
Università degli studi di Pisa - COREcore.ac.uk/download/pdf/14694699.pdf · 2.1.2 Miceti...
156
Università degli studi di Pisa Facoltà di Agraria Corso di Laurea Specialistica in Agricoltura Biologica Tesi di Laurea: “Determinazione dell’ocratossina A in prodotti derivati da suini allevati con metodo biologico” CANDIDATA: RELATORI: Milena Tolomeo Prof. Alessandro Pistoia Dott. ssa Valentina Meucci Anno Accademico 2006/2007
Transcript of Università degli studi di Pisa - COREcore.ac.uk/download/pdf/14694699.pdf · 2.1.2 Miceti...

Università degli studi di Pisa
Facoltà di Agraria
Corso di Laurea Specialistica in Agricoltura Biologica
Tesi di Laurea:
“Determinazione dell’ocratossina A in prodotti derivati da suini allevati con metodo biologico”
CANDIDATA: RELATORI:
Milena Tolomeo Prof. Alessandro Pistoia
Dott. ssa Valentina Meucci
Anno Accademico 2006/2007

Ai miei genitori

INDICE
1. INTRODUZIONE 1
1.1 LE MICOTOSSINE 1 1.1.1 Aspetti generali 1 1.1.2 Cenni storici 2 1.1.3 Miceti produttori di micotossine 2 1.1.4 Contaminazione degli alimenti 5 1.1.5 Effetti sulla salute dell’uomo e degli animali 12 1.1.6 Prevenzione e decontaminazione 15 1.1.7 Legislazione 27
2. OCRATOSSINE 38
2.1 GENERALITA’ 38 2.1.1Cenni storici 38 2.1.2 Miceti produttori e condizioni di sviluppo 39 2.1.3 Alimenti contaminati 42 2.1.4 Caratteristiche chimiche e strutturali 45
2.2 TOSSICOCINETICA 47 2.2.1 Generalità 47 2.2.2 Assorbimento 47 2.2.3 Distribuzione 48 2.2.4 Biotrasformazione 51 2.2.5 Eliminazione 54
2.3 EFFETTI E MECCANISMO DI AZIONE 56
2.4 TOSSICITA’ 62 2.4.1 Tossicità nel topo e nel ratto 63 2.4.2 Tossicità nel suino 64 2.4.3 Tossicità nell’uomo 65
3. PREMESSA 68
4. MATERIALI E METODI 71
4.1 Prove di allevamento 71 4.1.1 Rilievi in vita 71 4.1.2 Rilievi post-mortem 72
4.2 Determinazione dell’ocratossina A con metodo HPLC 73 4.2.1 Materiali 73

4.2.2 Strumentazione e condizioni cromatografiche 74 4.2.3 Soluzioni standard e soluzione stock 74 4.2.4 Standard esterno 75 4.2.5 Retta di calibrazione 75 4.2.6 Campioni 75 4.2.7 Estrazione e purificazione dei campioni di mangime 76 4.2.8 Estrazione e purificazione dei campioni di carne 80 4.2.9 Conferma 82 4.2.10 Validazione del metodo 82 4.2.11 Vetreria 84 4.2.12 Analisi statistica 85
5. RISULTATI 86
5.1 Prove di allevamento 86
5.2 Determinazione dell’ocratossina A tramite HPLC 91 5.2.1 Condizioni cromatografiche e di estrazione 91 5.2.2 Retta di calibrazione 97 5.2.3 Analisi dei campioni 98 5.3.4 Analisi statistica 105
6. DISCUSSIONE 110
7. CONCLUSIONI 114
8. BIBLIOGRAFIA 116

INTRODUZIONE

1
1. INTRODUZIONE
1.1 LE MICOTOSSINE
1.1.1 Aspetti generali Il termine micotossine comprende numerosi metaboliti secondari ad elevata
tossicità, prodotti in opportune condizioni microclimatiche da funghi
microscopici e filamentosi, meglio noti con il termine di “muffe”, che
colonizzano le piante e/o le derrate alimentari nel corso del loro
accrescimento. Il termine metabolita secondario significa che non si è in
grado di attribuire loro alcun ruolo evidente nella crescita dell’organismo che
le produce. Queste tossine non costituiscono una classe chimica, ma hanno
strutture tra loro molto diverse, e mentre il metabolismo primario è
fondamentalmente lo stesso per i funghi, quello secondario dipende dalle
specie e talvolta dal ceppo fungino. Da ciò la grande diversità di molecole
prodotte, anche se per famiglie di prodotti simili. (Piva e Pietri, 2006). Lo
sviluppo dei funghi tossigeni e la successiva sintesi di micotossine, può
avvenire in qualunque fase di produzione e trasformazione di un prodotto
alimentare. In particolare, la produzione di micotossine, può essere favorita
già nella fase di coltivazione dei vegetali, da una serie di fattori che,
provocando stress alle piante, possono aumentare la loro suscettibilità alle
infezioni fungine. Le derrate alimentari, le granaglie ed i mangimi,
rappresentano i substrati ideali per l’accrescimento dei funghi produttori di
micotossine (Brera et al., 2002). Esse possono giungere alla nostra tavola
sia direttamente attraverso il consumo di prodotti derivati da derrate
vegetali contaminate (cerali, prodotti da forno, legumi, caffè, frutta
tropicale, frutta secca a guscio, spezie, piante infusionali ecc.), sia

2
indirettamente attraverso prodotti di origine animale derivati da bestiame
alimentato con mangimi contaminati, qualora non sussistano casi acuti di
micotossicosi tali da indurre l’allevatore a sopprimere l’animale.
1.1.2 Cenni storici
Le micotossine, pur risalendo a tempi remoti, sono state scientificamente
oggetto di studio, specie nel campo veterinario, solo a partire dal 1850
quando si è dimostrata l’associazione tra l’ingestione di segale contaminata
con sclerozi di Claviceps purpurea e la comparsa di casi di ergotismo.
Successivamente, nel 1913, fu descritta una malattia degli equini
denominata “blind staggers”, conseguente all’ingestione di grani ammuffiti
da varie specie di Penicillium responsabili della produzione di acido
penicillico e acido micofenolico. Prima degli anni ’60 erano note altre
patologie sostenute da micotossine quali ad esempio l’angina tossica, la
leucopenia alimentare tossica in Russia, dove fù descritta l’insorgenza di
una tossicosi alimentare correlata all’ingestione di cereali colonizzati da
Fusarium sporotrichioides e da F.poae, e la Yellow rice tossicosi in
Giappone. L’inizio della moderna micotossicologia è databile al 1960, anno
in cui vennero identificate le aflatossine, , prodotte da Aspergillus flavus e
Aspergillus parasiticus, ritenute responsabili dell’insorgenza in Inghilterra di
una particolare malattia, denominata “malattia X turkeys disease”, che
provocò la morte di migliaia di volatili di allevamento (Tiecco, 2001).
1.1.3 Miceti produttori di micotossine Attualmente sono note più di 300 micotossine, per circa 60 delle quali è
stata individuata una potenziale tossicità, anche se la maggior parte delle
ricerche sono concentrate su aflatossine, ocratossine, tricoteceni,
zearalenone, fumonisine e patulina (Miraglia e Brera, 1999). Solo il 7%
delle oltre 300 micotossine identificate si ritrovano negli alimenti a livelli

3
significativamente elevati tali da costituire un pericolo per la salute umana.
Numerosi, sono i generi fungini responsabili della produzione di tali sostanze
e appartengono generalmente alla categoria dei Deuteromiceti la quale
raggruppa tutti gli anamorfi e tutti i miceti nei quali la riproduzione è di tipo
agamico (Matta, 1996). Tra di essi, le specie note e che destano maggior
preoccupazione sono comprese nei generi Aspergillus, Penicillium e
Fusarium, anche se si possono riscontare ceppi dotati di elevata tossicità e
facilmente presenti nei nostri ambienti, nei generi Alternaria e Claviceps
(Causin, 2004). Essendo prodotte da un’ampia gamma di organismi, si
potrebbe ricavare l’impressione errata, che tutte le materie di origine
organica, comprese quelle destinate all’alimentazione umana e zootecnica,
possano essere facilmente contaminate da sostanze dannose, derivanti dallo
sviluppo su queste matrici di qualche tipo di muffa. Fortunatamente ciò è
vero solo in parte, poiché non tutti i funghi compresi nei generi
potenzialmente capaci più o meno di produrre tossine, sono in grado di
produrle. Infatti, solo specifici ceppi, all’interno di alcune specie, riescono a
produrre le sostanze in questione e ciò può avvenire solamente se le
condizioni ambientali sono favorevoli a tali processi. Tuttavia, quando tali
tossine sono prodotte, la loro pericolosità risulta elevata in quanto possono
provocare danni molto gravi alla salute, riescono ad agire a concentrazioni
molto basse, possono passare attraverso l’apparato digerente degli animali
andandosi a ritrovare, variamente modificate nei prodotti zootecnici e sono
difficilmente eliminabili dai prodotti contaminati, tant’è che possono
permanere anche dopo l’eliminazione del l’organismo produttore (Causin,
2004). La Tabella 1 riporta i principali funghi tossigeni responsabili della
produzione di micotossine di maggior interesse.

4
FUNGHI PRODUTTORI
MICOTOSSINE PRODOTTE
Genere Penicillum P. patulum P. expansum
Patulina
P. viridicatum
Citrinina, Ocratossina A
Genere Fusarium F. moniliforme F. proliferatum
Fumonisine
F. graminearum F. culmorum F. poae F. sporotrichioides
Tricoteceni, Zearalenone
Genere Aspergillus A. flavus Aflatossine B1, B2
Acido ciclopiazonico
A. parasiticus
Aflatossine B1, B2, G1, G2
A. versicolor A. nidulans
Sterigmatocistina
A. ochraceus
Ocratossina A, acido penicillico, Citrinina
A. clavatus
Patulina
Genere Claviceps
C. purpurea Alcaloidi
Tabella 1. Funghi tossigeni e relative micotossine.

5
1.1.4 Contaminazione degli alimenti
Per prevenire la contaminazione da micotossine delle derrate alimentari,
occorre impedire la crescita fungina, in quanto la formazione di micotossine
vi è strettamente connessa; senza la crescita del micete produttore,
generalmente la produzione di tossine non avviene. Questo non sta a
significare però, che la presenza del fungo tossigeno in un prodotto, indichi
automaticamente la presenza di micotossine; anzi esse possono persistere
per lungo tempo anche dopo il termine della crescita vegetativa o la morte
del fungo, riscontrandosi nel prodotto contaminato anche in assenza di unità
vitali di miceti. Inoltre, l’assenza di isolati fungini negli alimenti non indica
necessariamente l’assenza di micotossine (Haouet e Altissimi, 2003). Non
dobbiamo tralasciare il fatto che all’interno di una specie fungina, esistono
ceppi capaci di produrre una grande quantità di micotossine, ed altri con
minori capacità, senza presentare differenze significative né nello sviluppo,
né nei caratteri morfologici. Ne consegue che l’analisi micologica, basata
sulla numerazione delle unità vitali e l’identificazione delle specie fungine,
non permette di quantificare il rischio tossico proprio di una derrata
alimentare, in funzione della presenza dei miceti (Haouet e Altissimi, 2003).
Per tale ragione, il rischio non può che essere determinato attraverso
l’analisi chimico-fisica per l’identificazione e la quantificazione delle
micotossine note (Miraglia e Brera, 1999). Lo sviluppo degli agenti tossigeni
e la successiva sintesi di micotossine, possono avvenire in tutte le fasi della
catena produttiva, in funzione di molteplici fattori tra loro interdipendenti,
per tale ragione, il controllo dei parametri chimico-fisici, riveste
un’importanza primaria nel settore del controllo e della prevenzione dalla
contaminazione (Miraglia e Brera, 1999). I principali fattori che consentono
la tossinogenesi sono:
• fattori intrinseci legati al ceppo fungino;
• la specie fungina che determina le classi di micotossine prodotte;
• il potenziale tossigeno che può variare tra i diversi ceppi;

6
• il livello iniziale di contaminazione il quale influenza la quantità di
tossine sintetizzabili (più muffe = maggior quantità potenziale di
micotossine);
• fattori estrinseci che comprendono l’insieme di tutte le condizioni
ecologiche in grado di agire sullo sviluppo fungino e di conseguenza
sulla produzione delle micotossine;
• fattori biologici, quali gli insetti come Ostrinia nubilalis, conosciuta
più comunemente come la piralide del mais, rivestono importanza in
quanto possono essere vettori di spore fungine ed agenti di lesioni alle
cariossidi, favorendo così l’insediamento delle muffe; la microflora con
risultante competizione tra le specie fungine; eventuali stress della
pianta (stress idrico o nutrizionale) e la resistenza al substrato, intesa
quest’ultima sia come resistenza genetica che come integrità delle
cariossidi;
• fattori chimici e chimico-fisici, quali l’umidità, l’acqua libera (aw), la
temperatura, la composizione gassosa dell’ambiente, il pH, la natura
del substrato ed i danni meccanici alla cariosside, risultano poi
essenziali. (Haouet e Altissimi, 2003).
Per prevenire la contaminazione da micotossine delle derrate alimentari,
bisogna impedire la crescita fungina. Per far questo, occorre perciò,
prendere in considerazione un insieme di misure che scaturiscono dalle leggi
che regolano la vita delle muffe; i funghi hanno bisogno di acqua, ossigeno
(minimo 1-2%) e temperatura adeguata, quest’ultima variabile a seconda
della specie, infatti le temperature elevate favoriscono lo sviluppo del
genere Aspergillus mentre le basse il genere Fusarium (Haouet e Altissimi,
2003). Il parametro cui prestare maggiore attenzione è senza dubbio l’aw.
Bisogna tener presente, che l’attività ed il contenuto dell’acqua, non sono la
stessa cosa: l’aw esprime la parte attiva del contenuto di umidità, nei
confronti dell’umidità totale, la quale comprende anche l’acqua legata.
L’acqua contenuta in un alimento, in generale, sarà quindi legata in maniera
più o meno intensa a seconda del tipo di substrato e della presenza in

7
questo di gruppi idrofobi e idrofili. La colonizzazione fungina degli alimenti,
si verifica più frequentemente di quella batterica a livelli di aw <0,85;
questo non perché i funghi non possano crescere a tenori di aw più elevati,
ma piuttosto perché i batteri sono fortemente competitivi e diventano la
microflora predominante a valori di aw di 0,85-1,00 ed in particolare a
valori > 0,90 – 0,93. In base alle loro differenze di comportamento in
funzione delle disponibilità di acqua, le specie fungine sono state classificate
in:
• Igrofile, le cui spore germinano solo con valori di aw superiori a 0,90
ed hanno una crescita ottimale ad aw pari a 1,00.
• Mesofite (es. Penicillium cyclopium), le cui spore germinano a valori
di aw di 0,80 – 0,90 ed hanno un optimum a 0,95 – 1,00.
• Xerofile (es. Aspergiullus repens, Aspergillus versicolor), le cui spore
germinano ad un valore di aw inferiore a 0,80 e la crescita ottimale
si osserva intorno a 0,95 (Haouet e Altissimi, 2003).
Il valore di aw minimo al quale è stata osservata una crescita fungina è di
0,61. Tuttavia, non si conoscono specie tossigene in grado di crescere a
valori di aw<0,78. Inoltre, i livelli minimi richiesti per la sintesi delle
micotossine, sono generalmente superiori a quelli necessari per la crescita
fungina (Haouet e Altissimi, 2003). Infatti, mentre il valore minimo di aw
per la crescita dei funghi tossigeni è di 0,79, quello necessario per la
produzione delle tossine va dallo 0,80 per l’acido penicillico allo 0,95 per
l’ocratossina, allo 0,99 per la produzione di patulina. Importante è da
sottolineare che la disponibilità d’acqua, dipende da altri fattori ambientali
quali la temperatura; infatti, qualora le spore delle muffe riuscissero a
germinare in presenza di valori di aw ridotti, valori limite di temperatura
possono inibire la crescita del micelio; al contrario , per un dato valore di
aw, il suo aumento può comportare la riduzione della vitalità delle spore,
mentre l’abbassamento ne favorisce la longevità (Aureli, 1996). Più in
generale, le temperature ideali per lo sviluppo di una muffa sono comprese

8
tra 15 e 30 °C , con un optimum di 20-25°C. Per quanto riguarda il pH, le
muffe al contrario dei batteri, tollerano un ampio range, in quanto molte
crescono bene a valori compresi tra 3 ed 8 con un optimum di circa pH 5
(Aureli, 1996); tuttavia, alcuni miceti possono manifestarsi anche a valori
più bassi o più elevati, modificando gradualmente l’acidità del mezzo nel
corso del loro sviluppo. Esse competono poco e male con i batteri a pH 7
specialmente quando l’aw del substrato è alta, mentre a pH 5 l’attività
metabolica batterica si riduce e le muffe predominano rapidamente. Un altro
fattore condizionante la crescita fungina è la presenza di ossigeno, il quale
fa sì che i funghi si sviluppino generalmente sulla superficie dei substrati,
sebbene alcune specie, siano in grado di crescere anche in profondità, o in
mezzi liquidi a basso tenore di ossigeno oppure in atmosfera modificata con
CO2 o N2 (Haouet e Altissimi, 2003). Per quanto concerne le caratteristiche
dei funghi tossigeni più pericolosi e conosciuti si può affermare che:
� Il genere ASPERGILLUS
� È il più termofilo dato che vive tra 7 – 42 °C
� Resiste di più al secco, visto che si sviluppa su substrati con umidità
del 18 – 20% ed in atmosfera con umidità relativa dell’85%
� Predilige i climi caldi
� È diffuso nelle aree più meridionali
� Normalmente non è patogeno, ma un’eccezione è rappresentata dall’
A.flavus che è capace di comportarsi come patogeno a temperature
>30°C
� Il genere FUSARIUM
� Preferisce temperature più fresche
� Esige livelli di umidità più alti e nei nostri ambienti si sviluppa su
substrati aventi un’umidità del 20 – 22%
� E’ diffuso nelle aree settentrionali
� Predilige climi temperato – freschi ed umidi che nei nostri ambienti
corrispondono alla primavera o inizio autunno

9
� Comprende molte specie di patogeni (es. F. moniliforme, F.
graminearum, F. culmorum)
� Il genere PENICILLIUM
� Si adatta meglio di Aspergillus ad ambienti e climi più freschi umidi
anche se, alcuni ceppi hanno la capacità di crescere su substrati con
umidità inferiore
� Sebbene non sia presente nei nostri ambienti, può comportarsi
anche da patogeno di pieno campo (Causin, 2004).
Riveste poca importanza per lo sviluppo delle muffe sugli alimenti, la natura
del substrato, se sussistono le condizioni ambientali sopra descritte. Non è
invece il caso della produzione delle loro micotossine, dove il tipo di
substrato e le condizioni ambientali assumono dei limiti molto più delineati.
La tossinogenesi infatti, è favorita da livelli di aw dei substrati, superiori a
quelli richiesti per lo sviluppo fungino (in campo >0,95 ed in magazzino
0,80), l’Aspergillus flavus può iniziare la produzione di aflatossine già a
0,83, mentre A. ochraceus necessita di almeno 0,97 per produrre
ocratossine. L’umidità del substrato, espresso come aw, è pertanto il vincolo
principale per prevenire la tossinogenesi in un alimento, per cui il suo
controllo è diventato indispensabile, per esempio in mangimistica.
Relativamente alle condizioni termiche, queste sono estremamente variabili
a seconda della specie. Aspergillus flavus, produce aflatossine
preferibilmente intorno ai 25°C e non è mai stata evidenziata una
tossinogenesi a temperature inferiori a 10°C. A queste ultime temperature
sono state prodotte sì aflatossine, in latte in polvere umidificato e in
formaggi, ma da A. parasiticus. Il Fusarium graminearum produce
zearalenone intorno a 14°C e anche a temperature inferiori, come il
Fusarium tricinctum che è in grado di produrre la tossina T2 a temperature
comprese tra 1 e 4°C e fino a 15°C, mentre Aspergillus ochraceus produce
ocratossina in un intervallo di temperatura compreso tra 20 e 30°C, e
comunque mai al di sotto di 12°C; la stessa micotossina viene poi anche
prodotta da Penicillium viridicatum, ma a temperature completamente

10
diverse e comprese tra 4 e 31 °C. E’ pertanto difficile generalizzare dei limiti
di contenimento, se si esclude la produzione di aflatossine che non si
accumulano al di sotto di 10°C neanche in substrati fortemente ammuffiti
(Haouet e Altissimi, 2003). Per ogni muffa, tuttavia, esiste una temperatura
minima ed una massima oltre le quali l’attività produttiva cessa (Tiecco,
2001). Il tipo di substrato è invece in questo caso, l’elemento che più
probabilmente più di ogni altro, influenza la tossinogenesi. I vegetali sono
più facilmente contaminati rispetto ai prodotti animali; la presenza di amido
inoltre, sembra incrementare la tossinogenesi ed in particolare quella di
zinco, limitatamente alla sintesi di aflatossine. I cereali, i semi oleaginosi e
la frutta secca, sono al vertice degli alimenti più frequentemente
contaminati da aflatossine: mais, arachidi e semi di cotone rappresentano i
prodotti più a rischio. La frutta e i succhi derivati sono invece i principali
veicoli di patulina, mentre i cereali quelli di zearalenone e vomitossina
(deossinivalenolo), cereali, birra, vino, cacao e caffè quelli di ocratossina.
Tra gli alimenti di origine animale, il latte e il formaggio sono prodotti in cui
il passaggio di aflatossine è più evidente, qualora le vacche siano alimentate
con prodotti zootecnici contaminati. In tal caso, nel latte, compaiono
molecole idrossilate delle aflatossine B1 e B2, denominate M1 e M2. Per
quanto riguarda invece i prodotti carnei, risulta che applicando basse
temperature nella trasformazione e nella conservazione, il rischio di
micotossinogenesi è limitato; su prosciutti crudi stagionati è stata messa in
evidenza la possibilità di produrre ocratossina da parte di Aspergillus
ochraceus solo se la temperatura di conservazione raggiungeva i 25-30°C;
in tal caso, dopo 21 giorni di mantenimento a questa temperatura, la
micotossina era penetrata nella massa carnea fino a mezzo centimetro di
profondità, in quantità pari a due terzi della produzione totale della muffa
(Haouet e Altissimi, 2003) anche se è stata osservata in alcuni casi la
presenza di aflatossina in carne macinata di manzo (reni e fegato
soprattutto) e ocratossina in carne di suino. Evento raro è invece la
contaminazione delle uova, che può derivare solo dalla contaminazione dei
mangimi utilizzati per l’alimentazione delle galline ovaiole (Haouet e

11
Altissimi, 2003). Se ne deduce che la potenzialità di produrre tossina si
estrinseca raramente nella pratica e solo quando si realizza un insieme di
condizioni favorevoli. La produzione massima, in genere, si verifica
all’insorgere di condizioni stressanti per il micete, rappresentate da bruschi
abbassamenti di temperatura, condizioni di umidità e di substrato non più
favorevoli allo sviluppo ed alla vita del fungo, stadio vegetativo finale e
stadio di sporulazione che attivano il metabolismo secondario del fungo
(Tiecco, 2001).
Tabella 2. Valori ottimali per lo sviluppo fungino.
Oltre alla pericolosità dovuta alla possibile produzione di micotossine, lo
sviluppo delle muffe nelle derrate alimentari provoca conseguenze ben
precise. Possono verificarsi infatti:
� Modificazione dell’aspetto;
� Alterazione delle qualità organolettiche: Penicillium cyclopium
conferisce un gusto di terra, il deossinivalenolo prodotto da diverse
specie di Fusarium, provoca fenomeni di rifiuto soprattutto nella
specie suina;
� Alterazioni delle qualità tecnologiche: gli enzimi prodotti dai funghi,
idrolizzano i lipidi, l’amido e le proteine.
� Riduzione quantitativa e soprattutto qualitativa del valore
alimentare: produzione di calore, anidride carbonica, acqua; perdita
di aminoacidi essenziali e vitamine. Prove di laboratorio hanno
evidenziato che le perdite di sostanza secca dovute allo sviluppo
fungino (produzione di CO2), con conseguente riscaldamento della
FATTORE MINIMO OTTIMO MASSIMO Aw 0,61 0,80-0,95 1,00 Temperatura -21 20-25 60 PH 2,0 4,5-6,5 8,0 Ossigeno 0,14 >2,0 - Anidride carbonica
- <10,0 >15,0

12
massa, possono raggiungere il 5% e che la produzione di acqua
inoltre, può favorire un’ulteriore crescita delle muffe. Alcune ricerche
hanno dimostrato che, nel caso del mais, un prodotto fortemente
contaminato, subisce diminuzioni nel tenore di energia, proteine e
grassi del 5, 7 e 63% rispettivamente. Come si nota, la quota
lipidica è infatti quella più soggetta ad attacco fungino.
� Rischi di micosi e di allergie per gli animali, ma anche per gli
operatori (Piva e Pietri, 1996).
Pertanto, essendo la problematica delle micotossine estremamente
variegata e non risolvibile con una sola azione specifica, può essere
affrontata in maniera efficace ed efficiente solamente con un approccio
integrato, supportato da un’attenta analisi comparata del rischio e dei mezzi
disponibili per ridurlo il più possibile al di sotto del limite accettabile, che
gestisca tutta la filiera del prodotto.
1.1.5 Effetti sulla salute dell’uomo e degli animali La presenza di micotossine nelle derrate alimentari costituisce un rischio
per la salute sia dell’uomo sia degli animali in seguito all’ingestione di
alimenti contaminati. La contaminazione di alimenti di origine animale da
parte di micotossine può essere diretta o primaria, cioè derivante dallo
sviluppo di funghi tossigeni sugli alimenti, oppure indiretta o secondaria,
causata dall’assunzione da parte degli animali, di alimenti contaminati da
funghi tossigeni a causa di un fenomeno denominato “carry over” (Miraglia
e Brera, 1999). Per tale ragione, queste molecole rivestono importanza, in
quanto la loro ingestione tramite foraggi, mangimi e cereali da parte degli
animali in produzione zootecnica, comporta successivamente il loro
accumulo in vari organi e tessuti o la loro eliminazione attraverso il latte e
le uova. I residui presenti possono essere costituiti sia dalle micotossine
inalterate, originariamente presenti nel mangime, sia da micotossine

13
prodotte dal metabolismo dell’animale. Le interazioni esistenti tra uomo,
animali da allevamento e alimenti contaminati da micotossine sono
rappresentate nella figura 1 (Miraglia e Brera, 1999).
Ceppo fungino
Mangimi Animali destinati all’ alimentazione umana Micotossine Cereali e semi oleaginosi
Latte
Carne
Uova
Alimentazione umana
Figura 1. Interrelazione tra micotossine e uomo.
La gravità dei loro effetti, dipende dalla quantità assunta tramite gli
alimenti, dalla tossicità del composto, dal peso corporeo dell’individuo, dalla
presenza di altre micotossine (in quanto si possono verificare effetti
sinergici), dai fattori dietetici (Carratù e Cuomo, 2001) nonché dalla specie
animale interessata, in quanto non tutte le specie animali, presentano la
stessa sensibilità agli effetti delle micotossine. In particolare, il suino e gli
animali da compagnia sono le specie più sensibili alle micotossicosi, mentre
i ruminanti, grazie alla degradazione delle tossine svolta a livello ruminale,

14
tendono ad essere le specie più resistenti (Hussein e Brasel, 2001). Gli
effetti tossici osservati, consentono di classificare le patologie in
micotossicosi acute primarie, croniche primarie e croniche secondarie
(Tiecco, 2001). Le micotossine possono provocare:
• intossicazioni acute, conseguenti all’ingestione di unan singola dose
o di più dosi ingerite in un breve periodo di tempo; tali intossicazioni
sono però fenomeni che non coinvolgono mai prodotti di origine
animale in quanto, solo i prodotti di origine vegetale possono
contenere dosi così elevate di micotossine;
• intossicazioni croniche, conseguenti all’ingestione di piccole dosi
ripetute nel tempo e in genere, maggiormente legate all’assunzione
di prodotti di origine animale. Infatti, le derrate animali, sono
generalmente meno contaminate delle derrate vegetali, in quanto
l’organismo degli animali produttori di alimenti destinati all’uomo,
agisce da potente sistema di depurazione (Piva e Pietri, 1996).
Le micotossine, essendo molto diverse tra loro dal punto di vista chimico,
mostrano una notevole gamma di effetti biologici dovuti alla loro capacità di
interagire con diversi organi e/o sistemi bersaglio degli organismi viventi
(Miraglia e Brera, 1999). Per tale ragione, esse sono classificate in
immunotossine, dermatossine, epatotossine, nefrotossine e neurotossine;
oppure sulla base del loro effetto cronico in mutagene, cancerogene e
teratogene. In particolare tutte queste attività biologiche sono dovute alla
capacità delle micotossine e/o dei loro metaboliti, di interagire con il DNA,
l’RNA, le proteine funzionali, i cofattori enzimatici ed i costituenti di
membrana. Gli effetti tossici osservati raramente possono dare origine a
fenomeni patologici di tipo acuto ed il rischio maggiore risiede nel loro
accumulo che può originare sintomatologie di tipo cronico. (Miraglia e Brera,
1999).
A causa della loro tossicità, le micotossine possono causare seri danni alla
salute umana e possono provocare notevoli danni economici negli

15
allevamenti e negli impianti zootecnici dovuti ad un calo nelle fasi produttive
e riproduttive. La Tabella 3, riporta la classificazione delle principali
micotossine in base alla loro tossicità (Tiecco, 2001).
EFFETTO TOSSICO MICOTOSSINA Epatotossicità
Aflatossine, ocratossina A, rubratossina, sporidesmine, tossina PR
Azione irritante del derma
Psolareni, tricoteceni
Teratogenicità
Aflatossine, rubratossine, tricoteceni, ocratossina A
Cancerogenicità
Aflatossine, tricoteceni, patulina, ac. penicillico, stirigmatocistina, luteoschirina
Nefrotossicità
Ocratossina A, citrinina, aflatossine
Neurotossicità
Tremorgeni, alcaloidi dell’ergot, verrucolotossina, ac. ciclomizzomico, citroviridina, roquefortina
Mutagenicità
Aflaossine, patulina, ac. penicillio, ac. micofenolico, sterigmacistina, rubratossina
Azione endocrinomimetica
Zearalenone, penitrem
Azione radiomimetica Tricoteceni
Tabella 3. Classificazione di alcune micotossine secondo la loro tossicità.
1.1.6 Prevenzione e decontaminazione Il primo passo nella prevenzione della contaminazione da micotossine nelle
diverse colture ed in modo particolare nel mais, consiste nell’ applicazione di
tecniche di coltivazione attente al “benessere del vegetale” ovvero
finalizzate alla riduzione massima di tutti gli stress che possono favorire

16
l’insediamento dei funghi tossigeni e la relativa produzione di micotossine.
Premesso che ad oggi, è difficilmente ipotizzabile la completa eliminazione
delle contaminazioni da micotossine, è possibile attivare efficaci azioni di
prevenzione per contenere il rischio attraverso l’elaborazione e
l’applicazione di buone pratiche agricole (GAP) e buone pratiche di
lavorazione (GMP) (Miraglia e Brera,1999). E’ bene ricordare innanzitutto
che la proliferazione sia delle muffe “di campo”, sia “di magazzino” parte dal
campo e può proseguire per entrambe se si mantengono condizioni ottimali
che ne facilitano la proliferazione durante una non corretta conservazione;
per tale ragione quindi, la prevenzione deve partire dalla coltivazione e
concludersi nelle lavorazioni del prodotto, senza interruzioni di attenzione.
In particolare, i miceti del genere Fusarium sono comuni saprofiti e patogeni
delle piante per cui sono ricorrenti in campo, mentre gli Aspergillus ed i
Penicillium, si sviluppano prevalentemente nelle fasi di conservazione per la
loro elevata capacità di svilupparsi su substrati caratterizzati da bassa
umidità (Borreani et al., 2003). Le condizioni che favoriscono lo sviluppo dei
funghi da campo includono l’alto grado di umidità (>70%) e le forti
escursioni termiche (giornate calde seguite da notti fredde). I cereali
maggiormente colpiti risultano essere mais, frumento ed orzo (Haouet e
Altissimi, 2003). Nel nostro ambiente di coltivazione, la situazione, riguardo
la contaminazione da micotossine delle derrate, e in particolare della
granella di mais, è sotto controllo rispetto alle più importanti aree maidicole
mondiali, questo perché caratterizzate da condizioni climatiche, più
favorevoli allo sviluppo dei funghi tossigeni e alla conseguente produzione di
micotossine, rispetto alle nostre zone in cui solo raramente e in casi
eccezionali (estate 2003), si verificano. Tuttavia, nei nostri allevamenti la
componente mais nella razione degli animali ha un peso molto rilevante;
inoltre, gli elevati standard qualitativi richiesti dal mercato ed una
legislazione europea molto restrittiva in materia, consigliano proprio per
questo di adottare delle misure preventive, per abbattere la concentrazione
di queste micotossine nel prodotto finale (Verderio,2001). La soluzione a
questo problema, non può che passare attraverso una serie di interventi

17
realizzabili per ridurre la presenza di micotossine nel mais e in altri cereali
così da impedirne il loro assorbimento. Alcuni ricercatori sostengono che la
formazione delle micotossine nelle colture attaccate dai miceti in condizioni
di pre-raccolta, abbia valori nettamente superiori rispetto alla fase di post-
raccolta; per questo motivo quindi, risulterebbero più efficaci le azioni
preventive attuabili in campo rispetto a quelle applicabili durante lo
stoccaggio, anche se sono più difficilmente eseguibili a causa
dell’interferenza delle condizioni ambientali. La prevenzione della
contaminazione è attuabile in due fasi operative della filera di produzione
dell’alimento a base di mais, le quali sono qui di seguito elencate (Bertocchi
et al., 2004):
1. Fase di pre-raccolta o coltivazione
2. Fase di raccolta e post-raccolta
FASE DI PRE-RACCOLTA
In questa fase la prevenzione della contaminazione fungina rappresenta una
delle migliori strategie per ridurre i rischi di contaminazione da micotossine
e garantire un prodotto alimentare sicuro. Fermo restando che la resistenza
della pianta ospite rappresenta la migliore strategia in pre-raccolta per
prevenire l’attecchimento di funghi e l’accumulo di micotossine, le ricerche
fatte finora per selezionare o sviluppare varietà di mais naturalmente
resistenti alla colonizzazione da parte di funghi tossigeni e all’accumulo di
micotossine hanno portato, salvo rare eccezioni, a risultati poco
soddisfacenti. E’ stato verificato però, che alcune caratteristiche
morfologiche della spiga e della granella, quali la completa copertura della
spiga e brattee consistenti contro l’attacco di insetti ed altri patogeni,
portamento non eretto della spiga in fase di maturazione per evitare una
ritenzione dell’acqua piovana ed una reidratazione della granella ed infine
una granella meno suscettibile (per la forma e la durezza dell’endosperma)
alle rotture meccaniche, che si possono verificare nei processi di raccolta ed
essiccazione, si sono dimostrate un possibile vantaggio nel contenere lo

18
sviluppo del fungo (Verderio,2001). I monitoraggi condotti in questi ultimi
anni in diversi comprensori a mais dell’Emilia-Romagna, hanno spesso
mostrato che gli ibridi più precoci (classe FAO 300-400), sono quelli
maggiormente suscettibili alla contaminazione da aflatossine, mentre gli
ibridi più tardivi (classe FAO 600-700) sono maggiormente suscettibili alla
contaminazione da fumonisine (Agricoltura, 2007). L’infezione primaria e il
successivo sviluppo del fungo avvengono più facilmente e più
frequentemente, in corrispondenza di periodi più o meno lunghi nei quali la
pianta è in stato di stress evapotraspirativo, causato da temperature
eccessive ed inadeguato rifornimento di acqua. Con lo scopo di diminuire il
livello generale di stress evapotraspirativo, considerato come la condizione
più importante per l’infezione da parte del micete, viene richiesto da parte
dell’agricoltore un adeguato e regolare rifornimento di acqua alla coltura, e
nelle situazioni in cui il fattore acqua è difficilmente controllabile, o per
ridotta disponibilità o per alti costi, viene consigliata l’applicazione di diverse
procedure agronomiche quali l’anticipo dell’epoca di fioritura, ottenibile con
l’anticipo dell’epoca di semina o con l’utilizzo di ibridi con un ciclo precoce
ed, infine, l’adozione di investimenti moderati, perché colture fitte tendono
maggiormente ed anticipatamente a manifestare i sintomi (appassimento,
disseccamenti basali, proteandria ecc..), che di conseguenza influiscono
negativamente anche sulla produzione (Verderio, 2001). Condizione ad alto
rischio di infezioni in campo da A. flavus, agente delle aflatossine, è la
presenza di stress idrico successivo alla maturazione cerosa della granella.
Al contrario nelle annate fresche, quando lo stress idrico è molto contenuto
e la maturazione è ritardata, si presentano le condizioni per lo sviluppo di
Fusarium graminearum e delle tossine zearalenone e DON; in questi casi
risulta più efficace evitare apporti irrigui eccessivi, che rischiano solo di
aumentare l’insorgenza di fumonisine, senza peraltro determinare
significativi incrementi di resa (Agricoltura, 2007). Sebbene è riportato che
il danno provocato alle colture dagli insetti fitofagi, nella maggioranza dei
casi provocato da Ostrinia nubilalis, conosciuta più comunemente come
Piralide del mais, non rappresenta un requisito essenziale per la

19
contaminazione da micotossine; è anche noto però, che l’incidenza di
infezione da parte miceti tossigeni è significativamente più alta nelle
cariossidi danneggiate, rispetto a quelle sane (Verderio, 2001).
Figura 2. Ostrinia nubilalis.
Gli insetti, oltre a danneggiare i tegumenti esterni delle cariossidi e facilitare
l’ingresso e la colonizzazione da parte di funghi micotossigeni, possono
inoltre agire da vettori delle spore fungine o creare punti critici nella massa
delle derrate, ad alto contenuto di umidità, favorevoli alla crescita dei funghi
e alla produzione di tossine; proprio per questo motivo le procedure e gli
accorgimenti adottabili dall’agricoltore per contrastare l’attività della
piralide, possono essere quelli di cercare di non utilizzare degli ibridi più
suscettibili all’attacco dell’insetto, anticipare l’epoca di fioritura (con le
modalità descritte precedentemente) per sfasare gli stadi fenologici della
coltura con il ciclo di riproduzione dell’insetto, oppure proteggere la coltura
con trattamenti specifici in post fioritura (Verderio, 2001). In prospettiva, è
ipotizzabile l’adozione di mezzi di lotta biologica, mediante pre-infezione
delle colture con isolati fungini non tossigeni bio-competitivi, infatti proprio
a questo scopo, diversi microrganismi sono stati proposti quali agenti di bio-
controllo in pre-raccolta della contaminazione da micotossine; isolati non
tossigeni della stessa specie possono infatti essere ottimi agenti bio-
competitivi, che ben si adattano alle condizioni ambientali tipiche delle

20
specie tossigene (Verderio, 2001). E’ opportuno ricordare, che un’efficace
difesa della piralide sembra permettere una consistente riduzione dei tenori
di contaminazione delle fumonisine mentre non risulta avere analogo effetto
sulla contaminazione da aflatossine (Miraglia e Brera, 1999). Il mais è una
coltura definita a ciclo primaverile estivo, in quanto il suo ciclo colturale, nei
climi temperati, avviene nel periodo compreso tra la primavera e l’estate e
per questo viene seminato, nei nostri ambienti, verso la prima metà di
Aprile con possibilità di un certo anticipo nelle zone meridionali, e viene
raccolto, nel caso di produzione di granella secca, quando viene raggiunta la
cosiddetta “maturità commerciale”, che si verifica 10-15 giorni dopo la
“maturazione fisiologica”, nel mese di Settembre (Giardini e Vecchietti,
2000). Quest’ultima fase è riconoscibile dal completamento della formazione
del cosiddetto “strato nero, ed è caratterizzata da un’umidità della granella
che è intorno al 30-32%. La successiva fase di perdita di umidità in campo,
fino allo stadio di “maturazione commerciale”, può avere diversa durata, in
relazione all’epoca di comparsa dello strato nero ed all’andamento
stagionale; proprio in questo stadio, la granella di mais, diventa
estremamente suscettibile all’attacco da parte dei funghi i quali, in base al
“potenziale inoculo del fungo”, alle “condizioni di incubazione” (andamento
climatico) e al tempo in cui il substrato, è lasciata a disposizione del
patogeno, determinano un livello finale di concentrazioni di micotossine nel
prodotto più o meno elevato. Si deve quindi evitare tassativamente la
pratica, assai diffusa in alcuni areali, di lasciare in campo la coltura fino al
tardo autunno, al fine di ottenere un’ulteriore riduzione del tenore di
umidità, proprio per evitare che il fungo attacchi il substrato, determinando
così la contaminazione, poichè si è potuto constatare che l’anticipo della
raccolta può prevenire la fase più attiva dell’invasione fungina. Ugualmente
correlata sia all’intensità dell’infestazione, sia alla produzione di tossine da
parte del fungo, è la concimazione, che se corretta, fornisce alla pianta
migliori difese contro l’attacco del fungo tossigeno. L’agricoltore in questo
caso, deve soprattutto considerare l’importanza di assicurare un buon
bilanciamento del rapporto azoto/potassio, affidandosi per quest’ultimo a

21
buoni test di laboratorio ed inoltre, fornire un’adeguata quantità di azoto
prendendo in considerazione la quantità, in teoria assorbita e le perdite
dell’elemento stesso (Verderio, 2001). Nel caso del mais, l’elemento che
richiede maggiore attenzione è l’azoto: piante con evidenti sintomi di
carenza azotata (limitato sviluppo vegetativo e produzione al di sotto della
media), sono maggiormente predisposte alla contaminazione da aflatossine.
Sperimentazioni condotte in Emilia-Romagna hanno poi rilevato che, apporti
decisivamente superiori alle dosi da bilancio, possono incrementare
sensibilmente la contaminazione delle fumonisine, probabilmente a seguito
dello sviluppo di condizioni micro-climatiche più favorevoli alla diffusione dei
funghi (minore circolazione dell’aria con piante eccessivamente vigorose ed
il mantenimento di elevati livelli di umidità) (Agricoltura,2007). E’ stato
inoltre sperimentato se l’adozione della pratica di rotazione, la quale è stata
storicamente ed in modo meno obbligato tuttora un mezzo potente, per
ridurre l’impatto dei parassiti sulle coltivazioni, avrebbe potuto avere effetti
significativi sull’incidenza del patogeno, ma questa non ha portato a risultati
apprezzabili in grado di ridurre il potenziale di inoculo del fungo. Al contrario
è stato constatato che, le pratiche e gli interventi agronomici volti ad
aumentare il benessere della pianta hanno ridotto sensibilmente sia lo
sviluppo del fungo sulla pianta, sia il potenziale di inoculo sul terreno
(Verderio, 2001).
FASE DI RACCOLTA E POST-RACCOLTA
Durante la fase di raccolta del prodotto, è importante che vengano
considerate alcune pratiche fondamentali, in modo da poter svolgere in
modo attento questa delicata fase (Verderio, 2001). La raccolta è una delle
fasi ove è possibile intervenire maggiormente per il controllo delle
micotossine (Agricoltura, 2007).

22
Figura 3. Mietitrebbiatura del mais.
La formazione di aflatossine è favorita in campo da temperature elevate
(massima giornaliera superiore ai 30 °C) nel periodo compreso tra la
maturazione fisiologica della granella e la raccolta e dall’umidità della
granella stessa. Per tale ragione, una consistente riduzione del rischio
aflatossine può essere ottenuta raccogliendo la granella con umidità non
inferiore al 22-24%. Si segnala che valori di umidità inferiori al 20% sono da
considerarsi ad alto rischio, in quanto predispongono fortemente alla
contaminazione delle aflatossine, soprattutto in annate con andamento
stagionale caldo e asciutto. Analogamente, la raccolta anticipata della
granella consente di ridurre anche la contaminazione da fumonisine che,
negli ibridi più tardivi può raggiungere livelli elevati, in particolare in annate
caratterizzate da periodi di stress idrico e altri a decorso umido, soprattutto
in prossimità della raccolta. E’ dunque preferibile effettuare trebbiature
tempestive, anche se con qualche punto di umidità in più, in modo tale da
ridurre il tempo a disposizione dei funghi tossigeni per svilupparsi e
accumulare tossine nella granella (Agricoltura, 2007). Risulta poi di
particolare importanza una regolazione puntuale della mietitrebbia, la quale
permette di ridurre rotture e fessurazioni delle cariossidi, e di pre-pulire il
prodotto dalle parti a più basso peso specifico, questo perché una partita di

23
mais che presenti rotture e fessurazioni dei chicchi, costituisce un substrato
facilmente più attaccabile da parte dei funghi e di più difficile conservazione
durante la fase di stoccaggio. Per quanto riguarda, invece la pre-pulitura del
prodotto, molto frequentemente i granelli invasi dal fungo sono molto più
piccoli, più leggeri e di conseguenza più soggetti a rotture. Pertanto,
attraverso questa piccola operazione sulla mietitrebbia, è possibile ottenere
un prodotto finale teoricamente non contaminato (Verderio, 2001). Una
volta raccolto il prodotto, questo viene destinato agli essiccatoi. Già nelle
prime ore di attesa del prodotto umido sui carri o sui piazzali degli essiccatoi,
si possono attivare processi di ossidazione e di fermentazione che causano
una riduzione di sostanza secca ed un aumento della temperatura di massa,
che favoriscono una rapidissima proliferazione secondaria di funghi, i quali
sono dotati di una capacità di invasione proporzionale ai tempi di attesa,
all’umidità della granella, alla temperatura di attesa e all’altezza e
compressione dei cumuli. Proprio per questo motivo, diventa essenziale
ridurre l’intervallo di tempo tra la raccolta e l’ essiccazione onde evitare
proliferazioni secondarie. E’ risaputo, che durante la fase di conservazione
del prodotto, possono venire a crearsi condizioni favorevoli allo sviluppo dei
funghi tossigeni di magazzino. Riveste particolare importanza, come
condizione primaria per inibire ogni attività fungina durante questa fase,
l’umidità finale della granella, la quale deve essere adeguata non solo alle
caratteristiche del prodotto da conservare, ma anche alla tipologia
dell’impianto e alla durata dello stoccaggio. Risulta molto importante oltre al
controllo del parametro umidità, a cui l’essiccatore-stoccatore è soggetto,
anche le operazioni di eliminazione delle parti piccole o leggere presenti nel
prodotto, quali spezzati piccoli, polveri ecc.. e di riduzione delle
microfessurazioni delle cariossidi, sempre con l’unico scopo di permettere un
abbattimento diretto dei livelli di micotossine ed una migliore conservazione
al prodotto. Vista la frequente correlazione tra i livelli di “rotture” del
prodotto e i livelli di infestazione da funghi, come precedentemente
enunciato, i compiti e le responsabilità dell’essiccatore-stoccatore si vanno
riconfigurando come finalizzate, non esclusivamente alla conservazione di un

24
prodotto, ma al miglioramento, in funzione della qualità finale e delle
richieste del mercato. Per quanto riguarda la fase di pre-raccolta e la fase
raccolta e post-raccolta, gli aspetti principali dell’agrotecnica di sicura
attuabilità sono riassunti nella Tabella 4.
Agrotecnica ad alto rischio Agrotecnica a basso rischio Scelta di epoche di semina e ibridi con cicli colturali tali da condurre a raccolte in periodi molto caldi
Scelta di epoche di semina e ibridi con cicli colturali tali da condurre a raccolte al termine dell’estate o inizio autunno
Densità di semina elevate (8 piante/metro quadrato per ibridi a ciclo pieno)
Densità di semina equilibrate (6-6,5 piante/metro quadrato per ibridi a ciclo pieno)
Minima lavorazione o semina su sodo
Lavorazioni che assicurino l’interramento dei residui
Diserbo assente o poco efficace
Diserbo accurato
Concimazione: • Carente per potassio e azoto
(< 100 Kg/ha) • Eccessiva per azoto (>300
Kg/ha) • Alti apporti organici
Concimazione • Equilibrata tra azoto, fosforo
e potassio • Dosi di azoto pari a 180-240
Kg/ha • Apporti di azoto frazionati
Nessun controllo sulla piralide e sulla semina
Trattamento contro la piralide con insetticidi
Irrigazione: • Assente • Insufficiente (< 0,7 ETc) • Precocemente interrotta
Irrigazione: • Corretta (0,9-1,1 ETc) • Fino alla maturazione lattea
avanzata Raccolta Essiccazione prolungata in campo della granella Elevate rotture alla trebbiatura
Raccolta tempestiva soprattutto per maturazioni estive Ridotte rotture alla trebbiatura
Trasporto della granella umida non tempestivo Impiego di macchinari non puliti
Trasporto ed essiccazione tempestivi Pulitura attenta della mietitrebbia e dei carri
Tabella 4. Confronto tra agrotecniche ad alto e basso rischio.

25
Sebbene la prevenzione risulti essere la strategia principale, in alcuni casi è
necessario intervenire sulla granella già contaminata (Berocchi et al., 2004).
Figura 4. Chicchi di mais contaminati da muffe.
Possono essere utilizzati sistemi di decontaminazione e detossificazione,
aventi come principale scopo quello eliminare, per quanto possibile dalla
massa del prodotto, le micotossine. I principali metodi di decontaminazione
consistono in una pulitura e in una separazione meccanica tramite
vagliatura, molitura e ventilazione delle cariossidi; in una lavatura della
granella che normalmente viene effettuata successivamente alla vagliatura,
ed infine una macinazione ad umido soprattutto utilizzata per il mais in
associazione e prima della molitura ad umido. Tra i principali metodi di
detossificazione, possiamo distinguere tre diverse categorie quali: metodi
fisici, chimici e biologici comprendenti diversi sistemi riassunti nella tabella
5:

26
Metodi Fisici
Metodi Chimici
Metodi Biologici
Inattivazione termica: cottura ad alta pressione Utilizzo di adsorbenti (alluminosilicati, carboni attivi, bentonite, zeoliti) in grado di sequestrare le micotossine grazie alla loro struttura e le proprietà chimico-fisiche.
Utilizzo di acidi, basi, agenti riducenti, agenti cloruranti, sali, formaldeide.
Utilizzo di specifici agenti biotici (batteri, muffe, lieviti), capaci di degradare o trasformare enzimaticamente le micotossine.
Tabella 5. Metodi di detossificazione.
Dei sistemi elencati, quello di decontaminazione attraverso pulitura e
separazione meccanica è il più facilmente applicabile. Questa operazione è
eseguita mediante l’utilizzo di appositi strumenti, quali i setacci che hanno
lo scopo di rimuovere dalla granella di mais, le cariossidi più piccole e rotte,
in quanto rappresentano la parte di prodotto a più alto rischio di
contaminazione. L’effettuazione di studi preliminari derivanti dal settore
mangimistico ha permesso di notare che, utilizzando setacci aventi fori da 5
mm, si ottiene la riduzione dei livelli di inquinamento mediamente del 50%
e che, maggiore è il livello di contaminazione della partita, migliori saranno i
risultati (Bertocchi et al., 2004). Durante la fase di alimentazione del
bestiame, è possibile invece, utilizzare delle sostanze che riducano
l’assorbimento enterico delle tossine. L’utilizzazione degli additivi
“sequestranti”, possono rappresentare un aiuto per ridurre il rischio di
contaminazione da micotossine, se nell’immediato futuro dovessimo
comunque utilizzare per l’alimentazione degli animali mais o altri cereali
contaminati (nei limiti di legge), questo perché sono caratterizzati dal poter
impedire l’assorbimento gastrointestinale delle micotossine, riducendo di
conseguenza la loro eventuale presenza negli alimenti. In natura esistono

27
composti chimici capaci per la loro struttura, di adsorbire le micotossine
presenti nelle derrate alimentari. Inizialmente utilizzati per il loro potere
antiagglomerante, questi prodotti, sono stati successivamente utilizzati a
scopo detossificante, per ridurre i rischi di intossicazione da micotossine.
Qui di seguito vengono elencati i prodotti maggiormente (già
precedentemente accennati) utilizzati per tale scopo:
• Zeoliti naturali e sintetiche
• Alluminosilicati
• Carboni attivi
• Bentonite
• Argille
Va sottolineato però, che l’efficacia di molti dei prodotti citati, rimane ancora
dubbia in quanto, se esistono prove della loro validità in vitro, pochi sono i
lavori che ne dimostrano la stessa in vivo. Al momento le sostanze che in
letteratura sembrano avere maggior numero di ricerche con risultati positivi,
sono gli alluminosilicati e la bentonite, i quali devono essere miscelati
all’alimento non eccessivamente contaminato, affinché diano buoni risultati.
(Bertocchi et al., 2004).
1.1.7 Legislazione
Le recenti e numerose direttive dell’Unione Europea e di altri Organismi
Internazionali (es. FAO), e Nazionali (Ministero della Sanità) in materia di
qualità e sicurezza degli alimenti, impongono una sorveglianza non più
limitata alla semplice ispezione da eseguire esclusivamente nei punti
terminali delle filiere agro-alimentari, ma dei controlli da effettuare nel
corso di tutte le fasi che costituiscono la filiera produttiva. In questa ottica
di gestione totale di qualità, protesa all’ottenimento di standard qualitativi
sempre più elevati e quindi di maggiore sanità e sicurezza alimentare,

28
un’attenzione particolare è stata data anche al problema della presenza di
micotossine le quali, tra i contaminanti naturali dei prodotti alimentari,
rivestono un ruolo importante al punto di essere oggetto di specifica
legislazione prodotta a tutela della salute umana. Conseguentemente a tale
motivo e per la tutela anche della salute e del benessere animale, tale
argomento è da sempre di grande interesse per gli operatori del settore
agroalimentare-zootecnico. Fino ad oggi, l’UE è tra i paesi al mondo ad
avere il pacchetto normativo più completo in materia di micotossine. Gli
organi internazionali hanno promosso l’acquisizione di nuove conoscenze
attraverso studi e programmi di ricerca, con il fine di avere indicazioni
risolutive per la regolamentazione e limitazione del rischio per la salute
dell’uomo e degli animali. I risultati degli studi tossicologici ed
epidemiologici, infatti, non solo hanno portato ad una revisione della
concentrazione massima ammissibile di alcune micotossine in alimenti, ma
costituiscono la base della discussione in atto sulla regolamentazione da
avviare ex novo. La ricerca ha fornito indicazioni per la definizione dei limiti
di legge distinguendo specifiche categorie di consumo (es. bambini) e
specifiche classi di prodotto, il cui consumo può incrementare l’esposizione
dell’uomo al rischio di contaminazione. A livello europeo le norme preposte
alla limitazione dei livelli di micotossine negli alimenti destinati all’uomo si
sono susseguite rapidamente (Tecnoalimenti, 2006). In particolare, le
aflatossine presenti all’interno di alimenti destinati al consumo umano sono
sottoposte alle limitazioni imposte dai Regolamenti 472/2002 (CE/2002a),
2174/2003 (CE/2003b) e 683/2004 (CE/2004).

29
Aflatossine (ppb)
Prodotto
B1 B1, B2, G1, G2
M1
Spezie 5 10
Arachidi, frutta a guscio e frutta secca e relativi prodotti di lavorazione per il consumo umano diretto e l’utilizzo come ingredienti per la produzione di derrate alimentari
2 4 -
Arachidi da sottoporre a cernita o ad altri trattamenti fisici prima del consumo umano o dell’impiego come ingrediente di derrate alimentari
8 15 -
Frutta a guscio e frutta secca da sottoporre a cernita o ad altri trattamenti fisici prima del consumo umano o dell’impiego come ingrediente di derrate alimentari
5 10 -
Cereali e derivati destinati al consumo umano diretto
2 4 -
Cereali destinati alla cernita o ad altri trattamenti fisici prima del consumo umano o dell’impiego come ingrediente di derrate alimentari
2 4 -
Granturco da sottoporre a cernita o ad altri trattamenti fisici prima del consumo umano o dell’impiego come ingrediente di derrate alimentari
5 10 -
Latte (crudo, trattato termicamente e destinato alla fabbricazione di prodotti a base di latte)
- - 0,05
Alimenti per l’infanzia e alimenti a base di cereali destinati a lattanti e prima infanzia
0,1
0
- -
Alimenti per lattanti e alimenti di proseguimento, compresi il latte per lattanti e il latte per lo svezzamento
- - 0,025
Alimenti dietetici a fini medici speciali destinati in modo specifico ai lattanti
0,1
0
- 0,025
Tabella 6. Reg.472/2002 (CE/2002), 2174/2003 (CE/2003b) e
683/2004 (CE/2004).

30
Sempre per quanto riguarda l’aflatossina B1, è stata redatta un’ulteriore
regolamentazione finalizzata a limitarne la presenza nelle derrate alimentari
destinate agli animali. Essa è rappresentata dalla Direttiva 2002/32
(CE/2002b), il cui allegato I riguardante i quantitativi massimi delle
sostanze indesiderabili, è stato poi modificato con la Direttiva 2003/100
(CE/2003c). I dati sono relativi al contenuto massimo di micotossina in
mg/Kg (ppm) in mangime al tasso di umidità del 12%.
Aflatossina B1 Ppm
Tutte le materie prime per mangimi 0,02
Mangimi completi per bovini, ovini e caprini ad eccezione di:
• Mangimi completi per animali da latte • Mangimi completi per vitelli ed agnelli
0,02
0.005 0,01
Mangimi completi per suini e pollame, salvo animali giovani
0,02
Altri mangimi completi 0,01
Mangimi complementari per bovini, ovini e caprini, ad eccezione dei mangimi complementari per animali da latte, vitelli ed agnelli
0,02
Mangimi complementari per suini e pollame, salvo per animali giovani
0,02
Altri mangimi completi 0,05
Tabella 7. Direttiva 2003/100/CE.
L’articolo 5 di tale Direttiva inoltre, prescrive che “i prodotti destinati
all’alimentazione degli animali il cui contenuto di sostanze indesiderabili
supera il livello massimo fissato non possono essere mescolati, a scopo di
diluizione, con lo stesso prodotto o con altri prodotti destinati
all’alimentazione animale”. Per quanto riguarda la presenza di ocratossina A

31
nelle derrate alimentari destinate al consumo umano, i Regolamenti
472/2002 (CE/2002a), 683/2004 (CE/2004) e 123/2005 (CE/2005),
costituiscono la normativa vigente a livello europeo.
Prodotto Ocratossina
A (ppb)
Cereali non lavorati 5 Tutti i prodotti derivati dai cereali (compresi i prodotti lavorati a base di cereali destinati al consumo umano diretto)
3
Frutti essiccati della vite 10 Alimenti per l’infanzia, alimenti a base di cereali destinati ai lattanti e prima infanzia, alimenti dietetici a fini medici speciali destinati in modo specifico ai lattanti
0,50
Caffè torrefatto e caffè torrefatto macinato 5 Caffè solubile 10 Vino ed altri vini e bevande spiritose a base di mosto d’uva; succo d’uva, ingredienti a base di succo d’uva in altre bevande incluso il nettare d’uva e il succo d’uva concentrato e ricostituito; mosto d’uva e mosto d’uva concentrato ricostituito, destinati direttamente al consumo umano
2
Caffè crudo, frutta secca diversa dalle uve secche, birra, cacao e prodotti a base di cacao, vini liquorosi, prodotti a base di carne, spezie e liquirizia
-
Tabella 8. Reg. (CE) 472/2002, Reg. (CE) 683/2004, Reg (CE) 123/2005.
Il Regolamento 1425/2003 (CE/2003a), a livello comunitario, pone le basi
per il controllo della patulina negli alimenti.

32
Prodotto Patulina (ppb)
Succhi di frutta, nettare di frutta, succo di frutta concentrato dopo ricostituzione
50
Bevande spiritose, sidro e alre bevande fermentate derivate dalle mele o contenenti succo di mela
50
Prodotti a base di mele allo stato solido 25 Succo di mela, prodotti a base di mela per l’infanzia, altri alimenti per bambini
10
Tabella 9. Reg (CE) 1425/2003.
Il Regolamento 1126/2007 (CE/2007), modifica il Reg. CE 1881/2006
(CE/2006b) e definisce i tenori massimi di alcuni contaminanti nei prodotti
alimentari per quanto riguarda le Fusarium-tossine nel granturco e nei
prodotti a base di granturco.

33
Tabella 10. Reg. (CE) 1126/2007.
Prodotto Zearalenone (ppb)
Cereali non trasformati diversi dal granturco 100 Granturco non trasformato ad eccezione del granturco non trasformato destinato alla molitura ad umido
350
Olio di granturco raffinato 400 Pane (compresi piccoli prodotti da forno), prodotti della pasticceria, biscotteria, merende a base di cereali e cereali da colazione, esclusi le merende a base di granoturco e i cereali da colazione
50
Cereali destinati al consumo umano diretto, farina di cereali, crusca e germe come prodotto finito commercializzato per il consumo umano diretto eccetto i prodotti alimentari di seguito riportati
75
Granoturco destinato al consumo umano diretto, merende a base di granoturco e cereali da colazione a base di granoturco
100
Alimenti a base di cereali trasformati (esclusi quelli a base di granoturco) e altri alimenti destinati ai lattanti e ai bambini
20
Alimenti a base di granoturco trasformato destinati ai lattanti e ai bambini
20
Frazioni della molitura del granoturco di dimensioni > 500 micron di cui al codice e altri prodotti della molitura del granoturco non destinati al consumo umano diretto di dimensioni > 500 micron di cui al codice NC 1904 10 10
200
Frazioni della molitura del granoturco di dimensioni < 500 micron di cui al codice e altri prodotti della molitura del granoturco non destinati al consumo umano diretto di dimensioni < 500 micron di cui al codice NC 1904 10 10
300
34
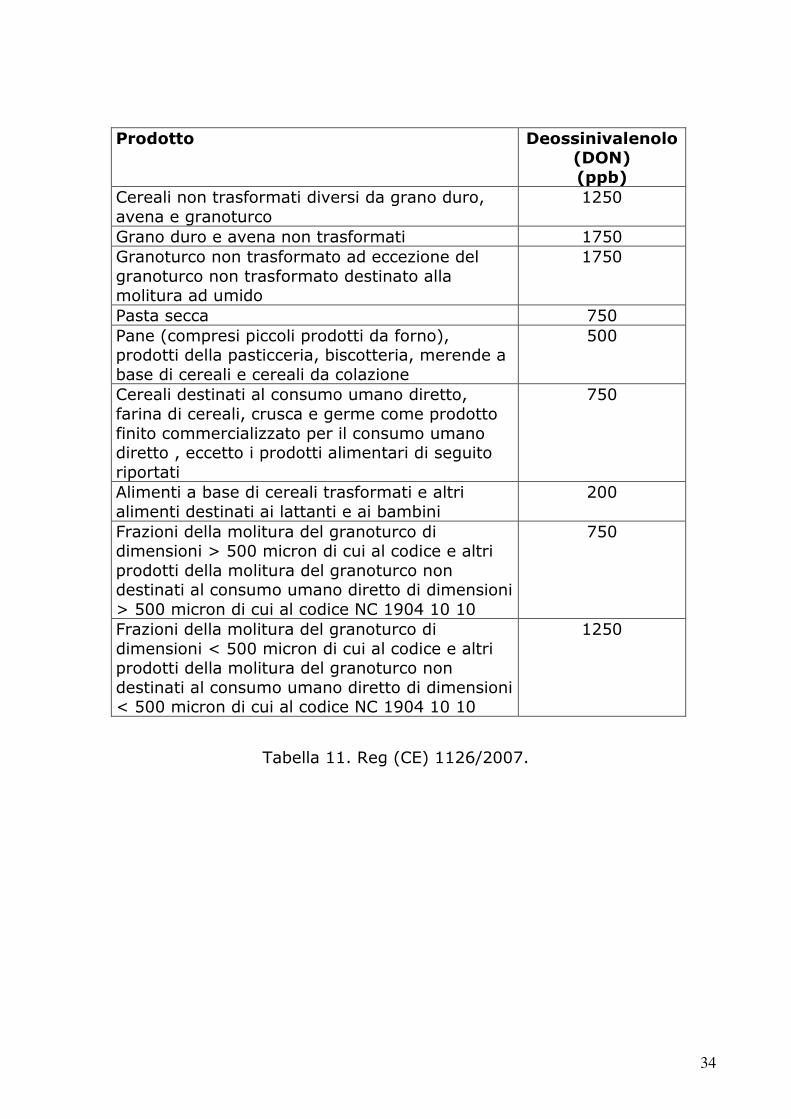
Prodotto Deossinivalenolo (DON) (ppb)
Cereali non trasformati diversi da grano duro, avena e granoturco
1250
Grano duro e avena non trasformati 1750 Granoturco non trasformato ad eccezione del granoturco non trasformato destinato alla molitura ad umido
1750
Pasta secca 750 Pane (compresi piccoli prodotti da forno), prodotti della pasticceria, biscotteria, merende a base di cereali e cereali da colazione
500
Cereali destinati al consumo umano diretto, farina di cereali, crusca e germe come prodotto finito commercializzato per il consumo umano diretto , eccetto i prodotti alimentari di seguito riportati
750
Alimenti a base di cereali trasformati e altri alimenti destinati ai lattanti e ai bambini
200
Frazioni della molitura del granoturco di dimensioni > 500 micron di cui al codice e altri prodotti della molitura del granoturco non destinati al consumo umano diretto di dimensioni > 500 micron di cui al codice NC 1904 10 10
750
Frazioni della molitura del granoturco di dimensioni < 500 micron di cui al codice e altri prodotti della molitura del granoturco non destinati al consumo umano diretto di dimensioni < 500 micron di cui al codice NC 1904 10 10
1250
Tabella 11. Reg (CE) 1126/2007.

35
Prodotto Fumonisine (somma di B1 e B2) (ppb)
Granoturco non trasformato, ad eccezione del granoturco non trasformato destinato alla molitura ad umido
4000
Granoturco destinato al consumo umano diretto, prodotti a base di granoturco destinati al consumo umano diretto, ad eccezione degli alimenti elencati di seguito
1000
Cereali da colazione e merende a base di granoturco 800 Alimenti a base di granoturco trasformato e altri alimenti destinati ai lattanti e ai bambini
200
Frazioni della molitura del granoturco di dimensioni > 500 micron di cui al codice e altri prodotti della molitura del granoturco non destinati al consumo umano diretto di dimensioni > 500 micron di cui al codice NC 1904 10 10
1400
Frazioni della molitura del granoturco di dimensioni < 500 micron di cui al codice e altri prodotti della molitura del granoturco non destinati al consumo umano diretto di dimensioni < 500 micron di cui al codice NC 1904 10 10
2000
Tabella 12. Reg (CE) 1126/2007.
Per quanto riguarda lo zearalenone, la sua presenza nelle derrate alimentari
destinate agli animali, costituisce anche una problematica di tipo legale in
quanto uno dei suoi metabolici, l’α-zearalanol, può essere utilizzato anche
come sostanza anabolizzante (Shier et al., 2001).
Anche per l’ocratossina A, il deossinivalenolo, lo zearalenone, le tossine T-2
e HT-2 e le fumonisine, è stata redatta una regolamentazione finalizzata a
limitarne la presenza nelle derrate alimentari destinate agli animali. Essa è
rappresentata dalla Direttiva 2006/576/CE (CE/2006a). I dati sono relativi
al contenuto massimo di micotossina in mg/Kg (ppm) di mangime al tasso
di umidità del 12%.

36
Micotossina Prodotti destinati all’alimentazione degli animali
ppm
Deossinivalenolo Materie prime per mangimi • Cereali e prodotti a base di cereali
fatta eccezione per sottoprodotti del granoturco
• Sottoprodotti del granoturco Mangimi complementari e completi ad eccezione di:
• Mangimi complementari e completi per suini
• Mangimi complementari e completi per vitelli (< 4 mesi), agnelli e capretti
8
12 5
0,9 2
Zearalenone Materie prime per mangimi • Cereali e prodotti a base di cereali
fatta eccezione per sottoprodotti del granoturco
• Sottoprodotti del granoturco Mangimi complementari e completi ad eccezione di:
• Mangimi complementari e completi per suinetti e scrofette (giovani scrofe)
• Mangimi complementari e completi per scrofe e suini da ingrasso
• Mangimi complementari e completi per vitelli, bovini da latte, ovini (inclusi agnelli) e caprini (inclusi capretti)
2 3
0,1
0,25
0,5
Ocratossina A Materie prime per mangimi • Cereali e prodotti a base di cereali
Mangimi complementari e completi • Mangimi complementari e completi
per suini • Mangimi complementari e completi
per pollame
0,25
0,05
0,1
Fumonisine B1 + B2
Materie prime per mangimi • Granoturco e prodotti derivati
Mangimi complementari e completi per: • Suini, equini, conigli e animali da
compagnia, • Pesci, • Pollame, vitelli (<4 mesi), agnelli e
capretti, • Ruminanti adulti (> 4 mesi)
60 5
10 20
50
Tabella 13. Direttiva (CE) 2006/576.

37
Anche in Italia sono stati adottati questi Regolamenti e tramite la Circolare
Ministeriale n° 10 del 9 Giugno 1999 sono stati forniti dei valori guida per le
autorità preposte al controllo ufficiale, cioè dei valori massimi di tossine
ammissibili nelle derrate alimentari di provenienza nazionale, comunitarie e
proveniente da Paesi Terzi.
Aflatossine (ppb)
Prodotto
B1 B1, B2, G1, G2
M1
Ocratossina A (ppb)
Caffè crudo - - - 8 Caffè tostato e solubile
- - - 4
Cacao e derivati - - - 0,5 Birra - - - 0,2 Carne suina e derivati
- - - 1
Piante infusionali 5 10 - -
Tabella 14. Valori guida della circolare n. 10, del 9 Giugno 99,
G.U n. 135.
Per quanto riguarda la Regolamentazione fuori dall’Europa, possiamo
prendere in considerazione il caso degli USA, i quali sono principalmente
sensibili alle contaminazioni di aflatossine legate al mais. Le condizioni
climatiche degli USA negli stati meridionali (clima caldo umido), fanno sì che
le derrate alimentari non possono che avere livelli di contaminazione da
micotossine decisamente superiori a quelli europei. In tal senso pertanto, la
normativa USA, risulta essere meno restrittiva di quella europea. E’ il caso
ad esempio dei limiti sulle aflatossine: per gli USA i valori regolati dalla UE,
sarebbero impossibili da ottenere viste le condizioni climatiche che
caratterizzano il paese; per tale ragione il loro livello di accettabilità per
legge è 10 volte superiore a quello indicato in Europa (Tecnoalimenti,
2006).

38
2. OCRATOSSINE
2.1 GENERALITA’
2.1.1Cenni storici
Nel Gennaio del 2006, il proprietario del più grosso impianto di molitura di
cereali in Italia, è stato arrestato per l’importazione di circa 58.000
tonnellate di frumento proveniente dal Canada, il quale risultava
contaminato da ocratossina A (OTA) a concentrazioni di 15 µg/kg (Hooper,
2006). Questo frumento è stato venduto e destinato direttamente al
consumatore finale e ai diversi processi di lavorazione alimentare. Questa
notizia, ha sconvolto i consumatori, ma l’esposizione al rischio ocratossine,
non è nuova. Si parla già di ocratossina nel 1750 in seguito ad una minore
mortalità in Gran Bretagna e in Francia dovuta in parte al miglioramento
delle condizioni igienico sanitarie e alimentari, ma anche alle micotossine, in
quanto, a quell’epoca, il consumo alimentare di patate, aumentò
drasticamente andando a sostituire alimenti come grano, orzo e riso,
maggiormente suscettibili rispetto alla patata, alla contaminazione di
micotossine (Stomer et al., 1992). Inoltre, si notò la correlazione tra gli
eventi di mortalità di massa e il clima favorevole alla formazione di
micotossine (Stomer et al., 1992). La scoperta dell’ocratossina però, risale a
tempi ancora più lontani; tant’è che, diversi archeologi studiosi delle antiche
tombe egizie, cercarono di far luce sulle cause di morte dei faraoni,
suggerendo come causa di morte un’insufficienza renale acuta provocata
dall’inalazione di spore fungine contenenti ocratossine (Di Paolo et al.,
1993).

39
2.1.2 Miceti produttori e condizioni di sviluppo
Le ocratossine sono un gruppo di metaboliti secondari strutturalmente simili
prodotti da funghi del genere Aspergillus e Penicillium, principalmente A.
ochraceus e P. verrucosum (Miraglia e Brera, 1999). La produzione di
ocratossine è dipendente da differenti fattori come la temperatura, l’acqua
libera ed altre condizioni che influiscono sulla fisiologia dei funghi produttori.
Nelle regioni caratterizzate da climi temperato-freddi, le ocratossine, sono
maggiormente prodotte da P. verrucosum (Pitt, 2000; Castella et al., 2002)
o P .nordicum (Larsen et al., 2001). P. verrucosum, contamina
principalmente piante come cereali, mentre P. nordicum è stato
maggiormente trovato in prodotti carnei e formaggio. Nelle regioni
caratterizzate invece, da climi tropicali e semitropicali, è stata rinvenuta la
presenza di A. ochraceus, il quale è stato ritrovato in diversi prodotti come
nocciole, arachidi, legumi, spezie, chicchi di caffè verde e frutta secca, ma
anche in processi di affumicatura e salatura di carne e pesce (WHO/FAO,
2001). Altre due specie di Aspergillus, rispettivamente, A.niger var niger
(Abarca et al., 2001; Belli et al.,2004) e A.carbonarius (Teren et al., 1996;
Mitchell et al., 2004), sono stati identificati anch’essi come produttori di
ocratossine. La contaminazione da ocratossine di substrati come i cereali e
le oleaginose nelle zone umide, si pensa sia dovuta a specie di A.niger var
niger in addizione ad A.ochraceus (Accensi et al., 2004), mentre
A.carbonarium sembra maggiormente coinvolto nella contaminazione di
uva, uva passa, e caffè (Sage et al., 2002; Cabanes et al., 2002). La
maggior parte delle muffe deputate alla sintesi delle ocratossine sono
xerofile. Ponendo attenzione sulle due specie maggiori produttrici di
ocratossine, possiamo dire che Aspergillus ochraceus sintetizza l’ocratossina
A quando l’aw è superiore a 0,80 e la produzione ottimale si osserva a valori
di aw 0,96-0,98 (Adebajo et al., 1994; Ramos et al., 1998); mentre
Penicillum verrucosum, sintetizza la micotossina quando l’aw è compresa tra
0,80 e 0,90 con un massimo di produzione a valori di aw compresi tra 0,95
e 0,99 (0,92 più specificatamente con riferimento al grano e all’orzo)
(Patterson e Darnogloul, 1986; Northolt e Bullerman, 1982; Northolt et al.,

40
1979). Alcune volte tali micotossine, sono sintetizzate durante la
conservazione o la commercializzazione stessa dei prodotti in precedenza
ben essiccati, quando per qualche causa i prodotti ritornano ad inumidirsi.
Questo in genere, accade nelle derrate conservate alla rinfusa in magazzino
o in silos per condensazione d’acqua sui pavimenti o su altre superfici
fredde. Piuttosto frequente, è anche l’eventualità di una condensazione di
acqua sui prodotti confezionati in buste di plastica quando il
confezionamento avviene ad elevate temperature ed umidità in quanto in un
secondo momento, se le buste vengono esposte a basse temperature,
l’umidità residua nella plastica si condensa e consente la crescita dei miceti
(Hesseltine, 1969). Per limitare la contaminazione degli alimenti, il tenore in
acqua deve essere contenuto durante la conservazione e mantenuto
inferiore al 13-13,5% per i cereali e al 7-8% per i semi oleosi. Per quanto
riguarda la temperatura, quelle ideali per lo sviluppo fungino sono comprese
tra 20 e 30°C con l’umidità del substrato del 30%. La produzione di tossine
invece, avviene a temperature lievemente più basse di alcuni gradi rispetto
a quelle ottimali per lo sviluppo del micelio fungino, ma che possono
oscillare in un range di 10-50°C (Osweiler, 1992). In particolar modo A.
ochraceus, sintetizza l’OTA a partire da una temperatura di 12°C, con un
massimo di produzione a 30°C (Bacon et al., 1973; Haggblom, 1982;
Northolt e Bullerman, 1982; Northolt et al., 1979; Ramos et al., 1998);
mentre A. nigri, cresce a temperature comprese tra 6 e 47°C con un
optimum di sviluppo a 35-37°C e sintetizza la micotossina in un range di
20-25°C. Infine A. carbonarius, si sviluppa a temperature comprese tra 10 e
40°C e produce l’OTA tra i 15 e i 35°C (Esteban et al., 2004). Il fattore
temperatura risulta essere inoltre, determinante per la sintesi di una
specifica tossina, se si considera che uno stesso fungo può elaborare tossine
diverse a temperature diverse (A. ochraceus A 25°C sintetizza l’OTA mentre
a 20°C sintetizza acido penicillio). I trattamenti termici classici di
sterilizzazione degli alimenti, permettono di distruggere le muffe, ma sono
per lo più, poco efficaci contro le ocratossine (termostabili come la maggior
parte delle micotossine) (Pasteiner, 1997). Anche la composizione gassosa

41
dell’atmosfera può influenzare la crescita delle specie tossigene e lo sviluppo
delle relative micotossine (Northolt e Bullerman, 1982; El-Halouat e
Debevere, 1997; Paster et al., 1983). Le muffe che sintetizzano ocratossine,
sono aerobie ma possono adattarsi, quando sussistono le altre condizioni
ottimali, all’ambiente in atmosfera modificata contenente più CO2 della
norma. Alcuni autori, hanno però dimostrato che, modificando la
composizione in CO2 dell’atmosfera (20%), era possibile ridurre
l’accrescimento fungino (Hesseltine, et al 1972) e che, inoltre un’atmosfera
contenente il 30% di CO2 era capace di inibire completamente la produzione
di OTA da parte di A. ochraceus (Paster et al., 1983). La mancanza di
ossigeno non comporta l’eliminzaione dei miceti, ma ne sospende l’attività e
quindi la sintesi di tossine. Anche il pH è un fattore importante che influenza
la crescita dei funghi ocratossigeni e la relativa sintesi di OTA. Lo sviluppo
del micelio, avviene a valori compresi tra 4 e 8. Il pH ottimale per la
produzione di OTA da parte di specie del genere Penicillium, è di 6,5
(Bullerman, 1985). Infine, il tipo di substrato, è l’elemento, come già
ricordato, che più probabilmente influenza la tossinogenesi. A tal proposito
(Madhyastha et al., 1990), è stato osservato che l’OTA viene sintetizzata da
P.verrucosum preferibilmente sui cereali rispetto alle leguminose; invece
avviene il contrario per l’A.ochraceus il quale colonizza principalmente le
leguminose, in particolare le arachidi e la soia. La presenza di miceti sui
cereali, potrebbe determinare variazioni della qualità nutrizionale degli
alimenti; è stato infatti dimostrato, come la contaminazione di grano e di
orzo da parte dei miceti, implica una grande riduzione della quantità di lipidi
e di amido rispetto alle colture non colonizzate, e che la composizione
aminoacidica subisce delle modifiche (Madhyastha et al., 1993). Sembra
infatti che le specie ocratossigne, sintetizzino più facilmente OTA sfruttando
proprio le riserve aminoacidiche dell’ospite; in particolare prolina ed acido
glutammico favoriscono la produzione di OTA nell’orzo (Haggblom e Ghosh,
1985). La presenza di ocratossine su specie vegetali, potrebbe poi essere
messa in relazione anche ad un diverso contenuto in microelementi, tra cui
zinco, ferro, boro, mobildeno e manganese. Bisogna puntualizzare invece

42
che, durante la conservazione, più che il substrato chimico, è lo stato fisico
dell’alimento che interviene nel favorire o meno la crescita dei miceti, in
quanto i semi macinati, verranno più velocemente contaminati poiché la
funzione protettiva offerta dal tegumento viene a mancare.
2.1.3 Alimenti contaminati
Grazie alla grande diffusione dei funghi produttori (Aspergillus ochraceus
nelle regioni a clima caldo e Penicillium verrucosum nei paesi a clima
freddo), l’ocratossina A presenta una distribuzione mondiale. Molteplici sono
gli alimenti che possono essere contaminati in primo luogo dalle diverse
specie fungine produttrici di ocratossine e secondariamente dagli stessi
metaboliti. I principali substrati contaminati da OTA sono: riso, segale,
mais, grano, sorgo, orzo, frumento, i cereali in genere (o per
contaminazione diretta dei cereali o per ammuffimento delle farine) ed i
prodotti da forno specialmente pane e biscotti. Inoltre, le noci, i pistacchi, le
arachidi ed i sottoprodotti delle loro rispettive lavorazioni (panelli e farine di
estrazione), come olive ma anche fagioli e legumi sono di frequente
contaminati dall’OTA (Campbell et al., 2000; Fukal, 1990; Hennigen e Dick;
1995; Hohler, 1998; Holmberg et al., 1991; Scott et al., 1972; Scudamore ,
1996; Wolff, 2000; Yoshizawa, 1991). In questi ultimi anni è stato messo in
evidenza come l’OTA contamini anche altri tipi di alimenti in particolare
vino, birra e caffè. Recenti studi hanno mostrato come la concentrazione di
OTA sia più elevata nei vini rossi , seguito da quelli rosati ed infine da quelli
bianchi (Burdaspal e Legarda, 1999; Soleas et al., 2001; Ueno, 1998;
Visconti et al., 1999; Zimmerli e Dick, 1996). Ciò è dovuto al diverso
processo di lavorazione dell’uva; infatti la produzione di vino rosso, prevede
la fermentazione del succo insieme alle bucce che potrebbero essere
contaminate nella parte esterna dalla micotossina. I vini dolci risultano
anche più contaminati dei vini rossi (Burdaspal e Legarda, 1999; Zimmerli e
Dick, 1996), in quanto, per ottenere un’uva più dolce, si effettua la
vendemmia più tardi e ciò favorisce lo sviluppo delle muffe ocratossigene e

43
delle relative micotossine. I vini che provengono dalle regioni del
Mediterraneo risultano più contaminati di quelli provenienti dai Paesi del
nord Europa (Hohler, 1998; Markaki et el., 2001; Zimmerli e Dick, 1996).
Anche alcuni succhi d’uva, possono contenere quantità di OTA, anche
superiori rispetto ai vini, risultando così, molto pericolosi in quanto destinati
principalmente al consumo dei bambini (Zimmerli e Dick, 1996).
Fortunatamente, la tossicità delle ocratossine nel vino, può essere
minimizzata con l’utilizzo di sostanze comunemente commercializzate
nell’industria alimentare, come carboni, bentonite e fibre vegetali dal potere
adsorbente, oltre che con l’ausilio di microrganismi come l’Acinetobacter
calcoaceticus che le degrada ad α-ocratossina (Carratù e Cuomo, 2001). La
presenza di OTA nel caffè è stata evidenziata per la prima volta nel 1974
(Bucheli e Taniwaki, 2002). La sua produzione nel caffè sembra sia dovuta
ad Aspergillus spp, principalemente A.niger, A.carbonarius ed A.ochraceus
(Bucheli et al., 2000; Bucheli e Taniwaki, 2002; Joosten et al., 2001; Tèren
et al., 1997). Dati recenti, indicano che l’80% dell’OTA è distrutta durante la
torrefazione industriale e che il caffè contaminato, venduto al dettaglio,
offre solo un contributo marginale all’assunzione quotidiana di OTA (Van Der
Stegen et al., 1997). Il caffè istantaneo però, risulta essere più pericoloso in
quanto contiene livelli di ocratossine significativamente più elevati rispetto
al caffè prodotto a partire da chicchi tostati (Bresch et al., 2000). La
contaminazione della birra da parte della micotossina invece, sembra sia
dovuta allo sviluppo di P.verrucosum durante la conservazione dell’orzo e
durante la produzione del malto (Baxter et al., 2001). E’ stata osservata
una percentuale di contaminazione del 42% nell’uva passa e nell’uva
sultanina proveniente dalla Turchia e dalla Grecia (con alti livelli di
contaminazione in un range di 4-53,6 µg/kg). Ugualmente alte
concentrazioni sono state riscontrate in Inghilterra sugli stessi substrati con
incidenza dell’88% dei campioni esaminati (MacDonald et al., 1999). E’
stata riscontrata una notevole presenza di OTA anche sulla frutta sottoposta
a procedimenti di essiccazione quale prugne, albicocche e fichi (Zohri e
Abdel-Gawad, 1993). Le oleaginose ed i semi di girasole, di arachidi e di

44
soia sono spesso invasi da funghi, però l’estrazione ed i processi industriali
cui sono sottoposti comportano la quasi totale eliminazione delle
micotossine. Il cacao, le spezie in genere, le foglie di tè e le erbe medicinali
(Halt, 1998), le mandorle ed i pistacchi, possono evidenziare concentrazioni
discrete di ocratossine, tuttava, le radiazioni possono efficientemente
determinare una drastica riduzione della loro concentrazione su questi
prodotti di piccolo volume. L’OTA è stata ritrovata anche in alimenti di
origine animale, in particolare in prodotti a base di carne di maiale e di
specie avicole (Canela et al., 1994; Curtui et al., 2001; Gareis e Wolff,
2000; Gareis e Scheuer, 2000; Holmberg et al., 1991; Jimenez et al., 2001;
Jorgensen, 1998; Kuiper-Goodman e Scott, 1989) a causa, come dimostrato
sperimentalmente, dal fenomeno del carry-over della micotossina dal
mangime ai tessuti animali (Abramson et al., 1983). Questo si verifica
maggiormente in animali che vengono alimentati con mangimi contaminati
da OTA (Fukal, 1990; Greis e Wolff, 2000; Pohland et al., 1992; Kuiper-
Goodman e Scott, 1989; Speijers e Van Egmond, 1993). Per alcuni studiosi,
la presenza delle micotossine negli alimenti di origine animale, è tuttavia,
più verosimilmente da addebitare ad altri ingredienti del prodotto sottoposto
a lavorazione come ad esempio i pistacchi utilizzati per aromatizzare la
mortadella. Alcuni ricercatori, hanno contaminato la porzione esterna di un
prosciutto crudo, ed hanno evidenziato come la tossina contamini solo
superficialmente il prodotto entrando di pochi mm nella cotenna (Escher et
al 1973). E’ stata riscontrata la presenza di OTA anche in diversi prodotti
carnei sottoposti ad affumicamento (Pepeljnjack e Blozevic, 1982). Un’altra
pericolosa fonte di contaminazione, può essere quella derivante dalla
presenza dei miceti utilizzati nell’industria di lavorazione dei prodotti carnei
(ad es. salumi), quali vari ceppi di Penicillium ed Aspergillus per conferire al
prodotto qualità organolettiche apprezzabili ma che, se sono presenti le
idonee condizioni, sono anche produttori di ocratossina, citrinina,
citroviridina e sterigmatocistina. La presenza della micotossina, è stata
riscontrata anche nel latte di bovine alimentate con mangime contaminato
(Hohler, 1998; Skaug, 1999; Valenta e Goll, 1996), tuttavia sono ancora

45
discordanti i pareri relativi alla sua presenza in questa matrice biologica i cui
livelli risultano a concentrazioni tali da non destare pericolo.
Figura 5. Mais contaminato da muffe.
2.1.4 Caratteristiche chimiche e strutturali
Le ocratossine, eccetto l’ocratossina α (OTα), costituiscono un gruppo di
derivati dell’isocumarina strettamente correlati tra loro, legati al gruppo
amminico della L-β-fenilalanina e classificati in base alla loro origine
biosintetica come pentachetidi (Turner, 1971). Le ocratossine attualmente
conosciute sono l’ocratossina A (OTA), l’ocratossina B (OTB), l’ocratossina C
(OTC), l’ocratossina α (OTα), l’ocratossina β (OTβ), i due epimeri 4R/S-
idrossiocratossina A, la 10-idrossiocratossina A e la forma aperta di OTA
(OP-OTA) (Van der Merwe et al., 1965a,b; Steyn e Holzapfel, 1967; Xiao et
al., 1995; Xiao et al., 1996a). Tra tutte l’OTA è quella più studiata per la
sua elevata diffusione e per la sua importanza tossicologica (Miraglia e
Brera, 1999). L’OTA è stata isolata per la prima volta nel 1965 in Sud africa

46
da A.ochraceus (Van der Merwe et al., 1965a). L’OTA o 7(L-β-fenilalanina-
carbonil)-carbossil-5-cloro-8-idrossi-3,4diidro3R-metilisocumarina (Kuiper-
Goodman e Scott, 1989), presenta un atomo di cloro in posizione C5. L’OTA
negli alimenti e nei mangimi, è spesso accompagnata dall’OTB che si
differenzia dal non avere l’ atomo di cloro. Sebbene l’OTB, possa co-esistere
con l’OTA in alcuni prodotti naturalmente contaminati, in alcuni studi sugli
effetti tossici in animali, si è potuto constatare che le sue concentrazioni
sono generalmente più basse e che la sua tossicità, è notevolmente
inferiore rispetto all’OTA (Mally at al., 2005). Il potenziale tossico di
quest’ultima è incrementato dalla presenza di un gruppo OH (Chu et al.,
1972) che, permettendo la formazione di legami idrogeno con altri elementi,
determina la formazione di strutture secondarie (Bredenkamp et al., 1989).
In particolare, l’OTA è un composto cristallino incolore, altamente solubile in
solventi organici polari e in soluzioni acquose di bicarbonato ma poco
solubile in acqua (Betina, 1989). Ha inoltre proprietà debolmente acide
(Chu, 1974). Gli esteri dell’OTA, possiedono una tossicità simile a quella del
loro precursore, mentre diversa è la tossicità degli esteri dell’OTB in quanto
è pressochè nulla (Ueno, 1987). L’ OTα ed i derivati idrossilati dell’OTA
invece, non risultano essere tossici; al contrario, la forma aperta dell’OTA,
sembra possedere una tossicità simile a quella del suo precursore (Xiao et
al., 1996a).
Figura 6. Struttura chimica dell'ocratossina A.

47
2.2 TOSSICOCINETICA
2.2.1 Generalità
Considerando la notevole diffusione in natura delle micotossine nelle derrate
alimentari, assume particolare importanza capire la tossicologia e il destino
biologico di questi composti. Il destino di una tossina in un organismo
animale, è il frutto dei processi di assorbimento, distribuzione,
biotrasformazione e dei processi di eliminazione che, nel caso di animalI di
interesse zootecnico, comprendono le modalità di passaggio dei metaboliti
nella carne, uova e latte.
2.2.2 Assorbimento
L’assorbimento dell’OTA in molte specie, avviene inizialmente a livello dello
stomaco a causa della sua acidità (pKa =7,1) .Comunque, da studi condotti
su animali con anse intestinali legate, il piccolo intestino si è dimostrato il
maggior sito di assorbimento, specialmente a livello del digiuno, il cui
passaggio di OTA può avvenire anche contro gradiente e dipende dal pH
della superficie mucosale (Kumagai e Aibara, 1982; Kumagai, 1988). I
valori di pKa del gruppo carbossilico della fenilalanina (4,2-4,4) e del gruppo
idrossilico del fenolo dell’isocumarina (7,0-7,3), iocano un ruolo essenziale
nell’assorbimento della micotossina. A condizioni di pH fisiologico del chimo
duodenale sono presenti sia la forma monoanionica (OTA-) sia la dianionica
(OTA-2), mentre la forma totalmente protonata è presente principalmente in
soluzioni acide, come nella parte superiore del tratto gastrointestinale. In
seguito ad assorbimento si lega alle siero proteine, principalmente alle
albumine, e a seconda dell’affinità e del grado di legame con le proteine, si
riscontrano notevoli differenze nell’emivita nel siero. La persistenza dell’OTA

48
nel sangue infatti è più lunga nell’uomo, dove raggiunge un’emivita di 840
ore (Miraglia e Brera, 1999) e nel maiale (35 giorni) che sono le specie più
sensibili. I picchi di OTA riscontrati nel siero e nel contenuto intestinale sono
una conseguenza della circolazione entero-epatica, in quanto l’escrezione
biliare di questa tossina è molto efficiente (Fuchs et al., 1988; Roth et al.,
1988). Negli animali la concentrazione della tossina e dei suoi metaboliti nei
vari tessuti varia a seconda della dose di somministrazione, della forma
dell’OTA somministrata (cristallina o presente naturalmente nel cibo), dalla
composizione della dieta, dallo stato di salute dell’animale e dalla specie. In
quest’ultimo caso la percentuale di OTA assorbita è pari al 66% nei maiali,
56% nei ratti e conigli e del 40% nei polli (Galtier et al., 1981). La
percentuale risulta bassa invece nei ruminanti poiché la flora ruminale
(costituita principalmente da protozoi), trasforma la micotossina
rapidamente in OTα (Kuiper-Goodman e Scott, 1989).
2.2.3 Distribuzione
LEGAME CON LE PROTEINE DEL PLASMA
La biodisponibilità di OTA, stimata paragonando la concentrazione sierica
massima dopo la somministrazione orale o endovenosa, è scarsa nel pesce
ma risulta compresa tra il 44 e il 97% nei mammiferi studiati (Hagelberg et
al., 1989). In seguito all’ assorbimento l‘OTA, viene convogliata nel circolo
sanguigno e lì interagisce rapidamente con le proteine sieriche in particolare
con le albumine con altre macromolecole (Chu, 1971; Chu, 1974), mentre
gli eritrociti ne contengono solo in tracce (Galtier, 1978). L’OTA legata alle
albumine e alle altre molecole ematiche costituisce una riserva mobile di
micotossina che può essere ceduta facilmente ai tessuti per lungo tempo
(Galtier, 1978; Hult et al., 1982). Il ruolo delle albumine nella cinetica di
distribuzione dell’OTA, è stato illustrato attraverso uno studio condotto su
ratti deficienti di albumina in cui si è evidenziato come fossero capaci di
eliminare l’OTA dal torrente ematico, molto più velocemente rispetto ai ratti
normali. Questo fenomeno ha portato alla conclusione che, il legame OTA-

49
albumina, permette di ritardare la sua eliminazione limitandone il
trasferimento dal torrente ematico alle cellule epatiche e renali (Kumagai,
1985). La frazione di OTA libera è lo 0,02% nell’uomo e nel ratto, 0,08%
nella scimmia, 0,1 % nel topo e nel maiale e 22% nel pesce (Hagelberg et
al., 1989). Sono state riportate notevoli differenze riguardo l’emivita
dell’OTA nel siero delle diverse specie: 72-120 ore nel suino (Galtier et al.,
1981; Mortensen et al., 1983a), 72 ore nel vitello (Sreemannarayana et al.,
1988), 55-120 ore nel ratto (Galtier et al., 1979; Ballinger et al., 1986;
Hagelberg et al., 1989) e 4,1 ore nel pollo (Galtier et al., 1981). L’emivita
dell’ OTA nell’uomo dopo somministrazione orale è di 840 ore e, poiché essa
impiega circa otto volte questo periodo per essere eliminata, il livello di OTA
ematica è quantificabile per 280 giorni (Petzinger e Ziegler, 2000).
CIRCOLAZIONE ENTEROEPATICA
La circolazione enteroepatica dell’OTA, è stata dimostrata attraverso studi
effettuati su roditori (Kumagai e Aibara., 1982; Roth et al.,1988; Fuchs et
al., 1988) e su vitelli (Sreemannarayana et al, 1988). Tutti questi studi,
hanno mostrato una distribuzione di picchi secondari di OTA nel siero e nel
contenuto intestinale portando alla conclusione di una secrezione biliare
della tossina seguita dal suo riassorbimento da parte dell’intestino. Il
riassorbimento di OTA dall’intestino ritorna in circolazione, come
conseguenza del riciclaggio biliare, favorisce così la distribuzione di OTA nei
differenti tessuti.
DISTRIBUZIONE TISSUTALE
La concentrazione di OTA e dei suoi metaboliti nei tessuti e nel plasma
degli animali, dipende dalla specie animale, dalla dose somministrata, dalla
sua forma (cristallina o naturalmente presente nei mangimi), dalla
composizione della dieta come anche dallo stato di salute dell’animale.
Generalmente, la velocità di eliminazione dell’ OTA dal sangue, è più lunga
rispetto a quella di altri tessuti, questo dovuto in parte, all’alta affinità di
legame della tossina con le proteine del sangue (Hagelberg et al., 1989). La

50
distribuzione nei tessuti di maiale, ratto, pollo e capra, segue l’ordine:
reni>fegato>muscolo>tessuto adiposo (Harwing et al., 1983; Ferrufino-
Guardia et al., 2000). Differenza nella distribuzione nei diversi tessuti di
ratto, sono state osservate in uno studio dopo ingestione di piccole dosi di
OTA radioattiva (in ordine decrescente: polmone, fegato, reni, cuore, lardo,
intestino, testicoli, muscolo, milza e cervello) (Kane et al., 1986). L’OTA
inoltre, è stata ritrovata nel cervello, nel cervelletto, nella parte ventrale del
mesencefalo, nello striato e nell’ippocampo di ratti maschi ad una
percentuale di 0,022-0,028% della dose somministrata di 289 g/kg/giorno
durante 8 giorni di somministrazione tramite intubazione gastrica
(Belmadani et al., 1988). Altri studi mostrano un trasferimento attraverso la
placenta nei mammiferi, soprattutto in topi, ratti e maiali (Kuipper Goodman
et al., 1989; Palminger et al., 1998).Questi studi hanno dimostrato che il
trasferimento dipende dallo stadio di sviluppo della placenta, che si
considera completato dopo il 9° giorno di gestazione. Nell’uomo, la
concentrazione fetale di OTA riscontrata è due volte maggiore rispetto a
quella materna; questo stà ad indicare un attivo trasferimento placentare
(Zimmerli e Dick,1995). L’OTA somministrata nella dose di 0,38 mg/kg p.v.
a scrofe a 21-28 giorni di gravidanza invece, non attraversava la placenta
(Patterson et al, 1976), e nessun residuo di micotossina è stato ritrovato nei
suinetti nati da scrofe alimentate con mangimi contenenti livelli pari a 7-16
µg di OTA durante tutta la gravidanza (Mortensen et al., 1983b). Però,
secondo uno studio recente, in soggetti alimentati con cibi naturalmente
contaminati, l’OTA è stata trasmessa in utero e a sei maialini: il livello di
micotossina ematica nei piccoli è risultato pari a 0,075-0,12 ng/ml e di 0,20
ng/ml nella madre (Barnikol e Thalmann, 1988). E’ comunque ancora poco
conosciuto il trasferimento dell’OTA attraverso la placenta. La presenza di
OTA, è stata ritrovata anche nelle uova di diverse specie di volatili. In
galline, alimentate con mangimi contaminati con una concentrazione di OTA
di 0,3-1 mg/kg per 341 giorni, non è stata ritrovata nessuna traccia di OTA
nelle uova (Krogh et al., 1976), ma nel corso di altri studi, la micotossina è

51
stata trovata in uova di uccelli che ne avevano ingerito una quantità pari a
10 mg/kg di p.v. (Juszkiewicz et al., 1982).
2.2.4 Biotrasformazione La principale via metabolica dell’OTA, consiste nella sua idrolisi in OTα, un
composto molto meno tossico originatosi attraverso la rottura del legame
peptidico. Nel ratto questa via di detossificazione, appare principalmente a
carico dell’ azione della microflora presente nel cieco (Galtier, 1978). In
particolare gli enzimi responsabili della reazione di idrolisi nei roditori e nel
bovino sono la carbossipeptidasi A e la chimotripsina (Pitout, 1969a, 1969b;
Pitout e Nel, 1969). Altre micotossine come l’acido penicillico, impediscono
la reazione di idrolisi (Parker et al., 1982), mentre l’inibizione della flora
dell’ultimo tratto intestinale da parte della neomicina, riduce la produzione
di OTα aumentando il livello di OTA ematica (Madhysta et al., 1992). Studi
condotti su omogenati di tessuto di ratto, hanno mostrato che tale reazione
può avvenire anche nel duodeno, nell’ileo, e nel pancreas, mentre è
risultata scarsa nel rene e nel fegato (Suzuki et al., 1977). Tale meccanismo
di detossificazione, avviene anche nei bovini grazie all’azione della frazione
protozoaria presente nel liquido ruminale. L’OTA in questo caso, viene
idrolizzata nella sua forma atossica anche nel secondo stomaco e
nell’omaso, mentre questo non succede nell’abomaso (Hult et al., 1976). E’
stato stimato che possono essere degradati fino a 12 mg di OTA/kg di
alimento (Hult et al., 1976; Petterson et al., 1982). Proprio per tale ragione,
i ruminanti sono meno sensibili alla tossina rispetto ai monogastrici. Da
alcuni studi è emerso inoltre che, esiste una correlazione tra la
concentrazione di protozoi ed il contenuto di amido nel rumine; infatti la
popolazione protozoaria, è significativamente influenzata dal contenuto nella
razione e quindi nel rumine, di carboidrati facilmente fermentescibili quali
l’amido (Abe e Iriki, 1978). Un incremento di amido nella dieta, porta ad
aumento di energia disponibile e ad un aumento della densità dei protozoi
nel rumine (Nakamura e Kanegasaki, 1969). Come il bovino, anche la

52
pecora si è mostrata capace di possedere una buona attività di
detossificazione dell’OTA, prima che questa raggiunga il circolo ematico
(Kiessling et al., 1984). Ricerche svolte sul topo, hanno mostrato che l’OTA
dal fegato, per mezzo della bile, viene riversata nell’intestino dove viene
degradata ad OTα (Moroi et al., 1985). Circa il 25-27% dell’OTA,
somministrata sia per via orale che per via intraperitoneale nel ratto, si
ritrova sotto forma di OTα nelle urine in quanto riassorbita dall’intestino
(Storen et al.,1982). Altri metaboliTi urinari dell’ OTA di minore importanza
sono la (4R)-4-idrossiocratossina A e la (4S)-4-idrossiocratossina A prodotte
dal fegato di ratto e di coniglio (Stormer et al., 1981) e dal rene di ratto
(Stein et al., 1985) per azione del citocromo P450 (Stormer et al., 1981;
1983). Il primo epimero, considerato meno tossico dell’OTA, è il principale
metabolita prodotto dal sistema microsomale epatico dell’uomo e del ratto
(Stormer et al., 1981), mentre l’altro, è principalmente formato dai
microsomi epatici di suino e non sono disponibili dati riguardanti la sua
tossicità (Moroi et al., 1985). Altri prodotti del metabolismo dell’OTA sono la
10-idrossiocratossina, identificata dopo incubazione di OTA con microsomi
epatici di coniglio (Stormer et al., 1983), e l’ocratossina C, prodotta nel
fluido ruminale e di tossicità paragonabile a quella del suo precursore
(Galtier et al., 1981). E’ stata dimostrata inoltre, la formazione in vivo di
una forma di OTA caratterizzata da un lattone aperto (OP-OTA). Questa
forma è stata ritrovata in particolare nella bile e nelle urine di ratti ai quali è
stata somministrata l’OTA per via endovenosa; ancora però non è chiaro
quale sia il meccanismo cellulare che ne determina la sua sintesi (Li et al.,
2000). Questa forma è inoltre risultata altamente tossica quando
somministrata per via endovenosa al ratto (Xiao et al., 1996b). Infine,
anche l’OTB, pur essendo spesso presente nei cereali e derivati insieme
all’OTA, può esserne uno dei suoi metaboliti in quanto è stata ritrovata nel
corso di uno studio in vitro in seguito all’incubazione dell’OTA in cellule
renali di scimmia (Grosse et al., 1995).
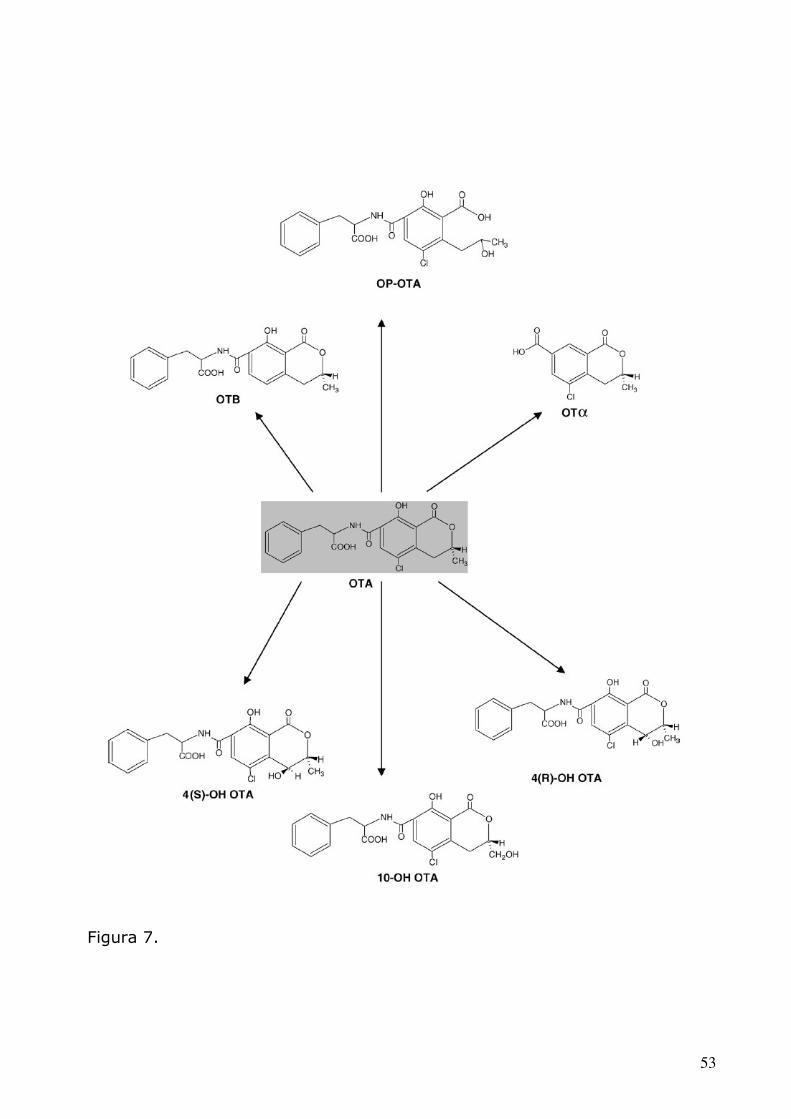
53
Figura 7.

54
2.2.5 Eliminazione L’ OTA è un composto altamente tossico con un relativo rapido
assorbimento ed una lenta eliminazione. In tutte le specie, l’OTA e i suoi
metaboliti vengono escreti fondamentalmente per via fecale ed urinaria. Il
differente contributo di ciascuna via di escrezione, dipende dalla quantità di
micotossina e dalla sua modalità di somministrazione (Kuipper et al., 1989).
Inoltre, in tutte le specie questo è influenzato dalla stabilità del legame tra
l’OTA e le proteine sieriche e dalla diversa intensità della circolazione
enteroepatica (Hagelberg et al., 1989). Sia l’escrezione biliare che la
filtrazione glomerulare, giocano un ruolo importante nell’eliminazione
dell’OTA contenuta nel plasma di ratto. In questi animali, i prodotti di
escrezione più rappresentati sono l’OTA stessa, l’OTα, (presente sia nelle
feci che nelle urine), e la (4R)-4-idrossiocratossina A, che nelle urine si
ritrovano rispettivamente per il 6%, 25-27%, e 1-1,5% della dose
somministrata (Storen et al., 1982). Il 33% dell’OTA somministrata per os
nel ratto è stata riscontrata nella bile dopo 6 ore, mentre solo piccole
quantità di OTα sono state ritrovate nelle urine (Suzuki et al., 1977). In
vitelli e bovini invece, l’80-90% dell’OTA somministrata per via orale, viene
secreta soprattutto con le urine sotto forma di OTα (Sreemannarayana et
al., 1988). E’ stato poi dimostrato che l’OTA viene secreta a livello
gastrointestinale (Suzuki et al., 1977; Kumagai e Ai bara, 1982; Berger et
al., 2003). Tramite un esperimento condotto sul ratto, si è potuto verificare
che la quantità di OTA eliminata dall’intero tubo gastroenterico è pari a
quella secreta con la bile (Suzuki et al., 1977). Sembra che tale secrezione,
è più lenta in presenza del contenuto intestinale e più abbondante nei tratti
di intestino con minore mobilità (Kumagai e Aibara, 1982). Uno studio
condotto sulla capra, ha mostrato che di una dose di OTA pari a 0,5 mg/kg
somministrata per via orale, solo il 53% della tossina veniva secreta nelle
feci (Nip e Chu, 1979), mentre nelle pecore, solo il 10% di OTA
somministrata per via orale veniva escreta nelle feci mentre la rimanente
parte veniva idrolizzata in OTα. La via fecale ed urinaria giocano un ruolo

55
importante nella cinetica plasmatici della tossina in tutte le specie ma anche
l’escrezione attraverso il latte nei mammiferi sembra essere rilevante.
Questa via di escrezione, riveste molta importanza in quanto il latte,
rappresenta l’alimento maggiormente consumato dai bambini, più sensibili
agli effetti tossici di tali sostanze. In uno studio con ratti in allattamento è
stato riscontrato un trasferimento di OTA dose dipendente nel latte in
seguito ad una somministrazione singola, Il rapporto tra concentrazione nel
latte e nel sangue alla 24° e 72° ora, era rispettivamente di 0,4 e 0,7. Lo
stesso rapporto latte/sangue è stato ritrovato in uno studio condotto su ratti
in allattamento esposti ad Ota per otto settimane con intubazione gastrica
(Breitholz-Emanuelsson et al., 1993a). E’ stato determinato
successivamente, un rapporto di concentrazioni latte/plasma più basso,
nutrendo conigli in allattamento ripetutamente con una dieta a base di
alimenti contaminati naturalmente. Questo basso rapporto, è stato correlato
alla modalità di esposizione alla tossina (Ferrufino-Guardia et al., 2000).
Diversi autori hanno riscontrato livelli di OTA nel latte umano (Breitholtz-
Emanuelsson et al., 1993b; Miraglia et al., 1995). Da questi studi è emerso
che l’OTA presenta significative variazioni interindividuali e geografiche.
Recentemente in altri studi, è stata mostrata una correlazione tra la
contaminazione da OTA nel latte materno e la sua introduzione attraverso la
dieta (Skaug et al., 2001).

56
2.3 EFFETTI E MECCANISMO DI AZIONE
L’OTA ha proprietà cancerogene, genotossiche, nefrotossiche,
immunosoppressive e teratogene (Kuiper-Goodman e Scott, 1989; Genkle e
Silbernagl, 1996). Il rene, è considerato essere il maggior organo bersaglio
sul quale l’OTA esplica i suoi effetti. Elevate dosi di OTA, infatti, ne alterano
la sua funzionalità e la sua morfologia, soprattutto danneggiando la parte
prossimale dei tubuli prossimali, deputati al riassorbimento (Berndt e
Hayes, 1979). Nel ratto, gli effetti dell’OTA sulla morfologia e sulla
funzionalità del rene sono indicati da un incremento del suo peso, del
volume delle urine, del glucosio urinario, della proteinuria e dal
danneggiamento del trasporto urinario degli anioni organici (Munro et al.,
1974; Berndt e Hayes, 1979) situato a livello dell’orletto a spazzola delle
cellule del tubulo prossimale e delle membrane basolaterali (Endou et al.,
1986; Sokol et al., 1988). Ed è proprio a quest’ultimo effetto che è legata
la nefrotossicità dell’OTA. Alcuni studi, hanno dimostrato che questo sistema
di trasporto, è anche quello responsabile dell’ingresso dell’OTA nelle cellule
del tubulo prossimale (Friis et al., 1988; Sokol et al., 1988), in particolare
per mezzo del trasportatore OAT1 (Tsuda et al., 1999). E’ stato dimostrato
che alcuni inibitori del OAT1 sono capaci di inibire il trasporto, ipotizzando
così, che questo meccanismo possa spiegare la capacità di questi composti
di prevenire gli effetti nefrotossici dell’OTA (Tsuda et al., 1999). Le parti
maggiormente sensibili alla tossicità dell’OTA, come dimostrato da una
significativa riduzione dell’ATP cellulare e mitocondriale, sono la parte
intermedia e quella terminale del tubulo prossimale (Jung e Endou, 1987).
E’ stato possibile valutare gli effetti dell’OTA dalla liberazione di enzimi
nell’urina a partire dal parenchima renale, risalendo ai diversi danni renali
dalla presenza di diversi enzimi e proteine nell’urina (Stonard et al., 1987).
Per esempio, somministrando a ratti 0,14 mg/kg p.v ogni 48 ore per 8-12
settimane, si è osservata una significativa riduzione dell’attività della lattato
deidrogenasi, della fosfatasi alcalina, della leucina aminopeptidasi e della
gamma-glutamil transferasi e la loro concomitante presenza nelle urine.

57
Questi ultimi tre enzimi sono localizzati proprio a livello dei tubuli prossimali
ed indicano pertanto un danno a questo livello. L’OTA inoltre, determina
l’innalzamento del pH nell’insterstizio della papilla renale e l’alterazione
dell’acidificazione delle urine (Kuramochi et al., 1997a), oltre che l’aumento
del pH e della concentrazione degli ioni bicarbonato nel fluido tubulare e nei
vasa recta senza modificare la pCO2. E’ stato ipotizzato che lo squilibro
dell’omeostasi del pH può contribuire alla tossicità dell’OTA sui reni
(Kuramochi et al., 1997b). Diverse ipotesi sul meccanismo di interazione
dell’OTA e dei suoi metaboliti con molecole endogene, sono portati avanti
per cercare di spiegare la sua tossicità. Esse fanno riferimento a specifiche
interazioni, basate sull’alta specificità di legame con particolari siti specifici
su molecole bersaglio e, interazioni di tipo non specifico, basate sulla
reattività chimica dell’OTA e dei suoi metaboliti e sulla loro vicinanza con le
molecole bersaglio. Sono stati identificati così, alcuni meccanismi molecolari
alla base degli effetti tossici indotti dalla micotossina. La disfunzione
mitocondriale è un primo evento durante la tossicità dell’OTA (Aleo et al.,
1991). E’ stato mostrato come l’OTA inibisce la respirazione nei mitocondri
di fegato dei ratti (Meisner e Chan, 1974; Wei et al, 1985) e ne altera la
morfologia dopo somministrazione in vivo (Suzuki et al., 1975). Questo
processo, correlato con la deplezione dell’ ATP, è considerato come una
conseguenza dell’inibizione del fosfato intramitocondriale attraverso
l’inibizione competitiva delle proteine carrier localizzate sulla membrana
interna del mitocondrio (Aleo et al., 1991; Meisner e Chan, 1974), e/o come
un effetto diretto sulla catena di trasporto di elettroni, attraverso l’inibizione
dell’attività del succinato. L’importanza del meccanismo mitocondriale però,
non è ancora del tutto chiaro, in quanto l’OTα, pur non avendo effetti
tossici, è capace di inibire anch’essa la respirazione mitocondriale (Meisner
e Chan, 1974). L’OTA possiede inoltre la capacità di interferire sui processi
metabolici coinvolgenti la fenilanina. Poiché l’OTA deriva dal legame
dell’isocumarina con la L-β-fenilalanina la componente fenilalaninica può
interagire con tutti i sistemi metabolici che coinvolgono l’amminoacido che è
un suo anologo strutturale. Infatti l’OTA, influisce principalmente sulla

58
sintesi proteica, ma anche sulla sintesi del DNA e dell’RNA in diversi
organismi (Marquardt e Frohlich, 1992; Fink-Gremmels et al., 1995). L’OTA
ha quindi un ruolo importante nell’inibizione della Fen-tRNA Sintetasi e della
Fen-idrolasi. Inoltre, poiché interrompe la sintesi proteica , indirettamente
altera l’attività di molti enzimi cellulari, ed in particolar modo l’attività
dell’enzima fosfoenolpiruvato carbossichinasi, un’ enzima chiave della
gluconeogenesi. Perciò una conseguenza tossicologica indiretta dell’OTA è
anche l’alterazione della via metabolica dei carboidrati. E’ stato verificato sia
in vitro che in vivo, e sia negli organismi procarioti che eucarioti, che la
fenilalanina tRNa- sintetasi viene inibita dall’OTA durante la reazione di
amminoacilazione della fenilalanina, determinando così, l’interruzione della
sintesi proteica (Creppy et al., 1983). Altri esperimenti in vitro svolti su
colture di lievito (Saccharomyces cerevisiae), mostrano come oltre all’OTA,
anche la 4R-4-idrossiocratossina A, esercita un effetto analogo, mentre
l’OTα e l’OTB non espletano alcuna attività tossica (Dirheimer e Creppy,
1991). L’OTA inibisce anche la fenilalanina idrossilasi agendo come
substrato per questo enzima, il quale catalizza l’idrossilazione della
fenilalanina a tiroxina con conseguente blocco del metabolismo della
tiroxina (Creppy et al., 1990). L’OTA inoltre, altera l’azione di diversi enzimi
ed in particolare, l’attività dell’enzima fosfoenolpiruvato carbossichinasi,
enzima chiave della gluconeogenesi, il quale può essere completamente
ridotto nei ratti (Meisner e Meisner, 1981) e nei maiali (Meisner e Krogh,
1986), degradando l’mRNA che codifica per questa molecola (Meisner et al.,
1983). La tossicità dell’OTA, non è legata solo all’alterazione della sintesi
proteica, ma anche ad altri effetti come la perossidazione dei lipidi, il
danneggiamento del DNA e l’alterazione dell’omeostasi del calcio. Altri studi
suggeriscono anche il coinvolgimento di stress ossidoriduttivo nella tossicità
e nella carcinogenicità. Lo stress ossidoriduttivo indotto da xenobiotici può
causare dirette lesioni cellulari ossidative, dovute alla produzione di forti
ossidanti come radicali superossidi (O2-), H2O2, e radicali idrossilici (HO)
provoca anche deterioramento del segnale di traduzione e regolazione
dell’espressione genica attraverso meccanismi redox-sensibili. Studi in vivo

59
e in vitro dimostrano che l’OTA potenzia la per ossidazione lipidica in quanto
stimola la per ossidazione nei microsomi sia NADPH-dipendente che
ascorbato-dipendente, con Fe+3 come co-fattore (Rahimtula et al., 1988;
Omar et al., 1990). L’aumentata perossidazione lipidica da parte dell’OTA,
influisce sulla permeabilità della membrana plasmatica al Ca+2, andando
così ad alterare l’omeostasi di questo elemento (Khan et al., 1989). Studi
effettuati suI ratti hanno dimostrato che, somministrando una singola dose
o dosi multiple più basse di OTA, si ha a livello renale, un incremento
dell’attività della pompa del calcio del reticolo endoplasmatico. L’OTA
potrebbe così alterare tutte le funzioni cellulari che sono sotto il controllo dei
livelli di concentrazione del calcio (Rahimtula e Chong, 1991). Per quanto
riguarda l’impatto sul genoma, l’OTA è in grado di determinare la
formazione di addotti con il DNA in diversi organi. Il meccanismo di azione
genotossica, è stato presupposto soprattutto sui risultati di alcuni
esperimenti che mostrano proprio come il trattamento con OTA a topi e
ratti, induce una formazione dose e tempo dipendente, di addotti con il DNA
in diversi organi (Pfohl-Leszkowicz et al., 1993; Pfohl-Leszkowicz et al.,
2002). La maggior parte di questi addotti dopo 5 giorni dalla
somministrazione di OTA, scompaiono dal fegato e dalla milza, mentre nel
rene, alcuni addotti persistono fino a 16 giorni (Pfohl-Leszkowicz et al.,
1993). I ratti e i topi trattati con 0,4-2,5 mg/kg p.v per un periodo da 1-16
giorni a 2 anni, hanno mostrato un numero di addotti a livello renale
compreso tra 1 e 200/109 nucleotidi (Pfohl-Leszkowicz et al., 1991; 1993;
Grosse et al., 1995; Pfohl-Leszkowicz et al., 1998). Ciò può essere causato
dal danno ossidativo dovuto alla produzione di radicali liberi (Grosse et al.,
1997), ma questo non è sicuramente l’unico meccanismo possibile, in
quanto altri addotti sono stati ottenuti anche in vitro a partire da DNA e
mononucleotidi purificati ed incubati con microsomi di rene o fegato di topo
NADPH o acido arachidonico come cofattori (Obrecht-Pflumio e Dirheimer,
2000). Nel 1993 l’International Agency for Research on Cancer (IARC), ha
stabilito l’appartenenza dell’OTA al gruppo 2B, in quanto ritenuta una
possibile sostanza cancerogena per l’uomo (Pittet, 1998). L’OTA si è

60
dimostrata cancerogena provocando tumori renali in ratti e topi con marcate
differenze in base alla specie e al sesso (US-NTP, 1989); tant’è che i
maschi, sono risultati maggiormente sensibili rispetto alla femmine ed i ratti
maggiormente sensibili rispetto ai topi. Proprio nei ratti maschi, l’OTA si è
dimostrata una delle più potenti sostanze capaci di determinare la
formazione di tumori renali (Mantle, 2002). Il meccanismo di induzione
tumorale è controverso. Molti studi, attribuiscono questo ruolo alla
biotrasformazione dell’OTA in quanto, un metabolita attivo della
micotossina, sembrerebbe capace di legarsi al DNA e attraverso l’azione
catalizzante del citocromo P450, della perossidasi e del glutatione S-
transferasi, capace di innescare il meccanismo tumorale (Hietanem et al.,
1991; Henning et al., 1991; Wurgler et al., 1991; Malaveille et al., 1994;
Fink-Gremmels et al., 1995; Obrecht-Pflumio et al., 1999; El Adlouni et al.,
2000). I processi di produzione dei metaboliti dell’OTA, non sono
sicuramente coinvolti nella sua cancerogenicità, in quanto aumentando il
tasso di biotrasformazione dell’OTA tramite l’azione del citocromo P450, si
determina una riduzione della sua tossicità (Omar et al., 1996). Si pensa
che la formazione di idrossi radicali (Hoeheler et al., 1996; 1997) o di
addotti di DNA (Pfohl-Leszkowicz et al., 1991; Wurgler et al., 1991; Grosse
et al., 1995; 1997; Obrecht-Pflumio e Dirheimer, 2000), possano avere un
ruolo importante. La nefrotossicità, lo stress ossidativo dovuto
all’alterazione della respirazione mitocondriale (Aleo et al., 1991), la
formazione di perossidi di idrogeno (Omar et al., 1996) e la proliferazione
cellulare rappresenta un meccanismo alternativo nella determinazione dei
tumori renali, questo perché nel rene di ratto per esempio, si è osservato
come lo stress ossidativo e la tossicità renale a lungo termine, siano capaci
di svolgere un ruolo nella determinazione dei tumori (Swenberg e Maronpot,
1991; Dietrich e Swenberg, 1993; Hard, 1998). Attraverso diversi studi, si è
osservato come l’OTA possiede una tossicità anche a livello riproduttivo.
Studi condotti sui ratti e sui topi, mostrano come l’OTA, sia in grado di
attraversare la placenta ed esplicare effetti teratogeni e embriotossici
(FAO/WHO, 1991; 1996; 2001; EC, 1998). Somministrando concentrazioni

61
pari a 0, 0,125, 0,25, 0,50 e 0,75 mg OTA/kg p.v. a topi gravidi, si è
osservato un aumento dell’incidenza di anomalie del feto soprattutto nei
gruppi ai quali venivano somministrate le dosi di OTA maggiori. Le anomalie
comprendevano principalmente difetti scheletrici a livello del cranio, costole
e vertebre, dovuti essenzialmente ad una assenza o ad un’incompleta
ossificazione e malformazioni localizzate alle strutture craniofacciali, dovute
alla mancata chiusura del cranio, quali mesencefalo, microencefalo e
mascella iposviluppata (Wangikar et al., 2004a,b). Altri studi sull’effetto
teratogeno dell’OTA, sono stati condotti in Nuova Zelanda su conigli bianchi
gravidi somministrando dosi di 0, 0,025, 0,05, 0,10 mg di OTA/kg
p.v./giorno (Wangikar et al., 2005). Nei gruppi a cui è stata somministrata
la più alta dose, si è osservato un calo nel numero dei feti e del loro peso ed
un’incidenza delle malformazioni. Tra i diversi bersagli dell’OTA, il sistema
immunitario, è uno dei più sensibili. Attraverso studi in vitro con linfociti di
ratto e concentrazioni di OTA pari a 0, 0,5, 2, e 20 µM, si è osservata una
diminuzione dose-dipendente dell’ attività delle cellule natural killer e una
diminuzione dell’attività dei T-linfociti, già a basse concentrazioni; mentre
subiva solo qualche variazione l’attività batteriologica dei macrofagi
(Alvarez-Erviti et.al, 2005). L’OTA, induce mielotossicità, effetto evidenziato
da una marcata riduzione della massa timica e da una ipocellularità del
midollo osseo, con conseguente diminuizione delle cellule staminali
totipotenti e significativa riduzione della sintesi dei precursori degli eritrociti,
dei granulociti e dei macrofagi (Boorman et al., 1984).

62
2.4 TOSSICITA’
Le micotossicosi in genere (e quindi anche le ocratossicosi), sono
intossicazioni acute e croniche riscontrate in uomini ed animali imputabili
principalmente, all’ingestione di alimenti e mangimi contaminati.
L’esposizione dell’uomo alle ocratossine, come alle altre micotossine, può
verificarsi principalmente attraverso il consumo di alimenti di origine
vegetale contaminati o l’ingestione di residui di micotossine o suoi
metaboliti contenuti in derrate (latte e derivati, carni, insaccati ecc.)
derivanti da animali alimentati con mangimi contaminati; oppure in seguito
all’inalazione di spore fungine tossigene presenti in elevate quantità sia in
particolari ambienti di lavoro (in cui si generano per esempio polveri di
cereali contaminati), sia in ambienti domestici umidi e poco areati. E’ stato
dimostrato che cani, maiali e polli, sono le specie più sensibili agli effetti
dell’OTA rispetto ai topi e ai ratti (IARC, 1993; Marquardt e Frohlich, 1992).
In particolare, l’OTA, è in grado di provocare gravi disordini e sintomi di
tossicità acuta a livello di differenti organi e sistemi anatomici, quali
principalmente i reni, il fegato, il sangue ed il sistema immunitario. Il rene è
considerato l’organo più suscettibile a questa tossina, tant’è che in tutte le
specie testate, l’OTA ha indotto una tossicità a livello renale. Studi condotti
su topi, ratti, cani e maiali, hanno mostrato una correlazione tra il
progressivo sviluppo della nefropatia, la dose ed il tempo di
somministrazione di OTA. Inoltre sono state osservate differenze per quanto
riguarda l’effetto nefrotossico, a seconda della specie e del sesso. Oltre ad
una tossicità acuta o subacuta, tali micotossine sono responsabili anche di
una tossicità cronica come conseguenza di un’esposizione protratta nel
tempo a bassi livelli di contaminazione degli alimenti che comporta effetti
cronici quali cancerogenicità, genotossicità, mutagenicità, teratogenicità ed
immunosoppressione.

63
2.4.1 Tossicità nel topo e nel ratto
Topi e ratti sono stati largamente utilizzati come modelli per lo studio delle
micotossicosi. Per quanto riguarda le indagini inerenti gli effetti legati alla
tossicità acuta dell’OTA nel ratto, si è osservato che essa, somministrata per
via orale alle dosi di 0,24, 0,48, 0,96 e 2,4 mg/kg p.v./giorno per 14 giorni,
determina il ritardo dello sviluppo, un ridotto consumo di alimento e un
incremento dell’urea ematica. Con il dosaggio più elevato, è stato osservato
un aumento del peso del rene mentre, con tutti i dosaggi, si è verificata la
degenerazione dell’intero sistema tubulare, una diminuizione del volume
urinario e eosinofilia e cariomegalia a livello del tubulo prossimale
convoluto. Inoltre i topi maschi, sono stati trovati molto più sensibili rispetto
alle femmine (Munro et al., 1974; Berndt e Hayes, 1979). In un altro studio
(Munro et al., 1974), gruppi di 15 ratti appena svezzati, sono stati
alimentati con una dieta contenente OTA a concentrazioni equivalenti a 0,
15, 75 o 370 µg/kg p.v/giorno per 90 giorni, seguiti da un uguale periodo di
dieta di controllo. Al termine dello studio, alcuni animali sono morti, mentre
gli altri, al termine dell’intero intervallo di tempo, non presentavano
alterazioni del profilo urinario ed ematologico e i loro reni, ridotti di peso
nella prima parte dell’esperimento, sono successivamente rientrati nei valori
standard, tranne nei maschi trattati con dosaggi maggiori. Istologicamente
gli organi presentavano comunque cariomegalia e ispessimento della
membrana basale (Munro et al., 1974). Da altri studi è emerso che le
femmine di ratto più vecchie, sono molto più sensibili all’induzione della
cariomegalia tubulare e alla necrosi rispetto ai giovani adulti (Dortant et
al., 2001). Sono stati effettuati anche numerosi studi a lungo termine per
testare la tossicità e la cancerogenicità dell’OTA. Topi alimentati con 5,6
mg/kg p.v./giorno di OTA per 44 settimane hanno mostrato un’elevata
incidenza di tumori epatici, adenomi renali cistici e tumori renali rispetto al
gruppo di controllo (Kanisawa e Suzuki, 1978). Altri studi hanno mostrato
come il trattamento con melatonina (10-20 mg/kg p.v./giorno), può avere
un effetto di tipo preventivo sulla tossicità indotta dall’OTA a livello del
fegato e del rene (Ayadin et al., 2003). Anche l’aspartame, si è rilevato un

64
composto in grado di svolgere un’azione protettiva contro i maggiori effetti
nefrotossici indotti dall’OTA (Baudrimont et al., 2001). Recenti studi, hanno
inoltre mostrato come anche i flavonoidi presenti nel vino rosso, possono
esercitare un’azione di protezione contro gli effetti dell’OTA (Bertelli et al.,
2005).
2.4.2 Tossicità nel suino I suini sono generalmente considerati come la specie animale più sensibile
alla nefrotossicità dell’OTA. L’intossicazione da OTA si può presentare in
forma acuta o cronica. La forma acuta si osserva soprattutto in soggetti da
poco svezzati ed è dovuta all’assunzione di mangimi contenentiuna quantità
di OTA superiore a 4000 µg per kg di alimento. Dal punto di vista clinico
l’ocratossicosi acuta è caratterizzata da edema sottocutaneo, atassia e
incartamento del dorso, mentre all’esame post-mortem si riscontrano
edema perirenale, nefrosi e necrosi tubulare (Marcato, 1998). In una serie
di esperimenti sulla tossicità acuta dell’OTA, gruppi di 3-6 scrofe, sono state
trattate con 0, 0,008, 0,04 o 0,2 mg/kg p.v./giorno di OTA per periodi di 5
giorni o 12-16 settimane rispettivamente, o 2 anni: in tutti i casi si è
verificata una riduzione della funzionalità renale, nefropatia e una diminuita
attività degli enzimi renali. Nelle femmine trattate con 1 mg/kg di OTA al
giorno per 2 anni si è riscontrata una nefropatia progressiva, ma non il
mancato funzionamento del rene, mentre non è stato riportato alcun
risultato nei maschi (Krogh e Elling, 1977; Elling, 1979; 1983; Elling et al.,
1985). Non sono stati osservati nei 2 anni di studio, effetti sugli enzimi e
sulle funzioni renali (Krogh e Elling, 1977; Elling, 1979a,b, 1983; Elling et
al., 1985; Meisener e Krogh, 1986; Krogh et al., 1988; FAO/WHO, 2001).
L’intossicazione cronica invece, è frequente soprattutto nei suini all’ingrasso
alimentati per almeno tre settimane con mangimi contenenti 200-1000
µg/kg di OTA. Clinicamente si riscontrano la diminuizione dell’appetito,
poliuria e polidipsia dovute all’aumento della permeabilità del filtro
glomerulare ed all’alterazione della funzione di riassorbimento a livello del

65
tubulo prossimale, da cui consegue proteinuria e glicosuria. L’esame
anatomo-patologico mostra l’aumento del volume dei reni che hanno una
colorazione normale o pallida. Istologicamente si notano lesioni atrofico-
degenerative degli epiteli dei tubuli prossimali, dilatati, fibrosi interstiziale e
ialinosi glomerulare (Marcato, 1998). In recenti studi, gruppi di maiali, sono
stati alimentati con una dieta contaminata da ceppi di Aspergillus
ochraceus, produttore sia di OTA che di acido penicillico (PA) e contenente
90, 130, o 180 µg OTA/kg per tre mesi. A fine trattamento, tutti i gruppi
hanno riportato lesioni renali microscopiche e variazioni di alcuni parametri
biologici ed ematologici. Aumentando i livelli di OTA nella dieta fino a
concentrazioni pari a 130, 305 o 790 µg OTA/kg per altri due mesi, l’esame
istologico ha mostrato a livello delle cellule epiteliali dei tubuli prossimali,
dei cambiamenti degenerativi principalmente nei primi stadi della malattia
e secondariamente cambiamenti a carico dell’intestino (Stoev et al., 2001).
A causa della sua tossicità, l’OTA è stata identificata come il possibile agente
causale della nefropatia micotossica suina, riscontrata per la prima volta
all’inizio degli anni ’70 in Danimarca.
2.4.3 Tossicità nell’uomo
Sebbene nell’uomo non siano stati riportati chiari casi di nefropatia
micotossica, è abbastanza verosimile che questa potente nefrotossina, che
determina danni renali ingenti in diverse specie animali, può anche indurre
alterazioni renali negli uomini esposti. E’ stata infatti riscontrata una
connessione tra l’insorgenza di tumori del tratto urinario e patologie renali
croniche nell’uomo e l’elevata incidenza di nefropatie nel suino (Olsen et al.,
1993), in zone con forte contaminazione di OTA negli alimenti (Miraglia e
Brera, 1999). Per tale ragione, si ritiene che la micotossina sia alla base di
patologie renali anche nell’uomo. In particolae si pensa che l’OTA svolga un
ruolo importante nella patogenesi della “nefropatia endemica dei Balacani”
(BEN). La BEN è una patologia renale dall’esito fatale che è stata osservata
prevalentemente nelle popolazioni rurali della Bulgaria, Romania, Serbia,

66
Bosnia, Erzegovina, Croazia e Yugoslavia; infatti, è stato stimato che circa
20.000 persone soffrono di tale patologia in queste regioni (Peraica et al.,
1999; 2001). Nel passato, si è cercato di individuare i possibili fattori
eziologici di tale nefropatia (batteri, virus, metalli tossici, fattori genetici)
senza tuttavia giungere a risultati convincenti; fino a che nel 1974 è stata
proposta come causa determinante dello sviluppo della patologia una
micotossina, concentrando l’attenzione soprattutto sull’ OTA (Krogh e Elling,
1976). Diversi studi epidemiologici hanno mostrato la correlazione esistente
tra la presenza di OTA negli alimenti e la maggiore incidenza di casi di BEN
(Krogh e Elling., 1977). Numerosi campioni di alimenti destinati al consumo
umano ed animale, prodotti nelle regioni endemiche, sono risultati
contaminati da OTA. La nefropatia, interessa maggiormente gli abitanti delle
regioni rurali, ma limitatamente anche quelli delle regioni urbane. Ciò è
stato spiegato considerando che le popolazioni contadine di questi paesi,
consumano alimenti prodotti in proprio e conservati, spesso in condizioni
non idonee e quindi altamente contaminate da OTA, mentre le popolazioni
urbane consumano prodotti a livello industriale. La BEN è una nefropatia
che colpisce più frequentemente i soggetti tra i 30 e i 50 anni con maggiore
incidenza di casi nei soggetti di sesso femminile, per le quali anche il tasso
di mortalità è superiore (Chernoremsky et al., 1977). Non è stata osservata
una fase acuta della malattia, ed i primi sintomi sono risultati aspecifici; essi
includono affaticamento, anemia, proteinuria, ingiallimento della pelle,
dolore di testa, perdita di peso, anoressia ed uremia. All’esame autoptico, i
reni appaiono notevolmente ridotti di volume con una diffusa fibrosi
corticale, spesso senza alcun segno di infiammazione (Vukelic et al., 1992).
L’esame istologico mostra la presenza di lesioni croniche della corteccia
renale con fibrosi interstiziale, ialinizzazione dei glomeruli, degenerazione
dell’epitelio tubulare e perdita dell’orlo a spazzola del tubulo renale. Nelle
regioni endemiche della Croazia, Bulgaria e Yugoslavia, è stata riscontrata
anche un’elevata incidenza di tumori uroteliali delle pelvi e dell’uretere
(UTT) (Ceovic et al., 1992; Chermozemsky, 1997). E’ stato suggerito, che
l’OTA potrebbe essere l’agente scatenante sia della nefropatia endemica sia

67
dei tumori del tratto urinario (Castegnaro et al., 1991). A tal proposito lo
IARC ha classificato l’OTA come una possibile sostanza cancerogena per
l’uomo. L’esposizione delle popolazioni balcaniche all’OTA è stata inoltre,
supportata da un’elevatissima presenza di residui della micotossina in
campioni di sangue (oltre 1-2 µg/kg) e di urine delle famiglie esposte
(Castegnaro et al., 1991). Il riscontro della presenza di OTA nel sangue e
nel latte (Miraglia et al., 1995), viene da alcuni anni utilizzato come
parametro di valutazione dei livelli di esposizione della popolazione anche di
aree non particolarmente a rischio.

PARTE SPERIMENTALE

68
3. PREMESSA
I sistemi di allevamento estensivi, nei quali i suini ptrascorrono all’aperto
gran parte della loro vita, hanno una lunga tradizione in molti paesi europei.
Tuttavia, la necessità dettata da motivi economici di incrementare
l’efficienza biologica della produzione della carne e di contenere i costi di
gestione degli animali, ha determinato il declino di questi sistemi di
allevamento e la loro evoluzione e trasformazione verso il sistema intensivo
al chiuso. La tecnica dell’allevamento razionale di suini all’aperto ha radici
antiche in Europa. Già nel 1934 Bonadonna e Scattolin scrivevano
“L’allevamento dei suini all’aperto: criteria e tecnica.” e successivamente
anche Stanga (1946) e Vezzani (1948) hanno pubblicato articoli su questo
argomento. Negli anni ’50 tale tecnica si è cominciata a diffondere in
Inghilterra; in Toscana si sono avuti alcuni esempi in provincia di Arezzo
(Savaglio). Ma la vera diffusione si ebbe negli anni ’80, sia nell’Europa del
Nord con razze migliorate (Nilzèn et al., 2001; Hogberg et al; 2001) che in
quella del Sud con razze rustiche (Diaz et al., 1996; Lopez-Bote, 1998). La
recente rinascita degli allevamenti all’aperto è stata guidata da una
combinazione di fattori come il basso valore del capitale fondiario di alcune
zone marginalizzate; gli aumentati costi delle strutture, della gestione e
delle attrezzature; l’attuazione di rigide normative circa lo stoccaggio e la
distribuzione dei liquami zootecnici; le pressioni delle organizzazioni
animaliste per un allevamento più consono al benessere animale ed eco-
compatibile; il bisogno di diversificare la produzione rendendola più attenta
all’impatto sull’ambiente e soprattutto la crescente richiesta di prodotti
“genuini” da parte dei consumatori. I principali vantaggi dell’allevamento
all’aperto stanno nella possibilità di contenere le spese di investimento
iniziali, che ammontano a circa il 20-25% di quello convenzionale nel
risparmio alimentare, qualora l’allevamento all’aperto è condotto con

69
l’utilizzazione del pascolo; nell’ottenimento di prodotti ad elevata
caratterizzazione organolettica. Tuttavia l’impatto ambientale, specie se
l’allevamento si svolge in terreni boschivi, resta uno degli aspetti critici
dell’allevamento all’aperto e va valutato opportunamente in quanto è molto
variabile in funzione del rapporto capi/superficie e della possibilità di turnare
le superfici utilizzate. Sulla scia degli ottimi risultati ottenuti in Spagna con il
suino iberico ed in Toscana con la Cinta Senese, l’allevamento macchiatico
del suino è stato preso in considerazione come elemento trainante per un
progressivo recupero economico ed ambientale dei territori marginali.
Questo è stato possibile grazie alla grande disponibilità di terreni soprattutto
boschivi, alla sempre crescente richiesta di prodotti tipici di qualità da parte
dei consumatori, soprattutto se provenienti da animali allevati all’aperto.
Tale forma di allevamento costituisce infatti, una valida alternativa alle
attuali attività legate all’utilizzo del bosco grazie anche alle elevate capacità
che il suino ha di valorizzare sia i prodotti del bosco e del sottobosco, che
integrano la sua alimentazione, sia questa particolare tecnica di allevamento
che lo rende meno soggetto a fattori causa di stress, presenti
nell’allevamento di tipo tradizionale. Durante questa fase, infatti, i suini
sono in grado di dare libero sfogo ai loro istinti naturali avendo la possibilità
di grufolare liberamente ed usufruire di appositi spazi quali zone fangose
per far fronte ai periodi di eccessiva calura e bagni di sabbia e di acqua per
liberarsi da eventuali parassiti. Gli animali così allevati, sono infatti, in grado
di fornire carni e prodotti caratterizzati da aromi e peculiarità organolettiche
strettamente correlate alle essenze e ai frutti del sottobosco che vanno ad
integrare la loro dieta, presentando inoltre una bassissima incidenza di
miopatie (PSE; DFD) derivanti perlopiù da stress delle fasi di allevamento.
Particolare importanza, riveste la qualità igienico-sanitaria di questi prodotti
ottenuti da suini allevati allo stato semi-brado. In questo caso, i suini sono
meno soggetti al controllo da parte dell’uomo soprattutto per quanto
riguarda l’alimentazione e possono quindi cibarsi liberamente di qualsiasi
prodotto che trovano. Per questo motivo, ci è sembrato interessante
effettuare questo studio che ha per obiettivo quello di determinare

70
l’ocratossina A nelle carni e nei prodotti carnei stagionati, di suini allevati
allo stato semi-brado nel bosco, rispetto a quelli ottenuti da animali in
allevamento stabulato.

71
4. MATERIALI E METODI
4.1 Prove di allevamento
4.1.1 Rilievi in vita
La prova è stata effettuata nell’azienda agricolo-forestale situata in località
Cambertano di Tinvegna nel comune di Follo (SP). La zona oggetto di
questa sperimentazione rispecchia le peculiarità del territorio dell’entroterra
ligure, caratterizzato dalla quasi totale assenza di zone pianeggianti e con
una orografia per lo più caratterizzata da terrazzamenti sorretti da tipici
muri a secco e da una copertura vegetale costituita prevalentemente da
bosco. Per la prova sono stati impiegati 10 suini maschi castrati derivanti
dall’incrocio di Large White X Duroc acquistati dall’azienda agricola
“Montone” sita a Villanova di Ravenna. Questi animali, acquistati ad un peso
vivo di circa 30 kg, sono stati immediatamente suddivisi in due differenti
gruppi da sei individui omogenei per peso; un primo gruppo denominato
“indoor” (stabulato) è stato allevato all’interno di un ricovero di 16 m2 ;
mentre il secondo denominato “outdoor” (brado), è stato lasciato libero di
pascolare all’interno di un recinto di 5000 m2 situato in una porzione di
bosco in evidente stato di abbandono, sede di un antico castagneto.
Entrambi i gruppi di animali sono stati allevati con metodo biologico ed
alimentati con una razione composta da una miscela di mangimi fioccati
(Pisello proteico, mais, grano ed orzo), tutti quanti certificati e garantiti
come prodotti O.G.M FREE. Il razionamento è stato di gruppo ed in base al
peso vivo degli animali, che sono stati pesati ad inizio e fine prova ed a
intervalli regolari di circa 2 mesi. Il mangime è stato somministrato una
volta al giorno alla mattina e periodicamente sono stati pesati i residui nelle
mangiatoie, per valutare l’effettiva quantità di mangime assunta dagli

72
animali. Il piano di razionamento ha visto crescere la quantità di alimento
somministrato e contemporaneamente diminuire la percentuale di pisello
proteico fioccato, ed aumentare quella di mais fioccato, mentre quelle di
grano fioccato e orzo fioccato sono state mantenute agli stessi livelli
(Tabella 15). A partire dalla fruttificazione delle castagne (mese di Ottobre),
al gruppo “outdoor” è stato somministrato 1 kg/capo/d di castagne in
sostituzione di un 1 kg/capo/d di mais, mentre per il gruppo “indoor” la
composizione della dieta è rimasta invariata.
Peso vivo kg
Mangime somministrato (kg/TQ)
Pisello proteico fioccato %
Mais fioccato %
Grano fioccato %
Orzo fioccato %
30-50
1,50 35 30 20 15
50-70
2,00 30 35 20 15
70-90
2,50 25 40 20 15
90-110
3,00 20 45 20 15
>110
3,50 15 50 20 15
Tabella 15. Composizione percentuale mangimi sulla razione.
4.1.2 Rilievi post-mortem
Ognuno dei due gruppi di animali, è stato poi macellato ad un peso
medio/capo di circa 140 kg; gli animali appartenenti al medesimo gruppo
sono stati macellati lo stesso giorno. Le macellazioni sono state effettuate
presso il macello e salumificio Savani situato in Località Borgotaro (PR) dove
è stato possibile compiere anche la sezionatura della carcassa, la
lavorazione e la stagionatura dei prodotti trasformati. Il gruppo “indoor” è
stato macellato il giorno 22-02-06 mentre il gruppo “outdoor” il 29-03-06.
Dopo la macellazione, le carcasse dei suini sono state sezionate per la
produzione di salumi (pancette, coppe e prosciutti); le restanti parti
anatomiche, sono state lavorate separatamente per i due gruppi per la
produzione di insaccati tipici (salami e mortadelle). Durante le fasi di

73
macellazione, sono stati prelevati da ciascun soggetto campioni freschi di
muscolo Longissimus lomborum e grasso. Successivamente, da ciascun
suino sono stati prelevati campioni di muscolo e campioni di grasso
sottocutaneo (lardo), e dopo la stagionatura dei prodotti trasformati, sono
stati prelevati campioni stagionati dello stesso tipo di muscolo e grasso.
Infine sono stati prelevati 3 campioni di mortadelle e 3 di salame ottenuti da
ciascun gruppo dei suini in prova. Tutti i campioni prelevati, sono stati
quindi sottoposti all’analisi per la determinazione dell’OTA mediante metodo
HPLC.
4.2 Determinazione dell’ocratossina A con metodo HPLC
4.2.1 Materiali
I reagenti utilizzati sono stati acquistati dalle comuni fonti commerciali, in
particolare l’OTA dalla Sigma Chemical Co. (St. Louis, MO, USA) ed i
solventi, tutti di grado HPLC, dalla Labscan (Hasselt, Belgium). L’acqua di
grado analitico impiegata per portare a concentrazione la fase mobile per il
sistema HPLC è stata prefiltrata mediante filtri di acetato di cellulosa
impermeabilizzati con silicone della PS Whatman® (Millipore Corporation,
Maid Stone, UK). Per l’estrazione sono state usate colonnine di
immunoaffinità NeoColumn for Ochratoxin A prodotte dalla Neogen Europe
Ltd (Diessechem, Milano, Italia). Le colonnine sono state conservate ad una
temperatura di 4 °C.
Il PBS utilizzato per la diluizione dei campioni da passare in colonna era
costituito da 8 g NaCl, 0,2 g KH2PO4, 1,16 g Na2HPO4*2H2O, 0,2 g KCl in 1 l
di H2O, tutto a pH 7,4.

74
4.2.2 Strumentazione e condizioni cromatografiche
Le analisi di laboratorio sono state condotte utilizzando un sistema HPLC
costituito da una pompa JASCO 880 PU (JASCO, Tokio, Japan) a flusso
variabile e da un rilevatore fluorimetrico JASCO 821-FP (JASCO, Tokio,
Japan) settato ad una lunghezza d’onda di eccitazione ed emissione di 380
nm e 420 nm, rispettivamente. Lo strumento è stato interfacciato ad un
personal computer tramite interfaccia Hercule 2000 (JMBS Inc., Bear, USA)
e l’integrazione dei picchi è stata effettuata tramite software Jasco Borwin
(JASCO, Tokio, Japan).
E’ stata utilizzata una colonna cromatografica Gemini ODS2, 150x4,60 mm
(Phenomenex®, Torrance, CA, USA), con granulometria delle particelle di 5
µm ed una precolonna Waters Guard-PakTM (Waters, Milford, MA, USA),
entrambe impaccate con gel di silice derivatizzato con gruppi alchilici C18.
Per l’analisi cromatografica è stata utilizzata una fase mobile costituita da
una miscela di tampone fosfato a pH 7,5 (Na2HPO4 0,03 M e Na2H2PO4
0,007M) e metanolo in rapporto 48/52 % v/v. Il flusso della fase mobile è
stato regolato a 1 ml/minuto ed è stato iniettato un volume di campione di
100 µl.
4.2.3 Soluzioni standard e soluzione stock
La soluzione stock di OTA (PM di 403,8), è stata ottenuta sciogliendo 1 mg
di OTA in 5 ml di una miscela costituita da toluene-acido acetico nelle
proporzioni 99/1 % v/v, in modo da ottenere una soluzione finale con una
concentrazione pari a 200 µg/ml. Le soluzioni standard necessarie per la
creazione della curva di calibrazione e per l’aggiunta ai campioni da cui
ricavare il recupero sono state ottenute riprendendo 100 µl della soluzione
stock, portati a secco sotto flusso di azoto, con 10 ml di fase mobile, in
modo da ottenere una soluzione madre con una concentrazione di 2 µg/ml.
A partire da questa, per mezzo di diluizioni successive, sono state ottenute

75
le soluzioni standard con concentrazioni di OTA di 0,2-0,5-1-2,5-5-10 e 20
ng/ml. Le diverse soluzioni sono state conservate a –20°C e, data la
fotosensibilità dell’OTA, sono state tenute al riparo dalla luce.
4.2.4 Standard esterno
Poiché non esiste uno standard interno per la micotossina analizzata, è
stato fatto riferimento a quello esterno, rappresentato dall’OTA stessa.
Infatti, sia i campioni di mangime che di carne, sono stati analizzati dopo
l’aggiunta di una concentrazione nota di OTA (10 µl di soluzione stock) della
quale è poi stato calcolato il recupero.
4.2.5 Retta di calibrazione
La retta di taratura è stata creata mediante l’iniezione delle soluzioni
standard precedentemente preparate. Ogni soluzione è stata iniettata tre
volte, e dalla media delle tre determinazioni è stata ottenuta l’equazione
della retta di calibrazione tramite il programma Graph Pad Prism® (Graph
Pad Software Inc., IL, USA). Come parametro quantitativo, è stata utilizzata
l’area sottesa ai picchi del tracciato cromatografico misurata tramite il
software Jasco Borwin (JASCO, Tokio, Japan).
4.2.6 Campioni
Sono stati analizzati in totale 60 campioni di cui 8 di mangimi e 52 di carne
suina. Per quanto riguarda i mangimi, più specificatamente, sono stati
analizzati campioni di mais, fioccato di orzo, fioccato di grano, pisello
proteico, castagne intere, castagne sbucciate, bucce di castagne e ghiande,
mentre per quanto riguarda le carni i campioni analizzati sono stati:
muscolo (Longissumus lomborum, muscolo fresco), lardo fresco, salami,

76
mortadelle, coppa (Longissimus lomborum dopo stagionatura, muscolo
stagionato) e pancetta (lardo stagionato).
4.2.7 Estrazione e purificazione dei campioni di mangime
Per ogni tipologia di mangime (fioccato d’orzo, fioccato di grano, mais,
pisello proteico, castagne intere, castagne sbucciate, bucce di castagne e
ghiande) preventivamente macinato finemente, sono stati utilizzati 5
grammi che sono stati posti in una provetta da 45 ml e addizionati con 40
ml della miscela estrattiva CH3OH e H2O. Il campione è stato poi vortexato
per 3 minuti, messo in oscillatore a 180 oscillazioni/minuto per 20 minuti e
successivamente sottoposto a filtrazione utilizzando filtri di carta Whatman.
Per l’estrazione dei campioni, sono state utilizzate delle colonnine di
immunoaffinità Neocolumn, che contengono anticorpi monoclonali anti-
Ocratossina A adsorbiti su particelle di gel. Le estrazioni sono state eseguite
con un collettore sottovuoto (Vacuum manifold, J.T. Baker, Deventer, The
Netherland), con il quale è possibile estrarre contemporaneamente fino a 12
campioni (Figura 8).
Figura 8. Vacuum manifold per l'estrazione dei campioni
con le colonnine di immunoaffinità.

77
La vaschetta, in vetro, presenta sul coperchio una serie di valvole di
connessione sulle quali vengono montate le colonnine. All’interno della
vaschetta, vi è una rastrelliera in grado di alloggiare le provette di raccolta.
L’intero sistema è connesso ad una pompa da vuoto elettrica e la
depressione è controllata all’uscita della camera da un manometro, mentre
ciascun alloggiamento delle colonnine è dotato di una valvola integrale di
controllo di flusso, regolabile indipendentemente per ogni campione,
cercando di assicurare un flusso costante di 1-2 gocce al minuto.
L’estrazione del campione con le colonnine è stata condotta con la seguente
procedura (Figura 9):
1. passaggio del campione attraverso la colonnina, per gravità o per
filtrazione sotto vuoto;
2. lavaggio della colonnina con 20 ml di PBS;
3. eluizione dell’ocratossina con la miscela metanolo e acido acetico.
Figura 9. Fasi di estrazione con colonnine di immunoaffinità.
Del campione filtrato sono stati prelevati 4 ml e vi sono stati aggiunti 46 ml
di PBS. La soluzione è stata fatta passare attraverso la colonnina. Agendo
sull’apposita valvola di connessione e regolando il vuoto all’interno della

78
vaschetta è stato assicurato un flusso costante di 1-2 gocce al secondo,
assicurandosi di non lasciare mai asciugare la colonna. E’ stato quindi
effettuato un lavaggio della colonna con 20 ml di PBS , mantenendo sempre
lo stesso flusso. Al termine del lavaggio, praticando un leggero vuoto, è
stata eliminata tutta l’acqua dalla colonna senza però farla seccare.
L’eluizione è stata eseguita utilizzando 4 aliquote da 0,75 ml di metanolo e
acido acetico 98/2 %, e l’eluato è stato raccolto in provette di vetro da 10
ml. La miscela di eluizione è stata lasciata all’interno della colonna per circa
3 minuti prima di essere fluita al fine di favorire il distacco dell’OTA dagli
anticorpi. Dopo aver aggiunto l’ultima aliquota della miscela di eluizione, è
stato praticato un vuoto molto spinto per far fluire tutta la fase liquida
rimasta nella colonna. L’eluato così ottenuto, è stato portato a secco sotto
flusso di azoto, ripreso con 250 µl di fase mobile, vortexato per qualche
minuto e iniettato in HPLC.
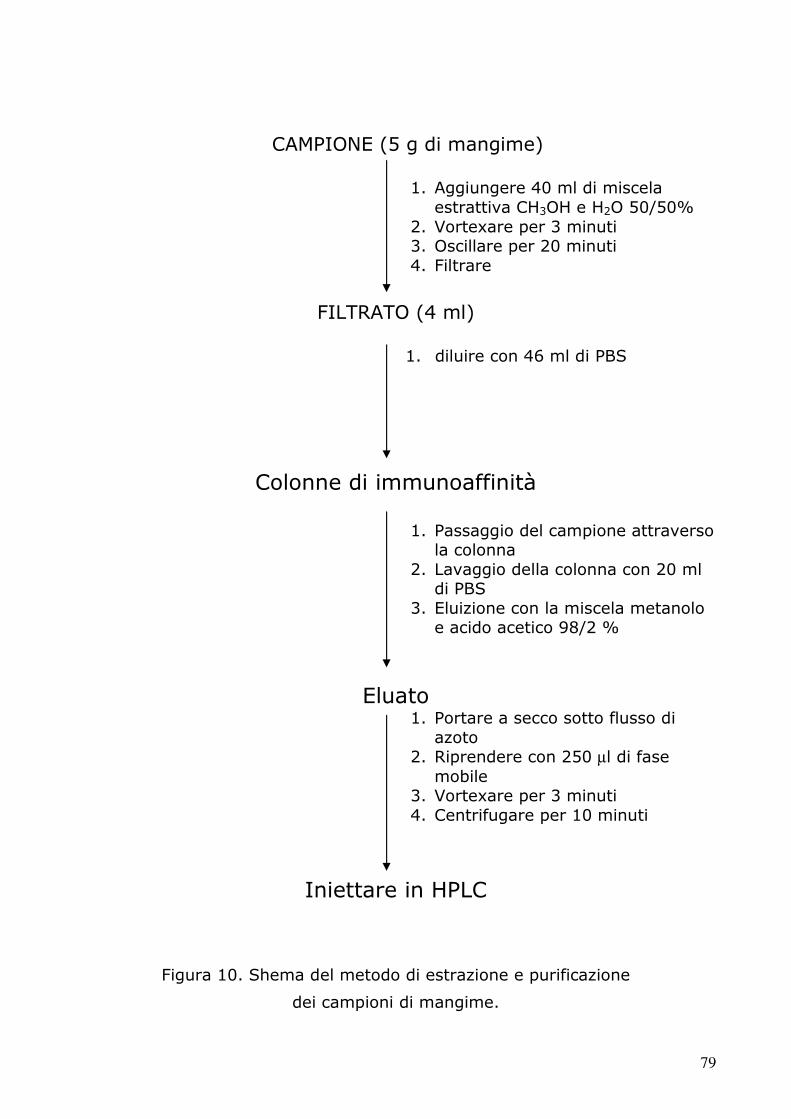
79
CAMPIONE (5 g di mangime) 1. Aggiungere 40 ml di miscela
estrattiva CH3OH e H2O 50/50% 2. Vortexare per 3 minuti 3. Oscillare per 20 minuti 4. Filtrare
FILTRATO (4 ml)
1. diluire con 46 ml di PBS
Colonne di immunoaffinità
1. Passaggio del campione attraverso
la colonna 2. Lavaggio della colonna con 20 ml
di PBS 3. Eluizione con la miscela metanolo
e acido acetico 98/2 %
Eluato
1. Portare a secco sotto flusso di azoto
2. Riprendere con 250 µl di fase mobile
3. Vortexare per 3 minuti 4. Centrifugare per 10 minuti
Iniettare in HPLC
Figura 10. Shema del metodo di estrazione e purificazione
dei campioni di mangime.

80
4.2.8 Estrazione e purificazione dei campioni di carne
Per ogni matrice (muscolo fresco, lardo fresco, salami, mortadelle, muscolo
stagionato e lardo stagionato) sono stati utilizzati 5 grammi che sono stati
posti in una provetta di vetro da 15 ml, addizionati con 5 ml di H3PO4 1M e
omogenati utilizzando l’apparecchio ultraturrax Pabish (IKA, LABORTCHIK,
Staufen, Germany). Dell’omogenato così ottenuto, sono stati prelevati 2,5
grammi e sono stati trasferiti in un’altra provetta di vetro da 15 ml, in cui
sono stati aggiunti 5 ml di etilacetato saturo di NaCl, che rappresenta il
solvente di estrazione. La provetta è stata poi vortexata per 3 minuti,
messa in oscillatore a 180 oscillazioni/minuto per 20 minuti e centrifugata a
3000 rpm per 10 minuti. Al termine di queste operazioni, è stato prelevato il
sovranatante (fase organica), mentre il residuo è stato sottoposto per una
seconda volta alle stesse fasi precedentemente illustrate. Il sovranatante
ottenuto dalla seconda estrazione, è stato prelevato e aggiunto alla fase
organica risultante dalle precedenti operazioni. La fase organica totale (10
ml), è stata successivamente concentrata sotto flusso di azoto fino ad un
volume di 5 ml. In seguito, vi sono stati aggiunti 5 ml di NaHCO3 0,5 M a pH
8,4. La provetta è stata nuovamente vortexata per 3 minuti, oscillata e
centrifugata per 10 minuti. A questo punto, è stata prelevata la fase
acquosa, acidificata con H3PO4 all’85% fino a pH 2,5, sonicata per 5 minuti e
nuovamente addizionata con 5 ml di etilacetato. Il tutto è stato vortexato
per ulteriori 3 minuti e centrifugata per 10 minuti a 3000 rpm. La fase
organica è stata prelevata, portata a secco sotto flusso di azoto e il residuo,
ripreso con 250 µl di fase mobile, è stato iniettato in HPLC. Solamente per
quanto riguarda i campioni di pancetta, dopo aver portato a secco la fase
organica, il residuo è stato ripreso con 500 µl di fase mobile.
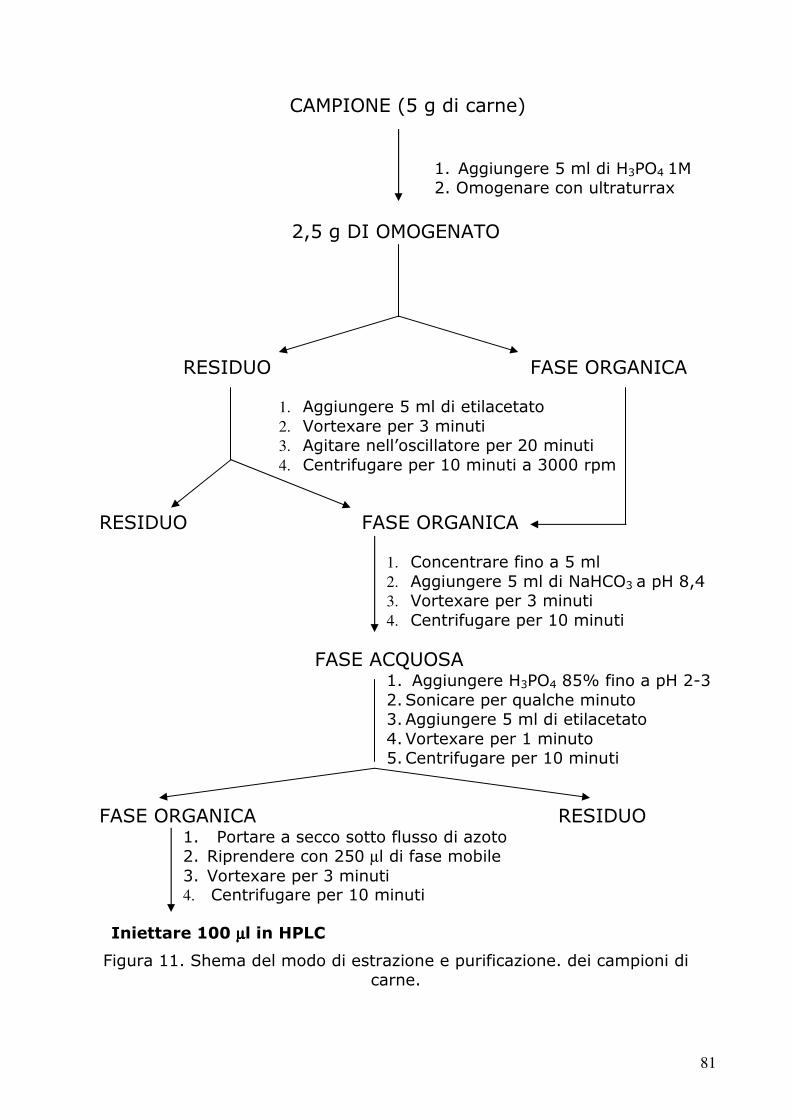
81
CAMPIONE (5 g di carne)
1. Aggiungere 5 ml di H3PO4 1M
2. Omogenare con ultraturrax
2,5 g DI OMOGENATO RESIDUO FASE ORGANICA
1. Aggiungere 5 ml di etilacetato
2. Vortexare per 3 minuti 3. Agitare nell’oscillatore per 20 minuti 4. Centrifugare per 10 minuti a 3000 rpm
RESIDUO FASE ORGANICA
1. Concentrare fino a 5 ml 2. Aggiungere 5 ml di NaHCO3 a pH 8,4
3. Vortexare per 3 minuti 4. Centrifugare per 10 minuti
FASE ACQUOSA 1. Aggiungere H3PO4 85% fino a pH 2-3 2. Sonicare per qualche minuto 3. Aggiungere 5 ml di etilacetato 4. Vortexare per 1 minuto 5. Centrifugare per 10 minuti
FASE ORGANICA RESIDUO 1. Portare a secco sotto flusso di azoto 2. Riprendere con 250 µl di fase mobile 3. Vortexare per 3 minuti 4. Centrifugare per 10 minuti
Iniettare 100 µµµµl in HPLC
Figura 11. Shema del modo di estrazione e purificazione. dei campioni di carne.

82
4.2.9 Conferma
La conferma dei campioni positivi è stata eseguita attraverso la
derivatizzazione dell’ OTA per metilazione e successiva analisi in HPLC
(Shephard et al., 2003). Il campione ottenuto da processi di estrazione o
l’OTA standard (150 µl) sono stati portati a secco e il residuo è stato poi
disciolto in una soluzione al 12,5% di trifluoruro di boro in metanolo (0,5
ml). L’esterificazione è avvenuta riscaldando la provetta chiusa per 15
minuti a 50-60°C. Il solvente è stato poi fatto evaporare e, dopo
raffreddamento della provetta, il residuo è stato ridisciolto in 250 µl di fase
mobile ed iniettato in HPLC. La conferma della presenza di OTA è mostrata
dalla scomparsa del picco corrispondente all’OTA e la comparsa del picco
corrispondente all’estere metilico dell’OTA con diverso tempo di ritenzione.
4.2.10 Validazione del metodo
La specificità è stata valutata mediante l’analisi HPLC di soluzioni standard
di OTA.In questo modo è stato possibile dimostrare quale fosse il il
cromatogramma della molecola in esame. La conferma che i picchi
cromatografici ottenuti fossero dovuti proprio a tale sostanza si è anche
avuta dalla constatazione che, aumentando la concentrazione delle soluzioni
iniettate, l’area sottesa al picco aumentava.
La linearità è stata valutata dall’analisi del grafico ottenuto riportando i
valori dell’area sottesa ai picchi cromatografici in funzione della
concentrazione.
L’intervallo di concentrazioni in cui il metodo è risultato valido, chiamato
range, deriva dagli studi sulla linearità. E’ stato stabilito in base al fatto che
i risultati ottenuti dall’analisi di un campione contenente una concentrazione
di analita compresa nel range specifico per quella procedura, devono
discostarsi di poco dalla linearità e devono essere precisi ed attendibili.
L’accuratezza del metodo è stata dimostrata facendo delle prove in bianco,
ossia iniettando degli estratti di mangime e carne bianchi. E’ stato così

83
dimostrato che ai tempi di ritenzione corrispondenti all’OTA, non vi erano
picchi relativi ad impurezze che potessero alterare l’esito dell’analisi.
La precisione è data dalla ripetibilità, valutata analizzando un minimo di
nove concentrazioni della sostanza in esame comprese nel range di
linearità, e dalla riproducibilità. Il coefficiente di variazione intra-day e inter-
day è stato determinato mediante analisi HPLC di soluzioni contenenti 2, 10
e 20 ng/ml di OTA. La variabilità intra-day è stata ottenuta iniettando tre
volte ciascuna concentrazione in tempi diversi dello stesso giorno. La
variabilità inter-day è stata ottenuta iniettando ciascuna soluzione tre volte
al giorno per cinque giorni consecutivi (Tabella 16).
Il limite di determinazione (LOD) ed il limite di quantificazione (LOQ) sono
stati calcolati analizzando concentrazioni progressivamente più basse delle
soluzioni di OTA. Il LOD è la concentrazione minima di sostanza in
corrispondenza della quale si riesce a vedere il picco corrispondente ma non
a quantificarlo; invece il LOQ è la minima concentrazione che si riesce anche
a quantificare. Sia il limite di determinazione che quello di quantificazione
sono stati valutati basandosi sulla deviazione standard del valore
sperimentale e sulla pendenza della curva di taratura. Essi possono essere
espressi come:
• LOD = 3 σ/S
• LOQ = 10 σ/S
dove σ rappresenta la deviazione standard del valore sperimentale e S la
pendenza della curva di taratura. Riferendosi alla curva di calibrazione,
come valore di deviazione standard può essere preso la deviazione standard
dell’intercetta con l’asse delle y o la deviazione standard della linea di
regressione (Tabella 17).
La robustezza del metodo HPLC è stata valutata prendendo in
considerazione tutti i parametri in grado di influenzarlo. Essi comprendono
la stabilità in soluzione dell’analita, il suo tempo di estrazione e le variazioni
di pH, composizione e temperatura della fase mobile.

84
OCRATOSSINA Concentrazione Numero CV (%) 2 ng/ml 3 6,9 10 ng/ml 3 2,3
Intra-day
20 ng/ml 3 5,1
2 ng/ml 3 8,9 10 ng/ml 3 2,8
Inter-day
20 ng/ml 3 1,6
Tabella 16. Coefficiente di variazione intra-day e inter-day.
LOD (ng/g) LOQ (ng/g) Mangimi
0,0625 0,125
Carni
0,0125 0,025
Tabella 17. Valori di LOD e LOQ dei campioni di mangime e di carne.
4.2.11 Vetreria
Tutta la vetreria utilizzata, deve essere lavata accuratamente per evitare
che eventuali residui di OTA possano falsare le analisi. Per questo motivo, le
provette sono state prima decontaminate tenendole a lungo in ammollo con
ipoclorito di sodio, poi lavate con sapone per impedire l’adsorbimento della
tossina al vetro. La vetreria deve poi essere sciacquata con attenzione
affinché il vetro risulti privo di residui di sapone alcalino o detergente che
potrebbe determinare la perdita della micotossina durante l’analisi a causa
della formazione di sali, della precipitazione o dell’adsorbimento al vetro..
per evitare questi inconvenienti si è effettuato un lavaggio con metanolo
prima di ogni analisi.

85
4.2.12 Analisi statistica
I risultati sono stati espressi come media ± deviazione standard. Le
differenze statisticamente significative tra i due diversi gruppi di suini
analizzati sono stati determinati con il T-Test e con l’analisi della varianza
(ANOVA) seguita dal test di Tukey-Kramer. Un valore di p<0,05 è stato
considerato statisticamente significativo. Tutte le analisi sono state
effettuate mediante il programma Prism (GraphPad software, San Diego,
CA, USA).

86
5. RISULTATI
5.1 Prove di allevamento
Dall’analisi della composizione chimica dei mangimi utilizzati nella dieta di
entrambi i gruppi (Tabella 18), si nota come questi presentino buone
caratteristiche qualitative e del tutto in linea con quelle riscontrate in
letteratura. Il pisello proteico, in particolare, ha mostrato un elevato
quantitativo di proteine (circa il 26%). Anche l’apporto nutritivo ottenuto
dalle sostanze boschive è risultato più che soddisfacente. Le castagne
presentano infatti, una composizione molto favorevole all’alimentazione dei
suini grazie agli elevati apporti di sostanze amilacee, anche se il contenuto
di ADL (lignina) presenta un valore piuttosto alto. Ciò è dovuto alle
caratteristiche del tegumento esterno che però, a causa dell’elevato
contenuto di tannini, viene scartato dagli animali. Per quanto concerne le
ghiande, pur presentando anch’essi elevati apporti amilacei, invece, sono
caratterizzati da un alto contenuto in sostanze fibrose (ADF, ADL) non
digeribili dai monogastrici.
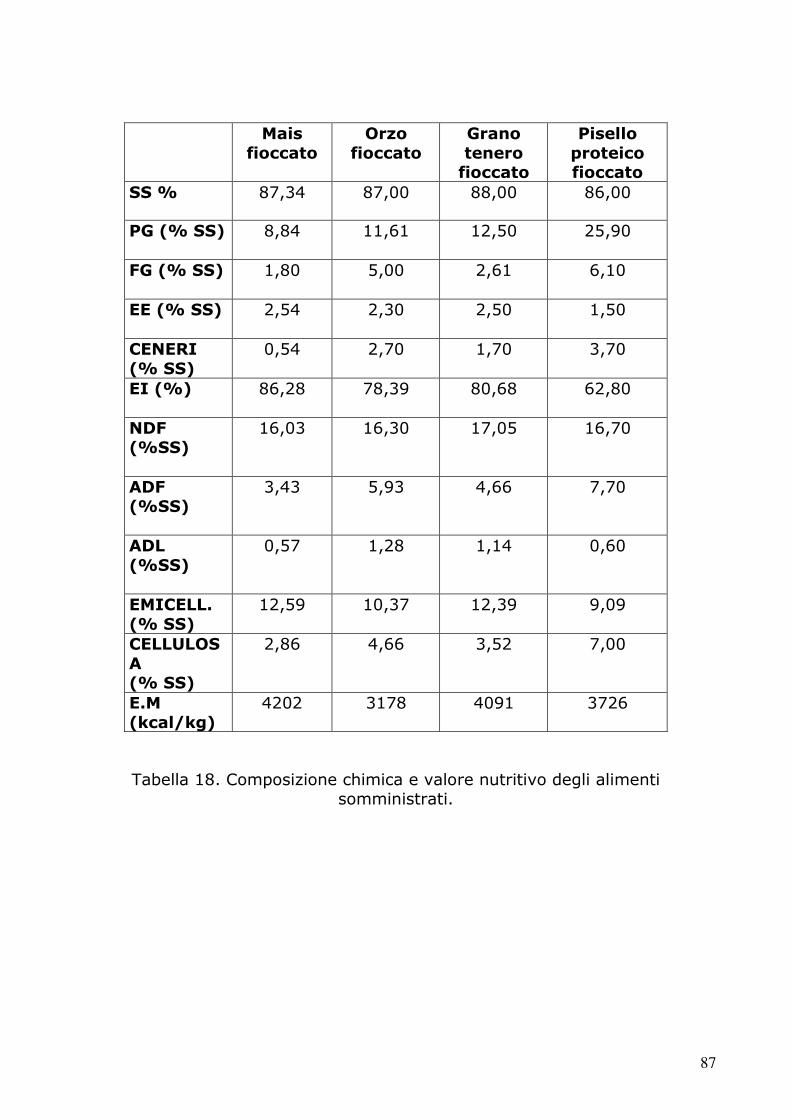
87
Mais fioccato
Orzo fioccato
Grano tenero fioccato
Pisello proteico fioccato
SS %
87,34 87,00 88,00 86,00
PG (% SS)
8,84 11,61 12,50 25,90
FG (% SS)
1,80 5,00 2,61 6,10
EE (% SS)
2,54 2,30 2,50 1,50
CENERI (% SS)
0,54 2,70 1,70 3,70
EI (%)
86,28 78,39 80,68 62,80
NDF (%SS)
16,03 16,30 17,05 16,70
ADF (%SS)
3,43 5,93 4,66 7,70
ADL (%SS)
0,57 1,28 1,14 0,60
EMICELL. (% SS)
12,59 10,37 12,39 9,09
CELLULOSA (% SS)
2,86 4,66 3,52 7,00
E.M (kcal/kg)
4202 3178 4091 3726
Tabella 18. Composizione chimica e valore nutritivo degli alimenti somministrati.

88
Castagne
intere
Castagne decorticate
Bucce di castagna
Ghiande intere
SS %
51,75 50,31 62,19 51,85
PG (% SS)
5,72 4,92 2,55 4,29
FG (% SS)
9,14 4,68 31,14 17,89
EE (% SS)
2,86 2,74 0,96 1,74
CENERI (% SS)
2,66 2,48 1,40 2,84
EI (%)
79,62 85,18 63,95 71,24
AIA (% SS)
0,00 0,05 0,16 0,02
NDF (% SS)
34,75 36,90 48,85 52,18
ADF (% SS)
18,75 8,47 46,12 20,84
ADL (%)SS)
9,00 3,00 31,69 8,88
EMICELL. (% SS)
16,00 28,43 2,73 31,35
CELLULOSA (% SS)
9,75 5,42 14,43 11,94
E.M (kcal/kg)
3374 3245 2838 2518
Tabella 19. Composizione chimica e valore nutritivo dei prodotti del bosco.

89
Relativamente ai principali parametri ottenuti durante il ciclo di allevamento
(Tabella 19), è stato evidenziato come il gruppo “indoor” abbia ottenuto un
miglior accrescimento (IMG 0,394 vs 0,364 g/capo/d) che ha permesso di
raggiungere il peso di macellazione prefissato a 140 kg circa, un mese
prima rispetto al gruppo “outdoor”. Dalla comparazione dei valori dell’indice
di conversione dell’ alimento (ICA), si nota un valore leggermente più alto
per il gruppo “outdoor”, a causa presumibilmente del maggior dispendio
energetico da parte degli animali allevati in libertà e dallo stress di
adattamento subito nei periodi iniziali dell’allevamento. Confrontando i
consumi medi giornalieri, è possibile notare un aumento dei consumi da
parte degli animali “indoor” rispetto a quelli “outdoor”; ciò è dovuto al fatto
che questi ultimi utilizzavano anche dei prodotti del bosco. Per quanto
riguarda infine, i consumi medi totali di mangime durante l’intero ciclo,
emerge una superiorità (circa 50 kg) per gli animali “outdoor”. Questa
differenza non deve attribuirsi ai maggiori consumi giornalieri medi, ma alla
maggior durata del ciclo di allevamento (35 giorni) degli animali allevati allo
stato brado.

90
Fase di allevamento
Indoor Outdoor
Peso inizio prova (kg)
30,00 30,00
Peso fine prova (kg)
138,40 140,80
Durata ciclo di allevamento (gg)
276 304
Consumo di mangime medio giornaliero (kg)
1,960 1,880
Incremento ponderale medio giornaliero IMG (g/capo/d)
0,394 0,364
Indice di conversione dell’alimento ICA (kg di alimento SS/kg PV)
4,840 5,280
Tabella 20. Principali parametri "in vita" relativi all'intero ciclo.
Riguardo i risultati post-mortem riportati in un altro elaborato, è stato
evidenziato come nei due gruppi in prova, non vi siano state differenze di
rilievo riguardo alla resa di macellazione (circa 80%) e le caratteristiche
tissutali della carcassa. Gli animali del gruppo outdoor hanno mostrato,
invece, un maggiore stato di ingrassamento con un più elevato spessore di
grasso sottocutaneo (29,2 vs 26,2 mm) e un più alto tenore di grasso
intramuscolare (9,35 vs 4,70%).
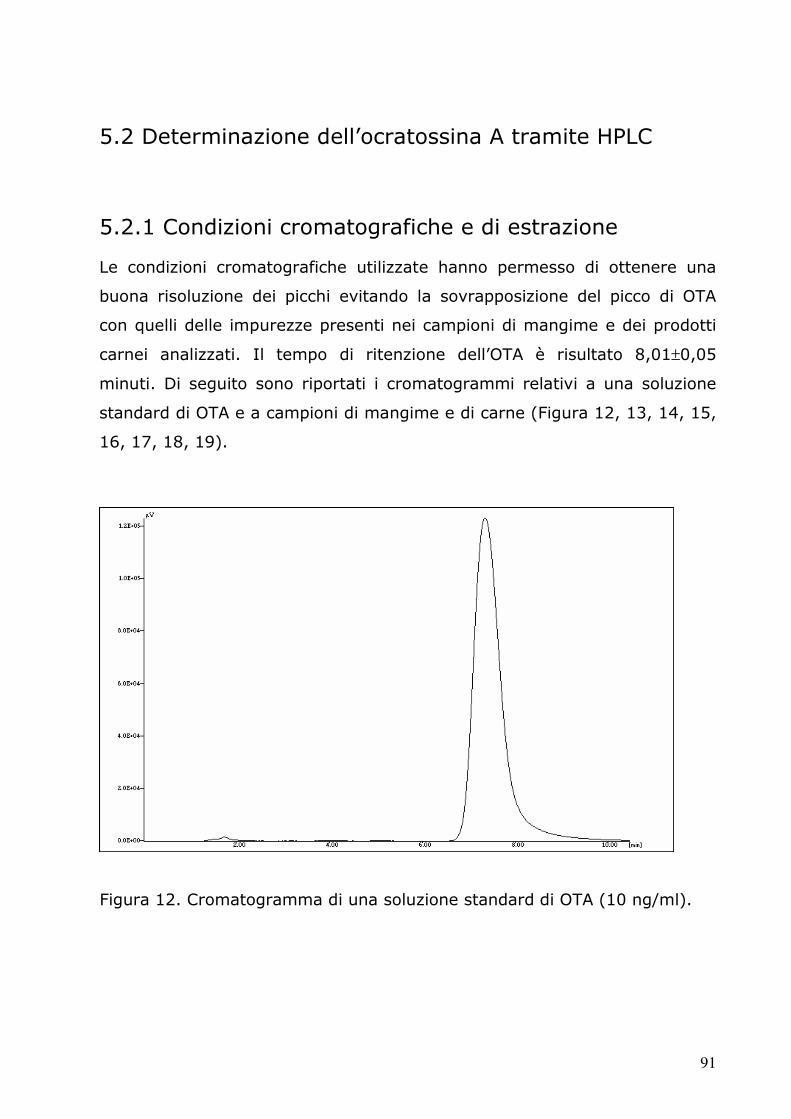
91
5.2 Determinazione dell’ocratossina A tramite HPLC
5.2.1 Condizioni cromatografiche e di estrazione
Le condizioni cromatografiche utilizzate hanno permesso di ottenere una
buona risoluzione dei picchi evitando la sovrapposizione del picco di OTA
con quelli delle impurezze presenti nei campioni di mangime e dei prodotti
carnei analizzati. Il tempo di ritenzione dell’OTA è risultato 8,01±0,05
minuti. Di seguito sono riportati i cromatogrammi relativi a una soluzione
standard di OTA e a campioni di mangime e di carne (Figura 12, 13, 14, 15,
16, 17, 18, 19).
Figura 12. Cromatogramma di una soluzione standard di OTA (10 ng/ml).

92
Figura 13. Cromatogramma di un campione di pisello proteico contaminato da
OTA.
Figura 14. Cromatogramma di un campione di orzo contaminato da OTA.
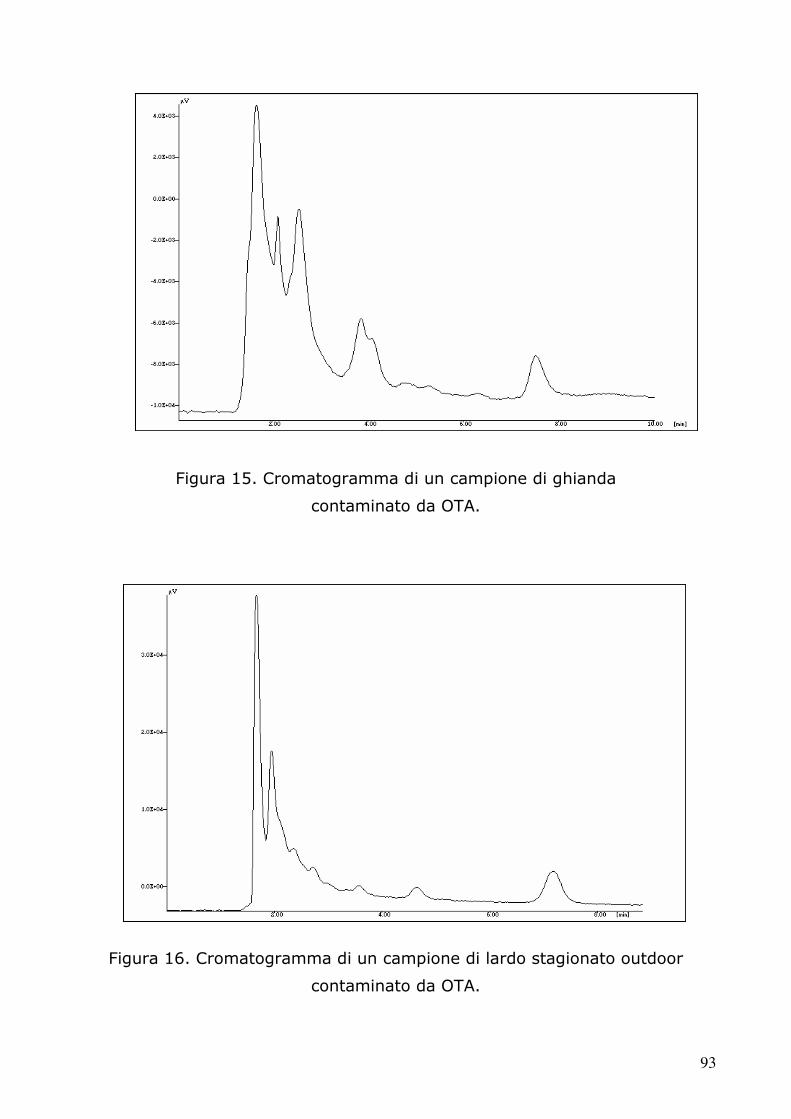
93
Figura 15. Cromatogramma di un campione di ghianda
contaminato da OTA.
Figura 16. Cromatogramma di un campione di lardo stagionato outdoor
contaminato da OTA.

94
Figura 17. Cromatogramma di un campione di lardo fresco indoor contaminato da OTA.
Figura 18. Cromatogramma di un campione di muscolo fresco outdoor
contaminato da OTA.

95
Figura 19. Cromatogramma di un campione di mortadella outdoor
contaminato da OTA.
I recuperi percentuali dell’estrazione dell’OTA dai diversi campioni analizzati
sono mostrati nelle tabelle 21 e 22.
Campione
Recupero % ±±±± DS
Mangimi 76,5 ± 2,1 Essenze del bosco 65,5 ± 5,1
Tabella 21. Recuperi relativi alla metodica di estrazione ottimizzata.

96
Campione
Recupero % ±±±± DS
Muscolo fresco
89,0 ± 3,0
Muscolo stagionato
95,3 ± 6,0
Lardo fresco
98,0 ± 6,0
Lardo stagionato
63,5 ± 4,3
Salame
74,3 ± 2,7
Mortadella
84,0 ± 5,0
Tabella 22. Recuperi relativi alla metodica di estrazione ottimizzata.

97
5.2.2 Retta di calibrazione La linearità del metodo è mostrata dai parametri della retta di calibrazione
ottenuta per l’OTA, il cui coefficiente di correlazione (r2) è risultato > di
0,99.
0 5 10 15 20 250
2.5×105
5.0×105
7.5×105
1.0×106
1.3×106
OTA (ppb)
are
a
Figura 20. Retta di taratura dell'OTA.
La retta di taratura ottenuta (y= ax+c) presentava i seguenti parametri:
• Pendenza (a) = 51780
• Intercetta con l’asse delle y (c) = -12010
• Coefficiente di correlazione (r2) = 0,9929.

98
5.2.3 Analisi dei campioni
Sono stati analizzati 8 campioni di mangime, tutti ottenuti mediante metodo
biologico. I risultati ottenuti sono indicati nelle Tabelle 23 e 24.
TIPO DI MANGIME
VALORI OTA (ng/g di mangime)
Mais
2,11
Pisello proteico
2,35
Fioccato di grano
1,06
Fioccato d’orzo
7,17
Tabella 23. Contenuto di OTA nei campioni di mangime.
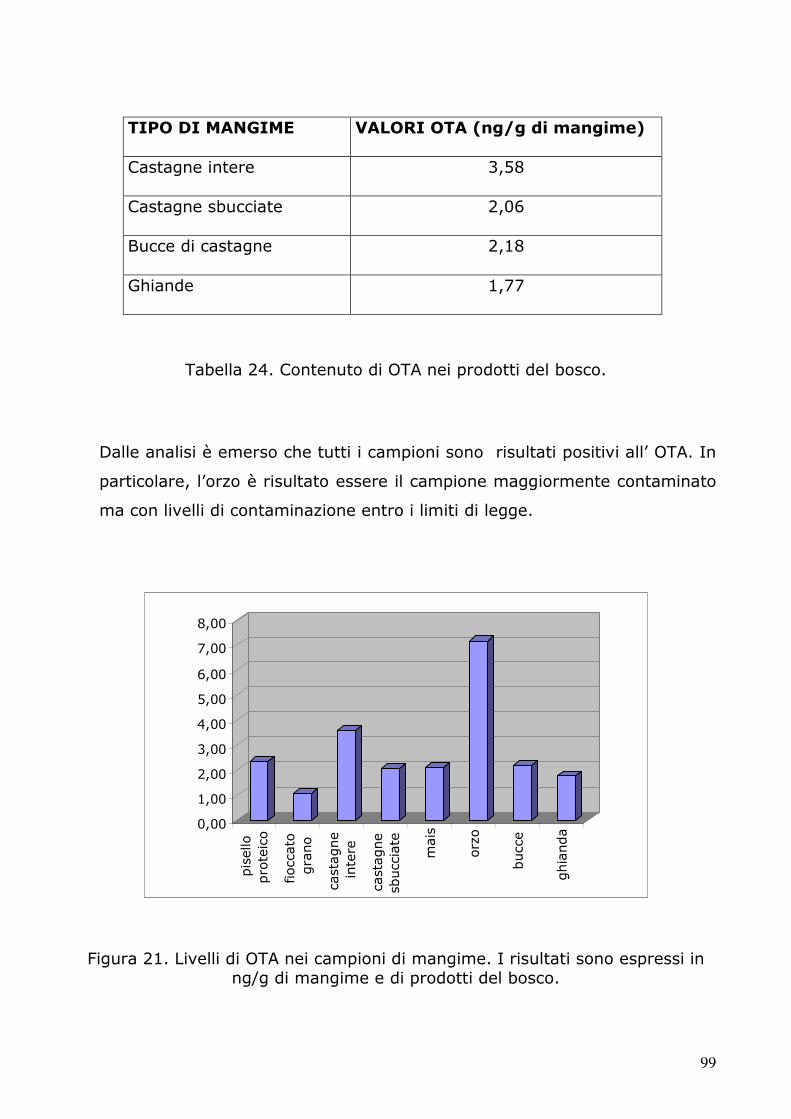
99
TIPO DI MANGIME
VALORI OTA (ng/g di mangime)
Castagne intere
3,58
Castagne sbucciate
2,06
Bucce di castagne
2,18
Ghiande
1,77
Tabella 24. Contenuto di OTA nei prodotti del bosco.
Dalle analisi è emerso che tutti i campioni sono risultati positivi all’ OTA. In
particolare, l’orzo è risultato essere il campione maggiormente contaminato
ma con livelli di contaminazione entro i limiti di legge.
0,00
1,00
2,00
3,00
4,00
5,00
6,00
7,00
8,00
pisello
pro
teico
fiocc
ato
gra
no
castagne
inte
re
castagne
sbucc
iate
mais
orz
o
bucc
e
ghianda
Figura 21. Livelli di OTA nei campioni di mangime. I risultati sono espressi in ng/g di mangime e di prodotti del bosco.

100
Sono stati analizzati un totale di 52 campioni di carne suina, di cui 5
campioni di muscolo e lardo freschi, muscolo e lardo stagionati e 3 campioni
di mortadelle e salami per ciascun gruppo (outdoor e indoor). Tutti i
campioni analizzati sono risultati positivi all’OTA. Il campione meno
contaminato è risultato il muscolo sia per quanto riguarda il gruppo
“outdoor” sia per quello “indoor”. In particolare, in tutti i campioni
analizzati, è stata riscontrata una maggiore presenza di OTA nei campioni
“outdoor”. Le Tabelle 25, 26, 27, 28, 29, 30 riportano le concentrazioni di
OTA per ognuno dei diversi campioni.
CAMPIONI DI MUSCOLO FRESCO
VALORI (ng/g di omogenato ±±±± DS)
0,092 0,067 0,068 0,080 0,085
Media 0,078
GRUPPO OUTDOOR
DS 0,011
0,071 0,042 0,061 0,066 0,036
Media 0,055
GRUPPO INDOOR
DS 0,015
Tabella 25. Livelli di OTA nei campioni di muscolo fresco. I risultati sono espressi in ng/g di omogenato.

101
Tabella 26. Livelli di OTA nei campioni di muscolo stagionato. I risultati sono espressi in ng/g di omogenato.
CAMPIONI DI MUSCOLO STAGIONATO
VALORI (ng/g di omogenato ±±±± DS)
0,172 0,182 0,228 0,151 0,154
Media 0,178
GRUPPO OUTDOOR
DS 0,031
0,153 0,040 0,057 0,047 0,042
Media 0,068
GRUPPO INDOOR
DS 0,048

102
CAMPIONI DI LARDO FRESCO
VALORI (ng/g di omogenato ±±±± DS)
0,078
0,129 0,078 0,065 0,078
Media 0,085
GRUPPO OUTDOOR
DS 0,025
0,074
0,080 0,055 0,080 0,105
Media 0,079
GRUPPO INDOOR
DS 0,018
Tabella 24. Livelli di OTA nei campioni di lardo fresco. I risultati sono espressi in ng/g di omogenato.

103
CAMPIONI DI LARDO STAGIONATO
VALORI (ng/g di omogenato ±±±± DS)
0,220 0,155 0,264 0,112 0,269
Media 0,204
GRUPPO OUTDOOR
DS 0,069
0,138 0,176 0,076 0,211 0,239
Media 0,170
GRUPPO INDOOR
DS 0,064
Tabella 28. Livelli di OTA nei campioni di lardo stagionato. I risultati sono espressi in ng/g di omogenato.

104
Tabella 29. Livelli di OTA nei campioni di salame. I risultati sono espressi in ng/g
Tabella 30. Livelli di OTA nei campioni di mortadella. I risultati sono espressi in ng/g di omogenato.
CAMPIONI DI SALAME
VALORI (ng/g di omogenato ±±±± DS)
0,060 0,068 0,063
Media 0,064
GRUPPO OUTDOOR
DS 0,004
0,074 0,054 0,045
Media 0,058
GRUPPO INDOOR
DS 0,015
CAMPIONI DI MORTADELLA
VALORI (ng/g di omogenato ±±±± DS)
0,570 0,601 0,578
Media 0,590
GRUPPO OUTDOOR
DS 0,016
0,517 0,889 0,206
Media 0,537
GRUPPO INDOOR
DS 0,342

105
5.3.4 Analisi statistica
Dall’analisi statistica eseguita per paragonare i livelli di OTA nei campioni di
muscolo fresco e stagionato, sono state evidenziate differenze
statisticamente significative tra i campioni del gruppo outdoor rispetto al
gruppo indoor. I livelli di OTA riscontrati nei campioni del gruppo outdoor
sono risultati significativamente più alti per il muscolo sia fresco (p< 0,05)
sia stagionato (p< 0,01) (Tabella 31).
TIPO DI CAMPIONE
VALORI (ng/g ±±±± DS) GRUPPO OUTDOOR
VALORI (ng/g ±±±± DS) GRUPPO INDOOR
Muscolo fresco
0,078 ± 0,011 0,055 ± 0,015*
Muscolo stagionato 0,178 ± 0,031 0,068 ± 0,048**
Lardo fresco
0,085 ± 0,025 0,079 ± 0,018
Lardo stagionato 0,204 ± 0,069 0,170 ± 0,064
Tabella 31. Livelli di OTA nei campioni di muscolo e lardo freschi e stagionati dei due gruppi di suini analizzati. I risultati sono espressi come media ±
deviazione standard. *significativamente differente per p < 0,05; **significativamente differente per p < 0,01.
Dall’analisi statistica eseguita per paragonare i livelli di OTA nei diversi
campioni di lardo fresco e stagionato, non sono state evidenziate differenze
significative (p>0,05) tra i due gruppi esaminati. Tuttavia i livelli di OTA
riscontrati nei campioni dei suini outdoor sono risultati solo di poco più alti
rispetto a quelli del gruppo indoor per entrambe le tipologie di campioni
analizzati. (Tabella 31).
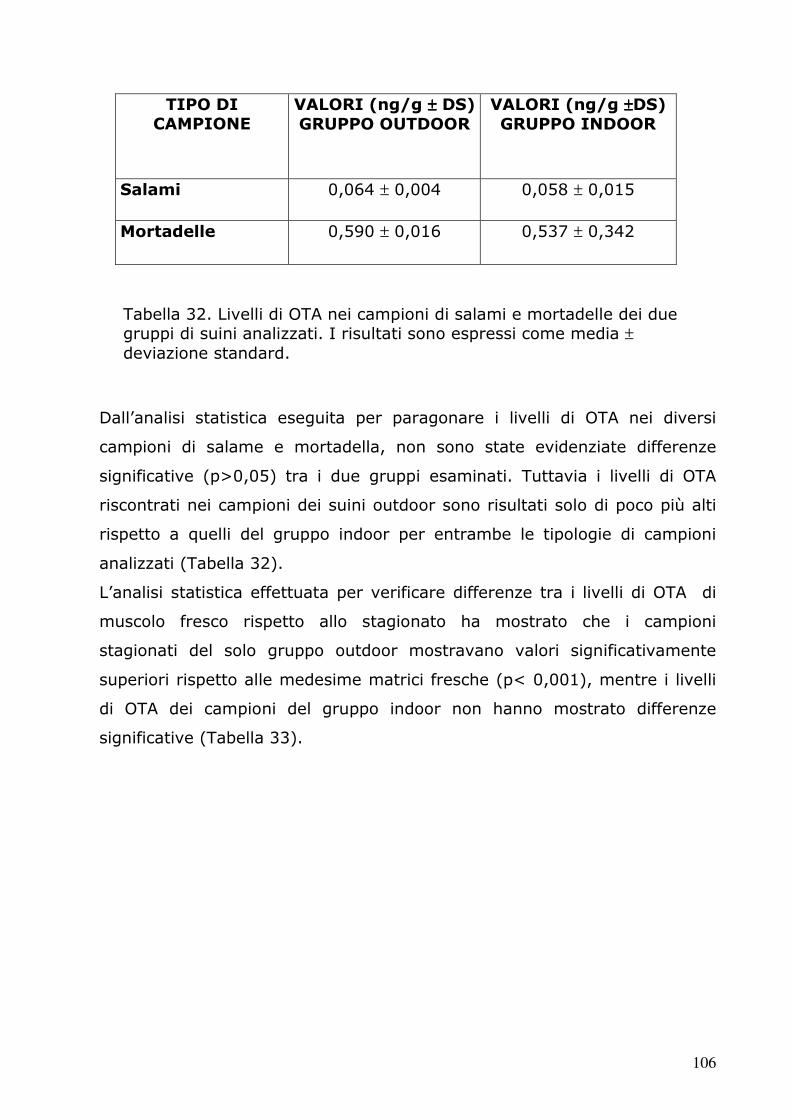
106
TIPO DI CAMPIONE
VALORI (ng/g ±±±± DS) GRUPPO OUTDOOR
VALORI (ng/g ±±±±DS) GRUPPO INDOOR
Salami
0,064 ± 0,004 0,058 ± 0,015
Mortadelle
0,590 ± 0,016 0,537 ± 0,342
Tabella 32. Livelli di OTA nei campioni di salami e mortadelle dei due gruppi di suini analizzati. I risultati sono espressi come media ± deviazione standard.
Dall’analisi statistica eseguita per paragonare i livelli di OTA nei diversi
campioni di salame e mortadella, non sono state evidenziate differenze
significative (p>0,05) tra i due gruppi esaminati. Tuttavia i livelli di OTA
riscontrati nei campioni dei suini outdoor sono risultati solo di poco più alti
rispetto a quelli del gruppo indoor per entrambe le tipologie di campioni
analizzati (Tabella 32).
L’analisi statistica effettuata per verificare differenze tra i livelli di OTA di
muscolo fresco rispetto allo stagionato ha mostrato che i campioni
stagionati del solo gruppo outdoor mostravano valori significativamente
superiori rispetto alle medesime matrici fresche (p< 0,001), mentre i livelli
di OTA dei campioni del gruppo indoor non hanno mostrato differenze
significative (Tabella 33).

107
MUSCOLO FRESCO
MUSCOLO STAGIONATO
Valori (ng/g ±±±± DS) gruppo outdoor
0,078 ± 0,011
0,178 ± 0,031***
Valori (ng/g ±±±± DS) gruppo indoor
0,055 0,015±
0,068 ± 0,048
Tabella 33. Livelli di OTA nei campioni di muscolo frescho e stagionato dei due gruppi di suini analizzati. I risultati sono espressi come media ± deviazione standard. ***significativamente differente per p < 0,001;
L’analisi statistica effettuata per verificare differenze tra i livelli di OTA di
lardo fresco rispetto allo stagionato ha mostrato che i campioni stagionati
del gruppo sia outdoor sia indoor mostravano valori significativamente
superiori rispetto alle medesime matrici fresche (grupo outdoor p< 0,01;
gruppo indoor p< 0,05) (Tabella 34).
LARDO FRESCO
LARDO STAGIONATO
Valori (ng/g ±±±± DS) gruppo outdoor
0,085 ± 0,025
0,204 ± 0,069**
Valori (ng/g ±±±± DS) gruppo indoori
0,079 ± 0,018
0,170 ± 0,064*
Tabella 34. Livelli di OTA nei campioni di lardo fresco e stagionato dei due gruppi di suini analizzati. I risultati sono espressi come media ± deviazione standard. *significativamente differente per p < 0,05; **significativamente
differente per p < 0,01.

108
L’analisi statistica effettuata per verificare differenze tra i livelli di OTA di
salame rispetto alla mortadella ha mostrato che i campioni di mortadella del
gruppo sia outdoor sia indoor mostravano valori significativamente superiori
al salame (gruppo outdoor p< 0,001; gruppo indoor p< 0,05) (Tabella 35).
SALAMI
MORTADELLE
Valori (ng/g ±±±± DS) gruppo outdoor
0,064 ± 0,004
0,590 ± 0,016***
Valori (ng/g ±±±± DS) gruppo indoor
0,058 ± 0,015
0,537 ± 0,342*
Tabella 35. Livelli di OTA nei campioni di salami e mortadelle dei due gruppi
di suini analizzati. I risultati sono espressi come media ± deviazione standard. * significativamente differente per p < 0,05;
***significativamente differente per p < 0,001.
Dall’analisi statistica effettuata all’interno del singolo gruppo, è emerso che i
campioni di mortadella hanno presentato livelli di contaminazione da OTA
significativamente superiori, in entrambi i gruppi, rispetto agli altri campioni
analizzati. Per quanto riguarda il gruppo outdoor, i campioni di muscolo
stagionato e di lardo stagionato, hanno presentato valori significativamente
superiori rispetto alle medesime matrici fresche (Figura 22, 23).

109
MF
MS LF LS SA M
O0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
a aa
bb
c
OTA (ng/g)
Figura 22. Livelli di OTA nei diversi campioni di carne del gruppo OUTDOOR. I risultati sono espressi come media ± deviazione standard. Differenti lettere
indicano medie che sono significativamente diverse (ANOVA, p<0,05)
MF
MS LF LS SA M
O
0.00
0.25
0.50
0.75
a a a
a
a
b
OTA (ng/g)
Figura 23. Livelli di OTA nei diversi campioni di carne del gruppo INDOOR. I risultati sono espressi come media ± deviazione standard. Differenti lettere
indicano medie che sono significativamente diverse (ANOVA, p<0,05).

DISCUSSIONE E CONCLUSIONI

110
6. DISCUSSIONE
La contaminazione da micotossine appare sempre più un fenomeno diffuso,
essendo influenzato da molteplici fattori, sia di tipo ambientale che
agronomico, che sono difficilmente controllabili. In particolare i cambiamenti
delle condizioni ambientali, con rapide escursioni termiche, accompagnate
da umidità elevate, ha comportato un microclima particolarmente adatto al
loro sviluppo. La presenza di micotossine nelle derrate alimentari costituisce
un rischio per la salute sia dell’uomo sia degli animali in seguito
all’ingestione di alimenti contaminati (Miraglia e Brera, 1999). Attualmente
le micotossine rappresentano uno dei problemi più critici proprio a causa dei
noti e pericolosi effetti tossici sulla salute. Pertanto, essendo la problematica
delle micotossine estremamente variegata e non risolvibile con una sola
azione specifica, può essere affrontata in maniera efficace ed efficiente
solamente con un approccio integrato, supportato da un’attenta analisi
comparata del rischio e dei mezzi disponibili per ridurlo il più possibile al di
sotto del limite accettabile, in modo da gestire tutta la filiera del prodotto.
L’OTA è una delle micotossine più comuni e diffuse (Miraglia e Brera, 1999),
della quale è ormai provato il potere nefrotossico, cancerogeno, teratogeno,
mutageno ed immunotossico (Kuiper-Goodman e Scott, 1989; Stormer,
1992; Gekle e Silbernagl, 1996; Kuiper-Goodman, 1996). I cereali
rappresentano la maggiore fonte alimentare di OTA sia per gli animali che
per l’uomo, per il quale l’assunzione di questa micotossina dipende in larga
misura da prodotti di origine animale. L’esposizione dell’uomo all’OTA è
stata chiaramente dimostrata tramite il suo ritrovamento nel sangue e nel
latte materno (Breitholtz et al., 1993; Miraglia et al., 1995), pertanto la sua
presenza negli alimenti costituisce un pericolo concreto. Molteplici sono gli
alimenti che possono essere contaminati in primo luogo dalle diverse specie
fungine produttrici di ocratossine e secondariamente dagli stessi metaboliti.
I principali substrati contaminati da OTA sono: riso, segale, mais, grano,

111
sorgo, orzo, frumento e i cereali in genere. In questi ultimi anni è stato
messo in evidenza come l’OTA può contaminare anche altri tipi di alimenti in
particolare vino, birra e caffè. L’OTA è stata ritrovata anche in alimenti di
origine animale, in particolare in prodotti a base di carne di maiale e di
specie avicole (Canela et al., 1994; Curtui et al., 2001; Gareis e Wolff,
2000; Gareis e Scheuer, 2000; Holmberg et al., 1991; Jimenez et al., 2001;
Jorgensen, 1998; Kuiper-Goodman e Scott, 1989) a causa, come dimostrato
sperimentalmente, del fenomeno del carry-over della micotossina dal
mangime ai tessuti animali (Abramson et al., 1997). Le principali vie di
escrezione sono quella urinaria e fecale, tuttavia un ruolo importante nella
clearance plasmatica della tossina è rivestito, nei mammiferi dall’escrezione
attraverso il latte (Kuipper et al., 1989). Nei ruminanti la percentuale di OTA
espulsa con il latte è limitata grazie all’azione della frazione protozoaria
presente nel liquido ruminale, capace di idrolizzare l’OTA nella sua forma
atossica OTα. E’ stato stimato che possono essere degradati fino a 12 mg di
OTA/kg di alimento (Hult et al., 1976; Petterson et al., 1982). Proprio per
tale ragione, i ruminanti sono meno sensibili alla tossina rispetto ai
monogastrici. I dati relativi al livello di contaminazione da OTA di suini
macellati in Italia e più in generale di prodotti a base di carne suina sono
piuttosto scarsi. Per tale ragione, con la presente tesi, è stato creato un
quadro rappresentativo della sua presenza in un totale di 60 campioni di cui
8 di mangimi e 52 di carne suina fresca e suoi derivati prodotti da suini
allevati con metodo biologico ma appartenenti a due diversi gruppi: indoor
(stabulati) allevati all’interno di un ricovero, e outdoor (brado) lasciati
pascolare liberamente in un recinto situato in una porzione di bosco. Per
quanto riguarda i mangimi, più specificatamente, sono stati analizzati
campioni di mais, fioccato di orzo, fioccato di grano, pisello proteico,
castagne intere, castagne sbucciate, bucce di castagne e ghiande, mentre
per quanto riguarda le carni sono stati analizzati campioni di muscolo
(Longissumus lomborum), lardo, salami, mortadelle, coppa (Longissimus
lomborum dopo stagionatura) e pancetta (lardo dopo stagionatura). Il
metodo utilizzato si è confermato semplice e valido, ed in particolare le

112
condizioni cromatografiche utilizzate hanno permesso di ottenere una corsa
cromatografica abbastanza breve (8 min) e picchi corrispondenti all’OTA
stretti, risolti e ben distinguibili da quelli delle impurezze dei campioni
esaminati. Il metodo di estrazione e purificazione adottato per l’analisi dei
campioni di mangime, ha previsto l’utilizzo di colonne di immunoaffinità che
hanno permesso di ottenere valori di recupero molto buoni. L’OTA è stata
determinata in tutti i campioni di mangime analizzati con valori compresi tra
1,06-7,17 ng/g. Il campione maggiormente contaminato è risultato il
fioccato d’orzo contenente 7,17 ng/g di OTA; tuttavia dall’analisi della
composizione chimica dei mangimi utilizzati nella dieta di entrambi i gruppi,
si è potuto notare come questi abbiano presentato buone caratteristiche
qualitative. Il motivo per cui tali mangimi sono stati analizzati è legato al
fenomeno del carry-over della micotossina dal mangime ai tessuti animali,
maggiormente evidenziato negli animali alimentati con prodotti contaminati.
Per quanto riguarda invece i campioni di carne, è stato necessario l’utilizzo
di una metodica estrattiva seguita da un doppio passaggio per la
purificazione dei campioni a causa della notevole quantità di grasso e di
impurità contenute negli estratti. Dall’analisi dei valori, in tutti i campioni è
stata riscontrata una maggiore presenza di OTA nelle carni dei suini
appartenenti al gruppo outdoor rispetto al gruppo indoor. Ciò è
probabilmente dovuto alla possibilità dei suini allevati allo stato semi-brado,
di alimentarsi liberamente anche di prodotti contaminati da muffe che, in un
allevamento di tipo stabulato, con un’ alimentazione più controllata da parte
dell’uomo non vengono somministrati. Sono state evidenziate differenze
statisticamente significative tra il muscolo fresco e il muscolo stagionato e
tra il lardo fresco e quello stagionato di entrambi i gruppi. L’aumento
dell’OTA dopo la stagionatura è dovuto probabilmente ad un aumento della
concentrazione di tale tossina, in seguito alla perdita di acqua a cui i
prodotti vanno incontro durante il processo di stagionatura. Confrontando i
valori di OTA nei salami e nelle mortadelle di entrambi i gruppi, è emersa
una contaminazione significativamente più alta della mortadella rispetto al
salame, dovuto probabilmente al maggior contenuto di grasso presente

113
nella mortadella poiché l’OTA è una molecola lipofila preferibilmente
accumulata nel tessuto adiposo. Dall’analisi statistica effettuata all’interno
del singolo gruppo di tipologia di allevamento, è emerso che i campioni di
mortadella hanno presentato livelli di contaminazione di OTA più alti, per
entrambi i gruppi, rispetto agli altri campioni analizzati. Ciò è probabilmente
legato alle caratteristiche di produzione della mortadella, in quanto risulta
costituita non solo da una maggiore quantità di grasso, ma anche da una
miscelazione di quest’ultimo con il muscolo già contaminati e appartenenti
ad ogni singolo individuo; fattori probabilmente responsabili dell’elevato
livello di contaminazione riscontrato. A tale proposito, in questa prova è
emerso come in un campione di mortadella il valore di OTA è risultato molto
vicino (0,886 ppb) ai limiti di legge (1 ppb). I muscoli presentano valori di
poco superiori rispetto ai livelli di OTA riscontrati in altri studi condotti sulla
stessa tipologia di campione (Matrella et al., 2006; Guillamont et al., 2005).
Per quanto riguarda i campioni di salame i valori riscontrati nella presente
tesi, sono risultati simili ad uno studio condotto sul salami di origine italiana
(Monaci et al., 2005). I dati derivati dal presente studio, pur essendo
inferiori ai limiti di legge stabiliti, mettono in evidenza quanto l’esposizione
all’OTA sia ormai un fenomeno comune per la cui limitazione è
indispensabile incrementare i controlli sulle derrate alimentari curando ogni
fase della filiera produttiva, data la crescente preoccupazione per la salute
del consumatore. Il problema più grande nella gestione del rischio da
micotossine, è rappresentato sia dalla difficoltà di individuare precisamente
le fasi nelle quali può avvenire la contaminazione, sia dalla grande varietà di
alimenti sui quali questa può verificarsi. Negli ultimi anni sono comunque
aumentati gli studi volti a controllare i livelli di contaminazione da OTA in
varie tipologie di alimenti proprio a testimonianza del fatto che questo è
riconosciuto come un rilevante problema di sanità pubblica. Solamente una
visione globale del problema che coinvolga competenze di ogni genere, di
tipo agronomico, biologico, fisiopatologico, chimico, zootecnico e molte altre
potrebbe portare ad una migliore valutazione e gestione del rischio da
micotossine e limitare la presenza dell’OTA nei nostri alimenti.

114
7. CONCLUSIONI
Dai risultati ottenuti si può concludere che:
� Gli animali bradi, hanno mostrato accrescimenti più bassi rispetto a
quelli confinati, pur ricevendo la stessa razione. Pertanto si ritiene
che, tali differenze, siano dovute più che alla maggiore attività
motoria, alla difficoltà di adattamento iniziale degli animali trasferiti
nei recinti esterni; in quanto provenivano da allevamento di tipoi
tradizionale e quindi non abituati a vivere all’aperto.
� I soggetti del gruppo outdoor sono stati macellati ad una età più
avanzata (circa 1 mese), e ciò ha determinato un maggior stato di
ingrassamento delle carcasse e delle carni.
� Il metodo utilizzato per la determinazione dell’ocratossina A è risultato
valido, semplice e sensibile ed ha permesso di ottenere recuperi
elevati ed una corsa cromatografica breve con picchi corrispondenti
all’OTA ben distinguibili da quelli delle impurezze dei campioni
esaminati.
� La presenza di OTA nella carne, è indice di contaminazione da OTA nei
mangimi a causa del fenomeno del carry over della micotossina dal
mangime ai tessuti animali, maggiormente evidenziato negli animali
alimentati con prodotti contaminati; tuttavia dall’analisi della
composizione chimica dei mangimi utilizzati nella dieta di entrambi i
gruppi, si è potuto notare come questi abbiano presentato buone
caratteristiche qualitative. Tra quest’ultimi, l’orzo è risultato essere il
campione maggiormente contaminato, ma con valori inferiori ai limiti
di legge stabiliti dalla UE. Si rende però necessario attuare piani di

115
prevenzione e di detossificazione al fine di ridurre il rischio di
contaminazione.
� Dall’analisi dei valori di tutti i campioni di carne, è stata riscontrata
una maggiore presenza di OTA nelle carni dei suini appartenenti al
gruppo outdoor. Ciò può probabilmente essere dovuto, alla possibilità
dei suini allevati allo stato semi-brado, di alimentarsi liberamente
anche di alimenti ammuffiti che in un allevamento di tipo stabulato
con alimentazione più controllata da parte dell’uomo, che non
vengono somministrati. Una seconda ipotesi potrebbe essere derivata
dalla maggior presenza di grasso intramuscolare nella carne dei suini
outdoor.
� Particolarmente importante riuslta l’aumento di OTA sia nel muscolo
che nel grasso di entrambi i gruppi durante la stagionatura. Tali
differenze sono probabilmente dovute ad un aumento della
concentrazione di tale tossina, in seguito alla perdita di acqua a cui i
prodotti vanno incontro durante il processo di stagionatura.
� Riguardo i prodotti trasformati è stato osservato come nelle
mortadelle di entrambi i gruppi, è emersa una contaminazione
significativamente più alta rispetto al salame. Tale diiffereza è
probabilmente legata al fatto che la mortadella contiene un maggior
tenore in grasso che rappresenta il tessuto maggiormente
contaminato da OTA.
� La presenza dell’OTA nei mangimi e di conseguenza nelle carni,
costituisce infine un concreto pericolo per la salute dell’animale e
dell’uomo. Per tale ragione, diventa indispensabile incrementare i
controlli sulle derrate alimentari curando ogni fase della filiera
produttiva, per ridurre così la quantità di OTA assunta con la dieta.

BIBLIOGRAFIA

116
8. BIBLIOGRAFIA
� Abarca ML., Accensi F., Bragulat MR., Cabanes FJ., 2001. “Current
importance of ochratoxin A-producing Aspergillus spp.” J. Food Protect. 64,
903-906.
� Abe M., Iriki T., 1978. “Effect of diet on the protozoa population in
permeable continuous cultures of rumen contents.” Br. J. Nutr. 39, 255-
264.
� Abramson D., Mills JT., Boycott BR., 1983. “Mycotoxins and mycoflora
in animal feedstuffs in western Canada.” Can. J. Comp. Med. 47, 23-26.
� Accensi F., Abarca ML., Cabanes FJ., 2004. “Occurence of Aspergillus
species in mixed feeds and component raw materials and their ability to
produce ochratoxin A.” Food Microbiol. 21, 623-627.
� Adebajo LO., Bamgbelu OA., Olowu RA., 1994. “Mould contamination
and the influence of water activity and temperature on mycotoxin
production by two aspergilli in melon seed.” Nahrung. 38, 209-217.
� Agricoltura, 2007. “Mais: Il rischio micotossine” In: “Agricoltura” pp
119-132.
� Aleo MD., Wyatt RD., Schnellmann RG., 1991. “Mitochondrial
dysfunction in an early event in ochratoxin A but not oosporein toxicity to
rats’ renal proximal tubules.” Toxicol. Appl. Pharmacol. 107, 73-80.

117
� Alvarez-Erviti L., Leache C., Gonzales-Penas E., De Cerain AL., 2005.
“Alterations induced in vitro by ochratoxin A in rat lymphoid cells.” Hum.
Exp. Toxicol. 24, 459-466.
� Aureli P., 1996. “Fattori di rischio per l’uomo derivati dalla
contaminazione fungina: modalità di prevenzione.” In: “Quaderni di igiene
pubblica veterinaria, infezioni, micosi e micotossicosi di interesse medico e
veterinario” pp 199-206. Regione Toscana Giunta Regionale, Dipartimento
del diritto alla salute e delle politiche solidarietà.
� Ayadin G., Ozcelik N., Cicek E., Soyoz M., 2003. “Histopathologic
changes in liver and renal tissues induced by ochratoxin A and melatoninn
in rats.” Hum. Exp. Toxicol. 22, 383-391.
� Bacon CW., Sweeney JG., Robbins JD., Burdick D., 1973. “Production
of penicillic acid and ochratoxin A on poultry feed by Aspergillus ochraceus:
temperature and moisture requirements.” Appl. Microbiol. 26, 155-160.
� Ballinger MB., Phillips TD., Kubena LF., 1986. “Assessment of the
distribution and elimination of ochratoxin A in the pregnant rat.” Food Saf.
8, 11-24.
� Barnikol H., Thalmann A., 1988. “Clinical observation in the pig in
relation to the mycotoxin ochratoxin A and zearalenone.” Tierartzl. Umsch.
43, 74-82.
� Baudrimont I., Sostaric B., Yenot C., Betbeder AM., Dano-Djedje S.,
Sanni A., Steyn PS., Creppy EE., 2001. “Aspartame prevents the
Karyomegaly induced bay ochratoxin A in rat kidney.” Arch. Toxicol. 75,
176-183.

118
� Baxter ED., Slaiding IR., Kelly B., 2001. “Behavior of ochratoxin A in
brewing.” J. Am. Soc. Brew. Chem. 59, 98-100.
� Belli N., Marin S., Sanchis V., Ramos AJ., 2004. “Influence of water
activity and temperature on growth of isolates of Aspergillus section nigri
obtained from grapes.” Int. J. Food Microbiol. 96, 19-27.
� Belmadani A., Tramu G., Betbeder AM., Steyn PS., Creppy EE., 1988.
“Regional selectivity to ochratoxin A, distribution and ciytotoxicity in rat
brain.” Arch. Toxicol. 72, 656-662.
� Berger V., Gabriel AF., Sergent T., Trouet A., Larondelle Y., Schneider
YJ., 2003. “Interaction of ochratoxin A with human intestinal CaCo-2cells:
possible implication of a multidrug resistance-assciated proteins (MRP2).”
Toxicol. Lett. 140-141, 456-476.
� Berndt WO., Hayes AW., 1979. “In vivo and in vitro changes in renal
function caused by ochratoxina A in the rat.” Toxicology 12, 5-17.
� Bertelli AA., Migliori M., Filippi C., Gagliano N., Donetti E., Panichi V.,
Scalori V., Colombo R., Mannari C., Tillement JP., Giovannini L., 2005.
“Effect of ethanol and red wine on ochratoxin A-induced experimental acute
nephrotoxicity.” J. Chromatrogr. A 1054, 397-401.
� Bertocchi L., Biancardi A., Boni P., Bonacina C., 2004. “Emergenza
aflatossine nella provincia di Brescia: esperienza di campo.” L’Osservatorio
n.1 Febbraio 2004, 9-13.
� Bonadonna T., Scattolin L., 1934. “L’allevamento dei suini all’aperto:
criteri e tecnica.” Nuovo Ercolani pp 3-37.

119
� Boorman GA., Hong HL., Dieter MP., Hayes HT., Pohland AE., Stack
M., Luster MI., 1984. “Myelotoxicity and macrophage alteration in mice
exposed to ochratoxin A.” Toxicol. Appl. Pharmacol. 72, 304-312.
� Borreani G., Tabacco E., Cavallarin L., 2003. “Contaminazione da
micotossine negli insilati di mais.” L’informatore Agrario 31, 49-55.
� Bredenkamp MW., Dillen JLM., Van Rooyen PH., Steyn PS., 1989.
“Crystal structures and conformational analysis of ochratoxin A and B:
Probing the chemical structure using toxicity.” J. Chem. Soc. Perk. T. 2,
1835-1839.
� Breitholz-Emanuelsson A., Palminger-Hallen I., Wohlin PO., Oskarsson
A., Hult K., Olsen M., 1993a. “Transfer of ochratoxina A from lactating rats
to their offspring: A short-term study.” Nat. Toxins. 1, 347-352.
� Breitholz-Emanuelsson A., Olsen M., Oskarsson A., Palminger I., Hult
K., 1993b. “Ochratoxin A in cow’s milk and in human milk with
corresponding human blood samples.” J. AOAC Int. 76, 842-846.
� Brera C., Caputi R., Miraglia M., Iavicoli I, Salerno A., Carelli G., 2002.
“Exposure assessment to mycotoxins in workplaces: aflatoxin and
ochratoxin A in airborne dust and human sera.” Microchem. J. 73, 167-173.
� Bresch H., Urbanek M., Hell K., 2000. “Ochratoxin A in coffe, tea and
beer.” Arch. Lebensmittelhyg. 51, 89-94.
� Bucheli P., Kanchanomai C., Meyer I., Pittet A., 2000. “Development
of ochratoxin A during Robusta (Coffea canephora) coffee cherry drying.” J.
Agr. Food Chem. 48, 1358-1362.

120
� Bucheli P., Taniwaki MH., 2002. “Research on the origin, and on the
impact of post-harvest handling and manufacturing on the presence of
ochratoxin A in coffee.” Food Addit. Contam. 19, 655-665.
� Bullerman LB., 1985. “Interactive effects of temperature and pH on
mycotoxin production.” Lebensm. Wiss. Technol. 18, 197-200.
� Burdaspal PA., Legarda TM., 1999. “Ocratoxina A en vinos, mostos y
zumos de uva elaborados en España y en otros paises europeos.”
Alimentaria 36, 107-111.
� Cabañes FJ., Accensi F., Bragulat MR., Abarca ML., Castellà G.,
Minguez S., Pons A., 2002. “What is the source of ochratoxin A in wine?.”
Int J. Food Microbiol. 79, 213-215.
� Campbell H., Choo TM., Vigier B., Underhill L., 2000. “Mycotoxin in
barley and oat sample from eastern Canada.” Can. J. Plant Sci. 80, 977-
980.
� Canela R., Viladrich R., Velazquez CA., Sanchis V., 1994. “A survey of
porcine kidneys and chicken liver for ochratoxin A in Spain.” Mycopathologia
125, 29-32.
� Carratù MR., Cuomo V., 2001. “Micotossine negli alimenti a rischio
tossicologico.” In: “La tossicologia per la qualità e la sicurezza alimentare”,
pp 39-44, Hrelia P., Cantelli Forti G., (Eds). Patron editore, Bologna, Italia.
� Castegnaro M., Barek J., Fremy J.M., La Fontane M., Miraglia M.,
Sansone EG., Telling GM., 1991. “Laboratory decontamination and
destruction of carcinogens in Laboratori Wastes: Some mycotoxins”
International Agency for research on cancer, Lyon, France.

121
� Castella G., Larsen TO., Cabanes J., Schmidt H., Alberesi A., Niessen
L., Farber P., Geisen R., 2002. “Molecular characterization of ochratoxin A
producing strains of the genum Penicillum.“ Syst. Appl. Microbiol. 25, 74-
83.
� Causin R., 2004. “Micotossine: chi le produce, cosa sono e cosa
fanno.” Bollettino settimanale di informazione Assincer. 04/03/2004.
� CE (Comunità europea), 2002a. Regolamento n. 472, 12 Marzo.
Gazzetta ufficiale delle comunità europee.
� CE (Comunità europea), 2002b. Direttiva n.32, 7 Maggio. Gazzetta
ufficiale delle comunità europee.
� CE (Comunità europea), 2003a. Regolamento n. 1425, 8 Agosto.
Gazzetta ufficiale delle comunità europee.
� CE (Comunità europea), 2003b. Regolamento n. 2174, 12 Dicembre.
Gazzetta ufficiale delle comunità europee.
� CE (Comunità europea), 2003c. Direttiva n. 100, 31 Ottobre. Gazzetta
ufficiale delle comunità europee.
� CE (Comunità europea), 2004. Regolamento n. 683, 13 Aprile.
Gazzetta ufficiale delle comunità europee.
� CE (Comunità europea), 2005. Regolamento n. 123, 26 Gennaio.
Gazzetta ufficiale delle comunità europee.
� CE (Comunità europea), 2006a. Direttiva n.576, 17 Agosto. Gazzetta
ufficiale delle comunità europee.

122
� CE (Comunità europea), 2006b. Regolamento n. 1881, 19 Dicembre.
Gazzetta ufficiale delle comunità europee.
� CE (Comunità europea), 2007. Regolamento n. 1126, 28 Settembre.
Gazzetta ufficiale delle comunità europee.
� Ceovic S., Hrabar A., Saric M., 1992. “Epidemiology of Balkan
endemic nephropaty.” Food Chem. Toxicol. 30, 183-188.
� Chernoremsky IN., Stoyanov IS., Petkova-Bocharova TK., Nicolov IG.,
Draganov IV., Stoichev I., Tanchev Y., Naidenov D., Kalcheva ND., 1977.
“Geographic correlation between the occurrence of endemic nephopathy and
urinary tract tumours in Vratza district, Bulgaria.” J. Cancer 19, 1-11.
� Chu FS., 1971. “Interaction of ochratoxin A with bovine serum
albumine, arch.” Biochem. Biophys. 147, 359-366
� Chu FS., Noh I., Chang CC., 1972. “Structure requirements for
ochratoxin A intoxication.” Life Sci. 11, 503-508.
� Chu FS., 1974. “A comaprative study of the interaction of ochratoxins
with bovine serum albumine.” Biochem. Pharmacol. 23, 1105-1107.
� Creppy EE., Kern D., Steyn PS., Vleggaar R., Dirheimer G., 1983.
“Comparative study on the effect of Ochratoxin A analogues on yeast
aminoacil-t-RNA synthetases and on the growht and protein synthesis of
hepatoma cells.” Toxicol. Lett. 19, 217-224
� Creppy EE., Chakor K., Fisher MJ., Dirheimer G., 1990. “The
mycotoxin ochratoxin A is a substrate for phenylallanine hydroxylase in
isolated rat hepatocystes and in vivo.” Arch. Toxicol. 64, 279-284.

123
� Curtui V., Gareis M., Usleber E., Märtlbauer E., 2001. “Survey of
Romanian slaughtered pigs for the occurrence of mycotoxins ochratoxins A
and B, and zearalenone.” Food Addit. Contam. 18, 730-738.
� Di Paolo N., Guarnieri A., Loi F., Sacchi G., Mangiarotti AM., Di Paolo
M., 1993. “Acute renal failure from inhalation of mycotoxins.” Nephron 64,
621-625.
� Diaz I., Gracia Reguiero JA., Casillas M., De Pedro E., 1996.
“Trilyceride composition of fresh ham fat from Iberian pigs produced with
different system of animal nutrition.” Food Chemistry. 55, 383-387.
� Dietrich DR., Swenberg JA., 1993. “Renal carcinogenesis.” In:
“Toxicolgy of the kidney.” pp 495-537. Hook JB, Golstein RS., (Eds) Raven
Press, New York, NY, USA.
� Dirheimer G., Creppy EE., 1991. “Mechanism of action of ochratoxin
A.” IARC Sci. Publ. 115, 171-186.
� Dortan PM., Peters-Volleberg GWM., VanLoveren H., Marquardt RR.,
Speijers GJA., 2001. “Age-related differences in the toxicity of ochratoxin A
in female rats.” Food Chem. Toxicol. 39, 55-65.
� EC (European Community), 1998. “Opinion on of the Scientific
Committee on Food (SCF) on ochratoxin A.
� El Adlouni C., Pinelli E., Azemar B., Zaoui D., Beaune P., Leszkowicz
AP., 2000. “Phenobarbital increases of DNA adduct and metabolites formed
by ochratoxin A: role of CYP 2C9 and microsomal glutathione-S-
transferase.” Environ. Mol. Mutagen. 35, 123-131.

124
� El-Halouat A., Debevere JM., 1997. “Effect of water activity, modified
atmosphere packaging and storage temperature on spore germination of
moulds isolated from prunes.” Int. J. Food Microbiol. 35, 41-48.
� Elling F., 1979. “Ochratoxin A-induced mycotoxin porcine
nephropathy: Alterations in enzyme activity in tubular cells.” Acta Pathol.
Microbiol. Scand. 87, 237-243.
� Elling F., 1983. “Feeding experiments with ochratoxin A-contaminated
barley to bacon pigs. IV. Renal lesions.” Acta Agric. Scand. 33, 153-159.
� Elling F., Nielsen JP., Lillehoj EB., Thomassen MS., Stormer FC., 1985.
“Ochratoxin A-induced porcine nephropathy: Enzyme and ultrastructure
changes after short-term exposure.” Toxicology 23, 247-254.
� Elling F., 1979a. “Ochratoxin A-induced mycotoxin porcine
nephropaty: Alterations in enzyme activity in tubular cells.” Acta Pathol.
Microbiol. Scand. 87, 237-243.
� Elling F., 1979b. “Enzyme histochemical studies of ochratoxin A-
induced mycotoxin porcine nephropaty.” In: 6th International Symposium
Animal, Plant Microbial Toxin. Volume 17.
� Endou H., Koseki C., Yamada H., Obara T., 1986. “Evaluation of
nephrotoxicity using isolated nephron segments.” Dev. Toxicol. Environ. Sci.
14, 207-216.
� Escher FE., Koehler PE., Ayres JC., 1973. “Production of ochratoxin A
and B on country cured ham.” Appl. Microbiol. 26, 27-30.

125
� Esteban A., Abarca ML., Bragulat MR., Cabañes FJ., 2004. “Effects of
temperature and incubation time on production of ochratoxin A by black
aspergilli.” Microbiology 155, 861-866.
� FAO/WHO (Food and Agricolture Organisation/World Health
Organisation), 1991. “Evaluation of certain food additives and contaminants
(Thirty-seventh report of the Joint FAO/WHO Expert Committee on Food
Additives). WHO Technical Report Series, No 806, 1991, and corrigenda.
World Health Organisation, Geneva, Switzerland.
� FAO/WHO (Food and Agricolture Organisation/World Health
Organisation), 1996. “Toxicological evaluation of certain food additives and
contaminants.” Joint FAO/WHO Expert Committee on Food Additives Series
47, pp 281-387. World Health Organisation, Geneva, Switzerland.
� FAO/WHO (Food and Agricolture Organisation/World Health
Organisation), 2001. “Ochratoxin A.” In: Safety evaluation of certain
mycotoxins in food, prepared by the 56th Meeting of the Joint FAO/WHO
Expert Committee on Food Additives (JECFA). Food Additives Series 47, pp
281-387. World Health Organisation, Geneva, Switzerland.
� Ferrufino-Guardia EV., Tanghi EK., La rondelle Y., Ponchaut S., 2000.
“Transfer of ochratoxin A and its metabolites in rats.” Toxicol. Appl.
Pharmacol. 145, 82-90.
� Fink-Gremmels J., John A., Blom MJ., 1995. “Toxicity and metabolism
of ochratoxin A.” Nat. Toxins 3, 214-220.
� Friis C., Brinn R., Hald B., 1988. “Uptake of ochratoxin A by slices of
pig kidney cortex.” Toxicology 52, 209-217.

126
� Fuchs R., Radic B., Peraica M., Hult K., Plestina R., 1988.
“Enterohepatic circulation of ochratoxin A in rats.” Period. Biol. 90, 39-42.
� Fukal L., 1990. “A survey of cereals, cereal products, feedstuffs and
porcine kidneys for ochratoxin A by radioimmunoassay.” Food Addit.
Contam. 7, 253-258.
� Galtier P., 1978. “Contribution of pharmacokinetic studies to
mycotoxicology.Ochratoxin A.” Vet. Sci. Commun. 1, 349-358.
� Galtier P., Charpenteau Jl., Alvinerie M., Labouche C., 1979. “The
pharmacokinetic profile of ochratoxin A in the rat after oral and intravenus
administration.” Drug Metab. Dispos. 7, 429-434.
� Galtier P., Alvinerie M., Charpenteau JL., 1981. “The pharmacokinetic
profiles of ochratoxin A in pigs, rabbits and chickens.” Food Cosmet. Toxicol.
19, 735-738.
� Gareis M., Scheuer R., 2000. “Ochratoxin A in meat and meat
products.” Arch. Lebensmittelhyg. 51, 102-104.
� Gareis M., Wolff J., 2000. “Relevance of mycotoxin contaminated feed
for farming animals and carry over of mycotoxins in fodd of animal origin.”
Mycoses 43, 79-83.
� Gazzetta Ufficiale della Repubblica italiana, 1999. N. 35, 11 giugno.
“Direttive in materia di controllo ufficiale sui prodotti alimentari: valori
massimi ammissibili di micotossine nelle derrate alimentari di origine
nazionale, comunitaria e Paesi terzi”, Circolare N. 10, 9 giugno. Roma,
Italia.

127
� Genkle M., Silbernagl S., 1996. “Renal toxicodynamics of ochratoxin A:
a phatophysiological approach.” Kidney Blood Press. R. 19, 225-235.
� Giardini A., Vecchietti M., 2000. “Mais o Granoturco (Zea Mays L.).”
In: “Coltivazioni erbacee Cereali e Proteaginose.” pp 168-169. Baldoni R.,
Giardini L., (Eds). Patron Editore, Bologna, Italia.
� Grosse Y., Baudrimont I., Castegnaro M., Betbeder AM., Creppy EE.,
Dirheimer G., Pfohl-Leszkowicz A., 1995. “Formation of ochratoxin A
metabolites and DNA-adducts in monkey kidney cells.” Chem. Biol.
Interactions 95, 175-187.
� Grosse Y., Chekir-Ghedira L., Huc A., Obrecht-Pflumio S., Dirheimer
G., Bacha H., Pfohl-Leszkowicz A., 1997. “Retinol, ascorbic acid and alpha-
tocpherol prevent DNA adduct formation in mice treated with the
mycotoxins ochratoxin A and zearalenone.” Cancer Lett. 114, 225-229.
� Guillamont EM., Lino CM., Baeta ML., Pena AS., Silveira MI., Vinuesa
JM., 2005. “A comparative study of extraction apparatus in HPLC analysis of
ochratoxin A in muscle.” Anal. Bioanal. Chem. 383, 570-575.
� Hagelberg S., Hult K., Fuchs R., 1989. “Toxicokinetics of ochratoxin A
in several species and its plasma-binding properties.” J. Appl. Toxicol. 9,
91-96.
� Haggblom P., 1982. “Production of ochratoxin A in barley by
Aspergillus ochraceus and Penicillium viridicatum: effect on fungal growth,
time, temperature, and inoculum size.” Appl. Environ. Microbiol. 43, 1205-
1207.

128
� Haggblom PE., Ghosh J., 1985. “Postharvest production of ochratoxin
A by Aspergillus ochraceus and Penicillium viridicatum in barley with
different protein levels.” Appl. Environ. Microb. 49, 787-790.
� Halt M., 1998. “Moulds and mycotoxins in herb tea and medicinal
plants.” Eur. J. Epidemiol. 14, 269-74.
� Haouet NM., Altissimi SM. 2003. “Micotossine negli alimenti e
micotossicosi animale umana.” Webzine Sanità Pubblica Veterinaria, 18:
Febbraio 2003.
� Hard GC., 1998. “Mechanisms of chemically induced renal
carcinogenesis in the laboratory rodent.” Toxicol. Pathol. 26, 104-112.
� Harwing J., Kuiper-Goodman T., Scott PM., 1983. “Microbial food
toxicants: Ochratoxin.” In: “Handbook of Foodborne Disease of Biological
Origin.” pp 193-238. Rechcigl M., (Ed). CRC Press, Boca Raton, FL, USA.
� Hennigen MR., Dick T., 1995. “Incidence and abundance of
mycotoxins in maize in Rio Grande do Sul, Brazil.” Food Addit. Contam. 12,
677-681.
� Henning A., Fink-Gremmels J., Leistener L., 1991. “ Mutagenicity and
effects of ochratoxin A on the frequency of sister chromatid exchanged after
metabolic attivation.” In: “Mycotoxins, Endemic Nephropathy and Urinary
Tract Tumours.” pp 141-148. Castegnaro M., Plestina R., Dirheimer G.,
Chernozemsky IN., Bartsch H., (Eds). IARC Press, Lyon, France.
� Hesseltine CW., 1969. “Mycotoxins.” Mycopathol. Mycol. Appl. 39,
371-383.

129
� Hesseltine CW., Vandergraft EE., Fennell DI., Smith ML., Shotwell
O.L., 1972. “Aspergilli as ochratoxin producers.” Mycologia 64, 539-550.
� Hietanen E., Bartsch H., Bèrèziat JC., Castegnaro M., Michelon J.,
1991. “Characterization of the cytochrome P450 isozyme that metabolizes
ochratoxina A, using metabolic inducers, inhibitors, and antibodies.” In
“Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours.” pp 141-
148. Castegnaro M., Plestina R., Dirheimer G., Chernozemsky IN., Bartsch
H., (Eds). IARC Press, Lyon, France.
� Hoehler D., Marquardt RR., Mcintosh AR., Hatch GM., 1997.
“Induction of free radicals in hepatocystes, mitochondria and microsomes of
rats by ochratoxin A and its analogues.” Biochim. Biophys. Acta 1357, 225-
233.
� Hogberg A., Pickova J., Dutta PC., Babol J., Bylund AC., 2001. “Effect
of rearing system on muscle lipids of gilts and castrated male pigs.” Meat
Sci. 58, 223-229.
� Hohler D., 1998. “Ochratoxin A in food and feed: occurrence,
legislation and mode of action.” Z. Ernahrungswiss. 37, 2-12.
� Holmberg T., Breitholtz-Emanuelsson A., Haggblom P., Schwan O.,
Hult K., 1991. “Penicillium verrucosum in feed of ochratoxin A positive
swine herds.” Mycopathologia 116, 169-176.
� Hooper J., 2006 “Polluted pasta caused toxin alarm in Italy.” Guardian
Unlimited 1/12/2006.
http://www.guardian.co.uk/food/Story/0,1684524,00.html
� Hult K., Teiling A., Gatenbeck S., 1976. “Degradation of ochratoxin A
by a ruminant.” Appl. Environ. Microbiol. 38, 772-776.

130
� Hult K., Plestina R., Habazin-Novak V., Radic B., Ceovic S., 1982. “
Ochartoxin A in human blood and Balkan endemic nephropathy.” Arch.
Toxicol. 51, 313-321.
� Hussein SH., Brasel MJ., 2001. “Toxicity, metabolism, and impact of
mycotoxins on humans and animals.” Toxicology, 167, 101-104.
� IARC., 1993. “Ochratoxin A” “Some naturally occurring substances:
Food items and constituents, heterocyclic, aromatic amines and
mycotoxins.” IARC Monographs on the Evolution of Carcinogenic Risks to
human pp 489-521. Geneva: International Agency for Research on Cancer.
� Jiménez AM., Lopez de Cerain A., Gonzalez-Penas E., Bello J., 2001.
“Determination of ochratoxin A in pig liver-derived pates by
highperformance liquid chromatography.” Food Addit. Contam. 18, 559-
563.
� Joosten HLMJ., Goetz J., Pittet A., Schellenberg M., Bucheli P., 2001.
“Production of ochratoxin A by Aspergillus carbonarius on coffee cherries.”
Int. J. Food Microbiol. 65, 39-44.
� Jorgensen K., 1998. “Survey of pork, poultry, coffee, beer and pulses
for ochratoxin A.” Food Addit. Contam. 15, 550-554.
� Jung KY., Endou H., 1987. “Nephrotoxicity assessment by measuring
cellular ATP content. II. Intranephron site of ochratoxin A nephrotoxicity.”
Toxicol. Appl. Pharmacol. 100, 383-390.
� Juszkiewicz T., Piskiorska-Pliszczynska J., Wisniexska H., 1982.
“Ochrtaoxin A in laying hens: Tissue deposition and passage into eggs.” In:

131
“Mycotoxin and Phycotoxin. Proceedings of the V International IUPAC
Symposius.” pp 122-125. Techinical University, Vienna, Austria.
� Kane A., Creppy EE., Roschenthaler R., Dirheimer G., 1986. “Changes
in urinary and renal tubular enzymes caused by subchronic adiministration
of ochratoxin A in rats.” Toxicology 42, 233-243.
� Kanisawa M., Suzuki S., 1978. “Induction of ranal and hepatic tumors
in mice by ochratoxin A, a mycotoxin.” Gann. 69, 599-600.
� Khan S., Martin M., Bartsh H., Rahimtula AD., 1989. “Perturbation of
liver microsomial calcium homeostasis by ochratoxin A.” Biochem.
Pharmacol. 38, 67-72.
� Kiessling KH., Patterson H., Sandholm K., Olsen M., 1984.
“Metabolism of aflatoxin, ochratoxin, zearalenone, and three Trichothecenes
by intact rumen fluid, rumen protozoa, and rumen bacteria.” Appl. Environ.
Microbiol. 47, 1070-1073.
� Krogh P., Elling F., Hald B., Jylling B., Petersen VE., Skadhauge E.,
Svendsen CK., 1976. “Experimental avian nephropathy.” Acta Pathol.
Microbiol. Scand. A. 84, 215-221.
� Krogh P, Elling F, 1977. “Mycotoxin nephropathy.” Vet. Sci. Commun.
1, 51-63.
� Kuiper-Goodman T., Scott PM., 1989. “Review: Risk assessment of
the mycotoxin ochratoxin A.” Biomed. Environ. Sci. 2, 179-248.
� Kuiper-Goodman T., 1996. “Risk assessment of ochratoxin A: an
update.” Food Addit. Contam. 13, 53-57.

132
� Kumagai S., Aibara K., 1982. “ Intestinal absorption and secretion of
ochratoxin A in the rat.” Toxicol. Appl. Pharmacol. 64, 94-102.
� Kumagai S., 1985. “Ochratoxin A: Plasma concentration and excretion
into bile and urine in albumin-deficient rats.” Food Chem. Toxicol. 23, 941-
943.
� Kumagai S., 1988. “ Effects of plasma ochratoxin A and luminal pH on
the jejunal absorption of ochratoxin A in the rats.” Food Chem. Toxicol. 26,
753-758.
� Kuramochi G., Gekle M., Silbernagl S., 1997a. “Derangement of pH
homeostatis in the renal papilla: Ochratoxin A increase pH in vasa recta
blood.” Nephron. 76, 472-476.
� Kuramochi G., Gekle M., Silbernagl S., 1997b. “Ochratoxin A disturbs
pH homeostasis in the kidney: Increases in pH and HCO3 in the tubules and
vasa recta.” Pflugers Arch. Eur. J. Physiol. 434, 392-397.
� Larsen TO., Svendsen A., Smedsgaard J., 2001. “Biochemical
characterization of ochratoxin A producing strains of the genus Penicillium.”
Appl. Environ. Microb. 67, 3630-3635.
� Li S., Marquardt RR., Froholich AA., 2000. “Identification of
ochratoxins and some of their metabolites in bile and urine of rats.” Food
Chem. Toxicol. 38, 141-152.
� Lopez-Bote CJ., 1998. “Sustain utilization of Iberian pig breed.” Meat
Sci. 49, 17-27.

133
� MacDonald S., Wilson P., Barnes K., Damant A., Massey R., Mortby E.,
Shepherd MJ., 1999. “Ochratoxin A in dried vine fruit: method development
and survey.” Food Addit. Contam. 16, 253-60.
� Madhyastha SM., Marquardt RR., Frohlich AA., Platford G., Abramson
D., 1990. “Effects of different cereals and oilseed substrates on the growth
and production of toxins by Aspergillus alutaceus and Penicillium
verrucosum.” J. Agr. Food Chem. 38, 1506-1510.
� Madhyastha MS., Marquardt RR., Frolich AA., 1992. “Hydrolysis of
ochratoxin A by the microbial activity of digesta in the gastrointestinal tract
of rats.” Arch. Envirion. Contam. Toxicol. 23, 468-472.
� Madhyastha SM., Marquardt RR., Abramson D., 1993. “Effect of
ochratoxin producing fungi on the chemical composition of wheat and
barley.” J. Food Quality 16, 287-299.
� Malaveille C., Brun G., Bartsch H., 1994. “Structure-activity studies in
E.coli strains on ochratoxin A (Ochratoxin A) and its analogues implicate a
genotoxic free radical and a cytotoxic thiol derivative as reactive
metabolites metabolites.” Mutat. Res. 307, 141-147.
� Mally A., Keim Heusler H., Ambreg A., Kurtz M., Zepnik H., Mantle P.,
Volkel W., Hard GC., Dekant W., 2005. “Biotransformation and
nephrotoxicity of ochratoxin B in rats.” Toxicol Appl. Pharmacol. 206, 43-53.
� Mantle PG., 2002. “Risk assessment and importance of ochratoxins.”
Int. Biodet. Biodegr. 50, 143-146.
� Marcato PS., 1998. “Patologia suina.” Ed. agricole, Bologna, Italia.

134
� Markaki P., Delpont-Binet C., Grosso F., Dragacci S., 2001.
“Determination of ochratoxin A in red wine and vinegar by immunoaffinity
high-pressure liquid cromatography.” J. Food Protect. 64, 533-537.
� Marquardt RR., Frohlich AA., 1992. “A review of recent advances in
understanding ochratoxicosis.” J. Anim. Sci. 70, 3968-3988.
� Matrella R., Monaci L., Milillo MA., Palmisano F., Tantillo MG., 2006.
“Ochratoxin A determination in paired kidneys and muscle samples from
swines slaughtered in southern Italy.” Food Control. 17, 114-117.
� Matta A., Luisoni E., Surico G., 1996. “Malattie Fungine.” In:
“Fondamenti di patologia vegetale.” Patron Editore, Bologna, Italia.
� Meisner H., Chan S., 1974., “Ochratoxin A, an inhibitor of
Mitochondrial transport systems.” Biochemistry 13, 2795-2800.
� Meisner H., Meisner P., 1981. “Ochratoxin A an in vivo inhibitor of
renal phosphoenolpyruvate carboxykinase.” Arch. Biochem. Biophys 208,
146-153.
� Meisner H., Cimbala MA., Hanson RW., 1983. “Decrease of renal
phospho-enolpyruvate carboxykinase RNA and poly(A) RNA level by
ochratoxin A.” Arch. Biochem. Biophys. 223, 264-270.
� Meisner H., Krogh P., 1986. “Phosphoenolpyruvate carboxukinase as a
selective indicator of ochratoxin A induced nephropathy.” Dev. Toxicol.
Environ. Sci. 14, 199-206.
� Miraglia M., Brera C., 1999. “Le micotossine.” In: “Tossicologia degli
alimenti.” pp 22-28. Captano A, Dugo G, Restani P (Eds). UTET, Torino,
Italia.

135
� Miraglia M., De Dominicis A., Brera C., Corneli S., Cava E., Menghetti
E., Miraglia E., 1995 “Ochratoxin A levels in human milk and related food
samples: an exposure assessment.” Nat. Toxins 3, 436-444.
� Mitchell D., Parra R., Aldred D., Magan N., 2004. “Water and
temperature relations of growth and ochratoxin A production by Aspergillus
carbonarius starins from grapes in Europe and Israel.” J. Appl. Microbiol. 97,
439-445.
� Monaci L., Pamisano F., Martella R., Tantillo G., 2005. “Determination
of ochratoxin A at part-per-trillion level in Italian salami by immunoaffinity
clean-up and high-performance liquid chromatography with fluorescence
detection.” J. Chromatogr. A 1090, 184-187.
� Moroi K., Suzuki S., Kuga T., Yamazaki M., Kanisawa M., 1985.
“Reduction of ochratoxin A toxicity in mice treated with phenylalanine and
phenobarbital.” Toxicol. Lett. 25, 1-5.
� Mortensen HP., Hald B., Madsen A., 1983a. “Feeding experiments with
ochratoxin A contaminated barley for bacon pigs. 5. Ochratoxin A in pig
blood.” Acta Agric. Scand. 33, 235-239.
� Mortensen HP., Hald B., Larsen AE., Mdsen A., 1983b. “Ochratoxin A-
contaminated barley for sows and piglets. Pig performance and residues in
milk and pigs.” Acta Agric. Scand. 33, 349-352.
� Munro IC., Moodie CA., Kuiper-Goodman T., Scott PM., Grice HC.,
1974. “Toxicologic changes in rats fed graded dietary levels of chratoxin A.”
Toxicol. Appl. Pharmacol. 28, 180-188.

136
� Nakamura K., Kanegasaki S., 1969. “Densities of ruminal protozoa of
sheep established under different dietary conditions.” J. Dairy Sci. 52, 250-
255.
� Nilzèn V., Babol J., Dutta PC., Lundeheim N., Enfalt AC., Lundstrojm
K., 2001. “Free range rearing of pigs with acces to patsure grazing effect on
fatty acid composition and lipid oxidation products.” Meat Sci. 58, 267-275.
� Nip WK., Chu FS., 1979. “The fate of ochratoxin A in goats.” J.
Environ. Sci. Health B 14, 319-333.
� Northolt MD., Van Egmond HP., Paulsch WE., 1979. “Ochratoxin A
production by some fungal species in relation to water activity and
temperature.” J. Food Protect. 42, 485-490.
� Northolt MD., Bullerman LB., 1982. “Prevention of mold growth and
toxin production througt control of environmental conditions.” J. Food
Protect. 45, 519-526.
� Obrecht-Pflumio S., Chassat T., Dirheimer G., Marzin D., 1999.
“Genotoxicity of ochratoxin A by Salmonella mutagenicity test after
bioactivation by mouse kidney microsomes.“ Mutat. Res. 446, 95-102.”
� Obrecht-Pflumio S., Dirheimer G., 2000. “In vitro DNA and dGMP
adducts formation caused by ochratoxin A.” Chem. Biol. Interactions. 127,
29-44.
� Olsen JH., Hald B., Thorup I., Carstensen B., 1993. “Distribution in
Denmark of porcine neprhopathy and cronic disordes of the urinary tract in
humans.” In: “Human Ochratoxicosis and its Pathologies.” pp 209-215.
Creppy EE., Castegnaro M., Dirheimer G., (Eds). Colloqui INSERM/J Libbery
Eurotext LDT, Montrougue, France.

137
� Omar RF., Hasinoff BB., Mejilla F., Rahimtula AD., 1990. “Mechanism
of ochratoxin A-stimulated lipid preoxidation.” Biochem. Pharmacol. 40,
1183-1191.
� Osweiler GD., 1992. “Mycotoxins.” In : “Disease of Swine.” pp 735-
743. Leman AD., Straw BE, Mengling WL., D’Allaire S., Taylor DJ., (Eds).
Wolfe Publishing, London, UK.
� Palminger I., Hallen A., Breitholtz-Emanuelssonn A., Hult K., Olsen M.,
Okarsson A., 1998. “Placental and lactional transfer of ochratoxin A in rats.”
Nat. Toxins 6, 43-49.
� Parker RW., Phillips TD., Kubena LF., Russell LH., Heidelbaugh ND.,
1982. “Inhibition of pancreatic carboxypeptidase A: A possible mechanism
of interaction between penicillic acid and ochratoxin A.” J. Environ. Sci.
Health 17, 77-91.
� Pasteiner S., 1997 “Coping with mycotoxin contaminated feedstuffs.”
Feed International pp 12-18.
� Paster N., Lisker N., Chet I., 1983. “Ochratoxin A production by
Aspergillus ochraceus Wilhelm grown under controlled atmospheres.” Appl.
Environ. Microb. 45, 1136-1139.
� Patterson DSP., Roberts BA., Small BJ., 1976. “Metabolism of
ochratoxins A and B in the pig during early pregnancy and the accumulation
in body tissue of ochratoxin A only.” Food Cosmet. Toxicol. 14, 439-442.
� Patterson M., Darnoglou AP., 1986. “The effect of water activity and
pH on the production of mycotoxins by fungi growing on a bread analogue.”
Lett. Appl. Microbiol. 3, 123-125.

138
� Pepeljnjack S., Blozevic N., 1982. “Contamination with moulds and
occurrence of ochratoxin A in smoked meat products from endemic
nephropathy regions in Yugoslavia.” In: “Proceedings of the 5th IUPAC
Symposium on Mycotoxins and Phycotoxins.” pp 102-105 Czedick-
Eysenberg (Vienna Austrian Chemical Society).
� Peraica M., Domijan AM., Fuchs R., Lucic A., Radic B., 1999. “The
occurrence of ochratoxin A in blood in general population of Croatia.”
Toxicol. Lett. 110, 105-112.
� Peraica M., Domijan AM., Matasin M., Lucic A., Radic B., Delas F., Horvat
M., Bosanac I., Balia M., Grgicevic D., 2001. “Variations of ochratoxin A
concentration in the blood of healthy populations in some Croatian
cities.” Arch. Toxicol. 75, 410-414.
� Pettersson H., Kiessling KH., Ciszuk P., 1982. “Degradation of
ochratoxin A in rumen.” In: “Proceedings, of 5th International IUPAC
symposium Mycotoxins Phycotoxins.” pp 313-316. Austrian Chemical
Society, Vienna, Austria.
� Petzinger E., Ziegler K., 2000. “Ochratoxin A from a toxicological
perspective.” J. Vet. Pharmacol. Ther. 23, 91-98.
� Pfohl-Leszkowicz A., Chakor K., Creppy E., Dirheimer G., 1991. “DNA
adducts formation in mice treated with ochratoxin A.” In: “Mycotoxins,
Endemic Nephropathy and Urinary Tract Tumours.” pp 141-148. Castegnaro
M., Plestina R., Dirheimer G., Chernozemsky IN., Bartsch H., (Eds). IARC
Press, Lyon, France.
� Pfohl-Leszkowicz A., Grosse Y., Castegnaro M., Nicolov IG.,
Chernozemsky IN., Bartsch H., Betbeder AM., Creppy EE., Dirheimer G.,

139
1993. “Ochratoxin A-related DNA adducts in urinary tract tumours of
Bulgarian subjecrs.” In: “Mycotoxins, Endemic Nephropathy and Urinary
Tract Tumours.” pp 141-148. Castegnaro M., Plestina R., Dirheimer G.,
Chernozemsky IN., Bartsch H., (Eds). IARC Press, Lyon, France.
� Pfohl-Leszkowicz A., Pinelli E., Bartsch H., Mohr U., Castegnaro M.,
1998. “Sex and strani-specific expression of cytochrome P450s in
ochratoxin A-induced genotoxicity and carcinogenicity in rats.” Mol.
Carcinog. 23, 76-85.
� Pfohl-Leszkowicz A., Bartsch H., Azemar B., Mohr U., Esteve J.,
Castegnaro M., 2002. “MESNA protects rats against nephrotoxicity but no
carcinogenicity induced by ochratoxin A, implicating two separate
pathways.” Facta Universitatis 9, 37-43.
� Pitout MJ., 1969. “A rapid spectrophotometric method for the assay of
carboxypeptidase A.” Biochem. Pharmacol. 18, 1829-1836.
� Pitout MJ., 1969a. “The hydrolysis of ochratoxin A by some proteolytic
enzymes.” Biochem. Pharmacol. 18, 485-491.
� Pitout MJ., Nel W., 1969b. “The inhibitory effect of ochratoxin A on
bovine carboxypeptidase A in vitro.” Biochem. Pharmacol. 18, 1837-1843.
� Pitt JI., 2000. “Fungi and food spoilage.” 2° Ed. Chapman and Hall,
Londra.
� Pittet A., 1998. “Natural occurrence of mycotoxins in foods and fedds-
an update review.” Rev. Med. Vet. 149, 479-492.
� Piva G., Pietri A., 1996. “Micotossine negli alimenti.” In: “Quaderni di
igiene pubblica veterinaria, infezioni, micosi e micotossicosi di intersse

140
medico e veterinario.” pp 109-115. Regione Toscana Giunta Regionale,
Dipartimento del diritto alla salute e delle politiche di solidarietà.
� Pohland AE., Nesheim S., Friedman L., 1992. “Ochratoxin A: a
review.” Pure Appl. Chem. 64, 1029-1046.
� Rahimtula AD., Bèreziat JC., Bussacchini-Griot V., Bartsch H., 1988.
“Lipid preoxidation as a possible cause of ochratoxin A toxicity.” Biochem.
Pharmacol. 37, 4469-4477.
� Rahimtula AD., Chong X., 1991. “Alterations in calcium homeostasis
as a possible cause of ochratoxin A nephrotoxicity.” In:“Mycotoxin, Endemic
Nephropathy and Urinary Tract Tumours.” pp 207-214. Castegnaro M.,
Plestina R., Dirheimer G., Chernozemsky IN., Bartsch H., (Eds). IARC Press,
Lyon, France.
� Ramos AJ., Labernia N., Marin S., Sanchis V., Magan N., 1998. “Effect
of water activity and temperature on growth and ochratoxin production by
three strains of Aspergillus ochraceus on a barley extract medium and on
barley grains.” Int. J. Food Microbiol. 44, 133-140.
� Roth A., Chakor K., Creppy EE., Kane A., Roschenthaler R, Dirheimer
G, 1988. “ Evidence for an enterohepatic circulation of ochratoxin A in
mice.” Toxicology. 48, 293-308.
� Sage L., Krivobok S., Delbons E., Seigle-Murandi F., Creppy EE.,
2002. “Fungal flora and ochratoxin A in production in grapes and musts
from France.” J. Agric. Food Chem. 50, 1306-1311.
� Scott PM., Van Walbeek W., Kennedy B., Anyeti D., 1972. “Mycotoxins
(ochratoxin A, citrinin, and sterigmatocystin) and toxigenic fungi in grains
and other agricultural products.” J. Agric. Food Chem. 20, 1103-1109.

141
� Scudamore KA., 1996. “Ochratoxin A in animal feed effects of
processing.” Food Addit. Contam. 13, 39-42.
� Shephard GS., Fabiani A., Stockenstrom S., Mschicileli N., Sewram V.,
2003. “Quantitation of Ochratoxin A in South African wines.” J. Agric. Food
Chem. 51, 1102-1106.
� Shier WT., Shier AC., Xie W., Mirocha CJ., 2001. “Structure-activity
relationship for human estrogenic activity in zearalenone mycotoxins.”
Toxicon. 39, 1435-1438.
� Skaug MA., 1999. “Analysis of norwegian milk and infant formulas for
ochratoxin A.” Food Addit. Contam. 16, 75-78.
� Skaug MA., Helland I., Solvoll K., Saugstad OD., 2001. “Presence of
ochratoxin A in human milk in relation to dietary intake.” Food Addit.
Contam. 18, 321-327.
� Sokol PP., Ripich G., Holohan PD., Ross CR., 1988. “Mechanism of
ochratoxin A transport in kidney.” J. Pharmacol. Exp. Ter. 246, 460-465.
� Soleas GJ., Yan J., Goldberg DM., 2001. “Assay of ochratoxin A in
wine and beer by high-pressure liquid cromatography photodiode array and
gas cromatpgraphy mass selective detection.” J. Agr. Food Chem. 49, 2733-
2740.
� Speijers GJA., Van Egmond HP., 1993. “Worldwide ochratoxin A levels
in food and feeds.” Human Ochratoxicosis and its Pathologies. Colloque
INSERM, Creppy EE., Castegnaro M., Dirheimer G., (Eds). 231, 85-100.

142
� Sreemannarayana O., Frohlich AA., Vitti TG. Marquardt RR.,
Abramson D., 1988. “Studies of the tolerance and disposition of ochratoxin
A in young calves.” Anim. Sci. 66, 1703-1711.
� Stanga I., 1946. “Suinocoltura semibrada secondo il metodo Stanga.”
Società editoriale “Cremona nuova”, Cremona, Italia.
� Stein AF., Phillips TD., Kubena LF., Harvey RB., 1985. “Renal tubular
secretion and reabsorption as factors in ochratoxicosis: Effects of
probenecis on nephrotoxicity.” J. Toxicol. Environ. Health. 16, 593-605.
� Steyn PS., 1984. “Ochratoxins and related dihydroisocoumarins.” “In:
Mycotoxins-Production, Isolation, Separation and Purification.” pp. 183-216.
� Stoev SD., Vitanov S., Anguelov G., Petkova-Bocharova T., Creppy
EE., 2001. “Experimental mycotoxic nephropathy in pigs provoked by a diet
containing ochratoxin A and penicillic acid.” Vet. Res Commun. 25, 205-223.
� Stomer FC., Bhatnagar D., Lillehoj EB., Arora DK., (Eds), 1992.
“Ochratoxin A- a mycotoxin of concern.” In: “Handbook of Applied
Mycology: Micotoxins in Ecological System.” pp 403-432. Marcel Dekker,
Inc, New York, NY, USA.
� Stonard MD., Gore CW., Oliver GJA., Smith IK., 1987. “Urinary
enzymes and protein patterns as indicators of injury to different regions of
the kidney.” Fund. Appl. Toxicol. 9, 339-351.
� Storen O., Holm H., Stormer FC., 1982. “Metabolism of ochratoxin A
by rats.” Appl. Environ. Microbiol. 44, 785-789.

143
� Suzuki S., Kozuka Y., Satoh T., Yamazaki M., 1975 “Studies on the
nephrotoxicity of ochratoxin A in rats.” Toxicol. Appl. Pharmacol 34, 479-
490.
� Suzuki S., Satoh T., Yamazaki M., 1977. “The pharmacokinetics of
ochratoxin A in rats.” Jpn. J. Pharmacol. 27, 735-744.
� Swenberg JA., Maronpot RR., 1991. “Chemically induced cell
preoliferation as a criterion in selecting doses for long-term bioassays.” In:
“Chemically Induced Cell Proliferation: Implications for Risk Assessment.”
pp 245-251. Wiley-Liss, New York, NY, USA.
� Tecnoalimenti 2006. “Le micotossine: modalità di contaminazione, i
rischi per la salute, gli strumenti per la prevenzione e la regolamentazione
nel mondo a tutela del consumatore.” Tecnoalimenti 21 Dicembre 2006, pp
3-12.
� Téren J., Varga J., Hamari Z., Rinyu E., Kevei F., 1996.
“Immunochemical detection of ochratoxin A in black Aspergillus strains.”
Mycophatologia 134, 171-176.
� Téren J., Palagyi A., Varga J. 1997. “Isolation of ochratoxin producing
Aspergilli from green coffee beans of different origin.” Cereal Res. Commun.
25, 303-304.
� Tiecco G, 2001. “Malattie da agenti chimici.” In: “Igiene e Tecnologia
alimentare.” pp 543-738. Calderin EdAgricole, Bologna, Italia.
� Tsuda M., Sekine T., Takeda M., Cha SH., Kanai Y., Kimura M., Endou
H., 1999. “Transport of ochratoxin A by renal multispecific organic anion
transporter 1.” J. Pharmacol. Exp. Ther. 289, 1301-1305.

144
� Turner WB., 1971. “Fungal Metabolites.” Academic Press, New York.
NY, USA.
� Ueno Y., 1987. “Mycotoxins.” In: “Toxicological Aspect of Food.” pp
139-204. Miller K. (Ed). Elsevier, London, UK.
� Ueno Y., 1998. “Residue and risk of ochratoxin A in human plasma
and beverages in Japan.” Mycotoxins 47, 25-32.
� US-NTP (United States-National Toxicolgy Program), 1989.
“Toxicology and Carcinogenesis Studies of Ochratoxin A in F344/N rats.”
� Valenta H., Goll M., 1996. “Determination of ochratoxin A in regional
samples of cow’s milk from Germany.” Food Addit. Contam. 13, 669-676.
� Van der Merwe KJ., Steyn PS., Fourie L., Scott DB., Theron JJ. 1965a
“Ochratoxin A, a toxic metabolite produced Aspergillus ochraceus Wilh.”
Nat. 205, 1112-1113.
� Van der Merwe KJ., Steyn PS., Fourie L., 1965b. “Micotoxins. II. The
constitution of ochratoxins A, B, C, metabolites of Aspergillus ochraceus
Wilh.” J. Chem. Soc. 7083-7088.
� Van Der Stegen G., Jorissen U., Pittet A., Saccon M., Steiner W.,
Vincenzi M., Winkler M., Zapp J., Schlatter C., 1997. “Screening of European
coffee final products for occurrence of ochratoxin A (OTA).” Food Addit.
Contam. 14, 211-216.
� Verderio A., 2001. “Linee guida per minimizzare la presenza di
aflatossine nella granella di mais.” In: “Quaderni della ricerca-Rischio di
aflatossine nel latte: linee guida per la produzione e l’acquisto di alimenti
zootecnici.” Regione Lombardia.

145
� Visconti A., Pascale M., Centonze G., 1999. “Determination of
ochratoxin A in wine by means of immunoaffinity column clean-up and
highperformance liquid chromatography.” J. Chromatogr. A 864, 89-101.
� Vukelic M., Sostaric B., Belicza M.,1992. “Pathomorphological of
Balkan endemic nephropathy.” Food Chem. Toxicol. 30, 193-200.
� Wangikar PB., Dwivedi P., Sinha N., 2004a. “Effect in rats of
simultaneus prenatal exposure to ochratoxin A and aflatoxin B1 I. Maternal
toxicity and fetal malformations.” Birth Defects Res B Dev. Reprod Toxicol.
71, 343-351.
� Wangikar PB., Dwivedi P., Sharma AK., Sinha N., 2004b. “Effect in
rats of simultaneous prenatal exposure to ochratoxin A and aflatoxin B1 II.
Histopathological features of teratological anomalies induced in fetuses.”
Birth Defects Res B Dev. Reprod Toxicol. 71, 352-358.
� Wangikar PB., Dwivedi P., Sharma AK., Sinha N., Telang AG., 2005.
“Teratogenic effects in rabbits of simultaneous exposure to ochratoxin A and
aflatoxin B1 with special reference to microscopic effects.” Toxicology 215
(1-2), 37-47.
� Wei YH., Lin TN., Wie RD., 1985. “Effect of ochratoxin A on rat liver
mithocondrial respiration and oxidative phosphorilation.” Toxicology 36,
119-130.
� WHO/FAO, 2001. “Ochratoxin A.” In: “Safety Evaluation of Certain
Mycotoxins in Food.” WHO Food Additives Series. pp 281-415.
� Wolff J., 2000. “Ochratoxin A in cereals and cereal products.” Arch.
Lebensmittelhyg 51, 85-88.

146
� Wurgler FE., Friedrich U., Schlatter J., 1991. “Lack of mutagenicity of
ochratoxin A and B, citrinin, patulin and conestine in Salmonella
typhimurium TA102.” Mutat. Res. 261, 209-216.
� Xiao H., Marquardt RR., Frohlich AA., Ling Y.Z., 1995. “Synthesis and
structural elucidation of analogs of ochratoxin A.” J. Agric. Food Chem. 43,
524-530.
� Xiao H., Madhyastha S., Marquardt RR., Li S., Vodela JK., Frohlich
AA., Kemppainen BW., 1996a. “Toxicity of ochratoxin A, its opened lactone
form and several of its analogs: Structure-activity relationships.” Toxicol.
Appl. Pharmacol. 137, 182-192.
� Xiao H., Marquardt RR., Abramson D., Frohlich AA., 1996b.
“Metabolites of ochratoxins in rat urine and in a culture of Aspergillus
ochraceus.” Appl. Environ. Microbiol. 62, 648-655.
� Yoshizawa T., 1991. “Natural occurrence of mycotoxins in small grain
cereals (wheat, barley, rye, oats, sorghum, millet, rice). In: “Mycotoxins
and animal foods.” pp 301-324. Smith JE., Henderson RS., (Eds). CRC
Press, London, UK.
� Zimmerli B., Dick R., 1995. “Determination of ochratoxin A at the ppt
level in human blood serum, milk and some foodstuffs by hight-
performance liquid chromatography with enhanced fluorescence detection
and immunoaffinity column cleanup: methodology and Swiss data.” J.
Chromatogr. B 666, 85-99.
� Zimmerli B., Dick R., 1996. “Ochratoxin A in table wine and grape-
juice: occurence and risk assessment.” Food Addit. Contam. 13, 655-668.

147
� Zohri AA., Abdel-Gawad K.M., 1993. “Survey of mycoflora and
mycotoxins of some dried fruits in Egipt.” J. Basic. Microbiol. 33, 279-88.

148
E dopo 154 pagine riguardanti sempre e solo l’Ocratossina A, mi sembra
doveroso dedicare questa pagina alle persone che in questo periodo rihanno
aiutato e mi sono state vicine. Innanzitutto ringrazio il Prof. Alessandro
Pistoia per la sua disponibilità e per la possibilità di realizzare questa tesi.
Un ringraziamento speciale va alla Dott.ssa Valentina Meucci per la sua
enorme pazienza (e con me, assicuro che ce n’è voluta) e per la sua
impareggiabile competenza che mi ha permesso di imparare qualcosa da
questa esperienza. Non dimentico di ringraziare anche Elisabbetta e le
bimbe del laboratorio che ho conosciuto perché hanno fatto sì da rendere
più allegre e più spensierate le mie giornate tra le provette. Un
ringraziamento ricco di amore va a Nicola che, a modo suo, tra alti e bassi,
è riuscito a starmi sempre vicino aiutandomi e cercando di capire o miei
momenti di gioia e quelli di sconforto. Infine il ringraziamento più grande va
però ai miei genitori, ultimi nella lista ma primi nella mia vita, perché è solo
grazie a loro se sono riuscita ad arrivare fino alla fine di questo percorso e a
diventare quella che sono.


















