






Le lingue
Pagine
Legale


Insieme di ceppi isolati in habitat diversi ed in tempi diversi che posseggono:
•una elevata similarità fenotipica, con almeno una proprietà distintiva nei confronti delle altre specie
•una elevata omologia genetica (riassociazione molecolare DNA/DNA >70%)
•una elevata omologia •una elevata omologia filogenetica (omologia di sequenza del gene codificante per rRNA 16S >98%)
Per ogni specie batterica descritta esiste un ceppo di riferimento o ceppo
type, mantenuto in collezioni internazionali

Il processo di denaturazione e riassociazione del DNA, in condizioni controllate è REVERSIBILE
La denaturazione è favorita a T > Tm e a bassa conc. salina
La riassociazione è favorita a T < Tm e ad alta conc. salina
Allora possiamo calcolare indirettamente la % di omologia tra i DNA di due ceppi attraverso una DNA di due ceppi attraverso una riassociazione molecolare per via spettrofotometrica

Metodologia di ibridazione per via spettrofotometrica
Condizioni operative e strumentali:
Tampone di ibridazione a elevata forza ionica (es. 5x, cioè 5 volte concentrato)
T di riassociazione Tr=Tm – 25°C (alle condizioni di forza ionica del saggio)
Spettrofotometro con lettura simultanea di almeno 4 cuvette, dotato di riscaldamento programmato e software specifico
Calcolo del Tr in tampone 5x (media dei due Tm ottenuti nelle condizioni del saggio)
Estrazione, purificazione e dosaggio del DNA dai ceppi A e B
Rottura del DNA in frammenti
1
2
3
4 Allestimento del saggio
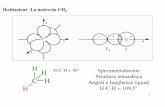
•Introdurre nella cuvetta 1 75 µg (corrispondenti a 1,5 OD260nm ) di DNA A
•Introdurre nella cuvetta 2 75 µg di DNA B
•Introdurre nella cuvetta 3 37,5 75 µg di DNA A e 37,5 75 µg di DNA B
•Step di denaturazione: impostare allo spettrofotometro una T di 98°C per 5-7 min
•Step di riassociazione: impostare Tr calcolato per 30-40 min
% riassociazioneper 30-40 min
OD260 nm
h
% riassociazione
OD iniz.
OD Tr=0.
50%

5 Calcolo e interpretazione dei risultati
Dal tabulato bisogna calcolare per ogni campione:
•Cot ½ = ½ OD (al 50% riassociazione) x t (h) impiegato a raggiungere tale valore
•Cot100 ½ = media Cot ½ dei DNA omologhi
•Cot0 ½ = somma Cot ½ dei DNA omologhi
Formula:
1-Cotmix ½ + Cot100 ½ - Cot0 ½
X 100
% riassociazione
1-Cotmix ½ + Cot100 ½ - Cot0 ½
Cot100 ½X 100
∆OD = ODTr (t=0) – ODiniziale
Al 50% riassociazione: OD50 = ODiniziale + 1/2 ∆∆∆∆OD
Cercare sul tabulato il tempo richiesto per raggiungere OD50 e trasformarlo in h
Calcolare il Cot ½ = t (h) x 1/2 OD50
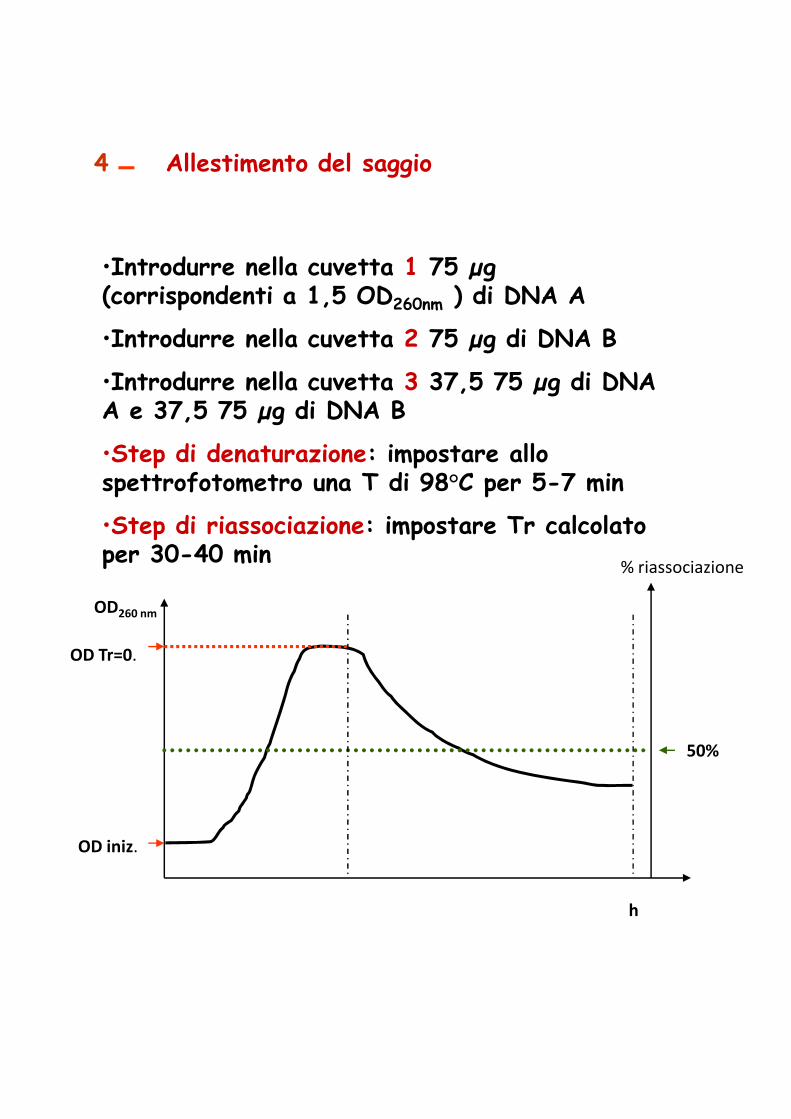
Il DNA plasmidico
StudioPresenza e numeroPeso molecolareMappa di restrizioneFunzioni codificate
Impiego come vettori di clonaggio
Per la loro estrazione si segue il protocollo di lisi del DNA a cui si deve aggiungere una fase di DENATURAZIONE del DNA cromosomale, prima della precipitazione (aggiunta di NaOH 3 N, e miscelazione per 10 min), e poi di neutralizzazione (aggiunta di Tampone 2 M pH 7)miscelazione per 10 min), e poi di neutralizzazione (aggiunta di Tampone 2 M pH 7)
Per la loro separazione dal DNA cromosomale si effettua una ULTRACENTRIFUGAZIONE in gradiente di cloruro di cesio (CsCl) ed in presenza di bromuro di etidio
DNA cromosomale
DNA plasmidico

Per valutare la presenza di DNA plasmidico in una preparazione di DNA totale, si esegue una gel elettroforesi: il DNA plasmidico più piccolo ed in forma superavvolta migra più velocemente del DNA cromosomale
Per valutare il peso molecolare delle bande elettroforetiche corrispondenti al DNA plasmidico bisogna utilizzare un marker di una miscela di DNA plasmidici in forma CCC di PM noto
Per valutare il numero di diverse molecole plasmidiche è necessario ricorrere ad una elettroforesi bidimensionale poiché durante elettroforesi bidimensionale poiché durante l’estrazione i plasmidi nativi in forma CCC, possono essersi trasformati nella corrispondente forma OC, che essendo in parte disavvolta migra più lentamente

Elettroforesi bidimensionale
Caricare in elettroforesi un campione di DNA plasmidico.
Al termine della corsa acquisire l’immagine delle bande di DNA plasmidico ottenute
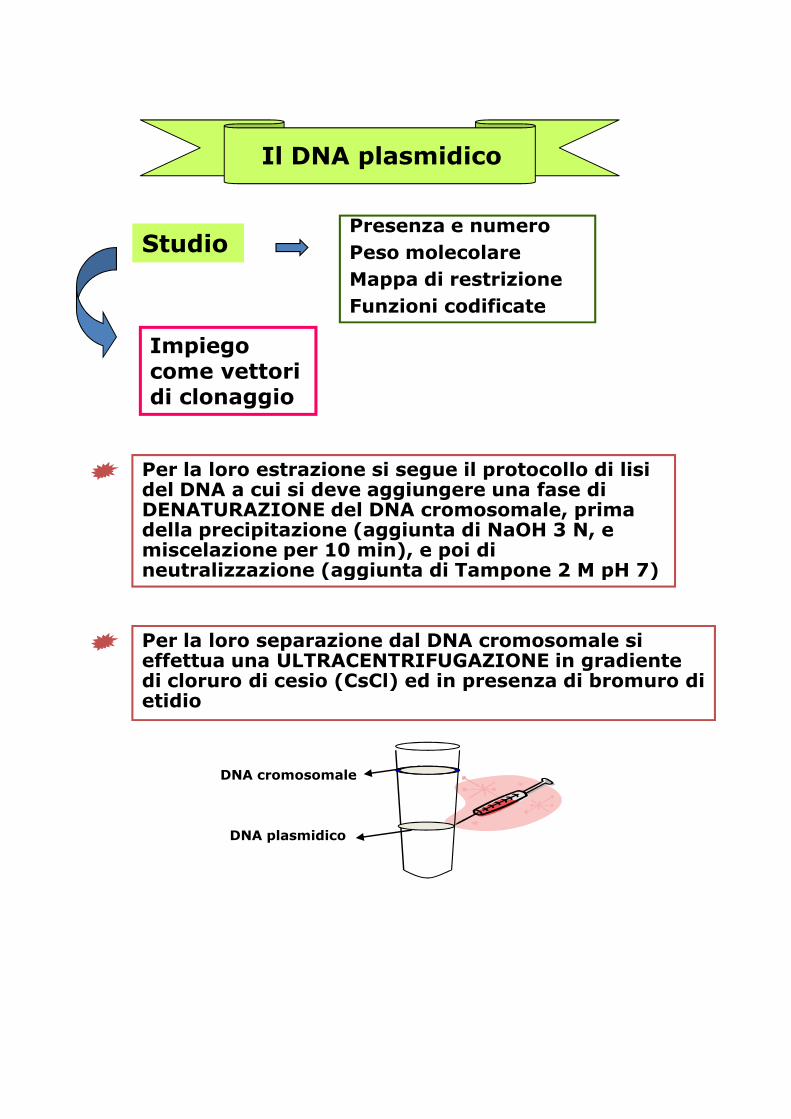
Quante, in realtà, delle bande ottenute rappresentano diversi plasmidi in forma CCC e quante possono essere le corrispondenti forme OC?
Trattare il gel con una soluzione concentrata di forme OC? soluzione concentrata di bromuro di etidio ed esporre agli UV per alcuni minuti.
Effettuare una seconda elettroforesi, ponendo il gel nella vaschetta ruotato di 90°
Direzione di migrazione Direzione di migrazione
Come interpreto i risultati?
Solo le bande che si sono sdoppiate nella seconda corsa elettroforetica sono da considerare plasmidi in forma CCC
inizialmente presentiinizialmente presenti
Perché? Il bromuro di etidio e i raggi UV hanno dato luogo alla rottura di un filamento della molecola con conseguente disavvolgimento dalla forma CCC alla OC. La seconda corsa elettroforetica permette di separare le due forme ottenute a partire dalla singola banda iniziale. Là dove la banda iniziale era già costituita da molecole plasmidiche nella forma OC, non si osserva sdoppiamento
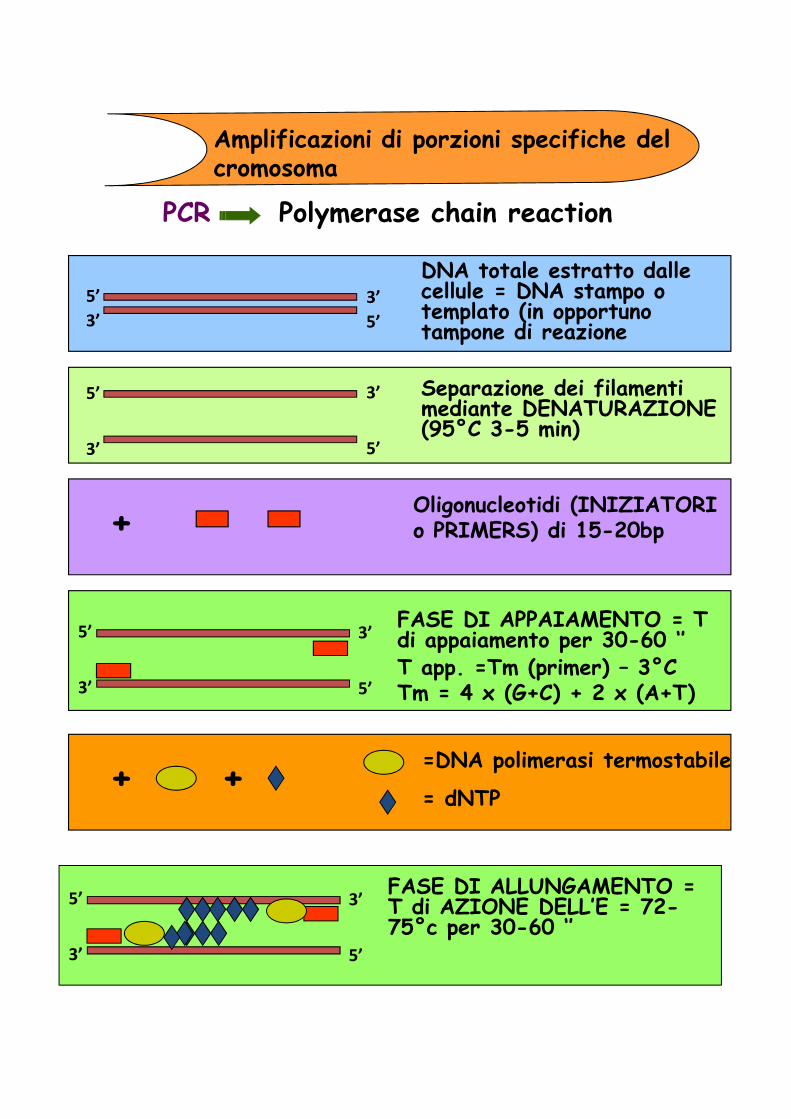
Amplificazioni di porzioni specifiche del cromosoma
PCR Polymerase chain reaction
DNA totale estratto dalle cellule = DNA stampo o templato (in opportuno tampone di reazione
5’
3’
3’
5’
Separazione dei filamenti mediante DENATURAZIONE (95°C 3-5 min)
5’
3’
3’
5’
Oligonucleotidi (INIZIATORI o PRIMERS) di 15-20bp+
3’
5’
5’
3’
FASE DI APPAIAMENTO = T di appaiamento per 30-60 ‘’ T app. =Tm (primer) – 3°CTm = 4 x (G+C) + 2 x (A+T)
=DNA polimerasi termostabile
= dNTP + +
3’
5’
5’
3’
FASE DI ALLUNGAMENTO = T di AZIONE DELL’E = 72-75°c per 30-60 ‘’
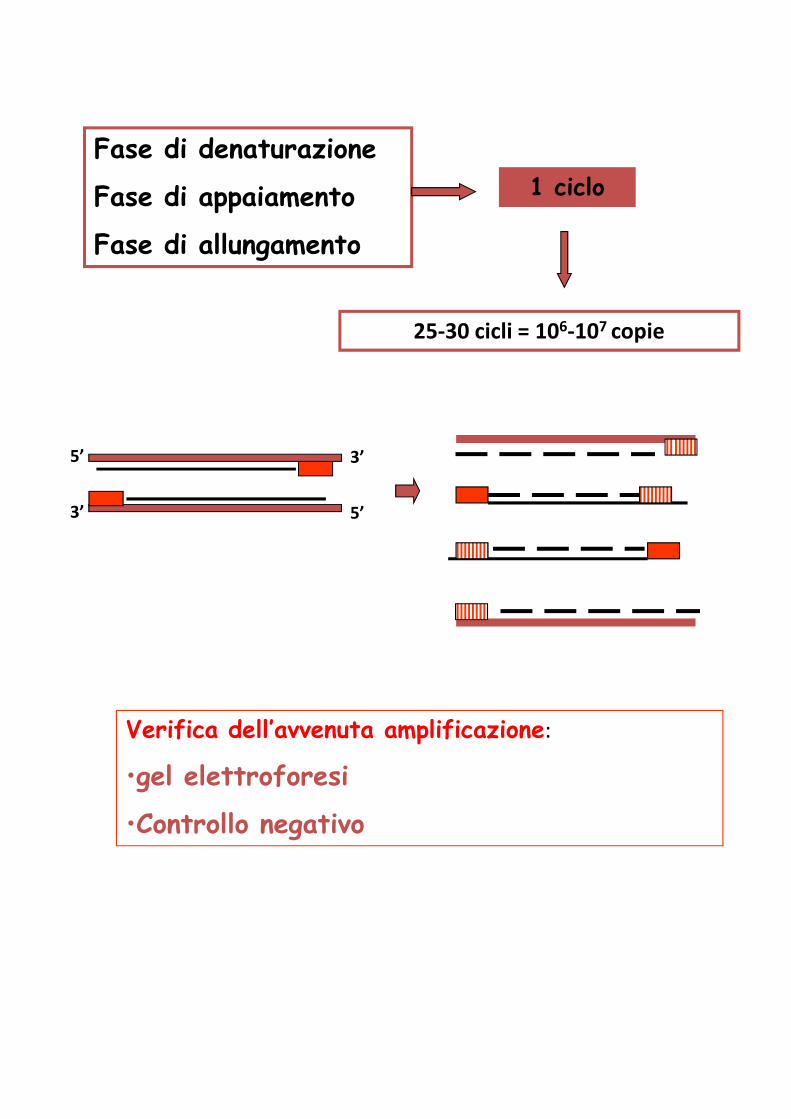
Fase di denaturazione
Fase di appaiamento
Fase di allungamento
1 ciclo
25-30 cicli = 106-107 copie
3’
5’
5’
3’
Verifica dell’avvenuta amplificazione:
•gel elettroforesi
•Controllo negativo
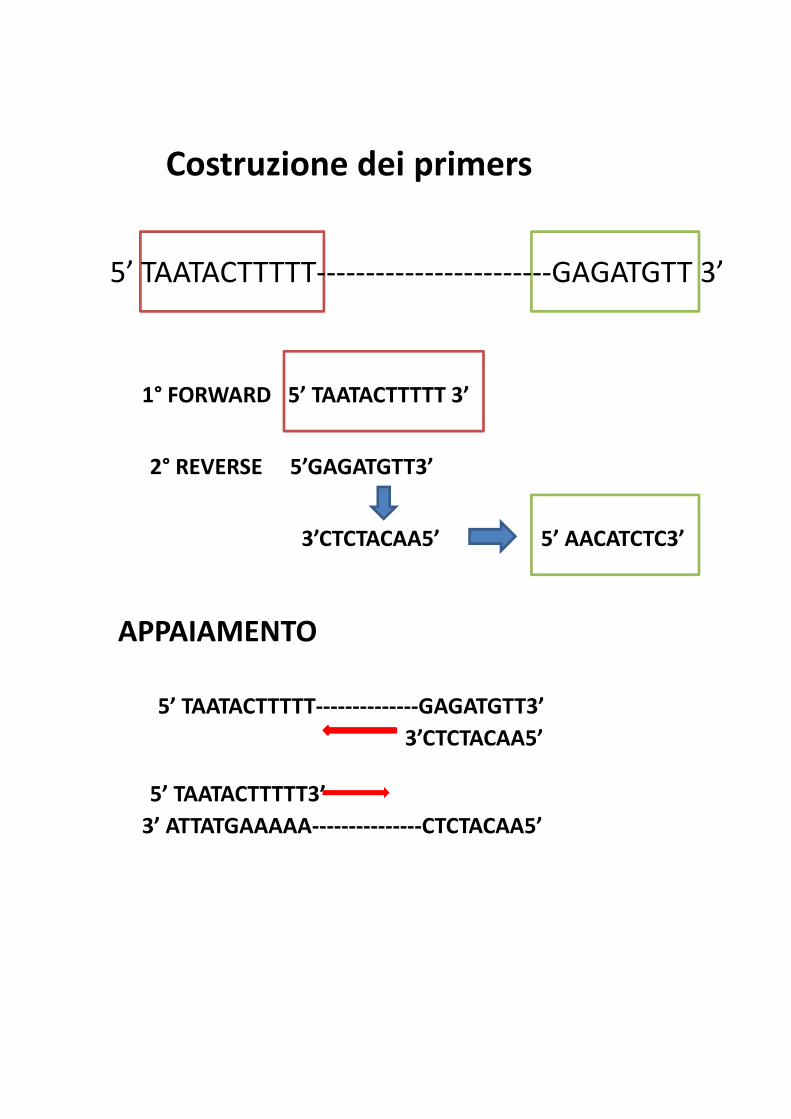
Costruzione dei primers
5’ TAATACTTTTT------------------------GAGATGTT 3’
1° FORWARD 5’ TAATACTTTTT 3’
2° REVERSE 5’GAGATGTT3’
3’CTCTACAA5’ 5’ AACATCTC3’
APPAIAMENTO
5’ TAATACTTTTT--------------GAGATGTT3’
3’CTCTACAA5’
5’ TAATACTTTTT3’
3’ ATTATGAAAAA---------------CTCTACAA5’

Riconoscimento a livello di specie in funzione dello studio molecolare delle porzioni amplificate
Se una specie batterica possiede nel suo cromosoma un gene o una regione specifici, allora posso costruire una
•Devo conoscere la sequenza nucleotidica agli estremi del gene o regione specifica
•Devo costruire i primers idonei
•Devo estrarre il DNA batterico totale dal campione in cui devo ricercare la presenza di quella determinata specie
•Devo condurre una amplificazione specie-specifica
•Devo conoscere il PM dell’amplificato che mi aspetto e l’eventuale numero di copie presenti lungo il cromosoma
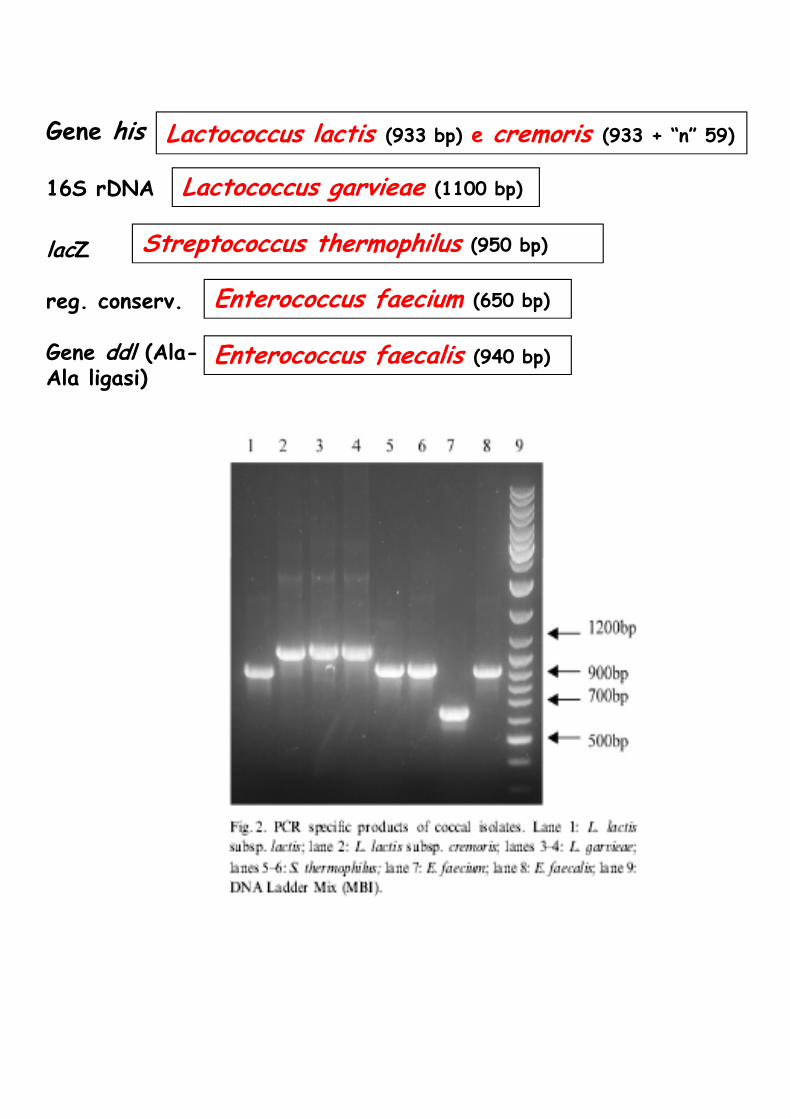
Lactococcus lactis (933 bp) e cremoris (933 + “n” 59)
lactis
Lactococcus garvieae (1100 bp)
Gene his
16S rDNA
lacZ
reg. conserv.
Gene ddl (Ala-Ala ligasi)
Streptococcus thermophilus (950 bp)
Enterococcus faecium (650 bp)
Enterococcus faecalis (940 bp)
Se devo identificare un numero elevato di isolati, le tecniche di indagine genetica mi possono aiutare?
studiando ad esempio il polimorfismo presente in operoni conservati e/o in geni conservati (housekeeping)
•Permettono di ottenere dei cluster omogenei di isolati
•In molti casi si hanno indicazioni già a livello di genere e specie
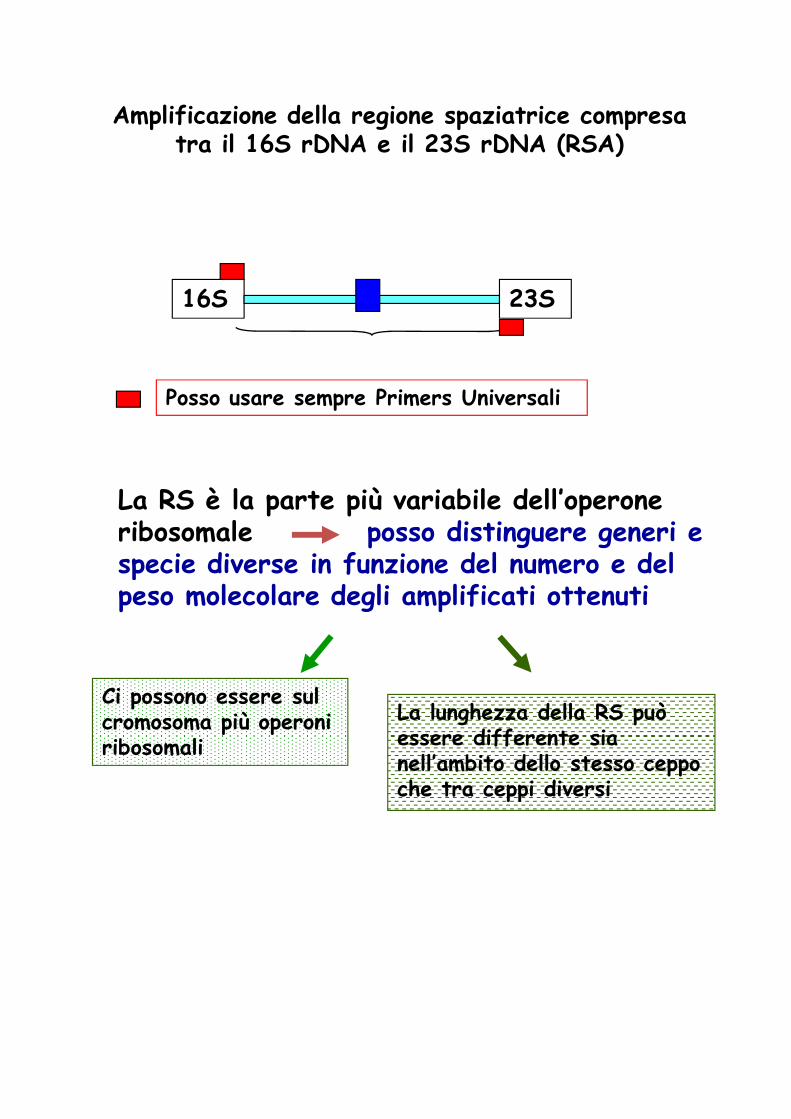
Amplificazione della regione spaziatrice compresa tra il 16S rDNA e il 23S rDNA (RSA)
16S 23S
Posso usare sempre Primers Universali
La RS è la parte più variabile dell’operone ribosomale posso distinguere generi e specie diverse in funzione del numero e del specie diverse in funzione del numero e del peso molecolare degli amplificati ottenuti
Ci possono essere sul cromosoma più operoni ribosomali
La lunghezza della RS può essere differente sia nell’ambito dello stesso ceppo che tra ceppi diversi

E. sulfureus
E. avium
E. gallinarum
nell’ambito lattiero-caseario:
1 380 bp L. lactis
2 410 bp L. garvieae
3 350 bp S. thermophilus
4 500 bp ??
5 a) b) c) d) 2 amplificati 300-500 bp Enterococcus spp.
1 2 3 4 5a 5b 5c 5d MRSA
Enterococcus sp.E. faecalis
E. durans
E. faecium
I risultati vanno verificati con sonde specie-specifiche o con riassociazione molecolare
DNA/DNA o con……..
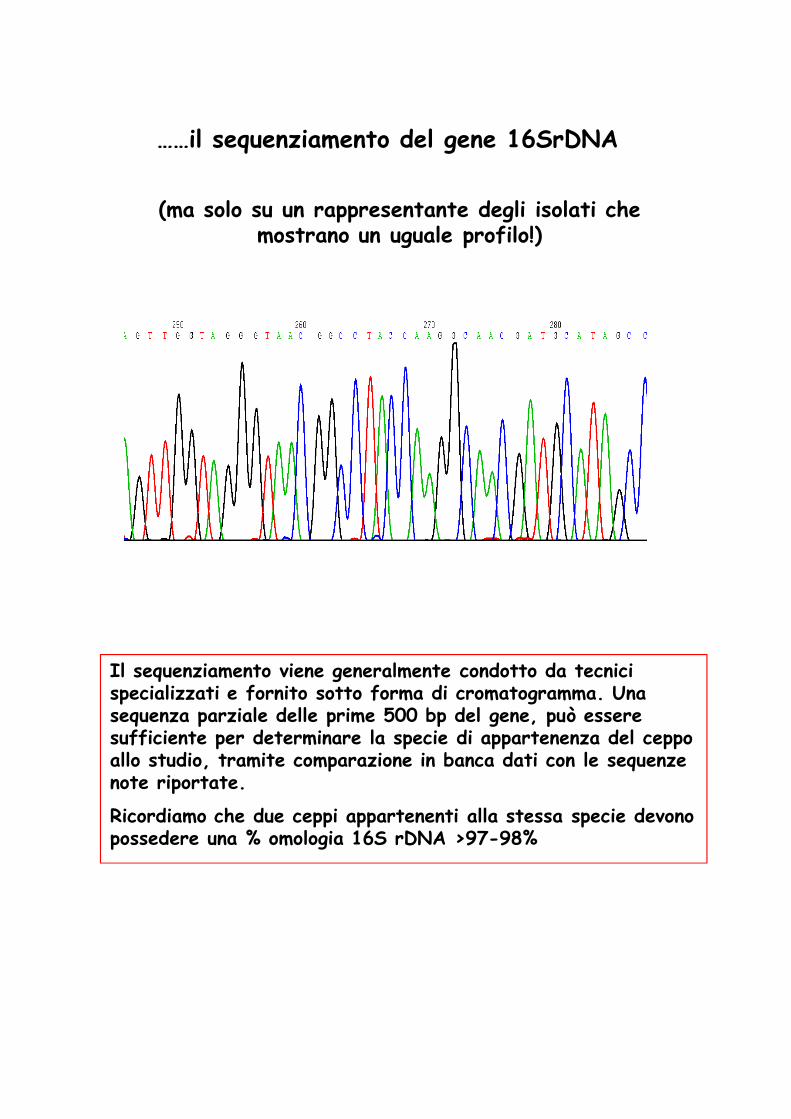
……il sequenziamento del gene 16SrDNA
(ma solo su un rappresentante degli isolati che mostrano un uguale profilo!)
Il sequenziamento viene generalmente condotto da tecnici specializzati e fornito sotto forma di cromatogramma. Una sequenza parziale delle prime 500 bp del gene, può essere sufficiente per determinare la specie di appartenenza del ceppo allo studio, tramite comparazione in banca dati con le sequenze note riportate.
Ricordiamo che due ceppi appartenenti alla stessa specie devono possedere una % omologia 16S rDNA >97-98%

Per valutare i rapporti filogenetici tra ceppi è necessario disporre dell’intera sequenza del gene 16S rDNA. La comparazione con altre specie note, consente di valutare la distanza evolutiva tra le specie, e viene solitamente riportata in un dendrogramma
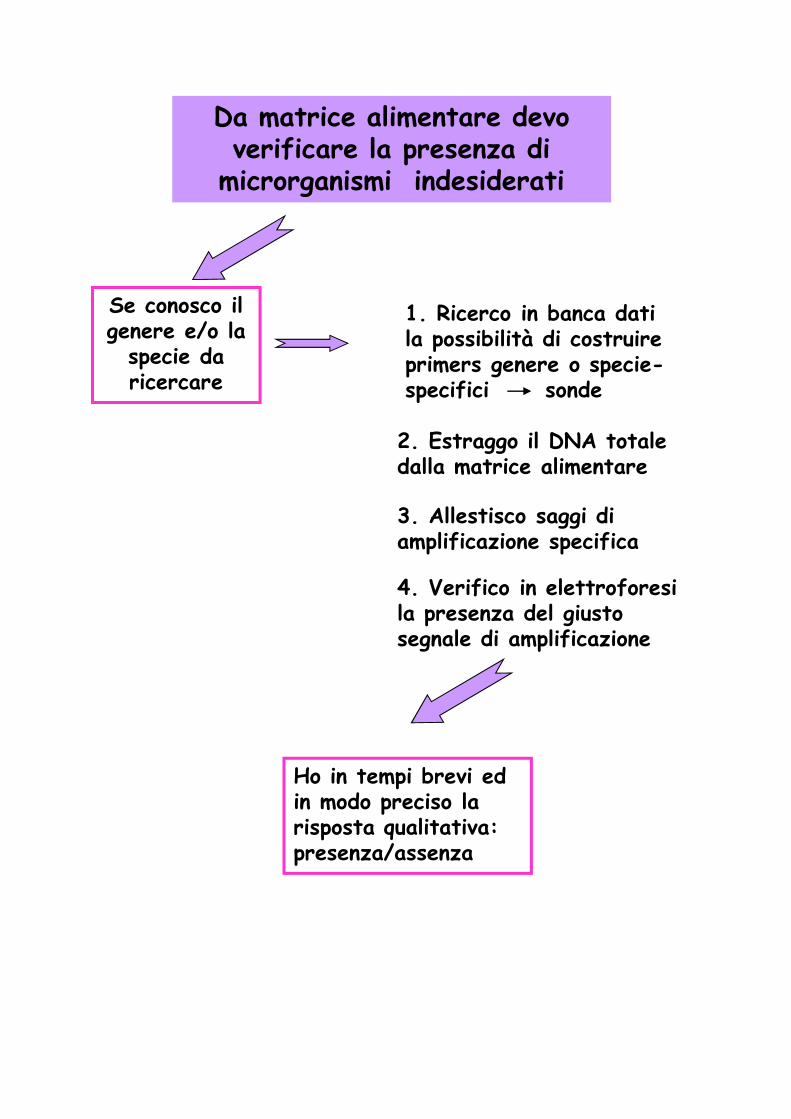
Da matrice alimentare devo verificare la presenza di
microrganismi indesiderati
Se conosco il genere e/o la
specie da ricercare
1. Ricerco in banca dati la possibilità di costruire primers genere o specie-specifici sonde
2. Estraggo il DNA totale dalla matrice alimentare
3. Allestisco saggi di amplificazione specifica
4. Verifico in elettroforesi la presenza del giusto segnale di amplificazione
Ho in tempi brevi ed in modo preciso la risposta qualitativa: presenza/assenza
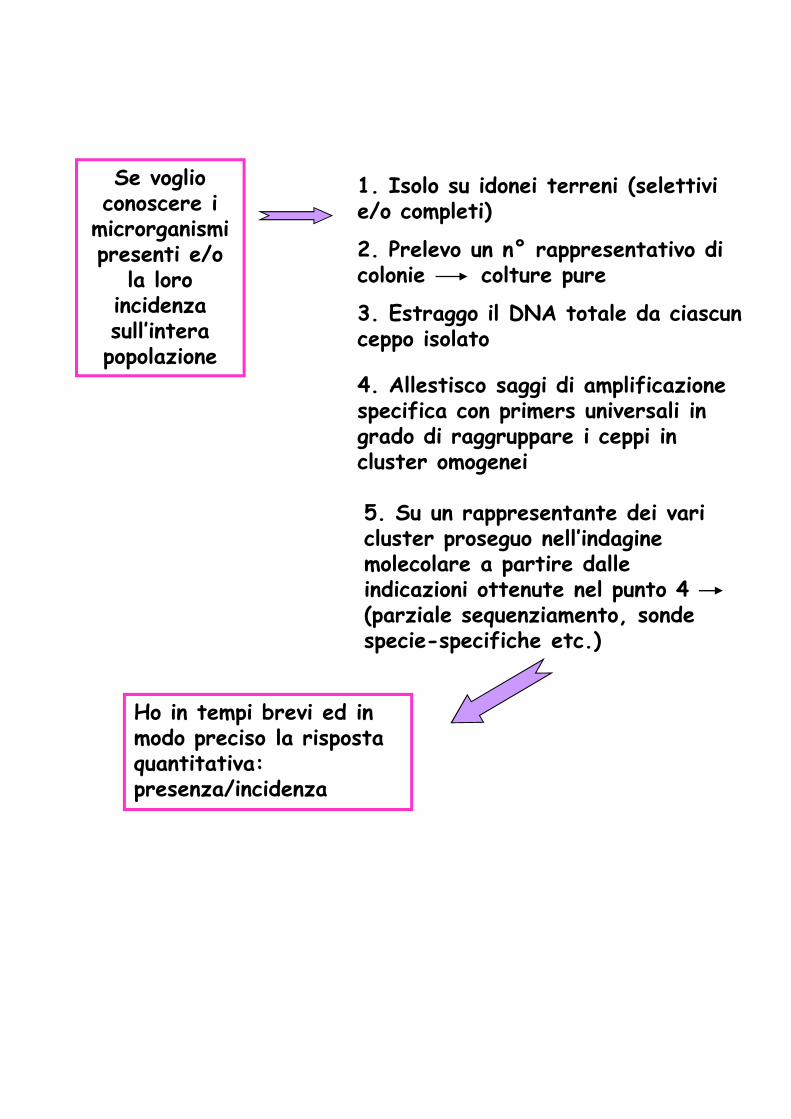
Se voglio conoscere i
microrganismi presenti e/o
la loro incidenza sull’intera popolazione
1. Isolo su idonei terreni (selettivi e/o completi)
3. Estraggo il DNA totale da ciascun ceppo isolato
4. Allestisco saggi di amplificazione specifica con primers universali in grado di raggruppare i ceppi in cluster omogenei
5. Su un rappresentante dei vari cluster proseguo nell’indagine molecolare a partire dalle
2. Prelevo un n° rappresentativo di colonie colture pure
molecolare a partire dalle indicazioni ottenute nel punto 4 (parziale sequenziamento, sonde specie-specifiche etc.)
Ho in tempi brevi ed in modo preciso la risposta quantitativa: presenza/incidenza
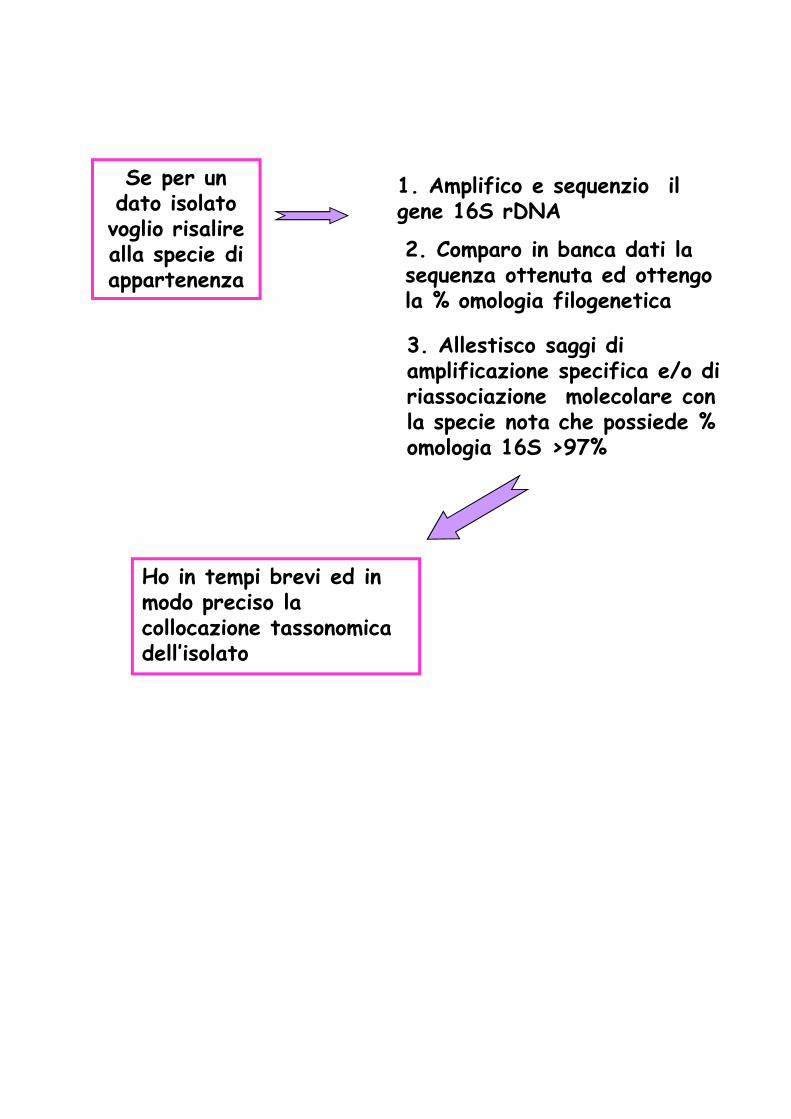
Se per un dato isolato voglio risalire alla specie di appartenenza
2. Comparo in banca dati la sequenza ottenuta ed ottengo la % omologia filogenetica
3. Allestisco saggi di amplificazione specifica e/o di riassociazione molecolare con la specie nota che possiede % omologia 16S >97%
1. Amplifico e sequenzio il gene 16S rDNA
Ho in tempi brevi ed in modo preciso la collocazione tassonomica dell’isolato
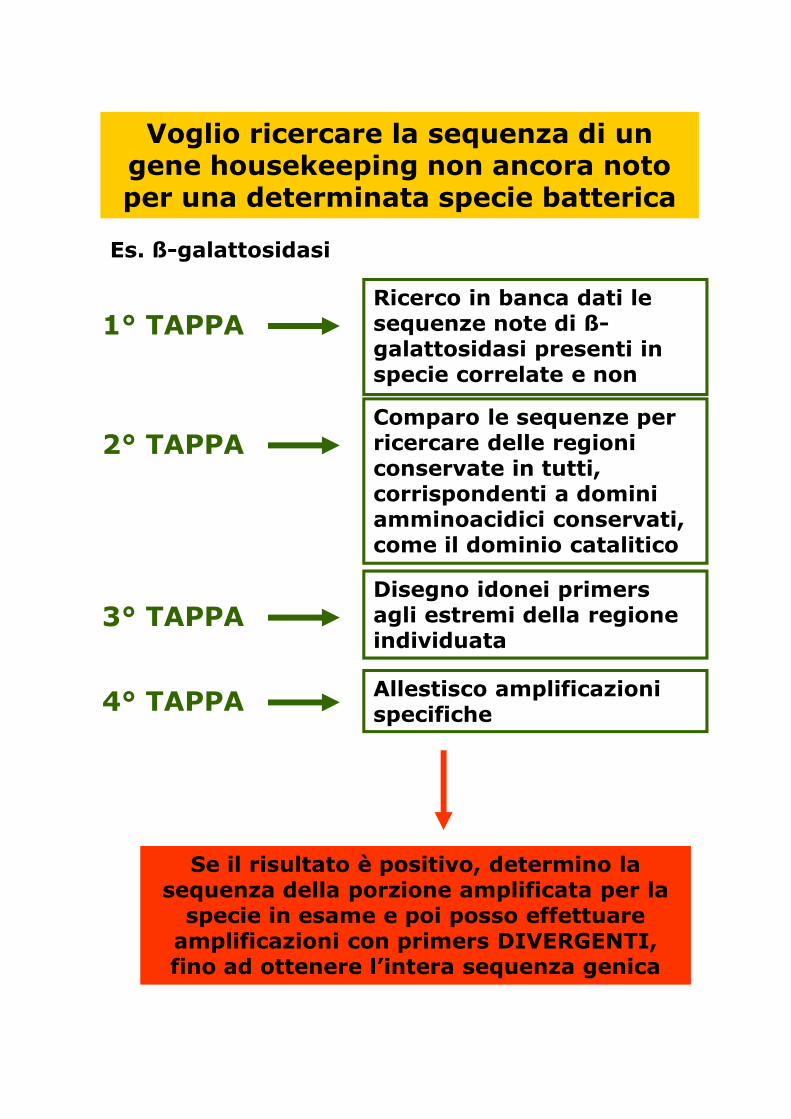
Voglio ricercare la sequenza di un gene housekeeping non ancora noto per una determinata specie batterica
Es. ß-galattosidasi
1° TAPPARicerco in banca dati le sequenze note di ß-galattosidasi presenti in specie correlate e non
2° TAPPAComparo le sequenze per ricercare delle regioni conservate in tutti, corrispondenti a domini amminoacidici conservati, come il dominio catalitico
3° TAPPADisegno idonei primers agli estremi della regione individuata
4° TAPPA Allestisco amplificazioni specifiche
Se il risultato è positivo, determino la sequenza della porzione amplificata per la specie in esame e poi posso effettuare amplificazioni con primers DIVERGENTI, fino ad ottenere l’intera sequenza genica

Se il risultato è negativo
Posso allestire esperimenti di IBRIDAZIONE SU MEMBRANA o IBRIDAZIONE SOUTHERN
1: amplifico il gene di interesse da una specie nota e correlata SONDA
2:effettuo una marcatura della sonda
3: utilizzo una membrana di nitrocellulosa su cui deposito per capillarità pochi su cui deposito per capillarità pochi microgrammi del DNA totale estratto dai ceppi in esame, previamente denaturato termicamente
4: fisso il DNA sulla membrana, mediante esposizione agli UV per qualche minuto
5:denaturo termicamente la sonda
6: preparo un tampone idoneo ad alta forza ionica a cui aggiungo la giusta concentrazione di sonda

7: metto a contatto la membrana con la sonda marcata ad una idonea temperatura di ibridazione per un minimo di 3-4 h
8: elimino la soluzione contenente la sonda ed effettuo dei lavaggi della membrana con tamponi a diversa forza ionica e a diverse temperature
Stringenza della reazione
Alta specificità Bassa specificità
* Forza ionica decrescente
* Temperatura crescente
* Forza ionica media
* Temperatura media
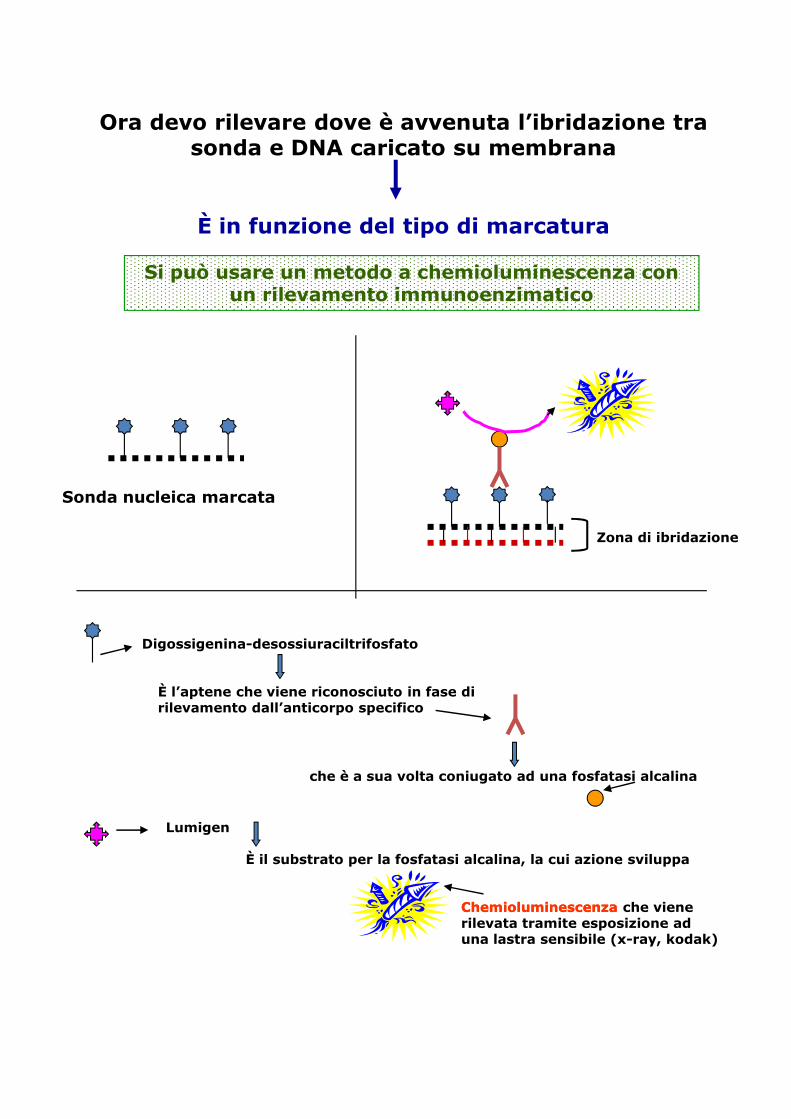
Ora devo rilevare dove è avvenuta l’ibridazione tra sonda e DNA caricato su membrana
È in funzione del tipo di marcatura
Si può usare un metodo a chemioluminescenza con un rilevamento immunoenzimatico
Sonda nucleica marcata
Zona di ibridazione
Digossigenina-desossiuraciltrifosfato
È l’aptene che viene riconosciuto in fase di rilevamento dall’anticorpo specifico
che è a sua volta coniugato ad una fosfatasi alcalina
È il substrato per la fosfatasi alcalina, la cui azione sviluppa
Lumigen
ChemioluminescenzaChemioluminescenza che viene rilevata tramite esposizione ad una lastra sensibile (x-ray, kodak)

In base al tipo di marcatura scelta:
•la sonda viene marcata prima dell’inizio dell’ibridazione
•al termine dell’ibridazione la membrana viene posta in contatto con idonei tamponi in cui sono sciolti l’anticorpo e poi il lumigen
•al termine dell’esposizione la lastra viene sviluppata
L’esperimento citato dove il DNA totale viene depositato come spot sulla membrana si
chiama:chiama:
Ibridazione DOT BLOT
Mi permette di rilevare la presenza della sonda sul DNA totale
Presenza/assenza

Se voglio sapere la localizzazione della regione nucleica allo studio (sonda)
Se voglio conoscere il numero di copie della regione nucleica lungo il DNA
Cromosoma o plasmide?
Devo separare per gel elettroforesi i due tipi di DNA
Devo procedere ad una restrizione del DNA con idonei ER e poi alla separazione elettroforetica
A questo punto è necessario seguire la metodica di SouthernA questo punto è necessario seguire la metodica di Southern
Il gel viene trattato con una soluzione di:
HCl
NaOH
Tampone pH 7
depurinazione
denaturazione
neutralizzazione
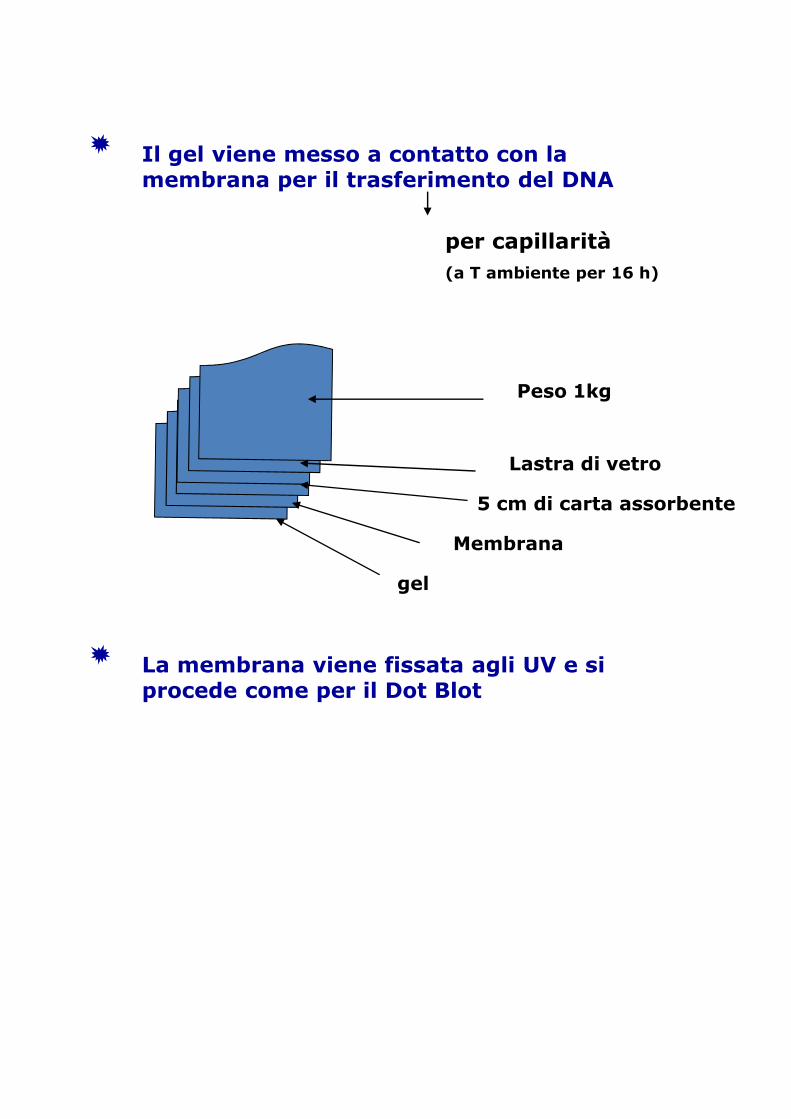
Il gel viene messo a contatto con la membrana per il trasferimento del DNA
per capillarità(a T ambiente per 16 h)
Peso 1kg
Lastra di vetro
5 cm di carta assorbente
Membrana
gel
La membrana viene fissata agli UV e si procede come per il Dot Blot
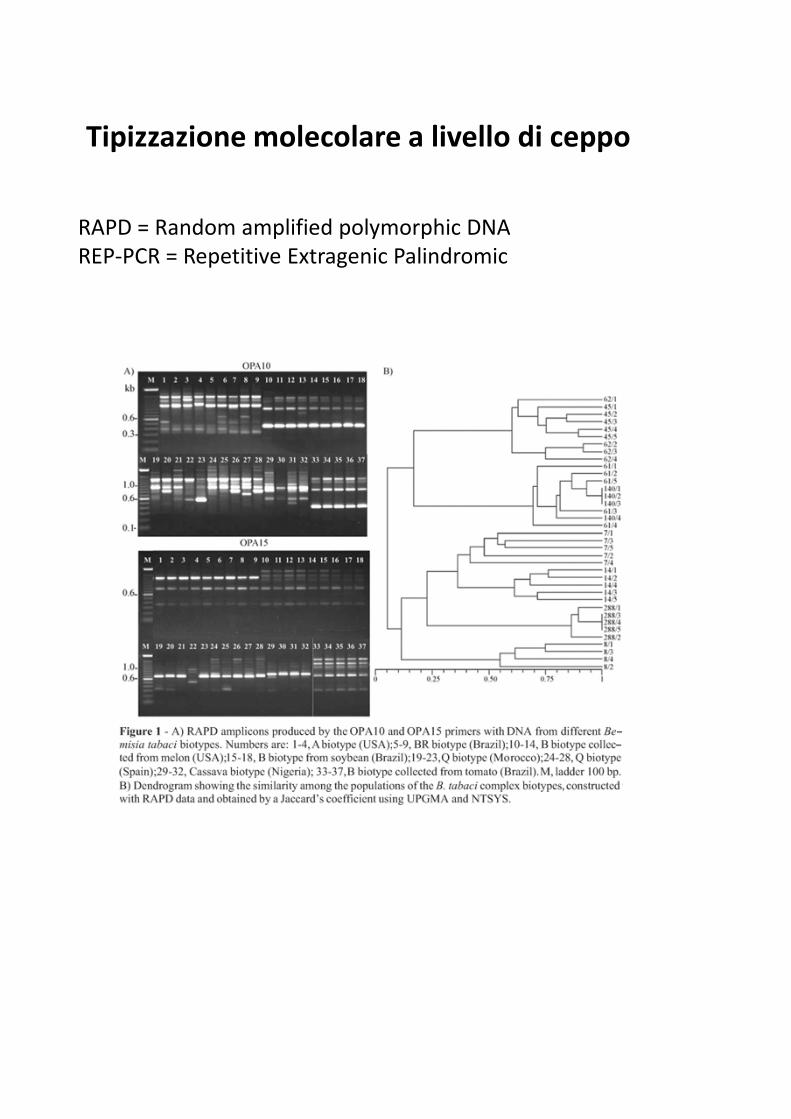
Tipizzazione molecolare a livello di ceppo
RAPD = Random amplified polymorphic DNA
REP-PCR = Repetitive Extragenic Palindromic
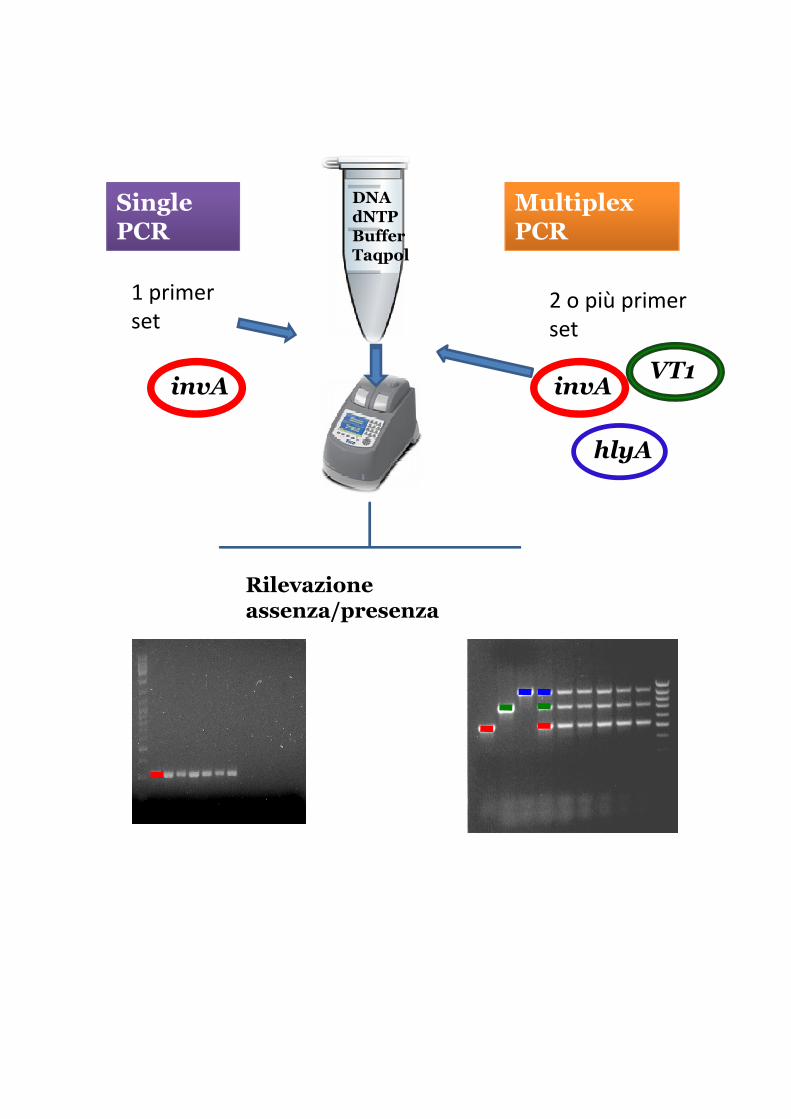
Single PCR
Multiplex PCR
DNAdNTPBufferTaqpol
1 primer
set2 o più primer
set
invA
hlyA
VT1invA
Rilevazione assenza/presenza

impiego di marcatori fluorescenti il cui accumulo segue la
stessa cinetica della reazione PCR. La misurazione della
fluorescenza dà in tempo reale la quantità dello specifico prodotto
di PCRTermociclatorecon detector per fluorocromi e software per analisi dati
Analisi PCR quantitativa
RealtimePCR
Real timemultiplex
Analisi PCR quantitativa
Linea di base (baseline):valore al di sopra del quale inizial’accumulo di un amplificato
Linea soglia (Threshold): scelta dall’operatore in modo da intersecare le curve di tutti i campioni nella fase esponenziale
Ciclo soglia: E’ il ciclo della reazione di amplificazione in cui il segnale di fluorescenza del campione è maggiore rispetto a quello della Threshold
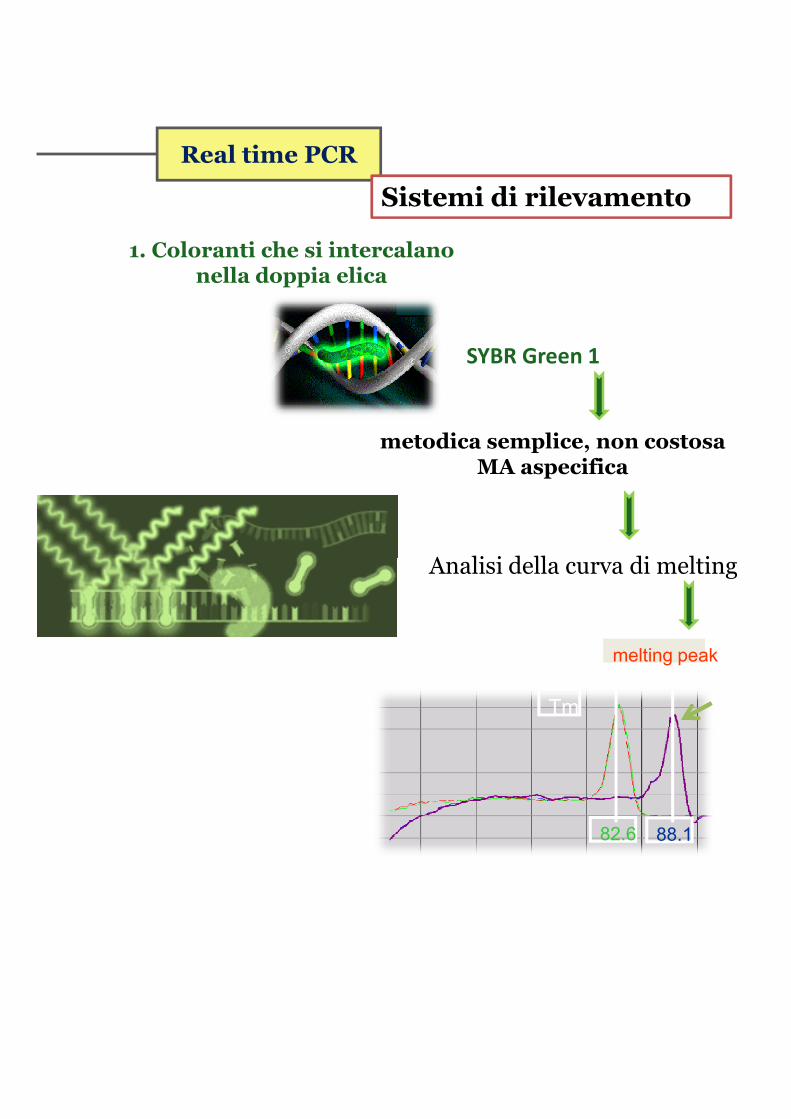
Real time PCR
Sistemi di rilevamento
1. Coloranti che si intercalano nella doppia elica
SYBR Green 1
metodica semplice, non costosa MA aspecifica
Analisi della curva di meltingAnalisi della curva di melting
Tm
82.6 88.1
melting peak
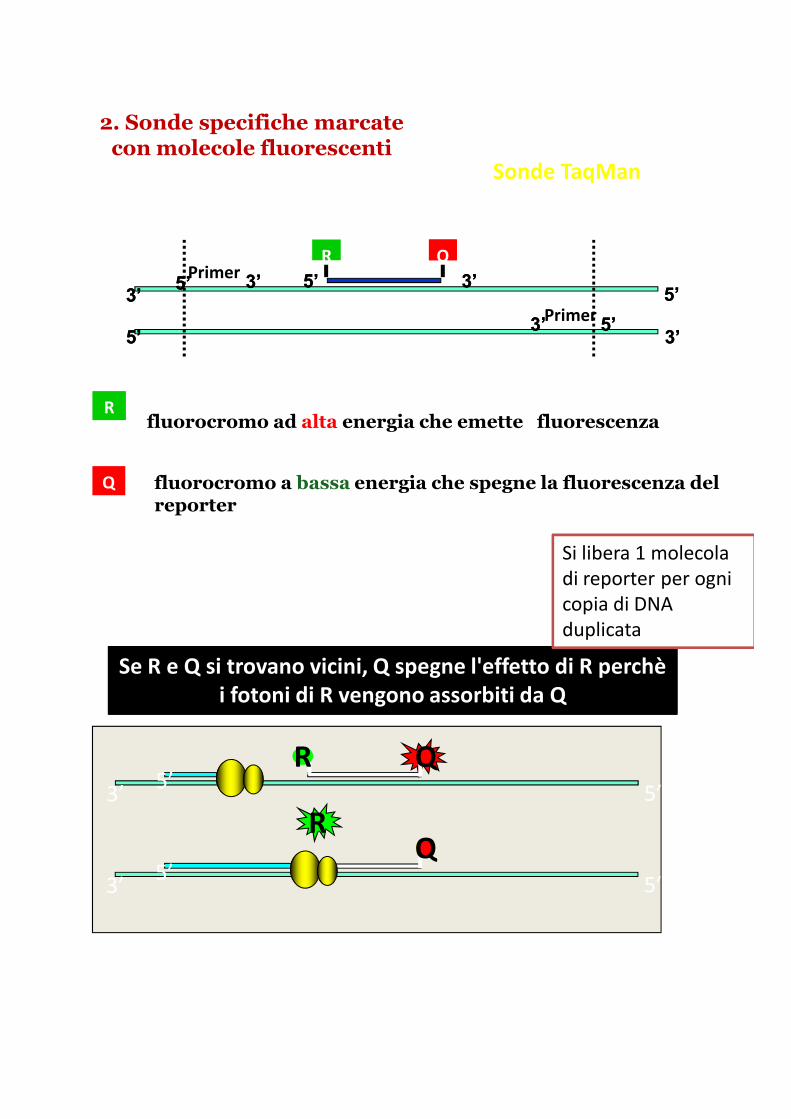
2. Sonde specifiche marcate con molecole fluorescenti
Primer
Primer3’3’
3’3’
3’3’3’3’
5’5’
5’5’5’5’
5’5’
R QQ
5’5’ 3’3’
Sonde TaqMan
Rfluorocromo ad alta energia che emette fluorescenza
fluorocromo a bassa energia che spegne la fluorescenza del reporter
Q
Si libera 1 molecola
Se R e Q si trovano vicini, Q spegne l'effetto di R perchèi fotoni di R vengono assorbiti da Q
R Q3’
3’5’
5’
RQ
3’ 5’5’
Si libera 1 molecola
di reporter per ogni
copia di DNA
duplicata

Estrarre l’RNA totale dal campione biologico che si vuoleesaminare
Trasformare il pool di RNA (che conterrà anche l’mRNAtrascritto dal gene di interesse) in cDNAtramite la reazione ditrascrizione inversa
Evidenziare il cDNA di interesse tramite la
=REVERSE TRANSCRIPTASE PCR=REVERSE TRANSCRIPTASE PCR=REVERSE TRANSCRIPTASE PCR=REVERSE TRANSCRIPTASE PCR
La RT-PCR amplifica un RNA convertendolo prima in DNA (oDNA omplementare all’RNA) tramite l’enzima trascrittasi inversa, e poi amplificandolo tramite PCR con inneschi specifici per la sequenza di interesse.
Evidenziare il cDNA di interesse tramite la reazione di PCR

Ingegneria genetica o tecnologia del Ingegneria genetica o tecnologia del
DNA ricombinanteDNA ricombinantepermette di isolare geni, clonarli, introdurli e
esprimerli in un ospite eterologo (differente
dall'ospite originale). Queste tecniche permettono
di conferire caratteristiche nuove alle cellule
riceventi. Le cellule così prodotte sono chiamate
ricombinanti.
Il fine è quello di esprimere una proteina all'interno
di un organismo diverso da quello di origine (la
proteina è detta per questo eterologa), sfruttando i
ribosomi e la sintesi proteica dell'organismo ospite.
Tale processo è noto come espressione eterologa.Tale processo è noto come espressione eterologa.•produzione di linee di cereali resistenti agli erbicidi •produzione di insulina attraverso batteri
l'insulina si ottiene con la tecnologia del DNA ricombinante dal 1982, quando negli Stati Uniti fu messo a punto un sistema batterico in E. coli. L'insulina è collegata al primo brevetto e al primo farmaco biotecnologico, messo in commercio.
.

Vettori di clonaggio
un'origine di replicazione compatibile con l'organismo ospite,
in modo da permettere al vettore di replicarsi attraverso il
replisoma dell'organismo
origine di replicazione indipendente, in modo tale che la
replicazione del plasmide nella cellula proceda
indipendentemente dal controllo diretto del cromosoma
uno o più marker di selezione, che permette di selezionare
I plasmidi hanno proprietà utili per il clonaggio e possono fungere da veicolo
molecolare per geni che occorre
esprimere in una cellula ospite:
uno o più marker di selezione, che permette di selezionare
dall'esterno le cellule contenenti il vettore (ad esempio
regioni che conferiscono resistenza ad antibiotici e
permettono di selezionare le cellule)
una regione detta multi-cloning site (MCS), al’interno della
quale è possibile inserire gli specifici geni di interesse
attraverso l'utilizzo di enzimi di restrizione.
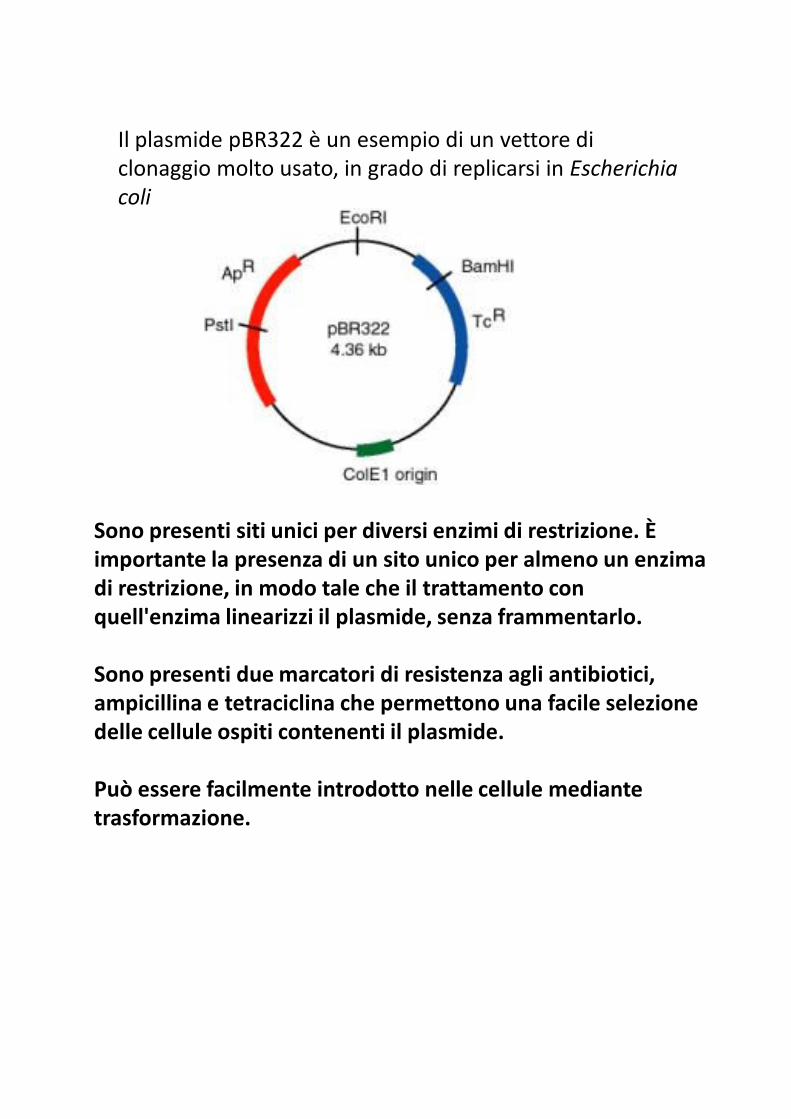
Il plasmide pBR322 è un esempio di un vettore di
clonaggio molto usato, in grado di replicarsi in Escherichia
coli
Sono presenti siti unici per diversi enzimi di restrizione. È importante la presenza di un sito unico per almeno un enzima importante la presenza di un sito unico per almeno un enzima di restrizione, in modo tale che il trattamento con quell'enzima linearizzi il plasmide, senza frammentarlo.
Sono presenti due marcatori di resistenza agli antibiotici, ampicillina e tetraciclina che permettono una facile selezione delle cellule ospiti contenenti il plasmide.
Può essere facilmente introdotto nelle cellule mediante trasformazione.

il sito BamHI è all'interno del gene per la resistenza alla
tetraciclina e il sito PstI in quello per la resistenza all'ampicillina.
Se un frammento di DNA esogeno viene inserito in uno di questi
siti, la resistenza all'antibiotico conferita dal gene localizzato in
quel punto viene persa con un fenomeno detto inattivazione
inserzionale. Questa inattivazione è utilizzata per identificare la
presenza di DNA esogeno nel plasmide.
Quindi, se pBR322 viene digerito con BamHI, legato al DNA da
inserire e successivamente vengono isolati i cloni batterici
trasformati, i cloni selezionati che risultano resistenti sia
all'ampicillina che alla tetraciclina sono quelli che non hanno
inserito il DNA esogeno (il plasmide incorporato nella cellula è il
vettore che si è richiuso senza introduzione di DNA esogeno). Al
contrario, quelle cellule che sono ancora resistenti all'ampicillina,
ma sensibili alla tetraciclina, contengono il plasmide nel quale si
è inserito DNA esogeno. Poiché la resistenza a questi due
antibiotici può essere valutata su piastre di terreno agarizzato, è
possibile individuare facilmente i batteri contenenti il clone
desiderato ed eliminare le cellule che non contengono il
plasmide.

Miscela di sequenziamento
• Primer
– Due amplificazioni parallele usando in
ciascuna un solo primer (per i filamento
forward o reverse)
• Tampone • Tampone
• Polimerasi
• Nucleotidi
• Nucleotidi terminator
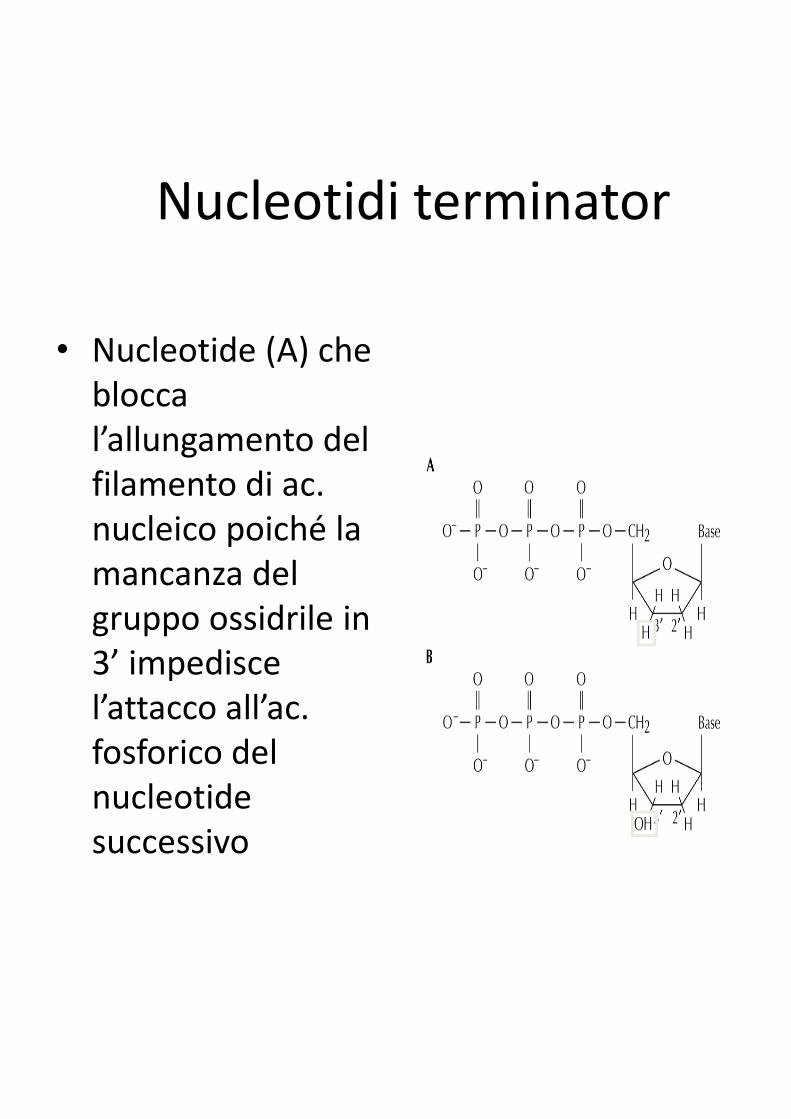
Nucleotidi terminator
• Nucleotide (A) che
blocca
l’allungamento del
filamento di ac.
nucleico poiché la
mancanza del mancanza del
gruppo ossidrile in
3’ impedisce
l’attacco all’ac.
fosforico del
nucleotide
successivo

PCR di sequenziamento (metodo manuale)
• Si esegue in quattro provette diverse
– Tutte e quattro le provette contengono: polimerasi, nucleotidi normali e primer
– Ciascuna delle quattro provette contiene inoltre un diverso tipo di nucleotide terminator (adenina-terminator, guanina-terminator, . . .)
• Annealing del primer
• Allungamento del primer:• Allungamento del primer:
– Legame di un nucleotide normale: l’allungamento prosegue
– Legame di un nucleotide terminator: l’allungamento si blocca
• In ciascuna delle 4 provette contenente nucleotidi terminator con una diversa base azotata si formano soltanto filamenti che terminano con tale base. Tali filamenti avranno tutte le lunghezze possibili a seconda che il nucleotide terminator si sia legato alla 1°, 2°, . . , ennesima, posizione possibile

Il principio del
sequenziamento (metodo manuale)
• Si esegue una elettroforesi disponendo i prodotti di amplificazione delle 4 provette in 4 corsie parallele così che in ognuna di esse siano presenti filamenti che terminano con una diversa base
• Si usa un gel ad altissima risoluzione, capace di separare frammenti di DNA che differiscono fra loro per un solo nucleotide. Il gel (0,3-0,4 mm di spessore) è interposto fra due lastre di vetro di notevole lunghezza (40 cm circa ).

Il principio del
sequenziamento (metodo manuale)
• La migrazione è
inversamente
proporzionale alla
lunghezza del filamento
• La posizione della • La posizione della
banda indica la
posizione occupata
all’interno della
sequenza, mentre il tipo
di base è indicato dalla
corsia in cui tale banda
si trova
A TA C A G C T G T T . . . . . .
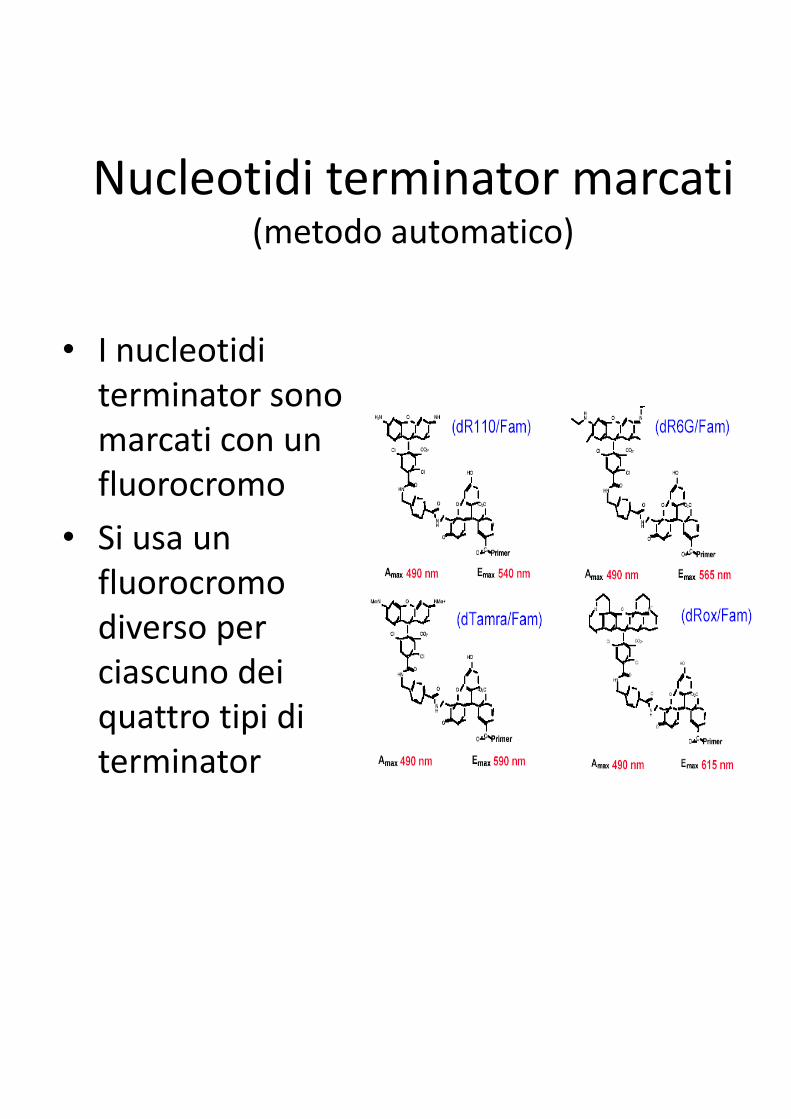
Nucleotidi terminator marcati(metodo automatico)
• I nucleotidi
terminator sono
marcati con un
fluorocromo
• Si usa un
fluorocromo fluorocromo
diverso per
ciascuno dei
quattro tipi di
terminator

PCR di sequenziamento (metodo automatico)
• Si esegue in una singola provetta
• Tutte le provette contengono: polimerasi, nucleotidi normali, nucleotidi terminator e primer
• I nucleotidi terminator sono marcati con fluorocromi
• Annealing del primer
• Allungamento del primer:• Allungamento del primer:– Legame di un nucleotide normale: l’allungamento
prosegue
– Legame di un nucleotide terminator: l’allungamento si blocca
• Nella provetta si formano contemporaneamente filamenti che avranno tutte le lunghezze possibili a seconda che il nucleotide terminator si sia legato alla 1°, 2°, . . , ennesima, posizione della sequenza. Si avranno quindi contemporaneamente filamenti terminanti con ciascuna delle quattro basi

Il prodotto della PCR (sequenziamento automatico)
• Se il bersaglio è composto da nnucleotidi si avranno filamenti di tutte le lunghezze possibili, da 1 ad n
• Indipendentemente dalla lunghezza, tutti i filamenti terminanti con adenina emetteranno fluorescenza di adenina emetteranno fluorescenza di tipo A ; quelli terminanti con citosina, di tipo B; quelli terminanti con guanina, di tipo C, e quelli terminanti con timina, di tipo D

Il sequenziatore automatico
• E’ un apparecchio che, dopo aver prelevato il prodotto della PCR di sequenziamento, lo sottopone ad elettroforesi all’interno di un capillare
• Un raggio laser colpisce il capillare • Un raggio laser colpisce il capillare eccitando la fluorescenza dei fluorocromi che lo attraversano
• Un cellula fotoelettrica rileva i segnali fluorescenti che vengono memorizzati
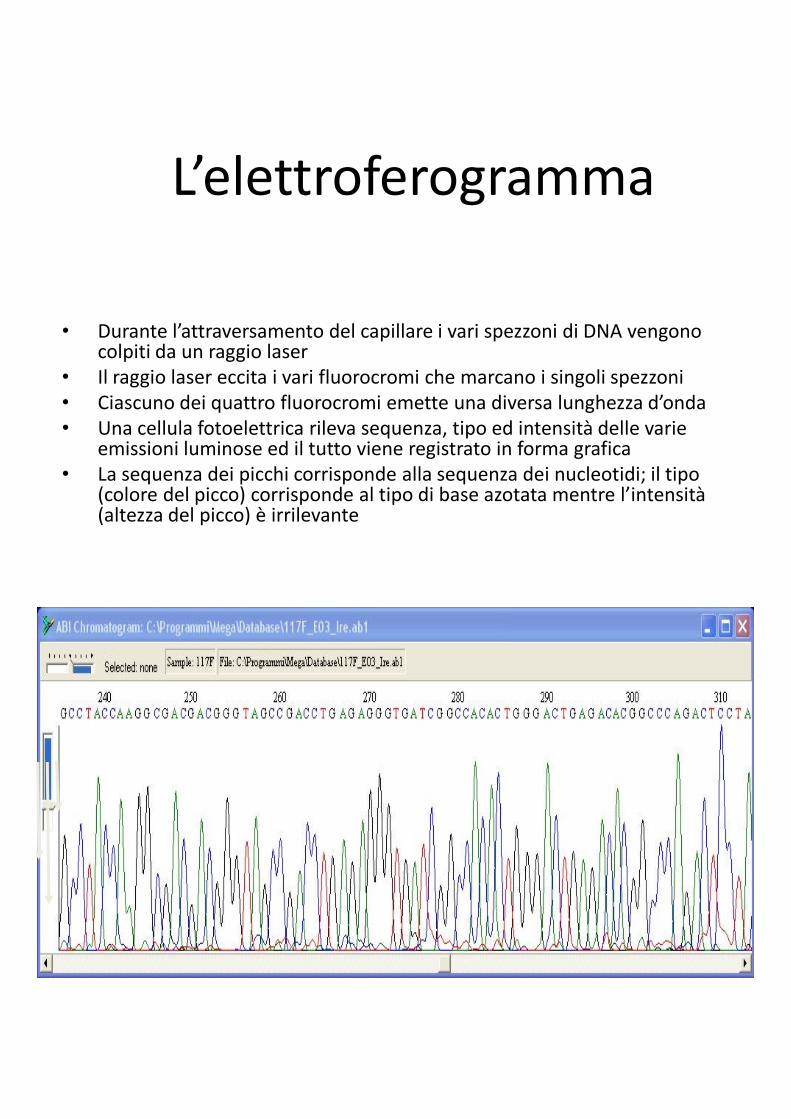
L’elettroferogramma
• Durante l’attraversamento del capillare i vari spezzoni di DNA vengono colpiti da un raggio laser
• Il raggio laser eccita i vari fluorocromi che marcano i singoli spezzoni
• Ciascuno dei quattro fluorocromi emette una diversa lunghezza d’onda
• Una cellula fotoelettrica rileva sequenza, tipo ed intensità delle varie emissioni luminose ed il tutto viene registrato in forma grafica
• La sequenza dei picchi corrisponde alla sequenza dei nucleotidi; il tipo (colore del picco) corrisponde al tipo di base azotata mentre l’intensità (altezza del picco) è irrilevante
Top Related