
Università degli Studi di Roma “La Sapienza” Tesi di Dottorato di ... · Università degli...
103
Università degli Studi di Roma “La Sapienza” Tesi di Dottorato di Ricerca in Biofisica XIV Ciclo Rapporto fra struttura nativa e processi dinamici in due emoproteine ALESSANDRO ARCOVITO Relatori Prof. Maurizio Brunori Prof. Alfredo Colosimo DICEMBRE 2001
Transcript of Università degli Studi di Roma “La Sapienza” Tesi di Dottorato di ... · Università degli...

Università degli Studi di Roma “La Sapienza”
Tesi di Dottorato di Ricerca in Biofisica XIV Ciclo
Rapporto fra struttura nativa e processi dinamici in due emoproteine
ALESSANDRO ARCOVITO
RelatoriProf. Maurizio Brunori Prof. Alfredo Colosimo
DICEMBRE 2001

2

3
Istituto Pasteur – Fondazione Cenci-Bolognetti
IL PRESENTE DOTTORATO È STATO INTERAMENTE FINANZIATO
DALL’ISTITUTO PASTEUR – FONDAZIONE CENCI-BOLOGNETTI
DELL’UNIVERSITÀ DI ROMA “LA SAPIENZA”.

4

5
Ad Alessandra

6

7
Indice
Capitolo 1 – Introduzione 9
1.1 Premessa 9
1.2 Il problema termodinamico ed il problema Cinetico 11
1.3 Un primo elemento unificante: il Molten Globule 13
1.4 Il nuovo modello teorico: il panorama energetico delle proteine 15
1.5 Topologia dello Stato Nativo 19
1.6 Le cavità nelle proteine 22
1.7 Folding e moti funzionalmente importanti: due processi dinamici nelle
proteine 25
Capitolo 2 - Scopo della Tesi 27
2.1 Linee guida 27
2.2 Aspetti metodologici 30
Capitolo 3 - Materiali e Metodi 33
3.1 Principi di fotochimica di base 33
3.2 Il sistema di acquisizione a larga banda 33
3.3 Il sistema di acquisizione a singola lunghezza d’onda e il flow-flash 37
3.4 Il citocromo c: proprietà e caratteristiche 41
3.5 L’assorbimento infrarosso 44
3.5.1 Cinetiche a singola lunghezza d’onda 44
3.5.2 Misure in gradiente di temperatura 47
3.6 La mioglobina: proprietà e caratteristiche 48

8
Capitolo 4- Risultati e Discussione 51
4.1 Premessa 51
4.2 Il citocromo c in condizioni denaturanti 51
4.3 Dalla proteina ai sistemi modello 55
4.3.1 Considerazioni cinetiche 55
4.3.2 I sistemi modello 56
4.3.3 Esperimenti mirati a singola lunghezza d’onda 57
4.3.4. Un estensione del modello 63
4.4 Il rapporto tra topologia e folding nel citocromo c551 65
4.4.1 Esperimenti di mescolamento a flusso interrotto 66
4.4.2 Esperimenti di flow-flash 70
4.5 La mioglobina, una proteina modello 72
4.6 Processi dinamici all’interno della matrice proteica 74
4.7 Relazione fra proprietà spettroscopiche e dinamica strutturale 81
Capitolo 5 - Conclusioni e Prospettive 87
5.1 I problemi biologici e lo sviluppo strumentale 87
5.2 Possibili sviluppi 90
Bibliografia 91
Ringraziamenti 97
Allegati 99

9
Capitolo 1
Introduzione
1.1 Premessa
Le proteine svolgono nelle cellule degli organismi viventi un gran numero difunzioni, che vanno dal semplice ruolo di costituente strutturale, al trasporto eall’immagazzinamento di piccole molecole e ioni fino ai più complessi e sofisticatiprocessi enzimatici necessari alla vita. Il motivo per il quale sono in grado disvolgere in modo efficace queste funzioni così diverse, pur avendo come elementifondamentali gli stessi 20 aminoacidi, è dovuto alla possibilità che hanno le catenepolipeptidiche di creare strutture tridimensionali in grado di soddisfare in modoefficiente la funzione biologica assegnata.
Non è realistico ipotizzare una semplice assegnazione sequenza-struttura, inquanto è noto che proteine che condividono solo un basso valore di omologia disequenza possono avere un struttura tridimensionale comune. Inoltre, i recentiprogressi nello studio del genoma umano indirizzano il numero di proteinepresenti nell’uomo ad un valore di circa 105 e consentono di stimare ad un valoredi circa 1011 il numero totale di proteine funzionalmente diverse presenti in tuttigli organismi viventi. Nonostante questi numeri abbiano una notevoleindeterminazione, forniscono comunque l’impressione corretta del numeroenorme di queste macromolecole presenti in natura. A fronte di questo, unarecente classificazione (Thornton et al.) ha evidenziato che, da un punto di vistatopologico, il numero di conformazioni strutturali osservate (il fold o strutturaterziaria della proteina) è minore di 700, mentre per i singoli domini proteici sonostati evidenziati solo 32 motivi strutturali diversi. In altre parole la complessitànecessaria allo svolgimento delle più diverse funzioni biologiche si realizzaattraverso la combinazione di un numero relativamente modesto di elementisemplici, quali sono i due elementi principali di struttura secondaria presenti nelleproteine, ovvero: la catena α e il foglietto β.
Il rapporto tra sequenza aminoacidica e struttura tridimensionale delleproteine si è rivelato complesso e non formalizzabile in regole semplici; inparticolare il meccanismo di avvolgimento della catena polipeptidica (folding),mediante il quale una proteina assume in condizioni fisiologiche la sua strutturatridimensionale funzionalmente attiva, costituisce un passaggio fondamentale econclusivo del processo di trasferimento dell’informazione genetica dal DNA alsuo prodotto finale, come rappresentato nel seguente diagramma di flusso:

10
DNA → RNA → catena polipeptidica ( ? folding ?) → proteina attiva
L’apparente semplicità nasconde la reale portata del problema; infatti, iprogressi enormi della genetica molecolare hanno portato alla caratterizzazionecompleta del genoma di 12 organismi fra i quali l’uomo, rendendo disponibile perintero il primo elemento del processo di trasferimento dell’informazione genetica,il DNA. La conoscenza della sequenza nucleotidica, non porta automaticamente aconoscere le proteine che vengono codificate ed anzi il problema principale dellabiologia post-genomica è quello di decodificare ed utilizzare questa enormequantità di informazione cercando una possibile chiave di lettura proprio nellastruttura tridimensionale funzionalmente attiva. È infatti la strutturatridimensionale, più della sequenza, a definire la funzione ed a consentirel’intervento biotecnologico.
Tabella 1 – proteine amiloidogeniche e le corrispondenti patologie
Sindrome Clinica Precursore Componente della Fibrilla
Morbo di Alzheimer Proteina precursore amiloide β-peptide 1-40 e 1-43
Amiloidosi primaria sistemica Catena leggera dell’immunoglobulina Il precursore o suoi frammenti
Amiloidosi senile sistemica Amiloide A di siero AmiloideA (frammento di 76 residui)
Amiloidosi secondaria sistemica Transtiretina Il precursore o suoi frammenti
Polineuropatia amiloide familiare I Transtiretina Circa 45 varianti
Angiopatia amiloide ereditaria cerebrale Cistatina C Cistatina C meno 10 residui
Amiloidosi Hemodialysis-related β2-Microglobulina β2-Microglobulina
Polineuropatia amiloide familiare III Apolipoproteina A1 Frammenti di apolipoproteina A1
Amiloidosi ereditaria sistemica finnica Gelsosina 71-residui di gelsosina
Diabete di tipo II Polipeptide amiloide Islet (IAPP) Frammenti di precursore
Carcinoma Medullary della tiroide Calcitonina Frammenti di calcitonina
Encefalopatia Spongiforme Prione Prione o frammenti
Amiloidosi atriale Fattore atriale natriretico Fattore atriale natriretico
Amiloidosi Lisozima Il precursore o suoi frammenti
Amiloidosi Injection-localized Insulina Insulina
amiloidosi ereditaria renale Fibrinogeno Frammenti di fibrinogeno
Il problema dell’interpretazione del genoma, la cui soluzione richiedel’individuazione dei singoli geni e delle proteine da essi codificate, costituisce unpunto di incontro e di interesse di numerose discipline scientifiche, poiché una

11
comprensione approfondita dei meccanismi molecolari che sono alla base delfolding delle proteine, oltre a rappresentare un traguardo della biologia moderna,avrebbe una ricaduta evidente nella possibilità di curare quelle patologie che sonolegate alla formazione di corpi inclusi intracellulari dovuti alla presenza diaggregati amiloidi stabili di proteine denaturate o frammenti di esse. Infatti, comeevidenziato nella tabella 1, esistono 16 proteine amiloidogeniche che possonodare origine in determinate condizioni alla formazione di fibrille di diametro paria 60-100 Å e di lunghezza variabile e caratterizzate da una struttura a croce βripetuta (Kelly, 1996). Fra le patologie associate, meritano un attenzioneparticolare per la loro immediata correlazione con il meccanismo di folding, leencefalopatie spongiformi; esse infatti, si possono originare a seguito di disordinigenetici di tipo sporadico od infettivo, che coinvolgono un cambiamentoconformazionale di una proteina (il prione). Tale proteina la cui funzione rimaneignota, è presente nella cellula nella sua conformazione normale PrPc e siconverte nella forma patologica PrPsc a seguito di un cambiamentoconformazionale nel quale parte delle α eliche della struttura nativa si trasformain foglietti β. Il meccanismo dell’azione infettiva del prione sembra essere quellodi agire da stampo per la conversione di altre particelle sane in molecolepatologiche (Prusiner, 1999): dunque, proteine patologiche sono in grado diindurre in proteine sane un cambiamento conformazionale che produce unriarrangiamento tridimensionale non nativo (misfolding).
1.2 Il problema termodinamico ed il problema cinetico
L’approccio ad un problema così vasto ed impegnativo trova un suo primocaposaldo nel lavoro di Anfinsen (Anfinsen et al., 1961), nel quale l’autoredimostrò che in vitro alcune proteine possono andare incontro ad un processo didenaturazione reversibile, nel corso del quale è possibile svolgere la strutturatridimensionale attraverso l’introduzione di agenti chimici quali la guanidina ol’urea. Rimovendo questi agenti denaturanti, attraverso successive dialisi, siottiene nuovamente una proteina attiva e caratterizzata da una strutturatridimensionale compatta. Questa semplice osservazione consente di affermareche l’informazione necessaria per ottenere la conformazione nativa (N) di unaproteina in una data condizione fisiologica è contenuta nella sua sequenzaaminoacidica. Da un punto di vista termodinamico questa osservazione si traducenell’affermare che, nelle condizioni fisiologiche, lo stato N costituisce un minimodell’Energia libera di Gibbs ed indica in un controllo di tipo termodinamico ilpossibile meccanismo di folding come evidenziato dallo stesso autoresuccessivamente (Anfinsen, 1973). E’ importante osservare che, in alcune

12
proteine, la denaturazione è irreversibile; questi casi termodinamicamente piùcomplessi sono stati anche meno studiati e non saranno ulteriormente discussi inquesta tesi.
Le osservazioni di Anfinsen vennero ulteriormente ampliate e discusse daLevintahl il quale si pose il problema del tempo necessario affinché un sistema diquesto genere raggiungesse il suo stato di equilibrio (Levintahl, 1968). Infatti,anche assumendo che il numero di conformazioni accessibili al singoloaminoacido sia soltanto 2, per un catena polipeptidica di 100 aminoacidi,otteniamo che il numero totale di conformazioni è pari a 2100, ovvero più di 1030.Se infine il tempo minimo di interconversione per un singolo residuo fra unaconformazione e la sua alternativa è 10-11 secondi , il tempo necessario affinché laconformazione totale, corrispondente allo stato nativo di minima energia N, siaraggiunto attraverso un campionamento stocastico dello spazio delle fasiaccessibile al sistema è pari a 1011 anni. Dato che i tempi di folding spaziano daqualche secondo ad alcuni minuti è evidente che l’evoluzione ha trovato unasoluzione efficace a questo problema combinatoriale. La soluzione di Levintahl aquesto paradosso, e che venne ampliata e portata avanti anche da altri autori(Wetlaufer 1973), fu che il meccanismo di folding era sottoposto ad un controllodi tipo cinetico, ovvero che esistevano dei veri e propri percorsi definiti checonducevano dalla struttura completamente casuale e lineare (U) alla strutturanativa e funzionale (N).
Questa visione, nota come visione classica, si basava su l’utilizzo di semplicimodelli cinetici di tipo fenomenologico per cercare di spiegare i risultatisperimentali che, dai primi anni ’70, vennero ottenuti su diverse proteine globularie monomeriche, tali quindi da costituire dei modelli semplici per l’analisi delfolding. Gli esperimenti classici analizzavano processi di rilassamento seguendol’andamento nel tempo di proprietà ottiche collegate a proprietà strutturali dellaproteina, a seguito di un repentino cambiamento nelle condizioni sperimentalitale da indurre processi di folding o unfolding. Infatti, per individuare eventualiintermedi stabili la cui analisi potesse dare informazioni sul processo diripiegamento, si perturbava con agenti denaturanti chimici (guanidina idrocloruro,urea, acido) o fisici (temperatura) l’equilibrio fra lo stato nativo e quellodenaturato. Di fronte a processi cinetici di tipo monoesponenziale, risultavaevidente proporre un semplice sistema a due stati (U ∆ N), mentre l’osservazionesperimentale di cinetiche di folding multiesponenziali veniva spiegata ricorrendo amodelli via via più complessi che prendevano in considerazione l’esistenza diintermedi stabili nel processo di rinaturazione della proteina.
Fin dai primi risultati ottenuti in questo senso ( Wong & Tanford, 1973;Holladay et al., 1974), emerse un’importante problematica legata alla difficoltà dicomprendere se gli stati parzialmente strutturati che venivano evidenziatirappresentassero degli intermedi produttivi (on pathway; Tsong et al., 1971),

13
ovvero delle tappe fondamentali nel percorso di folding, oppure degli intermediimproduttivi (off pathway; Ikai & Tanford, 1971), ovvero delle strutture formate aldi fuori del percorso stesso, e come tali non direttamente utilizzabili per ladecifrazione del codice del folding. Durante il decennio vennero individuatiintermedi stabili all’equilibrio per diverse proteine globulari (Wong & Hamlin,1974; Kuwajima et al., 1976; Robson & Pain, 1976; Nozaka et al., 1978), ma sidovette arrivare ai primi anni ’80 per poter cominciare a considerare almenoalcuni di essi come reali intermedi di folding, accostandoli agli intermediidentificati come specie transienti in esperimenti cinetici.
I modelli classici, sono riportati di seguito: il modello on-pathway nel qualel’intermedio si forma lungo il cammino che porta dallo stato denaturato allo statonativo attraverso un processo che può essere costituito da uno o più intermedisequenziali;
U ∆ Xi ∆ N con i=1… n (Eq. 1.1)
il modello off-pathway, nel quale il passaggio dallo stato N allo stato U è modificatoper la presenza di uno stato intermedio X che funge da trappola cineticarallentando di fatto il processo di folding della proteina;
X ∆ U ∆ N (Eq. 1.2)
Un primo elemento di confusione in questo tipo di approccio nasceva peròdal fatto che il termine percorso di folding veniva utilizzato sia per indicare unaproprietà microscopica del sistema legata alla diminuzione consecutiva delnumero di gradi di libertà presenti per la singola molecola proteica nel suoprocesso di rinaturazione, sia per indicare una proprietà macroscopica ove siintendeva per percorso di folding il passaggio attraverso gli intermedi evidenziatinei meccanismi cinetici proposti (vedi eq. 1.1-1.2). Il primo tentativo di superarequesto problema fu il tentativo di cercare negli intermedi caratterizzati per diverseproteine delle caratteristiche strutturali comuni.
1.3 Un primo elemento unificante: il Molten Globule
All’inizio degli anni ’80 si evidenziò che gli intermedi on pathway, mostravanouna struttura secondaria in grado di formare un nucleo piuttosto compatto, mapiù espanso della proteina nativa a causa dell’assenza di specifiche interazioniterziarie (figura 1.1). Sulla base di queste osservazioni, Ohgushi & Wada (1983) e

14
Dolgikh et al. (1983) proposero che simili intermedi all’equilibrio potesseroappartenere ad uno stato fisico comune delle proteine globulari, per il qualevenne scelto il nome di molten globule (letteralmente: globulo fuso). Subito dopo,Kuwajima et al. (1985) ed Ikeguchi et al. (1986) mostrarono che l’intermedio didenaturazione all’equilibrio (il molten globule) dell’α-lattalbumina (α-LA) èidentico all’intermedio transiente di folding, evidenziato mediante misure didicroismo circolare (CD) durante esperimenti cinetici di rinaturazione dallo statodenaturato in guanidina.
Precedentemente si era osservato (Kuwajima, 1977) che l’intermedio diquesta proteina ha una stabilità compresa tra quella dello stato nativo e quelladello stato denaturato, e che la transizione tra stato denaturato ed intermedio èmolto più veloce di quella fra stato denaturato e stato nativo. Queste osservazionirappresentano probabilmente la prima evidenza che il molten globule possacostituire uno stato intermedio metastabile in cui la catena polipeptidica deveripiegarsi prima di assumere la sua struttura terziaria compatta. Successivamentevennero riportati risultati simili per diverse proteine, fra cui la β-lactoglobulina(Kuwajima et al., 1987), l’Rnasi A (Labhardt, 1984), la β-lattamasi (Carrey & Pain,1987) e l’interleuchina 1-β (Craig et al., 1987). Basandosi su questi ed altri studipresenti in letteratura, ed integrandoli con dati sperimentali su cinque diverseproteine, Ptitsyn et al. (1990) rinnovarono l’ipotesi che il molten globule fosse unintermedio di folding di tutte le proteine globulari, corroborando in questo modola tesi proposta anni prima da Ogushi & Wada.
In questo contesto, sorsero modelli di folding alternativi che cercarono dirazionalizzare e generalizzare i concetti che venivano man mano evidenziati dagliesperimenti. Kim & Baldwin (1982) proposero il modello, detto framework,secondo il quale gli elementi di struttura secondaria venivano formati per primi,costituendo uno scheletro iniziale (framework, appunto): la loro coalescenza aseguito dell’espulsione del solvente (collasso idrofobico) avrebbe permesso
Molten-globule
Figura 1.1: Rappresentazione schematicadel molten globule a confronto con lastruttura dello stato nativo.
Stato Nativo

15
l’interazione fra le catene laterali, e la formazione dello stato nativo. Altri autorisostennero invece l’ipotesi che fosse il collasso idrofobico l’evento iniziale ingrado di guidare la proteina verso il suo stato nativo e che solo successivamente siformassero gli elementi di struttura secondaria (Dill, 1985). Infine Harrison eDurbin (1985), posero l’accento sul fatto che il folding potesse avvenireattraverso percorsi multipli in grado di giungere ad un’unica soluzione. Inaccordo con questa ipotesi l’identificazione di intermedi nel folding aveva unarappresentazione cinetica, più che strutturale, come di un’eterogeneità di specie inrapido equilibrio fra loro. Semplici considerazioni di cinetica chimica,suggeriscono inoltre che i percorsi di folding alternativo non possono essereinfiniti e nemmeno molto numerosi: infatti, se gli intermedi si interconvertonorapidamente e se un meccanismo è significativamente più rapido degli altri,quest’ultimo sarà preferito e diverrà dominante. Come conseguenza, affinché unpercorso di folding sia effettivamente utilizzato deve realizzarsi almeno una delledue seguenti condizioni:
1) le sue costanti cinetiche non sono troppo inferiori a quelle dei percorsialternativi.
2) uno dei suoi intermedi non è in rapido equilibrio con quelli degli altripercorsi di folding, ed “isola” efficacemente il percorso considerato.
Queste due condizioni limitano il numero dei percorsi di folding possibilie degli intermedi effettivamente popolati ed aggirano il paradosso diLevintahl.
1.4 Il nuovo modello teorico: il panorama energetico delle proteine
Negli ultimi dieci anni, grazie alla combinazione di esperimenti innovativi e diapprocci teorici più sofisticati che fanno largo uso di concetti cari alla meccanicastatistica, è stato possibile elaborare una visione di insieme più approfondita edefficace per il problema del folding nelle proteine giungendo al superamento dellavisione classica a fronte di un nuovo modello più complesso e più generale. Icontributi teorici di riferimento nello sviluppo di questa nuova visione sono statielaborati da numerosi autori (Wolynes, et al., 1995, Sali et al. 1994, Shakhnovic etal. 1996, Dill et al. 1997, Dobson et al. 1999) i quali hanno fatto uso di unmodello semplificato di molecola proteica per dedurre e calcolare la superficieenergetica corrispondente allo spazio delle fasi accessibile al sistema. Talemodello, noto come proteina giocattolo (toy protein), possiede solo alcune delleproprietà caratteristiche delle proteine quali l’eterogeneità intrinseca dei residui(idrofobici o polari) e la capacità di formare interazioni a lunga distanza.

Figura 1.2: Profiloparametro d’ordine h
In due dimencatena polipeptidiresidui polari nonformare contatti, figura, è rappresesituazione in cui proteina giocattolrappresentano lo conformazione inrappresenta lo stridimensionale, Montecarlo le conconsentito di ottpseudo proteina. funzione del numnon nativi, (C). Ucubo completamee Q0 = 28. La traper ogni valore diiniziata da una corossa) illustrano ivalore medio, tali
16
energetico della proteina giocattolo in due dimensioni in funzione del, che rappresenta il numero di contatti fra i residui idrofobici
sioni si può rappresentare, come mostrato nella figura 1.2, laca come una serie di bastoncelli intervallati da palline bianche (i interagenti) e palline nere (i residui idrofobici in grado disia nativi che non nativi). Nel caso esaminato nella suddettantato il profilo energetico corrispondente al passaggio da unail numero di contatti idrofobici h è pari a zero (ovvero la
o può assumere un numero elevatissimo di conformazioni chestato completamente denaturato del sistema); fino all’unica grado di produrre h=5 contatti idrofobici, situazione chetato nativo della proteina giocattolo. L’estensione al casoeffettuata simulando mediante campionamenti di tipoformazioni accessibili ad un reticolo cubico di 27 elementi, ha
enere la rappresentazione del panorama energetico di questaInfatti, in figura 1.3 l’energia libera (F) è stata riportata in
ero di contatti nativi (Q0) e del numero di contatti totali, nativi ena catena completamente estesa ha C = 0 e Q0=0, mentre unnte ripiegato corrispondente a l’unica struttura nativa ha C = 28iettoria gialla mostra il cammino medio tracciato campionando Q0 la variabile C(Q0) a partire da 1000 prove differenti ognunanformazione iniziale casuale. Le altre due traiettorie (verde e percorsi corrispondenti a due deviazioni standard intorno alda contenere dunque ~95% delle traiettorie possibili.

17
Figura 1.3: Rappresentazione dei percorsi di folding paralleli calcolati a partire da diverseconfigurazioni iniziali di un reticolo cubico di 27 elementi.

18
Nonostante l’apparente semplicità del modello, si può osservare come apartire da una delle 1016 possibili conformazioni iniziali, la catena collassirapidamente verso un globulo disordinato. Quindi, attraverso una lenta ricerca frale 1010 conformazioni semicompatte si diriga verso uno dei 103 stati di transizioneche conducono rapidamente verso l’unica struttura nativa. In questa nuovavisione, il concetto di percorso di folding costituito da eventi sequenziali vienesostituito dal concetto di imbuto (funnel) di eventi paralleli, rappresentato megliograficamente nei diagrammi energetici a tre dimensioni della Figura 1.4. L’asseverticale rappresenta l’energia libera interna di ogni specifica conformazione,ovvero la somma di legami idrogeno, coppie ioniche, interazioni idrofobiche edenergie di solvatazione. Gli assi orizzontali rappresentano le coordinateconformazionali necessarie per specificare ogni singola conformazione (peresempio, gli angoli diedri φ e ψ per ognuno dei residui della catena), il che rifletteil grande numero di gradi di libertà di una catena polipeptidica. Larappresentazione tridimensionale è perciò una semplice riduzione ad unadimensione ancora graficabile del complesso e multidimensionale panoramaenergetico (energy landscape) che è realmente accessibile alla proteina.
La forma ad imbuto descrive la progressiva riduzione dello spazioconformazionale accessibile, a partire dai molti gradi di libertà disponibili per lecatene denaturate, fino ad arrivare alla proteina nello stato nativo che ècaratterizzata, almeno in prima approssimazione da un unico sottostatoconformazionale.
Figura 1.4. Rappresentazione schematica attraverso diagrammi energetici a tre dimensioni(‘folding funnels’) del processo di folding a due stati (pannello a) e multistato (pannello b).
ba

19
Da un punto di vista della meccanica statistica, lo stato denaturato di unaproteina non è un singolo punto nel panorama energetico, ma una superficie,virtualmente infinita costituita da tutti i punti eccetto lo stato nativo. In questotipo di rappresentazione, il panorama energetico non è altro che l’energia libera diogni conformazione in funzione dei gradi di libertà, ed ogni configurazione èrappresentata da un punto su questa superficie energetica multidimensionale, sullaquale conformazioni geometricamente simili si trovano una vicina all’altra.Appena è in condizioni di ripiegarsi, la proteina tende a cambiare conformazionein modo da diminuire la propria energia, ma è anche costantemente spinta damoti Browniani ad esplorare altre conformazioni in tutte le altre dimensionidell’imbuto, finché tutte le conformazioni trovano la strada per la stessa (unica,corrispondente al minimo di energia) struttura nativa. Nella figura 1.4a èrappresentato l’imbuto che descrive il panorama energetico più semplice, ovveroquello di una reazione a due stati, in cui non si misura l’accumulo di alcuno statointermedio. Prendendo in considerazione la possibilità di formazione diintermedi, di trappole cinetiche e la presenza di barriere energetiche, il panoramadiviene più vario e corrugato, come illustrato in figura 1.4b. Anche complicando ilsistema, appare chiaro però che il modello ad imbuto supera il paradosso diLevinthal in quanto, pur ammettendo un gran numero di diversi camminimicroscopici alcuni dei quali possono dare origine ad intermedi inizialmenteimproduttivi, non consente un campionamento completamente casuale ma spingeil sistema verso il suo minimo di energia favorendo via via in modo più marcatoquei riarrangiamenti che portano verso lo stato nativo e rendendo estremamenteimprobabili dei percorsi che risalgano l’imbuto energetico.
1.5 Topologia dello stato nativo
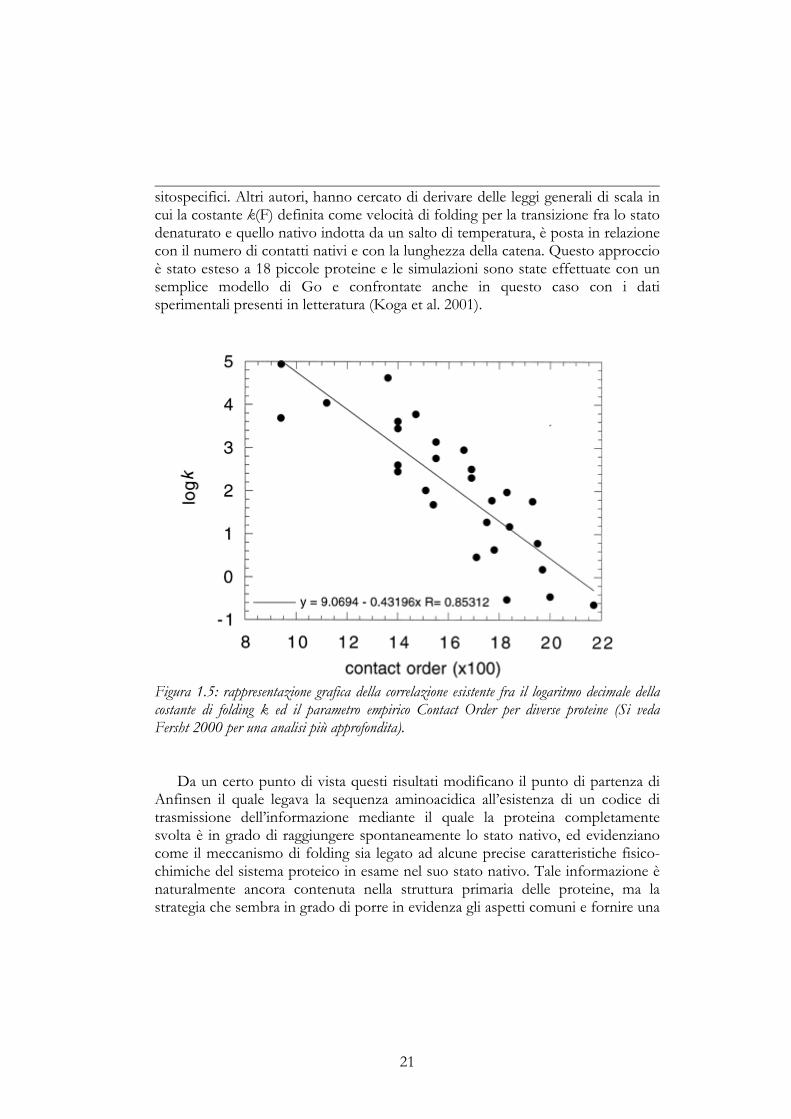
La nuova visione sul folding ha fornito un nuovo spunto per cercare,attraverso esperimenti mirati, di estrarre le informazioni necessarie a determinareuna comune chiave di lettura. Per quanto riguarda, ad esempio, semplici reazionidi folding a due stati numerosi risultati sono stati ottenuti mediante esperimenticinetici per diverse proteine globulari (Fersht, 1995; Jacob et al., 1997; Jackson,1998). In particolare l’attenzione è stata volta alla comprensione della relazioneesistente fra le caratteristiche intrinseche di una proteina e la sua velocità difolding, nello sforzo di identificare un parametro che permettesse in qualchemodo di predire l’una in base alle altre. Da un’analisi di 14 proteine appartenenti afamiglie non omologhe, per le quali era disponibile un’ampia quantità di datiprovenienti da studi strutturali e cinetici, è emersa una forte correlazione fra ladistanza media, nella sequenza, fra residui interagenti nello stato nativo e lavelocità di folding (Plaxco et al., 1998). Questo tipo di parametro, puramente

20
empirico, può essere facilmente calcolato per ogni proteina in base alla suastruttura, ed è stato definito contact order (ordine dei contatti). La sua espressione èla seguente:
∑=N
i,j∆ZLN
CO 1
dove N è il numero totale dei contatti nella proteina, ∆Zi,j è il numero di residuiche separano i due residui interagenti i e j, ed L è la lunghezza della proteina inresidui. In una proteina in cui il valore di contact order è basso, i residuiinteragiscono mediamente con altri che si trovano vicini nella sequenza.Viceversa, un valore alto di contact order indica che nella proteina ci sono unnumero cospicuo di interazioni a lungo raggio, ovvero che molti dei residuiinteragiscono con altri che si trovano lontani nella sequenza. Applicando questoparametro, oltre che alle proteine sopra citate, a diverse proteine modelloutilizzate in studi cinetici, è emersa una correlazione statisticamente significativatra la costante cinetica di folding ed il contact order, ovvero all’aumentaredell’uno diminuisce l’altra; questa evidenza indica che la topologia dello statonativo è un fattore determinante per la velocità di formazione di una proteina.Nella figura 1.5 è riportato un recente grafico pubblicato in un lavoro di rassegnada parte di Fersht (Fersht, 2000), nel quale ogni punto rappresenta una proteinadiversa per la quale è stato calcolato il contact order e che fornisce un’idea delgrado di affidabilità di questo parametro empirico. Il successo di questo tipo diapproccio sperimentale ha indotto numerosi altri autori a cercare di caratterizzareaspetti diversi della topologia delle proteine che possano fornire un ulterioreparametro in grado di complementare il parametro empirico contact order. Ai lavorisperimentali che sono stati citati, si sono perciò affiancati una serie di lavoriteorici di Dinamica Molecolare (DM), che hanno cercato mediante Hamiltonianemodello di ottenere delle evidenze indipendenti sull’importanza della topologiadello stato nativo nel meccanismo di folding. Si è messo in evidenza che, per dueproteine molto diverse, la deidrofolato reduttasi e la 1β interleuchina, è possibilesimulare le proprietà strutturali fondamentali degli intermedi di folding che siformano e che tali proprietà sono direttamente correlate a proprietà topologichedello stato nativo (Clementi et al., 2000). Inoltre, è stato determinato anche ilruolo giocato dalla posizione geometrica chiave di alcuni amino acidi nel processodi folding del dominio variabile di una importante classe di proteine, leImmunoglobuline (Settanni et al., 2001). In questo modo è stato possibileidentificare quali parti della proteina siano per ragioni geometriche maggiormentesuscettibili a modificare sostanzialmente la stabilità e i risultati ottenuti sono staticonfrontati con i dati sperimentali presenti in letteratura su determinati mutanti
(Eq. 1.3)
21
sitospecifici. Altri autori, hanno cercato di derivare delle leggi generali di scala incui la costante k(F) definita come velocità di folding per la transizione fra lo statodenaturato e quello nativo indotta da un salto di temperatura, è posta in relazionecon il numero di contatti nativi e con la lunghezza della catena. Questo approccioè stato esteso a 18 piccole proteine e le simulazioni sono state effettuate con unsemplice modello di Go e confrontate anche in questo caso con i datisperimentali presenti in letteratura (Koga et al. 2001).
Figura 1.5: rappresentazione grafica della correlazione esistente fra il logaritmo decimale dellacostante di folding k ed il parametro empirico Contact Order per diverse proteine (Si vedaFersht 2000 per una analisi più approfondita).
Da un certo punto di vista questi risultati modificano il punto di partenza diAnfinsen il quale legava la sequenza aminoacidica all’esistenza di un codice ditrasmissione dell’informazione mediante il quale la proteina completamentesvolta è in grado di raggiungere spontaneamente lo stato nativo, ed evidenzianocome il meccanismo di folding sia legato ad alcune precise caratteristiche fisico-chimiche del sistema proteico in esame nel suo stato nativo. Tale informazione ènaturalmente ancora contenuta nella struttura primaria delle proteine, ma lastrategia che sembra in grado di porre in evidenza gli aspetti comuni e fornire una

22
chiave di lettura generale per il meccanismo di folding, passa attraverso unacomprensione approfondita degli elementi caratteristici della topologia dello statonativo in grado di influenzare sensibilmente il ripiegamento tridimensionale. Aquesto proposito si pensi all’importanza di quei sistemi nei quali lo stato N è inuna configurazione metastabile, ovvero non corrisponde ad un minimo assolutodell’energia libera, in quanto un’estrema flessibilità conformazionale risultaessenziale per la funzione biologica. Appartengono a questa categoria alcuneproteine di fusione di membrana virali, o le serpine del plasma (gli inibitori delleserinoproteasi). Sebbene i dettagli del meccanismo mediante il quale si ottiene laflessibilità strutturale non sono completamente chiariti, alcuni autori hannomesso in evidenza come la presenza di elementi topologici energicamentesfavorevoli, quali l’introduzione di gruppi polari all’interno di residui polari nonaccessibili al solvente, la presenza di cavità o al contrario di elementi disovraimpaccamento delle catene laterali, possano essere la causa di questepeculiarità sia funzionali che strutturali (Bullough et al. 1994, Lee et al. 2000).Solo di recente, ed in un sistema più collaudato quale la mioglobina è statopossibile correlare in modo diretto la presenza di cavità all’interno della molecolaproteica con il suo ruolo funzionale.
1.6 Le cavità nelle proteine
La presenza nei sistemi proteici di difetti di impaccamento era nota aglistudiosi da numerosi anni, fin da quando grazie alla possibilità di osservarestrutture tridimensionali ad alta risoluzione (Richards, 1977) era stata posta inevidenza la presenza di cavità con volumi variabili fra i 30 e i 100 Å3. Inproposito, occorre citare il lavoro di Tilton et al. (1984) nel quale si era mostratoche la mioglobina di capodoglio è in grado di alloggiare al suo interno in quattrocavità preesistenti (con raggio>5Å), altrettanti atomi di Xenon (figura 1.6).
La presenza di queste cavità, in questa semplice proteina globulare emonometrica, e la contemporanea assenza di un canale che mettesse incomunicazione la tasca dell’eme, il sito attivo, con il solvente sono stati per lungotempo un punto oscuro nella chiarificazione dei dettagli molecolari relativi alprocesso di migrazione del ligando. Alcuni autori hanno messo in evidenza larelazione fra le cavità evidenziate da Tilton e i possibili siti occupatitransientemente dai ligandi della mioglobina (Scott & Gibson 1997). Inparticolare in un lavoro successivo (Brunori et al 1999), sono state misurate lecinetiche di ricombinazione a temperatura ambiente di tre diversi ligandi NO, O2e CO, sia con tecniche di mescolamento interrotto che di laser fotolisi per untriplo mutante di mioglobina (Leu(B10) → Tyr, His(E7) → Gln e Thr(E10) →Arg) denominato Mb-YQR.

23
Figura 1.6: Struttura tridimensionale della Mioglobina con evidenziati in rosso gli atomi diXenon alloggiati nelle cavità presenti nella proteina
L’emergere di alcune peculiarità, fra cui una ricombinazione geminata (ovverodall’interno della matrice proteica) del NO insolitamente lenta e bifasica misurataseguendo la variazione di assorbimento ottico nel tempo a seguito di un impulsodi luce laser, hanno suggerito agli autori di ripetere tale esperimento in presenzadi 12 atmosfere di Xenon. Il risultato, riportato nella figura 1.7, mostra che inpresenza di concentrazioni di Xenon saturanti, la ricombinazione del NOprocede attraverso un processo praticamente monofasico in cui la fase più lenta èstata annullata. Tale risultato, anche alla luce di simulazioni di dinamicamolecolare era stato interpretato ipotizzando che in condizioni di assenza diXenon, il ligando fotolizzato avesse accesso ad un sito alternativo e più distantedi quello immediatamente prossimale nel quale avesse una probabilitàsignificativa di migrare, e dal quale la ricombinazione fosse di conseguenza piùlenta. Tale sito alternativo era stato ipotizzato essere quello identificato come Xe4(si veda la figura 1.6 per il dettaglio topologico). Successivamente, (Brunori et al.2000) è stato possibile catturare la struttura tridimensionale del fotoprodotto, nelmedesimo mutante YQR di mioglobina, di un altro ligando il CO, ottenutoilluminando un cristallo a bassa temperatura (20 K) in modo da intrappolare la

24
molecola di CO proprio nel sito identificato come Xe4. Parallelamente altri autori(Ostermann et al. 2000) hanno dimostrato su un altro mutante di mioglobinaL29W (Leu 29 → Trp) che è possibile popolare in modo selettivo le due cavitàXe4 e Xe1, utilizzando diversi protocolli di fotolisi ed hanno risolto le strutturecristallografiche dei diversi fotoprodotti. I risultati sembrano definire un possibilepercorso migratorio fra l’eme ed il solvente, da parte del ligando e conferisconoalle cavità evidenziate da Tilton lo status di componenti cataliticamente attive delreattore macromolecolare che rappresenta la singola proteina.
Figura 1.7: Ricombinazione geminata del NO alla mioglobina mutante Mb-YQR (L29Y;H64Q; T67R). L’aumento di densità ottica ai tempi molto brevi è dovuto alla dissociazionedel ligando ottenuta con un impulso di laser di 9 ns. In assenza di xenon (pallini vuoti) laricombinazione procede con un andamento bifasico; in presenza di un’alta pressione di xenon(pallini pieni), la ricombinazione diviene più veloce e omogenea per la scomparsa della fase piùlenta (l’ampiezza totale si dimezza, ma questo dato non è visibile in questa rappresentazione).L’interpretazione dei risultati si accorda con un possibile sito secondario (la cavità denominataXe4) che può essere popolato in alternativa all’uscita del ligando nel solvente. In entrambi i casila ricombinazione passa per il sito primario.

25
Il ruolo strutturale di queste cavità è più difficile da definire ed occorreconsiderare due parametri: se la loro presenza si associ ad un costo o ad unguadagno energetico (ovvero se vi sia un aumento o una diminuzione dellastabilità della configurazione nativa); e se le cavità comportino specificheconseguenze nel meccanismo di folding. Una relazione diretta della presenza dicavità con la stabilità delle proteine è stata rivelata, in quanto si è valutato chemantenere tali cavità ha un costo energetico ben preciso come è stato dimostratoin particolare utilizzando la mutagenesi per riempire i vuoti di alcune piccoleproteine (ad esempio il lisozima T4 Anderson et al., 1993 e il citocromo c552Hasegawa et al., 1999), mimando in questo modo il comportamento delleproteine termofile che sono generalmente più stabili e meno flessibili dei lorocorrispondenti mesofili (Jaenike, 1999). Studi recenti inoltre hanno cercato diassegnare alle cavità nelle proteine anche un ruolo di determinante topologico nelmeccanismo di folding. In particolare il lavoro di Kocher et al (1996), haevidenziato che la tendenza a lasciare dei vuoti ha un costo energetico minore aparità di volume di vuoto per liquidi semplici quali l’acqua o l’esano piuttosto cheper il caso di due proteine quali il lisozima T4 e la barnasi. Di conseguenza èlecito ipotizzare che la formazione delle cavità possa essere anche da un punto divista del meccanismo di ripiegamento, il collo di bottiglia ovvero che lacaratterizzazione degli intermedi di folding possa avere come controparte delnumero di contatti nativi che si formano anche le cavità che tali contatti nativisottintendono. Queste considerazioni, hanno trovato una conferma nei lavorisperimentali che hanno studiato in modo sistematico il ruolo delle cavità neiprocessi di denaturazione indotti da salti di pressione (Fryer et al. 1998,Woenckhaus et al. 2001). E’ stato dimostrato in particolare che il contributopreponderante al cambiamento di volume indotto dal passaggio dallo stato N allostato U per la nucleasi di stafilococco è dovuta alla perdita del volume di vuotocontenuto nelle cavità della proteina. Tale risultato è stato ottenuto variando sia lecondizioni del solvente, per evidenziare eventuali contributi dovuti alla differenteidratazione della superficie, sia utilizzando mutazioni puntiformi per variare ilvolume di vuoto della proteina stessa.
1.7 Folding e moti funzionalmente importanti: due processi dinamici nelleproteine
In questo capitolo introduttivo, si è cercato di fornire un quadro ampio delproblema biologico connesso con il processo di folding delle proteine a partiredalla sua formulazione fino ai più recenti lavori volti alla comprensione delle basimolecolari che lo caratterizzano. In particolare, è stato posto in evidenza come losviluppo teorico e sperimentale che ha percorso questo ambito di ricerca negli

26
ultimi dieci anni, abbia avvicinato lo studio del meccanismo di folding con lostudio della dinamica delle proteine. Infatti, la comprensione dettagliata deirapporti fra i processi dinamici connessi con le transizioni conformazionali ed icorrispondenti elementi della struttura che sono coinvolti in tali processi,rappresenta una chiave di lettura comune sia nello studio del meccanismo difolding, sia nello studio più in generale della dinamica delle proteine. D’altraparte, un panorama energetico corrugato rappresenta un elemento di similitudinefra quanti vi immaginano avvenire quei moti funzionalmente importanti che sonole transizioni fra due conformazioni della stessa molecola proteica e quantiutilizzano le stesso schema per rappresentare il processo mediante il quale lacatena polipeptidica completamente svolta raggiunge la conformazione nativa.
Alcuni autori spingono queste considerazioni ad un livello più approfondito,infatti come mostrato in alcuni lavori citati nei paragrafi precedenti, nel casoparticolare delle serpine che hanno una struttura nativa metastabile, è statopossibile mostrare che i determinanti strutturali del folding sono gli stessi cherisultano legati ai moti funzionalmente importanti della proteina (Lee at al. 2000).Ma anche nel lavoro di Clementi (Clementi et al 2000), in cui sono posti inevidenza le interazioni fra la topologia dello stato nativo e i meccanismi di foldingdi due proteine molto diverse l’interleuchina 1β e la deidrofolato reduttasi, glistessi autori concludono chiedendosi se i vincoli topologici che sembranoindirizzare il meccanismo di ripiegamento tridimensionale non siano anche glistessi che la Natura usa per espletare la funzione biologica specifica, legando inquesto modo struttura, funzione e folding delle proteine.

27
Capitolo 2
Scopo della Tesi
2.1 Linee guida
Alcune caratteristiche strutturali delle proteine hanno un rapporto diretto epeculiare con la stabilità termodinamica della conformazione nativa: legami salini,interazioni a lunga distanza, cavità, etc., anche se non è sempre possibile dire apriori, prima di misure sperimentali dirette quali siano in generale questecaratteristiche. Altre caratteristiche strutturali hanno invece una direttaconnessione con il ruolo funzionale della proteina stessa e non di rado risultanoneutre o addirittura sfavorevoli alla stabilità dello stato nativo (Bullough et al.1994, Lee et al. 2000). Infatti, le proteine rappresentano dei sistemi biologiciestremamente complessi e dinamici, nei quali la topologia dello stato nativocombina in una soluzione di compromesso due principali esigenze: da una parte,la possibilità che la proteina eserciti la sua funzione biologica, che spesso richiedeuna certa flessibilità della struttura; dall’altra, che il suo stato funzionalmenteefficace sia stabile e possa essere raggiunto in modo rapido e senza errori diripiegamento. Il problema scientifico affrontato in questa tesi è la ricerca di uneventuale rapporto fra quegli elementi strutturali dello stato nativo che hanno unaconnessione diretta con la stabilità della proteina, e il ruolo che essi esercitano nelmeccanismo di ripiegamento tridimensionale e nella funzione biologica.
Nel capitolo precedente, sono stati messi in luce i progressi fatti nellosviluppo della conoscenza delle basi molecolari del processo di folding delleproteine. In particolare, è stato messo in evidenza il processo logico che haportato la comunità scientifica a ritenere che la topologia dello stato nativocostituisca un punto di partenza per ulteriori sviluppi nella comprensione delmeccanismo di ripiegamento tridimensionale. Occorre però determinare quali, fragli elementi che concorrono a determinare la struttura tridimensionale nativa,abbiano un ruolo chiave nei processi di formazione di intermedi efficaci difolding con particolare riferimento a quelli che determinano il collasso rapidodella catena polipeptidica. Da questo punto di vista, un limite intrinseco agliesperimenti classici di denaturazione e rinaturazione effettuati con la tecnica delmescolamento a flusso interrotto (stopped flow), è costituito dal tempo mortonecessario affinché le soluzioni a diversa concentrazione di denaturante chimico(guanidina od urea) si mescolino dando inizio al processo di unfolding o refoldingrispettivamente. Tale tempo morto non è mai inferiore ai 2-3 millisecondi e

28
costituisce un limite nello studio di quei processi rapidi che coinvolgonointermedi precoci o che comunque portano alla formazione dei primi elementi distruttura tridimensionale in grado di guidare la proteina al suo stato nativo. Èimportante considerare che la dinamica strutturale delle proteine è in genererapida ed anche riarrangiamenti strutturali importanti e su larga scala, possonooccorrere in tempi brevi, largamente inferiori a 1 ms.
Vi sono vari metodi sperimentali per cercare di superare questo problemasperimentale ed uno studio pionieristico compiuto da William Eaton e dai suoicollaboratori (Jones et al. 1993), ha fornito il punto di partenza di questo lavorodi tesi. Gli autori, infatti, hanno mostrato che un approccio possibile per unavasta classe di emoproteine, i citocromi, era suggerito dalla seguente osservazione:il citocromo ridotto (che nel lavoro originale è il citocromo c di cuore di cavallo),presenta un equilibrio di denaturazione in funzione della concentrazione diguanidina, che risulta dipendere dalla presenza del monossido di carbonio (CO).In particolare, come mostrato nella figura 2.1, ove è riportata la curva dititolazione eseguita misurando il quenching di fluorescenza del triptofano inpresenza ed in assenza di CO, esiste un intervallo di concentrazione didenaturante all’interno del quale la proteina in presenza del ligando risultacompletamente denaturata mentre in sua assenza risulta prevalentemente nel suostato nativo.
Figura 2.1: curva di titolazione all’equilibrio fra la specie N e la specie U del citocromo di cuoredi cavallo, in funzione della concentrazione di guanidina. L’intensità di fluorescenza che vieneriportata normalizzata fra 0 e 1 corrisponde al picco di emissione del triptofano nell’UV (350nm).
Dato che il complesso eme-CO è fotosensibile, utilizzando un laser impulsatodi breve durata è possibile rimuovere in modo praticamente istantaneo il ligando;nell’ipotesi originale, questo dovrebbe consentire alla molecola proteica di

29
raggiungere o almeno di approssimare lo stato termodinamicamente piùfavorevole nelle condizioni di assenza di CO, quello nativo. Il processo di foldingcosì iniziato viene nuovamente inibito una volta che il CO si ricombina, dunquegli intermedi che si popolano prima della ricombinazione del ligando sarebberoresponsabili del collasso precoce del citocromo c.
La tecnica sperimentale utilizzata da Eaton e dai suoi collaboratori percaratterizzare i presunti intermedi precoci del folding, è la spettroscopia diassorbimento nella regione del Soret, in quanto questa risulta estremamentesensibile alla presenza di diversi ligandi dell’eme e si presta ad un’analisi accuratadei diversi intermedi coinvolti. Infatti, il presupposto di questo esperimento è chela formazione del legame nativo Fe-metionina distale sia uno degli elementi ingrado di guidare il processo di rinaturazione corretto del citocromo c; mentre, lapresenza nella catena polipeptidica di residui di istidina o metionina diversi dailigandi fisiologici dell’eme produrrebbe delle trappole cinetiche capaci di impediretemporaneamente il corretto riavvolgimento tridimensionale, in quantoconsentirebbe la formazione di stati esacoordinati non fisiologici(miscoordinazione).
Il progetto di ricerca nel quale si inserisce questa tesi inizia pertanto con ilvolere perseguire lo stesso tipo di approccio elaborato da Eaton e collaboratori suun diverso citocromo, il citocromo c551 da Pseudomonas aeruginosa. Il citocromo c dacuore di cavallo e il citocromo c551 condividono un riarrangiamentotridimensionale comune, come appare evidente confrontando le due strutturemostrate nella figura 2.2:
Figura 2.2: Struttura tridimensionale del citocromo c551 (a) e del citocromo c di cuore dicavallo (b).
a b

30
Il citocromo batterico presenta alcune caratteristiche peculiari che lo rendonoun sistema ideale nel quale studiare la relazione fra il folding e il legame distale frail ferro dell’eme e la metionina, la cui importanza nella stabilità della specie nativarisulta un elemento comune all’intera classe dei citocromi. Infatti in questaproteina sono assenti istidine diverse da quella prossimale in grado di ostacolare ilcorretto processo di rinaturazione, formando miscoordinazioni; di conseguenza cisi aspetta che il panorama energetico che caratterizza il folding di questocitocromo presenti una superficie meno “corrugata” e quindi un meccanismocinetico semplificato.
La dinamica strutturale delle proteine native costituisce un campo di studioparallelo ma distinto da quello del folding. Le transizioni strutturali in questo casosono in genere piccole rispetto a quelle dovute ai processi di denaturazione erinaturazione e rapidamenete reversibili. Grazie alla collaborazione con ilDipartimento di Biofisica dell’Università di Ulm (Germania), è stato quindiavviato un secondo progetto, volto a determinare l’importanza di un altroelemento caratteristico della topologia dello stato nativo delle proteine, ovvero lapresenza di cavità. In questo caso si è scelto di studiare la relazione fra questielementi e i processi dinamici legati allo svolgimento della funzione biologica.Anche per questo studio è stata scelta la tecnica della fotolisi a luce laser inragione dell’elevata risoluzione temporale, accoppiata a protocolli di acquisizioneeffettuati in un ampio intervallo di temperature criogeniche. La spettroscopiautilizzata per mettere in evidenza le proprietà strutturali e funzionali delle cavitàinterne alla proteina, è stato l’assorbimento nella regione dell’infrarosso che èlegato ai moti vibrazionali del monossido di carbonio. Variazioni di questi motisono dovute ai diversi ambienti e siti di legame all’interno della matrice proteicache sono in grado di ospitare transientemente il CO. In particolare sono statistudiati due mutanti sitospecifici progettati e costruiti allo scopo di evidenziare sepossibile, le caratteristiche dovute ai diversi percorsi effettuati dal CO nel suoprocesso di ricombinazione a diverse temperature. Infatti, è possibile selezionaredelle condizioni particolari in cui il CO è forzato a rimanere confinato all’internodella matrice proteica ed è in grado di comportarsi come una vera e propria sondamolecolare, capace di fornire informazioni dinamiche e strutturali in grado digettare nuova luce sul ruolo delle cavità nelle proteine.
2.2 Aspetti Metodologici
Da un punto di vista metodologico, occorre spiegare la scelta di utilizzare dueproteine distinte per studiare il meccanismo del folding ed il ruolo e la dinamicadelle cavità interne alla molecola proteica. Infatti, per realizzare con metodifotochimici misure di folding è requisito necessario che il ligando fotolabile sia un

31
denaturante, come accade nel citocromo c. Al contrario quando si voglia studiarela dinamica dello stato nativo con la stessa tecnica, è necessario che il ligandofotolabile si combini reversibilmente con la proteina nativa, esattamente ciò cheavviene nella mioglobina.
Inoltre, il citocromo c è in grado di compiere cicli di denaturazione erinaturazione reversibili, senza perdita del gruppo prostetico eme che restacovalentemente legato alla catena polipeptidica e si presta pertanto agli studi dellarelazione fra topologia e folding. La mioglobina sarebbe del tutto inadatta perquesto tipo di esperimento perché, nello stato denaturato, perde il gruppo emeche non è covalentemente legato alla catena polipeptidica. Per control’esplorazione delle cavità interne alla struttura nativa della proteina utilizzandocome sonda un ligando esterno dell’eme è un metodo di indagine ideale a porre inrisalto la relazione fra topologia e funzione nella mioglobina, ma non è estendibilealla classe dei citocromi, nei quali i ligandi fisiologici dell’eme sono entrambiintramolecolari. Nonostante notevoli differenze nella struttura tridimensionale,sia la mioglobina che il citocromo c possiedono una struttura globulare compatta,delle cavità all’interno e il medesimo gruppo prostetico e si prestano pertanto adessere studiate come sistemi modello nei quali testare il rapporto fra funzione,folding e struttura.
Infine, un risultato ottenuto a margine di questo lavoro di tesi è stato la messaa punto di un sistema sperimentale di laser fotolisi accoppiato a due diversisistemi di rivelazione dell’assorbimento ottico, uno a singola lunghezza d’onda el’altro mediante l’acquisizione di spettri di assorbimento su un’ampia scala ditempi. Inoltre, come verrà evidenziato meglio nel capitolo successivo, è statopossibile grazie alla versatilità del sistema ottenuto, realizzare dei protocolli diacquisizione originali di flow flash unendo le caratteristiche di un sistema a flussointerrotto con le potenzialità del sistema di rivelazione di laser fotolisi.

32

33
Capitolo 3
Materiali e Metodi
3.1 Principi di fotochimica di base
Alcuni legami chimici di interesse biologico presentano la peculiarità di esserefotosensibili, nel senso che un impulso di radiazione luminosa di opportunafrequenza, è in grado di scinderli. A seconda del tipo di reazione chimicacoinvolta, il processo può essere reversibile o irreversibile. In entrambi i casi periniziare la reazione in modo quasi istantaneo, si utilizzano degli impulsi di laseraventi un’intensità estremamente elevata ed una durata estremamente breve. Inquesto modo è possibile promuovere il passaggio da una specie legata A ad unaspecie non legata A*, la quale si definisce specie eccitata; se quest’ultima a seguitodi una serie di reazioni chimiche è in grado di tornare alla specie legata A, allora ilprocesso risulta reversibile altrimenti la specie A* rilasserà verso una nuova speciechimica B ed il processo sarà irreversibile. Questo protocollo sperimentale difotolisi, può essere utilizzato con diverse tecniche spettroscopiche e consentepertanto di studiare con metodologie diverse i processi dinamici che seguono laformazione della specie eccitata.
In questa tesi, la fotolisi mediante un impulso laser di intensità e duratavariabili del legame Fe-CO sarà utilizzata per generare specie eccitate moltodiverse in grado di dare reazioni sia reversibili che irreversibili. Le tecnichespettroscopiche utilizzate per rivelare questi processi saranno l’assorbimentoottico nella regione del visibile e del vicino ultravioletto (UV-Vis) el’assorbimento nel medio infrarosso (IR).
3.2 Il sistema di acquisizione a larga banda
Per quanto riguarda l’assorbimento UV-Vis, parte del lavoro di tesi è stata lamessa a punto un sistema sperimentale per misurare le variazioni di assorbimentoindotte da un impulso di luce monocromatica intensa e di breve durataproveniente da una sorgente laser. In particolare, sono state realizzate due diverseconfigurazioni, una per acquisire spettri di assorbimento su di un intervallo dilunghezze d’onda di 150-300 nm ad un ritardo predefinito rispetto all’impulso diluce laser ed una in grado di seguire delle cinetiche a singola lunghezza d’onda sudiverse scale di tempi.

Figura 3.1: Schema del sistema di acquisizione a largabanda
SPETTROGRAFO
IMPULSADELLA
Specchio
Compu
Trigger IN
Trigge
I
34
Campione
TE
STA
DE
L L
ASE
R
Lampada
TORECCD
ter
r OUT
F1
F2
L1
L2
ALIMENTATOREDEL LASER
Q-switch
P

35
Lo schema completo della prima di queste configurazioni è riportato nellafigura 3.1. La sorgente luminosa è data da una lampada ad incandescenzastabilizzata in continua con un’emissione significativa nell’intervallo 300-700 nmed una potenza massima di 300 W che attraversa un pinhole (P) di meno di unmillimetro di diametro in modo da approssimare una sorgente puntiforme. Unfiltro passabanda (F1) nell’intervallo desiderato (tipicamente 350-500 nm, per lemisure nella regione del Soret) esclude la radiazione luminosa che non interessa.La lente (L1) raccoglie la radiazione luminosa proveniente dalla sorgentepuntiforme e la dirige, con una geometria parallela verso il campione solitamentecontenuto in un tubo di Thunberg saldato ad una cuvetta per fluorescenza delledimensioni ottiche 1cm x 1cm, in grado di contenere una soluzione di volume 2-3ml, in atmosfera controllata. La radiazione trasmessa dal campione viene raccoltada una seconda lente (L2) e focalizzata in uno spot delle dimensioniapprossimative della sorgente puntiforme su di una slitta all’ingresso di unospettrografo Princeton Instrument, in grado di raccogliere il segnale luminosocon un reticolo di passo variabile ed indirizzarlo alla video camera impulsata(ICCD). Di fronte allo spettrografo è inserito un filtro olografico (F2), conassorbimento selettivo per la lunghezza d’onda del laser, in modo da proteggere ilsensore della videocamera dalla luce diffusa a seguito dei flash intensi provocatidal laser stesso.
Il laser è un modello a stato solido Nd-YAG della Quanta System, in grado difornire sulla seconda armonica a λ=532 nm circa 80 mJ per pulso di durata fissa epari a 5 ns (FWMH), con una frequenza di ripetizione ottimale di 2 Hz regolabileper passi discreti fino a 0.2 Hz. Un segnale elettrico viene prelevato attraverso uncavo coassiale dal Q-switch del laser stesso, ovvero dall’interruttore che comandaalla frequenza stabilita (nel range 0.2-2 Hz) la scarica della lampada sul mezzoattivo e restituisce con un ritardo fisso e pari a 50 ns un impulso di luce laser, eviene diretto sull’impulsatore della video camera (Trigger IN). Il generatore diimpulsi, modello FG100 della Princeton Instrument, è in grado di fornire inuscita, con un ritardo modulabile fino ad un massimo di 1.7 µs, un segnaleelettrico (Trigger OUT) che può essere accoppiato mediante un successivo cavocoassiale ad una video camera impulsata (ICCD 576 x 384, Princeton Instrument)per comandare elettronicamente l’apertura della stessa, per un tempo di letturafissato e molto breve (fra i 3 e 50 ns). Il ritardo fra il laser e l’acquisizione puòessere esteso fino ai secondi mediante l’aggiunta di un ulteriore elemento, ungeneratore di funzioni programmabile Tektronix AFG 310 posto fra il segnale diuscita Trigger OUT e l’ingresso della video camera.
Il protocollo sperimentale di una misura tipica, prevede l’acquisizione di unsegnale di luce trasmessa attraverso l’aria o una soluzione di solo buffer, in mododa determinare l’intensità della lampada T0; quindi si procede all’acquisizione diun segnale di buio, Tb, ottenuto chiudendo il pinhole e misurando di conseguenza

36
la corrente di buio dovuta ad una somma di fattori elettrici ed ottici. Infine, adogni intervallo di tempo predefinito rispetto al momento dell’impulso di lucelaser, si acquisiscono due spettri di trasmissione. Il primo Tnf(t), in cui la luce dirivelazione proveniente dalla lampada attraversa il campione senza che questivenga eccitato dal laser; in questo caso come per l’acquisizione di T0 e Tb il laserviene bloccato da un otturatore metallico posto prima dello specchio e nonraggiunge il campione, in altre parole fornisce solo la temporizzazione ma nonl’eccitazione. Il secondo, Tf(t), in cui il laser effettivamente colpisce il campione eproduce lo stato eccitato. Ciascuno spettro è la media di 100 impulsi laser ealtrettanti acquisizioni della video camera e l’assorbimento in funzione del tempoper la specie eccitata Af(t) e per la specie non eccitata Anf(t) sono dati dalleformule seguenti:
−−
−=b
bnfnf
TTTtTtA
0
)(log)( ,
−−
−=b
bff
TTTtTtA
0
)(log)( Eq. 3.1
In generale l’assorbimento della specie non eccitata dovrebbe essere costantenel tempo, in realtà possono avvenire dei cambiamenti dovuti ad unadegradazione del campione per l’esposizione alla luce del laser. In ogni caso, lagrandezza analizzata è lo spettro di assorbimento differenziale in funzione deltempo che per le proprietà dei logaritmi è dato da:
−−
−=−=∆bnf
bfnff
TtTTtTtAtAtA
)()(log)()()( Eq. 3.2
L’analisi dei dati procede attraverso l’utilizzo del formalismo delle matrici, chesi rivela estremamente conveniente per estrarre le informazioni significative.
In pratica, i dati sperimentali ∆A(t) sono disposti in una matrice A( n x m ), lacui i-esima colonna (con i=1,…,m) rappresenta il singolo spettro differenzialepreso ad un certo istante di tempo e la i+1-esima , lo spettro differenziale acquisitoall’istante successivo. Di conseguenza le n righe sono cinetiche a singolalunghezza d’onda. La matrice A, rappresenta la convoluzione di tre matrici:
A = E x C + N Eq. 3.3
dove la prima è data dalla matrice dei coefficienti di assorbimento delle k speciechimiche popolate durante l’esperimento E( n x k ); la seconda dalla matrice cherappresenta la concentrazione delle k specie ad ogni istante di tempo misurato

37
C(k x m ) ed infine una matrice che rappresenta l’errore strumentale presente inogni esperimento N( n x m ).
Dato che le matrici E e C non possono essere direttamente calcolate da A, siapplica l’algoritmo di deconvoluzione SVD (singular value decomposition) chefornisce le tre matrici U, S e V attraverso il calcolo della seguente espressione:
A = U x S x VT Eq. 3.4
Questa deconvoluzione è unica in quanto esistono dei vincoli interni, ovverole matrici U( n x m ) e V( m x m ) sono ortogonali, mentre la matrice S è diagonaleed i suoi elementi significativi sono posti in ordine decrescente. U, S e V nonhanno un immediato significato chimico-fisico, ovvero non esiste una proceduraindipendente dal modello chimico che si vuole verificare per determinare le duematrici desiderate E e C. Però, dato che il prodotto U x S rappresental’assorbimento dei diversi componenti spettroscopici rivelati dall’algoritmo e datoche il peso dato da S dei diversi componenti è rapidamente decrescente, si puòridurre l’analisi ai soli componenti statisticamente significativi. Le corrispondenticolonne di V ne caratterizzeranno i rispettivi andamenti temporali e unaprocedura di minimizzazione non linere della devianza ( fit ) identifica sia lecostanti cinetiche dei diversi processi in atto, sia le corrispondenti ampiezze.Infine, siano j ( j < m ) i componenti giudicati significativi, è possibile calcolare lospettro differenziale di ogni specie chimica coinvolta E ( n x j ) moltiplicando lematrici U ( j x j ) x S ( j x j ) per le ampiezze ottenute dal fit delle componenti diV, Vfit ( j x j ):
E = U x S x VfitT Eq 3.5
Il confronto, quando possibile, fra gli spettri calcolati a partire dalle speciepopolate transientemente e gli spettri differenziali ottenuti da esperimentiindipendenti all’equilibrio, costituisce il metodo ottimale per discriminare fradiversi modelli chimici proposti per spiegare un determinato esperimento.
3.3 Il sistema di acquisizione a singola lunghezza d’onda e il flow - flash
Accanto al sistema d’acquisizione descritto nel paragrafo precedente, è statoanche sviluppato un sistema a lunghezza d’onda singola che a scapito del minoregrado di informazione, fornisce una versatilità molto maggiore, tale da permetteredi eseguire esperimenti combinando le tecniche di mescolamento interrotto efotolisi. Nella figura 3.2 è rappresentato lo schema del sistema di acquisizione.

38
In generale ad ogni esperimento vengono collezionate diverse tracce sudifferenti scale di tempi utilizzando due amplificatori, uno veloce ma conun’amplificazione di circa 30 volte, per l’intervallo [25 ns -1 µs]/divisione; l’altropiù lento da utilizzare nell’intervallo [2.5 µs - 100 ms]/divisione ma con unamplificazione pari a 100. Successivamente in sede di analisi queste vengono
dal laser
specchio
monocromatore
amplificatore
lampada
fotodiodo
fototubo
trigger INsegnale
Figura 3.2: Schema del sistema diacquisizione a singola lunghezzad’onda

39
combinate insieme tenendo conto di eventuali differenze dovute ai diversiamplificatori usati. Infatti, il fototubo è in grado di raccogliere segnali anchemolto veloci e di trasferirli sotto forma di segnale di tensione V(t) ad unamplificatore e da questo ad un oscilloscopio Tektronix TDS 360 da 200 MHz. Ilpassaggio da tensione di uscita emessa dal fototubo, ad una differenza diassorbimento in funzione del tempo è data, per amplificatori invertenti,dall’espressione seguente:
+
=∆),(
log),(λ
λtVV
VtAoff
offEq 3.6
dove Voff rappresenta la tensione di soglia alla quale viene fatto lavorare ilfototubo, misurata illuminando il campione con la lampada di rivelazione masenza eccitarlo con la luce laser.
Il campionamento effettuato dall’oscilloscopio raccoglie 1000 punti per ognitraccia dei quali 250 vengono utilizzati come segnale di pre-flash e forniscono illivello zero sul quale vengono azzerate le diverse tracce prese su scale di tempodifferenti. La possibilità di mediare in breve tempo le tracce dovute a numerosicolpi di laser, produce in molti casi dei segnali dai quali è possibile estrarre conestrema precisione le costanti cinetiche dei processi di rilassamento che seguonol’eccitazione fotochimica.
Infine, è possibile accoppiare a questo sistema anche un apparato di flussointerrotto come illustrato nella figura 3.3. In questo caso l’acquisizione vienecomandata dalla siringa di stop che con un tempo di ritardo di circa 50 ms dalmescolamento chiude l’interruttore del fotodiodo consentendo all’oscilloscopio diiniziare la raccolta del segnale dopo l’impulso laser. Di conseguenza l’iniziodell’acquisizione cade ad un istante di tempo sconosciuto posto nell’intervallo 50- 500 ms dopo il mescolamento, dove il primo tempo è dato dal tempo mortodovuto al mescolamento a mano e l’ultimo tempo dal tempo di ripetizioneminimo del colpo di laser.
Essendo la miscela eccitata in uno stato di non equilibrio non è possibileeseguire in linea di principio accumulazioni successive, in quanto si ecciterebberovia via diversi stati termodinamici della soluzione presente nella camera diosservazione. Per questo motivo, un esperimento di flow-flash è estremamentedelicato, sia nella realizzazione sia nell’interpretazione. Nel capitolo successivosarà descritto in quale caso si è scelto di adottare questo approccio sperimentale.

40
dal laser
monocromatore
amplificatore
fotodiodo
fototubo
trigger INsegnale
Figura 3.3: Schema del sistema diacquisizione flow-flash
siringa di stop
camera di mescolamento

41
3.4 Il citocromo c: proprietà e caratteristiche
Come illustrato nel capitolo 2, allo scopo di legare proprietà topologiche conintermedi precoci di folding, si è scelto di studiare il citocromo c in quantopresenta caratteristiche peculiari da un punto di vista delle proprietàspettroscopiche. Infatti, la presenza del gruppo prostetico eme legatocovalentemente alla catena polipeptidica consente di denaturare la proteinamediante l’introduzione di alte concentrazioni di urea o guanidina idrocloruro(Gdn), senza che tale gruppo prostetico sia rilasciato nel solvente. Il gruppo emepresenta un notevole assorbimento nella regione dello spettro compresa fra 260 e600 nm e dissipa termicamente l’eccitazione luminosa; non soltanto non emettefluorescenza ma, se posto in prossimità di fluorofori quali i residui di Trp, Tyr ePhe delle proteine, ne riduce o elimina l’emissione grazie ad un processo ditrasferimento energetico non radiativo (quenching). E’ pertanto possibile titolarecon concentrazioni crescenti di denaturante la frazione di molecole che passadallo stato nativo N allo stato denaturato U, seguendo l’aumento del picco difluorescenza. Parallelamente all’incremento di fluorescenza causatodall’allontanamento degli aminoacidi aromatici dall’eme, il processo didenaturazione causa la perdita di struttura secondaria che può essere rivelatamediante spettroscopia di dicroismo circolare (CD). Si veda in proposito la figura3.4.
Figura 3.4: Pannello A; spettro di fluorescenza misurato per il citocromo c551 ferrico incondizioni native ovvero in assenza di guanidina (curva rossa) e in presenza di unaconcentrazione 3M di guanidina (curva nera). Pannello B; Frazione di molecole denaturatemisurate seguendo il picco di fluorescenza (pallino nero) o il segnale di dicroismo circolare(pallino rosso), sempre per il citocromo c551 ferrico.

42
Un importante contributo alla stabilità della conformazione nativa delcitocromo c è dovuto al legame di coordinazione distale che si instaura fra il Fe ela Met80 (la numerazione riferisce al citocromo di cavallo, ma il residuo dimetionina è conservato in tutti i citocromi di classe I). Inoltre, il passaggio dallostato di ossidazione +3 allo stato di ossidazione +2 per il ferro dell’eme produceun aumento di stabilità a seguito dell’interazione elettronica più favorevole che siinstaura fra l’atomo di ferro ed il residuo distale di metionina. Nella forma ridottail citocromo c può inoltre legare molecole di CO con una probabilità cheaumenta all’aumentare della concentrazione di denaturante presente in soluzione(si veda l’inserto presente nella figura 2.1). Il CO compete con la Met distale epertanto agisce come un denaturante aggiunto diminuendo la stabilità della specieridotta e creando le condizioni per esperimenti di fotochimica. In figura 3.5 èriportata la titolazione in funzione della concentrazione di Gdn per il citocromoc551 ferrico, ridotto e ridotto in presenza di CO.
3210
1,2
1
0,8
0,6
0,4
0,2
0
[GdnHCl] (M)
Fluo
resc
enza
nor
mal
izza
ta (3
54 n
m)
Fig 3.5: curve di titolazione in fluorescenza della frazione di molecole nello stato denaturato infunzione della concentrazione di guanidina per il citocromo c551 da Ps. aeruginosa. Proteinaferrica (pallini neri), ferrosa (triangoli vuoti) e ferrosa in presenza di CO (pallini bianchi).
Tutti i derivati ferrosi delle emoproteine sono fotosensibili e l’assorbimento diun fotone ha una probabilità finita (nel caso del CO pari al 100%) di promuoverela dissociazione del ligando. Nel caso dei citocromi in condizioni denaturanti ci si

43
aspetta che questa rimozione fotochimica del ligando induca un processotransitorio di rinaturazione. Gli esperimenti di assorbimento ottico dopo lafotolisi del laser, sono stati eseguiti su due citocromi, il citocromo c da cuore dicavallo ed il citocromo c551 da Pseudomonas aeruginosa, oltre che sui sistemi modellomicroperossidasi (MP11) ed eme. Nel primo caso è stato utilizzato quelloprodotto dalla Sigma Aldrich, così come per l’eme e per la microperossidasi (unpeptide protelitico, costituito da soli 11 residui del citocromo c di cavallo e dalgruppo prostetico eme); mentre per quanto riguarda il citocromo batterico, èstato purificato secondo la procedura descritta da Cutruzzolà et al. 1997. Gliesperimenti sulla MP11 e sui citocromi sono stati eseguiti a diverseconcentrazioni di guanidina in un tampone fosfato 100 mM a pH 7. L’eme è statoinvece disciolto in una soluzione al 5% di Sodio-dodecil-solfato (SDS) e iltampone utilizzato è stato il Tris 50 mM a pH 7.4. In queste condizionisperimentali il detergente forma micelle e queste dissolvono l’eme mantenendoloin forma prevalentemente monomerica.
Nel caso del citocromo c551 sono stati eseguiti anche numerosi esperimentiutilizzando un apparato classico di mescolamento interrotto. Per gli esperimentidescritti in questa tesi è stato utilizzato sia un apparato simmetrico con le siringhedi spinta uguali e aventi un volume pari a 200 µl, sia asimmetrico che permette ilmescolamento rapido di due soluzioni (nel rapporto di 10 a 1), contenute in duesiringhe di spinta una del volume di 200 µl e l’altra del volume di 20 µl (AppliedPhotophysics MV17). Da queste siringhe i reagenti vengono trasferiti nellacamera di mescolamento sotto la spinta di un pistone pneumatico, e da qui lasoluzione giunge nella cella d’osservazione e viene bloccata dalla siringa d’arresto.La registrazione del fenomeno parte nel momento in cui il pistone della siringad’arresto colpisce l’interruttore posto a fine corsa. Una lampada fornisce lasorgente di luce, la cui radiazione luminosa, opportunamente selezionata da unmonocromatore, attraversa la camera d’osservazione; il sistema è in grado dilavorare sia in fluorescenza che in assorbimento. Nel primo caso, l’emissione difluorescenza conseguente ad un’eccitazione a 290 nm viene misurata edamplificata da un fotomoltiplicatore posto ortogonalmente rispetto alla direzionedi eccitazione del campione. Un filtro appropriato, posto fra la cellad’osservazione ed il fotomoltiplicatore, permette di registrare unicamentel’emissione di fluorescenza al di sopra dei 320 nm. Il segnale così rivelato èinviato ad un calcolatore elettronico su cui si può analizzare direttamentel’andamento temporale della variazione nell’emissione di fluorescenza associataalla reazione in esame. Nel secondo caso, il segnale trasmesso attraverso ilcampione viene raccolto da un fototubo posto in linea con la lampada enormalizzato rispetto al segnale di buio e fornisce la variazione di densità otticaconseguente al mescolamento. La massima risoluzione temporale di questo

44
strumento, che ha un tempo morto di ∼ 3 ms è di 400 punti sperimentali in undecimo di secondo (una lettura ogni 250 µs). Un esperimento di mescolamentorapido a flusso interrotto è composto da due parti distinte: i) misura della cineticadi denaturazione (unfolding), che avviene mescolando la proteina nello stato nativocon soluzioni contenenti diverse quantità di denaturante, così da ottenere ladipendenza della velocità di denaturazione dalla concentrazione di denaturante; ii)misura della cinetica di rinaturazione (refolding), che avviene mescolando laproteina denaturata con tampone e denaturante in quantità moderate e diverse, inmodo da ottenere la dipendenza della velocità di rinaturazione dallaconcentrazione di denaturante. Il grafico che si produce è definito chevron plot, perla caratteristica forma a V, e consente di ottenere le due costanti cinetiche didenaturazione e rinaturazione, kunf e kref, attraverso un’estrapolazione lineare aconcentrazione di denaturante nulla
3.5 L’assorbimento infrarosso
Come evidenziato nel paragrafo 2.2, parallelamente agli esperimenti di fotolisilaser in assorbimento visibile-UV, si è deciso di ampliare lo studio deideterminanti topologici del folding, utilizzando la spettroscopia di assorbimentoinfrarosso per caratterizzare in modo dinamico alcune proprietà funzionali estrutturali legate alla presenza di cavità nelle proteine.
Questi studi sono stati svolti in collaborazione con il Dipartimento diBiofisica dell’Università di Ulm (Germania), nel laboratorio diretto dal ProfessorUlrich Nienhaus. I campioni, costituiti da due diversi mutanti di mioglobina (siveda il paragrafo 3.6 per ulteriori dettagli), sono stati preparati dissolvendo laproteina liofilizzata ad una concentrazione pari a ~15 mM in presenza di untampone e di un crioprotettante, il glicerolo (75% glicerolo/25% fosfato dipotassio (v/v), pH 7) e riducendo successivamente il tutto mediante l’aggiunta diun eccesso di ditionito di sodio sotto un’atmosfera di CO. Su ognuno deicampioni studiati sono state eseguite, in un ampio intervallo di temperature (20 K– 300 K), misure di cinetica a seguito di un singolo impulso di laser; la variazionedel picco principale di assorbimento infrarosso dovuto alla presenza dei motivibrazionali associati al doppio legame del CO, rappresenta infatti una tecnicaclassica nello studio della dinamica della mioglobina (Austin et al., 1975, ). Inoltre,utilizzando dei protocolli sperimentali di acquisizione in gradiente di temperatura,sono state eseguite anche misure di assorbimento su larga banda, utilizzando unospettrometro per infrarosso a trasformata di Fourier (FTIR).
3.5.1 Cinetiche a singola lunghezza d’ondaNelle misure mostrate in questa tesi, un volume di circa 20 microlitri della
soluzione preparata come illustrato in precedenza, è stato depositato fra due
45
finestre di mylar di spessore pari a 75 µm, chiuse in un porta campioni a tenuta divuoto posto sull’estremità del dito freddo di un criostato a ricircolo di elioliquido. Il porta campioni risultava ruotato di 45° rispetto alla direzione dellasorgente infrarossa, fornita da un diodo laser a sale di piombo (Laser Photonics).Sulla direzione ortogonale della finestra, e su entrambe la facce, giungevasimultaneamente l’impulso della durata di 6 ns (30 mJ per pulso) di un laser astato solido Nd-YAG separato da un beam splitter, in modo da ottenere unafotolisi completa del campione. Il segnale raccolto da un rivelatore fotovoltaicoInSb (SAT Infrared Detectors), veniva trasferito ad un digitalizzatore in grado diacquisire due diverse scale di tempo simultaneamente, ovvero 20ns-400 µs e 1µs-100s.
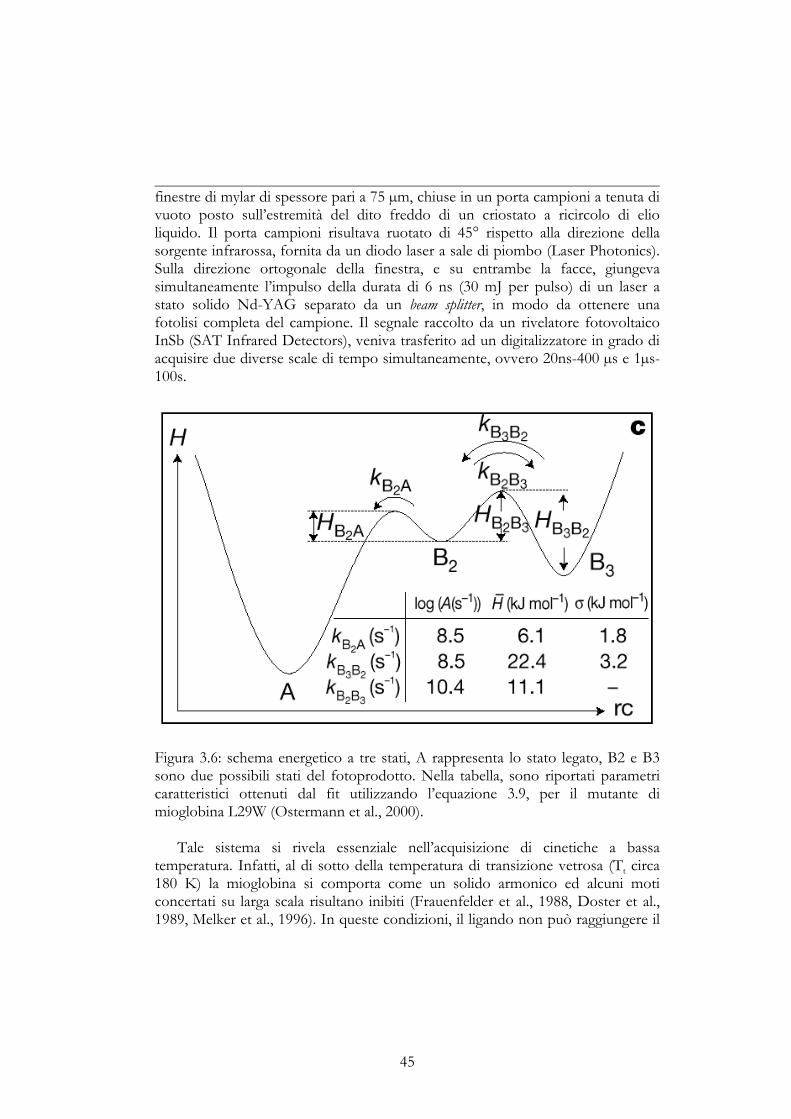
Figura 3.6: schema energetico a tre stati, A rappresenta lo stato legato, B2 e B3sono due possibili stati del fotoprodotto. Nella tabella, sono riportati parametricaratteristici ottenuti dal fit utilizzando l’equazione 3.9, per il mutante dimioglobina L29W (Ostermann et al., 2000).
Tale sistema si rivela essenziale nell’acquisizione di cinetiche a bassatemperatura. Infatti, al di sotto della temperatura di transizione vetrosa (Tt circa180 K) la mioglobina si comporta come un solido armonico ed alcuni moticoncertati su larga scala risultano inibiti (Frauenfelder et al., 1988, Doster et al.,1989, Melker et al., 1996). In queste condizioni, il ligando non può raggiungere il

46
solvente e per acquisire una seconda traccia da mediare, occorre effettuare ancheun ciclo di salita e discesa in temperatura in modo da far ricombinare il sistema eriportarlo alla condizione pre-flash. Una tipica traccia alla temperatura Tf risultaquindi dall’accumulazione della cinetica osservata dopo almeno tre impulsi dilaser, ognuno intervallato da un ciclo di riscaldamento a T> Tt e raffreddamentoa T = Tf < Tt.
L’analisi delle diverse curve ottenute in un ampio intervallo di temperature(20K – 300K) viene effettuata separando i processi al di sotto e al di sopra di Tt.In particolare, per T< Tt , il fit dei dati è stato eseguito risolvendo analiticamentel’equazione di un modello a tre stati, e calcolando globalmente le tracce a diversetemperature normalizzate in modo da fornire la frazione di molecole che ad uncerto istante t non hanno ancora ricombinato. Siano perciò lo stato A, lo statolegato e gli stati B2 e B3 due diversi stati del fotoprodotto,come evidenziato nellafigura 3.6. Immediatamente dopo la dissociazione si popola lo stato B2, il qualedecade con una cinetica governata da tre costanti principali: kB2A, kB2B3, kB3B2 (kAB2può essere trascurata in prima approssimazione). Se ciascun processo fosseesponenziale varrebbe l’equazione 3.7:
( )[ ]tNkNtN
kNtN fBBsf
ABf ∗−∗+
−∗= 23
2 expexp)( Eq. 3.7
dove Nf rappresenta la frazione che ricombina più velocemente e Ns quella piùlenta Nf /Ns = kB2A/kB2B3 e Nf + Ns = 1.
Dato che i processi osservati sono non-esponenziali, si considera unafunzione di distribuzione gaussiana delle barriere entalpiche HB2A e HB3B2, tale che
= 2
2ii1/2-2
2)H - (H-exp )(2g(Hi)
σπσ Eq 3.8
e quindi l’Eq. 3.7 diventa:
dHtNkHgNdHtN
kHgNtN fBBBBsf
ABABf ∫ ∗−+∫ −=
23232
2 exp)(exp)()( Eq 3.9
Al di sopra dei 40 K, si possono trascurare gli effetti di tunnelling quantistico ele costanti di velocità ki seguono la relazione di Arrhenius data da:

47
−=
RTHAk i
ii exp Eq. 3.10
dove Ai ed Hi rappresentano rispettivamente il fattore pre-esponenziale el’entalpia dell’i-esima barriera R è la costante dei gas e T la temperatura assoluta.
Per quanto riguarda l’analisi delle curve ottenute per T ≥ Tt, si utilizza unmetodo di calcolo differente che viene indicato con il termine di Metododell’Entropia Massima (MEM). Un descrizione approfondita di questo calcolo èpresente su Steinbach et al., 1992, mi limiterò pertanto a fornire il solo significatofisico di questa analisi. Al di sopra della temperatura di transizione anche ilsolvente diviene accessibile alla molecola del ligando, perciò l’approssimazionedel modello a tre stati diviene insufficiente. Il MEM è un metodo di calcolonumerico che estrae le costanti di velocità k relative ai diversi processi cheavvengono dopo la fotolisi, come quei valori che massimizzano l’entropia S,definita nell’accezione di Shannon – Jaynes come la funzione seguente:
−−= ∑
=
1)()(ln)(
1 j
jj
J
j FffS
λλλ Eq. 3.11
dove f(λj) rappresenta la funzione di distribuzione delle velocità che verràdeterminata numericamente, mentre F(λj), contiene le condizioni al contorno,note per ogni insieme di misure fatte. La funzione di distribuzione f(λj) e lafrazione di molecole che non ha ancora rilegato N(t) sono legati dalla seguenteequazione:
)log()(log)()(
tdtNdtNf −=λ Eq. 3.12
il processo iterativo del programma di calcolo trova la f(λ) che consente il fittingdei dati sperimentali massimizzando l’entropia S. I valori delle costanti k sono imassimi della funzione di distribuzione f(λ) per S=Smax
3.5.2 Misure in gradiente di temperaturaAccanto alle cinetiche di ricombinazione eseguite in corrispondenza del
numero d’onda del picco della banda A massimamente popolata, sono stateeseguite anche delle misure in gradiente di temperatura (Temperature DerivativeSpectroscopy, TDS). Dalla stessa soluzione preparata per le misure precedentivengono prelevati 20 microlitri e posti fra due finestre di CaF2 (diametro pari a25.4 mm) separate da uno spaziatore in mylar di 75 µm di spessore. Queste, sono

48
poi chiuse in un alloggiamento in rame ad alta conducibilità termica e poste sullaterminazione del dito freddo di un criostato a riciclo di elio (SRDK-205AW,Sumitomo), in grado di lavorare fra 3 e 320 K, inserito in uno spettrometro FTIR(IFS 66v/S, Bruker) capace di acquisire spettri di trasmissione nel medioinfrarosso fra 1800 and 2400 cm-1, ad una risoluzione di 2 cm-1. Un laser continuoal Nd-YAG, con emissione a 532 nm e potenza di 300 mW, viene usato perilluminare il campione ed è in grado di ottenere una fotolisi completa a bassatemperatura con un coefficiente kL=20 s-1.
In una misura di TDS, il campione viene perturbato dal suo stato diequilibrio, nel caso in questione da un laser, e successivamente la temperaturaviene aumentata in modo lineare con una velocità di riscaldamento di α = 5mK/s. Spettri di assorbimento sono acquisiti in modo continuo con unafrequenza di uno spettro per ogni grado Kelvin di aumento di temperatura.Vengono calcolati gli spettri differenziali ottenuti da due spettri aventitemperature successive e il risultato viene graficato in una superficie di livelli(contour plot) ottenuta riportando sulle ascisse la temperatura e sulle ordinate ilnumero d’onda e in cui la distanza fra una curva e la successiva è scalata neltempo con passo lineare (o logaritmico).
Il protocollo sperimentale è ideale per caratterizzare le proprietà cinetichedegli stati fotoeccitati termicamente attivi, che dipendono da barriere entalpichedistribuite (Berendzen & Braunstein, 1990). Infatti, dato che è possibile seguire inmodo indipendente, gli spettri di assorbimento caratteristici dello stato legato (lebande A) e dello stato fotoeccitato (le bande B), si possono realizzare differentiesperimenti volti a popolare in modo selettivo determinati tipi di stati fotoeccitati(Steinbach et al., 1991, Mourant et al., 1993 Agmon et al., 1994, Nienhaus et al.,1994). Questi differenti stati generano dei contour plot molto diversi fra loro, neiquali è possibile seguire i diversi processi di ricombinazione dovuti alle frazioni dimolecole di ligando, che a seconda dell’energia di attivazione termica chepossiedono, sono in grado di migrare all’interno della matrice proteica. Ilparallelismo dimostrato tra gli indicatori spettroscopici ottenuti con questatecnica e delle specifiche posizioni della molecola del ligando all’interno dellaproteina, rappresentano un metodo innovativo per studiare il rapporto trastruttura tridimensionale e dinamica delle proteine (Ostermann et al. 2000).
3.6 La mioglobina: proprietà e caratteristiche
La mioglobina di capodoglio è un’emoproteina largamente studiata, la cuiconoscenza approfondita dei dettagli strutturali e cinetici la rende un sistemaideale nel quale tentare di comprendere la relazione fra elementi della topologiadello stato nativo, funzione e folding. Nella figura 1.6 è mostrata la struttura

49
tridimensionale della proteina. La mioglobina è composta da 153 aminoacidi, hauna struttura globulare molto compatta formata dall’arrangiamento nello spaziodi otto catene α (identificate a partire dal residuo amino terminale dalle lettere A-H). Presenta al suo interno, in una tasca isolata dal solvente, il gruppo prosteticoeme oltre ad altre quattro cavità identificate come Xe1, Xe2, Xe3 e Xe4 per lacapacità di alloggiare degli atomi di Xenon all’interno di esse (Tilton et al., 1984).I mutanti utilizzati per lo studio mediante la spettroscopia IR del rapporto fraqueste cavità e la dinamica funzionale, sono il mutante denominato Mb-YQR.(Leu(B10) → Tyr, His(E7) → Gln e Thr(E10) → Arg) e il mutante Mb-YQRF(in cui risulta modificato l’ulteriore residuo Ile (G8) → Phe). Il primo è lo stessomutante sul quale è stato possibile intrappolare il fotoprodotto a bassatemperatura e verificare che la molecola di CO risultava occupare il sito indicatocome Xe4 (Brunori et al., 2000). Il secondo è stato realizzato specificamente permodificare il processo di migrazione del ligando all’interno della matrice proteica,inserendo un residuo di maggiore ingombro sterico, la fenilalanina, allo scopo diverificare la possibilità di modulare il percorso mediante il quale il ligando stessoraggiunge il solvente.
Entrambi i mutanti sono stati sintetizzati e purificati nel laboratorio delDipartimento di Scienze Biochimiche di Roma diretto dal Professor MaurizioBrunori, seguendo la metodologia indicata in Travaglini-Allocatelli et. al, 1994. Icampioni sono stati successivamente liofilizzati e trasportati in Germania, dovesono stati preparati seguendo la procedura indicata nel paragrafo precedente.

50

51
Capitolo 4
Risultati e Discussione
4.1 Premessa
I risultati ottenuti nello studio del citocromo c saranno trattati e discussiseparatamente da quelli ottenuti nello studio della mioglobina; questa scelta èstata operata allo scopo di chiarire i diversi problemi che sono stati affrontati,dato che i sistemi studiati sono differenti, come differenti sono le tecnichesperimentali scelte per cercare di evidenziare le proprietà dinamiche di possibilideterminanti topologici del folding. Questa apparente dicotomia sarà chiarita nelcapitolo conclusivo, nel quale mostreremo il punto di convergenza di questi duelavori paralleli.
4.2 Il citocromo c in condizioni denaturanti
Allo scopo di caratterizzare gli intermedi precoci del folding del citocromoc551, mediante l’approccio sperimentale elaborato da Eaton e dai suoicollaboratori, (Jones et al., 1993) è stato per prima cosa studiato il processo didenaturazione all’equilibrio per questa proteina batterica nella sua forma ridotta enella sua forma ridotta in presenza di CO (vedi figura 3.6).
Il primo dato che emerge è che il citocromo c551 risulta meno stabile delcitocromo di cavallo e di conseguenza il processo di unfolding si realizza su di unintervallo di concentrazione del denaturante ridotto. Questo fatto produce ancheuna minore separazione fra la curva all’equilibrio con il CO e quella senza illigando. Perciò, una condizione favorevole esplorata per il citocromo c551corrisponde al caso di [Gdn]=2M, in tampone fosfato a pH = 7, T=25 °C e[CO]= 1 mM. L’esperimento condotto in queste condizioni riproduce quellocondotto sul citocromo di cavallo a [Gdn]=4.6M, T=40 °C e [CO]= 1 mM;ovvero in entrambi i casi la condizione pre-flash corrisponde ad una speciedenaturata per almeno il 90% delle molecole, mentre a seguito della rimozione delligando, la specie più stabile è quella nativa. Il risultato dell’esperimento di fotolisinelle condizioni appena citate, per il citocromo c551 è mostrato nella figura 4.1.Nel pannello A è riportato un grafico tridimensionale che mostra la variazionedello spettro differenziale in funzione del tempo, mentre nel pannello B sonomostrati gli andamenti temporali delle prime due colonne della matrice V,ottenuta da un’analisi mediante SVD dei dati mostrati nel pannello A. Il minimo

modello capace di descrivere in modo esauriente i dati è stato ottenuto mediantela somma di 3 esponenziali.
52
D
CA

53
Figura 4.1: Pannelli A,C,E: esperimenti di laser fotolisi sul citocromo c551 a T=25°C, pH=7[CO]=1 atm in presenza di guanidina alle concentrazioni rispettive di [Gdn]=2,3,4 M;pannelli B,D,F le corrispondenti colonne v1 (cerchi blu) e v2 (stelle rosse) di V ed i relativi fit(linee continue).
Questo risultato sembrava incoraggiante in quanto il citocromo di cavallomostra almeno quattro fasi temporalmente risolte: dunque questo dato siaccordava con l’ipotesi che, in assenza di istidine in grado di comportarsi datrappole cinetiche, il citocromo c551 presenti una cinetica semplificata. Allo scopodi assegnare in maniera univoca le fasi associate alla ricombinazione del CO edistinguerle dalle fasi direttamente correlate con il processo di collasso ocomunque di coordinazione distale, lo stesso esperimento è stato ripetuto incondizioni completamente denaturanti, ovvero per [Gdn]=3, 4, 5 e 6 M. Nella
log t (s)
log t (s)λ (nm)
Col
onne
di V
(u.a
.)E F

54
figura 4.1 (pannelli C-F), sono riportati i casi corrispondenti a [Gdn]= 3 e 4 M.Come è facile osservare, vi è un incremento del segnale differenziale che produceun migliore rapporto segnale/rumore, tale da consentire a partire dal caso di[Gdn]= 4 M di riuscire ad apprezzare una ulteriore fase molto veloce ed aventeuna ampiezza molto piccola. Nella tabella 4.1, sono riportati i valori ottenuti per ilfit con 3 e 4 esponenziali a seconda della concentrazione di guanidina presenteper il citocromo c551.
Tabella 4.1
[Gdn] Fit per una somma di 3 esponenzialiτ 1 τ 2 τ 3
2 M 300 µs 1 ms 7 ms3 M 120 µs 800 µs 7 ms
Fit per una somma di 4 esponenzialiτ 1 τ 2 τ 3 τ4
4 M 5 µs 100 µs 900 µs 6 ms5 M 6 µs 340 µs 1 ms 7 ms6 M 10 µs 620 µs 2 ms 20 ms
Il tempo caratteristico di una reazione è definito come τ=t1/e=1/k
Il risultato ottenuto alle più elevate concentrazioni di denaturante èinaspettato: infatti, poiché la proteina è denaturata sia in presenza che in assenzadi ligando (fig. 3.6), ci si aspetterebbe che la rimozione fotochimica del CO nondebba essere seguita altro che dalla sua ricombinazione, in una cineticabimolecolare semplice che non popola intermedi (ed in particolare intermedi difolding). La complessità del fenomeno osservato è difficilmente interpretabile allaluce del modello cinetico proposto da Eaton e dai suoi collaboratori nel lavorooriginale (Jones et al., 1993), e in sviluppi successivi (Hagen et al., 1997).
Questo modello, che definiremo da ora in poi come il meccanismo dicoordinazione/miscoordinazione, assegna a determinati processi chimici le fasicinetiche calcolate e può essere schematizzato come segue:
Schema 4.1a b c
X – Fe + COX – Fe – CO X – Fe – Y + COhν

55
Secondo il modello della coordinazione/miscoordinazione, la specie dipartenza (a) presenta una geometria nella quale il monossido di carbonio siaffaccia in posizione distale, mentre in posizione prossimale è sempre presente ilresiduo di istidina (X). A seguito dell’impulso di luce laser (hν), viene popolata laspecie pentacoordinata (b), la quale può coordinare transientemente nel sitodistale un ligando intramolecolare (Y) dando luogo alla specie (c). Se essocorrisponde ad un residuo di istidina (nel citocromo c di cavallo) tale intermedio èimproduttivo da un punto di vista del folding, mentre se coordina un residuo dimetionina può andare incontro ad un processo di rinaturazione transiente primache il CO si leghi nuovamente, riportando la molecola alla condizione pre-flash.Nel citocromo c di cavallo, nelle condizioni di massima differenza della curva diequilibrio (ovvero [Gdn]=4.6 M, T=40°C, [CO]=1 atm), le quattro fasi cinetichepresentano i seguenti tempi caratteristici τ=2 µs, 60 µs, 500 µs e 7 ms. Il modellodi coordinazione/miscoordinazione assegna al legame di una metionina allaspecie (b), la fase con τ=2 µs (sia essa la metionina nativa M80, o la suapossibile antagonista M65). La fase con τ=60 µs è associata al legame della specie(b) con una delle due istidine H26 e H33 presenti nella catena polipeptidica,mentre la fase con τ= 500 µs corrisponde al legame bimolecolare del CO. La fasepiù lenta, τ=7 ms, rappresenterebbe quella quota di proteina che, avendo legatola metionina nativa, è andata incontro ad un processo di folding parziale e diconseguenza rilega il CO molto lentamente in quanto questo ligando devespiazzare il residuo distale nativo e riportare la molecola alla condizionedenaturata.
Secondo questa interpretazione, se si esegue un esperimento in condizionicompletamente denaturanti, il sistema dovrebbe presentare solo unaricombinazione del CO, eventualmente in competizione con la coordinazionetransiente di qualche residuo di istidina o metionina, ma dovrebbe avere abolitoper lo meno la fase più lenta. Questa predizione è falsa, in quanto sia per ilcitocromo di cavallo (i dati acquisiti a [Gdn]=8 M non sono mostrati) che per ilcitocromo c551, gli esperimenti hanno mostrato che in nessun caso scompare lafase più lenta a τ ≅ 7 ms (si vedano i pannelli E e F per il caso del citocromo c551).4.3 Dalla proteina ai sistemi modello
4.3.1 Considerazioni cinetichePoichè la spettroscopia di assorbimento ottico nella regione del Soret è
sensibile alle transizioni elettroniche a carico del gruppo prostetico eme, occorredeterminare se lo schema 4.1 non sia insufficiente a descrivere i dati in quantotrascura alcune reazioni caratteristiche che possono avere luogo fra il ferrodell’eme ed i suoi ligandi. È noto infatti che, dissolvendo l’eme in solventiorganici o in micelle, è possibile studiare la sua reattività al variare di ligandi

56
aggiunti senza interferenze da parte della matrice proteica. In particolare, è statodimostrato che, in presenza di Imidazolo (Im) o 2-metil imidazolo (MeIm), l’emelega il CO con più alta affinità che in assenza di questi ligandi dando luogo ad uncomplesso esacoordinato del tipo (Im o MeIm)-emeFe-CO; e viceversa anche laforza del legame nella configurazione trans di ognuna di queste basi organicheaumenta in presenza del CO (Rougee & Brault, 1975, Momentau et al., 1976). Acausa di questa relazione fra il CO ed il ligando prossimale del ferro dell’eme,quando si illumina il complesso (Im o MeIm)-emeFe-CO, si ha la rottura dellegame fotolabile emeFe-CO, e la ricombinazione di quest’ultimo procedeattraverso un meccanismo definito “a eliminazione della base” (White et al.,1979), rappresentato nello schema seguente:
Schema 4.2a b
d c
Nello schema 4.2, X indica la base prossimale (sia Im o MeIm) ed èformalmente analogo all’istidina prossimale dello schema 4.1. A causa dell’affinitàridotta per la base in assenza di CO, la specie (b), dissocia facilmente producendola specie (c), in cui il ferro tetracoordinato reagisce velocemente per dare luogoad una specie (d), la quale, legando nella quinta posizione di coordinazione unamolecola di CO e successivamente rilegando la base X restituisce la specie diequilibrio (a). Questo schema produce tre intermedi di reazione, duepentacoordinati ed uno tetracoordinato; quest’ultimo essendo estremamentereattivo non si accumula ma, a causa della maggiore reattività nei confronti delCO, fa si che, a partire dalla specie (b), il ciclo sia percorso con maggiore efficaciain senso orario. Il meccanismo di eliminazione della base prevede che laricombinazione del CO sia seguita da quella della base e non costituisca quindil’ultimo evento cinetico osservabile; l’importanza di questa precisazione saràchiarita fra breve.
Si noti inoltre che il meccanismo qui descritto opera efficacemente nel casodel CO, che è poco reattivo nei confronti del ferro eminico pentacoordinato epresumibilmente con minore efficacia nel caso di ligandi più reattivi.
4.3.2 I sistemi modello
X – Fe + COX – Fe – CO
X + Fe – CO X + Fe + CO
hν

57
Per stabilire se anche nel caso del citocromo in condizioni denaturanti,potesse agire un meccanismo analogo a quello descritto nello schema 4.2, sonostati realizzati una serie di esperimenti su alcuni sistemi modello, in parallelo conil citocromo c di cavallo e il citocromo c551.
Un punto di partenza è stata l’osservazione che, denaturando il citocromo dientrambe le specie con alte concentrazioni di guanidina od urea, la formadenaturata e ridotta non presentava uno spettro di assorbimento caratteristico diuna specie pentacoordinata. In particolare, nella regione delle bande Q delvisibile, lo spettro non mostrava un unico picco, caratteristico di una speciepentacoordinata bensì due picchi. Dato che, sia la specie tetracoordinata che laspecie esacoordinata presentano due picchi in questa regione dello spettro,mentre la specie pentacoordinata uno solo, risulta estremamente probabile chetutte e tre le specie siano presenti nella soluzione in condizioni denaturanti.Ancora più sorprendente è lo spettro ottico della microperossidasi-11, un peptideformato da 11 residui ottenuti dalla digestione proteolitica del citocromo dicavallo, in cui è presente l’eme legato covalentemente e il solo residuo di istidinaprossimale coordinato con l’atomo di ferro; sono assenti sia la metionina nativasia altri residui di istidina in grado di coordinare il ferro in posizione distale.Nonostante ciò lo spettro della specie ridotta presenta due picchi nella regionedel visibile, dovuti probabilmente alla presenza anche in questo caso di diversistati di coordinazione per il gruppo prostetico, eventualmente provocati dafenomeni di aggregazione in cui può incorrere la microperossidasi (Jehanly et al.,1976, Laberge et al., 1998).
Volendo ottenere degli spettri modello per i differenti stati di coordinazionedell’eme, in presenza di diversi tipi di ligando, è stato disciolto il gruppoprostetico in una soluzione di SDS al 5% e sono stati acquisiti gli spettri otticimostrati nella figura 4.2. Si noti in particolare che negli spettri mostrati nelpannello inferiore esiste una specie data dall’eme in presenza di CO 1mM che èesacoordinata avendo probabilmente legata in posizione distale una molecola diacqua; l’importanza dello spettro di questa specie sarà chiarito in seguito(paragrafo 4.3.4)

58
0
0.5
1
1.5
2
2.5
350 400 450 500 550 600 650
Abs
wavelength (nm)
0
0.5
1
1.5
2
2.5
Abs
Figura 4.2: Pannello superiore: spettri di assorbimento ottico dell’eme da solo (specietetracoordinata linea continua), in presenza di 0.1M 2-metil imidazolo (specie pentacoordinatalinea per punti) e in presenza di 0.2 M imidazolo (specie esacoordinata linea a tratti). Pannelloinferiore: eme in presenza di CO 1mM (specie esacoordinata con una molecola di acqua inposizione prossimale, linea per punti) e eme in presenza di CO 1mM e 2-metil imidazolo 0.1M (specie esacoordinata con una base prossimale linea continua).
4.3.3 Esperimenti mirati a singola lunghezza d’ondaDall’analisi degli spettri della figura 4.2 si evidenzia che esistono alcune
lunghezze d’onda caratteristiche alle quali il processo di ricombinazione del COed il riarrangiamento tra la specie tetracoordinata e quella pentacoordinata hannovariazioni spettrali opposte e di conseguenza rappresentano degli indicatori idealiove seguire questo processo cinetico. A questo scopo, utilizzando il sistema diacquisizione a singola lunghezza d’onda (paragrafo 3.3), sono stati realizzati alcuniesperimenti specifici in parallelo per i sistemi modello e per i citocromi allalunghezza d’onda λ=406 nm (figura 4.3).
Abs x 5
Abs x 5
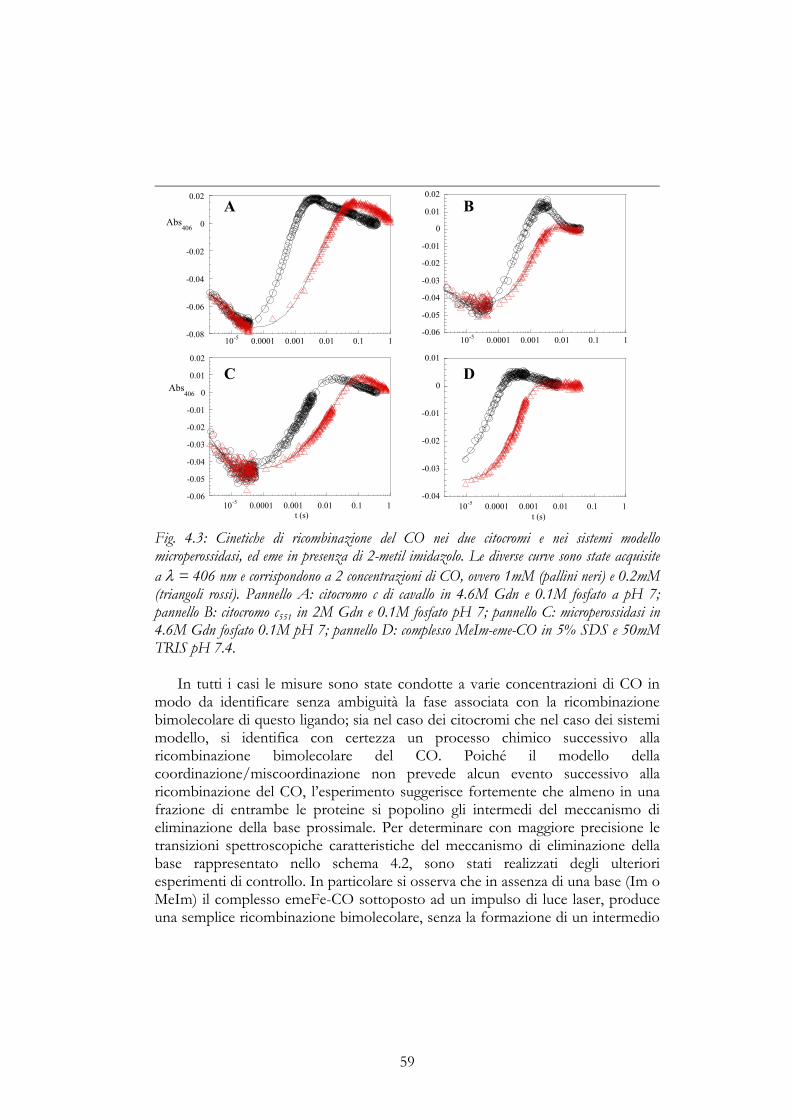
59
-0.08
-0.06
-0.04
-0.02
0
0.02
10-5 0.0001 0.001 0.01 0.1 1
Abs406
-0.06
-0.05
-0.04
-0.03
-0.02
-0.01
0
0.01
0.02
10-5 0.0001 0.001 0.01 0.1 1
-0.06
-0.05
-0.04
-0.03
-0.02
-0.01
0
0.01
0.02
10-5 0.0001 0.001 0.01 0.1 1t (s)
Abs406
10-5 0.0001 0.001 0.01 0.1 1-0.04
-0.03
-0.02
-0.01
0
0.01
t (s)
Fig. 4.3: Cinetiche di ricombinazione del CO nei due citocromi e nei sistemi modellomicroperossidasi, ed eme in presenza di 2-metil imidazolo. Le diverse curve sono state acquisitea λ = 406 nm e corrispondono a 2 concentrazioni di CO, ovvero 1mM (pallini neri) e 0.2mM(triangoli rossi). Pannello A: citocromo c di cavallo in 4.6M Gdn e 0.1M fosfato a pH 7;pannello B: citocromo c551 in 2M Gdn e 0.1M fosfato pH 7; pannello C: microperossidasi in4.6M Gdn fosfato 0.1M pH 7; pannello D: complesso MeIm-eme-CO in 5% SDS e 50mMTRIS pH 7.4.
In tutti i casi le misure sono state condotte a varie concentrazioni di CO inmodo da identificare senza ambiguità la fase associata con la ricombinazionebimolecolare di questo ligando; sia nel caso dei citocromi che nel caso dei sistemimodello, si identifica con certezza un processo chimico successivo allaricombinazione bimolecolare del CO. Poiché il modello dellacoordinazione/miscoordinazione non prevede alcun evento successivo allaricombinazione del CO, l’esperimento suggerisce fortemente che almeno in unafrazione di entrambe le proteine si popolino gli intermedi del meccanismo dieliminazione della base prossimale. Per determinare con maggiore precisione letransizioni spettroscopiche caratteristiche del meccanismo di eliminazione dellabase rappresentato nello schema 4.2, sono stati realizzati degli ulterioriesperimenti di controllo. In particolare si osserva che in assenza di una base (Im oMeIm) il complesso emeFe-CO sottoposto ad un impulso di luce laser, produceuna semplice ricombinazione bimolecolare, senza la formazione di un intermedio
A
DC
B

60
alla lunghezza d’onda caratteristica di λ=406 nm (figura 4.4 pannello A). Lacostante di secondo ordine calcolata è pari a k=2.8x108 s-1 M-1. Tale valore è inottimo accordo con quello determinato precedentemente per la coordinazione delCO ad un eme tetracoordinato (White et al., 1979).
-0.15
-0.1
-0.05
0
10-5 0.0001 0.001
Abs406
-0.08
-0.06
-0.04
-0.02
0
0.02
10-5 0.0001 0.001 0.01 0.1
10-5 0.0001 0.001 0.01 0.1-0.04
-0.03
-0.02
-0.01
0
0.01
t (s)
Abs406
10-5 0.0001 0.001 0.01 0.1 1-0.04
-0.03
-0.02
-0.01
0
t (s)
Figura 4.4: Cinetiche di ricombinazione del CO nel caso dell’eme in presenza di diverseconcentrazioni di Im o MeIm. Le diverse curve sono state acquisite a λ = 406 nm ecorrispondono a 2 concentrazioni di CO, ovvero 1mM (pallini neri) e 0.2mM (triangoli rossi).Pannello A: nessuna base aggiunta; pannello B 25 mM MeIm, pannello C 100mM MeIm,pannello D 200 mM Im.
Viceversa introducendo il 2-metil imidazolo 25 mM, il processo rallenta e siha la formazione di un intermedio (figura 4.4 pannello B). Aumentando laconcentrazione del MeIm a 100 mM il processo rallenta ulteriormente, ma anchel’ampiezza dell’intermedio diminuisce (figura 4.4 pannello C). Per far scomparirela fase cinetica lenta è però necessario introdurre un’elevata concentrazione dellabase a più alta affinità ovvero 200 mM imidazolo (figura 4.4 pannello D). Perciòla formazione dell’intermedio che decade nell’ultima fase dipende dallaconcentrazione della base aggiunta ed è trascurabile per valori elevati o in assenzadi essa, mentre ha il suo massimo per valori intermedi di MeIm (o Im).
D
B
C
A

61
Questo comportamento è in accordo con il meccanismo cinetico dieliminazione della base mostrato nello schema 4.2, in quanto mostra che, se laconcentrazione e l’affinità della base sono tali da saturare il ferro dell’eme anchein assenza di CO, la specie pentacoordinata X-emeFe, non riequilibra con laspecie tetracoordinata e la ricombinazione del CO dopo la fotolisi procedelentamente. Viceversa, in assenza di base il processo di ricombinazione divienemolto più veloce poiché a seguito dell’impulso di laser si popola integralmente lostato tetracoordinato, molto più reattivo. Alle concentrazioni intermedie lafotolisi del CO è associata ad una parziale dissociazione del MeIm (o Im); diconseguenza, la ricombinazione del CO stesso coinvolge in modo sostanziale laspecie tetracoordinata e si ha la formazione di una specie transientepentacoordinata legata al CO che, lentamente, rilega la base per dare la specie diequilibrio MeIm (o Im) – emeFe-CO.
Il processo cinetico più lento (τ ≅ 7 ms) osservato nei due citocromi esisteanche in sistemi modello quali la microperossidasi, ed il complesso MeIm-emeFe-CO. Tale processo è presente anche in condizioni completamente denaturanti(ovvero [Gdn]= 8 M per il citocromo di cavallo e [Gdn] =3-6 M per il citocromoc551); di conseguenza questo processo è caratteristico del gruppo prostetico eme epuò essere spiegato all’interno del meccanismo di eliminazione della base (schema4.2) e non ha alcuna connessione diretta con il folding del citocromo. Il modellodella ccordinazione/miscoordinazione che assegna questo processo allaricombinazione del CO con specie rinaturate del citocromo c , è pertantoinsoddisfacente.
Inoltre, appare chiaro che il sistema MeIm-emeFe-CO è un ottimo candidatoper mimare le proprietà intrinseche connesse con la ricombinazione del CO alcitocromo c in condizioni denaturanti. In particolare, volendo assegnare anche levariazioni spettroscopiche connesse con i processi rapidi del meccanismo dieliminazione della base è stato realizzato l’esperimento mostrato in figura 4.5.Mantenendo il CO costante e pari a 1 mM è stata variata la concentrazione diMeIm nell’intervallo 0-100 mM; sono state scelte due lunghezze d’ondacaratteristiche, λ =436 nm e λ=406 nm e sono state seguite con particolareattenzione le fasi che precedono il processo bimolecolare di ricombinazione delCO.
Al variare della concentrazione del MeIm appaiono due fasi rapide le qualisono entrambe presenti a [MeIm]=20mM (curva con quadrati verdi nel grafico4.5) ed hanno le seguenti costanti di tempo τ = 300 ns e τ = 6 µs.

62
-0.05
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
10-8 10-7 10-6 10-5 0.0001t (s)
Abs436nm
A
-0.25
-0.2
-0.15
-0.1
-0.05
0
0.05
10-8 10-7 10-6 10-5 0.0001 0.001 0.01t (s)
Abs406nm
B
Figura 4.5: Evoluzione temporale della ricombinazione del CO al complesso eme disciolto in5% SDS and 50 mM TRIS pH 7.4, acquisito a λ = 436 nm (pannello A) and λ = 406nm (pannello B) in funzione della concentrazione del 2-metil imidazolo. Cerchi neri: senzaaggiunta di base; triangoli rossi: 5 mM MeIm; quadrati verdi: 20 mM MeIm; rombi azzurri:100 mM MeIm
La fase più rapida (τ = 300 ns) deve probabilmente essere assegnata alriarrangiamento fra la specie pentacoordinata e la specie tetracoordinata cherappresenta il passaggio fra la specie (b) e la specie (c) del meccanismo descrittonello schema 4.2. La fase successiva (τ = 6 µs), è di secondo ordine rispetto allaconcentrazione di CO e rappresenta il legame del suddetto ligando alla specietetracoordinata (passaggio dalla specie (c) alla specie (d) nello schema 4.2). Dopoil processo cinetico con τ = 6 µs la concentrazione dell’intermediotetracoordinato rimane molto bassa e si osserva la scomparsa della specie (b)

63
dello schema 4.2 attraverso i due percorsi alternativi disponibili ( (b)→ (a)oppure (b) → (c) → (d) → (a) ).
Infine, volendo osservare l’effetto del MeIm sui processi cinetici successivialla fotolisi del CO nel citocromo c denaturato è stato realizzato l’esperimentomostrato nella figura 4.6. Il citocromo scelto è il citocromo c di cavallo e lecondizioni sperimentali sono:[Gdn]= 5 M, T=25°C, pH=7, [CO]=1mM.

64
-0.1
-0.08
-0.06
-0.04
-0.02
0
0.02
10-6 10-5 0.0001 0.001 0.01 0.1 1t (s)
Abs406nm

65
Figura 4.6: Evoluzione temporale della ricombinazione del CO a λ=406 nm nel citocromo cdi cavallo in 5M Gdn, pH 7 (pallini neri); in presenza di 1 mM MeIm (triangoli rossi), 50mM MeIm (quadrati verdi) e 250mM MeIm, (rombi azzurri)
In accordo con quanto mostrato per il gruppo prostetico in presenza di MeIme CO, anche nel caso del citocromo c di cavallo si ha che l’intermedio chescompare con il processo a τ ≅ 7ms non si accumula se l’esperimento è compiutoa concentrazioni di MeIm pari o superiori a 20 mM. A concentrazioni più bassel’intermedio si forma e la sua popolazione massima si ha in assenza di MeIm.Come osservato per l’eme disciolto in micelle, l’aggiunta di MeIm rallenta anchela ricombinazione bimolecolare del CO e a concentrazioni elevate, (250 mM,rombi azzurri nella figura 4.6) è in grado di interferire anche con le fasi rapide delprocesso di evoluzione temporale che segue la fotolisi. Anche questo esperimentoè compatibile con il modello cinetico dell’eliminazione della base prossimale;infatti l’aggiunta di MeIm mantiene il Fe eminico del citocromo cpentacoordinato dopo la fotolisi del CO (nelle due forme alternative His-emeFe oMeIm-emeFe), rallentando di conseguenza la ricombinazione del CO edeliminando gli intermedi (c) e (d) dello schema 4.2.
4.3.4. Un estensione del modelloI risultati ottenuti consentono di affermare che gli esperimenti condotti sul
citocromo c di cavallo e sul citocromo c551 utilizzando la fotolisi del CO perindurre un collasso transiente di queste proteine, hanno dimostrato l’esistenza diintermedi di reazione che non hanno una diretta correlazione con il folding masono dovuti a reazioni intrinseche dell’eme. I nostri dati spiegano inoltre ladiscrepanza fra le significative variazioni di assorbimento ottico che seguono lafotolisi del CO nel caso del citocromo c nelle condizioni opportune e le modesteriposte di indicatori specifici del riarrangiamento terziario della proteina quali lafluorescenza (Chan et al., 1996) e il dicroismo circolare (Chen et al., 1998). Diconseguenza, si possono escludere processi di folding parziale o di significativocollasso della proteina, mentre, specialmente nel caso del citocromo c di cavallonel quale sono presenti diversi residui di metionina e di istidina, si puòimmaginare che agisca un meccanismo chimico costituito da un’estensione delmeccanismo ad eliminazione della base, nel quale vengano popolatitransientemente degli stati denaturati che hanno una coordinazione distale di tipointramolecolare.
Inoltre, alcune osservazioni sperimentali (Jones et al., 1993) mostrano che nelcitocromo di cavallo il CO è in grado di legarsi in condizioni di bassaconcentrazione di denaturante, formando una specie “nativa”. Questa specie hauna struttura tridimensionale collassata e presenta gli spettri di fluorescenza e

66
dicroismo circolare caratteristici del citocromo nativo ma in essa il ferro dell’emecoordina una molecola di CO al posto della metionina distale. Se sottoposta afotolisi, questa specie si comporta come la mioglobina, ovvero ricombina illigando fotodissociato in un processo semplice di secondo ordine. Aconcentrazioni intermedie di denaturante (2.5 –3.5 M per il citocromo di cavallo),sono perciò presenti due diverse popolazioni che hanno il CO legato nella sestaposizione di coordinazione, una avente una struttura terziaria nativa ed unadenaturata. Perciò entrambe vengono normalmente eccitate negli esperimenti difotolisi e di conseguenza complicano ulteriormente il modello. Il meccanismocinetico generale da noi proposto è pertanto il seguente:
Schema 4.3
Dove U o N rappresentano lo stato del riarrangiamento terziario dellaproteina sia esso denaturato (U unfolded) o nativo (N), il numero in pedice indicalo stato di coordinazione del ferro dell’eme e la presenza del suffisso CO indica lapresenza di questo ligando nella posizione di coordinazione distale. La specie (a),può essere costituita interamente da citocromo c denaturato oppure da unamiscela contenente anche citocromo c nativo ma avente legato nella sestaposizione di coordinazione il CO; entrambe le specie sono fotosensibili. Aseconda della specie chimica di partenza U6CO o N6CO, l’evoluzione temporalesuccessiva alla fotolisi procede in modo completamente diverso. Nello schema4.3 sono stati perciò evidenziati graficamente in rosso i percorsi a carico dellaspecie N6CO mentre in nero quelli relativi a U6CO. Partiamo con il descrivere iprocessi cinetici a carico di quest’ultima specie. A seguito del flash del laser, sipopola la specie pentacoordinata U5 che reagisce lentamente con il CO (White etal., 1979), ma a causa della mancanza dei vincoli strutturali imposti dal fold dellaproteina, è in rapido equilibrio con una specie tetracoordinata U4 ((b) ↔ (c)).
U5 COa
b
d
c
e
f
hν
U6 CO + N6 CO
U5 + COU4 + CO U6
N5 + COhν

67
Quest’ultima, non si accumula significativamente e decade rapidamente con unprocesso di secondo ordine rispetto al CO, verso la specie pentacoordinata U5CO(d). Questa specie si accumula formando l’intermedio che decade lentamente (τ ≅7ms) mediante la coordinazione del ligando prossimale. La specie (f) si comportada trappola cinetica per questo meccanismo, in quanto a seguito dellacoordinazione distale di un ligando intramolecolare, quale un residuo dimetionina o istidina, può dare luogo a partire dallo stato U5 ad uno statoesacoordinato U6 che può accumularsi significativamente o meno, a seconda dellecondizioni di concentrazione di denaturante. Viceversa, la specie N6CO presentesolo a basse concentrazioni di guanidina, segue un percorso cinetico semplificatoperchè grazie alla favorevole disposizione spaziale dell’istidina prossimale non vaincontro ad un meccanismo ad eliminazione della base e popola uno stato N5, (laspecie (e)), il quale decade con un semplice processo di secondo ordine rispetto alCO alla specie di partenza.
L’analisi dei dati alla luce del modello presentato nello schema 4.3, consentedi generalizzare il comportamento del citocromo c e di spiegare l’intermediolento, il quale è presente ad ogni concentrazione di denaturante ed era statoerroneamente assegnato ad una proteina transientemente collassata. Inoltre loschema 4.3 descrive in modo esaustivo il comportamento del citocromo c incondizioni di basso denaturante, dove si ha la comparsa della specie N6CO.Infatti, consideriamo ad esempio l’esperimento mostrato nei due pannelli A e Bdella figura 4.1, che si riferiscono al caso del citocromo c551 in [Gdn]=2M,[CO]=1mM e pH 7. Le componenti v1 e v2 di V mostrate nel pannello B sonostate analizzate mediante un procedimento globale di minimizzazione, e hannofornito le tre costanti cinetiche riportate nella tabella 4.1; dall’ampiezza dellecomponenti ottenute mediante il fit è possibile ricostruire con il procedimentoillustrato nel paragrafo 3.1, gli spettri differenziali associati alle tre transizionievidenziate.

68
350 400 450 500-0.1
-0.08
-0.06
-0.04
-0.02
0
0.02
0.04
0.06
350 400 450 500-0.1
-0.08
-0.06
-0.04
-0.02
0
0.02
0.04
0.06
Figura 4.7: Pannello A: spettri differenziali ricostruiti per le specie corrispondenti alle trediverse fasi determinate nell’esperimento di fotolisi su citocromo c551 in [Gdn]=2M[CO]=1mM pH 7. Curva blu, differenziale associato a τ =100 µs, curva verde differenzialeassociato a τ =1 ms curva rossa differenziale associato a τ =7 ms. Pannello B: curva nera è lasomma di rossa e blu del pannello A, la verde è la stessa del pannello precedente.
Nella figura 4.7 pannello A, sono riportati i tre spettri differenziali ottenuti; inparticolare lo spettro blu si riferisce alla fase avente un τ =100 µs, quello verdealla fase successiva avente τ =1 ms ed infine quello rosso al processo più lentocon τ =7 ms. Alla luce del modello proposto, la fase rapida corrisponde allascomparsa della specie U5, attraverso un meccanismo analogo a quello dieliminazione della base che porta all’accumulazione di una specie U5CO; poichéin queste condizioni l’intermedio tetracoordinato non si accumula il primodifferenziale corrisponde alla conversione U5 → U5CO. Il processo più lento èassegnato alla scomparsa di questa specie e rappresenta pertanto il differenzialeU5CO-U6CO. La fase intermedia risulta invece dal percorso parallelo condottodalla specie N5 che si popola dopo l’impulso di laser e corrisponde pertanto aldifferenziale N5- N6CO. Sommando la curva blu e quella verde si ottienecorrettamente lo spettro differenziale dato dalla somma dei due processi, ovvero:
U5 - U5CO + U5CO - U6CO = U5 - U6CO Eq. 4.1
λ (nm) λ (nm)
∆Abs ∆Abs
A B

69
Nel pannello B della figura 4.7 sono riportati in verde lo stesso spettrodifferenziale riportato nel pannello A e assegnato alla transizione N5-N6CO ed innero la somma delle curve blu e rossa del pannello A che rappresentano latransizione evidenziata dall’equazione 4.1, ovvero U5-U6CO. Come appareevidente le due curve sono praticamente coincidenti, a testimonianza del fatto chedal punto di vista dello spettro di assorbimento dell’eme gli stati N6CO e U6CO,sono molto simili (come simili sono i rispettivi stati fotolizzati), mentre bendiverso è il processo cinetico che compete alle due specie dopo la fotolisi.
4.4 Il rapporto tra topologia e folding nel citocromo c551
Da un punto di vista della topologia dello stato nativo, gli esperimentimostrati nei paragrafi precedenti, sembrano assegnare al legame Fe-Metioninadistale un ruolo secondario nel meccanismo di rinaturazione dei due citocromi edin particolare nel citocromo c551 ridotto. Infatti il presupposto che gli esperimentidi fotolisi del CO in condizioni denaturanti dovessero produrre un intermedioefficace di folding si basava sull’assunzione che tale legame fosse anche da unpunto di vista cinetico un determinante del processo di formazione della strutturanativa.
4.4.1 Esperimenti di mescolamento a flusso interrottoPer verificare la validità di questa affermazione è stato realizzato un
esperimento di rinaturazione utilizzando l’apparato classico di mescolamento aflusso interrotto (stopped flow), con un protocollo di misura studiato specificamenteper caratterizzare in fluorescenza il processo di folding del citocromo c551 ridotto.Infatti, una difficoltà sperimentale che si incontra quando si lavora con la specieridotta e denaturata di un generico citocromo, è che per mantenere lo stato diossidazione +2 del Fe occorre porre in soluzione un largo eccesso di ditionito disodio. Dato però che lo spettro di assorbimento del ditionito presenta un largopicco nella regione di emissione del triptofano (fra 320 e 360 nm), ciòvirtualmente impedisce di seguire l’evoluzione temporale del picco difluorescenza presente nella specie denaturata U. Una soluzione strumentale èstata realizzata mediante l’utilizzo di camere di misura di volume ridotto (cammino ottico < 1mm), accompagnate da un aumento delle concentrazioni dilavoro fino a valori dell’ordine di 40-50 µM dopo mescolamento (Bhuyan et al.,2001). Tale sistema anche se ingegnoso, presenta lo svantaggio di non potereescludere la formazione di aggregati nel processo di rinaturazione a causa deglielevati valori di concentrazione di proteina usati. Viceversa, è stato messo a puntoun protocollo diverso che consente di lavorare con le concentrazioni usuali di

70
proteina dopo il mescolamento, che sono dell’ordine di 2-4 µM. Tale protocolloprevede l’introduzione nella siringa di spinta di volume ridotto, di una soluzionecontenente il citocromo c551 a pH 7 in presenza di guanidina (o urea) ad altaconcentrazione, tale da garantire che la proteina sia denaturata. Inoltre per unaconcentrazione di proteina di 20-40 µM è presente anche una concentrazione di10 mM ascorbato e 1 mM CO. L’ascorbato da solo non è in grado di mantenereridotta la specie denaturata, mentre può ridurre facilmente la proteina nativa. Lacontemporanea presenza di ascorbato e monossido di carbonio porta allaformazione della specie ridotta denaturata e avente il CO legato in posizionedistale U6CO. Mescolando questa soluzione contro l’altra siringa di spinta divolume molto maggiore (10:1) contenente il tampone in presenza di ascorbato econcentrazioni differenti di guanidina, è possibile seguire il processo dirinaturazione in funzione della concentrazione di denaturante. Allo scopo diridurre al minimo la contaminazione da O2, tutte le soluzioni sono stateprecedentemente equilibrate in azoto; nella siringa di diluizione è inoltre presenteuna concentrazione nM di glucosio-ossidasi e 10 mM glucosio, in modo daridurre ulteriormente la possibilità che la proteina denaturata e ridotta per lapresenza di ascorbato e CO possa andare incontro ad un’autossidazione.
Come mostrato nella figura 4.8, seguendo l’esperimento di mescolamentomediante misure di fluorescenza e di assorbimento è possibile separare i dueeventi; ovvero nel pannello A su una scala di tempi di 1 s è possibile osservare ilprocesso di collasso, ovvero il passaggio dalla specie U6CO alla specie N6CO. Nelpannello B, è mostrato invece il processo di rilascio del CO dalla specie collassatache conduce alla specie finale avente la metionina coordinata in posizione distale,ottenuto seguendo l’assorbimento ottico a λ=415 nm, su una scala di tempi di500 s.

71
0
0.2
0.4
0.6
0.8
1
0 0.2 0.4 0.6 0.8 1
fluor
esce
nza
norm
aliz
zata
(u.a
.)
t (s)
0
0.05
0.1
0.15
0.2
0 100 200 300 400 500
∆Αbs415 nm
t (s)
Figura 4.8: Pannello A, cinetica di rinaturazione del citocromo c551seguita in fluorescenza conun sistema a flusso interrotto. Pannello B: evoluzione temporale a seguito del rilascio del COdalla specie nativa N6CO, seguito in assorbimento ottico alla lunghezza d’onda λ=415 nm conlo stesso sistema a flusso interrotto; le condizioni sperimentali dopo il mescolamento in entrambii casi sono: [Gdn]= 0.9 M, [CO]=0.1 mM, Ascorbato 10 mM, fosfato 0.1M pH 7.
Eseguendo questo esperimento per diversi valori di concentrazione finale didenaturante, si possono riportare su di un grafico le velocità osservate influorescenza ed associate al processo di rinaturazione in funzione dellaconcentrazione di guanidina, ricostruendo in questo modo il braccio di refoldingdello Chevron plot del citocromo c551 ridotto e legato al CO (si veda il paragrafo3.4 per una descrizione delle caratteristiche di uno Chevron plot).
In figura 4.9 è riportato il confronto fra i risultati ottenuti con questoprotocollo (quadrati bianchi) e lo chevron plot completo della specie ossidata(pallini bianchi). Dall’osservazione di questo grafico si deduce che, il processo difolding del citocromo c551 procede alla stessa velocità indipendentemente dallo

72
stato di ossidazione del ferro; inoltre, dato che il CO blocca virtualmente lapossibilità di legarsi alla metionina, solo in un secondo tempo e a collasso giàavvenuto, è possibile per il ligando fisiologico spiazzare il ligando esterno.
Figura 4.9: (pallini bianchi): Chevron plot del citocromo c551 ossidato a pH 7 ottenuto damisure in fluorescenza, in rosso è riportato il fit dei dati; (quadrati bianchi): braccio di refoldingdel citocromo c551 ridotto e in presenza di CO seguito in fluorescenza, in blu è riportato il fit deidati; (quadrati neri): braccio di refolding del citocromo c551 seguito in assorbimento, in nero èriportato il fit dei dati.
Inoltre, si deduce che il collasso iniziale non coinvolge effettivamente illegame distale fra il ferro dell’eme e la metionina, nonostante il fatto che talelegame sia il principale responsabile dell’aumento di stabilità fra il citocromoossidato e ridotto (Moore & Pettigrew, 1990). È per altro interessante osservareche i risultati qui presentati confermano l’aspettativa che la dinamica del processodi folding dipenda più dalla topologia dello stato nativo (che è la stessa nei duestati ossidato e ridotto del citocromo c) che dalla sua stabilità termodinamica(molto maggiore nel citocromo c ridotto).
Infine in analogia a quanto mostrato da Bhuyan et al. 2001, è stato ricostruitoil braccio di refolding della specie ridotta in assenza di ligandi esterni, seguendo ilcambiamento di assorbimento ottico nel Soret, fra la specie denaturata e la specie
6420
1000
100
10
1
0,1
0,01
[GdnHCl] (M)
k obs
(s-1)

73
nativa. Tale esperimento è stato condotto in eccesso di ditionito e utilizzando alteconcentrazioni di proteina dopo il mescolamento (circa 50 µM ), perciò nonpossono essere esclusi fenomeni di aggregazione. Analogamente al caso delcitocromo di cavallo (Bhuyan et al. 2001), anche in questo caso si ottengono deivalori sistematicamente più veloci di quelli ottenuti seguendo il processo influorescenza. Questo dato riportato in figura 4.9 con i quadrati neri, si accordacomunque in modo piuttosto convincente con i dati in fluorescenza e fornisce unulteriore supporto all’ipotesi che il legame fra il ferro e la metionina non sia ildeterminante topologico significativo nella cinetica di rinaturazione deicitocromi.
4.4.2 Esperimenti di flow-flashI dati presentati nel paragrafo precedente si prestano ad un ulteriore
approfondimento, infatti le misure di fluorescenza partendo dalla specie U6COdel citocromo c551 hanno mostrato che il legame del CO persiste anche dopo cheil sistema è collassato in una struttura compatta, che dal punto di vista del foldterziario possiamo definire nativa, N6CO (figura 4.8). Questa specie, su una scaladi tempi più lenta, che dipende soltanto dalla costante di dissociazione del CO, èin grado di scambiare la sesta posizione di coordinazione con il ligandofisiologico per dare la specie nativa N6. Se questa miscela viene illuminata da unimpulso di laser è possibile accelerare il processo di coordinazione dellametionina distale in quanto si promuove la dissociazione del CO e si può studiarecon un apparato di flow flash (si veda il paragrafo 3.3 per una descrizione piùapprofondita) l’ultimo evento del processo di folding. Infatti, da un punto di vistadel folding, dopo il mescolamento ma prima che il laser ecciti il campione, laproteina ha percorso l’imbuto conformazionale (si veda il paragrafo 1.5) che apartire da un numero elevatissimo di configurazioni denaturate e attraversopercorsi paralleli, ha prodotto un numero estremamente ridotto di specie chehanno una distanza piccola da un punto di vista energetico e configurazionaledalla specie nativa.
Questo esperimento è stato condotto sul citocromo c di cavallo e sulcitocromo c551 e i risultati sono riportati nella figura 4.11. Per entrambe leproteine sono state seguite diverse lunghezze d’onda su di una stessa scala ditempi, quindi gli andamenti temporali sono stati analizzati minimizzandoglobalmente i dati ottenuti. Si tenga presente che ognuna delle tracce è raccoltamediando due soli flash di laser successivi, che avvengono con un ritardo fra loronoto e pari a 500 ms a partire da un istante di tempo dopo il mescolamento checade fra i 50 e i 500 ms. Le condizioni sperimentali precedenti al mescolamentosono: il citocromo di cavallo è nella forma ridotta in presenza di [CO]=1 mM e[Gdn]=5M a pH7, il citocromo c551 anche esso ridotto ed in presenza di

74
[CO]=1mM e [Gdn]=4M. Queste condizioni sono denaturanti per entrambe leproteine; la diluizione avviene mescolando le soluzioni precedenti controtampone fosfato a pH 7 in volume pari a 5:1.
-0.1
-0.08
-0.06
-0.04
-0.02
0
0.02
-0.2 0 0.2 0.4 0.6 0.8
∆Abs
t (ms)
-0.06
-0.05
-0.04
-0.03
-0.02
-0.01
0
0.01
-0.2 0 0.2 0.4 0.6 0.8
∆Abs
t (ms)
Figura 4.11: Pannello A: evoluzione temporale dopo flow-flash del citocromo c di cavallo nellecondizioni sperimentali:[Gdn]=1M [CO]=0.2mM dopo il mescolamento; rombi azzurriλ=417nm, quadrati verdi λ=415 nm, triangoli rossi λ=413 nm, cerchi neri λ=410 nm;pannello B: evoluzione temporale dopo flow-flash del citocromo c551 nelle condizionisperimentali:[Gdn]=0.8M [CO]=0.2mM dopo il mescolamento; triangoli rossi λ=415 nm,quadrati verdi λ=413 nm, cerchi neri λ =410 nm.
In entrambi i casi si osserva che il processo cinetico presenta un andamentobifasico e che non restituisce la specie pre-flash, come appare evidente a diverselunghezze d’onda. Per il citocromo di cavallo i tempi caratteristici dei due

75
processi sono: τ= 40 µs e τ= 400 µs, per il citocromo c551 si ha τ = 40 µs e τ =200 µs.
Le due fasi testimoniano l’eterogeneità del campione che viene illuminato dallaser, ma alla luce degli esperimenti di fotolisi all’equilibrio sono state assegnate adue processi diversi. Da una parte infatti la fase più rapida (τ= 40 µs) è associataalla ricombinazione del ligando fisiologico, la metionina, ad una specie nativa. Lafase più lenta (τ= 200/400 µs a seconda dei citocromi) rappresenta la quota diproteina che nelle condizioni di denaturante finale (rispettivamente [Gdn]=1 Mper il citocromo di cavallo e [Gdn]=0.8 M per il citocromo c551) si trova adequilibrio nello stato N6CO e pertanto si ricombina con il gas in un processobimolecolare. Poiché alle due specie N6 e N6CO è assegnata la stessa strutturatridimensionale ed un’energetica comparabile nelle presenti condizionisperimentali, riteniamo che nel processo con τ=40 µs la metionina distale siricombini con tutte le molecole nello stato N5 popolate dal flash; mentre, nellafase con τ= 200/400 µs si abbia la parziale sostituzione dell’aminoacido con ilCO come nello schema seguente:
Schema 4.4
N6 CO N5 N6* N6
Il dato ottenuto è in ottimo accordo con i dati presenti in letteratura cheassociano il legame del ferro dell’eme pentacoordinato con una specieintramolecolare ad un valore compreso fra 10 e 50 µs, (Pascher et al. 1996, Hagenet al., 1997). Alla luce di quanto mostrato nei paragrafi precedenti, la formazionedi questo legame non induce il processo di collasso nei citocromi, bensì ha unruolo significativo solo nella successiva stabilizzazione dello stato nativo, in modoparticolare per la specie ridotta.
4.5 La mioglobina, una proteina modello
Come già discusso nel capitolo 2, le fluttuazioni strutturali che occorrononelle proteine allo stato nativo, sono state studiate sulla mioglobina di capodoglio,ed in particolare sui suoi mutanti YQR e YQRF. Questa scelta è stata fatta inconsiderazione del fatto che questa proteina, la cui struttura cristallografica è statarisolta per la prima volta da Kendrew (Kendrew et al., 1960), è nota in grandedettaglio e rappresenta un modello sul quale studiare il rapporto fra struttura,sequenza e funzione. Anche dal punto di vista funzionale la mioglobina sicombina reversibilmente con i suoi ligandi quali O2 , CO e NO in reazionichimiche semplici. A partire dalla specie di equilibrio legata, sia essa ad esempio

76
MbCO, è possibile fotolizzare il ligando e seguire di conseguenza la reazione diricombinazione Mb+CO→ MbCO, su un ampio intervallo di temperature e convarie tecniche spettroscopiche, fra cui l’assorbimento IR si rivela estremamenteinformativo. Con questa tecnica infatti, è possibile dimostrare diversi intermedi direazione i quali producono una schema cinetico sequenziale dato da:
Schema 4.5
A B C D SMbCO← Mb*⋅CO ↔ Mb*⋅⋅CO ↔ Mb⋅⋅CO ↔ Mb+CO
A partire dallo stato legato di equilibrio MbCO (A), è possibile popolare condiversi protocolli di fotolisi differenti intermedi distinguibili per la struttura dellaproteina e per il sito di stazionamento del ligando (docking site, gli stati B,C e D). IlCO fotodissociato dal Ferro migra inizialmente in un sito poco distante,adagiandosi sul piano dell’eme (Mb*⋅CO); di qui può ricombinarsi o allontanarsiulteriormente, esplorando le cavità interne alla macromolecola (Mb*⋅⋅CO,Mb⋅⋅CO). La proteina invece si trova immediatamente dopo la fotolisi del ligandoin una struttura prossima a quella dello stato legato di equilibrio; questa strutturaè indicata con l’asterisco, Mb* (stati B e C) ed è distinta da quella non legata diequilibrio, indicata come Mb, alla quale rilassa gradualmente. Solo se latemperatura a cui è condotto l’esperimento consente moti su larga scala confluttuazioni concertate della struttura terziaria il ligando raggiunge il solvente(stato S).
La presenza di diverse bande nello stato legato A è uno degli indicatoriprincipali dell’esistenza di sottostati conformazionali nelle proteine, le quali graziealla presenza di questi diversi stati funzionalmente attivi e separati da barriereenergetiche molto piccole, sono in grado di modulare in modo estremamentesofisticato i processi dinamici necessari a svolgere la funzione biologica. È statoinfatti dimostrato da Frauenfelder e dai suoi collaboratori (Austin et al., 1975),che le cinetiche di ricombinazione del CO dei diversi sottostati presentano dellecostanti di tempo differenti. Inoltre, al di sotto della temperatura di transizionevetrosa (Tt=180 K) nella quale il solvente risulta inaccessibile al ligando, questaricombinazione è non-esponenziale, a testimonianza del fatto che a bassatemperatura è possibile isolare all’interno del sottostato principale unsottoinsieme ulteriore di conformazioni distinte, separate da barriere energeticheancora più piccole.
4.6 Processi dinamici all’interno della matrice proteica
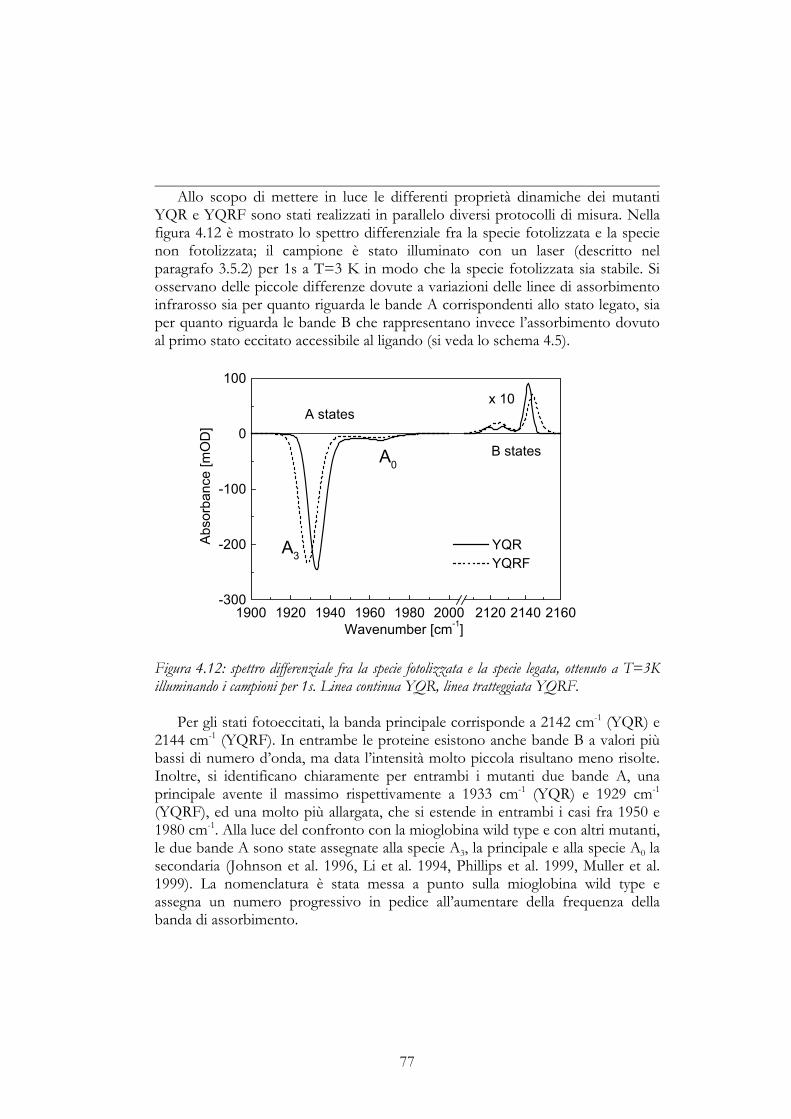
77
Allo scopo di mettere in luce le differenti proprietà dinamiche dei mutantiYQR e YQRF sono stati realizzati in parallelo diversi protocolli di misura. Nellafigura 4.12 è mostrato lo spettro differenziale fra la specie fotolizzata e la specienon fotolizzata; il campione è stato illuminato con un laser (descritto nelparagrafo 3.5.2) per 1s a T=3 K in modo che la specie fotolizzata sia stabile. Siosservano delle piccole differenze dovute a variazioni delle linee di assorbimentoinfrarosso sia per quanto riguarda le bande A corrispondenti allo stato legato, siaper quanto riguarda le bande B che rappresentano invece l’assorbimento dovutoal primo stato eccitato accessibile al ligando (si veda lo schema 4.5).
1900 1920 1940 1960 1980 2000 2120 2140 2160-300
-200
-100
0
100
A0
A3
A states
B states
x 10
YQR YQRF
Abso
rban
ce [m
OD
]
Wavenumber [cm-1]
Figura 4.12: spettro differenziale fra la specie fotolizzata e la specie legata, ottenuto a T=3Killuminando i campioni per 1s. Linea continua YQR, linea tratteggiata YQRF.
Per gli stati fotoeccitati, la banda principale corrisponde a 2142 cm-1 (YQR) e2144 cm-1 (YQRF). In entrambe le proteine esistono anche bande B a valori piùbassi di numero d’onda, ma data l’intensità molto piccola risultano meno risolte.Inoltre, si identificano chiaramente per entrambi i mutanti due bande A, unaprincipale avente il massimo rispettivamente a 1933 cm-1 (YQR) e 1929 cm-1
(YQRF), ed una molto più allargata, che si estende in entrambi i casi fra 1950 e1980 cm-1. Alla luce del confronto con la mioglobina wild type e con altri mutanti,le due bande A sono state assegnate alla specie A3, la principale e alla specie A0 lasecondaria (Johnson et al. 1996, Li et al. 1994, Phillips et al. 1999, Muller et al.1999). La nomenclatura è stata messa a punto sulla mioglobina wild type eassegna un numero progressivo in pedice all’aumentare della frequenza dellabanda di assorbimento.

78
Figura 4.13. Pannello superiore: grafico in scala doppiologaritmica della frazione di molecole diCO che non ha ancora rilegato in funzione del tempo N(t) per YQR; sono rappresentate ogni20 K le temperature nell’intervallo 60 K-160 K. Nell’inserto in piccolo vi è un ingrandimentoin scala semilogaritmica della prima parte del processo. Pannello inferiore: analogo al pannellosuperiore perYQRF; sono rappresentate ogni 20 K le temperature nell’intervallo 60 K-140 KInoltre alcune bande presenti nel wild type in certe condizioni, possono essereassenti nei mutanti, dove l’assegnazione risulta data in analogia ai valorideterminati nella proteina selvatica. In particolare la mancanza di una terza banda
60 K
160 K
140 K
60 K

79
(presente nel wild type) è dovuta al fatto che il residuo di istidina distale rimossopuò dare luogo a isoforme più pronunciate rispetto alla glutamina, mentre lapiccola differenza fra i due mutanti sembra indicare che per alloggiare il residuodi fenilalanina, vi sia un piccolo riarrangiamento dei residui distali. Perciò alloscopo di caratterizzare i processi dinamici coinvolti nella ricombinazione delligando, il primo passo è stato quello di eseguire degli esperimenti di fotolisi abassa temperatura ( T < Tt), seguendo il processo di ricombinazione sul massimodella banda A3 per entrambi i mutanti.
Nella figura 4.13, è mostrato il risultato di questo esperimento; in particolare, idati sono stati normalizzati in modo da rappresentare la variazione diassorbimento infrarosso, la quale risulta proporzionale alla frazione di molecoleche non ha ancora rilegato: DeltaA=[A(t=0)-A(t)]/A(t=0). Si osserva unadifferenza marcata fra i due mutanti. In YQR si possono identificare chiaramentetre processi: uno molto veloce a carico di circa un 5% delle molecole di COosservabile nei primi 10 µs (e che è possibile apprezzare nell’inserto dellasuddetta figura); un processo principale che raccoglie più del 90% delle molecoleed infine un processo che appare in modo evidente a partire da 120 K e cheraccoglie la restante frazione di molecole di ligando. In YQRF, sono visibiliancora tre processi, ma la frazione di molecole che ricombina con il processo piùrapido ha raggiunto ~ 50 % ed i due processi successivi sono meno distinguibilied hanno ampiezze molto ridotte rispetto al mutante YQR.
I soli risultati della fotolisi a bassa temperatura, pur fornendo una primaindicazione che i percorsi migratori nei due mutanti sono diversi non è in gradodi assegnare tali percorsi alla luce del modello che prevede l’occupazione da partedel ligando fotodissociato di cavità all’interno della matrice proteica. Combinandoquesti risultati con le misure in gradiente di temperatura si può ottenereun’indicazione più chiara del comportamento dinamico nei due mutanti. Inquesto caso, come illustrato nel paragrafo 3.5.2 si possono popolareselettivamente le diverse cavità utilizzando protocolli di fotolisi differenti. Perprima cosa il campione viene congelato al buio ad una temperatura di 3K, quindisi illumina per 1s con una sorgente laser e successivamente inizia il processo dilento riscaldamento (con un coefficiente α=5 mK/s) e si acquisisce uno spettrodi assorbimento per ogni grado di temperatura nell’intervallo 3 – 140 K. I risultatisono mostrati nella figura 4.14. I pannelli A e C della figura, sono separati fra laregione degli stati legati (i due pannelli superiori) e la regione degli statifotodissociati (i due pannelli inferiori) per entrambi i mutanti. Si osservaimmediatamente che la banda A0, associata al sottostato meno popolato, presentauna ricombinazione predominante nella regione al di sotto dei 40 K ed è analogain entrambe le proteine studiate.
YQR

80
YQRF

81
Figura 4.14.Pannello A (superiore e inferiore): mappa TDS per MbCO YQR dopoilluminazione per 1s a 3K e rampa 3-140 K. Linee continue aumento di assorbimento lineetrattegiate, diminuzione. Il pannello superiore riferisce agli stati legati, il pannello inferiore aglistati eccitati. Pannello B (superiore e inferiore): Segnale integrato fra le frequenze 1960-1980cm-1 per A0 e 1920-1940 cm-1 per A3. Le linee tratteggiate rappresentano il fit delladistribuzione dell’entalpia con delle gaussiane. Pannelli C e D: analoghi ad A e B per YQRF.
La figura 4.14 B e D, pannelli inferiori, mostra la predominanza di una barrieraenergetica con un massimo intorno ai 20K. Viceversa come atteso dalle misure diassorbimento a singola lunghezza d’onda, la ricombinazione del sottostato A3

82
risulta drasticamente diversa. In YQR, si estende sull’intero intervallo ditemperature fino a ~ 110 K, e la figura 4.14 B (pannello superiore), mostrachiaramente che esistono due barriere di energia aventi un massimocorrispondente alle temperature di circa 40K e 60K In YQRF invece, laricombinazione dallo stato A3 è massima a circa 10K e si estende con undecadimento graduale fino a 100K. Le barriere energetiche evidenti sono tre,anche se risultano molto meno risolte (figura 4.14 D pannello superiore einferiore). L’analisi dei dati seguendo lo schema di un modello a tre stati (comeillustrato nel paragrafo 3.6) ha prodotto i valori riportati nella tabella 4.2, nellaquale sono mostrati i valori estrapolati da un fit che utilizza distribuzionigaussiane per descrivere le barriere entalpiche distribuite che separano i diversisottostati.
Tabella 4.2 Picco 1 Picco 2 Picco 3
Campione/Sottostato
Pre-exp log(A/s)
H1 KJ/mol
σ1 KJ/mol
f1 (%)
H2 KJ/mol
σ 2 KJ/mol
f2 (%)
H3 KJ/mol
σ 3
KJ/mol
f3 (%)
YQR A0 8.75 0.7 4.7 91 - - - 15.5 3.0 9
YQR A3 8.75 - - - 7.8 2.1 34 13.5 3.1 66
YQRF A0 8.60 - - - 2.9 3.0 79 13.3 5.0 21
YQRF A3 8.60 1.0 1.7 24 5.9 3.8 70 14.2 2.8 6
Infine ciò che rende questo approccio sperimentale estremamente sofisticatoè la possibilità di confrontare la variazione di assorbimento dovuta alla comparsadei sottostati legati A, alla variazione sincrona degli stati fotodissociati B. Inparticolare si osserva che se l’unico processo in atto nella matrice proteica fosse laricombinazione del ligando, il contour plot che contraddistingue gli stati fotoeccitatidovrebbe contenere solo linee tratteggiate, a testimoniare la scomparsa deisuddetti stati B in favore degli stati legati A. Se osserviamo invece cosa avviene inYQR, è possibile immediatamente notare che il primo evento corrispondente aduna diminuzione della specie a 2118 cm-1 è accompagnato ad una piccolaricombinazione (si veda lo stato A), ma è concomitante con l’apparizione di unanuova specie eccitata a 2142 cm-1.

83
Figura 4.15. Pannello A (superiore e inferiore): Mappa TDS per YQR dopo lentoraffreddamento fra 140-3 K sotto illuminazione laser e successiva rampa fra 3-140 K. I datisono spaziati logaritmicamente. Pannello B (superiore e inferiore): analogo ad A ma perYQRF. Pannello C (superiore e inferiore): mappa TDS dopo illuminazione a 185 K ( 2 ore) eraffreddamento a 160K; rampa tra 160-220 K. Pannello D (superiore e inferiore): analogo aC ma per YQRF.
Al di sopra dei 20 K la banda a 2142 cm-1 scompare per due ragioni: i) si ha unaricombinazione pronunciata, come appare evidente confrontando il contour plotdei sottostati legati A; ii) vi è un nuovo trasferimento ad un doppietto di bande

84
centrate a 2127 cm-1 e 2135 cm-1. Al di sopra dei 60 K la banda a 2142 cm-1 èscomparsa e si ha il solo evento di ricombinazione dal doppietto. YQRF, mostraun contributo significativo alla ricombinazione che avviene nei primi 30 K, inparticolare la regione fra 2110 cm-1 e 2135 cm-1 contribuisce in modopredominante nei primi 10 K quindi si ha la scomparsa della banda a 2144 cm-1
fino a circa 50 K. Una piccola frazione di molecole, anche se con una resa moltopiù bassa che in YQR, è in grado di popolare uno stato eccitato diversoidentificato dal doppietto a 2123 e 2130 cm-1, il quale ricombina a partire dai 60K.
Da questa prima serie di esperimenti possiamo trarre due conclusioni; laprima è che in entrambi i mutanti esiste uno stato eccitato, che è presente conun’efficienza diversa, dovuto ad un trasferimento di popolazione da un primostato eccitato ad un altro ed è il principale indicatore del fatto che il ligando iniziaa migrare verso un sito di legame più distante dall’eme. La seconda è che ilmutante YQRF risulta avere un percorso migratorio reso più difficile dallapresenza del residuo di fenilalanina. Per cercare di chiarire ulteriormente ladinamica del ligando all’interno della mioglobina, sono stati eseguiti altri dueprotocolli di TDS. Il primo è ottenuto raffreddando lentamente il campione da140 K a 3 K sotto illuminazione continua da parte della sorgente laser, in mododa iniziare la nuova rampa da 3 K a 140 K avendo intrappolato una frazionesignificativa di molecole di CO negli stati eccitati a più alta energia. Il secondo siottiene fotolizzando per 2 ore il campione a 185 K (quindi al di sopra dellatemperatura di transizione vetrosa del solvente) e raffreddando quindi fino a 160K, dove si da inizio ad un gradiente di 5mK/s fra 160 e 220 K.
I risultati di queste due ulteriori esperimenti sono mostrati nella figura 4.15, inparticolare i pannelli A e B riferiscono al primo protocollo descritto e i pannelli Ce D al secondo. Per prima cosa occorre osservare che i dati mostrati nella figura4.15 sono drasticamente diversi da quelli mostrati nella figura 4.14, ma risultanopiù omogenei fra i due mutanti. In altre parole questi protocolli studiati perintrappolare il CO in regioni diverse della matrice proteica, mostrano che illigando risente sempre meno della mutazione aggiunta in YQRF, man mano cheesplora zone più distanti dal sito attivo. La mappa presentata nel pannello A dellafigura 4.15, mostra che la banda A3 a 1933 cm-1 presenta un picco prominente a60K e una nuova barriera centrata a 130K che contiene circa il 31% della frazionedi molecole di ligando; la barriera a 40 K che appariva nell’esperimento di fotolisia 3K è praticamente scomparsa. YQRF mostra una mappa relativa agli stati legatiA (pannello B), in cui esiste la barriera centrata a 130 K per la banda A3 mariguarda solo il 12% della popolazione di molecole di CO.
Per quanto riguarda gli stati fotodissociati si osserva che in YQR è scomparsala banda centrata a 2118 cm-1 mentre della banda a 2142 cm-1, sopravvive unapiccola popolazione che ha una barriera di attivazione centrata a 50 K. Questa

85
piccola frazione è data da quelle molecole di CO che rimangono intrappolatenegli stati a più alta energia della barriera identificata dall’esperimento mostratonella figura 4.14. La ricombinazione sincrona con le due barriere energeticheidentificate seguendo lo stato A3 proviene da due doppietti, il primo identificatodalle linee a 2127 e 2134 cm-1, il secondo dalle linee centrate a 2128 e 2133 cm-1.Al di sotto dei 30K si osserva anche una certa quota di scambio di popolazionefra i due picchi del primo doppietto, che mostra come l’iniziale aumento ditemperatura non è in grado di consentire una significativa ricombinazione masolo un riarrangiamento della stessa specie eccitata. YQRF presenta una dinamicadegli stati fotodissociati in apperenza più complessa; in realtà si ha un processoanalogo a YQR al quale si somma un processo di ricombinazione dovuto allaspecie fotodissiociata avente un picco centrato a 2144 cm-1 e che è responsabiledella larga quota di molecole di CO che presentano una ricombinazione al disotto dei 50 K. Inoltre un ulteriore differenza con il mutante YQR, risiede nelfatto che il doppietto che ricombina a circa 60K presenta una banda moltoallargata e centrata ai valori 2119 e 2135 cm-1 sensibilmente diversi sia dai valoridel doppietto corrispondente all’esperimento di fotolisi a 3K (2123 e 2130 cm-1)sia dai valori del doppietto che ricombina con la barriera energetica centrata a130K (2122 e 2128 cm-1).
Infine, resta da descrivere l’esperimento nel quale illuminando la proteina per2 ore a 185 K si vuole cercare di intrappolare degli stati eccitati presentiall’interno della matrice proteica, anche in presenza dei moti caratteristici dellamioglobina che rendono il solvente accessibile al ligando. I due mutanti sicomportano in modo analogo, ovvero mostrano una ricombinazione dovuta inmodo preponderante ad un’unica specie centrata a 2128 cm-1, alla qualecorrisponde l’apparizione di due stati legati A0 e A3 i quali vanno incontro inentrambi i mutanti ad un processo di interconversione per produrre il rapportocorretto della specie di equilibrio a 220 K. L’unica differenza riguarda il fatto chenel mutante YQRF il processo si estende complessivamente per 10 K in più diYQR a testimoniare che in qualche modo il quadruplo mutante ha unaricombinazione più difficile.
4.7 Relazione fra proprietà spettroscopiche e dinamica strutturale
I risultati presentati nel paragrafo precedente non hanno solo un significatocinetico importante, in quanto descrivono una serie di processi che avvengonoall’interno della matrice proteica (ovvero la ricombinazione del ligando o la suamigrazione verso altri siti), ma grazie alla conoscenza approfondita dei dettaglistrutturali della mioglobina, consentono una lettura dei risultati stessi alla luce diuno schema generale che può essere schematizzato nella figura 4.16 A.

86
In generale, gli esperimenti condotti hanno prodotto due risultati principali:da una parte hanno consentito attraverso l’osservazione dei processi a carico deglistati legati A, di mostrare che la dipendenza in temperatura dei processi cinetici diricombinazione del ligando risultano sfavoriti nel mutante YQR, mostrando inquesto modo che la mutazione addizionale I107F, presente nel quadruplomutante, ha prodotto una struttura del sito attivo che favorisce la ricombinazione.Dall’altra sono stati evidenziati una serie di stati fotoeccitati, caratterizzati ingenerale dalla presenza di due bande di assorbimento del CO, che possono essereidentificate dai loro numeri d’onda ed associati in modo estremamente puntualealla posizione della molecola del CO in una particolare regione della proteina.Nella tabella 4.3 sono stati riportati i valori corrispondenti ai picchi identificatiper i diversi stati fotoeccitati; la nomenclatura riflette l’assegnazione che è statopossibile fare fra diverse posizioni del CO nella molecola proteica e i suddettistati ottenuti dalla fotolisi del CO nelle diverse condizioni sperimentali. Perassegnare i diversi sottostati siamo partiti dall’osservazione di Anfinrud ecollaboratori (Lim et al., 1997) che sulla base dei loro esperimenti di fotolisi atemperatura ambiente utilizzando l’assorbimento IR ad una risoluzione temporaleprossima al femtosecondo, hanno assegnato al CO fotolizzato nella proteina wildtype, un sito transiente di approdo (docking site) al di sopra dell’anello pirrolico Cdell’eme con due possibili orientazioni (ruotate di 180° l’una rispetto all’altra).Questa interpretazione si accorda con i dati sperimentali acquisiti a bassatemperatura e mostrati in questa tesi; infatti, in YQR nell’esperimento di fotolisi a3K (figura 4.14) si osserva un trasferimento di popolazione fra la bande che sipopolano a seguito della fotolisi, attraverso uno spostamento di riga fra B2 (2118cm-1) e B1 (2142 cm-1). Questo processo si accorda con il fatto che a 3 K il ligandoè fotolizzato in uno stato metastabile molto vicino all’eme, ma possiede uneccesso di energia cinetica tale che, non appena l’energia termica fornita alsistema è sufficiente (~10 K), le due bande compiono un primo riarrangiamentoin accordo con la differenza di energia libera che le contraddistingue.Successivamente, al di sopra dei 20 K, il ligando può abbandonare il sito diapprodo B, sia a causa della ricombinazione oppure in quanto inizia a migrareverso un regione caratterizzata da un campo elettrico locale differente, comeevidenziato dal cambiamento delle righe del doppietto. Quest’ultima grazieall’esperimento di fotolisi su cristallo a 30K (Brunori et al., 1999) è assegnatasenza ambiguità alla cavità identificata come Xe4, e da un punto di vista dellanomenclatura degli stati fotoecciati dalla lettera C’.

87
Figura 4.16. Pannello A: Schema del sito attivo della mioglobina di capodoglio YQR (blu) eYQRF (rosso). Sono evidenziati i percorsi migratori del ligando attraverso i vari siti distazionamento e la loro relazione con le cavità presenti. Pannello B: Mappe crsistallografiche delsito attivo dei mutanti suddetti nello stato legato al CO. Le strutture sono state risolterispettivamente a 1.8 Å (YQR) e 1.5 Å (YQRF).
Tabella 4.3
# Dopo raffreddamento sotto illuminazione continua fra 140 e 3 K† Specie minoritarie‡ A/B1 (2142/2144 cm-1) soltanto+ fortemente dipendente dalla temperatura. Il rapporto passa da 45 (48) a 180 K a 68 (70) a 200K per YQR (YQRF).
ProteinaB1/B2
3 - 15 K
StatiC’
50 – 80 K
EccitatiC’#
50 – 80 K
C’’
110 – 130 K
D
180 – 200 K
Numerod’onda(cm-1)
21182142
21272135
(2132)†
21272134
21282133
2128(2135)†YQR
MbCORapporto di
assorbimento 42 + 3‡ 21 + 2 21 + 2 27 + 2 56 + 4+
Numerod’onda(cm-1)
(2112)†
214421232130
21192135
(2130)†
21222128
(2133)†
2128(2135)†
YQRFMbCO
Rapporto diassorbimento 40+2‡ 18 + 2 18 + 2 26 + 2 52 + 4+

88
Questo processo avviene in entrambi i mutanti, anche se l’ingombro stericoprodotto dal residuo di fenilalanina del mutante YQRF consente ad una frazioneminore di molecole di CO, di raggiungere il nuovo punto di approdo. Questasituazione può essere aggirata mediante il protocollo di fotolisi estensivacontemporaneo al raffreddamento fra 140 K e 3K, che produce le mappe TDSmostrate nella figura 4.15 (pannelli A e B). Per prima cosa si osserva che lo statoB2/B1, risulta non popolato o solo scarsamente popolato (in YQRF); inoltrenessun processo termicamente attivato, di trasferimento di popolazione avvienefra i due sottostati B2/B1 e C’. Il principale risultato è, invece, quello di produrrein entrambi i mutanti, un nuovo stato eccitato C” caratterizzato da unaricombinazione verso entrambi i sottostati conformazionali A0 e A3 che devesuperare una barriera energetica maggiore (~130 K). Inoltre pur presentando unprofilo energetico simile, questo protocollo di fotolisi provoca una differenzaimportante fra il doppietto che caratterizza lo stato C’ per YQR e il doppiettoche caratterizza C’ per YQRF (si veda la tabella 4.3 per i dettagli). Questaosservazione non è sorprendente alla luce del fatto che il residuo Phe 107 è incontatto con la cavità C’ e quindi può modificare le bande di assorbimento IR.Per quanto riguarda lo stato C” non esiste al momento alcuna informazionestrutturale diretta, capace di identificare senza ambiguità la posizione del ligandoin questo stato fotodissociato. Il fatto che la ricombinazione da C’ e C” versoentrambi i sottostati legati avvenga alla stesse temperature indica che la barrieraentalpica che governa questi processi non è in prossimità dell’eme; inoltre laseparazione netta nelle mappe TDS fra lo stato C’ e C” e il fatto che vi siano dellemodificazioni nei valori delle righe del doppietto che caratterizza ciascunsottostato, suggerisce che il CO migri dalla cavità Xe4, verso un sito di approdoulteriore. Come è possibile osservare dalla figura 4.16 A, un ovvio candidato èdato dalla cavità Xe2, che risulta lungo il cammino fra lo stato C’ e lo stato D.Allo stato attuale però non è possibile escludere che C” possa risultare dacambiamenti locali all’interno della tasca Xe4 e che questo nuovo statofotoeccitato non sia associato ad una posizione diversa all’interno della matriceproteica.
Infine utilizzando l’ultimo esperimento di TDS in cui il campione è statoilluminato estensivamente a 185 K è possibile intrappolare il ligando in unsottostato caratteristico, lo stato D (Ostermann et al., 2000). Quest’ultimo divieneaccessibile in quanto a queste temperature possono avvenire quei moti globalinella proteina, che sono in grado di aprire un canale di comunicazione fra la tascadell’eme e questo sito che si trova disposto sul lato prossimale nella cavità Xe1.Le bande caratteristiche di questo sottostato sono identiche fra YQR e YQRFcome ci si aspetta nel caso in cui la molecola di CO sia disposta lontano dallatasca distale che risulta modificata dalla mutazione (si osservi a questo propositoil dettaglio strutturale della figura 4.16 B). Inoltre come è riportato nella tabella

89
4.3, il rapporto tra l’assorbimento della banda IR nello stato legato e nello statofotodissociato nel sito D è 56(52) per YQR(YQRF). Tale rapporto è fortementedipendente dalla temperatura e aumenta in modo continuo all’aumentare dellastessa, suggerendo che la ricombinazione del CO al di sopra di 180 K includasempre di più molecole che siano prima scappate nel solvente divenendo silentiall’assorbimento IR.

90

91
Capitolo 5
Conclusioni e Prospettive
5.1 I problemi biologici e lo sviluppo strumentale
La struttura delle proteine è soggetta a fluttuazioni dinamiche rapide ereversibili, ben dimostrate nella letteratura scientifica (Austin et al., 1975,Frauenfelder et al 1988). La mutagenesi sitospecifica consente oggi di progettare erealizzare catene polipeptidiche selettivamente modificate rispetto a quelle wild-type e quindi di sottoporre a verifica sperimentale molte ipotesi sulla dinamicastrutturale delle proteine. È pertanto possibile oggi affrontare il problemabiologico, in precedenza elusivo, del ruolo strutturale e funzionale di singolielementi topologici della conformazione nativa delle proteine.
Nel corso del lavoro sperimentale descritto in questa tesi, che riguarda ladinamica strutturale di due emoproteine monometriche, l’una nello stato nativo,l’altra in quello denaturato, sono state affrontate difficoltà sia metodologiche chepiù direttamente biologiche. I risultati conseguiti dovrebbero pertanto esserevalutati separatamente, in relazione allo sviluppo strumentale, alla dinamica delfolding del citocromo c e alla dinamica strutturale della mioglobina nativa e deisuoi mutanti YQR e YQRF.
Poiché i fenomeni di dinamica strutturale delle proteine avvengono in tempibrevi, è necessario da un lato scegliere il sistema biologico che consental’applicazione di metodiche di cinetica biochimica rapida; dall’altro sviluppare latecnologia opportuna. È evidente che la scelta della proteina condiziona quelladel metodo. In questo lavoro si è deciso di studiare emoproteine con metodifotochimici e conseguentemente il primo passo è stato la messa a punto di unsistema sperimentale idoneo. Infatti, per studiare processi biologici rapidi occorrerisolvere i problemi strumentali connessi alla temporizzazione dell’esperimento.Se ad esempio il sistema compie dei processi che si esauriscono in 100 µs, ènecessario controllare l’inizio della reazione con una precisione che sia di almenodue ordini di grandezza superiore nella scala dei tempi. Un impulso di luce laser,che ha una durata di pochi nanosecondi, rappresenta un interruttore ideale perquelle reazioni che siano fotoattivabili. Il citocromo c e la mioglobina hanno lacapacità, nelle condizioni descritte nei capitoli precedenti, di legare il COformando il complesso emeFe-CO, che è estremamente fotosensibile erappresenta un derivato ideale per qualunque approccio fotochimico.

92
Superate le difficoltà metodologiche, il lavoro si è sviluppato parallelamentesui due sistemi sperimentali: da una parte cercando di identificare, nel citocromoc, i processi dinamici che avvengono nelle condizioni denaturanti in cui laproteina è in grado di legare il CO; dall’altra grazie alla collaborazione con ilDipartimento di Biofisica dell’Università di Ulm (Germania), cercando dimodulare il percorso migratorio del ligando nella mioglobina esplorandone inquesto modo le cavità interne, elementi strutturali altrimenti sordi.
Il filo conduttore comune in entrambi questi studi è stato quello di volereidentificare in modo rigoroso per quanto possibile, quali elementi della strutturanativa fossero coinvolti nei processi dinamici in entrambi i sistemi proteicistudiati. Infatti, se da una parte appare evidente che le proteine ripropongonostrategie vincenti già sperimentate, come risulta dal fatto che i fold noti ai qualipossono essere ricondotte le strutture tridimensionali sono limitati (si veda ilparagrafo 1.1), appare chiaro altresì che sia la struttura primaria che la funzione insistemi biologici che condividono un comune riarrangiamento tridimensionalenello spazio, possono essere drasticamente diverse.
In questo contesto si inquadra lo studio del legame fra il ferro e la metioninadistale che costituisce un elemento della struttura nativa comune ad una vastaclasse di citocromi e la cui importanza nello svolgimento della funzione ditrasferimento elettronico è primaria (Moore & Pettigrew, 1990). Questo aspetto èstato affrontato in modo sistematico, partendo dai risultati presenti in letteratura(Jones et al., 1993 Chen et al., 1997) che assegnavano a questo legame ancheun’importante ruolo come determinante topologico del folding. Una serie dirisultati non spiegati all’interno del modello cinetico proposto in precedenza ci hacondotto a studiare alcuni sistemi modello, quali la microperossidasi e l’emedisciolto in micelle. È emerso che il meccanismo proposto non teneva conto direazioni chimiche fondamentali a carico del gruppo prostetico eme. Questereazioni erano presenti anche negli esperimenti di fotolisi condotti sui citocromiin condizioni denaturanti, complicando considerevolmente la cinetica diricombinazione del CO. In particolare, alcuni intermedi assegnati ad un collassotransiente della proteina sono stati reinterpretati alla luce del nuovo modelloproposto, fornendo un’interpretazione in grado di spiegare le incongruenzeemerse in precedenza (Arcovito et al., 2001, si veda l’allegato A). Dal confrontofra i sistemi modello e le proteine studiate, ovvero il citocromo c di cavallo e ilcitocromo c551 da Pseudomonas aeruginosa, è emerso in modo convincente che laformazione del legame Fe-metionina, non è essenziale al processo dinamico difolding di queste proteine, pur essendo un elemento in grado di stabilizzarefortemente la struttura nativa, in particolare nella forma ridotta. Inoltre, conparticolare rilievo nel citocromo c551, si è dimostrato che la stabilità maggiore diquesta forma rispetto alla forma ossidata non è accompagnata da una sensibilevariazione nella velocità di rinaturazione della proteina. Alla luce di un modello

93
topologico che spieghi il meccanismo di folding, questo risultato si inquadraperfettamente e rappresenta un esempio in cui variando un parametro importantequale la carica netta del sistema (il Ferro passa da +3 a +2), non si ha un effettoimportante nel processo che intrappola gli intermedi efficaci in grado di guidare laproteina alla corretta struttura nativa.
Il secondo aspetto studiato e che si riallaccia al rapporto fra fold nativo edinamica delle proteine, è stato quello di identificare nelle cavità presenti nellamioglobina dei siti di stazionamento del ligando, i quali hanno una connessionediretta con la funzionalità della proteina stessa. Grazie alla possibilità di associareagli indicatori spettroscopici i corrispondenti sottostati identificati da un punto divista tridimensionale è stato possibile seguire e intuire il percorso completo cheuna molecola di ligando compie in determinate condizioni di temperatura esolvente. Il percorso individuato non è l’unico possibile, né il principale, per laproteina wild type in condizioni fisiologiche, in quanto, come mostrato da Gibsone collaboratori (Scott et al., 2001) la principale via di accesso dal solvente al sitoattivo è il canale controllato dall’istidina distale. Appare chiaro però, che lamodificazione di processi quali la ricombinazione geminata di ligandi fisiologiciper la presenza di alte pressioni di Xenon (Scott & Gibson, 1997) è direttamentecorrelata all’inibizione di possibili percorsi migratori all’interno della matriceproteica. Questi percorsi sono stati caratterizzati in modo puntuale in questolavoro di tesi grazie alla possibilità di modularli con la mutagenesi sitospecifica. Inparticolare il mutante YQR ha mostrato che è possibile favorire l’occupazione disiti alternativi a quello immediatamente prossimo al piano dell’eme, aumentandola probabilità che il ligando fotolizzato giunga a cavità più distanti quali adesempio Xe4. Viceversa, la mioglobina YQRF ha mostrato che cercaresemplicemente di bloccare la strada di accesso principale a questo nuovo sito distazionamento, attraverso un residuo di fenilalanina, riesce solo parzialmente.Infatti protocolli di fotolisi prolungata rendono i due mutanti più simili fra loroda un punto di vista della dinamica di ricombinazione. Inoltre, in entrambe leproteine, in condizioni nelle quali sono ammessi i moti concertati checoinvolgono tutta la struttura terziaria, è possibile popolare un sito alternativo,identificato come Xe1, sul lato prossimale del piano dell’eme (Lamb et al., 2002 siveda l’allegato B). La ricombinazione procede da questo sito al ferro dell’eme,attraverso un meccanismo che potrebbe coinvolgere l’apertura di canali dicomunicazione fra il lato distale ed il lato prossimale. In altre parole le cavitàpresenti nella mioglobina sembrano avere un ruolo funzionale di primo piano, nelcontrollo dei processi dinamici coinvolti nell’interazione con i ligandi, fornendo alsistema biologico delle vere e proprie camere, nelle quali alloggiaretransientemente questi ligandi gassosi.

94
5.2 Possibili sviluppi
Un primo elemento che sarebbe interessante poter studiare è dato senza alcundubbio dalla relazione fra le cavità presenti nei citocromi e il processo di folding.Questo tipo di approccio ha già fornito alcune indicazioni, (Hasegawa et al, 1999)ma anche alla luce dei risultati presentati in questi tesi sembra evidente che ilrapporto fra gli elementi che costituiscono gli snodi del sistema proteico e iprocessi dinamici che lo caratterizzano è di assoluto primo piano e merita unacaratterizzazione sistematica. In questo senso sarebbe ideale potere isolare lestrutture tridimensionali di intermedi funzionali per determinare quali elementidella struttura nativa sono collegati alla dinamica e alla funzione della proteina perselezionare tali residui come possibili determinanti anche del processo dirinaturazione. In alternativa, una strategia che ha fornito di recente alcuni successinella caratterizzazione degli intermedi di folding del citocromo c551 è stata quelladi studiare alcuni mutanti di specifici residui coinvolti in legami presenti nellastruttura nativa, in funzione del tipo di denaturante usato (Gianni et al. 2001). Èstato posto in evidenza dagli autori che alcune mutazioni che producono delledifferenze sostanziali nel processo di rinaturazione possono essere mascherate inguanidina ed esaltate in urea.
Per quanto riguarda lo studio sulla mioglobina sono in corso studi dicristallografia risolta in tempo a temperatura ambiente, proprio sui mutantioggetto di questa tesi. Tale progetto è volto a isolare gli intermedi di reazione chesono popolati durante il processo dinamico che avviene nelle condizionifisiologiche di tempo e temperatura e rappresenta perciò un progresso ideale diquesto lavoro di tesi nella caratterizzazione strutturale di intermedi di reazione.Inoltre, appare chiaro che la strategia di mutare i residui presenti in prossimitàdella cavità Xe2 la quale si è postulato venga popolata transientemente,rappresenta un ulteriore sviluppo che è in corso di perfezionamento.

95
Bibliografia
1. Agmon, N., Doster, W. & Post, F. (1994) The transition frominhomogeneous to homogeneous kinetics in CO binding to myoglobin.Biophys. J. 66, 1612–1622.
2. Anderson, D.E., Hurley, J.H., Nicholson, H., Basse, W.A. and Matthews,B.W. (1993). Hydrophobic core repacking and aromatic–aromaticinteraction in the thermostable mutant of T4 lysozyme Ser117∀Phe.Protein Sci. 2, 1285–1290.
3. Anfinsen, C.B. (1973). Principles that govern the folding of proteinchains. Science 181, 223-230.
4. Anfinsen, C.B., Haver, E., Sela, M. & White, F.H. Jr. (1961). The kineticsof formation of native ribonuclease during oxidation of the reducedpolypeptide chain. Proc. Natl. Acad. Sci. USA 47, 1309-1314.
5. Arcovito A., Gianni S., Brunori M., Travaglini-Allocatelli C., Bellelli A.(2001) Fast coordination changes in cytochrome C do not necessarilyimply folding. J. Biol. Chem. 276:41073-8.
6. Austin, R. H., Beeson, K. W., Eisenstein, L., Frauenfelder, H. &Gunsalus, I. C. (1975) Dynamics of ligand binding to myoglobin.Biochemistry 14, 5355–5373.
7. Berendzen, J. & Braunstein, D. (1990) Temperature-derivativespectroscopy: a tool for protein dynamics. Proc. Natl. Acad. Sci. USA 87,1–5.
8. Bhuyan A.K., & Udgaonkar, J.B. (2001) Folding of horse cytochrome c inthe reduced state. J Mol Biol 312, 1135-60
9. Brunori M, Vallone B, Cutruzzola F, Travaglini-Allocatelli C, BerendzenJ, Chu K, Sweet RM, Schlichting I. (2000). The role of cavities in proteindynamics: crystal structure of a photolytic intermediate of a mutantmyoglobin. Proc Natl Acad Sci USA 97, 2058-63
10. Bullough PA, Hughson FM, Skehel JJ, Wiley DC. (1994). Structure ofinfluenza haemagglutinin at the pH of membrane fusion. Nature 371, 37-43
11. Carrey E.A. & Pain, R.H. (1987). Conformation of a stable intermediatein the folding of Staphylococcus aureus penicillinase. Biochim. Biophys. Acta533, 12-22.
12. Chan, C.K., Hofrichter, J. and Eaton W.A. (1996) Optical triggers ofprotein folding. Science 274, 628-629.
13. Chen, E., Wood, M.J., Fink, A.L. and Kliger, D.S. (1998) Time-resolvedcircular dichroism studies of protein folding intermediates of cytochromec. Biochemistry 37, 5589-5598.

96
14. Clementi, C., Jennings, P. A. & Onuchic, J. N. (2000). How native-statetopology affects the folding of dihydrofolate reductase and interleukin-1β
Proc. Natl. Acad. Sci. USA 97, 5871-587615. Craig, S., Shmeissner, U., Wingfield, P. & Pain, R.H. (1987).
Conformation, stability, and folding of interleukin 1 beta. Biochemistry 26,3570-3576.
16. Cutruzzolà, F., Ciabatti, I., Rolli, G., Falcinelli, S., Arese, M., Ranghino,G., Anselmino, A., Zennaro, E., and Silvestrini, M.C. (1997) Biochem. J.322, 35-42.
17. Dill, K.A. & Chan, H.S. (1997). From Levinthal to pathways to funnels.Nat. Struct. Biol. 4, 10-19.
18. Dill, K.A. (1985). Theory for the folding and stability of globularproteins. Biochemistry 24, 1501-1509.
19. Dobson, C.M, & Kaplus, M. (1999). The fundamentals of protein folding:bringing together theory and experiment. Curr Opin Struct Biol. 9, 92-101.
20. Dolgikh, D.A., Abaturov, L.V., Brazhnikov, E.V., Lebedev, Y.O.,Chirgadze, I.N. & Ptitsyn, O.B. (1983). Dokl. Akad. Nauk. Sssr. 272,1481-1484.
21. Doster, W., S. Cusack, and W. Petry. 1989. Dynamical transition ofmyoglobin revealed by inelastic neutron scattering. Nature. 337:754-756
22. Fersht, A.R. (1995). Characterizing transition states in protein folding:An essential step in the puzzle. Curr. Opin. Struct. Biol. 5, 79-84.
23. Fersht, A.R. (2000). Transition-state structure as a unifying basis inprotein-folding mechanisms: Contact order, chain topology, stability, andthe extended nucleus mechanism. Proc. Natl. Acad. Sci. USA 97, 1525-1529.
24. Frauenfelder, H., F. Parak, and R. D. Young. 1988. Conformationalsubstates in proteins. Annu. Rev. Biophys. Biophys. Chem. 17:451-479
25. Frye, K.J. and Royer, C.A. (1998) Probing the contribution of internalcavities to the volume change of protein unfolding under pressure. ProteinSci. 7, 2217–2222
26. Hagen S. J., Hofrichter J. and Eaton W.A. (1997) J. Phys. Chem. B 1012352-2365
27. Harrison S.C. & Durbin R. (1985). Is there a single pathway for thefolding of a polypeptide chain?. Proc. Natl. Acad. Sci. USA 82, 4028-4030
28. Hasegawa, J. et al. (1999) Stabilization of Pseudomonas aeruginosacytochrome c551 by systematic aminoacid substitutions based on thestructure of thermophilic Hydrogenobacter thermophilus cytochromec552. J. Biol. Chem., 274, 37533–37537

97
29. Holladay, L.A., Hammonds, R.G. Jr. & Puett, D. (1974). Growthhormone conformation and conformational equilibria. Biochemistry 13,1653-1661.
30. Ikai, A. & Tanford, C. (1971). Kinetic evidence for incorrectly foldedintermediate states in the refolding of denatured proteins. Nature 230,100-102.
31. Ikeguchi, M., Kuwajima, K, Mitani, M. & Sugai, S. (1986). Evidence foridentity between the equilibrium unfolding intermediates: a comparativestudy of the folding reactions of alpha-lactalbumin and lysozyme.Biochemistry 25, 6965-6972.
32. Jackson, S.E. (1998). How do small single-domain proteins fold? FoldingDesign 3, R81-91.
33. Jacob, M., Schindler, T., Balbach, J. & Schmid, F.X. (1997). Diffusioncontrol in an elementary protein folding reaction. Proc. Natl. Acad. Sci.USA 94, 5622-5627.
34. Jaenike, R. (1999) Stability and folding of domain proteins. Progr. Biophys.Mol. Biol 71., 155–241.
35. Jehanly, A.M., Stotter, D.A. and Wilson, M.T. (1976) Magnetic studies onthe ferri-haem undecapeptide of cytochrome c. Eur. J. Biochem., 71, 613-616
36. Johnson, J.B., Lamb, D.C., Frauenfelder, H., Müller, J.D., McMahon,B.H., Nienhaus, G. U. & Young, R.D. (1996) Ligand binding to hemeproteins. VI. Interconversion of taxonomic substates in carbonmonoxymyoglobin. Biophys. J. 71, 1563–1573.
37. Jones, C.M., Henry, E.R., Chan, C.K., Luck, S.D., Bhuyan, A., Roder, H.,Hofrichter, J. and Eaton W.A. (1993) Proc. Natl. Acad. Sci., USA, 90,11860-11864.
38. Kelly, J.W. (1996), Alternative conformations of amyloidogenic proteinsgovern their behaviour. Current Opinion in Structural Biology, 6: 11-17
39. Kendrew, J. C., Dickerson, R. E., Strandberg, B. E., Hart, R. G., Davis,D. D., Phillips, D. C. & Shore, V. C. (1960) The three dimensionalstructure of myoglobin. Nature 185, 422–427.
40. Kim, P.S. & Baldwin, R.L. (1982). Specific intermediates in the foldingreactions of small proteins and the mechanism of protein folding. Ann.Rev. Biochem. 51, 459-490.
41. Kocher, J.P., Prevost M., Wodak S.J., Lee B. (1996). Properties of theprotein matrix revealed by the free energy of cavity formation. Structure 4,1517-29
42. Koga N, Takada S. (2001). Roles of native topology and chain-lengthscaling in protein folding: a simulation study with a go-like model. J MolBiol. 313, 171-80.

98
43. Kuwajima, K. (1977). A folding model of alpha-lactalbumin deducedfrom the three-state denaturation mechanism. J. Mol. Biol. 114, 241-258.
44. Kuwajima, K., Hiraoka, Y. , Ikeguchi, M. & Sugai, S. (1985). pH-jumptitration of the tyrosyl groups of bovine plasma albumin. Biochemistry 24,874-881.
45. Kuwajima, K., Nitta, K., Yoneyama, M. & Sugai, S. (1976). Three-statedenaturation of alpha-lactalbumin by guanidine hydrochloride. J. Mol. Biol.106, 359-373.
46. Kuwajima, K., Yamaya, H., Miwa, S., Sugai, S. & Nagamura, T. (1987).Rapid formation of secondary structure framework in protein foldingstudied by stopped-flow circular dichroism. FEBS Lett. 221, 115-118.
47. Laberge, M., Vreugdenhil, A.J., Vanderkooi, J.M and Butler, I.S. (1998)Microperoxidase-11: molecular dynamics and Q-band excited resonanceRaman of the oxidized, reduced and carbonyl forms J. Biomol. Struct. Dyn.15,1039-1050
48. Labhardt, A.M. (1984). Kinetic circular dichroism shows that the S-peptide alpha-helix of ribonuclease S unfolds fast and refolds slowly.Proc. Natl. Acad. Sci. USA 81, 7674-7678.
49. Lamb D. C., Nienhaus K., Arcovito A., Draghi F.,. Miele A. E, BrunoriM. e Nienhaus G. U. (2002) Structural dynamics of myoglobin: ligandmigration among protein cavities studied by FTIR-TDS spectroscopyaccettato per la pubblicazione su J. Biol. Chem.
50. Lee C, Park SH, Lee MY, Yu MH. (2000). Regulation of protein functionby native metastability. Proc Natl Acad Sci USA. 97(14):7727-31.
51. Levinthal, C. (1968). Are there pathways for protein folding? J. Chem.Phys. 65, 44-45.
52. Li, T., Quillin, M.L., G.N. Phillips, J. & Olson, J.S. (1994) Structuraldeterminants of the stretching frequency of CO bound to myoglobin.Biochemistry 33, 1433–1446.
53. Lim, M., Jackson, T. A. & Anfinrud, P. A. (1997) Ultrafast rotation andtrapping of carbon monoxide dissociated from myoglobin. Nature Struct.Biol. 3, 209–214.
54. Melkers, B., E. W. Knapp, F. Parak, L. Cordone, A. Cupane, and M.Leone. 1996. Structural fluctuations of myoglobin from normal modes,Mössbauer, Raman, and absorption spectroscopy. Biophys. J. 70:2092-2099
55. Momentau, M., Rougée, M. and Loock, B. (1976) Five-coordinate iron-porphyrin as a model for the active site of hemoproteins.Characterization and coordination properties Eur. J. Biochem. 71, 63-76.
56. Moore, G.R., and Pettigrew, G.W. (1990) Cytochrome c:Evolutionary,Structural and Physiochemical Aspects, Springer-Verlag,Berlin.

99
57. Mourant, J. R., Braunstein, D. P., Chu, K., Frauenfelder, H., Nienhaus, G.U., Ormos, P. & Young, R. D. (1993) Ligand binding to heme proteins:II. Transitions in the heme pocket of myoglobin. Biophys. J. 65, 1496–1507.
58. Müller, J.D., McMahon, B.H., Chien, E.Y.T., Sligar, S.G. & Nienhaus,G.U. (1999) Connection between the taxonomic substates andprotonation of histidines 64 and 97 in carbonmonoxy myoglobin. Biophys.J. 77, 1036–1051.
59. Nienhaus, G. U., Mourant, J. R., Chu, K. & Frauenfelder, H. (1994)Ligand binding to heme proteins. The effect of light on ligand binding inmyoglobin. Biochemistry 33, 13413–13430.
60. Nozaka, M. Kuwajima, K., Nitta, K. & Sugai, S. (1978). Detection andcharacterization of the intermediate of the folding pathway of humanalpha-lactalbumin. Biochemistry 17, 3753-3758.
61. Ohgushi, M. & Wada, A. (1983). ‘Molten-globule state’: a compact formof globular proteins with mobile side-chains. FEBS Lett. 164, 21-24.
62. Ostermann, A., Waschipky, R., Parak, F.G. and Nienhaus, G.U. (2000)Ligand binding and conformational motions in myoglobin. Nature, 404,205–208
63. Pascher, T., Chesick, J.P., Winkler, J.R. and Gray, H.B. (1996) Proteinfolding triggered by electron transfer Science 271, 1558-1560.
64. Phillips, G.N., Jr., Teodoro, M.L., Li, T., Smith, B. & Olson, J.S. (1999)Bound CO is a molecular probe of electrostatic potential in the distalpocket of myoglobin. J. Phys. Chem. B 103, 8817–8829.
65. Plaxco, K.M., Simons, K.T. & Baker, D. (1998). Contact order, transitionstate placement and the refolding rates of single domain proteins. J. Mol.Biol. 277, 985-994.
66. Prusiner, S.B. (1998), Prions (Nobel Lecture). Proc. Natl. Acad. of Sci.,USA, 95: 13363-13383
67. Ptitsyn, O.B., Pain, R.H., Semisotnov, G.V., Zerovnik, E. & Razgulyaev,O.I. (1990). Evidence for a molten globule state as a general intermediatein protein folding. FEBS Lett. 262, 20-24.
68. Richards, F.M. (1977) Areas, volumes, packing and protein structure.Annu. Rev. Biophys. Bioeng 6, 151–176.
69. Robson, B. & Pain, R.H. (1976). The mechanism of folding of globularproteins. Equilibria and kinetics of conformational transitions ofpenicillinase from Staphylococcus aureus involving a state of intermediateconformation. Biochem. J. 155, 331-344.
70. Rougée, M. and Brault, D. (1975) Biochemistry, 14, 4100-4105.71. Sali A, Shakhnovich E, Karplus M. (1994). How does a protein fold?
Nature 369, 248-51

100
72. Scott, E.E. & Gibson, Q.H., (1997) Ligand migration in sperm whalemyoglobin. Biochemistry 36, 11909-17.
73. Settanni G, Cattaneo A & Maritan A. (2001). Role of native-statetopology in the stabilization of intracellular antibodies. Biophys J. 81, 2935-45.
74. Shakhnovich, E., Abkevich, V. & Ptitsyn, O. (1996). Conserved residuesand the mechanism of protein folding. Nature 379, 96-98
75. Steinbach P.J., Chu K, Frauenfelder H., Johnson J.B., Lamb D.C.,Nienhaus G.U., Sauke T.B. and Young R.D. (1992) Determination of ratedistributions from kinetic experiments. Biophys J. 61, 235-245.
76. Steinbach, P. J., Ansari, A., Berendzen, J., Braunstein, D., Chu, K.,Cowen, B. R., Ehrenstein, D., Frauenfelder, H., Johnson, J. B., Lamb, D.C., Luck, S., Mourant, J. R., Nienhaus, G. U., Ormos, P., Philipp, R., Xie,A. & Young, R. D. (1991) Ligand binding to heme proteins: theconnection between dynamics and function. Biochemistry 30, 3988–4001.
77. Thornton, J.M., Orengo, C.A., Todd, A.E. and Pearl, F.M. (1999), Proteinfolds, functions and evolution. J. Mol. Biol., 293:333-342
78. Tilton, R.F., Kuntz, I.D. and Petsko, G.A. (1984) Cavities in proteins:structure of a metmyoglobin–xenon complex solved to 1.9 Å. Biochemistry23, 2849–2857.
79. Travaglini-Allocatelli, C., Cutruzzolà, F., Brancaccio, A., Vallone, B. &Brunori, M. (1994) Engineering Ascaris hemoglobin oxygen affinity insperm whale myoglobin: Role of tyrosine B10. FEBS Lett. 352, 63–66
80. Tsong, T.Y., Baldwin, R.L. & Elson, E.L. (1971). The SequentialUnfolding of ribonuclease A: Detection of a fast initial phase in thekinetics of unfolding. Proc. Natl. Acad. Sci. USA 68, 2712-2715.
81. Wetlaufer, D.B. & Ristow, S. (1973). Acquisition of the three dimensionalstructure of proteins. Annual Review of Biochemistry, 42, 135-158
82. White, D.K., Cannon, J.B. and Traylor, T.G. (1979) J. Am. Chem. Soc. 101,2443-2454.
83. Woenckhaus J, Kohling R, Thiyagarajan P, Littrell KC, Seifert S, RoyerCA, Winter R. (2001). Pressure-jump small-angle x-ray scattering detectedkinetics of staphylococcal nuclease folding. Biophys J 80, 1518-23
84. Wolynes, P.G., Onuchic J.N., Thirumalai, D., (1995) Navigating thefolding routes. Science 267, 1619-20.
85. Wong, K.P. & Hamlin, L.M. (1974). Acid denaturation of bovinecarbonic anhydrase B. Biochemistry 13, 2678-2683.
86. Wong, K.P. & Tanford, C. (1973). Denaturation of bovine carbonicanhydrase B by guanidine hydrochloride. A process involving separablesequential conformational transitions. J. Biol. Chem. 248, 8518-8523.

101
Ringraziamenti
Al termine di questo lavoro di tesi desidero per prima cosa ringraziare idocenti guida di questa tesi di dottorato. Per primo il prof. Maurizio Brunori, cheha coordinato e seguito il progetto, in gran parte realizzato nei laboratori da luidiretti nel Dipartimento di Scienze Biochimiche “A. Rossi-Fanelli”. Il suoincessante stimolo intellettuale mi è stato di grande aiuto in tutti i passaggi piùdifficili di questa tesi, fornendomi sempre un punto di partenza per nuovisviluppi nella comprensione dei fenomeni biologici indagati. Un ringraziamentosentito è inoltre indirizzato al prof. Alfredo Colosimo, per le intelligentidiscussioni che ha stimolato nei seminari di noi studenti di dottorato e per avereseguito con attenzione ed interesse i miei progressi.
Per gran parte di questo lavoro sono in debito con Andrea Bellelli, che mi hainsegnato quasi tutto quello che so, mentre il resto lo ha semplicemente corretto.Lavorare insieme a lui è stata un’esperienza scientifica ed umana di grande valore.Un grazie a Stefano, Carlo, Beatrice, Adriana e Federica con i quali ho potutolavorare in grande armonia e in modo proficuo. Desidero inoltre ringraziare tutticoloro che nel mio percorso di formazione mi hanno aiutato e incoraggiato, inmodo particolare il prof. Bruno Giardina, la professoressa Emilia Chiancone e ilprof. Gino Amiconi, insieme naturalmente a tutti gli amici e colleghi delDipartimento di Scienze Biochimiche e del Centro di Biologia Molecolare delCNR.
Ho avuto la fortuna di collaborare con il prof. Ulrich Nienhaus alDipartimento di Biofisica dell’Università di Ulm (Germania), che ringrazio perl’opportunità offertami e per l’ospitalità dimostrata nei miei confronti. Ringrazioinoltre Don Lamb, che mi è stato di grande aiuto nella preparazione erealizzazione di molti degli esperimenti condotti in Germania.
Infine ringrazio coloro che, pur non collaborando da un punto di vistascientifico alla realizzazione di questa tesi, mi sono stati vicini in questi anni diapprendistato; ovvero la mia famiglia, gli inseparabili “amichelli” e soprattuttomia moglie Alessandra.

102

103
Allegati
Di seguito sono riportati le due pubblicazioni scientifiche che sono stateattualmente ricavate dal lavoro di tesi. Durante il periodo di dottorato sono statocoinvolto anche in altri progetti che sono stati sviluppati nel Dipartimento diScienze Biochimiche dell’Università “La Sapienza” di Roma ed ho inoltrecollaborato con il Dr. Marco Girasole del CNR di Frascati e con il Dr. StefanoDella Longa dell’Università dell’Aquila. Il lavoro svolto ha prodotto le seguentipubblicazioni:
Girasole M., Cricenti A., Generosi R., Congiu Castellano A., Boffi F., ArcovitoA., Boumis G. and Amiconi G. Atomic force microscopy study of erythrocyteshape and membrane structure after treatment with a dihydropyridinic drugAppl. Phys. Lett., (2000) 76, 3650-3652
Giangiacomo L., Mattu M., Arcovito A., Bellenchi G., Bolognesi M., Ascenzi P.and Boffi A. (2001) Monomer-dimer equilibrium and oxygen binding propertiesof ferrous Vitreoscilla hemoglobin. Biochemistry. 40, 9311-6.
Della Longa S., Arcovito A., Girasole M., Hazemann J.L., Benfatto M. (2001)Quantitative analysis of x-ray absorption near edge structure data by a fullmultiple scattering procedure: the Fe-CO geometry in photolyzedcarbonmonoxy-myoglobin single crystal. Phys Rev Lett. 87,155501.
Miele A.E., Draghi F., Arcovito A., Bellelli A., Brunori M., Travaglini-AllocatelliC., Vallone B. (2001) Control of heme reactivity by diffusion: structural basis andfunctional characterization in hemoglobin mutants. Biochemistry. 40, 14449-58.













![Lucia 2008[Tesi dottorato-full]R06_low.pdf](https://static.fdocumenti.com/doc/165x107/5866862d1a28abe83f8b65fa/lucia-2008tesi-dottorato-fullr06lowpdf.jpg)




