
NotedelCorsodiTermodinamicaeMeccanica Statisticaolla/statmec/note.pdf · compendio di...
118
Note del Corso di Termodinamica e Meccanica Statistica Laurea Triennale in Fisica. Universit` a di Cagliari Piero Olla 29 novembre 2013
Transcript of NotedelCorsodiTermodinamicaeMeccanica Statisticaolla/statmec/note.pdf · compendio di...

Note del Corso di Termodinamica e Meccanica
Statistica
Laurea Triennale in Fisica. Universita di Cagliari
Piero Olla
29 novembre 2013

2

Indice
1 Introduzione 5
2 Richiami di probabilita e statistica 72.1 Definizioni fondamentali . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.1.1 Probabilita congiunte e condizionate . . . . . . . . . . . . . . . . . 82.1.2 Il concetto di media . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.2 Entropia ed informazione . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102.3 Somme di variabili aleatorie: il limite termodinamico . . . . . . . . . . . . 132.4 Entropia ed equilibrio termodinamico . . . . . . . . . . . . . . . . . . . . . 152.5 Fluttuazioni . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.6 Processi stocastici e campi random . . . . . . . . . . . . . . . . . . . . . . 18
2.6.1 Il teorema di Wiener-Khinchin . . . . . . . . . . . . . . . . . . . . . 222.7 Probabilita dipendenti dal tempo . . . . . . . . . . . . . . . . . . . . . . . 232.8 Coarse graining e limite continuo . . . . . . . . . . . . . . . . . . . . . . . 272.9 L’equazione master . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.9.1 Cammino random . . . . . . . . . . . . . . . . . . . . . . . . . . . . 302.9.2 Processo di estinzione . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.10 Esercizi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
3 La teoria cinetica 333.1 Alcune problematiche in teoria cinetica . . . . . . . . . . . . . . . . . . . . 333.2 Alcune questioni riguardo il calcolo dell’entropia . . . . . . . . . . . . . . . 37
3.2.1 Entropia, partizioni e paradosso di Gibbs . . . . . . . . . . . . . . . 383.2.2 Entropia in una singola realizzazione . . . . . . . . . . . . . . . . . 40
3.3 L’equazione di Boltzmann . . . . . . . . . . . . . . . . . . . . . . . . . . . 413.3.1 La distribuzione di Maxwell-Boltzmann . . . . . . . . . . . . . . . . 433.3.2 Entropia e spazio delle velocita . . . . . . . . . . . . . . . . . . . . 443.3.3 Il teorema H . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
3.4 Il limite fluido . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 463.5 Equazioni fluide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 483.6 Viscosita e diffusivita termica . . . . . . . . . . . . . . . . . . . . . . . . . 51
3.6.1 Diffusione in una miscela di gas . . . . . . . . . . . . . . . . . . . . 533.7 Dalla teoria cinetica alla termodinamica . . . . . . . . . . . . . . . . . . . 54
3

4 INDICE
3.7.1 Pressione e legge di stato . . . . . . . . . . . . . . . . . . . . . . . . 543.7.2 Energia termica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 553.7.3 Calore e lavoro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 563.7.4 Entropia e temperatura . . . . . . . . . . . . . . . . . . . . . . . . 57
3.8 Esercizi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4 Termodinamica 614.1 Processi reversibili e irreversibili . . . . . . . . . . . . . . . . . . . . . . . . 614.2 Diseguaglianze termodinamiche . . . . . . . . . . . . . . . . . . . . . . . . 634.3 Termodinamica: punto di vista macroscopico. . . . . . . . . . . . . . . . . 664.4 Lavoro massimo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 694.5 I potenziali termodinamici . . . . . . . . . . . . . . . . . . . . . . . . . . . 704.6 Lavoro massimo e potenziali termodinamici . . . . . . . . . . . . . . . . . . 734.7 Sistemi a numero di particelle variabile . . . . . . . . . . . . . . . . . . . . 744.8 Entropia e potenziali termodinamici in una miscela di gas . . . . . . . . . . 764.9 Reazioni chimiche in una miscela di gas . . . . . . . . . . . . . . . . . . . . 78
4.9.1 Entalpia di reazione . . . . . . . . . . . . . . . . . . . . . . . . . . . 794.10 Transizioni di fase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
4.10.1 Termodinamica dell’aria umida . . . . . . . . . . . . . . . . . . . . 834.11 Esercizi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
5 Meccanica statistica dell’equilibrio 855.1 Il punto di vista di Gibbs . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
5.1.1 Il concetto di ensemble . . . . . . . . . . . . . . . . . . . . . . . . . 875.1.2 Il problema ergodico . . . . . . . . . . . . . . . . . . . . . . . . . . 89
5.2 Il concetto di entropia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 905.3 Il metodo di massima entropia . . . . . . . . . . . . . . . . . . . . . . . . . 93
5.3.1 Cenni di statistica quantistica . . . . . . . . . . . . . . . . . . . . . 955.4 La distribuzione di Gibbs . . . . . . . . . . . . . . . . . . . . . . . . . . . . 985.5 Esercizi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
6 Cenni alla teoria delle fluttuazioni 1016.1 Fluttuazioni attorno all’equilibrio . . . . . . . . . . . . . . . . . . . . . . . 1016.2 Struttura temporale delle fluttuazioni . . . . . . . . . . . . . . . . . . . . . 105
6.2.1 Il moto Browniano . . . . . . . . . . . . . . . . . . . . . . . . . . . 1066.2.2 Relazioni fluttuazione-dissipazione . . . . . . . . . . . . . . . . . . . 108
6.3 Reversibilita temporale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1126.3.1 Relazioni di Onsager . . . . . . . . . . . . . . . . . . . . . . . . . . 1136.3.2 Produzione di entropia . . . . . . . . . . . . . . . . . . . . . . . . . 114
6.4 Esercizi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117

Capitolo 1
Introduzione
Un problema fondamentale della fisica teorica e come determinare le proprieta di un sistemamacroscopico note quelle dei suoi costituenti elementari. In linea di principio, la dinamicadel sistema potrebbe essere ottenuta da una conoscenza esatta dello stato delle sue parti.Un simile bagaglio di informazione, pero, oltre ad essere impossibile da ottenersi, e ancheassolutamente ridondante: lo stato del sistema e descritto di solito per mezzo di un numeroristretto di variabili, dette appunto macroscopiche, le quali non dipendono dai dettagli delsistema. (Per conoscere la pressione di un gas, non e necessario conoscere posizione evelocita di tutte le sue molecole). Sistemi piu o meno complicati possono venire descrittiper mezzo di un numero piu o meno alto di variabili, ma il principio generale che unadescrizione completa non e necessaria, rimane valido.
Il passaggio a un livello di descrizione macroscopico comporta evidentemente una per-dita di informazione, e non e detto che una simile operazione sia sempre possibile. Unaprima condizione e che le interazioni tra i costituenti fondamentali siano semplici (cosı e nelcaso delle molecole di un gas; non altrettanto, per dire, nel caso dei neuroni in un cervelloumano). Questo fa sı che le variabili macroscopiche siano esse stesse funzioni abbastanzasemplici di quelle microscopiche (tipicamente somme). Una seconda condizione e quelladi una rapida perdita di memoria dello stato microscopico iniziale del sistema. Come direche le dinamica microscopica diventa ininfluente a scala macroscopica. Questo presupponeche la dinamica microscopica sia almeno in parte caotica. Il tema e controverso. Il soddi-sfacimento di una tale condizione – ragionevole, ma difficile da dimostrare – implicherebbeuna perdita di informazione nel passaggio dal micro al macro, dovuta non solo a motivi di“coarse graining”, ma anche di tipo dinamico, e costituirebbe in se, una giustificazione inpiu per disinteressarsi dei gradi di liberta microscopici del sistema.
Il fatto che le variabili macroscopiche possano essere ottenute come somme (o in generefunzioni semplici) delle variabili microscopiche, fa sı che le prime possano essere interpre-tate in senso statistico. (Un esempio e l’energia interna di un gas ideale monoatomicoU = (3/2)NKT , in cui (3/2)KT e l’energia cinetica media delle molecole). I concetti diprobabilita e statistica giocano quindi un ruolo centrale. Due approcci sono sostanzial-mente possibili: quello della teoria cinetica, originato dal lavoro di L. Boltzmann, e quellodella meccanica statistica, originato dal lavoro di J.W. Gibbs. Anche se l’utilizzo di queste
5

6 CAPITOLO 1. INTRODUZIONE
teorie si estende al giorno d’oggi ben oltre il campo della fisica, il loro ambito originaledi applicazione era quello dei sistemi termodinamici. Semplificando molto, la teoria ci-netica si interessa di piu a situazioni di non-equilibrio, mentre la meccanica statistica sipuo vedere come un insieme di tecniche per ottenere la termodinamica di sistemi di tipogenerico. In entrambi i casi, l’aspetto centrale e la trattazione probabilistica dei gradi diliberta microscopici del sistema.
Un ruolo essenziale nello stabilire la connessione tra mondo macroscopico e mondomicroscopico e giocato dall’entropia. A volte si descrive l’entropia come una misura didisordine. Si tratta purtroppo di una caratterizzazione assai ambigua, visto che non echiaro cosa si intende con il termine disordine. Sarebbe d’altra parte un po’ riduttivo,qualificare ad esempio i processi in un ciclo biologico come un incremento di disordine,anche se essi invariabilmente comportano un aumento di entropia. Di fatto, il concetto dientropia assume significati diversi a seconda del contesto. Tutti questi significati possonoessere pero ricondotti alla quantita di informazione persa nel passaggio a una descrizionemacroscopica del sistema. Nelle note che seguono, particolare attenzione verra data airuoli diversi che l’entropia gioca nei tre approcci cinetico, termodinamico e di meccanicastatistica, sottolineando in tutti i casi il suo il suo significato dal punto di vista della teoriadell’informazione (entropia di Shannon).
Le pagine che seguono contengono una presentazione a livello introduttivo di comefunzionano i due approcci cinetico e di meccanica statistica. Si tratta in gran parte di uncompendio di rielaborazioni ed estratti da testi classici. Per la parte di termodinamica,per quella di meccanica statistica e per il capitolo finale sulle fluttuazioni, si e preso comeriferimento “Fisica Statistica” di Landau e Lifstits, in particolare i paragrafi 7, 19-20, 31,40, 83, 93, 101-103, 110, 112, 118-120. Per quanto riguarda la teoria cinetica, possono essereutili per approfondimento i capitoli 3-5 del testo “Statistical Mechanics” di K. Huang. Unriferimento per i preliminari di probabilita e statistica possono essere i capitoli 5-6 del testo“A modern course in statistical physics” di L.E. Reichl.

Capitolo 2
Richiami di probabilita e statistica
2.1 Definizioni fondamentali
La probabilita e uno di quei concetti estremamente familiari, che sono molto difficili dadefinire in maniera formale. La definizione comunemente accettata e quella assiomaticadovuta a Kolmogorov, che da le proprieta minime che il concetto di probabilita deve sod-disfare, ma non dice alcunche su cio che essa deve rappresentare. Quello che rappresenta,noi lo sappiamo benissimo: una misura della nostra aspettativa del verificarsi di un certorisultato in un esperimento. Questo non e pero sufficiente per costruirci sopra una teoria(per dirne una: come si definisce il concetto di aspettativa?)
La definizione assiomatica di probabilita si ottiene a partire dai concetti elementarinella maniera seguente.
• Si da l’insieme dei possibili risultati di un esperimento, detto lo spazio dei cam-pioni Ω. Esempi: l’insieme Ω = 1, 0, x dei risultati di una partita nella schedina;l’insieme delle velocita che puo assumere le molecole in un gas: Ω = R3. In que-sto secondo caso, la quantita continua che si misura nell’esperimento e detta unavariabile random oppure variabile aleatoria.
• Si definisce evento un insieme di possibili risultati di una misura. In altre parole,un evento e un sottoinsieme di Ω. (Nel caso continuo, non tutti i sottoinsiemi sonoammissibili; non andiamo ad approfondire la problematica. Per chi e interessato,dare un occhiata a Wentzel, Teoria delle probabilita). Si dice che due eventi A e Bsono mutualmente esclusivi se hanno intersezione vuota.
• La probabilita di un evento A e un numero P (A), 0 ≤ P (A) ≤ 1 che soddisfale proprieta: P (A ∪ B) = P (A) + P (B) se A e B sono mutualmente esclusivi, eP (Ω) = 1.
Non male come economia di mezzi, per una definizione.Vale la pena verificare le proprieta intuitive che ci si aspetta dovrebbe avere la proba-
bilita (per esempio P (∅) = 0). Notare come la scrittura P (A ∪ B) non sia altro che unanotazione stenografica per dire: “probabilita che si verifichi un risultato in A o in B”.
7

8 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
Nel caso di una variabile random x, gli eventi sono tipicamente intervalli o unioni diintervalli. In tal caso si definisce la funzione densita di probabilita (PDF) ρ tramitela formula
P ([a, b]) =
∫ b
a
ρ(x)dx. (2.1)
La PDF e una funzione non negativa (convincersene!)Notare che lo stesso evento puo essere rappresentato con variabili random diverse; per
esempio, se x = y/2, l’intervallo A = [a, b] per x, corrisponde all’intervallo [2a, 2b] per y.L’evento puo essere fisicamente lo stesso (un certo intervallo di velocita in due unita dimisura diverse). Per evitare ambiguita, si utilizza spesso il pedice sulla PDF per indicarela variabile aleatoria a cui di si riferisce. Quindi, la (2.1) diventa
P (A) =
∫ b
a
ρx(z)dz =
∫ 2b
2a
ρy(z)dz (2.2)
(x e y sono le variabili aleatorie; z sono i loro valori). Prendendo intervalli infinitesiminella (2.2), troviamo ρx(z)dz = 2ρy(2z)dz. In generale: ρx(x)dx = ρy(y(x))dy(x), che cida la formula per il cambio di variabili tra PDF:
ρx(x) = ρy(y(x))∣
∣
∣
dy(x)
dx
∣
∣
∣(2.3)
La formula puo essere generalizzata a un numero maggiore di dimensioni servendosi deldeterminante Jacobiano al posto della derivata y′(x).
Ci chiediamo se e possibile definire eventi per variabili aleatorie, che non sono intervalli,bensı punti isolati. Possiamo farlo servendoci della delta di Dirac. Esempio:
ρ(x) = aδ(x) + bθ(x) exp(−x), x ∈ R. (2.4)
Abbiamo evidentemente P (0) = a. Per avere P (Ω) = 1, dobbiamo imporre b = 1− a.
2.1.1 Probabilita congiunte e condizionate
Dati due eventi A e B, definiamo probabilita congiunta di A B la probabilita dell’in-tersezione P (A ∩B). Esempio importante nel caso Ω ⊂ R2: sia A = [x, x+ dx]⊗ Ωy(x) eB = Ωx(y)⊗ [y, y + dy]. In tal caso
P (A ∩ B) = P ([x, x+ dx]⊗ [y, y + dy]) := ρxy(x, y)dxdy, (2.5)
che definisce la PDF congiunta dei valori x e y delle variabili aleatorie x e y. (A menoche non sia strettamente necessario, da ora in poi tralasciamo i pedici sulle PDF e nonfacciamo distinzioni fra variabili aleatorie e loro valori). Possiamo chiaramente definireoggetti intermedi che sono probabilita rispetto a una variabile e PDF rispetto ad un’altra(ad esempio integrando ρ(x, y) rispetto a x in un intervallo A).

2.1. DEFINIZIONI FONDAMENTALI 9
Definiamo di seguito la probabilita condizionata a B dell’evento A:
P (A|B) = P (A ∩B)/P (B). (2.6)
Essenzialmente, si tratta della probabilita dell’evento A considerando come spazio deicampioni l’evento B. Notare che di nuovo possiamo generalizzare a PDF:
ρ(x|y) = ρ(x, y)/ρ(y); P ([x, x+ dx]|y) = ρ(x|y)dx. (2.7)
(dimostrare quest’ultimo risultato a partire dalla definizione di P ([x, x+dx]⊗ [y, y+dy]).Diciamo che due eventi A e B sono statisticamente indipendenti se P (A ∩ B) =
P (A)P (B). Il concetto di probabilita condizionata ci permette una comprensione intuitivadi questa definizione. Se due eventi A e B sono statisticamente indipendenti abbiamoinfatti:
P (A|B) = P (A); P (B|A) = P (B). (2.8)
In altre parole, il condizionamento da un evento statisticamente indipendente non cambiala probabilita. Nel caso di PDF: ρ(x|y) = ρ(x), cioe il fatto che x si verifichera o meno,indipendentemente che si sia ottenuto o no y. Se abbiamo ρ(x, y) = ρ(x)ρ(y) ∀x, y, diciamoche le variabili x e y sono statisticamente indipendenti.
Per ultimo notiamo la seguente relazione, che discende direttamente dalla definizione(2.5):
ρ(x) =
∫
Ωy(x)
ρ(x, y)dy; ρ(y) =
∫
Ωx(y)
ρ(x, y)dx. (2.9)
Le PDF ρ(x) e ρ(y) sono dette in questo contesto marginali.
2.1.2 Il concetto di media
Definiamo la media di una funzione f di variabile aleatoria x tramite la seguente formula:
〈f〉 =∫
ρ(x)f(x)dx. (2.10)
Vediamo subito che ritroviamo la definizione standard nel caso di variabili discrete:
〈f〉 =∑
i
P (xi)f(xi)
che puo essere ottenuta dalla (2.10) generalizzando in maniera opportuna la (2.4).Possiamo fare medie a partire da PDF condizionate, ottenendo quindi medie condi-
zionate:
〈f |y〉 =∫
ρ(x)f(x|y)dx (2.11)

10 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
(notare che 〈f |y〉 e ora una variabile aleatoria, dipendendo dal risultato di y).Un po’ di ripasso di terminologia e notazioni. Tipicamente µx ≡ 〈x〉 e detta media
della PDF e σ2 = 〈(x− µx)2〉 = 〈x2〉 − 〈x〉2 e detta varianza della PDF (σx e la deviazione
standard o RMS). La media 〈xn〉 e detto momento n-esimo della PDF. La quantita 〈fg〉e detta correlazione di f e g. Caso importante:
Cxy = 〈xy〉 =∫
ρ(x, y)xydxdy (2.12)
Notare che indipendenza statistica di x e y implica Cxy = 0 ma non il viceversa (provarea costruire un controesempio).
Un oggetto particolarmente importante di cui prendere la media e la cosı detta funzioneindicatrice di un evento:
δA(x) =
1 se x ∈ A,
0 altrimenti.(2.13)
Troviamo infatti subito
〈δA〉 =∫
A
ρ(x)dx = P (A). (2.14)
Il concetto si generalizza in dimensione maggiore di uno.Possiamo definire la funzione indicatrice del valore della variabile aleatoria x, che
e semplicemente la delta di Dirac:
δx(x) ≡ δ(x− x) ⇒ 〈δx〉 = ρ(x). (2.15)
Le Eqs. (2.13-2.14) [e in certo qual modo anche la (2.15)] costituiscono il punto di partenzaper la determinazione statistica delle probabilita (e delle PDF) a partire dal concetto difrequenza.
2.2 Entropia ed informazione
Se la probabilita di un evento parametrizza la nostra aspettativa che l’evento si verifichio meno, la domanda che e naturale porsi, e il contenuto di informazione dei risultati diun esperimento, portato dal conoscerne la funzione di probabilita. Per dire: dato unesperimento del tipo testa o croce, e evidente che ne sappiamo di piu se la probabilitae P (testa) = 1 e P (croce) = 0 (o viceversa), invece che se P (testa) = P (croce) = 1/2.Shannon ha trovato un modo naturale di parametrizzare questo contenuto di informazione,inventandosi una definizione di entropia, che non e l’entropia termodinamica, ne quella dellameccanica statistica, ma che in realta ha molto a che fare con entrambe. Come vedremo,l’entropia introdotta da Shannon, non parametrizza il contenuto di informazione di unadistribuzione, bensı quello di “ignoranza”, cioe la quantita di informazione che bisgnasupplire per sapere con esattezza il risultato di un esperimento.

2.2. ENTROPIA ED INFORMAZIONE 11
Ω
ΑΑ
ΑΑ
ΑΑ6
5
4
3
2
1
Figura 2.1: Partizione di un dominio bidimensionale Ω (spazio dei campioni).
Iniziamo subito con il chiarire che il contenuto di informazione dipende dal tipo diesperimento. Nel caso “testa o croce”, la questione puo sembrare ovvia, ma nel caso checi riguarda, quello di variabili random, non e cosı: non possiamo misurare una variabilerandom con infinita precisione. Di fatto possiamo misurare soltanto eventi. Introduciamosubito, quindi, il concetto di partizione di Ω (vedi Fig. 2.1) come una collezione di eventiAi mutualmente esclusivi, tali che la loro unione e Ω:
P = Ai, i = 1, . . . ; Ai ∩ Aj = ∅ se i 6= j; ∪iAi = Ω. (2.16)
Definiamo quindi l’entropia di Shannon della distribuzione P riferita alla partizione Pdi Ω tramite la formula
S[P ] = −∑
i
P (Ai) lnP (Ai) = −〈lnP 〉, (2.17)
dove la notazione S[P ] e una abbreviazione per S(Pi, i = 1, . . .). Vediamo subito che nelcaso “testa o croce” le cose tornano. In questo caso, banalmente P ≡ testa, croce. SeP (testa) = 1 oppure P (croce) = 1 (moneta truccata) abbiamo evidentemente S[P ] = 0(sappiamo il risultato; ignoranza minima; ma anche entropia minima, giacche 0 ≤ P ≤ 1 ⇒lnP ≥ 0 ⇒ S[P ] ≥ 0). Nel caso P (testa) = P (croce) = 1/2, abbiamo invece S[P ] = ln 2(massima ignoranza).
Di fatto le cose tornano molto bene. Cerchiamo di capire il perche.L’aspetto centrale e che l’entropia e nella forma
S[P ] =∑
i
g(Pi),
con g(P ) una funzione convessa dell’argomento. In altre parole vale la relazione
g(Pi) ≤ g(P ), (2.18)
dove x = 1N
∑Ni=1 xi e la media aritmetica degli xi. Il risultato e mostrato in Fig. 2.2, nel
caso di due eventi A1 e A2, aventi probabilita rispettivamente P1 e P2. Il loro contributoall’entropia e g(P1) + g(P2) = 2g(P ). Se le probabilita fossero uguali, mantenendo le altreprobabilita Pi, i = 3, . . . invariate (questo massimizza l’ignoranza solo rispetto ai primi due

12 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
g(P)
g(P)
P P1 2P
g
1 P
Figura 2.2: Rappresentazione grafica della relazione (2.18) nel caso N = 2.
eventi), avremmo P (A1) = P (A2) = (P1+P2)/2 = P . In tal caso il contributo all’entropiasarebbe 2g(P ) ≥ 2g(P ).
Questo esempio ci da un’illustrazione come l’entropia cresca con l’ignoranza, e sia ne-cessariamente massima quando tutti gli eventi hanno ugual probabilita. E come cio siaconseguenza del fatto che S[P ] e una somma di funzioni convesse della probabilita deglieventi.
L’informazione che deve essere supplita per conoscere lo stato del sistema dev’essere dinuovo aumentata se raffiniamo la partizione, per esempio se spacchiamo l’evento A6 in Fig.2.1 in due eventi A61 e A62 con A61 ∩ A62 = ∅ e A61 ∪ A62 = A6. Vediamo che di nuovol’entropia di Shannon si comporta bene ed aumenta in risposta al raffinamento. Infatti,il contributo alla entropia rimane g(A6) solo se tutta la probabilita di A6 si concentra inuno solo dei due nuovi sotto-eventi. In caso contrario, per quanto detto precedentemente,a causa della convessita di g, S dovra aumentare.
Un’altra proprieta, che ci immaginiamo debba godere un concetto di informazione pro-priamente definito, e che l’informazione riferita a due sistemi indipendenti sia la sommadelle informazioni riferite singolarmente a ciascuno. Quindi l’entropia deve essere additi-va se Ω = ΩA ⊗ ΩB, P = PA ⊗ PB e tutti gli eventi sono statisticamente indipendenti:P (Ai, Bj) = PA(Ai)PB(Bj). Da qui troviamo:
S[P ] = −∑
ij
P (Ai, Bj) ln(P (Ai, Bj) =
= −∑
ij
[PA(Ai)PB(Bj) lnPB(Bj) + PB(Bi)PA(Aj) lnPA(Aj)]
= −∑
i
[PB(Bi) lnPB(Bi) + PA(Ai) lnPA(Ai)] = S[PB] + S[PA]. (2.19)
Vediamo quindi che il logaritmo e importante...Nel tragitto a ritroso che compiremo dalla entropia di Shannon alla entropia termodi-
namica, vedremo che la proprieta di addittivita legata alla funzione logaritmica e essenzialeper aveve una termodinamica estensiva. La proprieta di convessita e in certo qual modopiu fondamentale ed ha a che fare con la possibilita di definire un concetto di equilibrio ter-modinamico. Un ambito di ricerca piuttosto attivo (e controverso) riguarda capire se per

2.3. SOMME DI VARIABILI ALEATORIE: IL LIMITE TERMODINAMICO 13
sistemi fortemente correlati (per cui una suddivisione in parti poco interagenti, al contrarioche in un gas, non e possibile), potrebbe aver senso una termodinamica “non estensiva”,costruita a partire da una definizione di entropia senza logaritmi.
Ritornando alla definizione originaria di entropia, Eq. (2.17), ci si potrebbe domandarese e possibile in qualche maniera definire una entropia non associata alla scelta di unapartizione. La risposta e “ni”. Esiste sı una particolare scelta di partizione uniforme (in1D sarebbe Ai = [xi, xi+1] con xi = i∆x), per cui
S[P ] ≃ −∫
ρ(x) ln(ρ(x)∆x)dx = −∫
ρ(x) ln ρ(x)dx− ln∆x (2.20)
e la funzione Sρ = −∫
ρ(x) ln ρ(x)dx e detta entropia di PDF. A parte la difficolta evidentenella definizione di S[P ] nel caso di una partizione infinitamente fine, una partizione involumetti infinitesimi, come vedremo ben presto, non e pero la piu appropriata per isistemi a cui siamo interessati. La scelta della partizione nella definizione dell’entropia none irrilevante.
2.3 Somme di variabili aleatorie: il limite termodina-
mico
Oggetto della statistica e la stima di medie e probabilita, a partire da medie campionariee frequenze. Di fatto, esiste una interpretazione empirica della probabilita come limite difrequenze e medie campionarie, considerata piu intuitiva (ma di sicuro meno soddisfacente)di quella assiomatica standard. Il concetto di media campionaria e definito nella manieraseguente: data una sequenza di esperimenti identici e indipendenti (tali cioe che i valoridella variabile aleatoria misurata in ciascun esperimento, siano statisticamente indipendentie identicamente distribuiti, abbreviato, i.i.d.), si definisce media campionaria 〈x〉N dellavariabile aleatoria x tramite la formula
〈x〉N =1
N
N∑
k=1
xk, (2.21)
dove xk e il risultato del k-esimo esperimento. La frequenza PN(A) dell’evento A eottenuta in maniera analoga a partire dalla definizione di funzione indicatrice (2.13) edalla (2.14)
PN(A) = 〈δA〉N . (2.22)
La connessione fra la definizione di media campionaria e lo studio dei sistemi termodina-mici risiede nel fatto che questi ultimi sono descritti da grandezze (ad esempio l’energia,oppure il numero di molecole in un volume), che sono somme delle corrispondenti variabilidelle singole molecole (energia della molecola; la funzione indicatrice riferita al volume dellamolecola). Se le molecole sono in prima approssimazione indipendenti, l’energia termica

14 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
nel volume, il numero di molecole, si comporteranno come somme di un gran numero di va-riabili aleatorie indipendenti, e, se il sistema e omogeneo, esse saranno anche identicamentedistribuite. Precisamente come la somma che appare a lato destro della (2.21).
Ci si riferisce di solito alla condizione che le fluttuazioni nel valore di una grandezzamacroscopica XN diventino trascurabili nel limite di N molto grande, come a una con-dizione di limite termodinamico. Sotto le ipotesi indicate sopra, i concetti di limitetermodinamico e di legge dei grandi numeri diventano equivalenti.
In realta, una media campionaria, cosı come una somma di varialbili aleatorie, sono an-cora variabili aleatorie. Il fatto che le fluttuazioni intorno alla media diventino trascurabiliper N grande e il contenuto della ben nota legge dei grandi numeri. Nel caso di unasomma di variabili aleatorie i.i.d. XN =
∑Ni=1 xi, con media e varianza finite:
µXN= Nµx; σXN
= N1/2σx (2.23)
e quindi σXN/µXN
= N−1/2σx/µx. In altre parole, la fluttuazione relativa δXN/XN eO(N−1/2) e XN cessa di essere aleatoria nel limite N → ∞. Se pensiamo a XN co-me a una variabile fisica macroscopica, abbiamo evidentemente una condizione di limitetermodinamico.
Verifichiamo la (2.23) in maniera esplicita.Abbiamo immediatamente per linearita della operazione di media µXN
= NµX . Calco-liamo la varianza di XN :
σ2XN
= 〈[N∑
i=1
(xi − µx)]2〉 =
N∑
ij=1
〈(xi − µx)(xj − µx)〉
Essendo pero le differenti xi statisticamente indipendenti, abbiamo per j 6= j 〈(xi−µx)(xj−µx)〉 = 〈(xi − µx)〉〈(xj − µx)〉 = 0 e quindi
σ2XN
=N∑
i=1
〈(xi − µx)2〉 = Nσ2
x,
che ci da la seconda delle (2.23). Notiamo che dividendo la (2.23) per N , ritroviamo lalegge dei grandi numeri nella sua formulazione standard:
〈〈x〉N〉 = 〈x〉; σ2〈x〉N
=σ2x
N. (2.24)
Un ulteriore risultato, detto teorema del limite centrale, ci dice che le deviazioni XN−µXN,
nel limite di grandi N , sono distribuite secondo una Gaussiana:
ρ(XN) =1
(2π)1/2σXN
exp(
− (XN − µXN)2
2σ2XN
)
. (2.25)
La dimostrazione generale della (2.25) puo essere trovata ad esempio nel libro di MeccanicaStatistica di Reichl, oppure nel capitolo sulle fluttuazioni di densita del testo “Fisica Sta-tistica” di Landau e Lifstits. Una delle conseguenze delle (2.23-2.25) e che per N grande,

2.4. ENTROPIA ED EQUILIBRIO TERMODINAMICO 15
una misura di XN dara quasi sempre un risultato XN ≃ 〈XN〉, il valore di equilibrio dellagrandezza macroscopica XN , cioe il valore piu probabile di XN .
Nota Vale il risultato generale che la somma di variabili Gaussiane e essa stessa una va-riabile Gaussiana. Il modo piu semplice di dimostrarlo e lavorando in spazio di Fourier. Sez = x+ y, con x e y variabili Gaussiane (e sufficiente considerare il caso in cui hanno en-trambe media nulla), possiamo scrivere ρz(z) = 〈δ(x+y− z)〉 =
∫
dx∫
dy ρx(x)ρy(y)δ(x+y − z) =
∫
dxρx(x)ρy(z − x). Abbiamo per le trasformate di Fourier di ρx e ρy: ρx(k) =
(2πσ2x)
−1/2∫ +∞
−∞exp(−x2/(2σ2
x) − ikx)dx = exp(−(1/2)σ2xx
2) e analoga espressione perρy(k). Da qui, sfruttando il fatto che la trasformata di Fourier di una convoluzione eun prodotto: ρz(k) = ρx(k)ρy(k) = exp[−(1/2)(σ2
x + σ2y)k
2], che mi dice che ρz(z) e unaGaussiana con varianza σ2
z = σ2x + σ2
y.
Come esempio di limite termodinamico, consideriamo il caso in cui la variabile aleatoriaassociata alla molecola k-esima e δVa(xk) con con xk la coordinata della molecola, e Va unsottovolume (macroscopico) del volume totale V occupato dal gas. La variabile d’interessesara percio il numero istantaneo Na di molecole nel volume Va (dal punto di vista proba-bilitico, il problema di determinare il numero di punti distribuiti a caso che cadono in uncerto dominio Va e detto problema dei punti di Poisson).
Supponendo il sistema spazialmente omogeneo (che e quanto ci si aspetta all’equilibriotermodinamico), la probabilita di trovare una molecola data in Va sara
P (xk ∈ Va) ≡ 〈δVa〉 = Va/V
e il numero medio di molecole in Va sara
Na = 〈Na〉 = N〈δVa〉 = NVa/V ≡ nVa,
dove n = N/V e la densita media del gas in V . Per calcolare la fluttuazione, dobbiamodeterminare σ2
δVa. Abbiamo
〈δ2Va〉 = 〈δVa〉 = Va/V
e pertantoσ2δVa
= 〈δ2Va〉 − 〈δVa〉2 = Va/V (1− Va/V ).
Se V viene mandato ad infinito e Va rimane fissato, possiamo trascurare il termine qua-dratico in Va/V e troviamo per la fluttuazione
σ2Na
= Nσ2δVa
≃ Na ⇒δNa
Na
= O(N−1/2a ). (2.26)
In particolare, la fluttuazione di densita in un volume Va sara δna = δNa/Va = (Van)−1/2n.
2.4 Entropia ed equilibrio termodinamico
La caratteristica piu importante dell’entropia termodinamica e quella di esprimere l’ap-proccio all’equilibrio termodinamico, come un processo di crescita di entropia. Vediamo

16 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
che l’entropia di Shannon ha tutte le carte in regola per svolgere un simile ruolo. Con-sideriamo come sistema un gas ideale di N particelle, posto in un recipiente di volumeV , in assenza di forze esterne. Interessiamoci del processo che porta da una situazioneiniziale in cui il gas era confinato in una parte del recipiente Va ⊂ V , a una condizione diequilibrio, in cui le molecole sono sparpagliate in tutto V . Cerchiamo di parametrizzarel’approccio all’equilibrio in modo piu quantitativo, e concentriamoci su una quantita comeil numero di molecole medio in Va, all’istante t: Na(t) ≡ 〈Na(t)〉. La quantita Na(t) indicail numero di occupazione istantaneo di Va, e immaginiamo la media effettuata attraversoun gran numero di esperimenti a partire dalla stessa configurazione iniziale, in cui tuttele molecole erano in Va. Cercheremo di essere piu precisi, nel seguito, su come definireconcetti quali medie e probabilita dipendenti dal tempo. Procedendo in maniera intuitiva,possiamo tuttavia provare a descrivere l’approccio all’equilibrio termodinamico attraversouna relazione come
limt→∞
Na(t) = Na = NVa/V.
Abbiamo visto nella sezione precedente che il numero istantaneo di molecole in Va puo esserescritto come Na(t) =
∑Ni=1 δVa(xi(t)). Possiamo quindi scrivere Na(t) = NP (Va; t), dove
P (Va; t) e la probabilita che una molecola data, si trovi all’istante t in Va. La transizioneall’equilibrio termodinamico puo essere quindi espressa in termini di probabilita attraversola relazione P (Va; t) → P (Va) = Va/V .
Questo ci da un suggerimento per ottenere dalla entropia di Shannon una entropiacon significato termodinamico. Suddividiamo il volume V in sottovolumi identici Va,a = 1, 2 . . . , K, che rispecchia la nozione che a priori punti diversi del recipiente devonoessere considerati equivalenti. Scopriamo subito che l’entropia di Shannon associata allapartizione uniforme Va, S[P ] = −∑
a P (Va; t) lnP (Va; t) sara massima all’equilibrio termo-dinamico S[P ] = lnK. Vedremo in seguito come la teoria cinetica (teorema H) ci permettedi mostrare che l’entropia raggiunge il suo valore di equilibrio attraverso un processo dicrescita monotona. Ci limitiamo a notare, per il momento, l’importanza della scelta dellapartizione nella procedura. La scelta di una partizione uniforme e cio che ci ha permesso divedere l’entropia di Shannon come una misura del grado di sparpagliamento delle molecolenel recipiente. Altre scelte avrebbero prodotto una definizione di entropia corretta dalpunto di vista formale, ma priva di significato dal punto di vista termodinamico. Vedremonella Sez. 3.2.1 un altro esempio di come una scelta appropriata della partizione risultifondamentale per ottenere una entropia dotata di significato termodinamico.
Cosı come abbiamo fatto con la distribuzione di probabilita P (Va, t), possiamo calco-lare l’entropia della distribuzione istantanea P (Va, t) = Na/N . Essa gioca nei confrontidi P (Va, t), lo stesso ruolo che la frequenza PN(A), definita nella Eq. (2.22), giocava neiconfronti della probabilita P (A): cosı come, per la legge dei grandi numeri, P (A) e lafrequenza piu probabile, P (Va, t) sara, nel limite termodinamico, la distribuzione istanta-nea P (Va, t) osservata con maggiore probabilita. Troviamo quindi che la distribuzione diequilibrio P (Va) e allo stesso tempo la distribuzione corrispondente al massimo di entropiae la distribuzione istantanea P (Va) osservata con maggiore probabilita all’equilibrio.

2.5. FLUTTUAZIONI 17
2.5 Fluttuazioni
Continuiamo a considerare l’esempio del gas in un volume V , analizzato nelle ultime duesezioni. La condizione di limite termodinamico ci permetteva di approssimare il numeroistantanteo di molecole Na(t) in un volumetto Va ⊂ V sufficientemente grande, con il suovalore medio Na(t) = 〈Na(t)〉. A causa del carattere casuale dei moti delle molecole, e dellapossibilita di moti macroscopici turbolenti, causati da eventuali condizioni di non equilibrio,una componente di fluttuazione Na−Na sara comunque presente. Per caratterizzare questefluttuazioni, e conveniente definire, a partire dai numeri di occupazione Na(t) ed Na(t),densita istantanee e medie, coarse grained a scala Va:
nVa(x, t) =Na(x, t)
Vae nVa(x, t) = 〈nVa(x, t)〉.
Concentriamoci sulla struttura spaziale delle fluttuazioni. Come si e detto, queste avran-no in condizioni di non equilibrio sia una componente microscopica che una macroscopica.Continuiamo a considerare il caso di un gas di molecole non interagenti (gas ideale). Consi-deriamo inizialmente un condizione di equilibrio, di modo che sicuramente solo fluttuazionimicroscopiche sono presenti. Abbiamo quindi n(x, t) = n = N/V e le fluttuazioni sonogovernate dalla legge (2.26). In termini di densita, possiamo scrivere
σ2n(Va) ≡ 〈(nVa − n)2〉 = n(v/Va)
1/2,
dove v = 1/n e il volume moleculare del gas. Vediamo quindi che la condizione per cui lefluttuazioni microscopiche possono essere trascurate e Va ≫ v, nel qual caso nVa(x, t) ≃ n.
In condizioni di non equilibrio, saranno in generale presenti fluttuazioni macroscopiche,la cui scala caratteristica l sara essa stessa, per definizione, macroscopica l ≫ v1/3. Sescegliamo v ≪ Va ≪ l3, avremo quindi che nVa(x, t) sara ancora una quantita fluttuante,ma la componente microscopica delle fluttuazioni non c’e piu. Abbiamo inoltre due fattiimportanti:
• La differenza di nVa(x, t) tra volumetti Va adiacenti e piccola (dell’ordine di V1/3a /l).
Questo implica che e possibile eseguire un limite continuo su nVa .
• Lo stesso fatto che nVa varia poco a scala Va, implica che la dipendenza sul parametrodi coarse graining Va e debole (a patto naturalmente che v ≪ Va ≪ l3).
Eseguendo la media troviamo quindi una densita indipendente da Va, n(x, t) = 〈n(x, t)〉, ilcui profilo avra una scala di variazione L sara tipicamente maggiore di l, e la cui scala divariazione temporale, che identifica il tempo di rilassamento all’equilibrio del sistema, saratipicamente piu lunga di quella delle fluttuazioni (sia microscopiche che macroscopiche).
Nota Osserviamo che la discussione che abbiamo effettuato e basata su diverse ipotesiche non sono in generale soddisfatte. In primo luogo, non e detto che una separazionetra fluttuazioni discrete e macroscopiche esista sempre. Condizioni di non equilibrio pos-sono benissimo verificarsi a scale dell’ordine della separazione tra le molecole. Se il gas

18 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
e sufficientemente rarefatto, un simile risultato potrebbe essere prodotto infatti anche dal-l’azione di un corpo macroscopico. In altre parole, non e detto che fluttuazioni generateda condizioni di non equilbrio siano necessariamente macroscopiche. Allo stesso tempo,non e detto che fluttuazioni microscopiche siano possibili nel sistema. La presenza di in-terazioni fra le molecole (in generale, tra i costituenti microscopici del sistema), potrebbeinfatti produrre correlazioni a scala macroscopica, tali che fluttuazioni di taglia inferiorealla lunghezza di correlazione siano inibite. Come si vedra nel Capitolo 6, la scala ener-getica delle fluttuazioni in un sistema a temperatura T e data da KT , con K la costantedi Boltzmann. Pertanto, se la presenza di correlazioni impone che ciascuna fluttuazionecoinvolga un numero macroscopico di molecole, queste fluttuazioni avranno ampiezza tra-scurabile. (Esempio banale: in un solido, in cui ciascuna molecola e bloccata nella suaposizione dalle forze esercitate dai suoi vicini, fluttuazioni di concentrazione saranno pra-ticamente assenti). Di nuovo pero ci sono eccezioni: in presenza di transizioni di secondaspecie (magneti al punto critico e simili), il contenuto energetico di fluttuazioni a scalamacroscopica puo tendere a zero, e anche l’ampiezza di tali fluttuazioni sara macroscopica.In altre parole, abbiamo un sistema che, sebbene all’equilibrio termodinamico, e caratteriz-zato da fluttuazioni macroscopiche, per cui il concetto stesso di limite termodinamico none ben definito.
2.6 Processi stocastici e campi random
Come si e detto, il passaggio da una descrizione microscopica di un sistema, ad una macro-scopica, sottointende una perdita di informazione. Operativamente, cio si concretizza inuna trattazione probabilistica dei gradi di liberta microscopici. La scelta diventa naturalese la dinamica alla microscala ha una componente intrinsecamente casuale, nel senso chee difficile prevedere l’evoluzione del sistema, a scala microscopica, a partire da una con-dizione iniziale data. Simili comportamenti casuali possono essere presenti anche a scalamacroscopica, lontano dall’equilibrio; un esempio sono le fluttuazioni turbolente. Diventaquindi necessario introdurre strumenti matematici in grado di descrivere grandezze fisicheche assumono istantaneamente valori casuali, che non saranno in generale indipendenti,come era nel caso degli esperimenti indipendenti considerati nella Sez. 2.3. In alternativa,potremmo desiderare descrivere l’andamento di campi fluttuanti sia nello spazio che neltempo, come ad esempio il campo di velocita in un flusso turbolento. Alcuni esempi diprocessi che potremmo voler descrivere:
1. L’uscita di un generatore di rumore; oppure la posizione di un batterio in una capsuladi Petri; oppure la velocita del vento misurata da un anemometro.
2. I valori della velocita del vento a un dato istante, misurati in una certa regionespaziale; oppure i valori di densita dei batteri nella capsula di Petri in questione aun dato istante, ma in diverse posizioni.
3. I valori della velocita del vento a diversi istanti di tempo e posizioni spaziali. Eccetera.

2.6. PROCESSI STOCASTICI E CAMPI RANDOM 19
Abbiamo a che fare con un insieme di valori casuali associati a diversi istanti di tempo(caso 1); diverse posizioni spaziali (caso 2); diverse posizioni spaziali e istanti di tempo(caso 3). Diamo pertanto le seguenti definizioni:
• Si chiama processo stocastico una sequenza di variabili aleatorie indicizzate con iltempo. Si ha un processo discreto se l’indicizzazione e discreta; per esempio, ψ(tn),tn = n∆t. Si ottiene un processo continuo, evidentemente, come limite ∆t → 0 diun processo discreto.
• Si parla di campo random, quando l’indicizzazione e fatta nello spazio (o nellospazio tempo). Per esempio: ψ(xn, tm).
Queste definizioni ci costringono a rivedere tutti i concetti di spazio dei campioni, risultatodi misura e probabilita, che abbiamo utilizzato sin’ora nel caso delle variabili aleatorie. Perfissare le idee, ci concentriamo sul caso di un processo stocastico, essendo la generalizzazioneal caso di un campo random abbastanza naturale.
Osserviamo in primo luogo che, mentre il risultato di una misura, nel caso di unavariabile aleatoria, era un numero reale, nel caso di un processo stocastico, e da intendersicome risultato l’intera sequenza di valori ψ(tn), n = 1, 2, . . . assunti da ψ agli istanti tn.Per dire, nel caso di un generatore di rumore, l’esperimento e da intendersi l’accensionedello strumento e la registrazione dei dati in un intervallo di tempo, e non la misura di unsingolo dato ψ a un dato istante t.
• Chiamiamo realizzazione del processo stocastico, la sequenza ψ(tn), n = 1, 2, . . .risultante da un dato esperimento.
Vediamo che, mentre nel caso di una variabile aleatoria, Ω era un’intervallo dei reali,nel caso di un processo stocastico, Ω e uno spazio di funzioni (di variabile discreta se ilprocesso discreto, di variabile reale se il processo e continuo).
Abbiamo inoltre che, mentre una variabile aleatoria x era caratterizzata da una PDFρ(x) che e una funzione di variabile reale, nel caso di un processo stocastico discreto,avremo una PDF nella forma ρ(ψ(tn), n = 1, 2 . . .), cioe una funzione che generalizzail caso di una PDF di un vettore random al caso in cui il vettore random puo avere unnumero arbitrario di componenti. Nel caso di un processo stocastico continuo, abbiamoaddirittura che la PDF, che in questo caso si indica solitamente utilizzando le parentesiquadre: ρ[ψ], e un “funzionale”, cioe una funzione che agisce da uno spazio di funzioni divariabile reale [le ψ(t)] al campo reale (i valori di ρ). Un esempio:
ρ[ψ] = N exp(
−∫ T
0
|ψ(t)|2dt)
. (2.27)
E in generale difficile lavorare con oggetti come la (2.27). Possiamo provare a ottenere unadescrizione “ridotta” della statistica di ψ, analoga a quella che si ottiene accontentandosinel dare media e varianza nel caso di una variabile aleatoria. Ecco alcuni esempi di oggettiche potrebbero fornirci una tale descrizione ridotta:
ρ(ψ(t)), ρ(ψ(t), ψ(t′)), . . . , (2.28)

20 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
cioe le PDF del valore di ψ a istanti dati, e
〈ψ(t)〉, 〈ψ(t)ψ(t′)〉, . . . , (2.29)
che sono dette funzioni di correlazione della ψ a un tempo, a due tempi, eccetera.L’utilita delle funzioni di correlazione e che ci permettono di parametrizzare in maniera
semplice il modo in cui la dipendenza statistica tra valori di ψ a tempi diversi diminuisceal crescere della separazione temporale. Abbiamo infatti per definizione:
〈ψ(t)ψ(t′)〉 =∫
dψ(t)
∫
dψ(t′) ρ(ψ(t), ψ(t′))ψ(t)ψ(t′)
e nel caso di variabili statisticamente indipendenti, cioe se ρ(ψ(t), ψ(t′)) = ρt(ψ(t))ρt′(ψ(t′)):
〈ψ(t)ψ(t′)〉 = 〈ψ(t)〉〈ψ(t′)〉.
Un indicatore del grado di dipendenza statistica e quindi fornito dalla quantita
C(t, t′) = 〈ψ(t)ψ(t′)〉 − 〈ψ(t)〉〈ψ(t′)〉, (2.30)
che sara massima per t = t′, con C(t, t) = σ2ψ(t), mentre ci si aspetta che tenda a zero per
|t−t′| → ∞. Possiamo introdurre un tempo di correlazione che ci dice per quali separazionitemporali le correlazioni possono iniziare a considerarsi trascurabili. Di solito si utilizza ladefinizione
τψ(t) =1
C(t, t)
∫ ∞
0
C(t, t+ τ)dτ (2.31)
[come dire, area sottesa da C(t, t + τ) uguale base, cioe τψ(t), per altezza, cioe C(t, t)].In termini di singola realizzazione del processo stocastico, il parametro τψ(t) ci dice essen-zialmente quanto bisogna aspettare prima che ψ(t + τ) differisca molto da ψ(t). In altreparole, il tempo di correlazione ci fornisce la scala di variazione temporale del processostocastico.
Le medie che compaiono nelle equazioni che abbiamo scritto sin’ora, possono esserestimate attraverso il calcolo di medie campionarie. Dobbiamo ricordarci pero che nel nostrocaso, queste medie campionarie sono di fatto medie su realizzazioni diverse (e indipendenti)dello stesso processo stocastico. Ora, ci sono molte situazioni in cui il processo stocastico,anche se descrive un qualche cosa che fluttua nel tempo, puo essere caratterizzato dauna statistica che e invece indipendente dal tempo; si parla in questo caso di statisticastazionaria. Intendiamo con cio che la PDF congiunta ρ(ψ(t1), ψ(t2), . . .) e invariante pertraslazioni temporali:1
ρ(ψ(t1), ψ(t2), . . .) = ρ(ψ(t1 + T ), ψ(t2 + T ), . . .), ∀ T.1Per contro, si dice spesso che un processo stocastico ha raggiunto uno stato stazionario, quando
la PDF condizionata ρ(ψ(t1), ψ(t2) . . . |ψ(t0)), t0 < t1 < t2 < . . ., di un processo stazionario, diventaindipendente dal condizionamento a t0. Tipicamente questo richiede che si prenda il limite t0 → −∞.Notare come il fatto che un processo sia stazionario non implica necessariamente che uno stato stazionariosia sempre raggiunto; vedere l’esempio del ping-pong a pagina 27.

2.6. PROCESSI STOCASTICI E CAMPI RANDOM 21
Questo implica naturalmente ρ(ψ(t)) = ρ(ψ), 〈ψ(t)〉 = 〈ψ〉 . . . e che le correlazioni di-pendono solo dalle differenze di tempo; in particolare C(t, t′) = C(t − t′). L’esempio piusemplice di processo stazionario che ci puo venire in mente, e quello che si ottiene mettendoin sequenza i risultati delle misure di una variabile aleatoria x: ψ(tn) ≡ x(n).
Nel caso di un processo stocastico costruito in questo modo, e abbastanza evidente checalcolare medie sulle realizzazioni come 〈ψ(tn)〉N = (1/N)
∑Nk=1 ψ
(k)(tn), [ψ(k)(tn) e il valore
di ψ(tn) nella n-esima realizzazione di ψ] diventa un’operazione inutilmente dispendiosa.Si puo infatti stimare 〈x〉, attraverso media campionaria 〈x〉N = (1/N)
∑Nk=1 x
(k), a partireda una sola realizzazione della ψ. Questo ci conduce a introdurre un nuovo tipo di media,la cosı detta media temporale:
〈ψ〉T =1
T
∫ T
0
ψ(t)dt, (2.32)
dove l’integrale, nel caso di un processo discreto, e sostituito dalla somma∑
n ψ(tn)∆t.Ovviamente, nel caso in cui il processo stocastico e il risultato di una sequenza di misure
indipendenti di una stessa variabile aleatoria, per costruzione, la media temporale e lamedia campionaria sulla variabile aleatoria coincidono. Ci domandiamo nel caso generale,quando una media temporale puo essere utilizzata per approssimare la media 〈.〉.
• Diciamo che un processo per cui la media temporale, per T → ∞, tende alla media〈.〉, gode della proprieta ergodica.
Ci aspettiamo che le condizioni che permettono il soddisfacimento della proprieta ergodicasiano:
• Che i valori di ψ ad istanti di tempo sufficientemente lontani diventino statisticamenteindipendenti.
• Che il processo sia stazionario a scale di tempo sufficientemente lunghe da conteneremolti intervalli di tempo tali che i valori di ψ nei diversi intervalli siano indipendenti.
• Che ovviamente siano soddisfatte le condizioni perche la legge dei grandi numeri siasoddisfatta, e cioe che la quantita di cui si calcola la media temporale abbia varianzae media finite.
A questo punto, la media temporale diventa essenzialmente una media campionaria tra ivalori di ψ nei diversi intervalli, dove ψ si puo considerare approssimativamente costante.
Verifichiamo questo risultato con un calcolo esplicito. Consideriamo il caso semplicein cui l’integrale nella Eq. (2.31) e finito (il risultato che segue puo essere generalizzatoal caso in cui τψ = ∞, ma la correlazione decresce lo stesso all’infinito). Supponiamo diapprossimare 〈ψ〉 con 〈ψ〉T , e, in analogia con la legge dei grandi numeri, calcoliamo 〈〈ψ〉T 〉e σ2
〈ψ〉T. Scambiando media e integrale nell’integrale per 〈ψ〉T , otteniamo subito.
〈〈ψ〉T 〉 = 〈〈ψ〉〉T = 〈ψ〉.

22 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
Definiamo quindi ψ(t) = ψ(t)−〈ψ〉 lo scarto (o fluttuazione rispetto alla media) e scriviamo:
σ2〈ψ〉T
=1
T 2
∫ T
0
dt
∫ T
0
dt′ 〈ψ(t)ψ(t′)〉 = 1
T 2
∫ T
0
dt
∫ T
0
dt′ C(t− t′).
Ora, per T ≫ τψ, possiamo approssimare l’integrale in dt′, per quasi tutti i valori di t, conun integrale tra ±∞ [a t′ = 0 e a t′ = T , se |t− t′| ≫ τψ, C(t− t′), e comunque gia nullo]:
σ2〈ψ〉T
≈ 1
T 2
∫ T
0
dt
∫ +∞
−∞
dτ C(τ) =2τψTC(0) ≡ 2τψ
Tσ2ψ (2.33)
ed ho utilizzato la (2.31). La varianza della media temporale decresce con τψ/T , che giocaquindi il ruolo del fattore 1/N nel caso della media campionaria.
2.6.1 Il teorema di Wiener-Khinchin
Nel caso di statistica stazionaria, una caratterizzazione alternativa delle proprieta di unprocesso stocastico (o di un campo random) puo essere ottenuta lavorando in spazio diFourier. Dato un processo stocastico x(t), possiamo definire la sua trasformata di Fourier:
xω =
∫ +∞
−∞
x(t)e−iωtdt. (2.34)
Dal punto di vista matematico, una simile espressione potrebbe avere senso solo se intesacome una distribuzione. Dal punto di vista pratico, ci accontentiamo di interpretare la(2.34) come una trasformata di Fourier in un dominio grande fissato [−T/2, T/2] e per unadiscretizzazione ∆t sufficientemente fine, ma anch’essa fissata. Cosı come abbiamo calco-lato le correlazioni del processo originario x(t), possiamo provare a calcolare le correlazionidella sua trasformata di Fourier. Otteniamo il seguente risultato:
〈xωxω′〉 =
∫ +∞
−∞
dt
∫ +∞
−∞
dt′〈x(t)x(t′)〉 exp[−i(ω′t′ + ωt)]
=
∫ +∞
−∞
dt
∫ +∞
−∞
dt′C(t− t′) exp[−iω(t− t′)− i(ω + ω′)t′],
dove si e sfruttato la statistica stazionaria per scrivere C(t, t′) = C(t− t′). Ora, l’integrale(2π)−1
∫ +∞
−∞dt′ exp[−i(ω + ω′)t′] non e altro che una delle rappresentazioni della delta di
Dirac δ(ω + ω′). Troviamo pertanto
〈xωxω′〉 = 2πCωδ(ω + ω′) (2.35)
dove Cω =∫ +∞
−∞dtC(t) exp(−iωt)dt, la trasformata di Fourier della funzione di correlazione
C(t), e il cosı detto spettro di potenza di x(t). Questo e il contenuto del teorema diWiener-Khinchin. Notiamo come l’antitrasformata a separazione temporale nulla
〈x2〉 ≡ C(0) =
∫ +∞
−∞
dω
2πCω

2.7. PROBABILITA DIPENDENTI DAL TEMPO 23
fornisca essenzialmente una decomposizione spettrale della varianza 〈x2〉. In svariati casi〈x2〉 puo essere interpretato come la potenza media di un segnale (x potrebbe essere adesempio il campo elettrico misurato vicino a un’antenna). Il significato di Cω come densitaspettrale di potenza del segnale e in questo caso evidente. Notiamo inoltre come per un
dominio finito abbiamo δ(0) → (2π)−1∫ T/2
−T/2dt = T/(2π),2, che permette di interpretare
〈|xω|2〉 = CωT
come la densita spettrale di energia a frequenza ω del segnale. (Si e utilizzata la proprietadelle trasformate di Fourier di funzioni reali: xω = x∗−ω).
2.7 Probabilita dipendenti dal tempo
Il passaggio da una descrizione microscopica di un sistema ad una macroscopica significain buona sostanza tralasciare una gran parte delle informazioni sullo stato istantaneo delsistema. Abbiamo visto che mediando a scale sufficientemente grandi, si potrebbe rag-giungere un limite termodinamico in cui il sistema (almeno vicino all’equilibrio) e descrittoda quantita che di nuovo obbediscono equazioni deterministiche (per esempio, la legge distato dei gas, oppure l’equazione del calore). Se pero ci limitiamo a considerare scale so-lo di poco superiori a quella di una piena descrizione microscopica, una tale descrizionediventa evidentemente impossibile. Analogamente se ci interessiamo a situazioni di fortenon-equilibrio in cui le fluttuazioni delle variabili macroscopiche diventano importanti.
Ci domandiamo come procedere per costruire un processo stocastico che modelli inmaniera soddisfacente situazioni come quelle considerate. Dal punto di vista dinamico, ilproblema e essenzialmente determinare l’evoluzione del processo (o meglio, determinarnela probabilita), data la storia pregressa del processo in questione. Il centro dell’analisisara necessariamente la probabilita delle realizzazioni del processo stocastico, che, comeabbiamo visto, fornisce una descrizione completa della statistica del processo in questione.
Per ragioni di semplicita (e altre ragioni di carattere concettuale che approfondiremofra breve), ci concentriamo sul caso di processi stocastici a valori discreti e a tempi discreti.Dato un processo stocastico ψk ≡ ψ(tk), utilizziamo quindi la notazione
P (ni; i = 0, 1, . . .) ≡ P (ψ(ti) ∈ [ψn, ψn+1], i = 0, 1, . . .); ti = i∆t, ψn = n∆ψ.
Chiamiamo gli intervalli [ψn, ψn+1] stati del sistema. Ci poniamo la seguente domanda:
• Qual’e la probabilita di un certo stato ni+1 al tempo ti+1 data una certa storiapregressa nk; k = 0, 1, . . . , i?
Questa e una probabilita condizionata, legata alla probabilita di realizzazione dalla rela-zione standard:
P (ni+1|nk; k = 0, 1, . . . i) = P (nk; k = 0, 1, . . . , i+ 1)P (nk; k = 0, 1, . . . , i) .
2Si ricorda che in un dominio finito la trasformata di Fourier viene rimpiazzata da una serie di Fourier.La distanza dei modi ∆ω ∼ T−1 e anche quindi la “larghezza” della delta di Dirac

24 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
Considereremo solo due casi chiave:
• Sistemi totalmente scorrelati (ad esempio il risultato del lancio di una moneta; seviene testa vale uno, se viene croce viene −1):
P (ni+1|nk; k = 0, 1, . . . i) = P (ni+1).
• Sistemi in cui conoscenza dello stato a un certo istante, rende inutile l’informazionesugli stati precedenti (esempio: il cosı detto camminatore random, che ad ogni istantefa un passo avanti o indietro a seconda del lancio della moneta. La posizione correntedel camminatore dipende solo dalla sua posizione precedente e dall’ultimo passoeffettuato; dove il camminatore si trovase in precedenza e completamente irrilevante).In formule:
P (ni+1|nk; k = 0, 1, . . . i) = P (ni+1|ni). (2.36)
Un processo con simili caratteristiche e detto un processo Markoviano.
I processi Markoviani ci forniscono uno strumento sufficientemente semplice per modellareil comportamento dei sistemi fisici di cui ci interesseremo lungo il corso. La probabilitaP (ni+1|ni) e detta una probabilita di transizione, e nel caso omogeneo nel tempo in cuiquesta e indipendente da i, utilizzeremo spesso la notazione
P (k → m) ≡ P (ni+1 = m|ni = k).
Consideriamo con un po’ piu di attenzione il cammino random, di cui abbiamo dato unesempio qui sopra. Nel caso in questione, ni rappresenta la casella dove si trova il cammina-tore all’istante ti e abbiamo evidentemente (se la moneta non e truccata) P (ni+1|ni) = 1/2se ni+1 = ni ± 1 e zero altrimenti. Ci domandiamo qual’e la probabilita P (ni|n0) delcamminatore di trovarsi in ni al tempo ti, se al tempo zero si trovava in n0.
Vediamo che abbiamo a che fare con un primo esempio di probabilita dipendentedal tempo, perche se P (n0 = k|n0 = j) = δkj, cioe la probabilita e concentrata in n0 = j,a un tempo successivo ti, il camminatore potra trovarsi in una qualsiasi casella compresatra j − i e j + i. Vediamo quindi che la distribuzione di probabilita del camminatoresi sparpaglia a passare del tempo. Il punto della questione, evidentemente, sta tutto neltrovare delle equazioni di evoluzione per queste probabilita dipendenti dal tempo, analoghealle equazioni del moto per un sistema deterministico.
Nel caso dei processi Markoviani, l’operazione e abbastanza semplice. Abbiamo infatti,dalla (2.36):
P (ni+1, ni, n0) = P (ni+1|ni, n0)P (ni|n0)P (n0) = P (ni+1|ni)P (ni|n0)P (n0)
e quindi
P (ni+1, ni|n0) = P (ni+1|ni)P (ni|n0),

2.7. PROBABILITA DIPENDENTI DAL TEMPO 25
da cui otteniamo, sommando sugli stati intermedi, la seguente equazione di Chapman-Kolmogorov:
P (ni+1|n0) =∑
ni
P (ni+1|ni)P (ni|n0). (2.37)
Detto in parole: la probabilita di trovare all’istante dato il sistema in un certo stato siottiene sommando sulle probabilita che questo si trovasse in tutti possibili stati all’istanteprecedente, pesate sulla probabilita di transizione dallo stato precedente a quello corrente.
Facciamo subito le seguenti osservazioni:
• Noi possiamo moltiplicare ambo i lati della equazione di Chapman-Kolmogorov (2.37)per una probabilita iniziale P0(n0) e sommare su P0(n0); otterremo la seguente varian-te della equazione di Chapman-Kolmogorov per una generica probabilita dipendentedal tempo:
P (ni) =∑
n0
P (ni|n0)P0(n0)
• E importante capire che la P0(n0) e arbitraria; nel caso del camminatore randompotremmo domandarci ad esempio come si distribuirebbe nel tempo un gruppo dicamminatori distribuiti inizialmente in maniera uniforme nelle caselle comprese trazero e dieci.
• Per determinare la probabilita che il sistema si trovi in un certo stato al tempo dato,dobbiamo quindi in genere assegnare una distribuzione di probabilita iniziale. Notabene che la scelta e nostra, non “appartiene” al processo stocastico: cio che e fissatodal processo stocastico e solo la probabilita di transizione.
• Notiamo infine che, se le probabilita di transizione sono indipendenti dal tempo,P (ni+1=m|ni=q) = P (q → m), e P0(n) e una soluzione stazionaria della equazionedi Chapman-Kolmogorov, P (ni) = P0(ni), allora la statistica del processo sara sta-zionaria: P (ni=m,nj=q, . . .) = P (ni+k=m,nj+k=q, . . .) ∀k (provare a dimostrarlo).
Ritorniamo ad interessarci al camminatore random, che, detto incidentalmente, sara unsistema con cui avremo a che fare per tutta la durata del corso. Supponiamo che il cam-minatore possa trovarsi in caselle n = 0, 1, . . . , N disposte ad anello, cosı che il puntoN coincide con zero. Supponiamo inoltre che il camminatore invece di essere costretto asaltare, sia caratterizzato da una probabilita PJ < 1/2 di salto avanti e di salto indietro euna probabilita 1 − 2PJ di restare nella casella corrente (questo serve per eliminare com-plicazioni irritanti del tipo che caselle pari sono raggiunte a partire da n0 pari solo a tempipari e viceversa).
Supponiamo che il nostro camminatore sia inizialmente in una certa casella n0. Abbia-mo visto che al passare del tempo, la distribuzione di probabilita, inizialmente concentratain n0, si sparpagliera. Una prima conseguenza evidente e che l’entropia del sistema, definitaa partire da P (t) cresce al passare del tempo.

26 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
Sappiamo pero che esiste una configurazione ad entropia massima, che e la distribuzioneequiprobabile P (n) = 1/N . Ci aspettiamo quindi che se prendessimo come distribuzioneiniziale P (n0) = 1/N , questa rimarrebbe costante nel tempo, il che e abbastanza compren-sibile, visto che non ci sono distribuzioni piu “sparpagliate” (cioe ad entropia piu alta)della distribuzione uniforme. In altre parole, la distribuzione uniforme e stazionaria. (Ineffetti, se distribuiamo un ugual numero di camminatori casella per casella, vediamo chese in media, dalla casella n a quella n+1, salta nel tempo ∆t una frazione PJ , nello stessotempo in media, un identica frazione saltera da n+ 1 a n).
Possiamo pero ottenere un risultato piu forte.Vediamo infatti che se due caselle vicine n ed n + 1 contengono numeri diversi Mn <
Mn+1 di camminatori, il numero di camminatori che si sposteranno da n + 1 a n sarapiu alto (in media) che il viceversa, e la differenza nel numero di camminatori tendera adiminuire. In altre parole, la distribuzione uniforme non e solo stazionaria, ma e anchequella secondo la quale tendono a disporsi i camminatori a tempi sufficientemente lunghi.
Riassumendo:
• La probabilita P (ni|n0) di trovare un camminatore in ni al tempo ti a partire dauna qualsiasi posizione iniziale n0 tende per i → ∞ alla distribuzione uniformeP (ni) = 1/N .
• Questa distribuzione risulta essere una soluzione stazionaria della equazione di Chapman-Kolmogorov (verificarlo!)
Abbiamo quindi la seguente definizione
• Quando la probabilita condizionata P (ni = m|n0) tende, per i → ∞, a un limiteP (m) indipendente dal tempo e dalla condizione iniziale, si dice che il sistema possiedeuno stato di equilibrio statistico e la P (m) e detta distribuzione d’equilibrio.
Nota bene che se il sistema possiede uno stato di equilibrio statistico, esso verra raggiuntoa partire da qualsiasi distribuzione iniziale P . Infatti:
limi→∞
P (ni = m) = limi→∞
∑
n0
P (n0)P (ni = m|n0) =∑
n0
P (n0)P (m) = P (m).
Come vedremo, i sistemi per cui ha senso pensare ad una descrizione termodinamica sonocaratterizzati da una legge di aumento di entropia e di solito sono anche caratterizzati dallapresenza di un unico stato di equilibrio (o perlomeno, per semplificarsi la vita, si supponedi solito che lo siano).
Va subito detto che esistono sistemi interessanti in cui l’entropia decresce a passaredel tempo. E analogamente, vi sono sistemi che non sono caratterizzati da un equilibriostatistico unico (e pero possibile dimostrare, che l’equazione di Chapman-Kolmogorov am-mette sempre una soluzione stazionaria; tutti i processi Markoviani posseggono quindi unadistribuzione stazionaria, anche se non e una distribuzione di equilibrio). Vediamo alcuniesempi:

2.8. COARSE GRAINING E LIMITE CONTINUO 27
• L’evoluzione di una popolazione sterile, la quale puo essere descritta da un processostocastico Ni, con Ni il numero di individui in vita all’istante i. Supponiamo cheinizialmente abbiamo N0 individui. Al passare del tempo questi iniziano a morire,con il risultato che, mentre inizialmente la distribuzione e concentrata in N0, peri > 0, altri valori N < N0 diventano via via possibili. Quindi inizialmente la P (Ni)si sparpaglia e l’entropia cresce. Per tempi lunghi, pero, la popolazione si estingue,e abbiamo la distribuzione d’equilibrio P (N) = δN0. L’entropia scende di nuovo azero.
• Il “ping-pong”: un sistema a due stati 0, 1 con probabilita di transizione P (0 → 1) =P (1 → 0) = 1. Quindi, la pallina salta da zero a uno senza interruzione. Ora, separto da 0, avro che P (0) sara per sempre 0 o 1 a seconda che i e dispari o pari, eviceversa se parto da 1. Quindi, il sistema non ammette equilibrio statistico. Notapero che se parto da una distribuzione iniziale P (n0) = 1/2 per n0 = 0, 1, avroP (ni) = 1/2 anche per i > 0 cosı che la P (ni) e stazionaria.
• Il camminatore random in una scatola con una parete in mezzo. E chiaro che seil camminatore parte da una meta di una scatola, la distribuzione di equilibrio sarauniforme da quel lato e zero nell’altro, e viceversa. Abbiamo quindi una distribuzionedi equilibrio che non e unica e dipende dalla condizione iniziale.
2.8 Coarse graining e limite continuo
Sino ad ora ci siamo mantenuti abbastanza sulle generali, senza farci domande sulla naturadello spazio degli stati ni su cui si evolve il processo stocastico, o sull’origine dinamica dioggetti, come la probabilita di transizione P (ni+1|ni). Una descrizione attraverso processistocastici di un sistema con una dinamica microscopica, e molto spesso il risultato di unaoperazione cosı detta di coarse graining, a partire da una dinamica microscopica, che inlinea di principio potrebbe anche essere deterministica (ma solitamente troppo difficile dadescrivere). La procedura puo essere esemplificata come segue:
• Identificazione di unamicroscala al di sotto della quale i dettagli della dinamica sonosupposti sconosciuti. Nel caso di un gas, potrebbe trattarsi della scala molecolare(descrizione attraverso equazioni di Newton delle molecole).
• Definizione (se necessario) di una mesoscala dove la dinamica e meno complicatache alla microscala. Esecuzione quindi di una operazione di coarse graining, in cuile proprieta di microscala sono mediate. Di nuovo nel caso del gas, la mesoscala po-trebbe essere individuata dal cammino libero medio (descrizione attraverso equazionedi Boltzmann per il gas).
Vediamo un esempio di come un’operazione di coarse graining puo portare a una sem-plificazione della dinamica. Immaginiamo di trovarci qualcosa di simile a un cammino

28 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
random, in cui la lunghezza dei passi puo variare, e in piu questi sono correlati nel tempo.Immaginiamo per semplificare che la correlazione temporale esista solo fra salti successivi:
〈∆ni∆ni±1〉 = cσ2∆n, 0 < c < 1; 〈∆ni∆ni±k〉 = 0, k ≥ 2.
Proviamo a fare coarse graining nel tempo e definiamo una mesoscala ∆t contenente mintervallini ∆t. Avremo quindi
∆ni =m∑
j=1
∆ni+j
Da qui, sfruttando c > 0, troviamo
σ2∆n
≥ mσ2∆n,
mentre〈∆ni ∆ni±1〉 = 〈∆ni∆ni±1〉 = cσ2
∆n
(solo l’ultimo contributo ∆ni in un intervallo ∆t e il primo nel successivo sono corre-lati). Vediamo quindi che l’importanza della correlazione, parametrizzata dal rapporto〈∆ni ∆ni±1〉/σ2
∆ndiventa trascurabile per m grande. In altre parole, il coarse graining
temporale ci riporta a una situazione di passi indipendenti, ed ni diventa un processoMarkoviano.
Il passaggio alla mesoscala comporta spesso la transizione da una dinamica di tipo di-screto a una di tipo continuo. Il passaggio al limite continuo, nel caso di processi stocastici,presenta pero difficolta sia di carattere procedurale che concettuale, e non detto in generaleche esista.
Un primo esempio di difficolta con il limite continuo, lo abbiamo visto nella definizionedi entropia di PDF [Eq. (2.20)]. Un secondo esempio lo troviamo, a livello di realizzazionedi processo stocastico, gia nel caso del cammino random. Iniziamo con il domandarci qual’elo spostamento tipico del camminatore in un tempo t≫ ∆t. Se PJ e la probabilita di salto(uguale in avanti e all’indietro), otteniamo subito
〈|x(t)− x(0)|2〉 = PJ
t/∆t∑
i=1
∆x2 =PJ∆x
2
∆tt := κt (2.38)
Un limite continuo si ottiene mandando simultaneamente ∆t e ∆x a zero, ma vediamosubito che, perche la varianza 〈|x(t) − x(0)|2〉 rimanga finita al limite, deve essere ∆x =O(∆t1/2). Questo ci crea un problema nella definizione di una velocita istantanea delcamminatore. Una stima “ragionevole” dalla (2.38) ci porta a un risultato infinito:
u(0) = limt→0
〈|x(t)− x(0)|2〉1/2t
=κ1/2
2limt→0
t−1/2 = ∞.
Vediamo quindi che la velocita istantanea non e definita. Il limite continuo per la velocitanon e ben definito.

2.9. L’EQUAZIONE MASTER 29
2.9 L’equazione master
I problemi nell’esecuzione di un limite continuo diminuiscono notevolmente se invece di la-vorare con le realizzazioni del processo, ci si interessa delle quantita medie, come ad esempiole probabilita degli stati del processo stocastico. Supponendo che queste quantita varinopoco sulla scala di discretizzazione (micro o mesoscala), possiamo provare ad approssimaredifferenze finite con derivate e somme con integrali. A questo punto, le separazioni t− t′ oψ − ψ′ possono essere mandate tranquillamente a zero, anche se originariamente ∆t e ∆ψerano finite.
Siamo interessati ad ottenere equazioni di evoluzione per la probabilita in forma dif-ferenziale, o al limite integro-differenziale (il fatto che si arrivi a equazioni di questo tipolineari, con coefficienti che dipendono lentamente dalle variabili, costituisce conferma aposteriori che il limite esiste). Ci focalizziamo sul caso di processi Markoviani. Il nostropunto di partenza sara pertanto l’equazione di Chapman-Kolmogorov (2.37).
Introduciamo la notazione P (xm, ti) ≡ P (m, i) ≡ P (ni = m). Continueremo ad indica-re la probabilita all’equilibrio con P (xm) ≡ P (m). L’equazione di Chapman-Kolmogorov(2.37) diventa:
P (m, i+ 1)− P (m, i) =∑
k
P (k → m)P (k, i)− P (m, i)
=∑
k 6=m
[P (k → m)P (k, i)− P (m→ k)P (m, i)], (2.39)
dove si e utilizzato∑
k P (m → k) = 1 e il passaggio∑
k →∑
k 6=m e giustificato dal fattoche i termine k = m nei due addendi in parentesi quadra si cancellano. In parole, laformula appena qui sopra ci dice che l’incremento nel numero di risultati in m al passaredel tempo e dato dal numero di risultati che passano da k a m meno quelli che da m se nevanno a k.
Per fare il limite continuo, e necessario che il cambio P (m, i+ 1)− P (m, i) sia piccoloconfrontato con i valori tipici di P (m, i). Questo richiede P (k → m) ≪ 1. Sarebbe belloa questo punto definire una probabilita di transizione per unita di tempo P (k → m) =P (k → m)/∆t e ottenere cosı una equazione differenziale. Vediamo in effetti che
P (k, t+ lδt|m, t) ≃ lP (k, t+∆t|m, t) +O([P (k, t+∆t|m, t)]2)
e quindiP (k, t+∆t|m, t)P (k, t+∆t|m, t) =
∆t
∆t⇒ P (m→ k) = P (m→ k)∆t,
con P (m→ k) indipendente da ∆t.Otteniamo finalmente la seguente cosı detta equazione master:
∂P (m, t)
∂t=
∑
k 6=m
[P (k → m)P (k, t)− P (m→ k)P (m, t)]. (2.40)

30 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
Il passo successivo e evidentemente P (m, t) ≃ ρ(xm, t)∆xm, P (k → m) ≃ ρ(xk → xm)∆xm,e la master equation diventa in generale un’equazione integro-differenziale:
∂ρ(x, t)
∂t=
∫
dy [ρ(y → x)ρ(y, t)− ρ(x→ y)ρ(x, t)].
Nel caso in cui le transizioni avvengano solo con i primi vicini, P (m→ m± 1), ci immagi-niamo che l’equazione master diventi una equazione differenziale alle derivate parziali. Unasituazione di questo genere si verifica sia nel caso del cammino random che dei processi dinascita e morte.
2.9.1 Cammino random
In questo caso abbiamo una probabilita di salto PJ = P (k → k + 1) = P (k → k − 1), cosıche P (k → k) = 1 − 2PJ . Dividendo per ∆t troviamo le probabilita per unita di tempoP (K → k + 1) = P (k → k − 1) = PJ/∆t, e l’equazione master (2.40) prende la forma
∂P (k, t)
∂t=PJ∆t
[P (k + 1, t) + P (k − 1, t)− 2P (k, t)] ≃ PJ∆x2
∆t
∂2P (xk, t)
∂x2k.
Definendo la diffusivita κ = PJ∆x2/∆t, otteniamo l’equazione del calore (o equazione
di diffusione):
∂ρ(x, t)
∂t= κ
∂2ρ(x, t)
∂x2. (2.41)
Notare il significato fisico della diffusivita:
〈|x(t)− x(0)|2〉 = Pj
t/∆t∑
k=1
∆x2 = κt,
che risulta essere quindi il tasso di crescita della varianza della posizione del camminatore(confrontare con la (2.38).
2.9.2 Processo di estinzione
Supponiamo in questo caso che un individuo abbia una probabilita di morte nell’intervallo∆t data da Γ∆t. Se al tempo ti ci sono Ni individui, la probabilita di avere una mortesara quindi NiΓ∆t (a patto sempre che NiΓ∆t ≪ 1). L’equazione master (2.40) diventaquindi:
∂P (N, t)
∂t= ΓD[(N + 1)P (N + 1, t)−NP (N, t)] ≃ ΓD
∂(NP (N, t))
∂N
e di nuovo possiamo passare alle PDF P (N, t) = ρ(N, t)∆N ≡ ρ(N, t)× 1 = ρ(N, t):
∂ρ(N, t)
∂t= ΓD
∂(Nρ(N, t))
∂N. (2.42)

2.10. ESERCIZI 31
Va notato come in entrambi i casi del cammino random (2.41) e del processo di estinzione(2.42), l’equazione di evoluzione ha la forma di un’equazione di continuita
∂ρ(N, t)
∂t+∂J(x, t)
∂x= 0,
dove nel primo caso J(x, t) = −κ∂ρ(x, t)/∂x, mentre nel secondo J(N, t) = −NΓDρ(N, t).E interessante osservare come nel secondo caso ΓDN e proprio una velocita di diminuzio-ne (e infatti ha segno negativo) della popolazione N . Nel caso della equazione del calore,abbiamo invece una cosı detta relazione flusso gradiente. Il flusso di probabilita va in di-rezione opposta al gradiente in ρ e agisce quindi nella direzione di appianare disomogeneitadella PDF.
Un’ultima osservazione concerne il legame tra l’ordine rispetto alla variabile stocastica(x o N) nella equazione differenziale, e il numero di siti vicini che sono richiamati a latodestro nella originaria equazione master. Vediamo che un primo vicino porta al piu a unaderivata prima, due vicini a una derivata seconda, eccetera. [“Porta al piu”; non “portasicuramente”. Abbiamo infatti P (k+1)−P (k) ≃ P ′(xk)∆xk, ma P (k+1)+P (k) ≃ 2P (xk)].Vediamo inoltre che l’operazione di coarse-graining descritta nella lezione precedente eequivalente a trascurare derivate di ordine successivo nella equazione differenziale che siottiene al limite continuo.
2.10 Esercizi
Esercizio 1 Supponiamo le variabili aleatorie x, y distribuite in modo equiprobabile neltriangolo con vertici (0, 0), (0, 1), (1, 1). Calcolare le PDF e probabilita condizionateρ(x|y > 0.5) e P (y > 0.5|x).
Esercizio 2 Un tiratore scarso, le rare volte che becca il bersaglio, lo colpisce in un punto acaso (supporre il bersaglio circolare e di raggio unitario). Si calcoli la PDF che il bersagliosia colpito a distanza r dal centro, condizionale al fatto che il bersaglio sia stato colpito. Dinuovo condizionale a che il bersaglio sia stato colpito, calcolare la probabilita che il colpovada a r < 1/2.
Esercizio 3 La PDF della velocita v = (vx, vy, vz) di una molecola monoatomica di un gasideale in un volume V , all’equilibrio termodinamico e:
ρ1(v) =( m
2πKT
)3/2
exp(
− mv2
2KT
)
(2.43)
dove m e la massa molecolare, T e la temperatura e K e la costante di Boltzmann (inun gas ideale le velocita delle differenti molecole sono approssimate come statisticamenteindipendenti ed all’equilibrio la densita del gas e uniforme: n = N/V dove N e il numerodi molecole nel gas).
• Si scrivano la PDF ρ1(r,v) e la PDF congiunta ρN(ri,vi).

32 CAPITOLO 2. RICHIAMI DI PROBABILITA E STATISTICA
• Si calcoli l’entropia del gas S = S(T, V,N) = −〈ln ρN〉.
• Stimare numericamente l’energia interna U =∑N
i=112m|vi|2 e l’ampiezza delle sue
fluttuazioni, note la costante di Boltzmann K ≃ 1.4 · 10−23J oK−1 e posto N ∼ 1020,V ≃ 1m3, T ∼ 300 oK, m ≃ 7 · 10−27Kg [ricordo che la varianza della (2.43) e:σ2vx = σ2
vy = σ2vz = KT/m].

Capitolo 3
La teoria cinetica
3.1 Alcune problematiche in teoria cinetica
Vogliamo descrivere la dinamica di un sistema macroscopico composto da un gran numerodi componenti microscopiche (molecole) non necessariamente indipendenti. La descrizionecinetica completa di un sistema con N gradi di liberta interagenti, e ottenuta assegnandola PDF congiunta ρN(yk, k = 1, . . . , N, t) del valore al tempo t di tutti gli N gradi diliberta. (Nel caso di molecole vere e proprie, la coordinata yi della molecola i-esima sarebbein realta il vettore a sei componenti (xi,vi)). La PDF ρN e definita su uno spazio a Ndimensioni, a cui viene dato in genere il nome di Γ-spazio. A partire dalla PDF sul Γ-spazio, possiamo ottenere le PDF marginali ρN−1, . . . definite su un sottoinsieme ristrettodi variabili, sino alla PDF a una particella ρ1(y, t), che e la PDF di trovare una molecoladata nel punto y al tempo t. A partire dalla ρ1, possiamo poi ottenere la densita media diparticelle f1 in un punto y dato:
f1(y, t) =N∑
i=1
ρ1(yi = y, t).
Pertanto, f1(y, t)∆y sara il numero medio di molecole al tempo t nell’intervallo [y, y+∆y].In maniera analoga, possiamo definire le densita di coppie f2(y, z; t) = N(N − 1)ρ2(y, z, t)e cosı via.
Va notato che se f1(y, t)∆y ≫ 1, abbiamo che questa quantita (che e una media)coincide di fatto con il numero effettivo (nella realizzazione) delle molecole nell’intervallino∆y. La quantita f puo quindi essere vista (a patto di fare un coarse-graining sufficiente iny) come una densita istantanea (vedere la discussione nella Sez. 2.3). Vediamo quindi che,al contrario della descrizione fornita dalla PDF ρ1, che e microscopica (stiamo chiedendodove si trova la particella data), quella fornita da f1 e macroscopica. Notiamo infine chesolo se supponiamo le particelle identicamente distribuite possiamo scrivere
f1(y, t) = Nρ1(yi = y, t), (3.1)
e le due descrizioni con f1 e con ρ1 diventano equivalenti.
33

34 CAPITOLO 3. LA TEORIA CINETICA
Abbiamo visto nella Sez. 2.3 come il concetto di entropia permetta di identificaresituazioni di equilibrio termodinamico in sistemi semplici come un gas in un recipienteisolato. In presenza di interazioni fra le molecole, sembrerebbe ragionevole utilizzare, comepunto di partenza per la definizione di una entropia termodinamica, l’entropia di Shannonassociata alla ρN su qualche partizione del Γ-spazio:
SN ≃ −∑
k
ρN(Γk)δΓk ln[ρN(Γk)δΓk],
dove δΓk indica la celletta centrata intorno al punto Γk del Γ-spazio (elemento k-esimo dellapartizione). Come vedremo ben presto, si tratta di una strada irta di ostacoli. Al contrariodella entropia S[PN ], introdotta nella Sez. 2.3, in cui PN(α) = Nα/N era la frazioneistantanea di molecole nel volumetto Vα, e difficile attribuire un significato macroscopicodiretto ad SN . La stessa interpretazione della ρN e problematica: punti diversi del Γ spaziopossono corrispondere tanto a condizioni di equilibrio che di non equilibrio termodinamico.L’entropia di Shannon SN e poi anche “troppo microscopica”, nel senso che tratta lemolecole come se fossero degli oggetti identificabili individualmente. Il problema rimaneanche nel caso di un sistema di molecole non-interagenti, per cui ρN(Γ) =
∏
k ρ1(yk), ed epossibile decomporre (supponiamo per semplicita una partizione uniforme):
SN = −〈ln(ρNδΓ)〉 ≃ −N〈ln ρ1δy〉 = NS1. (3.2)
Il fatto e che la PDF ρ1 e di per se una quantita riferita a una ben identificata molecola.Come si vedra nella Sez. 3.2.1, questo porta a una definizione di entropia priva di significatotermodinamico. L’entropia S[PN ] introdotta nella Sez. 2.3, associata a un numero dimolecole istantaneo in un volumetto, non soffriva chiaramente di questo problema.
Ritorniamo a considerare il problema originario del gas di particelle interagenti. Percapire come le interazioni modifichino la dinamica, consideriamo un modello giocattolo:un sistema di N particelle che si muovono lungo un anello di circonferenza V seguendoun cammino random. Supponiamo che le particelle siano in grado di interagire, se abba-stanza vicine, tramite modificazione delle proprieta del cammino random. Solo in seguitopasseremo all’analisi di sistemi fisici piu realistici.
In assenza di interazioni, notiamo subito che potremmo descrivere in modo completola dinamica del sistema. dando la equazione per la ρ1, che non e altro che l’equazione didiffusione (2.41). Volendo, uno puo scrivere anche l’equazione per la ρN :
∂ρN∂t
= κ
N∑
k=1
∂2ρN∂y2k
,
ma il contenuto addizionale di informazione, com’e ovvio, e nullo.Aggiungiamo ora le interazioni. Supponiamo che la molecola i-esima del nostro sistema
giocattolo, salti di ∆y con una probabilita che e funzione della presenza di molecole vicine:
PJ(y) = 〈∑
j 6=i
q(yj(t)− yi(t))|yi(t) = y〉,

3.1. ALCUNE PROBLEMATICHE IN TEORIA CINETICA 35
dove q(y) e una funzione definita positiva, che decade a zero su una scala microscopica λ.Quindi, quante piu molecole ci sono a distanza λ, tanto piu frequenti saranno i salti. Peril resto supponiamo che le molecole non interagiscano per niente e in particolare possanoattraversarsi senza problemi lungo V .
Riscrivendo la probabilita di salto nella forma
PJ(y) =∑
j 6=i
∫
V
dz q(y − z)〈δ(yj(t)− z)|yi(t) = z〉 = (N − 1)
∫
V
dz q(y − z)ρ2(z, t|y, t),
otteniamo l’equazione del tipo del calore
∂f1(y, t)
∂t=
∂2
∂y2
∫
dz q(y − z)f2(y, z; t), (3.3)
dove q = ∆y2q/∆t. Questa e la nostra equazione cinetica per f1. Vediamo subito peroche abbiamo un problema: per risolvere l’equazione per f1, dovremmo conoscere f2. Seprovassimo a risolvere l’equazione per f2, ci accorgeremmo che dovremmo conoscere f3 ecosı via. Ovviamente, visto che ci sono soloN molecole, solo l’equazione per fN non richiedeconoscenza di fN+1. Possiamo provare a scrivere la equazione per f2(y, z; t) tenendo contoche la molecola in y diffonde per effetto di quella in z piu tutte le altre N − 2 rimanenti, eviceversa. Il risultato e
∂f2(y, z; t)
∂t=
[ ∂2
∂y2+
∂2
∂z2
]
q(y − z)f2(y, z; t)
+
∫
dw ∂2
∂z2q(y − z) +
∂2
∂y2q(y − w)
f3(y, z, w; t).
Di nuovo possiamo scrivere l’equazione per ρN (o fN):
∂fN∂t
=∑
k 6=j
∂2
∂y2kq(yk − yj)fN ,
che e esatta, lineare, statisticamente chiusa (non richiede informazioni su fm con m > N),ma e impossibile da risolvere a causa del numero enorme di variabili da cui dipende.
Nota. Le equazioni per le correlazioni fk, k > 1 possono essere scritte in maniera si-stematica a partire dalla osservazione che la (3.3) puo essere scritta come media di unaequazione per densita fluttuanti nella forma
∂tf1(y, t) = ∂2y
∫
dz q(y − z)f2(y, z; t) (3.4)
dove f1(x, t) =∑
i δx(xi(t)), f2(y, z; t) =∑
i 6=j δy(yi(t))δz(yj(t)), con δy(yi(t)) ≡ δ(y −yi(t)) e la funzione indicatrice della coordinata della particella i. Una identica equazionedeterminera l’evoluzione della densita istantanee corse-grained a scala ∆y, ottenuta facendola sostituzione δy(yi) → (∆y)−1δ[y,y+∆y](yi).

36 CAPITOLO 3. LA TEORIA CINETICA
L’equazione per la correlazione a k + 1 particelle e quindi ottenuta moltiplicando la(3.4) per k funzioni indicatrici, sommando le permutazioni per scrivere la derivata tem-porale nella forma ∂t(δy1δy2 . . .), e prendendo la media. Questo ci da automaticamente ilrisultato che se l’interazione e a due particelle, l’equazione per la correlazione a k parti-celle richiamera la correlazione a k + 1 particelle (se l’interazione fosse a m corpi, ci sipuo convincere che quella che sarebbe richiamata, sarebbe una correlazione a k + m − 1particelle).
In pratica, l’unica opzione e risoluzione approssimata delle prime equazioni nella gerar-chia (si tratta di un esempio molto semplice di quella a cui ci si riferisce usualmente conil nome di gerarchia BBGKY; approfondimenti sulla gerarchia BBGKY e l’equazione diKlimontovich possono essere trovati sul testo di S. Ichimaru “Basic principles of plasmaphysics”). Una possibile soluzione approssimata e quella che si ottiene scrivendo la fk+1
che appare all’ordine k nella gerarchia in funzione di densita di ordine inferiore. Questoapproccio prende il nome di chiusura statistica. L’esempio piu semplice e fornito dallacosı detta approssimazione di campo medio:
f2(y, z; z) ≃ f1(y, t)f1(z, t), (3.5)
chiamata cosı appunto perche vengono trascurate le correlazioni associate con f2(y, z; t)−f1(x, t)f1(y, t). L’equazione per f1 diventa cosı, in luogo della (3.3)
∂f1(y, t)
∂t=
∂2
∂y2
∫
dz q(y − z)f1(y, t)f1(z, t)
e tenuto conto che f1 varia lentamente sulla scala di q:
∂f1(y, t)
∂t= α
∂2f 21 (y, t)
∂y2, α =
∫ +∞
−∞
q(y)dy (3.6)
(utilizziamo nella definizione di α la condizione V ≫ λ). Abbiamo quindi un’equazionedel calore non-lineare che puo essere risolta, per esempio, espandendo perturbativamenteattorno a qualche soluzione nota (per esempio la soluzione uniforme).
Nota. L’approssimazione di campo medio non si interessa di quella parte delle fluttuazioniassociate a discretezza delle molecole. Infatti, se ∆N e il contenuto istantaneo di molecolein un intervallo ∆y, possiamo scrivere
〈∆N2〉 =∑
i 6=j
〈δ∆y(yi)δ∆y(yj)〉+∑
i
〈δ∆y(yi)〉 =∫ y+∆y
y
dz
∫ y+∆y
y
dw f2(z, w)
+
∫ y+∆y
y
dz f1(z) ≃ f2(y, y)(∆y)2 + f1(y)∆y,
e il contributo associato a discretezza e quello dovuto alla somma∑
i〈δ∆y(yi)〉 ≃ f1(y)∆y.L’altro contributo f2(y, y) e la parte continua, che e la densita di coppie che entra nella

3.2. ALCUNE QUESTIONI RIGUARDO IL CALCOLO DELL’ENTROPIA 37
(3.3). La condizione di campo medio e quindi una condizione di trascurabilita della partenon discreta delle fluttuazioni, o, equivalentemente, di trascurabilita delle fluttuazioni in∆N , per una dimensione di coarse graining ∆y tale che 〈∆N〉 ≫ 1.
Un utile esercizio e dimostrare che l’entropia S1 nel nostro sistema giocattolo cresce neltempo. Abbiamo infatti immediatamente, dalla (3.6):
S1(t) = − d
dt
∫
V
dyρ1(y, t) ln ρ1(y, t) = −∫
V
dyρ1(y, t) ln ρ1(y, t). (3.7)
Usando l’equazione cinetica per ρ1 (che coincide con quella per f1):
S1(t) = −α∫
V
dy∂2ρ21(y, t)
∂y2ln ρ1(y, t)
e integrando per parti (sfruttando il fatto che V e un anello e il contributo agli estremi eassente):
S1(t) = α
∫
V
dy1
ρ(y, t)
∂ρ21(y, t)
∂y
∂ρ1(y, t)
∂y= 2α
∫
V
dy(∂ρ1(y, t)
∂y
)2
≥ 0.
L’entropia cresce, come ci aspettavamo.
3.2 Alcune questioni riguardo il calcolo dell’entropia
Come si e gia notato nella Sez. 3.1, l’entropia di Shannon SN = −〈ρN〉 ha un caratte-re fondamentalmente microscopico: ciascun punto nel Γ-spazio corrisponde infatti a unaconfigurazione microscopica. Proviamo ad approfondire un mimimo questo aspetto. Ab-biamo visto come punti di Γ possono corrispondere sia a configurazioni di equilibrio che aconfigurazioni di non equilibrio termodinamico. Dal punto di vista della PDF ρN , i duepunti nel Γ spazio individuano due delte di Dirac, e la corrispondente entropia microsco-pica SN (piu precisamente l’entropia definita con una partizione finita, e non l’entropia diPDF) in entrambi i casi sarebbe nulla. Vediamo quindi che l’entropia SN non e in gradodi distinguere condizioni di equilibrio e non-equilibrio termodinamico.
Potremmo tuttavia insistere su questa linea di ragionamento, e considerare una genericadistribuzione di punti in Γ (un cosı detto ensemble; vedi Sez. 5.1.1), descritta da una PDFρN . Se la dinamica del sistema fosse abbastanza “caotica”, ci si potrebbe aspettare chequesti punti si sparpaglino nel Γ-spazio, e il risultato finale (equilibrio statistico) sia una ρNdominata da configurazioni microscopiche, corrispondenti macroscopicamente a uno statodi equilibrio termodinamico. Al contempo, grazie allo sparpagliamento in Γ, SN tenderebbea un massimo. Il problema diventa a questo punto, quello di isolare nell’aumento dientropia SN , la componente associata al fatto che la gran parte dei punti staranno in zonedi Γ corrispondenti all’equilibrio termodinamico. L’approccio di Gibbs, che sara descrittoa grandi linee nella Sez. 5.2, consiste precisamente nel cercare di estrarre da SN la sua

38 CAPITOLO 3. LA TEORIA CINETICA
componente macroscopica. L’altrenativa, suggerita dall’analisi nella Sez. 2.3, consisteva nelcercare di riportarsi al caso in cui le molecole del sistema sono in prima approssimazioneindipendenti, e quindi lavorare con l’entropia S1. Vedremo pero ben presto, che anchelavorare con S1, indipendentemente dal fatto che le molecole si possano o meno considerarecome indipendententi, presenta problemi: l’entropia S1 e riferita infatti a una ben specificamolecola, ed e percio anch’essa, intrinsecamente, una quantita microscopica.
3.2.1 Entropia, partizioni e paradosso di Gibbs
Abbiamo visto che l’entropia e definita a partire da una partizione di Ω. Anche quandolavoriamo con una entropia di PDF, stiamo ipotizzando una partizione in volumetti didimensione uniforme del nostro spazio dei campioni. A seconda del sistema, la ragionedi una scelta invece che un’altra risiede nella nostra comprensione di quello che si verificaalla microscala. Nel caso delle molecole di un gas, questa scelta e fissata della meccanicaquantistica. Il principio di indeterminazione fissa infatti la scala δxδv ∼ ~/m, sotto laquale non ha senso distinguere volumetti di spazio (x,v).1
In generale, la scelta della partizione e fatta in modo di eliminare suddivisioni troppofini, tali che eventi diversi nella partizione siano indistinguibili dal punto di vista delladinamica.
Da questo punto di vista, notiamo subito un problema, trattando con un sistema diparticelle identiche. Vediamo infatti che scambiare fra loro le particelle e ininfluente dalpunto di vista della dinamica. Questo fa sı che la partizione dello spazio Γ che ci viene piunaturale, e cioe quella in un reticolo di cubi N -dimensionali di lato δx, non e appropriata.Vediamo infatti che il cubetto attorno al punto Γ = (y1, y2, . . . , yN ), yk ≡ (xk,vk), eassolutamente indistinguibile dal cubetto attorno al punto Γ′ = (y2, y1, y3, . . . , yN) ottenutoscambiando fra loro le particelle 1 e 2.
In definitiva, vediamo che la partizione a partire dalla quale va calcolata l’entropiadovrebbe essere quella in cui ciascun evento e l’unione di tutti gli eventi equivalenti perpermutazione di particelle. Se ciascuna particella si trova in un cubetto ∆y diverso, tuttele componenti yk nel vettore Γ (il cubetto nel Γ-spazio) saranno diverse. In questo caso, ilnumero delle permutazioni sara N !. Se δP e la probabilita che il punto di fase del sistemasi trovi in un dato cubetto δΓ nel Γ-spazio, la probabilita che si trovi in quel cubetto, o inun altro qualsiasi ottenuto tramite permutazione delle particelle sara quindi = N !δP .2
Supponendo una partizione uniforme di volumetti δΓ, e supponendo (consistentemntecon l’ipotesi di particelle indistinguibili) i volumetti equiprobabili, abbiamo quindi nei due
1Notare come la meccanica quantistica imponga naturalmente una partizione uniforme del Γ-spazio.Una tale scelta, nel caso della meccanica classica, puo essere giustificata in maniera agevole solo a posteriori,osservando che la PDF di equilibrio e uniforme (lavorando in coordinate canoniche qi, pi) in gusci adenergia costante del Γ-spazio (si veda la Sez. 5.1).
2Affinche questa condizione sia soddisfatta, e necessario che la partizione sia abbastanza fine, oppureche il gas sia sufficientemente diluito. Ritorneremo su questo punto nella Sez. 5.3.

3.2. ALCUNE QUESTIONI RIGUARDO IL CALCOLO DELL’ENTROPIA 39
casi di particelle distinguibili e non:
SdistN ≡ S[ρNδΓ] = −∫
dΓρN ln ρNδΓ (3.8)
SindistN = S[N !ρNδΓ] = −∫
dΓρN ln(N !ρNδΓ) = S[ρNδΓ]− lnN ! (3.9)
Nel caso di particelle indipendenti, per cui SdistN ≃ NS1, dove S1 ≡ −〈ρ1δy〉, possiamoscrivere, utilizzando l’approssimazione di Stirling lnN ! ≃ N(lnN − 1):
SindistN ≃ N(S1 − lnN + 1) = −N [〈ln(fδy)〉+ 1], (3.10)
da cui possiamo introdurre la entropia di PDF
Sf (t) = −∫
dy f(y, t) ln f(y, t) = −N〈ln f)〉. (3.11)
Vediamo quindi che la SindistN e la sua controparte di PDF Sf sono entropie macroscopiche,nel senso che possono essere determinate a partire da quantita macroscopiche come ladensita f .
Nota. Considerare una partizione che non distingue tra configurazioni che differiscono perpermutazioni di particelle, equivale a contradistinguere gli elementi della partizione solo peri numeri di occupazione delle singole celle di spazio delle fasi a una molecola. Fissare unmicrostato a meno di permutazioni fra le molecole, e equivalente quindi a conoscere ladistribuzione f(y) con il massimo livello di definizione: f = fδy. Se y ≡ (x,v), δy ∼ ~
3.La perdita di informazione nel passaggio da fδy alla f (o alla f∆y) generica, e quindi lostesso del passaggio da una ρN massimamente determinata (conosco il punto di fase a menodi permutazioni di molecole) alla ρN generica (sempre determinata a meno di permutazionidelle molecole). Non e percio sorprendente che Sf = Sindist.
Ci sono ragioni pratiche ben precise, per cui una definizione sensata di entropia dovrebbetenere conto della indistinguibilita delle molecole. Consideriamo di nuovo l’esempio di ungas di N molecole in equilibrio in un volume V . Abbiamo in questo caso ρN = 1/V N , doveV e il volume del recipiente, e quindi, per l’entropie corrispondenti alle (3.8-3.9):
SdistN = N ln(V/δV )
SindistN = N ln(V/(NδV ))
dove ho utilizzato la Eq. (3.10).Vediamo subito che, se prendessimo due recipienti di gas identici, di volumi V1 e V2,
uno con N1 = nV1, l’altro con N2 = nV2 particelle, ma per il resto identica temperaturaT e identica densita n, e li unissimo, troveremmo un’entropia, supponendo le particelledistinguibili
SdistN = (N1 +N2) ln(V1 + V2) 6= N1 lnV1 +N2 lnV2 = SdistN1,1+ SdistN2,2
,

40 CAPITOLO 3. LA TEORIA CINETICA
e cioe un’entropia non addittiva.Questo e un problema: dal punto di vista macroscopico ci aspetteremmo che le molecole
nelle due parti V1 e V2, anche dopo che i volumi sono stati messi in comunicazione, conti-nuino ad essere in prima approssimazione indipendenti. (Le due parti sono macroscopiche,e la condizione di indipendenza dovrebbe essere soddisfatta se un limite termodinamico epossibile). Il fatto e che evidentemente la componente di entropia associata a scambio trauna particella in V1 e una in V2 di cui si tiene conto nell’entropia considerando le particelledistinguibili, non ha una controparte dinamica e non e associata a correlazioni fra para-metri fisici delle due parti del sistema. (Nel caso di gas diversi, una variazione di entropiac’e ed e associata al fenomeno macroscopico del mescolamento).
Utilizzando una entropia basata su una partizione che non distingue tra configurazioniche differiscono solo per scambio di particelle, troviamo invece:
SindistN = (N1 +N2) lnn = SindistN1,1+ SindistN2,2
,
e la condizione di addittivita e soddisfatta.Queste considerazioni ci suggeriscono che la scelta di partizione effettuata nella (3.9) e
quella corretta per ottenere una entropia dotata di significato termodinamico. Per questaragione, d’ora in avanti, quando ci parleremo di entropia, ci riferiremo alla definizione (3.9)ponendo implicitamente SN ≡ SindistN .
3.2.2 Entropia in una singola realizzazione
Abbiamo visto che, tramite le equazioni (3.9-3.11), e stato possibile esprimere l’entropia infunzione della densita f(y, t). In analogia con quanto fatto nella Sez. 2.3, possiamo provarea definire una entropia a partire dalla densita istantanea f∆y(y, t) ottenuta dividendo ilnumero istantaneo ∆N di particelle nel volumetto ∆y ≡ (∆x∆v)3, per ∆y. Naturalmente,perche la densita istantanea abbia un senso, ∆y deve essere piu piccolo della scala divariazione tipica delle quantita medie Le due densita f∆y e f saranno legate dalla relazione
f = 〈f∆y〉,
e la dipendenza di f dalla scala di coarse graining ∆y scompare (f varia poco sulla scala∆y). Nel caso in cui la scala ∆y e macroscopica, (cioe sufficientemente grande che le flut-tuazioni in ∆N siano trascurabili) f∆y e f coincideranno. Quindi, per ∆y sufficientementegrande, f∆y → f ≃ f fornira una descrizione macroscopica del sistema. Possiamo a questopunto introdurre una nuova definizione di entropia
Sf∆y= −
∫
dy f∆y(y, t) ln f∆y(y, t), (3.12)
che descrivera la perdita di entropia dalla distribuzione massimamente definita fδy allaf∆y. Di fatto, la (3.12) corrisponde alla definizione originaria di entropia data da LudwigBoltzmann (entropia di Boltzmann); piu precisamente, la definizione data dal grande

3.3. L’EQUAZIONE DI BOLTZMANN 41
scienziato era H = −Sf∆y, da cui il nome di teorema H). In seguito, per non appesantire
la notazione, trascureremo di indicare la dipendenza di f∆y da ∆y. Essa continua pero ad
essere presente e determina di fatto la distanza da Sf . E importante notare come Sf siauna quantita fluttuante. Le fluttuazioni sono presenti anche all’equilibrio; esse diventanotrascurabili, e Sf ≃ Sf , solamente nel limite termodinamico di ∆y macroscopici.
3.3 L’equazione di Boltzmann
Il modello giocattolo di cinetica di un gas, che abbiamo considerato nella Sez. 3.1, hautilita limitata, visto che non e in grado di fornire informazioni sulla velocita delle molecole.Di fatto, essendo il moto delle molecole modellato tramite un cammino random, la lorovelocita istantanea risulterebbe infinita. Dobbiamo pertanto estendere il nostro spazio µin modo da includere le velocita. La nostra densita sara quindi nella forma f1 = f1(x,v; t).Supponiamo per semplicita di avere molecole monoatomiche e di massa uguale. Il fattoche le molecole siano monoatomiche serve a garantire che l’energie cinetica non contengatermini rotazionali. Trascuriamo anche gradi di liberta interni come vibrazioni elastichedella molecola.
E conveniente nella derivazione di una equazione cinetica per f1(x,v; t) separare con-tributi dovuti a interazioni microscopiche (per esempio urti), i quali hanno una naturaintrinsecamente stocastica, dai cambi di posizione e di velocita che non sono legati a pro-cessi microscopici. In effetti, sappiamo che la densita f1(y, t) per delle particelle che simuovono in uno spazio con coordinate y = (y1, y2, . . .) seguendo un campo di velocitau(y, t) secondo la legge y = u(y, t), evolvera secondo una equazione di continuita
∂ρ
∂t+∇y · (ρu) = 0. (3.13)
Una simile equazione presuppone pero che il campo u sia perlomeno differenziabile, cosanon verificata quasi per definizione nel caso di interazioni a livello microscopico. Nel casoin cui y = (x,v) (spazio delle fasi per una molecola), abbiamo
x = v, v = F(x,v; t)/m = [Fext(x,v; t) + Fmicr(x,v; t)]/m (3.14)
dove F e la forza che agisce sulla molecola in x in moto con velocita v, e abbiamo isolatol’effetto delle forze esterne (per esempio la gravita) dall’interazione con le altre molecole,tenuta in conto da Fmicr(x,v; t);m e evidentemente la massa delle molecole. Come abbiamodetto quest’ultimo termine non puo essere a priori trattato attraverso la divergenza di unaqualche corrente e otteniamo quindi dalle (3.13-3.14), una equazione di continuita nellaforma
∂f1∂t
+ v · ∇xf1 +1
m∇v · (F extf1) =
∂f1∂t
∣
∣
∣
coll(3.15)
dove il termine a lato destro dell’equazione contiene l’effetto di Fmicr(x,v; t), e ∇x e ∇v
sono i gradienti rispetto alle variabili x = (x1, x2, x3) e v = (v1, v2, v3) (notare che v escedal gradiente in x essendo le variabili x e v indipendenti).

42 CAPITOLO 3. LA TEORIA CINETICA
L’ipotesi fondamentale che viene fatta per determinare la forma del termine (∂f/∂t)coll,e quella di assenza di memoria nel processo collisionale: le velocita finali delle molecole chehanno partecipato ad un urto dipendono solo dalle loro velocita prima dell’urto, indipen-dentemente dalla loro storia di urti pregressa. Possiamo quindi rappresentare la velocitadelle molecole come un processo Markoviano, che sara descritto da una equazione master.Cio presuppone evidentemente una descrizione statistica degli urti, che puo essere vistacome il risultato di una microscala ∆x maggiore della scala delle molecole (e della distanzadi interazione fra queste). L’unica informazione che utilizziamo e che gli urti siano binari,elastici ed istantanei.
La struttura del termine ∂f1/∂t|coll puo essere determinata per analogia con il modellogiocattolo della sezione precedente. Indichiamo con ρ(v1,2 → v′
1,2|x1 − x2) la PDF perunita di tempo che una coppia di molecole a distanza x1 − x2 e aventi velocita v1,2, urtinofra loro e si ritrovino ad avere velocita v′
1,2. Questo ci da l’equazione master
∂f1(x1,v1; t)
∂t
∣
∣
∣
coll=
∫
d3x2d3v′1d
3v′2d3v2ρ(v1,2 → v′
1,2|x1 − x2)f2(x1,2,v′1,2; t)
− ρ(v1,2 → v′1,2|x1 − x2)f2(x1,2,v
′1,2; t)
Il carattere locale degli urti implica ρ(v1,2 → v′1,2|x1−x2) = ρ(v1,2 → v′
1,2)δ(x1−x2). Allostesso tempo, simmetria per inversione temporale delle leggi della meccanica (supponiamogli urti elastici) ci da ρ(v1,2 → v′
1,2) = ρ(v′1,2 → v1,2), e quindi
∂f1(x;v1; t)
∂t
∣
∣
∣
coll=
∫
d3v2d3v′1d
3v′2 ρ(v1,2 → v′1,2)[f2(x;v
′1,2; t)− f2(x;v1,2; t)].
Sostituendo nella equazione di continuita per f1 (3.15), vediamo che ci ritroviamo di nuovoil problema che di una equazione cinetica per f1 che a causa delle interazioni richiede perla sua risoluzione di conoscere f2. L’unica via d’uscita praticabile abbiamo visto esserein questi casi l’uso di una approssimazione di campo medio. Il risultato e la seguenteequazione di Boltzmann:
∂f1∂t
+ v · ∇xf1 +1
m∇v · (F extf1) =
∂f1∂t
∣
∣
∣
coll(3.16)
dove il termine di collisione e dato da
∂f1(x,v1; t)
∂t
∣
∣
∣
coll=
∫
d3v2d3v′1d
3v′2 ρ(v1,2 → v′1,2)
× [f1(x;v′1; t)f1(x;v
′2; t)− f1(x;v1; t)f1(x;v2; t)]. (3.17)
La giustificazione dell’approssimazione effettuata per raggiungere la (3.16) risiede princi-palmente nel successo delle (3.16-3.17) nel descrivere il comportamento dei gas. La ragioneper cui una tale approssimazione funziona ha probabilmente a che fare con l’effetto chehanno le collisioni nel distruggere correlazioni (ipotesi di caos molecolare). L’idea e che lemolecole 1 e 2, arrivino al punto x in seguito a collisioni distinte le quali rendono queste

3.3. L’EQUAZIONE DI BOLTZMANN 43
molecole essenzialmente scorrelate. Ottenere una dimostrazione quantitativamente precisadi questa asserzione e molto difficile, che, dal nostro punto di vista, puo essere vista co-me parte del bagaglio di informazione sulla dinamica delle molecole che trascuriamo allamicroscala.
Nota La derivazione che abbiamo effettuato della equazione di Boltzmann, e, in partico-lare, della forma del termine di collisione ∂f |coll ha sfruttato pesantemente la natura delladistribuzione f come una distribuzione media. Qualche precisazione e dovuta, a questopunto, sul tipo di media utilizzata. Se definissimo f attraverso una media statistica, ciritroveremmo infatti tra le mani un oggetto che non e in grado di descrivere l’evoluzioneper esempio di un sistema in cui sono presenti fluttuazioni macroscopiche (ad esempioturbolenza). Per descrivere l’evoluzione di queste fluttuazioni, dovremmo utilizzare la den-sita istantanea f(y, t). Come si e detto, pero, l’equazione di Boltzmann e il termine dicollisione ∂tf |coll, sono stati derivati per f e non per f . Non abbiamo a disposizione quindiuna equazione di evoluzione (semplice) per f .
Il problema puo essere ovviato, nel caso la scala l delle fluttuazioni macroscopiche siamolto maggiore del volume molecolare v, nel qual caso, a scala l, il limite termodinamico esoddisfatto. Possiamo quindi effettuare una media spaziale a una scala intermedia, o pos-sibilmente una media spazio-temporale a scale di tempi superiori al tempo di collisione (macomunque inferiori alle scale di variazione delle fluttuazioni). Sfruttando l’indipendenzatra le molecole e l’assenza di memoria degli urti, possiamo invocare la proprieta ergodicaed approssimare
f(x, t) ≃ 〈〈f(x, t)〉∆V 〉∆t ≡1
∆V∆t
∫
∆V
d3x′∫ ∆t
0
dt′f(x+ x′, t+ t′),
e ci ritroviamo una distribuzione f , definita attraverso una media locale, che sara in gradodi tenere in conto tanto l’evoluzione di quantita medie che di fluttuazioni macroscopiche.
3.3.1 La distribuzione di Maxwell-Boltzmann
Possiamo vedere che la distribuzione di Maxwell-Boltzmann
fMB(x,v) = n0
( m
2πKT
)3/2
exp(
− mv2
2KT
)
, (3.18)
dato un profilo uniforme di densita n(x, t) = n0 e di temperatura T (x, t) = T , in assenzadi forze esterne, e soluzione stazionaria della equazione di Boltzmann (d’ora in avantitralasceremo i pedici su f1).
Sostituendo la (3.18) nella (3.16) ci da la seguente equazione:
∂fBM(x,v1)
∂t
∣
∣
∣
coll= 0.
Per verificare che questa equazione sia effettivamente soddisfatta, notiamo che, in ognicollisione, essendo le collisioni elastiche:
v21 + v22 = v′12+ v′2
2,

44 CAPITOLO 3. LA TEORIA CINETICA
cioe, l’energia si conserva. (Stiamo supponendo che non ci siano interazioni con le pareti delrecipiente; in definitiva, il sistema e isolato sia dal punto di vista termico che meccanico).
Sostituendo f = fMB nella espressione di (∂f/∂t)coll (3.17), troviamo quindi, scrivendofMB = Ce−αv
2, con C = n0(m/(2πKT ))
3/2 e α = m/(2KT ):
[fMB(x;v′1)f
MB(x;v′2)− fMB(x;v1)f
MB(x;v2)]
= C[exp−α(v′12 + v′22) − exp−α(v12 + v2
2)] = 0,
cosı che (∂fMB/∂t)coll = 0, come ci aspettavamo. Verificheremo quando parleremo delmetodo di massima entropia, che la Gaussiana e la distribuzione di entropia massimaper una data varianza della distribuzione. In altre parole, la distribuzione di Maxwell-Boltzmann, oltre ad essere una soluzione stazionaria della equazione di Boltzmann, e anchela distribuzione di massima entropia per la data temperatura. Il teoremaH che vedremo piuavanti ci garantisce che sistemi descritti da una equazion di Boltzmann sono caratterizzatida crescita dell’entropia. Ci aspettiamo pertanto che fMB sia anche la distribuzione diequilibrio del sistema, sulla quale questo rilassa indipendentemente dalle condizioni iniziali(per data temperatura e volume del recipiente).
3.3.2 Entropia e spazio delle velocita
La dipendenza dalla velocita di f produce una nuova componente di informazione necessariaa conoscere lo stato del sistema. Succede nello spazio delle velocita qualcosa di analogoa quello che si verifica nello spazio delle x: in questo caso, l’entropia sara maggiore se leparticelle sono uniformemente distribuite, e se le dimensioni della scatola sono maggiori.
Nel caso della velocita, il ruolo delle dimensioni della scatola e giocato essenzialmentedalla temperatura, che fissa la varianza della distribuzione in v. Cosı come nel caso dellascatola, il gas potrebbe trovarsi fuori dall’equilibrio concentrato in una parte della scatolain questione, nel caso della velocita, il gas potrebbe trovarsi essere distribuito secondo delledelta di Dirac ρ1(v) =
12[δ(v−v0)+ δ(v+v0)] cosı che 〈v〉 = 0 e σ2
v = v20. La distribuzione(che corrisponde a due getti con velocita opposte ±v0) e chiaramente la distribuzione aentropia piu bassa per la data varianza v20. L’effetto degli urti fa sı che l’entropia aumentimentre la distribuzione rilassa su ρMB
1 con KT = (1/3)mv20.
3.3.3 Il teorema H
L’equazione di Boltzmann soddisfa a una condizione di crescita dell’entropia di BoltzmannSf (t) = −
∫
d3xd3vf(x,v; t) ln f(x,v; t), qualsiasi sia la forma iniziale della distribuzione.Dagli argomenti nelle Sez. 3.2.1 e 3.2.2, e questa la definizione di entropia che permetteuna connessione diretta con la termodinamica del sistema. (Notiamo tuttavia dalla (3.11),che crescita nel tempo di Sf implica comunque crescita anche dell’entropia di ShannonS1). Calcoliamo la quantita Sf esplicitamente. Procedendo come nel caso del modellogiocattolo, (3.7), troviamo subito:
Sf = −∫
d3xd3vf(x,v; t) ln f(x,v; t).

3.3. L’EQUAZIONE DI BOLTZMANN 45
Cominciamo subito con verificare che, come ci si potrebbe aspettare, l’unico contributoalla crescita di entropia e fornito dagli urti, e cioe da (∂f/∂t)|coll.
Abbiamo infatti, utilizzando il teorema della divergenza sul termine in ∇x della equa-zione di Boltzmann (3.16), e trascurando contributi di superficie all’infinito per x:
−∫
d3xd3v ln f∇x · (vf) =∫
d3xd3v ∇x · (vf) = 0.
Analogamente, per il contributo di forza esterna:
−∫
d3xd3v ln f∇v · (Fextf) =
∫
d3xd3v Fext · ∇vf = −∫
d3xd3v f∇v · Fext,
che e nullo sia nel caso di forze esterne indipendenti dalla velocita, che in presenza di campimagnetici (in quest’ultimo caso sfruttiamo la relazione ∇v · [v × B] = B · [∇v × v] − v ·[∇v ×B] = 0).
Troviamo in definitiva, come c’era d’aspettarsi, utilizzando la (3.17):
Sf = −∫
d3xd3v(∂f/∂t)|coll ln f(x,v; t)
=
∫
d3x
∫
dµvρ(v′1,2 → v1,2)[f(1
′)f(2′)− f(1)f(2)] ln f(1),
dove ho introdotto la notazione dµv = d3v1d3v2d
3v′1d3v′2, f(1) ≡ f(x,v1; t) e cosı via.
Vediamo subito che, grazie alla simmetria di dµvρ[. . .] il risultato nell’integrale non cambiase sostituisco nell’integrale ln f(1) con ln f(2). Posso quindi scrivere:
Sf = −1
2
∫
d3x
∫
dµvρ(v′1,2 → v1,2)[f(1
′)f(2′)− f(1)f(2)] ln(f(1)f(2)).
Un’altra simmetria e quella per inversione temporale in ρ, che mi permette di scrivere
Sf = −1
2
∫
d3x
∫
dµvρ(v′1,2 → v1,2)[f(1)f(2)− f(1′)f(2′)] ln(f(1′)f(2′)).
Sommando le due espressioni trovo
Sf =1
4
∫
d3x
∫
dµvρ(v′1,2 → v1,2)[f(1
′)f(2′)− f(1)f(2)][ln(f(1′)f(2′))− ln(f(1)f(2))],
dove l’integrando e sempre positivo. Troviamo in sostanza la condizione di crescita dientropia
Sf ≥ 0.
dove l’uguaglianza vale solo se la distribuzione e Maxwelliana. Non abbiamo ancora di-mostrato che la distribuzione deve essere spazialmente omogenea. Sappiamo pero (vediSez. 3.6) che una distribuzione Maxwelliana spazialmente non-uniforme evolve generandouna componente non Maxwelliana, che porta di nuovo a crescita dell’entropia. L’unicasoluzione che da S = 0 e evidentemente la Maxwelliana spazialmente omogenea.

46 CAPITOLO 3. LA TEORIA CINETICA
3.4 Il limite fluido
Abbiamo visto un volume di gas isolato raggiunge una condizione di equilibrio statisticodescritto da una distribuzione di Maxwell-Boltzmann in cui la temperatura e la densitasono costanti. Notare che in generale la distribuzione di Maxwell-Boltzmann puo non esserecentrata intorno allo zero:
fMB(x,v; t) = n( m
2πKT
)3/2
exp(
− m|v − u|22KT
)
,
cosı che la distribuzione sara determinata all’equilibrio dai parametri n, u e T .Per caratterizzare meglio il processo di rilassamento all’equilibrio, consideriamo piu in
dettaglio gli urti. Abbiamo essenzialmente due parametri chiave: il cammino libero medioλ e il tempo di collisione τcoll. Cerchiamo di stimarne l’ordine di grandezza in funzione dialtri parametri noti, quali la velocita termica vth ≡ σv, il raggio molecolare a e la densitan. Notiamo che fuori dall’equilibrio sia n che vth saranno dipendenti dal tempo e dallaposizione. Notiamo inoltre che parlare di raggio molecolare presuppone un’interazione“hard-core” tra le molecole, e non un’interazione a lungo raggio quale quella che si avrebbefra particelle cariche, ad esempio in un plasma.
Otteniamo subito le stime dimensionali
λ ∼ 1
na2, τcoll ∼
λ
vth. (3.19)
L’espressione di λ puo essere ottenuta a partire da considerazioni di tipo geometrico os-servando che la separazione tipica fra le molecole e l ∼ n−1/3. In ogni volume di lato l,ci sara quindi tipicamente una molecola. La probabilita per una molecola che attraversail volumetto di effettuare una collisione con la molecola contenuta in quest’ultimo saraP ∼ (a/l)2. Il numero di volumetti che bisogna attraversare perche si abbia una pro-babilita di collisione di ordine uno sara quindi 1/P ∼ (l/2)2 e la distanza percorsa saraλ ∼ l/P ∼ l3/a2 = 1/(na2).
Vediamo che per parametri tipici per un gas, quali
n ∼ 1020cm−3, a ∼ 10−8cm, vth ∼ 3 · 105cm/s,
abbiamo dalla (3.19):λ ∼ 10−4cm, τcoll ∼ 10−9s.
Questo e un tempo straordinariamente corto e definisce di fatto una microscala dal puntodi vista dei tempi per il nostro sistema. Ora, ciascun urto cambia in maniera drammatica lavelocita della molecola che lo subisce. Ne possiamo concludere che una distribuzione inizialedi velocita rilassera a una distribuzione di equilibrio (dal punto di vista delle velocita) inun numero non molto alto di urti. Questo processo di rilassamnto sara quindi esso stessoestremamente veloce. D’altra parte, e esperienza comune il fatto che due corpi in contattoraggiungono una identica temperatura in un tempo decisamente macroscopico.
Vediamo quindi che l’equilibrio in un recipiente di gas isolato e raggiunto in fasisuccessive:

3.4. IL LIMITE FLUIDO 47
• Equilibrio cinetico f(x,v; t) → fMB(x,v; t) in un tempo ∼ τcoll, dove fMB(x,v; t)
e una distribuzione di Maxwell-Boltzmann in cui i parametri di densita, temperaturae velocita media in generale non sono costanti: n = n(x, t), T = T (x, t), u = u(x, t).
• Equilibrio meccanico in cui i gradienti di pressione e di velocita media sono eli-minati. E il tempo necessario per far sı che cessino di esserci parti in movimentorelativo nel gas.
• Equilibrio termodinamico in cui anche i gradienti di temperatura (e gli ultimiresiduali gradienti di velocita e pressione) scompaiono. Questo e (come vedremo) ilprocesso piu lento.
Una volta che l’equilibrio cinetico viene raggiunto, abbiamo che per descrivere il sistema di-ventano sufficienti i campi n, u e T , dipendenti solo dalle coordinate, mentre la dipendenzada v e a questo punto nota e data dalla distribuzione di Maxwell-Boltzmann.
Le cose non sono pero mai cosı semplici; se fosse davvero f = fBM(x,v; t), il terminecollisionale sarebbe infatti identicamente nullo: (∂f/∂t)coll = 0 e l’entropia rimarrebbecostante. Vediamo quindi che una condizione di equilibrio (meccanico e termodinamico)non potrebbe mai essere raggiunta. La conclusione e che una situazione di non-equilibriorichiede sempre una componente non Maxwell-Boltzmann f = f − fMB, che potra esserepiccola una volta raggiunto l’equilibrio cinetico, ma che rimane non nulla ed e essenzialeper il raggiungimento dell’equilibrio termico e meccanico.
Proviamo a stimare la componente f , considerando f piccola rispetto a fMB, e linea-rizzando il termine collisionale (3.17) (approssimazione BGK):
∂f
∂t
∣
∣
∣
coll∼ −f − fMB
τcoll, (3.20)
che descrive un rilassamento esponenziale f → fMB su scale di tempi τcoll. Supponendoassenza di forze esterne, possiamo scrivere quindi:
( ∂
∂t+ v · ∇x
)
fMB ∼ − f
τcoll
e pertanto, se L e la scala spaziale di variazione di fMB:
f ∼ vthτcollfMB
L∼ λ
LfMB. (3.21)
La correzione sara piccola se la scala di variazione spaziale e molto maggiore del camminolibero medio.
Passiamo all’analisi del rilassamento meccanico. Notiamo che il tempo perche questovenga raggiunto e dell’ordine del tempo di spostamento relativo di parti di fluido a scalaL:
τmecc ∼L
u(x+ L)− u(x)∼ Lvth
λuτcoll, (3.22)

48 CAPITOLO 3. LA TEORIA CINETICA
che sara molto maggiore di τcoll se le velocita medie sono piccole rispetto alla velocitatermica e la scala L e macroscopica.
In definitiva vediamo che per tempi piu brevi di τmecc le quantita medie n, u e Trimangono praticamente costanti.
Notiamo che la condizione l ≪ λ ≪ L ci garantisce che il limite termodinamico esoddisfatto a scala L. Infatti, la condizione l ≪ L garantisce che il numero di molecolein un volume di lato L e molto grande. La condizione di caos molecolare ha d’altraparte la conseguenza che la lunghezza di correlazione del sistema e essenzialmente zero:le molecole sono praticamente indipendenti. La condizione di limite termodinamico equindi soddisfatta automaticamente se L ≫ l. In piu, notiamo il significato di λ comescala macroscopica di interazione: parti separate da distanze maggiori del cammino liberomedio sono caratterizzate da sequenze di urti distinte.
A questo punto, vediamo che e possibile scegliere una scala intermedia lm, con λ ≪lm ≪ L, dove il limite termodinamico e ancora soddisfatto, ma tale che le quantita medie n,T e u varino poco. Quindi, a tempi piu brevi di τmecc, (ma piu lunghi di τcoll, in modo chel’equilibrio cinetico sia gia raggiunto), i profili di n, u e T saranno localmente (a scala < lm)uniformi e stazionari, come all’equilibrio termodinamico. Ci si riferisce a una situazione diquesto genere con il termine di equilibrio termodinamico locale.
A tempi piu lunghi di τmecc, una descrizione in termini di equilibrio termodinamicolocale cessa evidentemente di valere e le quantita medie n, T e u iniziano a variare inmaniera sostanziale. Cio nonostante, il fatto che f ≪ fMB, garantisce che queste sianosufficienti, in prima approssimazione, a descrivere l’evoluzione del sistema. Inoltre, siccomea scala lm il limite termodinamico e soddisfatto, n, T e u non hanno solo il significato diquantita medie, ma anche quello di quantita istantanee misurate a scala lm, il che, nel casodi n e u, corrisponde alla nostra intuizione per i concetti di densita e velocita fluida.Notiamo che anche KT (x, t)/m prende un significato istantaneo e puo essere vista infatticome la varianza delle velocita molecolari misurata attraverso media campionaria sullemolecole in una scatola di lato lm intorno a x al tempo t.
A questa situazione, in cui n, u e T hanno un significato locale istantaneo (e nonstatistico) e si e in una condizione di equilibrio cinetico tale che la deviazione f − fMB siapiccola, e n, u e T sono sufficienti a descrivere lo stato del sistema, ci si riferisce con ilnome di limite fluido. Vedremo fra breve che in questo regime, n, u e T sono sufficientinon solo a determinare lo stato del sistema, ma anche la sua evoluzione futura.
3.5 Equazioni fluide
Equazioni per l’evoluzione delle quantita n, u e T possono essere ottenute prendendo imomenti rispetto a v della equazione di Boltzmann:
∫
d3v vk[∂f
∂t+ v · ∇xf +
1
m∇v · (Fextf)
]
=
∫
d3v vk∂f
∂t
∣
∣
∣
coll.

3.5. EQUAZIONI FLUIDE 49
Il senso di questo approccio e che le quantita fluide n, u e T , prima di essere parametridella distribuzione di Maxwell-Boltzmann fMB, sono momenti della distribuzione f :
n =
∫
d3v f ; u =1
n
∫
d3v fv; T =m
3Kn
∫
d3v f |v − u|2;
ci aspettiamo quindi che le equazioni per n, u e T non siano altro che le equazioni per imomenti di ordine zero, uno e due della equazione di Boltzmann.
Eseguiamo il calcolo in maniera esplicita nel caso k = 0. Otteniamo subito
∂
∂t
∫
d3v f +∇x ·∫
d3v fv +1
m
∫
d3v ∇v · (Fextf) =
∫
d3v∂f
∂t
∣
∣
∣
coll.
Utilizzando il teorema della divergenza, il terzo integrale a lato destro si converte in unintegrale sulla superficie a v → ∞ che possiamo porre uguale a zero (almeno se ammettiamoche f ≃ fMB). Rimaniamo quindi con l’equazione
∂n
∂t+∇x · (nu) =
∫
d3v∂f
∂t
∣
∣
∣
coll.
Possiamo vedere che il lato destro di questa equazione e identicamente nullo grazie aconservazione negli urti del numero di particelle. Abbiamo infatti, sfruttando la simmetriaρ(v′
1,2 → v1,2) = ρ(v1,2 → v′1,2), e indicando di nuovo dµv ≡ d3v1d
3v2d3v′1d
3v′2, f(1) ≡f(x,v1), etc:
∫
d3v1∂f(x,v1; t)
∂t
∣
∣
∣
coll=
∫
dµv ρ(v′1,2 → v1,2)[ρ1(1
′)ρ1(2′)− ρ1(1)ρ1(2)] = 0.
La densita n obbedisce pertanto la seguente equazione di continuita:
∂n
∂t+∇x · (nu) = 0. (3.23)
Diamo senza derivazione le equazioni per i momenti successivi u(x, t) = 〈v|x, t〉 e T (x, t) =m3Kσ2v|x,t (utilizziamo qui la notazione di somma sugli indici ripetuti di Einstein):
nm( ∂
∂t+ u · ∇x
)
ui +∂P
∂xi= n〈F ext
i 〉 − ∂πij∂xj
, (3.24)
n( ∂
∂t+ u · ∇x
)
KT +2
3P∇ · u = −2
3∇x · q− 2
3πij
∂ui∂xj
. (3.25)
(La derivazione esplicita delle (3.24-3.25) e delle (3.28-3.29) piu sotto, puo essere trovataad esempio nel capitolo 5 del testo “Statistical Mechanics” di K. Huang). Cosı come l’e-quazione di continuita impone la conservazione della massa (o del numero di particelle) alsistema, vediamo quindi le due equazioni per u e T impongono conservazione rispettiva-mente del momento lineare e dell’energia. Queste equazioni di conservazione sono detteequazioni fluide per il gas. Notare che abbiamo introdotto un numero di nuove quantita:

50 CAPITOLO 3. LA TEORIA CINETICA
• La pressione cinetica P = nKT .
• Il cosı detto tensore di pressione anisotropo: πij(x, t) = n(x, t)m〈(vivj−v2δij/3)|x, t〉,dove abbiamo indicato con v = v − u la fluttuazione termica della velocita.
• Il flusso di calore: q(x, t) = 12n(x, t)m〈v2v|x, t〉
Notare la media che rimane nel termine di forza esterna; il simbolo di media e necessario neicasi in cui la forza dipende dalla velocita come ad esempio in presenza di campi magnetici:
〈FEM |x, t〉 = eE(x, t) + 1
c[u(x, t)×B(x, t)]
Discuteremo piu sotto il significato fisico della pressione e del flusso di calore.Notiamo subito la presenza di una struttura gerarchica delle equazioni fluide, che richia-
ma quanto osservato nel caso delle equazioni per le distribuzioni ρ1, ρ2, eccetera. Vediamoinfatti che per risolvere l’equazione per n (momento zero), sarebbe necessario conoscere u(momento uno) e per risolvere l’equazione per u sarebbe necessario conoscere P e πij (mo-menti due). Analogamente, per risolvere l’equazione per T sarebbe necessario conoscere ilmomento tre q. Si potrebbe essere tentati a questo punto di seguire un approccio di tipochiusura statistica, fermandosi ad esempio al livello di campo medio. Vediamo subito peroche un simile approccio porterebbe a trascurare in una espressione come 〈v2〉 = u2 + v2th,la velocita termica vth, che in gran parte delle situazioni di interesse e tutt’altro che tra-scurabile (per dire, la velocita termica delle molecole d’aria a pressione e temperaturaatmosferica e dell’ordine della velocita del suono ∼ 330m/s). Questo suggerirebbe cautelaanche con chiusure di ordine piu alto, come trascurare il momento terzo q(x, t) (gia il nomeflusso di calore suggerisce che questo termine potrebbe essere invece importante).
Il problema quindi rimane, perche le equazioni dei momenti nella forma data qui soprarisultano essere non chiuse: ci mancano espressioni per il flusso di calore e per il tensore dipressione anisotropo. L’osservazione cruciale e che questi termini sono associati alla partenon Maxwelliana della distribuzione, che abbiamo indicato con f .
Il punto e importante e vale la pena verificarlo. Per far cio, scriviamo la distribuzionedi Maxwell-Boltzmann in funzione degli scarti:
fMB = C exp(−v2/(2σ2v)) ≡ C exp(−v21/(2σ2
v)) exp(−v22/(2σ2v)) exp(−v23/(2σ2
v)).
La dimostrazione che il flusso di calore si annulla e immediata (la distribuzione fMB e paririspetto a v, e q e la media di un oggetto dispari). Per quanto riguarda πij, consideriamoseparatamente i due casi i 6= j e i = j. Per i 6= j, troviamo subito, essendo le componentidello scarto indipendenti:
πij = nm〈vivj〉 = nm〈vi〉〈vj〉 = 0.
Per i = j abbiamo invece 〈v2〉 = 〈[v21 + v22 + v23]〉 = 3〈v2i 〉 e quindi:
πii = nm〈(v2i − v2/3)〉 = 0.
Ricordo che la componente f − fMB e quella associata agli urti e quindi in buona sostanzaquella che porta all’aumento di entropia e al rilassamento all’equilibrio.

3.6. VISCOSITA E DIFFUSIVITA TERMICA 51
• I termini πij e q nelle equazioni fluidi, associati alla deviazione f − fMB, sono dettipertanto termini non ideali, mentre le equazioni per i momenti n, u e T , in assenzadelle componenti πij e q descrivono la cosı detta fluidodinamica ideale.
3.6 Viscosita e diffusivita termica
I termini non ideali possono essere ottenuti tramite una espansione perturbativa dellaequazione di Boltzmann, in cui il parametro di espansione e il rapporto λ/L, approccio cheva sotto il nome di espansione di Chapman-Enskog. Alternativamente, si puo seguireun approccio euristico, accontentandosi di stime per ordine di grandezza delle quantita πije q, e focalizzandosi invece sul significato fisico di questi termini.
Cerchiamo di capire innanzitutto l’origine della componente non Maxwell-Boltzmann(cioe non-Gaussiana) della distribuzione di velocita, in presenza ad esempio di un gradientedi temperatura lungo x1.
Il fatto e che in un punto dato, passeranno molecole che arrivano da punti a x1 diversi.Quindi, se la distribuzione (almeno inizialmente) e Maxwelliana, ma con valori di T chevariano lungo x1, quello che si vedra dopo un po’ nel punto dato, sara una sovrapposizionedi Maxwelliane diverse, che non sara, in genere, una Maxwelliana. La presenza degliurti, attraverso il termine (∂f/∂t)coll, ha l’effetto che le molecole che arriveranno in x1,origineranno da urti avvenuti tipicamente a x1 ± λ. In altre parole, la distribuzione divelocita in x1 non sara una sovrapposizione di Maxwelliane arbitrarie, ma solo di quelleposte a distanza ∼ λ dal punto in questione.
Questo ci permette di stimare il flusso di calore in x1:
q =1
2nm〈v2v〉 ≡ m
2
∫
d3v v2vf ∼ Kn[vth(x1 − λ)T (x1 − λ)− vth(x1 + λ)T (x1 + λ)]e1
dove ±vth(x1∓λ) e la componente 1 della velocita delle molecole che arrivano in x1 a partireda urti in x1∓λ (ovviamente per arrivare in x1 da sinistra, la velocita deve essere positiva,e viceversa). Vediamo che abbiamo a che fare veramente con un flusso di energia termica:se moltiplichiamo per l’elemento d’area ∆x2∆x3, la quantita q∆x2∆x3 e proprio il numerodi molecole che passano attraverso ∆x2∆x3 da sinistra, moltiplicato la loro energia cinetica∼ KT (x1 − λ), meno il contributo da destra, in cui l’energia cinetica e ∼ KT (x1 − λ) asinistra. Siccome abbiamo preso λ≪ L, possiamo espandere in serie di Taylor e troviamoq(x1) ∼ −nλ(∂/∂x1)(vthKT )e1 ∼ nλvth(∂KT/∂x1)e1. Prendendo la variazione di T inuna direzione generica:
q = −nκT∇xKT, κT ∼ λvth ∼λ2
τcoll, (3.26)
dove il coefficiente κT e detto diffusivita termica del gas.Procedendo in maniera analoga a quanto fatto con il flusso di calore, possiamo vedere
che il tensore di pressione anisotropo e associato agli sforzi viscosi. Consideriamo il caso piusemplice in cui effetti di compressione nel gas sono trascurabili e gli unici moti presenti sono

52 CAPITOLO 3. LA TEORIA CINETICA
scivolamenti di masse di gas le una rispetto alle altre. Immaginiamo quindi che passandoda x1 − λ a x1 + λ, quello che succede sia un cambio nella componente u2 della velocitafluida.
In questo caso, le molecole che arrivano da x1±λ, quando arrivano in x1, si ritrovano conuna componente 2 della velocita media u2(x1 ± λ). Supponiamo per semplicita u(x) = 0,cosı che v = v in x1. Supponiamo poi n uniforme. Troviamo allora
π12(x1) = nm〈v1v2〉 ≡ m
∫
d3v v1v2f
∼ nm[vth(x1 − λ)u2(x1 − λ)− vth(x1 + λ)u2(x1 + λ)].
Di nuovo, ±vth(x1 ∓ λ) e la componente 1 della velocita delle molecole che arrivano in x1a partire da urti in x1 ∓ λ. In questo caso, abbiamo a che fare con un flusso di momentolineare, cioe il momento lineare in direzione 2 trasferito in direzione 1 per unita di tempoe unita di superficie nel piano x2x3. In altre parole e la forza in direzione 2 esercitata perunita di superficie dal gas a sinistra di x1 sul gas a destra di x1. In altre parole, si tratta diuno sforzo di taglio (o tangenziale). Notare che abbiamo parlato di pressione anisotropa;per confronto, la pressione in senso stretto e lo sforzo perpendicolare esercitato da unaparte del gas su un’altra adiacente. Di nuovo espandiamo in serie di Taylor per λ ≪ L,cosı da trovare π12(x1) ∼ −nλ(∂/∂x1)(vthu2) ∼ nλvth(∂u2/∂x1). Prendendo le variazionidi u in direzioni generiche.
πij = −nmν(∂ui∂xj
+∂uj∂xi
)
ν ∼ λvth ∼λ2
τcoll(3.27)
e il coefficiente ν e detto viscosita cinematica del gas, mentre la quantita µ = nmν echiamata viscosita dinamica. Nel caso di aria a temperatura e pressione atmosfericaabbiamo κT ∼ ν ∼ 0.1cm2/s.
A questo punto siamo finalmente in grado di scrivere le equazioni della fluidodinamicatenendo conto degli effetti non ideali. L’equazione per la conservazione del momento (3.24)diventa, utilizzando la (3.27), nel caso piu semplice di flussi non comprimibili (∇ · u = 0):
nm( ∂
∂t+ u · ∇x
)
u+∇xP = n〈Fext〉+ µ∇2xu, (3.28)
mentre l’equazione per la conservazione dell’energia diventa, nel caso piu semplice in cuile variazioni di densita sono piccole:
n( ∂
∂t+ u · ∇x
)
KT +2
3P∇ · u =
2
3nκT∇2
xKT − 2
3πij
∂ui∂xj
. (3.29)
Queste due equazioni sono dette l’equazione di Navier-Stokes (incomprimibile) e l’e-quazione del trasporto di calore.
Isoliamo nelle due equazioni l’effetto dei termini non ideali. Iniziamo dalla equazionedi Navier-Stokes e consideriamo il caso in cui le velocita sono piccole (cosı che il termine

3.6. VISCOSITA E DIFFUSIVITA TERMICA 53
quadratico puo essere trascurato), e non ci siano ne gradienti di pressione ne forze esterne.L’equazione diventa:
∂u
∂t= ν∇2
xu.
In maniera analoga, se supponiamo il fluido immobile, vediamo che l’equazione per iltrasporto del calore diventa
∂T
∂t= κT∇2
xT,
che non e altro che l’equazione di diffusione (2.41). Vediamo che in entrambi i casi, si hauna equazione del calore che descrive il rilassamento a un profilo di u (nel primo caso) edi T (nel secondo), uniformi su un tempo scala τtermo ∼ (L/λ)2τcoll. Questo e il tempodi rilassamento dovuto esclusivamente a effetti molecolari di diffusione del calore e delmomento e il risultato finale e precisamente lo stato di equilibrio termodinamico globaledel sistema isolato.
Il fatto che la componente di trasporto di calore e momento lineare dovuta a urti mo-lecolari sia descritta attraverso una equazione del calore, giustifica a posteriori l’utilizzo diun modello di gas basato su cammino random. Il cammino random e in pratica quello dellemolecole, caratterizzato da salti di lunghezza λ e probabilita di salto per unita di tempo∼ τ−1
coll (un salto per tempo di collisione). Vediamo quindi che il tempo di rilassamentocinetico individua la microscala del cammino random, mentre il tempo di rilassamentotermico non e altro che il tempo che impiega tipicamente il camminatore per percorrereuna distanza L.
3.6.1 Diffusione in una miscela di gas
E l’adattamento al caso di un gas, del classico problema della goccia di colorante che sidissolve in un liquido, anche se noi non mescoliamo. Evidentemente, fenomeni di diffusioneprodotti dalle collisioni molecolari, causano mescolamento anche se il liquido e fermo.
Consideriamo il caso di una miscela di due componenti a e b, di cui una molto diluita:a ≪ b ≃ , dove a,b = na,bma,b sono le densita di massa delle due specie e = a +b. Possiamo ottenere una velocita fluida u a partire dalla densita di momento lineareaua + bub = u e troviamo di nuovo u ≃ ub. Consideriamo una situazione di equlibriotermodinamico per tutte le variabili, eccetto la concentrazione na che e presa funzione dellaposizione. Quindi, n, T e u sono uniformi, e ci possiamo porre nel sistema di riferimentoin cui u = 0. La velocita ua diventa quindi la velocita di dispersione della specie anella miscela; la sua conoscenza ci permetterebbe di determinare l’evoluzione della densitana e di descrivere la dinamica del processo di mescolamento. Semplici considerazioni diconservazione di massa ci permettono di scrivere l’equazione di continuita (in assenza direazioni chimiche):
∂tna +∇ · (naua) = 0. (3.30)
La velocita ua e il risultato delle collisioni delle molecole a con le molecole b. Possiamostimarla attraverso considerazioni analoghe a quanto fatto nel caso della diffusivita termica.

54 CAPITOLO 3. LA TEORIA CINETICA
Supponendo un gradiente di na lungo x1, il flusso generato dall’arrivo in x1 di molecole chehanno termalizzato in posizioni x1 ± λa, sara:
na(x1, t))ua(x1, t) ∼ na(x1 − λa, t)vth,a − na(x1 + λa, t)vth,a ≃ −λavth,a∂na(x1)
∂x1, (3.31)
dove λa e il cammino libero medio delle molecole nel gas. Troviamo quindi una relazioneflusso gradiente naua = −κa∇na dove κa ∼ λavth,a e la diffusivita della specie a nellamiscela. Sostituendo nella (3.30), otteniamo l’equazione di diffusione:
∂tna = κa∇2na; (3.32)
l’analogia con il comportamento del camminatore random e evidente: in questo caso sonole molecole b che causano il moto random delle molecole a.
La presenza di una velocita relativa media tra una specie ed un’altra deve essere as-sociata evidentemente alla esistenza di una forza che agisce fra di loro. Cerchiamo dideterminarne la natura.
L’equazione di Navier-Stokes per la specie a avra la stessa espressione (3.28), eccettoper la forza dovuta alle collisioni con le molecole b, che puo essere stimata come fcoll ∼−aua/τcoll. Consideriamo la situazione semplificata in cui il profilo di densita a un certoistante e lineare, na(x, t) ≃ n0,a+a·x, cosı che ua e uniforme. Troviamo quindi la relazione3
∇Pa + aua/τcoll = 0,
dove Pa = naT e la pressione parziale della specie a. Risolvendo rispetto a ua, ritroviamola Eq. (3.31). Vediamo quindi che la velocita di dispersione non e altro che la reazionedella specie a al gradiente di pressione parziale prodotto dalla disomogeneita nel profilo dina.
3.7 Dalla teoria cinetica alla termodinamica
3.7.1 Pressione e legge di stato
Abbiamo chiamato la quantita P = nKT che entra nelle equazioni di Navier-Stokes e ditrasporto del calore pressione cinetica. Verifichiamo che si tratti effettivamente di unapressione.
Consideriamo allora un elemento di fluido V , cioe un volume di fluido sufficientementepiccolo che u sia approssimativamente costante al suo interno, e le cui pareti si muovanocon il fluido, di maniera tale che il contenuto di molecole del volumetto non vari nel tempo.Integrando l’equazione di Navier-Stokes (3.28) sull’elemento di fluido troviamo (vedi notasotto):
M( ∂
∂t+ u · ∇x
)
u(x, t) ≡Mdu(x(t), t)
dt= −
∫
V
d3x ∇xP +N(Fvisc + Fext), (3.33)
3L’assenza del termine a∂t, nella equazione, e dovuta alla Eq. (3.31), e al fatto che n e lineare, equindi, dalla (3.32), ∂tna = 0.

3.7. DALLA TEORIA CINETICA ALLA TERMODINAMICA 55
che non e altro che la seconda legge di Newton per l’elemento di fluido, con M la massatotale ed N il numero totale di particelle nell’elemento. Focalizziamoci sulla componente1 del contributo alla forza dovuto a P . Prendendo per V all’istante dato un cubo con lati∆xi, i = 1, 2, 3, abbiamo:
−∫
V
d3x∂P
∂x1≃ −∆x2∆x3[P (x1 +∆x1)− P (x1)].
Ma questa non e altro che la definizione di pressione come forza perpendicolare alla su-perficie diviso la superficie stessa. La forza totale in direzione 1 sul volumetto e quindiottenuta a partire dalla differenza tra le pressioni esterne sulle due facce ∆x2∆x3 poste ax1 e x1 +∆x1.
Notiamo per ultimo che la definizione della pressione cinetica, espressa in funzione delnumero di molecole totale in V , diventa la legge di stato dei gas ideali nella sua formastandard:
PV = NKT. (3.34)
Nota. Esaminiamo piu da vicino il passaggio nella Eq. (3.33) che ci ha permesso discrivere il termine (∂t + u · ∇x)u come una derivata totale. Abbiamo
M
m
du
dt≡ d
dt
∫
V (t)
d3x nu =
∫
V (t)
d3x∂nu
∂t+
1
δt
(
∫
V (t+δt)
d3xnu−∫
V (t)
d3xnu)
=
∫
V (t)
d3x∂nu
∂t+
∫
∂V (t)
dS · u nu =
∫
V (t)
d3x(∂nu
∂t+∂uinu
∂xi
)
D’altra parte possiamo scrivere ∂xi(uinu) = u∇x · (nu)+nu ·∇xu e utilizzando l’equazionedi continuita (3.23), troviamo la (3.33). Vediamo quindi che l’operatore (∂t + u · ∇x) checompare nelle equazioni fluide, descrive una variazione lungo una traiettoria del fluido. Iltermine u · ∇x che rappresenta la correzione a ∂t, dovuta al fatto che ci spostiamo con ilfluido, e detto termine di avvezione.
3.7.2 Energia termica
Abbiamo definito la temperatura cinetica a partire dalla distribuzione di Maxwell-Boltzmanne abbiamo esteso poi la definizione al caso di una generica distribuzione di non equilibriof tramite la relazione
T =m
3K〈v2|x, t〉.
L’energia media di una molecola e quindi ǫ = 32KT e l’energia termica di un gas ideale a
temperatura uniforme, contenente N molecole sara
U th =3
2NKT.

56 CAPITOLO 3. LA TEORIA CINETICA
Per ottenere l’energia interna totale U , dobbiamo aggiungere a U th l’energia dei legamichimici delle molecole U chem = Nǫ e l’energia cinetica dei moti macroscopici:
Umecc =m
2
∫
V
d3xn(x, t)u2(x, t).
Nel sistema di riferimento del volume V , il momento lineare totale del gas e nullo e quindi∫
Vd3xn(x, t)u(x, t) = 0. Nel caso di un sistema che non e un gas ideale, dovremmo
naturalmente includere anche l’energia di interazione U int fra le molecole (questo contributodiventa particolarmente importante in corrispondenza di transizioni di fase). L’energiainterna totale sara quindi U = U th + U chem + Umecc + U int. Imponendo conservazionedell’energia, otteniamo il primo principio della termodinamica:
∆U = ∆Q−∆R, (3.35)
dove δQ e il calore ricevuto dall’ambiente esterno, e δR e il lavoro eseguito dal corposull’ambiente esterno.
3.7.3 Calore e lavoro
Verifichiamo che i due termini 23P∇x·u e 2
3nκT∇2
xKT nella (3.29), permettono di descriverei cambi di energia termica del gas generati attraverso compressione e scambio di calore.Consideriamo allora un elemento di fluido V . Integrando l’equazione di trasporto del calore(3.29) sull’elemento di volume e utilizzando la definizione U th = (3/2)NKT , troviamo
dU th
dt= −
∫
V
d3x P∇x · u+ nκ
∫
∂V
dS · ∇xKT +dRvisc
dt, (3.36)
dove dRvisc/dt = (2/3)∫
Vd3x πij∂xjui. Supponiamo ora che il processo di compressione o
espansione si svolga abbastanza lentamente da far sı che il gas sia istantaneamente in unostato di quasi equilibrio dal punto di vista meccanico, e che quindi P sia uniforme e ugualealla pressione esterna. Abbiamo allora
−∫
V
d3x P∇x · u ≃ −P∫
V
d3x ∇x · u = −P∫
∂V
dS · u.
Ma∫
∂VdS · udt non e altro che il cambio di volume nell’intervallo di tempo dt e quindi
dU th
dt= −P dV
dt+ nκ
∫
∂V
dS · ∇xKT +dRvisc
dt.
Il termine PdV/dt e la potenza esercitata dal gas nel volume V sull’ambiente esterno,positivo in una espansione, negativo in una compressione. Notiamo che se considerassimol’equazione per l’energia dell’ambiente esterno, troveremmo la stessa equazione, solo condV ext/dt = −dV/dt. Quindi, se le pressioni esterne ed interna sono uguali, il che e verose il processo e sufficientemente lento, il lavoro eseguito dal volumetto corrisponde a una

3.7. DALLA TEORIA CINETICA ALLA TERMODINAMICA 57
perdita di energia termica dello stesso e a un aumento dell’energia dell’ambiente esterno.Quindi ∆R = P∆V . Al contrario, uno squilibrio di pressione implica accelerazione di partidel sistema e dell’ambiente esterno, e P∆V descrive la conversione di energia termica delsistema allo stesso tempo in energia termica dell’ambiente esterno ed in energia meccanicadel sistema e dell’ambiente esterno.
Vediamo che le stesse considerazioni sono associate al flusso totale di calore [secondotermine a lato destro della (3.36)] ΦT = nκ
∫
∂VdS · ∇xKT , dall’esterno al volumetto V
(ricordo che q entrava con segno meno a lato destro dell’equazione del calore). Abbiamoinfatti la relazione
ΦT = nκ
∫
∂V
dS · ∇xKT = −nκ∫
∂V ext
dSext · ∇xKT = −ΦextT ,
cioe il flusso totale dal volume all’esterno e pari a meno il flusso dall’esterno al volu-me. Quindi ∆Q =
∫
dtΦextT . Notiamo che se la temperatura all’esterno e maggiore che
all’interno, avremo dS · ∇xT < 0 e quindi c’e un flusso di calore positivo nel volumetto.Il risultato che il flusso di calore si verifica in direzione opposta al gradiente di tem-
peratura porta naturalmente all’enunciato di Clausius del secondo principio dellatermodinamica: Non esistono trasformazioni il cui unico risultato sia il trasferimento dicalore da un corpo freddo a un corpo caldo.
Rimane da considerare l’ultimo termine Rvisc. Questa e evidentemente il termine diconversione lavoro–calore dovuto a dissipazione viscosa dei moti interni del gas. Se il gasall’interno di V e in movimento, u(x, t) 6= 0, vi sara conversione di energia meccanicain energia termica, anche se, nel caso ∆Q = ∆R = 0, la variazione di U e zero. Dinuovo, se il gas e inizialmente fermo, e cambiamenti di volume e trasferimenti di caloresi svolgono abbastanza lentamente, i moti innescati saranno essi stessi sufficientementelenti e il contributo ∆Rvisc al cambio di U th diventa trascurabile. In parole povere, U th
soddisfarra una relazione del tipo della prima legge della termodinamica in forma analogaalla (3.35).
3.7.4 Entropia e temperatura
Abbiamo visto che il termine di flusso di calore nella equazione per il trasporto di caloreva in direzione opposta ai gradienti di temperatura e porta pertanto il gas ad assumereuna configurazione a temperatura uniforme. Ci farebbe piacere che l’entropia di Boltz-mann raggiungesse un massimo in questa che e evidentemente la condizione di equilibriotermodinamico per il gas. Vogliamo in buona sostanza stabilire l’identita fra l’entropiadi Boltzmann e l’entropia termodinamica del nostro sistema. Lavorando all’equilibrio, uncalcolo rapido ci da, utilizzando la (3.18):
SfMB = N(3
2lnT + ln(V/N) + ζ
)
:= S(T, V,N) (3.37)
dove ζ e una costante. Supponiamo quindi di avere due parti macroscopiche A e B di unsistema isolato, che le due parti siano internamente in equilibrio, anche se non in equilibrio

58 CAPITOLO 3. LA TEORIA CINETICA
fra di loro, e che l’unica maniera in cui esse comunichino sia attraverso scambio di calore.Abbiamo pertanto che U = UA + UB sara una quantita costante. In maniera analogapossiamo scrivere per l’entropia:
S = SA(UA) + SB(U − UA)
[la scelta (3.11) ci garantisce che l’entropia e additiva; ricordo inoltre che abbiamo ipotiz-zato che non ci sono scambi di molecole, fra le due parti, e quindi la dipendenza di S da ne irrilevante]. La condizione di massimo dell’entropia, data la condizione di costanza perUA + UB = U sara una condizione di massimo rispetto a UA:
dS
dUA= 0 ⇒ dSA
dUA=
dSBdUB
;d2SAdU2
A
+d2SBdU2
B
< 0.
Ora, utilizzando la relazione U = 32NKT , possiamo scrivere
dSAdUA
=dSAdTA
(dUAdTA
)−1
=1
KTA(3.38)
e similmente per SB, che ci dice TA = TB. Verifichiamo infine che TA = TB corrispondeeffettivamente a un massimo della entropia; utilizzando la relazione U = 3
2NKT , troviamo
infattid2SAdU2
A
=2N
3
d
dUA
1
UA= − 2N
3U2A
< 0,
e TA = TB corrisponde effettivamente a un massimo di S. Vediamo quindi che la tempera-tura cinetica definita a partire dalla varianza della velocita nella (3.18) ha il comportamen-to corretto di una temperatura termodinamica: l’equilibrio termodinamico corrisponde auguaglianza della temperatura dei due sistemi ed a al massimo della entropia.
La Eq. (3.38) puo essere invertita nella forma
dSA =dUAKTA
∣
∣
∣
VA=cost.,
ma questa (a meno di un fattoreK) non e altro che la definizione termodinamica di entropiadella parte A del sistema, essendo dUA uguale, nelle condizioni prese in esame, al calorericevuto. Possiamo quindi definire, per un generico sistema internamente in equilibriotermodinamico, che scambia calore con l’esterno:
dStermo =dQ
T, (3.39)
che e la definizione termodinamica di variazione di entropia nella sua forma piu familiare.Per evitare di portarci dietro costanti, poniamo subito K = 1, cosı che d’ora in poi S ≡Stermo.
L’associazione dell’entropia allo scambio di calore ci porta naturalmente alla seguenteespressione del primo principio della termodinamica, valida per sistemi vicini all’equilibrio:
dU = TdS − PdV, (3.40)

3.8. ESERCIZI 59
in cui l’energia e intesa come funzione di S e V , invece che di T (e in principio, per unsistema generico, anche di V ). Il primo principio della termodinamica diventa in questomodo equivalente a una definizione di temperatura e pressione a partire dall’energia:
T (S, V ) =∂U(S, V )
∂S, P (S, V ) = −∂U(S, V )
∂V(3.41)
(si ribadisce che il sistema deve essere supposto abbastanza vicino all’equilibrio, cosı cheeventuali variazioni di P e T al suo interno siano trascurabili). A questo punto, il lavoroadiabatico diventa essenzialmente il lavoro a entropia costante:
∆R = −∫ Vf
Vi
P (S, V )dV (3.42)
mentre
∆Q =
∫ Sf
Si
T (S, V )dS (3.43)
e il calore scambiato a volume costante.
3.8 Esercizi
Esercizio 1 Le amebe in una coltura hanno nell’intervallo di tempo dt probabilita ΓBdtdi dividersi in due, probabilita ΓDdt di morire, e probabilita 1− (ΓB+ΓD)dt di non moriree non dividersi (si trascura la probabilita di dividersi e poi morire nello stesso intervallo).Sia N(t) il numero di amebe nella coltura all’istante t.
• Scrivere l’equazione master per la PDF ρ(N, t).
• Derivare l’equazione di evoluzione per la media 〈N(t)〉 e per il secondo momento〈N2(t)〉. (Suggerimento: si provi a calcolare esplicitamente (d/dt)〈N(t)〉 e (d/dt)〈N2(t)〉;si consiglia di lavorare nel limite continuo in cui differenze finite in N diventanoderivate).
• Discutere il caso ΓB = ΓD.
Esercizio 2 E ragionevole supporre che la divisione cellulare sia bloccata da una popola-zione eccessiva. Supporre quindi che la probabilita di morte per unita di tempo sia nellaforma ΓD = ΓBN/N0.
• Scrivere di nuovo l’equazione master per la PDF ρ(N, t).
• Derivare l’equazione di evoluzione per la media 〈N(t)〉 e la varianza 〈(N(t))2〉 estimare i valori di queste quantita allo stato stazionario.

60 CAPITOLO 3. LA TEORIA CINETICA
• In base alla risposta al punto precedente, dire in quale range di parametri N0, cipotremmo aspettare che un’approssimazione di campo medio sia applicabile allo sta-to stazionario. Per far cio, conviene determinare allo stato stazionario la varianzaσ2N (supporre piccoli gli scarti ∆N = N − 〈N〉, e ottenere dalla equazione master,
lavorando a ordine piu basso in ∆N , e imponendo stato stazionario, l’equazione per〈(∆N)2〉).
Esercizio 3 Generalizziamo il problema di cui al caso precedente alla situazione in cui leamebe possono spostarsi avanti e indietro lungo un canale. Dividiamo il canale in settorisuccessivi i = 1, 2, ....M e supponiamo che un’ameba possa in un intervallo di tempo dt,oltre a riprodursi e morire, saltare avanti o indietro con probabilita Sdt (si trascuri dinuovo il caso di simultaneo salto e morte o divisione).
• Scrivere l’equazione master per ρM(Ni, t).
• Scrivere l’equazione per la distribuzione per il numero di amebe in un dato settoreρ1(N ; i, t).
Esercizio 4 Una superficie piana scorre a velocita costante e ad altezza costante su un’altrasuperficie parallela fissa. Calcolare la forza per unita di superficie che bisogna esercitaresulla superficie superiore per tenerla in moto. Si trascurari il termine avvettivo nellaequazione di Navier-Stokes (limite di bassi numeri di Reynolds). Supporre le superficiidealmente infinite e che non vi siano gradienti di pressione orizontali.
Esercizio 5 Modificare l’equazione di continuita per un gas nel caso ci siano due specie Ae B, con le molecole A che si trasformano in B con probabilita per unita di tempo Γ.
Esercizio 6 Lo space-shuttle e lungo una quarantina di metri. Calcolare l’altezza alla qualeuna descrizione fluida delle sue proprieta aerodinamiche risulterebbe inefficace (supporreper semplicita un profilo isotermo di temperatura: T = 300oK su tutta la colonna d’aria).Come dipende il risultato dalla velocita del veicolo?
Esercizio 7 Una massa M di un gas ideale monoatomico viene riscaldata in manieraisobara sino a passare da temperatura T a temperatura T +∆T . Sia V il volume inizialedel gas e m la massa atomica. Si calcoli la variazione di entropia di Boltzmann del gas nelpassare da uno stato all’altro (si considerino gli stati iniziali e finali di equilibrio). Il gas siespande nel processo; di quanto si espande?)
Esercizio 8 Ottenere dalla equazione di Navier-Stokes (3.28)), la potenza totale dissipatain un volume V dalle forze viscose, e dimostrare che e uguale al termine di conversionelavoro calore Rvisc che compare nella Eq. (3.36). Si consideri per semplicita un fluidoincomprimibile ∇ · u = 0.

Capitolo 4
Termodinamica
4.1 Processi reversibili e irreversibili
Ritorniamo al sistema isolato esaminato nella Sez. 3.7.4, formato da due sottosistemi A eB in equilibrio interno, che scambiano calore fra di loro a volume costante. Questo vuoldire ∆UA,B = ∆QA,B. Abbiamo visto che all’equilibrio l’entropia totale S = SA + SB haun massimo rispetto a come si distribuisce l’energia fra i due corpi. Questo fa sı che perpiccole deviazioni dall’equilibrio l’entropia differisca dal suo valore all’equilibrio per terminiquadratici nella deviazione dell’energia; se UA e l’energia del sottosistema A all’equilibrio:
∆S = S(UA)− S(UA) =1
2S ′′(UA)(UA − UA)
2 +O((UA − UA)3). (4.1)
Se guardiamo la variazione rispetto all’equilibrio dell’entropia dei due sottosistemi, vediamopero che questa e lineare nell’energia; a meno di termini quadratici in ∆UA = UA − UA:
∆SA = −∆SB = ∆U/T, (4.2)
dove ∆SA = SA(UA)− SA(UA) e ∆SB = SB(U − UA)− SA(U − UA).
• Chiamiamo reversibile (irreversibile) un processo, nel sistema isolato, in cuil’aumento di entropia globale si puo (non si puo) considerare trascurabile.
Possiamo precisare la condizione di reversibilita imponendo in maniera esplicita ∆SA,B ≫∆S. Utilizzando le due Eqs. (4.1) e (4.2), e utilizzando S ′
A(UA) = S ′B(UB) = 1/T insieme
a S ′′(UA) ∼ −T−2(∂UA/∂T )−1, troviamo la condizione
T∂UA∂T
≫ ∆UA, (4.3)
che, per un gas ideale, diventa semplicemente ∆TA < (TB − TA) ≪ TA: la differenza ditemperatura fra i due corpi in contatto doveva essere piccola. Una condizione analogaalla(4.3) vale per trasformazioni che coinvolgono cambi di volume, nel qual caso si richiede
61

62 CAPITOLO 4. TERMODINAMICA
SB
SA
U2∆
SA SB( + )/2
U/T∆S/2+
U/T∆S/2−
UA UB
A,B
A,B
S
S/2
UU/2
Figura 4.1: Profilo dell’entropia dei sottosistemi in funzione della loro energia interna.All’equilibrio S(U) = 2SA,B(U/2) = 2S. La pendenza della tangente in UA,B = UA,B = U/2e data dall’inverso della temperatura di equilibrio T
che la differenza di pressione tra i due corpi sia piccola rispetto alla pressione di ciascunodei due.
La proprieta di massimo dell’entropia all’equilibrio ha una interpretazione geometricanel caso i due sottosistemi A e B sono identici. Come nel caso dell’entropia di Shannon,un ruolo centrale e giocato dalle proprieta di convessita della funzione. In questo caso, lefunzioni entropia delle due parti, SA e SB, hanno identica forma e i valori di S possonoessere calcolati studiando una singola curva, come illustrato in Fig. 4.1. Vediamo dallafigura che i punti della curva giacciono al disotto della tangente. Questo fa sı che SA(UA)+SB(UB) ≤ S(U/2) con eguaglianza sodisfatta solo all’equilibrio UA,B = UA,B = U/2 (ilpunto di tangenza). Vediamo quindi che la condizione di massimo di entropia all’equilibrioe permessa dal fatto che S e una funzione crescente e convessa dell’energia (confrontarecon la discussione nella Sez. 2.2). Per contro abbiamo visto che queste proprieta sonoassociate alla positivita della temperatura ed al fatto che questa e a sua volta una funzionecrescente della energia del sistema. L’ambito delle trasformazioni reversibili si riferiscepertanto all’intervallo di valori di UA,B per cui vale l’approssimazione lineare SA,B(UA,B) ≃SA,B(UA,B) + ∆UA,B/T .
Abbiamo visto che in condizioni reversibili, il bilancio energetico in ciascuno dei duecorpi A e B puo essere espresso tramite la relazione dUA,B = TdSA,B − PdVA,B. Cidomandiamo come una relazione di questo genere possa essere generalizzata in condizioniirreversibili. E di particolare interesse il caso in cui uno dei due corpi e molto piu grandedell’altro e puo essere visto come un ambiente esterno.
Notiamo come una condizione di non-equilibrio fra ambiente e corpo, in cui le differenzefra la pressione e temperatura interne P e T e quelle esterne P0 e T0, non sono trascurabili,produrra comunque fenomeni di non equilibrio sia all’interno del corpo che al suo esterno,anche se entrambi erano inizialmente in equilibrio con se stessi. Quello che si verifica,

4.2. DISEGUAGLIANZE TERMODINAMICHE 63
tipicamente, sara la formazione di disomogeneita nella temperatura e nella pressione, chefaranno sı che il sistema e il corpo non possono piu essere descritti in maniera univoca dasingole variabili T, P e T0, P0. In generale non sara quindi possibile descrivere il bilancioenergetico tramite una singola equazione come la (3.40), e bisognerebbe determinare l’e-voluzione dei campi n(x, t), u(x, t) e T (x, t) all’interno del corpo per mezzo di equazionicome la (3.23), la (3.28) e la (3.29).
Una descrizione di tipo puramente termodinamico ridiventa possibile se l’ambienteesterno puo essere considerato in equilibrio. Questo significa in particolare che le pres-sione e la temperatura esterna P0 e T0 sono uniformi. Se P0 e T0 sono anche costanti,il lavoro eseguito sul corpo sara evidentemente P0∆V0 e il calore ricevuto dal corpo sara−T0∆S0. Avremo quindi alla fine del processo, sfruttando la condizione di crescita dientropia ∆S +∆S0 ≥ 0, e la relazione ∆V = −∆V0
∆U = −T0∆S0 + P0∆V0 ≤ T0∆S − P0∆V. (4.4)
Una volta che tutti i processi di rilassamento saranno terminati, si avra evidentemente P =P0 e T = T0. La Eq. (4.4) descrive quindi il rilassamento di un sistema termodinamico incondizioni iniziali generiche in un ambiente esterno a pressione e temperatura P0, T0 fissate.Notiamo come la crescita di entropia ∆S + ∆S0 contiene contibuti sia dal rilassamentointerno al corpo, sia al fatto che il trasferimento di calore si e verificato tra sistemi (ilcorpo e l’ambiente) a temperatura diversa. L’andamento dell’entropia del corpo e di fattouna somma di contributi diversi
∆S(t) = ∆Sril(t)− T0T (t)
∆S0(t)−∆Sdis(t)
dove ∆Sril e il contributo da una eventuale condizione di non equilibrio iniziale per il corpo;(T0/T )∆S0 e la variazione di entropia associata al trasferimento di calore T0∆S0, nel casoil corpo avesse temperatura uniforme T , e −∆Sdis e il contributo dalla disomogeneita delsistema durante il transiente. Notiamo quindi che, sebbene debba sempre valere ad ogniistante ∆S(t) ≥ −∆S0(t), ∆S(t) sara in generale minore di quanto predetto nel caso ilsistema fosse omogeneo.
Un caso di particolare interesse e quello di un corpo omogeneo (T e P uniformi), inquasi equilibrio con l’ambiente esterno, ma che e tuttavia in condizioni di non equilibriointerno, dal punto di vista chimico, o per la presenza di moti interni. Abbiamo in tal casoP ≃ P0, T ≃ T0, lo scambio di calore e di lavoro si svolge in modo reversibile, ma dScontiene una componente di rilassamento che fa sı che dS ≥ dQ/T . Il primo principioprende quindi la forma di una diseguaglianza
dU ≤ TdS − PdV. (4.5)
4.2 Diseguaglianze termodinamiche
Abbiamo visto che nel caso di un gas ideale, di cui la dipendenza dell’entropia da U e V eranota, l’equilibrio termodinamico viene raggiunto grazie al fatto che T = (∂S/∂U)−1 > 0 e

64 CAPITOLO 4. TERMODINAMICA
∂2S/∂U2 = −T−2(∂T/∂U) < 0. Ci domandiamo che cosa succederebbe se in un sistemagenerico queste diseguaglianze non fossero verificate.
Prima di rispondere a questa domanda, notiamo la simmetria fra il rapporto traentropia, calore e temperatura, da una parte, e volume, lavoro e pressione dall’altra:
dQ = TdS; dR = −PdV.Concentriamoci sugli effetti puramente meccanici e supponiamo che non ci sia trasferi-mento di calore del sistema con l’esterno. Vediamo subito che una pressione negativaimplicherebbe che il sistema sarebbe in grado di cedere energia comprimendosi: il sistemaimploderebbe senza bisogno di azione dall’esterno. Una pressione che diminuisce al de-crescere del volume, d’altra parte, renderebbe impossibile l’equilibrio in presenza di unapressione esercitata dall’esterno. La compressione causata dalla pressione esterna cause-rebbe una diminuzione (invece che un aumento) della pressione interna. La differenza dipressione fra esterno e interno aumenterebbe e il sistema sarebbe ulteriormente compressosino a portare di nuovo a una implosione.
L’analogia meccanica ci suggerisce che qualcosa di simile deve succedere con calore,temperatura ed entropia, e che le due condizioni T > 0 e ∂U(T, V )/∂T > 0 debbanosempre essere verificate in un sistema termodinamico.
Verifichiamo prima che la temperatura deve sempre essere positiva. In effetti, se aves-simo un sistema a temperatura negativa, vedremmo che questo sarebbe in effetti “piucaldo” di uno a temperatura positiva. Immaginiamo infatti di mettere a contatto un si-stema (A) con TA > 0 e uno (B) con TB < 0, e che continuino a valere le proprieta chel’entropia totale (nei pressi dell’equilibrio) e la somma delle due entropie, e che aumen-ti al passare del tempo. Abbiamo chiaramente ∆QA = −∆QB, con ∆SA = ∆QA/TAe ∆SB = ∆QB/TB = −∆QA/TB. Abbiamo pero, imponendo la condizione di crescitadell’entropia:
∆S = ∆SA +∆SB =( 1
TA− 1
TB
)
∆QA > 0
che implica ∆QA < 0: calore fluisce da B ad A.Una sistema con una temperatura positiva ma che decresce con l’energia avrebbe ca-
ratteristiche non meno paradossali. Supponiamo di mettere a contatto due sistemi A e Bidentici ma con energie interne diverse: UA < UB. Il profilo dell’entropia SA e SB e quelloraffigurato in Fig. 4.2: Vediamo che in questo caso, la funzione SA,B ha la convessitaverso il basso. I suoi punti giacciono pertanto al disopra della congiungente i due punticorrispondenti alla condizione iniziale del sistema, per energie minori di UA e maggiori diUB. L’entropia si massimizza per UA = 0 e UB = UA + UB (la perdita di informazione nelsistema B compensa l’aumento in A). In definitiva, per avere una termodinamica sensata,la pressione e la temperatura di un sistema devono sottostare alle seguenti condizioni:
P, T > 0,∂U
∂T
∣
∣
∣
V> 0,
∂P
∂V
∣
∣
∣
S< 0,
dove i pedici indicano che le due derivate sono eseguite rispettivamente a volume e aentropia costanti.
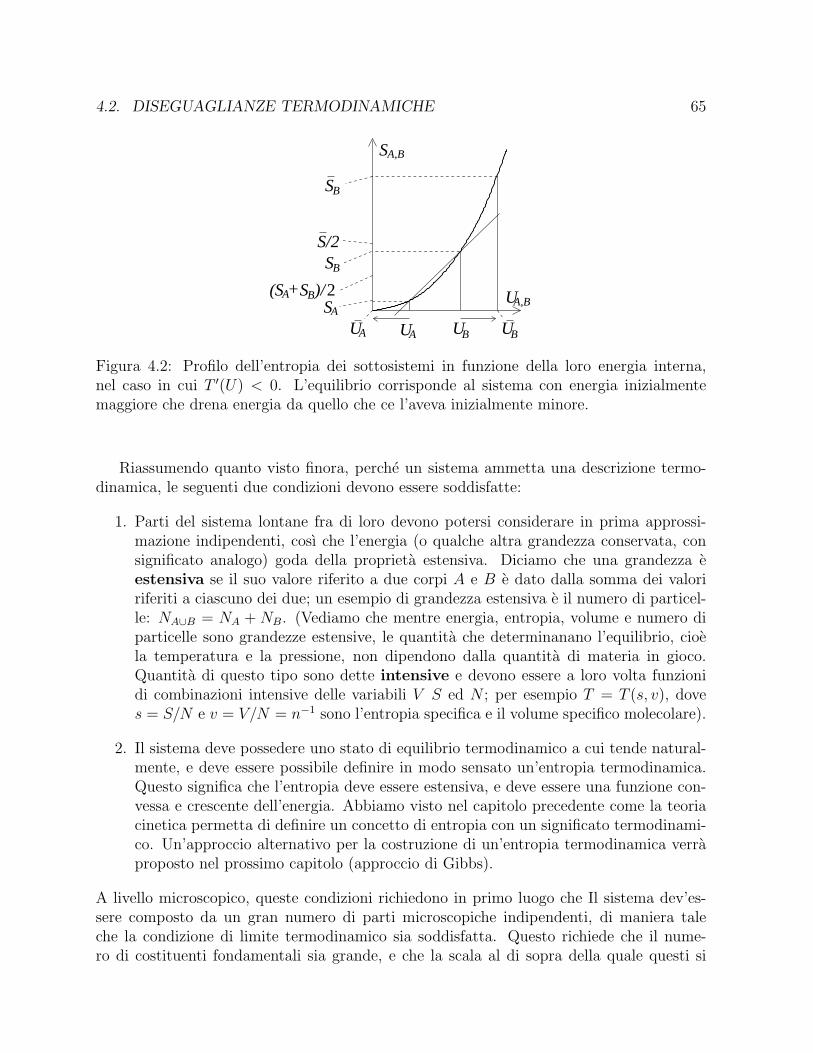
4.2. DISEGUAGLIANZE TERMODINAMICHE 65
UA UB
S/2
SA SB( + )/2
UA UB
SB
SA
SB
A,B
A,B
S
U
Figura 4.2: Profilo dell’entropia dei sottosistemi in funzione della loro energia interna,nel caso in cui T ′(U) < 0. L’equilibrio corrisponde al sistema con energia inizialmentemaggiore che drena energia da quello che ce l’aveva inizialmente minore.
Riassumendo quanto visto finora, perche un sistema ammetta una descrizione termo-dinamica, le seguenti due condizioni devono essere soddisfatte:
1. Parti del sistema lontane fra di loro devono potersi considerare in prima approssi-mazione indipendenti, cosı che l’energia (o qualche altra grandezza conservata, consignificato analogo) goda della proprieta estensiva. Diciamo che una grandezza eestensiva se il suo valore riferito a due corpi A e B e dato dalla somma dei valoririferiti a ciascuno dei due; un esempio di grandezza estensiva e il numero di particel-le: NA∪B = NA + NB. (Vediamo che mentre energia, entropia, volume e numero diparticelle sono grandezze estensive, le quantita che determinanano l’equilibrio, cioela temperatura e la pressione, non dipendono dalla quantita di materia in gioco.Quantita di questo tipo sono dette intensive e devono essere a loro volta funzionidi combinazioni intensive delle variabili V S ed N ; per esempio T = T (s, v), doves = S/N e v = V/N = n−1 sono l’entropia specifica e il volume specifico molecolare).
2. Il sistema deve possedere uno stato di equilibrio termodinamico a cui tende natural-mente, e deve essere possibile definire in modo sensato un’entropia termodinamica.Questo significa che l’entropia deve essere estensiva, e deve essere una funzione con-vessa e crescente dell’energia. Abbiamo visto nel capitolo precedente come la teoriacinetica permetta di definire un concetto di entropia con un significato termodinami-co. Un’approccio alternativo per la costruzione di un’entropia termodinamica verraproposto nel prossimo capitolo (approccio di Gibbs).
A livello microscopico, queste condizioni richiedono in primo luogo che Il sistema dev’es-sere composto da un gran numero di parti microscopiche indipendenti, di maniera taleche la condizione di limite termodinamico sia soddisfatta. Questo richiede che il nume-ro di costituenti fondamentali sia grande, e che la scala al di sopra della quale questi si

66 CAPITOLO 4. TERMODINAMICA
possono considerare indipendenti (lunghezza di correlazione) sia molto minore della scaladi variazione delle proprieta medie del sistema. Questo richiede in particolare che le di-mensioni del sistema siano maggiori della scala caratteristica di interazione delle sue partimicroscopiche.
In secondo luogo, ci si aspetta che la dinamica sia caratterizzata a scala microscopicada una di perdita di informazione, rispetto alle condizioni iniziali, che corre in paralleloalla la crescita di entropia a scala macroscopica. Questo corrisponde alla intuizione chegli stati di equilibrio termodinamico debbano corrispondere a configurazioni microscopichepiu “sparpagliata”. Diventa cosı naturale definire l’entropia come perdita di informazionenel passaggio da una descrizione pienamente determinata a scala microscopica, a quelladata dalla conoscenza dello stato macroscopico in cui si trova il sistema.
4.3 Termodinamica: punto di vista macroscopico.
E noto che la termodinamica fu derivata originariamente senza alcun riferimento alla strut-tura microscopica della materia. L’unica informazione disponibile in questo genere di de-scrizione, era quella fornita dalle cosı dette leggi di stato del sistema. Ci domandiamoquali condizioni devono soddisfare queste leggi di stato perche sia possibile una descrizionetermodinamica del sistema. Cominciamo con il presentare quelli che sono gli assiomi dibase della termodinamica.
Ci si ritrova tipicamente ad avere a che fare con un sistema su cui si puo esercitare dellavoro, e che puo a compiere lavoro sull’esterno. L’energia meccanica non e pero conservataed e necessario introdurre il concetto di calore per rendere conto del fenomeno. Il risultatoe il primo principio della termodinamica, che, nel caso di sistemi il cui stato e definitotramite pressione e volume puo essere scritto nella forma
dU = dQ− PdV
In analogia al caso di un sistema meccanico, si suppone che lo stato del sistema possaessere descritto dando la sua energia interna U (o alternativamente la pressione P ) e il suovolume V . L’afflusso di calore dQ non e invece associato a una funzione dello stato delsistema e dQ non e un differenziale esatto.
Si osserva che sistemi diversi messi in contatto possono scambiare o no calore; sistemiche non scambiano calore sono detti in equilibrio termodinamico. Si osserva poi che valeil cosı detto principio zero della termodinamica: se i sistemi A e B sono ciascuno inequilibrio con C, allora sono in equilibrio fra loro. Per caratterizzare l’equilibrio mutuoviene quindi introdotto il concetto di temperatura, come parametro che assume valoriuguali in sistemi all’equilibrio.
Si osserva a questo punto che e possibile ordinare le temperature in modo tale cheil calore si trasferisca sempre (fissati gli altri parametri, per esempio il volume) da corpia temperatura piu alta a quelli a temperatura piu bassa. Questo costituisce di fatto ilcontenuto del secondo principio della termodinamica. Notiamo pero che questo non e

4.3. TERMODINAMICA: PUNTO DI VISTA MACROSCOPICO. 67
sufficiente a definire la temperatura in maniera univoca: una qualsiasi funzione crescentedella temperatura T che abbiamo utilizzato sin’ora andrebbe ugualmente bene.
Si osserva infine che esistono delle leggi di stato che legano i parametri di stato delsistema, quali nel caso dei gas ideali U , V e P , alla temperatura. A questo punto, latemperatura e definita essenzialmente a partire dalla legge di stato dei gas ideali PV = NT ,dove in questo contesto N indica una quantita di materia (non necessariamente numero diparticelle) e K e scelto in modo da far sı che T sia misurata nei gradi che si preferiscono(K = 1 nel nostro caso).
Un punto di vista alternativo che evita una definizione della temperatura a partiredalle proprieta di un sistema specifico come un gas ideale, e quello di postulare da subitol’esistenza di una funzione di stato entropia. Questo e equivalente a postulare che, nel casodi un sistema in equilibrio interno, il primo principio della termodinamica si possa scriverenella forma
dU = TdS − PdV. (4.6)
Notiamo la simmetria tra la relazione fra entropia e temperatura, e quella fra volume epressione.
Lo stato del sistema puo quindi essere determinato a partire da P e V , oppure, utiliz-zando la legge di stato, da T e V , oppure (e questo e il postulato) a partire da S e V . Inaltre parole, S deve essere una funzione di stato, che deve potere essere scritta in funzionedegli altri parametri del sistema V e T .
Iniziamo con il verificare che nel caso di un gas ideale. l’entropia goda della proprieta ri-chiesta. Dobbiamo richiedere che dS = 1
T(dU+PdV ) sia un differenziale esatto. Dobbiamo
verificare che esista quindi una funzione S(V, T ) tale che
dS =∂S
∂TdT +
∂S
∂VdV.
D’altra parte, dal primo principio della termodinamica abbiamo (4.6), scrivendo tutti itermini come funzione di V e T :
dS =1
T(dU + PdV ) =
1
T
[∂U
∂TdT +
(∂U
∂V+ P
)
dV]
(4.7)
e utilizzando le due relazioni P (V, T ) = NT/V e U(V, T ) = (3/2)NT :
dS =3N
2TdT +
N
VdV.
La condizione che dS sia un differenziale esatto e che le derivate ∂V ∂TS e ∂T∂V S sianoidentiche, e infatti,
∂2S
∂V ∂T=
∂
∂V
3N
T= 0;
∂2S
∂T∂V=
∂
∂T
N
V= 0.
Pertanto, come richiesto (e come ci potevamo aspettare dalla teoria cinetica), nel caso diun gas ideale, la funzione di stato entropia esiste.

68 CAPITOLO 4. TERMODINAMICA
Chiaramente, vengono ritrovate le stesse proprieta dell’entropia ottenute a partire dalladerivazione cinetica. In particolare, che l’aumento di entropia globale nello scambio dicalore fra corpi diversi e positivo ed e trascurabile rispetto allo scambio di entropia quandola differenza di temperatura fra i due corpi e piccola. Infatti, supponendo TB > TA (equindi ∆QA > 0):
∆SA +∆SB = ∆QA
( 1
TA− 1
TB
)
> 0. (4.8)
Di nuovo, abbiamo un’interpretazione grafica di questa diseguaglianza (vedi la Fig. 1 dellalezione precedente). Le due temperatura inverse 1/TA e 1/TB sono infatti i valori delladerivate S ′
A,B nei due punti UA e UB. Il valore di ∆S = ∆SA +∆SB calcolato qui sopra, equindi quello che si ottiene scrivendo ∆S ≃ S ′′
A,B∆U2 e approssimando la derivata seconda
con la differenza finita [S ′A,B(UA)− S ′
A,B(UB)]/(2∆U).Vediamo che la condizione che l’entropia sia una funzione di stato nel caso di un sistema
generico impone delle condizioni sulla forma della legge di stato. Vediamo infatti che lacondizione di differenziale esatto, imposta sul lato destro della (4.7)
∂
∂V
1
T
∂U
∂T=
∂
∂T
(∂U
∂V+ P
)
conduce alla condizione, calcolando esplicitamente le derivate:
∂U
∂V= T
∂P
∂T− P.
Vale la pena a questo punto calcolare esplicitamente l’entropia di un gas ideale. Sfruttandol’indipendenza dell’integrale
∫ T,V
T0,V0dS dal percorso nel piano T, V , possiamo scomporlo in
due pezzi (T0, V0) → (T, V0) e poi (T, V0) → (T, V ):
S(T, V ) = S(T0, V0) +
∫ T
T0
dS(T ′, V0) +
∫ V
V0
dS(T, V ′).
Scrivendo TdS = dU + PdV = (3/2)NdT + (NT/V )dV :
S(T, V ) = S(T0, V0) +N[3
2
∫ T
T0
dT ′
T ′+
∫ V
V0
dV ′
V ′
]
= S(T0, V0) +N(3
2lnT/T0 + lnV/V0
)
,
che implica
S(T, V,N) = N(3
2lnT + lnV
)
+ C(N).
La forma di C(N) e determinata richiedendo la proprieta estensiva per l’entropia, e cioe,che se ho due sistemi A e B con identiche proprieta interne (tipo di molecole, densita etemperatura), deve valere SA∪B = SA + SB, dove SA∪B e l’entropia dei due sistemi messiin comunicazione. Questo ci da il risultato (3.37) che avevamo ottenuto in teoria cineticaa partire dalla distribuzione di Maxwell-Boltzmann (3.18):
S(T, V,N) = N(3
2lnT + ln(V/N)
)
+Nζ.

4.4. LAVORO MASSIMO 69
Nota. I termini in questa formula non sono ben definiti dal punto di vista dimensionale.Dalla costante ζ possono essere estratti i termini necessari per rendere l’argomento deilogaritmi adimensionale. Questo e possibile in maniera univoca invocando la meccanicaquantistica, nel qual caso l’entropia e ottenuta a partire da una partizione dello spaziodelle fasi in cellette di volume ∆x∆v = ~/m:
S(T, V,N) = N ln((mT )3/2V
N~3
)
+Nζ.
A questo punto, ζ prende le dimensioni di un’energia per grado di temperatura e descriveun contenuto interno di entropia delle molecole.
4.4 Lavoro massimo
Abbiamo visto (Eq. 4.8)) che l’entropia di un sistema isolato, a causa dei processi irrever-sibili che si svologono al suo interno, puo solo aumentare. Analogamente, la variazione dientropia in un sistema non isolato, oltre al contributo dQ/T associato a scambio di calore,avra una componente non negativa associata a processi di raggiungimento di equilibrio frale sue parti interne.
Continuando a lavorare con le variabili S e V , possiamo calcolare il lavoro massimoche puo essere estratto da un sistema isolato che sta rilassando all’equilibrio. Il fatto checonsideriamo un sistema isolato implica che trascuriamo quella componente di lavoro chepotrebbe risultare ad esempio da una espansione globale del sistema. Ci interessiamo soloalla componente che risulta dal movimento di parti interne. Il problema coincide pertantocon il calcolo dell’efficienza di un motore termico che estrae energia dal sistema tramiteuna trasformazione ciclica. (Le considerazioni che seguono sono tratte in gran parte dallaSez. 19 del testo “Fisica Statistica” di Landau e Lifsits).
Immaginiamo per esempio che nel sistema una parte A e a temperatura TA, una parteB a temperatura TB con TB > TA, e una parte ulteriore del sistema compia un ciclo, espan-dendosi prima isotermicamente a contatto con B, e poi adiabaticamente raffreddandosi atemperatura TA, eseguendo lavoro nelle due fasi sul motore. Nella fase inversa, il motoreesegue lavoro sul sottosistema che si contrae prima isotermicamente cedendo calore a A epoi adiabaticamente sino a ritornare nella condizione iniziale a temperatura TA. Questo epraticamente il ciclo di Carnot, ma il dettaglio del ciclo non e rilevante.
L’aspetto rilevante e che il lavoro estratto e la differenza tra il calore che il sottosistemaha ricevuto da B e quello che ha ceduto ad A:
∆R = −∆QB −∆QA
e che e pari allo stesso tempo alla variazione di energia interna del sistema in blocco (difatto solo quella parte costituita dai termostati; il sottosistema compie un ciclo e quindinon contribuisce):
∆R = −∆U = Ui − Uf .

70 CAPITOLO 4. TERMODINAMICA
Essendo l’energia dello stato finale Uf e una funzione crescente dell’entropia del sistema,il lavoro svolto risultera massimo quando l’entropia dello stato finale e minima, cioe setutti i processi nel frattempo si svolgono in maniera reversibile. Questo significa imporresimultanemente:
∆QA,B = TA,B∆SA,B, e ∆SA +∆SB = 0.
Sostituendo in ∆R = −∆QB −∆QA, troviamo il lavoro massimo che puo essere estrattodal sistema
∆RMAX = −(TB − TA)∆SB
Dividendo per il calore perso da B, otteniamo l’efficienza massima
ηMAX =∆R
−∆QB
= 1− TATB
,
che e consistente con l’enunciato di Kelvin del secondo principio della termodina-mica: e impossibile realizzare una trasformazione il cui unico risultato sia la conversionedi calore in lavoro, da un sistema a temperatura uniforme.
Nota. Puo essere interessante vedere in dettaglio cosa succede in condizioni irreversibili.In questo caso le variazioni di entropia in A e B non eguagliano quelle nel sottosistema.Abbiamo per esempio ∆QB < TB∆SB, dove ∆SB puo contenere contributi di riequilibriointerno di B. Nel sottosistema avremo in corrispondenza l’afflusso di calore ∆QSB =−∆QB e un corrispondente afflusso di entropia ∆SSB = ∆QSB/TSB, con TSB < TB e∆SSB, rispettivamente, la temperatura e la variazione di entropia del sistema, nel momentoin cui questo e in contatto con B. Una situazione analoga si verifichera durante il contattocon A, nel qual caso TSA > TA. In generale, in presenza di processio di riequilibrio internoal sottosistema potremmo avere ∆SSA + ∆SSB = ∆SS > 0 e quindi ∆SSA > −∆SSB.In una sequenza ciclica di trasformazioni, questo contributo e pero nullo e possiamo porre∆SA +∆SB = 0. Sostituendo nell’espressione dell’efficienza:
η =∆R
∆QSB
= 1 +TSA∆SSATSB∆SSB
= 1− TSATSB
.
Troviamo quindi, in definitiva:
η = 1− TSATSB
< 1− TATB
.
L’irreversibilita causata dal fatto che A e B non si trovano alla temperatura del sottosistemaquando vi entrano in contatto, fa sı che l’efficienza sia minore che nel caso di un processoreversibile.
4.5 I potenziali termodinamici
La relazione dU ≤ TdS − PdV permette di intepretare l’energia interna U come l’abilitadi svolgere lavoro e scambiare calore del sistema. In particolare, dU e il calore scambiato

4.5. I POTENZIALI TERMODINAMICI 71
reversibilmente a volume costante e −PdV il lavoro effettuato isoentropicamente (cioeattraverso una trasformazione adiabatica). I potenziali termodinamici sono introdotti alloscopo di descrivere l’attitudine di un sistema a svolgere lavoro in condizioni isoterme oscambiare calore in condizioni isobariche. Cosı come il fatto che l’energia U = U(S, V )descriva un attitudine a svolgere lavoro isoentropico (adiabatico) e scambiare calore avolume costante, e dovuta precisamente alla dipendenza da S e V della funzione, nel casodei potenziali termodinamici si cercheranno funzioni di S, P oppure T, V oppure T, P aseconda delle variabili su cui si vuole avere controllo nello scambio di calore o nel lavoro.Si vede subito che la condizione di tenere la pressione oppure la temperatura costante nelsistema significa che questo non e piu isolato e l’attitudine a svolgere lavoro o trasferirecalore non e piu solo contenuta in U . Il contributo aggiunto dal fatto che il sistema none piu isolato e determinato, come si vedra fra breve, per mezzo di una trasformazione diLegendre.
Il primo potenziale che introduciamo descrive l’attitudine a scambiare calore in condi-zioni isobariche ed e detto entalpia (o funzione termica):
W (S, P ) = U(S, V (S, P )) + PV (S, P ); dW = TdS + V dP, (4.9)
dove la seconda equazione vale per trasformazioni reversibili. Vediamo che la variazione diW a pressione costante coincide con il calore scambiato: dW = dU +d(PV ) = dU +PdV .Notiamo che mentre il sistema sta scambiando calore con qualche altro oggetto, perche lapressione del sistema di mantenga costante, su di esso l’ambiente esterno deve esercitareun lavoro P∆V . Il meccanismo e illustrato graficamente in Fig. 4.3. Il volume V (S, P ) edefinito dalla condizione di minimo rispetto a V per U(S, V ) + PV , cioe
P (S, V ) = −∂U(S, V )
∂V.
Attraverso questa condizione di minimo, possiamo vedere l’entalpia come la somma del-l’energia interna del sistema e di uno stantuffo a pressione P che spinge il volume delsistema ad assumere il valore V (S, P ) tale che la pressione interna controbilanci esatta-mente la pressione P dello stantuffo. Una volta che abbiamo a disposizione l’espressioneesplicita dell’entalpia, possiamo esprimere in maniera alternativa il volume in funzione dellapressione:
V (S, P ) =∂W (S, P )
∂P.
Il meccanismo con cui si effettua il cambio di variabili (S, V ) → (S, P ) e una trasformazionedi Legendre. E da notare il ruolo centrale della trasformata di Legendre nel cambio di va-riabili. Una semplice redifinizione della energia come funzione di S e P : U = U(S, V (S, P ))avrebbe fatto sı che la variazione di energia a pressione costante fosse dU = (T −P∂SV )dSche non e uguale al calore trasferito nella trasformazione.
Il secondo potenziale che introduciamo descrive l’attitudine a compiere lavoro a tem-peratura costante ed e detto energia libera, o energia libera di Helmholtz:
F (T, V ) = U(S(T, V ), V )− TS(T, V ); dF = −SdT − PdV (4.10)

72 CAPITOLO 4. TERMODINAMICA
A(S,P)=U(S,V(P))+PV(P)
.
−PV
U(S,V)
V
V=V(P)
U
Figura 4.3: Costruzione geometrica della trasformazione di Legendre per il passaggiodall’energia all’entalpia
e di nuovo la seconda vale solo in trasformazioni reversibili. La variazione di energialibera a temperatura costante dF = dU − d(TS) = −PdV non e altro che il lavorocompiuto sul sistema. In modo analogo all’entalpia, la dipendenza dell’entropia rispettoa (T, V ) e determinata tramite una trasformazione di Legendre di U rispetto a S. Inquesto caso la condizione di minimo e fatta rispetto a S su U(S, T ) − TS, la quale ci daS(T, V ) = ∂SU(S, V ), e poi invertendo, S(T, V ). Quando l’espressione esplicita di F (T, V )sia nota, possiamo utilizzare la seconda nella Eq. (4.10) per determinare S(T, V ):
S(T, V ) =∂F (T, V )
∂T.
Di nuovo, il sistema compie lavoro su qualche altro oggetto, mentre l’ambiente esternoagisce come bagno termico e fornisce il calore necessario per mantenere il sistema isotermo.
Possiamo definire un terzo potenziale termodinamico detto semplicemente potenzialetermodinamico o energia libera di Gibbs, funzione delle sole variabili intensive T e P :
Φ(T, P ) = F (T, V (T, P )) + PV (T, P ) dΦ = −SdT + V dP, (4.11)
dove, analogamente a quanto fatto per entalpia ed energia libera, V (T, P ) e ottenutoda inversione di P (T, V ) = ∂V F (T, V ), e la seconda nella (4.11) vale per trasformazionireversibili. Abbiamo quindi
S(T, P ) = −∂Φ(T, P )∂T
V (T, P ) =∂Φ(T, P )
∂P.
Di nuovo, possiamo interpretare il potenziale termodinamico di Gibbs come l’attitudi-ne a compiere lavoro su un oggetto esterno da parte di un sistema tenuto a pressione etemperatura costante da una sorgente di lavoro e di calore esterna.
Passando al caso delle trasformazioni irreversibili, la diseguaglianza (4.5), si traduce indiseguaglianze analoghe per i vari potenziali termodinamici. In particolare, per l’energia

4.6. LAVORO MASSIMO E POTENZIALI TERMODINAMICI 73
libera abbiamo che, se la temperatura e il volume del sistema rimangono costanti (quindidV = dT = 0):
dQ = dU + PdV ≤ TdS ⇒ dU − d(TS) = dF,≤ 0
cioe l’energia libera diminuisce in un processo irreversibile a temperatura e volume costan-te. In maniera analoga, vediamo che il potenziale termodinamico di Gibbs diminuisce intrasformazioni irreversibili a temperatura e pressioni costanti:
0 > dU + PdV − TdS = dU + d(PV )− d(TS) = dF − d(TS) = dΦ,
Pertanto, l’energia libera e il potenziale termodinamico di Gibbs saranno minimi in condi-zioni di equilibrio termodinamico.
4.6 Lavoro massimo e potenziali termodinamici
Cerchiamo di capire piu in dettaglio come l’energia libera sia in grado di descrivere l’atti-tudine di un sistema non isolato a compiere lavoro (la discussione segue da vicino quellacontenuta alle pagine 76 e 77 del testo di Landau e Lifsits “Meccanica Statistica”).
Ci domandiamo qual’e la capacita di un corpo immerso in un ambiente esterno a com-piere lavoro su un motore, dove il sistema composto dal corpo e dall’ambiente esterno esupposto isolato e il motore e supposto termoisolato. In queste condizioni abbiamo che:
• Cambiamenti ∆V0 di volume dell’ambiente esterno corrispondono a cambiamentiopposti ∆V = −∆V0 del volume del corpo (utilizziamo pedice zero per indicarel’ambiente esterno).
• Il motore non scambia calore con il corpo e con l’ambiente esteno, e scambia lavorosolo con il corpo (non con l’ambiente esterno).
• Il corpo riceve calore solo dall’ambiente esterno.
Tanto per cominciare, calcoliamo il cambio ∆U dell’energia del corpo, che avverrebbe inseguito all’esecuzione di un lavoro P0∆V0 dall’ambiente esterno e uno ∆Rin dal motore, eda un trasferimento di calore ∆Q = −∆Q0 = −T0∆S0 dall’ambiente. Troviamo subito,sfruttuando ∆V = −∆V0 e ∆S +∆S0 ≥ 0:
∆U = ∆Rin −∆Q0 + P0∆V0 ≤ ∆Rin + T0∆S − P0∆V,
cioe
∆Rin ≥ ∆U − T0∆S + P0∆V. (4.12)
Esiste quindi un lavoro minimo ∆Rminin = ∆U − T0∆S + P0∆V che deve essere eseguito
da un motore esterno per produrre un cambio ∆U,∆S,∆V nel corpo. Al contrario, illavoro eseguito dal corpo sul motore ∆Rout = −∆Rin, sara sempre minore di −(∆U −

74 CAPITOLO 4. TERMODINAMICA
T0∆S+P0∆V ). Troviamo quindi il lavoro massimo che il corpo puo eseguire su un motoreesterno in seguito a una trasformazione ∆U,∆S,∆V sara:
∆RMAXout = −(∆U − T0∆S + P0∆V ). (4.13)
Supponendo il corpo in equilibrio interno, possiamo esprimere la variazione di energiainterna attraverso la prima legge della termodinamica ∆U = T∆S − P∆V , e otteniamodalla (4.13):
∆RMAXout = (T0 − T )∆S − (P0 − P )∆V,
che sottolinea come in questo caso il lavoro ∆R origina da una condizione di non equilibriodel corpo rispetto all’ambiente circostante.
In generale, possono essere presenti processi irreversibili all’interno del corpo (ad esem-pio reazioni chimiche) che contribuiscono al lavoro ∆Rout. In tal caso il corpo puo eseguirelavoro anche se T = T0 e P = P0. Due situazioni sono di particolare interesse.
Se il corpo compie lavoro a temperatura e volume costante, e T = T0, vediamo che∆RMAX
out e dato dalla variazione della energia libera di Helmholtz del sistema. Combinandoinfatti la (4.10) con le condizioni ∆V = 0 e T = T0 = costante, e sostituendo nella (4.13),troviamo infatti:
∆Rout ≤ −(∆U − T0∆S + P0∆V ) = −∆(U − TS) = −∆F. (4.14)
In maniera analoga, se il corpo esegue lavoro a pressione e temperatura ambiente (quindi,ovviamente, ∆S,∆V 6= 0, ma P e T sono costanti), possiamo scrivere dalla (4.11):
∆Rout ≤ −(∆U − T0∆S + P0∆V ) = −∆(U − TS + PV ) = −∆Φ. (4.15)
Il lavoro massimo e dato in questo caso dalla variazione di potenziale di Gibbs.
4.7 Sistemi a numero di particelle variabile
Vi sono diverse situazioni in cui il numero di particelle puo non essere costante. Due esempiimportanti:
• Reazioni chimiche, che causano trasmutazioni di molecole da un tipo all’altro
• Transizione di fase, in cui molecole si trasferiscono da una fase ad un’altra.
In entrambi i casi, la dipendenza dell’energia interna dai numeri di molecole delle differentispecie diventa importante:
U = U(S, V, Ni); dU = TdS − PdV +∑
i
µidNi (4.16)

4.7. SISTEMI A NUMERO DI PARTICELLE VARIABILE 75
(trascuriamo d’ora in avanti contributi a dS provenienti da moti interni nel sistema, cosıche possiamo esprimere dU in funzione di S, V ed N attraverso una equazione, non unadisequazione). Abbiamo introdotto una nuova variabile intensiva
µi = µi(s, v, ci) =∂U(S, V, Ni)
∂Ni
s = S/N, v = V/N, ci = Ni/N, (4.17)
detta potenziale chimico, che giochera all’equilibrio nei confronti delle variabili Ni, lo stes-so ruolo giocato dalla temperatura nei confronti dell’entropia di corpi in contatto fra lo-ro. Analogamente, dovremo tenere conto della dipendenza dalle concentrazioni ci nellapressione P = P (s, v; ci) e nella temperatura T = T (s, v; ci).
La dipendenza dalle concentrazioni e importante nel caso di una miscela di gas e diventaessenziale per descrivere l’equilibrio chimico. Nel caso di transizioni di fase, invece, questadipendenza e ovviamente assente: la concentrazione di molecole in fase liquida la dove ilcorpo e in fase gassosa, e evidentemente nulla, e viceversa nella parte in fase liquida.
Possiamo generalizzare le espressioni dei potenziali termodinamici al caso in cui ilnumero di particelle puo variare. Abbiamo in particolare per l’energia libera di Gibbs(4.11):
Φ = Φ(T, P, Ni); dΦ = −SdT + V dP +∑
i
µidNi, (4.18)
dove ora
µi = µi(T, P, ci) =∂Φ(T, P, Ni)
∂Ni
. (4.19)
Prima di concentrarci sulle condizioni di equilibrio chimico (o delle fasi) del sistema, con-centriamoci un attimo sull’energetica del processo. Continuiamo a considerare il caso diun gas ideale monoatomico, o meglio, di una miscela di gas ideali monoatomici. Indicandocon ǫi la componente di energia media delle molecole della specie i, non associata a mototermico, possiamo scrivere l’energia interna della miscela nella forma
U(T, V, Ni) =3
2NT +
∑
i
Niǫi(T ). (4.20)
Considereremo in seguito per semplicita l’energia interna delle molecole indipendente dallatemperatura, ma e evidente che si tratta di un’approssimazione (in generale, gli urti mo-lecolari possono portare a immagazzinare energia in gradi di liberta interna, ad esempiovibrazionali).
Vediamo allora che una trasformazione che porta a un’aumento di molecole con energiaǫi bassa, a scapito di molecole con energia alta implichera una diminuzione della com-ponente
∑
iNiǫi(T ) di U . Se il sistema e isolato, perche U rimanga invariato, e quindinecessario che aumenti la quota di energia da moto termico, del sistema. Abbiamo quindiun esempio di reazione esotermica: l’energia dei legami chimici si trasforma in moto

76 CAPITOLO 4. TERMODINAMICA
termico. Per contro, una reazione in cui si ha un aumento di molecole con alto contenutodi ǫi richiedera una diminuzione della temperatura del sistema isolato e si parla in questocaso di una reazione endotermica.
Attraverso l’aumento del moto termico, una reazione esotermica porta a un contributopositivo all’entropia del sistema. La crescita di entropia in una reazione esotermica sugge-rirebbe che una reazione esotermica in una miscela di gas ideali dovrebbe procedere sinoall’esaurimento dei reagenti. Di fatto le cose non stanno cosı ne in un sistema isolato ne inuno in contatto con un ambiente esterno. Il fatto e che un contributo all’entropia viene pro-dotto anche dalla concentrazione delle varie specie nella miscela (il termine
∑
iNi lnV/Ni
nell’espressione dell’entropia di una miscela).Uno stesso fenomeno si verifica nel caso di una transizione di fase da gas a liquido:
l’energia delle molecole nella fase liquida e minore di quella nella fase gassosa. Quindila trasformazione da gas a liquido rilascia una quantita di calore positiva (il cosı dettocalore latente). Nel sistema isolato, il riscaldamento produce un contributo positivoall’entropia; cio nonostante, la transizione si verifica solo se e favorita nel bilancio globaledi entropia, che tiene conto di nuovo di termini di densita volumetrica nelle diverse fasi.Va quindi determinato il massimo della entropia S(U, V, Ni) per U e V costanti. Inmaniera analoga, nei casi in cui il sistema non e isolato ed e mantenuto in condizioni diT e V costanti, oppure di T e P costanti, va determinato il minimo dell’energia liberacorrispondente, rispetto a variazioni dei numeri di molecole Ni.
Ci limiteremo a considerare il caso interessante di reazioni chimiche e transizioni di faseche si svolgono a temperatura e pressioni costanti. Per far cio, sara necessario calcolareprima il potenziale chimico di una miscela di gas.
4.8 Entropia e potenziali termodinamici in una mi-
scela di gas
Come nel calcolo dell’energia di una miscela di gas, anche nel calcolo dell’entropia e ne-cessario tenere conto di possibili contributi “interni” delle molecole. Nel caso di gas ideali,questi contributi sono dovuti precisamente ai gradi di liberta interni di queste molecole.Per esempio, se le molecole di una specie potessero trovarsi in tre stati equiprobabili, indi-pendentemente dall’energia, avremmo un contributo addizionale di entropia per ciascunamolecola
∆ζ −3
∑
α=1
P (α) lnP (α) = ln 3,
rispetto a molecole in cui un solo stato e possibile. Quindi, in una transizione in cui ∆Nmolecole con un solo stato possibile si trasformano in altrettante molecole con tre statipossibili, si avrebbe un incremento di entropia nella miscela ∆S = ∆N∆ζ = ∆N ln 3.Come nel caso dell’energia interna, la componente di entropia interna per molecola sarauna funzione della temperatura (spesso, alte energie sono associate a un incremento nelnumero di stati accessibili da parte della molecola). Per semplicita, ci limiteremo pero

4.8. ENTROPIA E POTENZIALI TERMODINAMICI IN UNA MISCELA DI GAS 77
a considerare il caso in cui ζ e indipendente dalla temperatura. Il parametro ζi e dettocostante chimica della specie i-esima.
A questo punto, l’entropia per una miscela di gas monoatomici avra la forma, chegeneralizza la (3.37)
S(T, V, Ni) =3
2NT + (
∑
i
Ni lnV/Ni + ζi)
=3
2NT +N ln(V/N)−N
∑
i
ci(ln ci − ζi), (4.21)
dove ricordo che ci = Ni/N . Utilizzando la (4.10), otteniamo subito l’energia libera diHelmholtz:
F (T, V, Ni) =3
2NT +N
∑
i
ciǫi − TS(T, V, Ni)
=3
2NT (1− lnT )−NT ln(V/N) +
∑
i
Ni[ǫi + T (ln ci − ζi)]. (4.22)
Per ultimo, calcoliamo l’energia libera di Gibbs Φ = F + PV :
Φ(T, P, Ni) = F (T,NT/P, Ni) +NT
=5
2NT (1− lnT ) +NT lnP +
∑
i
Ni[ǫi + T (ln ci − ζi)], (4.23)
dove si e sfruttata la legge di stato PV = NT . A questo punto possiamo calcolare ilpotenziale chimico della specie i-esima e troviamo, dalla (4.19):
µi =∂Φ
∂Ni
=5
2T (1− lnT ) + T lnP + ǫi + T (ln ci − ζi). (4.24)
Notiamo che possiamo scrivere la (4.24) nella forma alternativa
µi =∂Φ
∂Ni
=5
2T (1− lnT ) + T lnPi + ǫi − Tζi. (4.25)
dove si e introdotta la pressione parziale Pi = ciP ≡ NiT . L’espressione del potenzialechimico per la componente i di una miscela di gas ideali, e per la stessa componente allostato puro, differiscono quindi solo per la sostituzione Pi → P . (Ricordiamo la legge distato per una miscela di gas, la cosı detta legge di Dalton:
∑
i PiV = NT ). Combinandole (4.23) e la (4.24), vediamo infine che possiamo scrivere
Φ(T, P, Ni) =∑
i
Niµi(T, P, ci). (4.26)

78 CAPITOLO 4. TERMODINAMICA
Vediamo quindi che il potenziale chimico della specie i della miscela e l’energia libera diGibbs per molecola della specie in questione. Notiamo inoltre, differenziando la (4.26), chepossiamo ottenere per le entropie e i volumi specifici delle molecole
si = −∂µi∂T
=5
2lnT − lnP + ζi + ln ci; vi =
∂µi∂P
=T
P. (4.27)
Da qui ritroviamo µi = ui − Tsi + Pvi, con ui = (3/2)T + ǫi l’energia media di unamolecola, che riproduce a livello molecolare la definizione di energia libera di Gibbs: Φ =U − TS + PV .
4.9 Reazioni chimiche in una miscela di gas
Immaginiamo di avere una miscela di specie chimiche gassose Ai, in cui le variazioni nelnumero di molecole di ciascuna specie sono fissate da rapporti stechiometrici:
νij =∆Ni
∆Nj
.
Per esempio, nel caso della reazione 2F2 +O2 → 2F2O, abbiamo i rapporti
νF2,O2 = 2, νF2O,O2 = −2.
In presenza di una singola reazione chimica, e quindi sufficiente dare la variazione di unaqualsiasi delle specie chimiche, per determinare le variazioni di tutte le altre. Prendendoper riferimento la specie 1, e scrivendo νi ≡ νi1, avremo quindi
∆Ni = νi∆N1. (4.28)
La condizione di equilibrio chimico a date condizioni di pressione e temperatura sara quindiottenuto dalla equazione
dΦ
dN1
=∑
i
dNi
dN1
∂Φ
∂Ni
=∑
i
νiµi = 0,
dove si sono sfruttate la (4.28) e la (4.18). Questa equazione ci permette di determinare ilrapporto fra le concentrazioni delle specie all’equilibrio.
Proviamo a fare questo calcolo nel caso piu semplice possibile di una reazione A1 → A2.In questo caso abbiamo ν1 = 1, ν2 = −1 e la condizione di equilibrio e semplicementeµ1 = µ2. Sostituendo l’espressione del potenziale chimico ottenuta nella sezione precedente,(4.24), troviamo subito
µ1 − µ2 = ǫ1 − ǫ2 + T (ln(c1/c2)− ζ1 + ζ2) = 0,
da cui otteniamo
c1c2
= exp(
ζ1 − ζ2 −ǫ1 − ǫ2T
)
. (4.29)

4.10. TRANSIZIONI DI FASE 79
Notiamo subito l’analogia con la distribuzione di Maxwell-Boltzmann: le concentrazionisono pesate da un fattore esponenziale nel rapporto fra energia molecolare e T . Notiamopero la presenza del contributo entropico associato alle costanti chimiche. Vediamo inparticolare che una reazione sfavorita da un’energia del prodotto di reazione piu alta diquella del reagente, puo essere lo stesso vantaggiosa dal punto di vista entropico se ζ2 > ζ1,e la reazione puo lo stesso portare all’esaurimento in pratica dei reagenti.
4.9.1 Entalpia di reazione
Il calore di reazione, che nelle condizioni isobare considerate, e di fatto un’entalpia direazione, puo essere a questo punto calcolato sfruttando la relazione (non indichiamo ladipendenza da Ni per non appesantire la formula):
W (S(T, P ), P ) = Φ(P, T ) + TS(T, P ) = Φ(P, T )− T∂Φ(T, P )
∂T= −T 2 ∂
∂T
Φ(P, T )
T,
dove ci si e serviti delle (4.9) e della (4.11). La variazione di entalpia che si avra nellatrasformazione di ∆N2 = −∆N1 > 0 molecole dalla specie A1 a quella A2 sara, per∆N2 ≪ N :
∆W ≃ ∆N1dW
dN1
= ∆N2T2 ∂
∂T
( 1
T
dΦ
dN1
)
= ∆N2T2 ∂
∂T
µ1 − µ2
T.
Dalla espressione per il potenziale chimico (4.24), ottenuta nella sezione precedente, tro-viamo pero
µ1 − µ2
T=ǫ1 − ǫ2T
+ ln(c1/c2)− ζ1 + ζ2
e quindi∆W = ∆N2(ǫ2 − ǫ1).
Vediamo che la variazione di entalpia e positiva se ǫ2 > ǫ1, cioe se la reazione e endotermica.Questo e corretto, perche ∆W e precisamente il calore che dovrebbe essere fornito (apressione costante) dall’ambiente esterno alla miscela di gas per mantenere costante latemperatura durante la reazione. Questo deve essere evidentemente positivo se la reazioneassorbe energia. Notiamo come in questo caso, rimanendo il numero totale di molecoleinvariato durante il processo, per mantenere la pressione costante nel sistema, e sufficientemantenerlo isotermo e a volume costante. In generale, il numero totale di molecole varierae il sistema dovra essere lasciato libero di espandersi o comprimersi (si veda l’esercizio 5).
4.10 Transizioni di fase
In questo caso, abbiamo per definizione che le molecole appartenenti a fasi differenti nonsono mescolate. Il valore di G in una regione del sistema in cui c’e solo una fase saraquindi dato dal potenziale chimico di quella fase, moltiplicato per il numero di molecole

80 CAPITOLO 4. TERMODINAMICA
nella regione. Il valore totale di G di tutto il sistema, essendo G estensiva, sara datodalla somma di G in tutte queste regioni. Troviamo pertanto dalla (4.26), supponendo lapresenza di due sole fasi (ad esempio liquido e gassoso):
Φ(P, T,N1, N2) = Nlµl(T, P ) +Nvµv(T, P ) (4.30)
e la condizione di equilibrio delle fasi e semplicemente
µl(T, P ) = µl(T, P ).
Il fatto che i potenziali chimici sono indipendenti dalle concentrazioni, fa sı che perche Graggiunga un minimo, se P e T sono fissati in modo che µ1 6= µ2, la fase con il valoremaggiore di µ deve scomparire (vedi Fig. 4.4a).
In condizioni di quasi equilibrio, la transizione di fase si verifichera quando la tempera-tura raggiungera il valore T (P ) (o alternativamente quando la pressione raggiunge il valoreP = P (T )), tale che µl(T, P ) = µl(T, P ). Nel caso di transizioni di fase di prima specie(vedi nota sotto), le due fasi del sistema sono caratterizzate, a temperatura e pressionefissate, da valori differenti dei due parametri estensivi volume ed entropia. Nel caso diuna transizione liquido vapore, la differenza di entropia tra le due fasi, moltiplicato perla temperatura a cui si effettua la trasformazione, fornisce l’entalpia di evaporazione delliquido. In analogia con quanto fatto nella sezione precedente, nel caso dell’entalpia direazione, possiamo calcolare l’entalpia di evaporazione a partire dalla relazione, al puntodi transizione µl = µv:
∆W = [µl − µv + (sl − sv)T ]∆Nl = (sv − sl)T∆Nv, (4.31)
dove sl,v = Sl,v/Nl,v indicano le entropie specifiche per una molecola nella fase liquida ein quella di vapore. Come ∆Nv = −∆Nl > 0 molecole passano nella fase di vapore, unaentalpia T (sv − sl)∆Nv viene fornita al sistema. La variazione di entalpia L = T (sv − sl)identifica il cosı detto calore latente di evaporazione per una molecola.
Nota. Le transizione di fase sono classificate di prima o di seconda specie a secondache il calore latente sia differente da zero o meno. Nel caso di transizioni di fase diseconda specie, la transizione e segnalata solo da una differenza di calore specifico tra lefasi. Durante una transizione di fase di prima specie, per esempio in una pentola d’acquain ebollizione, i passaggi della sostanza da una fase ad un’altra (la formazione delle bolle)necessitano un afflusso di calore dall’esterno. La presenza di zone di liquido e di vaporedistribuite casualmente a scala macroscopica, e quindi associata a una situazione di nonequilibrio (la pentola e sul fuoco). Nel caso di una transizione di fase di seconda specie (peresempio un magnete alla temperatura critica), simili fluttuazioni (la magnetizzazione delcorpo varia nello spazio e nel tempo in maniera casuale) si producono invece senza bisognodi sorgenti di calore esterne. Si ha quindi un sistema in equilibrio termodinamico per cuiuna delle condizioni che abbiamo fornito per una trattazione termodinamica vengono meno:quella che non ci siano fluttuazioni macroscopiche all’equilibrio. E possibile vedere che lacondizione che viene meno, e quella sulla presenza di una scala microscopica al disopra

4.10. TRANSIZIONI DI FASE 81
µ
T
P
Tc
v
a bPv(T)
P
µ
v
l
µ
Figura 4.4: Equilibrio delle fasi nel piano µP (a) e nel piano PT (b). Grafico a: perP < Pv, µv < µl, e solo la fase gassosa e possibile. Viceversa per P > Pv. All’equilibrio,avremo quindi Φ = Nµl,v a destra e a sinistra di Pv, rispettivamente. La discontinuitanella derivata e la variazione di volume nel cambio di fase N(vl− sv), illustrata in Fig. 4.5.Grafico b: il punto Tc indica la temperatura critica.
della quale parti separate del sistema si possono considerare scorrelate. In questo sistema,si puo vedere che questa scala non esiste e tutto le parti del sistema sono correlate fra diloro.
La condizione di equilibrio delle fasi µl(T, P ) = µv(T, P ) definisce una curva Pv =Pv(T ). Nel caso di una transizione liquido – vapore, la pressione Pv = Pv(T ) per cui lefasi sono all’equilibrio alla temperatura data, e detta pressione di vapore. (Nel casodell’acqua, la pressione di vapore corrispondente a 100oC e di una atmosfera). Il fatto chela condizione di equilibrio µl = µv sia indipendente dai numeri di molecole nelle singole fasi,fa sı che le fasi possono coesistere in rapporti di massa N1/N2 arbitrari. Questo corrispondeal quadro fornito in Fig. 4.5. Le porzioni orizzontali ad altezza Pk individuano differentipunti nella curva di equilibrio P = P (T ), e i corrispondenti cambi di volume isobarici sonoassociati al passaggio da una pura fase gassosa (estremo destro) a una pura fase liquida(estremo sinistro).
Possiamo determinare l’andamento della pressione di vapore differenziando la relazionedi equilibrio µl = µv rispetto a T e P . Sfruttando il fatto che µ1,2(T, P ) sono i potenzialitermodinamici specifici di una molecola nelle diverse fasi, possiamo scrivere, dalla (4.30):
dµ1,2(T, P ) =1
N1,2
dΦ1,2(T, P ) = −s1,2dT + v1,2dP, (4.32)
dove v1,2 = V1,2/N1,2 sono i volumi specifici delle molecole nelle due fasi. La condizione dispostarsi sulla curva di equilibrio sara dµl(T, P ) = dµv(T, P ), la quale ci da, sostituendoin dµl − dµv = 0:
dPvdT
=sv − slvv − vl
=L
(vv − vl)T.

82 CAPITOLO 4. TERMODINAMICA
T
P
3
P
T1
T2V
P
Pv2
v1
v3
Figura 4.5: Isoterme in presenza di una transizioni di fase liquido-gas, con indicazione dellerispettive pressioni di vapore
Per pressioni di tipo atmosferico, abbiamo vv ≫ vl e possiamo esprimere vv in funzione ditemperatura e pressione utilizzando la legge di stato PV = NT :
vv − vl ≃ vv =T
Pv.
Sostituendo nella espressione per dPv/dT , otteniamo la seguente equazione di Clausius-Clapeyron:
dPvdT
=LPvT 2
. (4.33)
L’equazione di Clapeyron puo essere risolta analiticamente, e, per piccole variazioni ditemperatura, ci da una dipendenza esponenziale della pressione di vapore dalla temperaturastessa:
Pv(T ) ≃ Pv(T0) exp(L(T − T0)
T 20
)
. (4.34)
Nota. Come illustrato in Fig. 4.4b e Fig. 4.5, la transizione di fase scompare al di sopradi una certa temperatura Tc, detta temperatura critica. Nel caso dell’acqua, Tc ≃ 374oC.In corrispondenza di Tc le differenze vv − vl e sv − sl vanno a zero e cessa di essercidifferenza tra le due fasi (piu precisamente, la transizione di fase diventa di secondo ordine).Avvicinandosi a Tc, l’approssimazione vv ≫ vl cessa di essere applicable e l’equazione diClausius-Clapeyron smette di essere valida.

4.10. TRANSIZIONI DI FASE 83
4.10.1 Termodinamica dell’aria umida
Le considerazioni che abbiamo effettuato sin’ora possono essere sfruttate per determinare lecondizioni di equilibrio tra una miscela di vapore acqueo e aria, e acqua allo stato liquido.Si parla in questo caso di una condizione di vapore saturo, che corrisponde infatti almassimo contenuto di vapore acqueo nell’aria alla temperatura data.
Consideriamo una condizione in cui il sistema e a pressione fissata e utilizziamo comeparametro libero il contenuto di umidita dell’aria cv = Nv/(Nv + Nd) ≃ Nv/Nd, dove Nv
e Nd sono rispettivamente i numeri di molecole di vapor acqueo ed aria secca. Possiamoin maniera equivalente considerare come parametro libero la pressione parziale di vaporePv = (Nv/Nd)P a pressione totale P costante. La condizione equilibrio delle fasi sara dinuovo
∂Φ
∂Nv
= µv − µl = 0,
dove ora Φ = Nlµl + Nvµv + Ndµd, ma la componente secca Ndµd evidentemente noncontribuisce. Esattamente come fatto per il calcolo della pressione di vapore, possiamocercare una equazione differenziale per la concentrazione di vapore all’equilibrio csatv (T ),differenziando la condizione µl − µv = 0 rispetto a T e Nv. In alternativa, sfruttando larelazione Pv = cvP , possiamo differenziare rispetto a T e Pv, che ci riporta precisamentealla (4.32):
0 =∂(µv − µl)
∂Nv
dNv +∂(µv − µl)
∂TdT =
∂µv∂Nv
dNv +LdTT
. (4.35)
[si e ipotizzato che la fase liquida sia pura, cosı che µl non contenga termini dipendenti daNi; confrontare con la (4.24)]. Sfruttando la (4.27), abbiamo d’altra parte
∂µv(T, P, cv)
∂Nv
dNv =∂µv(T, Pv)
∂PvdPv = vvdV,
dove, dalla (4.25), vv = T/Pv. Sostituendo nella Eq. (4.35), otteniamo la seguente variantedella equazione di Clausius-Clapeyron
dP satv
dT=
LP satv
T 2.
In altre parole, si ha saturazione quando la pressione parziale del vapore nell’aria Pv diventauguale alla pressione di vapore alla temperatura data Pv(T ). Da qui troviamo per l’umiditaalla saturazione
dcsatv
dT=
Lcsatv
T 2, (4.36)
e quindi, sfruttando la (4.34), crescita esponenziale con la temperatura, della umidita allasaturazione. Il valore di temperatura a cui la l’umidita di saturazione diventa uguale alcontenuto di umidita presente nell’aria e detto punto di rugiada.

84 CAPITOLO 4. TERMODINAMICA
4.11 Esercizi
Esercizio 1 Calcolare il lavoro eseguito da un gas che si espande isotermicamente davolume V a volume 2V . E possibile che durante l’espansione, il gas sia contenuto in unrecipiente isolato termicamente? Argomentare la risposta. Se no, il gas ha assorbito oceduto calore nel processo? Sempre se no, suggerire un approccio per il calcolo del calorescambiato.
Esercizio 2 L’energia interna di un certo sistema e
U(S, V,N) = CS3/(NV ) (4.37)
• Verificare che la (4.37) descrive in effetti un sistema termodinamico e che la pressionee la temperatura sono grandezze intensive.
• Trovare la legge di stato di questo sistema.
• Scrivere la legge adiabatica per questo sistema.
• Determinare i calori specifici a volume e a pressione costante.
• Trovare le espressioni dell’entalpia e delle energie libere di Helmholtz e di Gibbs diquesto sistema.
Esercizio 3 Abbiamo due gas ideali monoatomici identici, con identica temperatura, madiverse pressioni e numero di particelle. Supporre che i due recipienti che contengonoi gas vengano messi in comunicazione, cosı che le pressioni dei due gas si equilibrano.Determinare la variazione di entropia del sistema.
Esercizio 4 Un gas e racchiuso in un cilindro sigillato dalle pareti termoconduttrici, postoin un bagno termico a temperatura T . All’interno del cilindro e presente uno stantuffoa tenuta, bloccato da un fermo; siano Va,b, Na,b, Pa,b, i volumi, i numeri di molecole e lepressioni del gas ai due lati dello stantuffo. Calcolare la variazione di energia libera delsistema dopo che viene tolto il blocco allo stantuffo.
Esercizio 5 Una miscela di gas ideali A, B e C e caratterizzato inizialmente da valoricA = NA/N , cB = NB/N e cC = NC/N , N = NA + NB + NC delle concentrazioni.Supporre che le costanti chimiche ζi e le energie interne ǫi siano indipendenti da pressionee temperatura. I tre gas possono reagire chimicamente secondo il rapporto stechiometricoA+2B ↔ C. Supponendo che le reazioni si verifichino a temperatura e pressione costante,e che inizialmente cB = 2cA; cC = 0, calcolare le concentrazioni all’equilibrio cA, cB e cC ediscuterne la dipendenza da P, T e dalle costanti ζi ed ǫi.
Esercizio 6 Un certo liquido bolle a temperatura 95 oC in cima a una collina e a 105 oCalla sua base. Il calore latente e pari a 1000 cal/mole. Quanto e alta la collina?

Capitolo 5
Meccanica statistica dell’equilibrio
5.1 Il punto di vista di Gibbs
Nella trattazione di un sistema termodinamico abbiamo considerato essenzialmente dueapprocci:
• Approccio cinetico. Si cerca di derivare una equazione di evoluzione per la statisticamicroscopica del sistema. Quasi sempre si ipotizza che le componenti microscopichedel sistema (per esempio le molecole di un gas) siano in prima approssimazione scor-relate. Il risultato finale sono tipicamente equazioni di evoluzione per la distribuzionedegli stati della singola componente. Da queste equazioni, possono essere derivate inseguito tutte le piu importanti relazioni termodinamiche per il sistema.
• Approccio termodinamico. Si tralascia ogni informazione sulle caratteristiche micro-scopiche del sistema e ci si limita a postulare l’esistenza di una funzione di statoentropia e di una variabile intensiva temperatura che descrivono le proprieta di equi-librio termodinamico del sistema. Al contrario che nell’approccio cinetico, le leggi distato del sistema non vengono derivate e sono supposte date.
Vi e un terzo approccio, che e quello della meccanica statistica dell’equilibrio, o meccanicastatistica di Gibbs. In questo caso, come nell’approccio cinetico, si parte da un punto divista microscopico; viene pero saltata accuratamente ogni considerazione su come il sistemaraggiunge l’equilibrio. Quello che ci si limita a fare e postulare l’esistenza di un equilibriostatistico. Da questa condizione, unitamente ad alcune ipotesi importanti sulla strutturadella PDF di equilibrio ρ (la PDF nel Γ-spazio; tralasciamo d’ora in poi il pedice N peralleggerire la notazione) e sul tipo di partizione utilizzata nella definizione di entropia,viene poi derivata tutta la termodinamica all’equilibrio.
I vantaggi di quest’ultimo approccio risiedono nella sua potenza nel trattare sistemi condinamica complicata o in presenza di regimi correlati. Un esempio importante sono i feno-meni critici (magneti alla temperatura critica, teorie di campo). La meccanica statisticae in particolare in grado di trattare sistemi in cui gli effetti quantistici sono importanti.
85

86 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
Questa potenza va pero a scapito dell’ambito di applicabilita del metodo, che e ristretto asistemi la cui dinamica microscopica e di tipo Hamiltoniano.
Consideriamo un sistema Hamiltoniano isolato con N gradi di liberta. Un tale sistemasara descritto da 2N coordinate canoniche qi, pi; i = 1, . . . N (nel caso di un gas, perla particella i-esima avremmo tre coordinate qi1, qi2, qi3 ≡ xi1, xi2, xi3 e tre momenticoniugati pi1, pi2, pi3 ≡ mvi1, vi2, vi3, cioe 3N gradi di liberta per 6N coordinate cano-niche). La coordinate canoniche qi, pi; i = 1, . . . N definiscono il nostro Γ-spazio e su diesso definiamo la PDF ρ = ρ(qi, pi; t).
Il fatto che il sistema isolato abbia una dinamica di tipo Hamiltoniano ha alcuneimportanti conseguenze:
• L’energia e conservata. Quindi, se inizialmente l’energia del sistema e U , la PDF ρavra la forma ρ(qi, pi; t) ∝ δ(H(qi, pi) − U), dove H(qi, pi) e l’Hamiltonianadel sistema. La condizione H(qi, pi) = U individua la superficie a energia costantelungo la quale evolvono le traiettorie del sistema isolato.
• Il volume nello spazio delle fasi e conservato (teorema di Liouville). Questo haper conseguenza il fatto che una distribuzione ρ uniforme e stazionaria.
• Siccome le traiettorie evolvono sulle varieta a energia costante, individuate dallarelazione H(qi, pi) = U per diversi valori di U , anche una PDF uniforme in guscicompresi tra superfici con valori diversi di U sara stazionaria. Prendendo il limite digusci infinitamente sottili, otteniamo il risultato importante, valido per un sistemaisolato, che una PDF dipendente solo dalla energia del sistema sara stazionaria.
Ora, un sistema isolato avra sempre un’energia ben definita. Ha quindi senso considerareuna PDF che e non nulla solo in uno di questi gusci a energia fissata:
ρM(qi, pi) =
∆Γ−1, H(qi, pi) ∈ [U,U +∆U ]
0 altrimenti(5.1)
dove ∆Γ = ∆Γ(U) e il volume della regione di Γ-spazio compresa fra energie U e U+∆U e∆U e una scala energetica di coarse-graining arbitraria. La PDF indicata qui sopra gioca unruolo fondamentale nella meccanica statistica ed e detta distribuzione microcanonica.
• Si ipotizza di fatto che la distribuzione microcanonica sia la distribuzione di equilibrioper il sistema isolato, e che valga una proprieta ergodica per cui medie di ensembledescrivano il limite a tempi lunghi di medie temporali.
Come vedremo nella prossima sezione, la giustificazione di questa ipotesi non e banale. No-tiamo comunque che la distribuzione microcanonica corrisponde al massimo della entropiadi Shannon per ρ, fissato U . L’intuizione fisica ci suggerisce che la dinamica microscopi-ca sia in qualche senso caotica il che implica, in buona sostanza, crescita della entropia.(Notiamo come le condizioni che portano ad aspettarci crescita della entropia di Shannon

5.1. IL PUNTO DI VISTA DI GIBBS 87
−〈ln ρ〉 sono le stesse che ci portano ad aspettare crescita di entropie con significato ma-croscopico piu diretto, quali ad esempio l’entropia di Boltzmann Sf ). Allo stesso tempo,non ci aspettiamo patologie del tipo descritto alla fine della Sez. 2.7, e cioe presenza diregioni di Γ-spazio aventi lo stesso U , ma inaccessibili l’un l’altra. Questo risulta sufficiente(anche troppo!) a garantire che ρM sia la distribuzione di equilibrio per il sistema e chevalga la proprieta ergodica.
Nota. Diamo qui la dimostrazione che una distribuzione uniforme su gusci a U costantee stazionaria. Consideriamo una descrizione pienamente microscopica del sistema. Inquesta maniera, il dettaglio degli urti e completamente risolto e le variabili qi, pi; i =1, . . . N evolvono nel tempo in maniera continua. In queste condizioni, l’evoluzione dellaρ e descritta da una equazione di continuita, detta equazione di Liouville:
∂ρ
∂t+∇qi,pi · (ρVqi,pi) = 0, (5.2)
dove ∇qi,pi e Vqi,pi sono rispettivamente il gradiente e il campo di velocita nel Γ-spazio:
∇qi,pi ≡ (∂q1 , . . . ∂qN , ∂p1 , . . . ∂pN )
Vqi,pi ≡ (q1, . . . qN , p1, . . . pN)
Allo stato stazionario, l’equazione di Liouville (5.2) puo essere scritta nella forma
Vqi,pi · ∇qi,piρ = −ρ∇qi,pi ·Vqi,pi,
che ammettera una soluzione stazionaria uniforme nel Γ-spazio, a patto che il flusso nelΓ-spazio sia incomprimibile: ∇qi,pi ·Vqi,pi = 0. La condizione di incomprimibilita (cioedi conservazione del volume) nello spazio delle fasi e soddisfatta grazie alle equazioni diHamilton
qi =∂H∂pi
; pi = −∂H∂qi
;
da cui ottengo
∇qi,pi ·Vqi,pi =∑
i
(∂qi∂qi
+∂pi∂pi
)
=∑
i
( ∂
∂qi
∂H∂pi
− ∂
∂pi
∂H∂qi
)
= 0.
5.1.1 Il concetto di ensemble
L’identificazione della distribuzione microcanonica ρM con la distribuzione di equilibrio delsistema anche se intuitivamente ragionevole presenta una serie di problemi concettuali. Nediamo una rapida panoramica.
Una prima osservazione che e necessario fare riguarda la “misurabilita” di una PDFdel Γ-spazio. Una PDF nello spazio delle fasi (x,v) per una molecola puo essere ottenutacontando il numero di molecole che si trovano istantaneamente in una data celletta di

88 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
quello spazio. Ciascuna molecola e quindi un campione e la PDF e ottenuta a partire dallimite di una frequenza (ovvero media campionaria della funzione indicatrice della celletta).Alternativamente, possiamo focalizzarci su una molecola e calcolare la PDF a partire dauna media temporale, cioe come frazione di tempo in cui la molecola in questione si trovanella cella data (media temporale della funzione indicatrice della celletta).
Nel caso del Γ-spazio, le procedure descritte qui sopra diventano fisicamente irrealizza-bili. Ciascun punto nel Γ-spazio non corrisponde infatti alla coordinata e velocita di unamolecola, bensı allo stato dell’intero sistema isolato. I campioni che entrano nel calcolodella ρ a partire da media campionaria sono quindi stati microscopici del sistema. Iden-ticamente, il calcolo di una media temporale andrebbe fatto a priori osservando lo statodell’intero sistema.
L’insieme dei punti di fase corrispondenti ai diversi stati del sistema che entrano nelcalcolo delle medie campionarie vengono chiamate un ensemble. Per esempio, un insiemedi punti distribuiti come ρM e detto ensemble microcanonico.
Il fatto che entrambe le medie su Γ-spazio sono fisicamente irrealizzabili, diventa evi-dente contando le cellette che sarebbe necessario considerare per avere un campionamentominimo dello spazio in questione. Supponendo di dividere in due in ciascuna direzione delΓ-spazio (per dire, molecola 1 puo stare a sinistra o a destra, con velocita positiva o nega-tiva, e cosı via in ogni direzione dello spazio fisico e per ogni molecola), ci ritroveremmogia cosı un numero di cellette pari a 2N , dove N e dell’ordine del numero di Avogadro:N ∼ 1023.
Questo e il numero minimo di realizzazioni che sarebbero necessarie per campionaredecentemente il nostro Γ-spazio. Una determinazione alternativa tramite media temporalesoffre delle stesse limitazioni. 1 In questo caso, abbiamo un solo punto di fase (il nostrosistema) che deve esplorare 2N cellette e il tempo necessario cresce quindi come 2N∆t,dove ∆t e il tempo di permanenza in ciascuna celletta. Ora il tempo ∆t puo essere al piudell’ordine del tempo di collisione diviso per il numero di molecole (in un tempo di collisioneτcoll ciascuna molecola del sistema fara tipicamente una collisione, quindi ci saranno Ncollisioni nel sistema e il tempo fra ciascuna collisione sara ∼ τcoll/N). Un sistema, peresplorare tutto il Γ spazio, richiederebbe pertanto un tempo molto superiore alla durata divita dell’universo.
Di fatto, dai discorsi fatti a suo tempo sulla indistinguibilita delle particelle, nella Sez.3.2.1, ci rendiamo subito conto che il numero di cellette e comunque sovrastimato: un grannumero di esse (di fatto la maggioranza) corrisponde a scambi di particelle identiche. Inaltre parole, e assolutamente irrilevante se la distribuzione rilassa o meno all’interno divolumi di Γ composti da celle che differiscono esclusivamente per scambio di particelle.Eliminando suddivisioni di Γ che portano a celle che differiscono da permutazioni, rima-niamo con un numero di celle che e una frazione 1/N ! di quella iniziale e i problemi sullainterpretazione statistica della medie su Γ sono ridotti.
1Ricordiamo che un solo punto di fase esplora, lungo la sua traiettoria, tutte le possibili e piu impro-babili configurazioni di non-equlibrio del sistema. Questo ci fa capire fra le altre cose come l’ensemblemicrocanonico, anche se e la distribuzione di equilibrio statistico nel Γ-spazio, non e associato in modoesclusivo a configurazioni di equilibrio termodinamico del sistema (vedere nota al termine della Sez. 6.3).

5.1. IL PUNTO DI VISTA DI GIBBS 89
Raggiungiamo comunque le seguenti conclusioni:
• La forma esatta della distribuzione nel Γ-spazio non deve essere importante; di fattobasta che le proprieta macroscopiche del sistema (per dire, l’energia di una sua partemacroscopica) siano quasi costanti in gran parte del dominio in cui ρ e non nulla (laparte del dominio corrispondente a configurazioni di equilibrio termodinamico).
• Le medie temporali di grandezze macroscopiche non abbisognano quindi che le tra-iettorie su cui sono calcolate visitino tutto il Γ-spazio, per coincidere sostanzialmentecon le corrispondenti medie di ensemble.
• Il principale vantaggio di una ipotesi del tipo ρ = ρM e forse solo la facilita nei conti.
5.1.2 Il problema ergodico
Legato alle difficolta discusse qui sopra e il cosı detto problema ergodico: siamo sicuri chemedie di ensemble e medie temporali coincidono nel Γ-spazio? Abbiamo visto a suo tempoche una condizione per l’ergodicita di un processo stocastico x(t) e l’indecomponibilita delsuo spazio delle configurazioni: non devono esistere regioni del dominio di x inaccessibilise x parte da qualche altra regione dello stesso dominio.
Nel caso di un sistema conservativo isolato, tale proprieta di indecomponibilita eviden-temente non e soddisfatta: il sistema non puo spostarsi fra regioni ad energia diversa. Larisposta a questo stato di cose e stato l’ensemble microcanonico. La questione ergodicariguarda proprio cosa succederebbe se esistessero altre quantita conservate oltre l’energia(ed eventualmente il momento lineare e il momento angolare del sistema).
Una risposta a questa domanda e stata fornita da Khinchin (A.I. Khinchin, “Ma-thematical foundations of statistical mechanics”) e si puo riassumere (forse in manieraleggermente tautologica) nelle seguenti due osservazioni:
• Se la quantita conservata ha un significato macroscopico, l’ensemble microcanonicova generalizzato per tenere conto della presenza di tale quantita.
• Se la quantita non ha un significato macroscopico, allora la scelta della distribuzio-ne dei possibili suoi valori puo essere effettuata in maniera arbitraria e il risultatodelle medie delle grandezze macroscopiche, per definizione, non deve variare. Inparticolare, la scelta in cui i valori di questa quantita sono distribuiti uniformente-mente, corrispondente alla distribuzione microcanonica originaria, e perfettamenteaccettabile.
In altre parole, la proprieta ergodica per la generica quantita macroscopica m deve conti-nuare ad essere soddisfatta anche in presenza di quantita microscopiche conservate. Se Iindica il valore della quantita conservata nel sistema all’istante iniziale, avremo comunque
limT→∞
〈m|I〉T = 〈m|I〉 = 〈m〉M (5.3)

90 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
dove l’ultima media e calcolata rispetto alla distribuzione microcanonica. Notiamo che lascala dei tempi su cui la distribuzione ρ(m) varia, al contrario di quella per ρ(Γ), e la scaladei tempi di rilassamento del sistema: al contrario del caso della misura diretta di ρ(Γ), ilcalcolo di medie temporali per m ha senso.
5.2 Il concetto di entropia
La discussione che abbiamo portato avanti sin’ora, sembra suggerire che l’unica maniera didare un significato fisico a una PDF nel Γ-spazio e attraverso la media di grandezze macro-scopiche. In questo quadro, un ruolo centrale e svolto dal concetto di entropia. A questoscopo, introdurremo una nuova forma di entropia, espressa in termini di grandezze macro-scopiche (entropia di Boltzmann), che da un lato permettera di stabilire una connessionefra mondo microscopico (entropia di Shannon) e mondo macroscopico (entropia termodi-namica), e dall’altro permettera di chiarire il significato dell’ensemble microcanonico, comeun oggetto che descrive situazioni sia di equilibrio che di non-equilibrio termodinamico.
Per cominciare, un minimo di terminologia.
Sia m = ma, a = 1, . . .M il vettore dei parametri con cui vogliamo descrivere lo statomacroscopico (macrostato) del sistema. Chiamiamo lo spazio dove vivono i vettori m,lo spazio µ del sistema. Le componenti ma sono supposte conosciute con precisione ∆ma,cosı che ma assume di fatto valori discreti; come lo stato microscopico o microstato delsistema individua una celletta di dimensione N !δΓ ∼ N !~3N del Γ-spazio, il macrostatoindividuera una celletta di dimensione ∆m del µ-spazio. La scelta dei parametri chedeterminano il macrostato e arbitraria: m potrebbe essere il vettore le cui due componentisono il numero di molecole e l’energia interna di un volumetto del sistema. Oppure potrebbeessere il vettore con componenti f(xa,va)∆
3x∆3v dei numeri di molecole nella celletta dispazio delle fasi (per una molecola) ∆3x∆3v, centrata intorno a (xa,va). In ogni caso,e evidente che per avere un significato macroscopico, i parameteri ma devono riferirsia parti macroscopiche del sistema, contenenti un numero di molecole tale che il limitetermodinamico sia soddisfatto.
Indichiamo con ∆Γ(m) la parte di Γ-spazio corrispondente al macrostato m. Persemplicita, ci limitiamo a considerare il caso in cui i ∆Γ(m) sono dei sottoinsiemi di∆Γ(U), di modo che ∪m∆Γ(m) = ∆Γ(U). Il soddisfacimento della proprieta ergodicaper m, Eq. (5.3), ci dice che la probabilita di osservare un certo valore della variabilemacroscopica m, non e altro che la probabilita, calcolata rispetto alla PDF di equilibriostatistico ρM , che il punto di fase Γ cada in ∆Γ(m). In altre parole, dalla Eq. (5.1):
P (m) =∆Γ(m)
∆Γ(U).
La restrizione della PDF microcanonica al dominio ∆Γ(m) e evidentemente ρM(Γ|m) =1/∆Γ(m), e possiamo scrivere, per Γ ∈ ∆Γ(m): ρM(Γ) = P (m)ρM(Γ|m). Da qui vedia-mo che l’entropia di Shannon associata al microcanonico, per un sistema composto di N

5.2. IL CONCETTO DI ENTROPIA 91
molecole identiche, puo essere scritta nella forma (3.9):
SN = −∫
∆Γ(U)
dΓ ρM(Γ) ln[ρM(Γ)N !δΓ] = ln[∆Γ(U)/(N !δΓ)]
=∑
m
P (m)S(m)−∑
m
P (m) lnP (m), (5.4)
dove −∑
m P (m) lnP (m) e l’entropia di Shannon della distribuzione della variabile ma-croscopica m, e
S(m) = −∫
∆Γ(m)
dΓ ρM(Γ|m) ln[ρM(Γ|m)N !δΓ] = ln[∆Γ(m)/(N !δΓ)] (5.5)
e l’entropia di Shannon ottenuta considerando come spazio dei campioni ∆Γ(m). Notiamocome la Eq. (5.5) ci permette di scrivere le PDF nel Γ-spazio in funzione dell’entropia:
ρM(Γ|m) ∼ exp(S(m)), (5.6)
e la PDF microcanonica ρM(Γ) e ottenuta ponendo m = U .
Avevamo gia incontrato un oggetto simile alla S(m) nel Capitolo 3: l’entropia diBoltzmann Sf = −
∫
d3xd3v f(x,v; t) ln f(x,v; t), definita a partire dalla PDF di nonequilibrio ρN(Γ, t). Anche in quel caso avevamo una entropia associata al contenuto diinformazione del macrostato individuato dalla distribuzione f(x, t; t). Vediamo qui la dif-ferenza fondamentale tra l’approccio della teoria cinetica e quello della meccanica sta-tistica. In teoria cinetica, il macrostato m e il risultato della scelta di una certa PDFdi non equilibrio ρN(Γ, t), tale che
∫
dΓ m(Γ)ρN(Γ, t) = m(t). Per sistemi debolmen-te interagenti, ρN(Γ, t) ≃ ∏
k ρ1(xk,vk, t), che fornisce immediatamente il macrostato mma = Nρ1(xa,va; t)∆
3x∆3v (l’indice k identifica la molecola; l’indice a, la casella ∆3x∆3vdi spazio delle fasi per la molecola). Nel caso della meccanica statistica, m individua laPDF condizionata ρM(Γ|m), come restrizione della PDF microcanonica al dominio ∆Γ(m).Lo stesso macrostato m puo essere quindi realizzato a partire da una PDF di non equlibrioρN(Γ, t) o a partire dalla PDF condizionata microcanonica ρM(Γ|m). La differenza piu im-portante fra le due e che ρN(Γ, t) potrebbe essere una PDF di non equilibrio rispetto ad altrevariabili macroscopiche m′
a non contenute in m, mentre ρM(Γ|m), essendo la restrizione diρM a ∆Γ(m), sara dominata dalle configurazioni di equilibrio per m′
a. Per il resto, l’unicadifferenza risiede nelle fluttuazioni (nel caso di ρM , quelle di m sono per costruzione assen-ti). In assenza di differenze sui valori tipici di m′
a, le due distribuzioni sono praticamenteuguali e l’entropie ad esse associate saranno esse stesse praticamente identiche. Verifi-cheremo esplicitamente questo fatto nella Sez. 5.3, nel caso ma = Nρ1(xa,va; t)∆
3x∆3v.Queste osservazioni ci motivano a chiamare in generale anche la S(m), una entropia diBoltzmann.
Osserviamo la seguente importante proprieta delle entropie di Shannon (5.4) e diBoltzmann (5.5), nel caso di un sistema descritto tramite il microcanonico:

92 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
• L’entropia corrispondente al macrostatom e pari al logaritmo del numero di microsta-ti con il quale si realizza il macrostato. Nel caso in cui il macrostato e semplicementel’energia U del sistema, ritroviamo l’entropia di Shannon del sistema, e il numero dimicrostati e quello nel guscio energetico U .
Ora, se il limite termodinamico per le parti del sistema identificate dai parametri ma, esoddisfatto, e se il parametro di discretizzazione ∆ma non e troppo piccolo, praticamentesolo un macrostato sara realizzato, quello piu probabile m ≃ 〈m〉.2 In altre parole, abbiamoP (m) ≃ 1, cosı che, combinando le Eq. (5.4) e (5.5), troviamo
SN ≃ S(m) ≃ ln[∆Γ(m)/(N !δΓ)], (5.7)
dove l’entropia di Boltzmann S(m) corrispondente al macrostato di equilibrio m e det-ta entropia di Gibbs. Notiamo come l’entropia di Boltzmann S(m) e una quantitafluttuante, mentre quella di Shannon e una quantita costante, approssimata dal valore diequilibrio di S(m). Di nuovo, la PDF microcanonica puo essere ottenuta sostituendo nella(5.6) l’entropia di Gibbs S(m).
Riassumendo, abbiamo trovato il seguente importante risultati:
• L’equilibrio termodinamico corrisponde al macrostato realizzato attraverso il numeromassimo di microstati.
• L’entropia del sistema termodinamica del sistema isolato all’equilibrio, coincide conl’entropia di Gibbs S(m), che approssima a sua volta l’entropia di Shannon delsistema.
L’approccio della meccanica statistica di Gibbs ci ripropone quindi in maniera diretta laproprieta, gia osservata nella Sez. 2.3, che l’equilibrio termodinamico non e altro che lostato del sistema che viene realizzato con probabilita piu alta, che corrisponde al massimodi entropia del sistema.
In alcuni casi, la suddivisione in parti del sistema individuata dal macrostato m corri-sponde in una effettiva suddivisione in sottosistemi Γa, ciascuno con Na molecole, di modoche il Γ-spazio si decompone in un prodotto di sottospazi Γa, ciascuno dei quali descrittoda una PDF ρa(Γa). Naturalmente, una simile identificazione non e possibile nel caso leparti del sistema sono in grado di scambiare molecole fra loro, come lo sono ad esempio ivolumetti di spazio delle fasi considerati nel caso in cui ma = f(xa,va)∆
3x∆3v. Nel casodi sottosistemi veri e propri, la natura macroscopica degli stessi permette di considerareindipendenti le molecole in essi contenuti, e di scrivere
ρM(Γ) =∏
a
ρa(Γa); S(m) =∑
a
Sa(ma).
2In condizioni di limite termodinamico, abbiamo per le fluttuazioni del parametroma: σma∼ maN
−1/2a .
La condizione P (m) ≃ 1 richiede evidentemente ∆ma ≫ σma.

5.3. IL METODO DI MASSIMA ENTROPIA 93
Un caso importante e quello in cui ma ≡ Ua e l’energia del sottosistema. In tal caso,ρa(Γa) ≡ ρMa (Γa), la Eq. (5.4) ci da immediatamente
Sa(Ua) = ln[∆Γa(Ua)/(Na!δΓa)] (5.8)
e ritroviamo l’identificazione dell’entropia di Boltzmann come logaritmo del numero dimicrostati con cui si realizza in macrostato dato.
5.3 Il metodo di massima entropia
Si tratta di un metodo piuttosto generale per la determinazione di distribuzioni di probabi-lita che soddifino a condizioni date. L’imposizione di una condizione di massima entropiafa sı che l’unico contenuto di informazione della distribuzione sia quello proveniente daivincoli che sono stati imposti.
Nel nostro caso, la condizione di massima entropia fissa i valori all’equilibrio dei para-metri macroscopici. Un caso in cui questi parametri macroscopici determinano di fatto unadistribuzione di probabilita e quello della distribuzione di Maxwell-Boltzmann. In questocaso, la suddivisione del sistema e fatta considerando intervalli di energia delle molecole[ǫk, ǫk+1], ǫk = k∆ǫ e il macrostato e individuato dai numeri di molecole Nk in ciascun in-tervallo. La distribuzione di equilibrio, corrispondente al massimo di entropia del sistemarisultera essere proprio la distribuzione di Maxwell-Boltzmann del sistema. Notiamo la po-tenza del metodo, che risiede nel fatto che non si utilizzano in alcuna maniera informazionisulla dinamica microscopica del sistema. In particolare, la dipendenza delle energia ǫ sullevariabili pi, qi del sistema puo essere qualsiasi.
Dobbiamo quindi determinare il massimo della entropia di Boltzmann
S(Nk) =∑
k
Sk(Nk); Sk(Nk) = ln∆Γk(Nk), ∆Γk(Nk) ≡ ∆Γk(Nk)/(N !δΓ),(5.9)
rispetto ai numeri di occupazione Nk. Vediamo che si tratta di un massimo condizionatoal fatto che il sistema contiene in tutto N molecole, ed essendo isolato, la sua energia haun valore fissato U . I vincoli sono pertanto
∑
k
Nk = N e∑
k
Nkǫk = U,
e la condizione di massimo condizionato e espressa, utilizzando moltiplicatori di Lagrangeα e β, nella forma
dS − αdN − βdU = 0.
Per potere procedere, e necessario a questo punto avere una espressione per i numeri dimicrostati ∆Γk(Nk).
Il problema, dal punto di vista concettuale, e piuttosto delicato. Il fatto che abbiamodefinito l’entropia a partire da una partizione che considera le molecole indistinguibili porta

94 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
a conseguenze non triviali se la scala di coarse-graining δΓ non e abbastanza fine. Comesi e gia accennato, la meccanica quantistica fissa una scala minima δΓ = (2π~)3N , e non edetto che le scale di lunghezza e di velocita tirate in ballo dalla costante di Planck ~ sianonecessariamente microscopiche.
Nota. La condizione per avere un sistema classico e sostanzialmente una condizione didiluizione. Se il gas e posto in un recipiente di volume V , ciascuna molecola avra a dispo-sizione evidentemente un volume V in cui muoversi. Analogamente, se m e la massa dellemolecole, il momento lineare delle molecole sara compreso in un intervallo la cui ampiezzae dell’ordine di mvth dove vth e la velocita termica. Quindi il volume di spazio delle fasidisponibile e V (mvth)
3 e il numero di cellette sara Ncell ∼ V (mvth)3/~3. La condizione
che ciascuna celletta e occupata al massimo (di solito) da una molecola, e evidentementeN ≪ Ncell, dove N e il numero di molecole nel volume. Cioe, in funzione della densitan = N/V :
n≪ (mvth/~)3
Il gas dev’essere sufficientemente diluito, oppure la temperatura deve essere sufficientemen-te alta.
Focalizziamoci per il momento sul caso in cui sia possibile considerare la scala di coarse-graining arbitrariamente piccola e scegliamola in maniera tale che il numero Gk di celle divolume δΓ corrispondenti all’intervallo di energia [ǫk, ǫk+1] sia molto piu grande del numerodi occupazione corrispondente Nk. Supponiamo che si abbia cioe:
Gk ≫ Nk ≫ 1.
In queste condizioni, le cellette occupate conterranno al massimo una molecola e il numerodi microstati corrispondenti al numero di occupazione Nk del guscio energetico Nk sarail numero di modi in cui si possono distribuire le Nk cellette occupate fra le Gk cellettedisponibili. Questo numero e
∆Γ(Nk) =Gk(Gk − 1) . . . (Gk −Nk)
Nk!≃ GNk
k
Nk!. (5.10)
Sostituendo la (5.10) nella (5.9), e utilizzando l’approssimazione di Stirling lnNk! ≃Nk(lnNk − 1), l’entropia diventa pertanto
S ≃ −∑
k
Nk[ln(Nk/Gk)− 1] (5.11)
e la condizione di massimo condizionato diventa
∂
∂Nk
(S − αN − βU) = − ln(Nk/Gk)− α− βǫk = 0.
Da qui otteniamo
Nk = Φk exp(−α− βǫk). (5.12)

5.3. IL METODO DI MASSIMA ENTROPIA 95
I valori dei moltiplicatori α e β possono essere determinati a questo punto imponendoesplicitamente
∑
kNk = N e∑
kNkǫk = U . E piu interessante notare la connessione frala condizione di estremo dS − αdN − βdU e l’espressione del differenziale dell’energia infunzione delle variabili estensive S e N : dU = TdS + µdN . Utilizzando queste relazioninella (5.12), otteniamo immediatamente
Nk = Φk exp(µ− ǫk
T
)
= exp(
ζk +µ− ǫkT
)
(5.13)
e notiamo la relazione fra il numero Gk di cellette di µ-spazio nell’intervallo [ǫk, ǫk+1], e lacostante (del tipo della costante chimica) ζk.
Una domanda che a questo punto sorge naturale e se esiste una relazione tra l’entropiadi Gibbs (5.11) e l’entropia di Boltzmann Sf (3.12). Tutte e due, dopo tutto, si riferisconoalla distribuzione dei numeri di occupazione di gusci di energia in un sistema di molecole.Concentriamoci sul caso del gas ideale [a cui si riferisce di fatto la (3.12)]. Per sempli-cita consideriamo D = 1. Le cellette del µ-spazio sono quindi intervalli di velocita peruna molecola [vk, vk+1], vk+1 − vk = ∆v ≡ ∆µ. Abbiamo visto che ∆v deve essere abba-stanza grande affinche il numero Nk sia abbastanza grande da potere ricercare un limitetermodinamico Nk ≃ 〈Nk〉. Abbiamo quindi
Φk = Φ = ∆v/δv = costante,
e possiamo riscrivere la (5.11) nella forma
S = −Φ∑
k
(Nk/G) ln(Nk/G) +N ≃ −∫
dv f ln(f δv/e). (5.14)
A meno di costanti abbiamo ritrovato l’entropia di Boltzmann Sf (3.12).
5.3.1 Cenni di statistica quantistica
Si e gia accennato a come la meccanica quantistica fissi una microscala al di sotto dellaquale punti diversi nello spazio delle fasi di un sistema sono indistinguibili. Lo spazio dellefasi risulta cosı suddiviso in cellette, ciascuna di dimensioni ~N , dove N e il numero di gradidi liberta (pi, qi) del sistema, ciascuna corrispondente a uno stato quantistico. La sceltadegli autostati e in generale arbitraria. Per il genere di problemi a cui siamo interessati,pero, (statistica stazionaria di un sistema isolato), la scelta naturale e quella degli autostatidell’energia, che sono infatti gli stazionari del sistema. Per fissare le idee, consideriamo ilproblema di un gas di N particelle identiche non interagenti, in un potenziale armonicomonodimensionale. Indichiamo con |a; k〉 l’autostato della particella a-esima corrisponden-te a energia Ek = (1/2+k)~ω, dove ω e la frequenza naturale di oscillazione della particellanel potenziale.
Classicamente, ci ritroveremmo un Γ-spazio che e il prodotto cartesiano degli spazi dellefasi di ciascuna particella; pertanto, Γ = (n1, n2, . . . nN) sarebbe il vettore degli stati diciascuna particella. L’osservazione che stati ottenuti permutando i valori dell’energia delle

96 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
E
E
a
b
E E
E
E
1
1
2
2α
β
γ
Figura 5.1: Possibili stati di due particelle identiche a e b. Classicamente α e β sonodue stati indistinguibili. Insieme formano un microstato di peso 2, mentre γ forma unmicrostato di peso 1.
diverse particelle [ad esempio Γ = (n1, n2, . . . , nN ) e Γ′ = (n2, n1, . . . , nN )] sono fisicamente
indistinguibile, ci aveva spinto a definire i microstati del sistema come unione di tutti ipunti Γ che differiscono per una permutazione delle componenti. Questo fa sı pero che ilpeso statistico di microstati in cui tutte le particelle hanno energie diverse e maggiore diquello in cui alcune hanno energia uguale. Il peso statistico e dato infatti dal numero dipunti Γ contenuti nel microstato, che sara dato dal fattore multinomiale N !/
∏
kNk!, doveNk e il numeri di particelle con energia Ek. La situazione e illustrata in Fig. 5.1 nel casoN = 2: i due punti α e β formano il microstato “una particella con energia E1 e una conenergia E2”. Questo microstato ha evidentemente peso doppio di quello in cui tutte le dueparticelle hanno energia identica.
Quantisticamente, lo stato di un sistema a N particelle e determinato da combinazionisimmetriche o antisimmetriche, a seconda che si tratti di bosoni o di fermioni, di prodottidi funzioni d’onda delle particelle nel sistema. Nel caso N = 2:
|E1, E2〉 =1√2[|a; 1〉|b; 2〉 ± |a; 2〉|b; 1〉]. (5.15)
In altre parole, i due punti α e β in Fig. 5.1 corrispondono allo stesso stato, non due statiindistinguibili che insieme pesano 2. Nel caso di bosoni, lo stato |E1, E2〉 con E1 6= E2 pesatanto quanto |E2, E2〉 = |a, 2〉|b, 2〉; nel caso di fermioni, lo stato |E2, E2〉 semplicementenon esiste, che e il contenuto del principio di esclusione di Pauli. Lo stato quantisticoa N particelle potra essere quindi scritto, sia nel caso di bosoni che di fermioni, nella forma|Γ〉 ≡ |N1, N2, . . .〉, dove Na indica il numero di particelle con energia Ea, e tutti questistati avranno peso identico (la normalizzazione dello stato) pari a 1. (Il passaggio finaleda meccanica quantistica a meccanica statistica, e ottenuto trascurando la fase dello stato|Γ〉 e utilizzando una descrizione del sistema in termini di matrice densita, cioe di sempliciprobabilita, e non di ampiezza degli stati).
La differenza tra statistica quantistica e statistica classica e resa evidente, nel caso difermioni, dal principio di esclusione di Pauli. Nel caso di bosoni, la differenza e piu sottile

5.3. IL METODO DI MASSIMA ENTROPIA 97
ed ha a che fare con la maniera in cui sono pesati gli stati a piu particelle, che fa sı, nel casodei bosoni, che vi sia una maggiore facilita, rispetto al caso classico, di trovare stati con piuparticelle. La distribuzione di Maxwell-Boltzmann ci dice che nel caso classico, gli stati adenergia piu bassa sono comunque quelli che, tanto piu la temperatura e bassa, tendono adessere piu occupati. Nel caso di fermioni, questi stati tenderanno ad essere meno occupati,in quello dei bosoni, piu occupati. Per questa ragione, un gas a basse temperatura, nel casodi fermioni avra un contenuto energetico maggiore, di quello di un gas classico, mentre nelcaso di bosoni, questo contenuto sara minore.
Nota. Abbiamo ottenuto la distribuzione di equilibrio Nk, nel caso di gas diluito classico,massimizzando l’entropia S = ln(∆Γ(Nk)), dove ∆Γ(Nk) e il numero di microstati concui viene realizzato il macrostato f . Il senso fisico di questa massimizzazione e quello ditrovare il macrostato piu probabile, cioe quello che occupa la porzione di volume ∆Γ(U)maggiore. E evidente quindi che la scelta della partizione di Γ deve essere ininfluente nelladeterminazione del macrostato piu probabile.3 In particolare, utilizzando una partizione di∆Γ(U) in volumetti δΓ ≡ δΓindist ∼ ~
3N , ci ritroviamo con una entropia Sdist(Nk) =ln(∆Γ(Nk)/δΓdist) ≡ ln[∆Γdist(Nk)] pari al logaritmo di un numero di microstati,indipendentemente dal fatto che il gas e diluito o meno. Possiamo calcolare il massimo diSdist, da qui determinare la distribuzione Nk di equilibrio, e, nel caso diluito, ritroviamola distribuzione di Maxwell-Boltzmann. I problemi iniziano a sorgere quando il gas non ediluito.
Il fatto e che stiamo contando degli stati del sistema differenti per permutazione di par-ticelle, che non solo non sono distinguibili, ma dal punto di vista quantistico semplicementenon esistono. Nel caso di un gas diluito, l’assenza di celle δ3xδ3v con piu di una particella,fa sı che il fattore δΓ/δΓdist = N ! e uguale per tutti i microstati, e scompare nel calcolodel massimo dell’entropia. Per gas non diluiti, questo non e piu vero e la distribuzioneottenuta massimizzando Sdist(Nk), non sara piu la distribuzione di equilibrio. Proviamocomunque ad eseguire il calcolo e vedere cosa viene fuori.
Nel caso di particelle distinguibili, il numero di microstati e semplicemente il numerodei modi in cui le Nk particelle nei gusci energetici [ǫk, ǫk+1], possono essere disposte nelleGk caselle δ
3xδ3v presenti nei gusci, per tutti i k. Ci sono N !/∏
kNk! modi di raggruppareN particelle in gruppi di Nk. Ciascuna delle Nk particelle in ciascun gruppo ha poi Gk
caselle a disposizione nel guscio k fra cui scegliere. Il risultato finale sono
∆Γdist(Nk) = N !∏
k
GNk
k
Nk!
modi di disporre le molecole. Che e precisamente N ! volte il numero ottenuto moltiplicando inumeri di microstati ∆Γ(Nk) ottenuti nella Eq. (5.11). Da qui, la procedura per individuareil macrostato piu probabile e identica a quella seguita nella Sez. 5.3. Il risultato e ladistribuzione di Maxwell-Boltzmann, indipendentemente dai numeri di occupazione delle
3Abbiamo gia visto come problemi del tipo del paradosso di Gibbs non precludono l’utilizzo di Sdist
per determinare l’equilibrio termodinamico di sistemi.

98 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
cellette δ3xδ3v. Anche per situazioni, quindi, dove la statistica di Maxwell-Boltzmann nonvale piu, ed effetti di tipo quantistico diventano dominanti.
5.4 La distribuzione di Gibbs
Un altro modo in cui il nostro sistema isolato puo essere suddiviso, e considerando unvolumetto VA contenente un numero fissato di molecole (un sottosistema separato dal restodel corpo da una parete che conduce il calore). Indichiamo con B il rimanente del sistemaisolato (il bagno termico) e supponiamo UA ≪ U , dove UA e U sono le energie internedel sottosistema e del sistema isolato. Vorremmo calcolare la distribuzione ρA(UA) delleenergie interne del sottosistema. Per far questo possiamo partire dalla PDF marginale nelΓ-spazio per il sistema A:
ρA(ΓA) =
∫
ρ(Γ)dΓB =∆ΓB(ΓA)
∆Γ(U),
in cui ∆Γ(U) e il volume di Γ-spazio corrispondente al sistema isolato, e ∆ΓB(ΓA) e ilvolume di Γ-spazio del bagno termico corrispondente al microstato del sottosistema A. Orase i sottosistemi A e B sono macroscopici, una proprieta macroscopica di B come il volume∆ΓB(ΓA) (legato all’entropia di Boltzmann di B) non potra dipendere dal microstato ΓA,se non attraverso il macrostato di A che esso individua. Nel nostro caso UA. In altre parole,∆ΓB(ΓA) e determinato dal macrostato in cui si trova B in corrispondenza a UA che non ealtri che l’energia UB = U −UA(ΓA). Abbiamo quindi che la marginale ρA(ΓA) e uniformeper ciascun valore di UA, e possiamo definire
ρA(ΓA) =∆ΓB(U − UA(Γ))
∆Γ(U).
Sfruttando la (5.8), il volume ∆ΓB(UB) puo pero essere scritto in funzione dell’entropia diBoltzmann del sottosistema B, e troviamo infine:
ρA(ΓA) =NB!δΓB∆Γ(U)
exp(SB(U − UA(ΓA))) ≃1
Zexp(−UA(ΓA)
T), (5.16)
dove si e utilizzata la relazione SB(U − UA) ≃ SB(U) − UAS′B(U) = SB(U) − UA/T , T
e la temperatura del bagno termico, e Z = ∆Γ(U)/∆ΓB(U) e un fattore di normalizza-zione, detto funzione di partizione, che, come si vedra, gioca un ruolo estremamenteimportante nella teoria.
La (5.16) e detta distribuzione di Gibbs (oppure distribuzione canonica) ed e laforma che assume la PDF nel Γ-spazio, per un sistema in un bagno termico a temperaturaT .
Va ribadito che ρA(UA) non e ancora la PDF per UA, il che e rivelato dal suo esserepiccata intorno a UA = 0 e non intorno a UA ∼ NAT . Lo spostamento del picco da

5.4. LA DISTRIBUZIONE DI GIBBS 99
0 a UA nella PDF ρA(UA) e prodotto dal fattore di volume nel cambio di variabili daΓA ∈ ∆ΓA(UA) a UA:
ρA(UA) = ρA(ΓA)∆ΓA(UA)
∆UA=NA!δΓAZ∆UA
exp(TSA(UA)− UA
T
)
(abbiamo utilizzato di nuovo per il calcolo di ∆ΓA(UA) la (5.8)). Vediamo subito cheρA(UA) possiede un picco di larghezza ∆UA ∼ T nel minimo rispetto a UA della differenzaUA − TSA(UA). Ma la condizione di minimo non e altro che
1
T=∂SA∂UA
,
che e verificata solo se UA e al suo valore all’equilibrio in un bagno termico a temperatureT . Ci rendiamo quindi conto che il minimo di TSA(UA)−UA rispetto a UA non e altro chel’energia libera di Helmholtz FA(T, VA) = TSA(UA, VA)− UA ≡ TSA(T, VA)− UA(T, VA).
Il legame fra energia libera di Helmholtz e distribuzione canonica ha delle importanticonseguenze. Questo legame diventa evidente se si effettua il calcolo della funzione dipartizione Z. Imponendo la condizione
∫
ρA(UA) ΓA = 1, troviamo subito, utilizzando la(5.16):
Z =
∫
dΓA exp(
− UA(ΓA)
T
)
, (5.17)
e vediamo che l’integrale e dominato dalla configurazione di equilibrio UA. Possiamopertanto scrivere
Z ≃ ∆ΓA(UA) exp(
− UAT
)
= NA!δΓA exp(TSA(UA)− UA
T
)
,
dove si e utilizzata ancora una volta la (5.8). Abbiamo gia visto pero che l’espressioneTSA(UA)−UA ≡ TSA(T )−UA(T ) non e altro che l’energia di Helmholtz per il sottosistemain un bagno termico a temperatura T . Troviamo quindi la relazione fondamentale
Z(T, VA) =
∫
dΓA exp(
− UA(ΓA)
T
)
= NA!δΓA exp(
− FA(T, VA)
T
)
, (5.18)
che ci fornisce un modo diretto di calcolare l’energia libera del sistema a partire da unamedia statistica sul Γ-spazio, nota la dipendenza della energia interna UA dai gradi diliberta microscopici ΓA.
Nota. Puo valer la pena ripetere il calcolo della energia libera di Helmholtz per un gasideale a partire dalla (5.18). Abbiamo in questo caso U = (2m)−1
∑
i p2i , cosı che
Z =
∫
d3Nqd3Np exp(
− 1
2mT
3N∑
i=1
p2i
)
= V N[
∫
dp exp(
− p2
2mT
)]3N
= V N(2πmT )3N2 = exp(N(lnV +
3
2lnT +
3
2ln(2mπ)))

100 CAPITOLO 5. MECCANICA STATISTICA DELL’EQUILIBRIO
Combinando con la (5.18), e utilizzando la formula di Stirling lnN ! ≃ N(lnN − 1),troviamo quindi
F (T, V,N) = NT(3
2(1− lnT )− ln(V/N)− ζ
)
,
dove, utilizzando δΓ = (δpδq)3N , abbiamo ζ = 5/2 − (3/2) ln(δq2δp2/(2πm)) (confrontarecon la (4.22)).
5.5 Esercizi
Esercizio 1 Considerare un sistema di quattro particelle distinguibili, ciascuna delle qualipuo avere energia ǫ = n∆, n = 0, 1, 2 . . ..
• Trovare i microstati corrispondenti al macrostato con energia E = 6∆. (Occhio acontare i microstati corrispondenti a scambio di particelle tra i diversi livelli energe-tici!)
• Supponendo i microstati a priori ugualmente probabili, determinare la distribuzionea una particella f1(ǫ) corrispondente al macrostato E = 6∆.
• Quanto vale l’entropia di Boltzmann di questo macrostato?
Problema 2 Supponiamo che in un dado truccato si abbia P (1, 2) = 1/3, P (3, 4) =1/2, P (5, 6) = 1/6 e che le vincite corrispondenti ai diversi risultati siano x(1) = x(2) =1; x(3) = x(4) = 0; x(5) = x(6) = −1. Pertanto, 〈x〉 = 1/6 (se il dado fosse non truccatosarebbe chiaramente 〈x〉 = 0).
• Calcolare per N ≫ 1, la probabilita P (x|〈x〉N = 0), cioe la distribuzione tipica diuscite degli x che farebbe sı che la media campionaria in una sequenza molto lungadi prove coincidesse con la media del caso non truccato. (Suggerimento: cercarel’analogia con il calcolo con il metodo di MAX entropia, della distribuzione di velocitadi un gas ideale a temperatura fissata).
Problema 3 Si consideri un gas ideale di molecole biatomiche. Si modelli l’interazione fragli atomi nelle molecole con un potenziale elastico, cosı che l’energia di una molecola e
ǫ =1
2m(p21 + p22) +
α
2|r1 − r2|2
dove p1,2 and r1,2 sono i momenti lineari e le coordinate degli atomi nella molecola.
• Trovare l’energia libera di Helmholtz del sistema
• Determinare il calore specifico a volume costante del sistema
• Determinare il diametro quadratico medio delle molecole 〈|r1 − r2|2〉.

Capitolo 6
Cenni alla teoria delle fluttuazioni
6.1 Fluttuazioni attorno all’equilibrio
(Gran parte delle considerazioni che seguono sono tratte dalle Sez. 110-112 del libro “FisicaStatistica” di Landau e Lifsits). Abbiamo visto che l’entropia di Boltzmann e il logaritmodel numero di microstati con cui puo essere realizzato un dato microstato. Essendo i micro-stati di un sistema isolato egualmente probabili (ensemble microcanonico), la probabilitadi un macrostato m deve essere proporzionale al numero di microstati, che e exp(S(m)).Verifichiamo il risultato in maniera esplicita. Indicando con ρ la PDF nello spazio deimacrostati, abbiamo infatti
ρ(m) = ρ(Γ)∆Γ(m)
∆µ(m)=
∆Γ(m)
∆µ(m)∆Γ(U),
dove ∆µ(m) e l’elemento di volume nel µ-spazio generato da m, Γ ∈ ∆Γ(m), e ρ(Γ) =1/∆Γ(U) e la PDF microcanonica per il sistema isolato (5.1). Scrivendo tramite la (5.6)in funzione di S(m):
ρ(m) = C exp(S(m)).
Abbiamo anche visto che l’equilibrio corrisponde al massimo di S(m). La funzione S(m)varia sulla scala del valore medio 〈S(m)〉, pertanto, per variazioni di m dell’ordine dellefluttuazioni termiche (che abbiamo detto tendono a zero nel limite termodinamico per isottosistemi), possiamo espandere in serie di Taylor
S(m) ≃ Seq −1
2αijzizj, (6.1)
dove Seq ≡ S(mk) e l’entropia all’equilibrio, zk = mk − 〈mk〉 sono le fluttuazioni rispettoall’equilibrio e abbiamo utilizzato la convenzione di somma sugli indici ripetuti.
• Vediamo quindi che la PDF delle fluttuazioni e Gaussiana, e che la matrice dicorrelazione 〈zizj〉 e data dall’inversa della αij .
101

102 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
Nota. Verifichiamo che (α−1)ij e la matrice di correlazione. Notiamo subito che la parteantisimmetrica di αij non contribuisce nella somma αijzizj e puo quindi essere posta ugualea zero. Attraverso una trasformazione unitaria, la matrice αij puo essere quindi posta informa diagonale. Lo Jacobiano del cambio di variabili corrispondente nella PDF e ugualea uno ed abbiamo
ρz(z) = ρz(z(z)) = C exp(−1
2
∑
k
αkkz2k),
dove il cappello indica variabili nella rappresentazione diagonale. Ma (α−1)ik ≡ α−1kk δik =
〈zizk〉 definisce la matrice di correlazione nella rappresentazione diagonale e (α−1)ik e lasua forma nella rappresentazione originaria.
Abbiamo gia visto alcuni esempi di calcolo di ampiezze di fluttuazione. Per la fluttua-zione ∆N del numero di molecole in un volume V , abbiamo trovato il risultato tipico delprocesso di Poisson [vedi la (2.26)]
〈∆N2〉 = nV.
Nel caso dell’energia di un sottosistema che non scambia molecole ma scambia calore conil resto del sistema, l’ampiezza di fluttuazione ci era data dall’ensemble canonico (5.16):
〈∆U〉 ∼ T.
In quest’ultimo caso, il calcolo chiamava esplicitamente in conto l’espressione della proba-bilita di U in funzione dell’entropia. Il procedimento puo essere generalizzato a variabilitermodinamiche generiche, riferite a una parte macroscopica del sistema isolato, ricordan-do la relazione fra variazione di entropia e calore ceduto a un sistema a temperatura data:dS = dQ/T .
Per fissare le idee consideriamo una porzione del sistema isolato, che puo scambiare la-voro e calore con il resto del sistema, ma il cui numero di molecole rimane costante. Questaporzione sara caratterizzata da valori V e S del volume e della entropia che saranno quindiquantita fluttuanti. Come abbiamo visto, in corrispondenza delle fluttuazioni di V ed Sdel sottosistema, corrisponera un allontanamento dalla condizione di equilibrio del sistemaisolato e quindi una sua diminuzione di entropia ∆Stot < 0. Questa diminuzione di entropiatotale corrisponde a una dipartita dalla curva di equilibrio Stot = Stot(Utot), che potrebbeessere ottenuta in maniera alternativa eseguendo lavoro dall’esterno sul sistema isolato apartire da una condizione a energia minore (vedi Fig. 6.1). Ritorniamo per un attimo allaquestione del lavoro minimo che bisogna esercitare per provocare una certa modificazionein una parte di un sistema isolato. Ricordo il risultato [vedi la (4.13)], valido nel casoin cui il sistema e la sua parte sono inizialmente in equilibrio [da qui in poi, indichiamole variabili riferite al sottosistema con lettere senza indice, mentre contraddistinguiamo ilsistema isolato con il pedice tot; confrontare con la (4.12)]:
∆Rmin = ∆U − T∆S + P∆V. (6.2)

6.1. FLUTTUAZIONI ATTORNO ALL’EQUILIBRIO 103
Stot
Utot
Stot∆Wmin∆
Figura 6.1: La stessa variazione di entropia puo essere vista sia come il risultato di unafluttuazione, che (con il segno cambiato), come quello del raggiungimento dell’equilibrio,dopo che il sistema e stato spinto fuori dall’equilibrio da un azione esterna (lavoro ∆Rmin).
La modificazione veniva effettuata in maniera tale che il volume totale del sistema isolatonon variasse (per esempio una paletta che agita il gas in una parte di un recipente). Di fattoquindi, ∆Rmin e l’aumento di energia di tutto il sistema isolato. L’analogia con l’analisisvolta nella Sez. 4.6 puo essere spinta oltre se consideriamo come sottosistema un corpo acontatto con un bagno termico a temperatura fissata. Le equazioni (4.14) e (4.15) ci diconoinfatti che se il corpo e mantenuto a volume costante, per produrre una fluttuazione al suointerno il lavoro minimo sara pari alla variazione di energia libera di Helmholtz, mentre seil corpo e libero di espandersi o comprimersi, in un ambiente a pressione fissata, ∆Rmin
sara la variazione di energia libera di Gibbs.Ora, la modificazione associata a ∆V e ∆S, provochera una piccola variazione nella
pressione e nella temperatura del sottosistema, che sara quindi spinto leggermente fuoridall’equilibrio rispetto al resto. Dopo un po’, pero, l’equilibrio sara ritrovato e vediamoche l’aumento di energia ∆Utot = ∆Rmin ha lo stesso effetto della cessione di un ugualequantitativo di calore al sistema isolato. L’entropia di quest’ultimo crescera in risposta diun ammontare ∆Stot = ∆Rmin/T . Vediamo quindi che ∆Rmin/T e la crescita di entropianel raggiungimento dell’equilibrio, a partire da una condizione iniziale di non equilibrio,caratterizzata da parametri ∆V e ∆S del sottosistema (entrambi gli stati considerati nalvalore finale dell’energia totale del sistema isolato).
• Riassumendo, la probabilita di una fluttuazione associata a variazioni ∆V e ∆S nelsottosistema e data da, utilizzando la (6.2):
ρ(∆V,∆S) = C exp(−∆Rmin/T ) = C exp(
− ∆U − T∆S + P∆V
T
)
. (6.3)
Dobbiamo determinare a questo punto il lavoro minimo ∆Rmin. Vediamo subito che, svol-gendosi la modificazione all’equilibrio, le pressioni e le temperature interne ed esterne alsottosistema devono essere uguali. Il lavoro che e necessario esercitare, proviene pertantodalla dipartita della pressione e temperatura del sottosistema dai rispettivi valori all’equi-librio. Questo ci suggerisce che il lavoro minimo deve essere quadratico in ∆V e ∆S. In

104 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
effetti, e cosı che deve essere, se la trasformazione nel sistema isolato e effettuata attorno auna configurazione di equilibrio, che corrisponde quindi a un massimo di Stot rispetto all’e-nergia U della sua parte. La variazione di Stot rispetto a U deve quindi essere quadratica.Espandendo ∆U al secondo ordine in ∆V e ∆S:
∆U ≃ T∆S − P∆V +∂2U
∂S2(∆S)2 + 2
∂2U
∂S∂V∆S∆V +
∂2U
∂V 2(∆V )2
e vediamo che i termini lineari sono cancellati in ∆Rmin dai contributi che provengono dalresto del sistema isolato. Troviamo in definitiva
∆Rmin ≃ ∂2U
∂S2(∆S)2 + 2
∂2U
∂S∂V∆S∆V +
∂2U
∂V 2(∆V )2.
D’altra parte possiamo riscrivere
∂2U
∂S2(∆S)2 =
∂T
∂S(∆S)2 =
(
∆T − ∂T
∂V∆V
)
∆S
∂2U
∂V 2(∆V )2 = −∂P
∂V(∆V )2 = −
(
∆P − ∂P
∂S∆S
)
∆V
∂2U
∂S∂V=
∂T
∂V= −∂P
∂S
e percio, sostituendo di nuovo in ∆Rmin e poi in ρ(∆V,∆S): [Eq. (6.3)]:
ρ(∆V,∆S) = C exp(∆P∆V −∆T∆S
T
)
, (6.4)
dove ∆P e ∆T sono intese funzioni di ∆S e ∆V . Questa espressione sottolinea come ladeviazione dall’equilibrio deve essere associata a “forze di richiamo”, che sono appunto levariazioni ∆P e ∆T . Notiamo a questo punto che abbiamo quattro variazioni per dueparametri indipendenti. La coppia di parametri scelti come indipendenti sono anche quellidi cui e possibile calcolare esplicitamente l’ampiezza di fluttuazione. Consideriamo adesempio la coppia V, T . Dobbiamo quindi esprimere ∆S e ∆P nella (6.4) in funzione ∆Ve ∆T .
∆S =∂S
∂T∆T +
∂S
∂V∆V =
∂S
∂U
∣
∣
∣
V
∂U
∂T
∣
∣
∣
V∆T − ∂2H
∂V ∂T∆V =
CVT
∆T − ∂2H
∂V ∂T∆V
e
∆P =∂P
∂T∆T +
∂P
∂V∆V = − ∂2H
∂V ∂T∆T +
∂P
∂V∆V,
e quindi, sostituendo in ρ:
ρ(∆V,∆T ) = C exp(
− Cv2T 2
(∆T )2 +1
2T
∂P
∂V(∆V )2
)
(6.5)
da cui troviamo
〈(∆T )2〉 = T 2
Cv, 〈∆T∆V 〉 = 0; 〈(∆V )2〉 = −T
(∂P
∂V
)−1
. (6.6)
La stessa procedura che ha condotto alla (6.6) puo essere effettuata con coppie diverse divariabili termodinamiche, e si ottengono cosı le ampiezze di fluttuazione ad esempio perpressione ed entropia.

6.2. STRUTTURA TEMPORALE DELLE FLUTTUAZIONI 105
6.2 Struttura temporale delle fluttuazioni
Studiare la struttura temporale delle fluttuazioni ci porta necessariamente a considerare leproprieta dinamiche del sistema. Abbiamo visto a suo tempo, per lo meno nel caso di ungas ideale, che la teoria cinetica ci offre uno strumento per lo studio di queste proprieta.Ci rendiamo rapidamente conto, pero, che anche questa e insufficiente trattando anch’essaper definizione con quantita medie (sebbene riferite a una PDF in generale dipendentedal tempo). Un approccio possibile (ma molto difficile) potrebbe essere quello di esten-dere la teoria cinetica oltre il campo medio, derivare una equazione per la distribuzione adue-particelle f2(x1, x2;v1, v2; t), e riderivare da qui equazioni fluide che tengano in contol’effetto delle fluttuazioni.
La strategia alternativa (seguita essenzialmente da Einstein) e quella di supporre cheil comportamento delle quantita fluide “esatte” (che tengono conto cioe delle fluttuazioni)n(x, t), u(x, t), eccetera, differisca poco, all’equilibrio, da quello delle corrispettive medien(x, t), u(x, t) eccetera. In altre parole, si suppone che le quantita fluttuanti obbediscano,a meno di una correzione che dipendera da effetti microscopici da meglio definire, la stessaequazione fluida (o cinetica) cui obbedisce la corrispondente quantita media. Questa equa-zione, a causa della piccolezza delle fluttuazioni, sara necessariamente lineare in questeultime. Per esempio, l’equazione di trasporto del calore (3.29) avra ora la forma:
∂∆T
∂t+
2
3T∇ ·∆u− κ∇2∆T = ξ,
dove ξ(x, t) e il termine che descrive gli effetti microscopici responsabili per le fluttuazioni.(Per ottenere un sistema chiuso di equazioni, identiche formule possono essere introdotteper le fluttuazioni di velocita fluida e di densita).
Ora, gia osservando questo particolare esempio, ci rendiamo conto della situazione: inassenza del termine ξ, il termine di diffusivita (e il corrispondente termine viscoso nellaequazione di Navier-Stokes (3.28), attraverso la distruzione di ∆u) porterebbe alla distru-zione di qualsiasi disomogeneita in T . Il termine ξ e quindi a tutti gli effetti un forzante.Vediamo inoltre che, mentre il termine diffusivo agisce su una scala di tempi macroscopicaL2/κ, dove L e la scala spaziale a cui studiamo le fluttuazioni (per dire, L ∼ V 1/3), ξe una somma di contributi delle singole molecole che si cancellano fra loro (la media delcontributo deve essere ovviamente nulla). Il suo contributo fluttua quindi sul tempo scaladegli ingressi delle molecole nel sottosistema. Su scale di tempi macroscopiche, il forzanteξ si puo quindi considerare, oltre che a media nulla, scorrelato nel tempo. Un processostocastico con tali caratteristiche e chiamato comunemente rumore bianco.
In generale, troviamo quindi che le fluttuazioni di una variabile macroscopica in unsistema termodinamico saranno descritte da una equazione con un forzante random e untermine dissipativo. Nel caso semplice di una singola variabile macroscopica r, abbiamoquindi per z ≡ ∆r:
z(t) + Γz(t) = ξ(t).
Questo e il tipo di equazione che dobbiamo risolvere per determinare la struttura tem-porale della fluttuazione di r. Per situazioni lontane dall’equilibrio, l’equazione diventera

106 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
tipicamente nonlineare, ma anche se determinare la sua forma puo non essere in gereralepossibile, sappiamo che essa dovra comunque descrivere una situazione di avvicinamentoall’equilibrio. Sovrapposto a questo moto di avvicinamento, ci sara una parte fluttuante,che, relativamente trascurabile nella fase iniziale, che rappresentera il grosso della variazio-ne di z all’equilibrio. Nella stessa maniera, l’entropia di Boltzmann S(z), partendo da unacondizione lontano dall’equilibrio, crescera nel tempo, ma ad essa sara sempre sovrappostauna componente fluttuante, che rimarra l’unica dipendente dal tempo all’equilibrio.
Gran parte della informazione rilevante sulla struttura temporale delle fluttuazioni econtenuta nella funzione di correlazione C(t) = 〈z(t)z(0)〉, ed e ad essa che rivolgiamo lanostra attenzione. Possiamo scrivere la funzione di correlazione in funzione della mediacondizionata 〈z(t)|z(0)〉 tramite la relazione
C(t) = 〈〈z(t)|z(0)〉z(0)〉.
L’equazione per 〈z(t)|z(0)〉 si ottiene immediatamente prendendo per t > 0 la mediacondizionata della equazione per y:
(d
dt+ Γ)〈z(t)|z(0)〉 = 〈ξ(t)|z(0)〉 = 0,
dove ho sfruttato il fatto che ξ(t) e scorrelato e z(0), per causalita, puo dipendere solo davalori di ξ a tempi minori di zero. Moltiplicando l’equazione per la media condizionata perz(0) e prendendo la media, otteniamo infine
(d
dt+ Γ)C(t) = 0,
che ha soluzione C(t) = 〈z2〉e−Γt. Valendo per stazionarieta C(t) = C(−t), possiamoscrivere per t generico:
C(t) = 〈z2〉e−Γ|t|. (6.7)
Vediamo quindi che la scala temporale su cui la grandezza r fluttua, e data dal tempo didissipazione Γ−1.
6.2.1 Il moto Browniano
Un’applicazione importante (e in pratica il punto di partenza della teoria delle fluttua-zioni) e il problema del moto Browniano. Si tratta del cammino random che particellemicroscopiche in sospensione in un fluido (per esempio polline in acqua) effettuano a causadegli urti molecolari. Il problema e la determinazione del coefficiente di diffusione delleparticelle in funzione della loro massa, e dimensione e della temperatura del liquido.
Il punto di partenza e l’osservazione che il liquido e la particella in sospensione costi-tuiscono un sistema isolato in equilibrio. Allo stesso tempo, la particella in sospensionecostituisce un sottosistema macroscopico del sistema isolato. Un parametro che descrive

6.2. STRUTTURA TEMPORALE DELLE FLUTTUAZIONI 107
la nostra particella e la sua energia cinetica U = 12Mu2, che sara distribuita secondo la
distribuzione di Gibbs (5.16) (riutilizziamo per una volta la costante di Boltzmann):
dP = C exp(− U
KT)d3u.
Notiamo come interessandoci solo del movimento macroscopico della particella, il Γ-spaziodel sottosistema siano solo le tre componenti della velocita. Da qui otteniamo subito
〈u2〉 ∼ KT
M.
Allo stesso tempo pero, il moto della particella nel fluido deve essere descritto da unaequazione del tipo
u+ Γu = ξ,
dove MΓ e il coefficiente di attrito viscoso di una particella nel fluido. Dimensionalmentepossiamo scrivere
Γ
M∼ µνR−2,
dove ν e la viscosita cinematica del fluido (per l’acqua, ν ≃ 0.01cm2/s) e R e la dimensionedella particella. Sostituendo nella espressione per la correlazione temporale per u datadalla (6.7):
〈u(0)u(t)〉 = 〈u2〉e−Γ|t| ∼ KT
Mexp(−C(ν/R2)|t|).
A partire da questa informazione, possiamo ottenere il coefficiente di diffusione della parti-cella Browniana, che e il rapporto tra la varianza del suo spostamento e il tempo impiegatoad effettuarlo. Questo calcolo va effettuato per tempi lunghi rispetto al tempo di corre-lazione Γ−1, di maniera tale che il moto della particella sia a tutti gli effetti del tipo diun cammino random. Indicando con ∆x(t) lo spostamento della particella nel tempo t,abbiamo evidentemente 〈∆x(t)〉 = 0 e
〈(∆x(t))2〉 =∫ t
0
dτ
∫ t
0
dτ ′〈u(τ)u(τ ′)〉
e ponendo t≫ Γ−1 [confrontare con la (2.38)]:
κB =〈(∆x(t))2〉
t∼
∫ ∞
−∞
dτ〈u(0)u(τ)〉 ∼ 〈u2〉Γ
∼ KTR2
Mν. (6.8)
Notare come a partire da proprieta macroscopiche, tutte facilmente misurabili, sia possibiledeterminare un parametro puramente microscopico come la costante di Boltzmann K.
A questo punto pero, va spesa qualche parola sull’uso di termini come microscopicoe macroscopico riferiti alla particella Browniana. Nessuno dei due e in effetti corretto,e la particella andrebbe definita piu propriamente “mesoscopica”, visto che la sua scalae comunque superiore a quella delle molecole e la sua dinamica e descritta da parametri

108 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
macroscopici come viscosita e attrito. D’altra parte gli effetti dalla microscala non sonotrascurabili, visto che e da essi che dipende la sua mobilita. Puo essere interessante aquesto punto ridare un’occhiata alla relazione fra entropia di Boltzmann e di Shannon,in una situazione in cui, appunto, le fluttuazioni del macrostato non sono consideratetrascurabili.
Troviamo infatti un esempio in cui la scala di definizione del macrostato (in questocaso la velocita della particella Browniana) e necessariamente piu piccola dell’ampiezza difluttuazione: ∆u ≪ σu. Il contributo alla entropia di Shannon del sistema dai gradi diliberta macroscopici non e quindi trascurabile; in analogia con la Eq. (5.4):
S = −〈ln(ρN !δΓ)〉 =∑
u
P (u)S(u)−∑
u
P (u) ln P (u),
dove
S(u) = −∫
∆Γ(u)
dΓρ(u) ln(ρ(u)N !δΓ) = ln(∆Γ(u)/(N !δΓ))
e l’entropia di Boltzmann del macrostato u e S = −∑
u P (u) ln P (u) e l’entropia di Shan-non dei gradi di liberta macroscopici della particella. Notare che il Γ-spazio relativo ai gradimicroscopici del sistema comprende tanto le molecole di fluido, che quelle della particellaBrowniana.
Notiamo l’importante relazione fra l’entropia di Boltzmann, il termine di dissipazionee la fluttuazione della variabile termodinamica. Nel caso del moto Browniano, la variabiletermodinamica e u, e un suo aumento provoca un’aumento di energia cinetica della parti-cella e quindi un “raffreddamento” e diminuzione della entropia S(u) associata ai gradi diliberta termici del sistema isolato. Questa diminuzione di entropia e l’esponente negativo−U/T = −Mu2/(2T ) nella distribuzione di Gibbs, che guarda caso e quadratico nellavariabile fluttuante u (il valore di equilibrio della variabile e evidentemente 〈u〉 = 0). Lamolla di richiamo che spinge u al suo valore di equilibrio e la forza di attrito lineare −Γu.
Notiamo di passaggio, che anche il gradiente con cui decresce l’entropia al crescere diu, e lineare in u, e sarebbe interessante immaginare che la forza della molla di richiamo eil gradiente di entropia ∇uS(U) siano in qualche maniera connessi. Vedremo che questaconnessione e stabilita dal fatto che la soluzione della equazione u + Γu = ξ deve esseredistribuita secondo la ρ(u) = C exp(−Mu2/(2T )).
6.2.2 Relazioni fluttuazione-dissipazione
Abbiamo visto come le fluttuazioni attorno all’equilibrio siano il risultato del bilancio tral’azione di forze molecolari e forze di frenamento. Le prime agiscono come sorgente dellefluttuazioni. Le seconde agiscono come molla di richiamo. Il bilancio fra le due determinal’ampiezza delle fluttuazioni. La stessa quantita e determinata a partire dalla entropia delsistema. Possiamo quindi eliminare dalle tre quantita incognite: forza molecolare, forzadi frenamento e ampiezza di fluttuazione, la forza molelare, rimanendo cosı con una solarelazione tra forza di richiamo e ampiezza di fluttuazione. Abbiamo visto questa strategia

6.2. STRUTTURA TEMPORALE DELLE FLUTTUAZIONI 109
al lavoro nel caso del moto Browniano. Presentiamo qui un approccio leggermente diverso,che evita tirare in ballo le forze molecolari.
Supponiamo di isolare nel nostro sistema una parte (il sottosistema) descritta da unagrandezza macroscopica m(pi, qi), dove pi, qi ≡ Γ sono le coordinate canoniche del-la parte in questione. Supponiamo che l’equilibrio termodinamico corrisponda a m = 0.Vediamo subito che ci sono essenzialmente due tipi di grandezze macroscopiche con cuipossiamo avere a che fare. Il primo esempio e del tipo della velocita della particella Bro-wniana, cioe m ≡ u: per tenere la particella in moto dobbiamo agire con una forza che sioppone all’attrito Γu, generando una potenza media Γu2 nel sistema. Il secondo esempioe la coordinata della stessa particella, immaginata pero in una buca di potenziale (senzabuca di potenziale, evidentemente non ci sarebbe un valore di equilibrio per m). Quindim ≡ x e in questo caso la forza serve a controbilanciare la forza conservativa della buca dipotenziale. In questo secondo caso, in media, non si esercita potenza. Il concetto di forzadi frenamento e evidentemente diverso nei due casi: nel primo e una forza di attrito. Nelsecondo e una forza di richiamo. In entrambi i casi, se la forza e sufficientemente piccola,possiamo aspettarci una dipendenza lineare dalla forza esterna:
〈m〉f ≃ Γmf,
dove 〈.〉f indica media sul sistema effettuata in presenza della forza f . La risposta linearedel sistema e quindi definita dalla relazione
Γm =d〈m〉fdf
∣
∣
∣
f=0. (6.9)
Consideriamo prima il caso della forza conservativa. Sia U(Γ) l’energia del sottosistemaisolato. Per quanto abbiamo detto, U sara minimo in x = 0, dove m ≡ x ha il senso di unacoordinata macroscopica del sistema. La forza necessaria per tenere il sistema nel nuovostato sara evidentemente f = −∂U/∂x. Un sistema con Hamiltoniana U(Γ) − fx(Γ))avra pertanto equilibrio in x(Γ) = xf 6= 0. Consideriamo ora il nostro sistema immersonel bagno termico. Esso sara descritto dalla distribuzione di Gibbs (si vedano le (5.16) e(5.18)):
ρ(Γ) =1
N !δΓexp
(F (〈x〉f )− U(Γ) + fx(Γ)
T
)
e la media sara
〈x〉f =∫
dΓ x(Γ) exp(F (〈x〉f )− U(Γ) + fx(Γ)
T
)
(6.10)
dove dΓ ≡ dΓ/(N !δΓ). Per ottenere la funzione di risposta, sostituiamo la (6.9) nella(6.10), ponendo m = x. Otteniamo, sfruttando la condizione ∂xF |x=0 = 0 (l’energia liberae minima all’equilibrio):
Γx =1
T
∫
dΓ(x(Γ))2 exp(F (0)− U(Γ)
T
)
=〈x2〉T
. (6.11)

110 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
La (6.11) e il primo esempio di relazione fluttuazione-dissipazione, valida nel caso diuna dinamica dominata da forze conservative. Notiamo come attraverso le derivate dellafunzione di partizione in presenza di forze esterne, possiamo ottenere medie di funzionigeneriche delle variabili microscopiche pi, qi. La (6.10) e la (6.11) possono essere scritteinfatti nella forma:
〈x〉f =dZ(f)
dfe 〈x2〉 = d2Z(f)
df 2
∣
∣
∣
f=0,
il che mostra come il metodo delle funzioni di partizione puo essere adattato per il calcolodi funzioni correlazione.
Nel caso puramente dissipativo, il sottosistema non esercita forze di risposta e l’unicareazione ad f e quella esercitata dal bagno termico. In questo caso la variabile macroscopicadi interessem e la “velocita” u = x, dove x ha la proprieta ∇Γx·∇ΓU = 0; in parole povere,variazioni di x non influenzano U . (Nel caso della particella Browniana: x = 1
N
∑
i qi e lacoordinata del centro di massa della particella, e l’energia della particella U = 1
2m
∑
i p2i
e infatti indipendente da x). L’Hamiltoniana modificata Uf = U − fx adesso descrive unsistema in cui la forza f porta a un’accelerazione indefinita del sistema. Nel caso dellaparticella Browniana, infatti: pi = −∂qiUf = f/N , e quindi u(t) = x(t) = u(0) + ft. La(6.10) diventa:
〈u〉f =∫
dΓ u(Γ) exp(F (〈u〉f )− U(Γ) + fx(Γ)
T
)
, (6.12)
e la (6.9) produce il risultato, ponendo m = u:
Γu =1
T
∫
dΓu(Γ)x(Γ) exp(F (0)− U(Γ)
T
)
=1
T
∫
dΓ u(t; Γ)
∫ t
−∞
dt′〈u(t′)|Γ, t〉 exp(F (0)− U(Γ)
T
)
=1
T
∫ 0
−∞
dt〈u(t)u(0)〉. (6.13)
Questa relazione, detta formula di Green-Kubo, fornisce il corrispettivo, valido nel casodi sistemi puramente dissipativi, della relazione fluttuazione-dissipazione (6.11). E facilevedere come la (6.13), nel caso del moto Browniano, ci riporta alla (6.8).
Nota. Possiamo generalizzare le relazioni fluttuazione-dissipazione a situazioni dipendentidal tempo. Per far cio, modifichiamo leggermente il nostro punto di vista, domandandociqual’e la forza di risposta del sistema a una perturbazione esterna. Per esempio, invece didomandarci la posizione media 〈x〉f di una particella in una buca di potenziale, in rispostaa una forza esterna f , ci chiediamo qual’e la forza di reazione media 〈X〉x che agiscesu di noi se fissiamo la particella in una certa posizione x. Ci concentriamo sul problemaconservativo, cosı che l’effetto della perturbazione e una variazione dell’energia del sistema.Per piccole perturbazioni, possiamo linearizzare:
U → Ux ≃ U +Xx,

6.2. STRUTTURA TEMPORALE DELLE FLUTTUAZIONI 111
dove X = dU/dx = X(Γ) e la forza di reazione a x, che ora, a differenza di x, e unaquantita fluttuante. Supponiamo che la perturbazione x(τ) sia stata accesa al tempo τ = 0,e che il sistema fosse inzialmente in equilibrio con un bagno termico a temperatura T . Aτ = 0, il sistema era quindi descritto dalla distribuzione di Gibbs:
ρ(Γ, 0) = ρ0(Γ) =1
Ze−βU , β = 1/T.
L’evoluzione a tempi successivi e ottenuta risolvendo l’equazione di Liouville, che possiamoscrivere nella forma ∂tρ+Ux, ρ) = 0, dove ., . indicano parentesi di Poisson nello spazioΓ: f, g = ∂pf∂qg−∂qf∂pg. Sfruttando il fatto che la perturbazione x e piccola, possiamoespandere ρ ≃ ρ0 + ρ1. A ordine piu basso in x, abbiamo ρ = ρ0, che e la distribuzione diequilibrio, ed e soluzione stazionaria della equazione di Liouville ∂tρ0 = −U, ρ0 = 0. AO(x), troviamo invece:
∂tρ1 + U, ρ1 := ∂tρ1 + L0ρ1 = −xX, ρ0,
dove si e introdotto l’operatore parentesi di Poisson rispetto a U : L0 ≡ U, .. Possia-mo risolvere in maniera formale, imponendo la condizione iniziale ρ1(0) = 0 (condizioneiniziale di equilibrio):
ρ1(t) = −∫ t
0
dτ x(τ)e−L0(t−τ)X, ρ0 = β
∫ t
0
dτ x(τ)e−L0(t−τ)ρ0X,U
= −β∫ t
0
dτ x(τ)e−L0(t−τ)ρ0X, (6.14)
dove X = U,X ≡ Γ∂ΓX(Γ) e la velocita di variazione di X(Γ) indotta dal flusso Hamilto-niano di U(Γ). Da qui troviamo per la forza di risposta media 〈X(t)〉x ≃
∫
dΓρ1(Γ, t)X(Γ):
〈X(t)〉x = −β∫ t
0
dτ x(τ)
∫
dΓ Xe−L0(t−τ)ρ0X = −β∫ t
0
dτ〈X(t− τ)X(0)〉x(τ),
dove si e sfruttata la relazione X(Γ(0))e−L0(t−τ) = X(Γ(t − τ)) (si ricordi che per x =0, ∂tρ = −L0ρ, ma X = L0X). Abbiamo trovato una nuova relazione fluttuazione-dissipazione (dinamica) per le forze di risposta del sistema
〈X(t)〉x =∫ t
0
dτ χ(t− τ)x(τ), χ(t) = βd
dt〈X(t)X(0)〉.
Notiamo come in analogia con il caso statico, il fatto che ci si e mantenuti all’interno diun’approssimazione lineare per la risposta, fa sı che la correlazione 〈X(t)X(0)〉 e calcolataper x = 0. (Ulteriori approfondimenti possono essere trovati nel testo “Non-equilibriumStatistical Mechanics” di R. Zwanzig).

112 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
6.3 Reversibilita temporale
Abbiamo visto che le fluttuazioni di una grandezza macroscopica z in un sistema ter-modinamico isolato sono associate a fluttuazioni della entropia di Boltzmann S(z). Laformazione di una fluttuazione e associata a una diminuzione di S(z) (raffreddamento delsistema), la dissipazione della fluttuazione e associata a un aumento di S(z) (riscalda-mento del sistema). Il riscaldamento-raffreddamento del sistema puo essere visto come untrasferimento da gradi di liberta microscopici (termici) a gradi di liberta macroscopici (lafluttuazione). Nel caso di un sistema isolato, cosı come le fluttuazioni della grandezza ma-croscopica hanno per definizione media nulla, anche le fluttuazioni dell’entropia avrannomedia nulla. Questo risultato cessa di essere vero nel caso di un sistema non isolato, e lefluttuazioni risultano essere in pratica il meccanismo attraverso il quale si genera l’aumen-to di entropia del sistema. Questo e particolarmente evidente nel caso della turbolenza:energia viene pompata dall’esterno (un fluido viene agitato), ma la dissipazione in caloredi questa energia avviene attraverso il passo intermedio della formazione delle fluttuazioniturbolente.
La produzione di entropia in un sistema non isolato e associata al fatto che la dinamicamacroscopica cessa di essere reversibile. Va sottolineato che il significato assunto qui dalconcetto di reversibilita e differente da quello riferito ai processi di quasi equilibrio intermodinamica. In questo caso, si dovrebbe parlare piu correttamente di simmetria rispettoall’inversione temporale delle fluttuazioni all’equilibrio. Questa simmetria e conseguenzadi due fatti:
• Il carattere stazionario dal punto di vista statistico del regime di equilibrio.
• La natura simmetrica per inversione temporale della dinamica microscopica di unsistema isolato.
Una conseguenza di questa simmetria e il fatto che una evoluzione temporale in un inter-vallo [0, t] della variabile termodinamica z(τ) deve avere la stessa probabilita di verificarsi(all’equilibrio) della evoluzione ottenuta per inversione temporale z(τ) = z(t− τ). In altreparole, deve essere soddisfatta la relazione fra PDF di storie del processo stocastico z(τ):
ρ[z] = ρ[z].
Questo ha come conseguenza, la seguente simmetria tra PDF congiunte di valori dellavariabile z ad istanti diversi:
ρ[z(t1) = a, z(t2) = b] = ρ[z(t1) = b, z(t2) = a]. (6.15)
Questa condizione di simmetria ha il nome di bilancio dettagliato. Notiamo che questacondizione non significa affatto che la PDF di passare da a a b e da b ad a, agli istanti t1e t2, siano uguali. Queste due sono le PDF condizionate
ρ(z(t2) = b|z(t1) = a) e ρ(z(t2) = a|z(t1) = b),

6.3. REVERSIBILITA TEMPORALE 113
dove evidentemente la PDF di passare da uno stato di equilibrio a uno di non equilibriosara minore di quella del processo inverso. Il bilancio dettagliato ci dice un’altra cosa: chela PDF di vedere una transizione da un valore di z di equilibrio a uno di non equilibriodeve essere uguale a quella del processo inverso (cio e d’altra parte abbastanza intuitivo,se per ogni dipartita dall’equilibrio ci deve essere poi un ritorno; la condizione di bilanciodettagliato e che questo ritorno deve avvenire in maniera identica alla dipartita).
6.3.1 Relazioni di Onsager
La condizione di bilancio dettagliato ha una importante conseguenza per quanto riguardail modo in cui le fluttuazioni vengono dissipate in condizioni di equilibrio. Il soddisfaci-mento di questa condizione si traduce in termini di correlazioni temporali nella importantesimmetria
〈zi(0)zj(t)〉 = 〈zj(0)zi(t)〉,che implica
〈zi(0)zj(t)〉 = 〈zj(0)zi(t)〉. (6.16)
La generalizzazione della equazione z = Γz + ξ al caso di piu variabili fluttuanti e eviden-tente
zi = Γijzj + ξi, (6.17)
che, sostituita nella (6.16), mi da
Γjk〈zi(0)zj(t)〉 = Γik〈zj(0)zi(t)〉. (6.18)
Prendendo il limite t→ 0+ della (6.18) e utilizzando la relazione 〈zjzi〉 = (α−1)ij troviamoquindi la simmetria
Γjk(α−1)ki = Γik(α
−1)kj.
Ora, noi possiamo scrivere l’equazione per le fluttuazioni (6.17) nella forma alternativa
zi = γij∂S
∂zj+ ξi = −γijαjkzk + ξi, (6.19)
dove ho utilizzato la relazione S = Seq − 12αijzizj [Eq. (6.1)]. Confrontando con la (6.17),
troviamo pero γij = Γik(α−1)kj, da cui si ottiene la seguente relazione, detta relazione di
Onsager:
γij = γji. (6.20)
La diminuzione ha di entropia associata alla fluttuazione di zj ha lo stesso effetto sullavariabile zi che la diminuzione associata alla variazione di zi avrebbe su zj.
I coefficienti cinetici γij godono di una seconda importante proprieta che discende dallacondizione di crescita di entropia. Dalle (6.1) e (6.19) possiamo infatti ottenere l’equazione

114 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
di evoluzione per l’entropia. Prendendo una condizione iniziale per S sufficiente granderispetto all’ampiezza delle fluttuazioni, ma abbastanza piccola che la dinamica resti lineare,la dinamica sara dominata dal rilassamento:
S =∂S
∂zizi = γij
∂S
∂zi
∂S
∂zj
La condizione di aumento di entropia nell’approccio all’equilibrio, ci dice che γij deve essereuna matrice non solo simmetrica, ma anche definita positiva.
Nota. Di fatto, non e necessario invocare uno stato di equilibrio termodinamico, in cui levariabili termodinamiche fluttuano vicino ai loro valori di equilibrio, per avere reversibilitatemporale. La distribuzione microcanonica di un sistema tiene in conto della possibilita, inprincipio, di fluttuazioni di ampiezza arbitraria. Il problema, come abbiamo visto, e cheper osservare simili fluttuazioni sarebbe necessario attendere un tempo molto maggiore dellavita dell’universo. Ludwig Boltzmann a suo tempo ipotizzo che l’universo nel quale viviamoe la freccia del tempo che la caratterizza non sia altro che il rilassamento all’equilibriodi una fluttuazione cosmica. La condizione iniziale del nostro universo sarebbe l’istantedi ampiezza massima (e quindi minima entropia) della fluttuazione. A questo punto, lariconciliazione fra una dinamica microscopica simmetrica per inversione temporale ed unamacroscopica che non lo e, risiederebbe nel fatto che dall’altra parte (nel tempo) di questominimo dell’entropia, ci sarebbe una freccia del tempo con direzione opposta alla nostra.In altre parole, l’evoluzione dell’universo (secondo Boltzmann) sarebbe una sequenza diimprobabili fluttuazioni, presso le quali, ad ambo i lati delle stesse, esisterebbe una frecciadel tempo, mentre per il resto della durata di vita dell’universo una freccia del tempo nonesisterebbe.
6.3.2 Produzione di entropia
Vogliamo studiare a questo punto come il fatto che il sistema sia non isolato porta allarottura della simmetria e nel contempo a una crescita di entropia. Facciamo cio conun sistema semplificato: una particella sottoposta a una forza esterna dipendente dallaposizione F, e che sente la presenza del bagno termico attraverso un attrito viscoso el’effetto degli urti molecolari. Il sistema e supposto sovrasmorzato, nel senso che le forzedi attrito sono molto piu intense di quelle esterne. Indichiamo con x la posizione dellaparticella e supponiamo per semplicita l’attrito lineare in x. Abbiamo quindi il sistema
M x = −Γx+ F(x) + ξ, (6.21)
dove ξ e l’effetto degli urti molecolari ed M e la massa della particella. Limite sovrasmor-zato significa essenzialmente che il termine inerzialeM x puo essere trascurato e l’equazionedel moto diventa
Γx = F(x) + ξ. (6.22)

6.3. REVERSIBILITA TEMPORALE 115
Il calore somministrato al sistema in media non e altro che il lavoro medio esercitato dallaforza esterna sulla particella. La produzione media di entropia sara pertanto
∆S =1
T
∫ t
0
dτ〈F(x(τ)) · x(τ)〉dτ. (6.23)
Vediamo subito che se la forza e conservativa, non si ha produzione di entropia. Scrivendola forza in termini di un potenziale: F = −∇V , avremmo infatti, sostituendo nella (6.23):
S(t)− S(0) = − 1
T
⟨
∫ t
0
dτ 〈x(τ) · ∇V (x(τ))⟩
= − 1
T〈[V (x(t))− V (x(0))]〉,
che e zero in condizioni stazionarie. Confrontando con la (6.21), notiamo come in pre-senza di forze conservative, bilancio entropico nullo implica bilancio in media fra produ-zione di entropia Γx2 (distruzione delle fluttuazioni) e sua distruzione ξx (produzione difluttuazioni).
Vogliamo studiare la relazione che esiste fra produzione di entropia e asimmetria fratraiettorie in avanti e indietro nel tempo, in presenza di forze non conservative. L’oggettoche vogliamo studiare e la PDF della traiettoria ρ[x|x(0)] nell’intervallo [0, t] a partire dauna condizione iniziale a t = 0. Per far cio, discretizziamo l’equazione del moto (6.22), chediventa
xk+1 − xk − Fk∆τ = ∆Bk,
dove ∆B(τk) =∫ τk+1
τkξ(τ)dτ , xk ≡ x(τk), Fk = [F(x(τk+1)) + F(x(τk))]/2, e abbiamo
posto per semplicita Γ = 1. Notiamo subito che essendo ξ(τ) scorrelato nel tempo, anche i∆B(τk) a tempi diversi lo saranno. Questo ha fra l’altro come conseguenza il fatto che x(τk)e un processo Markoviano. Notiamo anche che, essendo ∆Bk la somma di infiniti contributiξdτ scorrelati fra loro e identicamente distribuiti, si puo supporre che valga il teorema dellimite centrale (2.25) e ∆B sia distribuita Gaussianamente. Supponendo che le diversecomponenti di ∆B siano identicamente distribuite, troviamo subito 〈(∆Bi)
2〉 = D∆τ .Possiamo pertanto scrivere:
ρ(∆B) ∼ exp(
− |∆B|22D∆t
)
. (6.24)
Utilizzando l’equazione del moto discretizzata, possiamo scrivere i ∆B(tk) che entrano inρ[∆B] in funzione di x(tk) e x(tk+1):
ρ(xk+1|xk) =∼ exp(
− |xk+1 − xk − Fk∆τ |22D∆τ
− Jk∆τ)
.
dove Jn e il contributo dallo Jacobiano nel passaggio da ρ∆B a ρxn+1 .
Nota Il contributo Jacobiano dipende dal fatto che abbiamo utilizzato una espressione“semi-implicita” per Fn. Abbiamo di nuovo un esempio di regole dell’analisi che smettono

116 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
di funzionare nel caso di processi stocastici. Se avessimo posto Fk = F(x(τk)), avremmoottenuto immediatamente Jk = 0, mentre con Fk = [F(x(τk+1)) + F(x(τk))]/2 abbiamo:
det[∂∆Bk
∂xk+1
]
= det[
δij −∂Fk,i∂xk+1,j
∆τ]
= 1− (∇xk+1· Fk)∆τ +O((∆τ)2)
= exp(−Jk∆τ) +O((∆τ)2),
dove Jk = ∇xk+1· Fk = (1/2)∇ · F(xk+1). L’approssimazione “esplicita” Fk ≃ F(x(τk))
rimane inaccurata anche nel limite ∆τ → 0.
Per ottenere la PDF della storia x(tk), k = 1, 2, . . ., sfruttiamo il fatto che x(tk) e unprocesso Markoviano:
ρ[x] ≃ ρ(xn,xn−1, . . . ,x0)
= ρ(xn|xn−1)ρ(xn−1|xn−2) . . . ρ(x1|x0)ρ(x0) (6.25)
(un simile oggetto e detto una catena di Markov). Sostituendo la (6.24) nella (6.25):
ρ[x|x(0)] ∼ exp[
−∑
k
( |xk+1 − xk − Fk∆τ |22D∆τ
+ Jk∆τ)]
. (6.26)
Nella stessa identica maniera possiamo scrivere la PDF della storia corrispondente al pro-cesso inverso ρ[x|x(0)], dove x(τ) = x(t − τ) e quindi la condizione iniziale del processoinverso e la condizione finale per il processo diretto: x(0) = x(t). Definendo xk = xn−k eFk = [F(xk+1) + F(xk)]/2:
ρ[x|x(0)] ∼ exp[
−∑
k
( |xk+1 − xk − Fk∆τ |22D∆τ
+ Jk∆τ)]
.
= exp[
−∑
k
( |xk+1 − xk + Fk∆τ |22D∆τ
+ Jk∆τ)]
. (6.27)
Consideriamo ora il logaritmo del rapporto delle PDF dei due processi diretto e inverso(6.25) e (6.26). Otteniamo:
ln(ρ[x|x(0)]ρ[x|x(0)]
)
=2
D∆t
∑
k
Fk · (xk+1 − xk)∆t
≃ 2
D
∫ t
0
F(x(τ))x(τ)dτ =2Q
D, (6.28)
dove Q e il calore trasferito al sistema per azione della forza F lungo la traiettoria x.Ponendo uguali le condizioni iniziali e finali x(0) ≡ x(t) = x(0), vediamo subito che, incondizioni stazionarie, dobbiamo avere ρ[x(0) = x0] = ρ[x(t) = x0]. Questo ci permette di

6.4. ESERCIZI 117
scrivere ρ[x|x(0)]ρ(x(0)) = ρ[x], e allo stesso tempo ρ[x|x(0)]ρ(x(0)) = ρ[x|x(0)]ρ(x(0)) =ρ[x]. Troviamo quindi dalla (6.28), nel caso di traiettorie cicliche:
ln(ρ[x]
ρ[x]
)
≃ 2Q
D, (6.29)
e questa espressione sara differente da zero nel caso di forze non conservative. Abbiamotrovato un esempio esplicito della connessione tra forze non conservative e violazione dellasimmetria per inversione temporale ρ[x] 6= ρ[x]. Troviamo in particolare il risultato che letraiettorie associate a trasferimento di calore al sistema, sono anche quelle la cui proba-bilita e maggiore di quella delle loro versioni invertite nel tempo. E abbastanza intuitivocomprendere cosa si sta verificando cnel caso di una particella costretta a muoversi su unanello da una forza non conservativa. Una traiettoria tipica x corrispondera alla particellache gira lungo l’anello nello stesso stesso della forza, mentre x sarebbe quella estremamenteimprobabile traiettoria in cui gli urti molecolari spingono la particella in verso opposto allaforza esterna. Il fatto che le traiettorie piu probabili siano quelle associate a un calorepositivo ceduto al sistema fa sı che la media del lato destro della (6.29) sia una quantitapositiva, che e pari a T volte la produzione di entropia da parte della forza esterna:
〈ln ρ[x]〉 − 〈ln ρ[x]〉 = 2T
D[S(t)− S(0)] ≥ 0.
(si ricorda che tutte le medie sono calcolate rispetto alla PDF ρ[x] e che x e una traiettoriaciclica).
6.4 Esercizi
Esercizio 1 Si immagini che una particella Browniana sia attaccata con una molla aun supporto fisso. Come si dovrebbe modificare l’equazione del moto della particella?Descrivere qualitativamente il comportamento della particella in funzione della forza dellamolla, dell’attrito del fluido e della temperatura. Determinare la distanza quadratica mediadella particella dal supporto.
Esercizio 2 Si consideri la parte fluttuante del campo di temperatura e la si esprima comeintegrale di Fourier (componenti spaziali).
• Calcolare la diminuzione di entropia in funzione dell’ampiezza delle componenti diFourier di T . Studiare quindi la dipendenza spaziale delle fluttuazioni.
• Si consideri l’equazione di trasporto del calore in assenza di contributi di compressionee di dissipazione viscosa ∂tT = κ∇2T . Da qui si ottenga l’equazione del moto per lecomponenti di Fourier di T e si scriva la funzione di correlazione temporale di questefluttuazioni.

118 CAPITOLO 6. CENNI ALLA TEORIA DELLE FLUTTUAZIONI
Esercizio 3 Considerare una particella Browniana immersa in un flusso di Couette u =(αy, 0, 0). Stimare la produzione d’entropia della particella (Suggerimento: in un fluidofermo, ogni collisione molecolare cede entropia alla particella, che la restituisce poi al fluidoattraverso le forze d’attrito. Nel fluido in moto, le collisioni spingono la particella in puntidel fluido dove questo si muove piu velocemente rispetto alla particella, cosı che l’attritodeve eseguire un lavoro maggiore per fermarla).


















