e/o per dis-sipazione viscosa tramite attrito meccanico tra i pelletoppure tra questi e le componenti interne dell’estru-sore. Il polimero fuso viene poi trasportato, con moda-lità che dipendono dal comportamento reologico delmateriale, dalla geometria della vite e dalle condizio-ni di esercizio, ed estruso nella parte finale, detta ‘testa’,attraverso un orifizio opportunamente calibrato; la suageometria determina infatti lo spessore e in generalela geometria dell’estruso.
Un’altra operazione fondamentale per conferire l’a-spetto desiderato al prodotto finito è la calandratura, chepermette al polimero di raffreddarsi e di assumere lospessore più consono alla destinazione finale. Comeriportato in tab. 5, le varie tipologie di manufatti pianiottenibili dai polimeri stirenici sono in genere suddivi-se in base al loro spessore. A parte qualche caso nel qualefilm o lastre costituiscono il prodotto finito, il materia-le proveniente dall’estrusione viene inviato a uno stadiodi lavorazione successiva, che con eccezioni trascurabi-li è quello noto come termoformatura.
Estrusione in direct gassingNegli ultimi due decenni è stata particolarmente svi-
luppata una tecnologia di trasformazione, denominataXPS (eXtruded PolyStyrene), che permette di ottenerelastre e foglie espanse a partire da granuli di polistirenecristallino. Dopo uno stadio di plastificazione analogo aquello dell’estrusione classica, nel polimero fuso vieneiniettato in pressione un agente espandente. All’uscitadalla testa, il materiale risulta quindi espanso e in formadi lastra o foglia. Rispetto al materiale stirenico espan-so più tradizionale, l’EPS, quello da direct gassing pre-senta vantaggi quali la maggiore resistenza alla com-pressione e la minore tendenza all’assorbimento di acqua,
856 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
visc
osit
à (P
a.s)
1
10
100
1.000
10.000
100.000
gradiente di velocità (1/s)
SANABS 20%
0,001 0,1 10 1.000 100.000
fig. 21. Viscosità del fuso in funzione del gradiente di velocità per un SAN e un ABS con matrice equivalente al SAN e 20% in volume di fase gommosa.
tramoggia di carico
vite testa
calandra
fig. 22. Processo di estrusione con calandratura.
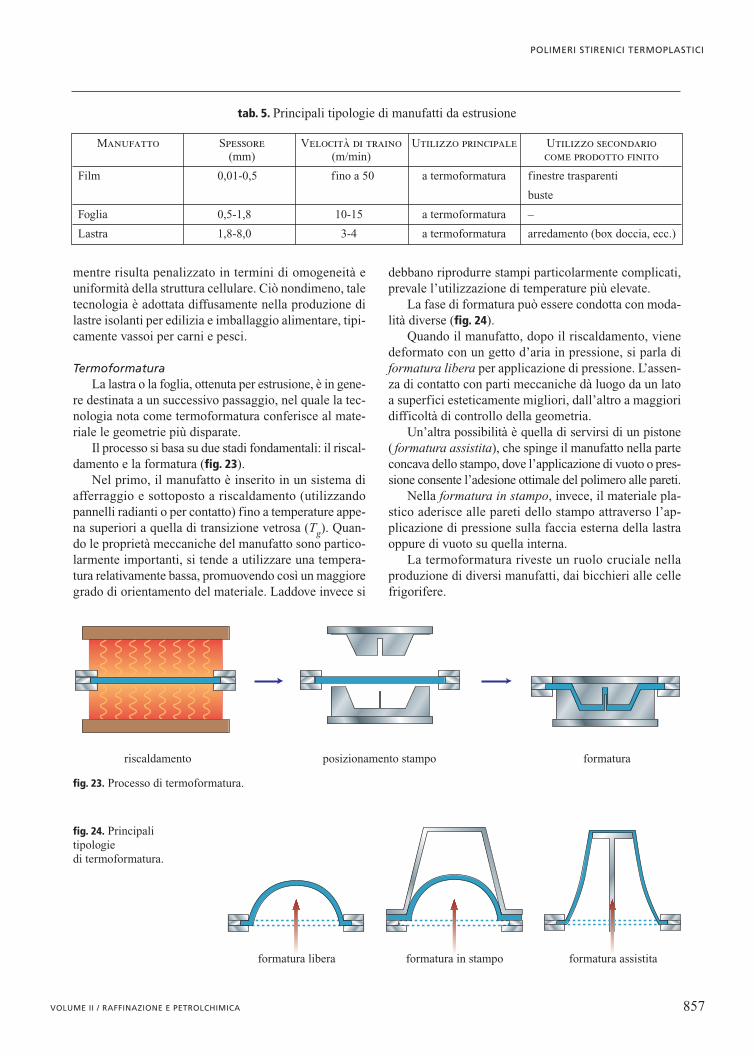
mentre risulta penalizzato in termini di omogeneità euniformità della struttura cellulare. Ciò nondimeno, taletecnologia è adottata diffusamente nella produzione dilastre isolanti per edilizia e imballaggio alimentare, tipi-camente vassoi per carni e pesci.
TermoformaturaLa lastra o la foglia, ottenuta per estrusione, è in gene-
re destinata a un successivo passaggio, nel quale la tec-nologia nota come termoformatura conferisce al mate-riale le geometrie più disparate.
Il processo si basa su due stadi fondamentali: il riscal-damento e la formatura (fig. 23).
Nel primo, il manufatto è inserito in un sistema diafferraggio e sottoposto a riscaldamento (utilizzandopannelli radianti o per contatto) fino a temperature appe-na superiori a quella di transizione vetrosa (Tg). Quan-do le proprietà meccaniche del manufatto sono partico-larmente importanti, si tende a utilizzare una tempera-tura relativamente bassa, promuovendo così un maggioregrado di orientamento del materiale. Laddove invece si
debbano riprodurre stampi particolarmente complicati,prevale l’utilizzazione di temperature più elevate.
La fase di formatura può essere condotta con moda-lità diverse (fig. 24).
Quando il manufatto, dopo il riscaldamento, vienedeformato con un getto d’aria in pressione, si parla diformatura libera per applicazione di pressione. L’assen-za di contatto con parti meccaniche dà luogo da un latoa superfici esteticamente migliori, dall’altro a maggioridifficoltà di controllo della geometria.
Un’altra possibilità è quella di servirsi di un pistone( formatura assistita), che spinge il manufatto nella parteconcava dello stampo, dove l’applicazione di vuoto o pres-sione consente l’adesione ottimale del polimero alle pareti.
Nella formatura in stampo, invece, il materiale pla-stico aderisce alle pareti dello stampo attraverso l’ap-plicazione di pressione sulla faccia esterna della lastraoppure di vuoto su quella interna.
La termoformatura riveste un ruolo cruciale nellaproduzione di diversi manufatti, dai bicchieri alle cellefrigorifere.
857VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
Manufatto Spessore Velocità di traino Utilizzo principale Utilizzo secondario(mm) (m/min) come prodotto finito
Film 0,01-0,5 fino a 50 a termoformatura finestre trasparenti
buste
Foglia 0,5-1,8 10-15 a termoformatura –
Lastra 1,8-8,0 3-4 a termoformatura arredamento (box doccia, ecc.)
tab. 5. Principali tipologie di manufatti da estrusione
riscaldamento posizionamento stampo formatura
fig. 23. Processo di termoformatura.
formatura libera formatura in stampo formatura assistita
fig. 24. Principalitipologie di termoformatura.

Stampaggio a iniezioneLa tecnologia dello stampaggio a iniezione (injec-
tion moulding) si basa sulla peculiare capacità dei poli-meri termoplastici, una volta riscaldati oltre la propriaTg, di riempire rapidamente uno stampo, anche di geo-metria complessa; dopo raffreddamento e quindi soli-dificazione del materiale plastico, il manufatto aventela forma dello stampo può essere estratto e utilizzato(fig. 25).
La parte iniziale del processo, nella quale avviene laplastificazione dei granuli, non è molto dissimile da quel-la già descritta per l’estrusione. A differenza di quantoavviene in quest’ultima, però, la vite di plastificazionenon è fissa, ma indietreggia sotto la spinta del flusso delpolimero durante la plastificazione. Al termine di que-sta fase la vite, dopo avere sospeso il moto rotatorio, simuove rapidamente in senso opposto, spingendo conun’azione simile a quella di un pistone il polimero fusoall’interno dello stampo.
Peculiare dello stampaggio a iniezione è anche lasezione comunemente definita gruppo di chiusura, checomprende una parte fissa, dove il polimero è convo-gliato in canali verso lo stampo, e una parte mobile, checonsente l’apertura dello stampo. Dopo l’iniezione e lesuccessive fasi di pressurizzazione e raffreddamento, ilmanufatto può essere estratto dallo stampo.
Rispetto alla termoformatura, lo stampaggio a inie-zione consente tempi di ciclo notevolmente più brevie l’ottenimento di manufatti con geometrie più com-plicate e a maggiore spessore. Gli svantaggi principa-li sono i costi di investimento legati alla progettazio-ne e costruzione degli stampi e la difficoltà a gestirestampi di grandi dimensioni. Lo stampaggio a inie-zione, in genere, non si presta all’utilizzazione di poli-meri dalla fluidità particolarmente bassa; l’alta visco-sità del materiale genera infatti un incremento dei costienergetici a causa della maggiore forza necessaria perla chiusura dello stampo. Per ottenere un tempo di cicloconvenientemente breve, si dovrebbe inoltre operare atemperature più alte, con conseguente impatto negati-vo sia sugli aspetti energetici sia sulla degradazionedel polimero.
Materiali espandibiliIl processo in sospensione permette, come già visto,
l’ottenimento di perle, dal diametro variabile, contenentidal 3 al 7% di espandente. La trasformazione delle perlein prodotto finito si articola tipicamente in tre fasi prin-cipali: espansione (o preespansione), maturazione, stam-paggio.
EspansioneDurante la prima fase, detta di espansione (o pre-
espansione), il materiale diminuisce la propria densitàfino a raggiungere quella dell’applicazione finale. All’in-terno di specifiche apparecchiature, le perle sono riscal-date a una temperatura di circa 100 °C, superiore quin-di sia alla Tg del polimero sia alla temperatura di ebol-lizione dell’espandente. L’ebollizione dell’espandentegenera così una pressione all’interno della perla di PS,causandone l’aumento di volume. Dal punto di vistatecnologico, il processo di preespansione può esserecondotto sia in continuo sia in discontinuo (fig. 26). I preespansori continui consentono una notevole pro-duttività specif ica, ma d’altro canto si rivelano, ingenere, meno efficienti nel controllo della densità. Ipreespansori discontinui moderni, pur penalizzati intermini di produttività, consentono invece un controllodi densità ottimale.
MaturazioneAl termine dell’espansione il prodotto viene raf-
freddato fino a temperatura ambiente. L’espandente e ilvapore presenti all’interno delle celle sono pertanto sot-toposti a una sensibile diminuzione di pressione legataalla loro condensazione. In questa fase le perle sono estre-mamente instabili: è sufficiente una lieve pressione ester-na per deformarle in modo permanente. Per riequilibra-re la pressione interna attraverso la diffusione di ariaall’interno della perla, è quindi necessario conservare leperle espanse per alcune ore in un silo con pareti per-meabili.
Il passaggio successivo, lo stampaggio, richiede lapresenza di una quantità di pentano residuo che dipen-de dalla densità, dal tipo di materiale e dalle condizioni
858 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
tramoggia di carico
stampo
partemobile
partefissa
gruppo di spintagruppo di iniezionegruppo di chiusura
fig. 25. Processo di stampaggio a iniezione.

ambientali. Dal momento che, oltre alla permeazionedell’aria, durante la maturazione si verifica anche un’ul-teriore perdita di espandente, la durata del processo nonpuò essere prolungata indefinitamente.
StampaggioLo stampaggio dell’EPS consiste in un’ulteriore
espansione in un volume definito: lo stampo è riempitodi perle preespanse e sottoposte a maturazione e suc-cessivamente è scaldato con vapore (fig. 27). L’espan-dente, che come già visto è ancora presente nelle perle,causa una nuova espansione del materiale all’internodello stampo. La pressione generata dall’espandente,inoltre, comprime le perle l’una contro l’altra, causan-done l’adesione e quindi la sinterizzazione; il grado diadesione tra le perle è detto livello di sinterizzazione.Al termine dello stampaggio, il manufatto viene sotto-posto a raffreddamento e successivamente a un ulteriore
stadio di maturazione. In generale, le perle a diametromedio-elevato sono stampate in apparecchiature di gran-di dimensioni, dette blocchiere, dalle quali si ottengonoappunto blocchi che possono essere poi tagliati in lastre.Le frazioni a dimensioni inferiori sono di norma trasfor-mate in stampi con la forma del manufatto finale.
12.4.8 Mercato
Ripartizione dei polimeri stirenici nel mondo e in Europa
I polimeri stirenici termoplastici occupano un ruolorilevante nel mercato mondiale dei materiali macromo-lecolari. Infatti circa il 12% della produzione di materie pla-stiche nel mondo è ascrivibile a tale categoria. La tab. 6riporta la ripartizione della produzione fra le varie tipo-logie (SAN, ABS, GPPS/HIPS, EPS) con riferimento siaal mercato mondiale sia al mercato europeo.
Principali settori applicativi dei polimeri stireniciI polimeri stirenici termoplastici presentano caratte-
ristiche generali, quali facilità di trasformazione (pro-cessabilità), buona stabilità dimensionale e di aspetto ebilancio di proprietà meccaniche, che li rendono partico-larmente adatti a una vasta gamma di settori applicativi.
859VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
preespansorecontinuo
agitatore
materiaprima
materia prima
cella di pesata
sensoredi livello
agitatore
vite senzafine
scaricoacqua
ingressovapore
uscitaperleespanse
uscitaperleespanse
preespansorediscontinuo
fig. 26. Processo di preespansione continuo e discontinuo(materiali espandibili).
carico delle perle ingresso e uscitadel vapore
raffreddamento edespulsione del blocco
fig. 27. Processo di stampaggio(materiali espandibili).
PolimeroMercato Mercatoeuropeo mondiale
SAN 2% 3%
ABS 24% 15%
GPPS/HIPS 57% 58%
EPS 17% 24%
tab. 6. Il mercato dei polimeri stirenici termoplastici

Di seguito, sono segnalati i settori più rilevanti, suddi-visi per tipologia di polimero utilizzato.
GPPS/HIPS. Le principali applicazioni di HIPS eGPPS sono: a) imballaggio: usa e getta (coppette, bic-chierini, ecc.), vassoi, vaschette per gelati e yogurt,vaschette espanse per carni, scatole per dolciumi e cosme-tici; b) elettrico/elettronico: scocche di computer e tele-visori, alloggiamenti e custodie per audiocassette e video-cassette, dischetti per computer, compact disc, CD ROMe DVD; c) elettrodomestici: cella e componenti internidi frigoriferi e congelatori, parti plastiche per TV, com-puter, lavatrici; d) edilizia: pannelli espansi isolanti; e)altro: profili e componenti per mobili, contenitori e arti-coli vari per cucine, box doccia, articoli da laboratorio,cancelleria, articoli medicali, giocattoli. La tab. 7 indi-ca la ripartizione per settori applicativi di GPPS e HIPSnel mercato europeo.
EPS. Le principali applicazioni di EPS sono: edili-zia, con pannelli isolanti, casseforme per cemento arma-to, alleggerimento per strutture portanti; imballaggio,
con contenitori per cibi caldi o freddi, inserti protettivi;altro, con caschi per sport, interno caschi per moto, animea perdere per fusione di metalli. La tab. 8 mostra comeil mercato europeo si ripartisce tra tali applicazioni.
ABS/SAN. Il campo applicativo dei copolimeri a baseacrilonitrile, riportato in tab. 9, è in genere simile a quel-lo già citato per GPPS/HIPS. Il vantaggio in termini diprestazioni, indotto dalla presenza di acrilonitrile, con-sente però utilizzazioni anche in altre applicazioni piùspecifiche: interni di auto, parti per motoscooter, fana-lerie e altri componenti esterni, corpi per batterie, pan-nelli di strumentazione; illuminotecnica, finestre percaravan, accendini, contenitori per batterie statiche, arre-do da bagno.
Bibliografia citata
Amos J.L. (1974) SPE international award address, 1973.Development of impact polystyrene. Review, «PolymerEngineering and Science», 14, 1-11.
Amos J.L. et al. (1954) US Patent 2694692 to Dow.Brandrup J., Immergut E.H. (edited by) (1989) Polymer
handbook, New York, John Wiley.Bucknall C.B. (1977) Toughened plastics, London, Applied
Science.Bucknall C.B. et al. (1989) Rubber toughening of plastics.
Part 12: Deformation mechanisms in toughened Nylon 6.6,«Journal of Materials Science», 24, 2255-2261.
Campbell J.D. et al. (2003) High temperature free radicalpolymerization. Investigation of continuous styrenepolymerisation, «Macromolecules», 36, 5491-5501.
Castellani L., Lomellini P. (1994) Phase volume and sizeeffects on the terminal relaxation of ABS melts, «RheologicaActa», 33, 446-453.
Cigna G. (1970) Dynamic mechanical properties, structure,and composition of impact polystyrene, «Journal of AppliedPolymer Science», 14, 1781-1793.
Claudy P. et al. (1983) Glass transition of polystyrene versusmolecular weight, «Polymer Bulletin», 9, 208-215.
Doi M., Edwards S.F. (1988) The theory of polymer dynamics,Oxford, Oxford University Press.
Donald A.M. (1994) Failure mechanisms in polymericmaterials, in: Collyer A.A. (editor) Rubber toughenedengineering plastics, London, Chapman & Hall, 1-28.
Donald A.M., Kramer E.J. (1982a) Craze initiation andgrowth in high-impact polystyrene, «Journal of AppliedPolymer Science», 27, 3279.
Donald A.M., Kramer E.J. (1982b) Effect of molecularentanglements on craze microstructure, «Journal of PolymerScience. Polymer Physics Edition», 20, 899-909.
Ferrando A., Longo A. (1997) Determination of reactivityratios from 13C NMR analysis of triads fractions for styrene-acrylonitrile copolymerisation as a function of temperature,«Polymer Preprints», 38, 798-799.
Ferry J.D. (1980) Viscoelastic properties of polymers, NewYork, John Wiley.
Georges M.K. et al. (1993) Narrow molecular weight resinsby a free-radical polymerization process, «Macromolecules»,26, 2987-2988.
860 ENCICLOPEDIA DEGLI IDROCARBURI
MATERIALI POLIMERICI
Settore Quota
Imballaggio 37%
Elettrico/elettronico 13%
Elettrodomestici 12%
Edilizia 11%
Altro (mobili, casalinghi, cancelleria, ecc.) 27%
tab. 7. Ripartizione per settori applicativi di GPPS/HIPS
Settore Quota
Isolamento ed edilizia 70%
Imballaggio 25%
Altro 5%
tab. 8. Ripartizione per settori applicativi di EPS
Settore Quota
Casalinghi/piccoli elettrodomestici 22%
Trasporti 19%
Estrusione 16%
Compound 15%
Elettrico/elettronico 11%
Altro 17%
tab. 9. Ripartizione per settori applicativi di ABS/SAN

Goodier J.N. (1933) Concentration of stress around sphericaland cylindrical inclusions and flaws, «Journal of AppliedMechanics», 55, 39.
Hauss A. (1969) Property characteristics of thermally producedpolystyrenes of different molecular weight distribution,«Die Angewandte Makromolekulare Chemie», 8, 73-86.
Haward R.N., Young R.J. (edited by) (1997) The physics ofglassy polymers, London, Chapman & Hall.
Huang N.J., Sundberg D.C. (1995) Fundamental studies ofgrafting reactions in free radical copolymerization. IV:Grafting of styrene, acrylate, and methacrylate monomersonto vinyl-polybutadiene using benzoyl peroxide and AIBNinitiators in solution polymerisation, «Journal of PolymerScience. Part A: Polymer Chemistry», 33, 2587-2603.
Ito K., Yamashita Y. (1965) Copolymer composition andmicrostructure, «Journal of Polymer Science. Part A:Polymer Chemistry», 3, 2165-2187.
Kramer E.J. (1983) Microscopic and molecular fundamentalsof crazing, «Advances in Polymer Science», 52/53, 1-56.
Kramer E.J., Berger L.L. (1990) Fundamental processes ofcraze growth and failure, «Advances in Polymer Science»,91/92, 1-68.
Krevelen D.W. van (1990) Properties of polymers,Amsterdam, Elsevier.
Lazzeri A., Bucknall C.B. (1995) Applications of a dilationalyielding model to rubber-toughened polymers, «Polymer»,36, 2895-2902.
Lomellini P. (1992a) Williams-Landel-Ferry versus Arrheniusbehaviour. Polystyrene melt viscoelasticity revised,«Polymer», 33, 4983-4989.
Lomellini P. (1992b) Effect of chain length on the networkmodulus and entanglement, «Polymer», 33, 1255-1260.
Lomellini P., Ferri D. (2001) Effetto del contenuto di gommasulla reologia estensionale di polimeri tenacizzati conelastomeri, in: Atti del 7° Convegno nazionale di reologiaapplicata, Faenza, 20-22 giugno.
Lomellini P., Rossi A.G. (1990) Effect of composition on theentanglement density in random copolymers, «MakromolareChemie», 191, 1729-1737.
McCormick H.W. et al. (1959) The effect of molecular-weightdistribution on the physical properties of polystyrene,«Journal of Polymer Science», 34, 87-100.
Maestrini C. et al. (1992) Particle entrapping by crazes inHIPS (high-impact polystyrene), «Polymer», 33, 1556-1558.
Matyjaszewsky K., Wang J.S. (1995) Controlled/’living’radical polymerization. Atom transfer radical polymerizationin the presence of transition-metal complexes, «Journal ofthe American Chemical Society», 117, 5614-5615.
Mayo F.R. (1968) The dimerization of styrene, «Journal of theAmerican Chemical Society», 90, 1289-1295.
Olaj O.F. (1971) Zur Kinetik der spontanen und der durchextrem kleine Starterkonzentrationen initiierten radicalischenPolymerisation des Styrols, «Monatshefte für Chemie»,102, 648-671.
Ramsteiner F., Heckmann W. (1985) Mode of deformationin rubber-modified polyamide, «Polymer Communication»,26, 199-200.
Rizzardo E. et al. (1999) Tailored polymers by free radicalprocesses, «Macromolecular Symposia», 143, 291-307.
Rossi A.G. (1984) Relazione tecnica 40/84, Polimeri Europa,Mantova.
Scheirs J., Priddy D.B. (edited by) (2003) Modern styrenicpolymers. Polystyrenes and styrenic copolymers, Chichester,John Wiley.
Staudinger H. (1932) Die hochmolekularen organischenVerbidungen. Kautschuk und Cellulose, Berlin, Springer.
Tsenoglou C. (2000) Molecular rheology of the non-newtonianbehaviour in polymer melts and concentrated solutions, in:Proceedings of the 13th International congress on rheology,Cambridge, 20-25 August, 50-52.
Walker I., Collyer A.A. (1994) Rubber toughenedmechanisms in polymeric materials, in: Collier A.A. (editor)Rubber toughened engineering plastics, London, Chapman& Hall.
Leonardo Castellani Aldo Longo
Francesco Pasquali
Polimeri EuropaMantova, Italia
861VOLUME II / RAFFINAZIONE E PETROLCHIMICA
POLIMERI STIRENICI TERMOPLASTICI
















![Prampolini MPPC29012015 [modalità compatibilità] · 2015-02-06 · polimeri sintetici vinilici o acrilici pvalcool pvacetato copolimeri acido/estere acrilico cere paraffiniche o](https://static.fdocumenti.com/doc/165x107/5e639ea8a9aa9d41c21676af/prampolini-mppc29012015-modalit-compatibilit-2015-02-06-polimeri-sintetici.jpg)


