2-Flogosi Tonsillari Ad Evoluzione Complicata-Lascessoperitonsillare(1)

UNIVERSITA’ DEGLI STUDI DI PISA
Facoltà di Medicina e Chirurgia
TESI DI LAUREA
Studio, mediante esame ecografico, dell’impegno
articolare della mano nei pazienti con sindrome di
Sjogren in fase precoce
Candidato Relatore
Niccolò Possemato Prof. Lucrezia Riente
Anno Accademico 2005-2006
1

INDICE
1. RIASSUNTO
2. INTRODUZIONE
DEFINIZIONE
EPIDEMIOLOGIA
ANATOMIA PATOLOGICA
PATOGENESI
CLINICA
ECOGRAFIA ARTICOLARE
3. SCOPO DELLO STUDIO
4. MATERIALI E METODI
5. RISULTATI
6. DISCUSSIONE
7. BIBLIOGRAFIA
2

Riassunto
La sindrome di Sjogren (SS) è una patologia autoimmune
caratterizzata da infiammazione cronica delle ghiandole
esocrine, in particolare delle ghiandole salivari e lacrimali.
Tra le numerose manifestazioni extraghiandolari che si
possono rilevare durante la malattia, riveste un ruolo di
primaria importanza l’interessamento articolare (artralgie,
rigidità mattutina, artrite intermittente o cronica), la cui
frequenza non è, tuttavia, ancora ben stabilita.
In passato, l’accertamento di tale coinvolgimento si è
sempre basato sulla raccolta dell’anamnesi e sull’esame
clinico dei pazienti, talvolta associati ad esami radiografici
standard, che peraltro sono ormai considerati poco sensibili
nel rilevare segni di artrite, soprattutto in fase iniziale.
E’ noto che l’ecografia consente una valutazione accurata e
riproducibile delle caratteristiche morfo-strutturali dei
tessuti molli e rappresenta una tecnica estremamente
sensibile per individuare i segni precoci della flogosi e del
danno articolare.
Nel presente studio, mediante esame ecografico, sono
state valutate la frequenza e le caratteristiche
3

dell’interessamento delle piccole articolazioni della mano in
pazienti affetti da SS di recente insorgenza.
In 18 soggetti con SS, 17 con sindrome sicca ed in 8
volontari sani sono stati ricercati, a livello delle
articolazioni metacarpo-falangee (MCF) ed interfalangee
prossimali (IFP), la presenza di versamento intrarticolare,
iperplasia sinoviale (con eventuale segnale power-doppler)
ed erosioni ossee.
I risultati evidenziano un’elevata frequenza (55%) di
artrite a carico delle piccole articolazioni delle mani nei
pazienti con SS già in fase iniziale. Le articolazioni più
frequentemente colpite sono le MCF (4 pazienti su 18 con
SS avevano interessamento multiplo – 3 o più - delle MCF
mentre nessuno dei pazienti con sindrome sicca presentava
un simile quadro; pvalue<0,039)
In conclusione, il nostro studio evidenzia come, mediante
l’esame ecografico, sia possibile dimostrare un impegno
articolare in un numero elevato di pazienti con SS già in
fase iniziale.
4

INTRODUZIONE
Definizione
La Sindrome di Sjogren (SS) è una malattia autoimmune
sistemica a carattere infiammatorio cronico che colpisce
tipicamente le ghiandole esocrine ed in particolare le
ghiandole salivari e lacrimali; oltre alle ghiandole esocrine
può interessare potenzialmente tutti gli organi ed
apparati.(1) La SS, può presentarsi in forma isolata (SS
idiopatica o primaria), oppure in associazione ad altre
malattie autoimmuni come l’Artrite Reumatoide, il Lupus
Eritematoso Sistemico o la Sclerodermia (SS secondaria) (2)
Epidemiologia
Dal punto di vista epidemiologico la malattia ha una
distribuzione ubiquitaria e colpisce preferenzialmente
soggetti di sesso femminile (rapporto femmine:maschi = 9:1)
con massima incidenza nella IV-V decade di vita. (3). La sua
prevalenza nella popolazione generale non è stata ancora
precisamente valutata: si ritiene che la forma primitiva si
5

presenti con una percentuale dello 0.3-1.5%. Nella
popolazione geriatrica è stata stimata una prevalenza del 3%
(1). L’incidenza della malattia è stimata in circa 1 nuovo caso
ogni 1000 abitanti\anno. (2,4)
Anatomia Patologica
La SS primitiva presenta un quadro istopatologico
caratterizzato da infiltrati linfocitari focali che interessano
generalmente la regione periduttale delle ghiandole esocrine
ma che potenzialmente possono colpire qualunque organo e
apparato ( ad es. epitelio tubulare renale, epitelio bronchiale,
epitelio dei colangioli epatici etc). Tali aggregati originano
essenzialmente dalle interazioni tra le cellule epiteliali e le
cellule immuni T e B. Gli aggregati linfocitari sono costituiti
prevalentemente da linfociti CD4 positivi che presentano
solitamente un fenotipo memoria (CD45 RO+), esprimono il
recettore TCR a/b e sono in grado si secernere INF-g e IL-
10.(5) In percentuale minore sono stati evidenziati anche
linfociti T CD8 positivi che esprimono l’integrina (CD 103) e
che si localizzano in prossimità di cellule epiteliali acinari E-
caderina positive.(6) Il rapporto CD4/CD8 a livello degli
infiltrati è di 3:1 - 5:1. I linfociti B costituiscono solo il 20%
6

degli infiltrati, e ancora più rari sono le cellule natural killer
ed i macrofagi (complessivamente < 5%).(7) Accanto agli
infiltrati T linfocitari, è possibile osservare frequentemente
anche delle strutture ectopiche che sono analoghe ai centri
germinativi e sono costituite essenzialmente da linfociti B,
con una piccola percentuale di linfociti T e di cellule
follicolari dendritiche. I linfociti B sono organizzati in una
‘dark zone’ centrale in attiva proliferazione (centroblasti) e
in una zona più periferica ‘light zone’ di centrociti che vanno
incontro a processi di selezione per l’espressione in
superficie di anticorpi ad alta affinità. La presenza di questi
centri germinativi ectopici è stata associata ad una più
elevata positività di autoanticorpi anti Ro/SSA ed anti
La/SSB, nonché ad un più alto rischio di trasformazione in
senso linfoproliferativo.(8)
PATOGENESI
Dal punto di vista patogenetico la SS è descritta come un
processo sequenziale “multistep” di natura autoimmunitaria.
Tale processo sembra essere di tipo multifattoriale e
originare dall’interazione tra fattori di predisposizione
genetica e agenti esogeni ambientali verosimilmente virali.
7

Fattori ambientali non meglio specificati, infatti, in soggetti
geneticamente predisposti innescherebbero risposte
autoimmunitarie abnormi sia da parte dei linfociti T che dei
linfociti B rivolte contro autoantigeni tissutali; tali risposte
sarebbero in grado di sostenere, perpetuare e amplificare il
danno tissutale portando ad uno stato infiammatorio cronico
e ad una progressiva perdita funzionale degli organi colpiti.
Predisposizione genetica
I fattori di predisposizione genetica suggeriti sono
essenzialmente rappresentati dai geni del sistema maggiore
di istocompatibilità (HLA). La SS è, infatti, strettamente
associata all’espressione dell’HLA-DR3 e dei geni ad esso
correlati B8 e DQ2. Tale associazione è particolarmente
forte per i soggetti con SS che producono anticorpi anti-
Ro/SSA. Oltre al sistema HLA gli studi più recenti hanno
focalizzato l’attenzione su geni polimorfici che codificano
per molecole coinvolte fisiologicamente nella risposta
immunitaria come il gene dell’IL-10 (aplotipo GCC), il gene del
TNFa (allele TNF-308A) e il gene della catena alfa del
recettore dell’IL4.(9, 10) Altri polimorfismi genici sono stati
osservati a carico del gene della lectina legante il mannosio e
dei recettori di alcune chemochine, in particolare del
recettore CCR5 che lega le chemochine chemoattrattanti
8

delle cellule mononucleari CCL3, CCL4 e CCL5.(11)
Agenti virali
Anche gli agenti virali legati alla patogenesi della SS sono
molteplici. In primis i virus scialotropi come il virus di
Epstein-Barr (EBV), il virus dell’epatite C (HCV) e i
retrovirus come il retrovirus –I della leucemia umana a
cellule T (HTLV-I). Negli ultimi anni, il gruppo di
Moutsopoulos ha identificato la presenza di RNA virale di
Coxsackie virus nelle biopsie delle ghiandole salivari minori di
pazienti con SS e un’omologia di sequenza dell’87% tra un
peptide derivato dalla proteina 2B dei Coxsackievirus e la
regione 222-229 dell’epitopo lineare maggiore
dell’autoantigene Ro60KD che è considerato uno degli
autoantigeni più specifici della SS.(5,12,13,14)
Nonostante le più recenti acquisizione la patogenesi della SS
rimane in larga parte sconosciuta e sono stati elaborati
diversi modelli patogenetici che possano spiegare il ruolo dei
diversi elementi cellulari nel danno ghiandolare che si
verifica in corso di malattia. Il modello patogenetico più
largamente condiviso è quello dell’”epitelite autoimmune”
secondo il quale le cellule epiteliali rivestirebbero un ruolo
cruciale nella formazione degli aggregati linfocitari
periduttali. Esse, infatti, una volta attaccate da un ipotetico
9

agente esogeno, si attiverebbero e inizierebbero a
secernere elevate quantità di citochine proinfiammatorie ed
ad esprimere alti livelli di HLA DR richiamando in situ le
cellule linfocitarie. Le cellule dell’epitelio ghiandolare
fungerebbero da cellule presentanti l’antigene (APC)
attivando i linfociti T che potrebbero a loro volta interagire
con i linfociti B innescando la risposta autoimmune.(15)
Accanto a questo modello istopatologico epiteliocentrico,
però, un secondo modello proposto per la formazione degli
aggregati, è quello di una disregolazione sistemica dei
linfociti T circolanti, ed in particolare dei linfociti Th1
positivi, che migrerebbero attivamente nelle ghiandole
esocrine con un processo di homing mediato dall’ adesione
alle venule ad endotelio alto (HEV), fin dalle prime fasi di
malattia. (5) I linfociti Th1, attraverso la liberazione di
citochine proinfiammatorie, ed in particolare di INF-gamma,
attiverebbero le cellule epiteliali e le indurrebbero ad
esprimere potenti chemoattrattanti in grado di richiamare
altri linfociti e di amplificare il processo infiltrativo. (5) I
chemoattranti implicati sarebbero le chemochine, proteine a
basso peso molecolare in grado di reclutare e attivare
specifiche sottopopolazioni leucocitarie, modificando
l’espressione e lo stato di affinità delle molecole di adesione
10

sulla superficie leucocitaria. Sulla scia di questa ipotesi,
studi recenti hanno focalizzato l’attenzione, in particolare,
su alcune chemochine INF-gamma-indotte come la CXCL10 e
la CXCL9, il cui mRNA è risultato up-regolato a livello delle
cellule epiteliali dei dotti salivari dei pazienti con SS
rispetto ai controlli. Tali chemochine sarebbero in grado a
loro volta di attrarre linfociti T CD3+, questi ultimi in grado
di esprimere in superficie il CXCR3, recettore specifico per
le chemochine suddette. Chemochine diverse come la
CXCL13, chemoattrattanti verso i linfociti B, e il BAFF,
citochina dalla superfamiglia del TNF, sarebbero invece
coinvolti nella formazione delle strutture ectopiche simil
germinative il cui ruolo nella successiva potenziale
trasformazione linfomatosa rimane da chiarire.(16,17)
Comunque abbiano origine gli infiltrati linfocitari, una volta
formatisi, essi comportano una progressiva distruzione
dell’epitelio ghiandolare. Il principale meccanismo di
distruzione dell’epitelio ghiandolare è rappresentato dalla
morte cellulare programmata o apoptosi mediata dal sistema
Fas/FasL e dal rilascio di perforina e grandina.(18,19) Anche
se è ancora da chiarire se lo stimolo iniziale per il processo
apoptotico sia intrinseco alle cellule epiteliali o sia mediato
dai linfociti T, la formazione di blebs di membrana durante
11

l’apoptosi porterebbe alla traslocazione a livello di membrana
di componenti autoantigeniche intracellulari che in questo
modo guadagnerebbero l’ambiente extracellulare e
potrebbero evocare la risposta autoanticorpale.(20,21) Un
altro ulteriore meccanismo di esposizione di autoantigeni
endocitari sarebbe la liberazione di exosomi da parte delle
cellule epiteliali ovvero di vescicole di membrana risultanti
dalla fusione di endosomi e lisosomi. Tale meccanismo è stato
osservato in vitro, ma non ancora in vivo.(22,23) I
meccanismi patogenetici descritti giustificano, in realtà, una
distruzione del 50-60% delle strutture acinari con una
riserva funzionale residua del 40%. L’entità della
compromissione della funzione secretoria è molto superiore
nella SS rispetto al danno organico, e ciò ha portato ad
ipotizzare che altri meccanismi siano coinvolti nella
patogenesi della malattia. A livello del tessuto ghiandolare
residuo, in particolare, alcuni autori hanno ipotizzato una
disregolazione del trasporto dei fluidi, sottolineando in
particolare una anomala distribuzione dei canali numero 5
dell’acquaporina nelle ghiandole salivari.
Tali canali regolano i movimenti dell’acqua attraverso le
membrane biologiche e sono modulati via M3 mAchR, ed una
loro ridotta densità potrebbe contribuire al deficit
12

secretivo dei fluidi. (24) In letteratura sono presenti dati
contrastanti in questo senso, secondo alcuni studiosi, infatti,
la distribuzione e la densità dell’acquaporina 5 nelle
ghiandole salivari non differirebbe nei pazienti con SS
rispetto ai controlli sani. (25) Altri autori, negli ultimi anni,
hanno indagato possibili alterazioni del circuito
neurosecretorio che regola il funzionamento delle ghiandole
esocrine. Sul parenchima ghiandolare sono presenti recettori
transmembrana di tipo muscarinico, in particolare gli M3,
che legano l’acetilcolina e che regolano il trasporto di ioni e
di acqua attraverso le membrane cellulari, nei dotti
ghiandolari. Il numero dei recettori M3 eccede quello delle
sinapsi neuronali cosicché l’acetilcolina rilasciata da una
terminazione nervosa può stimolare più acini adiacenti a
quello direttamente innervato. Le alterazioni funzionali a
carico del circuito neurosecretivo riguardano essenzialmente
il braccio efferente del circuito, mentre quello afferente
rimane integro, come è evidente, dal momento che il paziente
avverte i sintomi legati alla secchezza orale e oculare. (26)
Uno dei meccanismi potenzialmente responsabili
dell’iposecrezione potrebbe essere rappresentato da un
ridotto rilascio di neurotrasmettitori (Ach) a livello delle
giunzioni sinaptiche e/o da un’ alterata risposta ai
13

neurotrasmettitori a livello post-sinaptico. Durante il
processo infiammatorio, citochine pro-infiammatorie come
l’IL-1, il TNF-alfa o l’IL-6, possono interferire con il rilascio
di acetilcolina da parte delle terminazioni colinergiche,
inibendolo. Anche la produzione di secondi messaggeri a
livello post-sinaptico è allo stesso modo fortemente
condizionata in senso negativo dalla presenza di tali
citochine, e di conseguenza le cellule ghiandolari appaiono
meno responsive ai segnali efferenti. (27) Infine studi
recenti hanno evidenziato la presenza di elevati livelli di
anticorpi anti-M3R nei pazienti con SS in grado di fungere
da antagonisti muscarinici inibendo la contrazione della
muscolatura liscia. Il blocco del M3R potrebbe interferire
con la secrezione ghiandolare anche inducendo una
traslocazione dell’acquaporina.(28)
In conclusione la patogenesi della SS rimane ancora in larga
da parte da chiarire essendo legata sia ad un danno organico
che funzionale dei diversi parenchimi e solo acquisizioni
ulteriori consentiranno di mettere in atto strategie
terapeutiche sempre più specifiche e efficaci.
14

CLINICA
MANIFESTAZIONI GHIANDOLARI
Sintomatologia orale
La xerostomia è presente in oltre l’85% dei pazienti con SS
primaria e in oltre il 75% di quelli affetti da SS secondaria.
(29) L’iposecrezione salivare e le concomitanti alterazioni
qualitative della saliva prodotta comportano,
progressivamente, l’instaurarsi di una condizione
caratterizzata da iperviscosità salivare, danno a carico dei
processi di mineralizzazione dentaria e alterazione della
flora microbica orale con aumento relativo dei batteri
‘cariogeni’ e delle diverse specie di Candida.(30) I sintomi
soggettivi riferiti dal paziente sono quindi di difficoltà nella
masticazione, di disfagia, di modificazioni del gusto (es
‘sensazione metallica in bocca’, ‘bocca amara’), di tosse secca,
di difficoltà nell’eloquio, di disagio nel portare protesi
dentarie, e di necessità di bere molto frequentemente con
conseguente nicturia e disturbi del ritmo sonno-veglia. In
caso di sovrainfezione da Candida sono frequenti anche il
bruciore e la sensazione di fastidio con i cibi piccanti. (31)
All’esame obiettivo la mucosa orale appare spesso
15

eritematosa e asciutta e di solito si osserva la mancanza di
saliva sul pavimento della bocca e/o la presenza di saliva
particolarmente densa e viscosa in corrispondenza dello
sbocco dei dotti delle ghiandole salivari maggiori. Sono
frequenti, inoltre, le carie dentarie che hanno la peculiarità
di prendere origine dal colletto del dente anziché dall’apice
del dente. Carie apicali si ritrovano soprattutto sulle teste
degli incisivi. La superficie dorsale della lingua è spesso
anch’essa arrossata e può presentare atrofia delle papille
filiformi e fissurazioni laterali che possono essere sede di
sovrainfezioni fungine. La candidiasi orale si manifesta, oltre
che con le classiche lesioni a placca, con l’arrossamento e
l’assottigliamento della mucosa e con cheilite angolare.(31)
In un terzo circa dei pazienti le ghiandole salivari maggiori
possono presentare tumefazioni ricorrenti e bilaterali. La
tumefazione delle parotidi è la più evidente ma spesso è
preceduta da quella delle sottomandibolari che, tuttavia, può
essere misconosciuta sia dal paziente che dal medico.
All’esame obiettivo le ghiandole tumefatte hanno una
consistenza teso-elastica e di solito non sono dolenti alla
palpazione. Possono, inoltre, presentare delle oscillazioni
periodiche nelle loro dimensioni senza, tuttavia, cambiamenti
repentini.(30)
16

Xerostomia e tumefazione delle ghiandole salivari sono
sintomi di per sé aspecifici che nella valutazione clinica
richiedono un attenta diagnosi differenziale. (30,32)
La secchezza orale può dipendere da patologie ghiandolari o
da malattie extraghiandolari. Le affezioni responsabili
possono inoltre essere temporanee e reversibili o croniche.
Tra le cause non-ghiandolari di xerostomia la più frequente è
quella iatrogena. Farmaci anti-depressivi, neurolettici,
parasimpaticolitici (es anti-parkinson), anti-istaminici,
decongestionanti (es pseudoefedrina +triptrolidina ) e alcuni
anti-ipertensivi sono frequentemente chiamati in causa.
Nell’ambito delle patologie in grado di interferire con il
circuito neurosecretivo che regola la produzione salivare
sono inoltre da escludere disturbi d’ansia, disfunzioni
autonomiche e affezioni del SNC traumatiche, degenerative
o infiammatorie (es. morbo di Alzheimer, encefalite, tumori
cerebrali). (30) E’ verosimile che anche la sindrome sicca
descritta in corso di fibromialgia primitiva o di ‘sindrome da
fatica cronica’ possa essere la conseguenza di alterazioni
della funzione neurosecretiva a livello dei nuclei salivare e
lacrimale della ‘medulla’.(33)
Nell’ambito delle cause extra-ghiandolari di xerostomia sono
da includere anche cause sistemiche di disidratazione come
17

un diabete mellito non controllato, diabete insipido, stati di
ipertermia, vomito prolungato o diarrea, emorragie massive e
infine un ridotto introito di acqua. Generalmente queste
condizioni sono tuttavia acute e reversibili una volta rimossa
la causa scatenante. (34)
Le cause ghiandolari di xerostomia vengono, invece,
differenziate in condizioni in cui il danno ghiandolare è
limitato al parenchima ghiandolare e condizioni in cui
l’interessamento delle ghiandole salivari è secondario ad una
patologia sistemica. (34) Tra le prime riveste una particolare
importanza il danno parenchimale provocato dalla terapia
radiante effettuata in caso di tumori della testa e del collo.
Nell’ambito delle condizioni sistemiche nella diagnosi
differenziale con la sindrome di SS primaria e scondaria
vanno considerate altre patologie croniche come la
sarcoidosi o la amiloidosi, patologie virali come l’infezione da
HCV e da HIV o la ‘graft versus host disease’.(34) Infine è
dubbio se lo stesso invecchiamento fisiologico possa
comportare un decremento della funzione secretiva
sottomandibolare e sublinguale. L’aumentata prevalenza di
xerostomia nell’anziano sembra piuttosto dipendere dalla
maggiore incidenza delle altre condizioni responsabili di ‘dry
mouth’ come le affezioni del SNC (depressione, Parkinson,
18

Alzhaimer), l’uso di farmaci anticolinergici e la
disidratazione conseguente a patologie metaboliche ed
endocrine. (35)
Alcune delle cause sopra descritte come la sarcoidosi,
l’infezione da HIV o altre infezioni virali possono rendersi
responsabili anche di tumefazione delle ghiandole parotidi.
Anche in questi casi come nella SS la tumefazione è
generalmente bilaterale. Una tumefazione monolaterale,
meno comune in corso di SS, deve, invece, far escludere
un’infezione batterica retrograda dalla cavità orale, una
neoplasia epiteliale un linfoma non Hodgkin o, specie
nell’anziano, una forma di scialoadenite cronica ad esempio
secondaria a litiasi salivare.(30,32,35)
Xeroftalmia
La secchezza oculare è l’altro sintomo cardine della sindrome
sicca ed è legata ad una ridotta produzione del film
lacrimale.(36)
La principale funzione delle lacrime è quella di mantenere
umettata la superficie anteriore dell’occhio; in tal modo esse
creano un ambiente idoneo per le cellule dell’epitelio
corneale, proteggono il principale meccanismo rifrattivo
dell’occhio e facilitano il mantenimento di una superficie
corneale uniforme e libera da detriti per opera delle
19

palpebre. Il film lacrimale è composto da tre strati: uno
esterno, superficiale, di natura lipidica, prodotto dalle
ghiandole di Meibomio, che serve ad impedire l’evaporazione
dello strato acquoso sottostante; uno strato acquoso
intermedio che da solo costituisce il 90% dello spessore del
film lacrimale che è secreto dalla ghiandola lacrimale
principale e dalle ghiandole lacrimali accessorie;uno strato
mucoso profondo a contatto con la cornea, prodotto dalle
cellule caliciformi della congiuntiva, che serve a tenere lo
strato acquoso aderente all’epitelio corneale idrorepellente.
(36) In condizioni fisiologiche si realizza un equilibrio
ottimale tra la continua produzione di lacrime e il loro
smaltimento mediante l’evaporazione o attraverso le vie
lacrimali di deflusso. Tale equilibrio può essere alterato sia
per difetto di produzione che per eccesso di evaporazione.
In base alla definizione del ‘National Eye Institute/Industry
Workshop on dry eyes in Bethesda’ la definizione di ‘dry eye’
è quella di un disordine del film lacrimale legato ad un deficit
lacrimale o ad una eccessiva evaporazione.(37) Nella SS
l’iposecrezione è causata dalla prima condizione ovvero dalla
ridotta produzione della componente acquosa intermedia del
film lacrimale. Il film lacrimale risulta pertanto, più instabile
e può rompersi tra un ammiccamento e l’altro esponendo
20

all’evaporazione l’epitelio della congiuntiva e della cornea. I
sintomi che ne derivano sono rappresentati da sensazione di
sabbia nell’occhio, bruciore e arrossamento da irritazione
della congiuntiva e dei margini palpebrali. Alcuni pazienti
riferiscono la sensazione di avere occhi pesanti e gonfi. Nei
casi più gravi si possono formare erosioni della superficie
corneale che a loro volta possono trasformarsi in ulcere con
conseguenze anche pesanti sul visus.(33)
Le condizioni di iposecrezione del film lacrimale, oltre alla
SS, prevedono quadri di cherato-congiuntivite sicca non
infettiva attribuibili ad un’alterazione funzionale del circuito
riflesso neurosecretivo lacrimale, a ostruzione dei dotti
lacrimali o a patologie ‘mild’ di incerta eziologia delle
ghiandole lacrimali stesse. Tutte e tre le condizioni vengono
definite nel complesso cherato-congiuntiviti non-Sjogren e
considerate come forme fruste della malattia
autoimmune.(37) L’altra categoria è invece quella delle
‘cheratocongiuntiviti sicche’ da eccessiva evaporazione. Tra
le condizioni più comuni incluse in questo gruppo sono
compresi l'uso protratto di lenti a contatto, i disordini
tiroidei (esoftalmo e proptosi) e le affezioni delle ghiandole
di Meibomio la cui flogosi compromette la formazione dello
strato lipidico esterno del film lacrimale.(37,38)
21

MANIFESTAZIONI EXTRAGHIANDOLARI
Le manifestazioni extra-ghiandolari coinvolgono circa un
20% dei pazienti, ed hanno un decorso generalmente benigno
ed una prognosi favorevole.
• Impegno cutaneo
L’impegno cutaneo è considerato una delle manifestazioni
extraghiandolari caratteristiche della SS spaziando dalla
xerosi cutanea a quadri vasculitici; questi ultimi sono stati
recentemente descritti in uno studio sulle manifestazioni
cutanee in corso di SS e quantificate in circa il 10% dei
quadri presenti.(39)
La caratteristica principale delle vasculiti associate alla
SS è l’interessamento dei vasi di piccolo calibro con
quadri di vasculite leucocitoclastica che si manifesta
clinicamente con la porpora cutanea. Altri autori hanno
dimostrato l’associazione tra porpora cutanea e sviluppo
di linfoma.
Questi studi dimostrano l’importanza delle vasculiti
cutanee nella prognosi dei pazienti con SS primaria.
Oltre all’impegno cutaneo su base vasculitica vengono
22

riportate lesioni a coccarda, pallide al centro con bordi
eritematosi (eritema anulare), rilevati che si localizzano
tipicamente alle estremità superiori e al dorso, descritte
in origine nei pazienti Asiatici con SS e recentemente
descritte in pazienti di etnia Caucasica. (40)
Queste lesioni sono clinicamente identiche a quelle
descritte nei pazienti con Lupus cutaneo subacuto,
suggerendo una causa comune (in relazione con gli
anticorpi anti- Ro/SSA).
Nella tabella seguente sono riportate altre patologie
cutanee descritte nei pazienti con SS primitiva
Interessamento cutaneo nella SS primariaVasculiti cutanee Altre lesioni cutanee
• vasculiti dei piccoli vasi associate a SS
• vasculiti crioglobulinemiche
• vasculiti orticarioidi
• altrevasculiti leucocitoclastiche
• vasculiti dei vasi di mediodiametro
associate a SS
• eritema anulare
• eritema nodoso
• livedo reticularis
• porpora trombocitopenica
• lichen planus
• vitiligine
• amiloidosi cutanea
• granuloma anulare
• panniculite granulomatosa
23

• Impegno polmonare
Numerosi studi hanno recentemente ridimensionato
l’impegno interstiziale del parenchima polmonare
dimostrando come predomini, invece, l’interessamento dei
bronchi e dei bronchioli.(41,42) Franquet et al hanno
descritto l’interessamento bronchiolare in circa un terzo
dei pazienti con una frequenza più alta di “air trapping”
nei lobi inferiori.(43) Papiris et al hanno descritto
l’interessamento delle piccole vie aeree come il maggiore
disordine funzionale, mentre Tabuli et al hanno
dimostrato come un coinvolgimento delle vie aeree fosse
dimostrabile alla TC in oltre la metà dei loro pazienti.(44)
I test di funzionalità polmonare non sempre correlano con
quanto evidenziato alla TC suggerendo l’opportunità di
adoperare entrambe queste procedure diagnostiche nei
pazienti in cui si sospetti un impegno polmonare.
La storia naturale dei pazienti con interessamento
polmonare in corso di SS primaria è stata studiata da
Davidson et al il quale ha dimostrato come, benché
l’insorgenza dell’impegno d’organo sia precoce, la grande
maggioranza dei pazienti non svilupperà una patologia
polmonare progressiva.(45)
24

• Impegno renale
L’impegno renale in corso di SS viene solitamente
considerato essenzialmente tubulare benché diversi studi
abbiano descritto la presenza di una gromerulonefrite in
una percentuale rilevante di pazienti.(46)
Nei pazienti con glomerulonefrite è stata documentata in
ordine di frequenza:
glomerulonefrite membranoproliferativa;
glomerulonefrite con proliferazione mesangiale e
glomerulonefrite membranosa.
In questi studi è stata rilevata la presenza di
crioglobuline nel sangue di circa la metà dei pazienti con
una modesta percentuale (circa il 7%) di progressione
verso l’insufficienza renale.
Tanto l’mpegno tubulare che glomerulare collegato alla SS
hanno importanti implicazioni patogeniche, cliniche e
prognostiche.
L’interessamento tubulare viene considerata una epitelite
tubulare specifica, solitamente diagnosticata nei pazienti
più giovani e caratterizzata da un decorso clinico
indolente senza progressione verso l’insufficienza renale.
La glomerulonefrite invece, può essere considerata come
un’impegno renale severo, strettamente correlato alla
25

presenza di crioglobuline e all’ipocomplementemia, che
appare tardivamente nella storia clinica del paziente ed è
correlata ad una maggiore morbilità e mortalità.
Una biopsia renale non è verosimilmente necessaria in
pazienti con sospetto interessamento tubulare mentre la
glomerulonefrite implica una diagnosi precoce ed una
accurata gestione del paziente. (47)
• Impegno vascolare
La manifestazione vascolare più comune in corso di SS è
probabilmente il fenomeno di Raynaud, con una prevalenza
del 13%;(48) nel decorso della malattia il fenomeno di
Raynaud può avere un andamento polimorfico: nella
maggior parte dei casi rimane immodificato ma, può
andare incontro a remissione (circa il 15% dei casi) oppure
il numero degli attacchi può ridursi drasticamente (circa il
30% dei casi). (40) Il fenomeno di Raynaud può associarsi
alla presenza delle cosiddette “dita a salsicciotto” o alla
calcificazione dei tessuti molli ma, a differenza di quanto
avviene nella Sclerodermia, i pazienti non sviluppano
ulcere digitali o teleangectasie.
26

• Impegno neurologico
Recenti studi identificano l’impegno del Sistema Nervoso
Centrale (SNC) come frequente manifestazione
extraghiandolare in corso di SS primaria; la reale
prevalenza dell’interessamento neurologico in questi
pazienti è, però, difficilmente valutabile data l’ampia
gamma di possibili manifestazioni cliniche e la
sovrapposizione che queste possono avere con patologie
neurologiche non legate alla SS, soprattutto nel paziente
anziano (patologie cerebrovascolari, Alzheimer, demenza
multiinfartuale..).(49)
A livello periferico il quadro clinico più frequente è quello
della polineuropatia periferica sensitivo-motoria a
distribuzione distale e simmetrica. (50,51) Si tratta di un
quadro di neuropatia assonale caratterizzato dalla
degenerazione delle estremità distali degli assoni di
maggiore lunghezza, con secondaria degenerazione delle
guaine mieliniche demielinizzate. Dal punto di vista clinico
è di solito presente una ipoestesia globale, quasi
omogenea, anche se talora vi è un più marcato deficit
della sensibilità termo-dolorifica. La sintomatologia
d’esordio è spesso inizialmente costituita da disturbi
sensitivi irritativi e deficitari, con parestesie ed
27

ipoestesia a carico delle punte delle dita dei piedi. Solo
più tardivamente, con l’estensione prossimale dei deficit
sensitivi, si manifestano i disturbi motori, tipicamente con
distribuzione a calza.
Il quadro più peculiare di neuropatia periferica in corso di
SS, seppure non il più comune è quello della neuropatia
sensitiva (51) Per neuronopatia o degenerazione neuronale
primitiva, si intende la degenerazione del corpo della
cellula nervosa e la conseguente sofferenza dei
prolungamenti periferici e centrali. Nella neuronopatia
sensitiva istologicamente si osserva una infiltrazione
linfocitaria dei gangli delle radici dorsali e delle radici
stesse responsabile della loro progressiva
degenerazione.(50) La neuronopatia sensitiva è
caratterizzata da una alterata funzione delle fibre
nervose di grosso calibro che si manifesta clinicamente
con alterazioni della sensibilità propriocettiva e
chinestesica. (51) L’esordio della neuronopatia può essere
acuto o più frequentemente può avere un andamento
progressivo nell’arco di mesi o anni.(50) Il quadro clinico
è quello di una neuropatia asimmetrica che può coinvolgere
gli arti superiori, le gambe, la faccia o il tronco.(50) I
pazienti possono presentare: atassia ovvero quadri di
28

incoordinazione motoria sia nella statica che nella
dinamica, movimenti pseudoatetosici involontari, lenti,
aritmici e protratti nel tempo e areflessia a carico degli
arti colpiti. (51) La diagnosi differenziale è con le forme
di neuropatia paraneoplastica, con la variante sensitiva
della Guillain Barrè (Miller Fisher syndrome) e con i
quadri di intossicazione da arsenico, cisplatino e
piridoxina. (50)
Di comune riscontro nella SS come nelle altre
connetivopatie sono le sindromi da intrappolamento ed in
particolare la sindrome del tunnel carpale legata ad
infiltrazione infiammatoria dei tessuti costituenti il
tunnel carpale e dei tendini.(50)
Un altro quadro relativamente peculiare descritto nei
pazienti con SS è la neuropatia sensitiva del
trigemino.(50) Tale affezione causa intorpidimento e
disestesie in tutto il territorio di distribuzione delle tre
branche del V nervo cranico ed in particolare nel
territorio mascellare e mandibolare, spesso con
distribuzione asimmetrica. La funzione motoria del
trigemino è costantemente conservata. (50)
29

• Manifestazioni ematologiche
Alterazioni ematologiche in corso di SS sono tutt’altro
che rare ma generalmente rivestono uno scarso
significato clinico. (52)
Quadri di linfocitopenia assoluta o limitata ai linfociti T
CD4 + possono costituire una manifestazione d’esordio
della malattia benchè, solitamente, non comportino
l’instaurarsi di infezioni opportunistiche severe. (53, 54)
Forme di trombocitopenia autoimmune legate
all’espansione policlonale dei linfociti B sono ugualmente
spesso presenti ma, anche in questo caso, di regola non si
associano a rischio di sanguinamento per il paziente e
rispondono bene alla terapia steroidea a medio-basso
dosaggio. (53)
Per quanto concerne la linea eritroide vengono descritti in
corso di SS quadri di anemia a diversa patogenesi a volte
coesistenti (53). Possono costituire manifestazioni di
malattia, infatti, forme di anemia emolitica con test di
Coombs positivo come pure casi di anemia infiammatoria
cronica. (53). Più raramente a livello di case-reports sono
descritte forme di anemia perniciosa (55) o di anemia
aplastica, queste ultime spesso in associazione a infezioni
da parvovirus B19. (56)
30

Dal punto di vista ematologico le complicanze più temibili
nei pazienti con SS sono rappresentate dalle malattie
linfoproliferative che si manifestano nel 5-10% dei casi.
(57, 58) Nell’ambito delle malattie linfoproliferative
vanno distinti i quadri ad evoluzione benigna come la
gammopatia monoclonale (MGUS) o la linfoadenopatia
reattiva che sono tipiche delle malattie infiammatorie
croniche, da quadri di linfoma franco che possono
costituire complicanze life-threatening per il paziente.
(59)
• Apparato gastroenterico
L’apparato gastroenterico, considerata la ricchezza di
ghiandole esocrine minori e maggiori (fegato e pancreas)
di cui è fornito, rappresenta uno degli apparati più
frequentemente colpiti in corso di SS.
A carico del tratto faringo-esofageo il sintomo più spesso
riferito dai pazienti è la disfagia soprattutto per i cibi
solidi o secchi (60). La disfagia è in parte legata alla
mancanza di saliva e alla conseguente secchezza del
faringe e dell’esofago ed in parte è dovuta alle alterazioni
della cinesi esofagea (61).
A livello gastroenterico il quadro più frequentemente
31

descritto nei pazienti affetti da SS è la gastrite atrofica
responsabile della sintomatologia dispeptica e
dell’epigastralgia spesso lamentate dai pazienti. (62)
Dal punto di vista istologico la mucosa gastrica presenta i
diversi gradi della gastrite cronica. (62)
La gastrite atrofica inizia in genere dall’antro e si
estende in direzione prossimale verso il corpo e il fondo
gastrico. Molti pazienti sviluppano di conseguenza
alterazioni funzionali della mucosa gastrica con ipo-
acloridria, ipo-pepsinogenemia e iper-gastrinemia.(63)
Il coinvolgimento dell’intestino tenue in corso di SS è
controverso. In letteratura, in particolare, è stata
descritta un’aumentata incidenza di casi di celiachia tra
pazienti affetti da SS e viceversa.(60)
L’iperamilasemia è un altro rilievo relativamente
frequente nei pazienti con SS che si ritrova in circa Il
25% dei pazienti (59%). Tale rilievo non si traduce
clinicamente, se non in rari casi, in forme di pancreatite
acuta o cronica ma piuttosto sembra rispecchiare una
forma di impegno infiammatorio subclinico del pancreas.
(60, 61, 62, 64, 65)
Anche l’impegno epatico in corso di SS primitiva è raro e
spesso subclinico, sono frequenti, invece, forme di SS
32

secondaria in associazione a malattie epatiche croniche
come la cirrosi biliare primitiva. (65)
Nella SS primitiva i pazienti presentano per lo più una
modesta epatomegalia e un moderato rialzo degli enzimi di
necrosi e/o di colestasi. (60)
• Impegno muscolo-scheletrico
L’impegno dell’apparato muscoloscheletrico, nell’ambito
delle manifestazioni extra-ghiandolari della SS, riveste
un ruolo di primaria importanza per frequenza e
caratteristiche cliniche potendo manifestarsi fin
dall’esordio. Il coinvolgimento dell’apparato muscolo-
scheletrico, infatti, si verifica nel 60-70% dei pazienti e
nel 28% fin dall’esordio della malattia.(57, 3, 59)
La manifestazione clinica più frequente è l’artralgia a
carico delle piccole e grandi articolazioni (in particolare
del ginocchio). La prevalenza dell’artrite in letteratura
varia considerevolmente. In uno studio prospettico
finlandese, condotto su 110 pazienti seguiti per 3 anni, la
prevalenza rilevata è stata del 24%. (67) Altri studi
descrivono una prevalenza inferiore intorno al 10-11%.
(68, 69)
L’artrite ha le caratteristiche di una poliartrite
33

simmetrica che può coinvolgere grandi e piccole
articolazioni interessando con pari frequenza sia le
metacarpofalangee e le interfalangee prossimali che le
caviglie, le spalle, le ginocchia e i gomiti. La simmetricità
dell’impegno articolare è analoga a quella che occorre in
corso di Artrite Reumatoide e di Lupus Eritematoso
Sistemico. A differenza della prima si tratta, tuttavia,
generalmente di un’artrite non erosiva che si caratterizza
per una risposta favorevole alla terapia di fondo.
Rispetto ai quadri di artrite descritti in corso di Lupus
Eritematoso Sistemico è, invece, più frequente l’impegno
delle articolazioni degli arti inferiori e delle caviglie in
particolare (70, 69) Sono, inoltre, rare le deformità
articolari anche se sono descritti casi di artropatia di
Jaccoud. (71) Il quadro poliartritico è, infine simile a
quello osservabile in altre patologie come la sarcoidosi
(specie nel caso del coinvolgimento delle caviglie) e le
artriti virali, anche in considerazione del modesto rialzo
degli indici aspecifici di flogosi che si associa alla fase
acuta dell’impegno articolare.(72)
I diversi studi della letteratura non hanno documentato
correlazioni clinico-serologiche positive tra la presenza di
impegno articolare e la positività di specifici subset
34

autoanticorpali. (72) Non sono state descritte, infatti, a
differenza di quanto avviene per altre manifestazioni
extraghiandolari, correlazioni tra la presenza di
artralgie/artrite e la positività di fattore reumatoide,
anticorpi antinucleari, anticorpi contro antigeni nucleari
estraibili (Ro/SSA; La/SSB) e ipergammaglobulinemia
che rappresentano i più importanti markers sierologici di
malattia.(72).
35

DIAGNOSI
La diagnosi di SS si avvale di elementi clinici, sierologici,
strumentali e istologici secondo un algoritmo validato che
comprende la somministrazione di un questionario per il
rilievo della sintomatologia soggettiva oculare e orale, test
funzionali di studio del film lacrimale (test di Schirmer e
Rosa Bengala), test morfologici/funzionali di studio delle
ghiandole salivari maggiori (scialografia, scialometria e
scintigrafia), valutazione istologica delle ghiandole salivari
minori e infine, ricerca di autoanticorpi non organo specifici
(Ro/SSA e La/SSB in particolare) (59). Il quadro
sierologico della SS è estremamente variegato
caratterizzandosi per la positività di un’ampia gamma di
autoanticorpi non organo specifici (anticorpi antinucleari
presenti in circa l’80% dei pazienti; fattore reumatoide
positivo nel 90%, ENA Ro/SSA e La/SSB: 50-90%) (1, 3)
Data l’aspecificità della xerostomia come sintomo soggettivo
l’iter diagnostico della SS prevede che la sintomatologia
orale venga obiettivata sia dal punto di vista istologico che
strumentale.(59)
La biopsia delle ghiandole salivari minori è considerata ad
oggi il criterio diagnostico più specifico di SS (82-95%). (73)
Ciononostante non ha validità assoluta in quanto la
36

percentuale dei falsi positivi è intorno al 6-8% e uno score
uguale a 1 (Chisolm e Mason) si ritrova in soggetti sani ed in
altre patologie come la miastenia e le scialoadeniti la
sensibilità dell’esame è più bassa (75-90%) e la percentuale
dei falsi negativi è intorno al 20%.(74,75)
I limiti principali dell’esame sono legati alla sua invasività,
all’andamento fluttuante del grado di infiltrazione nelle
ghiandole, alla necessità di prelevare un campione di tessuto
sufficientemente grande considerando la variabilità inter-
ghiandolare dell’infiltrato ed al fatto che il danno a carico
delle ghiandole salivari minori e di quelle maggiori può non
essere identico. Nonostante questa possibile discrepanza la
biopsia delle ghiandole salivari maggiori non rientra tra i test
diagnostici routinari e viene praticata solo nel sospetto di
una patologia tumorale.
Oltre all’istologia l’obiettivazione della sintomatologia orale
avviene mediante: scialometria, scialografia o
scintigrafia.(59)
La scialometria misura il flusso salivare e può valutarne
l’intera produzione o quella selettiva di una ghiandola.
L’esame può essere inoltre eseguito in condizioni basali o
dopo stimolo. I ‘Revised Criteria’ prevedono che l’esame
debba essere eseguito in condizioni basali e considerano
37

patologico un flusso salivare < a 1,5 min /15 minuti.(59) Il
test basale rispetto a quello condotto dopo stimolo ha il
vantaggio di cogliere eventuali alterazioni fin dagli stadi più
precoci di malattia, non è invasivo ed è riproducibile.(76)
La scialografia è un’indagine radiografica che consente di
valutare l’architettura e la configurazione del sistema
duttale. Il reperto caratteristico della SS è quello della
cosiddetta tempesta di neve ‘snow-storm’ o ad albero di
Natale. Quando il danno è severo può essere osservata una
distruzione completa delle strutture. Lo studio richiede
l’instillazione di mezzo di contrasto nel dotto escretore che
deve essere reperito e incannulato. Da qui la relativa
invasività dell’esame che ne controindica l’esecuzione nei casi
gravi per la potenziale persistenza indefinita del mezzo di
contrasto all’interno della ghiandola. Dopo la biopsia delle
ghiandole salivari minori la scialografia rimane comunque il
test diagnostico più accurato nella diagnosi di SS (sensibilità
72-86%9 e specificità del 78-100%).(74)
La scintigrafia fornisce una valutazione funzionale delle
ghiandole salivari in maniera non invasiva. L’indagine consiste
nel somministrare del pertecnato di sodio marcato con Tc
99m e nel valutare la velocità e l’entità della captazione del
mezzo di contrasto. Il test non è invasivo ha una alta
38

sensibilità ma è poco specifico (sensibilità 75-87%,
specificità 54-79%).(74)
I test diagnostici che consentono una valutazione obiettiva
della sindrome sicca oculare sono il test di Schirmer e il
Rosa Bengala.(4)
Il test di Schirmer fornisce una valutazione quantitativa
della produzione delle lacrime e consiste nella misurazione
della secrezione lacrimale in un tempo prefissato.(59) Le
estremità di due piccole strisce di carta bibula sterile
vengono piegate e poste nel fornice congiuntivali inferiore
tra il bordo palpebrale (1/3 esterno) e la sclera e lasciate in
situ per 5 minuti. Il test è positivo se dopo 5 ‘ sono bagnati <
di 5 mm della striscia di carta bibula.
L’altra modalità con cui viene eseguito il test è il cosiddetto
test di Schirmer II che permette di misurare l’output
massimo delle ghiandole lacrimali maggiori e minori dopo
stimolo. Con un cotton-fiock viene provocato il riflesso naso-
lacrimale, in assenza di anestesia. ll test è più specifico ma
meno sensibile in special modo nelle fasi precoci di malattia.
(59) La valutazione qualitativa del film lacrimale è invece
ottenuta con il test al Rosa Bengala. Il Rosa Bengala è un
colorante specifico per cellule devitalizzate e mucina. Una
goccia di colorante viene instillata nell’occhio e lavata con
39

lacrime artificiali. In presenza di un danno dell’epitelio
congiuntivale o corneale il colorante si fissa sulle cellule e le
lesioni epiteliali possono essere osservate con
l’oftalmoscopio o, meglio, con la lampada a fessura. In
alternativa al Rosa Bengala viene descritto anche l'impiego
del verde di lissamina che risulta meno irritante perché non
fototossico.(77) Nell’ambito dei test oculari, infine, non è
contemplato nei test validati per la classificazione della
malattia ma è di comune uso clinico anche il break-up time
che valuta il tempo di rottura del film lacrimale tra un
ammiccamento e l’altro valutato mediante l’impiego di
fluoresceina. Un tempo di rottura troppo breve del film
lacrimale indica anomalie nella mucina o nello strato
lipidico.(77)
40

Criteri classificativi
Stabilire dei criteri classificativi per la SS che fossero
universalmente accettati, ha costituito, negli ultimi venti
anni, una delle impegni principali per gli esperti di SS di
tutto il mondo. Gli attuali criteri classificativi per la SS
primitiva e secondaria sono stati elaborati e validati solo
recentemente, nell’ambito dell’ American-European
Consensus Group nel 2002 (tabella 1). (59) I criteri
classificativi data la loro alta sensibilità e specificità
possono avere significato diagnostico, anche se, soprattutto
quando la malattia è in fase iniziale, una quota parte dei
pazienti può essere misconosciuta. E’ stato così creato un
algoritmo diagnostico decisionale (‘classification tree
procedure’) che appresenta una valida alternativa ai criteri
presentati. Questi ultimi rimangono tuttavia lo strumento più
utile nella standardizzazione dei pazienti da includere nei
trial clinici ed epidemiologici. (59)
Sono attualmente in fase di elaborazione criteri di
valutazione dell’ “outcome” di malattia che saranno in grado
di consentire un migliore inquadramento dell’attività di
malattia e del danno d’organo ad essa correlato
direttamente e indirettamente.
41

Tabella 1: Revised International Classification Criteria
Per SS primario:
Presenza di 4 criteri su sei positivi di cui almeno uno sia rappresentato dall’istopatologia o
dalla sierologia
Presenza di 3 dei 4 criteri “obiettivi” (punti 3,4,5,6)
Per SS secondario: criteri 1 e 2 + almeno due tre 3, 4 e 5
Esclusion criteria: radioterapia, HCV, AIDS, linfoma pre-esistente, sarcoidosi,
GVHD, uso di farmaci anticolinergici
1. Sintomi oculari
Una risposta positiva almeno ad una delle seguenti domande:
a. Ha sensazione quotidiana di occhi secchi da oltre tre mesi?
b. Ha sensazione di sabbia/corpo estraneo negli occhi?
c. Utilizza lacrime artificiali per più di tre volte al giorno?2. 2. Sintomi orali
Una risposta positiva almeno ad una delle seguenti domande:
a. Ha sensazione quotidiana di bocca secca da oltre tre mesi?
b. Ha tumefazione ricorrente o persistente delle ghiandole salivari?
c. Deve bere molto per inghiottire cibi secchi?3. Segni Oculari
Schirmer test (<5 mm in 5 min)
Rosa Bengala positivo4. 4. Istopatologia
Focus score > 1 (Focus: aggregato di almeno 50 cellule mononucleate
adiacente ad acino intatto, Focus score numero di foci per 4 mmq)5. 5. Impegno delle ghiandole salivari
Scintigrafia salivare
Scialografia parotidea
Riduzione del flusso salivare(< 1,5 ml in 15 min)6. Autoanticorpi
Ro-SSA e/o La-SSB
42

L’ecografia nelle patologie di interesse reumatologico
Nel corso dell’ultimo decennio, si è registrato un interesse
sempre crescente da parte dei reumatologi nei confronti
dell’ecografia, per le sue indubbie potenzialità nello studio
delle caratteristiche morfostrutturali dei tessuti molli. I
progressi tecnologici in tale settore hanno reso possibile la
realizzazione di apparati di ecografia ad elevato potere di
risoluzione che consentono uno studio accurato dei tessuti
superficiali (tendini, piccole articolazioni, cute, nervi
periferici, vasi sanguigni). Appare ormai evidente che
l’ecografia riveste un ruolo fondamentale nella pratica clinica
reumatologica integrandosi utilmente nel percorso
diagnostico a cui il paziente con problemi articolari è
sottoposto. L’ecografia ha rivelato un indubbio valore nello
studio di un’ampia gamma di affezioni quali artrite
reumatoide, artrite psoriasica, artropatie da microcristalli,
artrosi, borsiti, tendinite, sindrome del tunnel carpale, SS,
arterite temporale, amiloidosi, sindrome di Tietze, fratture
costali.
Come detto precedentemente, l’ecografia consente una
valutazione accurata e riproducibile delle caratteristiche
morfo-strutturali dei tessuti molli. In particolare l’ecografia
43

può essere considerata la metodica di elezione per lo studio
dei tendini, le cui caratteristiche morfologiche e strutturali
risultano agevolmente valutabili. A livello tendineo, le
espressioni del processo flogistico possono essere
documentate nei diversi stadi evolutivi. La distensione della
guaina tendinea, le alterazioni della caratteristica
ecostruttura fibrillare del tendine, la perdita di definizione
dei margini tendinei sono le anomalie elementari di più
frequente osservazione. Anche a livello delle grandi e piccole
articolazioni, i processi di sinovite acuta e cronica possono
essere documentati in modo rapido ed accurato perfino nel
caso di un interessamento subclinico. Backhaus et al., (78) in
uno studio su 60 pazienti con artrite, hanno evidenziato,
mediante l’ecografia, un numero maggiore di articolazioni
delle mani e dei polsi colpite da sinovite rispetto a quanto
rilevato dall’esame clinico associato alla radiografia
convenzionale, con risultati comparabili a quelli che si
possono ottenere con l’uso della Risonanza Magnetica.
Attualmente le applicazioni dell’ecografia nelle patologie di
interesse reumatologico si sono estese in maniera
progressiva dalle poliartriti croniche alle connettiviti
sistemiche fino alle vasculiti. Le alterazioni muscolo-
scheletriche rappresentano un problema di grande rilevanza
44

nel Lupus Eritematoso Sistemico (LES) dal momento che
colpiscono oltre il 90% dei pazienti(79) con quadri clinici che
vanno dalla rigidità e dolore (persistenti o temporanee)
articolare fino all’artrite. Sinoviti ricorrenti sono descritte
in circa il 10-30% dei pazienti mentre le deformità articolari
si ritrovano in una percentuale modesta. Le erosioni ossee
sono rare (80) mentre più frequenti sono le patologie a
carico dei tendini, dovute sia all’infiammazione che all’uso
protratto dei corticosteroidi (80,81).
Studi recenti hanno utilizzato l’esame ecografico per
rilevare le alterazioni muscolo-scheletriche nel LES e poter
così impostare più correttamente la terapia .
Recentemente è stato valutato l’impiego dell’ecografia anche
per lo studio dell’arterite temporale: gli studi compiuti
riportano con l’uso del color Doppler una sensibilità elevata
che che arriva al 95%.
45

Ecografia interventistica
E’ stato dimostrato come il 50% delle iniezioni intra-
articolari eseguita senza l’assistenza dell’esame ecografico
non sia correttamente effettuata(82); l’ecografia permette,
invece, un corretto posizionamento dell’ago sia nelle terapie
iniettive che nell’aspirazione di raccolte liquide,
decompressione di cisti, drenaggi di ascessi o ematomi e
biopsie. (83,84,85).
La guida ecografica aumenta, quindi, l’accuratezza delle
procedure invasive e riduce il rischio di iniezioni a livello di
tendini, tessuto adiposo, muscoli, nervi o cute che
determinerebbero un danno ai tessuti e/o renderebbero
inefficace la terapia.
Scopo dello studio
Lo studio si propone di valutare, mediante esame ecografico,
in pazienti affetti da SS primitiva di recente insorgenza, la
frequenza e le caratteristiche dell’interessamento delle
piccole articolazioni della mano.
46

Materiali e metodi
Pazienti
Abbiamo studiato 35 pazienti (33 femmine e 2 maschi, età
media±SD 59±13,75 anni) afferenti all’ambulatorio dell’U.O.
di Reumatologia dell’Università di Pisa e 8 volontari sani
reclutati tra il personale sanitario (7 femmine e 1 maschio
età media±SD 45,5±8,17 anni).
Tutti i pazienti lamentavano xerostomia ed xeroftalmia
insorte da non più di 12 mesi e da non meno di 3 mesi; 33/35
soggetti riferivano artralgie diffuse (mani, spalle, ginocchia).
Nessuno dei pazienti in esame presentava segni o sintomi
suggestivi per LES od altre connettiviti né franca artrite. In
tutti i soggetti è stata eseguita la visita oculistica con test
di Schirmer e Rosa Bengala, la ricerca del fattore
reumatoide, degli anticorpi diretti verso i peptidi citrullinati,
degli anticorpi anti-nucleo, degli anticorpi diretti verso gli
antigeni nucleari estraibili ed il DNA nativo e gli esami
ematochimici di base (velocità di eritrosedimentazione,
proteina-C reattiva, fibrinogenemia, protidogramma,
emocromo, creatininemia, transaminasi). Inoltre tutti i
pazienti sono stati sottoposti a biopsia ed esame istologico
di una ghiandola salivare minore. Sulla base dei criteri
47

internazionali del 2002 la SS è stata diagnosticata in 18
pazienti; in 17 soggetti è stata posta diagnosi di sindrome
sicca.
Metodi
In tutti i pazienti è stata eseguita la valutazione ecografica
delle articolazioni metacarpo-falangee (MCF), ed
interfalangee prossimali (IFP). L’esame ecografico in scala di
grigi è stato effettuato immediatamente prima di eseguire
la biopsia della ghiandola salivare da un unico operatore (a
cui non erano noti i dati clinici e sierologici del paziente)
mediante apparecchio Toshiba Power Vision 6000 (Toshiba
Corporation, Giappone) dotato di una sonda lineare di ultima
generazione ad ampia banda di frequenza (8-15 MHz). La
frequenza utilizzata in tutti i pazienti è stata di 15 MHz. Lo
studio con tecnica power doppler è stato condotto con lo
stesso apparecchio.
48

Sono stati ricercati:
1) il versamento intrarticolare definito come materiale ipo
od anecogeno, all’interno dell’articolazione, mobile e
comprimibile e che non presenta segnale power doppler.
2) l’iperplasia sinoviale definita come presenza, a livello
intrarticolare, di tessuto ipoecogeno non comprimibile, non
mobile e che può presentare segnale power doppler
3) le erosioni ossee definite come discontinuità intrarticolari
del profilo osseo visualizzabili in almeno due scansioni su
piani perpendicolari.
49

Risultati
Pazienti affetti da SS
Articolazioni MCF
Il versamento a livello delle articolazioni MCF era presente
in 9/18 pazienti (50%). In un soggetto erano interessate 8
articolazioni, in un altro 6, in un altro 4, in uno 3 ed, infine, in
5 pazienti era coinvolta solo una articolazione. In un paziente
oltre al versamento si osservava iperplasia sinoviale in 6
articolazioni, in due pazienti la sinovia era iperplastica solo in
1 articolazione; l’iperplasia sinoviale non si associava in
nessun caso a segnale power doppler. In una paziente era
presente una piccola erosione a livello della testa
metacarpale del II dito (vedi foto).
Erosione della MCF
50

Articolazioni IFP
Si evidenziava versamento a livello delle articolazioni IFP in
3/18 (16%) pazienti. Di questi 3 pazienti, 1 presentava
versamento in 2 articolazioni, 2 in 1 articolazione. In nessun
soggetto l’esame power doppler risultava positivo né erano
evidenziate erosioni ossee.
In 2 pazienti erano interessate contemporaneamente sia le
articolazioni MCF che le IFP.
In 5 pazienti con versamento a carico di MCF od IFP erano
presenti irregolarità dei profili ossei su base osteoartrosica
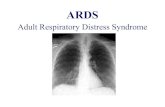
Nella Tabella A sono riassunte le alterazioni ecografie
rilevate nei pazienti con SS.
Versamento a livello dell’IFP
51
*
Versaento
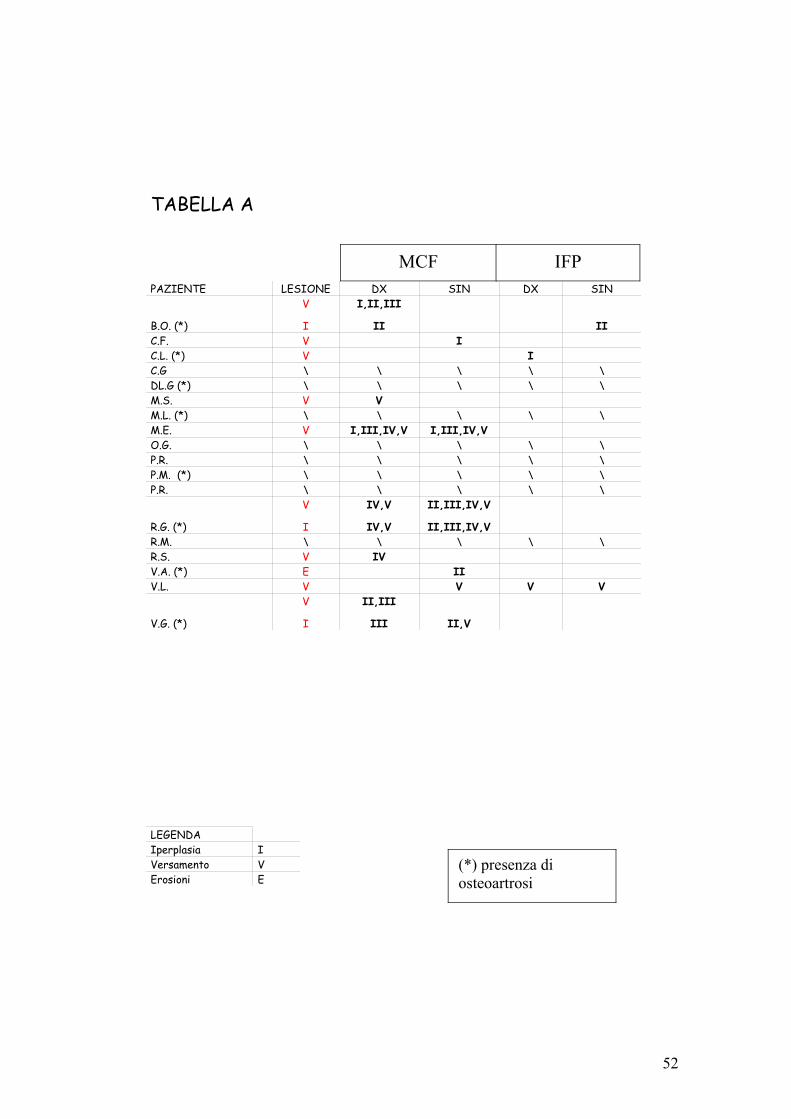
TABELLA A
PAZIENTE LESIONE DX SIN DX SIN
B.O. (*)
V
I
I,II,III
II IIC.F. V IC.L. (*) V IC.G \ \ \ \ \DL.G (*) \ \ \ \ \M.S. V VM.L. (*) \ \ \ \ \M.E. V I,III,IV,V I,III,IV,VO.G. \ \ \ \ \P.R. \ \ \ \ \P.M. (*) \ \ \ \ \P.R. \ \ \ \ \
R.G. (*)
V
I
IV,V
IV,V
II,III,IV,V
II,III,IV,VR.M. \ \ \ \ \R.S. V IVV.A. (*) E IIV.L. V V V V
V.G. (*)
V
I
II,III
III II,V
LEGENDAIperplasia IVersamento VErosioni E
52
MCF
(*) presenza di osteoartrosi
IFP

Pazienti affetti da sindrome sicca
Articolazioni MCF
Il versamento a livello delle articolazioni MCF era presente
in 6/17 (35%) pazienti. In un 1 paziente con sindrome sicca
si evidenziava versamento a carico di 3 articolazioni; in 1
paziente erano interessate 2 articolazioni e in 4 soggetti era
rilevabile versamento in 1 sede articolare. In 1 paziente al
versamento si associava iperplasia sinoviale a carico di 2
articolazioni mentre in 1 soggetto si è riscontrata iperplasia
sinoviale in 1 articolazione; in entrambi i casi l’esame power
doppler risultava negativo e non erano evidenziate erosioni
ossee
Articolazioni IFP
Si osservava versamento intrarticolare in 2/17 (11%)
pazienti ma non iperplasia sinoviale, segnale power od
erosioni ossee. In 2 pazienti erano interessate
contemporaneamente sia le MCF che le IFP.
In 3 soggetti con versamento intrarticolare erano presenti
anche alterazioni di tipo artrosico.
Nella Tabella B sono riassunte le alterazioni ecografiche
presenti nei pazienti con sindrome sicca
53

TABELLA B
PAZIENTE LESIONE DX SIN DX SINA.R. \ \ \ \ \B.D. V I
B.R. (*)
V
I
II
II
C.C. (*) \ \ \ \ \C.G. (*) \ \ \ \ \C.A. V IV IV IVF.V. (*) \ \ \ \ \G.T. (*) V V IV IVG.A. \ \ \ \ \G.S. \ \ \ \ \L.L. V IP.E. \ \ \ \ \P.M.N. (*) \ \ \ \ \R.P.A. \ \ \ \ \
S.A. (*)
V
I
II,IV
II,IV
IV
IV
S.E. \ \ \ \ \T.C. (*) \ \ \ \ \
LEGENDAIperplasia IVersamento VErosioni E
54
MCF
(*) presenza di osteoartrosi
IFP

Gruppo di controllo volontari sani
Le uniche alterazioni ecografiche riscontrabili
ecograficamente nel gruppo dei volontari sani sono state
segni indicativi di artrosi rilevati in 4 pazienti su 8.
Tabella CNOME LESIONE DX SIN DX SINP.C. \ \ \ \ \G.S.* \ \ \ \ \
M.A,* \ \ \ \ \Z.K. \ \ \ \ \R.M. \ \ \ \ \B.P.* \ \ \ \ \L.L.* \ \ \ \ \T.D. \ \ \ \ \
LEGENDAIperplasia IVersamento VErosioni E
55
MCF IFP
(*) presenza di osteoartrosi
Risultati complessivi
Irregolarità dei profili ossei su base osteoartrosica erano
presenti in 20 pazienti, 8 affetti da SS, 8 da sindrome sicca
e 4 nel gruppo di volontari.
Sindrome diSjogren
Sindrome sicca Controllo
44,50% 47% 50%
0,00%
10,00%
20,00%
30,00%
40,00%
50,00%
60,00%
70,00%
80,00%
90,00%
100,00%
Segni di osteoartrosi all'ecografia
56
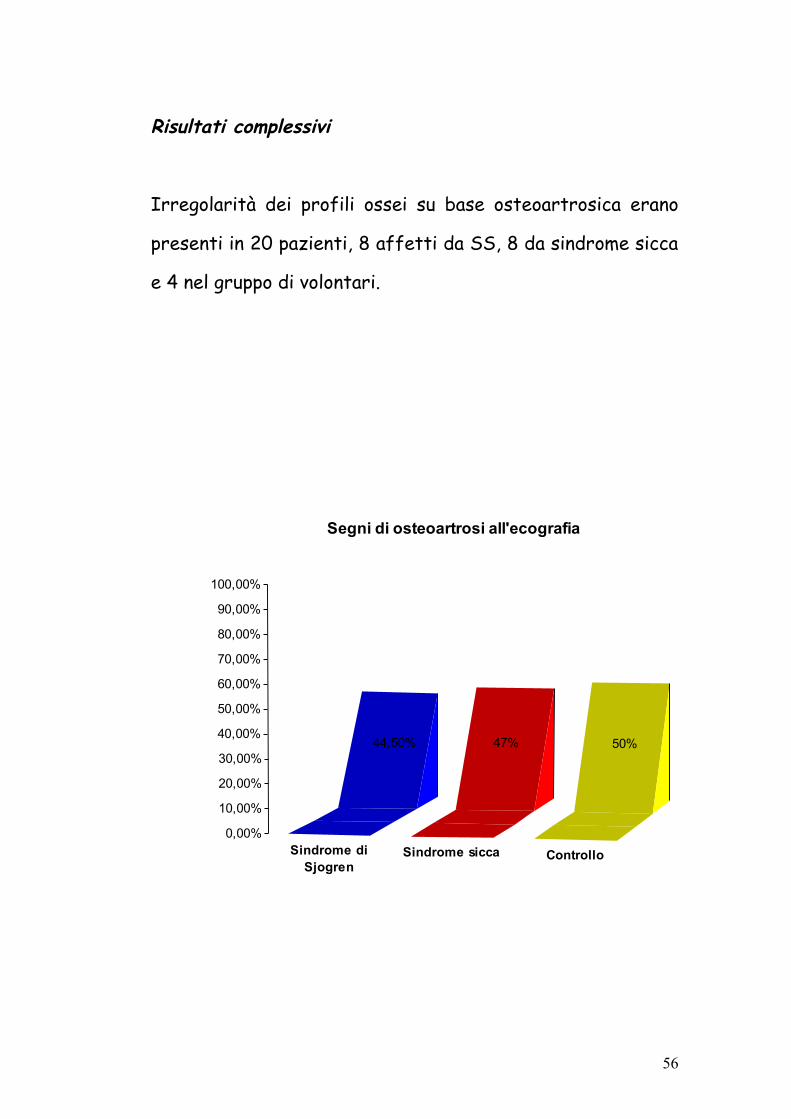
Complessivamente segni di artrite sono stati evidenziati più
frequentemente nei pazienti con SS rispetto ai pazienti con
sindrome sicca (55% versus 35%) anche se con una
differenza non statisticamente significativa (pvalue<0,23).
Sindrome diSjogren
Sindrome sicca Controllo
55,50%
35,30%
0%0,00%
10,00%
20,00%30,00%40,00%50,00%
60,00%
70,00%
Segni di artrite all'ecografia (iperplasia, versamento, erosione)
57
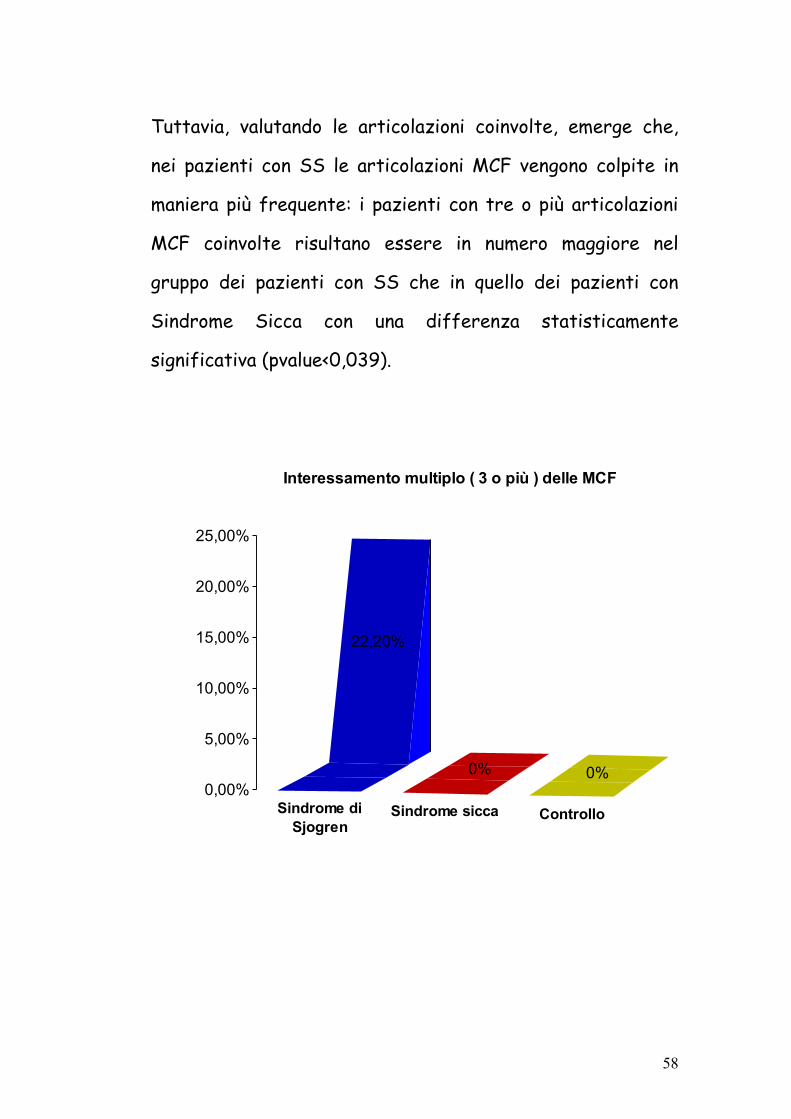
Tuttavia, valutando le articolazioni coinvolte, emerge che,
nei pazienti con SS le articolazioni MCF vengono colpite in
maniera più frequente: i pazienti con tre o più articolazioni
MCF coinvolte risultano essere in numero maggiore nel
gruppo dei pazienti con SS che in quello dei pazienti con
Sindrome Sicca con una differenza statisticamente
significativa (pvalue<0,039).
Sindrome diSjogren
Sindrome sicca Controllo
22,20%
0% 0%0,00%
5,00%
10,00%
15,00%
20,00%
25,00%
Interessamento multiplo ( 3 o più ) delle MCF
58

Discussione
La SS è una malattia autoimmune caratterizzata da
infiammazione cronica delle ghiandole esocrine, in
particolare delle ghiandole salivari e lacrimali, che può
presentare numerose manifestazioni extraghiandolari, tra
cui l’interessamento articolare (artralgie, rigidità mattutina,
artrite intermittente o cronica). La frequenza del
coinvolgimento articolare non è, tuttavia, ancora ben
stabilita. La maggior parte degli studi riportano nei pazienti
con SS una alta prevalenza di artralgie ma una bassa
frequenza di artrite. Nel 1993 Pease et al studiando 48
pazienti con SS primitiva riportavano una prevalenza di
artrite e/o artralgie del 54%. In un terzo di questi pazienti
il coinvolgimento articolare rappresentava uno dei sintomi di
esordio della malattia e precedeva la sindrome sicca.
L’artropatia tendeva ad essere poliarticolare, spesso
simmetrica e l’articolazione più frequentemente colpita era il
ginocchio(70). Uno studio prospettico condotto da Kruize et
al nel 1996 su 31 pazienti con SS primitiva, seguiti per 10-12
anni, non ha, invece, evidenziato artrite in nessun
paziente(86). Da una successiva analisi di una casistica più
59

ampia, 110 soggetti con SS primitiva diagnosticata nel
periodo 1977-1992, sottoposti ad una rivalutazione del loro
stato di malattia negli anni compresi fra il 1994-97, la
frequenza dell’artrite risulterebbe, invece, del 22%(67).
Garcia-Carrasco et al riportano, inoltre, nei pazienti anziani
con SS primitiva esordita in tarda età (dopo 70 anni) un
interessamento articolare nel 29% dei casi.
Va, comunque, precisato che negli studi citati l’accertamento
del coinvolgimento articolare si è sempre basato sulla
raccolta dell’anamnesi e sull’esame clinico dei paziente
associati, molto raramente, ad esami radiografici standard,
una forma di imaging ormai considerato decisamente poco
sensibile nel rilevare segni di artrite, soprattutto in fase
iniziale. In letteratura è presente solo uno studio su pazienti
con SS primitiva che valuta la presenza di sinovite a livello
del ginocchio mediante esame ecografico ed i cui risultati
indicano che segni di lieve sinovite sono frequenti in corso di
tale affezione.
Oltre a non disporre di dati certi sulla prevalenza
dell’impegno articolare in corso di SS, anche le
caratteristiche di tale impegno non sono state fino ad ora
indagate in maniera approfondita perché, come
precedentemente detto, nella maggior parte dei casi gli
60

studi sono stati condotti senza ricorrere a tecniche in grado
di visualizzare anche le forme più lievi, subcliniche di artrite
e di definire le caratteristiche dell’interessamento
articolare (ad es. la presenza o meno di proliferazione della
sinovia, il grado di vascolarizzazione del panno sinoviale).
Il nostro studio ha evidenziato una frequenza elevata (55%)
di artrite a carico delle piccole articolazioni delle mani nei
pazienti con SS già in fase iniziale, seppure con una
differenza non statisticamente significativa sia rispetto ai
soggetti con sindrome sicca che ai soggetti sani . E’ possibile
ipotizzare che la concomitante presenza di osteoartrosi
della mani in alcuni pazienti possa giustificare la presenza di
versamento intrarticolare. Va però precisato che il numero
di soggetti con segni di osteoartrosi è esattamente uguale
nel gruppo con SS e nel gruppo con sindrome sicca; inoltre,
come noto, l’artrosi colpisce prevalentemente le articolazioni
interfalangee prossimali e distali e molto più raramente le
articolazioni metacarpo-falangee, che invece risultano le
articolazioni più frequentemente colpite nei nostri pazienti
(4 pazienti su 18 nel gruppo della SS avevano interessamento
multiplo – 3 o più - delle MCF mentre nessuno dei pazienti
con Sindrome Sicca presentava un simile quadro;
pvalue<0,039)
61

Tale risultato è stato raggiunto, utilizzando per la prima
volta nello studio della mano del paziente con SS una
metodica di indagine, l’ecografia, che permette una
valutazione accurata e riproducibile delle caratteristiche
morfo-strutturali dei tessuti molli. Questa tecnica è in
grado di monitorare l’evoluzione della malattia articolare e di
valutare l’efficacia del trattamento. Il coinvolgimento
articolare osservato nel gruppo con sindrome sicca, sebbene
interessi un numero esiguo di soggetti, è, al momento,
difficilmente spiegabile e sarà solo il follow up clinico e
strumentale ad aiutarci per arrivare ad un più corretto
inquadramento.
In conclusione, il nostro studio evidenzia come, mediante
l’esame ecografico, sia possibile dimostrare nei pazienti con
SS, già in fase iniziale, un impegno articolare in un numero
elevato di soggetti.
La nostra ricerca è preliminare ed è stata condotta su un
numero ristretto di pazienti: l’ampliamento della casistica ed
uno stretto monitoraggio dei pazienti nel tempo potranno
chiarire ulteriormente la frequenza e le caratteristiche
dell’interessamento articolare in corso di SS.
62

BIBLIOGRAFIA
1. Gerli R et al. Quantitative assessment of salivary gland inflammatory
infiltration in primary SS: its relationshio to different demographic,
clinical and serological features of the disorders. Br Journ Rheum
1997; 36:969-975
2. Nusair S. The use of oral pilocarpine in xerostomia and SS. Seminars
in Arthritis Rheum 1999; 28(6):360-367
3. Kelley’s. Text Book of Rheumatology Edition VI, 2001 chap 68,
pp1027-1038
4. Stanley R. Sjogren’s syndrome.Primer on the Rheumatic Disease.
Edition XII, chap 20, pp 377-383
5. Yamamoto K. Pathogenesis of Sjogren’s syndrome. Autoimmunity
Reviews 2003; 2:13-18
6. Fujihara T, Fujita H, Tsubota , Saito K, Tsuzaka K, Abe T, Takeuchi T.
Preferential localization of CD8+ alpha E beta 7+ T cells around acinar
epithelial cells with apoptosis in patients with in Sjogren’s Syndrome.
J Immunol 1999;163:2226-35
7. Arnett FC, Hamilton RG, Reveille JD, Bias WB, Harley JB, Reichlin M.
Genetics studies of Ro(SS-A) and La(SS-B) autoantibodies in families
with systemic lupus erythematosus and primary Sjogren’s syndrome.
Arthritis Rheum 1989; 32:413-9.
8. Ramos-Casals M, Font J. Primary Sjogren’s syndrome:current and
emergent aetiopathogenic concepts. Rheumatology 2005; 44:135-
1367.
9. Hulkkonen J, Pertovaara M, Antonen J, Lahdenpohja N, Pasternack A,
Hrme M. Genetic association between interleukin-10 promoter resion
polymorphisms and primary Sjogren’s syndrome. Arthritis Rheum
2001; 44:176-9
63

10. Font J, Garcia-Carrasco M, Ramos-Casals M et al. The role of
interleukin-10 promoter polymorphisms in the clinical expression of
primary Sjogren’s syndrome. Rheumatology 2002;41:1025-30.
11. Petrek M, Cermakova Z, Hutyrova B et al. CC chemokine receptor 5
and interleukin-1 receptor antagonist gene polymorphisms in patients
with primary Sjogren’s syndrome. Clin Exp Rheumatol 2002;20:701-3.
12. Haddad J, Deny P, Munz-Gotheil C, Ambrosini JC, Trinchet JC,
Pateron D et al. Lymphocytic sialadenitis of Sjogren’s syndrome
associated with chronic hepatitis C virus liver disease. Lancet
1992;339:321-3.
13. Vernant JC, Buisson G, Magdeline J, De Thore J, Jouannelle A,
Neisson-Vernant C, et al. T-lymphocyte alveolitis, tropical spastic
paresis, and Sjogren’s syndrome. Lancet 1988,1:177
14. Terada K, Katamine S, Eguchi K, Moriuchi R, Kita M, Shimada H, et al.
Prevalence of serum and salivary antibodies to HTLV-1 in Sjogren’s
syndrome. Lancet 1994;344:1116-9.
15. Mitsias DI, Kapsogeorgou EK, Moutsopoulos HM. The role of epithelial
cells in the initiation and perpetuation of autoimmune lesions: lesson
from Sjogren’s syndrome (autoimmune epithelitis). Lupus
2006;15:255-261.
16. Amft N, Curnow Sj, Scheel-Ttoellner D et al. Ectopic expression of
the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells
and within lymphoid follicles contributes to the establishment of
germinal centre-like structures in Sjogren’s syndrome. Arthritis
Rheum 2001;44:2633-2641.
17. OgawaN, Ping L, Zhenjun L, Takada Y, Sugai S. Involvement of the
interferon-gamma-induced T cell-attracting chemokines, interferon-
gamma-inducible 10-Kd protein (CXCL10) and monokine induced by
64

interferon-gamma (CXCL9), in the salivary gland lesions of patients
with Sjogren’s syndrome. Arthritis Rheum 2002; 46: 2730-2741.
18. Polihronis M, Tapinos NI, Theocharis SE, Economou A, Kittas C,
Moutsopoulos HM. Modes of epithelial cell death and repair in
Sjogren’s syndrome (SS). Clin Exo Immunol 1998; 114:485-490.
19. Manganelli P, Fietta P. Apoptosis and Sjogren’s syndrome. Semin
Arthritis Rheum 2003; 33:49-65.
20. McArthur C, Wang Y, Veno P, Zhang J, Fiorella R, Intracellular
trafficking and surface expression of SS-A (Ro), SS-B (La),
poly(ADP-ribose) polymeraseand alpha-fodrin autoantigens
duringapoptosis in human salivary gland cells induced by tumor
necrosis factor-alpha. Arch Oral Biol 2002; 47:433-448.
21. Ohlsson M, Jonsson R, Brokstad KA. Subcellular redistribution and
surface exposure of the Ro52, Ro60 and La48 autoantigens during
apoptosis in human ductal epithelial cells: a possible mechanism in the
pathogenesis of Sjogren’s syndrome. Scand J Immunol 2002; 56: 456-
469.
22. Thery C, Zitvogel L, Amigorena S. Exosomes: compositions, biogenesis
and function. Nat Rev Immunol 2002; 2:569-579.
23. Kapsogeorgou EK, Abu-Helu RF, Moutzopoulos HM, Manoussakis MN.
Salivary gland epithelial cell exosomes: a source of autoantigenic
ribonucleoproteins. Arthritis Rheum 2005; 52: 1517-1521.
24. Steinfeld SD, Appelboom T, Delporte C. Treatment with infliximab
restores normal aquaporin 5 disribution in minor salivary glands of
patients with Sjogren’s syndrome. Arthritis Rheum 2002; 46:2249-51.
25. Beroukas D, Hiscock J, Jonsson R, Watermann SA, Gordon TP.
Subcellular distributions of aquaporin 5 in salivary glands in primary
Sjogren’s syndrome. Lancet 2001; 358:1875-6.
26. Hakala M, Niemela RK. Does autonomic nervous impairment have a role
65

in pathophysiology of Sjogren’s syndrome. Lancet 2000; 355:1032-3.
27. Zoukhri D, Kublin CL. Impaired neurotransmitter release from
lacrimal and salivary glands nerves of a murine model of Sjogren’s
syndrome. Invest Ophthalmol Vis Sci 2001; 42:925-32.
28. Waterman SA, Gordon TP, Rishmuller M. Inhibitory effects of
muscarinic receptor autoantibodies on parasympathetic
neurotrasmission in Sjogren’s Syndrome. Arthritis Rheum 2000;
43:1647-54.
29. Daniels TE. Association of patterns of labial salivary gland
inflammation with Keratoconjunctivitis sicca.Analysis of 618 patients
with suspected SS.Arthritis Rheum 1994; 37: 869-77
30. Daniels TE. Evaluation, Differential diagnosis and treatment of
xerostomia. Journal Rheum 2000;27 (S61): 6-10
31. Soto-Rojas AE et al. The oral side of SS: diagnosis and treatment. A
review. Archives of Medical Research 2002;33:95-106
32. Hamburger J. SS as seen by an oral physician. Scand J Rheum 2001;
(S115):34-9
33. Fox RI. Approaches to the treatment of Sjogren’s Syndrome. Journ
Rheum 2000;27(s61):15-21
34. Sreebny LM. Treatment of Drug-induced xerostomia in “Saliva and
Oral Health”. Second Edition.1996
35. Benjamin A et al.Dry mouth and nose in the older patient. Geriatrics
2002; 57(6):22-35
36. Tzioufas A, Moutsopoulos HM. Sjogren’s syndrome. In Klippel JH eds
Rheumatology London: Mosby-Year Book Europe Lid 2001, volume 2,
chap 7, pp32.1-32.11
37. Lemp MA. Evaluation and Differential Diagnosis of Kerato-
conjunctivitis Sicca. Journ Rheum 2000; 27(61):11-14
38. Rolando M. SS as seen by an ophthalmologist. Scand J Rheum
66

2001(115):27-33
39. Al-Hashimi I, Khuder S, Haghighat N, Zipp M. Frequency and
predictive value of the clinical manifestation in Sjogren’s syndrome. J
Oral Pathol Med. 2001;30:1-6
40. Bernacchi E, Amato L, Parodi A, Cottoni F, Rubegni P, De Pita O, Papini
M,Rebora A, Bombardieri S, Fabbri P.Sjogren's syndrome: a
retrospective review of the cutaneous features of 93 patients by the
Italian Group of Immunodermatology. Clin Exp Rheumatol. 2004 Jan-
Feb;22(1):55-62
41. Wrigth SA, Convery RP, Ligget N. Pulmonary involvment in Sjogren’s
syndrome. Rheumatology (Oxford) 2003; 42:697-8.
42. Koyama M, Johkoh T, Honda O, Mihara N, Kozuka T, Tomiyama N, et
al. Pulmonary involvement in primary Sjogren’s syndrome:spectrum of
pulmonary abnormalities and computed tomography findings in 60
patients. J Thorac Imaging 2001; 16:290-6.
43. Franquet T, Diaz C, Domingo P, gimenez A, Geli C. Air trapping in
primary Sjogren’s syndrome: correlation of expiratory CT with
pulmonary function tests. J Comput Assist Tomogr 1999;23:169-73.
44. Papiris SA, Maniati M, Constantopoulos SH, Roussos C, Moutsopoulos
HM, Skopouli FN. Lung involvement in primary Sjogren’s syndrome is
mainly related to the small airway disease. Ann Rheum Dis 1999;
58:61-4.
45. Davidson BK, Kelly CA, Griffiths ID. Ten year follow up of pulmonary
function in patients with primary Sjogren’s syndrome. Ann Rheum Dis
2000; 59:709-12.
46. Bossini N, Savoldi S, Franceschini F, Mombelloni S, Baronio M,
Cavazzania I, et al. Clinical and morphological features of kidney
involvement in primary Sjogren’s syndrome. Nephrol Dial Transplant
2001;16:2328-36.
67

47. Goules A, Masouridi S, Tzioufas AG, Ioannidis JP, Moutsopoulos HM.
Clinically significant and biopsy-documented renal involvement in
primary Sjogren’s syndrome. Medicine (Baltimore) 2000;79:241-9.
48. Garcia-Carrasco M, Siso A, Ramos-Casals M, Rosas J, De la Red J, Gil
V, et al. Raynaud’s phenomenon in primary Sjogren’s syndrome.
Prevalence and clinical characteristics in a series of 320 patients. J
Rheumatol 2002; 29:726-30.
49. Coates T, Slavotinek Jp, Rishmuller M, Shultz D, Anderson C,
Dellamelva M, et al. Cerebral white metter lesions in primary
Sjogren’s syndrome: a controlled study. J Rheumatol 1999; 26:1301-5.
50. Rosenbaum R. Neuromuscolar complications of CTD. Muscle and Nerve
2001. 24(2):154-169
51. Hess D. Identifiying the Spectrum of Neurologic complications in SS.
Advances in Immunotherapy 2001.18(2):12-16
52. Aoki A. Hematological abnormalities of 1°SS. Nhon Rinsho Meneki
Gakkai Kaishi 2000.23(2):124-8
53. Schattner A.Immune cytopenias as the presenting finding in 1°SS.
QJM 200093(12): 825-9
54. Ferraccioli GF et al CD4 cytopenia and occasional expansion of
CD4+CD8+ lymphocytes in SS. Clin Exp Rheumatol 1996.14(2). 125-30
55. Rodriguez A. SS and pernicious anemia . Scand J Rheumatol
1998.27(1): 83-5
56. Ramakhrisna R. Haematological manifestations of 1° SS: a
clinicopatological study. QJM 1992. 83(303):547-54
57. Primer on the Rheumatic Disease. Edition XII. 2001, cap20 pag 377-
383
58. Ioannidis. SS: too many associations, too limited evidence. The
enigmatic example of CNS Involvement. Seminars Arthritis Rheum
1999 29(1):1-3
68

59. Klippel 2001 Volume 2, cap7 pag 32.1-32.11
60. Constantopoulos SH et al Pulmonary and GI manifestations of SS. Rh
Dis of N Am 1992 18 (3): 617-631
61. Sheikn SH. The GI manifestations of SS. Am J Gastroenterol 1995.
90(1): 9-14
62. Tsianos EB The GI involvement in 1° SS. Scand J Rheumatol Suppl
1986. 61: 151-5
63. Rosztoczy A. Manometric assessment of impaired esophageal motor
function in 1° SS. Clin Exp Rheumatol 2001. 19(2):147-152
64. Ostuni PA. Pancreatic exocrine involvement in 1° SS. Scand J
Rheumatol 1996; 25(1):47-51
65. Sumii T. Chronic pancratitis and pancreatic dysfunction associated
with SS. Nippon Rinsho 1995; 53 (10) 2525-9
66. Skoupouli FN et al. Liver involvement in 1° SS . Br J Rheumatol 1994.
33(8): 745-8
67. Pertovaara M., PuKKala E, Laippala P, Miettinen A, Pasternack A. A
longitudinal cohort study of Finish patients with primary sjogren’s
sindrome: clinical, immunological and epidemiological aspects Ann
Rheum Dis 2001; 60: 467-472
68. Haga HJ, Jonsson R. The influence of age on disease manifestations
and serological characteristics in
primary Sjogren’s sindrome. Scand J Rheumatol 1999; 28: 227-232
69. Ramos-Casals M, Cervera R, Font J, Garcia-Carrasco M, Espinosa G.,
Reino S, Pallares L., Ingelmo. Young onset of primary Sjogren’s
syndrome: clinical and immunological characteristics. Lupus 1998; 7(3):
202-6
70. Pease Ct. The arthropathy of SS. Br J Rheumatol 1993, 32 (7):609-13
71. Arrago JP et al. Scintigraphy of the salivary glands in SS. J Clin
Pathol 1987, 40:1463
69

72. Locht H., Pelck R. Manthorpe R. Clinical manifestations correlated to
the prevalence of autoantibodies in a large (n =321) cohort of patients
with primary Sjogren’s syndrome. A comparison of patients initially
diagnosed according to the Copenaghen classification criteria with the
American –European consensus criteria. Autoimmunity Reviews 2005;
4:276-281
73. Daniels T.E et al. Salivary and oral components of SS. Rheum Dis Cl of
North America 1992, 18 (3) pag 571-588
74. Daniels TE. Evaluation, Differential diagnosis and treatment of
xerostomia. Journal Rheum 2000;27 (S61): 6-10.
75. Skopouli FN, Dafni U, Ioannidis JP, Moutsopoulos HM.Clinical
evolution, and morbidity and mortality of primary Sjogren's
syndrome.Semin Arthritis Rheum. 2000 Apr;29(5):296-304.
76. Haga HJ, Hulten B, Bolstad AI, Ulvestad E, Jonsson R.J
Rheumatol.Reliability and sensitivity of diagnostic tests for primary
Sjogren's syndrome.1999 Mar;26(3):604-8.
77. Rolando M. SS as seen by an ophthalmologist. Scand J Rheum
2001(115):27-33
78. Backhaus M, Kamradt T, Sandrock D et al.: Arthritis of the finger
joints. A comprehensive approach comparing conventional radiography,
scintigraphy, ultrasound, and contrast-enhanced magnetic resonance
imaging. Arthritis Rheum 1999; 42: 1232-45.
79. Iagnocco A, Ossandon A, Coari G, Conti F, Priori R, Alessandri C,
Valesini G: Wrist joint involvement in systemic lupus erythematosus.
An ultrasonographic study. Clin Exp Rheumatol 2004; 22:621-624
80. Wallace DJ: The musculoskeletal system. In J and HAHN BH (Eds.):
Dubois’ Lupus Erythematosus, Baltimore, Williams & Wilkins, 1997:
635-51.
81. Furie RA, Chartash EK: Tendon rupture in systemic lupus
70

erythematosus. S e m i n Arthritis Rheum 1988; 18: 127.
82. Jones A, Regan M, Ledingham J, Pattrick M, Manhire A, Doherty M.
Importance of placement of intra-articular steroid injections. Br Med
J 1993; 307:1329-30.
83. McGonagle D, Gibbon W, O’Connor P, et al. A preliminary study of
ultrasound aspiration of bone erosion in early rheumatoid arthritis.
Rheumatology 1999; 38:329-31.
84. Kane D, Greaney T, Shanahan M, et al. The role of ultrasonography in
the diagnosis and management of idiopatic plantar fascitis.
Rheumatology 2001; 40:1002-8.
85. Koski JM. Ultrasoun guided injections in rheumatology. J Rheumatol
2000; 27:2131-8
86. Kruize AA, Henè RJ, van der Heide A, Bodeitsch C, de Wilde PCM, van
Bijsterveld OP, de Jong J, Feltkamp TEW, Kater L, Bijlsma WJ: Long-
term followup of patients with Sjogren’s sindrome. Arthritis and
Rheumatism 1996; 39(2):297-303.
71