
Lezione 4 I numeri quantici allopera..gli atomi diventano plurielettronici.

1. Introduzione.
1.1 Classificazione delle caspasi.
La famiglia delle caspasi (acronimo di cysteinil aspartate-specific proteases)
presenta almeno 14 membri tra i mammiferi, di cui 11 sono presenti nell’uomo (figura 1)
[1].
Figura 1 Le caspasi si suddividono in due principali sottofamiglie filogenetiche (ICE, CED-3). In base alle
loro specificità proteolitiche, si suddividono ulteriormente in tre gruppi: gli enzimi del I gruppo (in blu)
mediano la maturazione delle citochine; gli enzimi del II gruppo (in rosso) sono gli effettori del processo
apoptotico; infine, gli enzimi del III gruppo (in verde) sono gli attivatori a monte dell’apoptosi.
In particolare, le caspasi sono un gruppo di proteasi con cisteina nel sito attivo, le
quali operano, su altre proteine, un taglio specifico subito dopo un residuo di acido
aspartico: tale capacità non è comune ad alcun altro gruppo di proteasi [2].
Un’analisi filogenetica indica che questa famiglia è composta da due principali
sottofamiglie che sono correlate alla caspasi-1 o ICE (interleukin-1β converting enzyme)
oppure agli omologhi del gene CED-3 nei mammiferi [1]. In particolare, è stato
ampiamente documentato che il gene CED-3 è coinvolto in un programma di morte
cellulare nel nematode Caenorhabditis elegans [3]. Successivamente, il clonaggio ed il
sequenziamento di questo gene hanno permesso di individuare nel sito attivo della proteina
1

una elevata omologia con l’enzima ICE di mammifero. Ciò supporta fortemente il
coinvolgimento di ICE in un processo apoptotico analogo nei mammiferi [4]. In seguito a
queste osservazioni sono stati ricercati e clonati i geni di ulteriori membri della famiglia di
ICE come: Ich-1/Nedd-2; ICErel II; Mch2; CPP32/apopaina/YAMA, recentemente inclusi
tra le caspasi. Tutti i membri della famiglia condividono un’omologia di sequenza con ICE
e contengono un pentapeptide conservato “QACRG” (Gln, Ala, Cys, Arg, Gly) in cui la
cisteina partecipa all’azione catalitica [5]. La sottofamiglia delle caspasi correlate ad ICE
include le caspasi -1, -4, -5, -11, -12, -13, -14, le quali sono coinvolte nella maturazione
delle citochine e, quindi, nei processi infiammatori [6]. La sottofamiglia correlata al gene
CED-3 include le caspasi -3, -6, -7, -8, -9, -10, coinvolte nel processo di morte cellulare
programmata, nota come apoptosi. La caspasi-2 sembra costituire una sottofamiglia a sè
[5].
1.2 Struttura e meccanismo d’attivazione delle caspasi.
Le caspasi esistono sotto forma di zimogeni, cioè precursori enzimatici inattivi che
necessitano di una modificazione biochimica per convertirsi nella loro forma matura.
Infatti, tali zimogeni o procaspasi sono costituiti da un prodominio N-terminale seguito da
una subunità maggiore di circa 20 kDa, p20, e da una subunità minore di circa 10 kDa,
p10. Queste due subunità costituiscono il dominio catalitico [7]. In un determinato numero
di procaspasi, le subunità p20 e p10 sono separate da una piccola sequenza linker costituita
da circa 10 amminoacidi . In ragione della struttura del prodominio e della loro funzione, le
caspasi sono suddivise in tre gruppi principali (figura 2). Le caspasi costituite da un lungo
prodominio sono caspasi mediatrici dell’infiammazione ed iniziatrici del programma
apoptotico, appartenenti al I ed al II gruppo, rispettivamente. Le caspasi che presentano un
corto prodominio, costituito da circa 20-30 amminoacidi, sono invece definite caspasi
effettrici dell’apoptosi ed appartengono al III gruppo [8]. I lunghi prodomini delle
procaspasi presentano temi strutturali appartenenti alla superfamiglia del cosiddetto
dominio di morte o death domain (DD): i domini di morte, costituiti da 80-100 residui
amminoacidici, sono coinvolti nella trasduzione del segnale apoptotico. Questa
superfamiglia consta di un dominio di morte (DD), di un dominio effettore di morte (DED)
e, di un dominio di reclutamento delle caspasi (CARD) [9]. Ciascuno di questi gruppi di
amminoacidi interagisce con altre proteine attraverso interazioni omotipiche. Infatti, tutti i
membri della superfamiglia death domain sono caratterizzati da strutture similari che
comprendono 6 o 7 α-eliche anfipatiche antiparallele [10]. Le caspasi attive sono state
2

Figura 2 La famiglia delle caspasi. I tre gruppi principali di caspasi. Gruppo I: caspasi infiammatorie;
gruppo II: caspasi iniziatrici dell’apoptosi; gruppo III: caspasi effettrici dell’apoptosi. Sono indicati i domini
CARD, DED e le subunità catalitiche p20 e p10.
definite come “omodimeri di eterodimeri” [11]. Più in dettaglio, il cleavage di una
procaspasi dopo specifici residui di acido aspartico, causando il rilascio del prodominio,
conduce alla forma matura della caspasi, la quale è costituita dall’eterotetramero p202-p102
(figura 3) [8]: la presenza di aspartato al sito di maturazione è coerente con la capacità
delle caspasi di auto-attivarsi o di essere attivate da altre caspasi in un processo a cascata
[4]. Le subunità di ogni eterodimero (p20-p10) sono ripiegate in un cilindro compatto
dominato da un foglietto β centrale, sei volte pieghettato, e da cinque α-eliche che sono
distribuite sui lati opposti del piano, a sua volta, costituito da foglietti β (figura 4) [12].
Nell’eterotetramero caspasi, due di questi cilindri (eterodimeri) sono allineati secondo una
configurazione testa-coda. Di conseguenza, i due siti attivi sono posizionati alle estremità
opposte della molecola [4]. Tale cosiddetto ripiegamento della caspasi rappresenta una
3

struttura quaternaria unica fra le proteasi ed è caratteristica esclusiva delle caspasi e della
gingipaina R, proteasi cisteinica del batterio Porphyromonas gingivalis [13]. Tuttavia, la
presenza di due siti attivi non fornisce alcuna evidenza riguardo ad una eventuale
collaborazione o modulazione allosterica fra questi ultimi. L’intera configurazione del
tetramero e l’orientamento delle singole subunità al suo interno suggeriscono un
affascinante meccanismo di attivazione delle proteasi. In questo modello, due proenzimi si
associano e, in seguito ad un processo di maturazione, formano un tetramero nel quale
ciascuno dei due domini catalitici eterodimerici è formato da una subunità, a sua volta,
derivante da ciascun proenzima. Tale modello è supportato dalla vicinanza dell’estremità
C-terminale della subunità maggiore di un eterodimero, all’estremità N-terminale della
subunità minore nell’eterodimero opposto; però, non sono escluse altre possibilità [4].
L’architettura del sito attivo comprende residui amminoacidici derivanti da
entrambe le subunità. In particolare, “l’apparato” catalitico è costituito da un gruppo
sulfidrilico della Cys285 e da un anello imidazolico dell’His237: entrambi sono localizzati
nella subunità p20 [14]. Lo stato di transizione tetraedrico delle caspasi è stabilizzato
attraverso il legame a idrogeno con i protoni ammidici della catena principale della Cys285 e
della Gly238. Invece, l’asparagina del substrato sembra essere stabilizzata da quattro residui
amminoacidici: l’Arg179 e la Gln283 della subunità p20 e, l’Arg341 e la
Figura 3 Schema di attivazione di una procaspasi. Il cleavage della procaspasi a livello di specifici legami
Asp-X porta alla formazione della caspasi matura, costituita dall’eterotetramero p202-p102.
4

A B
Figura 4 (A) Struttura tridimensionale dell’eterotetramero caspasi 3. Ogni eterodimero è caratterizzato da
interazioni idrofobiche che determinano la formazione di foglietti β per lo più paralleli, composti, a loro
volta, da sei pieghe β antiparallele. I due eterodimeri “combaciano” formando un foglietto β dodici volte
pieghettato, il quale è circondato da α-eliche. In figura sono indicate le estremità N- e C-terminali di
entrambe le subunità. (B) Struttura cristallografica a raggi-x di una caspasi (PDB ID:1PAU). Il tetramero
caspasi è costituito da due subunità maggiori (la più esterna a sinistra in blu e la più esterna a destra in
bronzo) e da due subunità minori (la più interna a sinistra in bronzo e la più interna a destra in blu). La
struttura della caspasi-3 è mostrata con il suo inibitore, Ac-DEVD-CHO (CH3CO-Asp-Glu-Val-Asp-CHO; in
giallo), in ciascuno dei due siti attivi.
Ser339 della subunità p10 (figura 5) [8].
Figura 5 Principali interazioni all’interno del sito attivo della caspasi.
5

1.3 Il sito attivo delle caspasi: descrizione del processo catalitico.
Le caspasi riconoscono una sequenza tetrapeptidica molto corta all’interno dei
propri substrati polipeptidici; per tale motivo questa sequenza bersaglio costituisce la base
per la sintesi di inibitori e di nuovi substrati: il core tetrapeptidico dominante corrisponde
ai quattro residui amminoacidici sul lato N-terminale del legame peptidico scissibile (P4-
P3-P2-P1), cioè del sito di cleavage (vedi figura 5) [4]. Quindi, queste proteasi cisteiniche
riconoscono quattro amminoacidi, che occupano le tasche S4-S3-S2-S1: per convenzione, la
tasca di legame delle caspasi per il residuo amminoacidico N-terminale al legame scissibile
è denominata tasca S1; il corrispondente residuo amminoacidico del substrato, che occupa
tale tasca, è denominato P1. I residui amminoacidici (del substrato) adiacenti sul lato N-
terminale sono identificati come P2, P3 e P4 [15].
Le caspasi hanno un assoluto bisogno di aspartato in P1, preferiscono un residuo di
istidina o isoleucina in P2, un residuo di glutammato in P3, ma mostrano diverse preferenze
in P4 [4]. Nonostante l’apparente semplicità di queste “esigenze” appena elencate, le
caspasi presentano una specificità molto rigorosa, in quanto il contesto tridimensionale e la
superficie di presentazione appropriati sono fattori chiave, che determinano se la presenza
di una determinata sequenza, all’interno di un polipeptide, risulta altresì idonea al processo
proteolitico [4]. Ciò implica che la specificità di substrato mostrata dalle caspasi, in vitro,
può differire da quella mostrata in vivo. Questa elevata specificità delle caspasi permette
loro di compiere dei tagli selettivi, spesso in regioni limitate di proteine target, che
possono condurre all’attivazione ed anche all’inattivazione delle stesse proteine [16].
La cavità del sito attivo della caspasi è ben definita e si estende lungo la superficie
dell’enzima (figura 6). Più in dettaglio, la catena laterale carbossilata dell’aspartato in P1 si
adatta in un alloggiamento altamente specifico e si lega tramite legami a idrogeno con tre
residui conservati in tutte le caspasi (Arg179, Gln283, Arg341): le strette dimensioni fisiche di
S1 giustificano la presenza necessaria dell’aspartato in questa posizione. I siti di legame di
P2 e P3 (S2 ed S3) sono caratteristici, ma sono “tolleranti” ad un buon numero di sostituzioni
amminoacidiche. La catena peptidica principale del substrato legato forma legami ad
idrogeno con la Ser339 (conservata nella maggior parte delle caspasi) e con l’Arg341
(conservata in tutte le caspasi). Il sito di legame di P4 (S4), che è quello che conferisce la
specificità per il substrato, varia considerevolmente fra i differenti membri della famiglia
delle caspasi. Tale variabilità si evince, ad esempio, dal confronto tra le caspasi-1 e -3,
nelle quali il sottosito S4 varia radicalmente nella geometria e nella natura chimica (vedi
6

figura 6): nel caso della caspasi-1, S4 è una depressione, larga e poco profonda, sulla
superficie della proteasi, che ospita, senza difficoltà, residui idrofobici stericamente
ingombranti, quali ad esempio tirosina o triptofano. Tale sito accoglie anche altri residui
amminoacidici; ciò conferisce una relativa promiscuità a questo enzima. D’altra parte, la
caspasi-3 possiede una tasca S4, ben definita e poco profonda, che avvolge la catena
laterale dell’aspartato P4: l’intricato network di interazioni polari e la geometria fisica del
sito S4 giustificano la forte preferenza per l’amminoacido aspartato e, quindi, il profilo di
specificità globale delle caspasi. La conformazione fisica del sito è determinata, in parte,
dal Trp348 e da un loop superficiale, derivante dalla subunità minore, che determina un
capovolgimento irregolare del sito attivo contribuendo alla formazione di S4.
Complessivamente, queste informazioni consentono una comprensione “molecolare” delle
caratteristiche alla base della specificità di questi enzimi e della loro relativa variabilità o
A B
Figura 6 Topologia dei siti attivi della caspasi-1 (A) e della caspasi-3 (B). La superficie accessibile al
solvente è mostrata in verde. Gli inibitori legati sono mostrati in giallo (gli atomi di azoto sono di colore blu,
gli atomi di ossigeno sono di colore rosso). La caspasi-1 (A) è mostrata con il proprio inibitore (di natura
aldeidica) Ac-WEHD-CHO (Ac-Trp-Glu-His-Asp-CHO; PDB ID:1IBC), laddove la caspasi-3 (B) è mostrata
con il proprio inibitore (di natura aldeidica) Ac-DEVD-CHO (Ac-Asp-Glu-Val-Asp-CHO; PDB ID:1PAU).
Sono visibili i principali sottositi (S4, S3, S2). L’Asp P1 penetra nel piano della figura per cui non è visibile.
Da notare la principale differenza nel sottosito S4, il quale è una depressione larga ed aperta nella caspasi-1,
invece, nella caspasi-3, è una tasca più piccola e stretta.
rigorosità [17]. In seguito al legame del substrato all’enzima, la catalisi “si serve” di un
meccanismo tipico delle proteasi cisteiniche, che coinvolge una diade catalitica costituita, a
7
S4
S3
sua volta, dalla Cys285 e dall’His237, ed anche un ossianione , costituito dagli atomi di azoto
della Gly238 e della Cys285 [17]: il “buco ossianionico” costituisce una tasca che, durante la
catalisi, forma legami ad idrogeno con l’ossigeno carbonilico del residuo amminoacidico
P1 del substrato [12].
La somiglianza tra i gruppi chimici coinvolti nella catalisi e le strutture
cristallografiche delle caspasi attive in complesso con inibitori suggerisce che la reazione
catalitica segue un percorso simile in tutte le caspasi, così come è stato documentato per le
proteasi seriniche. Il meccanismo catalitico proposto può essere brevemente schematizzato
in cinque fasi (figura 7): durante la fase di acilazione (1), l’ossigeno carbonilico del residuo
P1 legato in maniera non covalente è ancorato, tramite legami ad idrogeno, agli atomi di
azoto della Cys285 e della Gly238 (buco ossianionico). Tale ancoraggio aumenta la
polarizzazione del legame carbonio-ossigeno (C-O) e, quindi, facilita l’attacco nucleofilo
dell’atomo di zolfo della Cys285 al carbonio carbonilico del residuo P1, altamente elettrofilo.
Il risultato è un addotto covalente enzima-substrato, l’intermedio tetraedrico ad alta energia
(2). In questa fase della catalisi, la porzione imidazolica dell’His237 si comporta da acido di
Bronsted protonando il gruppo α-amminico del prodotto peptidico uscente ed evitando così
la ricostituzione del legame peptidico. La successiva deacilazione del complesso acil-
enzima avviene in una maniera simile: la catena laterale deprotonata dell’His237 sottrae un
protone ad una molecola d’acqua, l’acqua idrolitica (3), rendendola attiva per l’attacco al
legame tioesterico. La deacilazione procede attraverso un secondo intermedio tetraedrico
(4), formato dall’attacco nucleofilo del gruppo idrossilico al carbonio carbonilico. La
conseguente rottura del legame carbonio-zolfo (C-S) rigenera l’enzima sotto forma di
complesso non covalente con il prodotto peptidico N-terminale (5). Per analogia con le
proteasi seriniche, è plausibile che i movimenti dei loops di legame del substrato, 341 e/o
381, siano accoppiati a quest’ultima reazione, determinando conseguentemente la
distruzione dei legami ad idrogeno catena principale-catena principale con i residui P1/P3, e
dei legami ad idrogeno tra l’atomo di ossigeno carbonilico in P1 ed il buco ossianionico. In
sintesi, l’idrolisi del tioestere ed il rilascio successivo del prodotto potrebbero essere
sincronizzati garantendo un’alta efficienza della catalisi [12].
In alternativa, è stato proposto che l’atomo di azoto dell’His237 stabilizzi la carica
che si sviluppa sul gruppo uscente [12, 18]. Inoltre, è stato postulato che una molecola
d’acqua, che forma legami ad idrogeno con l’ammide della Gly238, doni un protone al
gruppo amminico uscente. Questo meccanismo capovolge completamente i ruoli “classici”
del buco ossianionico e dell’His237. La controversia si basa su una caratteristica peculiare
8
delle caspasi che le distingue dalle proteasi seriniche e dalle altre proteasi cisteiniche: la
catena laterale dell’His237 è localizzata ad una distanza di oltre 5 Å dall’atomo di zolfo
della Cys285. A tale distanza inusuale, l’His237 non può accettare il protone tiolico della
Cys285, la quale, di conseguenza, non potrà essere pre-polarizzata. Ciò implica che la specie
nucleofila potrebbe scaturire da una reazione di coordinazione [19].
Figura 7 Meccanismo catalitico di idrolisi del substrato.
9

2. Inibizione della Caspasi-1 o ICE (Interleukin-1β converting enzyme).
2.1 Caspasi-1 o ICE (Interleukin-1β converting enzyme): struttura, meccanismo
d’attivazione, e funzioni.
La caspasi-1 (o ICE) fu identificata, per la prima volta, nel 1989 in cellule
monocitiche [20, 21]. La sua scoperta permise di definire una nuova classe di proteasi
cisteiniche, che si distingueva dalle altre famiglie di proteasi cisteiniche per due
peculiarità: 1) l’organizzazione strutturale; 2) l’assoluta necessità di aspartato nella
posizione P1 del legame scissibile dei substrati per il riconoscimento dei medesimi [3, 22,
23]. L’enzima ICE attivo è composto da due subunità, p10 e p20, di massa molecolare,
10248 Da e 19866 Da, rispettivamente. Diverse studi suggeriscono che entrambe le
subunità sono necessarie per l’attività catalitica. Ciò è stato confermato dalle strutture
cristallografiche dell’enzima legato ad inibitori tetrapeptidici; tali strutture mostrano che le
subunità p20 e p10 sono intimamente associate, in quanto entrambe contribuiscono con
residui amminoacidici “chiave” alla formazione del sito attivo (figura 8). Gli amminoacidi
catalitici, Cys285 e His237, si trovano nella subunità p20; invece, la tasca di legame per
l’aspartato P1 è costituita da residui amminoacidici di entrambe le subunità [21].
Come mostrato nella figura 8, i due eterodimeri p20/p10 si associano nel cristallo
formando un tetramero: è stato proposto che questa è la forma cataliticamente attiva
dell’enzima in soluzione. Tale ipotesi è sostenuta da tre importanti evidenze: 1) i contatti
fra gli eterodimeri nel cristallo sono estesi (coprono un’area di ~ 5200 Å2); 2) i residui
amminoacidici localizzati all’interfaccia del tetramero sono altamente conservati; 3)
mutazioni di uno di questi residui annulla l’attività di ICE [14].
La forma matura di ICE, p20/p10, deriva da un proenzima di 45 KDa (p45) tramite
la rimozione proteolitica di un propeptide di 11.5 KDa e di un peptide di 2 KDa che funge
da linker fra le due subunità [21].
L’esame, ad alta risoluzione, del sito attivo di ICE mostra differenze sostanziali con le
proteasi cisteiniche appartenenti alla superfamiglia della papaina, come la catepsina B.
Inoltre, le strutture, secondaria e quaternaria, di ICE non mostrano analogie con le strutture
omologhe appartenenti ad altre classi di proteasi seriniche e cisteiniche. Tuttavia, le
interazioni molecolari degli inibitori peptidici di ICE con i residui del sito attivo
dell’enzima sono simili allo schema di riconoscimento, tipo foglietto-β, osservato in
complessi inibiti di proteasi seriniche come la chimotripsina [24, 25, 14]. Di contro, la
machinery catalitica del sito attivo di ICE è costituita da una diade cisteina-istidina, la
10

Figura 8 Struttura cristallografica (tetramerica) di ICE legato ad un suo inibitore tetrapeptidico (PDB
ID:1ICE). Le subunità p20 (verde e blu) e le subunità p10 (rossa e oro) formano due domini catalitici:
(verde/rosso) e (blu/oro). L’inibitore tetrapeptidico Ac-YVAD-CHO (Ac-Tyr-Val-Ala-Asp-CHO) è mostrato
in color porpora.
quale differisce sia dalla triade catalitica cisteina-istidina-asparagina della papaina e della
catepsina B, sia dalla triade serina-istidina-aspartato delle proteasi seriniche [26]. La
struttura di ICE fornisce altresì una sorta di “intelaiatura” che consente di comprendere la
sua selettività di substrato, tramite studi di cinetica su piccoli substrati peptidici [27, 28].
Due residui di arginina sono accostati nella tasca S1. Questi residui amminoacidici basici
conferiscono all’enzima una elevatissima affinità di legame verso substrati peptidici o
verso inibitori con un residuo di aspartato in posizione P1. Mutazioni a carico dei due
residui di arginina o di uno dei due componenti della diade catalitica inibiscono l’attività
enzimatica di ICE [14]. A differenza della tasca di legame S1, le tasche S2 ed S3 sono
maggiormente esposte ai solventi, per cui “tollerano” una grande varietà di residui
amminoacidici, di substrati o di inibitori, [29, 25, 30] o di mutazioni [26]. La tasca P4 (S4)
11

prevede un certo numero di contatti con substrati o inibitori importanti, sebbene ICE possa
accettare sostituenti S4 sia idrofobici che carichi elettrostaticamente [31].
La scoperta degli omologhi di ICE negli uomini e nei roditori [32, 33, 34, 35, 36]
ha documentato che la machinery catalitica del sito attivo di ICE è completamente
conservata in tutta la famiglia degli omologhi di questa caspasi [33], suggerendo come
queste proteasi utilizzino un meccanismo catalitico comune [29, 14]. D’altra parte, se si
eccettuano i residui di arginina della tasca S1, che “dettano” la specificità per l’aspartato in
P1, gli altri residui amminoacidici nelle tasche di legame S2, S3 ed S4 differiscono tra gli
omologhi di ICE, fornendo una base razionale alle diverse preferenze di substrato di questi
ultimi [34].
Nonostante siano state determinate numerose strutture cristallografiche di ICE
complessato con inibitori recanti aspartato in P1, il meccanismo di attivazione di questa
caspasi non è stato completamente chiarito. Di recente, un gruppo di ricercatori californiani
ha tentato di comprendere, in maniera più profonda, il ruolo svolto dall’elemento cruciale
di riconoscimento del substrato, l’aspartato, nel controllare la conformazione del sito attivo
dell’enzima. A tale scopo sono state utilizzate tre strutture cristallografiche: 1) ICE privo di
ligando; 2) ICE complessato con una molecola di malnato; 3) ICE privo di ligando dopo la
rimozione del malonato. L’analisi della struttura di ICE, privo di ligando, ha suggerito che
l’enzima, in soluzione, mantiene il sito attivo in una conformazione chiusa, in virtù del
movimento del loop 3 della subunità p10. Tale movimento copre la cavità catalitica della
proteina rendendola inaccessibile allo scheletro di qualsiasi substrato. Tuttavia, ICE
subisce un significativo riarrangiamento strutturale quando transita dallo stato libero allo
stato legato (figura 9). La modificazione conformazionale più significativa avviene
all’interno del loop 3 nella subunità p10, a livello dell’Arg341: il riarrangiamento consente
alla caspasi-1 di trasferire l’Arg341 dalla superficie della proteina alla regione S1 del sito
attivo e, di rendere gli atomi della catena laterale di questo amminoacido, disponibili per
effettuare i necessari legami ad idrogeno con l’aspartato del substrato. Oltre al
riarrangiamento del loop 3, si verifica una riorganizzazione dei legami ad idrogeno che
stabilizza la conformazione dell’enzima libero da ligando [37]. La struttura del complesso
ICE-malonato si è mostrata, invece, virtualmente identica alla struttura dell’enzima
complessato con un inibitore tetrapeptidico, come Ac-Tyr-Val-Ala-Asp (figura 10) [14].
Ogni atomo di ossigeno (O) nella molecola di malonato partecipa alla formazione di
legami ad idrogeno con gli atomi della catena laterale o del backbone di residui
12
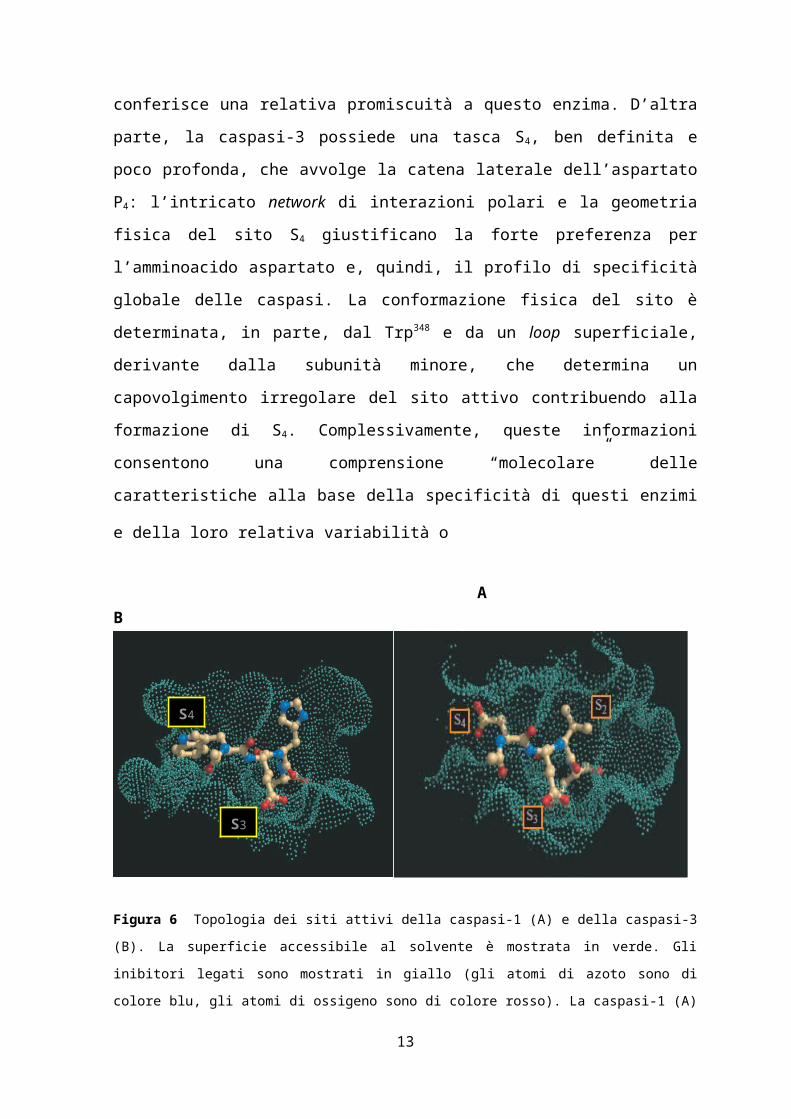
Figura 9 Analisi dei riarrangiamenti strutturali nel sito attivo della caspasi-1 durante la transizione dalla
conformazione libera da ligando alla conformazione legata al ligando. Le proteine legate all’inibitore sono
mostrate con disegni o bastoncini verdi, le forme libere sono porpora o rosa. Le linee gialle tratteggiate
indicano la distanza tra i corrispondenti carboni carbonilici in α.
(A) Raffigurazione della caspasi-1 legata all’inibitore peptidico (PDB ID:1ICE) e libera da ligando
(PDB ID:1SC1).
(B) Raffigurazione della caspasi-1 in complesso con un inibitore peptidico (PDB ID:1ICE) e
dell’enzima cristallizzato in presenza di malonato dopo rimozione del ligando (PDB ID:1SC4).
(C) Loop 2 (L2) della caspasi-1 nelle due conformazioni, legata all’inibitore e libera da ligando.
(D) Loop 3 (L3) della caspasi-1 nelle due conformazioni, legata all’inibitore e libera da ligando. L3 si
sposta approssimativamente di 4.9 Å al di là del sito attivo in assenza di un inibitore.
(E) Loop 4 (L4) della caspasi-1 nelle due conformazioni, legata all’inibitore e libera da ligando.
13

A B
Figura 10 Veduta all’interno della regione S1 del sito attivo della caspasi-1 umana: sono mostrati i contatti
fra gli atomi della molecola di malonato o del residuo di aspartato di un inibitore e gli atomi della proteina
enzimatica.
La regione S1 del sito attivo mostra la conservazione degli amminoacidi primari completi e tridimensionale
dei residui catalitici. I residui amminoacidici conservati, coinvolti nelle interazioni ad idrogeno con gli atomi
di ossigeno del malonato e dell’aspartato sono evidenziati. Le linee gialle tratteggiate indicano le distanze tra
gli atomi dell’enzima e quelli dell’inibitore.
(A) Caspasi-1 in complesso con una molecola di malonato (PDB ID:1SC3).
(B) Caspasi-1 in complesso con un inibitore aldeidico aspartato-mimetico (PDB ID:1ICE).
amminoacidici appartenenti ad entrambe le subunità, p20 (Arg179, His237, Gly238, Gln283,
Ala285) e p10 (Arg341), nella tasca S1. Tale network di interazioni appare stabilizzare il
complesso dimerico della proteina: il malonato riproduce i legami ad idrogeno osservati in
strutture cristallografiche di complessi covalenti enzima-inibitori. Infine, è stata analizzata
la struttura della caspasi-1 cristallizzata, nella forma ligando-legata, in presenza di
malonato, da cui, in seguito, il malonato è stato rimosso: la cavità catalitica è risultata
parzialmente chiusa (vedi figura 9, pannello B). Ciò suggerisce che solo alcune parti
dell’enzima hanno subito un riarrangiamento strutturale nel cristallo. In particolare, la
struttura ha rivelato che i loops, che definiscono il sito attivo dell’enzima, non si
riposizionano completamente come erano posizionati nell’originaria struttura
cristallografica dell’enzima libero. Infatti, essi trovano la loro energia minima occupando
all’interno del cristallo uno spazio tetragonale vincolante. Il successivo confronto tra le
strutture cristallografiche della caspasi-1 complessata con una molecola di malonato e
dell’enzima libero ha portato ad un’importante conclusione: l’aspartato, che ha l’essenziale
14

funzione di aprire il sito attivo di ICE ai substrati, in quanto elemento di riconoscimento
obbligatoriamente presente sui ligandi di ICE, potrebbe essere il principale responsabile
dei cambiamenti conformazionali che caratterizzano la transizione dell’enzima dalla forma
libera alla forma legata. Questa considerazione suggerisce un nuovo approccio
nell’identificazione di nuovi inibitori di ICE aspartato-mimetici [37].
L’enzima ICE rappresenta il prototipo della famiglia delle caspasi [29]: è coinvolto
sia nella maturazione delle citochine pro-infiammatorie che nell’apoptosi delle cellule
infettate da determinati agenti patogeni [38]. In particolare, ICE è la caspasi responsabile
della maturazione proteolitica della pro-interleuchina-1β (pro-IL-1β) [29, 39]: ICE opera
un taglio sul precursore pro-IL-1β, di 31 KDa, a livello del sito Asp116-Ala117, generando la
forma matura, biologicamente attiva, dell’interleuchina-1β (IL-1β), di 17.5 KDa.
Similmente, la forma matura attiva dell’enzima ICE umano deriva da un precursore (p45),
costituito da 404 amminoacidi, che subisce un cleavage proteolitico agli amminoacidi
Asp103, Asp119, Asp297 ed Asp316 [40]. Studi recenti hanno documentato come la caspasi-1
possa anche stimolare la biogenesi della membrana cellulare, favorendo così la riparazione
dell’eventuale danno causato da tossine batteriche perforanti. Dunque, la caspasi-1
potrebbe promuovere la sopravvivenza della cellula ospite, esplicando una sorta di
meccanismo di resistenza nei confronti dei batteri patogeni [38].
L’IL-1β è una citochina pro-infiammatoria prodotta prevalentemente dai monociti e
dai macrofagi in risposta ad una varietà di stimoli [41]. Essa riveste un ruolo fondamentale
in molte e varie patologie infiammatorie ed autoimmuni [23], quali l’artrite reumatoide
[42], l’osteoartrite [43], l’arteriosclerosi [44], lo shock settico [45], la sindrome
infiammatoria delle viscere [46], il diabete mellito [47] e la pancreatite [48, 49]. Inoltre,
tale citochina è coinvolta in diversi altri processi fisiologici e patologici, che includono, ad
esempio, la cicatrizzazione delle ferite e lo sviluppo di determinate forme di leucemia,
rispettivamente [44]. Diversi disordini neurodegenerativi quali le patologie di Alzheimer e
di Parkinson, la sclerosi multipla ed il danno cerebrale di origine traumatica e/o ischemica
potrebbero essere, verosimilmente, accelerati dal rilascio di IL-1β dalle cellule della
microglia [47]. Peraltro, tali condizioni neurodegenerative ed ischemiche potrebbero essere
anche favorite dal processo apoptotico a carico dei tessuti cerebrali coinvolti: l’apoptosi
prevede la proteolisi intracellulare di substrati cosiddetti apoptotici ad opera di ICE o dei
suoi omologhi [50, 51]. Questo quadro sintetico delle molteplici implicazioni fisio-
patologiche dell’IL-1β suggerisce come il suo antagonismo e/o la sua neutralizzazione
possano migliorare sensibilmente le condizioni patologiche da essa sostenute [52]. Per
15

questo motivo, l’inibizione di ICE rappresenta un “attraente” target terapeutico per il
controllo dei livelli di IL-1β e, di conseguenza, potrebbe costituire un efficace “tool” per il
trattamento delle svariate patologie mediate da questa citochina [53, 23].
2.2 Gli inibitori della Caspasi-1.
Sin dalla sua identificazione, l’enzima ICE rappresentò, immediatamente, un
affascinante target terapeutico per due ragioni principali. In primo luogo, era pensiero
comune che la caspasi-1, in quanto proteasi, fosse più funzionale del recettore delle
citochine alla scoperta di una piccola molecola con potenzialità terapeutiche.
Secondariamente, la desueta specificità di substrato dell’enzima, che operava “il taglio”
dopo un residuo di aspartato nella pro-IL-1β, suggerì la possibilità di sintetizzare dei suoi
inibitori, altrettanto selettivi [21].
Studi cinetici, strutturali e di mutagenesi hanno documentato che il residuo tiolico
della cisteina catalitica nell’enzima ICE, cioè la Cys285, è un centro nucleofilo attivo [29,
14]. Di conseguenza, non sorprende che tutti gli inibitori di ICE, riportati in letteratura,
incorporino nella propria struttura, precisamente all’estremità C-terminale, un centro
elettrofilo (aldeidi, nitrili o chetoni), che subisce l’attacco da parte del tiolato nucleofilo
della Cys285. Tale attacco nucleofilo può essere reversibile o irreversibile [29, 54]; dunque,
gli inibitori di ICE e dei suoi omologhi possono essere, convenientemente, distinti in
reversibili ed irreversibili [26]. In particolare, sono state impiegate con successo due
strategie generali finalizzate allo sviluppo di inibitori reversibili ed irreversibili di ICE
(figura 11) [55, 56]. Le aldeidi (VI), i nitrili (VIII) ed i chetoni (X) di natura peptidica sono
inibitori reversibili che subiscono l’addizione nucleofila della cisteina catalica formando
tioemiacetali (VII), tioimmine (IX) e tioemichetali (XI), rispettivamente. La potenza di
questi composti è stata attribuita alla somiglianza strutturale esistente fra questi addotti
tiolici e gli intermedi tetraedrici (II e IV) e acil-enzima (III) che si formano durante
l’idrolisi del substrato. Il secondo approccio coinvolge chetoni peptidici α-sostituiti di
struttura generale R-CO-CH2-X (XII), dove X rappresenta il gruppo uscente, il quale può
essere un alogeno, uno ione diazonio o un carbossilato. Quindi, a seconda del gruppo
uscente, si ottiene un alometilchetone, un diazometilchetone o un acilossimetilchetone,
rispettivamente. Queste tre classi di composti inibiscono irreversibilmente le proteasi
cisteiniche, ICE compresa, tramite l’espulsione del gruppo uscente e la successiva
formazione di un tiometilchetone con la cisteina del sito attivo dell’enzima (XIV) [21].
16

Idrolisi del legame ammidico
Inibitori Reversibili
Inibitori Irreversibili
Chetoni α-sostituiti
Figura 11 Strategie usate nello sviluppo di inibitori di proteasi cisteiniche.
17
Aldeidi peptidiche
Nitrili peptidici
Chetoni peptidici

Entrambe queste strategie sono state applicate con successo allo sviluppo di
inibitori di ICE. Gli inibitori più potenti e selettivi contengono la sequenza tetrapeptidica,
Ac-Tyr-Val-Ala-Asp (Ac=CH3CO-), coerentemente con la specificità di substrato
dell’enzima. Sebbene la natura peptidica di questi composti limiti la loro utilità nelle
applicazioni terapeutiche, essi si sono dimostrati preziosi per studi riguardanti il
meccanismo catalitico, la struttura e la biologia cellulare [21].
Il più potente inibitore reversibile sviluppato per ICE è l’aldeide tetrapeptidica Ac-
Tyr-Val-Ala-Asp-CHO (Ac-YVAD-CHO; Ac=CH3CO-) (figura 12) [57, 29]. In passato,
Figura 12 Struttura chimica dell’aldeide tetrapeptidica Ac-Tyr-Val-Ala-Asp-CHO.
questo tetrapeptide è stato utilizzato per dimostrare che l’inibizione di ICE, nell’intero
sistema circolatorio umano, può bloccare il rilascio dell’IL-1β matura da parte dei
monociti. Ac-YVAD-CHO, come altri inibitori peptidici (figura 13), si lega
reversibilmente ad ICE; tuttavia, poiché mostra un’affinità per l’enzima prossima a 100
pM [Ki (costante di inibizione)=0.76 nM], i processi di formazione e di dissociazione del
complesso enzima-inibitore sono lenti [29]. Ac-YVAD-CHO è anche uno dei più selettivi
inibitori di ICE, prova ne sia la misura della sua affinità per l’omologo di ICE CPP32, la
quale risulta essere più bassa di almeno sei ordini di grandezza [34]. Un altro importante
inibitore reversibile dell’omologo di ICE CPP32, connesso ad Ac-YVAD-CHO, è l’aldeide
tetrapeptidica Ac-Asp-Glu-Val-Asp-CHO (Ac-DEVD-CHO) [58]. Anche questo composto
inibisce la caspasi-1 con un’affinità dell’ordine del nanomolare. In particolare, la struttura
cristallografica di Ac-DEVD-CHO legata al sito attivo di ICE illustra la capacità
dell’enzima di formare favorevoli interazioni elettrostatiche con substrati peptidici aventi
residui acidi carichi in P3 ed in P4 (figura 14) [26]. Considerando la struttura chimica
dell’inibitore Ac-YVAD-CHO, la sostituzione della funzione aldeidica con un gruppo
18

Figura 13 Inibitori peptidici di ICE
Figura 14 Rappresentazione della superficie molecolare del sito attivo della caspasi-1 con la superficie
dell’enzima colorata secondo i potenziali elettrostatici: le regioni positive e negative sono individuate dal blu
e dal rosso, rispettivamente. L’inibitore è raffigurato con un modello a bastoncini e colorato in funzione del
tipo di atomo.
19

ciano (-CN) dà luogo alla formazione del corrispondente nitrile (2, figura 15), il quale ha
una Ki pari a 57 nM, in accordo con la sua più bassa elettrofilicità intrinseca, che giustifica
la sua minore affinità per ICE [21].
È noto da tempo che le aldeidi peptidiche inibiscono le proteasi cisteiniche
formando un tioemiacetale con la cisteina del sito attivo. Tuttavia, resta ancora da capire se
le aldeidi peptidiche sono analoghi dello stato di transizione [59], oppure inibiscono
l’enzima tramite la formazione di un tioemiacetale legato in uno stato conformazionale di
non-transizione [60, 61, 62, 63]. La struttura cristallografica di ICE in complesso con Ac-
YVAD-CHO mostra chiaramente l’aldeide tetrapeptidica legata secondo una
conformazione di non-transizione, in cui l’ossianione dell’inibitore è stabilizzato
dall’istidina del sito attivo [14].
Un’altra classe di inibitori competitivi e reversibili di ICE, sorprendentemente
potenti, è costituita dai fenilalchil chetoni non sostituiti di natura peptidica. Questo
procedimento di sintesi offre il vantaggio di “accogliere”, su entrambi i lati del gruppo
carbonilico, sostituenti che aumentano l’affinità e la specificità degli stessi inibitori. Il
miglior inibitore di questa classe di composti è Ac-YVAD-CO-(CH2)5-Ph (3, figura 15) che
possiede una Ki pari a 19 nM [64]. La potenza di questi inibitori si deve, almeno in parte,
all’abilità dell’enzima di accogliere residui idrofobici nei sottositi S1-S2. Nel tentativo di
aumentare la potenza di questi inibitori attraverso l’aggiunta di sostituenti contenenti atomi
di fluoro, elettron-attrattori, si è giunti alla sintesi del composto fenilpropionil-Val-Asp-
CO-CF2-(CH2)3-Ph; tuttavia tale composto risulta essere un inibitore più debole (K i= 6µM)
rispetto alla sua controparte non sostituita (Ki= 42 nM) [65]. In alternativa, l’attivazione
del carbonile, tramite sostituzione del carbonio in β con un eteroatomo, determina un
significativo aumento della potenza, come dimostra il fenossimetilchetone peptidico Ac-
Tyr-Val-Asp-CO-CH2-O-Ph (4, figura 15), il quale ha una Ki pari a 3 nM [65].
Tra gli inibitori irreversibili di ICE, più potenti e selettivi, si distinguono gli
acilossimetilchetoni α-sostituiti peptidici di struttura generale, Ac-Tyr-Val-Ala-Asp-CH2-
O-CO-Ph (6, figura 15). L’inattivazione della caspasi-1 procede attraverso l’espulsione del
gruppo uscente carbossilato e la successiva formazione di un tiometilchetone con la Cys285
del sito attivo. Questi inibitori sono relativamente inerti nei confronti dei “bionucleofili”;
ciò li rende ottimi candidati per lo studio, in vivo, dell’inibizione enzimatica [66, 67].
Recentemente, gli inibitori peptidomimetici di ICE sono stati al centro di una nuova
strategia di sintesi: limitare la porzione Val-Ala nel tetrapeptide, conservando l’interazione
20

1 Not Determined
Figura 15 Inibitori tetrapeptidici di ICE.
cruciale dello scheletro peptidico. Ciò determina l’appropriato orientamento delle regioni
di riconoscimento P1 e P4 della molecola nella tasca enzimatica. Un noto esempio è
costituito dal Pralnacasan® (figura 16) [68], il pro-farmaco di un inibitore reversibile di
ICE che consente a quest’ultimo di essere somministrato per via orale. In particolare, la
molecola Pralnacasan® possiede un core peptidomimetico biciclico che costringe la regione
P2-P3 a mantenere l’inibitore in una conformazione che favorisca il legame con l’enzima
[68, 23]. Attualmente, questo composto ha raggiunto la fase III degli studi clinici [68].
Poiché la sintesi di inibitori biciclici, Pralnacasan-correlati, è piuttosto lenta [23],
Ellis e collaboratori hanno valutato l’utilità delle tiazepine come inibitori peptidomimetici
21

Figura 16 Struttura chimica del Pralnacasan: pro-farmaco (R=Et) e farmaco libero (R=H).
monociclici di ICE: la loro sintesi risulta più rapida rispetto a quella di strutture bicicliche.
In particolare, essi hanno teorizzato che lo zolfo della tiazepina può orientarsi nella tasca S3
dell’enzima in maniera da occupare uno spazio simile a quello occupato dalla valina di Ac-
YVAD-CHO. Sulla base della struttura cristallografica di questo inibitore legato ad ICE
(figura 17), è stato costruito il modello di inibitore tiazepinico riportato in figura 18 (la
struttura chimica è riportata in figura 19). I legami, liberi di ruotare, delle regioni P1 e P4 di
questo modello sono stati orientati in conformazioni a bassa energia in maniera da
sovrapporsi ai corrispondenti sostituenti P1 e P4 nella struttura di Ac-YVAD-CHO
all’interno del cristallo. Infine, il modello è stato agganciato nel sito attivo della struttura
cristallografica della proteina ICE in sostituzione della struttura di Ac-YVAD-CHO [68].
Figura 17 Struttura cristallografica (visione ai raggi-X) di Ac-YVAD-CHO complessata con ICE (PDB ID:1ICE).
22

Figura 18 Modello molecolare dell’inibitore tiazepinico.
In seguito, sono stati sintetizzati altri composti a scheletro tiazepinico, i quali hanno
mostrato di possedere una buona potenza ed un’ottima selettività nel saggio di inibizione di
ICE: gli analoghi con sostituenti biciclici fusi nella regione P4, compreso l’inibitore in
figura 19, si sono mostrati più attivi rispetto ai composti con sostituenti aromatici
monociclici. Inoltre, il saggio enzimatico ha evidenziato una riduzione dell’attività, da
quattro a cinque volte, quando si passa da solfuri a sulfoni. Alla luce di queste
considerazioni, il lavoro di Ellis dimostra che lo scheletro tiazepinico monociclico
conserva le interazioni a idrogeno cruciali tipiche della struttura peptidomimetica ed, al
tempo stesso, orienta i sostituenti P1 e P4 nella conformazione necessaria al legame con
l’enzima, in maniera simile ad Ac-YVAD-CHO. Quindi, i composti tiazepinici, sebbene
Figura 19 Struttura chimica dell’inibitore tiazepinico di ICE.
23

meno attivi del Pralnacasan®, hanno dimostrato la potenziale utilità delle strutture
monocicliche come “tools” per un’inibizione efficace di ICE [68].
Utilizzando come termine di paragone la struttura cristallografica di ICE
complessato con l’inibitore Ac-YVAD-CHO, sono stati condotti ulteriori studi finalizzati a
superare i problemi di sintesi legati ai composti biciclici, Pralnacasan-correlati. Tali studi
hanno documentato che i composti peptidomimetici con anelli lattamici monociclici
insaturi (figura 20), a sette ed a otto membri, sono ottimi inibitori di ICE. Tuttavia, sono
necessari ulteriori studi farmacocinetici per un loro utilizzo futuro nella pratica clinica
[23].
Figura 20 Struttura generale dei composti lattamici monociclici insaturi: potenti inibitori di ICE.
Nonostante la loro notevole utilità in sistemi in vitro, gli inibitori peptidici di ICE, a
causa della loro natura, hanno una scarsa biodisponibilità che ne limita fortemente
l’utilizzo terapeutico [21]. Questo problema ha indotto ad elaborare nuove strategie
sintetiche per sviluppare inibitori non-peptidici di ICE che offrino una maggiore
biodisponibilità. In particolare, si è pensato di sostituire il tripeptide Ac-Tyr-Val-Ala con
un gruppo benzilossicarbonilico (CBZ) nella nota struttura fenilalchil-chetonica, Ac-
YVAD-CO-(CH2)5-Ph, ottenendo il composto 3 (tabella 1). Successivamente, utilizzando
la struttura cristallografica del fenilalchil-chetone di partenza, è stato realizzato un modello
molecolare del composto 3, il quale, dopo essere stato agganciato al sito attivo della
struttura cristallografica di ICE, ha suggerito due ipotetiche conclusioni: 1) il gruppo CBZ
probabilmente occupa il sito S2; 2) il gruppo fenilpentilico è diretto verso la tasca della
parte originaria della molecola. Tali ipotesi sono state basate sulla compatibilità sterica del
ligando con la regione di binding dell’enzima. Nel tentativo di aumentare la potenza del
composto 3, sono stati sintetizzati gli analoghi 5, 7, e 10. Inizialmente, il β-metilene nel
composto 3 è stato sostituito con un atomo di zolfo, ottenendo il composto 5. Quest’ultimo
si è mostrato cinque volte più potente di 3. L’inibitore 7 è derivato dalla sostituzione in 3
24

Tabella 1 Inibitori irreversibili e relativi valori del saggio di inibizione di ICE.
del gruppo CBZ con una funzionalità fenilsulfonammidica, che è capace di interagire con
ICE formando legami ad idrogeno con la catena laterale dell’Arg341 ed il carbonile del
backbone. Tale sostituzione ha prodotto un aumento dell’affinità di legame di cinque volte
rispetto al composto 3. Infine, si è cercato di aumentare ulteriormente la potenza del
composto 7 combinando la funzionalità fenilsulfonammidica con una porzione
tiometilchetonica, anch’essa capace di aumentare la potenza di inibizione. Tuttavia, il
composto 10 risultante si è mostrato equipotente con i composti 5 e 7. Allo scopo di
interpretare il mancato effetto sinergico tra i due gruppi, fenilsulfonammidico e
tiometilchetonico, sono state realizzate le strutture cristallografiche di 7 e 10 (figura 21).
La struttura del composto 7 complessato con ICE ha mostrato che entrambi gli atomi di
ossigeno della fenilsulfonammide presentano legami ad idrogeno con ICE: il gruppo N-H
sulfonammidico forma un legame ad idrogeno con il carbonile della Ser339; l’atomo di
ossigeno del sulfonile, nel piano, forma un legame ad idrogeno con il gruppo NH2
guanidinico dell’Arg341, mentre l’altro ossigeno sulfonilico forma un legame ad idrogeno
con l’ammide della catena principale dell’Arg341. Queste interazioni orientano l’anello
fenilico del gruppo arilsulfonammidico, esposto al solvente, all’interno del contatto
idrofobico dell’anello indolico del Trp340 e della catena polimetilenica principale. Inoltre,
l’esame ai raggi-X della struttura cristallografica del composto 7 ha indicato che un atomo
25

di idrogeno in posizione orto nel gruppo fenilsulfonammidico è molto vicino alle unità
metileniche della catena laterale fenilpentilica, che si estende dall’aspartato alla
funzionalità chetonica. Probabilmente, quest’ultima è una modalità di legame favorevole,
pre-organizzata dal “collasso idrofobico”. Tuttavia, l’attento esame ai raggi-X della
struttura cristallografica del composto 10 complessato con ICE ha evidenziato che l’atomo
di zolfo, nella catena laterale fenil-tiometilica, è orientato in maniera tale da trovarsi più
lontano dal gruppo fenilico rispetto alla corrispondente unità metilenica nel composto 7,
probabilmente per evitare un conflitto sterico. Sulla base di queste osservazioni, è stato
supposto che se la catena laterale C-terminale del composto 10 fosse stata troncata
ottenendo un’aldeide, la successiva sostituzione della posizione orto del gruppo
Figura 21 Raffigurazione delle strutture cristallografiche dei composti 7 (linee spesse verdi) e 10 (linee
spesse arancioni) legati al sito attivo di ICE. Gli inibitori sono colorati secondo il tipo di atomo [blu per
l’azoto, rosso per l’ossigeno, giallo per lo zolfo, verde (7) e arancione (10) per gli atomi di carbonio]. I
residui dell’enzima sono raffigurati in rosa. Sono mostrati i legami ad idrogeno tra gli inibitori
solfonammidici (SO2NH) e l’Arg341 (NH2CNH), la Ser339 (CO), e tra l’acido (CO2) e l’Arg179 (NH2CNH).
L’His237 è mostrata in grigio-azzurro.
26

fenilsulfonammidico con un anello fenilico avrebbe fornito un composto analogo dotato di
maggiore affinità verso ICE. Infatti, il prodotto orto-bifenil-sulfonammidico (composto 12,
tabella 2) risultante da questa strategia sintetica funge da “cappello” idrofobico per l’anello
imidazolico dell’His237 del sito attivo, ostruendo parzialmente il “buco” ossianionico al
passaggio dell’acqua e, al tempo stesso, instaurando favorevoli interazioni di Van der
Waals con la proteina. Successivi indagini sulle relazioni struttura-attività (SARs) hanno
documentato che questo nuovo inibitore bifenil-sulfonammidico è quindici volte più
potente dell’inibitore 10 [40].
L’ulteriore esame del composto 12 complessato con ICE, tramite cristallografia a
raggi-X (figura 22), ha consentito di dimostrare che l’incremento di potenza è da ascrivere
alle modalità di legame ed alle interazioni dell’inibitore stesso con il sito attivo [40].
Tabella 2 Valutazione di una nuova classe di inibitori non-peptidici, a basso peso molecolare, di ICE.
27

Tuttavia, altre indagini del complesso enzima-inibitore 12 hanno evidenziato una
seconda conformazione della catena laterale dell’His237: la catena laterale di questo residuo
catalitico ruota dalla conformazione gauche (+), precedentemente osservata, ad una
conformazione trans, creando una larga tasca adiacente al sito S1 [69].
In seguito, nel tentativo di incrementarne la potenza, il composto 12 è stato
variamente modificato a livello del sistema bifenilico nell’esteso sito S1, ottenendo i
composti 13, 14, 15, 16, 17, 18 (vedi tabella 2). Tuttavia, questi analoghi dell’inibitore 12,
Figura 22 Struttura cristallografica a raggi-X dell’inibitore 12 nel sito attivo di ICE. L’inibitore è colorato
secondo il tipo di atomo (blu per l’azoto, rosso per l’ossigeno, giallo per lo zolfo, e bianco per il carbonio).
Sono mostrati i legami ad idrogeno tra l’inibitore sulfonammidico (SO2NH) e l’Arg341 (NH2CNH), la Ser339
(CO), e tra l’acido (CO2) e l’Arg179 (NH2CNH). L’His237 è mostrata in grigio-azzurro.
sebbene più potenti degli inibitori iniziali 3, 5, 7, e 10 (strutture non troncate ad aldeidi),
hanno mostrato una potenza inferiore al composto di origine. Quindi, la prossima sfida sarà
28

quella di aumentare la potenza di questa nuova ed utile classe di inibitori non-peptidici di
ICE [40].
2.3 Inibitori macromolecolari della caspasi-1.
Un noto inibitore macromolecolare di ICE è la tossina CrmA (cytokine response
modifier A), codificata dal virus del vaiolo bovino [70]. Questa macromolecola è un
membro degli inibitori delle proteasi seriniche della superfamiglia delle serpine [71].
Confrontata con le strutture note di altre serpine (inibitore dell’α1-proteinasi, α1-
antichimotripsina), la struttura cristallografica di CrmA (figura 23), clivata a livello del
loop del sito reattivo (a livello del legame peptidico P4-P3), mostra un elevato livello di
conservazione nonostante l’identità di sequenza relativamente bassa tra questi membri
della famiglia (20-35% per queste serpine). In particolare, questa serpina differisce dagli
altri membri della famiglia poiché il loop del suo centro reattivo è più corto di un solo
amminoacido [72].
CrmA inibisce rapidamente la caspasi-1 con una costante di associazione (Kon) di
1.7 ∙ 107 M-1 ∙ s -1, formando uno stretto complesso con una costante di inibizione (K i)
inferiore a 4 ∙ 10-12 M. Questi dati indicano che CrmA è un potente inibitore di ICE. CrmA
è l’unico membro della superfamiglia delle serpine capace di inibire una proteasi
cisteinica, piuttosto che una proteasi serinica [71]. La struttura cristallografica di questa
tossina suggerisce che essa può inibire le proteasi cisteiniche attraverso un meccanismo
analogo a quello utilizzato dalle altre serpine per l’inibizione delle proteasi seriniche [72].
Infatti, l’analisi del processo di formazione del complesso ICE-CrmA ha evidenziato la
presenza di un intermedio tio-acilico, equivalente all’intermedio acilico che caratterizza i
complessi serpina-proteasi seriniche [70]. Probabilmente, tale insolita capacità inibitoria è
giustificata dal fatto che ICE possiede una geometria di legame del substrato simile a
quella di una proteasi serinica [71]: esiste una stretta somiglianza tra la sequenza Leu-Val-
Ala-Asp presente nel loop del sito reattivo di CrmA e la sequenza tetrapeptidica di
substrato preferita da ICE, Tyr-Val-Ala-Asp (P4-P1) [73]. Dunque, la selettività di CrmA
per ICE è presumibilmente dovuta alla presenza dell’aspartato P1 nel loop del sito reattivo
[21]. Per quanto concerne il meccanismo d’azione, pare che la formazione del complesso
serpina-ICE determini la dissociazione del tetramero caspasi ed il successivo distacco della
subunità minore dalla proteasi stessa [70].
29

In sintesi, l’inibizione di ICE da parte di CrmA costituisce un tipico esempio di
interazione “cross-class”, in cui una serpina inibisce una proteasi non serinica [71].
A
B
Figura 23 (A) Rappresentazione a nastri della struttura cristallografica della serpina CrmA nella sua forma
clivata al loop del centro reattivo (PDB ID:1F0C). (B) Rappresentazione tridimensionale a nastri del mutante,
privo di cisteina, di CrmA nella sua forma clivata (PDB ID:1C8O). I foglietti β (s4A, s1’A) sono mostrati in
30

grigio-azzurro; le α-eliche (hA, hE) sono mostrate in magenta ed in giallo. Il loop del centro reattivo clivato è
completamente inserito nel corpo della molecola come s4A.
31

Recentemente, diversi studi biochimico-clinici hanno documentato che una
proteina, la cui mutazione è alla base della “Febbre Mediterranea Familiare” (FMF),
modula negativamente l’attivazione della caspasi-1. Tale proteina, denominata pirina, è
costituita da 781 amminoacidi ed è espressa soprattutto nei granulociti, nei monociti
attivati da citochine, e nei fibroblasti del siero e del liquido sinoviale [74, 75]. L’elemento
principalmente coinvolto nell’inibizione di ICE è il dominio C-terminale B30.2
(PRYSPRY), costituito da circa 200 amminoacidi: ~ 139 amminoacidi compongono il
segmento SPRY C-terminale, i rimanenti 61 residui costituiscono il subdominio PRY N-
terminale. All’estremità N-terminale della proteina è localizzato un altro dominio di circa
90 amminoacidi, denominato PYRIN, che assume una struttura a 6 α-eliche simile quella
descritta per i domini di morte (DDs), i domini effettori di morte (DEDs), ed i domini di
reclutamento delle caspasi (CARDs), caratteristici delle procaspasi. Tramite quest’ultimo
dominio la pirina stabilisce un’interazione omotipica con la proteina adattatrice
infiammatoria ASC (apoptosis-associated speck-like protein with a CARD), anch’essa
munita di un dominio di morte strutturalmente simile al dominio PYRIN, cioè CARD [76,
77]. ASC, a sua volta, si assembla in complessi macromolecolari denominati
“infiammasomi” e, tramite il “proprio dominio di morte PYRIN”, interagisce con alcune
proteine di attivazione della procaspasi-1; inoltre, essa stabilisce interazioni omotipiche
dirette con la stessa procaspasi-1 a livello dei domini CARDs di entrambe. In seguito a tali
interazioni a livello dell’infiammasoma, due molecole di procaspasi-1 vengono a trovarsi
vicine e danno luogo ad un processo autocatalitico che conduce al rilascio delle subunità
catalitiche attive, p20 e p10, della caspasi-1 [78].
Il ruolo della pirina nel modulare l’attività della caspasi-1 è controverso: è stato
osservato che, in funzione delle condizioni sperimentali, essa può inibire o accentuare
l’attività di ICE attraverso l’interazione eterotipica del dominio PYRIN con il dominio
CARD di ASC [77]. Probabilmente, la pirina modula negativamente la caspasi-1 in quanto
compete, per il legame ad ASC, con le proteine pro-infiammatorie deputate all’attivazione
di questa proteasi [79]. Recenti studi hanno altresì rivelato che la pirina può inibire
l’attività di ICE interagendo direttamente con quest’ultimo: il dominio PYRIN N-terminale
di questa proteina è un membro della superfamiglia dei domini di morte che include anche
il dominio CARD della caspasi-1; di conseguenza, è plausibile che il legame della pirina
alla caspasi-1 sia mediato da un’interazione eterotipica tra i rispettivi domini di morte.
Tuttavia, successivi esperimenti di co-immunoprecipitazione hanno chiarito che il dominio
coinvolto nell’interazione diretta pirina/caspasi-1 non è quello PYRIN, bensì il dominio C-
32

terminale B30.2. Infatti, questo dominio stabilisce un’interazione con i domini catalitici
della caspasi. Inoltre, è emerso che l’effetto finale dell’interazione diretta pirina/caspasi-1 è
di tipo inibitorio [80].
Poiché molte delle principali mutazioni associate alla FMF interessano proprio il
dominio B30.2 della pirina, l’indagine si è orientata anche verso i possibili effetti di queste
mutazioni sul legame alla caspasi-1. In particolare, la pirina wild-type e le forme mutate,
M680I ed M694V, sono state confrontate relativamente alla capacità di interagire con la
caspasi-1, tramite Western blotting e successiva analisi densitometrica: il legame della
caspasi-1 con i mutanti è risultato sostanzialmente ridotto rispetto a quello con la proteina
wild-type; di conseguenza, è stato ipotizzato che queste varianti non inibiscono ICE con la
stessa efficacia [80].
Allo scopo di analizzare più dettagliatamente l’interazione pirina/caspasi-1, le
strutture cristallografiche della proteasi e del dominio B30.2 della pirina [14, 81] sono state
utilizzate per produrre un modello computazionale dell’interazione di queste due strutture
(figura 24). L’analisi di questo modello ha mostrato che i residui amminoacidici
equivalenti alle mutazioni M694V ed M680I, nel dominio B30.2 della pirina, cadono in
distinti loops caratterizzati da una comune superficie di legame. Infatti, gli studi
Figura 24 Un modello per lo studio dell’interazione della pirina con la caspasi-1. Il modello del dominio
B30.2 della pirina (PDB ID:2FNJ) (colorato in blu) è stato agganciato alla struttura cristallografica della
caspasi-1 (PDB ID:1SC1) (subunità p20, in verde; subunità p10, in marrone). Due mutazioni comuni che
causano la FMF (M694V e M680I; in giallo) sono localizzate all’interfaccia di legame.
33

computazionali relativi al processo di “docking” pirina/caspasi-1 hanno indicato, da un
lato, che questa superficie del dominio B30.2 della pirina può interagire con la caspasi-1 e,
dall’altro, che le mutazioni associate alla FMF alterano tale interazione. L’esame
dell’orientazione della caspasi-1 nel modello ha altresì rivelato che entrambe le subunità,
p20 e p10, contribuiscono all’interazione con la pirina. Ciò corrobora ulteriormente i dati
sperimentali precedentemente descritti: 1) il dominio B30.2 della pirina lega le subunità
p20 e p10 della caspasi-1 inibendone l’attività catalitica; 2) due mutazioni del dominio
B30.2, associate alla FMF, attenuano questo effetto [80]. Globalmente, questi dati
supportano un modello in base al quale la forma naturale della pirina inibisce l’attivazione
di ICE [80].
2.4 Inibitori allosterici della caspasi-1.
Nonostante le importanti conseguenze cliniche, i siti attivi delle caspasi
costituiscono dei targets molto difficili da raggiungere con le grosse molecole a
disposizione. Tale difficoltà è dovuta alle preferenze chimiche dell’enzima: le caspasi
preferiscono composti chimici con centri elettrofili in grado di subire l’attacco nucleofilo
da parte della cisteina catalitica [82].
Al fine di individuare piccole molecole dirette al sito attivo della caspasi-7 è stato
utilizzato un approccio noto come “trappola disolfuro” o “disulfide trapping”. In
particolare, una serie di cisteine naturali o ingegnerizzate, presenti sulla superficie
enzimatica, sono state esaminate con una “libreria” di piccole molecole contenenti gruppi
disolfuro in un tampone redox [83]. Questo screening ha rivelato una importante novità:
molti dei composti tiolici, pur non interagendo efficacemente con la cisteina del sito attivo
della caspasi-7, hanno dimostrato di essere ottimi inibitori di un’altra cisteina, collocata in
una cavità all’interfaccia del dimero. Successivi studi strutturali della caspasi-7, contenente
inibitori tiolici all’interfaccia dimerica, hanno confermato che questi composti sono capaci
di revertire la transizione di questa proteasi dalla forma di zimogeno alla forma matura,
operando una modulazione, di tipo allosterico, della caspasi-7 [84].
La caspasi-1 non presenta una cisteina “allosterica” nella stessa posizione della
caspasi-7, tuttavia ne possiede una vicina nella cavità centrale, lontana 15 Å dal sito attivo,
la Cys331. Utilizzando questo sito come ancoraggio per la “trappola disolfuro”, Scheer ed i
suoi colleghi hanno provato ad identificare piccole molecole, capaci di legarsi
all’interfaccia del dimero, da poter utilizzare per verificare se anche ICE è soggetto ad una
34

regolazione allosterica. Inizialmente, con l’ausilio della spettrometria di massa, sono stati
individuati dei composti tiolici capaci di coniugarsi alla Cys331 della caspasi-1 (tramite
legami disolfuro) e di inibire l’attività enzimatica. In seguito, tra questi composti tiolici, è
stato scelto un tienopirazolo (figura 25) ed è stata determinata la struttura cristallografica
Figura 25 Composto tienopirazolico selezionato per lo studio.
della caspasi-1 in complesso con questo composto. L’analisi di questa struttura purificata
ha mostrato due molecole tienopirazoliche legate, in maniera simmetrica, all’interfaccia del
dimero (figura 26). Queste due molecole appaiono abbastanza vicine da dar luogo a
reciproche interazioni di Van der Waals ed aromatiche. Ciascuna di esse ha un
orientamento trans ed è incatenata alla Cys331 in una subunità, ma stabilisce gran parte delle
sue interazioni non covalenti con i residui amminoacidici della subunità vicina. Il
A B
Figura 26 Struttura di un inibitore allosterico legato alla caspasi-1 (PDB ID:2FQQ). (A) Due molecole del
composto tiolico in esame sono mostrate come sfere nella cavità centrale all’interfaccia del dimero della
caspasi-1. (B) I residui amminoacidici, coinvolti nella formazione della tasca di legame per il composto
tiolico, sono raffigurati mediante sfere. I residui appartenenti alla subunità maggiore (Glu241, Gln257, Arg286)
sono colorati in blu; i residui appartenenti alla subunità minore (Thr388, Glu390, Arg391) sono colorati in
marrone rossiccio.
35

gruppo trifluorometilico del composto tienopirazolico è strettamente “pressato” contro la
catena laterale dell’Arg391 ed è, presumibilmente, stabilizzato da interazioni polari. D’altra
parte, il gruppo pirazolico è “pressato” contro la catena laterale della Leu258, localizzata
nella subunità maggiore del dimero adiacente. Il bordo della tasca di legame è composto
dai residui amminoacidici della subunità maggiore, Gln257, Glu241, e Arg286; invece, la parte
inferiore è delineata dai seguenti amminoacidi della subunità minore, Thr288, Glu390, e
Arg391 (vedi figura 26, pannello B). È stata osservata anche un’intima interazione tra la
porzione ammidica linker del tienopirazolo ed il carbossilato del Glu390 nella subunità
minore del monomero [85].
In definitiva, la coniugazione del composto tienopirazolico in esame con la caspasi-
1, paragonata alla forma attiva della caspasi-1 marcata con un inibitore del sito attivo, Z-
VAD-FMK (benzilossicarbonil-Val-Ala-Asp-fluorometil-chetone), mostra tre conseguenze
molto importanti dal punto di vista funzionale: 1) la Cys285 catalitica subisce una rotazione
di ~ 5 Å lontano dalla sua normale posizione a causa dell’idrolisi dei legami peptidici; 2) i
loops, che definiscono il sito di legame per il substrato, subiscono un’alterazione della loro
posizione tale per cui non sono più capaci di interagire produttivamente con un substrato;
3) la catena laterale dell’Arg286 ruota verso la tasca di legame del substrato conservando,
però, un elevato grado di flessibilità (figura 27, pannello A) [85].
Poiché il legame del ligando ad entrambi i siti, attivo e allosterico, determina
cambiamenti drammatici e concertati, è stato condotto un ulteriore esperimento finalizzato
a capire se tali cambiamenti si escludono a vicenda. In particolare, è stato osservato che la
reazione della Cys285 del sito attivo con un suo inibitore irreversibile, come Z-VAD-FMK,
nella subunità maggiore impedisce il successivo legame del composto tienopirazolico al
sito allosterico nella subunità minore (figura 27, pannello B). Di converso, il legame del
tienopirazolo alla Cys331 del sito allosterico dell’enzima si è mostrato capace di prevenire il
legame dell’inibitore Z-VAD-FMK al sito attivo dell’enzima. Ciò suggerisce che,
probabilmente, i due siti lavorano in una modalità reciproca e mutualmente esclusiva [85].
Questi studi, considerati in toto, hanno consentito di identificare nuovi composti, a
basso peso molecolare, che si legano specificamente al sito allosterico della caspasi-1
inibendone efficacemente l’attività enzimatica. Questi composti tiolici compromettono
l’integrità di un network di interazioni che attraversa il sito attivo all’interfaccia del
dimero. Tale meccanismo si può osservare anche nell’inibizione allosterica della caspasi-7;
ciò suggerisce che queste proteasi cisteiniche conservano caratteristiche strutturali comuni,
36

A
B
Figura 27 Confronto del sito attivo della caspasi-1 nella struttura complessata al sito attivo e nella struttura
complessata al sito allosterico.
(A) Le regioni a quattro loops, che modificano la loro posizione fra la conformazione in cui è complessato il
sito attivo e la conformazione in cui è complessato il sito allosterico, sono raffigurati da fiocchi. La Cys 285 del
sito attivo e l’adiacente Arg286 sono rappresentati dai bastoncini. (Lato sinistro) La struttura in cui il sito
attivo è complessato, è mostrata in complesso con l’inibitore del sito attivo Z-VAD-FMK (PDB ID:2HBR)
(bastoncini verdi). (Lato destro) La struttura in cui il sito allosterico è complessato, è mostrata in complesso
con l’inibitore allosterico tienopirazolico (PDB ID:2FQQ).
(B) Analisi, tramite spettrometria di massa, del legame competitivo dei siti, attivo ed allosterico, con Z-VAD-
FMK e con il composto tienopirazolico sulle subunità maggiore e minore, rispettivamente.
37
nonostante la loro diversità per quanto concerne l’identità di sequenza e la funzione
biologica. La conservazione del meccanismo inibitorio si riflette nelle strutture delle
conformazioni inattive delle caspasi che includono la struttura priva di ligando e quella
dello zimogeno. Parimenti interessante risulta il confronto tra le strutture della caspasi-1
nelle conformazioni, allostericamente inibita e libera da ligando: i composti allosterici
“intrappolano” l’enzima nella conformazione naturalmente inattiva, osservata anche
nell’enzima privo di ligando (figura 28). Questa scoperta suggerisce che la conformazione
“a loop inattivo” potrebbe essere energeticamente favorita. Inoltre, tale conformazione
potrebbe avere due importanti funzioni: 1) proteggere la cisteina del sito attivo da
un’ossidazione irreversibile e/o; 2) consentire un maggiore grado di regolazione
sull’attività dell’enzima opponendo una barriera conformazionale alla catalisi [85].
Figura 28 Confronto della superficie di tre strutture della caspasi-1. La struttura della caspasi-1 (in cui il sito
attivo è legato da ligando) complessata con l’inibitore del sito attivo Z-VAD-FMK (PDB ID:2HBR), la
struttura della caspasi-1 (in cui il sito allosterico è legato da ligando) complessata con l’inibitore allosterico
tienopirazolico prescelto (PDB ID:2FQQ) ed, infine, la struttura dell’enzima libero da ligando (PDB
ID:1SC1) sono mostrate in acqua. La cavità allosterica fra i due monomeri è indicata in rosso.
38

3. Prospettive future: implicazioni farmacocinetiche.
La vasta letteratura relativa alla chimica ed alla biochimica degli inibitori delle proteasi
maschera il fatto che pochissime proteasi sono state mirate, con successo, dai farmaci
attualmente a disposizione in terapia. Infatti, gli inibitori di proteasi sono utilizzati,
esclusivamente, per il trattamento clinico dell’ipertensione e dell’infezione da HIV, avendo
come targets l’enzima di conversione dell’angiotensina e l’HIV aspartil-proteasi,
rispettivamente [26].
In quest’ultimo decennio, la struttura ed il meccanismo catalitico della caspasi-1
sono stati compresi più profondamente [8]; tuttavia, la maggior parte degli inibitori di ICE,
sintetizzati e studiati, è di natura peptidica. Sebbene le più potenti tra queste molecole
siano capaci di inibire efficacemente l’enzima in sistemi in vitro, esse mostrano diversi
limiti farmacocinetici, in sistemi in vivo, proprio a causa del loro scheletro peptidico. Tali
limiti sono rappresentati da una scarsa selettività, da una breve emivita plasmatica e da una
notevole instabilità metabolica che, di fatto, rendono difficoltoso un efficace utilizzo di
questi composti nella terapia clinica. Dunque, per le applicazioni cliniche di natura cronica,
occorre sintetizzare inibitori di ICE che presentino un maggiore selettività, una maggiore
stabilità metabolica ed una migliore biodisponibilità orale. In particolare, il trattamento
clinico iniziale può riguardare patologie in cui, brevi cicli di somministrazione del farmaco
potrebbero offrire benefici effetti antiinfiammatori. Inoltre, coniugando una migliore
biodisponibilità orale con un’elevata potenza cellulare, si potranno, in futuro, ottenere
composti che potrebbero essere molto utili nel trattamento di patologie croniche [26].
Recentemente, sono stati sintetizzati inibitori delle caspasi, a basso peso molecolare, che
non hanno origine peptidica. Uno di questi inibitori di caspasi, IDN-6556 (figura 29), ha
già raggiunto la fase II degli studi clinici: è un inibitore ad ampio spettro che forma addotti
irreversibili con i residui di cisteina del sito attivo delle caspasi, quindi, potrebbe essere un
ottimo candidato per il trattamento delle patologie conseguenti a danni tissutali acuti,
caratterizzate da un’apoptosi eccessiva [86]. Un altro inibitore di caspasi, a basso peso
molecolare, è la molecola VX-740 (Pralnacasan). Essa è un inibitore selettivo e reversibile
della caspasi-1, sviluppato per il trattamento dell’artrite reumatoide [87].
Un’altra affascinante sfida futura per la chimica “medico-farmaceutica” riguarda la
progettazione di nuovi “drug delivery systems” capaci di veicolare i farmaci inibitori di
ICE, ma anche di altre proteasi, sul proprio target intracellulare. Infatti, a livello
intracellulare, la selettività e l’affinità elevate degli inibitori verso l’enzima non
39

garantiscono che questi composti penetrino efficacemente attraverso la membrana
plasmatica o si localizzino nel compartimento subcellulare desiderato. Prova ne sia
l’inibitore di ICE, DEVD-H, il quale, a dispetto della sua elevata affinità di legame (bassi
valori di Ki, dell’ordine del nanomolare) verso molteplici omologhi di ICE, non è in grado
di bloccare l’apoptosi, caspasi-mediata, a concentrazioni inferiori a 100 µM.
Generalmente, risulta improbabile un rilascio, terapeuticamente sicuro ed efficace, di un
inibitore di proteasi, a concentrazioni ematiche proporzionalmente elevate. Peraltro, il
trattamento di patologie neurodegenerative, quali l’Alzheimer, richiede necessariamente la
penetrazione della barriera emato-encefalica, e ciò rappresenta un altro significativo
ostacolo per il buon esito di un eventuale drug design [26, 8].
Queste considerazioni e speculazioni di natura farmacocinetica orientano,
inevitabilmente, la ricerca futura verso la scoperta di inibitori non-peptidici, potenti e
selettivi, con caratteristiche farmacologiche adeguate all’esecuzione di ulteriori studi in
vivo. Infatti, inibitori di questo tipo potrebbero rivelarsi elementi fondamentali per
rispondere a tutti gli interrogativi, che ancora restano, circa i ruoli biologici di ICE e dei
suoi analoghi nei processi infiammatori e di morte cellulare [21, 8].
Figura 29 Struttura chimica dell’inibitore IDN-6556.
40

4. Bibliografia.
1. Alnemri, E. S.; Livingston, D. J.; Nicholson, D. W.; Salvesen, G.; Thornberry, N.
A.; Wong, W. W. Juan, J. Human ICE/CED-3 protease nomenclature. Cell 1996,
87, 171.
2. Yuan, J.; Horvitz, H. R. A first insight into molecular mechanisms of apoptosis.
Cell 2004, 116 (2 Suppl), S53-S56.
3. Ellis, R. E.; Yuan, J. Y.; Horvitz, H. R. Mechanisms and functions of cell death.
Annu. Rev. Cell. Biol. 1991, 7, 663-698.
4. Nicholson, D. W. Caspase structure, proteolytic substrates, and function during
apoptotic cell death. Cell Death Differ. 1999, 6, 1028-1042.
5. Cohen, G. M. Caspases: the executioners of apoptosis. Biochem. J. 1997, 326, 1-16.
6. Cornelis, S.; Kersse, K.; Festjens, N.; Lamkanfi, M.; Vandenabeele, P.
Inflammatory caspases: targets for novel therapies. Curr. Pharm. Des. 2007, 13,
367-385.
7. Thornberry, N. A.; Lazebnik, Y. Caspases: enemies within. Science 1998, 281,
1312-1316.
8. Lavrik, I. N.; Golks, A.; Krammer, P. H. Caspases: pharmacological manipulation
of cell death. J. Clin. Invest. 2005, 115, 2665-2672.
9. Fesik, S. W. Insights into programmed cell death through structural biology. Cell
2000, 103, 273-282.
10. Hofmann, K. The modular nature of apoptotic signaling proteins. Cell. Mol. Life
Sci. 1999, 55, 1113-1128.
11. Walker, N. P.; Talanian, R. V.; Brady, K. D.; Dang, L. C.; Bump, N. J.; Ferenz, C.
R.; Franklin, S.; Ghayur, T.; Hackett, M. C.; Hammill, L. D.; et al. Crystal structure
of the cysteine protease interleukin-1 beta-converting enzyme: a (p20/p10)2
homodimer. Cell 1994, 78, 343-352.
12. Fuentes-Prior, P.; Salvesen, G. S. The protein structures that shape caspase activity,
specificity, activation and inhibition. Biochem. J. 2004, 384, 201-232.
13. Eichinger, A.; Beisel, H. G.; Jacob, U.; Huber, R.; Medrano, F. J.; Banbula, A.;
Potempa, J.; Travis, J.; Bode, W. Crystal structure of gingipain R: an Arg-specific
bacterial cysteine proteinase with a caspase-like fold. EMBO J. 1999, 18, 5453-
5462.
41

14. Wilson, K. P.; Black, J. A.; Thomson, J. A.; Kim, E. E.; Griffith, J. P.; Navia, M.
A.; Murcko, M. A.; Chambers, S. P.; Aldape, R. A.; Raybuck, S. A.; et al. Structure
and mechanism of interleukin-1 beta converting enzyme. Nature 1994, 370, 270-
275.
15. Thornberry, N. A.; Rano, T. A.; Peterson, E. P.; Rasper, D. M.; Timkey, T.; Garcia-
Calvo, M.; Houtzager, V. M.; Nordstrom, P. A.; Roy, S.; Vaillancourt, J. P.;
Chapman, K. T.; Nicholson, D. W. A combinatorial approach defines specificities
of members of the caspase family and granzyme B. Functional relationships
established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907-17911.
16. Chang, D. W.; Ditsworth, D.; Liu, H. T.; Srinivasula, S. M.; Alnemri, E. S.; Yang,
X. L. Oligomerization is a general mechanism for the activation of apoptosis
initiator and inflammatory procaspases. J. Biol. Chem. 2003, 278, 16466-16469.
17. Talanian, R. V.; Quinlan, C.; Trautz, S.; Hackett, M. C.; Mankovich, J. A.; Banach,
D.; Ghayur, T.; Brady, K. D.; Wong, W. W. Substrate specificities of caspase
family proteases. J. Biol. Chem. 1997, 272, 9677-9682.
18. Brady, K. D.; Giegel, D. A.; Grinnell, C.; Lunney, E.; Talanian, R. V.; Wong, W.;
Walker, N. A catalytic mechanism for caspase-1 and for bimodal inhibition of
caspase-1 by activated aspartic ketones. Bioorg. Med. Chem. 1999, 7, 621-631.
19. Stennicke, H. R.; Salvesen, G. S. Catalytic properties of the caspases. Cell Death
Differ. 1999, 6, 1054-1059.
20. Kostura, M. J.; Tocci, M. J.; Limjuco, G.; Chin, J.; Cameron, J.; Hillman, A. G.;
Chartrain, N. A.; Schmidt, J. A. Identification of a monocyte specific pre-
interleukin 1 beta convertase activity. Proc. Natl. Acad. Sci. USA 1989, 86, 5227-
5231.
21. Thornberry, N. A.; Molineaux, S. M. Interleukin-1 beta converting enzyme: a novel
cysteine protease required for IL-1 beta production and implicated in programmed
cell death. Protein Sci. 1995, 4, 3-12.
22. Laufersweiler, M. C.; Wang, Y.; Soper, D. L.; Suchanek, M. K.; Fancher, A. N.;
Lu, W.; Wang, R. L.; Oppong, K. A.; Ellis, C. D.; Baize, M. W.; O’Neil, S. V.;
Wos, J. A.; Demuth, T. P. Jr. Synthesis and evaluation of tricyclic
pyrrolopyrimidinones as dipeptide mimetics: inhibition of interleukin-1beta-
converting enzyme. Bioorg. Med. Chem. Lett. 2005, 15, 4322-4326.
42

23. Wang, Y.; O’Neil, S. V.; Wos, J. A.; Oppong, K. A.; Laufersweiler, M. C.; Soper,
D. L.; Ellis, C. D.; Baize, M. W.; Fancher, A. N.; Lu, W.; Suchanek, M. K.; Wang,
R. L.; Schwecke, W. P.; Cruze, C. A.; Buchalova, M.; Belkin, M.; De, B.; Demuth,
T. P. Jr. Synthesis and evaluation of unsaturated caprolactams as interleukin-1beta
converting enzyme (ICE) inhibitors. Bioorg. Med. Chem. 2007, 15, 1311-1322.
24. Dolle, R. E.; Singh, J.; Rinker, J.; Hoyer, D.; Prasad, C. V. C.; Graybill, T. L.;
Salvino, J. M.; Helaszek, C. T.; Miller, R. E.; Ator, M. A. Aspartyl alpha-((1-
phenyl-3-(trifluoromethyl)-pyrazol-5-yl)oxy)methyl ketones as interleukin-1 beta
converting enzyme inhibitors. Significance of the P1 and P3 amido nitrogens for
enzyme-peptide inhibitor binding. J. Med. Chem. 1994, 37, 3863-3866.
25. Mullican, M. D.; Lauffer, D. J.; Gillespie, R. J.; Matharu, S. S.; Kay, D.; Porritt, G.
M.; Evans, P. L.; Golec, J. M. C.; Murcko, M. A.; Loung, Y. P.; Raybuck, S. A.;
Livingston, D. J. The synthesis and evaluation of peptidyl aspartyl aldehydes as
inhibitors of ICE. Bioorg. Med. Chem. Lett. 1994, 4, 2359-2364.
26. Livingston, D. J. In vitro and in vivo studies of ICE inhibitors. J. Cell. Biochem.
1997, 64, 19-26.
27. Sleath, P. R.; Hendrickson, R. C.; Kronheim, S. R.; March, C. J.; Black, R. A.
Substrate specificity of the protease that processes human interleukin-1 beta. J.
Biol. Chem. 1990, 265, 14526-14528.
28. Howard, A. D.; Kostura, M. J.; Thornberry, N. A.; Ding, G. J.; Limjuco, G.,
Weidner, J.; Salley, J. P.; Hogguist, K. A.; Chaplin, D. D.; Mumford, R. A.;
Schmidt, J. A.; Tocci, M. J. IL-1-converting enzyme requires aspartic acid residues
for processing of the IL-1 beta precursor at two distinct sites and does not cleave
31-kDa IL-1 alpha. J. Immunol. 1991, 147, 2964-2969.
29. Thornberry, N. A.; Bull, H. G.; Calaycay, J. R.; Chapman, K. T.; Howard, A. D.;
Kostura, M. J.; Miller, D. K.; Molineaux, S. M.; Weidner, J. R.; Aunins, J.;
Elliston, K. O.; Ayala, J. M.; Casano, F. J.; Chin, J.; Ding, G. J.; Egger, L. A.;
Gaffney, E. P.; Limjuco, G.; Palyha, O. C.; Faju, S. M.; Rolando, A. M.; Salley, J.
P.; Yamin, T.; Lee, T. D.; Shively, J. E.; MacCross, M.; Mumford, R. A.; Schmidt,
J. A.; Tocci, M. J. A novel heterodimeric cysteine protease is required for
interleukin-1 beta processing in monocytes. Nature 1992, 356, 768-774.
43

30. Prasad, C. V. C.; Prouty, C. P.; Hoyer, D.; Ross, T. M.; Salvino, J. M.; Awad, M.;
Graybill, T. L.; Schmidt, S. J.; Osifo, I. K.; Dolle, R. E.; Helaszek, C. T.; Miller, R.
E.; Ator, M. A. Structural and stereochemical requirements of the time-dependent
inactivators of the interleukin-1β converting enzyme. Bioorg. Med. Chem. Lett.
1995, 5, 315-318.
31. Ator, M. Peptide and non-peptide inhibitors of interleukin-1β converting enzyme:
Paper presented at Inflammatory Cytokine Antagonists-Targets, Strategies and
Indications. Cambridge Healthtech Institute (Walthman, M.A.), Philadelphia. 1994.
32. Fernandes-Alnemri, T.; Litwack, G.; Alnemri, E. S. CPP32, a novel human
apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3
and mammalian interleukin-1 beta-converting enzyme. J. Biol. Chem. 1994, 269,
30761-30764.
33. Fernandes-Alnemri, T.; Litwack, G.; Alnemri, E. S. Mch2, a new member of the
apoptotic Ced-3/Ice cysteine protease gene family. Cancer Res. 1995, 55, 2737-
2742.
34. Fernandes-Alnemri, T.; Takahashi, A.; Armstrong, R.; Krebs, J.; Fritz, L.;
Tomaselli, K. J.; Wang, L.; Yu, Z.; Croce, C. M.; Salveson, G.; Earnshaw, W. C.;
Litwack, G.; Alnemri, E. S. Mch3, a novel human apoptotic cysteine protease
highly related to CPP32. Cancer Res. 1995, 55, 6045-6052.
35. Wang, L.; Miura, M.; Bergeron, L.; Zhu, H.; Yuan, J. Ich-1, an Ice/ced-3-related
gene, encodes both positive and negative regulators of programmed cell death. Cell
1994, 78, 739-750.
36. Faucheu, C.; Diu, A.; Chan, A. W. E.; Blanchet, A. M.; Miossec, C.; Herve, F.;
Collard-Dutilleul, V.; Gu, Y.; Aldape, R.; Lippke, J.; Rocher, C.; Su, M. S.;
Livingston, D. J.; Hercend, T.; Lalanne, J. L. A novel human protease similar to the
interleukin-1 beta converting enzyme induces apoptosis in transfected cells. EMBO
J. 1995, 14, 1914-1922.
37. Romanowski, M. J.; Scheer, J. M.; O’Brien, T.; McDowell, R. S. Crystal structures
of a ligand-free and malonate-bound human caspase-1: implications for the
mechanism of substrate binding. Structure 2004, 12, 1361-1371.
38. Saleh, M. Caspase-1 builds a new barrier to infection. Cell 2006, 126, 1028-1030.
39. Cerretti, D. P.; Kozlosky, C. J.; Mosley, B.; Nelson, N.; Van Ness, K.; Greenstreet,
T. A.; March, C. J.; Kronheim, S. R.; Druck, T.; Cannizzaro, L. A.; et al. Molecular
cloning of the interleukin-1 beta converting enzyme. Science 1992, 256, 97-100.
44

40. Shahripour, A. B.; Plummer, M. S.; Lunney, E. A.; Albrecht, H. P.; Hays, S. J.;
Kostlan, C. R.; Sawyer, T. K.; Walker, N. P.; Brady, K. D.; Allen, H. J.; Talanian,
R. V.; Wong, W. W.; Humblet, C. Structure-based design of nonpeptide inhibitors
of interleukin-1β converting enzyme (ICE, Caspase-1). Bioorg. Med. Chem. 2002,
10, 31-40.
41. Dinarello, C. A. Inflammatory cytokines: interleukin-1 and tumor necrosis factor as
effector molecules in autoimmune diseases. Curr. Opin. Immunol. 1991, 3, 941-
948.
42. Kay, J.; Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid
arthritis. Rheumatology (Oxford) 2004, 43 Suppl 3, iii2-iii9.
43. Goldring, M. B.; Goldring, S. R. Osteoarthritis. J. Cell. Physiol. 2007, 213, 626-
634.
44. Dinarello, C. A.; Wolff, S. M. The role of interleukin-1 in disease. N. Engl. J. Med.
1993, 328, 106-113.
45. Fisher, C. J. J.; Dhainaut, J. F.; Opal, S. M.; Pribble, J. P.; Balk, R. A.; Slotman, G.
J.; Iberti, T. H.; Rackow, E. C.; Shapiro, M. J.; Greenman, R. L. Recombinant
human interleukin 1 receptor antagonist in the treatment of patients with sepsis
syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase
III rhIL-1ra Sepsis Syndrome Study Group. JAMA 1994, 271, 1836-1843.
46. Casellas, F.; Papo, M.; Guarner, F.; Antolin, M.; Segura, R. M.; Armengol, J. R.;
Malagelada, J. R. Intracolonic release in vivo of interleukin-1 beta in chronic
ulcerative colitis. Clin. Sci. 1995, 89, 521-526.
47. Holden R. J.; Mooney, P. A. Interleukin-1 beta: a common cause of Alzheimer's
disease and diabetes mellitus. Med. Hypoth. 1995, 45, 559-571.
48. Bamba, T.; Yoshioka, U.; Inoue, H.; Iwasaki, Y.; Hosoda, S. Serum levels of
interleukin-1 beta and interleukin-6 in patients with chronic pancreatitis. J.
Gastroenterol. 1994, 29, 314-319.
49. Norman, J. G.; Franz, M. G.; Fink, G. S.; Messina, J.; Fabri, P. J.; Gower, W. R.;
Carrey, L. C. Decreased mortality of severe acute pancreatitis after proximal
cytokine blockade. Ann. Surg. 1995, 221, 625-634.
50. Miura, M.; Zhu, H.; Rotello, R.; Hartwieg, E. A.; Yuan, J. Induction of apoptosis in
fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C.
elegans cell death gene ced-3. Cell 1993, 75, 653-660.
45

51. Gagliardini, V.; Fernandez, P. A.; Lee, R. K.; Drexier, H. C.; Rotello, R. J.;
Fishman, M. C.; Yuan, J. Prevention of vertebrate neuronal death by the crmA
gene. Science 1994, 263, 826-828.
52. Ichikawa, K.; Inagaki, T.; Kachi-Tonai, H.; Kojima, Y.; Nakamura, T.; Nishida, H.;
Ueno, Y.; Binding, P.; Gabel, C. A.; Lucas, V.; McNiff, P. A.; Kojima, N. LL-
Z1271alpha: an interleukin-1beta production inhibitor. Biochem. Biophys. Res.
Commun. 2001, 286, 697-700.
53. Soper, D. L.; Sheville, J.; O’Neil, S. V.; Wang, Y.; Laufersweiler, M. C.; Oppong,
K. A.; Wos, J. A.; Ellis, C. D.; Fancher, A. N.; Lu, W.; Suchanek, M. K.; Wang, R.
L.; De, B.; Demuth, T. P. Jr. Synthesis and evaluation of novel 1-(2-
acylhydrazinocarbonyl)-cycloalkyl carboxamides as interleukin-1beta converting
enzyme (ICE) inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4233-4236.
54. Thornberry, N. A. Interleukin-1 beta converting enzyme. Methods Enz. 1994, 244,
615-631.
55. Rich, D. H. Inhibitors of cysteine proteases. In: Proteinase inhibitors; Barrett, A. J.;
Salvesen, G., Eds.; Amsterdam: Elsevier. 1986, 153-178.
56. Shaw, E. Cysteinyl proteinases and their selective inactivation. Adv. Enzymol.
Relat. Areas Mol. Biol. 1990, 63, 271-347.
57. Chapman, K. T. Synthesis of a potent, reversible inhibitor of interleukin-1β
converting enzyme. Bioorg. Med. Chem. Lett. 1992, 2, 613-618.
58. Nicholson, D. W.; Ali, A.; Thornberry, N. A.; Vaillancourt, J. P.; Ding, C. K.;
Gallant, M.; Gareau Y.; Griffin, P. R.; Labelle, M.; Lazebnik, Y. A.; Munday, N.
A.; Raju, S. M.; Smulson, M. E.; Yamin, T. T.; Yu, V. L.; Miller, D. K.
Identification and inhibition of the ICE/CED-3 protease necessary for mammalian
apoptosis. Nature 1995, 376, 37-43.
59. Westerik, J. O.; Wolfenden, R. Aldehydes as inhibitors of papain. J. Biol. Chem.
1972, 247, 8195-8197.
60. Frankfater, A.; Kuppy, T. Mechanism of association of N-acetyl-L-
phenylalanylglycinal to papain. Biochemistry 1981, 20, 5517-5524.
61. Mackenzie, N. E.; Grant, S. K.; Scott, A. I.; Malthouse, J. P. G. 13C NMR study of
the stereospecificity of the thiohemiacetals formed on inhibition of papain by
specific enantiomeric aldehydes. Biochemistry 1986, 25, 2293-2298.
46

62. Ménard, R.; Carrière, J.; Laflamme, P.; Plouffe, C.; Khouri, H. E.; Vernet,
T.;Tessier, D. C.; Thomas, D. Y.; Storer, A. C. Contribution of the glutamine 19
side chain to transition-state stabilization in the oxyanion hole of papain.
Biochemistry 1991, 30, 8924-8928.
63. Schröder, E.; Phillips, C.; Garman, E.; Harlos, K.; Crawford, C. X-ray
crystallographic structure of a papain-leupeptin complex. FEBS Lett. 1993, 315, 38-
42.
64. Mjalli, A. M. M.; Chapman, K. T.; MacCoss, M.; Thornberry, N. A. Phenyl-alkyl
ketones as potent reversible inhibitors of interleukin-1β converting enzyme. Bioorg.
Med. Chem. Lett. 1993, 3, 2689-2692.
65. Mjalli, A. M. M.; Chapman, K. T.; MacCoss, M. Synthesis of a peptidyl 2,2-
difluoro-4-phenylbutyl and its evaluation as an inhibitor of interleukin-1β
converting enzyme. Bioorg. Med. Chem. Lett. 1993, 3, 2693-2698.
66. Dolle, R. E.; Hoyer, D.; Prasad, C. V. C.; Schmidt, S. J.; Helaszek, C. T.; Miller, R.
E.; Ator, M. A. P1 aspartate-based peptide alpha-((2,6-dichlorobenzoyl)oxy)methyl
ketones as potent time-dependent inhibitors of interleukin-1 beta-converting
enzyme. J. Med. Chem. 1994, 37, 563-564.
67. Thornberry, N. A.; Peterson, E. P.;Zhao, J. J.; Howard, A. D.; Griffin, P. R.;
Chapman, K. T. Inactivation of interleukin-1 beta converting enzyme by peptide
(acyloxy)methyl ketones. Biochemistry 1994, 33, 3934-3940.
68. Ellis, C. D.; Oppong, K. A.; Laufersweiler, M. C.; O’Neil, S. V.; Soper, D. L.;
Wang, Y.; Wos, J. A.; Fancher, A. N.; Lu, W.; Suchanek, M. K.; Wang, R. L.; De,
B.; Demuth, T. P. Jr. Synthesis and evaluation of thiazepines as interleukin-1beta
converting enzyme (ICE) inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4728-
4732.
69. Okamoto, Y.; Anan, H.; Nakai, E.; Morihira, K.; Yonetoku, Y.; Kurihara, H.;
Sakashita, H.; Terai, Y.; Takeuchi, M.; Shibanuma, T.; Isomura, Y. Peptide based
interleukin-1 beta converting enzyme (ICE) inhibitors: synthesis, structure activity
relationships and crystallographic study of the ICE-inhibitor complex. Chem.
Pharm. Bull. 1999, 47, 11-21.
70. Dobò, J.; Swanson, R.; Salvesen, G. S.; Olson, S. T.; Gettins, P. G. W. Cytokine
response modifier a inhibition of initiator caspases results in covalent complex
formation and dissociation of the caspase tetramer. J. Biol. Chem. 2006, 281,
38781-38790.
47

71. Komiyama, T.; Ray, C. A.; Pickup, D. J.; Howard, A. D.; Thornberry, N. A.;
Peterson, E. P.; Salvesen, G. Inhibition of interleukin-1 beta converting enzyme by
the cowpox virus serpin CrmA. An example of cross-class inhibition.
J. Biol. Chem. 1994, 269, 19331-19337.
72. Simonovic, M.; Gettins, P. G. W.; Volz, K. Crystal structure of viral serpin crmA
provides insights into its mechanism of cysteine proteinase inhibition. Protein Sci.
2000, 9, 1423-1427.
73. Bajusz, S.; Fauszt, I.; Nemeth, K.; Barabas, E.; Juhasz, A.; Patthy, M.; Bauer, P. I.
Peptidyl beta-homo-aspartals (3-amino-4-carboxybutyraldehydes): new specific
inhibitors of caspases. Biopolymers 1999, 51, 109-118.
74. Centola, M.; Wood, G.; Frucht, D. M.; Galon, J.; Aringer, M.; Farrell, C.; Kingma,
D. W.; Horwitz, M. E.; Mansfield, E.; Holland, S. M.; O’Shea, J. J.; Rosenberg, H.
F.; Malech, H. L.; Kastner, D. L. The gene for familial Mediterranean fever,
MEFV, is expressed in early leukocyte development and is regulated in response to
inflammatory mediators. Blood 2000, 95, 3223-3231.
75. Diaz, A.; Hu, C.; Kastner, D. L.; Schaner, P.; Reginato, A. M.; Richards, N.;
Gumucio, D. L. Lipopolysaccharide-induced expression of multiple alternatively
spliced MEFV transcripts in human synovial fibroblasts: a prominent splice
isoform lacks the C-terminal domain that is highly mutated in familial
Mediterranean fever. Arthritis Rheum. 2004, 50, 3679-3689.
76. Richards, N.; Schaner, P.; Diaz, A.; Stuckey, J.; Shelden, E.; Wadhwa, A.;
Gumucio, D. L. Interaction between pyrin and the apoptotic speck protein (ASC)
modulates ASC-induced apoptosis. J. Biol. Chem. 2001, 276, 39320-39329.
77. Chae, J. J.; Komarow, H. D.; Cheng, J.; Wood, G.; Raben, N.; Liu, P. P.; Kastner,
D. L. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity
to endotoxin and a defect in macrophage apoptosis. Mol. Cell. 2003, 11, 591-604.
78. Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: a molecular platform
triggering activation of inflammatory caspases and processing of proIL-beta. Mol.
Cell. 2002, 10, 417-426.
79. Yu, J. W.; Wu, J.; Zhang, Z.; Datta, P.; Ibrahimi, I.; Taniguchi, S.; Sagara, J.;
Fernamdes-Alnemri, T.; Alnemri, E. S. Cryopyrin and pyrin activate caspase-1, but
not NF-kappaB, via ASC oligomerization. Cell. Death Differ. 2006, 13, 236-249.
48

80. Chae, J. J.; Wood, G.; Masters, S. L.; Richard, K.; Park, G.; Smith, B. J.; Kastner,
D. L. The B30.2 domain of pyrin, the familial Mediterranean fever protein,
interacts directly with caspase-1 to modulate IL-1beta production. PNAS 2006, 103,
9982-9987.
81. Masters, S. L.; Yao, S.; Willson, T. A.; Zhang, J. G.; Palmer, K. R.; Smith, B. J.;
Babon, J. J.; Nicola, N. A.; Norton, R. S.; Nicholson, S. E. The SPRY domain of
SSB-2 adopts a novel fold that presents conserved Par-4-binding residues. Nat.
Struct. Mol. Biol. 2006, 13, 77-84.
82. O’Brien, T.; Lee, D. Prospects for caspase inhibitors. Mini Rev. Med. Chem. 2004,
4, 153-165.
83. Erlanson, D. A.; Braisted, A. C.; Raphael, D. R.; Randal, M.; Stroud, R. M.;
Gordon, E. M.; Wells, J. A. Site-directed ligand discovery. Proc. Natl. Acad. Sci.
USA 2000, 97, 9367-9372.
84. Hardy, J. A.; Lam, J.; Nguyen, J. T.; O’Brien, T.; Wells, J. A. Discovery of an
allosteric site in the caspases. Proc. Natl. Acad. Sci. USA 2004, 101, 12461-12466.
85. Scheer, J. M.; Romanowski, M. J.; Wells, J. A. A common allosteric site and
mechanism in caspases. PNAS 2006, 103, 7595-7600.
86. Hoglen, N. C.; Chen, L. S.; Fisher, C. D.; Hirakawa, B. P.; Groessl, T.; Contreras,
P. C. Characterization of IDN-6556 (3-[2-(2-tert-butyl-phenylaminooxalyl)-
amino]-propionylamino]-4-oxo-5-(2,3,5,6-tetrafluoro-phenoxy)-pentanoic acid): a
liver-targeted caspase inhibitor. J. Pharmacol. Exp. Ther. 2004, 309, 634-640.
87. Leung-Toung, R.; Li, W.; Tam, T. F.; Karimian, K. Thiol-dependent enzymes and
their inhibitors: a review. Curr. Med. Chem. 2002, 9, 979-1002.
49

50

51


















