
11739 In t r c y Medicine - simi.it · Medicina interna, complessità biologica, complessità...
72
123 EDITOR-IN-CHIEF Domenico Prisco GUEST EDITOR Francesco Perticone Volume 11 · Supplemento · Ottobre 2016 Atti 117° Congresso Nazionale della Società Italiana di Medicina Interna Roma, 14-16 Ottobre 2016
Transcript of 11739 In t r c y Medicine - simi.it · Medicina interna, complessità biologica, complessità...

123
E D I T O R- I N - C H I E F
Domenico Prisco
G U E S T E D I T O R
Francesco Pert icone
Internal and Emergency M
edicineVolum
e 10 - Supplement - O
ctober 2015pp. 1-107
11739
Volume 11 · Supplemento · Ottobre 2016
Atti 117° Congresso Nazionale della Società Italiana di Medicina Interna
Roma, 14-16 Ottobre 2016


Internal and Emergency Medicine
Official Journal of the Italian Society of Internal Medicine
Volume 11 · Supplemento · Ottobre 2016
LETTURA
Medicina interna, complessità biologica, complessità clinicaGino Roberto Corazza • Donatella Padula • Marco Vincenzo Lenti S1
RELAZIONE
La Medicina di Precisione: basi molecolari e applicazioni clinicheGianluca Gaidano S8
Precision Medicine: dagli slogan all’applicazione clinicaPier Mannuccio Mannucci S15
Per accertamenti e cure: il ricovero è sempre utile? ProPaolo Cavallo Perin • Paolo Fornengo • Cristina Amione S19
Per accertamenti e cure: il ricovero è sempre utile? ControRodolfo Sbrojavacca S24
IL DIBATTITO
L’origine dell’obesità: un colpo al centro e uno alla periferia. Parte I: Un colpo al centroPaolo Sbraccia S26
L’origine dell’obesità: un colpo al centro e uno alla periferia. Parte II: Un colpo alla periferiaRoberto Vettor S31
MEDICINA IN CORSIA
La medicina perioperatoria: l’interazione medico-chirurgo nella gestione del rischio Fabrizio Fabris • Irene Di Pasquale S39
Il delirium nei reparti di Medicina InternaGiuseppe Bellelli S43
MEET THE EXPERT
L’angioedema acquisitoMarco Cicardi • Roberto Castelli • Maddalena A. Wu • Andrea Zanichelli S47
Le porfirie: incroci pericolosi fra cute, fegato e midollo osseoPaolo Ventura S50
FOCUS ON
Le nuove frontiere della cardiologia interventistica strutturaleMarco Barbanti • Corrado Tamburino S57
IL PUNTO SU
Sicurezza e appropriatezza degli inibitori di pompa protonicaCorrado Blandizzi S61


Internal and Emergency Medicine
Official Journal of the Italian Society of Internal Medicine
Editor-in-ChiefD. PriscoDepartment of Experimental and Clinical Medicine, University of Florence and Interdisciplinary Internal Medicine Unit AOU Careggi, Florence, Italy
Co-EditorP. Rosen, Boston, USA
Advisory Board
E. Agabiti Rosei, Brescia, ItalyM. Alderman, New York, USAR. Barkin, Cambridge, MA, USA G. Berni, Florence, ItalyJ. Bousquet, Paris Cedex, FranceF. Caligaris Cappio, Milan, ItalyN. Carulli, Modena, Italy G. Davì, Chieti, ItalyM.M. d’Elios, Florence, ItalyG. Di Minno, Naples, ItalyA.F. Dominiczack, Glasgow, UKL. Fabbri, Modena, ItalyL. Gesualdo, Bari, ItalyP.H.R. Green, New York, USAA.M. Heagerty, Manchester, UKA. Iorio, Hamilton, ON, CanadaR.J. Johnson, Gainsville, FL, USAR. Landolfi , Rome, ItalyR. Lauro, Rome, ItalyG. Licata, Palermo, Italy G.D.O. Lowe, Glasgow, UKL. Luzzatto, Florence, ItalyT. Macdonald, Dundee, UKG. Mancia, Milan, ItalyN. Marchionni, Florence, ItalyG. Masotti, Florence, ItalyH. Meislin, Tucson, AZ, USAG. Palareti, Bologna, ItalyP. Pignatelli, Rome, ItalyE. Ritz, Heidelberg, GermanyS. Romagnani, Florence, ItalyB. Trimarco, Naples, ItalyA. Tuttolomondo, Palermo, ItalyA. Tyndall, Basel, SwitzerlandG. Valesini, Rome, Italy
Editorial BoardChairmanD. Prisco, Florence, ItalyEditorsW. Ageno, Varese, ItalyG. Agnelli, Perugia, ItalyA. Bartoloni, Florence, Italy S. Basili, Rome, Italy
C. Borghi, Bologna, ItalyM.D. Cappellini, Milan, ItalyA. Carolei, L’Aquila, ItalyE.M. Clini, Modena, ItalyG.R. Corazza, Pavia, ItalyJ. Douketis, Hamilton, ON, CanadaP. Gresele, Perugia, ItalyG. Laffi, Florence, Italy M. Levi, Amsterdam, NetherlandsA. Licata, Palermo, ItalyE. Maggi, Florence, Italy P.M. Mannucci, Milan, ItalyE. Manzato, Padua, ItalyR. Marcucci, Florence, ItalyP. Martelletti, Rome, Italy P.A. Modesti, Florence, Italy N. Montano, Milan, ItalyM. Muscaritoli, Rome, ItalyL. Pagliaro, Palermo, ItalyV. Pengo, Padua, ItalyF. Perticone, Catanzaro, ItalyR. Pontremoli, Genoa, ItalyP. Portincasa, Bari, ItalyP. Prandoni, Padua, Italy R. Polosa, Catania, ItalyP. Sbraccia, Rome, ItalyV. Toscano, Rome, ItalyB. Vaira, Bologna, ItalyA. Vannucchi, Florence, ItalyF. Violi, Rome, Italy
Emergency Medicine Chairman B. Adams, San Antonio, TX, USAEditorsP. Biddinger, Boston, MA, USAW. Brady, Charlottesville, PA, USAE. Brandler, New York, USA D. Coen, Milan, ItalyG.P. Conners, Kansas City, MO, USAS. di Somma, Rome, ItalyM.W. Donnino, Boston, MA, USAF. Franceschi, Rome, Italy J.N. Goldstein, Boston, MA, USAJ.E. Hipskind, Visalia, CA, USAC. Kabrhel, Boston, MA, USA L. Kass, Hershey, PA, USAA.M. Landry, Boston, MA, USAW.B. Lau, Philadelphia, PA, USAD. Lauque, Toulouse, FranceG. Morabito, Rome, ItalyJ. Mosier, Tucson, AZ, USAT. Osborn, Saint Louis, Missouri, USAR. Pini, Florence, ItalyS.R. Rezaie, San Antonio, TX, USAP. Rosen, Boston, MA, USAL. Sanchez, Boston, MA, USA
R. Sbrojavacca, Udine, ItalyD. Wampler, San Antonio, TX, USA R.E. Wolfe, Boston, MA, USA
Clinical Evidence and HealthTechnology AssessmentChairmanG.F. Gensini, Florence, ItalyDeputy ChairmenG. Costantino, Milan, ItalyG. Casazza, Milan, ItalyEditorsS. Corrao, Palermo, ItalyL. Moja, Milan, ItalyG.M. Podda, Milan, Italy S. Ricci, Perugia, ItalyA. Squizzato, Varese, ItalyG. Tripepi, Reggio Calabria, ItalyG. Virgili, Florence, Italy
Aims and Scope
Internal and Emergency Medicine (IEM) is an independent, international, English-language, peer-reviewed journal designed for internists and emergency physicians. IEM publishes a variety of manuscript types including Original investigations, Review articles, Letters to the Editor, Medical Illustrations and Invited Editorials, Points of view and Commentaries. Occasionally IEM accepts unsolicited Reviews or Editorials. The journal is divided into three sections, i.e., Internal Medicine, Emergency Medicine and Clinical Evidence and Health Technology Assessment, with three separate editorial boards. In the Internal Medicine section, invited Case records and, occasionally, Physical examinations, devoted to underlining the role of a clinical approach in selected clinical cases, are also published. Occasionally, the Emergency Medicine section includes a Morbidity and Mortality Report and an Airway Forum concerning the management of diffi cult airway problems. Finally, in the Clinical Evidence and Health Technology Assessment section brief discussions of topics of evidence-based medicine (Cochrane’s corner) and Research updates are published. IEM encourages letters of rebuttal and criticism of published articles whereas submission of case reports is not of interest for the journal. Topics of interest include all subjects that relate to the science and practice of Internal and Emergency Medicine. IEM is the offi cial journal of the Italian Society of Internal Medicine and is published eight times per year starting with 2013.

Copyright InformationFor AuthorsAs soon as an article is accepted for publication, authors will be requested to assign copyright of the article (or to grant exclusive publication and dissemination rights) to the publisher (respective the owner if other than Springer). This will ensure the widest possible protection and dissemination of information under copyright laws.
More information about copyright regulations for this journal is available at www.springer.com/11739
For ReadersWhile the advice and information in this journal is believed to be true and accurate at the date of its publication, neither the authors, the editors, nor the publisher can accept any legal responsibility for any errors or omissions that may have been made. The publisher makes no warranty, express or implied, with respect to the material contained herein.
All articles published in this journal are protected by copyright, which covers the exclusive rights to reproduce and distribute the article (e.g., as offprints), as well as all translation rights. No material published in this journal may be reproduced photographically or stored on microfi lm, in electronic data bases, on video disks, etc., without fi rst obtaining written permission from the publisher (respective the copyright owner if other than Springer). The use of general descriptive names, trade names, trademarks, etc., in this publication, even if not specifi cally identifi ed, does not imply that these names are not protected by the relevant laws and regulations.
Springer has partnered with Copyright Clearance Center’s RightsLink service to offer a variety of options for reusing Springer content. For permission to reuse our content please locate the material that you wish to use on
link.springer.com or on springerimages.com and click on the permissions link or go to copyright.com and enter the title of the publication that you wish to use. For assistance in placing a permission request, Copyright Clearance Center can be contacted directly via phone: +1-855-239-3415, fax: +1-978-646-8600 or e-mail: [email protected]
© SIMI, Società Italiana di Medicina Interna Viale dell’Università, 25, 00185 Rome, Italy
Journal Websitewww.springer.com/11739Electronic edition: link.springer.com/journal/11739
Subscription InformationInternal and Emergency Medicine is published 8 times a year. Volume 11 (8 issues) will be published in 2016.
ISSN: 1828-0447 print ISSN: 1970-9366 electronic
For information on subscription rates please contact Springer Customer Service Center:[email protected]
The Americas (North, South, Central America and the Caribbean)Springer Journal Fulfi llment,233 Spring Street, New York, NY 10013-1578, USATel.: 800-SPRINGER (777-4643); 212-460-1500 (outside North America)
Outside the AmericasSpringer Customer Service Center GmbH,Haberstr. 7, 69126 Heidelberg, GermanyTel.: +49-6221-345-4303
Advertisements E-mail contact: [email protected]
DisclaimerSpringer publishes advertisements in this journal in reliance upon the responsibility of the advertiser to comply with all legal requirements relating to the marketing and sale of products or services advertised. Springer and the editors are not responsible for claims made in the advertisements published in the journal. The appearance of advertisements in Springer publications does not constitute endorsement, implied or intended, of the product advertised or the claims made for it by the advertiser.
Offi ce of PublicationSpringer-Verlag Italia S.r.l. Via Decembrio, 28, 20137, Milan, Italy
Direttore ResponsabileCarlotta d’Imporzano, Milan, Italy
Printforce
Printed on acid-free paper
Registrazione del Tribunale di Milano N. 195 del 26 Marzo 2007
Distribution in Italy occurs according to the Italian Law 196/2003
Springer is part of Springer Science+Business Media

© SIMI 2016
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
LETTURA
Gino R. Corazza (•)Clinica Medica I Fondazione IRCCS Policlinico San MatteoPiazzale Golgi 19, 27100 Pavia E-mail: [email protected] [email protected]
D. Padula • M.V. LentiClinica Medica I Fondazione IRCCS Policlinico San Matteo, Pavia
Medicina interna, complessità biologica, complessità clinica
Gino Roberto Corazza • Donatella Padula • Marco Vincenzo Lenti
Carico e tipologia delle malattie sono in continua trasformazio-ne e i sistemi sanitari debbono prontamente adeguarsi a questi cambiamenti [1]. Recentemente, il progressivo invecchiamento della popolazione ha fatto sì che un numero crescente di perso-ne convivano con malattie croniche multiple (MCM) e, di con-seguenza, quello del paziente “complesso” è allo stato attuale uno dei temi più impegnativi in campo sanitario [2-4].
Secondo una recente definizione “la medicina interna è la specialità che applica le conoscenze scientifiche e le capacità cliniche al paziente adulto che passa dallo stato di salute alla malattia complessa” [5]. L’internista, quindi, è una figura di riferimento per i pazienti con condizioni cliniche complesse.
Come definire la complessità?
Studiare la complessità è complesso [6-8] e, a tutt’oggi, non è stata ancora accettata una definizione univoca di paziente complesso [9]. Più in generale, il concetto di complessità, o meglio di sistema complesso, ha determinato un cambia-mento di paradigma della conoscenza, segnando la fine del razionalismo classico, lineare e riduzionista, e l’avvento di una nuova concezione [10]. Al semplice e consueto oggetto di studio si sostituisce il sistema, cioè una serie di elementi dal-la cui interrelazione dinamica, negativa o positiva, emergono proprietà nuove del sistema stesso [11]. Esempi di sistema complesso sono il sistema immunitario e nervoso, l’indivi-
duo, i mercati finanziari, le aggregazioni sociali. Le sue pro-prietà comuni sono rappresentate dalla capacità di adattare il comportamento dei singoli elementi e di tutto il sistema all’ambiente e di co-evolvere con esso. Qualsiasi sistema complesso ha, di conseguenza, un andamento non lineare, nel senso che piccole modifiche delle variabili iniziali possono ri-percuotersi amplificate su altre e portare, quindi, a variazioni sproporzionate e a comportamenti difficilmente predittibili.
Non vi è dubbio che la maggioranza degli attuali pazienti internistici, anziani e con MCM, si ponga, nel classico dia-gramma di Stacey (Figura 1) [12], in una zona grigia, non evi-dence-based, intermedia tra razionalità e caos, caratterizzata da scarse certezze e bassi livelli di consenso. In un paziente con coliche biliari recidivanti l’indicazione alla colecistecto-mia laparoscopica cade nella zona della razionalità, regolata da ragionamenti meccanicistici basati sul semplice rapporto causa/effetto. Tuttavia, gran parte dei problemi che dobbiamo quotidianamente affrontare sono diversi: in un paziente dia-betico la glicemia ha spesso un andamento non lineare, livelli glicemici e fabbisogno insulinico non procedono in maniera parallela e il loro andamento può farsi francamente caotico nel caso di infezioni intercorrenti [13]. Tali considerazioni
Fig. 1 Diagramma certezza/accordo [12].
AREA DEL CAOS AREA DELLA
COMPLESSITÀ
AREA DELLA RAZIONALITÀ
Livello di Certezza
Live
llo d
i Acc
ordo
Alto Basso
Alto
Ba
sso

S2
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
suggeriscono che l’approccio meccanicista, pervicacemente fissato sull’organo o la malattia, sia del tutto inadeguato alla soluzione di problematiche cliniche complesse.
Come affrontare la complessità medica?
Finora il problema della complessità medica è stato affrontato attraverso una logica istintivamente riduzionista, basata sulla scomposizione in problemi sempre più piccoli da analizzare separatamente attraverso processi di tipo ipotetico-deduttivo, e sulla loro successiva, artificiosa ricomposizione. In altre paro-le, riferendoci al diagramma di Stacey, si è tentato di trasferire quei problemi propri dell’area complessa, caratterizzati quindi da bassi livelli di certezza e consenso, nei porti più sicuri del pensiero razionale. L’inadeguatezza dell’analisi riduzionista è già stata discussa nell’ambito della nostra Società. Nel 1982 Dioguardi e coll. [14], trattando la fisiopatologia del fegato in termini di sistema, sottoposero a una critica stringente le re-gole cartesiane di affrontare la complessità. L’approccio cau-sale ha limiti precisi: la soluzione di un singolo problema ne genera nuovi altri, producendo così un’evoluzione delle cono-scenze ad andamento progressivamente divergente. Per realtà altamente parametrate, come il fegato appunto, si impone un approccio sistemistico che ponga in primo piano la teleologia del sistema stesso, intesa come il suo contributo al manteni-mento dell’omeostasi generale. In questi casi, il pensiero deve muoversi in direzione del tutto speculare alla precedente, uti-lizzando il principio dell’aggregazione delle singole comples-sità attraverso l’identificazione in ogni situazione complessa di un numero di spiegazioni relativamente semplici. La malattia nasce ed evolve all’interno delle interazioni tra i vari elementi del sistema e non per il cedimento di uno di essi e, quindi, il problema della complessità va affrontato attraverso un pensiero flessibile, basato sull’intuizione, sull’esperienza e su approcci multipli non vincolati a rigidi protocolli [15]. A volte non è necessario risolvere tutti i problemi del paziente ma solo alcuni di essi, valutando poi se dal nuovo pattern emergente scaturi-scano possibili soluzioni agli altri.
Perché la medicina interna per il paziente complesso?
Suona certo autoreferenziale definire l’internista come il me-dico della complessità, ma non vi è dubbio che la comples-sità rappresenti una caratteristica intrinseca alla disciplina. A differenza delle altre specialità mediche, quello che meglio differenzia e caratterizza la medicina interna non è l’oggetto dello studio, quanto l’atteggiamento nello studio: il pensare appunto in termini di sistema, di elementi in interazione, nella consapevolezza che tale interazione sia più importante dei sin-
goli elementi e che il comportamento del tutto sia qualcosa di più e di diverso dalla somma del comportamento delle singole parti [16].
Gli attributi fondamentali della disciplina sono tradizio-nalmente ritenuti l’attitudine olistica, che mette al centro non organi o malattie ma il paziente come persona, e l’attitudine metodologica, più elusiva nella sua definizione, ma che può ricondursi alla capacità di riordinare il quadro clinico del pa-ziente in modo da porre in una prospettiva più chiara ed inter-pretabile proposizioni apparentemente oscure e convolute. Non pensiamo che tali attributi possano essere considerati come esclusivo patrimonio della medicina interna, ma, per certo, si sono particolarmente sviluppati nell’internista come reazione e compenso ad un corpo dottrinale particolarmente vasto e in continua espansione. In effetti, la complessità clinica può es-sere affrontata solo attraverso una configurazione “allargata” di questo approccio olistico. E d’altra parte, anche sul piano metodologico, gli attuali orientamenti privilegiano nel ragiona-mento clinico, al posto del tradizionale metodo analitico, ridu-zionista e fondato sull’impiego deliberato di algoritmi e alberi decisionali basati sull’evidenza, forme di ragionamento sinteti-camente orientate all’intero quadro clinico nella sua sistemica complessità [17]. Tali strategie, costruite su processi di pattern recognition automaticamente rapportati alla precedente espe-rienza del medico, si sono dimostrate superiori per efficacia, e soprattutto per efficienza, a quelle analitiche [18].
Come formare i medici alla gestione del paziente complesso?
La formazione medica è stata finora finalizzata alla compe-tenza, cioè a quello che si deve sapere o si deve saper fare. Tutto ciò è importante, ma lo studio dei sistemi complessi ci sta insegnando che è soprattutto la “capacità di adattamento” e di far fronte a contesti in continua trasformazione a gene-rare nuove conoscenze e migliorare la performance [19]. Il maggior nemico dell’innovazione è rappresentato dalla “re-sistenza al cambiamento” che è propria di ambiti culturali particolarmente elevati, quali quello dell’insegnamento uni-versitario [20]. Ad essa, tuttavia, si oppone una forza eguale e contraria, una vera e propria “pulsione al cambiamento” che caratterizza aree a basso livello di accordo e certezza, come appunto quelle della formazione medica. È necessario un nuo-vo approccio educazionale, in grado di adattare competenze classiche a circostanze nuove che si verificano in un contesto di relazioni ad andamento non lineare (Figura 2). La forma-zione alla complessità non si impara dai libri, ma deve avve-nire nell’ambito della complessità stessa, cioè in un ambiente necessariamente clinico, anche se il bed-side rappresenta un posto notoriamente più scomodo del bench-side. Scienza e

S3
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
pratica clinica, anche se hanno a che fare con lo stesso corpo di conoscenze, sono cose diverse, che hanno obiettivi diversi, regole diverse e diversi criteri di successo [21].
La nuova expertise consiste, in particolare, nella capaci-tà di individuare connessioni tra fenomeni apparentemente non correlati, mentre la didattica corrente è ancora fondata su competenze libresche, eventi formali rigidamente pianificati con obiettivi prefissati. Metodi non lineari di apprendimento danno valore alla risoluzione di problemi affrontati da pic-coli gruppi, che discutono e condividono, con la guida di un tutore, idee, piani di azione ed eventuali conclusioni [22]. Vi sono evidenze, infatti, che la conoscenza medica è depositata nella nostra memoria sotto forma di storie di pazienti (illness scripts) anziché come nozioni disgiunte [23]. In conclusione, progetti fondati su requisiti minimi sono da preferire a piani didattici troppo dettagliati. Facilitano l’innovazione e l’azio-ne condivisa e, in quanto prodotti di un’interazione organiz-zativa, non nascono perfetti ma evolvono nel tempo.
Quali sono le componenti di un paziente complesso?
Fino a un decennio fa, la complessità medica veniva identifica-ta con la presenza di MCM e/o con la maggiore utilizzazione di risorse [24]. Fattori demografici hanno favorito la maggiore prevalenza di malattie croniche neurodegenerative, metaboli-che ed autoimmuni e determinato una “epidemia” di pazienti con MCM. Si stima che nel 2030 più di 170 milioni di Ameri-cani saranno afflitti da malattie croniche e che la metà di essi sarà portatrice di tre o quattro condizioni associate [2]. Uno studio trasversale scozzese, effettuato su un vastissimo cam-pione di pazienti di tutte le età, ha confermato che la prevalenza di multimorbilità aumenta come atteso con l’avanzare dell’età, ma, che in termini assoluti, sono soprattutto i pazienti al di sot-to dei 65 anni a presentare due o più malattie [25].
La Figura 3 mostra la rappresentazione schematica di un paziente con multimorbilità. Alla malattia indice si associa-
no due comorbilità, configurando un quadro di multimorbi-lità che rappresenta l’intero carico patologico del paziente e che prescinde dall’identificazione di una malattia indice [26]. Altri fattori che incidono sul suo stato di salute sono rappre-sentati dal sesso, dall’età, dalla fragilità, intesa come ridotta riserva funzionale dei sistemi omeostatici, e dalla severità di ciascuna delle tre malattie associate. Tutto questo carico pa-tologico caratterizza la “complessità biologica” del paziente, alla quale immancabilmente si implementano fattori inerenti al suo contesto socio-ambientale e che sono parte integrante della risultante “complessità clinica”.
Complessità biologica
Già nel più ristretto ambito della comorbilità, l’identificazio-ne della “malattia indice”, importante per stabilire una gerar-chia relazionale e per il successivo decision-making, costi-tuisce un problema difficilmente risolvibile. In un paziente con diabete, ipertensione e depressione potrebbe, infatti, va-riare in rapporto allo specialista a turno coinvolto nelle cure, né, d’altra parte, la cronologia di comparsa delle tre malattie rappresenta un criterio affidabile di priorità [27]. A volte la situazione è più chiara, qualora vi sia un nesso eziologico evi-dente (emorragia cerebrale in corso di terapia anticoagulante prescritta per fibrillazione atriale) o per la condivisione di fat-tori di rischio (broncopneumopatia cronica ostruttiva e car-diopatia ischemica in paziente tabagista). In altri casi, come per due condizioni relativamente frequenti nella popolazione generale, l’associazione può essere casuale e la sua probabili-tà è pari al prodotto delle singole prevalenze o, in altri ancora, essere legata a “bias” di selezione, nel senso che pazienti con MCM si sottopongono a più frequenti controlli e hanno mag-giori probabilità di identificare ulteriori condizioni associate.
Un importante meccanismo di associazione nasce da re-centi evidenze che proteine malate, codificate da geni mutati, non si distribuiscono a caso, ma tendono ad aggregarsi tra loro dando origine a moduli patologici [28]. Questo meccani-
Fig. 2 Rappresentazioni grafiche di traiettorie razionali, complesse e caotiche.
Fig. 3 Rappresentazione schematica delle componenti della complessità biologica e clinica.
A B Linea della razionalità
Linea della complessità
Linea del caos
Complessità Clinica
FATTORI EXTRA-BIOLOGICI
Complessità Biologica
SEVERITÀ DI CIASCUNA MALATTIA
SESSO ETÀ FRAGILITÀ
Multimorbilità
DIABETE (malattia indice)
Comorbilità alla malattia indice
IPERTENSIONE DEPRESSIONE

S4
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
smo contribuirebbe a conferire una suscettibilità generale, più che specifica, alle malattie e trova una indiretta validazione in un recente studio che ha esplorato longitudinalmente i pattern di multimorbilità in una coorte di pazienti che presentavano più di dieci affezioni croniche [29]. I risultati mostrano il coinvolgimento cumulativo di una serie di organi e non di uno specifico sito, quale risultato di una serie di pressioni interne ed esterne all’organismo.
La multimorbilità comporta gestioni cliniche più difficili, costi sanitari più elevati, prescrizioni incongrue di farmaci, minore compliance terapeutica, tempi di degenza più lunghi, prognosi peggiori [29-32] e tassi di mortalità più elevati [33]. La medicina basata sulle evidenze si è focalizzata più sulla malattia individuale che sul paziente individuale e pazienti con MCM sono stati sistematicamente esclusi dai trials cli-nici randomizzati [34,35]. In altre parole, non disponiamo di linee guida validate per pazienti complessi con MCM [36] e nei loro confronti quelle attuali possono addirittura essere “cattive maestre”. Applicandole alla lettera per ogni singola patologia in un paziente con cinque diverse malattie, bisogne-rebbe prescrivere 19 dosi di 12 farmaci diversi da assumere in cinque diversi momenti della giornata, aumentando espo-nenzialmente il rischio di interazioni o eventi avversi [3]. C’è urgente necessità, quindi, di studi che affrontino in maniera pragmatica il problema della multimorbilità [37].
Qual è quindi, allo stato attuale, la strategia migliore nei confronti di questa tipologia di pazienti? È stato proposto di valutare se il tempo necessario al beneficio cumulativo di un farmaco compari, favorevolmente o meno, con il suo rischio cumulativo [38], anche se l’efficacia di tale approccio non è mai stata testata per decidere su singoli trattamenti [39]. Un paradigma alternativo è quello di concordare le cure con il paziente stesso, tenendo conto delle sue personali prefe-renze, dei suoi bisogni e delle sue priorità [40]. Si tratta di una novità importante. Finora la medicina si è impegnata a trattare al meglio ogni singola malattia, con risultati molto spesso inferiori alle attese. A 357 pazienti, il 69% dei quali portatore di quattro o più malattie croniche, è stato chiesto di indicare le loro priorità in termini di risultati attesi dalle cure. La larga maggioranza di essi (76%) si è espressa a favore del mantenimento dell’autosufficienza fisica e cognitiva, mentre solo l’11% ha scelto il prolungamento della sopravvivenza in quanto tale [41]. Un anziano con MCM potrebbe, ad esem-pio, ritenere eccessivo (e potenzialmente nocivo) un interven-to di protesi d’anca, se la sua unica aspirazione è quella di due passi sotto casa. Tenere presenti le priorità del paziente, anziché perseguire ad ogni costo temerari progetti di guari-gione, è ora considerata una strategia efficace ed economica-mente vantaggiosa di affrontare la multimorbilità [42]. Le sue aspettative costituiscono la lente attraverso la quale filtrare qualsiasi decisione.
Complessità clinica
Si capisce, da quanto si è detto, che la multimorbilità costi-tuisce di per se stessa un sistema complesso e rappresenta, allo stesso tempo, l’elemento più qualificante della comples-sità biologica. Non riflette, tuttavia, da sola la complessità clinica del paziente. A tale ultima concorrono una serie di variabili non biologiche operanti nel suo più immediato con-testo [7,8,43]. Fattori socio-economici (solitudine, nutrizione carente, mancanza di trasporti autonomi, dei moderni mezzi di comunicazione, di polizze assicurative), culturali (barriere linguistiche, differenze etniche, scolarità insufficiente), am-bientali (distanza dai servizi sanitari, difficile accesso all’as-sistenza territoriale, inquinamento, esposizione a tossine), comportamentali (tabagismo ed altre dipendenze, sedentarie-tà, dieta incongrua) interagiscono con la complessità biologi-ca nel determinare la globale complessità clinica (Figura 4). Non è facile per il medico, concentrato sull’assioma diagno-si-trattamento-guarigione, riconoscere una valenza clinica ad una serie di fattori extra-biologici, anche se è proprio la gestione di tali variabili contestuali ad assorbire quotidiana-mente una parte consistente del suo tempo e delle sue energie.
Che la complessità clinica richieda un approccio multi-dimensionale è confermato da una serie di studi. A Boston, in un setting di cure primarie, i pazienti venivano classificati come complessi sulla base del giudizio globale ed empirica-mente soggettivo dei loro curanti [44]. A differenza che in studi precedenti, fattori sociali e comportamentali contribui-vano a tale classificazione che, quindi, correlava scarsamen-te con indici di complessità focalizzati essenzialmente sulla multimorbilità. Sebbene questo studio abbia evidenti lacune (la possibilità di scambiare per complessità l’insufficiente conoscenza di una specifica malattia, l’incontrollata - ma intuitivamente alta - variabilità tra osservatori, l’assenza di un gold standard, la non estrapolabilità dei risultati ad altri setting), i pazienti definiti complessi andavano incontro ad un outcome clinico più sfavorevole. Questi risultati sono stati confermati dall’osservazione longitudinale, per quattro anni, della stessa coorte: la definizione di complessità clinica che,
Fig. 4 Rappresentazione grafica delle variabili che concorrono alla com-plessità clinica.
COMPLESSITÀCLINICA
Complessitàbiologica
Fattoriambientali
Fattoricomportamentali
Fattoriculturali
Fattorisocioeconomici

S5
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
come si è detto, incorporava oltre a quelli biologici anche fat-tori socio-comportamentali, si confermava, rispetto agli altri indici precedentemente in uso, più predittiva nei confronti di neoplasie, diabete, ipercolesterolemia, visite ambulatoriali, ricoveri e riammissioni a 30 giorni [45]. L’importanza di tali risultati, confermati da quelli di altri studi [46,47], consiste nella dimostrazione che è la complessità clinica dell’intero sistema, e non solo quella biologica, a determinare la stra-tificazione del rischio e a rappresentare il reale indicatore prognostico sul quale ricollocare risorse e redistribuire in-terventi mirati.
La natura dinamica della complessità clinica è stata analiz-zata da un semplice modello centrato sul paziente e basato sul rapporto tra carico di lavoro (malattie e correlati, attività quo-tidiane, eventi stressanti di varia natura) e capacità (condizio-ni fisiche e cognitive, risorse economiche ed organizzative) di far fronte a tali richieste [48]. Tale rapporto, inevitabilmente, tende nel tempo a squilibrarsi per aumento del primo e ridu-zione della seconda, ma, trovandoci in un sistema complesso, ciò non si verifica in maniera linearmente progressiva. Dif-ferenze biologiche (malattie croniche o neoplastiche, insor-genza o meno di complicanze, diversa risposta alle terapie), embricandosi con una serie di fattori contestuali, determinano pattern evolutivi ad andamento irregolare. Un’ulteriore varia-bile della quale tener conto è la cosiddetta resilienza, cioè la capacità del paziente, non solo fisica ma anche psicologica, di adattarsi, di reagire, di rimbalzare in alto a seguito di eventi stressogeni di varia natura. Tale capacità non dipende dalla te-
rapia, è del tutto inerente al paziente, non rappresenta un trat-to statico e può essere, quindi, proficuamente coltivata [49].
Come misurare la complessità clinica?
Sebbene le difficoltà nel definire e caratterizzare la comples-sità clinica siano evidenti, il poter disporre di uno score globa-le per misurare il paziente complesso sarebbe importante, in particolare in ambito internistico. Inizialmente, quando com-plessità e multimorbilità erano considerati sinonimi, l’indice di Charlson [50] ha rappresentato per semplicità e ripetibilità la metodica più usata, i cui risultati correlavano con i dati di mortalità e di consumo di risorse sanitarie. L’indice consiste nella somma di punteggi preassegnati ad ogni singola pato-logia corretti per l’età anagrafica. Il CIRS [51] (Cumulative Illness Rating Scale) introduce un grading di severità che si rapporta a 14 predefiniti cluster patologici.
I successivi studi, volti alla quantizzazione della comples-sità clinica, sono riportati nella Tabella 1. Lo studio di Saf-ford e coll. [55] merita qualche ulteriore commento. Prende in considerazione tutte le variabili riportate nella Figura 3, ciascuna delle quali è espressa da un vettore, che si interseca con gli altri in un punto centrale e che registra la forza e la direzione di quella variabile in quel particolare momento. At-traverso la somma di ogni singolo vettore è possibile costruire un vettore finale, espressione della complessità clinica glo-bale di quel paziente. L’intersecazione centrale è importante
Tabella 1 Sintesi degli studi volti alla quantizzazione della complessità clinica.
Bibliografia Descrizione Setting, n. pazienti
N. items
Finalità Variabili biologiche
Variabili non biologiche
Commenti
Flugelman e coll. [52]
Questionario da somministrare al ricovero
Geriatrico, 70 pazienti
7 Previsione, durata, degenza e mortalità ospedaliera
Età, ulcere da decubito, autonomia funzionale, stato cognitivo
Fabbisogno assistenziale
Introduce variabili non biologiche, semplice utilizzazione, non considera multimorbilità
De Jonge e coll. [53] (INTERMED)
Griglia di 4 domini, ciascuno analizzato da 20 variabili, punteggio 0-60
Psichiatria, medicina interna
80 Previsione, variabilità assistenziale passata, presente e futura
Complessità del paziente (dominio biologico)
Complessità della cura (dominio psico-sociale, sanitario)
Difficile utilizzazione in pratica clinica, risultati semi-quantitativi
Huyse e coll. [54] (COMPRI)
Questionario per medici e paramedici su durata e andamento del ricovero
Medicina interna, multicentro 2158 pazienti
13 Valutazione richiesta assistenziale
Polifarmacologia, neoplasie
Percezione dello stato di salute
Valutazione soggettiva, scarsa riproducibilità, variabili dicotomizzate
Safford e coll. [55]
Modello vettoriale che considera 5 determinanti della salute
Modello teorico
5 – Vettore biologico
Vettore socio-economico culturale, comportamentale, ambientale
Modello solo teorico, manca il sistema metrico validato
Bernabeu-Wittel e coll. [56] (PROFUND)
Questionario per medici
Medicina interna, geriatria, multicentro 1632 pazienti
9 Previsione mortalità
Demografiche, clinico-laboratoristiche
Psico-funzionali, sociali, familiari
Centrato più sulla mortalità del paziente che sulla sua complessità assistenziale

S6
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
e differenzia questo modello dalle precedenti concettualizza-zioni di complessità, rimarcando la dinamica interrelazione tra variabili biologiche e non biologiche e la possibilità che il peggioramento di una determini il peggioramento dell’intero sistema. Il limite attuale, ma tuttavia risolvibile, del modello consiste nella mancanza di un sistema metrico validato che correli un punto x del vettore a un grado y di compromissione.
Conclusioni
Il continuo aumento di prevalenza della multimorbilità, e quindi della complessità biologica, è stato più volte con-fermato [3]. Ciò comporterà un drammatico aumento della spesa sanitaria dal momento che già attualmente il 68% dei programmi di spesa di Medicare è assorbito da pazienti con MCM [57]. In assenza di una reale presa di coscienza del problema della complessità clinica e di ciò che ne consegue (ridistribuzione di risorse e personale, adeguati programmi di formazione, piani di assistenza coordinata, riorganizzazione territoriale), c’è il rischio consistente che questi pazienti rice-vano cure di bassa qualità, a costi elevati ed evitabili.
La Medicina Interna, essendo naturalmente orientata in senso generalistico, privilegiando il ragionamento clinico sull’effettuazione di esami strumentali costosi e invasivi, avendo dato prova negli anni di duttilità concettuale e or-ganizzativa [58], possiede i connotati ideali per “adattarsi” ai sistemi complessi. Occorre evitare ospedalizzazioni non strettamente necessarie o difensive [59], indagini dannose e/o non utili [60] e terapie potenzialmente conflittuali e favorire, al contrario e in maniera ragionata, la compliance a terapie essenziali e i flussi di informazione tra diverse figure profes-sionali [61].
Da quanto detto ne consegue che una corretta politica sa-nitaria nei confronti della complessità clinica non possa pre-scindere dal parallelo potenziamento della Medicina Interna, oltreché di quella territoriale. Purtroppo, nel nostro Paese, i segnali che è possibile cogliere hanno direzione diametral-mente opposta: il numero di posti letto di Medicina Interna è stato significativamente decurtato e dal rapporto tra flussi pensionistici e nuovi contratti di formazione è possibile pre-vedere, nei prossimi anni, un consistente saldo negativo di specialisti internisti. Occorre invertire questa tendenza ed è possibile che ciò si possa realizzare proprio attraverso una maggiore consapevolezza della rilevanza della complessità clinica. Sebbene sia stato autorevolmente ribadito che il si-stema di remunerazione delle prestazioni di assistenza ospe-daliera debba tenere in debito conto la complessità clinica [62,63], negli Stati Uniti, come da noi, il DRG per protesi articolare viene pagato circa il doppio di quello per scompen-so cardiaco con complicanze o comorbilità [64].
Dal momento che la complessità clinica può essere ora mi-surata, l’avvio di un progetto - senz’altro impegnativo - che porti ad implementare la complessità agli attuali sistemi di remunerazione, non è più rinviabile.
Bibliografia
1. Jones DS, Podolsky SH, Greene JA (2012). The burden of disease and the changing task of medicine. New Engl J Med 366(25):2333-8
2. Anderson G, Horvath J (2004). The growing burden of chronic dis-ease in America. Public Health Rep 119(3):263-70
3. Boyd CM, Darer J, Boult C et al (2005). Clinical practice guidelines and quality of care for older patients with multiple comorbid dis-eases: implications for pay for performance. JAMA 294(6):716-24
4. Parekh AK, Goodman RA, Gordon C, Koh HK; HHS Interagency Workgroup on Multiple Chronic Conditions (2011). Managing mul-tiple chronic conditions: a strategic framework for improving health outcomes and quality of life. Public Health Rep 126(4):460-71
5. Weinberger SE (2015). Challenges for internal medicine as the American College of Physicians celebrates its 100th anniversary. Ann Intern Med 162(8):585-6
6. Whittle J, Bosworth H (2007). Studying complexity is complex. J Gen Intern Med 22 Suppl 3:379-81
7. Turner BJ, Cuttler L (2011). The complexity of measuring clinical complexity. Ann Intern Med 155(12):851-2
8. Safford MM (2015). The complexity of complex patients. J Gen In-tern Med 30(12):1724-5
9. Peek CJ, Baird MA, Coleman E (2009). Primary care for patient complexity, not only disease. Fam Syst Health 27:287-302
10. Nicolis G, Prigogine I (2001). Exploring complexity: an introduc-tion. New York, WH Freeman
11. Plsek PE, Greenhalgh T (2001). Complexity science: the challenge of complexity in health care. Br Med J 323(7313):625-8
12. Stacey RD (1999). Strategic management and organizational dy-namics: the challenge of complexity. London, Financial Times
13. Kroll MH (1999). Biological variation of glucose and insulin includes a deterministic chaotic component. Biosystems 50(3):189-201
14. Dioguardi N, Di Padova C, Pietrogrande M (1982). Il sistema epati-co, la sua funzione ed i differenti quadri della sua insufficienza. Atti 83° Congresso SIMI. Pozzi, Roma
15. Wilson T, Holt T, Greenhalgh T (2001). Complexity science: com-plexity and clinical care. Br Med J 323(7314):685-8
16. Federspil G, Scandellari C (1994). La natura della medicina interna. Ann Ital Med Int 9(2):74-81
17. Eva KW (2004). What every teacher needs to know about clinical reasoning. Med Educ 39(1):98-106
18. Coderre S, Mandin H, Harasym PH, Fick GH (2003). Diagnostic rea-soning strategies and diagnostic success. Med Educ 37(8):695-703
19. Fraser SW, Greenhalgh T (2001). Coping with complexity: educat-ing for capability. Br Med J 323(7316):799-803
20. Plsek PE, Wilson T (2001). Complexity, leadership, and manage-ment in healthcare organisations. Br Med J 323(7315):746-9
21. Cox K (1995). Clinical practice is not applied scientific method. ANZ J Surg 65:553-7
22. Bligh J (1995). Problem-based, small group learning. Br Med J 311(7001):342-3
23. Cox K (2011). Stories as case knowledge: case knowledge as sto-ries. Med Educ 35(9):862-6
24. Friedman B, Jiang HJ, Elixhauser A, Segal A (2006). Hospital inpa-tient costs for adult with multiple chronic conditions. Med Care Res Rev 63(3):327-46

S7
123
Intern Emerg Med (2016) 11 (Suppl):S1-S7
25. Barnett K, Mercer SW, Norbury MN et al (2012). Epidemiology of multimorbidity and implications for health care, research and medi-cal education: a cross sectional study. Lancet 380(9836):37-43
26. Valderas JM, Starfield B, Sibbad B et al (2009). Defining comorbid-ity: implications for understanding health and health services. Ann Fam Med 7(4):357-63
27. John R, Kerby DS, Hennessy CH (2003). Patterns and impact of comorbidity and multimorbidity among community-resident Amer-ican Indian elders. Gerontologist 43(5):649-60
28. Menche J, Sharma A, Kitsak M et al (2015). Disease networks. Un-covering disease-disease relationships through the incomplete inter-actome. Science 347(6224):1257601
29. Vos R, van den Akker M, Boesten J et al (2015). Trajectories of mul-timorbidity: exploring patterns of multimorbidity in patients with more than ten chronic health problems in life course. BMC Fam Pract 16:2
30. Atun R (2015). Transitioning health systems for multimorbidity. Lancet 386(9995):721-2
31. Vogeli C, Shields AE, Lee TA et al (2007). Multiple chronic condi-tions: prevalence, health consequences, and implications for quality, care management, and costs. J Gen Intern Med 22 Suppl 3:391-5
32. Mannucci PM, Nobili A; REPOSI Investigators (2014). Multimor-bidity and polypharmacy in the elderly: lessons from REPOSI. In-tern Emerg Med 9(7):723-34
33. Lee TA, Shields AE, Vogeli C et al (2007). Mortality rates in vet-erans with multiple chronic conditions. J Gen Intern Med 22 Suppl 3:403-7
34. Fortin M, Dionne J, Pinho G et al (2006). Randomized controlled trials: do they have external validity for patients with multiple co-morbidities? Ann Fam Med 4(2):104-8
35. Tinetti ME (2014). The gap between clinical trials and the real world. Extrapolating treatment effects from younger to older adults. JAMA 174(3):397-8
36. Tinetti ME, Bogardus ST Jr, Agostini JV (2004). Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 351(27):2870-4
37. Mangin D, Heath I, Jamoulle M (2012). Beyond diagnosis: rising to the multimorbidity challenge. Brit Med J 344:1-3
38. Braithwaite RS, Fiellin D, Justice AC (2009). The payoff time: a flexible framework to help clinicians decide when patients with co-morbid disease are not likely to benefit from practice guidelines. Med Care 47(6):610-7
39. Guthrie B, Payne K, Alderson P et al (2012). Adapting clinical guidelines to take account of multimorbidity. Brit Med J 345:e6341. doi: 10.1136/bmj.e6341
40. Reuben DB, Tinetti ME (2012). Goal-oriented patient care--an al-ternative health outcomes paradigm. N Engl J Med 366(9):777-9
41. Fried TR, Tinetti ME, Iannone L et al (2011). Health outcome pri-oritization as a tool for decision making among older persons with multiple chronic conditions. Arch Intern Med 171(20):1856-8
42. Tinetti ME (2012). The retreat from advanced care planning. JAMA 307(9):915-6
43. Nardi R, Scanelli G, Corrao S et al (2007). Co-morbidity does not reflect complexity in internal medicine patients. Eur J Intern Med 18(5):359-68
44. Grant RW, Ashburner JM, Hong CC et al (2011). Defining patient complexity from the primary care physician’s perspective. Ann In-tern Med 155(12):797-804
45. Hong CS, Atlas SJ, Ashburner JM et al (2015). Evaluating a model to predict primary care physician-defined complexity in a large ac-
ademic primary care practice-based research network. J Gen Intern Med 30(12):1741-7
46. Moran WP, Zhang J, Gebregziabher M et al (2015). Chaos to com-plexity: leveling the playing field for measuring value in primary care. J Eval Clin Pract doi: 10.1111/jep.12298. [Epub ahead of print]
47. Safford MM, Brimacombe M, Zhang Q et al (2009). Patient com-plexity in quality comparisons for glycemic control: an observation-al study. Implement Sci 4:2
48. Shippee ND, Shah ND, May CR et al (2012). Cumulative complex-ity: a functional, patient-centered model of patient complexity can improve research and practice. J Clin Epidemiol 65(10):1041-51
49. Zullig LL, Whitson HE, Hastings SN et al (2016). A systematic review of conceptual frameworks of medical complexity and new model development. J Gen Intern Med 31(3):329-37
50. Charlson ME, Pompei P, Ales KL, MacKenzie CR (1987). A new method of classifying prognostic comorbidity in longitudinal stud-ies: development and validation. J Chronic Dis 40(5):373-83
51. Linn BS, Linn MW, Gurel L (1968). Cumulative Illness Rating Scale. J Am Geriatr Soc 16(5):622-6
52. Flugelman MY, Ben David Y, Harats N, Eliakim M (1986). A simple prognostic index for hospitalized geriatric patients. A prospective study of 70 patients. Gerontology 32(5):272-6
53. de Jonge P, Huyse FJ, Stiefel FC et al (2001). INTERMED--a clin-ical instrument for biopsychosocial assessment. Psychosomatics 42(2):106-9
54. Huyse FJ, de Jonge P, Slaets JP et al (2001). COMPRI--an instru-ment to detect patients with complex care needs: results from a Eu-ropean study. Psychosomatics 42(3):222-8
55. Safford MM, Jeroan JA, Kiefe CI (2007). Patient complexity: more than comorbidity. The vector model of complexity. J Gen Intern Med 22 Suppl 3:382-90
56. Bernabeu-Wittel M, Ollero-Baturone M, Moreno-Gaviño L et al (2011). Development of a new predictive model for polypathologi-cal patients. The PROFUND index. Eur J Intern Med 22(3):311-17
57. Anderson GF (2005). Medicare and chronic conditions. N Engl J Med 353(3):305-9
58. Corazza GR, Lenti MV, Di Sabatino A (2014). Trusting internal medicine in hard times. Intern Emerg Med 9(2):121-22
59. Sbrojavacca R, Pietrangelo A, Fenoglio L et al (2015). Reducing the risk of hospital admission: a call to action from the Italian Society of Internal Medicine. Eur J Intern Med 26(7):476-7
60. Morden NE, Colla CH, Sequist TD, Rosenthal MB (2014). Choos-ing wisely--the politics and economics of labeling low-value ser-vices. N Engl J Med 370(7):589-92
61. Luck J, Parkerton P, Hagigi F (2007). What is the business case for improving care for patients with complex conditions? J Gen Intern Med 22 Suppl 3:396-402
62. Larson EB, Fihn SD, Kirk LM et al (2004). The future of general internal medicine. Report and recommendations from the Society of General Internal Medicine (SGIM) Task Force on the Domain of General Internal Medicine. J Gen Intern Med 19(1):69-77
63. Corrocher R, Amodio P, Pini R et al (2010). La complessità clinica della medicina interna e le relative problematiche. In: L’innovazione in Medicina Interna. Errebi Grafiche, Falconara, pp 5-21
64. Mechanic R (2014). Post-acute care--the next frontier for con-trolling Medicare spending. N Engl J Med 370(8):692-4
RingraziamentiGli autori sono profondamente grati al Prof. Nicola Dioguardi, Istituto Humanitas Milano, per i preziosi suggerimenti e commenti.

123
© SIMI 2016
RELAZIONE
Gianluca Gaidano (•)Struttura Complessa a Direzione Universitaria di Ematologia Dipartimento di Medicina TraslazionaleUniversità del Piemonte OrientaleVia Solaroli 17, 28100 NovaraE-mail: [email protected]
La Medicina di Precisione: basi molecolari e applicazioni cliniche
Gianluca Gaidano
Definizione e razionale molecolare della Medicina di Precisione
La definizione di Medicina di Precisione prevede lo sviluppo di terapie sempre più mirate e personalizzate che sfruttino le informazioni genetiche per curare ogni paziente con farmaci specifici. Lo sviluppo di questo settore della medicina è il risultato della crescente disponibilità di database biologici, dell’aumentata accessibilità ed affidabilità di nuove tecnolo-gie per caratterizzare molecolarmente le malattie (tra cui la genomica, la transcrittomica, la proteomica, la metabolomica e, più in generale, le varie “-omics”) e di strumenti informati-ci per analizzare la mole di dati (comunemente definiti come “big data”) generati dai vari approcci “-omics”.
Il termine “Medicina di Precisione” è relativamente recen-te ed è stato storicamente preceduto dalla locuzione “Medi-cina Personalizzata” per indicare una medicina fondata sulle caratteristiche molecolari della malattia. Tuttavia, i medici lamentarono che l’approccio clinico da sempre era stato “per-sonalizzato” pur in assenza di dati molecolari, e che quindi la definizione di “Medicina Personalizzata” come medicina basata sulla genomica era inappropriata. Ne è conseguita l’in-troduzione del termine “Medicina di Precisione” [1-3].
Nell’ambito delle varie branche della medicina, la Medici-na di Precisione ha finora avuto un particolare sviluppo in on-cologia ed oncoematologia. Ciò, naturalmente, non significa che la Medicina di Precisione sia ristretta a questi ambiti, ma piuttosto che l’oncologia e l’oncoematologia hanno rappre-sentato dei modelli per meglio comprendere le potenziali ap-plicazioni cliniche e i potenziali benefici, nonché le potenzia-
li problematiche, della Medicina di Precisione. Lo sviluppo preferenziale della Medicina di Precisione nell’ambito delle malattie neoplastiche trova fondamento storico nel primo esempio, in senso temporale, di terapia molecolare, rappre-sentato dalla leucemia mieloide cronica. L’utilizzo dell’ini-bitore delle tirosin-chinasi, imatinib, nella leucemia mieloide cronica ha fornito la proof-of-concept che la caratterizzazio-ne molecolare di un tumore può condurre ad una diagnosti-ca differenziale su base genetica e, soprattutto, può fornire il razionale biologico per disegnare nuove terapie altamente mirate e selettive contro l’alterazione molecolare driver di un tumore [4].
Il concetto di terapia mirata (target therapy) richiede a pri-ori l’identificazione di rilevanti anomalie molecolari associa-te a tumori specifici. Questi biomarcatori tumorali, che inclu-dono sia varianti della linea germinale sia mutazioni somati-che, possono essere classificati come prognostici (associati all’outcome della malattia in coorti di pazienti) o predittivi (associati alla risposta ad una determinata terapia in un singo-lo paziente) [5-8]. La distinzione fra biomarcatori prognostici e biomarcatori predittivi riveste una sostanziale importan-za: la ricerca biomedica ha generato una grande quantità di biomarcatori prognostici, mentre il numero di biomarcatori predittivi, clinicamente più utili, è ancora oggi limitato [5-8]. La selezione di una particolare terapia antitumorale è basata sulla presenza di un bersaglio actionable (termine non facil-mente traducibile, ma che implica la possibilità di mettere in atto una “azione” contro il bersaglio stesso) e sulla possibi-lità di interferire con la funzione del bersaglio nella crescita, progressione ed evoluzione clonale neoplastica. Informazioni sulle principali alterazioni genomiche, tra cui le mutazioni somatiche, le alterazioni del numero di copie alleliche e poli-morfismi che influenzano il metabolismo dei farmaci, hanno già contribuito a definire lo sviluppo e l’uso di alcuni dei più nuovi trattamenti mirati contro determinate neoplasie, sottoli-neando l’importanza della genomica dei tumori nel far avan-zare la Medicina di Precisione [3-8].
Intern Emerg Med (2016) 11 (Suppl):S8-S14

S9
123
Intern Emerg Med (2016) 11 (Suppl):S8-S14
genesi [13,14]. La determinazione dello status di HER2 si basa sull’utilizzo dell’immunoistochimica per valutare l’i-perespressione della proteina o sull’utilizzo di ibridazione in situ per analizzare l’amplificazione genica [15,16]. Quattro farmaci sono approvati come terapia mirata contro HER2 (trastuzumab, pertuzumab, ado-trastuzumab emtansine e la-patinib), insieme ai propri companion kits diagnostici [17].
Inattivazione di TP53 nella leucemia linfatica cronica. Il gene onco-soppressore TP53 mappa sulla banda cromoso-mica 17p13 e codifica per un regolatore centrale della via di risposta ai danni al DNA. L’attivazione di TP53 porta all’ar-resto del ciclo cellulare cui consegue riparazione del DNA, apoptosi oppure senescenza cellulare [18]. Nella leucemia linfatica cronica, che rappresenta la leucemia più frequente della popolazione adulta, TP53 può essere inattivato trami-te delezioni, mutazioni puntiformi o una combinazione di entrambi [7,19-21]. L’inattivazione di TP53 si riscontra nel 3-8% dei casi di leucemia linfatica cronica al momento della diagnosi e nell’8-12% dei casi al momento della richiesta di trattamento di prima linea [7,19-21]. Nei pazienti refrattari alla terapia di prima linea, la prevalenza di inattivazione di TP53 sale al 30-40%, e nei casi di trasformazione a linfoma aggressivo (sindrome di Richter) sale fino al 60% [7,19-21]. L’importanza clinica delle anomalie di TP53 nella leucemia linfatica cronica è strettamente legata alla prognosi sfavore-vole segnata da questa lesione genetica ed alla sua stretta as-sociazione con la chemorefrattarietà ai farmaci genotossici [7,19-22]. Pertanto, l’inattivazione di TP53 nella leucemia linfatica cronica rappresenta non solo un biomarcatore pro-gnostico, ma un vero biomarcatore predittivo di chemiore-frattarietà che guida la scelta del trattamento [23]. Le linee guida attuali per la leucemia linfatica cronica raccomandano l’esecuzione dell’analisi molecolare di TP53 prima di avviare un paziente, in prima linea o in recidiva, alla terapia [20,24]. L’analisi molecolare, in base alle linee guida, deve essere ese-guita sia tramite FISH per esplorare la presenza di delezioni 17p13, sia tramite sequenziamento del DNA per esplorare la presenza di mutazioni. In caso di evidenza di lesioni inatti-vanti di TP53, è indicata la terapia con farmaci molecolari innovativi che inibiscono il B cell receptor (cosiddetti BCR inhibitors) che, non essendo farmaci genotossici come i che-mioterapici convenzionali, aggirano la chemiorefrattarietà associata all’inattivazione di TP53 [24].
Biomarcatori predittivi di tossicità. Gli esempi di HER2 e di TP53 esemplificano due biomarcatori predittivi utilizzati nella scelta del trattamento di uno specifico paziente in on-cologia ed oncoematologia. Altri biomarcatori tumorali sono associati al metabolismo ed alla tossicità di un farmaco. Poli-morfismi di popolazione, presenti nella linea germinale, che
Applicazioni cliniche di Medicina di Precisione nelle malattie neoplastiche
La Medicina di Precisione in oncologia è il risultato di una crescente consapevolezza che le caratteristiche cliniche di un determinato tumore sono dovute, almeno in parte, alle spe-cifiche lesioni molecolari del tumore stesso. Quindi, definire l’assetto genomico di un tumore rappresenta il presupposto per lo sviluppo di strategie di diagnostica e di terapia mirate. I termini companion diagnostics e target therapy sono quin-di entrati nel linguaggio clinico comune per intendere, da un lato, kit diagnostici di accompagnamento alla scelta di uno specifico farmaco molecolare e, dall’altro, strategie di terapia propriamente mirate a lesioni molecolari actionable [8]. Po-limorfismi o mutazioni germinali, clinicamente rilevanti, in enzimi e trasportatori (ad esempio TPMT, DPYD) che meta-bolizzano i farmaci chemioterapici, hanno dimostrato di ave-re un impatto nella risposta ai farmaci, fornendo il razionale per un dosaggio individualizzato al fine di ottimizzare il trat-tamento chemioterapico. Saggi di espressione di molti geni, derivati dall’approccio di gene expression profiling, servono per analizzare simultaneamente molteplici biomarcatori ed hanno dimostrato di poter contribuire alle decisioni terapeu-tiche in alcuni casi, in particolare nel carcinoma della mam-mella [8]. Più di recente, l’introduzione del Next Generation Sequencing, che ha permesso il sequenziamento ad alta pro-cessività degli acidi nucleici, ha condotto all’identificazione di molte alterazioni molecolari potenzialmente actionable nei tumori [9-11]. La complessità genetica dei tumori, quindi, in-duce la transizione dal paradigma “un gene, un farmaco” (one gene-one drug) ad un approccio multigenico [12].
Le autorità regolatorie, sia in Europa sia negli Stati Uniti, hanno approvato diverse target therapies basate su mutazioni specifiche essenziali per l’efficacia (e quindi per l’indicazio-ne clinica) del farmaco insieme ad un companion kit, il cui utilizzo è approvato per definire il profilo molecolare del tu-more. Metodologie di uso comune includono l’ibridazione in situ fluorescente (FISH), l’immunoistochimica, la polymerase chain reaction (PCR) e il sequenziamento del DNA; questi ul-timi due metodi sono utilizzati per rilevare la presenza o l’as-senza di una specifica mutazione. Qui di seguito sono descritti alcuni esempi di applicazioni cliniche consolidate della Me-dicina di Precisione nel contesto delle malattie neoplastiche.
Iperespressione/Amplificazione di HER2 nel carcinoma della mammella. Il gene che codifica per lo human epidermal growth factor receptor (HER2) è iperespresso nel 15-20% dei carcinomi della mammella e in altri tipi di tumore [13,14]. HER2 forma eterodimeri con altri recettori tirosin-chinasici transmembrana (EGFR, HER3, HER4), e l’attivazione di queste vie di trasduzione del segnale promuove la tumori-

S10
123
Intern Emerg Med (2016) 11 (Suppl):S8-S14
ottimale delle metodologie basate sulle CTC per guidare in modo efficace il trattamento antineoplastico.
Il ctDNA è rappresentato da acidi nucleici solubili ri-lasciati nel sangue periferico in pazienti neoplastici come conseguenza di apoptosi e necrosi delle cellule tumorali nel microambiente [26]. Già nel 1977, Leon et al. [32] avevano identificato ctDNA nel plasma di pazienti affetti da cancro del polmone. Il DNA circolante (cell free DNA, cfDNA) è stato identificato come fenomeno fisiologico anche nel sangue pe-riferico di individui sani; tuttavia pazienti affetti da tumori hanno maggiori quantità di cfDNA, la cui rilevazione è asso-ciata a prognosi peggiore [28,33]. Nel contesto dei tumori, le cellule tumorali rilasciate sono di solito fagocitate dai macro-fagi che digeriscono le cellule e ne rilasciano il DNA nel mi-croambiente tessutale [33,34]. Inoltre, i tumori di regola sono composti da una miscela di diversi cloni di cellule neoplasti-che a cui si deve l’eterogeneità genomica ed epigenomica dei tumori umani; le cellule neoplastiche poi sono mescolate ad altri tipi di cellule normali, quali le cellule stromali [26,35]. Così, durante lo sviluppo del tumore, il cfDNA può essere rilasciato nel sangue sia da parte di cellule neoplastiche sia da parte di cellule normali [26]. Di conseguenza, la proporzione di cfDNA che origina dalle cellule tumorali (vero e proprio ctDNA) varia a seconda dello stato e dimensione del tumore. Questo aspetto deve essere considerato con attenzione nell’a-nalisi del ctDNA ai fini della biopsia liquida.
Complessivamente, con appositi accorgimenti pre-analiti-ci, analitici e interpretativi, il ctDNA può essere sottoposto ad analisi con Next Generation Sequencing per ottenere il qua-dro dell’assetto mutazionale del tumore. Questo approccio ha numerosi aspetti positivi, tra cui: – l’assetto mutazionale del ctDNA riflette la composizione
genetica di tutto il tumore, quindi anche di sedi distanti dal sito della biopsia originale. Ciò permette, quindi, di identi-ficare potenziali marcatori molecolari di chemiorefrattarie-tà presenti in una sede differente rispetto alla sede sottopo-sta a biopsia e inizialmente valutata molecolarmente;
– la non invasività di questa strategia diagnostica conferisce la possibilità di monitorare l’assetto genetico del tumore in maniera seriata nel tempo con prelievi intervallati longitu-dinalmente. Ciò permette, ad esempio, l’identificazione di nuove mutazioni, insorte de novo o selezionate dalla tera-pia, che possono costituire predittori di chemiorefrattarie-tà. Alcuni trial clinici hanno dimostrato chiaramente che il ctDNA è uno specifico biomarcatore altamente sensibile per i tumori, ad esempio in pazienti con carcinoma della mammella [28,36].
In ambito ematologico, il linfoma diffuso a grandi cellule B ha rappresentato un modello pilota per applicare lo studio del ctDNA [37,38]. Nell’esperienza del nostro gruppo, il Next
modulano il metabolismo di farmaci chemioterapici inclu-dono i geni che codificano per tiopurina-S-metil transferasi (TPMT), diidropirimidina deidrogenasi (DPYD) e uridin-di-fosfato-glucuronosiltransferasi 1A1 (UGT1A1) [8]. Le linee guida del Clinical Pharmacogenomics Implementation Con-sortium (CPIC) assistono il clinico nell’ottimizzazione della dose del farmaco in base al profilo molecolare del pazien-te [25]. Il ruolo di questi polimorfismi è anche riconosciuto dalle autorità regolatorie (ad esempio, Food and Drug Admi-nistration, FDA) che prescrivono una modulazione della dose del farmaco in base al genotipo. I farmaci in questione sono la 6-mecaptopurina e la tioguanina nel caso di TPMT, il 5-flo-rouracile nel caso di DPYD e irinotecan nel caso di UGT1A1.
Il potenziale della biopsia liquida per la Medicina di Precisione Oncologica
La Medicina di Precisione Oncologica può richiedere la pos-sibilità di analisi molecolari seriate durante la storia clinica di un paziente, da cui deriva la necessità di poter disporre di materiale biologico prelevabile con approcci non invasivi. Inoltre, l’analisi molecolare di una biopsia singola può non essere sufficiente per ricapitolare l’eterogeneità clonale e la complessità molecolare del tumore da cui un paziente è af-fetto. In quest’ottica, due biomarcatori circolanti importanti sono rappresentati dallo studio delle cellule tumorali circo-lanti (CTC) e dal DNA tumorale circolante (circulating tu-mor DNA, ctDNA) [26]. Entrambi questi marcatori sono stati “promossi” come possibilità concreta di biopsia liquida, ov-vero biomarcatori minimamente invasivi in grado di fornire un quadro globale dell’assetto genetico di un tumore in base ad un semplice prelievo di sangue periferico [27,28].
Le CTC sono rare cellule rilasciate dal tumore primario o metastatico che possono essere isolate dal sangue periferico di pazienti con tumori solidi [29]. La rarità e breve emivita delle CTC, probabilmente misurata in ore, rende l’isolamento di queste cellule una sfida sperimentale; quindi una metodica ottimale di dosaggio delle CTC dovrebbe idealmente essere in grado di processare una grande quantità di cellule in un lasso di tempo relativamente breve, ed al contempo dovrebbe poter specificamente enumerare e catturare le cellule maligne nel contesto del background eterogeneo del sangue. In prati-ca, ciò richiede la capacità di rilevare una CTC per 105-107 cellule mononucleate [26]. Le CTC sono state caratterizzate molecolarmente tramite diversi approcci, compreso l’uso di Next Generation Sequencing [26]. Questi studi hanno rivelato alcuni meccanismi di resistenza ai farmaci nel carcinoma del-la mammella e possono contribuire a caratterizzare meglio le cellule tumorali capaci di metastatizzare [30,31]. Rimangono ancora molte sfide nell’identificazione, isolamento e utilizzo

S11
123
Intern Emerg Med (2016) 11 (Suppl):S8-S14
ne alterazioni genomiche rispondono ai farmaci da cui sono bersagliate. Per esempio, la mutazione V600 → E del gene BRAF risponde ai farmaci che inibiscono BRAF nel melano-ma e nella leucemia a cellule capellute (hairy cell leukemia), mentre questi stessi farmaci hanno bassi tassi di risposta nei tumori del colon con la stessa mutazione se il farmaco è som-ministrato come agente singolo [40].
I cosiddetti umbrella trials sono progettati per analizzare l’impatto di diversi farmaci destinati a diverse mutazioni o in un singolo sottotipo di cancro o in una varietà di sottotipi tu-morali [41]. Dopo l’analisi del profilo molecolare del tumore di ciascun paziente, un algoritmo guidato molecolarmente è for-mulato per stabilire un piano di trattamento individualizzato.
Il National Cancer Institute (NCI) statunitense ha recente-mente proposto un nuovo disegno di trial, altamente innova-tivo in linea di principio, per esaminare l’utilità della target therapy sulla base dell’informazione genomica. Il trial NCI-MATCH (Molecular Analysis for Therapy Choice) intende sottoporre a screening 3.000 pazienti ed arruolare 1.000 adul-ti con tumori solidi avanzati e linfomi refrattari alla terapia o per i quali non vi sia alcuna terapia standard [40]. Strutturato come un trial di fase II multi-braccio, lo studio analizzerà 4000 diverse varianti in 143 geni. Utilizzando i risultati geno-mici, i pazienti saranno assegnati al trattamento con uno dei 20 farmaci diretti contro mutazioni actionable. Insita nel pro-tocollo è la possibilità di ri-biopsia e di trasferire il paziente ad un altro braccio dello studio sulla base di nuove alterazioni genomiche che si siano eventualmente rese presenti con l’e-voluzione clonale. L’endpoint primario per ogni braccio è il tasso di risposta globale, con l’obiettivo degli sperimentatori di ottenere almeno una risposta parziale al trattamento in 5 su 31 pazienti (16%) [40,42,43]. È probabile che lo studio NCI-MATCH ed altri con disegno simile possano fornire molti spunti utili per la Medicina di Precisione. Auspicabilmente, i risultati di questi studi forniranno evidenze scientifiche per la validazione clinica e per la reale utilità clinica del principio di utilizzare informazioni molecolari per guidare la terapia. Per il futuro della Medicina di Precisione, infatti, è importante raggiungere un consenso sul livello di evidenza necessario per scegliere di utilizzare un’anomalia molecolare al fine di scegliere un determinato trattamento [40,42,43].
Il progetto della Precision Medicine Initiative
Nel discorso “State of the Union” del 2015, il Presidente Barack Obama ha dichiarato: «Doctors have always recog-nized that every patient is unique, and doctors have always tried to tailor their treatments as best they can to individuals. You can match a blood transfusion to a blood type — that was an important discovery. What if matching a cancer cure to
Generation Sequencing ad altissima profondità (Ultra-deep Next Generation Sequencing) applicato al cfDNA plasmatico di pazienti affetti da linfoma diffuso a grandi cellule B ha correttamente identificato la presenza di mutazioni presenti in almeno il 20% degli alleli della biopsia istologica del lin-foma con una sensibilità del 96% e una specificità di circa il 100% [37]. La genotipizzazione del cfDNA plasmatico del linfoma diffuso a grandi cellule B ha permesso anche di “re-cuperare” mutazioni che non erano state rilevabili nella biop-sia del tessuto perché, a causa dell’eterogeneità spaziale del tumore, tali mutazioni erano ristrette a cloni di linfoma anato-micamente distanti dal sito di biopsia. L’analisi longitudinale dei campioni di cfDNA plasmatico, raccolti in corso di tera-pia con immunochemioterapia rituximab-CHOP, ha mostrato la rapida clearance dal cfDNA di mutazioni del linfoma nei pazienti che hanno risposto alla terapia [37]. Al contrario, tra i pazienti resistenti alla terapia, le mutazioni basali non sono scomparse dal cfDNA, e nuove mutazioni sono state acquisite nel cfDNA consistentemente con l’emergere di cloni resisten-ti selezionati durante l’evoluzione clonale [37]. I risultati di questo studio pilota dimostrano che la genotipizzazione del cfDNA nel linfoma diffuso a grandi cellule B è sovrapponibi-le alla genotipizzazione della biopsia diagnostica per rilevare mutazioni tumorali somatiche clonalmente rappresentate, e fornisce un approccio non invasivo in tempo reale per moni-torare l’evoluzione clonale e la comparsa di cloni resistenti al trattamento nei linfomi.
Il ctDNA può rappresentare un’opzione di monitoraggio molecolare della malattia minima residua per i tumori solidi e per i linfomi; in quest’ottica, tuttavia, la sensibilità delle metodiche molecolari di analisi mutazionale devono ancora essere ottimizzate per raggiungere la sensibilità di 10-5-10-6.
I trial clinici di Medicina di Precisione
Un processo di disegno ottimale per gli studi clinici basati sulla genomica rimane un aspetto critico per il futuro. I cosid-detti basket trials (anche detti “bucket trials”) sono trial clini-ci focalizzati sul genotipo tumorale e tesi a valutare l’efficacia di un singolo farmaco su una mutazione (o più mutazioni) in vari tipi di tumore [39]. Nell’ambito di tali trial indipendenti dall’istologia (anche definiti histology-agnostic), pazienti con diversi tipi di tumore possono essere raggruppati in bracci di studio (baskets), consentendo un’analisi separata delle ri-sposte nei pazienti con ogni tipo di tumore ed al contempo permettendo di valutare l’impatto del farmaco sull’intero gruppo dei pazienti nel suo complesso. Una sfida importante per la progettazione dei basket trials è il fatto che tali disegni sperimentali non possono ignorare del tutto l’istologia del tu-more. Il tipo di tumore, infatti, sembra influenzare come alcu-

S12
123
Intern Emerg Med (2016) 11 (Suppl):S8-S14
our genetic code was just as easy, just as standard? What if fi-guring out the right dose of medicine was as simple as taking our temperature?», letteralmente: «I medici hanno sempre riconosciuto che ogni paziente è unico, e hanno sempre cer-cato di adattare i loro trattamenti come meglio possono agli individui. È possibile abbinare una trasfusione di sangue a un gruppo sanguigno - ed è stata una scoperta importante. Che cosa succederebbe se far corrispondere una cura per il can-cro al nostro codice genetico fosse altrettanto facile, proprio come se fosse la regola? Che cosa succederebbe se capire la giusta dose di farmaco da assumere fosse altrettanto semplice che misurare la temperatura corporea?» [44].
Con questo discorso presidenziale, è stata lanciata la Pre-cision Medicine Initiative, cui corrisponde un importante investimento nel budget presidenziale 2016 per i National Institutes of Health (NIH) e la FDA [44,45]. Il progetto si propone di: – definire una coorte di 1 milione o più di volontari per mi-
gliorare la comprensione di salute e malattia e definire un nuovo modo di fare ricerca tramite “engaged participants” e condivisione aperta e responsabile dei dati;
– rafforzare l’identificazione delle lesioni molecolari driver dei tumori e in tal modo sviluppare approcci terapeutici più efficaci;
– favorire lo sviluppo di database di alta qualità; – definire gli standard che, nel rispetto della privacy, permet-
tano lo scambio in sicurezza di dati tra differenti sistemi operativi.
La Precision Medicine Initiative si propone, nel breve tem-po, di focalizzarsi sulle patologie neoplastiche come model-lo, per poi esportare quanto appreso in questa fase anche alle malattie non tumorali. La scelta dell’oncologia per la prima fase delle Precision Medicine Initiative si fonda su vari razio-nali. I tumori sono malattie comuni; nel complesso, sono tra le cause principali di morte e la loro incidenza è in aumento con l’invecchiamento della popolazione. I tumori sono anche particolarmente temuti dall’opinione pubblica a causa della loro letalità, dei loro sintomi e della tossicità delle terapie. La ricerca ha già rivelato molte delle lesioni molecolari che guidano i tumori, mostrando che ogni tumore ha una propria “firma” genomica, che comprende sia alcune caratteristiche specifiche del tumore sia alcune caratteristiche comuni a più tipi. Anche se i tumori sono in gran parte una conseguenza dell’accumulo di danni genomici durante la vita, e quindi di natura somatica, alcune varianti genetiche ereditarie posso-no contribuire a volte profondamente al rischio di neoplasie. Questa nuova comprensione dei meccanismi oncogenetici ha cominciato a influenzare la valutazione del rischio, delle ca-tegorie diagnostiche e delle strategie terapeutiche, con l’uso crescente di farmaci a tipo small molecules e di anticorpi de-
stinati a contrastare l’influenza dei driver molecolari specifici delle cellule neoplastiche.
Per quanto attiene l’espansione della Precision Medicine Initiative alle malattie non neoplastiche, un aspetto partico-larmente innovativo è la costituzione di una coorte di 1 milio-ne o più di volontari, che scaturisce dal desiderio sempre più forte della popolazione statunitense di essere partner attiva nella ricerca medica [44,45]. I partecipanti saranno chiamati a dare il consenso per un’ampia caratterizzazione dei campioni biologici (popolazioni cellulari, proteine, metaboliti, RNA e DNA - tra cui tutto il sequenziamento del genoma) e per la raccolta di dati comportamentali in cartelle cliniche elettro-niche. Ricercatori qualificati provenienti da molte organizza-zioni, con adeguata protezione della riservatezza del paziente, avranno accesso ai dati della coorte, in modo che ricercatori di tutto il mondo possano contribuire con intuizioni e analisi. Questi dati consentiranno studi osservazionali di farmaci e dispositivi, e potranno potenzialmente indurre studi interven-tistici più rigorosi che affrontino questioni specifiche.
Naturalmente, la Precision Medicine Initiative pone anche problematiche etiche e bioetiche che dovranno essere studia-te con attenzione in parallelo alla generazione dei dati. Sarà anche importante mettere in atto tutti i mezzi a disposizione affinché le acquisizioni della Precision Medicine Initiative di-ventino veramente utili clinicamente per “all those in need”, cioè per tutti coloro che ne abbiano un bisogno clinico, in-dipendentemente dal contesto geografico e socio-economico del singolo paziente.
Conclusioni
La rapida espansione delle conoscenze molecolari delle ma-lattie umane resa possibile dalle nuove tecnologie “-omics”, tra cui in particolare negli ultimi anni il Next Generation Sequencing, rappresenta il razionale indispensabile per una Medicina di Precisione che porti alla transizione dalla terapia “one-size-fits-all” (“una taglia per tutti”) ad una “taylored the-rapy” o “terapia sartoriale su misura”. Il campo dell’oncologia e dell’oncoematologia ha fornito un modello di Medicina di Precisione, grazie alla dettagliata caratterizzazione genomica dei tumori di cui oggi disponiamo. L’evoluzione rapida delle nuove tecnologie, sia in termini di semplificazione delle me-todiche sia in termini di maggiore processività e di riduzione dei costi, potrà condurre ad una ulteriore accelerazione delle conoscenze di Medicina di Precisione. La Precision Medici-ne Initiative lanciata dagli Stati Uniti nel 2015 rappresenta un paradigma di ricerca ad ampio raggio, che verosimilmente porterà ad acquisizioni importanti per una diagnostica e stra-tificazione terapeutica più precise, e potrà rappresentare un modello per ulteriori iniziative pubbliche in questo ambito.

S13
123
Intern Emerg Med (2016) 11 (Suppl):S8-S14
In un’ottica di Medicina di Precisione, anche nuovi disegni di trial clinici si stanno affacciando e, in parte, già sono in atto. A fronte di questi investimenti massivi di risorse pubbliche nel settore della Medicina di Precisione e a fronte di interessi privati sempre crescenti nel settore della diagnostica e terapia di precisione, sarà importante salvaguardare i principi di sa-nità pubblica per tutti ed un’etica della ricerca rigorosamente scevra da conflitti di interessi. I ricercatori biomedici e clini-ci impegnati nell’ambito della Medicina di Precisione sono quindi chiamati a rispondere alle grandi attese generate negli stakeholders ed alle nuove problematiche che potranno emer-gere con senso profondo di responsabilità del bene comune, presupposto irrinunciabile della buona scienza.
Bibliografia
1. Arora N. Varmus encourages provocative questions. https://sites.duke.edu/dukeresearch/2012/04/16/varmus-encourages-provoca-tive-questions/ (Accessed on June 3, 2016)
2. Adashi EY, Varmus H. NCI’s Varmus changes the metaphor: there is no war on ‘cancer’. http://www.medscape.com/viewarti-cle/755368#vp_2 (Accessed on June 3, 2016)
3. Prasad V, Fojo T, Brada M (2016). Precision oncology: origins, optimism, and potential. Lancet Oncol 17(2):e81-6. doi: 10.1016/S1470-2045(15)00620-8
4. Mughal TI, Radich JP, Deininger MW et al (2016). Chronic myeloid leukemia: reminiscences and dreams. Haematologica 101(5):541-58. doi: 10.3324/haematol.2015.139337
5. Kohn EC, Ivy SP (2016). Confronting the care delivery challenges arising from Precision Medicine. Front Oncol 6:106. doi: 10.3389/fonc.2016.00106
6. Niroula A, Vihinen M (2016). Variation interpretation predictors: principles, types, performance, and choice. Hum Mutat 37:579-97. doi: 10.1002/humu.22987
7. Rossi D, Gaidano G (2016). The clinical implications of gene muta-tions in chronic lymphocytic leukaemia. Br J Cancer 114(8):849-54. doi: 10.1038/bjc.2016.78
8. Schmidt KT, Chau CH, Price DK, Figg WD (2016). Precision Oncology Medicine: the clinical relevance of patient specific bio-markers used to optimize cancer treatment. J Clin Pharmacol. doi: 10.1002/jcph.765 [Epub ahead of print]
9. Wagle N, Berger MF, Davis MJ et al (2012). High-throughput detec-tion of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov 2(1):82-93. doi: 10.1158/2159-8290
10. Frampton GM, Fichtenholtz A, Otto GA et al (2013). Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-31. doi: 10.1038/nbt.2696
11. Pritchard CC, Salipante SJ, Koehler K et al (2014). Validation and implementation of targeted capture and sequencing for the detec-tion of actionable mutation, copy number variation, and gene rear-rangement in clinical cancer specimens. J Mol Diagn 16:56-67. doi: 10.1016/j.jmoldx.2013.08.004
12. Pant S, Weiner R, Marton MJ (2014). Navigating the rapids: the development of regulated next-generation sequencing-based clini-cal trial assays and companion diagnostics. Front Oncol 4:78. doi: 10.3389/fonc.2014.00078
13. Moasser MM (2007). The oncogene HER2: its signaling and trans-forming functions and its role in human cancer pathogenesis. Onco-gene 26:6469-87
14. Moasser MM, Krop IE (2015). The evolving landscape of HER2 targeting in breast cancer. JAMA Oncol 1:1154-61. doi: 10.1001/jamaoncol.2015.2286
15. Gutierrez C, Schiff R (2011). HER2: biology, detection, and clinical implications. Arch Pathol Lab Med 135:55-62. doi: 10.1043/2010-0454-RAR.1
16. Wolff AC, Hammond ME, Hicks DG et al; American Society of Clinical Oncology; College of American Pathologists (2014). Rec-ommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med 138:241-56. doi: 10.5858/arpa.2013-0953-SA
17. Myers MB (2016). Targeted therapies with companion diagnostics in the management of breast cancer: current perspectives. Pharmge-nomics Pers Med 9:7-16. doi: 10.2147/PGPM.S56055
18. Aubrey BJ, Strasser A, Kelly GL (2016). Tumor-suppressor func-tions of the TP53 pathway. Cold Spring Harb Perspect Med 6(5) pii: a026062. doi: 10.1101/cshperspect.a026062
19. Rossi D, Khiabanian H, Spina V et al (2014). Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 123:2139-47. doi: 10.1182/blood-2013-11-539726
20. Pospisilova S, Sutton LA, Malcikova J et al; European Research Initiative on CLL (ERIC) (2016). Innovation in the prognostica-tion of chronic lymphocytic leukemia: how far beyond TP53 gene analysis can we go? Haematologica 101:263-5. doi: 10.3324/hae-matol.2015.139246
21. Rossi D, Gaidano G (2016). Richter syndrome: pathogenesis and management. Semin Oncol 43(2):311-9. doi: 10.1053/j.seminon-col.2016.02.012
22. Rossi D, Terzi di Bergamo L, De Paoli L et al (2015). Molecular prediction of durable remission after first-line fludarabine-cyclo-phosphamide-rituximab in chronic lymphocytic leukemia. Blood 126:1921-4. doi: 10.1182/blood-2015-05-647925
23. Stilgenbauer S (2015). Prognostic markers and standard management of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2015:368-77. doi: 10.1182/asheducation-2015.1.368
24. Eichhorst B, Robak T, Montserrat E et al; ESMO Guidelines Com-mittee (2015). Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 26 Suppl 5:v78-84. doi: 10.1093/annonc/mdv303
25. Caudle KE, Klein TE, Hoffman JM et al (2014). Incorporation of pharmacogenomics into routine clinical practice: the Clinical Phar-macogenetics Implementation Consortium (CPIC) guideline devel-opment process. Curr Drug Metab 15(2):209-17
26. Forte VA, Barrak DK, Elhodaky M et al (2016). The potential for liq-uid biopsies in the precision medical treatment of breast cancer. Can-cer Biol Med 13:19-40. doi: 10.28092/j.issn.2095-3941.2016.0007
27. Pantel K, Alix-Panabières C (2013). Real-time liquid biop-sy in cancer patients: fact or fiction? Cancer Res 73:6384-8. doi: 10.1158/0008-5472. CAN-13-2030
28. Dawson SJ, Tsui DW, Murtaza M et al (2013). Analysis of circulat-ing tumor DNA to monitor metastatic breast cancer. N Engl J Med 368:1199-209. doi: 10.1056/NEJMoa1213261
29. Alix-Panabières C, Pantel K (2016). Clinical applications of circu-lating tumor cells and circulating tumor DNA as liquid biopsy. Can-cer Discov 6:479-91. doi: 10.1158/2159-8290.CD-15-1483
30. Yu M, Bardia A, Aceto N et al (2014). Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized test-ing of drug susceptibility. Science 345:216-20. doi: 10.1126/sci-ence.1253533

S14
123
Intern Emerg Med (2016) 11 (Suppl):S8-S14
31. Kang Y, Pantel K (2013). Tumor cell dissemination: emerging bi-ological insights from animal models and cancer patients. Cancer Cell 23:573-81. doi: 10.1016/j.ccr.2013.04.017
32. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ (1977). Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37:646-50
33. Sidransky D (1997). Nucleic acid-based methods for the detection of cancer. Science 278:1054-9
34. Schwarzenbach H, Hoon DS, Pantel K (2011). Cell-free nucleic ac-ids as biomarkers in cancer patients. Nat Rev Cancer 11:426-37. doi: 10.1038/nrc3066
35. Lipinski KA, Barber LJ, Davies MN et al (2016). Cancer evolution and the limits of predictability in Precision Cancer Medicine. Trends Cancer 2:49-63
36. Murtaza M, Dawson SJ, Tsui DW et al (2013). Non-invasive analy-sis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497:108-12. doi: 10.1038/nature12065
37. Rasi S, Monti S, Zanni M et al (2015). Liquid biopsy as a tool for monitoring the genotype of diffuse large B-cell lymphoma. https://ash.confex.com/ash/2015/webprogram/Paper85201.html (Accessed on June 3, 2016)
38. Scherer F, Kurtz DM, Newman AM et al (2015). Noninvasive ge-notyping and assessment of treatment response in diffuse large B cell lymphoma. https://ash.confex.com/ash/2015/webprogram/Pa-per84301.html (Accessed on June 3, 2016)
39. Redig AJ, Janne PA (2015). Basket trials and the evolution of clini-cal trial design in an era of genomic medicine. J Clin Oncol 33:975-7. doi: 10.1200/JCO.2014.59.8433
40. McNeil C (2015). NCI-MATCH launch highlights new trial design in precision-medicine era. J Natl Cancer Inst 107(7). doi: 10.1093/jnci/djv193
41. Kummar S, Williams PM, Lih CJ et al (2015). Application of mo-lecular profiling in clinical trials for advanced metastatic cancers. J Natl Cancer Inst 107(4). doi: 10.1093/jnci/djv003
42. Mullard A (2015). NCI-MATCH trial pushes cancer umbrella trial paradigm. Nat Rev Drug Discov 14(8):513-5. doi: 10.1038/nrd4694
43. Brower V (2015). NCI-MATCH pairs tumor mutations with match-ing drugs. Nat Biotechnol 33:790-1. doi: 10.1038/nbt0815-790
44. The precision medicine initiative. https://www.whitehouse.gov/pre-cision-medicine (Accessed on June 3, 2016)
45. Collins FS, Varmus H (2015). A new initiative on precision medi-cine. N Engl J Med 372:793-5. doi: 10.1056/NEJMp1500523

© SIMI 2016
123
RELAZIONE
Precision Medicine: dagli slogan all’applicazione clinica
Pier Mannuccio Mannucci
Pier Mannuccio Mannucci (•)Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico e Università di MilanoVia Pace 9, 20122 MilanoE-mail: [email protected]
Introduzione
Gianluca Gaidano ha esaurientemente e chiaramente spiegato cosa sia la Precision Medicine Initiative lanciata il 20 Gennaio 2015 dal Presidente degli Studi Uniti d’America Barack Oba-ma. È un’iniziativa dirompente ed esaltante per vari motivi. Non si può non essere colpiti dal fatto che il leader carismati-co di una grande nazione, dopo aver affrontato con successo il problema della mancanza di assistenza sanitaria per molti cit-tadini americani (20 milioni di nuovi assistiti), abbia presentato un’iniziativa di ricerca scientifica traslazionale in un’occasione così significativa e simbolica per gli Stati Uniti come il discor-so sullo Stato dell’Unione. Pronunciato annualmente dal Presi-dente di fronte al Congresso riunito, il discorso viene trasmesso in tutto il Paese dalla maggior parte dei canali televisivi e ra-diofonici e dai social network, ed è seguito in diretta da più di 50 milioni di Americani. Non ricordo sia mai successo che un rapporto alla nazione così simbolico come lo Stato dell’Unione sia stato centrato sull’importanza della ricerca scientifica, e in particolare di quella biomedica. Inoltre, non può non colpire, noi che in Italia gioiamo (o ci lamentiamo) per lo stanziamento promesso dal Presidente del Consiglio per Human Technopo-le, l’entità della Precision Medicine Initiative: 215 milioni di dollari per l’anno 2016, ma soprattutto quasi 100 milioni in più per il 2017 (309 milioni) [1]. Inizialmente la Precision Medi-cine Initiative avrà come bersaglio il cancro: come abbiamo appreso da Gaidano, le malattie oncoematologiche fanno già la parte del leone nell’implementazione pratica dell’iniziativa. L’iniziativa prevede anche di affrontare presto malattie come il diabete, il Parkinson e l’Alzheimer: intenzioni testimoniate dal fatto che, oltre al milione di cittadini americani di tutte le età,
etnie e strati sociali che rappresenteranno la coorte iniziale del progetto di ricerca, verranno utilizzati anche i dati già dispo-nibili del Million Veteran Program del Department of Veteran Affairs. Semmai vi è chi ha criticato che la Precision Medicine Initiative sia dedicata alla diagnosi e alla cura delle malattie (e nello specifico dei tumori), ignorando il grande problema della prevenzione e delle diseguaglianze sociosanitarie tra i cittadini americani [2].
Con queste premesse, è chiaro che chi scrive è senz’altro entusiasta della Precision Medicine Initiative. La scelta del cancro come primo bersaglio è logica, anche se risponde all’e-mozione che sempre circonda questa patologia presso l’opinio-ne pubblica e quindi presso i politici. Non vi è infatti dubbio, come detto sopra, che l’oncologia e l’oncoematologia siano le discipline che per prime ci hanno già portato esempi concreti di medicina di precisione. Sono anche convinto che un approccio più individualizzato al cancro e alle emopatie fornirà modelli per adottare la medicina di precisione in altre malattie, attra-verso una migliore valutazione del rischio individuale, l’au-mentata comprensione dei meccanismi patogenetici e quindi l’implementazione di terapie individuali più ottimizzate. Quin-di, il mio intervento non è diretto a criticare e a limitare, ma piuttosto a vedere positivamente e praticamente come la Pre-cision Medicine Initiative possa essere applicata a quello che è il maggiore problema globale di salute pubblica del Terzo Millennio: l’invecchiamento della popolazione. Un problema che riguarda il mondo ad alto reddito come pure quello dei Pa-esi a medio e basso reddito; un problema che sta attualmente aumentando il grado di diseguaglianza nello stato di salute dei cittadini, in Italia come ovunque.
Il problema dell’invecchiamento globale e l’Italia che invecchia
Qualche numero come premessa e come sfondo. I cittadini del mondo di età superiore ai 65 anni saranno il 30% nel 2050: ciò
Intern Emerg Med (2016) 11 (Suppl):S15-S18

S16
123
Intern Emerg Med (2016) 11 (Suppl):S15-S18
sta avvenendo anche in Africa e in America Latina! In Italia, uno dei Paesi caratterizzati maggiormente da questa rivolu-zione demografica (nonostante il recentissimo piccolo passo indietro), gli ultrasettantenni (attualmente 9,7 milioni su una popolazione di 59,7) saranno nel 2050 16,3 milioni su una po-polazione totale di 56 milioni e gli ultraottantenni saranno 8 milioni contro i 4,1 milioni del 2015. Quindi, dalla metà di questo secolo un Italiano su sei avrà più di 80 anni, di gran lun-ga guidando la corrispondente classifica europea davanti alla Germania e alla Francia. Infine, per fare un esempio clamoroso dell’invecchiamento con successo, i centenari: nel 2050 a spe-gnere cento candeline saranno in tutto 3,7 milioni di persone nel mondo, in Italia 228.000 (sono attualmente 24.000).
L’Italia che invecchia è una buona notizia, ma…siamo pron-ti? Un grande cambiamento sociale sta avvenendo nel nostro Paese, che non solo invecchia ma è caratterizzato da un lento ri-cambio di popolazione dovuto alla bassa natalità: per esempio, sarà molto più frequente la convivenza di quattro generazioni (nipoti, genitori, nonni e bisnonni), cambiando drasticamente il ruolo dei singoli membri delle famiglie estese, già in atto. L’e-splosione del problema demografico non ha però permesso, da noi come altrove, l’elaborazione di contenuti organizzativi (e soprattutto culturali) indispensabili per sviluppare sistemi assi-stenziali e previdenziali adeguati. La gestione clinica e sociale degli anziani è attualmente il principale problema non risolto sia del Servizio Sanitario Nazionale (SSN) che del sistema pre-videnziale/assistenziale dell’Istituto Nazionale della Previden-za Sociale (INPS): due capisaldi fondamentali del welfare, di cui si è finora giovata (e vantata) l’Italia.
Un altro aspetto di inadeguatezza è legato alla formazione degli operatori, che si applica ai Corsi Universitari di Medici-na e Chirurgia, Infermieristica e alle discipline paramediche, nonché alle Scuole di Specializzazione e ai Corsi Regionali di formazione dei medici di medicina generale. L’addestramen-to e la pratica clinica dei medici e infermieri generalisti che si dovranno occupare più di altri dei cittadini anziani malati (geriatri, internisti, medici di famiglia) sono stati finora foca-lizzati su pazienti relativamente giovani, con in genere singo-le malattie acute: ma l’invecchiamento porta inevitabilmente con sé la multimorbilità e la presenza nello stesso individuo di più di una malattia cronica, apparentemente non connesse tra di loro.
Multimorbilità e polifarmacoterapia
Nel 2050 gli anziani con multiple malattie croniche saranno almeno il 10% della popolazione dell’Europa (75 milioni) e dell’Italia (6 milioni). E consumeranno sempre più farma-ci, oltre ad altre risorse del SSN e dell’INPS. Secondo i dati emanati nel 2013 dall’Agenzia Italiana del Farmaco (AIFA),
1,3 milioni di anziani oltre i 65 anni assumono 6 o più farmaci al giorno, assorbendo il 70% del costo totale per i farmaci del SSN, quasi 20 miliardi di euro [3]. E non è solo un problema di costi. La terapia con farmaci multipli (polifarmacoterapia) aumenta di per sé la mortalità, l’ospedalizzazione, i ricoveri in residenze sanitarie e l’invalidità. Garfinkel e Mangin [4] hanno scritto testualmente “la polifarmacoterapia deve esse-re considerata una malattia: con conseguenze e complicazio-ni anche più gravi delle multiple malattie per cui i differenti farmaci sono stati prescritti”.
Perché la polifarmacoterapia è così frequente? Perché ai pazienti anziani e complessi con malattie multiple e croniche vengono prescritti per ciascuna di esse i farmaci raccomandati dalle linee guida specifiche, che a loro volta sono basate sui risultati di sperimentazioni cliniche randomizzate, i randomi-zed clinical trials (RCT). Ma i RCT studiano la malattia, non il paziente! E arruolano casi completamente diversi dagli an-ziani con malattie croniche multiple: con una singola malattia, più giovani, con basso rischio di eventi avversi e di interazioni farmacologiche, e scelti con elevata probabilità di dare risultati positivi nella valutazione del farmaco oggetto delle sperimen-tazioni. Quindi, i RCT danno informazioni di efficacy: non di effectiveness in una situazione di mondo reale come quella che riguarda l’anziano con comorbilità. Di questo problema si è accorta anche un’agenzia regolatoria come la European Medi-cines Agency (EMA) [5], affermando che le agenzie regolatorie devono assicurarsi che nel presente e nel futuro lo sviluppo e la valutazione dei farmaci tengano conto dei cambiamenti demo-grafici globali, affinché farmaci efficaci e sicuri raggiungano anche i pazienti che più li usano, cioè gli anziani.
I limiti della evidence based medicine
Come conseguenza di tutto ciò, la medicina basata sulle evi-denze (e quindi su RCT) non è idonea a fornire indicazioni sulle combinazioni di farmaci multipli utili per curare l’an-ziano con multimorbilità. Come scegliere? L’ovvietà della cura diminuisce per l’anziano. Per essere accettata, la cura deve rispondere alle sue preferenze, cosicché l’esplicitazione e condivisione del perché della cura sono dimensioni fonda-mentali per la successiva aderenza alla terapia. La valutazione della complessità dell’individuo anziano malato deve guida-re le scelte terapeutiche più oculate, tenendo conto dei reali bisogni e problemi del singolo e, soprattutto, che le sue pre-ferenze siano integrate nelle scelte: limitando almeno la pre-scrizione a farmaci che è più probabile non nuocciano e diano un reale beneficio ai problemi e bisogni reali del singolo pa-ziente piuttosto che alla malattia. La scelta dei farmaci dovrà sempre più ancorarsi all’identità corporea, personale, sociale, culturale, relazionale e affettiva della persona malata. Si ri-

S17
123
Intern Emerg Med (2016) 11 (Suppl):S15-S18
torna quindi alla medicina personalizzata dei nostri maestri, che i RCT ci avevano fatto vituperare e abbandonare dagli anni ’70 in avanti. Queste indicazioni conservative basate sul vecchio adagio “primum non nocere” e quello più moderno di “choosing wisely” hanno un approccio valido da un punto di vista pragmatico ma non scientifico! Bisognerebbe infatti arrivare a capire con metodologia scientifica quali sono per il cittadino anziano malato le più rilevanti malattie multiple di cui soffre. E soprattutto se malattie apparentemente indipen-denti l’una dall’altra non abbiano in realtà dei legami patoge-netici comuni, permettendo così un approccio meccanicistico e razionale alla scelta dei farmaci. Ed è qui che nuovi approc-ci come la Precision Medicine potrebbero aprire nuove strade di ricerca scientifica sul grande problema globale dell’invec-chiamento, ma anche la Systems Medicine [6].
L’invecchiamento e i Big Data
L’approccio scientifico alle malattie multiple dell’anziano po-trebbe avere un rapido e concreto sviluppo se vi fosse la pos-sibilità di interrogare e unificare i tanti dati che i ricercatori, i clinici e gli amministratori sanitari già hanno in loro possesso. Precision Medicine Initiative è stata guidata dall’eterogeneità delle malattie, e in particolare dei tumori. Jamieson e Longo [7] ci ricordano che la tassonomia delle malattie è caratterizzata da chi cerca di raggruppare insieme entità nosologiche apparen-temente diverse (lumping), e da chi invece vuole individuare entità sempre più discrete (splitting). Nel campo della multi-morbilità dell’anziano vi è una necessità di lumping piuttosto che di splitting, che è invece l’attuale approccio basato sui RCT e sulla medicina basata sulle evidenze. Le varie tecnolo-gie delle cosiddette scienze omiche possono da una parte agire come lumpers nel mettere in evidenza ciò che vi è in comune in malattie apparentemente diverse, ma d’altra parte forniscono delle firme (signatures) individuali, che distinguono un pazien-te da un altro sulla base di caratteristiche peculiari di geni e di biomarcatori. Ciò sottolinea come i recenti progressi della tecnologia sono e saranno i protagonisti della Precision Me-dicine Initiative: la convergenza della genetica, dell’informa-tica e dell’imaging (insieme ad altre tecnologie in espansione come il cell sorting, l’epigenetica, la proteomica e la metabo-lomica) potrebbero aiutarci a raggiungere la tassonomia della multimorbilità, con importanti implicazioni sulla terapia con i farmaci di questi casi.
L’opportunità di Watson Health
Come ciò può avvenire? I ricercatori dedicati alla Precision Medicine devono avere accesso alla grande massa di dati già
disponibili su salute e malattie: la disponibilità di questi dati è critica per lo sviluppo e la validazione di una nuova tassono-mia. Per applicare i sistemi cognitivi alla salute e alle malat-tie, IBM (la multinazionale americana leader nel mondo) ha sviluppato Watson Health, che prende il nome dal fondatore di IBM. Il sistema informatico Watson Health aumenta in ma-niera gigantesca il limite della quantità di dati che un sistema cognitivo è in grado di elaborare da solo, ed è anche capace di rispondere ai quesiti che vengono posti al sistema stesso. Si dice che tra 3-4 anni Watson Health potrà di gestire 44 mila miliardi di giga bytes (quando un compact disc ne contiene 0,8, tanto per dare un’idea). Questa che sembra fantascienza arriverà in Italia a Milano nell’area di EXPO, attraendo spe-rabilmente ricercatori da tutto il mondo. È stato già firmato un accordo preliminare e per l’estate del 2016 è prevista l’intesa definitiva tra il governo Italiano e IBM, che ha già messo a di-sposizione 150 milioni di dollari di investimenti, sui 6 miliar-di per cinque anni che promette di destinare al programma.
Watson Health dovrebbe essere capace di integrare su scala gigantesca i dati clinici e di ricerca, le linee guida, le informa-zioni sull’ambiente e tanto e tutto di più, potendo incorporare nel supercomputer (e rendere gestibili in maniera integrata) anche articoli di giornali scientifici, percorsi diagnostici-te-rapeutici di singoli ospedali e consorzi medici, nonché dati di RCT e anche libri di testo. Watson Health sarebbe persino capace di interfacciarsi con cartelle cliniche elettroniche e di leggere e capire le semplici note di diario clinico, processan-do il linguaggio nativo.
Cosa si potrebbe fare in Italia per dare attraverso Watson Health un contributo significativo al problema gigantesco dell’invecchiamento, della multimorbilità e della costosa e dannosa polifarmacoterapia? Bisognerebbe innanzitutto contribuire a creare un patrimonio di big data italiano: con l’orgoglio di appartenere alla Società Scientifica che lo ha creato e lo gestisce insieme all’Istituto di Ricerca Farma-cologica Mario Negri e all’IRCCS Fondazione Ca’ Gran-da di Milano, penso a REPOSI, che contiene già ora tanti dati su 5000 anziani ospedalizzati con età media di quasi 80 anni [8]. Il registro continua, e quindi non è irrealistico pensare di poter arrivare in 4-5 anni a 10.000 casi. Ma non basta. Vi sono in Italia tanti altri database tipo REPOSI, per esempio quello delle cure intensive. La senatrice Cattaneo poi ha lanciato nella più recente legge di stabilità il Progetto Genomi Italia: non se ne conoscono ancora i contorni (salvo gli stanziamenti pubblici: 15 miliardi all’anno per 3 anni), ma s’immagina che potrà assomigliare come disponibilità di dati al programma inglese varato ormai da parecchi anni da Rory Collins a Oxford.
La Danimarca e Taiwan ci insegnano che a livello centrale è possibile creare un enorme database sulla salute e le malat-tie dei loro cittadini, i cui dati hanno già fornito importanti

S18
123
Intern Emerg Med (2016) 11 (Suppl):S15-S18
informazioni sulle associazioni tra diverse malattie e soprat-tutto sui vantaggi e i rischi dei farmaci nel mondo reale, al di là dell’atmosfera rarefatta dei RCT. Anche la Regione Lom-bardia ha raccolto questi dati sui suoi cittadini e ha sviluppa-to programmi come DENALI, da cui sono nate significative pubblicazioni real life, purtroppo recentemente e inopinata-mente bloccate dall’intervento del Garante della Privacy per un malinteso eccesso di tutela.
Commenti conclusivi
È entusiasmante che IBM abbia scelto l’Italia, Milano e l’area EXPO per implementare Watson Health. Nella conferenza stampa di lancio l’Amministratore Delegato di IBM ha di-chiarato che questa scelta è stata basata sull’eccellenza del si-stema sanitario del nostro Paese, nonché sulla particolare lon-gevità dei cittadini italiani. È compito della comunità scien-tifica, e in particolare di quella degli internisti italiani, non deludere IBM e quindi contribuire a dimostrare con i fatti che meritiamo questa scelta. Sono convinto che Watson Health possa essere uno strumento eccezionale per porre l’Italia nel-
la frontline di coloro che affrontano l’invecchiamento, uno dei più grandi problemi planetari.
Bibliografia
1. Collins FS, Varmus H (2015). A new initiative on precision medi-cine. N Engl J Med 372:793-95
2. Bayer R, Galea S (2015). Public health in the precision-medicine era. N Engl J Med 372:499-501
3. AIFA (2013). http://www.agenziafarmaco.gov.it/sites/default/files/COMUNICATO%20AIFA%20-%20N.%20313%20-%20STU-DIO%20WG%20GERIATRICO%20AIFA.pdf.
4. Garfinkel D, Mangin D (2010). Feasibility study of a systematic ap-proach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 170:1648-54
5. Cerreta F, Eichler HG, Rasi G (2012). Drug policy for an aging population--the European Medicines Agency’s geriatric medicines strategy. N Engl J Med 367:1972-4
6. Bousquet J, Anto JM, Sterk PJ et al (2011). Systems medicine and integrated care to combat chronic noncommunicable diseases. Ge-nome Medicine 3:43
7. Jameson JL, Longo DL (2015). Precision medicine--personalized, problematic, and promising. N Engl J Med 372: 2229-34
8. Mannucci PM, Nobili A (2014). Multimorbidity and polypharmacy in the elderly: lessons from REPOSI. Intern Emerg Med 9:723-34

© SIMI 2016
123
RELAZIONE
Intern Emerg Med (2016) 11 (Suppl):S19-S23
Paolo Cavallo Perin (•)Dipartimento di Scienze Mediche, Università di TorinoCorso A.M. Dogliotti 14, 10126 Torino E-mail: [email protected]
P. Fornengo • C. Amione Dipartimento di Scienze Mediche, Università di Torino
Per accertamenti e cure: il ricovero è sempre utile? Pro
Paolo Cavallo Perin • Paolo Fornengo • Cristina Amione
Introduzione
Fin dalla sua istituzione, il Servizio Sanitario Nazionale (SSN) si è dimostrato solidale ed efficace e ha mantenuto importanti risultati sulla salute dei cittadini. A fronte di tali punti di forza, il SSN presenta tuttavia problemi di sostenibilità ed efficienza legati al suo finanziamento che destano preoccupazioni e in-ducono alla ricerca di proposte migliorative e innovative per poter far fronte ad uno scenario in continua evoluzione.
L’invecchiamento della popolazione si associa ad un au-mento dell’incidenza delle malattie croniche e impone di ri-pensare al modello di cura del malato, spostandolo verso il territorio. Per il riequilibrio ospedale-territorio assumono un ruolo chiave i seguenti fattori: il sistema di cura della croni-cità e l’istituzione di nuove figure professionali, come il Case Manager; i nuovi modelli del sistema di emergenza-urgenza, come l’Osservazione Breve Intensiva (OBI); alcuni model-li organizzativi della medicina convenzionata, come l’Unità Territoriale di Assistenza Primaria (UTAP), l’Unità di Cure Primarie (UCP), l’Assistenza Domiciliare Integrata (ADI), il Servizio Infermieristico Domiciliare (SID); l’integrazione “in rete” tra le diverse strutture socio-sanitarie; un’adeguata formazione del personale del SSN.
Dall’insieme di questi elementi dipendono in larga misura la necessità e l’appropriatezza dei ricoveri ospedalieri al di fuori del contesto delle emergenze. Inoltre, la disponibilità di strutture extra-ospedaliere per sub-acuti o a bassa intensità di cure è inadeguata ad ospitare i malati dimissibili dai reparti ospedalieri con problemi sociali (vivono soli, non sono in gra-
do di procurare/cucinare i cibi, hanno limitata autonomia); ne deriva un aumento dei tempi di degenza nei reparti per acuti con riduzione della disponibilità dei posti letto per accogliere nuovi malati e aumento ingiustificato dei costi. Ad esempio, il 25% dei pazienti ricoverati in una struttura di Medicina In-terna di una Azienda Ospedaliera-Universitaria per acuti ha un ritardo di dimissione di durata superiore a 10 giorni per motivi non-clinici, ma di tipo organizzativo o sociale [1].
Nell’ambito della cronicità, nuove figure sono diventate emergenti nell’ultimo decennio con i loro bisogni di salute, come l’anziano “fragile”, il malato neoplastico, i migranti, i soggetti senza-tetto o senza fissa dimora. Per far fronte all’evo-luzione della domanda di salute, la risposta organizzativa delle strutture ospedaliere e del territorio risulta oggi ancora caren-te e con marcate differenze geografiche a causa di disservizi, liste d’attesa, complessità burocratica, difficoltà di accesso alle cure [2,3]. Ad esempio, l’utilizzo dell’ADI non è ancora adeguato a soddisfare le esigenze socio-sanitarie dei cittadini. Infatti, si tende a considerare prevalentemente la componente “medica” dell’ADI, trascurando altre componenti fondamen-tali di tale assistenza, quali la fisioterapia, l’assistenza sociale ecc. Analoghe considerazioni possono essere fatte per le strut-ture residenziali e per l’ospedalizzazione a domicilio, numeri-camente insufficienti e con distribuzione territoriale inadeguata alle necessità della popolazione urbana e rurale [4].
Il numero di ricoveri ospedalieri per condizioni croniche gestibili a livello ambulatoriale (Ambulatory Care Sensitive Conditions, ACSC), utilizzato come indice di efficienza della sanità territoriale a livello di popolazione, risulta geografica-mente variabile e non fornisce criteri utili a livello individuale per operare una decisione nel singolo paziente distinguendo il ricovero necessario da quello prevenibile [5].
In quali casi il ricovero per accertamenti e cure è utile nella realtà attuale e con quali modalità deve essere effettuato? La risposta banale a questo interrogativo potrebbe essere: «Se l’organizzazione sanitaria ospedaliera e territoriale funzio-nasse secondo quanto previsto dalle attuali norme e disposi-

S20
123
Intern Emerg Med (2016) 11 (Suppl):S19-S23
zioni regionali e nazionali senza discrepanze geografiche, il bisogno di ricovero per accertamenti e cure potrebbe essere soddisfatto nella maggior parte dei casi senza ricorrere ai re-parti ospedalieri per acuti». Tuttavia, la realtà italiana attuale presenta un quadro complesso, composto da innovazione, ma anche da disfunzioni e ritardi organizzativi che hanno riper-cussioni variabili nelle diverse Regioni e talora nelle diverse aree della stessa Regione.
Qui di seguito saranno considerati alcuni esempi utili a comprendere le ragioni per le quali è oggi ancora giustificato ricorrere al ricovero per accertamenti e cure.
A. Malato cronico
La prevalenza delle malattie croniche in Italia è crescente e nella popolazione anziana le malattie croniche tendono ad aggregarsi nello stesso soggetto. La gestione di questi casi risulta complessa con difficoltà nella continuità delle cure, resa ulteriormente critica nei pazienti con bassi livelli socio-economici e in condizioni di isolamento [6].
Nello scenario della cronicità, il diabete tipo 2, la sindrome metabolica e le malattie cardiovascolari rappresentano pato-logie in crescita preoccupante, anche nell’adulto non anziano, a causa della crescente diffusione dell’obesità, a sua volta ri-feribile a iperalimentazione e a scarsa attività fisica nel sog-getto geneticamente predisposto.
Diabete tipo 2. I casi noti di diabete in Italia erano 1,5 milioni nel 1985 e si avvicinano ora ai 4 milioni, ai quali deve essere aggiunto 1 milione di casi di diabete misconosciuto. La prevalenza media è circa il 6%, ma raggiunge il 20% oltre i 75 anni di età, ed è in continua crescita. L’età media alla diagnosi del diabete tipo 2 è attualmente 50-55 anni, ma in molti soggetti avviene prima dei 30 anni. Il diabete è la prima causa di cecità, la seconda causa di insufficienza renale termi-nale, la prima causa di amputazione non traumatica degli arti inferiori, una concausa di metà dei casi di infarto miocardico e ictus. La spesa sanitaria per la persona con diabete è circa 2,5 volte quella di una persona non-diabetica ed è per oltre il 60% assorbita dai ricoveri ospedalieri dettati dalle compli-canze e dalla comorbilità [7]. Sebbene la necessità di ricove-ro nel paziente diabetico si sia ridotta nell’ultimo decennio verosimilmente in rapporto al miglioramento dell’assistenza ambulatoriale [8], il tasso di ricoveri per tutte le cause risulta aumentato di 1,7-3 volte nella popolazione diabetica rispetto a quella non diabetica [9].
Sindrome metabolica. Presenta in Italia una prevalen-za superiore al 30% [10] e coinvolge anche la fascia di età infanto-giovanile con valori del 10-15% [11], aumentando in
modo esponenziale il rischio di varie patologie anche in età non avanzata e vanificando l’aumento dell’aspettativa di vita ottenuto negli ultimi decenni. Infatti, risulta associata a iper-glicemia e aumentato rischio di diabete tipo 2, ipertensione arteriosa, dislipidemia, steato-epatite non-alcolica, sindrome dell’ovaio policistico, sindrome delle apnee ostruttive not-turne, disfunzione sessuale; questi fattori sono implicati sia nell’aumento di episodi cardiovascolari sia di alcuni tumori maligni [12].
Malattie cardiovascolari. Rappresentano la principale causa di morte in Italia, essendo responsabili del 44% di tutti i decessi. In particolare, la cardiopatia ischemica è la prima causa di morte, mentre l’ictus è al terzo posto, dopo i tumo-ri. Chi sopravvive ad un episodio cardio- o cerebrovascolare acuto diventa un malato cronico e richiede nel tempo rivalu-tazioni cliniche utili a modificare la terapia [13,14]. Anche un pregresso episodio di attacco ischemico transitorio (TIA) o l’essere portatori di una stenosi carotidea pone al medico difficoltà decisionali di fronte alla comparsa di quadri clini-ci sfumati o aspecifici con necessità di un approfondimento diagnostico [15].
La diffusione della sindrome metabolica, del diabete tipo 2 e della cardiopatia ischemica associata all’aumento del-la durata della vita ha generato un’aumentata incidenza di scompenso cardiaco, che richiede interventi transitori extra-ospedalieri [16], ospedalizzazione a domicilio [17], ricoveri in unità di diagnosi rapida o reparti di medicina interna per definizione diagnostica e/o aggiustamenti terapeutici [18]. Tale necessità persiste oggi al di fuori delle emergenze, nono-stante il numero di ricoveri per complicanze acute di malattie croniche risulti sempre più ridotto [8,19].
B. Deficit cognitivo
Il deficit cognitivo lieve ha una prevalenza in Italia che su-pera il 10% nella popolazione adulta e raddoppia nell’età su-periore a 80 anni [20]. Questa condizione si associa ad un incremento dell’indice di ospedalizzazione del 17-20%, che raggiunge livelli del 40% nei soggetti che vivono in famiglia rispetto a quelli che vivono soli. Quest’ultimo è un aumento paradosso rispetto ad altre condizioni croniche in cui il sup-porto socio-familiare riduce la necessità di ricovero offrendo un sostegno organizzativo per risolvere a domicilio un pro-blema clinico di nuova insorgenza. Quindi, chi vive solo ha una minore probabilità di essere identificato per la necessità di un approfondimento diagnostico e, quando ciò accade, la necessità di ricovero risulta elevata anche per la soluzione di problemi diagnostico-terapeutici non impegnativi [21].
Nei casi di deficit cognitivo più grave, la frequenza di

S21
123
Intern Emerg Med (2016) 11 (Suppl):S19-S23
ospedalizzazione per tutte le cause risulta più elevata e si prevede tenda ad aumentare nel prossimo futuro. Anche le ospedalizzazioni ritenute “prevenibili” non risultano ridu-cibili nonostante il miglioramento degli interventi sanitari extra-ospedalieri utili ad evitare gli effetti negativi del ricove-ro, come il delirio, le cadute, le complicazioni conseguenti a procedure e i costi elevati [22].
C. Insufficienza renale cronica
La prevalenza dell’insufficienza renale cronica lieve (eGFR < 60 ml/min) presenta in Italia valori del 6-13% che raggiun-gono il 30% nei soggetti con età > 75 anni [23] ed è in con-tinuo aumento. La forma di grado lieve/moderato si associa ad un aumento del rischio cardiovascolare di 1,5-2 volte, che aumenta ulteriormente in caso di comorbilità [24]. L’insuf-ficienza renale cronica richiede attenzione nella prescrizio-ne dei farmaci, sia per il rischio di progressione nel caso di farmaci nefrotossici, sia per l’insorgenza di reazioni avverse. L’insufficienza renale presenta variazioni transitorie o per-manenti che rimangono misconosciute con importanti con-seguenze sull’effetto dei farmaci: ad esempio, nel paziente anziano gli episodi di disidratazione o l’assunzione di FANS possono ridurre la filtrazione glomerulare che deve essere all’occorrenza valutata. In tale contesto, soprattutto nei pa-zienti in polifarmacoterapia (anticoagulanti, diuretici, far-maci cardiovascolari, analgesici, antidiabetici) può rendersi necessario un breve ricovero per il ripristino del bilancio idro-elettrolitico e la correzione della terapia [25].
D. Malato con polipatologia
Il soggetto anziano spesso presenta comorbilità (diabete, iper-tensione, scompenso cardiaco, anemia, osteoporosi, insuffi-cienza renale), disabilità e isolamento sociale configurando un quadro di fragilità. Stime della comorbilità forniscono va-lori del 60% che superano il 90% nella popolazione con età > 75 anni e nelle fasce socio-economicamente più povere. Il paziente con comorbilità è solitamente trattato con polifar-macoterapia che lo espone al rischio di uso inappropriato di farmaci, di errori terapeutici (dosi e intervalli scorretti, scarsa aderenza alla terapia), di interazioni tra farmaci e di reazioni avverse. Talora accade che, invece di sospendere il farmaco responsabile della reazione avversa, un nuovo farmaco sia im-propriamente aggiunto alla terapia creando un circolo vizioso: la prescrizione a cascata. Comorbilità e polifarmacoterapia li-mitano il ricorso alla medicina basata sulle prove di efficacia, per scarsità di studi di intervento disponibili. Questi pazienti richiedono una valutazione multidimensionale (clinica, nutri-
zionale, funzionale, cognitiva, psicologica, socio-economica) e una particolare attenzione alla comunicazione nell’ambito dell’assistenza extra-ospedaliera (medico, caregiver, ambula-tori) e ospedaliera (day hospital, unità di ricovero breve, re-parti) durante la degenza e all’atto del trasferimento o della dimissione; tale attenzione deve coinvolgere anche la pianifi-cazione dell’assistenza del periodo finale della vita [26]. Co-morbilità e polifarmacoterapia possono generare la necessità di ricovero sia per il paziente che vive in famiglia, sia per quel-lo ospitato nelle Residenze Sanitarie per Anziani (RSA) e nel-le Residenze Sanitarie per Disabili (RSD) [27]. In alcuni casi, la necessità di ricovero è percepita dal paziente che si presenta direttamente al dipartimento di emergenza, senza l’invio da parte del medico, per ottenere una risposta immediata con va-lutazione multidisciplinare e alta tecnologia [28].
Di fronte alla complessità del paziente con comorbilità, in-fluenzata dalle condizioni socio-economiche e dalla disponi-bilità dei servizi territoriali, il ricorso al ricovero ospedaliero nelle unità di diagnosi rapida, di degenza breve o nei reparti di Medicina Interna risulta oggi utile e giustificato. Tale ne-cessità può essere indirettamente quantificata considerando che sul totale dei pazienti con polipatologia che giungono spontaneamente in ospedale, il ricovero è giudicato evitabile solo nel 15% dei casi [29].
E. Malato neoplastico
Le malattie neoplastiche hanno una diffusione crescente, so-prattutto nella popolazione anziana, che è portatrice di poli-patologia. Il numero di malati neoplastici è destinato ad au-mentare come conseguenza dell’efficacia della terapia che ha in molti casi aumentato la sopravvivenza con effetti variabili sulla qualità della vita. Nonostante la migliorata organizza-zione della rete ambulatoriale oncologica, la risposta ai biso-gni assistenziali del paziente oncologico non risulta soddisfa-cente per il controllo dei sintomi soprattutto se refrattari alla terapia: il dolore è motivo di ricovero nel 30% dei casi, l’a-stenia grave nel 18%, la dispnea nel 14%, la febbre nel 12%; si tratta di ricoveri spesso ripetuti, che in un terzo dei casi sono prolungati fino al decesso o al trasferimento presso una struttura per cure palliative o presso l’hospice per il periodo di fine vita. Il ricovero non è pianificabile nella maggior parte dei casi e si rende necessario quando il sovrapporsi dei biso-gni del paziente (sintomi, insufficienza d’organo, sofferenza psicologica, fragilità sociale) non è compatibile con una ge-stione ambulatoriale; infatti, i ricoveri giudicati evitabili non superano il 20% del totale. La “dipendenza” dall’ospedale non si riduce al crescere della qualità delle cure palliative domiciliari e il ricovero non costituisce necessariamente una spia di terapie troppo aggressive; perciò, il ricovero deve es-

S22
123
Intern Emerg Med (2016) 11 (Suppl):S19-S23
sere serenamente considerato come una tappa inevitabile e ri-petuta del percorso terapeutico del paziente oncologico [30]. Solo nel periodo di fine vita, la necessità di ricovero può note-volmente essere ridotta da fattori socio-economici favorevoli e dalla disponibilità di caregiver e di familiari per la gestione domiciliare [31].
F. Migranti, minoranze etniche, soggetti senza fissa dimora
Da circa 30 anni in Italia il numero dei migranti è crescente ed ha raggiunto nel 2011 il 9% della popolazione. Fin dal 1990, le regole burocratico-amministrative consentono ai regolari cittadini non-italiani l’accesso al SSN e l’assistenza viene ero-gata anche a coloro che risultano temporaneamente irregolari. Ciononostante, l’eterogeneità inter-regionale nell’applicazione di queste norme e il basso livello socio-economico e culturale costituiscono fattori sfavorenti l’accesso alle cure dei migranti e delle minoranze etniche che, nonostante la giovane età, pre-sentano uno stato di salute più deteriorato a causa del mancato trattamento dei fattori di rischio e delle malattie croniche [3]. In questo contesto, il numero di ricoveri ospedalieri risulta crescente anche nel paziente stabile in rapporto alla difficoltà di gestione sul territorio di quadri clinici che richiedono una messa a punto diagnostico-terapeutica [32]. Analogamente, la crescente popolazione dei senza-tetto e senza fissa dimora, che nel 30% dei casi ha un’età inferiore a 25 anni, presenta condi-zioni di salute molto scadenti, con frequenti accessi ospedalieri e scarso accesso alle comunità assistenziali.
La necessità di ricovero potrebbe forse essere contenuta dal miglioramento dei servizi sanitari e socio-assistenziali sul territorio anche ad opera del volontariato, ma nelle condizioni attuali il ricorso all’ospedale costituisce una risorsa ancora indispensabile in questi soggetti [33].
Conclusioni
Il ricorso al ricovero di pazienti in condizioni non-acute in reparti ospedalieri per acuti è formalmente inappropriato e può risultare rischioso per il paziente; negli ultimi anni si è progressivamente ridotto in rapporto al migliorameno dell’as-sistenza ambulatoriale. Tuttavia, la recente crescita numerica di alcuni gruppi di pazienti (cronici con comorbilità, deficit cognitivo, neoplasie, migranti e senza fissa dimora) richiede di considerare il ricovero per la definizione diagnostico-tera-peutica come strumento necessario; infatti esso non può esse-re sostituito dal ricorso a strutture sanitarie extra-ospedaliere che non sono in grado di rispondere ai bisogni di questi pa-zienti o risultano di difficile accesso per ragioni organizzative o amministrative [34].
La consapevolezza che il ricovero rappresenti uno stru-mento appropriato in queste condizioni deve essere condivisa dagli operatori sanitari e dagli organi di governo, per una più efficace gestione di questi pazienti svantaggiati nella situazio-ne attuale e per pianificare interventi di politica sanitaria. Le Società scientifiche possono fornire un importante contributo a mantenere l’attenzione sul problema e a proporre soluzioni migliorative.
Bibliografia
1. Panero F, Gruden G, Zucco C et al (2013). Delayed discharge: a rising cause of concern in general internal medicine wards. Minerva Med 104:113-5
2. Busby J, Purdy S, Hollingworth W (2015). A systematic review of the magnitude and cause of geographic variation in unplanned hos-pital admission rates and length of stay for ambulatory care sensitive conditions. BMC Health Serv Res 15:324. doi: 10.1186/s12913-015-0964-3
3. de Waure C, Bruno S, Furia G et al (2015). Health inequalities: an analysis of hospitalizations with respect to migrant status, gender and geographical area. BMC Int Health Hum Rights 15:2. doi: 10.1186/s12914-014-0032-9
4. Leff B, Burton L, Mader SL et al (2009). Comparison of function-al outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatr Soc 57:273-8. doi: 10.1111/j.1532-5415.2008.02103.x
5. Longman JM, Passey ME, Ewald DP et al (2015). Admissions for chronic ambulatory care sensitive conditions − a useful measure of potentially preventable admission? BMC Health Serv Res 15:472. doi 10.1186/s12913-015-1137-0
6. Napolitano F, Napolitano P, Garofalo L et al (2016). Assessment of continuity of care among patients with multiple chronic con-ditions in Italy. PLoS One 11(5):e0154940. doi: 10.1371/journal.pone.0154940
7. Bonora E, Sesti G (2016). Il diabete in Italia. SID. Bononia Univer-sity Press, Forlì
8. Lombardo F, Maggini M, Gruden G, Bruno G (2013). Temporal trend in hospitalizations for acute diabetic complications: a nation-wide study, Italy, 2001-2010. PLoS One 8(5):e63675. doi:10.1371/journal.pone.0063675
9. Sinclair AJ, Rodriguez-Mañas L (2016). Diabetes and frailty: two converging conditions? Can J Diabetes 40(1):77-83. doi: 10.1016/j.jcjd.2015.09.004
10. Tocci G, Ferrucci A, Bruno G et al (2015). Prevalence of meta-bolic syndrome in the clinical practice of general medicine in It-aly. Cardiovasc Diagn Ther 5(4):271-9. doi:10.3978/j.issn.2223-3652.2015.07.03
11. Martino F, Puddu PE, Pannarale G et al (2013). Arterial blood pressure and serum lipids in a population of children and adoles-cents from Southern Italy: the Calabrian Sierras Community Study (CSCS). Int J Cardiol 168:1108-14
12. Forti P, Pirazzoli GL, Maltoni B et al (2012). Metabolic syndrome and all-cause mortality in older men and women. Eur J Clin Invest 42(9):1000-9. doi: 10.1111/j.1365-2362.2012.02688.x
13. Volpe M, Mastromarino V, Battistoni A (2015). Integrated pre-clinical cardiovascular prevention: a new paradigm to face grow-ing challenges of cardiovascular disease. Am J Cardiovasc Drugs 5(3):163-70. doi: 10.1007/s40256-015-0114-7

S23
123
Intern Emerg Med (2016) 11 (Suppl):S19-S23
14. Oliveira GB, Avezum A, Roever L (2015). Cardiovascular disease burden: evolving knowledge of risk factors in myocardial infarc-tion and stroke through population-based research and perspectives in global prevention. Front Cardiovasc Med 2:32. doi: 10.3389/fcvm.2015.00032
15. Sacco RL, Rundek T (2016). The value of urgent specialized care for TIA and minor stroke. N Engl J Med 374:1577-9. doi: 10.1056/NEJMe1515730
16. Vedel I, Khanassov V (2015). Transitional care for patients with congestive heart failure: A systematic review and meta-analysis. Ann Fam Med 13(6):562-71. doi: 10.1370/afm.1844
17. Qaddoura A, Yazdan-Ashoori P, Kabali C et al (2015). Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PLoS One 10(6):e0129282. doi: 10.1371/jour-nal.pone.0129282
18. Strøm C, Rasmussen LS, Rasmussen SW et al (2016). Admission of elderly medical patients to fast track or standard hospitalisation: protocol for a randomised trial. Dan Med J 63(3). pii: A5189
19. Freund T, Campbell SM, Geissler S et al (2013). Strategies for reducing potentially avoidable hospitalizations for ambulatory care-sensitive conditions. Ann Fam Med 11(4):363-70. doi:10.1370/afm.1498
20. Sachdev PS, Lipnicki DM, Kochan NA et al; Cohort Studies of Memory in an International Consortium (COSMIC) (2015). The prevalence of mild cognitive impairment in diverse geographical and ethnocultural regions: the COSMIC collaboration. PloS One 10(11):e0142388. doi:10.1371/journal.pone.0142388
21. Callahan KE, Lovato JF, Miller ME et al (2015). Associations of mild cognitive impairment with hospitalization and readmission. J Am Geriatr Soc 63(9):1880-5. doi:10.1111/jgs.13593
22. Phelan EA, Debnam KJ, Anderson LA, Owens SB (2015). A sys-tematic review of intervention studies to prevent hospitalizations of community-dwelling older adults with dementia. Med Care 53:207-13. doi: 10.1097/MLR.0000000000000294
23. Bruck K, Stel V, Hallan S et al; European CKD Burden Consortium (2015). CKD prevalence varies across the European general pop-ulation. J Am Soc Nephrol Dec 23. pii: ASN.2015050542. [Epub ahead of print]
24. Zambon S, Maggi S, Zanoni S et al (2012). Association of single measurement of estimated glomerular filtration rate and non-quan-
titative dipstick proteinuria with all-cause and cardiovascular mortality in the elderly. Results from the Progetto Veneto Anziani (Pro.V.A.) Study. Atherosclerosis 220(1):201-7. doi: 10.1016/j.ath-erosclerosis. 2011.09.023
25. Eppenga WL, Wester WN, Derijks HJ et al (2015). Fluctuation of the renal function after discharge from hospital and its effects on drug dosing in elderly patients--study protocol. BMC Nephrol 16:95. doi: 10.1186/s12882-015-0095-4
26. Nobili A, Marengoni A, Tettamanti M et al (2011). Association be-tween clusters of diseases and polypharmacy in hospitalized elderly patients: results from the REPOSI study. Eur J Intern Med 22:597-602. doi: 10.1016/j.ejim.2011.08.029
27. Renom-Guiteras A, Uhrenfeldt L, Meyer G, Mann E (2014). Assess-ment tools for determining appropriateness of admission to acute care of persons transferred from long-term care facilities: a system-atic review. BMC Geriatrics 14:80. doi: 10.1186/1471-2318-14-80
28. Barbadoro P, Di Tondo E, Menditto VG et al (2015). Emergency department non-urgent visits and hospital readmissions are asso-ciated with different socio-economic variables in Italy. PLoS One 10(6):e0127823. doi: 10.1371/journal.pone.0127823
29. Bosch X, Jordán A, López-Soto A (2013). Quick diagnosis units: avoiding referrals from primary care to the ED and hospitalizations. Am J Emerg Med 31:114-23. doi: 10.1016/j.ajem.2012.06.013
30. Elsayem AF, Elzubeir HE, Brock PA, Todd KH (2016). Integrating palliative care in oncologic emergency departments: challenges and opportunities. World J Clin Oncol 7(2):227-33. doi: 10.5306/wjco.v7.i2.227
31. Numico G, Cristofano A, Mozzicafreddo A et al (2015). Hospital admission of cancer patients: avoidable practice or necessary care? PLoS One 10(3):e0120827. doi: 10.1371/journal.pone.0120827
32. Montesi L, Turchese Caletti M, Marchesini G (2016). Diabetes in migrants and ethnic minorities in a changing world. World J Diabe-tes 7(3):34-44. doi: 10.4239/wjd.v7.i3.34
33. Mackelprang JL, Qiu Q, Rivara FP (2015). Predictors of Emer-gency Department visits and inpatient admissions among home-less and unstably housed adolescents and young adults. Med Care 53(12):1010-7. doi: 10.1097/MLR.0000000000000436
34. Afilalo M, Soucy N, Xue X et al (2015). Hospital stay on acute care units for non-acute reasons: effects of patient pre-hospitalization and admission factors. Healthc Manage Forum 28(1):34-9

© SIMI 2016
123
RELAZIONE
© SIMI 2016
Rodolfo Sbrojavacca (•)Dipartimento di Medicina, ASUI di Udine Piazzale Santa Maria della Misericordia 1, 33010 Udine E-mail: [email protected]
*Questa relazione è ripresa dal seguente articolo:Sbrojavacca R, Pietrangelo A, Fenoglio L, Violi F, Perticone F, Corazza GR (2015). Reducing the risk of hospital admission: A call to action from the Italian Society of Internal Medicine. Eur J Intern Med 26:476-7
Per accertamenti e cure: il ricovero è sempre utile? Contro*
Rodolfo Sbrojavacca
Qualsiasi ricovero ospedaliero implica in tutte le sue fasi, dall’accettazione alla dimissione, un’assunzione di responsa-bilità nei confronti del paziente.
Interrogarsi sul significato del ricovero comporta una ri-flessione sulla sua utilità e sicurezza. Sfortunatamente, la consapevolezza che la permanenza in ospedale può costituire di per se stessa, come accade per ogni altra procedura medi-co-sanitaria, un rischio per i degenti non è diffusa né tra gli utenti né tra gli operatori sanitari. Quanti sono consapevoli della possibilità che diversi pazienti potrebbero essere me-glio curati a casa o potrebbero essere dimessi molto prima di quanto avvenga di fatto, se non si dovessero adattare a tempi, ritmi e logiche costruiti non su di loro ma sugli operatori? Dovremmo inoltre prendere coscienza che la decisione di ri-coverare un paziente è talvolta parte di una strategia difensiva e risponde ad una esigenza di sicurezza più per il medico che per il malato.
La letteratura scientifica offre ormai un’ampia e circostan-ziata documentazione dei danni che l’ospedalizzazione può produrre [1-3]. Nel bilancio tra vantaggi e svantaggi del rico-vero, raramente si tiene conto dell’impatto che la degenza di per se stessa ha sul benessere del paziente. In una società che va invecchiando, l’ospedale può divenire, suo malgrado, un ambiente ostile per il paziente complesso e fragile. L’allon-tanamento dal proprio mondo, lo sconvolgimento delle abitu-dini, il disorientamento che deriva dal variare delle figure di
riferimento, l’immobilizzazione e la frequente malnutrizione, la deprivazione del sonno, l’aggiunta di nuovi farmaci, il ben noto rischio di infezioni ospedaliere [4] e di altri danni iatro-geni come il delirium sono sottostimati nella loro incidenza reale [5]. Alcuni di questi fattori si traducono in importanti fattori di stress, che rappresentano le cause della cosiddetta “post hospital syndrome”, un periodo di vulnerabilità acqui-sito e transitorio scarsamente dipendente dalla severità della patologia iniziale, frequentemente alla base della riospedaliz-zazione a 30 giorni [6].
Riconsiderare criticamente il ricovero è un modo per ra-gionare non solo sul ruolo dell’ospedalizzazione ma anche su quelli che sono i corretti paradigmi di ricaduta sulla sa-lute. Per quanto attiene al primo, che è solo una tappa anche se talora molto influente del percorso del malato, ad oggi la sua valutazione critica si è limitata alla sola appropriatezza, nei fatti un mantra declinabile in modi molto diversi (clinico, organizzativo, economico) fino a poter risultare addirittura conflittuale con il reale interesse del malato. Un esame inuti-le, un trattamento non giustificato o subottimale, un ricovero inappropriato non sono solo costi per il Servizio Sanitario Nazionale, ma costi, disagi e danni per il paziente e per ogni singolo cittadino. Per ciò che riguarda le ricadute sulla salute, fino ad ora la medicina ha ruotato intorno ad outcomes spe-cifici per la singola malattia attribuendo poca attenzione ad un approccio centrato sul malato, meno oneroso in termini di consumo di risorse e basato sul recupero funzionale e sui valori dei pazienti, non solo su una guarigione non sempre realizzabile [4].
La Medicina Interna affronta la maggior parte dei ricove-ri di pazienti fragili e complessi e per questo è la disciplina che per prima deve interrogarsi sul ruolo dell’ospedale per proporre una call to action rivolta a stakeholders, medici di altre specialità e a tutti gli operatori sanitari, in particolare l’infermiere, la figura professionale più costantemente in con-tatto con il paziente ed i suoi bisogni. La Medicina Interna può avere un ruolo chiave nella trasformazione del sistema,
Intern Emerg Med (2016) 11 (Suppl):S24-S25

S25
123
Intern Emerg Med (2016) 11 (Suppl):S24-S25
ma potrà svolgerlo solo se finalmente gli amministratori e gli stessi operatori del Servizio Sanitario Nazionale avranno la piena consapevolezza di quanto sia cambiato il contesto cli-nico epidemiologico (aumento delle comorbilità, invecchia-mento della popolazione, politerapia) e di quanto sia conse-guentemente necessario un adeguamento strutturale [7].
Non ci sono evidenze che il potenziamento dei servizi ter-ritoriali possa ridurre il numero o la durata dei ricoveri ospe-dalieri [8]. È tuttavia necessario che ci si impegni a costru-ire anche percorsi alternativi al ricovero che consentano al paziente di curarsi restando quanto più a lungo possibile al proprio domicilio, dove minore è il rischio di complicanze ia-trogene e più facile la personalizzazione delle scelte assisten-ziali. Ovviamente, da una tale prospettiva, la medicina ospe-daliera non deve risultare esclusa a favore di una medicina territoriale dai contorni non ben definiti. È invece necessario che si lavori a soluzioni organizzative che ne riconoscano un ruolo centrale nella programmazione e nella valutazione delle indagini e della terapia anche a livello territoriale.
Il ricovero ospedaliero deve essere giustificato sul piano clinico o assistenziale, offrendo al malato percorsi diagnostici e cure appropriate con garanzie di sicurezza o sostegno dei bi-sogni essenziali se essi non possono essere altrimenti assicu-rati. Esso resta insostituibile in molti casi, in considerazione della loro complessità o della preesistenza di percorsi iniziati in ospedale che non sia opportuno interrompere.
Un problema culturale complesso non può avere solo solu-zioni organizzative. Ridurre il rischio del ricovero presuppone un cambiamento culturale radicale, condiviso da tutti gli ope-ratori, un sapere comune fondato su basi razionali, prove di efficacia e rispetto per i valori dei pazienti. Ne conseguirebbe un ripensamento da parte di tutti del proprio lavoro, ponendo
finalmente attenzione nella pratica quotidiana al controllo di quelle condizioni elementari che possono tradursi in fattori di stress negativi per i pazienti. Per le sue caratteristiche la Me-dicina Interna deve farsi promotrice di una nuova cultura del ricovero anche attraverso la formazione di medici non proni a una medicina difensiva, maggiormente attenti ai reali bisogni dei pazienti e non schiavi della compulsività diagnostica e te-rapeutica indotta dalla disponibilità di attrezzature altamente tecnologiche. La prima e più efficace cura al paziente è l’at-tenzione che il medico e il personale sanitario tutto sapranno prestare alla persona e ai suoi bisogni.
Bibliografia
1. Leape LL, Brennan TA, Laird N et al (1991). The nature of adverse events in hospitalized patients. Results of the Harvard Medical Prac-tice Study II. N Engl J Med 324(6):377-84
2. Yun W, Eldridge N, Metersky ML et al (2014). National trends in patient safety for four common conditions, 2005-2011. N Engl J Med 370(4):341-51
3. National Institute for Health and Clinical Excellence (2007). Acute-ly ill patients in hospital. Recognition of and response to acute illness in adults in hospital. NICE clinical guideline 50. London: NICE 2007
4. Reuben DB, Tinetti ME (2012). Goal-oriented patient care — an alternative health outcomes paradigm. N Engl J Med Mar 1;366(9):777-9
5. McIntyre H (2013). Admission to hospital could be considered a disease. BMJ 346:f3242
6. Krumholz HM (2013). Post-hospital syndrome--an acquired, tran-sient condition of generalized risk. N Engl J Med 368(2):100-2
7. Jones DS, Podolsky SH, Greene JA (2012). The burden of disease and the changing task of medicine. N Engl J Med 366(25):2333-8
8. D’Souza S, Guptha S (2013). Preventing admission of older people to hospital. BMJ 346:f3186

© SIMI 2016
123
IL DIBATTITO
© SIMI 2016
Paolo Sbraccia (•)Dipartimento di Medicina dei SistemiUniversità di Roma “Tor Vergata”Via Montpellier 1, 00133 RomaE-mail: [email protected]
L’origine dell’obesità: un colpo al centro e uno alla periferia.Parte I: Un colpo al centro
Paolo Sbraccia
Introduzione
Secondo l’Organizzazione Mondiale della Sanità (OMS) “l’o-besità rappresenta per l’Europa una sfida sanitaria pubblica senza precedenti, finora sottostimata, scarsamente valutata e non perfettamente accettata come problema governativo stra-tegico associato a notevoli implicazioni economiche”. Essa è al contempo problema clinico, con le sue molteplici complicanze (diabete tipo 2, ipertensione arteriosa, dislipidemia aterogena, malattie cardiovascolari, molti tumori specie dell’apparato ga-stroenterico, sindrome delle apnee notturne, artropatie, depres-sione) e sfida socio-culturale, problematica psicologico-psichia-trica e disturbo nutrizionale, problema riabilitativo e problema di sanità pubblica. L’obesità è anche certamente il principale contributore all’aumento incontrollato delle malattie non tra-smissibili, vera sfida dei sistemi sanitari nel prossimo futuro.
L’obesità si manifesta a causa di uno squilibrio tra introi-to calorico e spesa energetica con conseguente accumulo dell’eccesso di calorie in forma di trigliceridi nei depositi di tessuto adiposo. È una patologia eterogenea e multifattoriale, al cui sviluppo concorrono sia fattori ambientali che genetici, e il contributo relativo di ognuno dei due fattori varia da indi-viduo a individuo.
Cosa si intende per centro e periferia che, vis-à-vis, si conten-derebbero il primato di responsabili dell’obesità in questa che, speriamo, non sia una mera dissertazione scolastica? E qual è la cogenza di tale argomentare, visto poi che centro (sistema ner-voso centrale) e periferia (il resto del nostro organismo) sono indissolubilmente legati? Prima di rispondere faccio un piccolo passo indietro. I nostri pazienti, noi medici, il resto di chi vive
al di fuori degli ospedali hanno sempre dibattuto sulle cause dell’obesità imputandole banalmente al “mangiar troppo” o al “consumare poco”. La maggior parte dei nostri pazienti ritiene quasi sempre di appartenere a quest’ultima categoria; quante volte abbiamo sentito frasi del tipo: «Mangio esattamente come mio marito, ma il mio metabolismo è lento e ingrasso mentre lui è magrissimo». È davvero così? Gli anglosassoni hanno co-niato da tempo il termine mouth-eyes gap, per indicare appunto la condizione di chi è convintissimo di mangiare poco perché non vedrebbe quello che in realtà ingerisce. Posto naturalmente che le definizioni di “troppo” e di “poco” riferiti al mangia-re sono sempre molto (troppo!) relative. Infatti se l’omeostasi energetica funzionasse, il basso consumo dovrebbe condurre ad accumulo energetico e quindi, con meccanismo di feedback, a riduzione dell’appetito; e quindi sarebbe sempre il centro ha non svolgere correttamente il proprio lavoro.
Ma tornando al punto iniziale, la rilevanza di questa tema-tica ha due risvolti molto importanti:– il primo è relativo a quale sia la direzione che la ricerca
farmacologica debba prendere in relazione, ad esempio, allo sviluppo di un farmaco anoressizzante rispetto ad un farmaco che acceleri il metabolismo;
– il secondo è legato alla questione del libero arbitro: gli obe-si, come purtroppo qualcuno ancora oggi sostiene, sono in-dividui privi di forza di volontà che creano problemi a loro e agli altri, oppure sono affetti da alterazioni molecolari di circuiti ancestrali che in presenza di libero accesso al cibo rendono di fatto impossibile non ingrassare?
Nelle pagine che seguono sosterrò la tesi, supportata da una mole ponderosa di dati, che negli obesi è il centro ad essere in qualche modo difettoso, con conseguente progressiva, e non ne-cessariamente marcata, iperalimentazione in relazione al consu-mo energetico; ciò perpetua una condizione di bilancio energeti-co cronicamente positivo con sviluppo di obesità di grado sem-pre maggiore. Questa alterazione “centrale” imprime, inoltre, uno stimolo incoercibile (anche nella maggior parte dei pazienti
Intern Emerg Med (2016) 11 (Suppl):S26-S30

S27
123
Intern Emerg Med (2016) 11 (Suppl):S26-S30
non affetti da disturbi del comportamento alimentare), che rende vano qualsiasi ragionevole ricorso alla cosiddetta forza di volon-tà. Questo è il vero motivo per il quale l’obesità va considerata una vera e propria malattia e non una condizione che interessa una categoria di persone da relegare nel girone dei golosi.
Controllo omeostatico del bilancio energetico
Sebbene l’estrema variabilità interindividuale del peso cor-poreo appaia poco compatibile con uno stretto controllo omeostatico dei depositi energetici, la forza con la quale in ogni individuo l’organismo reagisce ad un calo di peso ren-de ragione dell’esistenza di un sistema di controllo attento ai flussi di energia, sebbene più indulgente per ragioni evolutive a quelli in entrata.
Che esistesse un sistema omeostatico di controllo del bilan-cio energetico lo si è ipotizzato da più di mezzo secolo, ma i meccanismi molecolari che sottendono il dialogo tra le scorte energetiche ed il cervello sono stati progressivamente chiariti a iniziare dalla scoperta della leptina nel 1994 [1]. A livello del nucleo arcuato dell’ipotalamo vi sono i due nuclei neuronali, che esprimono rispettivamente il neuropeptide-Y (NPY) e la pro-opiomelanocortina (POMC); essi rappresentano il cuore della centralina omeostatica e controllano l’introito calorico e la spesa energetica in base a segnali ormonali (principalmente la leptina), metabolici e meccanici che ricevono dalla perife-ria. La leptina inibisce i neuroni NPY ad azione oressizzante e stimola i neuroni POMC dal quale, per clivaggio, origina l’a-MSH che, interagendo con il recettore melanocortinico-4 (MC4R), veicola il principale segnale anoressizzante [2].
Questo sistema omeostatico, come detto, non è tarato in modo stretto come i sistemi che controllano, ad esempio, il pH ematico, l’equilibrio idro-elettrolitico o l’osmolalità pla-smatica. Esso si è evoluto in centinaia di migliaia di anni ca-
ratterizzati da un ambiente molto povero di cibo ed è regolato in modo asimmetrico: è in grado di reagire in modo efficiente in caso di riduzione ponderale e, al contrario, tende ad essere più permissivo nei confronti del guadagno di peso.
I nuclei ipotalamici, inoltre, sono influenzati da molti altri fattori, quali la vista, l’olfatto, il gusto, la sfera emotiva ed il co-siddetto sistema edonico (regolato dal nucleo accumbens e dal sistema dopaminergico): il sistema edonico influenza fortemente l’assunzione di cibo palatabile indipendentemente dalle esigenze energetiche. L’intero sistema è arcaico e deputato all’assunzione regolata della fonte primaria di sopravvivenza, i nutrienti. Sia la teoria del genotipo economo [3] che la teoria della predazione [4] spiegano la progressiva tendenza nell’evoluzione della nostra specie di alleli “permissivi” nei confronti dell’accumulo energe-tico. Vi è naturalmente una notevole variazione interindividuale spiegabile con l’ereditarietà del tratto “adiposità”.
L’ereditarietà dell’obesità è stata chiaramente dimostrata dai ponderosi studi effettuati nei gemelli monozigoti, valutati rispetto a quelli dizigoti [5-7] e, nell’ambito dei monozigoti, tra quelli vissuti assieme rispetto a quelli adottati e cresciuti in famiglie diverse [8]. Da questi studi è stato possibile calco-lare che il contributo dell’ereditarietà allo sviluppo di obesità varia dal 40 al 70% [9].
Le obesità monogeniche e quelle sindromiche vedono coinvolti geni ipotalamici che regolano appetito e sazietà
È singolare che tutte le obesità monogeniche fino ad oggi identificate (Tabella 1 [10]) riguardino esclusivamente geni che codificano per proteine ipotalamiche coinvolte principal-mente nella regolazione dell’appetito [11]. Mutazioni omozi-goti o eterozigoti composte di cinque geni coinvolti nelle vie di trasmissione del segnale nell’ambito del sistema leptina-melanocortina (Figura 1 [12]) sono responsabili di obesità
Tabella 1 Obesità monogeniche e sindromiche umane [10].
Sindrome con obesità Gene Caratteristiche distintive principali
Deficit di LEP LEP Ipogonadismo, infezioni frequenti, leptina sierica non rilevabile
Deficit di LEPR LEPR Ipogonadismo
Deficit di POMC POMC Ipopigmentazione, deficit isolato di ACTH
Deficit di PCSK1/3 PCSK1 Ipoglicemia post-prandiale, ipogonadismo, elevati valori plasmatici di proinsulina e proinsulina split 32-33
Deficit di MC4R MC4R Crescita accelerata, statura finale aumentata
Deficit di BDNF BDNF Ritardo di sviluppo, iperattività, memoria compromessa, alterata sensibilità al dolore
Deficit di TrkB NTRK2 Ritardo di sviluppo, iperattività, memoria compromessa, alterata sensibilità al dolore
Deficit di SIM1 SIM1 Ritardo dello sviluppo ad ampio spettro
BBS BBS1-16 Polidattilia, distrofia retinica, ipogonadismo, alterazioni renali, ritardo di sviluppo
Sindrome di Alström ALMS1 Fotofobia, nistagmo, compromissione del visus, sordità, grave insulino-resistenza
AHO GNAS1 Bassa statura, difetti scheletrici, resistenza ormonale
AHO: osteodistrofia ereditaria di Albright; BBS: sindrome di Bardet-Biedl

S28
123
Intern Emerg Med (2016) 11 (Suppl):S26-S30
murine e delle corrispettive forme nell’uomo. Inoltre, i pa-zienti con aploinsufficienza del gene SIM1 (un fattore di tra-scrizione necessario per il normale sviluppo ipotalamico [13] e del gene BDNF (brain-derived neurotrophic factor [14], così come coloro che hanno una mutazione del suo recettore TrkB [15], sviluppano obesità grave in associazione con altre alterazioni dello sviluppo cerebrale (Tabella 1).
Sulla base di questi dati si può quindi affermare che:1. l’obesità può derivare da un semplice difetto genetico che,
nel caso della leptina, può essere curato con terapia ormo-nale sostitutiva;
2. tutti i difetti genetici descritti finora, che causano forme monogeniche di obesità, agiscono alterando i meccanismi ipotalamici che regolano l’appetito e soprattutto la sazietà.
Le varianti genetiche comuni, identificate attraverso approcci genome-wide, appartengono anch’esse al sistema di regolazione centrale del bilancio energetico
Negli ultimi dieci anni, in modo progressivamente più avan-zato dal punto di vista tecnologico, sono stati pubblicati i risultati di diversi studi cosiddetti genome-wide association study (GWAS), in grado cioè di setacciare l’intero genoma di un ampio numero di soggetti (fino ad alcune centinaia di migliaia negli ultimi studi), per individuare varianti geneti-che associate all’indice di massa corporea (BMI). I primi stu-di hanno identificato una variante genica del gene FTO che
quando presente in omozigosi aumenta in media di 3 kg il peso corporeo [16]. Questo risultato è stato poi replicato in molti studi con una forza statistica schiacciante. Successiva-mente è stato dimostrato che il gene FTO codifica per una proteina appartenente alla famiglia delle ossigenasi 2-ossi-glutarato dipendenti [17] e il grado più elevato di espressione di questa proteina è a livello del nucleo arcuato dell’ipotala-mo, dove la sua espressione è regolata dal digiuno e dall’a-limentazione [15]. Un’altra proteina, il cui gene è emerso in alcune metanalisi (utilizzate per raggiungere una numerosità del campione che desse maggiore forza statistica) come in grado di influenzare il BMI, è quella del MC4R (melanocor-tin 4 receptor) [18,19]. Quest’ultimo risultato non sorprende visto che mutazioni di questo gene sono già state descritte in forme monogeniche di grave obesità.
Ma i risultati più interessanti sono stati ottenuti recente-mente dalla metanalisi più ampia sia in termini di soggetti studiati (quasi 340.000) che di polimorfismi (SNP, single nucleotide polymorphisms), dalla quale sono emersi 97 loci associati al BMI [20]. Questi loci interessano geni espressi a livello del sistema nervoso centrale (Figura 2) [20].
Ruolo del metabolismo basale
Una domanda che ci si è sempre posti per tentare di spiegare lo sviluppo di obesità è se un metabolismo basale ai limiti bassi della norma possa causare obesità. Nell’ultimo mezzo
NPY/AgRP POMC
Sistema leptina/melanocortina
Sistema del nucleo paraventricolare
Tessuto adiposo
Consumo energetico
Introito alimentare
BDNF
TrkB
SIM1
αMSH βMSH
Consumo energetico
Introito alimentare
LEPR
+
+
+–
–
+
Leptina
TUB
MRAP2
CPE
PC1/3
MC4R MC3R
SH2B1
JAK
STAT3
Fig. 1 Geni coinvolti nel sistema “leptina-melanocortina” (rettangoli grigi) le cui mutazioni sono responsabili delle forme di obesità monogeniche e sindromiche umane [12].
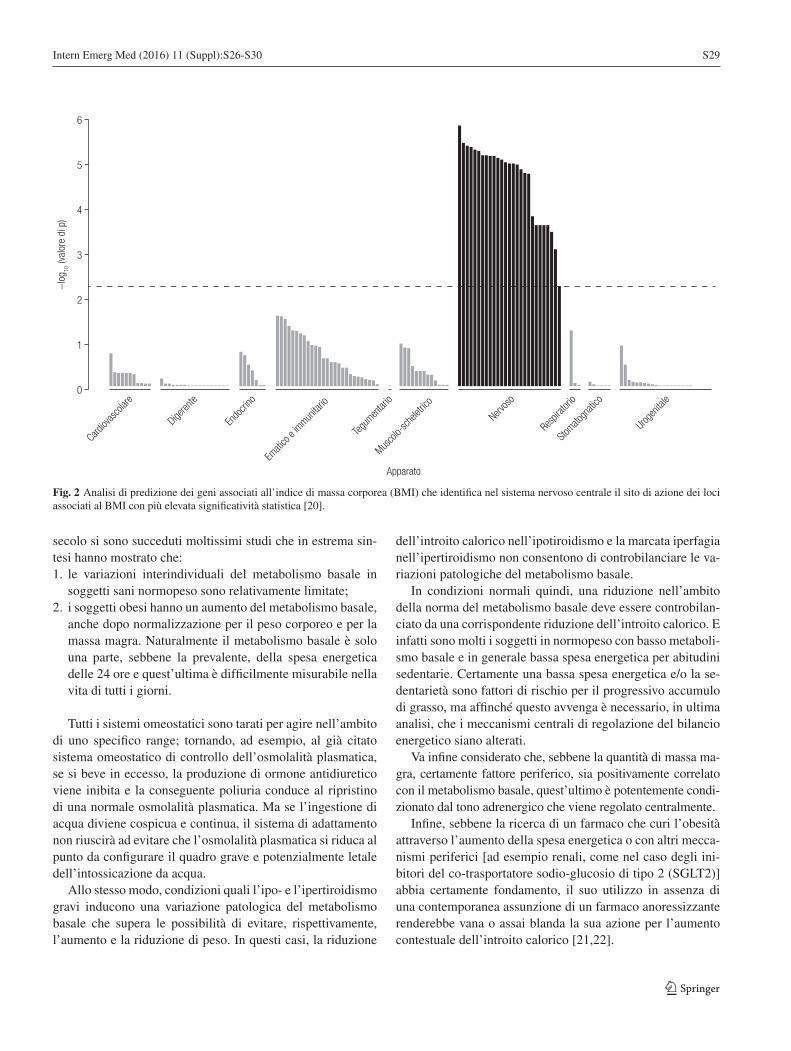
S29
123
Intern Emerg Med (2016) 11 (Suppl):S26-S30
secolo si sono succeduti moltissimi studi che in estrema sin-tesi hanno mostrato che: 1. le variazioni interindividuali del metabolismo basale in
soggetti sani normopeso sono relativamente limitate; 2. i soggetti obesi hanno un aumento del metabolismo basale,
anche dopo normalizzazione per il peso corporeo e per la massa magra. Naturalmente il metabolismo basale è solo una parte, sebbene la prevalente, della spesa energetica delle 24 ore e quest’ultima è difficilmente misurabile nella vita di tutti i giorni.
Tutti i sistemi omeostatici sono tarati per agire nell’ambito di uno specifico range; tornando, ad esempio, al già citato sistema omeostatico di controllo dell’osmolalità plasmatica, se si beve in eccesso, la produzione di ormone antidiuretico viene inibita e la conseguente poliuria conduce al ripristino di una normale osmolalità plasmatica. Ma se l’ingestione di acqua diviene cospicua e continua, il sistema di adattamento non riuscirà ad evitare che l’osmolalità plasmatica si riduca al punto da configurare il quadro grave e potenzialmente letale dell’intossicazione da acqua.
Allo stesso modo, condizioni quali l’ipo- e l’ipertiroidismo gravi inducono una variazione patologica del metabolismo basale che supera le possibilità di evitare, rispettivamente, l’aumento e la riduzione di peso. In questi casi, la riduzione
dell’introito calorico nell’ipotiroidismo e la marcata iperfagia nell’ipertiroidismo non consentono di controbilanciare le va-riazioni patologiche del metabolismo basale.
In condizioni normali quindi, una riduzione nell’ambito della norma del metabolismo basale deve essere controbilan-ciato da una corrispondente riduzione dell’introito calorico. E infatti sono molti i soggetti in normopeso con basso metaboli-smo basale e in generale bassa spesa energetica per abitudini sedentarie. Certamente una bassa spesa energetica e/o la se-dentarietà sono fattori di rischio per il progressivo accumulo di grasso, ma affinché questo avvenga è necessario, in ultima analisi, che i meccanismi centrali di regolazione del bilancio energetico siano alterati.
Va infine considerato che, sebbene la quantità di massa ma-gra, certamente fattore periferico, sia positivamente correlato con il metabolismo basale, quest’ultimo è potentemente condi-zionato dal tono adrenergico che viene regolato centralmente.
Infine, sebbene la ricerca di un farmaco che curi l’obesità attraverso l’aumento della spesa energetica o con altri mecca-nismi periferici [ad esempio renali, come nel caso degli ini-bitori del co-trasportatore sodio-glucosio di tipo 2 (SGLT2)] abbia certamente fondamento, il suo utilizzo in assenza di una contemporanea assunzione di un farmaco anoressizzante renderebbe vana o assai blanda la sua azione per l’aumento contestuale dell’introito calorico [21,22].
Cardiov
asco
lare
−lo
g 10 (v
alor
e di
p)
0
6
5
3
4
2
1
Apparato
Endo
crino
Digeren
te
Emati
co e
immun
itario
Tegum
entar
io
Musco
lo-sch
eletric
o
Resp
irator
io
Nervos
o
Uroge
nitale
Stomato
gnati
co
Fig. 2 Analisi di predizione dei geni associati all’indice di massa corporea (BMI) che identifica nel sistema nervoso centrale il sito di azione dei loci associati al BMI con più elevata significatività statistica [20].

S30
123
Intern Emerg Med (2016) 11 (Suppl):S26-S30
La periferia ha un qualche ruolo nella patogenesi dell’obesità?
La periferia ha un ruolo fondamentale nella patogenesi delle complicanze metaboliche dell’obesità, ma il suo ruolo nella genesi dell’obesità appare marginale. Shift dei substrati verso il tessuto adiposo, sensibilità insulinica, attività mitocondria-le, tessuto adiposo bruno, microbiota intestinale, adipochine, sarcopenia e moltissimi altri “attori” della complessa mac-china metabolica che ossida, trasforma e deposita i macro-nutrienti al fine di avere sempre a disposizione l’energia ri-chiesta dal nostro organismo, possono certamente influenzare l’accumulo di trigliceridi nel tessuto adiposo, ma in assenza di alterazioni che superino per entità il range di azione omeo-statica, è sempre il centro che fallisce nel non adeguare appe-tito e sazietà ad una condizione di surplus energetico.
Conclusioni
Negli ultimi vent’anni abbiamo assistito ad un enorme amplia-mento delle nostre conoscenze sia nell’ambito dei meccani-smi neuroendocrinologici che regolano il bilancio energetico, sia per quanto riguarda la genetica dell’obesità. L’ereditarietà contribuisce per il 40-70% allo sviluppo di obesità. Tutte le forme monogeniche di obesità murine e umane coinvolgono geni ipotalamici. Tutti i polimorfismi che si associano al BMI con alta significatività statistica, interessano geni ipotalamici.
Quindi:1. tutti i dati in nostro possesso ci spingono a sostenere che
l’obesità è geneticamente determinata;2. l’impatto genetico sembra esercitarsi esclusivamente a li-
vello del sistema nervoso centrale;3. l’obesità origina da un’alterazione predeterminata e indi-
pendente dal libero arbitrio che, agendo su appetito e sa-zietà, induce uno stato di relativa (rispetto alla spesa ener-getica) iperalimentazione.
In conclusione, l’obesità è una malattia neuro-comporta-mentale il cui “sintomo” principale è rappresentato dall’ec-cessiva ingestione di cibo [23,24].
Bibliografia
1. Zhang Y, Proenca R, Maffei M et al (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372:425-32
2. Cone RD (2005). Anatomy and regulation of the central melanocor-tin system. Nature Neurosci 8:571-8
3. Neel JV (1962). Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14:353-62
4. Speakman JR (2007). A nonadaptive scenario explaining the genetic predisposition to obesity: the “predation release” hypothesis. Cell Metab 6:5-12
5. Maes HH, Neale MC, Eaves LJ (1997). Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 27:325-51
6. Allison DB, Kaprio J, Korkeila M et al (1996). The heritability of body mass index among an international sample of monozygotic twins reared apart. Int J Obes Relat Metab Disord 20:501-6
7. Allison DB, Heshka S, Neale MC et al (1994). A genetic analysis of relative weight among 4,020 twin pairs, with an emphasis on sex effects. Health Psychol 13:362-5
8. Stunkard AJ, Harris JR, Pedersen NL, McClearn GE (1990). The body-mass index of twins who have been reared apart. N Engl J Med 322:1483-7
9. Bell CG, Walley AJ, Froguel P (2005). The genetics of human obe-sity. Nature Rev 6:221-34
10. Ramachandrappa S, Farooqi IS (2011). Genetic approaches to un-derstanding human obesity. J Clin Invest 121:2080-6
11. Farooqi IS, O’Rahilly S (2005). Monogenic obesity in humans. Annu Rev Med 56:443-58
12. Yazdi FT, Clee SM, MeyreD (2015). Obesity genetics in mouse and human: back and forth, and back again. Peer J Mar 24;3:e856
13. Holder JL Jr, Butte NF, Zinn AR (2000). Profound obesity associat-ed with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 9:101-8
14. Gray J, Yeo GS, Cox JJ et al (2006). Hyperphagia, severe obesi-ty, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55:3366-71
15. Yeo GS, Connie Hung CC, Rochford J et al (2004). A de novo mu-tation affecting human TrkB associated with severe obesity and de-velopmental delay. Nat Neurosci 7:1187-9
16. Frayling TM, Timpson NJ, Weedon MN et al (2007). A common variant in the FTO gene is associated with body mass index and pre-disposes to childhood and adult obesity. Science 316:889-94
17. Gerken T, Girard CA, Tung YC et al (2007). The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demeth-ylase. Science 318:1469-72
18. Heid IM, Vollmert C, Kronenberg F et al (2008). Association of the MC4R V103I polymorphism with the metabolic syndrome: the KORA Study. Obesity 16:369-76
19. Young EH, Wareham NJ, Farooqi IS et al (2007). The V103I polymor-phism of the MC4R gene and obesity: population based studies and meta-analysis of 29 563 individuals. Int J Obes (Lond) 31:1437-41
20. Locke AE, Kahali B, Berndt SI et al (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518(7538):197-206
21. Piaggi P, Thearle MS, Krakoff J, Votruba SB (2015). Higher dai-ly energy expenditure and respiratory quotient, rather than fat-free mass, independently determine greater ad libitum overeating. J Clin Endocrinol Metab 100:3011-20
22. Ferrannini G, Hach T, Crowe S et al (2015). Energy balance after so-dium-glucose cotransporter 2 inhibition. Diabetes Care 38: 1730-5
23. O’Rahilly S, Farooqi IS (2008). Human obesity: a heritable neuro-behavioral disorder that is highly sensitive to environmental condi-tions. Diabetes 57:2905-10
24. Sharma AM, Padwal R (2010). Obesity is a sign – over-eating is a symptom: an aetiological framework for the assessment and man-agement of obesity. Obesity Rev 11:362-70

© SIMI 2016
123
IL DIBATTITO
Intern Emerg Med (2016) 11 (Suppl):S31-S38
Roberto Vettor (•)Clinica Medica 3, Dipartimento di Medicina−DIMEDUniversità di PadovaVia Giustiniani 2, 35128 Padova E-mail: [email protected]
L’origine dell’obesità: un colpo al centro e uno alla periferia.Parte II: Un colpo alla periferia
Roberto Vettor
Introduzione
Il mantenimento a lungo termine di un peso corporeo stabile richiede un fine sistema di controllo, la cui ridondanza è ga-ranzia del mantenimento dell’omeostasi energetica. Nell’uo-mo si sono sviluppati complessi meccanismi che tendono a mantenere un perfetto equilibrio tra l’introito calorico e la spesa energetica e un loro squilibrio porta a variazioni del contenuto di adipe. Perciò un efficace sistema di regolazione del peso corporeo implica il fatto che la quantità di energia immagazzinata possa tradursi in risposte adeguate al mante-nimento del peso e della composizione corporei.
Numerose teorie sono state proposte a sostegno del con-trollo del bilancio energetico immaginando sensori ed effetto-ri del sistema. La teoria glucostatica (glucosio), quella glico-genostatica (glicogeno epatico) ed energostatica (ATP) hanno lasciato il posto alla teoria adipostatica del controllo del peso che è stata avvalorata dalla scoperta della leptina, localizzata e secreta dal tessuto adiposo; essa costituisce un segnale di natura ormonale per il sistema nervoso centrale (SNC) sullo stato delle riserve lipidiche. Infatti a livello del SNC sono sta-ti identificati recettori specifici leganti la leptina.
Quanto appena detto introduce la possibilità che una primitiva alterazione funzionale dell’organo adiposo possa costituire gerarchicamente il primum movens nella genesi dell’obesità e delle sue complicanze. È possibile peraltro pensare che la genesi dell’obesità sia multifattoriale e che più centri, piuttosto che un solo centro e una sola periferia, debbano essere presi in considerazione come elementi pri-mari nella spiegazione fisiopatologica dell’obesità e delle sue complicanze.
In questo contesto, l’apologo di Menenio Agrippa che diede una spiegazione metaforica dell’organizzazione so-ciopolitica romana, sottolineando che in un corpo umano l’insieme è costituito da parti strettamente connesse l’una con l’altra e gli organi sopravvivono solo se dialogano tra loro attivamente, è adeguato al contesto. Se le braccia (il popolo) si rifiutassero di lavorare, lo stomaco (il senato) non avrebbe più cibo a sua disposizione e in tal caso tutto il cor-po, ma anche le braccia, soffrirebbe per la mancanza di cibo. Se il SNC e l’ipotalamo con il controllo neuroendocrino del metabolismo non dialogassero con gli altri organi e l’orga-no adiposo come elemento centrale nella gestione periferica dell’equilibrio energetico non inviasse segnali al SNC, il sistema sarebbe inesorabilmente destinato a morire. Deve essere comunque sottolineato che il livello di complessità eziopatogenetica dell’obesità considera che modificazioni antropologiche e sociologiche e in particolare del comporta-mento sociale della specie umana presenti in epoca contem-poranea, attraverso una aumentata disponibilità e consumo di cibo e una drastica riduzione dell’attività fisica comples-siva, hanno portato a un progressivo aumento del contenuto di grasso corporeo rispetto alla massa magra. Poiché l’e-nergia sotto forma di depositi energetici dell’organismo è il frutto di una fine regolazione di quanto viene introdotto rispetto a quanto viene consumato, è possibile pensare che una riduzione della capacità ossidativa del muscolo possa essere la causa prima di un preferenziale accumulo di sub-strati energetici nel tessuto adiposo (Figura 1). La sedenta-rietà, al pari o forse in maniera superiore all’iperalimenta-zione, può condizionare lo sviluppo dell’obesità.
Le ragioni del dialogo centro-periferia nella regolazione del bilancio energetico
Un meccanismo attraverso cui la leptina, l’ormone prodot-to dal tessuto adiposo, regola l’apporto alimentare e la spesa

S32
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
energetica è costituito principalmente dall’inibizione sulla sintesi e sul rilascio di neuropeptide Y (NPY), ma anche attra-verso l’intervento di altri neuropeptidi quali la pro-opiomela-nocortina (POMC), l’ormone melanocito-stimolante (aMSH, a-melanocyte stimulating hormone), l’ormone melanocito-concentrante (MCH, melanocyte concentrating hormone), la corticotropina (CRF, corticotropin releasing factor) e l’orexi-na. La leptina rappresenta un ormone strettamente correlato con l’entità della massa adiposa, ma è il digiuno, indipen-dentemente da apprezzabili variazioni ponderali, la situazio-ne che induce un rapido decremento dei livelli di leptina che può rappresentare un precoce segnale di deficit energetico al SNC. La riduzione dei livelli di leptina potrebbe quindi essere responsabile del decremento della spesa energetica che normalmente si accompagna alla riduzione ponderale. Nell’uomo la leptina conserva una piena valenza fisiologica solo in condizioni di deprivazione energetica come il digiuno. Questo fenomeno offre una spiegazione della difficoltà nel conseguire un calo ponderale con la sola modifica dello stile di vita e la restrizione calorica.
Oltre alla leptina, sono note alcune centinaia di adipochi-ne che hanno effetto non solo sul tessuto adiposo, ma anche su pancreas, fegato, cuore, vasi, SNC, sistema immunitario e altri organi [1].
Il ruolo centrale dell’organo adiposo e la regolazione periferica del metabolismo energetico
Due importanti concetti hanno profondamente cambiato la fisiopatologia dell’obesità: 1. il dialogo tra tessuto adiposo e SNC attraverso la leptina;2. l’esistenza dell’organo adiposo come elemento anatomico
dissecabile, in perfetta continuità anatomica tra una sua porzione viscerale e sottocutanea, dotato di una peculiare innervazione e vascolarizzazione e costituito da almeno due stipiti cellulari diversificati che sono stati recentemen-te ampliati e rivisitati: si descrivono attualmente tre tipi principali di cellula adiposa, caratterizzati da differenti lo-calizzazioni anatomiche, morfologia cellulare, espressione genica e funzione.
Il tessuto adiposo bianco
Gli adipociti bianchi in vivo appaiono come cellule sferiche, di grandi dimensioni, uniloculate, con contenuto di mitocon-dri relativamente basso; contribuiscono a formare i depositi di tessuto adiposo sottocutaneo e viscerale. Il tessuto adiposo bianco (WAT) è preposto al deposito dei nutrienti sotto forma di trigliceridi, alla secrezione di ormoni specifici, denominati “adipochine”, coinvolti nell’omeostasi energetica e nella re-golazione dell’appetito/sazietà.
Il tessuto adiposo bruno
Il tessuto adiposo bruno (BAT) prende il proprio nome dal suo colore macroscopico caratteristico conferitogli dalla ricca vascolarizzazione e dal gran numero di mitocondri; è forma-to da cellule multivacuolate ricche di mitocondri a morfolo-gia caratteristica (grandi dimensioni, creste molto sviluppate che si estendono anche da un’estremità all’altra). I deposi-ti di BAT sono caratteristicamente localizzati nelle regioni interscapolari, paravertebrali e perirenali (roditori) [2]. La loro funzione termogenica si esplica grazie all’uncoupling protein1 (UCP1), codificata nel DNA nativo e localizzata a livello della membrana mitocondriale interna, che permette il disaccoppiamento della catena mitocondriale e di conse-guenza la dissipazione di energia sotto forma di calore [3,4].
Dal punto di vista embriologico, gli adipociti bruni derive-rebbero da precursori esprimenti i geni paired box 7 (Pax7) e myogenic factor 5 (Myf5), considerati fino a pochi anni fa geni muscolo-specifici [5,6]. Nel differenziamento dei precursori bruni, un ruolo fondamentale è svolto dall’early B cell fac-tor-2 (Ebf-2) [7] che, insieme a PPARγ, promuove l’espressio-ne del gene PRDM16 (PR domain containing protein 16) che permette la maturazione della cellula bruna e l’attivazione di PGC1a (peroxisome proliferator-activated receptor-gamma
Fig. 1 Schema rappresentativo di come un aumento dell’apporto di sub-strati energetici al muscolo ne determini una ridotta sensibilità all’insu-lina e una riduzione dell’ossidazione dei substrati, con un preferenziale indirizzamento degli stessi verso il tessuto adiposo dove vengono im-magazzinati producendo un aumento della dimensione e, in seguito, del numero delle cellule adipose. Uno stesso fenomeno può essere evocato dalla prolungata sedentarietà o da primitive alterazioni del muscolo.
Capacità di riserva energetica(insulinodipendente)
Capacità ossidativa(insulinodipendente)
Substrati energetici

S33
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
coactivator 1 a), che a sua volta regola l’espressione dei geni della mitocondriogenesi, della termogenesi e del metaboli-smo ossidativo, prima fra tutte UCP1 [8,9]. In risposta inoltre all’esposizione al freddo, che agisce sul BAT indirettamente tramite l’attivazione del sistema ortosimpatico [10], o ad ago-nisti β-adrenergici, il programma termogenetico viene indotto ulteriormente per il mantenimento della temperatura corporea.
Anche nell’uomo adulto si hanno evidenze sempre più con-sistenti della presenza di tessuto adiposo a capacità termoge-netica, che può essere identificato come area captante il 18-F-deossi-glucosio alla PET a livello delle regioni interscapolare, peri-renale, cervicale, periaortica e carotidea [11-14]. La carat-terizzazione di tale tessuto ha suscitato il vivo interesse della ricerca attuale. È stato dimostrato che la quantità di tessuto adi-poso termogenetico individuato alla PET correla inversamente con l’indice di massa corporea (BMI) [11,15], risultando nei soggetti obesi meno presente e con ridotta capacità di indurre termogenesi in risposta all’esposizione al freddo; inoltre l’e-stensione e l’intensità dell’uptake delle zone captanti il 18-F-deossi-glucosio alla PET correlano nell’uomo con la presenza di diabete mellito, oltre che con l’età [16]. A conferma di quan-to detto, vi sono anche gli studi di espressione molecolare su biopsie umane prelevate durante interventi chirurgici a livello della regione sopraclavicolare e nucale [12], che hanno dimo-strato la presenza nell’uomo di BAT che aumenta man mano che ci si sposta verso i piani profondi.
Il tessuto adiposo beige
Dopo l’esposizione al freddo [17,18] o allo stimolo β-adre-nergico [19] ci può essere, nel contesto di alcuni depositi di tessuto adiposo bianco, la comparsa di cellule multivacuolate positive per UCP1 e altri geni coinvolti nella mitocondrioge-nesi (PRDM16, PGC1a), con caratteristiche intermedie tra gli adipociti bianchi e bruni, e denominate pertanto adipociti beige o “brite” (brown in white) o tessuto adiposo bruno “reclutabi-le” [20].
Gli adipociti beige deriverebbero per trans-differenziazio-ne da adipociti bianchi maturi in risposta allo stimolo, come suggerito dalla presenza di forme di transizione con carat-teristiche morfologiche intermedie tra cellule bianche e bru-ne [21,22] dopo l’esposizione al freddo o il trattamento con β-agonisti. L’idea alternativa è che le cellule beige derivino per adipogenesi de novo a partire da precursori residenti nei depositi di WAT reclutati al bisogno. Dal punto di vista mo-lecolare, gli adipociti beige sembrano presentare un pattern di espressione distinto dagli adipociti bruni “classici”. Sono state proposte come beige-specifici le molecole di superficie CD137 e la transmembrane protein 26 (Tmem 26), T-box 1 (TBX1); Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 1 (Cited1) [23]; l’homeobox
C9 (Hoxc9) [24]. Dal punto di vista funzionale, il tessuto adi-poso beige sembrerebbe contribuire alla termogenesi dell’or-ganismo in risposta a stimoli esterni.
Il sistema nervoso simpatico e in particolare l’attivazione dei recettori β-3 costituiscono lo stimolo principale per l’at-tivazione del tessuto adiposo bruno o per l’avvio della tra-sformazione bianco-bruno. A tal riguardo, è la stimolazione cronica con agonisti del PPARγ [25], acidi biliari, fibroblast growth factor 21 (FGF21) [26], atrial e ventricular natriu-retic peptides (ANP, BNP) [27], bone morphogenetic pro-teins (BMP4 e BMP7) [28] che induce la differenziazione di adipociti beige secondo diversi meccanismi. Una miochina scoperta di recente, denominata irisina, è stata riconosciuta come una potente attivatrice del processo di browning [29], così come il micro-RNA26 [30]. Recentemente il nostro gruppo di ricerca ha confermato che l’esercizio fisico è uno stimolo per la biogenesi mitocondriale non solo a livello del muscolo, ma anche a livello del tessuto adiposo sottocutaneo. Abbiamo inoltre chiaramente identificato nell’ossido nitrico un altro elemento fondamentale nel processo di biogenesi mitocondriale e di browning del tessuto adiposo sottocuta-neo [31], ma l’aspetto più rilevante e concettualmente desti-nato a far prevalere l’idea che la malattia del tessuto adiposo possa essere un elemento fondamentale nella genesi e pro-gressione dell’obesità e delle sue complicanze è stato fornito dagli esperimenti in cui si è trapiantato, in animali obesi e diabetici, del tessuto adiposo di animali sottoposti ad eserci-zio fisico prolungato che aveva acquisito tutte le qualità del tessuto adiposo bruno. Tale manipolazione ha condotto a un importante miglioramento del fenotipo [32], suggerendo la possibilità che un intervento terapeutico mirato a migliorare la qualità del tessuto adiposo possa essere utilizzato per la terapia dell’obesità e del diabete di tipo 2.
Obesità ovvero la malattia dell’organo adiposo (adiposopatia)
Come già accennato nel paragrafo precedente, l’adiposopa-tia, ovvero le alterazioni morfologiche e funzionali dell’or-gano adiposo, è paradigmatica dell’obesità come malattia ed è alla base del processo che lega l’obesità alle sue com-plicanze. L’adiposopatia rappresenta il passaggio a una de-finizione qualitativa della malattia obesità, abbandonando il suo inquadramento nosografico solo dovuto al superamento di una soglia quantitativa: il BMI. Essa è caratterizzata da cambiamenti della distribuzione dell’adipe con maggior propensione all’accumulo addominale viscerale, dalla trans-differenziazione del tessuto adiposo bruno in bianco o da un deficit di reclutamento della cellula adiposa bianca verso un fenotipo bruno (browning), da alterazioni del numero e della

S34
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
funzione mitocondriale, da alterazioni della staminalità del tessuto adiposo, dall’aumento delle dimensioni della cellula adiposa e dalla progressiva riduzione della sensibilità all’in-sulina, dall’incapacità del tessuto adiposo bianco di accoglie-re adeguatamente il flusso di substrati dovuta ad una incapa-cità di ulteriore espandibilità, dall’infiltrazione da parte dei macrofagi con l’avvio di una infiammazione cronica di basso grado, dalla formazione di tessuto fibroso con un’ulteriore limitazione all’espansione dell’organo con deviazione del flusso di substrati dal tessuto adiposo verso gli altri organi dando avvio al fenomeno della lipotossicità cioè la presen-za di tessuto adiposo bianco all’interno di altri parenchimi, e da alterazioni qualitative e quantitative della secrezione delle adipochine (Figura 2).
L’obesità come malattia primariamente mitocondriale
Uno degli elementi ritenuti oggi più rilevanti nella biologia dell’organo adiposo è costituito dal ruolo esercitato dal nu-mero e dall’attività ossidativa dei mitocondri, le cui alterazio-ni sono ritenute cruciali nella genesi dell’insulino-resistenza, nello sviluppo dell’obesità e della sua progressione verso il
diabete di tipo 2. Tuttavia, se la disfunzione mitocondriale nel tessuto adiposo preceda o sia conseguenza dell’obesità è an-cora oggetto di dibattito. Numerosi studi, condotti sia in mo-delli animali che nell’uomo, hanno riportato un’associazione tra la presenza di insulino-resistenza e riduzione della massa e della funzione ossidativa mitocondriali, e numerosi interventi terapeutici sullo stile di vita o farmacologici, che producono una riduzione del peso e un miglioramento della sensibilità all’insulina, sono spesso accompagnati da un incremento della funzione mitocondriale e della termogenesi.
Uno degli aspetti più controversi è se il danno mitocon-driale preceda o sia secondario allo sviluppo dell’obesità, sostenendo la secondarietà del danno cellulare periferico rispetto a determinanti genetici prevalentemente centrali. A sostegno di questa ipotesi sono i risultati ottenuti in gemel-li monozigoti discordanti per fenotipo ovvero con lo stesso patrimonio genetico, ma l’uno obeso e l’altro normopeso. In questo studio rispetto ai gemelli magri, nei gemelli obesi è emersa una ridotta espressione dei fattori che regolano l’atti-vità ossidativa mitocondriale associata a ridotti livelli di DNA mitocondriale. Infine, rispetto ai magri, nei gemelli obesi è emerso un maggiore livello di metilazione di PGC1a, danno epigenetico inattivante, che riduce di conseguenza l’attività
Fig. 2 Rappresentazione schematica del concetto di adiposopatia, con l’elenco delle principali alterazioni patologiche presenti nell’organo adiposo di un soggetto obeso.
OBESITÀ SOTTOCUTANEATessuto adiposo “sano”
Profilo metabolico nella norma Profilo metabolico alterato
↑ grasso epatico e funzionalità alterata
Scarso grasso epatico e funzionalità nella norma
Scarso grasso epicardico
Scarso grasso muscolare
↑ grasso epicardico
↑ grasso muscolare (↑lipidi intracellulari)
Rilascio di adipochine alterato
Metabolismo degli acidi grassi
liberi alterato
OBESITÀ VISCERALETessuto adiposo “disfunzionale”
Assenza di tessuto adiposo ectopico Iperafflusso lipidico,tessuto adiposo ectopico
1. Obesità viscerale
2. Transdifferenziazione del tessuto adiposo bruno in bianco
3. Aumento del volume della cellula adiposa
4. Ridotta sensibilità all’insulina delle cellule adipose
5. Incapacità del tessuto adiposo di compiere la funzione di deposito energetico
6. Lipotossicità periferica
7. Variazioni quali- e quantitative della secrezione delle adipochine
8. Danno della funzione e della biogenesi mitocondriale
9. Infiammazione e infiltrazione macrofagica
10. Anomalie delle cellule staminali
11. Fibrosi
12. Alterazioni dell’angiogenesi

S35
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
di questo fattore trascrizionale sulla biogenesi mitocondriale. Questi dati sottolineano con forza che un aumento dell’at-tività mitocondriale nel tessuto adiposo sottocutaneo possa condurre a un miglioramento dei danni metabolici prodotti dall’obesità [33].
Sono peraltro proprio gli studi su geni candidati in relazio-ne allo sviluppo dell’obesità, con cui la teoria di una genesi esclusivamente centrale della malattia può essere confutata. L’obesità appare infatti frequentemente legata a varianti po-limorfiche di differenti geni, espressi o agenti a livello delle strutture del SNC implicate nella regolazione del controllo dell’appetito e della spesa energetica. Tra questi, sicuramente il gene FTO presenta la più forte associazione con l’obesità. È stato riconosciuto, mediante un approccio di studio geno-me-wide, che la principale variante di rischio è rappresentata dall’allele rs1421085 che presenta una variante polimorfica di un singolo nucleotide (T–C), che produce una alterazione su una sequenza intronica che interagisce con il gene repres-sore ARID5B, producendo una derepressione di un attivatore preadipocitario risultante in un marcato incremento dell’e-spressione dei geni IRX3 e IRX5 durante l’adipogenesi. La presenza dell’allele di rischio rs1421085 del gene FTO asso-ciato all’obesità determina una repressione della termogenesi mitocondriale. La manipolazione genetica della pathway in-dividuata, attraverso knockdown o iperespressione del rego-latore ARID5B, la modifica genica della variante rs1421085 ed il knockdown o l’iperespressione dei geni bersaglio IRX3 e IRX5, ha mostrato un importante effetto sul controllo del fe-notipo cellulare. Nel tessuto adiposo murino, il silenziamento di IRX3 ha determinato una riduzione del peso corporeo e una maggiore dissipazione di energia senza variazioni dell’atti-vità fisica o appetito. Questo studio è di fondamentale im-portanza perché assegna un ruolo cruciale al controllo della termogenesi adipocitaria nello sviluppo dell’obesità. L’idea di un intervento di terapia genica mediante un editing gene-tico su adipociti umani non appare così lontano dall’essere proposto come una valida strategia terapeutica [34].
Obesità e infiammazione cronica di basso grado
Le modificazioni morfologiche e metaboliche che avvengo-no a livello del tessuto adiposo si accompagnano a maggio-re espressione di citochine ad azione proinfiammatoria, quali ad esempio interleuchina-6 (IL-6) e il tumor necrosis factor-alpha (TNF-a), e a ridotta produzione di molecole antinfiam-matorie, come l’adiponectina [35], con comparsa di uno stato di infiammazione cronica di basso grado, che sembra avere un ruolo fisiopatologico importante nello sviluppo e nella pro-gressione delle complicanze legate all’obesità. I fattori d’in-nesco di questa condizione non sono stati ancora totalmente
compresi, ma sembrano coinvolgere l’ipossia [36], lo stress reticolo-endoplasmatico a seguito di un eccessivo carico di li-pidi intracellulari [37], la lipotossicità [38] e l’attivazione dei toll-like receptors mediata dagli acidi grassi liberi (FFA) [39].Lo stato di ipossia che si verrebbe a sviluppare nel tessuto adiposo del soggetto obeso è uno dei principali meccanismi proposti [35]. All’aumentare delle dimensioni dei deposi-ti adiposi, l’ossigenazione e la vascolarizzazione tissutale e dell’adipocita diventano insufficienti [40]. La cellula adipo-sa aumenta di dimensioni raggiungendo un diametro massi-mo stimato di 140-180 μm, definito come dimensione critica, che è geneticamente determinato e specifico per ogni depo-sito [41]. Tale accrescimento arriverebbe a superare il limite di diffusibilità dell’ossigeno all’interno delle cellule (stimato di circa 100-200 μm), creando uno stato di ipossia cellulare che determinerebbe una condizione di stress per il reticolo en-doplasmatico [41]. Nel complesso questi fattori stimolano gli adipociti a secernere in maniera anormale adipochine e cito-chine pro-infiammatorie, tra cui il TNF-a [42] e il monocyte chemo attractant protein-1 (MCP-1) [43], che a loro volta esercitano la loro azione sulle cellule immunitarie residenti nel tessuto adiposo, con alterazione nel numero e nel fenotipo di tali cellule. Si genera di conseguenza un quadro flogistico in cui i macrofagi circondano gli adipociti morti in strutture a forma di corona, proprie dell’infiammazione cronica [44,45]. A loro volta, i leucociti attivati secernono ulteriori citochine pro-infiammatorie, creando un circolo vizioso che perpetua lo stato di infiammazione cronica di basso grado e la disfunzione del tessuto adiposo, nel cui contesto la produzione di adipo-chine è alterata e contribuisce alle complicanze dell’obesità.
Un’altra importante periferia: il tratto gastrointestinale
In tempi relativamente recenti è divenuta evidente l’impor-tanza del ruolo svolto dall’asse neuroendocrino intestino-cervello, e in particolare di alcuni ormoni gastrointestinali tra cui sicuramente il più importante è il glucagon-like pep-tide-1 (GLP-1). Questo ormone è un potente regolatore dello svuotamento gastrico, ma questo effetto è soggetto a rapida tachifilassi dopo stimolazione continuata [46]. Si ritiene che il GLP-1 circolante possa agire come un fisiologico fattore di sazietà [47]. A livello del SNC, GLP-1 è un neurotrasmet-titore nelle vie coinvolte nella sazietà [48]. Gli analoghi del GLP-1, come liraglutide, sono in grado di attraversare la barriera ematoencefalica, anche se non è noto in dettaglio attraverso quali meccanismi e in quali aree agiscano [49]. Recentemente è stato dimostrato che nell’animale la sommi-nistrazione periferica di liraglutide è in grado di interagire con i recettori per il GLP-1 localizzati nel nucleo arcuato, attivando direttamente i neuroni POMC/CART (cocain- and

S36
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
amphetamine-regulated transcript) e inibendo indirettamente i neuroni NPY/AgRP tramite attivazione dei neuroni GABA. È stato ipotizzato che l’agonista periferico del GLP-1 rag-giunga il SNC tramite trasporto GLP-1r mediato in aree cir-cumventricolari e forse in altre zone dell’ipotalamo attraverso capillari fenestrati [50].
Il dialogo con il diverso dal sé genomico: ruolo della flora batterica intestinale
Nel tratto gastrointestinale umano è presente una popolazio-ne microbica detta “microbiota gastrointestinale”. Esso è co-stituito da un’enorme quantità di microrganismi che supera di gran lunga il numero delle cellule dell’organismo ospite. L’intestino è colonizzato sin dalla nascita da una microflora che cresce al crescere dell’età e che è peculiare di ogni indivi-duo in dipendenza dei suoi comportamenti alimentari, lo stile di vita, le eventuali malattie e le conseguenti terapie.
Lo sviluppo di questa flora è sicuramente determinato dall’alimentazione del neonato che se allattato al seno mo-stra una prevalenza di Bifidobacterium, con una un’inibizione della flora putrefattiva a vantaggio di quella fermentante. Nei neonati nutriti invece con formule lattee specifiche, la flora microbica si presenta mista e complessa, molto simile a quel-la di un soggetto adulto, ovvero contenente Bifidobatteri, En-terobatteri, Lattobacilli, Bacteroides, Clostridi, Enterococchi e Streptococchi [51]. L’iniziale colonizzazione risulta sicura-mente importante per definire la flora batterica definitiva in età adulta, infatti, una volta costituitasi, essa rimane stabile ad eccezione di possibili variazioni a seguito di diversi fattori di varia natura come ad esempio un cambiamento delle abitudi-ni alimentari o l’insorgenza di patologie.
Nell’adulto la maggior parte dei batteri d’origine fecale ap-partiene a due maggiori famiglie: Bacteroidetes e Firmicutes.
I primi costituiscono circa il 25% del microbiota intestina-le. Il Bacteroidetes thetaiotaomicron è il principale compo-nente della normale flora batterica intestinale; ha una grande capacità di digestione dei polisaccaridi in quanto presenta due proteine di membrana che legano ed importano amido, 226 glicosidasi contro le 98 dell’uomo, 64 polisaccaridasi contro una dell’uomo. Il Bacteroidetes thetaiotaomicron ha svilup-pato la capacità di favorire nell’organismo ospite l’utilizzo di molti carboidrati della dieta, otre ai glicani del muco [52]. I Bacteroidetes possiedono inoltre un complesso sistema per metabolizzare i carboidrati inutilizzati dall’organismo ospite.
I Firmicutes sono suddivisi in tre classi: Clostridi, Bacilli e Mollicuti. I Clostridi sono anaerobi obbligati in grado di produrre endospore; se coltivati in ambiente con basso po-tenziale redox sono estremamente attivi dal punto di vista fermentativo: metabolizzano sostanze organiche (carboidra-
ti e proteine) con produzione di alcoli, acido acetico, acido butirrico, acido succinico e sostanze volatili come anidride carbonica, idrogeno e acido solfidrico.
La microflora è in grado di influenzare il bilancio energeti-co dell’organismo ospite, come dimostrato da diversi studi su animali germ-free in cui è richiesto un 30% in più di energia nella normale dieta per mantenere il peso ideale [53]. I batteri intestinali traggono l’energia necessaria attraverso il metabo-lismo di zuccheri e proteine (processo noto col termine di fer-mentazione). La trasformazione di polisaccaridi non digeribili della dieta (cellulosa, pectine, amido non digeribile) avviene a opera di enzimi batterici che trasformano il materiale derivante dagli alimenti in sostanze volatili (anidride carbonica, idrogeno solforato etc.) e acidi grassi a corta catena (SCFA) quali l’acido acetico, che viene riassorbito dalla parete intestinale, riversato nel circolo periferico e utilizzato come substrato energetico dai vari tessuti tra cui il tessuto adiposo per la lipogenesi, l’acido butirrico e l’acido propionico che viene riassorbito a livello in-testinale e utilizzato dal fegato come substrato gluconeogeni-co. La fermentazione saccarolitica, ovvero la sintesi degli acidi grassi, determina un’acidificazione del pH intestinale che rap-presenta un efficiente sistema di difesa contro i microrganismi patogeni. La produzione di SCFA, inoltre, consente la crescita delle cellule intestinali epiteliali, favorendone la proliferazione e la differenziazione [54]. La flora batterica intestinale rappre-senta uno stimolo per lo sviluppo e la regolazione del sistema immunitario dell’ospite e inoltre costituisce una barriera fisi-ca, insieme alla produzione di sostanze antimicrobiche come le batteriocine, determinando un efficace sistema di difesa nei confronti della flora patogena.
Il microbiota intestinale interviene in fondamentali processi quali il bilancio energetico e il controllo della risposta immu-nitaria e flogistica. Esso facilita inoltre l’estrazione di calorie dagli alimenti, favorendo l’afflusso di acidi grassi nel tessu-to adiposo e nello stesso tempo fornendo energia e nutrien-ti per la stessa crescita microbica. Le differenze individuali nel recupero dell’energia da parte del microbiota intestinale hanno suggerito una spiegazione fisiopatologica dell’obesità in soggetti che non assumono elevate quantità di alimenti. Molti studi, infatti, hanno dimostrato che la flora intestinale di ciascun individuo possiede una propria efficienza metabolica. Quest’ultima sarebbe correlata a una diversa composizione del microbiota stesso. A Bäckhed [55] va il merito di aver sug-gerito, per primo, una possibile relazione tra la composizione della flora microbica intestinale e l’obesità, focalizzando l’at-tenzione sulle proporzioni delle due principali divisioni batte-riche componenti il microbiota: i Firmicutes e i Bacteroidetes, con una prevalenza dei primi rispetto ai secondi nel soggetto obeso. Questo fenotipo obeso è incredibilmente trasmissibile perché la colonizzazione di topi germ-free con una flora bat-terica di derivazione obesa determina un aumento del grasso

S37
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
totale pari al 60% in più rispetto a topi colonizzati con la flora batterica di un topo normopeso. L’aumento di tessuto adipo-so, inoltre, avviene indipendentemente dalla quantità di flora batterica acquisita o dalla diminuzione del dispendio energeti-co [56]. La flora batterica fecale di soggetti obesi sottoposti ad un programma di riduzione del peso corporeo subisce notevoli variazioni. Prima dell’inizio della dieta ipocalorica, ristretta in grassi e carboidrati, i pazienti mostravano elevati livelli di Fir-micutes. Al termine dello studio si è verificato un aumento nel numero di Bacteroidetes, dal 3% iniziale al 15% finale [57]. Non è stato ancora chiarito il motivo per cui questo avviene, probabilmente la grande varietà di specie presenti in questo phylum contribuisce a una maggiore efficienza nell’estrazione di energia a livello intestinale.
Da quanto riportato emerge come, da un lato, l’obesità sia caratterizzata da un microbiota differente rispetto al normale e, dall’altro, lo stesso microbiota, insieme al genotipo dell’o-spite e al suo stile di vita, potrebbe contribuire allo sviluppo di questa disfunzione metabolica.
Quindi: 1. l’ambiente influenza in maniera rilevante il fenotipo;2. l’obesità origina sicuramente da un apporto calorico su-
periore rispetto alla spesa energetica, ma la riduzione dell’output calorico (sedentarietà) gioca un ruolo almeno paragonabile all’incremento dell’assunzione di calorie;
3. l’impatto genetico spiega perlopiù le sindromi caratteriz-zate da obesità iperfagica. Una delle più recenti acquisizio-ni dell’impatto di alcune varianti genetiche del gene FTO mostra che la sua azione non si esercita esclusivamente a livello del SNC, ma anche a livello del tessuto adiposo;
4. la mancanza della leptina (ormone periferico) o del suo recettore determina un fenotipo obeso;
5. l’adiposopatia definisce lo stato di vera malattia: è tempo di passare da una definizione solo quantitativa a una defi-nizione qualitativa della malattia;
6. l’alterazione del dialogo tra muscolo e tessuto adiposo è fondamentale per contrastare la genesi dell’obesità e la sua progressione verso il diabete di tipo 2;
7. gli ormoni prodotti dal tratto gastrointestinale non solo potenziano la secrezione insulinica in risposta al pasto (azione incretinica), ma hanno un impatto rilevante sulla regolazione del bilancio energetico;
8. la flora batterica intestinale influenza in maniera rilevante molti processi metabolici;
9. l’efficienza della macchina ossidativa mitocondriale è essenziale per contrastare lo sviluppo dell’obesità e del diabete di tipo 2;
10. il mantenimento di un tessuto adiposo bruno è il prerequi-sito per contrastare lo sviluppo dell’obesità, il manteni-mento di un tessuto adiposo bianco sano è il prerequisito per contrastare lo sviluppo delle sue complicanze.
In conclusione, l’obesità è una malattia complessa, a gene-si complessa in cerca di una migliore definizione fisiopatolo-gica, clinica e nosografica. L’influenza di meccanismi centrali e periferici nel governo della spesa energetica e nella genesi ed evoluzione della malattia è senza dubbio secondaria all’in-terruzione del dialogo tra di essi e ciò è sicuramente la condi-tio sine qua non per lo sviluppo della patologia.
Bibliografia
1. Blüher M, Mantzoros CS (2015). From leptin to other adipokines in health and disease: facts and expectations at the beginning of the 21st century. Metabolism 64:131-45
2. Bartness TJ, Vaughan CH, Song CK (2010). Sympathetic and senso-ry innervation of brown adipose tissue. Int J Obes (Lond) 34 Suppl 1:S36-42
3. Cannon B, Hedin A, Nedergaard J (1982). Exclusive occur-rence of thermogenin antigen in brown adipose tissue. FEBS Lett 150(1):129-32
4. Rial E, Gonzalez-Barroso MM (2001). Physiological regulation of the transport activity in the uncoupling proteins UCP1 and UCP2. Biochim Biophys Acta 1504(1):70-81
5. Lepper C, Fan CM (2010). Inducible lineage tracing of Pax7-de-scendant cells reveals embryonic origin of adult satellite cells. Gen-esis 48(7):424-36
6. Seale P, Bjork B, Yang W et al (2008). PRDM16 controls a brown fat/skeletal muscle switch. Nature 454(7207):961-7
7. Rajakumari S, Wu J, Ishibashi J et al (2013). EBF2 determines and maintains brown adipocyte identity. Cell Metab 17(4):562-74
8. Giguere V (2008). Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev 29(6):677-96
9. Seale P, Kajimura S, Yang W et al (2007). Transcriptional control of brown fat determination by PRDM16. Cell Metab 6(1):38-54
10. Li S, Yu Q, Wang GX, Lin JD (2013). The biological clock is reg-ulated by adrenergic signaling in brown fat but is dispensable for cold-induced thermogenesis. PLoS One 8(8):e70109
11. Cypess AM, Lehman S, Williams G et al (2009). Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360(15):1509-17
12. Cypess AM, White AP, Vernochet C et al (2013). Anatomical local-ization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med 19(5):635-9
13. Orava J, Nuutila P, Lidell ME et al (2011). Different metabolic re-sponses of human brown adipose tissue to activation by cold and insulin. Cell Metab 14(2):272-9
14. Virtanen KA, Lidell ME, Orava J et al (2009). Functional brown adipose tissue in healthy adults. N Engl J Med 360(15):1518-25
15. Vijgen GH, Bouvy ND, Teule GJ et al (2011). Brown adipose tissue in morbidly obese subjects. PLoS One 6(2):e17247
16. Ouellet V, Routhier-Labadie A, Bellemare W et al (2011). Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-de-tected BAT in humans. J Clin Endocrinol Metab 96(1):192-9
17. Loncar D, Bedrica L, Mayer J et al (1986). The effect of intermittent cold treatment on the adipose tissue of the cat. Apparent transfor-mation from white to brown adipose tissue. J Ultrastruct Mol Struct Res 97(1-3):119-29
18. Young P, Arch JR, Ashwell M (1984). Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett 167(1):10-4
19. Cousin B, Cinti S, Morroni M et al (1992). Occurrence of brown

S38
123
Intern Emerg Med (2016) 11 (Suppl):S31-S38
adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci 103 (Pt 4):931-42
20. Rosen ED, Spiegelman BM (2014). What we talk about when we talk about fat. Cell 156(1-2):20-44
21. Himms-Hagen J, Melnyk A, Zingaretti MC et al (2000). Multilocu-lar fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol 279(3):C670-81
22. Vitali A, Murano I, Zingaretti MC et al (2012). The adipose organ of obesity-prone C57BL/6J mice is composed of mixed white and brown adipocytes. J Lipid Res 53(4):619-29
23. Sharp LZ, Shinoda K, Ohno H et al (2012). Human BAT possess-es molecular signatures that resemble beige/brite cells. PLoS One 7(11):e49452
24. Walden TB, Hansen IR, Timmons JA et al (2011). Recruited vs non-recruited molecular signatures of brown, “brite”, and white adipose tissues. Am J Physiol Endocrinol Metab 302(1):E19-31
25. Ohno H, Shinoda K, Spiegelman BM, Kajimura S (2012). PPAR-gamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab 15(3):395-404
26. Fisher FM, Kleiner S, Douris N et al (2012). FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive ther-mogenesis. Genes Dev 26(3):271-81
27. Villarroya F, Vidal-Puig A (2013). Beyond the sympathetic tone: the new brown fat activators. Cell Metab 17(5):638-43
28. Elsen M, Raschke S, Tennagels N et al (2013). BMP4 and BMP7 in-duce the white-to-brown transition of primary human adipose stem cells. Am J Physiol Cell Physiol 306(5):C431-40
29. Bostrom P, Wu J, Jedrychowski MP et al (2012). A PGC1-alpha-de-pendent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481(7382):463-8
30. Karbiener M, Pisani DF, Frontini A et al (2014). MicroRNA-26 family is required for human adipogenesis and drives characteristics of brown adipocytes. Stem Cells 32(6):1578-90
31. Trevellin E, Scorzeto M, Olivieri M et al (2014). Exercise training induces mitochondrial biogenesis and glucose uptake in subcutane-ous adipose tissue through eNOS-dependent mechanisms. Diabetes 63(8):2800-11
32. Stanford KI, Middelbeek RJ, Goodyear LJ (2015). Exercise effects on white adipose tissue: beiging and metabolic adaptations. Diabe-tes Jul;64(7):2361-8
33. Heinonen S, Buzkova J, Muniandy M et al (2015). Impaired mito-chondrial biogenesis in adipose tissue in acquired obesity. Diabetes 64(9):3135-45
34. Claussnitzer M, Dankel SN, Kim KH et al (2015). FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med 373(10):895-907
35. Arita Y, Kihara S, Ouchi N et al (1999). Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79-83
36. Halberg N, Khan T, Trujillo ME et al (2009). Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29:4467-83
37. Ozcan U, Cao Q, Yilmaz E et al (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457-61
38. Unger RH, Scherer PE (2010). Gluttony, sloth and the metabolic syn-drome: a roadmap to lipotoxicity. Trends Endocrinol Metab 21:345-52
39. Shi H, Kokoeva MV, Inouye K et al (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015-25
40. Wood IS, de Heredia FP, Wang B, Trayhurn P (2009). Cellular hypoxia and adipose tissue dysfunction in obesity. Proc Nutr Soc 68:370-7
41. Hosogai N, Fukuhara A, Oshima K et al (2007). Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56:901-11
42. Hotamisligil GS, Shargill NS, Spiegelman BM (1993). Adipose ex-pression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87-91
43. Kanda H, Tateya S, Tamori Y et al (2006). MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116:1494-505
44. Cinti S, Mitchell G, Barbatelli G et al (2005). Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46:2347-55
45. Weisberg SP, McCann D, Desai M et al (2003). Obesity is associ-ated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796-808
46. Nauck MA, Kemmeries G, Holst JJ, Meier JJ (2011). Rapid tachy-phylaxis of the glucagon-like peptide 1-induced deceleration of gas-tric emptying in humans. Diabetes 60(5):1561-5
47. Williams DL, Baskin DG, Schwartz MW (2009). Evidence that in-testinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 150(4):1680-7
48. Turton MD, O’Shea D, Gunn I et al (1996). A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379(6560):69-72
49. Kastin AJ, Akerstrom V, Pan W (2002). Interactions of gluca-gon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neu-rosci 18(1-2):7-14
50. Secher A, Jelsing J, Baquero AF et al (2014). The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest 124(10): 4473-88
51. Matsuki T, Watanabe K, Tanaka R et al (1999). Distribution of bi-fidobacterial species in human intestinal microflora examined with 16S rRNA-gene-targeted species-specific primers. Appl Environ Microbiol 65(10):4506-12
52. Bäckhed F, Ley RE, Sonnenburg JL et al (2005). Host-bacterial mu-tualism in the human intestine. Science 307(5717):1915-20
53. Hooper LV, Midtvedt T, Gordon JI (2002). How host-microbial in-teractions shape the nutrient environment of the mammalian intes-tine. Annu Rev Nutr 22:283-307
54. Guarner F (2006). Enteric flora in health and disease. Digestion 73 Suppl 1:5-12
55. Bäckhed F, Ding H, Wang T et al (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101(44):15718-23
56. Tilg H, Moschen AR, Kaser A (2009). Obesity and the microbiota. Gastroenterology 136(5):1476-83
57. Ley RE, Bäckhed F, Turnbaugh P et al (2005). Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102(31):11070-5

© SIMI 2016
123
MEDICINA IN CORSIA
Intern Emerg Med (2016) 11 (Suppl):S39-S42
Fabrizio Fabris (•)Università di Padova, Dipartimento di Medicina-DIMEDVia Giustiniani 2, 35128 Padova E-mail: [email protected]
I. Di PasqualeUniversità di Padova, Dipartimento di Medicina-DIMED, Padova
La medicina perioperatoria: l’interazione medico-chirurgo nella gestione del rischio
Fabrizio Fabris • Irene Di Pasquale
Il metodo di valutazione clinica preoperatoria è in fase di evoluzione in considerazione dei progressi della chirurgia e delle tecniche di anestesia, dei cambiamenti demografici e di un approccio più individualizzato del paziente [1]. Il cam-biamento dei modelli organizzativi ha portato ad un deciso accorciamento dei tempi di degenza pre e postoperatoria ren-dendo ancora più indispensabile l’interazione tra internista, anestesista e chirurgo nella determinazione del rischio opera-torio dei pazienti.
L’inquadramento clinico resta senza dubbio importante associato ad uno screening di laboratorio sulla base delle co-morbilità di ogni singolo paziente [2]: – emocromo, – funzionalità renale,– test di gravidanza in tutte le donne in età fertile [3], – ECG in pazienti sottoposti a chirurgia a rischio interme-
dio-alto, – Rx torace nei pazienti di età > 60 anni.
Nessun test di laboratorio deve essere ripetuto se i risultati sono normali nei 4 mesi precedenti la chirurgia, salvo novità cliniche.
Per quanto riguarda il paziente geriatrico con età > 65 anni, la valutazione preoperatoria pone attenzione anche al consenso informato, che sottintende la stima della capacità decisionale del paziente, lo status mentale, lo stato nutrizio-nale, le patologie preesistenti cardiovascolari, renali, metabo-liche e neurologiche [4].
Valutazione ematologica
Una valutazione efficace dell’anemia nella fase preoperatoria si basa su anamnesi, esame fisico e un approccio sistematico alle indagini successive. La presenza di anemia, così come l’uso di trasfusioni perioperatorie, ha potenziali conseguenze sull’outcome chirurgico [5].
La raccolta anamnestica dovrebbe indagare una preceden-te storia di anemia, il tipo di esordio, i valori precedenti di emoglobina ed eventuali terapie quali precedenti trasfusioni di sangue, splenectomia e donazioni di sangue. L’anemia può essere trattata agendo sulle cause reversibili laddove possibi-le, o mediante trasfusione. Le linee guida dell’American So-ciety of Anesthesiology (ASA) raccomandano la trasfusione se il livello di emoglobina (Hb) è < 6 g/dl, mentre la trasfu-sione è raramente necessaria quando il livello è > 10 g/dl. Quando la concentrazione di Hb è compresa tra 6 e 10 g/dl, la decisione di trasfondere dovrebbe essere basata sulla presen-za di ischemia d’organo.
Per quanto riguarda gli aspetti coagulativi, è stato condotto un recente studio retrospettivo su 316.644 pazienti sottoposti a chirurgia non cardiaca, che non avevano indicazioni cliniche ad eseguire test di coagulazione preoperatori. I pazienti con piastrinopenia allo screening preoperatorio sono associati a un più alto tasso di trasfusione di sangue e a una mortalità tanto maggiore quanto più grave è l’entità della trombocitopenia [6].
Le coagulopatie, congenite e acquisite, richiedono una valutazione ematologica specialistica per la preparazione di protocolli di copertura coagulativa individuali, definiti sulla base del difetto e in considerazione del tipo di intervento chi-rurgico.
Valutazione dell’insufficienza renale
Una ridotta funzionalità renale, solitamente valutata sulla base dei livelli di creatinina sierica, rappresenta un fattore

S40
123
Intern Emerg Med (2016) 11 (Suppl):S39-S42
di rischio indipendente per outcome sfavorevole; una valu-tazione più accurata può essere ottenuta tramite la stima del-la clearance. Un aumento dei livelli di creatininemia correla con un aumento della mortalità perioperatoria; una riduzione della clearance della creatinina di 10 ml/min è associata ad un aumento del 40% del rischio di mortalità postoperatoria e un suo deterioramento postoperatorio rappresenta un fattore prognostico sfavorevole a lungo termine [7,8].
Nei pazienti a rischio di sviluppare nefropatia da mezzo di contrasto, si raccomanda, a scopo profilattico, di idratare il paziente con soluzione fisiologica a base di cloruro di sodio, in associazione o meno ad N-acetilcisteina, prima dell’esame con mezzo di contrasto. Infine, i pazienti con insufficienza re-nale richiedono un aggiustamento della dose di molti farmaci.
Valutazione del paziente diabetico
Il diabete determina un aumento della morbilità ospedaliera ed un allungamento della degenza con conseguente aumen-to dei costi. Le ragioni sono multifattoriali e comprendono l’aumentato rischio ipo e iperglicemico, la presenza di co-morbilità multiple, la presenza di complicanze croniche del diabete, la complessità della terapia compresi i rischi della terapia insulinica, le infezioni [9,10].
Gli obiettivi del controllo glicemico preoperatorio sono: ridurre la mortalità e la morbilità a breve e a lungo termine, evitare l’iperglicemia, la chetoacidosi, l’ipoglicemia. In caso di intervento chirurgico programmato, è necessaria un’attenta valutazione preoperatoria del compenso glicemico, dello sta-to delle complicanze e della necessità di modificare la terapia ipoglicemizzante [11]. È consigliabile inoltre programmare l’intervento al mattino per non interferire sul controllo glice-mico, mantenere il regime alimentare ipoglucidico abituale, organizzare il monitoraggio della glicemia in rapporto alle necessità del paziente, istruire il personale alla gestione di eventuali crisi ipoglicemiche o iperglicemiche.
Valutazione del paziente con patologia cardiovascolare
Le complicanze cardiache correlate alla chirurgia non cardia-ca dipendono sia dai fattori di rischio del paziente sia dal-la natura della procedura chirurgica. I fattori chirurgici che condizionano il rischio cardiovascolare sono legati a urgenza, entità, natura e durata dell’intervento, così come alle varia-zioni della temperatura corporea, alle perdite ematiche e alla dislocazione di liquidi. L’atto chirurgico può anche provocare alterazioni dell’equilibrio tra fattori protrombotici e fibrinoli-tici tali da favorire uno stato di ipercoagulabilità e possibile trombosi coronarica [12,13]. Sotto il profilo del rischio car-
diovascolare, gli interventi chirurgici possono essere raggrup-pati in tre categorie - a basso, intermedio ed alto rischio - a cui corrisponde un’incidenza di eventi cardiaci, a 30 giorni, rispettivamente < 1, 1-5 e > 5%.
La determinazione della capacità funzionale rappresenta uno step fondamentale nella valutazione preoperatoria del ri-schio cardiovascolare. Questa viene misurata in equivalenti metabolici (METs), dove 1 MET corrisponde al tasso meta-bolico basale: salire due piani di scale richiede 4 METs, men-tre un’attività sportiva strenua come il nuoto richiede oltre 10 METs. La difficoltà di salire due piani di scale o percorrere una breve distanza di corsa (4 METs) è indicativa di una scar-sa capacità funzionale, associata ad un’aumentata incidenza di eventi cardiaci postoperatori. Quando la capacità funzio-nale è elevata, la prognosi è eccellente anche in presenza di cardiopatia ischemica stabile o di fattori di rischio [14].
Negli ultimi 30 anni sono stati elaborati diversi indici di ri-schio cardiovascolari, di cui i più noti sono quelli di Goldman (1977), Detsky (1986) e Lee (1999). L’indice di Lee, che di fatto rappresenta una modifica di quello di Goldman, è con-siderato attualmente il miglior indice predittivo di rischio cardiovascolare nella chirurgia non cardiaca. L’indice di Lee comprende 5 determinanti cliniche indipendenti di eventi car-diaci maggiori perioperatori:– storia di cardiopatia ischemica, – storia di vasculopatia cerebrale,– scompenso cardiaco, – diabete mellito insulino-dipendente, – insufficienza renale, nonché un sesto fattore rappresentato dalla chirurgia ad alto rischio [15].
In ambito perioperatorio, i biomarker possono essere utili nella valutazione del rischio suddividendoli in indicatori di ischemia miocardica o danno miocardico, di infiammazione e di funzione del ventricolo sinistro.
Le troponine cardiache T e I [16] forniscono informazioni prognostiche indipendenti e complementari a quelle di altri indicatori cardiaci di rischio importanti (tratto ST e funzione del ventricolo sinistro) validate sia in studi di comunità che in trial clinici [17].
I marker di infiammazione possono contribuire ad identi-ficare quei pazienti con aumentato rischio di instabilità della placca, così il peptide natriuretico cerebrale (BNP) può essere analizzato in casi particolari [18].
Gli esami preoperatori di tipo non invasivo mirano a va-lutare la disfunzione del ventricolo sinistro, l’ischemia mio-cardica e le anomalie valvolari, ciascuno dei quali è un de-terminante maggiore di outcome postoperatorio sfavorevole. L’ECG è parte integrante della valutazione preoperatoria, è predittivo dell’outcome a lungo termine indipendentemente

S41
123
Intern Emerg Med (2016) 11 (Suppl):S39-S42
dai rilievi clinici e dall’ischemia perioperatoria [19] ed è ob-bligatorio in tutti i pazienti.
Il test da sforzo al cicloergometro rappresenta un aiuto nell’ulteriore definizione del rischio: l’insorgenza di una ri-sposta ischemica a bassi carichi di lavoro è associata ad un aumento significativo del rischio di eventi cardiaci periope-ratori e a lungo termine; viceversa, se l’ischemia si manifesta ad elevati carichi di lavoro è associata ad un rischio significa-tivamente inferiore [14].
La scintigrafia miocardica perfusionale o l’ecocardiogra-fia con stress farmacologico sono maggiormente indicate nei pazienti che presentano una limitata capacità all’esercizio e permettono una ulteriore stratificazione preoperatoria del ri-schio operatorio [20].
L’ecocardiografia consente di ottenere un insieme di in-formazioni sulla funzione ventricolare sinistra a riposo, sulle anomalie dell’apparato valvolare e sulla presenza ed esten-sione di ischemia inducibile, e ha un elevato valore predittivo negativo.
Valutazione del paziente con pneumopatia
I fattori di rischio correlati al paziente sono rappresentati da età e obesità, che provoca una riduzione del volume polmo-nare, mismatch ventilo-perfusorio e relativa ipossiemia che si accentua dopo l’intervento chirurgico. Nei casi più gravi l’obesità è associata a ipertensione polmonare, cuore pol-monare e insufficienza respiratoria ipercapnica (sindrome di Pickwick). Altri fattori sono: la presenza di altre comorbilità, il tabagismo attivo, la BPCO, l’asma, l’ipertensione polmona-re [21], le interstiziopatie, la sindrome delle apnee notturne. Nei pazienti che possono avere apnee, la diagnosi deve essere confermata e la gravità deve essere valutata in fase preope-ratoria con uno studio formale del sonno (polisonnografia). I fattori di rischio legati alla procedura includono il sito chirur-gico, la durata dell’intervento, il tipo di anestesia. Gli esami preoperatori includono l’anamnesi, le prove di funzionalità respiratoria con emogasanalisi, che hanno scarso valore pre-dittivo per le complicanze postoperatorie [22] e la radiografia del torace che, pur essendo l’indagine più richiesta prima di un intervento, non è raccomandata in individui sani [23].
Tra i vari score di rischio polmonare elaborati in questi anni, quello più utilizzato è la classificazione ASA basata su criteri clinici semplici e facile da quantificare [24].
Bibliografia
1. Dhesi JK, Swart M (2016). Specialist pre-operative assessment clin-ics. Anaesthesia 71(Suppl 1):3-8
2. Böhmer AB, Wappler F, Zwissler B (2014). Preoperative risk assess-ment—from routine tests to individualized investigation. Deutsch Ärztebl Int 111:437-46
3. American Society of Anesthesiologists Task Force on Preanesthesia Evaluation (2002). Practice advisory for preanesthesia evaluation: a report by the American Society of Anesthesiologists Task Force on Preanesthesia Evaluation. Anesthesiology 96(2):485-96
4. Kim S, Brooks AK, Groban L (2014). Preoperative assessment of the older surgical patient: honing in on geriatric syndromes. Clin Interv Aging 16;10:13-27
5. Shander A (2014). Preoperative anemia and its management. Trans-fus Apher Sci 50(1):13-5
6. Glance LG, Blumberg N, Eaton MP et al (2014). Preoperative thrombocytopenia and postoperative outcomes after noncardiac sur-gery. Anesthesiology 120(1):62-75
7. Kertai MD, Boersma E, Bax JJ et al (2003). Comparison between serum creatinine and creatinine clearance for the prediction of post-operative mortality in patients undergoing major vascular surgery. Clin Nephrol 59(1):17-23
8. Welten GM, Schouten O, Chonchol M et al (2007). Temporary worsening of renal function after aortic surgery is associated with higher long-term mortality. Am J Kidney Dis 50:219-28
9. Bruno A, Gregori D, Caropreso A et al (2008). Normal glucose val-ues are associated with a lower risk of mortality in hospitalized pa-tients. Diabetes Care 31:2209-10
10. Sampson MJ, Brennan C, Dhatariya K et al (2007). A national sur-vey of in-patient diabetes services in the United Kingdom. Diabetic Med 24:643-9
11. Marks JB (2003). Perioperative management of diabete. Am Fam Physic 67:93-100
12. Mangano DT (2004). Perioperative medicine: NHLBI working group deliberations and recommendations. Cardiothorac Vasc Anesth 18:1-6
13. Poldermans D et al; Task Force della Società Europea di Cardiolo-gia (ESC) e della Società Europea di Anestesiologia (ESA) (2010). Linee guida per la valutazione preoperatoria del rischio cardiaco e la gestione perioperatoria del paziente cardiopatico nella chirurgia non cardiaca. G Ital Cardiol 11(10 Suppl 2):e136-e181
14. Morris CK, Ueshima K, Kawaguchi T et al (1991). The prognostic value of exercise capacity: a review of the literature. Am Heart J 122:1423-31
15. Lee TH, Marcantonio ER, Mangione CM et al (1999). Derivation and prospective validation of a simple index for prediction of cardi-ac risk of major noncardiac surgery. Circulation 100:1043-9
16. Maisel AS, Bhalla V, Braunwald E (2006). Cardiac biomarkers: a contemporary status report. Nat Clin Pract Cardiovasc Med 3:24-34
17. Sabatine MS, Morrow DA, Giugliano RP et al (2004). Implica-tions of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Cir-culation 109:874-80
18. Tsimikas S, Willerson JT, Ridker PM (2006). C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol 47(8 Suppl):C19-C31
19. Jeger RV, Probst C, Arsenic R et al (2006). Long-term prognos-tic value of the preoperative 12-lead electrocardiogram before major noncardiac surgery in coronary artery disease. Am Heart J 151:508-13
20. Elhendy A, Valkema R, van Domburg RT et al (1998). Safety of dobutamine-atropine stress myocardial perfusion scintigraphy. J Nucl Med 39:1662-6

S42
123
Intern Emerg Med (2016) 11 (Suppl):S39-S42
21. Sweitzer BJ, Smetana GW (2009). Identification and evaluation of the patient with lung disease. Anesthesiol Clin 27(4):673-86
22. Fisher BW, Majumdar SR, McAlister FA (2002). Predicting pulmo-nary complications after nonthoracic surgery: a systematic review of blinded studies. Am J Med 112(3):219-25
23. Joo HS, Wong J, Naik VN, Savoldelli GL (2005). The value of screening preoperative chest x-rays: a systematic review. Can J Anaesth 52(6):568-74
24. Barnett S, Moonesinghe SR (2011). Clinical risk scores to guide perioperative management. Postgrad Med J 87(1030):535-41

© SIMI 2016
123
MEDICINA IN CORSIA
Intern Emerg Med (2016) 11 (Suppl):S43-S46
Giuseppe Bellelli (•)Dipartimento di Medicina e Chirurgia Università degli Studi Milano-BicoccaClinica Geriatrica AO S. Gerardo, MonzaVia G.B. Pergolesi 33, 20052 MonzaE-mail: [email protected]
Il delirium nei reparti di Medicina Interna
Giuseppe Bellelli
Introduzione
Perché un internista moderno dovrebbe occuparsi di deli-rium? La medicina moderna ci ha insegnato che spesso le patologie acute, in particolare nel soggetto anziano, decor-rono con una sintomatologia atipica, che sfugge ai canoni della semeiotica tradizionale. Non è infrequente, infatti, nel soggetto anziano, che i sintomi “classici” che il medi-co generalmente ricerca quando sospetta una diagnosi siano soltanto parzialmente presenti o del tutto assenti. Si pensi ad esempio all’infarto del miocardio, che solo raramente si presenta con angor irradiato al giugulo e al braccio sinistro, e alla polmonite, che molto spesso decorre senza febbre, tosse e non sempre con dispnea. Tra i sintomi atipici, il delirium è certamente uno dei più frequenti e merita dunque di essere trattato in modo specifico.
La clinica del delirium
Il delirium è un disturbo neuropsichiatrico caratterizzato dall’alterazione di molteplici funzioni cognitive (principal-mente l’attenzione e le funzioni esecutive), scatenato dall’in-sorgenza di un problema clinico acuto (o cronico riacutizza-to), come espressione di una sofferenza metabolica cerebra-le [1]. Il decorso è fluttuante (tipicamente il soggetto affetto da delirium alterna fasi di relativa lucidità a fasi di confusio-ne, specialmente notturne), motivo per cui il riconoscimento della sindrome non è sempre facile. Clinicamente esso può presentarsi in 3 differenti varianti:
1. forma ipercinetica o iperattiva: è caratterizzata da agitazio-ne psicomotoria, aumentata sensibilità agli stimoli esterni, stato di iperallerta; si sviluppano spesso illusioni e alluci-nazioni;
2. forma ipocinetica o ipoattiva: si presenta con rallentamen-to psicomotorio, tendenza all’assopimento, ridotta risposta agli stimoli esterni, apatia;
3. forma mista: in questo caso si registra un’alternanza di fasi di tipo ipercinetico ad altre di tipo ipocinetico.
Il delirium si associa a molteplici outcomes avversi, tra cui un incremento del rischio di sviluppare limitazioni funzionali e disabilità, di essere ricoverati in strutture residenziali e di an-dare incontro a decesso nel breve-medio termine [2-4]. Tanto maggiore è la durata del delirium, tanto minore è la probabili-tà di sopravvivenza nel medio-breve periodo [3]. Studi recenti hanno inoltre dimostrato che il delirium accelera la velocità di progressione del declino cognitivo nelle persone affette da de-menza e ne favorisce la comparsa nei soggetti non dementi [5]. Il rapporto è bidirezionale, per cui tanto maggiore è la gravità del deficit cognitivo, tanto maggiore è il rischio di sviluppare delirium [5]. Il delirium è inoltre associato a un incremento dei costi sanitari, dovuto in larga parte all’aumento delle gior-nate di degenza [6], e a un aumento dello stress del paziente, del personale sanitario e dei caregivers. Parte di questo stress è correlata all’impreparazione del personale e dei caregivers nel-la gestione dei comportamenti del paziente affetto da delirium, e parte è correlata a sentimenti di angoscia che colpiscono que-ste stesse persone quando il proprio congiunto (o il proprio as-sistito) sviluppa tale condizione clinica [7,8].
L’eziologia del delirium
L’eziologia del delirium è generalmente multifattoriale, moti-vo per cui è quasi sempre inefficace l’approccio farmacologico rivolto a un’unica causa. In generale si ritiene che il delirium

S44
123
Intern Emerg Med (2016) 11 (Suppl):S43-S46
insorga per un’interazione fra fattori predisponenti e precipi-tanti. Tali fattori hanno fra loro un rapporto di proporzionalità inversa. In un individuo fragile, quale sovente è un paziente an-ziano ricoverato in ospedale, sarà suffi ciente un insulto di lieve entità (ad esempio un’infezione delle vie urinarie) per scatena-re il delirium. Al contrario, in un individuo “fi t” (non fragile), sarà necessario il concorrere di eventi clinici molteplici e più severi. Tale modello interattivo richiama fortemente il clinico a sviluppare competenze di assessment multidimensionale ge-riatrico, fi nalizzate a cogliere il grado di “fragilità biologica” del soggetto a rischio di sviluppare delirium [9]. Nel lavoro originale di Inouye, che per prima teorizzò il “modello inte-rattivo”, furono individuati quattro fattori predisponenti (defi -cit sensoriale, deterioramento cognitivo, gravità della malattia acuta e disidratazione) e cinque fattori precipitanti (mezzi di contenzione fi sica, malnutrizione, introduzione di tre o più far-maci in corso di degenza, cateterismo vescicale, eventi iatroge-ni di qualunque natura) [10]. Fra i fattori predisponenti vanno annoverati anche precedenti episodi di delirium, disabilità, sto-ria di depressione, attacco ischemico transitorio (TIA) e stro-ke, abuso di alcool, nonché il carico delle comorbilità [4]. Per quanto riguarda i fattori precipitanti, vanno aggiunte all’elenco originario di Inouye le anomalie laboratoristiche, in particola-re quelle che riguardano gli elettroliti, il glucosio, la funzione renale e l’albumina, la ritenzione acuta di urine e la coprostasi. Interventi chirurgici e traumi sono anch’essi da annoverare tra i fattori scatenanti [4]. I farmaci, e soprattutto quelli con attività psicoattiva, rappresentano un altro potenziale fattore di precipi-tazione del delirium. Il delirium farmaco-indotto dipende non soltanto dalle possibili interazioni tra farmaci, ma anche dal fatto che la farmacocinetica e la farmacodinamica nell’anziano sono spesso alterate ed è dunque possibile un effetto iatrogeno pur in presenza di normali concentrazioni ematiche. In gene-rale è opportuno ricordare che tutti i farmaci dotati di attività anticolinergica sono potenzialmente a rischio ma in particolare benzodiazepine e neurolettici sono da evitare il più possibile, come recentemente raccomandato anche dall’American Geria-tric Society, nel paziente anziano fragile [11].
Diagnosi
Il gold standard per la diagnosi è rappresentato dai criteri del Diagnostic and Statistical Manual dell’American Psychiatric Association (DSM), 5a edizione [1], secondo cui il delirium è defi nito dalla coesistenza di 5 criteri: 1. disturbo dell’attenzione (con ridotta capacità di fi ssare,
mantenere e spostare l’attenzione) e dello stato di consa-pevolezza di sé nell’ambiente;
2. insorgenza acuta o subacuta (vi deve essere un cambiamen-to rispetto alle condizioni di base) e decorso fl uttuante;
3. coesistenza di altri defi cit cognitivi (disorientamento tem-poro-spaziale, disturbi del linguaggio, della memoria o della percezione);
4. assenza di severa compromissione del livello di coscienza (ad esempio il coma);
5. evidenza di patologia acuta in atto, intossicazione da far-maci, sindrome da astinenza, esposizione a tossine o mul-tieziologia.
Usando i criteri diagnostici del DSM, è stato dimostrato che la prevalenza di delirium nei reparti medici oscilla fra il 18% e il 35% [4]. Tra i molto anziani (> 80 anni), fi no al 50% può presentare delirium durante la degenza in ospedale [4]. Tali criteri, tuttavia, non sono di facile utilizzo, soprattutto perché possono prestarsi a interpretazioni soggettive. Parimenti anche l’impressione clinica è fallace: a questo proposito un recente studio, basato sui dati del registro REPOSI (comprendente 80 reparti di medicina e geriatria italiani), ha dimostrato che il me-dico di reparto tende a sottostimare ampiamente la prevalen-za del delirium: su una casistica di 2521 pazienti, soltanto nel 2,9% dei casi infatti veniva registrata la diagnosi di delirium in cartella clinica a fronte della coesistenza di disturbi dell’atten-zione, dell’orientamento e della memoria in quasi il 20,0% del-la popolazione [12]! Il problema della sottodiagnosi riguarda soprattutto la variante ipocinetica del delirium, che non attira l’attenzione dello staff, sebbene sia la variante più frequente e associata a una prognosi peggiore [13].
Per una buona accuratezza diagnostica è dunque necessario l’utilizzo di strumenti di screening validati [14]. In letteratura sono state descritte più di ventiquattro scale di valutazione. La più utilizzata è la CAM (Confusion Assessment Method), che in buona parte ricalca i criteri del DSM: per la diagnosi di de-lirium sono richiesti l’insorgenza acuta, l’andamento fl uttuante e un chiaro defi cit attentivo in associazione alla disorganizza-zione del pensiero e/o all’alterato stato di coscienza [15]. La CAM è stata validata in diversi setting e ha un’ottima sensi-bilità (94%) e specifi cità (89%), ma ha il grande limite che la sua somministrazione richiede un training preliminare. A que-sto proposito, uno studio di Inouye ha dimostrato che l’uso di CAM da parte di un team infermieristico non preliminarmente addestrato era in grado di cogliere la presenza di delirium solo nel 31% dei casi reali [16]. Il 4AT è un nuovo strumento di screening (Figura 1), di rapido utilizzo e che non richiede un training particolare, e che quindi può essere facilmente utiliz-zato nella pratica clinica da medici e infermieri [17].
Conclusioni
Il delirium è una sindrome di grande impatto nei reparti di medicina interna e geriatria, in ragione della sua inciden-

S45
123
Intern Emerg Med (2016) 11 (Suppl):S43-S46
Fig. 1 4AT: strumento di screening del delirium.Il 4AT è uno strumento di screening ideato per un assessment rapido del delirium. Gli items 1-3 sono valutati sulla base dell’osservazione del pazi-ente al momento dell’assessment. L’item 4 richiede informazioni da una o più sorgenti (ad es. la vostra conoscenza del paziente, altri membri dello staff, relazioni del medico di famiglia, caregivers etc.). L’esaminatore dovrebbe tenere in conto la presenza di defi cit della comunicazione (ipoacusia, afasia, barriere linguistiche) nel portare a termine il test ed interpretare lo score. Allerta: un alterato stato di allerta è molto indicativo di delirium in ambito ospedaliero. Se il paziente mostra variazioni notevoli dell’allerta durante la valutazione, cerchiate lo score 4. Cambiamento acuto o decorso fl uttuante: Una fl uttuazione dello stato cognitivo può avvenire anche in assenza di delirium nei pazienti affetti da demenza, ma una fl uttuazione marcata generalmente indica delirium. Per elicitare la presenza di allucinazioni o sintomi psicotici porre al paziente domande tipo: “È preoccupato/a di qualcosa in questo momento?”; “Si sente spaventato da qualcosa o qualcuno?”; “Ha sentito/visto qualcosa di strano qui?”. Generalmente i sintomi psicotici in ambiente ospedaliero rifl ettono più frequentemente delirium che non una patologia psichiatrica funzionale (come la schizofrenia).
4AT
Nome del paziente: Data di nascita: Numero del paziente:
Data: Compilatore:
Cerchiare la risposta [1] ALLERTA Riguarda pazienti che possono essere considerati in stato soporoso (per esempio pazienti per il quale sia difficile svegliarsi e/o che sono evidentemente soporosi durante questo test) oppure agitati/iperattivi. Osservare il paziente. Se dorme, provare a svegliarlo, parlandogli, o con un leggero tocco sulla spalla. Chiedere ai pazienti di dichiarare il proprio nome e l’indirizzo della propria abitazione per valutare il livello di collaborazione Normale (completamente attento, ma non agitato durante tutta la valutazione) 0
Moderata sonnolenza per meno di 10 secondi dopo il risveglio, poi normale 0 Livello di vigilanza evidentemente anomalo 4
[2] AMT4 Età, data di nascita, luogo (nome dell’ospedale e dell’edificio), anno corrente
Nessun errore 0
1 errore 1 2 o più errori / non è possibile somministrare il test al paziente 2
[3] ATTENZIONE Chiedere al paziente: “per favore, mi dica i mesi dell’anno in ordine contrario, partendo da dicembre” Per aiutare la comprensione della domanda, è consentito inizialmente un suggerimento come: “qual è il mese prima di dicembre?” Mesi dell’anno al contrario nomina senza errori 7 mesi o più 0
inizia, ma nomina meno di 7 mesi / si rifiuta di iniziare 1 test non effettuabile (poiché il paziente è indisposto, assonnato o disattento) 2
[4] ACUTO CAMBIAMENTO O DECORSO FLUTTUANTE Dimostrazione di un evidente cambiamento o di un andamento fluttuante nei seguenti domini: attenzione, comprensione o altre funzioni mentali (ad esempio ossessioni e/o allucinazioni) che sono comparse nelle ultime 2 settimane e che sono ancora presenti nelle ultime 24 ore No 0 Sì 4
4 o più: possibile delirium +/− deterioramento cognitivo 1-3: possibile deterioramento cognitivo 0: improbabile il delirium e/o deterioramento cognitivo (ma il delirium può essere presente se il punto 4 è incompleto)
PUNTEGGIO 4AT
Il 4AT è uno strumento di screening ideato per un assessment rapido del delirium. Gli items 1-3 sono valutati sulla base dell’osservazione del paziente al momento dell’assessment. L’item 4 richiede informazioni da una o più sorgenti (ad es. la vostra conoscenza del paziente, altri membri dello staff, relazioni del medico di famiglia, caregivers etc.). L’esaminatore dovrebbe tenere in conto la presenza di deficit della comunicazione (ipoacusia, afasia, barriere linguistiche) nel portare a termine il test ed interpretare lo score. Allerta: un alterato stato di allerta è molto indicativo di delirium in ambito ospedaliero. Se il paziente mostra variazioni notevoli dell’allerta durante la valutazione, cerchiate lo score 4. Cambiamento acuto o decorso fluttuante: Una fluttuazione dello stato cognitivo può avvenire anche in assenza di delirium nei pazienti affetti da demenza, ma una fluttuazione marcata generalmente indica delirium. Per elicitare la presenza di allucinazioni o sintomi psicotici porre al paziente domande tipo: “È preoccupato/a di qualcosa in questo momento?”; “Si sente spaventato da qualcosa o qualcuno?”; “Ha sentito/visto qualcosa di strano qui?”. Generalmente i sintomi psicotici in ambiente ospedaliero riflettono più frequentemente delirium che non una patologia psichiatrica funzionale (come la schizofrenia).
4AT
Nome del paziente: Data di nascita: Numero del paziente:
Data: Compilatore:
Cerchiare la risposta [1] ALLERTA Riguarda pazienti che possono essere considerati in stato soporoso (per esempio pazienti per il quale sia difficile svegliarsi e/o che sono evidentemente soporosi durante questo test) oppure agitati/iperattivi. Osservare il paziente. Se dorme, provare a svegliarlo, parlandogli, o con un leggero tocco sulla spalla. Chiedere ai pazienti di dichiarare il proprio nome e l’indirizzo della propria abitazione per valutare il livello di collaborazione Normale (completamente attento, ma non agitato durante tutta la valutazione) 0
Moderata sonnolenza per meno di 10 secondi dopo il risveglio, poi normale 0 Livello di vigilanza evidentemente anomalo 4
[2] AMT4 Età, data di nascita, luogo (nome dell’ospedale e dell’edificio), anno corrente
Nessun errore 0
1 errore 1 2 o più errori / non è possibile somministrare il test al paziente 2
[3] ATTENZIONE Chiedere al paziente: “per favore, mi dica i mesi dell’anno in ordine contrario, partendo da dicembre” Per aiutare la comprensione della domanda, è consentito inizialmente un suggerimento come: “qual è il mese prima di dicembre?” Mesi dell’anno al contrario nomina senza errori 7 mesi o più 0
inizia, ma nomina meno di 7 mesi / si rifiuta di iniziare 1 test non effettuabile (poiché il paziente è indisposto, assonnato o disattento) 2
[4] ACUTO CAMBIAMENTO O DECORSO FLUTTUANTE Dimostrazione di un evidente cambiamento o di un andamento fluttuante nei seguenti domini: attenzione, comprensione o altre funzioni mentali (ad esempio ossessioni e/o allucinazioni) che sono comparse nelle ultime 2 settimane e che sono ancora presenti nelle ultime 24 ore No 0 Sì 4
4 o più: possibile delirium +/− deterioramento cognitivo 1-3: possibile deterioramento cognitivo 0: improbabile il delirium e/o deterioramento cognitivo (ma il delirium può essere presente se il punto 4 è incompleto)
PUNTEGGIO 4AT
Il 4AT è uno strumento di screening ideato per un assessment rapido del delirium. Gli items 1-3 sono valutati sulla base dell’osservazione del paziente al momento dell’assessment. L’item 4 richiede informazioni da una o più sorgenti (ad es. la vostra conoscenza del paziente, altri membri dello staff, relazioni del medico di famiglia, caregivers etc.). L’esaminatore dovrebbe tenere in conto la presenza di deficit della comunicazione (ipoacusia, afasia, barriere linguistiche) nel portare a termine il test ed interpretare lo score. Allerta: un alterato stato di allerta è molto indicativo di delirium in ambito ospedaliero. Se il paziente mostra variazioni notevoli dell’allerta durante la valutazione, cerchiate lo score 4. Cambiamento acuto o decorso fluttuante: Una fluttuazione dello stato cognitivo può avvenire anche in assenza di delirium nei pazienti affetti da demenza, ma una fluttuazione marcata generalmente indica delirium. Per elicitare la presenza di allucinazioni o sintomi psicotici porre al paziente domande tipo: “È preoccupato/a di qualcosa in questo momento?”; “Si sente spaventato da qualcosa o qualcuno?”; “Ha sentito/visto qualcosa di strano qui?”. Generalmente i sintomi psicotici in ambiente ospedaliero riflettono più frequentemente delirium che non una patologia psichiatrica funzionale (come la schizofrenia).

S46
123
Intern Emerg Med (2016) 11 (Suppl):S43-S46
za, degli outcomes negativi cui si associa e dei costi elevati che impone in termini di salute e monetari. Ciò nonostante tale condizione clinica è ancora largamente sottostimata e sottodiagnosticata. Il delirium deve essere quindi “insegna-to” ai giovani medici, inducendoli a considerarlo una sorta di “sesto segno vitale” alla stessa stregua, in termini di im-portanza, della pressione arteriosa, della frequenza cardiaca, della saturazione periferica di ossigeno e della temperatura corporea [18]. La sensibilità nei confronti di questa sindrome dovrebbe poi essere promossa mediante iniziative di sensi-bilizzazione all’interno degli ospedali e dei luoghi di cura, ad esempio istituendo una giornata di valutazione sistemati-ca del delirium tra tutti i pazienti degenti (un delirium day), così da diffondere la cultura del delirium e avvicinare i clinici all’uso di strumenti di valutazione standardizzati.
Bibliografia
1. American Psychiatric Association, APA (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th ed, DSM-5. American Psychiatric Association
2. Witlox J, Eurelings LS, de Jonghe JF et al (2010). Delirium in elder-ly patients and the risk of postdischarge mortality, institutionaliza-tion, and dementia: a meta-analysis. JAMA 304:443-51
3. Bellelli G, Mazzola P, Morandi A et al (2014). Duration of post-operative delirium is an independent predictor of 6-month mortality in older adults after hip fracture. J Am Geriatr Soc 62:1335-40
4. Inouye SK, Westendorp RG, Saczynski JS (2014). Delirium in el-derly people. Lancet 383:911-22
5. Davis DH, Skelly DT, Murray C et al (2015). Worsening cognitive impairment and neurodegenerative pathology progressively increase risk for delirium. Am J Geriatr Psychiatry 23:403-15
6. Leslie DL, Marcantonio ER, Zhang Y et al (2008). One-year health
care costs associated with delirium in the elderly population. Arch Intern Med 168:27-32
7. Morandi A, Lucchi E, Turco R et al (2015). Delirium superimposed on dementia: a quantitative and qualitative evaluation of infor-mal caregivers and health care staff experience. J Psychosom Res 79:272-80
8. Morandi A, Lucchi E, Turco R et al (2015). Delirium superimposed on dementia: a quantitative and qualitative evaluation of patient ex-perience. J Psychosom Res 79:281-7
9. Bellelli G, Morandi A, Trabucchi M (2013). The meaning of deliri-um. JAMA Intern Med 173:597-8
10. Cole MG (2004). Delirium in elderly patients. Am J Geriatr Psychi-atry 12:7-21
11. American Geriatric Society (AGS) Choosing Wisely Workgroup (2013). American Geriatrics Society identifies five things that healthcare providers and patients should question J Am Geriatr Soc 61:622-31
12. Bellelli G, Nobili A, Annoni G et al (2015). Under-detection of delirium and impact of neurocognitive deficits on in-hospital mor-tality among acute geriatric and medical wards. Eur J Intern Med 26:696-704
13. Bellelli G, Speciale S, Barisione E, Trabucchi M (2007). Delirium subtypes and 1-year mortality among elderly patients discharged from a post-acute rehabilitation facility. J Gerontol A Biol Sci Med Sci 62:1182-3
14. National Institute of Clinical Excellence (NICE) (2010). Delirium: diagnosis, prevention and management. Clinical guideline 103, London
15. Inouye SK, van Dyck CH, Alessi CA et al (1990). Clarifying confu-sion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med 113:941-8
16. Inouye SK, Foreman MD, Mion LC et al (2001). Nurses’ recog-nition of delirium and its symptoms: comparison of nurse and re-searcher ratings. Arch Intern Med 161:2467-73
17. Bellelli G, Morandi A, Davis DH et al (2014). Validation of the 4AT, a new instrument for rapid delirium screening: a study in 234 hospi-talised older people. Age Ageing 43:496-502
18. Bellelli G, Trabucchi M (2008). Delirium as the sixth vital sign. J Am Med Dir Assoc 9:279; author reply 279-80

© SIMI 2016
123
MEET THE EXPERT
Intern Emerg Med (2016) 11 (Suppl):S47-S49
Marco Cicardi (•)Dipartimento di Scienze Biomediche e Cliniche Luigi Sacco Università di Milano ASST Fatebenefratelli Sacco, MilanoVia Giovanni Battista Grassi 74, 20157 Milano E-mail: [email protected]
R. Castelli • M.A. Wu • A. ZanichelliDipartimento di Scienze Biomediche e Cliniche Luigi Sacco Università di MilanoASST Fatebenefratelli Sacco, Milano
L’angioedema acquisito
Marco Cicardi • Roberto Castelli • Maddalena A. Wu • Andrea Zanichelli
Il termine angioedema acquisito viene riservato a quelle for-me di angioedema ricorrente, non secondarie ad uno specifi-co fattore scatenante e non accompagnate da manifestazioni orticarioidi, che non ha una trasmissione familiare e neppure un’eziologia geneticamente trasmessa. Sulla base della clas-sificazione degli angioedemi senza orticaria pubblicata nel 2014 [1], possono essere identificate 4 forme diverse di an-gioedema acquisito.
La prima di queste forme viene identificata come angio-edema acquisito idiopatico istaminergico (IH-AAE). L’e-ziologia non è determinabile e quindi la diagnosi è posta dopo aver escluso le diverse cause di angioedema e dopo aver veri-ficato che l’angioedema cessa di ricorrere quando il paziente è posto in trattamento continuativo con antistaminici [2]. La dose di antistaminico necessaria per la prevenzione delle ri-correnze è superiore a quella prevista per le patologie aller-giche per cui è indicato. Generalmente è sufficiente una dose doppia, ma talvolta può servire un aumento fino a 4 volte, come previsto nell’orticaria cronica spontanea [3]. Tra le for-me di angioedema senza orticaria è quella più frequente e si pone, in un certo senso, a ponte con l’orticaria stessa. Le ma-nifestazioni sono circoscritte e con una discreta componente di eritema, come di solito si riscontra nelle lesioni pomfoidi dell’orticaria e poco o nulla negli angioedemi tipici. Anche la localizzazione è prevalentemente periorale o periorbitaria.
L’altra forma di angioedema acquisito che ricorre con ele-
vata frequenza è quella correlata al trattamento con inibi-tori dell’angiotensina (ACEi-AAE). È definita dal fatto che un soggetto manifesta i suoi primi episodi di angioedema ricorrente nel corso di trattamento con ACEi. Si genera per accumulo di bradichinina secondario all’inibizione del suo principale enzima di degradazione che è l’enzima di con-versione dell’angiotensina [4]. Un’importante percentuale di pazienti che sviluppano angioedema ricorrente durante trattamento con ACEi continua ad avere saltuarie ricorrenze anche dopo la sospensione [5]. Questo è probabilmente lega-to al fatto che i soggetti con ACEi-AAE sono geneticamen-te caratterizzati da una ridotta capacità di catabolismo della bradichinina [6].
Parallelo all’IH-AAE è l’angioedema ad eziologia sco-nosciuta che non è ereditario e continua a ricorrere in cor-so di terapia antistaminica (idiopatico non istaminergico, InH-AAE). È diagnosticato dopo aver escluso ogni possibile causa nota di angioedema e dopo un ciclo di antistaminici adeguato per dimostrare che le ricorrenze non si riducono con questa terapia. Oltre a non conoscersi le cause, non si conosce neppure il meccanismo con cui si generano questi angioedemi. In analogia ad altre forme, si ritiene che alla base possa esserci sempre un eccesso di bradichinina [7]. Nella nostra esperienza e in quella di altri, l’acido tranexamico, un antifibrinolitico che inibisce la plasmina, si dimostra alta-mente efficace nel prevenire le ricorrenze in questa forma di angioedema, quando somministrato in maniera continuativa [8,9]. L’efficacia dell’acido tranexamico tende a supportare l’ipotesi che la plasmina abbia un ruolo nella patogenesi degli angioedemi. Studi recenti hanno dimostrato che gli angioe-demi che si verificano nei soggetti portatori di mutazioni del fattore XII della coagulazione (FXII) sarebbero determinati dal fatto che queste mutazioni modificano la struttura di FXII, facilitandone la conversione in FXII attivato (FXIIa) da par-te della plasmina. FXIIa determina l’attivazione della fase di contatto con liberazione di bradichinina ed angioedema [10]. Questo meccanismo viene inibito, in vitro, dall’aggiunta di

S48
123
Intern Emerg Med (2016) 11 (Suppl):S47-S49
acido tranexamico ma non dall’aggiunta di C1inibitore (C1-INH). Estrapolando da questi dati, si può ipotizzare che an-che l’InH-AAE possa generarsi attraverso un meccanismo plasmina-dipendente.
La prima forma di angioedema identificata come angioe-dema acquisito è stata quella legata alla carenza, non su base genetica, di C1-INH. Questa forma è oggi più correttamente definita come angioedema acquisito da deficit di C1-INH (C1-INH-AAE). Clinicamente è identica alla forma ereditaria (C1-INH-HAE) con episodi ricorrenti di angioedema senza orticaria che interessano la cute, le mucose gastrointestinali con importante sintomatologia dolorosa e le mucose delle pri-me vie aeree con possibilità di morte per asfissia [11]. Ciò che tipicamente distingue il deficit acquisito è l’inizio delle manifestazioni dopo i 40 anni di età. L’altro elemento distin-tivo è l’altissima frequenza di associazione con patologie dei linfociti B. Questo aspetto permette di speculare sull’origine di questo deficit, alla cui base sta un aumentato catabolismo che in forme diverse viene ricondotto alla patologia dei B lin-fociti. L’analisi dei 77 pazienti finora diagnosticati nella nostra struttura a partire dal 1975, ci ha infatti portati a vedere i di-versi aspetti con cui questa patologia può estrinsecarsi come momenti successivi di un unico fenomeno rappresentato dallo sviluppo di cloni di linfociti B in grado di riconoscere C1-INH. Pazienti con C1-INH-AAE presentano, per definizione, deficit di C1-INH che in alcuni può essere la sola alterazio-ne visibile, in altri si associa alla presenza di autoanticorpi contro C1-INH e/o banda monoclonale di incerto significato (MGUS) e/o linfoma non Hodgkin (NHL). In tutte queste pre-sentazioni è sempre invariabilmente presente un’iperattivazio-ne della via classica del complemento e un iperconsumo di C1-INH che sono riconducibili agli autoanticorpi, alla com-ponente monoclonale o al tessuto neoplastico [12-15]. Dal momento che queste diverse patologie possono sia coesistere che succedersi nello stesso paziente, riteniamo che alla base di tutto vi sia l’espansione di cloni anti-C1-INH [16]. Que-sti cloni possono secernere anticorpi in quantità tale da essere talvolta visibili nel plasma e arrivare anche a un’espansione clonale in grado di determinare la formazione di componenti monoclonali anti-C1-INH rilevabili all’elettroforesi. La proli-ferazione dei cloni B può infine evolvere in senso neoplastico con formazione di NHL. In una recente revisione della nostra casistica abbiamo messo in evidenza come questi linfomi, che si sviluppano in circa un terzo degli affetti da C1-INH-AAE, siano per oltre il 70% linfomi marginali splenici che rappre-sentano invece un’esigua minoranza (nettamente meno del 10%) tra i NHL [17]. I linfociti che originano dalla zona mar-ginale sono peraltro linfociti che sono già entrati in contatto con l’antigene. In accordo con ciò, originano dalla zona mar-ginale anche i NHL che si trovano nel contesto di patologie caratterizzate da cronica immunostimolazione antigenica o
autoreattività [18,19]. I portatori di C1-INH-AAE vanno quin-di attentamente sorvegliati per la presenza di segni indicativi di linfoproliferazione, anche se la maggior parte dei linfomi che si sviluppano in questi pazienti è ad andamento indolente e solo tre dei 23 pazienti in cui noi li abbiamo rilevati sono deceduti a causa della progressione del linfoma. Da un pun-to di vista clinico abbiamo quindi una situazione di rischio duplice, inerente le ricorrenze di angioedema e lo sviluppo di neoplasia aggressiva. Le ricorrenze di angioedema bene-ficiano degli stessi farmaci usati nell’angioedema da deficit ereditario di C1-INH, anche se, in assenza di registrazione per questa specifica patologia, il loro impiego rimane off label. Per gli attacchi acuti si può utilizzare il concentrato plasmatico di C1-INH. Alcuni rari pazienti, probabilmente a causa di un ca-tabolismo di C1-INH straordinariamente elevato, rispondono poco a questo trattamento e beneficiano invece dell’icatibant, antagonista del recettore B2 della bradichinina [20]. Per i pa-zienti nei quali la frequenza delle ricorrenze è pari o maggio-re di due al mese e la risposta alla terapia al bisogno non è soddisfacente, si può prendere in considerazione un intervento di profilassi degli attacchi. Anche in questi pazienti, come in quelli con InH-AAE, la somministrazione di acido tranexami-co dà buoni risultati [11]. Questo farmaco, sulla scorta di uno studio controllato su piccola casistica, è entrato nell’uso per la profilassi degli attacchi nei portatori del deficit ereditario di C1-INH. In questa indicazione trova però oggi scarso impiego per la presenza di farmaci, come gli androgeno attenuati e il C1-INH plasmatico, che sono nettamente più efficaci [21,22]. Curiosamente, nei pazienti con C1-INH-AAE gli androgeno attenuati sono efficaci in meno del 50% dei casi e anche il C1-INH, per i motivi sopra menzionati, non rappresenta l’ap-proccio ideale alla profilassi. L’acido tranexamico riduce inve-ce marcatamente la frequenza delle ricorrenze in oltre il 70% dei trattati [9]. Il motivo di questa maggiore efficacia è pro-babile che risieda, anche in questa situazione, in un eccessivo coinvolgimento della plasmina [10].
Bibliografia
1. Cicardi M, Aberer W, Banerji A et al; HAWK under the patronage of EAACI (European Academy of Allergy and Clinical Immunolo-gy) (2014). Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema In-ternational Working Group. Allergy 69:602-16
2. Faisant C, Boccon-Gibod I, Mansard C et al (2016). Idiopathic his-taminergic angioedema without wheals: A case series of 31 patients. Clin Exp Immunol. doi: 10.1111/cei.12789 [Epub ahead of print]
3. Zuberbier T, Aberer W, Asero R et al; European Academy of Aller-gy and Clinical Immunology; Global Allergy and Asthma European Network; European Dermatology Forum; World Allergy Organiza-tion (2014). The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision and update. Allergy 69:868-87

S49
123
Intern Emerg Med (2016) 11 (Suppl):S47-S49
4. Agostoni A, Cicardi M, Cugno M et al (1999). Angioedema due to an-giotensin-converting enzyme inhibitors. Immunopharmacology 44:21-5
5. Beltrami L, Zanichelli A, Zingale L et al (2011). Long-term fol-low-up of 111 patients with angiotensin-converting enzyme inhibi-tor-related angioedema. J Hypertens 29:2273-7
6. Byrd JB, Adam A, Brown NJ (2006). Angiotensin-converting en-zyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 26:725-37
7. Nussberger J, Cugno M, Cicardi M (2002). Bradykinin-mediated angioedema. N Engl J Med 347:621-2
8. Wintenberger C, Boccon-Gibod I, Launay D et al (2014). Tranexamic acid as maintenance treatment for non-histaminergic angioedema: anal-ysis of efficacy and safety in 37 patients. Clin Exp Immunol 178:112-7
9. Mansi M, Zanichelli A, Coerezza A et al (2015). Presentation, diag-nosis and treatment of angioedema without wheals: a retrospective analysis of a cohort of 1058 patients. J Intern Med 277:585-93
10. de Maat S, Björkqvist J, Suffritti C et al (2016). Plasmin is a natu-ral trigger for bradykinin production in patients with hereditary an-gioedema with factor XII mutations. J Allergy Clin Immunol Apr 6. pii: S0091-6749(16)30006-9. doi: 10.1016/j.jaci.2016.02.021
11. Zingale LC, Castelli R, Zanichelli A, Cicardi M (2006). Acquired deficiency of the inhibitor of the first complement component: pre-sentation, diagnosis, course, and conventional management. Immu-nol Allergy Clin North Am 26:669-90
12. Hauptmann G, Petitjean F, Lang JM, Oberling F (1979). Acquired C1 inhibitor deficiency in a case of lymphosarcoma of the spleen. Reversal of complement abnormalities after splenectomy. Clin Exp Immunol 37:523-31
13. Schreiber AD, Zweiman B, Atkins P et al (1976). Acquired an-
gioedema with lymphoproliferative disorder: association of C1 in-hibitor deficiency with cellular abnormality. Blood 48:567-80
14. Jackson J, Sim RB, Whelan A, Feighery C (1986). An IgG autoanti-body which inactivates C1-inhibitor. Nature 323:722-4
15. Cicardi M, Beretta A, Colombo M et al (1996). Relevance of lymph-oproliferative disorders and of anti-C1 inhibitor autoantibodies in acquired angio-oedema. Clin Exp Immunol 106:475-80
16. Wu MA, Castelli R (2016). The Janus faces of acquired angioede-ma: C1-inhibitor deficiency, lymphoproliferation and autoimmuni-ty. Clin Chem Lab Med 54:207-14
17. Castelli R, Wu MA, Arquati M et al (2016). High prevalence of splenic marginal zone lymphoma among patients with acquired C1 inhibtor deficiency. Br J Haematol 172:902-8
18. Vannata B, Arcaini L, Zucca E (2016). Hepatitis C virus-associated B-cell non-Hodgkin’s lymphomas: what do we know? Ther Adv He-matol 7:94-107
19. Dias C, Isenberg DA (2011). Susceptibility of patients with rheu-matic diseases to B-cell non-Hodgkin lymphoma. Nat Rev Rheuma-tol 7:360-8
20. Zanichelli A, Bova M, Coerezza A et al (2012). Icatibant treatment for acquired C1-inhibitor deficiency: a real-world observational study. Allergy 67:1074-7
21. Sheffer AL, Austen KF, Rosen FS (1972). Tranexamic acid therapy in hereditary angioneurotic edema. N Engl J Med 287:452-4
22. Cicardi M, Bork K, Caballero T et al; HAWK (Hereditary An-gioedema International Working Group) (2012). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 67:147-57

© SIMI 2016
123
MEET THE EXPERT
Intern Emerg Med (2016) 10 (Suppl):S50-S56
Paolo Ventura (•)Dipartimento di Scienze Medico-Chirurgiche Materno-Infantili e dell’AdultoCentro di Riferimento Regionale per la Diagnosi e la Cura delle PorfirieUniversità di Modena e Reggio Emilia, AOU Policlinico di ModenaLargo del Pozzo 71, 41124 ModenaE-mail: [email protected]
Le porfirie: incroci pericolosi fra cute, fegato e midollo osseo
Paolo Ventura
Biosintesi dell’eme
L’eme è il gruppo prostetico delle emoproteine (emoglobina, mioglobina, citocromi, catalasi etc.). È prodotto e utilizzato per l’85% dal midollo eritroide per la sintesi di emoglobi-na; del rimanente, la maggior parte è prodotta dagli epatoci-ti per la sintesi dei citocromi (citocromo P
450). La biosintesi
dell’eme si sviluppa lungo 8 tappe enzimatiche successive: 2 composti lineari (glicina/succinil-coenzima A) sono progres-sivamente convertiti in strutture tetrapirroliche cicliche (por-firine) il cui ultimo prodotto, la protoporfirina IX, comples-sandosi con Fe++, viene convertito in eme (Figura 1) [1,2]. In condizioni normali, questo processo è estremamente efficace e strettamente regolato: non determina alcun accumulo signi-ficativo di intermedi metabolici (precursori non porfirinici e/o porfirine). A livello epatico, l’espressione e l’attività dell’i-soenzima responsabile della prima tappa metabolica (ALA sintetasi, ALAS1) sono regolate dalla disponibilità stessa di eme: un incremento del “pool libero” di eme ha effetto inibi-torio/repressorio su ALAS1, e viceversa (Figura 1). ALAS1 può essere indotto sia direttamente che indirettamente (con-sumo di eme libero) da molti fattori (farmaci, steroidi, stati di iponutrizione, infezioni, stress etc.). L’isoenzima eritroide di ALA sintetasi (ALAS2) è invece regolato mediante l’attiva-zione di una iron regulatory protein: è indotto dalla disponibi-lità di ferro e inibito in caso di sideropenia [1,3,4].
Le porfirie: aspetti generali
Il termine porfirie comprende un gruppo eterogeneo di ma-lattie metaboliche che conseguono a una disfunzione su base congenita (raramente acquisita) di uno degli enzimi della biosintesi dell’eme (Tabelle 1 e 2). Il blocco che ne deriva provoca un accumulo anomalo di intermedi metabolici (pre-cursori non porfirinici e/o porfirine), la cui diversa tossicità è responsabile dei quadri clinici peculiari di ciascuna di queste malattie [5-8]. In base alla sede prevalente del difetto enzima-tico, le porfirie sono distinte (classificazione fisiopatologica) in epatiche o eritropoietiche; sulla base delle loro principali manifestazioni (classificazione clinica), sono anche distinte in porfirie con sintomi neuroviscerali (porfirie acute) e por-
Fig. 1 Biosintesi dell’eme.
Glicina + SuccinilCoA
ALA sintetasi
ALA deidrasi
PBG deaminasi
Uropor�rinogeno IIIcosintetasi
Uropor�rinogeno decarbossilasi
Copropor�rinogenoossidasi
Protopor�rinogenoossidasi
Ferrochelatasi
Acido d-aminolevulinico (ALA)
Porfobilinogeno (PBG)
Idrossimetibilano
Uropor�rinogeno IIIControllo
a feed-backnegativo(ALAS1)
Copropor�rinogeno III
Protopor�rinogeno IX
Protopor�rina IX
ALA = Acido d-Aminolevulinico; PBG = Porfobilinogeno.
Eme
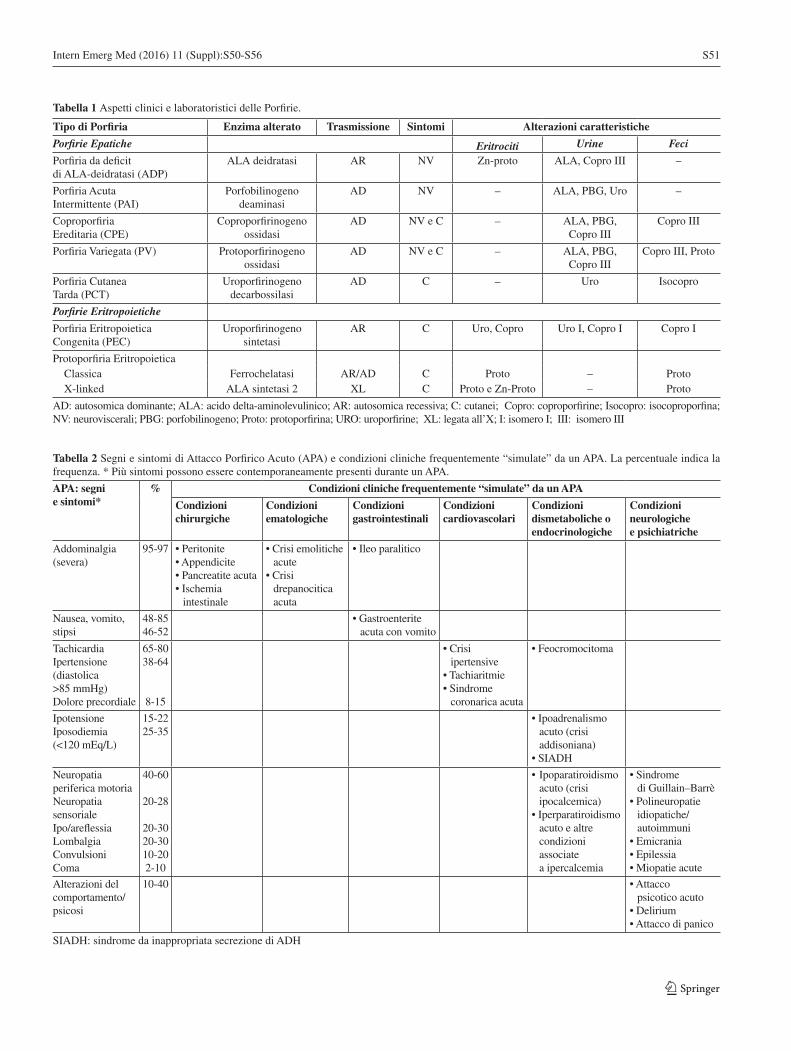
S51
123
Intern Emerg Med (2016) 11 (Suppl):S50-S56
Tabella 1 Aspetti clinici e laboratoristici delle Porfirie.
Tipo di Porfiria Enzima alterato Trasmissione Sintomi Alterazioni caratteristiche
Porfirie Epatiche Eritrociti Urine Feci
Porfiria da deficit di ALA-deidratasi (ADP)
ALA deidratasi AR NV Zn-proto ALA, Copro III –
Porfiria Acuta Intermittente (PAI)
Porfobilinogeno deaminasi
AD NV – ALA, PBG, Uro –
Coproporfiria Ereditaria (CPE)
Coproporfirinogeno ossidasi
AD NV e C – ALA, PBG, Copro III
Copro III
Porfiria Variegata (PV) Protoporfirinogeno ossidasi
AD NV e C – ALA, PBG, Copro III
Copro III, Proto
Porfiria Cutanea Tarda (PCT)
Uroporfirinogeno decarbossilasi
AD C – Uro Isocopro
Porfirie Eritropoietiche
Porfiria Eritropoietica Congenita (PEC)
Uroporfirinogeno sintetasi
AR C Uro, Copro Uro I, Copro I Copro I
Protoporfiria EritropoieticaClassica Ferrochelatasi AR/AD C Proto – ProtoX-linked ALA sintetasi 2 XL C Proto e Zn-Proto – Proto
AD: autosomica dominante; ALA: acido delta-aminolevulinico; AR: autosomica recessiva; C: cutanei; Copro: coproporfirine; Isocopro: isocoproporfina; NV: neuroviscerali; PBG: porfobilinogeno; Proto: protoporfirina; URO: uroporfirine; XL: legata all’X; I: isomero I; III: isomero III
Tabella 2 Segni e sintomi di Attacco Porfirico Acuto (APA) e condizioni cliniche frequentemente “simulate” da un APA. La percentuale indica la frequenza. * Più sintomi possono essere contemporaneamente presenti durante un APA.
APA: segni e sintomi*
% Condizioni cliniche frequentemente “simulate” da un APA
Condizioni chirurgiche
Condizioni ematologiche
Condizioni gastrointestinali
Condizioni cardiovascolari
Condizioni dismetaboliche o endocrinologiche
Condizioni neurologiche e psichiatriche
Addominalgia (severa)
95-97 • Peritonite• Appendicite• Pancreatite acuta• Ischemia
intestinale
• Crisi emolitiche acute
• Crisi drepanocitica acuta
• Ileo paralitico
Nausea, vomito, stipsi
48-85 46-52
• Gastroenterite acuta con vomito
Tachicardia Ipertensione (diastolica >85 mmHg) Dolore precordiale
65-80 38-64
8-15
• Crisi ipertensive
• Tachiaritmie• Sindrome
coronarica acuta
• Feocromocitoma
IpotensioneIposodiemia (<120 mEq/L)
15-2225-35
• Ipoadrenalismo acuto (crisi addisoniana)
• SIADH
Neuropatia periferica motoria Neuropatia sensorialeIpo/areflessiaLombalgiaConvulsioniComa
40-60
20-28
20-3020-3010-202-10
• Ipoparatiroidismo acuto (crisi ipocalcemica)
• Iperparatiroidismo acuto e altre condizioni associate a ipercalcemia
• Sindrome di Guillain–Barrè
• Polineuropatie idiopatiche/ autoimmuni
• Emicrania• Epilessia• Miopatie acute
Alterazioni del comportamento/psicosi
10-40 • Attacco psicotico acuto
• Delirium• Attacco di panico
SIADH: sindrome da inappropriata secrezione di ADH

S52
123
Intern Emerg Med (2016) 11 (Suppl):S50-S56
firie con sintomi cutanei (porfirie non-acute). Nessuna di tali classificazioni ha valore assoluto (esistono punti di sovrappo-sizione) [6,8-10] (Tabella 1). La caratteristica clinica fonda-mentale delle porfirie acute sono gli Attacchi Porfirici Acuti (APA), manifestazioni ricorrenti di coinvolgimento neurovi-scerale acuto di varia intensità [6,9,11-15]. Le porfirie non-acute sono caratterizzate soprattutto da manifestazioni cuta-nee, conseguenti a fenomeni di fotosensibilità [5,6,8,16-25]. In entrambi i casi, la qualità di vita dei pazienti risulta grave-mente compromessa [26-29].
Porfirie con sintomatologia neuroviscerale (Porfirie acute)
Quattro varianti di porfiria possono presentarsi clinicamen-te con APA (porfirie acute) [10,11]. Fra queste, la forma più frequente è la Porfiria Acuta Intermittente (PAI, incidenza annuale in Europa 0,1-0,2/milione, prevalenza 1-2:100.000) [30-32]. È stato descritto che gli APA che caratterizzano la PAI siano clinicamente più gravi, benché qualitativamente indistinguibili da quelli che caratterizzano le varianti più rare di porfiria acuta: Porfiria Variegata (PV, incidenza annuale 0,05-0,1/milione) [7,33-36], Coproporfiria Ereditaria (CPE, 0,02/milione) [7,9,37,38] e la rarissima Porfiria da deficit di ALA-deidratasi (ALAd-P, descritti una decina di casi) [39]. Una precisa definizione epidemiologica di queste malattie è però piuttosto difficile, in quanto malattie a bassa penetran-za clinica (solo il 10-30% dei soggetti portatori di mutazione risulta sintomatico). Manifestazioni simili agli APA possono verificarsi in corso di intossicazione da piombo (che blocca l’attività dell’ALA-deidratasi): il saturnismo (o plumbopor-firia) può quindi essere considerato un disturbo acquisito del metabolismo dell’eme (Figura 1) [40,41]. Grazie ai recenti miglioramenti in campo diagnostico e terapeutico, la letalità degli APA (inizialmente descritta fino al 50% dei casi) si è molto ridotta [26,42]. Secondo dati americani, tuttavia, il tas-so di mortalità rimane circa 3 volte più alto nei soggetti con PAI rispetto alla popolazione generale [43].
Clinica delle porfirie acute: l’attacco porfirico acuto. La sindrome dell’attacco porfirico acuto si caratterizza per modalità di presentazione multiformi, in cui manifestazioni neuropsichiche, neuroendocrine e cardiovascolari risultano variamente associate fra loro (Tabella 2) [7]. La neuropatia autonomica si presenta con la classica triade addominale acuta: dolore, vomito e stipsi. Il dolore addominale è descrit-to di solito come lancinante e può assumere caratteristiche suggestive di “addome acuto”; ciononostante, i sintomi ap-paiono spesso sproporzionati rispetto all’esame obiettivo (i segni propri di peritonismo sono per lo più assenti) [10,15]. Alla neuropatia autonomica e a una iperincrezione di cate-
colamine (riflessa?) sono invece ascrivibili le manifestazioni cardiovascolari: tachicardia sinusale e ipertensione arteriosa instabile, con ipotensione posturale. La neuropatia periferi-ca sensitivo-motoria è caratterizzata da parestesie, ipostenia, paresi e paralisi flaccide ai muscoli prossimali degli arti, fino alla tetraplegia e alla paralisi respiratoria (interessamento dei muscoli intercostali, addominali e del diaframma) [14,15]. L’interessamento respiratorio è una possibile causa di exitus in corso di APA. La neuropatia centrale può interessare sedi diverse: convulsioni (interessamento corticale); disfagia, re-flusso gastro-esofageo, oftalmoplegia, disfonia e paresi del facciale inferiore (nuclei bulbari dei nervi cranici); sindrome da inadeguata secrezione di ADH, con iponatriemia e ipoma-gnesiemia (ipotalamo) [10,14,15]. Relativamente frequenti anche i disturbi della sfera psichica, che variano dalla sem-plice instabilità emotiva con alterazioni del comportamen-to, a quadri più complessi: irritabilità, angoscia, insonnia, depressione o agitazione psico-motoria con allucinazioni [6,7,10,11,14,44,45].
Fisiopatologia. Le evidenze più recenti ascrivono la pa-togenesi degli APA alla neurotossicità dell’intermedio me-tabolico non porfirinico acido delta-aminolevulinico (ALA) [46-48]. Alla base dell’accumulo di ALA vi è l’iperespres-sione dell’ALAS1 epatico: fattori in grado di determinare una riduzione del pool di eme libero epatocitario riducono il feed-back negativo da questo esercitato su ALAS1, che, ipe-respresso, in presenza di un deficit enzimatico di alcuni punti chiave della via biosintetica dell’eme, conduce all’accumulo di ALA (Figura 1). Diversi fattori esogeni o endogeni in gra-do di provocare una iperespressione diretta di ALAS1 o di determinare una brusca riduzione del pool di eme libero sono quindi fattori scatenanti di APA in un paziente portatore di porfiria acuta [5,7,10,11].
Diagnosi. La diagnosi di porfiria acuta si basa sul sospet-to clinico di APA (caratterizzato però da una tale variabilità clinica da risultare spesso non immediatamente identifica-bile), specie se ricorrenti o associati a fattori scatenanti noti [5,49]. Urine color rosso-borgogna [o che assumono questo aspetto a breve distanza dall’emissione] rappresentano un segno suggestivo. La diagnosi biochimica di APA si basa sulla dimostrazione, contestualmente ai segni dell’APA, di elevati livelli urinari di precursori non porfirinici [ALA e porfobilinogeno (PBG)]. Sono disponibili metodiche quali-tative per la determinazione “bed-side” del PBG, utilizzabili in condizioni di urgenza [50,51]. La successiva determina-zione del pattern completo di escrezione urinaria (e fecale) delle diverse frazioni porfiriniche può portare alla defini-zione della variante specifica di porfiria acuta sottostante (Tabella 1) [49,51]. In caso di PV e CPE, porre attenzione

S53
123
Intern Emerg Med (2016) 11 (Suppl):S50-S56
a una storia o a segni suggestivi di lesioni cutanee da foto-sensibilità [49,51]. L’analisi genetica conferma la diagnosi e identifica la mutazione sottostante, anche se l’associazione fenotipo-genotipo non è sempre immediata [52]. La determi-nazione dell’attività enzimatica (eritrociti o fegato) è esegui-ta solo in casi particolari [7,49].
Terapia. Il trattamento specifico di un APA non può pre-scindere dall’identificazione e dalla rimozione di qualsiasi po-tenziale fattore scatenante (stress psico-fisico, restrizione die-tetica, infezioni, assunzione di farmaci etc.). Si basa sull’infu-sione endovenosa di eme (Eme Arginato, 2-4 mg/kg/die per 3-4 giorni). L’eme esercita un feed-back negativo su ALAS1, determinando una rapida riduzione dell’accumulo di ALA: l’infusione in genere risolve gli APA nel giro di pochi giorni. Il glucosio (infusione di 1500-2000 cc/die di soluzioni al 10% o 20%) ha un effetto simile su ALAS1. L’eme è però dotato di un’efficacia assai maggiore ed è raccomandato in presenza di APA severi: in caso di manifestazioni neuropatiche importan-ti, un suo utilizzo ritardato è stato associato a una più lenta e, a volte, incompleta remissione [10,27,53]. L’infusione di eme può provocare sovraccarico marziale e complicanze locali (trombosi venosa e tromboflebite). Nei pazienti affetti da gra-vi forme di porfiria acuta, a rischio di sviluppare complicanze a lungo termine (renali e neuropatiche), il trapianto di fegato è un’opzione terapeutica curativa [46,54]. Gli ottimi risultati di alcuni recenti trials sperimentali, basati su approcci innovativi di modulazione molecolare diretta di ALAS1, stanno fornen-do nuove opportunità di trattamento nelle porfirie acute, sia in acuto che in profilassi [55,56].
Porfirie con sintomatologia cutanea (Porfirie non-acute)
Tre varianti di porfirie vengono annoverate fra le forme con (prevalente) sintomatologia cutanea. Fra queste, la forma di gran lunga più frequente è la Porfiria Cutanea Tarda (PCT, la forma più frequente di porfiria con una prevalenza stimata, in Europa, intorno a 1:5-10.000 [57]), seguita dalla Protoporfi-ria Eritropoietica (PPE, 1:75.000-200.000) e dalla rarissima Porfiria Eritropoietica Congenita (PEC, qualche centinaio di casi decritti [58]) (malattia di Gunther) [5,6,7,21].
Clinica delle porfirie con sintomatologia cutanea: re-azioni da fotosensibilità. Le porfirie non-acute sono carat-terizzate dalla comparsa di manifestazioni cutanee, conse-guenti a fenomeni di fotosensibilità, indotte dall’accumulo cutaneo di porfirine. Le diverse espressioni cliniche dipen-dono sia dall’entità che dal tipo di porfirina accumulantesi [es. diversa idrosolubilità (uroporfirine > coproporfirine > protoporfirine)]; tutte le lesioni sono però caratterizzate da
interessare esclusivamente le aree fotoesposte (dorso delle mani, viso, collo, capo, orecchie) [25]. Oltre alle manifesta-zioni cutanee, in relazione alla sede di produzione (midollo per CPE e PPE; fegato per PCT) e/o di eliminazione delle porfirine (fegato per PPE) possono associarsi altri sintomi. PCT e CPE (accumulo di uroporfirine e di uro/coproporfi-rine, rispettivamente) si presentano con fragilità cutanea (fe-rite ai minimi traumi), eruzioni eritemato-vescicolo-bollose contenenti liquido sieroso/siero-ematico (fluorescente in luce di Wood) che tendono a rompersi, lasciando ulcere do-lenti evolventi in cicatrici atrofiche e discromiche; iperpig-mentazione (tempie, zigomi e dorso delle mani) e ipertricosi (malare, padiglioni auricolari). Mentre la PCT si presenta in età avanzata (> 40 anni), la CPE compare subito dopo la na-scita, ma ha un decorso assai più grave e devastante, poiché le ulcerazioni coinvolgono sottocutaneo, cartilagini e osso, portando a vere e proprie mutilazioni (falangi, cartilagini na-sali e auricolari) e gravi deturpazioni del viso. Alla PCT si associano alterazioni epatiche a carattere evolutivo [epatopa-tia cronica, aumentata incidenza di carcinoma epatocellulare (HCC)] e con frequenza tale da suggerire un rapporto causa-effetto con la malattia [57]. Alla CPE, causa affinità dell’u-roporfirina per il fosfato di calcio, si associano colorazione spontanea rosso-scura dei denti (e delle ossa) (eritrodonzia) e alterazioni ematologiche [iperplasia con eritroblasti e nor-moblasti fluorescenti (porfiroblasti), reticolocitosi, eritrociti policromatofili fluorescenti (porfirociti) e anemia emolitica e/o da eritropoiesi inefficace] [7,9].
La PPE si presenta nell’infanzia/adolescenza, con eritema ed edema acuto con intenso bruciore (con o senza prurito) a breve distanza dall’esposizione al sole (orticaria solare). Tali manifestazioni regrediscono completamente entro ore dalla cessazione dell’esposizione alla luce e non esitano in cicatri-ci. Frequenti i segni di coinvolgimento epatico: coliche bilia-ri recidivanti, epatopatia cronica avanzata o (più raramente) insufficienza epatica acuta a evoluzione rapidamente fatale (epatite fulminante). Si può riscontrare anemia ipocromica o emolitica [7,9].
Fisiopatologia. Le manifestazioni comuni a tutte le por-firie “cutanee” si sviluppano con un meccanismo fotodina-mico, dipendente dalla capacità dei composti porfirinici di assorbire energia radiante in una zona dello spettro tra l’UV e il visibile (400-410 nm), che viene poi liberata portando alla formazione di radicali liberi dell’ossigeno, responsabili del-la reazione flogistica e del danno tessutale [7,9,25,32]. Nella PEC e nella PPE la sede primaria del difetto è il midollo osseo, con accumulo di porfirine negli eritroblasti e negli eri-trociti circolanti e successiva diffusione al microcircolo der-mico [7,9,59]. Assai recentemente è stata descritta una forma sovrapponibile alla PPE “classica” non associata a mutazioni

S54
123
Intern Emerg Med (2016) 11 (Suppl):S50-S56
del gene FECH. Tale variante di PPE (indicata come PPE do-minante X-linked) consegue a una mutazione responsabile di iperattività (“gain of function” mutation) di ALAS2 (variante eritroide di ALA-S) [60]. Nella PPE la protoporfirina viene captata elettivamente dagli epatociti ed escreta nella bile. Di recente è stato dimostrato però che anche il fegato condivide con il midollo eritroide il deficit enzimatico. L’escrezione bi-liare della protoporfirina in eccesso (prodotta da reticolociti + eritrociti + epatociti) rende instabile la bile e predispone alla colelitiasi (calcoli di protoporfirina) ed è responsabile della epatopatia cronica evolutiva e del danno epatico acu-to (tossicità da protoporfirina) [32,59]. Nella PCT, affinché i sintomi si manifestino, è necessario che l’attività enzimatica [uroporfirinogeno-decarbossilasi (URO-D) epatica] risulti ≤ 20% del normale: diversi fattori ambientali, contribuendo a ridurre l’attività enzimatica (già geneticamente compromes-sa), risultano spesso determinanti nella espressione fenotipica [61-67]. La frequente associazione (> 70% dei casi) di PCT conclamata con l’infezione da HCV e siderosi epatica (alcool, emocromatosi) sarebbe spiegata dalla formazione, in presen-za di queste epatopatie, di un composto porfirinico anomalo prodotto dall’ossidazione delle uroporfirine in eccesso (por-fometene), che inibisce l’attività URO-D [68,69]. Evoluzione a cirrosi epatica è stata descritta nel 30-40% dei pazienti con PCT, come un maggiore rischio di HCC [70].
Diagnosi. Si basa sull’osservazione clinica delle tipiche ma-nifestazioni cutanee, esclusive delle aree fotoesposte [9]. L’età di esordio e il tipico pattern urinario, fecale ed eritrocitario delle porfirine consentono in genere una diagnosi corretta (Tabella 1) [32,59,66]. La valutazione genetica completa l’iter [71].
Terapia. Gli interventi terapeutici sono diretti alla foto-protezione, evitando l’esposizione alla luce solare [vestiti, schermatura delle finestre (tende o vetri appositi), lampade a incandescenza] o al contenimento della reazione fotodinami-ca [assunzione orale di antiossidanti (β-carotene 150 mg/die, fino a 600 mg/die) con risultati variabili; l’effetto migliora associando vitamina E] [7,9,59,72,73]. Recentemente, in pazienti con PPE, l’impianto sottocutaneo di formulazioni a lento rilascio di un MSH-analogo (alfa-melanotide) ha forni-to risultati molto positivi [74]. Nel caso della PCT, il tratta-mento della siderosi epatica (salassoterapia/terapia ferroche-lante), dell’infezione da HCV e la cessazione della consuetu-dine etilica inducono spesso completa remissione del quadro clinico [9,75]; un’alternativa è l’idrossiclorochina (125-250 mg/ogni 3 giorni), che, complessandosi con le uroporfirine, favorisce l’eliminazione renale [9,76].
La prevenzione dell’epatopatia in corso di PPE può esse-re ottenuta mediante modulazione dell’ALAS1 [glucosio o eme arginato (vedi sopra)] o interrompendo il circolo entero-
epatico della protoporfirina, con colestiramina (4 g × 3/die). Il trapianto di fegato, praticato nei casi di epatite fulminante, ha efficacia parziale: la recidiva di epatopatia è frequente (tossi-cità da protoporfirina midollare) e tale da rendere opportuno un doppio trapianto midollo-fegato [7,9,77].
Bibliografia
1. Ponka P (1999). Cell biology of heme. Am J Med Sci 318(4):241-562. Ajioka RS, Phillips JD, Kushner JP (2006). Biosynthesis of heme in
mammals. Biochim Biophys Acta 1763(7):723-363. Ponka P (1997). Tissue-specific regulation of iron metabolism and
heme synthesis: distinct control mechanisms in erythroid cells. Blood 89(1):1-25
4. Ponka P, Schulman HM (1993). Regulation of heme biosynthesis: distinct regulatory features in erythroid cells. Stem Cells 11 Suppl 1:24-35
5. Puy H, Gouya L, Deybach JC (2010). Porphyrias. Lancet 375(9718):924-37
6. Kauppinen R (2005). Porphyrias. Lancet 365(9455):241-527. Ventura P (2011). Le Porfirie. In: Brunetti P, Santeusanio F (eds).
Trattato di Medicina Interna, vol Malattie delle Ghiandole Endo-crine, del Metabolismo e della Nutrizione. vol III. Piccin Nuova Li-braria, Padova, pp 931-52
8. Desnick RJ (2002). Porfirie. In: Braunwald E, Fauci AS, Kasper DL et al (eds). Harrison’s: Principi di Medicina Interna. 15th edn. Mc-Graw-Hill, Milan, pp 2261-302
9. Ventura E, Rocchi E (2001). Le Porfirie. In: Guarini G, Fiorelli G, Malliani A et al (eds). Teodori 2000. Trattato di Medicina Interna, vol 2. 6th edn. Società Editrice Universo, Roma, pp 2301-34
10. Elder GH, Hift RJ, Meissner PN (1997). The acute porphyrias. Lan-cet 349(9065):1613-7
11. Ventura P, Cappellini MD, Rocchi E (2009). The acute porphyrias: a diagnostic and therapeutic challenge in internal and emergency medicine. Intern Emerg Med 4(4):297-308
12. Menegueti MG, Gil Cezar AT, Casarini KA et al (2011). Acute in-termittent porphyria associated with respiratory failure: a multidis-ciplinary approach. Crit Care Res Pract 2011:283690
13. Bylesjo I, Wikberg A, Andersson C (2009). Clinical aspects of acute intermittent porphyria in northern Sweden: a population-based study. Scand J Clin Lab Invest 69(5):612-8
14. Solinas C, Vajda FJ (2008). Neurological complications of porphyr-ia. J Clin Neurosci 15(3):263-8
15. Pischik E, Kauppinen R (2009). Neurological manifestations of acute intermittent porphyria. Cell Mol Biol (Noisy-le-grand) 55(1):72-83
16. Thunell S (2000). Porphyrins, porphyrin metabolism and porphyr-ias. I. Update. Scand J Clin Lab Invest 60(7):509-40
17. James MF, Hift RJ (2000). Porphyrias. Br J Anaesth 85(1):143-5318. Gross U, Hoffmann GF, Doss MO (2000). Erythropoietic and hepat-
ic porphyrias. J Inherit Metab Dis 23(7):641-6119. Chemmanur AT, Bonkovsky HL (2004). Hepatic porphyrias: diag-
nosis and management. Clin Liver Dis 8(4):807-3820. Dombeck TA, Satonik RC (2005). The porphyrias. Emerg Med Clin
North Am 23(3):885-9921. Poblete-Gutierrez P, Wiederholt T, Merk HF, Frank J (2006). The
porphyrias: clinical presentation, diagnosis and treatment. Eur J Dermatol 16(3):230-40
22. Canavese C, Gabrielli D, Guida C, Cappellini MD (2002). Nephrol-ogists and porphyrias. G Ital Nefrol 19(4):393-412

S55
123
Intern Emerg Med (2016) 11 (Suppl):S50-S56
23. Thunell S, Pomp E, Brun A (2007). Guide to drug porphyrogenicity prediction and drug prescription in the acute porphyrias. Br J Clin Pharmacol 64(5):668-79
24. Gorchein A (1997). Drug treatment in acute porphyria. Br J Clin Pharmacol 44(5):427-34
25. Deybach JC (2001). Porphyria. Painful photosensitivity. Lancet 358 Suppl:S49
26. Stein PE, Badminton MN, Barth JH et al (2012). Acute intermittent porphyria: fatal complications of treatment. Clin Med 12(3):293-4
27. Elder GH, Hift RJ (2001). Treatment of acute porphyria. Hosp Med 62(7):422-5
28. Herrick AL, McColl KE, Moore MR et al (1989). Controlled trial of haem arginate in acute hepatic porphyria. Lancet 1(8650):1295-7
29. Herrick A, McColl KE, McLellan A et al (1987). Effect of haem arginate therapy on porphyrin metabolism and mixed function oxy-genase activity in acute hepatic porphyria. Lancet 2(8569):1178-9
30. Anderson KE, Sassa S, Kappas A (1981). Acute intermittent por-phyria. Ann Intern Med 95(6):784-5
31. Grandchamp B (1998). Acute intermittent porphyria. Semin Liver Dis 18(1):17-24
32. Anderson KE, Sassa S, Bishop DF et al (2001). Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet A, Sly WS et al (eds). The metabolic and molecular basis of inherited diseases. McGraw-Hill, New York, pp 2991-3062
33. Hift RJ, Peters TJ, Meissner PN (2012). A review of the clinical presentation, natural history and inheritance of variegate porphyria: its implausibility as the source of the ‘Royal Malady’. J Clin Pathol 65(3):200-5
34. Kirsch RE, Meissner PN, Hift RJ (1998). Variegate porphyria. Se-min Liver Dis 18(1):33-41
35. Di Pierro E, Ventura P, Brancaleoni V et al (2009). Clinical, bio-chemical and genetic characteristics of Variegate Porphyria in Italy. Cell Mol Biol (Noisy-le-grand) 55(2):79-88
36. Hift RJ, Meissner PN (2005). An analysis of 112 acute porphyric attacks in Cape Town, South Africa: evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and sever-ity. Medicine (Baltimore) 84(1):48-60
37. Martasek P (1998). Hereditary coproporphyria. Semin Liver Dis 18(1):25-32
38. Kuhnel A, Gross U, Doss MO (2000). Hereditary coproporphyria in Germany: clinical-biochemical studies in 53 patients. Clin Biochem 33(6):465-73
39. Sassa S (1998). ALAD porphyria. Semin Liver Dis 18(1):95-10140. Warren MJ, Cooper JB, Wood SP, Shoolingin-Jordan PM (1998).
Lead poisoning, haem synthesis and 5-aminolaevulinic acid dehy-dratase. Trends Biochem Sci 23(6):217-21
41. Herman DS, Geraldine M, Venkatesh T (2007). Evaluation, diagno-sis, and treatment of lead poisoning in a patient with occupational lead exposure: a case presentation. J Occup Med Toxicol 2:7
42. Waldenstrom J, Haeger-Aronsen B (1963). Different patterns of hu-man porphyria. Br Med J 2(5352):272-6
43. Jeans JB, Savik K, Gross CR et al (1996). Mortality in patients with acute intermittent porphyria requiring hospitalization: a United States case series. Am J Med Genet 65(4):269-73
44. Kauppinen R (1998). Management of the acute porphyrias. Photo-dermatol Photoimmunol Photomed 14(2):48-51
45. Crimlisk HL (1997). The little imitator--porphyria: a neuropsychia-try disorder. J Neurol Neurosurg Psychiatry 62(4):319-28
46. Yasuda M, Erwin AL, Liu LU et al (2015). Liver transplantation for acute intermittent porphyria: biochemical and pathologic studies of the explanted liver. Mol Med 21:487-95
47. Bissell DM, Lai JC, Meister RK, Blanc PD (2015). Role of del-
ta-aminolevulinic acid in the symptoms of acute porphyria. Am J Med 128(3):313-7
48. Dowman JK, Gunson BK, Bramhall S et al (2011). Liver transplan-tation from donors with acute intermittent porphyria. Ann Intern Med 154(8):571-2
49. Ventura P, Cappellini MD, Biolcati G et al (2014). A challenging diagnosis for potential fatal diseases: recommendations for diagnos-ing acute porphyrias. Eur J Intern Med 25(6):497-505
50. Puy H, Aquaron R, Lamoril J et al (1997). Acute intermittent por-phyria: rapid molecular diagnosis. Cell Mol Biol (Noisy-le-grand) 43(1):37-45
51. Sandberg S, Elder GH (2004). Diagnosing acute porphyrias. Clin Chem 50(5):803-5
52. Grob U, Puy H, Jacob K et al (2006). Biochemical compared to molecular diagnosis in acute intermittent porphyria. J Inherit Metab Dis 29(1):157-61
53. Pischik E, Kauppinen R (2015). An update of clinical management of acute intermittent porphyria. Appl Clin Genet 8:201-14
54. Singal AK, Parker C, Bowden C et al (2014). Liver transplantation in the management of porphyria. Hepatology 60:1082-9
55. Yasuda M, Gan L, Chen B et al (2014). RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc Natl Acad Sci U S A 111(21):7777-82
56. Paneda A, Lopez-Franco E, Kaeppel C et al (2013). Safety and liver transduction efficacy of rAAV5-cohPBGD in nonhuman primates: a potential therapy for acute intermittent porphyria. Hum Gene Ther 24(12):1007-17
57. Yachimski P, Shah N, Chung RT (2007). Porphyria cutanea tarda. Clin Gastroenterol Hepatol 5(2):e6
58. Prasad AN (2006). Congenital erythropoeitic porphyria. Indian Pe-diatr 43(8):740
59. Thunell S, Harper P, Brun A (2000). Porphyrins, porphyrin metab-olism and porphyrias. IV. Pathophysiology of erythyropoietic pro-toporphyria--diagnosis, care and monitoring of the patient. Scand J Clin Lab Invest 60(7):581-604
60. Seager MJ, Whatley SD, Anstey AV, Millard TP (2014). X-linked dominant protoporphyria: a new porphyria. Clin Exp Dermatol 39(1):35-7
61. Christoph S, Huttmann A, Duhrsen U, Röth A (2010). Porphyria cutanea tarda. Br J Haematol 148(4):493
62. Lambrecht RW, Thapar M, Bonkovsky HL (2007). Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 27(1):99-108
63. Sams H, Kiripolsky MG, Bhat L, Stricklin GP (2004). Porphyria cutanea tarda, hepatitis C, alcoholism, and hemochromatosis: a case report and review of the literature. Cutis 73(3):188-90
64. Fargion S, Fracanzani AL (2003). Prevalence of hepatitis C virus infection in porphyria cutanea tarda. J Hepatol 39(4):635-8
65. Lambrecht RW, Bonkovsky HL (2002). Hemochromatosis and por-phyria. Semin Gastrointest Dis 13(2):109-19
66. Thunell S, Harper P (2000). Porphyrins, porphyrin metabolism, por-phyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda--suggestions for a handling programme. Scand J Clin Lab In-vest 60(7):561-79
67. Frank J, Poblete-Gutierrez P (2010). Porphyria cutanea tarda--when skin meets liver. Best Pract Res Clin Gastroenterol 24(5):735-45
68. Ryan Caballes F, Sendi H, Bonkovsky HL (2012). Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 32(6): 880-93
69. Phillips JD, Bergonia HA, Reilly CA et al (2007). A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cuta-nea tarda. Proc Natl Acad Sci U S A 104(12):5079-84
70. Lee KG, Hyun JJ, Seo YS et al (2010). Liver cirrhosis induced

S56
123
Intern Emerg Med (2016) 11 (Suppl):S50-S56
by porphyria cutanea tarda: a case report and review. Gut Liver 4(4):551-5
71. Ged C, Moreau-Gaudry F, Richard E et al (2009). Congenital erythro-poietic porphyria: mutation update and correlations between genotype and phenotype. Cell Mol Biol (Noisy-le-grand) 55(1):53-60
72. Desnick RJ, Astrin KH (2002). Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol 117(4):779-95
73. Dawe SA, Peters TJ, Du Vivier A, Creamer JD (2002). Congeni-tal erythropoietic porphyria: dilemmas in present day management. Clin Exp Dermatol 27(8):680-3
74. Mullard A (2015). Clinuvel’s tanning agent nears approval for por-phyria. Nat Biotechnol 33(1):7
75. Rocchi E, Gibertini P, Cassanelli M et al (1986). Serum ferritin in the assessment of liver iron overload and iron removal therapy in porphyria cutanea tarda. J Lab Clin Med 107(1):36-42
76. Sarkany RP (2001). The management of porphyria cutanea tarda. Clin Exp Dermatol 26(3):225-32
77. Harada FA, Shwayder TA, Desnick RJ, Lim HW (2001). Treatment of severe congenital erythropoietic porphyria by bone marrow trans-plantation. J Am Acad Dermatol 45(2):279-82

© SIMI 2016
123
FOCUS ON
Intern Emerg Med (2016) 11 (Suppl):S57-S60
Corrado Tamburino (•)Divisione di Cardiologia, Ospedale Ferrarotto Università di CataniaVia Salvatore Citelli 6, 95124 CataniaE-mail: [email protected]
M. BarbantiDivisione di Cardiologia, Ospedale Ferrarotto Università di Catania, Catania
Le nuove frontiere della cardiologia interventistica strutturale
Marco Barbanti • Corrado Tamburino
Introduzione
La cardiologia interventistica strutturale rappresenta un folto insieme di trattamenti innovativi, che negli ultimi 10-15 anni sono cresciuti da un ristretto numero di interventi ad una rosa vastissima di approcci terapeutici a patologie cardiache fino-ra di stretta pertinenza cardiochirurgica o della sola terapia medica. Alcune procedure hanno l’obiettivo di migliorare la sopravvivenza e di prevenire manifestazioni cliniche (ictus, scompenso cardiaco etc.), mentre altre sono più votate a mi-gliorare la sintomatologia clinica e la qualità di vita. Si tratta di una disciplina relativamente giovane, che sta suscitando un grande interesse scientifico, ed è considerata da molti come la “terza grande ondata” della cardiologia interventistica dopo quelle dell’angioplastica coronarica e vascolare [1]. Negli ul-timi anni la crescita di questa disciplina è stata trainata prin-cipalmente dal trattamento percutaneo delle due valvulopatie più frequenti nei Paesi industrializzati: la stenosi aortica (SA) e l’insufficienza mitralica (IM). L’impianto transcatetere della valvola aortica (TAVI, Transcatheter Aortic Valve Implanta-tion) e il trattamento transcatetere dell’insufficienza valvolare mitralica (riparazione e sostituzione) monopolizzano infatti gran parte degli argomenti trattati nei principali congressi del settore [2,3]. Questo avviene alla luce della continua produ-zione di evidenze scientifiche e dell’incredibile numero di nuovi dispositivi e tecniche che vengono introdotti nell’arena.
Tuttavia, numerose altre tecniche di cardiologia interven-tistica sono oggi. Tra queste, le più diffuse sono la chiusura
percutanea dell’auricola sinistra in pazienti affetti da fibrilla-zione atriale con controindicazioni alla terapia anticoagulante; il trattamento di cardiopatie congenite (difetto interatriale ed interventricolare, pervietà del forame ovale e del dotto di Bo-tallo, coartazione aortica); la chiusura dei leak periprotesici valvolari; il partizionamento del ventricolo sinistro; il tratta-mento delle valvulopatie del cuore destro e delle stenosi dei rami principali dell’arteria polmonare [1]. Va detto però che, a differenza della TAVI e del trattamento dell’insufficienza mi-tralica, queste ultime sono procedure più consolidate e utiliz-zate e fanno parte della regolare pratica clinica da diversi anni.
Trattamento percutaneo della stenosi aortica
Nel corso degli ultimi 50 anni, l’intervento di sostituzione valvolare aortica chirurgica si è evoluto fino ad affermarsi come il gold standard terapeutico in pazienti affetti da SA se-vera [4]. Nonostante gli innumerevoli progressi ottenuti dalla cardiochirurgia, esistono condizioni (soprattutto in soggetti anziani) che incrementano esponenzialmente il rischio ope-ratorio e rendono l’intervento chirurgico ad alto rischio o, in certi casi, quasi proibitivo. Ne deriva che, paradossalmente, nella popolazione in cui vi è la maggiore prevalenza della patologia valvolare aortica, l’intervento convenzionale non viene eseguito nonostante esistano chiare indicazioni.
La TAVI è nata pertanto per rispondere ad una necessità cli-nica irrisolta e nell’ultimo decennio tale metodica ha subito so-stanziali miglioramenti, è stata studiata in studi clinici rigorosi, ed è stata messa a disposizione della comunità clinica di tutto il mondo. Dai primi impianti pionieristici eseguiti da Alain Cribier nel 2002 [5] sono trascorsi poco più di 15 anni, ma la TAVI è stata già completamente integrata nell’armamentario terapeutico per la gestione dei pazienti con SA severa [4]. Sulla base delle attuali linee guida internazionali (ultimo aggiorna-mento nel 2014), la TAVI deve essere riservata a pazienti ino-perabili (classe di raccomandazione A, linea di evidenza B) o

S58
123
Intern Emerg Med (2016) 11 (Suppl):S57-S60
ad alto rischio chirurgico (classe di raccomandazione IIa, linea di evidenza B) [4]. Queste indicazioni si basano sui risultati di diversi Registri europei e di due trial randomizzati che han-no utilizzato protesi valvolari di prima generazione: lo studio PARTNER (coorti A e B) [6,7] e il CoreValve U.S. [8]. Que-sti ultimi hanno infatti dimostrato che la TAVI garantisce una migliore sopravvivenza rispetto alla terapia medica in pazienti inoperabili (PARTNER coorte B) e garantisce risultati a medio termine sovrapponibili (PARTNER coorte A) o superiori (Co-reValve U.S.) rispetto alla chirurgia tradizionale in pazienti ad alto rischio. Sin dalle prime procedure, è emerso che la TAVI, per quanto si tratti di una tecnica mini-invasiva, è gravata da diverse complicanze [9]. Tra tutte, quelle più studiate, in con-siderazione del loro rilevante impatto su prognosi, qualità di vita e costi, sono gli eventi cerebrovascolari, le complicanze vascolari, i disturbi di conduzione, l’insufficienza renale acuta ed il rigurgito paravalvolare aortico residuo [9].
Oggi sono disponibili e approvati per utilizzo clinico in Europa otto dispositivi TAVI denominati di “nuova genera-zione” [3]. Questi, rispetto alle precedenti generazioni, hanno incorporato numerose caratteristiche (minore profilo, mem-brane esterne che consentono di ridurre i rigurgiti periprotesi-ci, possibilità di essere ricatturate e riposizionate) che hanno consentito di ridurre drasticamente molte delle complicanze legate alla procedura. È fuor di dubbio tuttavia che il miglio-ramento dei risultati clinici sia stato ottenuto anche grazie a una maggiore esperienza degli operatori e a una selezione cli-nica e anatomica sicuramente più avanzate.
Questi aspetti, unitamente alle evidenze crescenti sulla du-rata delle protesi percutanee, hanno spinto la comunità scien-tifica a prevedere un ampliamento delle indicazioni a TAVI anche in popolazioni a più basso rischio. In questo contesto, gli studi randomizzati PARTNER 2 e NOTION e lo studio OBSERVANT hanno recentemente dimostrato che la TAVI non è inferiore alla sostituzione chirurgica anche in pazienti a rischio intermedio [10-12]. Una sottoanalisi predetermi-nata del trial PARTNER ha inoltre mostrato che l’approccio transarterioso femorale (utilizzato in circa il 90% dei casi) ha consentito alla TAVI di ottenere risultati anche superiori rispetto a quelli della chirurgia tradizionale [10].
La Food and Drug Administration (FDA) ha recentemente approvato l’inizio di due ulteriori trial randomizzati che con-fronteranno la TAVI con la sostituzione valvolare chirurgica anche in pazienti a basso rischio.
Negli ultimi mesi, la discussione sull’estensione delle in-dicazioni della TAVI non si limita solamente alle categorie di rischio più basse, ma anche ai diversi scenari clinici o ana-tomici che possono riscontrarsi in pazienti con SA. Ad oggi infatti, esistono diverse condizioni in cui le evidenze a favore della TAVI sono meno robuste. Tra queste, quelle che hanno una maggiore rilevanza clinica sono:
1. la bicuspidia aortica: generalmente esclusa dai grandi trial e associata a risultati meno soddisfacenti quando la TAVI è eseguita con protesi di prima generazione, esistono ancora poche evidenze che i nuovi dispositivi possano garantire risultati più favorevoli [13,14];
2. la patologia coronarica concomitante: per quanto sia stata ampiamente dimostrata la fattibilità e la sicurezza di un approccio percutaneo combinato con angioplastica e TAVI (stadiato o nella stessa seduta), resta da definire se que-sta strategia di trattamento possa essere paragonabile con l’intervento chirurgico di sostituzione valvolare aortica e bypass coronarico in termini di efficacia a medio e lungo termine [15];
3. l’insufficienza mitralica concomitante: anche in questo caso esistono poche e contrastanti evidenze su quale possa essere il migliore approccio terapeutico in questa categoria di pazienti [16-18].
Una volta data una risposta ai quesiti e quando avremo più dati su pazienti a basso rischio, è probabile che l’utilizzo della TAVI non sarà vincolato da una stratificazione del rischio, ma sarà influenzato più da specifici fattori anatomici e clinici in cui un “heart team” multidisciplinare giocherà un ruolo sempre più centrale.
Trattamento percutaneo dell’insufficienza mitralica
L’insufficienza mitralica (IM) è la seconda valvulopatia più frequente nella popolazione occidentale [19]. Nella sua forma severa, essa è associata a una ridotta aspettativa di vita e a un aumento del rischio di insufficienza cardiaca [19]. Per anni l’intervento chirurgico di sostituzione o riparazione valvolare è stato l’unico approccio percorribile [4]. Negli ultimi anni sono stati sviluppati numerosi dispositivi per la riparazione della valvola mitralica per via transcatetere [20]. La gran parte di questi si basa su principi e tecniche già ampiamen-te utilizzati dalla cardiochirurgia. Si tratta di una tecnologia in grandissimo fermento, tuttavia le popolazioni di pazienti che oggi possono beneficiare di questa tipologia di interventi è molto ristretta a causa della scarsa mole di dati clinici a supporto. Inoltre, la valvola mitrale è significativamente più complessa rispetto alla valvola aortica. Essa infatti è solo uno dei componenti di un apparato ben più complesso di cui fanno parte anche il ventricolo sinistro stesso, oltre ai lembi, l’anel-lo mitralico, i muscoli papillari e le corde tendinee. Questa complessità è alla base di gran parte degli insuccessi di molti dispositivi di riparazione percutanea della valvola mitralica che si sono succeduti negli ultimi anni.
La conoscenza della patologia e del meccanismo che sot-tendono il rigurgito mitralico risultano pertanto fondamentali

S59
123
Intern Emerg Med (2016) 11 (Suppl):S57-S60
per la selezione di quei sistemi di riparazione che possono garantire i migliori risultati.
Oggi sono disponibili una serie di diverse tecnologie. Esse possono essere classificate in base al target anatomico e fisio-patologico in: – procedure di riparazione dei lembi e delle corde tendinee; – procedure di anuloplastica indiretta o diretta [21].
Attualmente la MitraClip (Abbott Vascular, USA) è il di-spositivo di gran lunga più utilizzato nella pratica clinica con circa 20.000 pazienti trattati in tutto il mondo [22]. Tale dispo-sitivo si è mostrato particolarmente versatile e può essere uti-lizzato con successo in pazienti sia con insufficienza mitralica funzionale sia degenerativa. La procedura consiste nel posi-zionamento di una “clip”, che ancora i lembi anteriore e po-steriore mitralici creando una valvola a doppio orifizio (questo approccio mima una tecnica chirurgica introdotta nei primi anni ‘90 da Ottavio Alfieri e definita tecnica “edge-to-edge”). La MitraClip ha ottenuto il marchio CE nel 2008 e l’approva-zione da parte della FDA nel 2013 per il trattamento di pazien-ti sintomatici con insufficienza mitralica degenerativa ad alto rischio chirurgico. Dal punto di vista tecnico, la maggior parte delle tecnologie di riparazione mitralica percutanea richiede competenze avanzate e specifiche. Alcune tecniche sono più facili da eseguire: ad esempio l’anuloplastica indiretta, esegui-ta mediante il posizionamento di due “ancore” nel seno coro-narico, si basa sulla vicinanza anatomica del seno coronarico con l’anulus posteriore mitralico. Questo approccio è partico-larmente interessante perché la cannulazione del seno coro-narico è una tecnica di accesso venoso facile e riproducibile. Anche se questa procedura ha comportato un discreto miglio-ramento clinico dei pazienti trattati, questo approccio è poco adottato, principalmente a causa della sua limitata efficacia (il seno coronarico e l’anulus mitralico hanno rapporti anatomici estremamente variabili) e al rischio di compressione dell’arte-ria coronaria e dislocazione periferica [20].
Più di recente si è affermata una tecnica di anuloplastica diretta transcatetere che riproduce molto fedelmente il posi-zionamento di un anello chirurgico. La procedura prevede un approccio transettale e l’innesto di diversi “tasselli” sul lato atriale dell’anello mitralico, da trigono a trigono, che permet-tono la riduzione delle dimensioni dell’anello, favorendo la coaptazione dei lembi (Cardioband). Pur trattandosi di una procedura complessa, i primi risultati si sono dimostrati pro-mettenti, sia in termini di riduzione dell’insufficienza mitra-lica sia di miglioramento della sintomatologia clinica [23].
Oggi, il limite principale di tutte le tecniche di riparazione mitralica percutanea mediante anuloplastica diretta è la loro complessità tecnica, che potrebbe limitare la loro diffusione nella pratica clinica.
Oltre le tecniche di anuloplastica indiretta e diretta, sono
in sperimentazione altre tecniche di rimodellamento dell’a-nulus mitralico (compressione esterna del solco atrio-ventri-colare, impianto di dispositivi che permettono una plicatura dell’anulus e l’impiego di fonti di energia a radiofrequenza o ultrasuoni per ridurre il collagene anulare) [23]. Tuttavia, la riproducibilità, l’efficacia e la sicurezza di queste tecnologie devono essere ancora dimostrate.
Negli ultimi anni, la tecnica che sta suscitando maggior-mente l’interesse scientifico è l’impianto transcatetere di protesi mitraliche [24]. Questa procedura è stata utilizzata con successo per il trattamento di protesi mitraliche chirur-giche degenerate, o per recidive di insufficienza mitralica in seguito a riparazione mitralica con anello mediante l’impian-to di protesi percutanee, nate per il trattamento della stenosi aortica. L’impianto di protesi transcatetere, disegnate per il trattamento di valvole mitraliche native, rappresenta l’ultima sfida per il cardiologo interventista. Queste protesi sono auto-espandibili ed impiantate attraverso l’accesso transapicale o transettale. Rispetto alla riparazione, la sostituzione mitralica può offrire alcuni vantaggi teorici, come una maggiore ripro-ducibilità e applicabilità in popolazioni di pazienti più vaste. Tuttavia, la riparazione transcatetere della mitrale, anche se più complessa e tecnicamente impegnativa, è più fisiologica e quindi associata a un profilo di sicurezza superiore rispetto alla sostituzione, in quanto non comporta un impianto di tes-suto eterologo e non richiede terapia anticoagulante. Il saldo complessivo tra i vantaggi e gli svantaggi dei due approcci in-troduce pertanto la necessità di selezionare una tecnica sulla base delle specifiche caratteristiche di ogni singolo paziente. Infatti, è verosimile che in futuro la riparazione e la sostitu-zione mitralica transcatetere abbiano un ruolo complementa-re, piuttosto che competitivo.
Bibliografia
1. Feldman T, Hellig F, Möllmann H (2016). Structural heart interven-tions: the state of the art and beyond. EuroIntervention May 17;12 Suppl X:X6
2. Barbanti M, Immè S, Grasso C (2015). Transcatheter mitral valve repair: a brief review. EuroIntervention Sep;11 Suppl W:W42-4
3. Vahl TP, Kodali SK, Leon MB (2016). Transcatheter aortic valve replacement 2016: A modern-day “through the looking-glass” ad-venture. J Am Coll Cardiol 67:1472-87
4. Nishimura RA, Otto CM, Bonow RO et al; ACC/AHA Task Force Members (2014). 2014 AHA/ACC Guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 129:e521-643
5. Cribier A, Eltchaninoff H, Bash A et al (2002). Percutaneous tran-scatheter implantation of an aortic valve prosthesis for calcific aortic stenosis: first human case description. Circulation 106:3006-8
6. Mack MJ, Leon MB, Smith CR et al; PARTNER 1 trial investigators (2015). 5-year outcomes of transcatheter aortic valve replacement

S60
123
Intern Emerg Med (2016) 11 (Suppl):S57-S60
or surgical aortic valve replacement for high surgical risk patients with aortic stenosis (PARTNER 1): a randomised controlled trial. Lancet 385:2477-84
7. Kapadia SR, Leon MB, Makkar RR et al; PARTNER trial investiga-tors (2015). 5-year outcomes of transcatheter aortic valve replace-ment compared with standard treatment for patients with inoperable aortic stenosis (PARTNER 1): a randomised controlled trial. Lancet 385:2485-91
8. Deeb GM, Reardon MJ, Chetcuti S et al; CoreValve US Clinical Investigators (2016). Three-year outcomes in high-risk patients who underwent surgical or transcatheter aortic valve replacement. J Am Coll Cardiol Mar 22. [Epub ahead of print]
9. Généreux P, Head SJ, Van Mieghem NM et al (2012). Clinical out-comes after transcatheter aortic valve replacement using valve aca-demic research consortium definitions: a weighted meta-analysis of 3,519 patients from 16 studies. J Am Coll Cardiol 59:2317-26
10. Leon MB, Smith CR, Mack MJ et al; PARTNER 2 Investigators (2016). Transcatheter or surgical aortic-valve replacement in inter-mediate-risk patients. N Engl J Med 374:1609-20
11. Thyregod HG, Steinbrüchel DA, Ihlemann N et al (2015). Tran-scatheter versus surgical aortic valve replacement in patients with severe aortic valve stenosis: 1-year results from the all-comers NO-TION randomized clinical trial. J Am Coll Cardiol 65:2184-94
12. Tamburino C, Barbanti M, D’Errigo P et al; OBSERVANT Research Group (2015). 1-Year outcomes after transfemoral transcatheter or surgical aortic valve replacement: results from the Italian OBSER-VANT study. J Am Coll Cardiol 66:804-12
13. Mylotte D, Lefevre T, Søndergaard L et al (2014). Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 64:2330-9
14. Perlman GY, Blanke P, Dvir D et al (2016). Bicuspid aortic valve stenosis: favorable early outcomes with a next-generation transcath-eter heart valve in a multicenter study. JACC Cardiovasc Interv 9:817-24
15. Goel SS, Agarwal S, Tuzcu EM et al (2012). Percutaneous coronary intervention in patients with severe aortic stenosis: implications for transcatheter aortic valve replacement. Circulation 28;125:1005-13
16. Barbanti M, Dvir D, Tan J, Webb JG (2013). Aortic stenosis and mitral regurgitation: implications for transcatheter valve treatment. EuroIntervention Sep 10;9 Suppl:S69-71
17. Barbanti M, Webb JG, Hahn RT et al; Placement of Aortic Tran-scatheter Valve Trial Investigators (2013). Impact of preoperative moderate/severe mitral regurgitation on 2-year outcome after tran-scatheter and surgical aortic valve replacement: insight from the Placement of Aortic Transcatheter Valve (PARTNER) Trial Cohort A. Circulation 128:2776-84
18. Nombela-Franco L, Eltchaninoff H, Zahn R et al (2015). Clinical impact and evolution of mitral regurgitation following transcatheter aortic valve replacement: a meta-analysis. Heart 101:1395-405
19. Enriquez-Sarano M, Avierinos JF, Messika-Zeitoun D et al (2005). Quantitative determinants of the outcome of asymptomatic mitral regurgitation. N Engl J Med 352:875-83
20. Taramasso M, Candreva A, Pozzoli A et al (2015). Current challeng-es in interventional mitral valve treatment. J Thorac Dis 7:1536-42
21. Feldman T (2016). The explosion of percutaneous mitral valve ther-apies. J Cardiovasc Surg (Torino) 57:350-1
22. Deuschl F, Schofer N, Lubos E et al (2015). MitraClip-data analysis of contemporary literature. J Thorac Dis Sep;7(9):1509-17
23. Maisano F, Taramasso M, Nickenig G et al (2016). Cardioband, a transcatheter surgical-like direct mitral valve annuloplasty system: early results of the feasibility trial. Eur Heart J 37:817-25
24. Figulla HR, Webb JG, Lauten A, Feldman T (2016). The transcath-eter valve technology pipeline for treatment of adult valvular heart disease. Eur Heart J May 8. pii: ehw153. [Epub ahead of print]
Disclosures: Marco Barbanti è consulente per Edwards Lifesciences; Corrado Tamburino ha ricevuto onorari da Abbott Vascular, St Jude Medical e Medtronic Inc.

© SIMI 2016
123
IL PUNTO SU
Intern Emerg Med (2016) 11 (Suppl):S61-S64
Corrado Blandizzi (•)Divisione di FarmacologiaDipartimento di Medicina Clinica e SperimentaleUniversità di PisaVia Roma 55, 56126 PisaE-mail: [email protected]
Sicurezza e appropriatezza degli inibitori di pompa protonica
Corrado Blandizzi
Introduzione
I farmaci inibitori di pompa protonica (PPI) disponibili per l’uso clinico comprendono omeprazolo, lansoprazolo, pan-toprazolo, rabeprazolo ed esomeprazolo. A causa del loro potente effetto inibitore sulla secrezione acida, i PPI sono impiegati nel trattamento di varie condizioni patologiche comprendenti soprattutto la malattia da reflusso gastro-eso-fageo (GERD), la malattia peptica associata all’infezione da Helicobacter pylori (HP), il danno gastro-duodenale indotto da farmaci anti-infiammatori non steroidei (FANS) e alcune forme di dispepsia funzionale.
L’impiego dei PPI continua a crescere in tutto il mondo, soprattutto come trattamento a lungo termine, ma le ragioni di questo fenomeno non sono ben chiare. La terapia cronica con PPI è di fatto indicata nei casi di GERD grave o come trattamento profilattico nei pazienti con fattori di rischio in trattamento con FANS a lungo termine. In tutti gli altri casi, soprattutto nei pazienti trattati con farmaci diversi dai FANS e per i quali le evidenze di azioni gastro-protettive da parte dei PPI sono inconsistenti o assenti, la terapia con PPI è inap-propriata [1].
I PPI sono generalmente prescritti a popolazioni di pa-zienti eterogenee, appartenenti a varie fasce di età, spesso in presenza di varie condizioni di comorbilità e, quindi, di poli-terapie farmacologiche. Nonostante tale eterogeneità, tutti i PPI hanno mostrato un buon profilo di tollerabilità nell’uso sia a breve che a lungo termine. Nel breve termine gli effet-ti avversi più comuni sono rappresentati da cefalea, astenia, sonnolenza, capogiri, ansia, depressione, dispepsia, nausea,
vomito, fastidio addominale, diarrea, stipsi, gonfiore addo-minale [2]. Vengono riportate anche alterazioni dei parametri ematochimici, tra i quali aumento delle transaminasi, abbas-samento del sodio, variazioni dei leucociti e aumento della gamma-glutamiltransferasi [3,4].
Le osservazioni a lungo termine indicano un profilo di tollerabilità dei PPI sovrapponibile a quello riscontrato nei trattamenti a breve termine. Tuttavia, la crescente tendenza a somministrare i PPI per periodi estesi ha fatto emergere il rischio di effetti avversi dei quali si sta cercando di com-prendere l’effettiva prevalenza e rilevanza clinica [3,4], e che sembrano in larga misura associati alla prolungata inibizione della secrezione gastrica acida.
Gli eventi avversi associati alla terapia a lungo termine con PPI si possono suddividere in due categorie maggiori: - conseguenze dell’inibizione della secrezione acida sulla
morfologia, trofismo e funzioni assorbitive della mucosa digestiva;
- relazione tra inibizione della secrezione acida e rischio in-fettivo.
Gli eventi che rientrano nella prima categoria comprendo-no: polipi ghiandolari del fondo gastrico; malassorbimento di vitamina B12, malassorbimento di calcio e rischio di fratture ossee; malassorbimento di ferro; ipomagnesemia. Nella se-conda categoria rientrano: diarrea e colite da Clostridium dif-ficile; iperproliferazione batterica nell’intestino tenue (SIBO); polmonite acquisita in comunità o in ospedale [4].
Effetti dei PPI sul trofismo e le funzioni della mucosa digestiva
La soppressione della secrezione acida indotta dai PPI de-termina una condizione di ipergastrinemia, che, a sua volta, può causare iperplasia delle cellule ECL della mucosa gastri-ca, ma non sembra in grado di promuovere la formazione di

S62
123
Intern Emerg Med (2016) 11 (Suppl):S61-S64
tumori carcinoidi o di altre neoplasie maligne. Una recente revisione sistematica della letteratura ha valutato gli effetti di terapie a lungo termine (3,5-15 anni) con PPI su livelli ematici di gastrina e morfologia della mucosa gastrica. Gli autori hanno osservato incrementi della gastrinemia da uno a tre volte rispetto al normale, in concomitanza con iperplasia delle cellule ECL, aumento del rischio di displasia delle cel-lule ECL e atrofia del corpo gastrico in pazienti HP positivi in assenza di cambiamenti di natura neoplastica [5]. Poiché l’iperplasia delle cellule ECL è stata riscontrata soprattutto nei pazienti HP positivi particolarmente in presenza di atrofia e infiammazione grave della mucosa gastrica, alcuni autori consigliano l’eradicazione di HP nei pazienti sottoposti a trattamenti cronici con PPI, ma questo orientamento non è unanimemente condiviso [3].
Pazienti in terapia a lungo termine con PPI possono svi-luppare formazioni polipoidi della mucosa del fondo-corpo gastrico. Uno studio caso-controllo su circa 600 pazienti ha evidenziato un aumento più elevato del rischio di lesioni po-lipoidi fundiche in pazienti trattati con PPI per oltre un anno rispetto a quelli con terapia a breve termine. Queste formazio-ni sembrano tuttavia prive della capacità di evolvere in lesioni neoplastiche maligne [6].
Un aspetto molto discusso concerne la possibile relazio-ne tra terapia a lungo termine con PPI, ipergastrinemia e rischio di carcinoma del colon-retto. La gastrina può infatti stimolare la proliferazione di diversi tipi di epiteli, compreso quello della mucosa colica, e studi su modelli preclinici han-no suggerito un’associazione tra ipergastrinemia indotta da omeprazolo e aumento dell’attività proliferativa di adenomi colo-rettali. Tuttavia, studi osservazionali che hanno valutato pazienti con un follow-up di oltre 15 anni hanno evidenziato che la terapia a lungo termine con PPI non è associata a un rischio significativo di carcinoma colo-rettale [3,4].
Relazione tra PPI, variazioni della vitamina B12 e del ferro e anemia
La vitamina B12 si trova negli alimenti in forma coniugata con proteine dalle quali, una volta raggiunto il lume gastrico, deve essere dissociata tramite l’azione del secreto acido-pep-tico per poter poi essere assorbita a livello intestinale. L’inibi-zione cronica della secrezione acida indotta dai PPI potrebbe quindi interferire con l’assorbimento della vitamina B12 fino a indurre condizioni patologiche. Alcuni studi hanno valutato i livelli ematici di vitamina B12 nei pazienti trattati con PPI ottenendo risultati contrastanti. Tuttavia, la misurazione della vitamina B12 da sola non è sufficiente a evidenziare uno stato di carenza, che di solito si associa a un parallelo aumento dei livelli di omocisteina e acido metilmalonico. Infatti, l’analisi
concomitante di questi tre parametri ha permesso di rilevare una carenza di vitamina B12 in circa il 30% dei pazienti trat-tati con PPI a lungo termine [7].
L’assorbimento del ferro presente negli alimenti è facili-tato dall’acidità gastrica, per cui periodi protratti di terapia con PPI potrebbero causare malassorbimento di ferro, con rischio di anemia. Stewart et al. [8] hanno valutato pazienti con sindrome di Zollinger-Ellison trattati a lungo termine con PPI, osservando che il trattamento con omeprazolo per 6 anni non ha causato carenza di ferro circolante. Il monitoraggio del metabolismo del ferro non sembra quindi indicato nei pa-zienti in terapia cronica con PPI.
Effetti dei PPI su calcio e metabolismo del tessuto osseo
La possibile relazione tra PPI e rischio di fratture è stata esa-minata da meta-analisi condotte su studi osservazionali che hanno evidenziato un modesto, ma significativo, grado di associazione [9]. I meccanismi biologici coinvolti non sono chiari, ma sono stati suggeriti effetti contrapposti dei PPI sul metabolismo osseo, quali: – inibizione delle pompe protoniche degli osteoclasti, con
riduzione del riassorbimento osseo; – inibizione dell’assorbimento di calcio; – stimolazione del riassorbimento osseo indotta dall’iperga-
strinemia [10].
Sembra comunque opportuna una certa cautela nei casi in cui sia necessario prescrivere un PPI a pazienti con rischio elevato di sviluppare fratture ossee: verifica accurata dell’ef-fettiva indicazione terapeutica, utilizzazione della minima dose efficace e monitoraggio dei fattori di rischio osseo.
PPI ed episodi acuti di ipomagnesemia, ipopotassiemia e ipocalcemia
Sono stati riportati casi di ipomagnesemia associati a terapia a lungo termine con PPI. Questi pazienti hanno sviluppato ipomagnesemia grave, spesso associata a ipocalcemia secon-daria, che ha richiesto l’ospedalizzazione, tendeva a persiste-re malgrado la somministrazione di magnesio e si è risolta in 1-2 settimane dopo sospensione della terapia con PPI. Una recente meta-analisi ha evidenziato un rischio moderato, ma significativo, nei pazienti trattati con PPI [11]. I meccanismi che correlano i PPI all’ipomagnesemia non sono chiari e an-che i fattori di rischio non sono noti. È stato tuttavia suggerito che i PPI possano interferire con l’assorbimento intestinale del magnesio, riducendo la sua affinità per i sistemi di tra-sporto attivo TRPM-6/7 [12]. I medici dovrebbero tenere in

S63
123
Intern Emerg Med (2016) 11 (Suppl):S61-S64
considerazione questa possibile associazione e interrompere il trattamento con PPI nei casi di insorgenza di ipomagnese-mia grave in assenza di disfunzioni renali o malassorbimento intestinale.
PPI e rischio infettivo
Un’importante funzione dell’acidità gastrica è quella di inat-tivare i microrganismi introdotti per via orale e impedire che raggiungano il lume intestinale. Pertanto, la marcata inibizio-ne acida indotta dai PPI può aumentare il rischio di infezioni enteriche (per esempio Vibrio cholerae, Shigella spp., Salmo-nella spp., Clostridium difficile) e di infestazioni parassitarie (per esempio giardiasi e amebiasi) [3,4].
L’impiego eccessivo di PPI, soprattutto nei pazienti anziani, è stato associato a episodi di infezione epidemica da Clostri-dium difficile, che si manifesta con diarrea grave [4]. Tale as-sociazione sembra dipendere dal fatto che elevati valori di pH nel lume gastrico favoriscono l’inizio della germinazione delle spore del Clostridium, seguita da una rapida proliferazione a livello del lume intestinale [2,4]. Valutazioni meta-analitiche, condotte su studi osservazionali, hanno valutato il rischio di in-fezione enterica da Clostridium difficile nei pazienti trattati con PPI e tutte hanno evidenziato associazioni significative [13].
L’inibizione della secrezione gastrica può promuovere pro-cessi di iperproliferazione batterica nell’intestino tenue (SIBO), che clinicamente si manifestano con gonfiore addominale, fla-tulenza, dolore addominale e diarrea. Di recente, un’associa-zione significativa tra terapia con PPI e rischio di insorgenza di SIBO è stata evidenziata da una meta-analisi, condotta su 11 studi, che ha stimato un odds ratio di 2,28 (1,24-4,20) [14]. In letteratura sono stati riportati anche casi di peritonite batterica spontanea in pazienti con cirrosi epatica trattati con PPI. Una recente meta-analisi ha effettivamente evidenziato una relazio-ne significativa tra terapia con PPI e insorgenza di peritonite batterica spontanea in pazienti anziani con cirrosi ricoverati, con un odds ratio di 3,15 (2,09-4,74) [15].
È stata suggerita una relazione causale tra trattamento con PPI e sviluppo di polmonite infettiva acquisita in ospedale o in comunità. Tuttavia gli studi osservazionali sull’argomento non sono riusciti a stabilire una chiara correlazione tra que-sti eventi avversi e la terapia a lungo termine con PPI. Una meta-analisi ha valutato 8 studi osservazionali evidenziando un rischio modesto di polmonite per i PPI, e 23 studi clinici randomizzati e controllati che non hanno confermato tale ri-schio [16]. Una successiva meta-analisi non ha evidenziato un rischio significativo di ospedalizzazione per polmonite acquisita in comunità nei pazienti in terapia con PPI [17]. Sebbene i meccanismi che correlano la polmonite ai PPI non siano chiari, è stato suggerito che la colonizzazione del tratto
digerente prossimale da parte dei batteri svolga un ruolo rile-vante in questo processo. Inoltre alcune popolazioni cellulari del sistema respiratorio, soprattutto quelle secernenti muco, sono munite di pompe protoniche. Pertanto è stato ipotizzato che le variazioni di pH delle secrezioni mucose delle vie re-spiratorie, indotte dal trattamento con PPI, potrebbero contri-buire allo sviluppo di processi infettivi polmonari [3].
Conclusioni
I dati attualmente disponibili non pongono specifiche con-troindicazioni al trattamento a lungo termine con PPI nei pazienti con una chiara indicazione terapeutica. Nei pazien-ti con GERD, la dose di mantenimento nel singolo pazien-te viene stabilita in base alla risposta in termini di efficacia e tollerabilità. I pazienti anziani, con necessità di assumere cronicamente FANS e rischio elevato di complicanze gastro-duodenali, sono candidati a lunghi cicli di terapia con PPI. In generale, i PPI mostrano un favorevole profilo di tollerabilità, sia a breve che a lungo termine. Tale tollerabilità, monito-rata per periodi di oltre 15 anni, ne consente l’utilizzazione in popolazioni eterogenee per età, presenza di comorbilità e terapia concomitante con altri farmaci. Tuttavia, l’uso scor-retto dei PPI può comportare un rischio di eventi avversi, ed è quindi importante evitare il più possibile che questi farmaci vengano prescritti in maniera inappropriata a pazienti per i quali non sussiste un’effettiva indicazione terapeutica.
Bibliografia
1. Masclee GM, Sturkenboom MC, Kuipers EJ (2014). A benefit-risk assessment of the use of proton pump inhibitors in the elderly. Drugs Aging 31:263-82. doi: 10.1007/s40266-014-0166-4
2. Boparai V, Rajagopalan J, Triadafilopoulos G (2008). Guide to the use of proton pump inhibitors in adult patients. Drugs 68:925-47. doi: 10.2165/00003495-200868070-00004
3. Savarino V, Di Mario F, Scarpignato C (2009). Proton pump inhib-itors in GORD. An overview of their pharmacology, efficacy and safety. Pharmacol Res 59:135-53. doi: 10.1016/j.phrs.2008.09.016
4. Ali T, Roberts DN, Tierney WM (2009). Long-term safety con-cerns with proton pump inhibitors. Am J Med 122:896-903. doi: 10.1016/j.amjmed.2009.04.014
5. Lundell L, Vieth M, Gibson F et al (2015). Systematic review: the effects of long-term proton pump inhibitor use on serum gastrin lev-els and gastric histology. Aliment Pharmacol Ther 42:649-63. doi: 10.1111/apt.13324
6. Freeman HJ (2008). Proton pump inhibitors and an emerging ep-idemic of gastric fundic gland polyposis. World J Gastroenterol 14:1318-20
7. Hirschowitz BI, Worthington J, Mohnen J (2008). Vitamin B12 de-ficiency in hypersecretors during long-term acid suppression with proton pump inhibitors. Aliment Pharmacol Ther 27:1110-21. doi: 10.1111/j.1365-2036.2008.03658.x

S64
123
Intern Emerg Med (2016) 11 (Suppl):S61-S64
8. Stewart CA, Termanini B, Sutliff VE et al (1998). Iron absorption in patients with Zollinger-Ellison syndrome treated with long-term gastric acid antisecretory therapy. Aliment Pharmacol Ther 12:83-98. doi: 10.1046/j.1365-2036.1998.00274.x
9. Ngamruengphong S, Leontiadis GI, Radhi S et al (2011). Proton pump inhibitors and risk of fracture: a systematic review and me-ta-analysis of observational studies. Am J Gastroenterol 106:1209-18. doi: 10.1038/ajg.2011.113
10. Yang YX, Metz DC (2010). Safety of proton pump inhibitor expo-sure. Gastroenterology 139:1115-27. doi: 10.1053/j.gastro.2010. 08.023
11. Park CH, Kim EH, Roh YH et al (2014). The association between the use of proton pump inhibitors and the risk of hypomagnesemia: a systematic review and meta-analysis. PLoS One 9:e112558. doi: 10.1371/journal.pone.0112558
12. Perazella MA (2013). Proton pump inhibitors and hypomagne-semia: a rare but serious complication. Kidney Int 83:553-6. doi: 10.1038/ki.2012.462
13. Furuya-Kanamori L, Stone JC, Clark J et al (2015). Comorbidities, exposure to medications, and the risk of community-acquired Clos-tridium difficile infection: a systematic review and meta-analysis. In-fect Control Hosp Epidemiol 36:132-41. doi: 10.1017/ice.2014.39.
14. Lo WK, Chan WW (2013). Proton pump inhibitor use and the risk of small intestinal bacterial overgrowth: a meta-analysis. Clin Gas-troenterol Hepatol 11:483-90. doi: 10.1016/j.cgh.2012.12.011
15. Deshpande A, Pasupuleti V, Thota P et al (2013). Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cir-rhotic patients: a meta-analysis. J Gastroenterol Hepatol 28:235-42. doi: 10.1111/jgh.12065
16. Eom CS, Jeon CY, Lim JW et al (2011). Use of acid-suppressive drugs and risk of pneumonia: a systematic review and meta-analy-sis. CMAJ 183:310-9. doi: 10.1503/cmaj.092129
17. Filion KB, Chateau D, Targownik LE et al; CNODES Investigators (2014). Proton pump inhibitors and the risk of hospitalisation for community-acquired pneumonia: replicated cohort studies with me-ta-analysis. Gut 63:552-8. doi: 10.1136/gutjnl-2013-304738


117°CONGRESSONAZIONALESocietà Italiana di Medicina Interna
Roma, 14/16 Ottobre 2016Congress Center Rome Cavalieri
WWW.ARISTEA.COM/SIMI
Segreteria Società Italiana di Medicina InternaScientifica Viale dell’Università, 25 • 00185 Roma
Tel. 06 44340373 • Fax 06 44340474Email [email protected] • Web www.simi.it
Segreteria Via Roma, 10 • 16121 GenovaOrganizzativa Tel. 010 553591 • Fax 010 5535970
Email [email protected] www.aristea.com
•PAG SPRINGER 210x279-1_Layout 1 10/11/15 12:00 Pagina 1


















