
Un gene , una malattia… - docente.unicas.it · Fosforilasi cinasi (VI A) 80% Fosforilasi (VI...
33
Un gene , una malattia…
Transcript of Un gene , una malattia… - docente.unicas.it · Fosforilasi cinasi (VI A) 80% Fosforilasi (VI...

Un gene , una malattia…

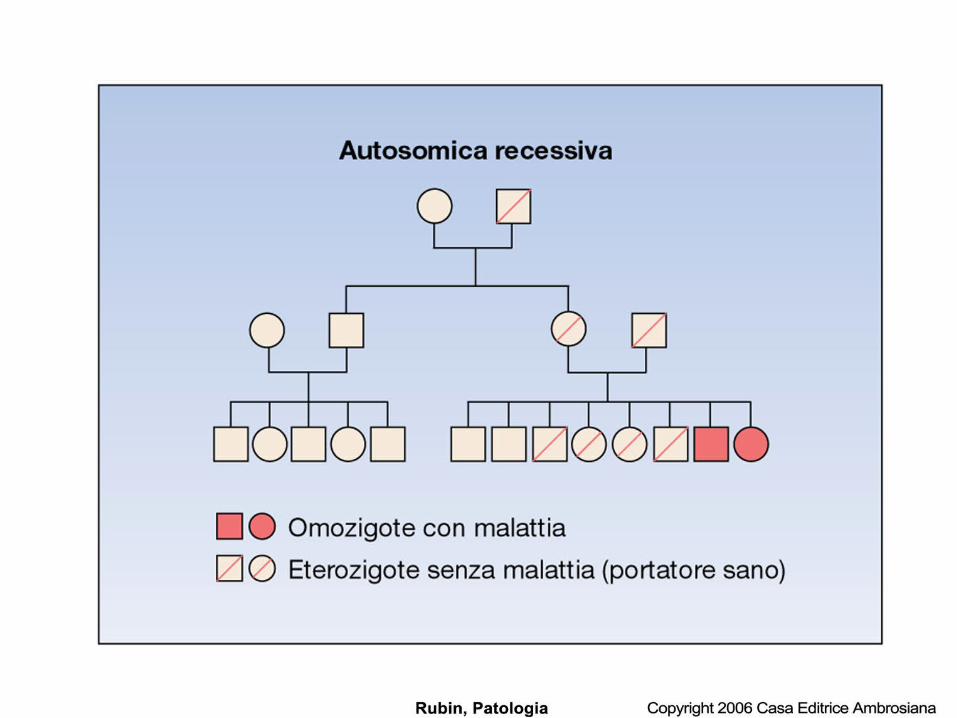
Malattia come errore congenito


Le Glicogenosi
Il glicogeno è il polisaccaride di riserva delle cellule animali (fegato e muscolo!) Polisaccaridi di riserva molto diffusi nel mondo animale/vegetale: mantengono bassa osmolarità nel citosol
α (1 → 4) e α (1 → 6)
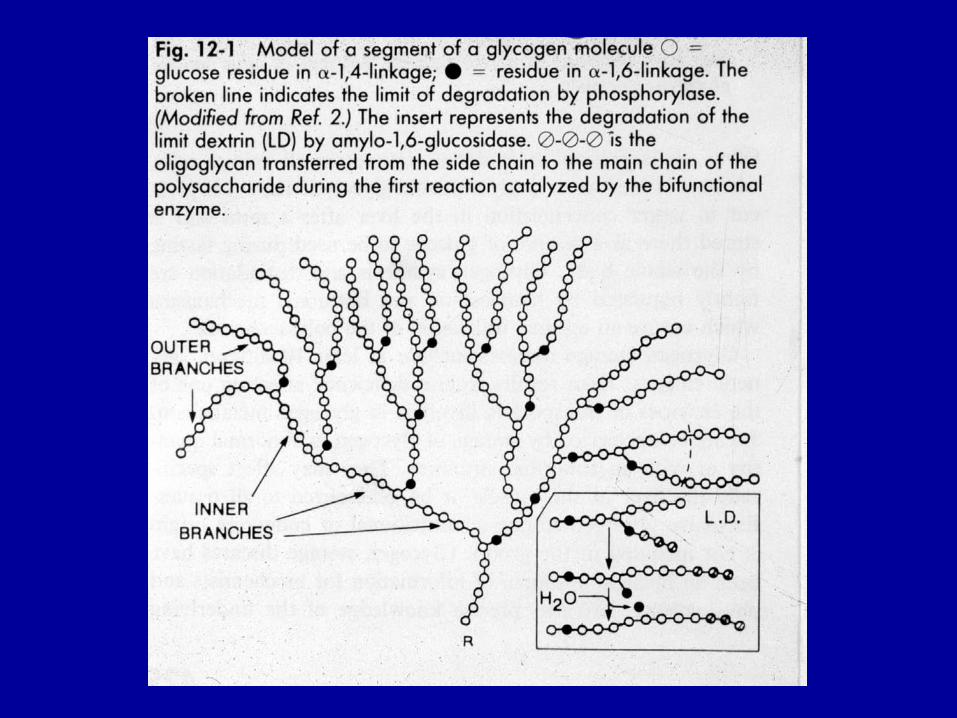
Polimero ramificato di glucoso: + ramif. dell’amilopectina (1 ramif. ogni 8-12 residui)
Glicogeno
β (1 → 4) Polimero lineare di glucoso Cellulosa
α (1 → 4) e α (1 → 6)
Polimero ramificato di glucoso Amilopectina
α (1 → 4) Polimero lineare di glucoso Amiloso
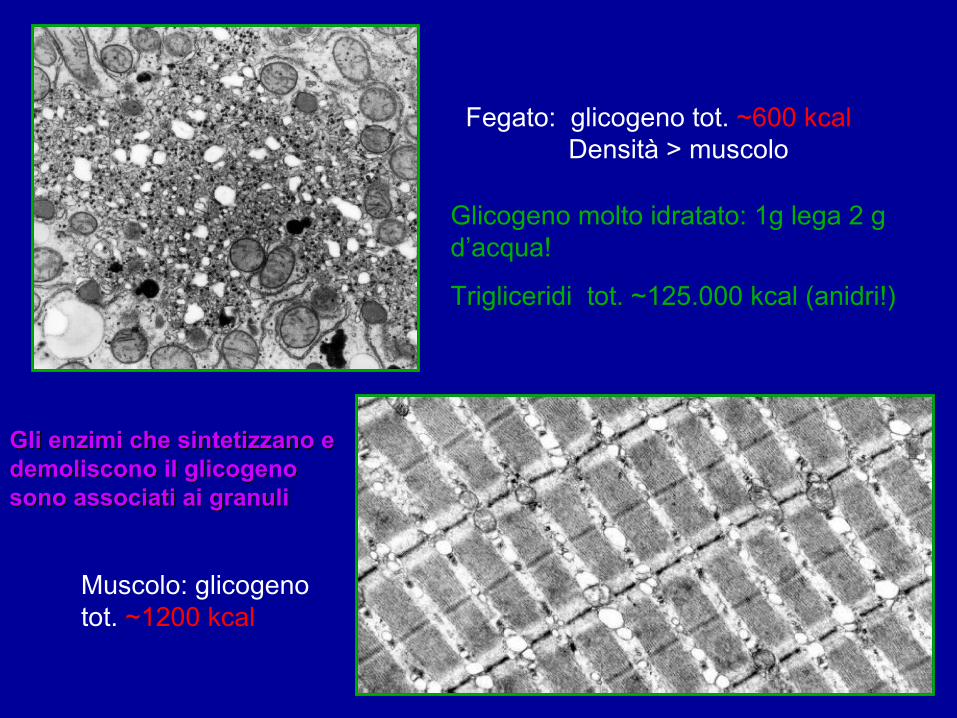
Fegato: glicogeno tot. ~600 kcal Densità > muscolo
Muscolo: glicogeno tot. ~1200 kcal
Glicogeno molto idratato: 1g lega 2 g d’acqua!
Trigliceridi tot. ~125.000 kcal (anidri!)
Gli enzimi che sintetizzano e Gli enzimi che sintetizzano e demoliscono il glicogeno demoliscono il glicogeno sono associati ai granulisono associati ai granuli

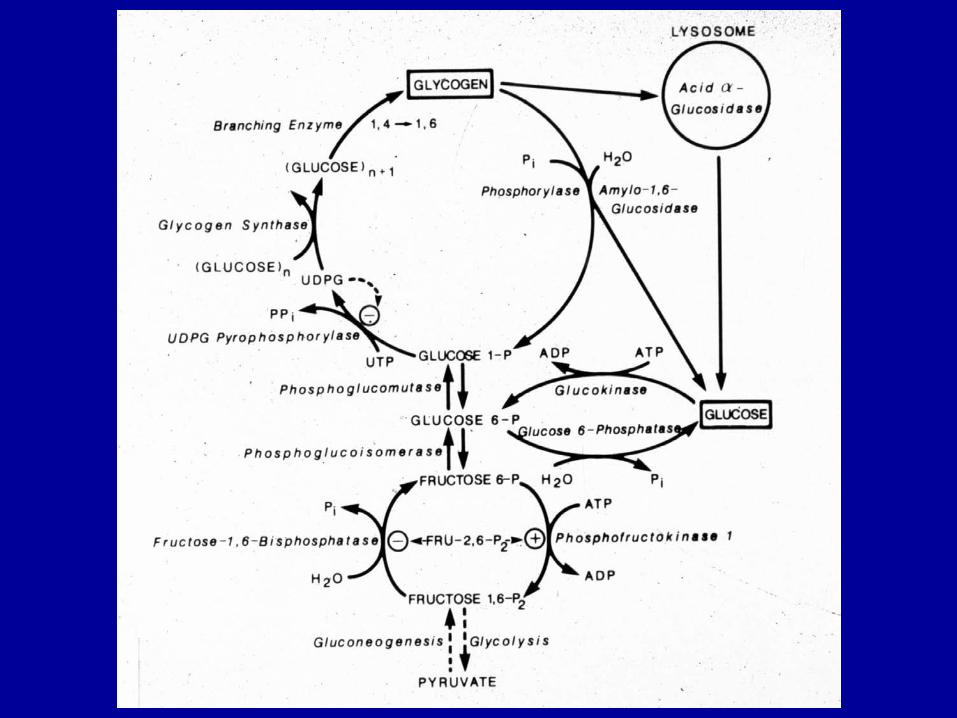
Glicogeno-lisi
Avviene a partire dalle estremità non riducenti.
Glicogenon + Pi ↔ Glicogenon-1 + glucoso 1-P glicogeno fosforilasi (fosforil-transferasi) tanto! poco! Pi/G1P ≥ 100 G1P ↔ G6P fosfoglucomutasi
G6P entra nel reticolo endoplasmatico (traslocasi) e viene defosforilato (solo nel fegato!) G6P + H2O → glucoso + Pi glucoso 6-fosfato fosfatasi (inducibile)
Glucoso rientra nel citosol attraverso trasportatore ad alta capacità ed esce nel sangue.
La glicogeno fosforilasi agisce fino a 4 residui da una ramificazione. Poi interviene enzima de-ramificante: bifunzionale
transferasi (su ultimi 3 residui) idrolasi su legame α (1 → 6)

Glicogeno-sintesi
Glicogenon + UDP-glucoso → Glicogenon+1 + UDP glicogeno sintasi (glucosil transferasi)
Glucosio-1-P + UTP ↔ UDP-glucoso + PPi UDP-glucoso pirofosforilasi
reazione resa irreversibile dall’idrolisi del PPi a 2Pi
Enzima ramificante Trasferisce ultimi 7 residui da una catena su un’altra catena, in posizione α (1 → 6), a distanza di almeno 4 residui da un’altra ramificazione È una transferasi Aumenta i punti di attacco sulla molecola del glicogeno per enzimi che lo sintetizzano e demoliscono → ↑ velocità turnover glicogeno
Glicogenina Proteina di 37 kDa, funziona da primer x la sintesi del glicogeno (-OH di Tyr) Catalizza autoglicosilazione di 8 residui (da UDP-G), poi subentra la glicogeno sintasi

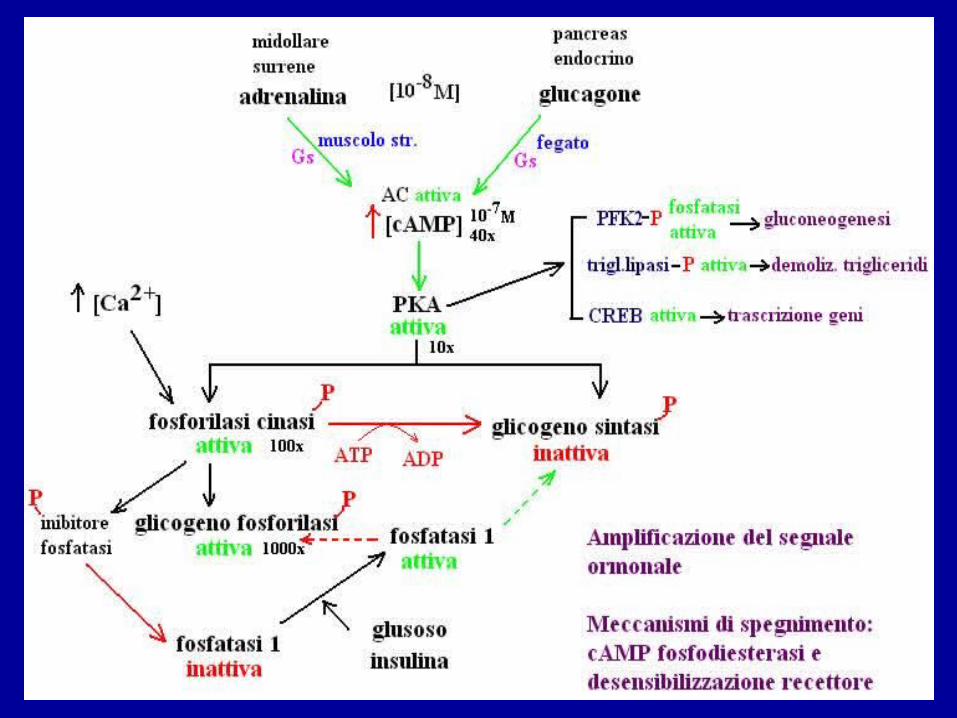
Regolazione del “ciclo del glicogeno” (sintesi-demolizione)
GLUCAGONEGLUCAGONEINSULINAINSULINA
G6PG6PGLUCAGONEGLUCAGONEINSULINA INSULINA
GLUCOSOGLUCOSOFegato
ADRENALINAADRENALINA G6PG6PADRENALINAADRENALINA AMPAMPMuscolo
Covalente Allosterica Covalente Allosterica
Glicogeno sintasiGlicogeno sintasi Glicogeno fosforilasiGlicogeno fosforilasi
Solo nel fegato la [glucoso] intracellulare rispecchia la [glucoso] sangue perché il fegato ha un trasportatore del glucoso ad alta capacità e insulino-indipendente (Glut-2)

Le glicogenosi
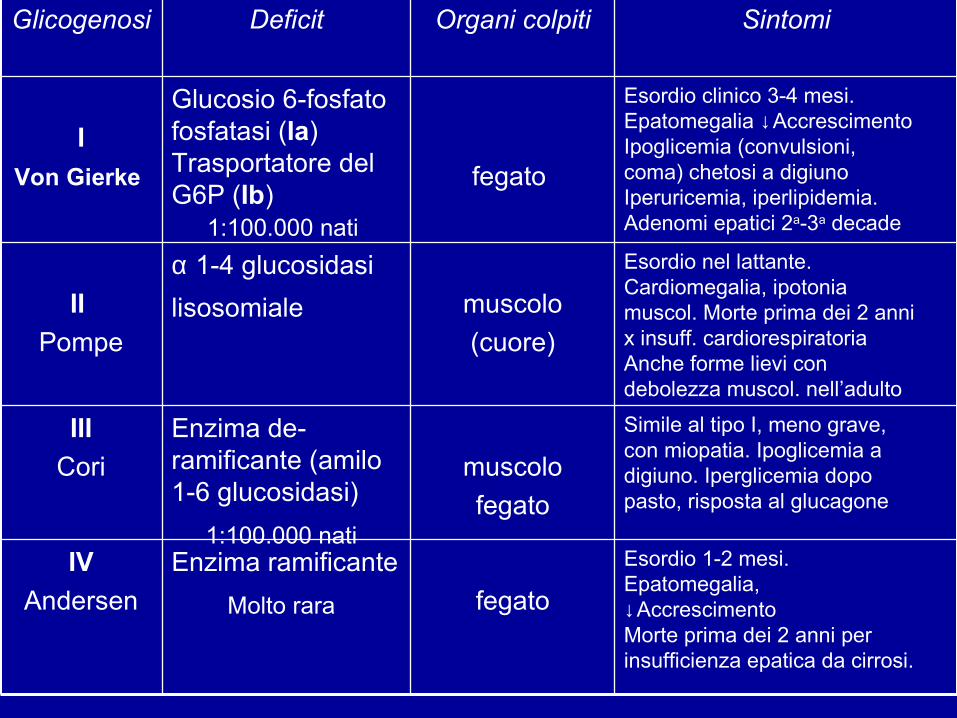
malattie congenite causate da difetti del metabolismo del glicogeno 8 forme (epatiche e muscolari) carattere autosomico recessivo incidenza complessiva 1:20-25.000 nati vivi tipi I-II-III-IX + frequenti
Esordio 1-2 mesi.Epatomegalia,↓AccrescimentoMorte prima dei 2 anni perinsufficienza epatica da cirrosi.
fegatoEnzima ramificante
Molto rara IV
Andersen
Simile al tipo I, meno grave,con miopatia. Ipoglicemia adigiuno. Iperglicemia dopopasto, risposta al glucagone
muscolofegato
Enzima de-ramificante (amilo 1-6 glucosidasi)
1:100.000 nati
IIICori
Esordio nel lattante.Cardiomegalia, ipotoniamuscol. Morte prima dei 2 annix insuff. cardiorespiratoriaAnche forme lievi condebolezza muscol. nell’adulto
muscolo(cuore)
α 1-4 glucosidasi lisosomiale II
Pompe
Esordio clinico 3-4 mesi.Epatomegalia ↓AccrescimentoIpoglicemia (convulsioni,coma) chetosi a digiuno Iperuricemia, iperlipidemia.Adenomi epatici 2a-3a decade
fegato
Glucosio 6-fosfato fosfatasi (Ia)Trasportatore del G6P (Ib)
1:100.000 nati
IVon Gierke
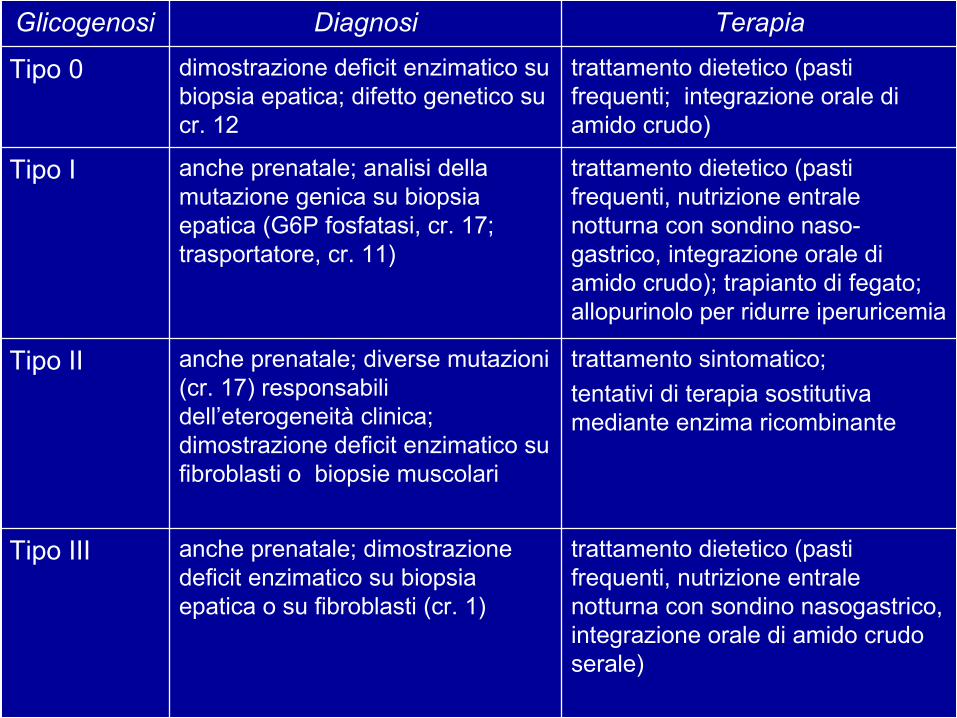
SintomiOrgani colpitiDeficitGlicogenosi
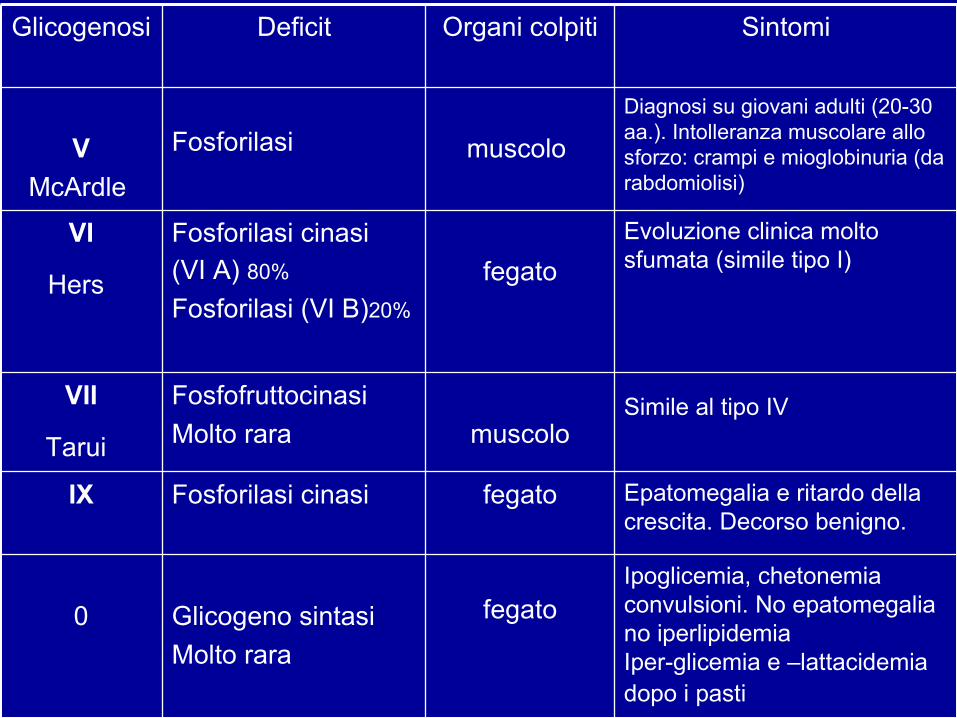
Epatomegalia e ritardo dellacrescita. Decorso benigno.
fegatoFosforilasi cinasiIX
Ipoglicemia, chetonemiaconvulsioni. No epatomegaliano iperlipidemiaIper-glicemia e –lattacidemiadopo i pasti
fegatoGlicogeno sintasiMolto rara
0
Simile al tipo IV muscolo
FosfofruttocinasiMolto rara
VII
Tarui
Evoluzione clinica moltosfumata (simile tipo I) fegato
Fosforilasi cinasi(VI A) 80%
Fosforilasi (VI B)20%
VI
Hers
Diagnosi su giovani adulti (20-30aa.). Intolleranza muscolare allosforzo: crampi e mioglobinuria (darabdomiolisi)
muscolo Fosforilasi VMcArdle
SintomiOrgani colpitiDeficitGlicogenosi
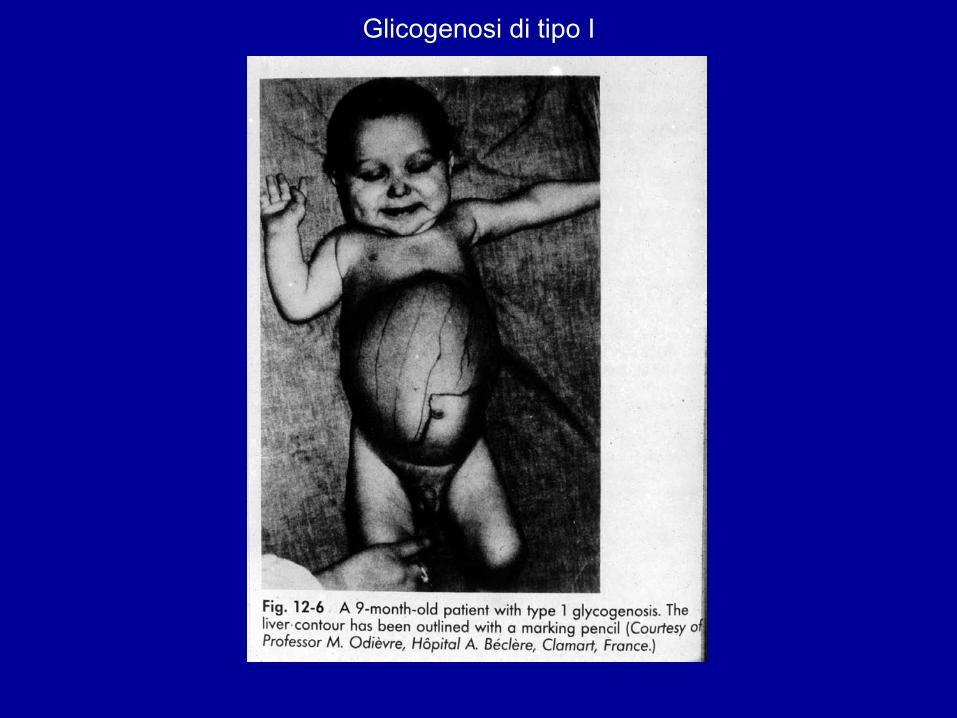
Glicogenosi di tipo I
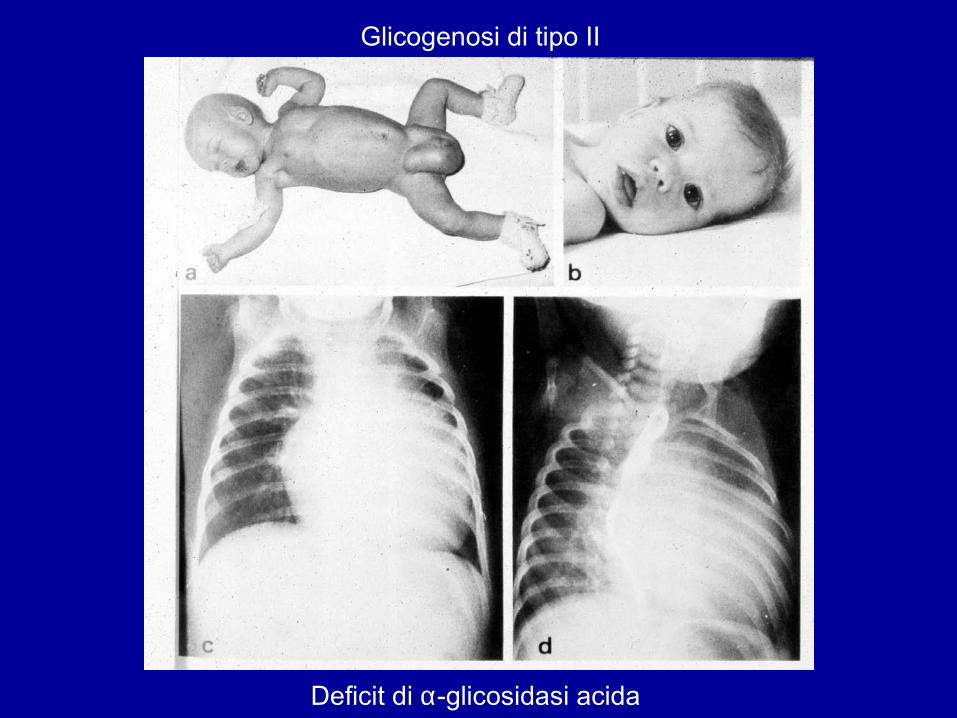
Glicogenosi di tipo II
Deficit di α-glicosidasi acida
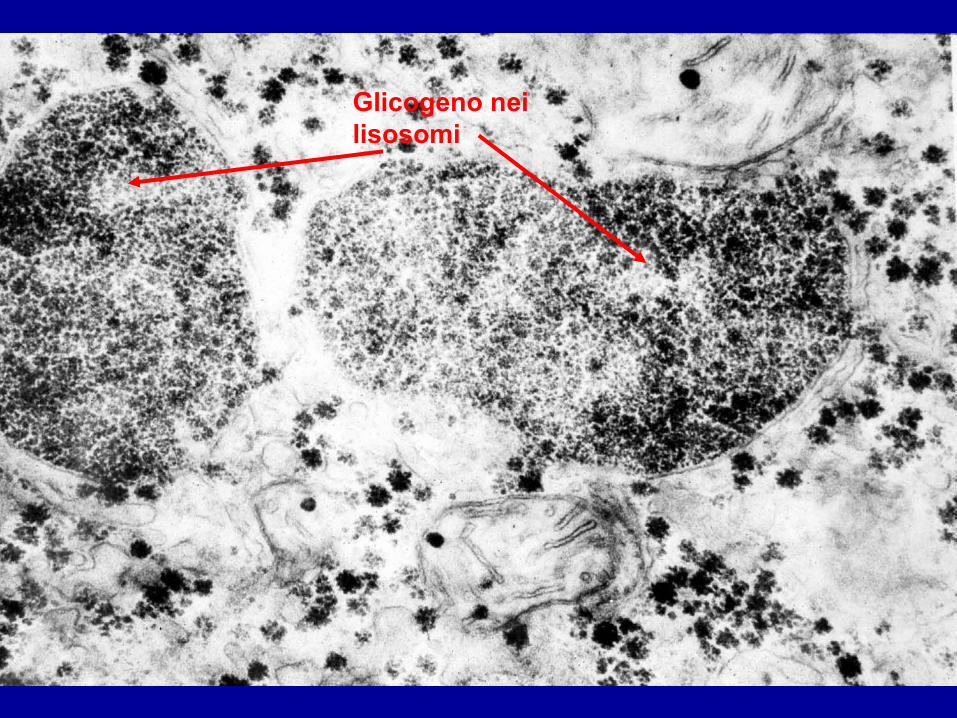
Glicogeno nei lisosomi
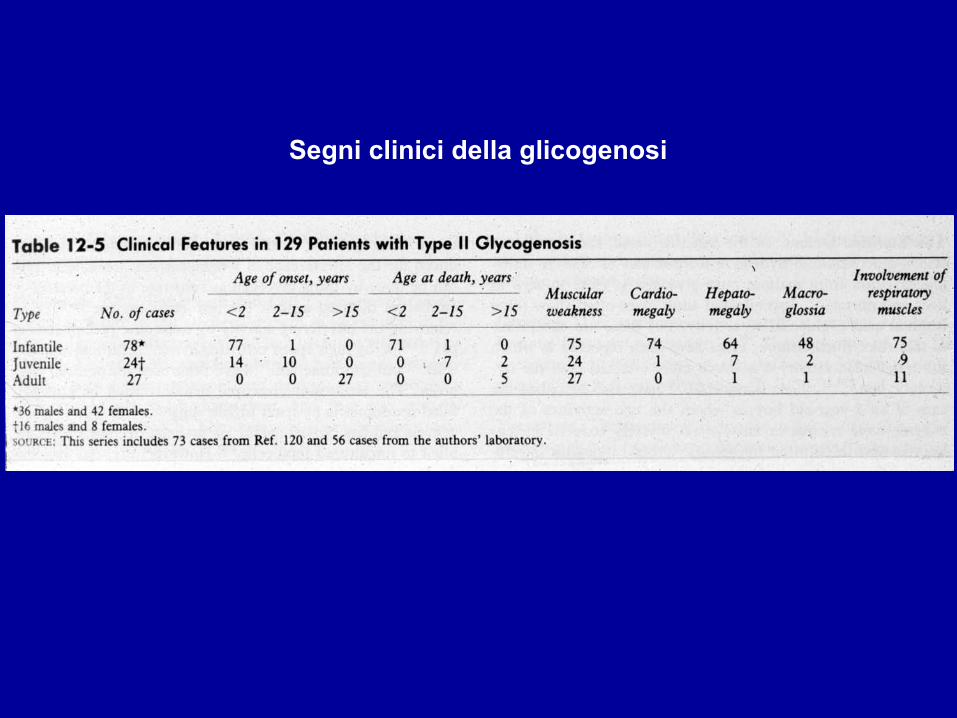
Segni clinici della glicogenosi

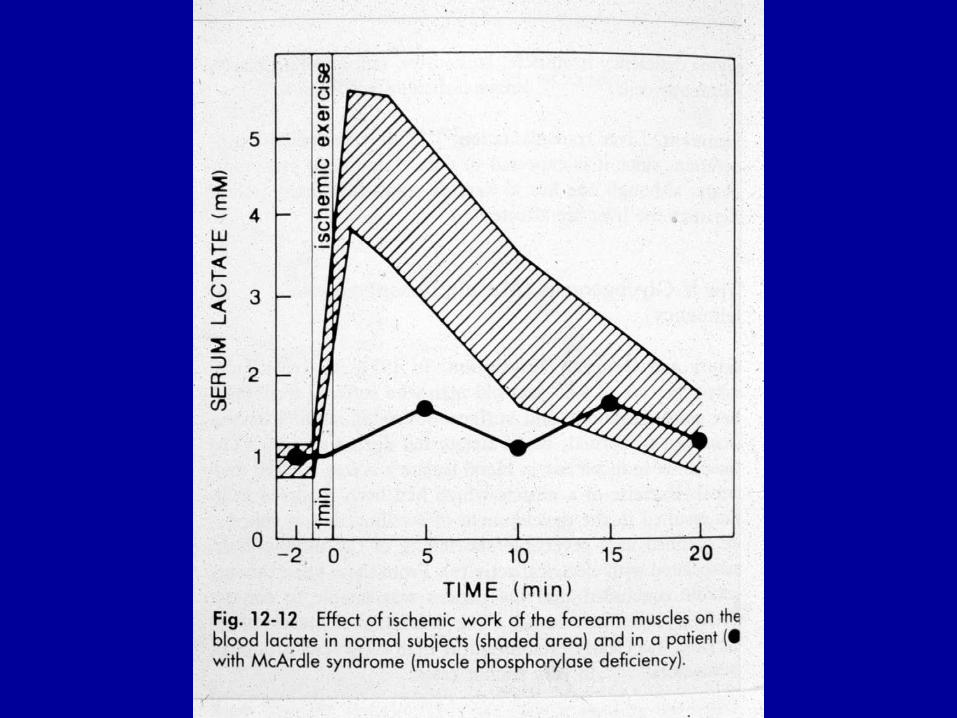
trattamento dietetico (pasti frequenti, nutrizione entrale notturna con sondino nasogastrico, integrazione orale di amido crudo serale)
anche prenatale; dimostrazione deficit enzimatico su biopsia epatica o su fibroblasti (cr. 1)
Tipo III
trattamento sintomatico; tentativi di terapia sostitutiva mediante enzima ricombinante
anche prenatale; diverse mutazioni (cr. 17) responsabili dell’eterogeneità clinica; dimostrazione deficit enzimatico su fibroblasti o biopsie muscolari
Tipo II
trattamento dietetico (pasti frequenti, nutrizione entrale notturna con sondino naso-gastrico, integrazione orale di amido crudo); trapianto di fegato; allopurinolo per ridurre iperuricemia
anche prenatale; analisi della mutazione genica su biopsia epatica (G6P fosfatasi, cr. 17; trasportatore, cr. 11)
Tipo I
trattamento dietetico (pasti frequenti; integrazione orale di amido crudo)
dimostrazione deficit enzimatico su biopsia epatica; difetto genetico su cr. 12
Tipo 0
TerapiaDiagnosiGlicogenosi
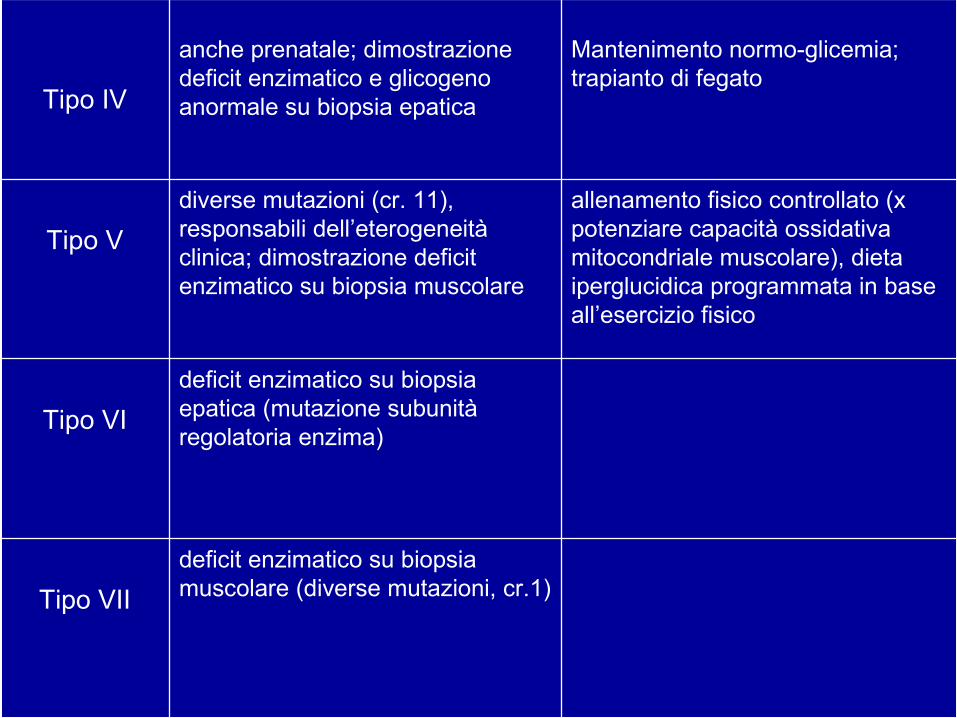
deficit enzimatico su biopsia muscolare (diverse mutazioni, cr.1)Tipo VII
deficit enzimatico su biopsia epatica (mutazione subunità regolatoria enzima)
Tipo VI
allenamento fisico controllato (x potenziare capacità ossidativa mitocondriale muscolare), dieta iperglucidica programmata in base all’esercizio fisico
diverse mutazioni (cr. 11), responsabili dell’eterogeneità clinica; dimostrazione deficit enzimatico su biopsia muscolare
Tipo V
Mantenimento normo-glicemia; trapianto di fegato
anche prenatale; dimostrazione deficit enzimatico e glicogeno anormale su biopsia epatica Tipo IV

Fruttosuria essenziale: deficit di fruttocinasi epatica.
Non avviene la reazione di fosforilazione del fruttoso nel fegato:
fruttoso + ATP → fruttoso 1-fosfato + ADP
Il fruttoso non viene trattenuto dentro le cellule epatiche e rimane nel sangue → nell’urina. Asintomatica.
Malattie genetiche da dismetabolismo dei disaccaridi
Intolleranza congenita al fruttoso: deficit di fruttoso 1-fosfato aldolasi epatica.
E’ l’enzima che scinde il F1P in gliceraldeide e diidrossiacetone-fosfato e consente l’ingresso del fruttoso 1-P nella glicolisi:
fruttoso-1P → gliceraldeide + diidrossiacetone-fosfato
Il fruttoso-1P si accumula nell’epatocita → ipoglicemia e danno epato-cellulare!
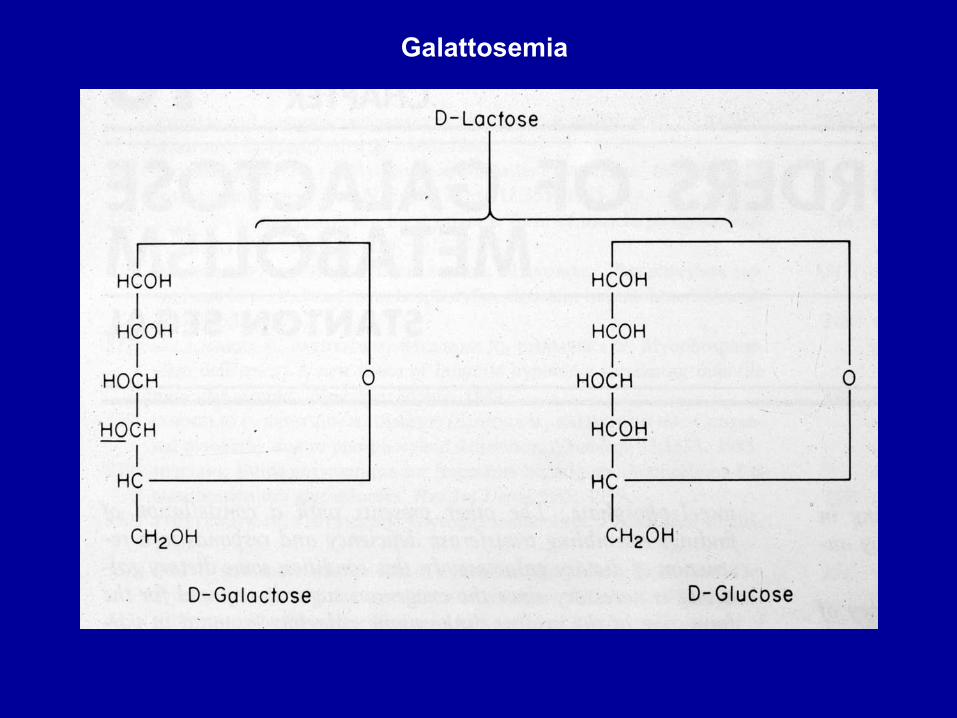
Galattosemia

Metabolismo del galattosio

Galattosemia: deficit di galattoso-1P uridil transferasi
Non avviene la reazione di trasferimento del gal-1P al posto dell’unità di gluc-1P sulla molecola dell’UDP-glucoso, che consente la trasformazione del gal-1P in gluc-1P: manca l’enzima gal-1P uridil transferasi.
Il galattoso-1P si accumula nelle cellule e causa danno cellulare (epatico e neuronale): deficit mentale permanente, se il galattoso non viene rimosso presto dalla dieta del lattante!
Screening diagnostico (elevata galattosemia e galattosuria) si basa sulla capacità del galattoso (riducente!) di ridurre ioni metallici (Cu2+) e indurne viraggio del colore.
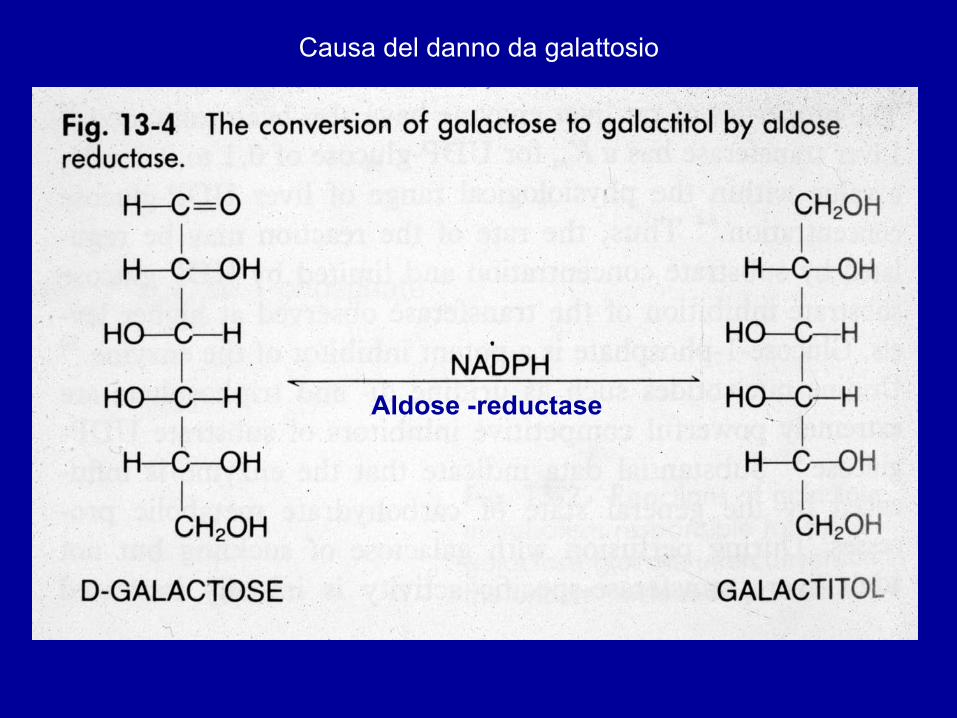
Aldose -reductase
Causa del danno da galattosio

Segni clinici della galattosemia
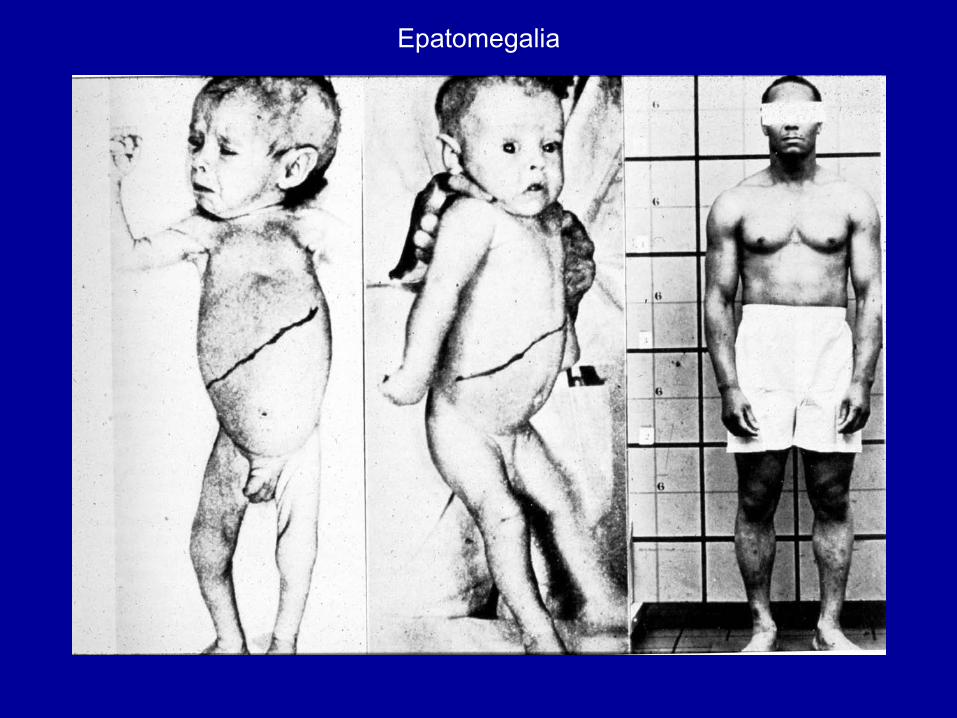
Epatomegalia
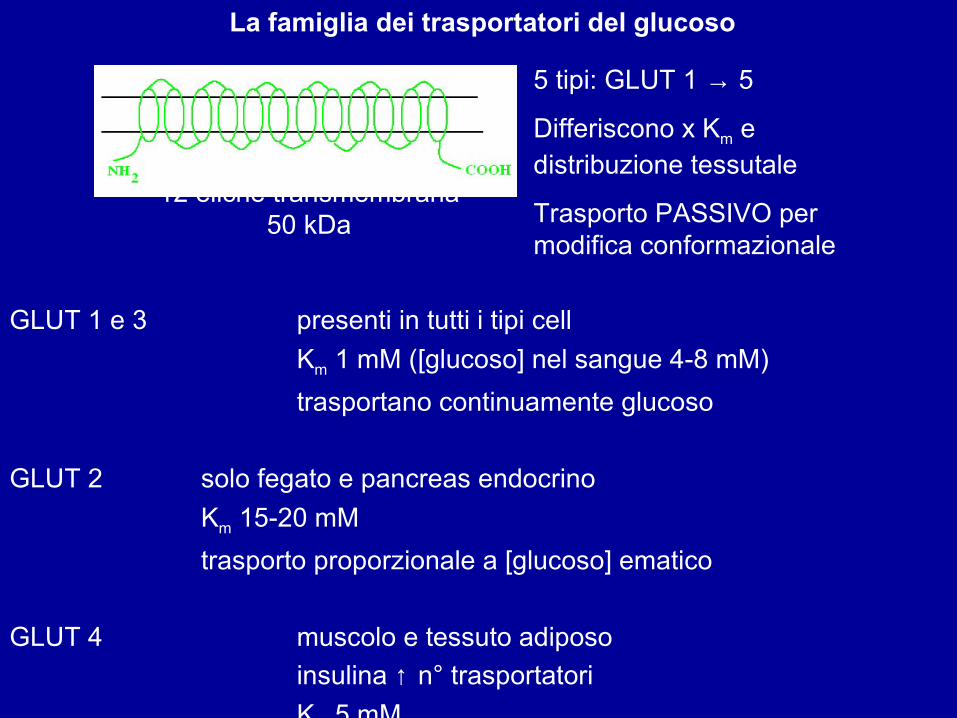
GLUT 1 e 3 presenti in tutti i tipi cellKm 1 mM ([glucoso] nel sangue 4-8 mM)trasportano continuamente glucoso
GLUT 2 solo fegato e pancreas endocrinoKm 15-20 mMtrasporto proporzionale a [glucoso] ematico
GLUT 4 muscolo e tessuto adiposoinsulina ↑ n° trasportatoriKm 5 mM
La famiglia dei trasportatori del glucoso
5 tipi: GLUT 1 → 5
Differiscono x Km e distribuzione tessutale
Trasporto PASSIVO per modifica conformazionale
12 eliche transmembrana50 kDa
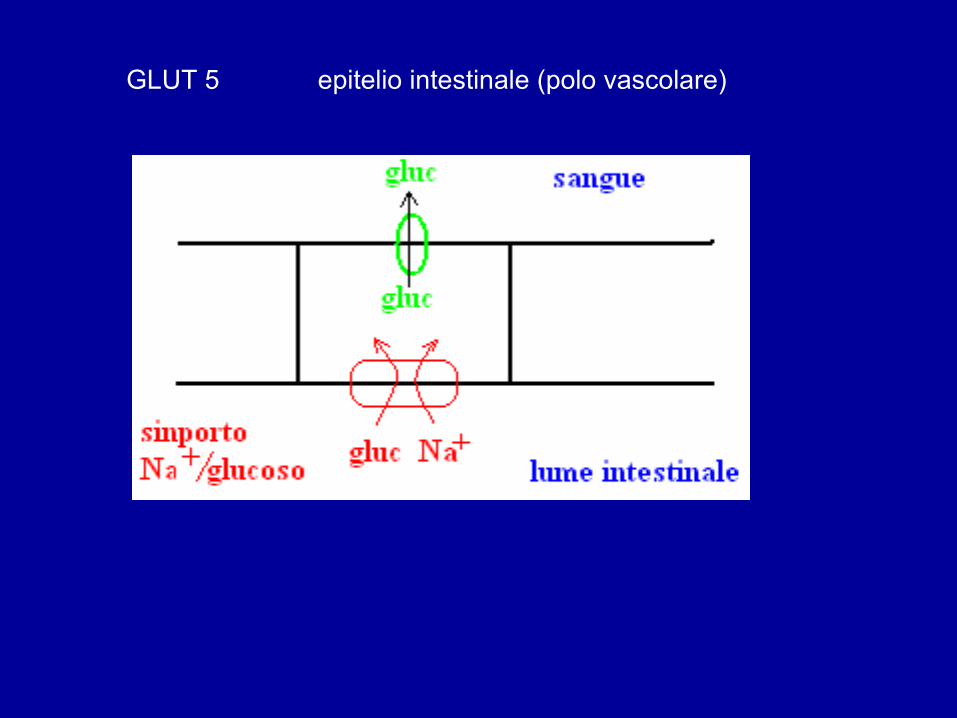
GLUT 5 epitelio intestinale (polo vascolare)

CFTR = Cystic fibrosis transmembrane conductance regulator











![· cantaluppi oreste colombo stefano botta christian grassi andrea mai-darell] dario lijppi massimo redaecli gianluca cat. vi vi vi vi vi vi vi vi vi vi vi vi vi vi vi vi vi vi vi](https://static.fdocumenti.com/doc/165x107/5c69360d09d3f242168cb2b8/-cantaluppi-oreste-colombo-stefano-botta-christian-grassi-andrea-mai-darell.jpg)





