POLITECNICO DI MILANO - politesi.polimi.it · La maggior parte delle proprietà meccaniche del...
107
1 POLITECNICO DI MILANO Scuola di Ingegneria dei Sistemi Corso di Laurea Magistrale in Ingegneria Biomedica STUDIO DELLA RISPOSTA NANOMECCANICA DEI GLICOSAMMINOGLICANI DELLA CARTILAGINE MEDIANTE MODELLI MOLECOLARI relatore: Ing. Simone Vesentini correlatore: Ing. Alfonso Gautieri Tesi di Laurea di: Andrea MEZZANZANICA Matr. 765835 Anno Accademico 2011 - 2012
Transcript of POLITECNICO DI MILANO - politesi.polimi.it · La maggior parte delle proprietà meccaniche del...

1
POLITECNICO DI MILANO
Scuola di Ingegneria dei Sistemi
Corso di Laurea Magistrale in
Ingegneria Biomedica
STUDIO DELLA RISPOSTA NANOMECCANICA DEI GLICOSAMMINOGLICANI
DELLA CARTILAGINE MEDIANTE MODELLI MOLECOLARI
relatore: Ing. Simone Vesentini
correlatore: Ing. Alfonso Gautieri
Tesi di Laurea di:
Andrea MEZZANZANICA Matr. 765835
Anno Accademico 2011 - 2012

2
A tutti quelli che mi vogliono bene

3
INDICE
pagina
INDICE DELLE FIGURE .............................................................................................................. 5
INDICE DELLE TABELLE ........................................................................................................... 6
Abstract .......................................................................................................................................... 7
Sommario ....................................................................................................................................... 8
Summary ...................................................................................................................................... 16
Introduzione ................................................................................................................................. 23
1.1 La Cartilagine ................................................................................................................. 23
1.1.1 Cartilagine ialina ................................................................................................. 23
1.1.2 Cartilagine fibrosa .............................................................................................. 24 1.1.3 Cartilagine elastica ............................................................................................. 24
1.2 Struttura molecolare della cartilagine articolare ....................................................... 24 1.2.1 La matrice extra cellulare della cartilagine articolare .................................... 26 1.2.2 .Il collagene ......................................................................................................... 27
1.2.3 I Proteoglicani ..................................................................................................... 27 1.2.4 L’aggrecano ......................................................................................................... 28
1.2.5 Lo Ialuronano ...................................................................................................... 29 1.2.6 I Glicosamminoglicani ........................................................................................ 29
1.2.7 L’acido ialuronico ................................................................................................ 29 1.2.8 GAG solfatati ....................................................................................................... 30
1.2.9 Il Condroitin Solfato ............................................................................................ 30
1.2.10 Altre molecole ................................................................................................... 32 1.3 Funzioni e proprietà della cartilagine articolare ....................................................... 32
1.4 Calcolo proprietà meccaniche ..................................................................................... 33 1.4.1 Pressione Osmotica ........................................................................................... 34
1.4.2 Lunghezza persistente....................................................................................... 36 1.4.3 Esperimenti di nanoindentazione ..................................................................... 38
1.5 Obiettivo.......................................................................................................................... 42
Materiali e metodi ........................................................................................................................ 43
2.1 Dinamiche molecolari ................................................................................................... 43
2.2 Funzione Energia potenziale ....................................................................................... 44 2.3. Termini di legame ........................................................................................................ 44
2.3.1 Bond ..................................................................................................................... 45 2.3.2 Angle o bending .................................................................................................. 46
2.3.3 Angoli diedrici ...................................................................................................... 46
2.4 Termini di non legame .................................................................................................. 47 2.4.1 Van der Waals..................................................................................................... 47
2.4.2 Legame a idrogeno ............................................................................................ 48 2.4.3 Energia elettrostatica ......................................................................................... 49

4
2.5 Algoritmo globale .......................................................................................................... 50
2.6 Cutoff ............................................................................................................................... 51
2.7 Solvente .......................................................................................................................... 52 2.7.1 Condizioni periodiche al contorno .................................................................... 52
2.8 Minimizzazione .............................................................................................................. 53
2.9 La simulazione si basa sulla teoria della Meccanica Classica .............................. 54 2.10 Ruolo degli elettroni .................................................................................................... 54
2.11 Software utilizzato ....................................................................................................... 55 2.12 Costruzione modello ................................................................................................... 55
2.12.1 Gags Builder ..................................................................................................... 56 2.13 Dinamica in vuoto ....................................................................................................... 57 2.14 Costruzione del box d'acqua ..................................................................................... 57
2.15 Ionizzazione del box ................................................................................................... 58 2.16 Calcolo proprietà meccaniche .................................................................................. 58
2.16.1 Pressione Osmotica ......................................................................................... 58 2.16.2 Lunghezza persistente .................................................................................... 60
2.16.3 Esperimenti di resistenza alla compressione .............................................. 62
Ristultati e Discussione .............................................................................................................. 66
3.1 Pressione Osmotica ..................................................................................................... 66
3.2 Lunghezza Persistente ................................................................................................. 67 3.2.1 Ottimizzazione della durata della dinamica .................................................... 67
3.2.2 Ottimizzazione della lunghezza della catena ................................................. 68 3.2.3 Kinking .................................................................................................................. 69
3.2.4 Conformazione a elica ....................................................................................... 70 3.2.5 Esperimenti .......................................................................................................... 70
3.2.6 Confronto tra diverse percentuali di solfatazione in catene isolate ............ 71
3.2.6.1 Molarità bassa ................................................................................................. 71
3.2.7 Confronto tra diverse percentuali di solfatazione in set di catene .............. 73
3.3 Esperimenti di resistenza alla compressione ........................................................... 74 3.3.1 Catene distanziate 1.5 nm ................................................................................ 74
3.3.1 Catene distanziate 3 nm .................................................................................... 74
Conclusioni e Sviluppi futuri ...................................................................................................... 76
Bibliografia ................................................................................................................................... 78
Appendice A - script di Gagsbuilder ........................................................................................ 83
Appendice B - topology del condroitin solfato ........................................................................ 93
Appendice C - script utilizzato per il calcolo della lunghezza persistente........................ 104

5
INDICE DELLE FIGURE
Figura 1 Illustrazione schematica dei costituenti molecolare della cartilagine e loro disposizione in aggregati multimolecolari. .......................................................... 26
Figura 2 Struttura dell’aggrecano ............................................................................................. 28
Figura 3 Struttura dele Keratan solfato e del Condroitin Solfato ......................................... 32
Figura 4 Pressione Osmotica: Confronto tra studio sperimentale e computazionale ............................................................................................................... 35
Figura 5 Morfologia di un filamento di actina in diversi istanti temporali ............................ 38
Figura 6 Setup sperimentale e grafico che rappresenta la relazione Forza-distanza ............................................................................................................................ 41
Figura 7 Confronto tra metodo armonico e metodo Morse .................................................. 45
Figura 8 Andamento del potenziale nell’agolo diedrico ........................................................ 46
Figura 9 Andamento del potenziale di vdW al variare della distanza di due atomi ................................................................................................................................. 47
Figura 10 Schema riassuntivo di una simulazione di dinamica molecolare ...................... 50
Figura 11 Confronto tra i diversi metodi di cut-off .................................................................. 51
Figura 12 Condizioni periodiche al contorno bidimensionali................................................ 53
Figura 13 Struttura dello script GagsBuilder .......................................................................... 56
Figura 14 Catena di 5 dimeri di CS “dritta” ............................................................................. 57
Figura 15 Catena di CS in box d’acqua e con concentrazione ionica pari a 0,15 M ............................................................................................................................... 58
Figura 16 Simulazione dell’aumento di concentrazione di catene di CS mediante “membrane semipermeabili” virtuali ........................................................... 59
Figura 17 Modello di C4S con in evidenza gli atomi di Ossigeno ....................................... 60
Figura 18 Atomi di Osssigeno in diversi istanti temporali ..................................................... 61
Figura 19 Test dello script con una molecola di collagene .................................................. 61
Figura 20 Vista dall’alto della piastra d’oro con le catene di CS ......................................... 62
Figura 21 Vista prospettica della piastra d’oro con le catene di CS ................................... 63

6
Figura 22 Densità dell’acqua nei sistemi ................................................................................ 64
Figura 23 Visione del sistema con la tip virtuale all’inizio dell’esperimento ...................... 65
Figura 24 Variazione della pressione osmotica ..................................................................... 67
Figura 25 Variazione della lunghezza persistente al variare del tempo ............................ 67
Figura 26 Variazione della lunghezza persistente al variare del numero di dimeri ................................................................................................................................ 68
Figura 27 Esempio di kinking .................................................................................................... 69
Figura 28 Variazione della lunghezza persistente con concentrazione ionica pari a 0,05 M .................................................................................................................... 72
Figura 29 Variazione della lunghezza persistente con concentrazione ionica pari a 0,15 M .................................................................................................................... 73
Figura 30 Forze agenti sulla tip. ............................................................................................... 75
INDICE DELLE TABELLE
Tabella 1 Valori di pressione osmotica al variare della concentrazione di CS ................. 35
Tabella 2 Risultati degli studi sulla lunghezza persistente del CS in diverse solfatazioni ....................................................................................................................... 37
Tabella 3 Confronto della pressione osmotica tra diversi sistemi in funzione della concentrazione ...................................................................................................... 66
Tabella 4 Esperimenti effettuati per il calcolo della lunghezza persistente ....................... 70
Tabella 5 Valori di lunghezza persistente ............................................................................... 71
Tabella 6 Lunghezza persistente di un set di catene distanziate reciprocamente 1,5 e 3 nm ............................................................................................ 73
Tabella 7 Esperimenti di resistenza a compressione di catene distanziate reciprocamente 1,5 e 3 nm ............................................................................................ 74

7
ABSTRACT
La maggior parte delle proprietà meccaniche del tessuto cartilagineo sono dovute alle
interazioni molecolari della sua matrice extracellulare con una fase fluida formata da
acqua e ioni. In questo studio ci si è focalizzati su un particolare costituente di questa
matrice, un proteoglicano (PG) denominato aggrecano. Questa molecola è composta da
una struttura di natura proteica composta da tre domini globulari e due domini dove, grazie
a proteine linker vengono innestati dei glicosamminoglicani (GAG), in particolare il
condroitin solfato (CS) e il keratan solfato (KS). È stato caratterizzato il comportamento
biomeccanico del CS, poiché esso contribuisce in maniera rilevante alla risposta a stimoli
meccanici di compressione dell’aggrecano, e quindi del tessuto cartilagineo. Sono state
misurate tre proprietà meccaniche (pressione osmotica, lunghezza persistente, resistenza a
compressione) e per poter ottenere uno studio preciso e completo sono stati utilizzati gli
strumenti di modellistica computazionale, senza i quali non sarebbe stato possibile ottenere
risultati dipendenti unicamente da una variabile. Infatti il comportamento di questa
molecola dipende da svariati parametri, e solamente attraverso uno studio computazionale
con un grado di dettaglio atomico si possono capire i meccanismi che stanno alla base del
tessuto cartilagineo. I risultati ottenuti hanno permesso comprendere meglio il
comportamento molecolare della catena di CS. Questo studio potrà servire alla
progettazione di molecole artificiali che possano mimare il comportamento dell’aggrecano.
Queste molecole potranno essere inserite in scaffolds in modo da aiutare l’ingegneria dei
tessuti nello sviluppo di tessuto cartilagineo partendo unicamente da una biopsia di
condrociti, operazione ad oggi molto faticosa.

8
SOMMARIO
INTRODUZIONE
La cartilagine è un tessuto
avascolarizzato e idratato che svolge
diverse funzioni; dal punto di vista
istologico viene divisa in tre tipi: la
cartilagine ialina, la quale è molto
elastica ed ha un’alta resistenza a
compressione; la cartilagine fibrosa,
più rigida e la cartilagine elastica,
molto flessibile. Questo studio si
concentrerà sulla cartilagine presente
nelle giunzioni articolari, chiamata
cartilagine articolare, la quale è
composta principalmente da
cartilagine ialina. Nonostante la
cartilagine si presenti e sia stata
studiata come se fosse un materiale
continuo omogeneo, è composta
principalmente da tre fasi che
svolgono funzioni differenti: una fase
è la componente cellulare, la quale è
responsabile della produzione e/o del
rimodellamento della seconda fase, la
matrice extracellulare (ECM). A
queste due fasi solide si aggiunge una
fase liquida, costituita dall’acqua con
elettroliti in soluzione. Dal punto di
vista meccanico, un ruolo
fondamentale nella risposta a stimoli
di natura fisica è dovuto
all’interazione della fase fluida con
l’ECM. L’ECM è composta da un
intricato groviglio di molecole. É
presente una rete formata da fibrille di
collagene, (in particolare il collagene
di tipo II) la quale sostiene la
cartilagine per quanto riguarda la
risposta a stimoli di compressione.
Un’ altra tipica molecola della
cartilagine è lo ialuronano, aggregato
molecolare formato dall’acido
ialuronico, un glicosamminoglicano
(GAG) ad altissimo peso molecolare,
coniugato grazie a proteine linker con
svariate molecole di aggrecano;
l’aggrecano è un proteoglicano (PG),
cioè una biomolecola formata da un
core proteico che presenta tre domini
globulari e due domini nei quali
vengono innestati dei GAG; nel caso
specifico questi GAG sono il keratan
solfato (KS) e il condroitin solfato
(CS). L’aggrecano viene considerato
come la molecola che in percentuale
maggiore contribuisce alla resistenza a
stimoli di compressone. Questo è
dovuto al fatto che le catene dei GAG
presentano cariche negative, le quali
se vengono avvicinate a causa di uno
sforzo di compressione, si oppongono

9
a questo movimento generando una
forza che tende a ristabilire la
conformazione originale. Per
conoscere e studiare al meglio il
comportamento di questa molecola, si
è scelto di focalizzarsi su una parte di
esso, cioè la zona in cui sono presenti
le catene di CS. Il CS è un GAG
composto dalla coniugazione di due
zuccheri,l’N-acetil-D- galattosammina
(GalNAc) e l’ acido D – glucuronico
(GlcUA). Questi due zuccheri
presentano già una carica negativa, ma
oltre a questa è stato riscontrato che
nel tessuto cartilagineo è presente una
modifica post-produzionale che solfata
lo zucchero GalNAc in posizione 4 o
in posizione 6. Per comprendere
ancora di più il comportamento della
cartilagine a stimoli di compressione e
potere in un futuro riprodurre alcune
caratteristiche di questo tessuto in un
materiale bioartificiale, è necessario
concentrarsi sulle proprietà
nanomeccaniche dei singoli costituenti
della ECM, poiché ormai è chiaro che
il comportamento macroscopico di
questo tessuto deriva dalla struttura
nanoscopica delle molecole che lo
compongono. In questo studio si è
voluta caratterizzare la risposta
nanomeccanica del CS attraverso
esperimenti virtuali in silico. È stato
scelto questo approccio perché la
modellistica computazionale permette
di calcolare le proprietà meccaniche al
variare di uno solo dei diversi
parametri, in modo da capire quanto
questa variabile condizioni la risposta
meccanica a definiti stimoli. Inoltre si
è scelto un modello full-atom, cioè un
modello con dettaglio atomistico che
possiede il maggior livello di dettaglio
che si possa avere se si considera una
simulazione che si basa sui principi
della meccanica classica.
MATERIALI E METODI
É stato utilizzato un modello
molecolare esistente, scritto da Cilpa
[1] e perfezionato da Marascio [2]. Per
generare le catene è stato utilizzato
uno script di Linux che permette di
costruire catene di diversa lunghezza e
solfatazione. Per la visualizzazione e
l’elaborazione dei vari esperimenti ci
si è affidati a programmi largamente
utilizzati e apprezzati negli studi di
modellistica molecolare come VMD,
NAMD e ACEMD. È importante
permettere alla catena di CS di
disporsi in una configurazione che
minimizzi il valore di energia interna ;
quindi tutte le dinamiche sono state
svolte per un tempo abbastanza lungo

10
da poter permettere di raggiungere un
valore di plateau nel valore
dell’RMSD (cioè lo spostamento
quadratico medio della molecola).
Tutti gli esperimenti sono stati svolti
in acqua perché l’acqua svolge un
ruolo fondamentale nella disposizione
della molecola e nella sua risposta a
stimoli fisici. Sono state esplorati
sistemi con due diverse concentrazioni
ioniche (0.05 M e 0.15M). Si è scelto
di analizzare tre particolari
caratteristiche meccaniche: la
pressione osmotica, la lunghezza
persistente e la resistenza a
compressione. Per il calcolo della
pressione osmotica è stato analizzato
un sistema che prevedeva un
confinamento delle catene di CS
attraverso particolari membrane
permeabili all’acqua e ai soluti ma non
ai GAG in un volume via via sempre
minore, in maniera da aumentare la
concentrazione del GAG e quindi
calcolare la pressione esercitata sulle
membrane da queste molecole (Fig.1).
Sono quindi state costruite set
contenenti 4 catene formate da 5
dimeri ciascuno, e sono stati svolti tre
set di esperimenti, uno con i dimeri
non solfatati, uno con dimeri solfatati
in posizione 4 e l’altro con dimeri
solfatati in posizione 6. Per la
lunghezza persistente è stata analizzata
Fig.1 Simulazione dell’aumento di concentrazione di catene di CS mediante “membrane
semipermeabili” virtuali

11
la traiettoria che una molecola
presenta in diversi istanti temporali.
Inizialmente è stata analizzata una
singola molecola. Nello studio è stata
considerata solamente la traiettoria
avuta dalla molecola in un intervallo
di tempo con una configurazione
energeticamente stabile (tratto
dell’andamento dell’RMSD piatto) e
per calcolare la lunghezza oltre la
quale i vettori tangenti della molecola
non sono più correlati tra loro è stato
sviluppato uno script di Matlab per
l’analisi delle traiettorie. Questo script
valuta il coseno formato dai vettori
tangenti la molecola in funzione della
distanza s. Quando questo valore
arriva a 0, si può decretare che a
quella distanza si è persa ogni
correlazione tra i vettori tangenti per
quanto riguarda l’istante temporale
analizzato. Il risultato finale sarà la
media di tutte le lunghezze persistenti
calcolate durante gli istanti temporali
nei quali la molecola ha raggiunto una
disposizione stabile. Questo studio è
stato svolto per diverse lunghezze
della catena (da 5 a 20 dimeri) e con
differenti percentuali di solfatazione
dei dimeri (0%, 30%, 50%, 100%) sia
in posizione 4 che in posizione 6.
Inoltre è stata valutata la lunghezza
persistente studiando la traiettoria che
la molecola mostra in un sistema
formato da più catene 0equidistanti tra
loro e innestate perpendicolarmente su
una superficie d’oro, per questo studio
sono state utilizzate catene composte
da 10 dimeri e si è deciso di analizzare
una catena contenente dimeri non
solfatati, una contenente dimeri
solfatati in posizione 4 e una con
dimeri solfatati in posizione 6. Per
quanto riguarda la resistenza a
compressione, si è modellizzato un
esperimento reale di
nanoindentazione tramite AFM
costruendo un set di catene di CS
Fig 2. Visione del sistema con la tip
virtuale all’inizio dell’esperimento

12
Tab 1. Confronto della pressione osmotica tra diversi sistemi in funzione della concentrazione.
C0S catena non solfatata, C4S-C6S catena solfatata in posizione 4 o 6;
disposte perpendicolarmente su
superficie d’oro e indentate da una tip
virtuale la quale simula l’estremità di
dimensioni atomiche di un
nanoindentatore sperimentale (Fig. 2).
Dopo questo esperimento si è
analizzata la forza agente sul tip in
relazione alla sua distanza dal
substrato ed è stato estrapolato il
valore di forza più alto.
RISULTATI E DISCUSSIONE
Il confronto con studi precedenti (i
quali hanno utilizzato metodi sia
sperimentali che computazionali) ha
permesso di comprendere meglio e
validare il modello. Per quanto
riguarda la pressione osmotica si è
evinto che il modello e la modalità di
esperimento portano ad avere risultati
(i valori sono in Tab. 1) confrontabili
con analisi precedenti [3,4] ma con
valori sempre superiori rispetto ai
suddetti studi. Inoltre, le catene
contenenti dimeri solfatati in posizione
4 e 6 in presenza di alte concentrazioni
esercitano una pressione osmotica
maggiore rispetto a catene composte
Conc (mg/ml) C0S C4S C6S Bathe [3] Ehrlich [4]
0 0 0 0 0
20 52,36 98,76 54,28 33,79
50,66 23,17
40 140,08 114,47 119,87 88,19
60 236,94 301,70 198,18 167,31
80 428,33 449,27 554,97 277,75
101,33 69,51
253,313 101,94
Fig 3. Esempio di kinking; nel dettaglio è
mostrato lo zucchero che subisce la modifica
conformazionale, “piegando” l’intera catena.
13
da dimeri non solfatati. Questo è più
evidente nella catena composta da
dimeri solfatati in posizione 6. La
discrepanza rispetto al precedente
studio computazionale di Bathe [3]
potrebbe essere legato al fatto che si
stanno confortando lavori che
utilizzano livelli di dettaglio differenti.
Quindi l’aver modellizzato il sistema
con un metodo ancora più preciso
porta alla conclusione che una
modellizzazione più rigorosa permette
di calcolare meglio le interazioni che i
singoli atomi hanno tra loro. Nello
studio della lunghezza persistente i
valori ottenuti sono confrontabili con
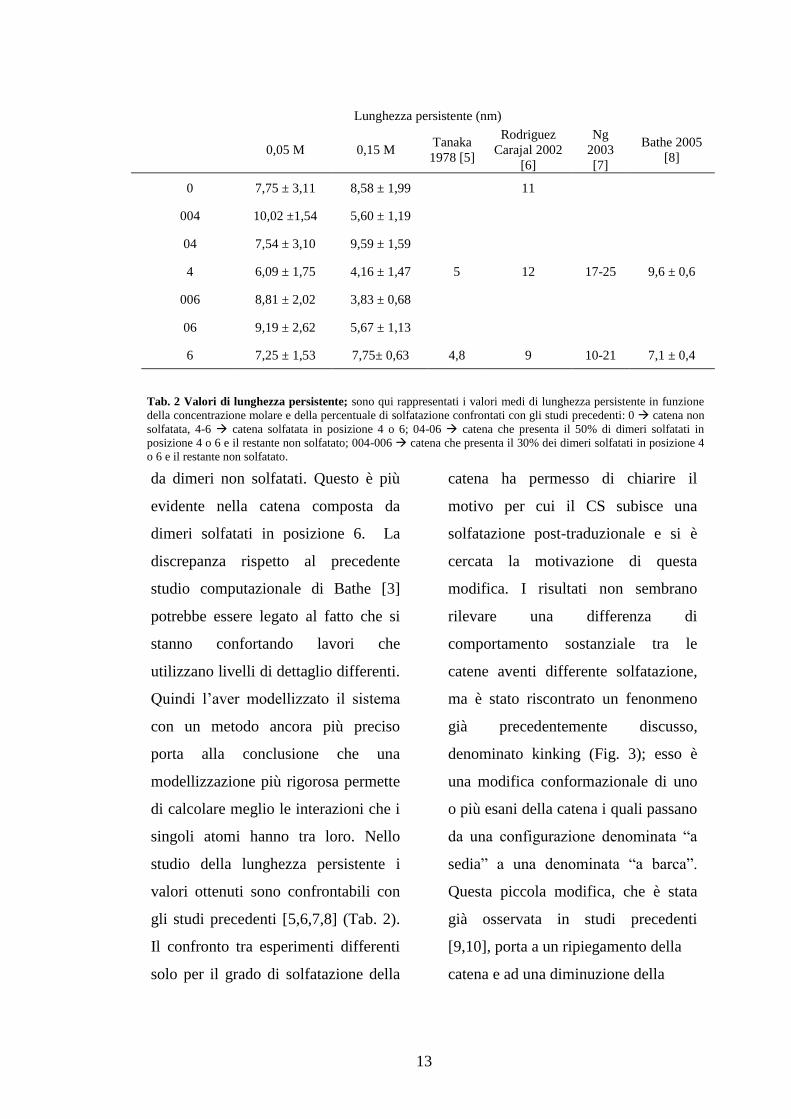
gli studi precedenti [5,6,7,8] (Tab. 2).
Il confronto tra esperimenti differenti
solo per il grado di solfatazione della
catena ha permesso di chiarire il
motivo per cui il CS subisce una
solfatazione post-traduzionale e si è
cercata la motivazione di questa
modifica. I risultati non sembrano
rilevare una differenza di
comportamento sostanziale tra le
catene aventi differente solfatazione,
ma è stato riscontrato un fenonmeno
già precedentemente discusso,
denominato kinking (Fig. 3); esso è
una modifica conformazionale di uno
o più esani della catena i quali passano
da una configurazione denominata “a
sedia” a una denominata “a barca”.
Questa piccola modifica, che è stata
già osservata in studi precedenti
[9,10], porta a un ripiegamento della
catena e ad una diminuzione della
Lunghezza persistente (nm)
0,05 M 0,15 M
Tanaka
1978 [5]
Rodriguez
Carajal 2002
[6]
Ng
2003
[7]
Bathe 2005
[8]
0 7,75 ± 3,11 8,58 ± 1,99 11
004 10,02 ±1,54 5,60 ± 1,19
04 7,54 ± 3,10 9,59 ± 1,59
4 6,09 ± 1,75 4,16 ± 1,47 5 12 17-25 9,6 ± 0,6
006 8,81 ± 2,02 3,83 ± 0,68
06 9,19 ± 2,62 5,67 ± 1,13
6 7,25 ± 1,53 7,75± 0,63 4,8 9 10-21 7,1 ± 0,4
Tab. 2 Valori di lunghezza persistente; sono qui rappresentati i valori medi di lunghezza persistente in funzione
della concentrazione molare e della percentuale di solfatazione confrontati con gli studi precedenti: 0 catena non
solfatata, 4-6 catena solfatata in posizione 4 o 6; 04-06 catena che presenta il 50% di dimeri solfatati in
posizione 4 o 6 e il restante non solfatato; 004-006 catena che presenta il 30% dei dimeri solfatati in posizione 4
o 6 e il restante non solfatato.
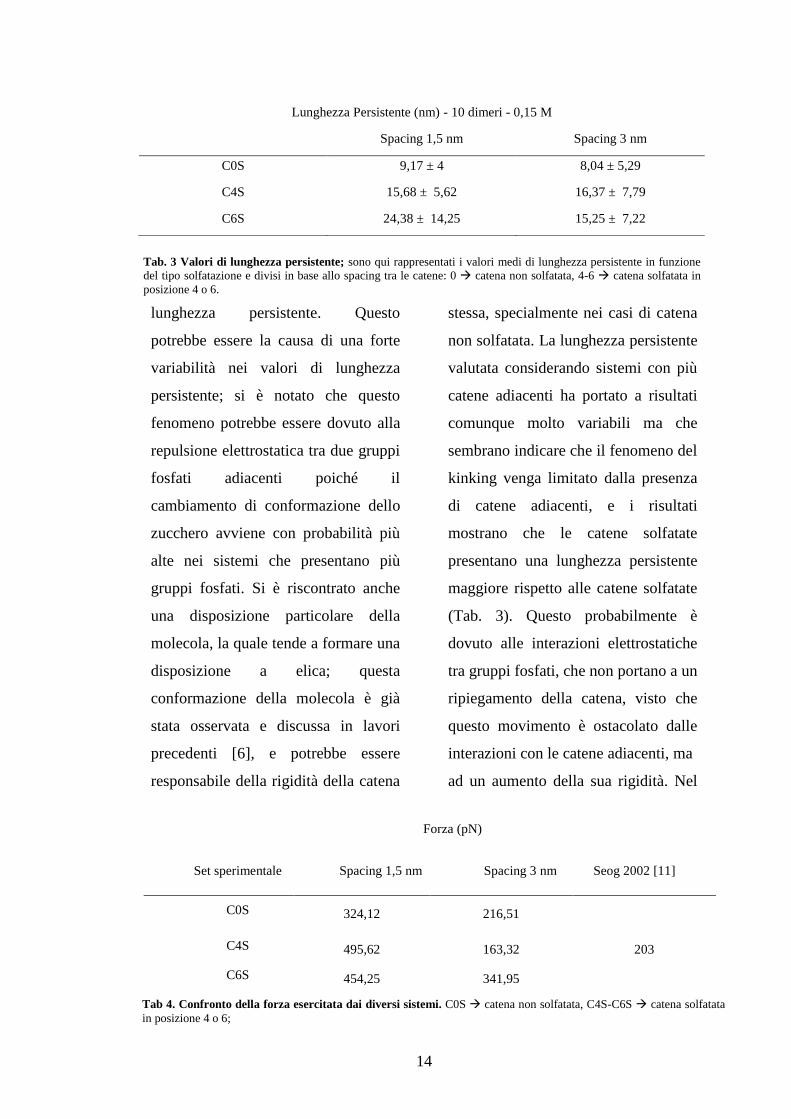
14
Tab 4. Confronto della forza esercitata dai diversi sistemi. C0S catena non solfatata, C4S-C6S catena solfatata
in posizione 4 o 6;
lunghezza persistente. Questo
potrebbe essere la causa di una forte
variabilità nei valori di lunghezza
persistente; si è notato che questo
fenomeno potrebbe essere dovuto alla
repulsione elettrostatica tra due gruppi
fosfati adiacenti poiché il
cambiamento di conformazione dello
zucchero avviene con probabilità più
alte nei sistemi che presentano più
gruppi fosfati. Si è riscontrato anche
una disposizione particolare della
molecola, la quale tende a formare una
disposizione a elica; questa
conformazione della molecola è già
stata osservata e discussa in lavori
precedenti [6], e potrebbe essere
responsabile della rigidità della catena
stessa, specialmente nei casi di catena
non solfatata. La lunghezza persistente
valutata considerando sistemi con più
catene adiacenti ha portato a risultati
comunque molto variabili ma che
sembrano indicare che il fenomeno del
kinking venga limitato dalla presenza
di catene adiacenti, e i risultati
mostrano che le catene solfatate
presentano una lunghezza persistente
maggiore rispetto alle catene solfatate
(Tab. 3). Questo probabilmente è
dovuto alle interazioni elettrostatiche
tra gruppi fosfati, che non portano a un
ripiegamento della catena, visto che
questo movimento è ostacolato dalle
interazioni con le catene adiacenti, ma
ad un aumento della sua rigidità. Nel
Forza (pN)
Set sperimentale Spacing 1,5 nm Spacing 3 nm Seog 2002 [11]
C0S 324,12 216,51
C4S 495,62 163,32 203
C6S 454,25 341,95
Lunghezza Persistente (nm) - 10 dimeri - 0,15 M
Spacing 1,5 nm Spacing 3 nm
C0S 9,17 ± 4 8,04 ± 5,29
C4S 15,68 ± 5,62 16,37 ± 7,79
C6S 24,38 ± 14,25 15,25 ± 7,22
Tab. 3 Valori di lunghezza persistente; sono qui rappresentati i valori medi di lunghezza persistente in funzione
del tipo solfatazione e divisi in base allo spacing tra le catene: 0 catena non solfatata, 4-6 catena solfatata in
posizione 4 o 6.

15
calcolo della resistenza a
compressione si è evinta una forte
disparità per quanto riguarda la forza
opposta all’indentazione tra i sistemi
composti da catene non solfatate
rispetto ai sistemi composti da dimeri
solfatati in posizione 4 o 6. Le catene
che presentano dimeri solfatati
rispondono con una forza maggiore
all’indentazione (Tab. 4). C’è inoltre
un comportamento differente se si
varia lo spacing reciproco delle catene.
CONCLUSIONI
Con questo studio si è potuto studiare
nel dettaglio una molecola che svolge
un ruolo fondamentale nella risposta a
stimoli fisici di compressione
all’interno del tessuto cartilagineo;
l’approccio modellistico
computazionale permette di svolgere
un’analisi precisa e completa delle
interazioni molecolari che causano le
risposte macroscopiche. Una volta
avuta chiarezza riguardo a queste
cause, si potrà valutare le modalità per
poter generare una molecola artificiale
che possa mimare il comportamento
dell’aggrecano. Inserendo queste
molecole in scaffolds per la
coltivazione di condrociti, si
permetterà a queste cellule di
svilupparsi in un ambiente che
somiglia molto a quello naturale.
Questo miglioramento tecnologico
potrà risolvere le maggiori difficoltà
incontrate dall’ingegneria dei tessuti
nel ricreare il tessuto cartilagineo, cioè
il limitato numero di cellule e la
perdita del fenotipo dei condrociti.
BIBLIOGRAFIA
1. Cilpa, G., et al., Atomistic insight into
chondroitin-6-sulfate glycosaminoglycan chain through quantum mechanics calculations and
molecular dynamics simulation. Journal of
computational chemistry, 2010. 31(8): p. 1670-80.
2. Marascio, M., Progettazione in silico di nuove molecole per la cartilagine ingegnerizzata, 2011,
Politecnico di Milano.
3. Bathe, M., et al., Osmotic pressure of aqueous
chondroitin sulfate solution: a molecular
modeling investigation. Biophysical journal, 2005. 89(4): p. 2357-71.
4. Ehrlich, S., et al., The osmotic pressure of chondroitin sulphate solutions: experimental
measurements and theoretical analysis.
Biorheology, 1998. 35(6): p. 383-97.
5. Tanaka, K., Physicochemical properties of
chondroitin sulfate. I. Ion binding and secondary structure. Journal of biochemistry, 1978. 83(3):
p. 647-53.
6. Rodriguez-Carvajal, M.A., A. Imberty, and S.
Perez, Conformational behavior of chondroitin
and chondroitin sulfate in relation to their physical properties as inferred by molecular
modeling. Biopolymers, 2003. 69(1): p. 15-28.
7. Ng, L., et al., Individual cartilage aggrecan
macromolecules and their constituent glycosaminoglycans visualized via atomic force
microscopy. Journal of Structural Biology, 2003.
143(3): p. 242-257.
8. Bathe, M., et al., A coarse-grained molecular
model for glycosaminoglycans: application to chondroitin, chondroitin sulfate, and hyaluronic
acid. Biophysical journal, 2005. 88(6): p. 3870-
87.
9. Haverkamp, R.G., A.T. Marshall, and M.A.
Williams, Model for stretching elastic biopolymers which exhibit conformational
transformations. Physical review. E, Statistical,
nonlinear, and soft matter physics, 2007. 75(2 Pt 1): p. 021907.
10. Haverkamp, R.G., M.A. Williams, and J.E. Scott, Stretching single molecules of connective tissue
glycans to characterize their shape-maintaining
elasticity. Biomacromolecules, 2005. 6(3): p. 64
11 . Seog, J., et al., Direct Measurement of
Glycosaminoglycan Intermolecular Interactions via High-Resolution Force Spectroscopy.
Macromolecules, 2001. 35: p. 5601-5615.1816-
8.

16
SUMMARY
INTRODUCTION
Cartilage is an avascular and hydrated
tissue that performs several functions.
It is divided into three types: hyaline
cartilage, which is very elastic and has
a high compressive strength; the
fibrous cartilage, more rigid; and
elastic cartilage, very flexible. This
study will focus on the articular
cartilage found in joints, called
articular cartilage, which is mainly
composed of hyaline cartilage. Despite
the cartilage has been often
characterized as a homogeneous
material, it is a complex material
which can be divided into three major
phases: a first phase is the cellular
component, which is responsible for
producing and/or remodeling of the
second phase, the Extracellular Matrix
(ECM). Finally, there is a liquid phase
constituted by water with electrolytes
in solution. From the mechanical point
of view, a fundamental role in the
response to physical stimuli is played
by the interaction of the fluid phase
with the ECM. The ECM is composed
of an intricate entanglement of
molecules: the major ones are collagen
type II fibrils and hyaluronan. The
latter is a molecular aggregate formed
of hyaluronic acid, an ultra-high
molecular weight glycosaminoglycan
(GAG), to which several molecules of
aggrecan are linked. Aggrecan, in turn,
is a proteoglycan (PG), i.e. a
biomolecule consisting of a protein
core to which GAGs are grafted. In the
specific case, these GAGs are the
keratan sulfate (KS) and the
chondroitin sulfate (CS). Aggrecan is
considered as the molecule that most
contributes to compressive
deformation resistance. This is due to
the fact that the GAG chains are
negatively charged, hence if they are
compressed, they generate a force that
tends to restore the original
conformation. To fully understand the
behavior of aggrecan and its role in
cartilage mechanics, i focused on a
part of it, i.e. the CS - region. CS is a
GAG composed by conjugation of two
sugars, N-acetyl-D-galactosamine
(GalNAc) and D-glucuronic acid
(GlcUA). These two sugars present a
negative charge and, in addition their
native charge, in cartilage tissue the
GalNAc sugar is sulfate in position 4
or 6, which adds a further negative
charge. To understand how cartilage
withstands high compressive loads and

17
transfer its features to a bioartificial
material, is important to study the
nanomechanical properties of the
individual constituents of the ECM,
since it is now clear that the
macroscopic behavior of this tissue is
derived from the nanoscopic structure
of the molecules that compose it. In
this study, we aim to characterize the
nanomechanical response of the CS
through virtual experiments. This
approach was chosen because
computational modeling allows the
calculation of mechanical properties
by varying one parameter at a time, in
order to understand how the different
variables determine the mechanical
response. In addition, we choose a
full-atom model, i.e. a model with
atomistic detail that has the highest
level of detail that we can have if we
consider a simulation that is based on
the principles of classical mechanics.
MATERIALS AND METHODS
This work is based on existing
molecular models, written by Cilpa [1]
and refined by Marascio [2]. To
generate the CS chains, it has been
used an in-house script that allows to
build chains of different lengths and
sulphation. For running and
Fig.1 Simulation of the increase in concentration of chains of the CS using virtual
semi-permeable membranes

18
processing the virtual experiments I
used software widely used in
molecular modeling filed as VMD,
NAMD and ACEMD. All virtual
experiments were carried out using
explicit solvent because water plays a
key role in the arrangement of the
molecules and in its response to
physical stimuli. Concerning the ion
concentration, two different values
have been considered (0.05 M and
0.15 M). Three mechanical behaviours
were analized: osmotic pressure,
persistent length and compressive
strength. For the calculation of the
osmotic pressure it was analyzed a
system which provided a confinement
of the chains of CS through a virtual
membrane permeable to water and
ions but not to GAGs and by
decreasing the volume available to
GAGs, so as to increase the
concentration of GAGs and then
calculate the pressure exerted on the
membrane (Fig.1). To calculate the
persistent length, the molecular
dynamics trajectory was analyzed and
the structure of the GAG at different
time frames was considered using a in-
house Matlab script to evaluate the
cosine of the angle formed by the
tangent vectors of molecule segments
as a function of the distance s between
the segments. When the cosine value
reaches 0, it implies that that distance
it is lost every correlation between the
tangent vectors. The final result will
be the average of all persistent lengths
calculated during different time
frames. This study was carried out for
different lengths of chains (5 to 20
dimers) and with different percentages
of sulphation of the dimers (0%, 30%,
50%, 100%), in position 4 and in
position 6. Furthermore, the persistent
length has been evaluated for chains
grafted on one end as a function of the
spacing between the chains.
Concerning the resistance to
compression, it is modeled a
nanoindentation experiment by
grafting a set of CS chains on gold

19
Tab 1. Comparison of osmotic pressure between different systems according to the concentration.
C0S unsulphated chain, C4S-C6S chain sulfated in position 4 or 6;
surface and by indenting the surface
by a virtual tip which simulates the
end of an Atomic Force Microscopy
indenter (Fig. 2). This experiment
provides the force acting on the tip
in relation to its distance from the
value of the highest force to
characterize the resistance to
compression of the different GAGs.
RESULTS AND DISCUSSION
The comparison with previous
studies (which have used both
experimental and computational
methods) has allowed better
understanding and validating the
proposed model.
Regarding the osmotic pressure, the
results (values are in Table 1) are
comparable with previous analyzes
[3,4] even if showing slightly higher
values than reference studies. In
particular, it is observed that chains
containing dimers sulfated in position
4 and 6 in the presence of high
Osmotic Pressure (kPa)
Conc (mg/ml) C0S C4S C6S Bathe [3] Ehrlich [4]
0 0 0 0 0
20 52,36 98,76 54,28 33,79
40 140,08 114,47 119,87 88,19
50,66 23,17
60 236,94 301,70 198,18 167,31
80 428,33 449,27 554,97 277,75
101,33 69,51
253,31 101,94
Fig 2. CS system with virtual tip at the
beginning of the experiment
20
concentrations exert an osmotic
pressure greater than chains composed
by unsulphated dimers. This is most
evident in the chain composed by
dimers sulfated in position 6. The
discrepancy with the previous
computational study of Bathe [3] may
be due to the different levels of detail:
Bathe et al. used a coarse-grain model,
whereas atomistic models are used in
the present work. In the study of the
persistent length, the values obtained
are comparable with previous studies
[5,6,7,8] (Table 2). The results do not
seem to detect a substantial difference
in behavior between the chains having
different sulphation. In addition, it has
been observed a previously discussed
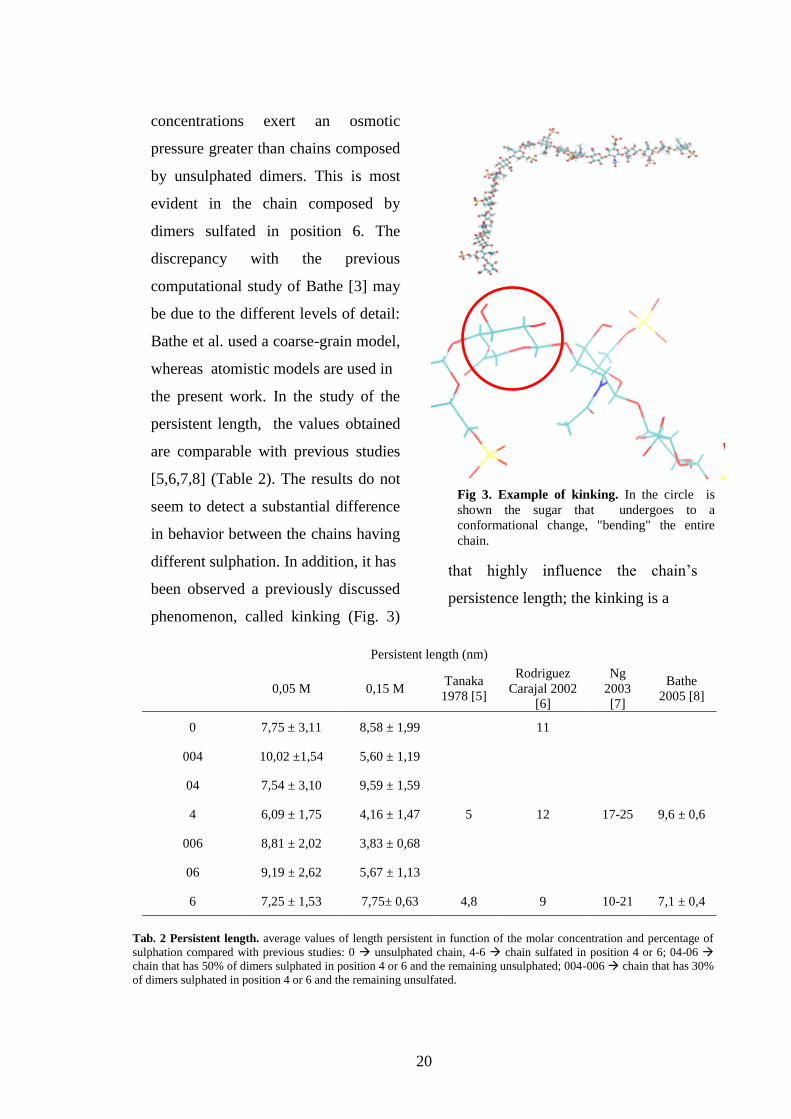
phenomenon, called kinking (Fig. 3)
that highly influence the chain’s
persistence length; the kinking is a
Persistent length (nm)
0,05 M 0,15 M
Tanaka
1978 [5]
Rodriguez
Carajal 2002
[6]
Ng
2003
[7]
Bathe
2005 [8]
0 7,75 ± 3,11 8,58 ± 1,99 11
004 10,02 ±1,54 5,60 ± 1,19
04 7,54 ± 3,10 9,59 ± 1,59
4 6,09 ± 1,75 4,16 ± 1,47 5 12 17-25 9,6 ± 0,6
006 8,81 ± 2,02 3,83 ± 0,68
06 9,19 ± 2,62 5,67 ± 1,13
6 7,25 ± 1,53 7,75± 0,63 4,8 9 10-21 7,1 ± 0,4
Fig 3. Example of kinking. In the circle is
shown the sugar that undergoes to a
conformational change, "bending" the entire
chain.
Tab. 2 Persistent length. average values of length persistent in function of the molar concentration and percentage of
sulphation compared with previous studies: 0 unsulphated chain, 4-6 chain sulfated in position 4 or 6; 04-06
chain that has 50% of dimers sulphated in position 4 or 6 and the remaining unsulphated; 004-006 chain that has 30%
of dimers sulphated in position 4 or 6 and the remaining unsulfated.

21 Tab 4. Comparison of the force exerted in different CS-systems C0S unsulphated chain, C4S-C6S
chain sulfated in position 4 or 6;
conformational change of one or more
hexanes in the chain which switch
from one configuration called "seat" to
one called "boat". This small change,
which has been already observed in
previous studies [9,10], leads to an
abrupt change in the overall
configuration of the chain and leads to
a decreased value of the persistent
length. This could be the cause of a
great variability in the values of
persistent length; it was noted that this
phenomenon might be due to the
electrostatic repulsion between two
adjacent phosphate groups since the
change in conformation of the sugar
occurs with probability higher in
systems that have multiple groups
phosphates. It was also found a
particular arrangement of the
molecule, which tends to form a
helical arrangement; this conformation
of the molecule has already been
observed and discussed in previous
work [6], and might be responsible for
the rigidity of the chain itself,
especially in cases unsulphated chain.
Persistent lengths assessed by
considering systems with grafted
chains show that the phenomenon of
kinking is limited by the presence of
adjacent chains, and the results show
that sulphated chains have a persistent
length longer than unsulphated chains
(Table 3). This is probably due to
electrostatic interactions between
Persistent Legth (nm) - 10 dimers - 0,15 M
Spacing 1,5 nm Spacing 3 nm
C0S 9,17 ± 4 8,04 ± 5,29
C4S 15,68 ± 5,62 16,37 ± 7,79
C6S 24,38 ± 14,25 15,25 ± 7,22
Force (pN)
Sperimental set Spacing 1,5 nm Spacing 3 nm Seog 2002 [11]
C0S 324,12 216,51
C4S 495,62 163,32 203
C6S 454,25 341,95
Tab. 3 Persistence length in different CS-systems; C0S unsulphated chain, C4S-C6S chain sulfated in
position 4 or 6;

22
phosphate groups which prevent the
chains bending, because this
movement is hindered by interactions
with adjacent chains, and lead to and
overall rigidity. When calculating the
compressive strength there is a
significant difference between systems
consist of chains not sulfated
compared to systems composed of
dimers sulfated in position 4 or 6. In
particular, sulphated chains present
larger forces in indentation tests
(Table 4). Also the spacing between
the chains influences the mechanical
behavior.
CONCLUSIONS
With this study it was possible to
investigate in details a molecule that
plays a key role in the response to
compressive stimuli within the
cartilage tissue; the modeling
approach allows to perform an
accurate and full study of molecular
interactions that cause the
macroscopic responses. Once had
clarity about these causes, it will be
possible to generate an artificial
molecule that can mimic the behavior
of aggrecan. The addition of these
molecules in scaffolds for the
cultivation of chondrocytes, will allow
these cells to grow in an environment
similar to natural tissue. This
technological improvement can solve
the major difficulties encountered
from engineering tissue in recreating
the cartilage tissue, i.e., the limited
number of cells and the loss of
phenotype of chondrocytes.
BIBLIOGRAPHY
1. Cilpa, G., et al., Atomistic insight into chondroitin-6-sulfate glycosaminoglycan chain
through quantum mechanics calculations and
molecular dynamics simulation. Journal of computational chemistry, 2010. 31(8): p. 1670-
80.
2. Marascio, M., Progettazione in silico di nuove
molecole per la cartilagine ingegnerizzata, 2011,
Politecnico di Milano.
3. Bathe, M., et al., Osmotic pressure of aqueous
chondroitin sulfate solution: a molecular modeling investigation. Biophysical journal,
2005. 89(4): p. 2357-71.
4. Ehrlich, S., et al., The osmotic pressure of
chondroitin sulphate solutions: experimental
measurements and theoretical analysis. Biorheology, 1998. 35(6): p. 383-97.
5. Tanaka, K., Physicochemical properties of chondroitin sulfate. I. Ion binding and secondary
structure. Journal of biochemistry, 1978. 83(3):
p. 647-53.
6. Rodriguez-Carvajal, M.A., A. Imberty, and S.
Perez, Conformational behavior of chondroitin and chondroitin sulfate in relation to their
physical properties as inferred by molecular
modeling. Biopolymers, 2003. 69(1): p. 15-28.
7. Ng, L., et al., Individual cartilage aggrecan
macromolecules and their constituent glycosaminoglycans visualized via atomic force
microscopy. Journal of Structural Biology, 2003.
143(3): p. 242-257.
8. Bathe, M., et al., A coarse-grained molecular
model for glycosaminoglycans: application to chondroitin, chondroitin sulfate, and hyaluronic
acid. Biophysical journal, 2005. 88(6): p. 3870-
87.
9. Haverkamp, R.G., A.T. Marshall, and M.A.
Williams, Model for stretching elastic biopolymers which exhibit conformational
transformations. Physical review. E, Statistical, nonlinear, and soft matter physics, 2007. 75(2 Pt
1): p. 021907.
10. Haverkamp, R.G., M.A. Williams, and J.E. Scott,
Stretching single molecules of connective tissue
glycans to characterize their shape-maintaining elasticity. Biomacromolecules, 2005. 6(3): p.
1816-8
11 . Seog, J., et al., Direct Measurement of
Glycosaminoglycan Intermolecular Interactions
via High-Resolution Force Spectroscopy. Macromolecules, 2001. 35: p. 5601-5615.1816-
8.

23
CAPITOLO 1:
INTRODUZIONE
1.1 La Cartilagine
Il tessuto cartilagineo è un tessuto connettivo di sostegno specializzato. È costituito
da cellule dette condrociti, immerse in un'abbondante sostanza amorfa extracellulare,
formata da fibre collagene e da una matrice amorfa gelatinosa. Le principali
caratteristiche di questo tessuto sono la solidità, la flessibilità e la capacità di
deformarsi. La cartilagine forma l'abbozzo per la maggior parte delle ossa dello
scheletro umano, nonché nelle metafisi durante l'accrescimento corporeo (cartilagine
di coniugazione), le quali successivamente verranno mineralizzate e sostituite da
tessuto osseo. Nell'adulto la cartilagine rimane in corrispondenza delle superfici
articolari, nei dischi intervertebrali, nello scheletro del padiglione dell'orecchio
esterno, partecipa alla formazione della trachea e dei bronchi, nella sinfisi pubica e
nei menischi. Si forma inoltre in seguito a fratture in qualsiasi fase della vita. In tutte
le zone in cui è localizzata, fatta eccezione per le superfici articolari, la cartilagine è
rivestita da un involucro costituito da tessuto connettivo denso fibroso detto
pericondrio. La cartilagine non è vascolarizzata e non è innervata, la diffusione dei
metaboliti avviene invece attraverso la matrice. Le cartilagini vengono classificate in
base alla quantità e alla costituzione della sostanza amorfa e in base alle fibre in essa
presenti. Si distinguono perciò tre tipi di cartilagine:
Cartilagine ialina
Cartilagine fibrosa
Cartilagine elastica
1.1.1 Cartilagine ialina
La cartilagine ialina è la varietà più diffusa di cartilagine. È una cartilagine piuttosto
elastica e con grande resistenza alla compressione. Le sue cellule (i condrociti) sono
collocate in cavità della sostanza amorfa dette lacune. La cartilagine ialina costituisce

24
l’abbozzo della maggior parte della maggior parte delle ossa, riveste le superfici
articolari, forma la trachea, la laringe e i bronchi, nonché la cartilagine del naso e
delle coste.
1.1.2 Cartilagine fibrosa
La cartilagine fibrosa può essere considerata una forma intermedia tra un tessuto
connettivo denso e la cartilagine ialina, con il primo tipo di tessuto ha in comune la
presenza di spessi fasci di fibre collagene di tipo I, con il secondo la matrice
cartilaginea, che tuttavia risulta piuttosto scarsa. Le fibre collagene raggiungono lo
spessore di 50-80 nm. Le cellule sono poche, disperse tra le fibre collagene o
caratteristicamente allineate. La cartilagine fibrosa è la costituente dell'anello fibroso
dei dischi intervertebrali, della sinfisi pubica, delle zone di inserzione tra tendini e
osso, dei menischi.
1.1.3 Cartilagine elastica
La cartilagine elastica è la tipologia di cartilagine più elastica e flessibile, ciò è
dovuto ad una prevalenza della componente fibrosa rispetto a quella della matrice
cartilaginea. Le fibre hanno struttura simile al connettivo denso elastico, per cui si
ramificano in molte direzioni, non si aggregano in fasci, ma sono talmente dense da
rendere invisibile la sostanza amorfa sottostante. La densità delle fibre è progressiva,
maggiore nelle parti interne, minore in quelle esterne. Forma il padiglione auricolare,
la tuba uditiva e l'epiglottide.
1.2 Struttura molecolare della cartilagine articolare
In questo studio ci concentreremo sulla cartilagine articolare, che in genere è
costituita da tessuto ialino. È possibile distinguere all’interno della cartilagine due
fasi principali: la fase solida, costituita principalmente da una matrice di fibre di
collagene e proteoglicani (PG) e la fase fluida, costituita da acqua con elettroliti in
soluzione. A queste due fasi si aggiungono le cellule, di un unico tipo che prendono
il nome di condrociti. L’acqua in peso costituisce il 60-80% del totale. Tra i tipi di

25
collagene il collagene II è presente tra il 10 e il 22% del peso umido della cartilagine,
mentre gli altri collageni, che sono di tipi I, V, VI, IX, XI sono presenti
complessivamente in proporzioni minori del 2%. Tra i PG l’aggrecano è presente tra
il 4 e il 7% e molto minori sono le percentuali di altri PG quali il versicano, il
blicano, la decorina e il perlecano. Minori percentuali ancora caratterizzano la
presenza di altre proteine, che pure hanno grande importanza quali elementi di
legame (matrilline e trombospondine). Le molecole di collagene e PG che
costituiscono la matrice solida della cartilagine interagiscono con le molecole
d’acqua e gli elettroliti. In particolare, i gruppi solfato delle catene dei
glicosamminoglicani (GAG) dei PG si idratano generando un’interazione di tipo
elettrostatico nei confronti delle molecole d’acqua e degli elettroliti che sono così
trattenuti all’interno della cartilagine. In base alla organizzazione e al contenuto di
colllagene e PG si possono distinguere nella cartilagine articolare quattro strati: lo
strato superficiale (~15% dello spessore), lo strato intermedio (~50% dello spessore),
lo strato profondo (~35% dello spessore) e, infine, uno strato di cartilagine calcificata
all’interfaccia con il tessuto osseo. Lo strato superficiale presenta la maggior quantità
di acqua (80% del peso) e di collagene (85% del peso secco) e la minor quantità di
PG. Le fibrille di collagene hanno piccolo diametro e sono orientate parallelamente
alla direzione del movimento della superficie articolare, caratteristica che rende
questo strato particolarmente resistente agli sforzi di taglio derivanti dal movimento
articolare. La percentuale di collagene diminuisce del 15% nello strato intermedio
dove le fibrille sono caratterizzate da diametri maggiori e non presentano un
orientamento preferenziale. Analogamente diminuisce il contenuto di acqua il cui
contributo diviene il 65% del peso, mentre i PG aumentano. Nello strato profondo il
contenuto di collagene e di acqua rimane costante e i PG raggiungono la massima
concentrazione. Le fibrille di collagene si orientano perpendicolarmente alla
superficie dell’osso sottostante col quale si fondono nella zona calcifica.

26
1.2.1 La matrice extra cellulare della cartilagine articolare
La matrice extracellulare (ECM) della cartilagine articolare è un reticolo intricato
contenente essenzialmente collageni fibrillari (20%), PG (PG) (5%) e acqua (75%), e
l'interazione di queste strutture influisce sul comportamento dei tessuti. Il collagene
di tipo II forma una struttura fibrosa che conferisce al tessuto la sua forma, resistenza
e rigidezza a trazione. Per quanto riguarda la seconda classe di componenti, il
proteoglicano predominante nella cartilagine articolare è l'aggrecano, il quale
determina principalmente le proprietà meccaniche a compressione della cartilagine.
La figura 1 riassume la struttura del tessuto cartilagineo.
Figura 1 Illustrazione schematica dei costituenti molecolare della cartilagine e loro disposizione in
aggregati multimolecolari. Nella figura a si osserva un vonfronto macroscopico tra tessuto cartilagineo
sano e tessuto cartilagineo osteoartritico. In figura b viene presentato uno schema della cartilagine che
mostra la dipendenza dalla profondità per quanto riguarda la forma delle cellule e la topologia del
collagene. Le cellule sono più piatte vicino alla superficie e diventano più rotonde all’aumentare della
profondità. In c è mostrtata la composizione molecolare della matrice e organizzazione nelle diverse
regioniextracellulari. In d sono presenti immagine ottenute tramite microscopia elettronica di nanostrutture
di differenti costituenti cellulari.

27
1.2.2 .Il collagene
Il collagene è un eteropolimero fibrillare, e nella cartilagine sono presenti i tipi II, IX
e XI: filamenti centrali di tipo XI sono circondati da fibrille di tipo II (che
rappresentano circa il 90% del collagene presente) e da uno strato esterno di fibrille
di tipo IX; questa struttura facilita l’interazione con gli altri costituenti dell’ECM [3].
Cross-links intra e intermolecolari contribuiscono alle proprietà meccaniche delle
fibrille di collagene [4]. In vivo, le fibrille di collagene hanno diametro di ~30-80nm
e sono distanziate di ~100 nm dai condrociti, molto impacchettate con i PG e
intrappolano in una rete le altre molecole dell’ECM. La rete di collagene è la
maggior responsabile delle proprietà di risposta a stimolo di trazione della
cartilagine; questo è dovuto a alla sua struttura altamente orientata e ai cross-link.
1.2.3 I Proteoglicani
I PG sono strutture proteiche che fungono da supporto a un numero variabile di
catene polisaccaridiche di GAG. La principale funzione biologica svolta dai PG è
intimamente connessa alle caratteristiche fisico-chimiche della componente di GAG
che è ancorata al core proteico. Il peso molecolare è ovviamente direttamente
funzione del numero di catene di GAG attaccate al core proteico. In relazione al
numero di catene di GAG i PG si dividono in PG a basso, alto e altissimo peso
molecolare. Tra i PG a basso peso molecolare si ricordano la decorina, la
fibromodulina, il biglicano. La decorina e la fibromodulina hanno un core proteico
vincolato ad una singola catena di GAG, mentre il biglicano ha due catene di GAG.
Questi PG hanno solitamente la funzione di ordinare la ECM. Tra i PG ad elevato
peso molecolare si ricordano i versicani che presentano fino a 15 catene di GAG e gli
aggrecani dove il numero di catene può arrivare ad alcune centinaia. Nel tessuto
cartilagineo, l’aggrecano è il PG contenuto in percentuali maggiori, e conferisce al
tessuto importanti caratteristiche biomeccaniche svolgendo la funzione di mantenere
idratato il tessuto.

28
1.2.4 L’aggrecano
L'aggrecano è un proteoglicano ad alto peso molecolare (1-3,5 MDa) che è composto
da un backbone proteico lineare (con lunghezza di contorno di circa 300 Å)
composto da diversi domini funzionali G1, IGD, G2, KS, CS e G3 (Fig. 2). I domini
globulari G1, G2 e G3 e il dominio interglobulare IGD non verranno considerati in
questo studio perché ad oggi non è stato riconosciuto loro un contributo determinante
per quanto riguarda la risposta allo stimolo di compressione della cartilagine
articolare. Invece, la regione contenente i domini Keratan Solfato (KS) e Condroitin
Solfato (CS) svolgono un ruolo fondamentale per quanto riguarda le proprietà
meccaniche della cartilagine articolare. In questi domini il backbone proteico forma
legami covalenti con numerose catene di GAG; il GAG presente in maniera
maggiore è il CS, il secondo GAG più diffuso è il KS. Una singola molecola di
aggrecano contiene circa 100 catene di CS che sono legati al core proteico con un
elevata densità (sono distanti ~2-4 nm l’una dall’altra) [5].L’aggrecano è spesso
aggregato in struttura inter-molecolari chiamati iauloronani. Gli aggregati formano
un gel che viene rinforzato con una rete di fibrille di collagene.
Fig2. Struttura dell’aggrecano

29
1.2.5 Lo Ialuronano
In vivo, le molecole di aggrecano sono intrappolate nella rete porosa del collagene.
Esse sono unite con legami non covalenti a molecole di un altro GAG, l’acido
ialuronico (HA); questo complesso prende il nome di Ialuronano. Questo legame è
stabilizzato da una proteina linker (una piccola proteina globulare sintetizzata dai
condrociti indipendentemente e contemporaneamente all’aggrecano e all’HA[6]). Lo
spazio tra le molecole di aggrecano adiacenti lungo la catena di HA è di ~20-50nm, e
questo molecole formano un largo aggregato di aggrecani. Il gel risultante (formato
da acqua e aggrecani) contribuisce a numerose e importanti caratteristiche della
cartilagine, come la pressione osmotica[7], la permeabilità idraulica[8], e resistenza a
sforzi di compressione[9, 10] e di taglio[11].
1.2.6 I Glicosamminoglicani
I membri della famiglia dei GAG si trovano sia disposti sulla superficie della
membrana cellulare sia all’interno della ECM. I GAG sono carboidrati costituiti dalla
ripetizione di un numero variabile di unità disaccaridi che possono andare incontro a
modifiche di natura chimica, una tra tutte la formazione di gruppi carichi
negativamente lungo la catena biopolimerica. Esistono due tipo principali di GAG:
l’acido ialuronico, che rappresenta una classe a se, e i GAG ricchi di gruppi
funzionali carichi. Diverse sono le differenze: quelle di natura chimica è che il primo
non subisce alcuna solvatazione mentre gli altri sono ricchi di gruppi SO3-, inoltre il
primo non si lega covalentemente ad altre strutture proteiche collaterali a formare i
PG, ed infine il primo ha dimensioni molto maggiori rispetto agli altri che sono
costituiti da un numero di disaccaridi variabile tra 20 e 200.
1.2.7 L’acido ialuronico
L’acido ialuronico (HA) è un GAG lungo, lineare e carico negativamente[12]. Venne
isolato già nella metà degli anni 30 nell’humor vitreo e rappresenta da solo una delle
strutture più importanti della ECM. Analogamente a tutti gli altri GAG ha una

30
struttura molto semplice dada dalla ripetizione di disaccaridi. L’acido ialuronico
consiste nella ripetizione di due unità disaccaridi [GlcAβ31-3GlcNAcβ1-4]n dove il
numero di unità ripetitive, n, può arrivare fino a 25000. L’acido ialuronico si trova
distibuito in molti tessuti e si trova in concentrazioni maggiori in tutti quei tessuti in
cui viene richiesta un’elevata capacità di reclutare acqua. Data la sua struttura e il
numero elevato di unità ripetute l’acido ialuronico ha la capacità, sotto particolari
condizioni chimico/fisiche, di trattenere o rilasciare le molecole d’acqua con cui si
coordina. In vivo, l’HA può legarsi fino con ~100 aggrecani a formare una molecola
denominata ialuronano. In aggiunta, HA si lega con la superfice di alcune cellule
attraverso il recettore CD44; questo legame può inibire l’adesione cellulare e quindi
promuovere la migrazione cellulare[13]. HA è inoltre presente in abbondanza nel
fluido sinoviale, il fluido presente nelle articolazioni, e quindi ha un ruolo nella
biolubrificazione della cartilagine. Studi passati hanno suggerito diversi ruoli
dell’HA, come il suo aiuto all’aumento della viscosità del fluido sinoviale[14] o
come trasportatore di fosfolipidi lubrificanti[15]. Comunque, non è chiaro in che
maniera l’HA contribuisce alla biolubrificazione della cartilagine[16].
1.2.8 GAG solfatati
I GAG solfatati possono essere divisi in tre gruppi sulla base della loro composizione
e quindi delle loro unità di disaccaridi che li formano: eparan solfato,CS e dermatan
solfato (che differiscono solo dalla posizione del gruppo carico SO3-) e KS. Questi
GAG difficilmente si trovano da soli ma si associano alle strutture proteiche dei PG
in catene che arrivano fino a 200 ripetizioni[17]. Nella cartilagine, il GAG più
diffuso è il CS.
1.2.9 Il Condroitin Solfato
Il CS è un GAG prodotto attraverso l’assemblaggio di unità ripetute di disaccaridi e
quindi presenta una catena alternata di zuccheri; N-acetil-D-galattosammina
(GalNAc) e acido D- glucuronico (GlcUA), alternativamente legati in siti di legame
glicosidici in posizione β1,3 e β1,4. Nell’aggrecano una catena di CS è generalmente

31
formata da 20-60 unità di disaccaridi, nei quali la N-acetilgalattosammina può essere
solfatata nelle posizioni 4 o 6 .Quindi nella cartilagine si trovano tre diverse forme di
condroitin solfato; condroitin (CH) condroitin-4-solfato (C4S) e condroitin-6-solfato
(C6S). I gruppi con carica negativa COO- e OSO3- generano un’alta densità di
cariche negative. Quando due catene di GAG vengono compresse generano alte forze
repulsive proprio grazie alla alta densità di cariche negative che tendono ad
allontanare l’intera catena. Queste cariche negative sono alla base della resistenza a
compressione dell’intero tessuto cartilagineo. Sia C4S che C6S hanno questi gruppi
negativi, ma negli adulti c’è una maggior concentrazione di C6S [18, 19]. La carica
presente su questo GAG, insieme a quella del collagene, è definita fissa. In
contrapposizione, la carica degli ioni mobili (sodio Na+, calcio Ca
2+, cloro Cl
- per
citare i principali) attirati dalla carica fissa è definita mobile. La presenza di H+ e OH
-
influenza in maniera importante i meccanismi di trasporto e le proprietà meccaniche
del tessuto. Questo è dovuto al fatto che le cariche fisse variano con il pH
dell'ambiente fisiologico. Uno dei fenomeni più interessanti a livello locale è il forte
gradiente osmotico che i PG sono in grado di creare all'interno del tessuto. In
condizioni fisiologiche, le cariche fisse richiamano ioni liberi costituiti
principalmente da ioni sodio Na+. Quest’accumulo di ioni all'interno della matrice
causa una forte pressione osmotica. Il richiamo di acqua all'interno del tessuto che ne
deriva espande la rete di collagene; le proprietà della cartilagine articolare derivano
dall'equilibrio tra la tensione intrinseca delle fibre di collagene e la tensione indotta
dal rigonfiamento dovuto ai PG. Tra le cariche fisse c'è un fenomeno di repulsione
sterica, tale da indurre le macromolecole di GAG a occupare il maggior volume
possibile all'interno dell’ ECM. Tuttavia, questo effetto dipende strettamente dalla
pressione osmotica: un aumento degli ioni mobili induce un effetto di schermatura
tra le cariche fisse, riducendo l'effetto di schermatura sopra descritto. Secondo gli
studi di Bathe [20], la rigidezza intrinseca della struttura caratteristica del CH
influenza in maniera significativa la pressione osmotica, a differenza del volume
sterico escluso, i cui effetti sono trascurabili. In particolare, il C4S aumenta la
rigidezza della molecola molto più rispetto al C6S e al CH, essendo la solfatazione
vicina alla regione di legame β (1->3). Gli studi su quale sia la struttura che presenta
la backbone più rigida tra il CH e il C6S danno pareri discordanti [20, 21].

32
Concentrazioni tipiche del CS nella cartilagine articolare sono pari a 20-80
mg/ml[22]. In figura 3 sono presentati il condroitin solfatato in diverse posizioni.
1.2.10 Altre molecole
Altri componenti molecolari presenti nell’ECM della cartilagine svolgono un ruolo
importante nell’assemblamento e nell’integrità del tessuto. Queste molecole che
agiscono come cross-linker per fomare una rete interconnessa di molecole di
collagene, includono: la famiglia delle matrilline[23], piccole proteine ricche di
Leucina (decorina, asporina, fibromodulina, lumicano, cherotacano e osteoaderina
[24-26]), e trombospondine, [4].
1.3 Funzioni e proprietà della cartilagine articolare
La cartilagine articolare ha la funzione di rivestimento delle superfici articolari ed ha
uno spessore che varia da 1 a 5 mm; essa riduce l’attrito durante i movimenti relativi,
produce un’ottimale distribuzione dei carichi e funge da ammortizzatore nelle
trasmissione dei carichi impulsivi. Durante il movimento delle articolazioni, la
Fig3. Struttura del Keratan solfato e del Condroitin Solfato solfatato in posizioni diverse.

33
cartilagine sostiene una complessa combinazione di stimoli compressivi fino a ~20
Mpa [27, 28] e può sostenere deformazioni in compressione fino al 40% [29]. Per
svolgere queste funzioni, il tessuto cartilagineo utilizza il fluido in essa contenuto; la
cartilagine articolare presenta infatti porosità tra loro connesse che sono riempite di
fluido sinoviale. Questa caratteristica attribuisce al tessuto proprietà meccaniche
marcatamente viscoelastiche. Quando la cartilagine viene sollecitata, questo fluido
può fuoriuscire dalla matrice cartilaginea nel caso di una compressione e rientrare al
cessare di questa sollecitazione. Inoltre, la cartilagine possiede una eccellente
lubrificazione che la porta ad avere un’ottima resistenza all’usura: infatti possiede un
coefficiente di frizione compreso tra ~0.0005 e 0.04 in presenza del fluido sinoviale
[30]. Questa caratteristica è stata fortemente discussa in letteratura ed è stata
attribuita alla pressurizzazione interstiziale [31, 32] e alla lubrificazione di contorno
dovuta a molecole cariche [33, 34]. La funzione biomeccanica della cartilagine è
quindi quasi completamente ascrivibile alla ECM poiché la degradazione e la
modifica strutturale dei componenti molecolari dell’ECM influenza in maniera
rilevante il comportamento generale del tessuto e può portare alla perdita della
funzionalità dello stesso. Per capire al meglio la funzione della cartilagine a livello
tissutale, le proprietà meccaniche della cartilagine sono state studiate attraverso
differenti configurazioni di carico differenti, per esempio attraverso compressione
confinata e non confinata, sforzo di taglio, pressione osmotica e indentazione.
1.4 Calcolo proprietà meccaniche
Tradizionalmente, il tessuto cartilagineo veniva considerato e studiato senza tener
conto della sua struttura molecolare, e quindi venivano effettuati esperimenti
considerando la cartilagine come un continuo macroscopico omogeneo. Negli anni
successivi si è cercato di affinare questa metodologia di esperimento e sono stati
effettuati esperimenti tenendo conto che la cartilagine è un materiale avente proprietà
dipendenti dalla profondità. Recenti modelli teorici a livello molecolare [20, 22, 35,
36] hanno mostrato invece un potenziale collegamento tra le interazioni molecolari
nell’ECM e le proprietà meccaniche macroscopiche della cartilagine. Quindi, per
conoscere completamente le funzioni meccaniche della cartilagine e i motivi che

34
portano alla sua disfunzione, è essenziale quindi realizzare prove su componenti del
tessuto a scale dimensionali dello stesso ordine di quelle delle macromolecole della
ECM. Recentemente si è osservata la comparsa di studi sperimentali volti ad
indagare strutture particolari (ben definite) della ECM articolare. Ancor più
recentemente alcuni lavori teorici hanno cercato di fornire presupposti ad un
particolare comportamento nanomeccanico. Per quanto riguarda gli studi
sperimentali, le deformazioni che avvengono al livello di scala delle molecole
dell’aggrecano possono modificare la densità di carica locale [22], permeabilità
idraulica [37], streaming potential [38] e altri mediatori biofisici della
meccanotrasduzione ai condrociti [39]. I metodi nanomeccanici hanno il vantaggio di
testare variazioni spaziali nelle proprietà meccaniche della cartilagine con un’alta
risoluzione. Queste variazioni sono direttamente collegate alla funzione del tessuto
nelle differenti regioni a livello microscopico, o alle proprietà dei diversi costituenti
molecolari alla nanoscala. Questi metodi inoltre permettono lo studio delle proprietà
individuali delle differenti molecole che compongono l’ECM nella forma di
aggregati molecolari o come singola molecola; entrambi questi studi sono
determinanti per comprendere il comportamento della cartilagine utilizzando una
prospettiva molecolare. Lo studio che utilizza modelli computazionali, però è
preferibile poiché in questi sistemi molecolari vi è un altissimo numero di parametri
che variano e che solo con un approccio molecolare possono essere variati e
considerati. In questi paragrafi riassumerò le procedure specificamente rilevanti per
lo studio di tre principali proprietà meccaniche di catene di CS; Pressione osmotica,
Lunghezza persistente e comportamento sforzo – deformazione.
1.4.1 Pressione Osmotica
La grande densità di cariche negative presente nelle catene di CS dell’aggrecano
genera una pressione osmotica che mantiene la cartilagine articolare in uno stato
idratato (60-80% di acqua in peso) e questo gioca un ruolo fondamentale nella
risposta del tessuto a uno stimolo di compressione elevato, determinando le sue
proprietà meccaniche [40, 41]. Quindi uno studio della pressione osmotica del CS è

35
molto utile per investigare le proprietà meccaniche del CS. Questi studi hanno
portato a risultati simili, come si evince dalla figura 4 e dalla tabella 1 .
Sperimentale [42] Computazionale [35]
Concentrazione (mg/ml) Pressione Osmotica
(Kpa) Concentrazione (mg/ml)
Pressione Osmotica (Kpa)
50,6625 23,168427 0 0
101,325 69,505281 20 33,7912
253,3125 101,9410788 40 88,1868
60 167,3077
80 277,7473
1.4.1.1 Metodi sperimentali
Ci sono state numerose misure sperimentali delle pressioni osmotiche generate dai
GAG di vari tipi di PG. I primi esperimenti [43] utilizzavano basse concentrazioni
simili a quelle trovate in tessuti connettivi sottoposti a carichi come la cartilagine. Il
range di concentrazioni è stato successivamente aumentato usando diverse varietà di
Tab.1 Valori di pressione osmotica al variare della concentrazione di CS
Fig4. Pressione Osmotica: Confronto tra studio sperimentale [42] e computazionale [35]
0
50,6625
101,325
253,3125
0
33,7912
88,1868
167,3077
277,7473
0
50
100
150
200
250
300
-10 10 30 50 70 90 110
Pre
ssio
ne
(kP
a)
conc (mg/ml)
Sperimental
Computational

36
metodi sperimentali, alcuni dei quali sono indiretti e utilizzano relazioni tra parametri
termodinamici [44]. L’approccio più utilizzato è quello che sfrutta l’equilibrio
dialitico contro soluzioni polimeriche delle quali è nota la pressione osmotica[45]. In
alcuni studi [46] vengono usate soluzioni di polietilen glycole come soluzioni di
riferimento.
1.4.1.2 Metodi Computazionali
Bathe et al [20], hanno sviluppato e analizzato un modello molecolare del CS. La
pressione osmotica è definita come la differenza in pressione tra il comportamento
contenente le catene di CS e il reservoir di acqua.
1.4.2 Lunghezza persistente
La lunghezza persistente è una proprietà caratteristica di una macromolecola
polimerica. Fornisce una misura della rigidità della catena. Tale rigidità si
contrappone ai moti termici che tendono a far curvare la catena, cioè a ridurre la
distanza tra i due estremi rispetto alla lunghezza complessiva del suo profilo. Il
valore della lunghezza persistente si può interpretare come la lunghezza oltre la quale
i moti termici divengono apprezzabili, tali cioè da competere efficacemente con le
interazioni tra i frammenti successivi della catena che tendono invece a conferirle
rigidità. Pertanto, se si considerano una catena o una porzione di catena più lunghe
della lunghezza persistente, le orientazioni delle due estremità saranno governate
essenzialmente dai moti termici casuali e quindi non correlate. All’interno della
lunghezza persistente, viceversa, i moti termici non riescono a prevalere sulle
interazioni e quindi la curvatura della catena in due punti successivi è fortemente
correlata. In termini geometrici, se si immagina la catena polimerica come un filo, la
lunghezza persistente esprime l’entità della correlazione orientazionale del vettore
tangente al filo lungo il suo profilo ovvero la lunghezza oltre la quale i vettori
tangenti alla molecola sono scorrelati [47]. In particolare, definendo con θ l’angolo
formato tra il vettore tangente in un punto qualsiasi della molecola e il vettore
tangente nella posizione iniziale, si può dimostrare che vale la reazione

37
Dove con s si indica la lunghezza di contorno della molecola ed Lp la lunghezza
persistente. I risultati sono esposti in tabella 2
1.4.2.1 Metodi sperimentali
Nei primi studi, vi era un forte utilizzo del microscopio elettronico per generare
immagini dell’aggrecano o del CS, visualizzando così proteolgicani fissati, deidratati
e metal-coated. Quando si riusciva ad avere una risoluzione tale da riuscire a
distinuere le catene di GAG, esse spesso apparivano come ammassamenti collassati,
rendendo difficile la determinazione del loro numero, dello spacing e della
conformazione[48-50]. Con l’avvento della microscopia a forza atomica ad alta
risoluzione (AFM), che può essere svolta in liquido e in condizioni fisiologiche, è
stata raggiunta una risoluzione fino alla scala nanometrica. Partendo quindi da
immagini ottenute grazie all’AFM (Fig. 5), attraverso uno software di analisi di
immagini è stato possibile ottenere la configurazione della catena di CS in diversi
istanti temporali, riuscendo in questo modo ad estrapolare il valore di lunghezza
persistente.
Tipo di solfatazione Lunghezza persistente [nm]
CS 11+
C4S 5*; 12+; 9,6 ±0,6 ˜; 17-25 ˚
C6S 4,8*; 9+; 7,1±0,4 ˜; 10-21 ˚
Tab.2 Risultati degli studi sulla lunghezza persistente del CS in diverse solfatazioni.
* Tanaka, 1978 [52] ; +
Rodriguez- Carajal, 2002 [51] ; ˚ Ng, 2003 [5]; ˜ Bathe, 2005 [20]

38
1.4.2.2 Metodi computazionali
Studi precedenti [20, 51, 52] hanno calcolato la lunghezza persistente su un modello
computazioale del CS; per farlo hanno svolto i calcoli attraverso due approcci:
- microscopicamente, utilizzando la formula già utilizzata da Flory [53]
Dove b1 è il primo vettore legame (virtuale) nella catena, b1 è il suo valore
quadratico medio e Rcc è il vettore che esprime la distanza dal primo punto
all’ultimo.
- macroscopicamente, utilizzando il wormlike chain model, e in particolare con
la seguente espressione
Dove a è la lunghezza persistente, L la lunghezza di contorno e s è il raggio
d’inerzia.
1.4.3 Esperimenti di nanoindentazione
Instrumented (depht-sensing) nanoindentation e nanoindentazione con microscopio a
forza atomica vengono utilizzate per misurare la forza di penetrazione rispetto alla
profondità dell’indentazione su campioni di cartilagine. Sono stati impiegate un certo
numero di geometrie per l’indenter , comprese quella sferica, conica, piramidale, e
Berkovich. Un parametro importante estratto dai dati della nanoindentazione è il
modulo di indentazione del campione, Eind, che rappresenta la resistenza alla
penetrazione locale durante il carico elastico multiassiale. Un approccio utilizzato per
Fig. 5 Morfologia di un filamento di actina in diversi istanti temporali

39
determinare Eind è il metodo di Oliver-Pharr, che si basa su un modello di contatto
elastico con un continuum isotropo e omogeneo per determinare il modulo ridotto, Er
dal tratto di scarico del grafico forza-profondità di penetrazione utilizzando la
formula
√ √
dove S è la pendenza del tratto iniziale della curva forza di scarico (F) - profondità
(D) e A è l'area di contatto dell’indenter. Eind del campione testato è calcolata tramite
dove è il rapporto di Poisson del campione e Ei e sono rispettivamente modulo di
Young e Rapporto di Poisson dell’indenter. Per l’indentazione della cartilagine
mediante indenter costruiti con materiali duri (ad esempio, diamante, nitruro di
silicio, silicio, e borosilicato), il secondo termine dell'equazione 2 in genere può
essere trascurato poiché Ei »Eind. Il metodo Oliver-Pharr [54] presuppone che il tratto
di scarico sia elastico e indipendente dal tempo. Una differenza tra l’indentazione
della cartilagine e il metodo Oliver-Pharr è che la cartilagine subisce danni
permanenti solo a tensioni molto elevate (deformazioni del 30-40% [55]). Queste
deformazioni sono tipicamente molto maggiori rispetto a quelle applicate tramite
nano o micro-indentazioni [56], e quindi ci si aspetta che la deformazione
permanente della cartilagine sia trascurabile in questi casi. Invece, la cartilagine
esibisce un comportamento meccanico tempo-frequenza-dipendente e subisce
rilassamento poroviscoelastico e creep [57], che influenza notevolmente sia i dati di
carico che quelli di scarico. Quindi, la stima di Eind utilizzando il metodo Oliver-
Pharr può incorporare gli effetti della resistenza elastica alla compressione
multiassiale combinato con gli effetti associati al rilassamento
poroviscoelastico/creep. Inoltre, alla scala di lunghezza di indentazione, può essere
presente un comportamento anisotropo a causa delle caratteristiche nano e
microstrutturali della cartilagine, come l'allineamento delle fibrille di collagene [58],
la presenza di condrociti, e le differenze locali nella densità dell’aggrecano. Date le

40
limitazioni del metodo Oliver-Pharr di interpretazione del carico sulla cartilagine ,
l’Eind calcolata è una misura efficace della rigidità dell’indentazione multiassiale e
può essere utilizzata per valutare le tendenze nella risposta meccanica della
cartilagine a diverse condizioni sperimentali ad una determinata velocità di
indentazione. Un secondo metodo per il calcolo della Eind si basa sulla teoria di
contatto di Hertz [59], che presuppone l’isotropia, l’omogeneità, e l’elasticità lineare
del materiale. Lin & Horkay [60] hanno ampiamente recensito l’indentazione in
tessuti biologici e gel. Questa recensione discute e riassume le formulazioni
analitiche sulla base della teoria di contatto elastico hertziane, nonché le modifiche
JKR (Johnson-Kendall-Roberts) [61] e DMT (Derjaguin-Muller-Toporov) [62] del
modello hertziano originale che incorporano adesione indotta da deformazione e le
variazioni corrispondenti dell'area di contatto. Algoritmi utili per la determinazione
del punto di contatto punta-campione per i set di dati irregolari, come la ricerca
sezione aurea, sono presenti nel secondo articolo. Esempi di set di dati irregolari
sono :rumore eccessivo, adesione e repulsione significativa, contatto imperfetto
punta-campione, grandi deformazioni. Nei modelli hertziani si presume siano
trascurabili plasticità e poroviscoplasticità tempo-dipendente, e Eind è approssimata
dalla porzione di carico della curva forza-profondità. Vengono comunemente
utilizzate due geometrie per l’indenter: sferica (colloidale) e piramidale (nitruro di
silicio standard). Per le punte delle sonde sferiche, si utilizza la seguente formula
dove F è la forza di indentazione, D la profondità di penetrazione, R il raggio della
punta, e ν il coefficiente di Poisson (ad esempio, ν = 0.1 per cartilagine bovina
giovane [63]). Per la sonda con punta piramidale (R « D),
Dove è l'angolo di apertura della faccia piramidale. L’AFM viene utilizzata per
quantificare interazioni a livello molecolare delle molecole dell’ECM in diverse
tipologie. Per quantificare queste interazioni, le catene di CS vengono fissate
chimicamente ad una superficie piana per formare una “spazzola” il cui
comportamento mima le funzioni fisiologiche dell’aggrecano. Le proprietà di questa
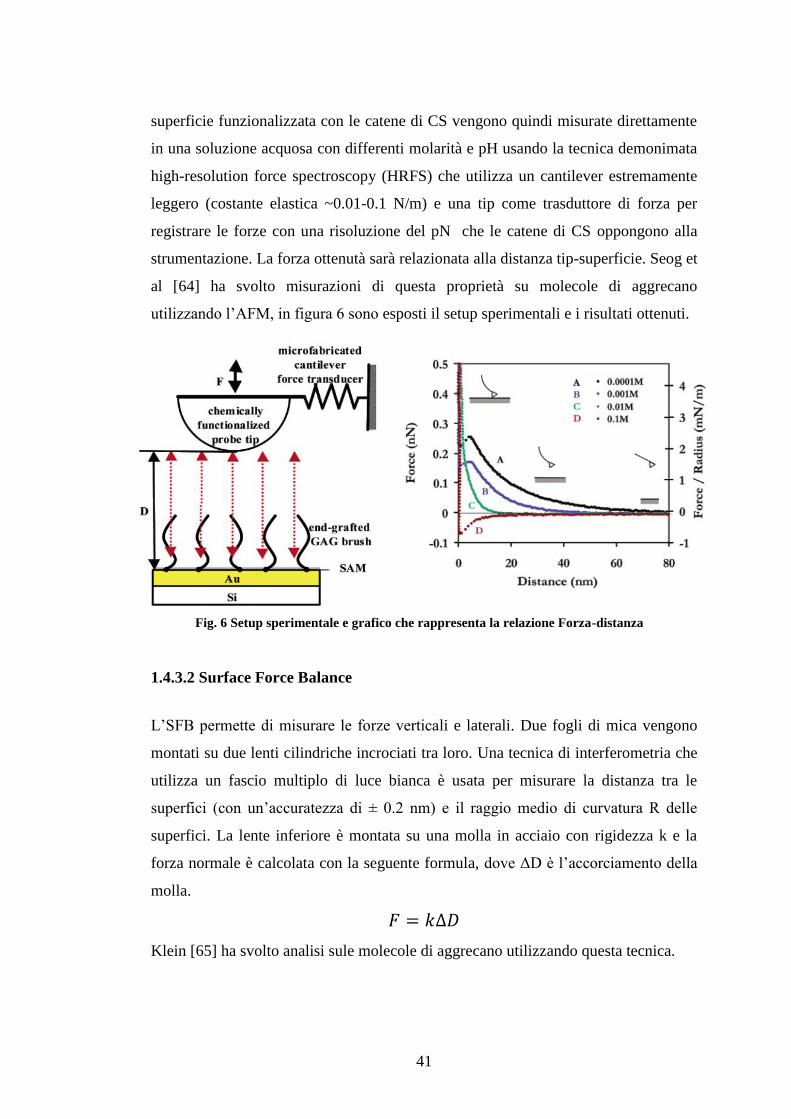
41
Fig. 6 Setup sperimentale e grafico che rappresenta la relazione Forza-distanza
superficie funzionalizzata con le catene di CS vengono quindi misurate direttamente
in una soluzione acquosa con differenti molarità e pH usando la tecnica demonimata
high-resolution force spectroscopy (HRFS) che utilizza un cantilever estremamente
leggero (costante elastica ~0.01-0.1 N/m) e una tip come trasduttore di forza per
registrare le forze con una risoluzione del pN che le catene di CS oppongono alla
strumentazione. La forza ottenutà sarà relazionata alla distanza tip-superficie. Seog et
al [64] ha svolto misurazioni di questa proprietà su molecole di aggrecano
utilizzando l’AFM, in figura 6 sono esposti il setup sperimentali e i risultati ottenuti.
1.4.3.2 Surface Force Balance
L’SFB permette di misurare le forze verticali e laterali. Due fogli di mica vengono
montati su due lenti cilindriche incrociati tra loro. Una tecnica di interferometria che
utilizza un fascio multiplo di luce bianca è usata per misurare la distanza tra le
superfici (con un’accuratezza di ± 0.2 nm) e il raggio medio di curvatura R delle
superfici. La lente inferiore è montata su una molla in acciaio con rigidezza k e la
forza normale è calcolata con la seguente formula, dove ΔD è l’accorciamento della
molla.
Klein [65] ha svolto analisi sule molecole di aggrecano utilizzando questa tecnica.

42
1.5 Obiettivo
L’ECM della cartilagine è un intricato reticolo di molecole, le quali hanno proprietà
meccaniche dipendenti da un altissimo numero di variabili. Per questo un approccio
di tipo computazionale, che permette di poter variare con facilità una sola delle
variabili e valutare come questa modifica comporta variazioni nella funzione della
molecola è preferibile ad un approccio di tipo sperimentale. Per esempio si possono
costruire modelli di catene di CS di lunghezza e solfatazione differenti, oppure
generare sistemi comprendenti più catene nelle quali si può valutare l’importanza
della distanza relativa tra due catene vicine. Fino ad ora non è stata svolta una analisi
full-atom del CS, poiché fino all’anno scorso non esisteva un modello
computazionale specifico che potesse descrivere la catena di CS comprendendo
anche le catene con i dimeri solfatati in posizione 4 e 6. Inoltre gli studi precedenti
non hanno svolto un’attenta e precisa analisi sul reale contributo dei gruppi solfati
alla risposta nanomeccanica del CS. Quindi l'obiettivo principale del lavoro di tesi è
quello di realizzare esperimenti virtuali in grado di ottenere informazioni sul loro
comportamento meccanico. In virtù della vastità dello spazio dei parametri di
interesse (ad esempio, il tipo di solfatazione, modello solfatazione, peso molecolare
CS e la spaziatura tra le catene CS) le simulazioni di modellistica molecolare sono in
grado di fornire uno strumento utile, un ambiente controllato, in cui poter svolgere
una indagine sistematica della caratteristiche del CS. Il lavoro di tesi si incentra sullo
svolgimento di simulazioni di dinamica molecolare con le catene di CS variando 3
parametri principali;
- Il grado di solfatazione delle catene (non solfatate, solfatate in posizione 4,
solfatate in posizione 6)
- Il peso molecolare delle molecole di CS
- La distanza laterale tra le diverse catene
In modo da capire come la modifica di questi parametri agisca sul comportamento a
compressione dell'intero sistema.

43
CAPITOLO 2
MATERIALI E METODI
2.1 Dinamiche molecolari
A causa del rapido incremento della velocità di calcolo dei computer, e constatando
che questo aumento delle prestazioni ha ormai un andamento esponenziale, le
simulazioni computazionali possono essere considerate un ottimo e affidabile
strumento per generare esperimenti e ottenere risultati. Il modello viene ancora
generato dalla teoria, ma i calcoli sono permessi da computer rispettando algoritmi
prestabiliti. Le dinamiche molecolari (MD) sono simulazioni computazionali dove
l’evoluzione temporale di un insieme di atomi che interagiscono tra loro è seguita
integrando le loro equazioni del moto[66]. Le dinamiche molecolari seguono la
meccanica classica, basata sulla celebre legge di Newton:
Per ogni atomo i in un sistema costituito da N atomi. Qui mi rappresenta la massa
dell’atomo,
la sua accelerazione e Fi la forza che agisce su di esso dovuta alle
interazioni con gli altri atomi. Fornendo un set iniziali di posizioni e velocità il
computer calcola una traiettoria in uno spazio di dimensione 6N (3N posizioni e 3N
velocità). Comunque, una traiettoria da sola serve a poco. Le dinamiche molecolari
utilizzano un metodo meccanico statistico: è un modo per ottenere un set di
configurazioni distribuite seguendo un certo insieme statistico. Una configurazione è
definita come un set di vettori a 3 dimensioni per ogni atomo in un preciso istante
temporale. Un insieme è una collezione di tutte le possibili configurazioni che hanno
differenti stati microscopici ma hanno identico stato macroscopico o termodinamico.
Le differenti definizioni degli stati termodinamici costituiscono una diversa
classificazione degli insiemi. L’unico ensemble utilizzato in questo studio è il

44
i
canonico (NVT) dove viene definito e rimane fisso il numero di atomi N, il volume
V e la temperatura T.
2.2 Funzione Energia potenziale
Per poter modellizzare un sistema fisico bisogna scegliere una funzione per il
potenziale. Questa è una funzione Φ(r1,….,rN) che descrive la posizione dei
nuclei rappresentando l’energia potenziale del sistema quando gli atomi sono in una
particolare configurazione. Questa funzione è solitamente costruita basandosi sulle
posizioni relative degli atomi, difficilmente con le posizioni assolute. Le forze sono
derivate dal gradiente della funzione potenziale
Fi r Φ(r1 ,..., r )
Questa forma implica che l’energia totale, E = K + Φ, sia conservata, dove K è
l’energia cinetica. La scelta più semplice per Φ la somma di interazioni pair-wise
Φ(r1 ,..., r ) ri r j
Ci sono diverse funzioni potenziali. La funzione più comune e più utilizzata prende il
nome di CHARMM (Chemistry at HARvard Macromolecular Mechanics) [67].
Questa funzione è abitualmente costituita da due termini: uno che descrive le
interazioni di legame, l’altro che espone le interazioni di non-legame.
2.3. Termini di legame
I termini di legame sono costituiti dalle iterazioni tra coppie, terne e quaterne di
atomi legati tra loro in catena. La dimensione della Funzione di energia potenziale è
3n, dove n è il numero di atomi del sistema. Inoltre, essendo funzione delle
coordinate dei punti, per ogni particolare stato energetico è possibile associare un
valore di energia.
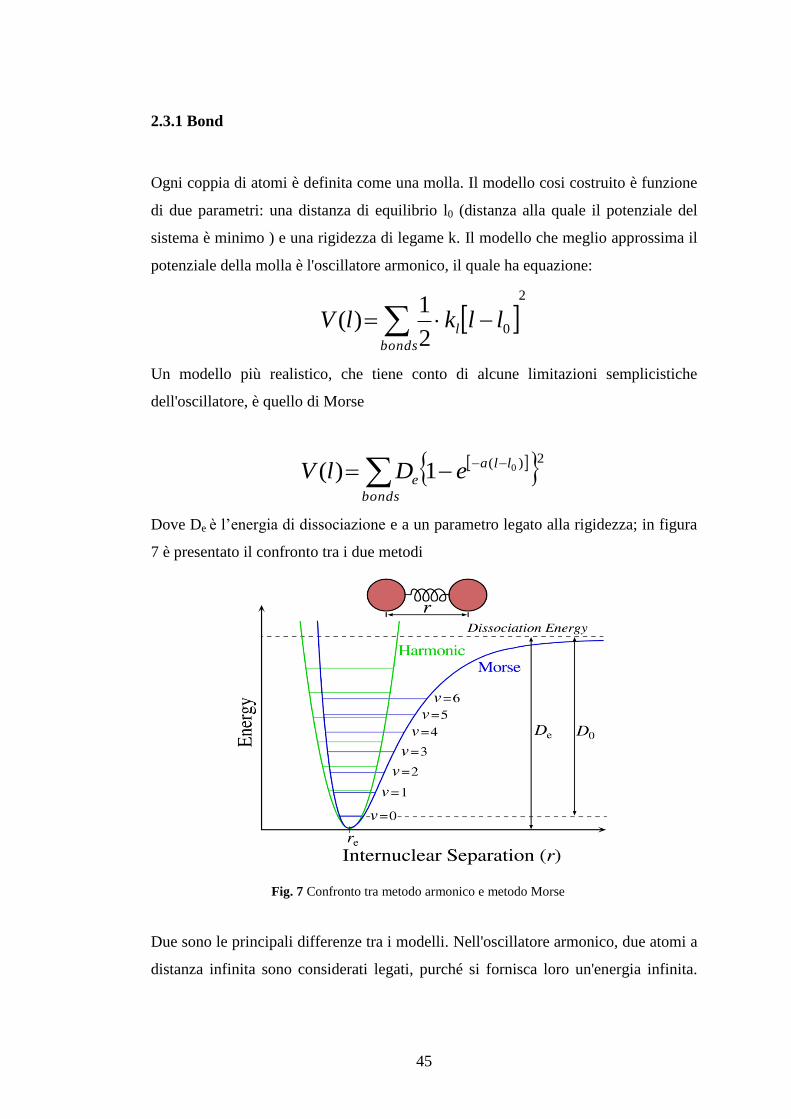
45
2.3.1 Bond
Ogni coppia di atomi è definita come una molla. Il modello cosi costruito è funzione
di due parametri: una distanza di equilibrio l0 (distanza alla quale il potenziale del
sistema è minimo ) e una rigidezza di legame k. Il modello che meglio approssima il
potenziale della molla è l'oscillatore armonico, il quale ha equazione:
2
02
1)(
bonds
l llklV
Un modello più realistico, che tiene conto di alcune limitazioni semplicistiche
dell'oscillatore, è quello di Morse
bonds
lla
e eDlV2)( 01)(
Dove De è l’energia di dissociazione e a un parametro legato alla rigidezza; in figura
7 è presentato il confronto tra i due metodi
Due sono le principali differenze tra i modelli. Nell'oscillatore armonico, due atomi a
distanza infinita sono considerati legati, purché si fornisca loro un'energia infinita.
Fig. 7 Confronto tra metodo armonico e metodo Morse

46
Questo paradosso è risolto in Morse, dove viene definito un potenziale di
dissociazione che indica l'energia da fornire per rompere il legame. La seconda
differenza è il costo del modello: mentre il primo è pienamente determinato da due
parametri, Morse ne richiede tre.
2.3.2 Angle o bending
Dati tre atomi consecutivi legati tra loro, un semplice modello che approssima il loro
comportamento energetico è quello dell'oscillatore armonico
2
02
1)(
angles
kV
2.3.3 Angoli diedrici
Si definisce dietro l'angolo formato da 4 atomi in successione. In particolare, è dato
dal piano passante per tre di loro e il quarto atomo. Esso esprime l'ingombro sterico
degli atomi. L'andamento del suo potenziale è mostrato in figura 8.
Fig. 8 Andamento del potenziale dell’angolo diedrico

47
La periodicità del picco, pari a 2π, si evince dall’espressione del potenziale:
n
dihedrals
nCV )cos()(
In alcuni casi, è possibile migliorare la qualità del modello andando a modificare
opportunamente i termini per rispettare delle evidenze sperimentali, come particolari
configurazioni della struttura. Modelli maggiormente complessi tengono conto dei
diedri impropri, cui la differenza rispetto i classici è la presenza di una biforcazione a
“y” nella catena.
2.4 Termini di non legame
I termini di non-legame comprendono tutte le interazioni tra le molecole (Van der
Waals e interazione elettrostatica), entrambe definite da opportuni modelli.
2.4.1 Van der Waals
Il potenziale riferito a Van der Waals esprime la possibilità di atomi non legati tra
loro di scambiarsi energia. In particolare, descrive le forze che nascono a causa della
distribuzione elettronica attorno al nucleo (Fig. 9). Il moto degli elettroni attorno al
Fig. 9 Andamento del potenziale di Van der Waals al variare della distanza tra due atomi

48
nucleo permette di considerare l'atomo come un dipolo indotto in grado di scambiare
energia con i dipoli indotti relativi ad altri atomi. Il potenziale di Van der Waals è
costituito da due termini: una componente attrattiva e una repulsiva, che impedisce
agli atomi di collassare uno sull'altro. La componente attrattiva è dominante a lungo
raggio (atomi distanti), mentre quella repulsiva aumenta di intensità avvicinando gli
atomi tra loro. La bontà del modello è verificata, in quanto:
La distanza a 3,8Å è un punto di minimo per la funzione. Il potenziale è nullo e
quindi anche la forza che i dipoli scambiano. Il sistema si può considerare in
equilibrio. A distanze maggiori dal punto di equilibrio, il potenziale tende a zero e
quindi la forza. I dipoli non scambiano energia e la componente dominante è
attrattiva (forza negativa). Per distanze inferiori a quella di equilibrio, il potenziale
aumenta e la forza è positiva. Domina la componente repulsiva. Il modello più
comunemente usato nella MD è quelli di Lennard-Jones o potenziale 12-6, in
notazione semplificata
Dalla formula, si nota come per r → +oo la componente positiva è dominante (forza
attrattiva), mentre per f → 0 lo è quella negativa (repulsiva). In forme più complesse
si tiene conto della distanza di equilibrio alla quale si ha un minimo di energia rm. La
sua relazione con , (diametro per cui l'energia è 0) si ottiene ponendo a zero la
derivata del potenziale rispetto ad r:
6
1
2mr
2.4.2 Legame a idrogeno
E' presente solo in alcuni modelli molecolari, dato che è possibile associare la quota
di energia dovuta al legame a idrogeno mediante considerazioni geometriche

49
(distanza e angolo di legame). Il potenziale è stato costruito su base sperimentale,
con la formula:
1012)(
r
C
r
ArV
2.4.3 Energia elettrostatica
E' il termine dovuto alle interazioni tra coppie di atomi non legati elettricamente
carichi, che si scambiano forze di tipo columbiano. La sua espressione deriva dalla
legge di Coulomb:
N
i
N
ij ijr
ji
r
1 1 0 )4(
La carica degli atomi può essere modellizzata in due modi:
2.4.3.1 Partial atomic charges
Tutta la carica è posta sul nucleo. L'interazione tra molecole o parti di esse sono
sommatorie di forze di Coulomb scambiate tra coppie di punti carichi.
2.4.3.2 Central multipole expansion
Ogni molecola è una singola unità, descritta come un multipolo. Si riduce
notevolmente il costo computazionale, dato che raggruppo gli atomi in macrounità –
le molecole, appunto. Il suo limite è che bisogna avere a disposizione uno studio
elettronico della molecola. Inoltre, essendo dei multipoli, l'energia decade tanto
velocemente quanto allontaniamo tra loro le cariche. L'insieme di tutti questi
parametri per una molecola prende il nome di campo di forza o force field.

50
2.5 Algoritmo globale
Per riassumere le informazioni date della sezione precedente, viene mostrato un
schema che riassume l’algoritmo generale utilizzato durante una MD (Fig 10).
L’algoritmo calcola il potenziale, la posizione e la velocità di tutti gli atomi del
sistema e ripete gli step 2,3 e 4 per il numero di step previsti. Durante gli steps 2,3 e
4 vengono calcolate le forze, aggiornato il potenziale, le posizioni e le velocità.
Fig. 10 Schema riassuntivo di una simulazione di dinamica molecolare

51
2.6 Cutoff
Il calcolo dei termini di non legame è la parte computazionalmente più massiccia di
una simulazione di dinamica molecolare. In teoria, l’interazione tra ogni coppia di
atomi dovrebbe essere calcolata, ma il numero di calcoli aumenta con il quadrato del
numero degli atomi. Per velocizzare questo calcolo, le interazioni tra due atomi
separati da una distanza più grande di un cutoff predeterminato vengono ignorate. Ci
sono alcune vie per determinare le interazioni di una coppia di atomi; sono presentati
in Figura 11.
Utilizzando il truncation metod tutte le interazioni sono imposte a 0 per una distanza
interatomica maggiore della distanza di cut-off. Il metodo Shift cutoff ivece modifica
l’intera superficie di enegia potenziale in modo che l’interazione potenziale alla
distanza di cutoff sia zero. La terza tecnica è chiamata switch cutoff. In questo
metodo il potenziale assume il suo andamento usuale fino a un primo cutoff, e viene
modificato in modo da annullare l’interazione potenziale nella zona che va dal primo
al secondo cutoff. Per le simulazioni in genere viene utilizzato lo switch method
Fig. 11 Confronto tra i diversi metodi di cutoff

52
perché è stato dimostrato in studi precedenti che è stabile è ha un’alta correlazione
con i dati sperimentali [68].
2.7 Solvente
Il solvente, tipicamente l’acqua, ha un’influenza fondamentale sulla struttura, sulla
dinamica e la termodinamica delle molecole biologiche. Una delle funzioni più
importanti del solvente è schermare le interazioni elettrostatiche. Nonostante l’acqua
sia una molecola neutra la sua forma crea un dipolo formato da una carica parziale
positiva e una carica parziale negativa. Questo causa interazioni elettrostatiche che
non possono essere ignorate. È molto importante includere gli effetti del solvente
nelle simulazioni di dinamica molecolare dei tessuti biologici, poiché molte delle
interazioni atomiche di queste molecole avvengono con le molecole d’acqua. Se
l’acqua non viene inclusa nelle simulazioni, è come se la molecola fosse simulata in
vuoto senza una costante dielettrica e nessuna schermatura di carica elettrostatica
[69]. Il metodo più semplice è di includere una costante dielettrica costante nel
termine elettrostatico della funzione di energia potenziale. Con questo metodo
implicito le molecole di solvente vengono incluse nel sistema ma viene utilizzata una
costante dielettrica. Spesso viene utilizzata una costante dipendente dalla distanza
(εeff = rijε) dove ε va da 4 a 20. Questa è una cruda approssimazione ma è comunque
meglio che simulare una molecola in vuoto. Invece, con il metodo esplicito le
molecole d’acqua vengono incluse nel sistema, e quindi la costante dielettrica viene
impostata a 1 nella funzione di energia potenziale. In questo trattamento molto più
dettagliato del solvente, bisogna impostare condizioni di boundary per evitare che le
molecole d’acqua diffondano lontano dal sistema. Un metodo molto popolare del
trattamento del boundary è chiamato periodic boundary conditions.
2.7.1 Condizioni periodiche al contorno
Le condizioni periodiche al contorno permettono di svolgere una simulazione
utilizzando un numero minore di particelle basandosi sul fatto che le particelle
provino forze come se fossero in costrette a rimanere in un cubo di determinate

53
dimensioni. Come esempio si guardi la figura 12, dove, intorno al box centrale di
interesse sono stati costruiti 8 altri box. Le particelle nei box vicini vengono
chiamate particelle immagine della particella originale. Le coordinate sono in
relazione con quelle del box primario e sono state ottenute con una semplice
traslazione. Il box periodico più semplice è cubico ma si possono creare box rombici,
dodecaedrici e ottaedrici troncati. Solitamente i box immagine sono impostati in un
formato tridimensionale, in modo che il box primario è circondato da 26 box vicini.
Le forze sulle particelle primarie vengono calcolate allo stesso modo nel box
primario come nei box immagine. Le interazioni atomiche vengono calcolate al di
sopra di una certa distanza chiamata distanza di cutoff scelta dall'utente. Il cutoff
viene scelto in modo che una particella nel box primario non possa “sentire” la
particella dei box vicini. La figura 12 riassume questi concetti
2.8 Minimizzazione
In molti casi è necessario, prima di lanciare le simulazioni d'interesse sul modello,
effettuare un'operazione di minimizzazione dell'energia, in modo da rendere univoco
Fig 12. Condizioni periodiche al contorno bidimensionali. Il box colorato in azzurro rappresenta il box
primario che è circondato da 8 box immagine dove x e y sono i vettori del box e r è la distanza tra un atomo e
la sua copia traslata

54
il punto di partenza ed eliminare tutte le componenti energetiche che potrebbero
sfalsare i risultati della simulazione. Si descrive questo tipo di simulazione nel
capitolo del protocollo di simulazione. Dopo aver esaminato la struttura della
funzione potenziale, è opportuno fare qualche considerazione riguardo la MD.
In primo luogo, la simulazione mediante MD non è l'unica tecnica applicabile: con il
Metodo Montecarlo è possibile determinare lo stato di equilibrio in maniera
semplice, senza passare dalla soluzione del sistema di equazioni definito per la MD.
Tuttavia il metodo non è utilizzabile per determinare gli stati di non equilibrio. Per
Inoltre, anche per la tecnica di MD, la più completa e soddisfacente, sono presenti
delle limitazioni cui è bene tenere conto sia in sede di modellizzazione che di analisi
dei risultati delle simulazioni.
2.9 La simulazione si basa sulla teoria della Meccanica Classica
L'uso dell'equazione di Newton per il moto approssima bene il comportamento di un
sistema atomico nel range di temperature fisiologiche, ma ci sono alcune eccezioni.
Ad esempio, non tiene conto dell'effetto di tunnelling dei protoni che può verificarsi
in un legame idrogeno. La meccanica statistica di un oscillatore armonico classico
differisce significativamente da quella di un oscillatore quantistico quando la
frequenza di vibrazione è pari a
A temperatura ambiente, tutti i legami e angoli di legame sono vicini a tale limite,
causando degli errori di simulazione. Quello che si può fare per ovviare a questa
situazione mantenendo la semplicità dell'oscillatore classico è di inserire un termine
di correzione nella funzione dell'energia del sistema o considerare legami e angoli di
legame costanti durante il moto.
2.10 Ruolo degli elettroni
I force field costruiti per la MD non tengono conto dello stato di eccitazione degli
elettroni, basandosi sull'approssimazione di Born-Oppenheimer (ogni elettrone

55
tornano allo stato di riposo quando cambia la posizione del suo atomo di
riferimento). Questa limitazione non permette la simulazione di tutti i processi che
coinvolgono il trasferimento di elettroni o la loro eccitazione.
2.11 Software utilizzato
Per questo lavoro di tesi sono stati utilizzati principalmente tre programmi:
- VMD: Visual Molecular Dynamics è progettato per la visualizzazione, la
modellizzazione e l’analisi dei sistemi biologici come proteine, acidi nucleici,
lipidi ecc. VMD provvede a una grande varietà di metodi per visualizzare la
molecola di interesse (punti, linee, sfere, cilindri) evidenziandone la struttura
principale o particolari residui o solo certi tipi di atomi ecc . Può essere
inoltre utilizzato per dar vita e analizzare la dinamica di una molecola che
percorre una determinata traiettoria, cioè è possibile osservare l’andamento
della simulazione di MD.
- NAMD (Not (Just) Another Molecular Dynamics): è un programma
sviluppato per effettuare simulazioni di dinamica molecolare (calcolo energia
di interazione, minimizzazione, equilibrazione di un sistema) e per l’analisi
delle traiettorie tramite l’utilizzo del software VMD. Lavora su CPU
- ACEMD : è un programma che processa dinamiche biomolecolari
specialmente ottimizzato per lavorare su graphics processing units (GPU)
alloggiate sulle schede grafiche. Questo gli permette di essere molto più
veloce di NAMD, e quindi è stato maggiormente utilizzato nel mio lavoro
2.12 Costruzione modello
Il primo passo sarà quello di definire i modelli atomistici di blocchi di CS.
Recentemente, Cilpa et al. [1] ha fornito i parametri di forza del campo GROMOS
per C6S. A partire dalla loro struttura e dai loro parametri, svilupperemo il modello

56
atomistico e i parametri per il C4S e per il C6S. Questo lavoro è stato svolto dal Dr.
Matteo Marascio, che nel suo lavoro di tesi triennale si è occupato di definire i
modelli atomistici del CS nelle forme non solfatato, solfatato in posizione 4 e
solfatato in posizione 6 [2]. Quindi, per ottenere una catena di CS composta anche da
dimeri con diversa solvatazione è stato utilizzato lo script GagsBuilder.
2.12.1 Gags Builder
Appena eseguito lo sctipt, ci viene chiesto se si vuole costruire una catena di CS
oppure di dermatan solfato (un GAG molto simile al CS).
Digitando “c” lo script ci chiede di inserire la sequenza della catena di CS
digitando una sequenza di numeri si può decidere la lunghezza e il tipo di
solvatazione di una catena di CS. Per esempio, digitando “00000” si otterrà una
catena composta da 5 dimeri di CS non solvatato, invece digitando “060606” si
otterrà una catena di 6 dimeri composta da 3 dimeri non solfatati e 3 dimeri solfatati
in posizione 6. In figura 13 sono presenti due snapshot del programma.
Lo script restituisce due file:
il file contenente tutte le coordinate della molecola, con estensione .pdb
il file contenente i vari tipi di atomi e legami, con estensione .psf
In appendice A è presente lo script Gagsbuilder e in appendice B la topologia
utilizzata per la costruzione delle molecole di interesse. In Figura 14 viene mostrata
una catena generata dallo script
Fig. 13. Struttura dello script GagsBuilder

57
2.13 Dinamica in vuoto
Lo script GagsBuilder restituisce una catena che non è equilibrata, infatti viene
costruita una catena “dritta”; Prima di poter effettuare la dinamica vera e propria, è
necessario svolgere una dinamica molto breve in vuoto, in modo da ottenere una
catena che presenta una disposizione con energia interna minore.
2.14 Costruzione del box d'acqua
Le molecole ottenute vengono quindi inserite in un box d’acqua, utilizzando lo
strumento di VMD “Solvate”. Per poter costruire il box d'acqua il programma
necessita del file .pdb e .psf della molecola. Si possono modificare diversi parametri,
ma in questo lavoro ci si è concentrati particolarmente su due:
Boundary, il quale rappresenta la distanza minima che le molecole d'acqua devono
avere dalla molecola, diminuendo questa distanza si avrà un sistema contenente più
molecole d'acqua.
Box size, il quale rappresenta le dimensioni del box d'acqua che circonda la
molecola: questa variabile è fondamentale per quanto riguarda il calcolo
computazionale perché la grandezza del box fa aumentare esponenzialmente i tempi
di calcolo, è quindi necessario dimensionare questo parametro in maniera da
minimizzare il numero di molecole di solvente per poter ottimizzare al meglio il
numero di ore di calcolo necessarie alla simulazione. Questo strumento genera un file
.pdb e un file .psf del box d’acqua contenente la molecola.
Fig.14 catena di 5 dimeri di CS “dritta”

58
2.15 Ionizzazione del box
Una volta costituito il box d'acqua, bisogna aggiungere al sistema un numero di ioni
sufficiente per ottenere la concentrazione ionica desiderata; per fare questo si utilizza
un altro strumento di VMD denominato “Autoionize”. Questa tool necessita il file
.pdb e .psf del box d’acqua contenente la molecola e può aggiungere gli ioni in modo
da neutralizzare la carica o in maniera da mantenere la concentrazione entro un dato
valore di molarità. In questo lavoro sono stati scelti due valori diversi di molarità,
pari a 0,05, per simulare un sistema povero di ioni e 0.15 M, che corrisponde alla
molarità fisiologica. In figura 15 è presentato un box d’acqua contenente una catena
di CS e degli ioni
2.16 Calcolo proprietà meccaniche
2.16.1 Pressione Osmotica
Per calcolare la pressione osmotica si è scelto di simulare il “confinamento” delle
catene di CS entro delle barriere semipermeabili; si è quindi costruito un box d’acqua
contenente le catene di CS (concentrazione ionica pari a 0,05 M) e, attraverso uno
script di NAMD, si è imposto alle catene di CS di rimanere confinate entro un
volume del box sempre più piccolo, in modo da aumentare la loro concentrazione. È
stata poi misurata la pressione che queste catene esercitavano sulle membrane. La
pressione osmotica è stata calcolata utilizzando un box contenente 4 catene di CS
composte da 5 dimeri ciascuna e lo studio è stato svolto concentrandosi su 3
Fig.15 Catena di CS in box d’acqua e con concentrazione ionica pari a 0,15 M

59
esperimenti; uno con le catene non solfatate, uno con le catene solfatate in posizione
4 e l'ultimo con le catene solfatate in posizione 6. Sono stati svolti 5 esperimenti, con
concentrazioni pari a 20, 40, 60, 80 mg/ml e durante le dinamiche è stata monitorata
la forza che le catene esercitavano sulle barriere. Calcolando il grafico dell’RMSD
delle catene si è concluso che una dinamica da 50ns è sufficiente per avere un
sistema energeticamente stabile. Per calcolare la pressione osmotica in simulazioni di
dinamica molecolare, si utilizza uno script in TCL (chiamato TCL Forces) che
permette di generare delle “barriere invisibili” all’interno del box d’acqua (Fig 16.), e
misurare le forze che la molecola esercita su queste superfici .
2.16.1.1 TCL Forces
L'idea di base di Tcl Forces è semplice: fornire a NAMD uno script in Tcl il quale
specifica alcune forze da esercitare su certi atomi. Per ottenere delle “barriere semi-
permeabili” basta obbligare solamente gli atomi delle molecole di CS a rimanere in
uno spazio delimitato, permettendo invece alle molecole d’acqua di muoversi
liberamente. Questo è possibile esercitando una forza verso il centro del box in
esame ad ogni atomo della molecola che supera il valore massimo (impostato a
priori) di distanza dal centro. In questo modo si possono confinare le molecole di CS
in uno spazio sempre più limitato aumentando così la concentrazione del GAG.
Calcolando la forza necessaria per contenere le molecole all’interno delle
Fig.16 Simulazione dell’aumento di concentrazione di catene di CS mediante
“membrane semipermeabili” virtuali

60
“membrane semipermeabili” si può ricavare la pressione esercitata dalle catene
poiché le dimensioni del box sono note.
2.16.2 Lunghezza persistente
Per calcolare la lunghezza persistente, quindi capire qual è la lunghezza oltre la quale
si perde una correlazione tra i vettori tangenti alla catena, è necessario svincolare la
catena generata dalla sua configurazione iniziale, e quindi bisogna sostenere una
dinamica in modo da permettere alla catena di disporsi in una conformazione che le
permette di minimizzare la propria energia interna. Sono state lanciate dinamiche
lunghe fino a 100 ns, e sono state utilizzati catene lunghe fino a 20 dimeri. La
procedura adottata per il calcolo della lunghezza persistente nel presente lavoro
consiste innanzitutto nella suddivisione della molecola di CS costituita da 20 dimeri
in diversi segmenti le cui estremità sono gli atomi di Ossigeno che collegano gli
esani tra loro (Fig17).
L’angolo θ è stato volta per volta calcolato come l’angolo formato tra un segmento
lungo la molecola di CS e il segmento iniziale. Sono state quindi effettuate
simulazioni di dinamica molecolare dalle quali sono state estratte le traiettorie della
molecola. In questo modo, sono state raccolte in diversi file di testo le posizioni
assunte dai diversi atomi di ossigeno in differenti istanti temporali (Fig. 18). È stata
poi valutata la deviazione quadratica media della molecola rispetto alla
conformazione inziale. All’inizio della simulazione, la molecola si trova inizialmente
in una configurazione non realistica, poiché la vera configurazione equilibrata è
raggiunta solo dopo un certo periodo di simulazione. Solo le traiettorie relative agli
Fig17. Modello di C4S con in evidenza (colorati in verde) gli atomi di Ossigeno utilizzati per il calcolo
della lunghezza persistente

61
istanti temporali in cui si è in presenza di una configurazione equilibrata sono state
quindi utilizzate nei calcoli successivi. Attraverso un codice sviluppato in Matlab
(disponibile in appendice C) è stato possibile calcolare i valori assunti dagli angoli θ
in ogni istante temporale, e i valori corrispondenti di lunghezza di contorno s,
definita come la somma delle lunghezze dei segmenti considerati. Infine, calcolando
il valore atteso di cosθ, è stato ottenuto un grafico da cui in ascissa è stata posta la
lunghezza di contorno s, mentre in ordinata è stato posto il log<cosθ> cambiato di
segno. Il risultato finale è dato dalla media di tutti gli istanti temporali analizzati. La
lunghezza persistente risulta quindi dall’inverso della pendenza della curva
interpolante i dati ottenuti. Questo script in Matlab è stato testato con un filamento di
collagene, del quale è nota la lunghezza persistente (Fig 19.).
Fig18. Atomi di ossigeno facenti parte della catena di CS in diversi istanti temporali
Fig19. Test dello script con una molecola di collagene

62
Inoltre, è stata calcolata la lunghezza persistente di catene fissate su una lastra d’oro
con distanza relativa di 1,5 e 3 nm (vedi paragrafo successivo).
2.16.3 Esperimenti di resistenza alla compressione
Per effettuare questo esperimento catene di CS sono state disposte su una superficie
d’oro ad una distanza reciproca di 1,5 e 3 nm. Si è scelto di utilizzare come “unità di
base” una superficie di oro contenente 12 catene di CS, composte da 10 dimeri
ciascuna. Per questo, la piastra contenente le catene distanziate 1,5 nm ha
dimensioni pari a 4,4 x 4,9 nm (Fig 20)., mentre la piastra contenente le catene
distanziate tra loro 3,5 nm ha dimensioni pari a 9,1 x 10,2 nm. Per quanto riguarda
l’altezza, la base aurea e le catene di CS raggiungono la quota di 11 nm.
Successivamente il sistema è stato solvatato e si è scelto di creare un box che avesse
un’altezza maggiore delle catene di CS, in modo da poter “alloggiare” più
Fig20. Vista dall’alto della piastra d’oro con le catene di CS
1,5 nm

63
comodamente la tip che poi verrà fatta scendere sul substrato, calcolando la forza che
resiste all’indentazione virtuale. Quindi le dimensioni finali del box sono: 4,4 x 4,9 x
16 nm e 9,1 x 10,2 x 16 nm (Fig 21). Durante le dinamiche ci si è accorti che si
generavano innnaturali “bolle” causate da una bassa concentrazione di molecole
d’acqua. Per questo, nella generazione del box d’acqua si è modificata la variabile
“boundary” che indica la distanza minima che ci deve essere tra le molecole d’acqua
e la molecola da solvatare. In questo modo è stato costruito un box contenente un
numero maggiore di molecole d’acqua. Per essere sicuri di non aver inserito troppe
molecole d’acqua, è stata calcolata la densità nello strato libero da catene di CS, per
11 nm
16 nm
Fig 21. Vista prospettica della piastra d’oro con le catene di CS

64
assicurarsi che questa densità sia pari al valore naturale dell’acqua, cioè 1000
Kg/m3(Fig. 22).
Dopo una necessaria equilibrazione di 10ns, la quale permette alle catene di disporsi
in una conformazione tale per cui l’energia interna alla molecola raggiunga il valore
minore possibile, è stato inserito nel sistema un atomo (denominato TIP) con un
raggio di vdW molto alto (10 Å) in modo da simulare la parte terminale estrema di
una ipotetica tip di un nanoindentatore AFM (Fig 23). Questa tip è stata fatta
scendere sulle catene di substrato, e attraverso uno script di Tcl Forces si è calcolata
la forza che la tip subisce dall’interazione con le catene di CS. Questa forza è stata
poi relazionata alla distanza tip-substrato.
Fig 22. Densità dell’acqua in funzione della distanza: si può notare che ad un altezza parti a 110
nm (dove sono presenti unicamente molecole d’acqua) la densità dell’acqua è pari al valore reale.
0
500
1000
1500
2000
2500
0 20 40 60 80 100 120 140
De
nsi
tà (
Kg/
m^3
)
Distanza (Å)
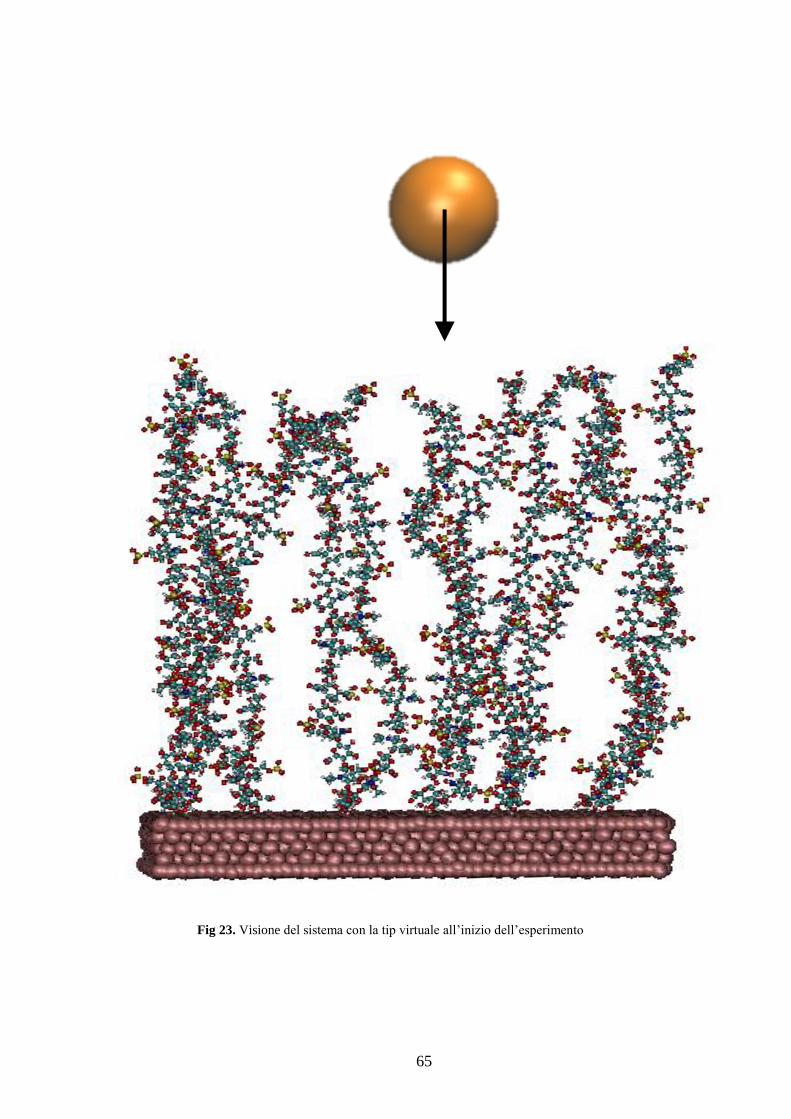
65
Fig 23. Visione del sistema con la tip virtuale all’inizio dell’esperimento

66
CAPITOLO TERZO
RISTULTATI E DISCUSSIONE
3.1 Pressione Osmotica
I risultati ottenuti sono stati confrontati con il precedente studio computazionale
svolto da Bathe [35] e con lo studio sperimentale di Ehrlich [42]. Il confronto
dimostra che il modello sviluppato produce valori di pressione osmotica che sono
dello stesso ordine di grandezza sia dei risultati sperimentali [42] sia del precedente
modello teorico [35]. Rispetto ad entrambi questi studi oltre a consentire lo studio
degli effetti della concentrazione ionica sulla pressione osmotica consente di
discriminare eventuali comportamenti diversi in funzione del tipo di solfatazione
(Fig 24 e Tab 3). I dati mostrano maggiori valori di pressione osmotica a parità di
concentrazione . Questo può essere dovuto al fatto che un modello full-atom è più
preciso nel tener conto dell’interazione che i gruppi carichi della catena di CS hanno
con gli ioni e con le molecole d’acqua e per questo approssima meglio il
comportamento di una catena carica che cerca di opporsi ad un aumento della sua
concentrazione. La distanza maggiore con gli studi precedenti si ha ad alte
concentrazioni. I calcoli fatti non mostrano differenze significative tra CS, C4S e
C6S, anche se a concentrazioni più alte, i sistemi che contengono dimeri solfatati
generano una pressione leggermente maggiore rispetto al sistema composto da
Pressione Osmotica (kPa)
Conc (mg/ml) C0S C4S C6S Bathe [35] Ehrlich [42]
0 0 0 0 0
20 52,36 98,76 54,28 33,79
40 140,08 114,47 119,87 88,19
50,66 23,17
60 236,94 301,70 198,18 167,31
80 428,33 449,27 554,97 277,75
101,33 69,51
253,31 101,94
Tab 3. Confronto della pressione osmotica tra diversi sistemi in funzione della concentrazione.
C0S catena non solfatata, C4S-C6S catena solfatata in posizione 4 o 6;
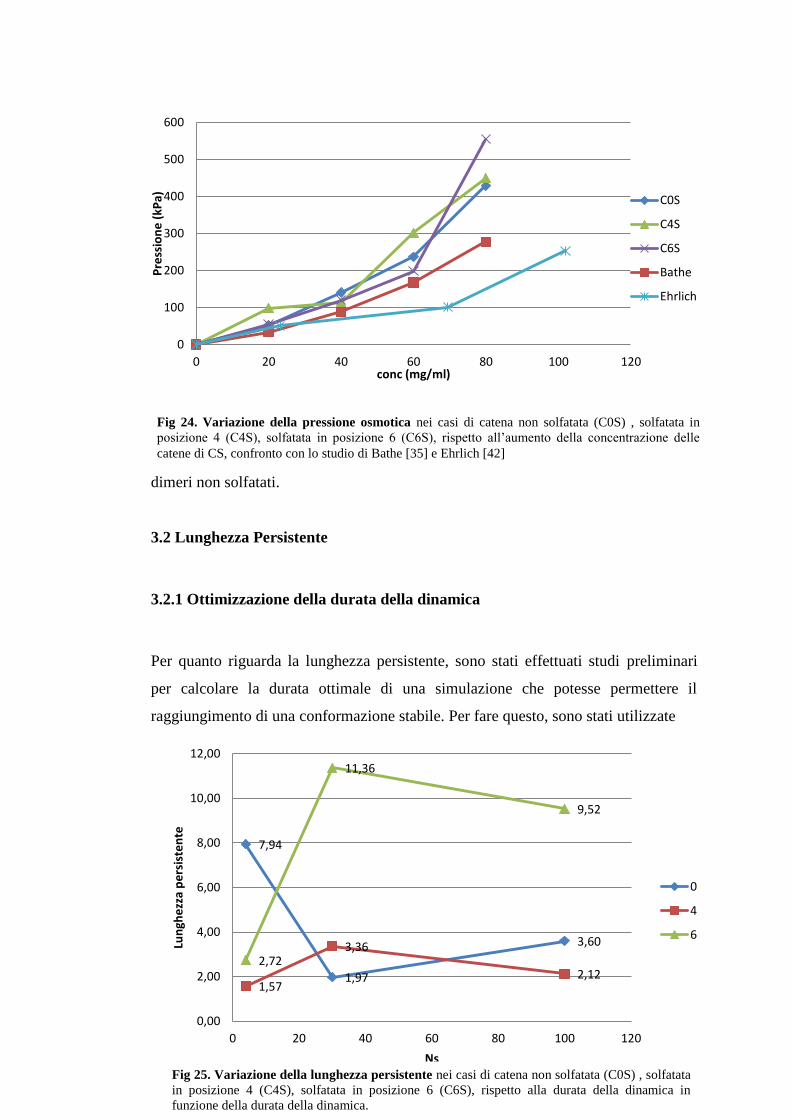
67
dimeri non solfatati.
3.2 Lunghezza Persistente
3.2.1 Ottimizzazione della durata della dinamica
Per quanto riguarda la lunghezza persistente, sono stati effettuati studi preliminari
per calcolare la durata ottimale di una simulazione che potesse permettere il
raggiungimento di una conformazione stabile. Per fare questo, sono stati utilizzate
7,94
1,97
3,60
1,57
3,36
2,12 2,72
11,36
9,52
0,00
2,00
4,00
6,00
8,00
10,00
12,00
0 20 40 60 80 100 120
Lun
ghe
zza
pe
rsis
ten
te
Ns
0
4
6
Fig 25. Variazione della lunghezza persistente nei casi di catena non solfatata (C0S) , solfatata
in posizione 4 (C4S), solfatata in posizione 6 (C6S), rispetto alla durata della dinamica in
funzione della durata della dinamica.
Fig 24. Variazione della pressione osmotica nei casi di catena non solfatata (C0S) , solfatata in
posizione 4 (C4S), solfatata in posizione 6 (C6S), rispetto all’aumento della concentrazione delle
catene di CS, confronto con lo studio di Bathe [35] e Ehrlich [42]
0
100
200
300
400
500
600
0 20 40 60 80 100 120
Pre
ssio
ne
(kP
a)
conc (mg/ml)
C0S
C4S
C6S
Bathe
Ehrlich

68
delle catene composte da 5 dimeri e sono state svolte dinamiche di 3 diverse
lunghezze; una da 4, una da 30 e una da 100 ns. Si è visto confrontando i risultati e il
grafico dell’RMSD che dopo 30ns non vi erano particolari modifiche nella struttura,
e quindi si è deciso di svolgere dinamiche da 50 ns. In Fig 25 si nota che dopo 30
nanosecondi non si hanno cambiamenti sostanziali per quanto riguarda il valore di
lunghezza persistente, poiché si tende a raggiungere un valore di plateau.
Probabilmente dopo 30 ns la molecola raggiunge una conformazione che le permette
di minimizzare la propria energia interna, e quindi è sufficiente considerare e
analizzare la traiettoria della catena solo dopo questo tempo per poter estrapolare il
valore della lunghezza persistente.
3.2.2 Ottimizzazione della lunghezza della catena
Sono stati svolti successivi studi per capire qual era la lunghezza ottimale per
simulare un comportamento il più possibile fisiologico della catena. Questo
parametro è importante poiché una catena troppo corta potrebbe portare a risultati
non fisiologici a causa di disturbi degli estremi con le molecole d’acqua, ma una
catena troppo lunga richiederebbe tempi computazionali esageratamente onerosi.
Quindi sono state eseguite delle dinamiche da 50 ns con catene lunghe 5, 10 e 20
dimeri. Come nella ricerca della lunghezza temporale ottimale, i risultati hanno detto
3,75 3,60
6,71
5,53
3,36
5,03
10,31
11,36
10,42
0,00
2,00
4,00
6,00
8,00
10,00
12,00
0 5 10 15 20 25
Lun
ghe
zza
pe
rsis
ten
te
numero dimeri
0
4
6
Fig 26. Variazione della lunghezza persistente nei casi di catena non solfatata (C0S) ,
solfatata in posizione 4 (C4S), solfatata in posizione 6 (C6S), rispetto al numero di
dimeri della catena

69
che un numero di dimeri pari a 20 minimizza i disturbi alle estremità (Fig. 26), ma
durante queste dinamiche sono stati riscontrati due comportamenti particolari della
dinamica, che di seguito analizzerò più nel dettaglio.
3.2.3 Kinking
Durante le dinamiche delle catene contenenti gruppi fosfati (C4S e C6S) si è notato
un deciso cambiamento conformazionale di uno o più esani (Fig 27), i quali sono
passati dalla conformazione chiamata “a sedia”, energeticamente più favorevole, alla
conformazione denominata “a barca”; probabilmente questo è dovuto alla repulsione
reciproca tra gruppi fosfati carichi; questo kinking ha quindi pesantemente ridotto il
valore di lunghezza persistente. Inoltre il mantenimento di questa conformazione nel
tempo può far pensare a uno stato molto stabile della catena, che una volta che si è
“piegata” difficilmente riesce a tornare a una configurazione più lineare. Questo
fenomeno è presente anche nelle catene di CS non solfatate, ma in numero minore.
Questo effetto è già stato studiato da Haverkamp[70, 71].
Fig 27. Esempio di kinking; nel dettaglio è mostrato lo zucchero che subisce la modifica
conformazionale, “piegando” l’intera catena

70
3.2.4 Conformazione a elica
Durante le dinamiche quasi tutte le catene hanno raggiunto una conformazione che
prevede una certa elicità, fatto già notato e calcolato da studi precedenti [51].
Probabilmente, è questa particolare conformazione che agisce in maniera
predominante nel raggiungimento della sua lunghezza persistente. Quindi la
lunghezza persistente della catena potrebbe essere dovuta a questa particolare
conformazione, e non tanto alla presenza di gruppi carichi negativamente che si
oppongono e mantengono dritta la catena.
3.2.5 Esperimenti
Come esperimenti principali ne sono stati scelti 28, rappresentati nella tabella 4 divisi
per concentrazione ionica e grado di solfatazione.
Conf1 0.05 M Conf2 0.05 M Conf1 0.15 M Conf2 0.15M
C0S C0S C0S C0S
C4S C4S C4S C4S
C6S C6S C6S C6S
C04S C04S C04S C04S
C06S C06S C06S C06S
C004S C004S C004S C004S
C006S C006S C006S C006S
“Conf 1” e “Conf 2” sono due diverse conformazioni della catena, ottenuta lanciando
due dinamiche in vuoto indipendenti. Gli esperimenti “C0S” “C4S” e “C6S”
prevedono una catena lunga 20 dimeri costituita interamente da dimeri non solfatati,
solfatati in posizione 4 e solfatati in posizione 6. Gli esperimenti “C04S” e “C06S”
sono costituiti da una catena lunga 20 dimeri composta dal 50% di dimeri non
solfatati e dal restante 50% da dimeri solfatati rispettivamente in posizione 4 e 6 .
Tab. 4 Esperimenti effettuati

71
Gli esperimenti “C004S” e “C006S” contengono catene lunghe 21 dimeri che hanno
una percentuale di solfatazione pari al 30%. Durante questi calcoli sono stati
riscontrati due diversi effetti fisiologici della catena, che quindi validano ancora una
volta il modello.Quindi, una volta deciso la lunghezza della catena e la durata della
dinamica, sono stati svolti 28 esperimenti nei quali varia la concentrazione ionica di
NaCL, e la percentuale di dimeri solfatati presenti nella catena. La tabella 5 riassume
i risultati di tutti gli esperimenti.
Lunghezza persistente (nm)
0,05 M 0,15 M
Tanaka
1978
[52]
Rodriguez
Carajal
2002 [51]
Ng
2003
[5]
Bathe
2005
[20]
0 7,75 ± 3,11 8,58 ± 1,99 11
004 10,02 ±1,54 5,60 ± 1,19
04 7,54 ± 3,10 9,59 ± 1,59
4 6,09 ± 1,75 4,16 ± 1,47 5 12 17-25 9,6 ± 0,6
006 8,81 ± 2,02 3,83 ± 0,68
06 9,19 ± 2,62 5,67 ± 1,13
6 7,25 ± 1,53 7,75± 0,63 4,8 9 10-21 7,1 ± 0,4
3.2.6 Confronto tra diverse percentuali di solfatazione in catene isolate
3.2.6.1 Molarità bassa
Con una concentrazione ionica bassa (0.05 M) non ci sono sostanziali differenze tra
la catena non solfatata e le catene solfatate (Fig. 28) ; questo probabilmente è dovuto
al fatto che ci sono pochi ioni che possono interagire con le catene per permettere ai
gruppi solfatati di respingersi e aumentare la rigidità della catena. Inoltre i fenomeni
del kinking e della conformazione a elica possono giustificare un comportamento
Tab. 5 Valori di lunghezza persistente; sono qui rappresentati i valori medi di lunghezza persistente in
funzione della concentrazione molare e della percentuale di solfatazione confrontati con gli studi precedenti: 0
catena non solfatata, 4-6 catena solfatata in posizione 4 o 6; 04-06 catena che presenta il 50% di
dimeri solfatati in posizione 4 o 6 e il restante non solfatato; 004-006 catena che presenta il 30% dei dimeri
solfatati in posizione 4 o 6 e il restante non solfatato.

72
così variabile e difficilmente prevedibile della lunghezza persistente delle catene in
esame.
3.2.6.1 Molarità fisiologica
Per quanto riguarda la molarità fisiologica invece, si nota che il condroitin solfatato
in posizione 4 possiede una lunghezza persistente maggiore rispetto agli altri (Fig.
29) Questo è vero quando la concentrazione dei gruppi solfatati è pari al 50%.
Probabilmente, il fatto che una concentrazione di gruppi solfatati porti a una
lunghezza persistente minore può sembrare una elemento di contraddizione, ma c’è
da tenere in conto il fatto che una presenza così massiccia di gruppi solfati aumenta
in maniera considerevole la possibilità che uno zucchero passi dalla conformazione
“a sedia” a quella “a barca”, facendo così crollare il valore di lunghezza persistente.
Comunque, in tutti e due i casi si è notata una forte variabilità per quanto riguarda i
valori di lunghezza persistente.
Fig 28. Variazione della lunghezza persistente: nei casi di catena non solfatata (C0S) , solfatata in
posizione 4 (C4S), solfatata in posizione 6 (C6S), rispetto alla percentuale di dimeri solfatati, con
concentrazione ionica pari a 0.05 M
7,75
10,02
7,54
6,09
8,81 9,19
7,25
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
0,5 M
C0S
C4S
C6S

73
3.2.7 Confronto tra diverse percentuali di solfatazione in set di catene
Per capire quanto il fenomeno del kinking potesse inficiare sulla lunghezza
persistente si è provata un’altra via. È stato calcolato il valore di lunghezza
persistente prendendo in esame i sistemi utilizzati per la nanoindentazione, che sono
formati da più catene. In questo caso però la catena non è completamente libera di
muoversi, poiché, per evitare lo sfaldamento del set di catene, è stato impedito il
movimento dell’atomo più vicino alla superficie d’oro. Quindi i valori ottenuti
utilizzando questo set sperimentale non possono essere confrontati con i precedenti,
ma si può sostenere che questo tipo di modellizzazione simuli il segmenti di CS
prossimale al core proteico dell’aggrecano. I risultati riassunti nella tabella 6
mostrano che c’è una forte differenza tra la catena non solfatata e quelle solfatate.
Guardando l’elevato valore della deviazione standard si evidenzia una forte
variabilità dei risultati.
Lunghezza Persistente (nm) - 10 dimeri - 0,15 M
Spacing 1,5 nm Spacing 3 nm
C0S 9,17 ± 4 8,04 ± 5,29
C4S 15,68 ± 5,62 16,37 ± 7,79
C6S 24,38 ± 14,25 15,25 ± 7,22
Tab 6. Lunghezza persistente di set di catene distanziate reciprocamente 1,5 nm e 3 nm nei casi
di catena non solfatata (C0S) , solfatata in posizione 4 (C4S), solfatata in posizione 6 (C6S)
8,58
5,60
9,59
4,16 3,83
5,67
7,75
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
0% 20% 40% 60% 80% 100%
0,15 M
C0S
C4S
C6S
Fig 29. Variazione della lunghezza persistente: nei casi di catena non solfatata (C0S) ,
solfatata in posizione 4 (C4S), solfatata in posizione 6 (C6S), rispetto alla percentuale di
dimeri solfatati, con concentrazione ionica pari a 0.15 M

74
3.3 Esperimenti di resistenza alla compressione
Il valori ottenuti sono stati confrontati col precedente valore di Seog [64], che però
effettua l’esperimento ad una molarità inferiore (0,001 M rispetto a 0,15)
3.3.1 Catene distanziate 1.5 nm
Per questo particolare set sperimentale si è notato che il valore massimo di forza si
ottiene appena la tip si avvicina alle estremità delle catene; per quanto riguarda i
valori massimi, si evince che il set composto da catene non solfatate oppone una
forza minore rispetto ai set composti da catene solfatate, e tra queste due, il set che
presenta catene con dimeri solfatati in posizione 4 ha un valore di forza maggiore.
Una volta che la tip ha “bucato” la superficie formata dagli estremi delle catene, la
forza scende rapidamente verso un valore nullo, probabilmente perché la tip scende
nello spazio tra le due catene, visto che è energeticamente favorevole.
3.3.1 Catene distanziate 3 nm
Per quanto riguarda i sistemi contenenti catene spaziate 3 nm il comportamento è
differente, infatti il valore massimo è inferiore rispetto al sistema che presenta uno
spacing tra le catene di CS di 1,5 nm. Inoltre si è riscontrato che le dimensioni della
tip non sono sufficienti per permettere una completa integrazione della tip con le
catene di CS spaziate 3 nm. I risultati sono in Tab 7 e in Fig 30 è descritto
l’andamento caratteristico del valore della forza agente sulla tip rispetto alla
coordinata Z (sempre della tip).
Forza (pN)
Spacing 1,5 nm Spacing 3 nm Seog 2002 [64]
C0S 324,12 216,51
C4S 495,62 163,32 203*
C6S 454,25 341,95
Tab 7. Esperimenti di resistenza alla compressione. Valori di forza ottenuti conspacing tra
catene pari a 1,5 nm nei casi di catena non solfatata (C0S) , solfatata in posizione 4 (C4S),
solfatata in posizione 6 (C6S). * valore ottenuto a molarità 0.001 M

75
a
b
-200
-100
0
100
200
300
400
500
600
700
0 20 40 60 80 100 120 140 160
F (p
N)
Z (Å)
Fig 30. Forze agenti sulla tip. Andamento della forza resistente alla tip della nanoindentazione
virtuale. Nel grafico si raggiunge un valore di massimo relativo quando la tip è in prossimità degli
estremi delle catene di CS. Inoltre si ha un valore massimo assoluto quando la tip è prossima alla
superficie in oro.

76
CAPITOLO QUARTO
CONCLUSIONI E SVILUPPI FUTURI
Per quanto riguarda lo studio del tessuto cartilagineo, è ormai chiaro che una
completa conoscenza della struttura e delle funzioni a livello molecolare è
fondamentale per poter procedere allo sviluppo di tecnologie da utilizzare per
risolvere patologie che attentano a un sereno stile di vita. Infatti il danneggiamento
del tessuto cartilagineo è un problema che affligge 36 milioni di persone solamente
negli USA [72]. Le cause principali delle lesioni e dei difetti articolari sono da
ricercarsi nei processi degenerativi dovuti all’invecchiamento, al manifestarsi di
patologie quali l’osteoartrite o conseguenti ad eventi traumatici. Il crescente sviluppo
dell’ingegneria dei tessuti potrebbe portare all’introduzione di nuove tecniche per
curare e risolvere queste patologie, sostituendo il tessuto degenerato non con
materiali artificiali ma con tessuto omologo, rigenerato partendo dalle cellule del
paziente, il che porterebbe a una completa e totale ripresa della struttura e della
funzionalità della cartilagine. Per poter indagare l'effetto di particolari nano-
caratteristiche sul comportamento a compressione di tutta la molecola, è necessario
un approccio che permetta di raggiungere un livello di dettaglio atomistico.
L’utilizzo di strumenti di modellistica molecolare per simulare lavori sperimentali
permette di utilizzare metodi consolidati ed efficaci avendo la possibilità di lavorare
su di un sistema che presenta valori fisici di partenza certi e che porta a risultati
estremamente precisi e che dipendono unicamente dalla variazione di uno solo tra gli
svariati parametri che vanno considerati quando si analizza una molecola. Questo
studio ha evidenziato come una analisi virtuale del comportamento delle catene di
CS sia uno strumento efficace per eseguire indagini a livello molecolare sia di
singole molecole sia di sistemi composti da più molecole. É importante quindi
continuare ad analizzare questo GAG sempre tenendo conto che le sue proprietà sono
date sì dalla struttura molecolare, ma anche dall’organizzazione generale del sistema
che le circonda. Infatti la struttura dell’aggrecano ci suggerisce che il CS svolge un
ruolo importante nella cartilagine proprio perché è organizzato in sistemi che
possono presentare fino a 100 catene. Dal punto di vista modellistico un sistema

77
formato da così tanti atomi richiede un investimento tecnologico (ad oggi)
decisamente importante. Questo fatto non deve spaventare poiché negli ultimi
trent’anni siamo stati testimoni di uno sviluppo tecnologico incredibile, che ha
portato a un grande miglioramento della velocità di calcolo. Visto che questo
progresso procede in maniera esponenziale, si può sostenere che in un futuro sarà
possibile analizzare e caratterizzare il comportamento meccanico di una molecola
molto complessa, come quella dell’aggecano. Se nel ‘59, Alder e Wainwright [73] si
fossero abbattuti poiché con gli strumenti del tempo era possibile per loro analizzare
unicamente il comportamento di un numero esiguo di atomi, forse non si sarebbe
partiti con lo sviluppo di metodi computazionali per l’analisi di molecole e non si
sarebbe potuto svolgere questo lavoro. Uno studio completo dell’aggrecano potrà
fornire criteri per progettare razionalmente nuovi sostituenti biomimetici per la ECM,
i quali forniranno proprietà strutturali e meccaniche simili a quelle dei PG presenti in
natura. Così facendo, si potranno generare scaffolds che siano simili come struttura e
composizione alla ECM naturale, in modo da poter inserire i condrociti in un
ambiente ottimale per il loro sviluppo, ed evitare la frequente e, per ora, difficilmente
evitabile, perdita del fenotipo. Per questi motivi, lo studio dei costituenti della ECM
potrà servire a sviluppare scaffolds che aiutino a evitare questi problemi, poiché
mimerebbero l’ambiente naturale in cui le cellule potranno svilupparsi in maniera
fisiologica e corretta.

78
BIBLIOGRAFIA
1. Cilpa, G., et al., Atomistic insight into chondroitin-6-sulfate
glycosaminoglycan chain through quantum mechanics calculations and
molecular dynamics simulation. Journal of computational chemistry, 2010.
31(8): p. 1670-80.
2. Marascio, M., Progettazione in silico di nuove molecole per la cartilagine
ingegnerizzata, 2011, Politecnico di Milano.
3. Eyre, D., Collagen of articular cartilage. Arthritis research, 2002. 4(1): p. 30-
5.
4. Heinegard, D., Proteoglycans and more--from molecules to biology.
International journal of experimental pathology, 2009. 90(6): p. 575-86.
5. Ng, L., et al., Individual cartilage aggrecan macromolecules and their
constituent glycosaminoglycans visualized via atomic force microscopy.
Journal of Structural Biology, 2003. 143(3): p. 242-257.
6. Hardingham, T.E. and H. Muir, The specific interaction of hyaluronic acid
with cartillage proteoglycans. Biochimica et biophysica acta, 1972. 279(2): p.
401-5.
7. Maroudas, A., et al., Studies of hydration and swelling pressure in normal
and osteoarthritic cartilage. Biorheology, 1985. 22(2): p. 159-69.
8. Maroudas, A., et al., [Biochemical and physico-chemical studies on
osteoarthrosis in relation to some clinical aspects]. Der Orthopade, 1983.
12(2): p. 109-18.
9. Ziv, I., et al., Human facet cartilage: swelling and some physicochemical
characteristics as a function of age. Part 2: Age changes in some biophysical
parameters of human facet joint cartilage. Spine, 1993. 18(1): p. 136-46.
10. Williamson, A.K., A.C. Chen, and R.L. Sah, Compressive properties and
function-composition relationships of developing bovine articular cartilage.
Journal of orthopaedic research : official publication of the Orthopaedic
Research Society, 2001. 19(6): p. 1113-21.
11. Zhu, W., et al., Viscoelastic shear properties of articular cartilage and the
effects of glycosidase treatments. Journal of orthopaedic research : official
publication of the Orthopaedic Research Society, 1993. 11(6): p. 771-81.
12. Cowman, M.K., M. Li, and E.A. Balazs, Tapping mode atomic force
microscopy of hyaluronan: extended and intramolecularly interacting chains.
Biophysical journal, 1998. 75(4): p. 2030-7.
13. Mikecz, K., et al., Anti-CD44 treatment abrogates tissue oedema and
leukocyte infiltration in murine arthritis. Nature medicine, 1995. 1(6): p. 558-
63.
14. Tadmor, R., N. Chen, and J.N. Israelachvili, Thin film rheology and lubricity
of hyaluronic acid solutions at a normal physiological concentration. Journal
of biomedical materials research, 2002. 61(4): p. 514-23.
15. Hills, B.A., Boundary lubrication in vivo. Proceedings of the Institution of
Mechanical Engineers. Part H, Journal of engineering in medicine, 2000.
214(1): p. 83-94.

79
16. Mori, S., M. Naito, and S. Moriyama, Highly viscous sodium hyaluronate and
joint lubrication. International orthopaedics, 2002. 26(2): p. 116-21.
17. Jarvelainen, H.T., et al., Differential expression of small
chondroitin/dermatan sulfate proteoglycans, PG-I/biglycan and PG-
II/decorin, by vascular smooth muscle and endothelial cells in culture. The
Journal of biological chemistry, 1991. 266(34): p. 23274-81.
18. Tovar, A.M., et al., Age-related changes in populations of aortic
glycosaminoglycans: species with low affinity for plasma low-density
lipoproteins, and not species with high affinity, are preferentially affected.
Arteriosclerosis, thrombosis, and vascular biology, 1998. 18(4): p. 604-14.
19. Bayliss, M.T., et al., Sulfation of chondroitin sulfate in human articular
cartilage. The effect of age, topographical position, and zone of cartilage on
tissue composition. The Journal of biological chemistry, 1999. 274(22): p.
15892-900.
20. Bathe, M., et al., A coarse-grained molecular model for glycosaminoglycans:
application to chondroitin, chondroitin sulfate, and hyaluronic acid.
Biophysical journal, 2005. 88(6): p. 3870-87.
21. Cordomi, A., et al., Study of the conformational profile of the norbornane
analogues of phenylalanine. Journal of peptide science : an official
publication of the European Peptide Society, 2002. 8(6): p. 253-66.
22. Buschmann, M.D. and A.J. Grodzinsky, A molecular model of proteoglycan-
associated electrostatic forces in cartilage mechanics. Journal of
biomechanical engineering, 1995. 117(2): p. 179-92.
23. Wiberg, C., et al., Complexes of matrilin-1 and biglycan or decorin connect
collagen VI microfibrils to both collagen II and aggrecan. The Journal of
biological chemistry, 2003. 278(39): p. 37698-704.
24. Hedbom, E. and D. Heinegard, Binding of fibromodulin and decorin to
separate sites on fibrillar collagens. The Journal of biological chemistry,
1993. 268(36): p. 27307-12.
25. Kalamajski, S. and A. Oldberg, Homologous sequence in lumican and
fibromodulin leucine-rich repeat 5-7 competes for collagen binding. The
Journal of biological chemistry, 2009. 284(1): p. 534-9.
26. Kalamajski, S., et al., Asporin competes with decorin for collagen binding,
binds calcium and promotes osteoblast collagen mineralization. The
Biochemical journal, 2009. 423(1): p. 53-9.
27. Morrell, K.C., et al., Corroboration of in vivo cartilage pressures with
implications for synovial joint tribology and osteoarthritis causation.
Proceedings of the National Academy of Sciences of the United States of
America, 2005. 102(41): p. 14819-24.
28. Maroudas, A., Adult Articular Cartilage. Physicochemical properties of
articular cartilage, ed. F. MAR1979.
29. Van de Velde, S.K., et al., Increased tibiofemoral cartilage contact
deformation in patients with anterior cruciate ligament deficiency. Arthritis
and rheumatism, 2009. 60(12): p. 3693-702.
30. Forster, H. and J. Fisher, The influence of loading time and lubricant on the
friction of articular cartilage. Proceedings of the Institution of Mechanical
Engineers. Part H, Journal of engineering in medicine, 1996. 210(2): p. 109-
19.

80
31. Gleghorn, J.P. and L.J. Bonassar, Lubrication mode analysis of articular
cartilage using Stribeck surfaces. Journal of biomechanics, 2008. 41(9): p.
1910-8.
32. Ateshian, G.A., The role of interstitial fluid pressurization in articular
cartilage lubrication. Journal of biomechanics, 2009. 42(9): p. 1163-76.
33. Raviv, U., et al., Lubrication by charged polymers. Nature, 2003. 425(6954):
p. 163-5.
34. Schmidt, T.A. and R.L. Sah, Effect of synovial fluid on boundary lubrication
of articular cartilage. Osteoarthritis and cartilage / OARS, Osteoarthritis
Research Society, 2007. 15(1): p. 35-47.
35. Bathe, M., et al., Osmotic pressure of aqueous chondroitin sulfate solution: a
molecular modeling investigation. Biophysical journal, 2005. 89(4): p. 2357-
71.
36. Nap, R.J. and I. Szleifer, Structure and interactions of aggrecans: statistical
thermodynamic approach. Biophysical journal, 2008. 95(10): p. 4570-83.
37. Reynaud, B. and T.M. Quinn, Anisotropic hydraulic permeability in
compressed articular cartilage. Journal of biomechanics, 2006. 39(1): p. 131-
7.
38. Eisenberg, S.a.G., AJ,, Electrokinetic micromodel of extracellular matrix and
other polyelectrolyte networks. PhysicoChemical Hydrodynamics, 1988(10):
p. 517-539.
39. Guilak, F., A. Ratcliffe, and V.C. Mow, Chondrocyte deformation and local
tissue strain in articular cartilage: a confocal microscopy study. Journal of
orthopaedic research : official publication of the Orthopaedic Research
Society, 1995. 13(3): p. 410-21.
40. Maroudas, A.I., Balance between swelling pressure and collagen tension in
normal and degenerate cartilage. Nature, 1976. 260(5554): p. 808-9.
41. Grodzinsky, A.J., Electromechanical and physicochemical properties of
connective tissue. Critical reviews in biomedical engineering, 1983. 9(2): p.
133-99.
42. Ehrlich, S., et al., The osmotic pressure of chondroitin sulphate solutions:
experimental measurements and theoretical analysis. Biorheology, 1998.
35(6): p. 383-97.
43. Comper, W.D. and T.C. Laurent, Physiological function of connective tissue
polysaccharides. Physiological reviews, 1978. 58(1): p. 255-315.
44. Williams, R.P. and W.D. Comper, Osmotic flow caused by polyelectrolytes.
Biophysical chemistry, 1990. 36(3): p. 223-34.
45. Peitzsch, R.M. and W.F. Reed, High osmotic stress behavior of hyaluronate
and heparin. Biopolymers, 1992. 32(3): p. 219-38.
46. Urban, J.P., et al., Swelling pressures of proteoglycans at the concentrations
found in cartilaginous tissues. Biorheology, 1979. 16(6): p. 447-64.
47. Chu, J.W. and G.A. Voth, Coarse-grained modeling of the actin filament
derived from atomistic-scale simulations. Biophysical journal, 2006. 90(5): p.
1572-82.
48. Buckwalter, J.A. and L.C. Rosenberg, Electron microscopic studies of
cartilage proteoglycans. Direct evidence for the variable length of the
chondroitin sulfate-rich region of proteoglycan subunit core protein. The
Journal of biological chemistry, 1982. 257(16): p. 9830-9.

81
49. Morgelin, M., et al., Cartilage proteoglycans. Assembly with hyaluronate and
link protein as studied by electron microscopy. The Biochemical journal,
1988. 253(1): p. 175-85.
50. Rosenberg, L., W. Hellmann, and A.K. Kleinschmidt, Macromolecular
models of proteinpolysaccharides from bovine nasal cartilage based on
electron microscopic studies. The Journal of biological chemistry, 1970.
245(16): p. 4123-30.
51. Rodriguez-Carvajal, M.A., A. Imberty, and S. Perez, Conformational
behavior of chondroitin and chondroitin sulfate in relation to their physical
properties as inferred by molecular modeling. Biopolymers, 2003. 69(1): p.
15-28.
52. Tanaka, K., Physicochemical properties of chondroitin sulfate. I. Ion binding
and secondary structure. Journal of biochemistry, 1978. 83(3): p. 647-53.
53. Flory, P.J., Statistical Mechanics of Chain Molecules. Oxford University
Press, New York, 1988.
54. Oliver, W.C. and G.M. Pharr, An improved technique for determining
hardness and elastic modulus using load and displacement sensing
indentation experiments. Journal of Materials Research, 1992. 7(06): p. 1564-
1583.
55. Loening, A.M., et al., Injurious mechanical compression of bovine articular
cartilage induces chondrocyte apoptosis. Archives of biochemistry and
biophysics, 2000. 381(2): p. 205-12.
56. Miller, G.J.M., E.F., Use of Microindentation to Characterize the Mechanical
Properties of Articular Cartilage: Comparison of Biphasic Material
Properties Across Length Scales. 2010. 18(8): p. 1051-1057.
57. Mak, A.F., The apparent viscoelastic behavior of articular cartilage--the
contributions from the intrinsic matrix viscoelasticity and interstitial fluid
flows. Journal of biomechanical engineering, 1986. 108(2): p. 123-30.
58. Lewis, J.L. and S.L. Johnson, Collagen architecture and failure processes in
bovine patellar cartilage. Journal of anatomy, 2001. 199(Pt 4): p. 483-92.
59. Hertz, H., Über die berührung fester elastischer Körper (On the contact of
rigid elastic solids). J. reine und angewandte Mathematik, 1896. 92: p. 156.
60. Lin, C.D.H.A., Nanomechanics of polymer gels and biological tissues: A
critical review of analytical approaches in the Hertzian regime and beyond
Soft Matter 2008. 4: p. 669-682.
61. Johnson , K.K., K; Roberts, AD, Surface Energy and the Contact of Elastic
Solids. Proc. R. Soc. Lond. A 1971. 324(1558): p. 301-313.
62. Derjaguin, B.V., V.M. Muller, and Y.P. Toporov, Effect of contact
deformations on the adhesion of particles. J. Colloid Interface Sci, 1975. 53:
p. 314-326.
63. Buschmann, M.D., et al., Stimulation of aggrecan synthesis in cartilage
explants by cyclic loading is localized to regions of high interstitial fluid flow.
Archives of biochemistry and biophysics, 1999. 366(1): p. 1-7.
64. Seog, J., et al., Direct Measurement of Glycosaminoglycan Intermolecular
Interactions via High-Resolution Force Spectroscopy. Macromolecules,
2001. 35: p. 5601-5615.
65. Seror, J., et al., Articular cartilage proteoglycans as boundary lubricants:
structure and frictional interaction of surface-attached hyaluronan and

82
hyaluronan--aggrecan complexes. Biomacromolecules, 2011. 12(10): p.
3432-43.
66. Ercolessi, F., A Molecular Dynamics Primer. Spring College in
Computational Physics ICTP, Trieste, 1997.
67. Brooks, B.R., et al., CHARMM: the biomolecular simulation program.
Journal of computational chemistry, 2009. 30(10): p. 1545-614.
68. Garemyr, R. and A. Elofsson, Study of the electrostatics treatment in
molecular dynamics simulations. Proteins, 1999. 37(3): p. 417-28.
69. Solmajer, T. and E.L. Mehler, Electrostatic screening in molecular dynamics
simulations. Protein engineering, 1991. 4(8): p. 911-7.
70. Haverkamp, R.G., A.T. Marshall, and M.A. Williams, Model for stretching
elastic biopolymers which exhibit conformational transformations. Physical
review. E, Statistical, nonlinear, and soft matter physics, 2007. 75(2 Pt 1): p.
021907.
71. Haverkamp, R.G., M.A. Williams, and J.E. Scott, Stretching single molecules
of connective tissue glycans to characterize their shape-maintaining
elasticity. Biomacromolecules, 2005. 6(3): p. 1816-8.
72. Temenoff, J.S. and A.G. Mikos, Review: tissue engineering for regeneration
of articular cartilage. Biomaterials, 2000. 21(5): p. 431-40.
73. Alder, B.J. and W. T.E., Studies in Molecular Dynamics. I. General Method
Journal of Chemical Physics 1959. 31(2).

83
APPENDICE A - SCRIPT DI GAGSBUILDER
#!/bin/bash
export LANG=en_US.UTF-8
rm -r Final
rm -r Working
mkdir Working
cd Working
clear
token=0
echo "Welcome in GAGsBuilder."
echo -n "
Do you want to build Chondroitin [c] or Dermatan [d] ? "
read std
clear
case $std in
'c')
echo -n "Enter chondroitin sequence (use 0,4,6): "
read sequence
len=$(expr length $sequence)
for((j=0;$j<$len;j=$(($j+1))));do
resid1=$(echo '1+('$j' * 2)' | bc)
resid2=$(echo '1+('$resid1')' | bc)
disacch=${sequence:$j:1}
case $disacch in
'0')

84
x=$(echo '('$j'*0.904)' | bc)
y=0
z=0
cp ../C0_Coordinates/CGlcn.gro secondC.gro
cp ../C0_Coordinates/CGalNAc.gro secondS.gro
editconf -f secondC.gro -translate $x $y $z -o a.gro
cat a.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid1'"GLC",$2,$3,$4,$5,$6}' > $resid1.gro
rm temp.out
editconf -f secondS.gro -translate $x $y $z -o b.gro
cat b.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid2'"GAX",$2,$3,$4,$5,$6}' > $resid2.gro
rm temp.out
;;
'4')
x=$(echo '('$j'*0.904)' | bc)
y=0
z=0
cp ../C4_Coordinates/CGlcn.gro secondC.gro
cp ../C4_Coordinates/CGalNAc.gro secondS.gro
editconf -f secondC.gro -translate $x $y $z -o a.gro
cat a.gro | sed -e '1,2d' -e '$d' >temp.out

85
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid1'"GLC",$2,$3,$4,$5,$6}' > $resid1.gro
rm temp.out
editconf -f secondS.gro -translate $x $y $z -o b.gro
cat b.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid2'"GA4",$2,$3,$4,$5,$6}' > $resid2.gro
rm temp.out
;;
'6')
x=$(echo '('$j'*0.904)' | bc)
y=0
z=0
cp ../C6_Coordinates/CGlcn.gro secondC.gro
cp ../C6_Coordinates/CGalNAc.gro secondS.gro
editconf -f secondC.gro -translate $x $y $z -o a.gro
cat a.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid1'"GLC",$2,$3,$4,$5,$6}' > $resid1.gro
rm temp.out
editconf -f secondS.gro -translate $x $y $z -o b.gro
cat b.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid2'"GA6",$2,$3,$4,$5,$6}' > $resid2.gro
rm temp.out
;;

86
*)
echo '*****************************'
echo '* Wrong sequence, quitting! *'
echo '*****************************'
exit;;
esac
done;;
'd')
echo -n "Enter dermatan sequence (use 0,4,6): "
read sequence
len=$(expr length $sequence)
for((j=0;$j<$len;j=$(($j+1))));do
resid1=$(echo '1+('$j' * 2)' | bc)
resid2=$(echo '1+('$resid1')' | bc)
disacch=${sequence:$j:1}
case $disacch in
'0')
x=$(echo '('$j'*0.904)' | bc)
y=0
z=0
cp ../D0_Coordinates/CIda.gro secondC.gro
cp ../D0_Coordinates/CGalNAc.gro secondS.gro
editconf -f secondC.gro -translate $x $y $z -o a.gro
cat a.gro | sed -e '1,2d' -e '$d' >temp.out

87
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid1'"GLC",$2,$3,$4,$5,$6}' > $resid1.gro
rm temp.out
editconf -f secondS.gro -translate $x $y $z -o b.gro
cat b.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid2'"GAX",$2,$3,$4,$5,$6}' > $resid2.gro
rm temp.out
;;
'4')
x=$(echo '('$j'*0.904)' | bc)
y=0
z=0
cp ../D4_Coordinates/CIda.gro secondC.gro
cp ../D4_Coordinates/CGalNAc.gro secondS.gro
editconf -f secondC.gro -translate $x $y $z -o a.gro
cat a.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid1'"GLC",$2,$3,$4,$5,$6}' > $resid1.gro
rm temp.out
editconf -f secondS.gro -translate $x $y $z -o b.gro
cat b.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid2'"GA4",$2,$3,$4,$5,$6}' > $resid2.gro
rm temp.out
;;

88
'6')
x=$(echo '('$j'*0.904)' | bc)
y=0
z=0
cp ../D6_Coordinates/CIda.gro secondC.gro
cp ../D6_Coordinates/CGalNAc.gro secondS.gro
editconf -f secondC.gro -translate $x $y $z -o a.gro
cat a.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid1'"GLC",$2,$3,$4,$5,$6}' > $resid1.gro
rm temp.out
editconf -f secondS.gro -translate $x $y $z -o b.gro
cat b.gro | sed -e '1,2d' -e '$d' >temp.out
cat temp.out | gawk '{printf "%8s%7s%5.0f%8.3f%8.3f%8.3f\n",
'$resid2'"GA6",$2,$3,$4,$5,$6}' > $resid2.gro
rm temp.out
;;
*)
echo '*****************************'
echo '* Wrong sequence, quitting! *'
echo '*****************************'
exit;;
esac
done;;
*)

89
echo '*****************************'
echo '* Error *'
echo '*****************************'
exit;;
esac
### Merge Chains
totres=$(echo $len'*2' | bc)
seq=$(seq 1 $totres)
for i in $(echo $seq) ; do
cat $i.gro >>tempmol.out
done
number=$(echo $(cat tempmol.out | wc -l))
echo Chain of Chondroitin Sulphate >name.out
echo $number >number.out
echo " 50.00000 50.00000 50.00000" >dim.out
cat name.out number.out tempmol.out dim.out >chain.gro
rm tempmol.out name.out dim.out number.out
editconf -f chain.gro -o chain.pdb
mkdir ../Final
mv chain.pdb ../Final/
cd ..
rm -r Working
### PSF generator

90
vmd Final/chain.pdb -dispdev text -e psfgen.tcl
clear
echo "
Your chain has been generated successfully.
May the forcefield be with you!
M.
"
echo
"*******************************************************************
****
!!! WARNING !!!
Since VMD does not recognize charges correctly, ions must be added manually
#Cl- = (#Conc * #Waters) / 55.5
#Na+ = #Cl- + |GAGCharge|
Note:
#Cl- = number of Cl- ions to be added
#Na+ = number of Na+ ions to be added
#Conc = 0.15 mol/L
#Waters = number of water molecules (not atoms!), to be read after solvation
#GAGCharge = Absolute value of GAG charge
"

91
charge=0
for((j=0;$j<$len;j=$(($j+1))));do
disacch=${sequence:$j:1}
case $disacch in
'0') charge=$(echo '1+('$charge')' | bc) ;;
'4') charge=$(echo '2+('$charge')' | bc) ;;
'6') charge=$(echo '2+('$charge')' | bc) ;;
esac
done
echo 'Calculated |GAG Charge| for this molecule= ' $charge
echo "
********************************************************************
***
"
echo "Do you want to visualize your chain in VMD now?"
echo "Type:"
echo " 1 for YES"
echo " 0 for NO "
read input
case $input in
'1')
vmd Final/chain_start.psf Final/chain_start.pdb;;
'0')
exit;;

92
*)
echo '**************************'
echo '* Wrong input, quitting! *'
echo '**************************'
clear
exit;;
esac
clear
exit

93
APPENDICE B - TOPOLOGY DEL CONDROITIN SOLFATO
* $Id: top_allxx_sugar.inp,v 1.98 2011-07-20 15:03:53 prabhu Exp
$
*>>>>>>>>>>>> All-hydrogen topologies used in the
<<<<<<<<<<<<<<<<
*>>>>> development of the CHARMM carbohydrate force
field<<<<<<<<
*>>>>>>>>>>>>>>>>>>>>>>>>> June 2009
<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
*>>>>>>>> Direct comments to Alexander D. MacKerell Jr.
<<<<<<<<<<
*>>>>>>>>>> via the CHARMM web site: www.charmm.org
<<<<<<<<<<<<<<
*>>>>>>>>>>>>>>> parameter set discussion forum
<<<<<<<<<<<<<<<<<<
*
32 1
! please reference the following:
! pyranose monosaccharides
!Guvench, O., Greene, S.N., Kamath, G., Brady, J.W., Venable,
R.M.,
!Pastor, R.W., MacKerell, Jr., A.D. "Additive empirical force
field for
!hexopyranose monosaccharides," Journal of Computational
Chemistry, 29:
!2543-2564, 2008. PMID: 18470966
! linear sugars, sugar alcohols, and inositol
!Hatcher, E., Guvench, O., and MacKerell, Jr., A.D. "CHARMM
Additive
!All-Atom Force Field for Acyclic Polyalcohols, Acyclic
Carbohydrates
!and Inositol," Journal of Chemical Theory and Computation, 5:
!1315-1327, 2009, DOI: 10.1021/ct9000608.
! hexopyranose glycosidic linkages
!Guvench, O., Hatcher, E. R., Venable, R. M., Pastor, R. W.,
MacKerell, Jr.,
!A. D. "Additive Empirical CHARMM Force Field for glycosyl linked
!hexopyranoses," Journal of Chemical Theory and Computation, 5,
!2353-2370, 2009, DOI: 10.1021/ct900242e
! furanose monosaccharides
!Hatcher, E. R.; Guvench, O. and MacKerell, Jr., A.D.
!"CHARMM Additive All-Atom Force Field for Aldopentofuranose
! Carbohydrates and Fructofuranose." Journal of Physical
Chemistry B.
! 113:12466-76, 2009, PMID: 19694450
! glycosidic linkages involving furanoses
!Raman, E. P., Guvench, O., MacKerell, Jr., A.D., "CHARMM
Additive All-Atom

94
!Force Field for Glycosidic Linkages in Carbohydrates Involving
Furanoses,"
!Journal of Physical Chemistry B, 114: 12981-12994, 2010, PMID:
20845956
! carbohydrate derivatives and glycosidic linkages for
glycoproteins
!Guvench, O.; Mallajosyula, S. S.; Raman, E. P.; Hatcher, E. R.;
!Vanommeslaeghe, K.; Foster, T. J.; Jamison, F. W. and MacKerell,
Jr., A.D.,
!"CHARMM additive all-atom force field for carbohydrate
derivatives and its
!utility in polysaccharide and carbohydrate-protein modeling,"
!Journal of Chemical Theory and Computation, (In Press, 2011),
DOI: 10.1021/ct200328p
!O-glycan linkages
!Mallajosyula, S. S. and MacKerell, Jr., A.D., "Influence of
Solvent and
!Intramolecular Hydrogen Bonding on the Conformational Properties
of O-Linked
!Glycopeptides," Journal of Physical Chemistry B, (In Press,
2011),
!DOI: 10.1021/jp203695t
! Phosphates and sulfates
! Mallajosyula, S. S.; Guvench, O.; Hatcher, E. R. and MacKerell,
Jr., A.D.,
!"CHARMM Additive All-Atom Force Field for Phosphate and Sufate
Linkages in
!Carbohydrates" Manuscript under preparation, 2011
! adm: Alex MacKerell
! sng: Shannon Greene
! og: Olgun Guvench
! erh: Elizabeth Hatcher
! pram: E. Prabhu Raman
! sai: Sairam S. Mallajosyula
! tip3p water
!MASS 1 HCTIP3 1.00800 H ! TIP3P water hydrogen
!MASS 2 OCTIP3 15.99940 O ! TIP3P water oxygen
! gold
MASS 85 O116 15.99940 O !SPC
MASS 86 H117 1.00800 H !SPC
MASS 87 AUB 196.97 Au
MASS 88 AUC 1.00800 Au ! Au image charge; increased from
0.5 to 1.0 to use bonds constrain
MASS 89 AUS 196.97 Au
MASS 90 AUI 196.97 Au
! C6H12O6 pyranose monosaccharide atom types
MASS 171 CC301 12.01100 C ! aliphatic C, no H's
MASS 172 CC311 12.01100 C ! generic acyclic CH carbon
MASS 173 CC312 12.01100 C ! CH carbon in linear polyols
MASS 174 CC3161 12.01100 C ! C2, C3, C4 CH bound to OH
MASS 175 CC3162 12.01100 C ! C1 (anomeric) CH bound to OH

95
MASS 176 CC3163 12.01100 C ! C5 CH bound to exocylic CH2OH
MASS 177 CC321 12.01100 C ! generic acyclic CH2 carbon
(hexopyranose C6)
MASS 178 CC322 12.01100 C ! CH2 carbon in linear polyols erh
MASS 179 CC3263 12.01100 C ! C5 in xylose
MASS 180 CC331 12.01100 C ! generic acyclic CH3 carbon (xyl
C6, glcna/galna CT)
MASS 181 CC2O1 12.01100 C ! sp2 carbon in amides, aldoses
MASS 182 CC2O2 12.01100 C ! sp2 carbon in carboxylates
MASS 183 CC2O3 12.01100 C ! sp2 carbon in acetone, ketoses
MASS 184 CC2O4 12.01100 C ! c22 CD
MASS 185 HCA1 1.00800 H ! aliphatic proton, CH
MASS 186 HCA2 1.00800 H ! aliphatic proton, CH2
MASS 187 HCA3 1.00800 H ! aliphatic proton, CH3
MASS 188 HCP1 1.00800 H ! polar H
MASS 189 HCR1 1.00800 H ! c22 HR1
MASS 191 OC311 15.99940 O ! hydroxyl oxygen
MASS 192 OC3C61 15.99940 O ! ether in six membered ring
MASS 193 OC301 15.99940 O ! generic linear ether
MASS 194 OC302 15.99940 O ! linear ether in 1-1 glycosidic
linkage
MASS 195 OC2D1 15.99940 O ! sp2 oxygen in amides, aldoses
MASS 196 OC2D2 15.99940 O ! sp2 oxygen in carboxylates
MASS 197 OC2D3 15.99940 O ! sp2 oxygen in acetone, ketoses
MASS 198 OC2D4 15.99940 O ! par22 O
MASS 200 NC2D1 14.00700 N ! peptide, NMA, IPAA nitrogen
(C=NHR)
! model compound atom types
MASS 201 CC321C 12.01100 C ! cyclohexane, thp CH2
MASS 202 HCA3M 1.00800 H ! alcohol aliphatic proton, CH3
MASS 203 HCP1M 1.00800 H ! EGLY hydroxyl H
MASS 204 OC311M 15.99940 O ! MEOH, ETOH, PRO2, EGLY hydroxyl
O
MASS 205 CC321D 12.01100 C ! cyclohexane, thp CH2 model for
1-1 linkage
MASS 206 CC311C 12.01100 C ! patch C1 in model compound
MASS 207 CC311D 12.01100 C ! patch C1 in model compound
! THF atom types
MASS 208 OC3C5M 15.99940 O ! thf ring oxygen
MASS 209 CC322C 12.01100 C ! cyclopentane, thf CH2
MASS 210 HCA2C2 1.00800 H ! cyclopentane, thp aliphatic
proton, CH2
MASS 211 CC312C 12.01100 C ! tf2m CH1
MASS 212 HCA1C2 1.00800 H ! tf2m aliphatic proton, CH1
! Furanose atom types; erh 10/24/07
MASS 213 OC3C51 15.99940 O ! furan ring oxygen
MASS 214 CC3152 12.01100 C ! furan ring carbon
MASS 215 CC3153 12.01100 C ! furan ring carbon
MASS 216 CC3251 12.01100 C ! furan ring carbon; C2 deoxy
MASS 217 CC3151 12.01100 C ! furan ring carbon
MASS 218 CC3051 12.01100 C ! furan ring carbon; C2 fructose
! pyranose derivatives
MASS 219 CC3062 12.01100 C ! C2 on NE5AC
MASS 220 CC3261 12.01100 C ! C3 on NE5AC
!dummy atom
!MASS 99 DUM 1.00800 H ! dummy atom

96
!Added by sai for modelling phosphate
MASS 221 OC312 15.99940 O ! OH in PO3H (phosphate) || OHL in
top_all27_lipid.rtf
MASS 222 OC30P 15.99940 O ! ester O in PO3H (phosphate) ||
OSL in top_all27_lipid.rtf
MASS 223 OC2DP 15.99940 O ! =0 in P03H (phosphate) || O2L in
top_all27_lipid.rtf
MASS 224 PC 30.974000 P ! phosphorus || PL in
top_all27_lipid.rtf
MASS 225 SC 32.060000 S ! Sulfate sulfur
!pram, furanosyl linkages
MASS 226 CC312D 12.01100 C ! from CC322C; THF anomeric carbon
MASS 227 OC303 15.99940 O ! from OC301; linear ether in P1-
>F3 pyranose-furanose glycosidic linkage
!ions
!MASS 111 SOD 22.989770 NA ! Sodium Ion
!MASS 112 CAL 40.080000 CA ! Calcium Ion
!Added for modelling sulphate
MASS 228 SO 32.060000 S ! sulfonate sulfur || from
Tani/Cilpa
MASS 230 OC20S 15.99940 O ! ester -O- in SO3- (sulfonate) ||
from Tani/Cilpa
MASS 231 OC2DS 15.99940 O ! -O in SO3- (sulfonate) || from
Tani/Cilpa
MASS 232 OC301 15.99940 O ! -O- for bond glcn-galnac
DECL -O12
DECL +O12
DEFA FIRS NTER LAST CTER
AUTO ANGLES DIHE
RESI GA4 -1.000 ! 2-acetyl-2-deoxy-beta-D-galactosamine-
4-sulphate
! (Modello di
Meziane-Tani)
!
GROU !
H30
ATOM C1 CC3162 0.340 !
|
ATOM H20 HCA1 0.090 !
H28---C8---H29
ATOM O12 OC301 -0.460 !
|
ATOM C5 CC3163 0.110 !
C7=O16
ATOM O14 OC3C61 -0.400 ! --------
---- |

97
ATOM H24 HCA1 0.090 !
\ N10---H27
GROU ! \
|
ATOM C2 CC3161 0.070 !
H22---C3---C2---H21
ATOM H21 HCA1 0.090 ! O17
/ \
ATOM N10 NC2D1 -0.470 ! | /
\
ATOM H27 HCP1 0.310 ! O18---S11---O13-
--C4---H23 C1---O12-------------
GROU ! |
\ / \
ATOM C3 CC3161 0.140 ! O19
\ / H20
ATOM H22 HCA1 0.090 !
H24---C5---O14
GROU !
|
ATOM C4 CC3161 0.140 !
H25---C6---O15---H33
ATOM H23 HCA1 0.090 !
|
ATOM O13 OC20S -0.430 ! H26
GROU !
ATOM C6 CC321 0.050 !
ATOM H25 HCA2 0.090 !
ATOM H26 HCA2 0.090 !
ATOM O15 OC311 -0.650 !
ATOM H33 HCP1 0.420 !
GROU !
ATOM S11 SO 1.240 !
ATOM O17 OC2DS -0.680 !
ATOM O18 OC2DS -0.680 !
ATOM O19 OC2DS -0.680 !
GROU
ATOM C7 CC2O1 0.510 !
ATOM O16 OC2D1 -0.510 !
GROU !
ATOM C8 CC331 -0.270 !
ATOM H28 HCA3 0.090 !
ATOM H29 HCA3 0.090 !
ATOM H30 HCA3 0.090 !
!
BOND C1 C2 C1 O12 C1 O14 C1 H20
C2 C3
BOND C2 N10 C2 H21 C3 C4 C3 H22
O12 +C4
BOND C4 C5 C4 O13 C4 H23 C5 C6
C5 O14
BOND C5 H24 C6 O15 C6 H25 C6 H26
C7 C8
BOND C7 N10 C7 O16 C8 H28 C8 H29 C8
H30

98
BOND N10 H27 S11 O13 S11 O17 S11 O18
S11 O19
BOND O15 H33
IMPR C7 C8 N10 O16
IMPR N10 C7 C2 H27
RESI GA6 -1.000 ! 2-acetyl-2-deoxy-beta-D-galactosamine-
6-sulphate
! (Modello di
Cilpa)
GROU !
ATOM C1 CC3162 0.340 !
H30
ATOM H20 HCA1 0.090 !
|
ATOM O12 OC301 -0.460 !
H28---C8---H29
ATOM C5 CC3163 0.110 !
|
ATOM O14 OC3C61 -0.400 !
C7=O16
ATOM H24 HCA1 0.090 !
|
GROU ! -----------
N10---H27
ATOM C2 CC3161 0.070 !
\ |
ATOM H21 HCA1 0.090 !
H22---C3---C2---H21
ATOM N10 NC2D1 -0.470 !
/ \
ATOM H27 HCP1 0.310 !
/ \
GROU ! H04---O13--
-C4---H23 C1---O12---------
ATOM C3 CC3161 0.140 !
\ / \
ATOM H22 HCA1 0.090 !
\ / H20
GROU !
H24---C5---O14
ATOM C4 CC3161 0.140 !
|
ATOM H23 HCA1 0.090 !
H25---C6---H26
ATOM O13 OC311 -0.650 !
|
ATOM H04 HCP1 0.420 !
O15
GROU ! |
ATOM C6 CC321 0.050 !
O18---S11---O19
ATOM H25 HCA2 0.090 !
|
ATOM H26 HCA2 0.090 !
O17

99
ATOM O15 OC20S -0.430 !
GROU !
ATOM S11 SO 1.240 !
ATOM O17 OC2DS -0.680 !
ATOM O18 OC2DS -0.680 !
ATOM O19 OC2DS -0.680 !
GROU !
ATOM C7 CC2O1 0.510 !
ATOM O16 OC2D1 -0.510 !
GROU
ATOM C8 CC331 -0.270 !
ATOM H28 HCA3 0.090 !
ATOM H29 HCA3 0.090 !
ATOM H30 HCA3 0.090 !
!
BOND C1 O12 C1 H20 C1 O14 C1 C2
C2 H21
BOND C2 N10 N10 H27 C2 C3 C3 H22
S11 O15
BOND C3 C4 C4 H23 C4 O13 O13 H04
C4 C5
BOND C5 H24 C5 C6 C6 H26 C6 H25
C6 O15
BOND C5 O14 N10 C7 C7 O16 C7 C8
O12 +C4
BOND C8 H28 C8 H29 C8 H30 S11 O17
S11 O18
BOND S11 O19
IMPR C7 C8 N10 O16
IMPR N10 C7 C2 H27
RESI GAX 0.000 ! 2-acetyl-2-deoxy-beta-D-
galactosamine
! (beta N-acetylgalactosamine or
GalNAc)
GROU !
ATOM C1 CC3162 0.340 ! O15---H33
ATOM H20 HCA1 0.090 ! |
ATOM O12 OC301 -0.460 ! H25---C6---H26
ATOM C5 CC3163 0.110 ! |
ATOM H24 HCA1 0.090 ! H24---C5---O14
ATOM O14 OC3C61 -0.400 ! HO4---O13 / \ O12----
------
GROU ! \ / \ /
ATOM C2 CC3161 0.070 ! C4 C1
ATOM H21 HCA1 0.090 ! / \ H22 H21/ \
ATOM N10 NC2D1 -0.470 ! H23 \| | / H20
ATOM H27 HCP1 0.310 ! C3---C2
GROU ! / |
ATOM C7 CC2O1 0.510 ! --------- N10---H27
ATOM O16 OC2D1 -0.510 ! /
GROU ! O16=C7 H28
ATOM C8 CC331 -0.270 ! \ /
ATOM H28 HCA3 0.090 ! H29---C8
ATOM H29 HCA3 0.090 ! \

100
ATOM H30 HCA3 0.090 ! H30
GROU !
ATOM C3 CC3161 0.140 !
ATOM H22 HCA1 0.090 !
GROU !
ATOM C4 CC3161 0.140 !
ATOM H23 HCA1 0.090
ATOM O13 OC311 -0.650
ATOM HO4 HCP1 0.420
GROU
ATOM C6 CC321 0.050
ATOM H25 HCA2 0.090
ATOM H26 HCA2 0.090
ATOM O15 OC311 -0.650
ATOM H33 HCP1 0.420
!
BOND C1 O12 C1 H20 C1 O14 C1 C2
C2 H21
BOND C2 N10 N10 H27 C2 C3 C3 H22
O12 +C4
BOND C3 C4 C4 H23 C4 O13 O13 HO4
C4 C5
BOND C5 H24 C5 C6 C6 H26 C6 H25
C6 O15
BOND O15 H33 C5 O14 N10 C7 C7 O16
C7 C8
BOND C8 H28 C8 H29 C8 H30
IMPR C7 C8 N10 O16
IMPR N10 C7 C2 H27
!----------------------------GLUCURONICO-------------------------
----------
RESI GLC -1.000 ! 4C1 beta-D-glucoronic acid
!
GROU ! O61(-)
ATOM C1 CC3162 0.340 ! |
ATOM H1 HCA1 0.090 ! O62=C6
ATOM O12 OC301 -0.460 ! |
ATOM C5 CC3163 0.110 ! H5---C5---O5
ATOM H5 HCA1 0.090 ! H4 / \ O12----
ATOM O5 OC3C61 -0.400 ! \ / \ /
GROU ! C4 C1
ATOM C2 CC3161 0.140 ! / \ H3 H2 / \
ATOM H2 HCA1 0.090 ! ------ \| | / H1
ATOM O2 OC311 -0.650 ! C3---C2
ATOM HO2 HCP1 0.420 ! | |
GROU ! HO3---O3 O2---HO2
ATOM C3 CC3161 0.140 !
ATOM H3 HCA1 0.090 !
ATOM O3 OC311 -0.650 !
ATOM HO3 HCP1 0.420 !
GROU !
ATOM C4 CC3161 0.140 !
ATOM H4 HCA1 0.090 !
GROU

101
ATOM C6 CC2O2 0.520 !
ATOM O61 OC2D2 -0.760 !
ATOM O62 OC2D2 -0.760 !
!
BOND C1 O12 C1 H1 O12 +C3 C1 O5
C1 C2
BOND C2 H2 C2 O2 O2 HO2 C2 C3
C3 H3
BOND C3 O3 O3 HO3 C3 C4 C4 H4
C6 O62
BOND C4 C5 C5 H5 C5 C6 C6 O61
C5 O5
IMPR C6 C5 O62 O61
!--------------------------IDURONICO-----------------------------
------
RESI IDA -1.000 ! alpha-L-iduronic acid
!
GROU ! H5
ATOM C1 CC3162 0.340 ! |
ATOM H1 HCA1 0.090 ! C5---O5
ATOM O12 OC301 -0.460 ! H4 /| \ O12---
ATOM C5 CC3163 0.110 ! \ / C6 HO3 \ /
ATOM H5 HCA1 0.090 ! C4 / C1
ATOM O5 OC3C61 -0.400 ! / \ O3 H2 / \
GROU ! ------ \| | / H1
ATOM C2 CC3161 0.140 ! C3---C2
ATOM H2 HCA1 0.090 ! | |
ATOM O2 OC311 -0.650 ! H3 O2-HO2
ATOM HO2 HCP1 0.420 !
GROU ! n.b.: O61 and O62 are
attached to C6
ATOM C3 CC3161 0.140 !
ATOM H3 HCA1 0.090 ! |
ATOM O3 OC311 -0.650 ! O62=C6
ATOM HO3 HCP1 0.420 ! |
GROU ! O61(-)
ATOM C4 CC3161 0.140 !
ATOM H4 HCA1 0.090 !
GROU
ATOM C6 CC2O2 0.520 !
ATOM O61 OC2D2 -0.760 !
ATOM O62 OC2D2 -0.760 !
!
BOND C1 O12 C1 H1 O12 +C3 C1 O5
C1 C2
BOND C2 H2 C2 O2 O2 HO2 C2 C3
C3 H3
BOND C3 O3 O3 HO3 C3 C4 C4 H4
C4 C5
BOND C5 H5 C5 C6 C6 O61 C6 O62
C5 O5
RESI TIP3 0.000 ! tip3p water model, generate using
noangle nodihedral

102
GROUP
ATOM OH2 OT -0.834
ATOM H1 HT 0.417
ATOM H2 HT 0.417
BOND OH2 H1 OH2 H2
ANGLE H1 OH2 H2
ACCEPTOR OH2
PATCHING FIRS NONE LAST NONE
RESI SPC 0.000 ! tip3p water model, generate using
noangle nodihedral
GROUP
ATOM OH2 O116 -0.82
ATOM H1 H117 0.41
ATOM H2 H117 0.41
BOND OH2 H1 OH2 H2
ANGLE H1 OH2 H2
ACCEPTOR OH2
PATCHING FIRS NONE LAST NONE
RESI AUB 0.000 | Gold Bluk
GROUP
ATOM AU AUB -0.3
ATOM AUC AUC 0.3
BOND AU AUC
PATCHING FIRS NONE LAST NONE
RESI AUS 0.000 | Gold Surface
GROUP
ATOM AU AUS -0.3
ATOM AUC AUC 0.3
BOND AU AUC
PATCHING FIRS NONE LAST NONE
RESI AUI 0.000 | Gold Surface
GROUP
ATOM AUI AUI 0.0
PATCHING FIRS NONE LAST NONE
!----------------------------TERMINI-----------------------------
------
PRES NTER 0.00 ! N-terminus (for GLC/IDA)
GROUP !
ATOM C4 CC3161 0.140 ! H4
ATOM H4 HCA1 0.090 ! |
ATOM O13 OC311 -0.650 ! HO4--O13--C4----
ATOM HO4 HCP1 0.420 !
!
BOND C4 O13 O13 HO4
IC O13 C3 *C4 C5 1.4163 111.43 -118.91 108.83
1.5171
IC C3 C4 O13 HO4 1.5102 111.43 47.58 105.89
0.9645

103
PRES CTER 0.000 ! C-terminus (for BGALNAxx)
GROUP !
ATOM C1 CC3162 0.340 !
ATOM H20 HCA1 0.090 !
ATOM O12 OC311 -0.650 ! |
ATOM C5 CC3163 0.110 ! H24--C5---O14
ATOM H24 HCA1 0.090 ! / \ O12-H12
ATOM O14 OC3C61 -0.400 ! \ /
ATOM H12 HCP1 0.420 ! C1
! / \
! H20
!
BOND H12 O12
IC O14 C1 O12 H12 1.4328 112.45 125.25 107.32
0.9601

104
APPENDICE C - SCRIPT UTILIZZATO PER IL CALCOLO DELLA
LUNGHEZZA PERSISTENTE
clear
%quanti frames esamino
numfiles = 100;
%da quale frame parto
nf= 83;
%quanti punti ho
np=40;
%costruzione celle
A = cell(1, numfiles);
C = cell(1, numfiles);
St= cell(1,numfiles);
D = cell(np-2,1);
E = cell(np-2,1);
vec= cell(1,numfiles);
v1= cell (1,length(A));
v2= cell (1,length(A));
vt= cell (1,numfiles);
k=1;
q=1;
%estrazione coordinate
for q = 1:numfiles
myfilename = sprintf('frame%d.dat', nf);
A{q} = importdata(myfilename);
%inversione coordinate (Nel caso Vmd inverta gli atomi)
%for x=1:2:length(A{q})-1
% A{q}([x x+1],:) = A{q}([x+1 x],:);
%end

105
j=1;
nf=nf+1;
%k identifica il passo col quale calcolo il coseno
for k=1:1:length(A{q})-1
%costruzione vettori e calcolo del coseno nei vari passi
for i=1:1:length(A{q})-1
v1= (A{q}(i+1,:) - A{q}(i,:));
if (i+k<np)
v2= (A{q}(i+k+1,:) - A{q}(i+k,:));
C{q}(j,2) = v1*v2'/(norm(v1)*norm(v2));
C{q}(j,1)=k;
j=j+1;
end
end
end
end
%calcolo lunghezza della catena
for q=1:numfiles
for o=1:1:np-1
vec2{q}(o,:)= (A{q}(o+1,:) - A{q}(o,:));
end
end
for q=1:numfiles
for o=1:np-1
vec{q}(o,1)=sqrt((vec2{q}(o,1))^2 + (vec2{q}(o,2))^2+ (vec2{q}(o,3))^2);
if (o<2)
vt{q}(o,1)=vec{q}(o,1);
elseif (o>1)
vt{q}(o,1)=vec{q}(o,1)+vt{q}(o-1,1);
end
end

106
end
vector=zeros(np-1,1);
%media delle lunghezze tra i vari frames
for r=1:numfiles
vector = vector + vt{r};
end
lung=vector/numfiles;
%costruzione matrice totale (la classe D contiene i passi dello stesso
%valore)
for s=np:1
D{s} = zeros(s*numfiles,1)
end
di = ones(np-2,1);
for m=1:numfiles
p=1 ;
for o=1:np-2
D{o}(di(o):(di(o)+np-o-2),1)=C{m}(p:(p+np-o-2),1);
D{o}(di(o):(di(o)+np-o-2),2)=C{m}(p:(p+np-o-2),2);
di(o)=di(o)+np-o-1;
p=p+np-o-1;
end
end
%Costruzione matrice finale (Tot contiene nella prima colonna la lunghezza
%del vettore, nella seconda la media dei coseni, nella terza la std, nella
%quarta la media del logaritmo dei coseni e nella quinta la std dei
%logaritmi dei coseni
Tot(:,1)= lung(:,1);
for l=1:np-2
E{l}(:,2) = -log(abs(D{l}(:,2)));
Tot(l,4) = mean(E{l}(:,2));
Tot(l,5) = std(E{l}(:,2));
Tot(l,3) = std(D{l}(:,2));

107
Tot(l,2) = mean(D{l}(:,2));
end
%creazione file Excel
xlswrite('Cos.xls',Tot)