
Neurologia Fazi0 cap26 Malattie Demielinizzanti
25
26. Malattie demielinizzanti G.L. Mancardi, A. Uccelli Molte malattie del sistema nervoso centrale (SNC) sono responsabili di una sofferenza della mielina, ma il termine “malattia demie- linizzante” si riferisce convenzionalmente ad un gruppo ben preciso e limitato di malattie di natura infiammatoria caratterizzate, sul piano neuropatologico, dalla perdita primiti- va della guaina mielinica cui può conseguire o meno la conservazione dell'assone. Non prenderemo in considerazione pertanto le malattie della mielina secondarie ad un pro- cesso infettivo, quali ad esempio la leucoen- cefalopatia multifocale progressiva e le leu- codistrofie in cui la degenerazione mielinica è secondaria ad un difetto genetico primitivo della mielina stessa. Saranno anche escluse tutte le malattie in cui una qualunque causa determina una degenerazione mielinica, se- condaria ad un danno del soma o dell'assone del neurone, come ad esempio accade nei traumi cranici o nelle malattie cerebrovasco- lari. Più rare sono le condizioni in cui la sof- ferenza del SNC, sotto forma di una demie- linizzazione periassiale compare nell'ambito di una malattia sistemica o di una malattia nota. Il termine “malattie demielinizzanti” viene pertanto convenzionalmente utilizzato per individuare un gruppo di malattie carat- terizzate da una sofferenza della mielina se- condaria ad un processo infiammatorio di probabile natura autoimmunitaria ed il cui de- corso può essere cronico, come nella sclero- si multipla, o acuto, come nella encefalomie- lite acuta disseminata, o iperacuto, come nella encefalomielite acuta emorragica. Sclerosi multipla Dopo i traumi cranici, la Sclerosi Multipla (SM) è la malattia più frequentemente respon- sabile di disabilità neurologica nell'età giova- nile-adulta. Per le difficoltà di comprensione dei meccanismi eziopatogenetici, per i proble- mi terapeutici e il profondo impatto che ha sui pazienti e sulla loro vita, la SM rappresenta una delle malattie con cui è più difficile con- frontarsi. Descritta inizialmente da Cruveilhier (1835), nel 1868 Charcot ne delineava già con precisio- ne gli aspetti clinici e neuropatologici quali le multiple aree di demielinizzazione sparse nel SNC, con predilezione per le vie lunghe e la sostanza bianca periventricolare, midollo spina- le, nervi ottici, tronco encefalico e cervelletto. Il decorso è variabile e imprevedibile, ma più spesso la malattia si manifesta con ricorrenti episodi di sofferenza focale del SNC che tendo- no inizialmente alla regressione spontanea, ma che con il passare del tempo sono responsabili di deficit neurologici non più reversibili. Dopo anni dall'esordio la malattia assume frequente- mente un andamento progressivo, con o senza esacerbazioni. Anche se la causa è a tutt'oggi sconosciuta, la SM viene considerata, sulla base di dati cli- nici, di laboratorio e sperimentali, una malattia autoimmune secondaria ad una risposta diretta contro un evento “trigger”, verosimilmente un patogeno ubiquitario, che determina il ricono- scimento a bassa affinità di auto-antigeni, pro- babilmente mielinici, da parte di linfociti T
-
Upload
andrea-torquati -
Category
Documents
-
view
22 -
download
5
description
cap 26 neuro
Transcript of Neurologia Fazi0 cap26 Malattie Demielinizzanti

1111Malattie demielinizzanti
26. Malattie demielinizzanti
G.L. Mancardi, A. Uccelli
Molte malattie del sistema nervoso centrale(SNC) sono responsabili di una sofferenzadella mielina, ma il termine “malattia demie-linizzante” si riferisce convenzionalmente adun gruppo ben preciso e limitato di malattiedi natura infiammatoria caratterizzate, sulpiano neuropatologico, dalla perdita primiti-va della guaina mielinica cui può conseguireo meno la conservazione dell'assone. Nonprenderemo in considerazione pertanto lemalattie della mielina secondarie ad un pro-cesso infettivo, quali ad esempio la leucoen-cefalopatia multifocale progressiva e le leu-codistrofie in cui la degenerazione mielinicaè secondaria ad un difetto genetico primitivodella mielina stessa. Saranno anche esclusetutte le malattie in cui una qualunque causadetermina una degenerazione mielinica, se-condaria ad un danno del soma o dell'assonedel neurone, come ad esempio accade neitraumi cranici o nelle malattie cerebrovasco-lari. Più rare sono le condizioni in cui la sof-ferenza del SNC, sotto forma di una demie-linizzazione periassiale compare nell'ambitodi una malattia sistemica o di una malattianota. Il termine “malattie demielinizzanti”viene pertanto convenzionalmente utilizzatoper individuare un gruppo di malattie carat-terizzate da una sofferenza della mielina se-condaria ad un processo infiammatorio diprobabile natura autoimmunitaria ed il cui de-corso può essere cronico, come nella sclero-si multipla, o acuto, come nella encefalomie-lite acuta disseminata, o iperacuto, come nellaencefalomielite acuta emorragica.
Sclerosi multipla
Dopo i traumi cranici, la Sclerosi Multipla(SM) è la malattia più frequentemente respon-sabile di disabilità neurologica nell'età giova-nile-adulta. Per le difficoltà di comprensionedei meccanismi eziopatogenetici, per i proble-mi terapeutici e il profondo impatto che ha suipazienti e sulla loro vita, la SM rappresentauna delle malattie con cui è più difficile con-frontarsi.
Descritta inizialmente da Cruveilhier (1835),nel 1868 Charcot ne delineava già con precisio-ne gli aspetti clinici e neuropatologici quali lemultiple aree di demielinizzazione sparse nelSNC, con predilezione per le vie lunghe e lasostanza bianca periventricolare, midollo spina-le, nervi ottici, tronco encefalico e cervelletto.Il decorso è variabile e imprevedibile, ma piùspesso la malattia si manifesta con ricorrentiepisodi di sofferenza focale del SNC che tendo-no inizialmente alla regressione spontanea, mache con il passare del tempo sono responsabilidi deficit neurologici non più reversibili. Dopoanni dall'esordio la malattia assume frequente-mente un andamento progressivo, con o senzaesacerbazioni.
Anche se la causa è a tutt'oggi sconosciuta,la SM viene considerata, sulla base di dati cli-nici, di laboratorio e sperimentali, una malattiaautoimmune secondaria ad una risposta direttacontro un evento “trigger”, verosimilmente unpatogeno ubiquitario, che determina il ricono-scimento a bassa affinità di auto-antigeni, pro-babilmente mielinici, da parte di linfociti T

1112 Malattie del sistema nervoso
autoreattivi che hanno eluso il processo di se-lezione negativa nel timo. La suscettibilità in-dividuale dipenderebbe altresì dal “background”genetico da cui potrebbero dipendere le moda-lità della risposta immune, forse in relazioneall'aplotipo MHC.
EPIDEMIOLOGIA
La SM è più frequente nel sesso femminile, essendoil rapporto femmine/maschi di circa 2:1 (variabile da 1,9a 3,1). L'esordio è raro prima della pubertà e dopo i 65anni, situandosi in genere fra i 20 e i 45 anni, con un piccodi incidenza per età intorno ai 30 anni, anche se ben do-cumentati sono casi in età infantile o tardivi. La preva-lenza più alta è nelle isole Orcadi (300/100.000), men-tre nel Nord Europa la prevalenza è intorno a circa 100casi/100.000 abitanti. La tesi che l'Italia rappresenti unazona a rischio relativamente basso rispetto al Nord Eu-ropa è stata rivista da studi epidemiologici accurati effet-tuati in questi ultimi anni, che hanno permesso di eviden-ziare tassi di circa 50-80 casi /100.000 abitanti, collocan-do quindi il nostro fra i paesi ad alto rischio di malattia.La maggior parte degli studi epidemiologici ha eviden-ziato un rapporto fra SM e latitudine: la malattia è moltorara intorno ai tropici (dal 23 N al 23 S), e aumenta difrequenza con l'aumentare della latitudine fino a diven-tare comune fra il 50 e il 60 N e in minor misura fra il50 e il 60 Sud, per poi diminuire nuovamente. Questi datisembrano suggerire l'esistenza di fattori ambientali legatiin modo particolare al “mondo occidentale”, con ungradiente di frequenza in rapporto al variare della latitu-dine. Tale ipotesi è stata tuttavia recentemente messa indubbio dagli studi di genetica che hanno dimostratoun'associazione tra la distribuzione geografica e fattorigenetici della popolazione. In Sardegna, per esempio,un'alta prevalenza della malattia (superiore a 100 casi/100.000 abitanti) si associa con un particolare aplotipodel sistema maggiore di istocompatibilità (DR4). In talsenso va interpretato il dato di una scarsa incidenza dimalattia in popolazioni quali i Giapponesi, gli asiatici chevivono in Gran Bretagna, gli zingari di Ungheria, gli afro-americani degli USA, i maori della Nuova Zelanda o gliafricani del Sud Africa, gruppi razziali che vivono in zonegeograficamente definite ad alto rischio. Le differenzeosservate nella distribuzione geografica sono quindi ingran parte dovute alle caratteristiche genetiche della po-polazione anche se altri fattori, per esempio ambientali,potrebbero giustificare la presenza di epidemie di SM,documentate nelle isole Faroe dopo la seconda guerramondiale probabilmente in seguito all'occupazione del-le truppe britanniche. Inoltre, studi di migrazione hannoevidenziato che le popolazioni migranti tendono a con-
servare il rischio della zona di origine quando la migra-zione avviene dopo il 15° anno di vita, mentre al contra-rio acquistano il rischio del nuovo paese di residenzaquando la migrazione avviene prima del 15° anno di vita.Questi ultimi dati suggerirebbero pertanto l'esistenza difattori ambientali che agirebbero in epoca infantile o nellaprima adolescenza.
EZIOPATOGENESI
Benché la causa della SM sia tuttora ignota numero-se evidenze suggeriscono la possibilità che si tratti di unamalattia autoimmunitaria in cui fattori immunologici,ambientali e genetici svolgono un ruolo. In tal senso, que-st'ipotesi si basa su evidenze cliniche e sperimentali chederivano da:
• studi di genetica ed in particolare dall'associazio-ne con molecole del MHC di classe II
• somiglianza con il modello sperimentale anima-le, encefalite autoimmune sperimentale (EAS)
• produzione di immunoglobuline clonali all'inter-no del SNC
• studi immunologici su pazienti con SM• neuropatologia della placca di demielinizzazione• risposta clinica a trattamenti immunomodulanti
o immunosoppressivi
Fattori genetici. - L'esistenza di una suscettibilitàgeneticamente determinata a contrarre la malattia è av-valorata da numerosi studi su gruppi etnici, su clusterfamigliari e sui gemelli: in alcune razze la malattia è rara,mentre in altre, in particolare nella razza caucasica, è par-ticolarmente frequente; è nota la possibilità di casi fa-miliari, supportata dal fatto che il rischio di contrarre lamalattia è più alto nei fratelli di soggetti con SM e gra-dualmente diminuisce nei figli e più ancora nei cugini.Si può ritenere che per i parenti di primo grado esista unrischio di circa l'1-3% di contrarre la malattia rispetto alrischio di 1/1000 -2000 nella popolazione generale. Studisu gemelli hanno rilevato una concordanza per la presen-za di SM tra il 25 ed il 30% nei monozigoti, contro il 2-5% nei dizigoti. Il concetto di ereditarietà per questamalattia non è basato sull'ipotesi di un singolo gene cau-sale, ipotesi in contrasto con le stime di concordanzafamigliare e con l'osservazione di una riduzione non li-neare del rischio nei soggetti geneticamente più lontanidal probando. È probabile invece che la suscettibilità siadeterminata da molteplici loci indipendenti l'uno dall'al-tro (ereditarietà poligenica), ciascuno dei quali contribu-isce in modo limitato al rischio complessivo. Studi dilinkage e di associazione hanno indicato il complessomaggiore di istocompatibilità (MHC) sul cromosoma 6come uno dei determinanti genetici della SM. Questocomplesso codifica per gli antigeni di istocompatibilità

1113Malattie demielinizzanti
(sistema HLA) che presentano gli antigeni proteici ailinfociti T. Gli antigeni di classe II sono stati più frequen-temente associati alla malattia ed in particolare, nella po-polazione caucasica, gli aplotipi DRB1 *1501, DQA1 *0102, DQB1 * 0602 dell'allele DR2. In altre razze tutta-via la malattia è associata a differenti aplotipi HLA, qualiad es., in Sardegna, l'HLA-DR4 e DQW3. È stata ripor-tata inoltre un'associazione fra la malattia e alcuni locidella catena Vb del recettore del linfocita T sul cromo-soma 7, delle immunoglobuline sul cromosoma 19 e nellapopolazione finlandese ad un gene legato alla proteinabasica della mielina sul cromosoma 18.
Agenti infettivi. - Studi epidemiologici suggerisconola possibilità che individui con SM siano stati espostinell'infanzia a fattori ambientali, probabilmente patogenivirali e non. Inoltre, un rischio aumentato di contrarre lamalattia è stato associato con migliori condizioni socio-economiche, cosa che potrebbe suggerire un migliora-mento delle condizioni igienico sanitarie della popolazio-ne e pertanto una ritardata esposizione a certi patogeni.Numerosi agenti infettivi sono stati correlati alla SM main nessuno caso si è arrivati ad una dimostrazione con-clusiva di associazione. Tra i molti agenti studiati vi è ilvirus del morbillo, l'Epstein Barr virus (EBV), il virusSimian 5 (SV5), gli Herpes virus tra cui l'HSV6, alcuniretrovirus compreso l'HTLV1 e recentemente la Chlamy-dia pneumoniae. La possibilità di una origine virale dellamalattia è suggerita anche da studi sperimentali sull'ani-male. Infatti, l'infezione con alcuni ceppi virali quali peresempio il virus Theiler è in grado di provocare in alcu-ne specie murine una forma di encefalomielite demieli-nizzante cronica somigliante alla SM alla cui patogenesicontribuiscono linfociti T CD8+ diretti verso l'agenteinfettante e le proteine mieliniche.
Una forte associazione tra agenti infettivi e SM sca-turisce da importanti dati ottenuti nell'EAS dove celluleautoreattive presenti nel repertorio periferico degli ani-mali possono essere attivate attraverso un meccanismo dimimetismo molecolare tra autoantigeni e proteine micro-biche o attraverso una stimolazione superantigenica.
In conclusione, numerosi dati sperimentali suggeri-scono la possibilità che, in soggetti geneticamente pre-disposti, un agente infettivo probabilmente non unico, siain grado di scatenare una risposta autoimmunitaria a se-guito di un meccanismo di mimetismo molecolare. Allapropagazione di questo meccanismo contribuirebbero poialtri fattori che amplificherebbero la risposta verso altriantigeni del SNC, provocando così un danno tissutale ela cronicizzazione della malattia (Fig. 26.1).
Fattori immunologici. - Molte delle nostre conoscen-ze sull'immunologia della SM derivano da studi speri-mentali sull'EAS, il modello animale della malattia.
L'EAS è una malattia autoimmunitaria del SNC carat-terizzata da infiltrati linfo-monocitari, gliosi e, in alcu-ne specie, da demielinizzazione e talora remielinizza-zione. Il tipo di malattia dipende dalla specie e dal cep-po animale (per esempio specie “inbred”, quali i rodi-tori utilizzati in laboratorio, animali geneticamente iden-tici tra loro, oppure specie “outbred”, quali i primati,animali geneticamente eterogenei e quindi simili all'uo-mo) utilizzato e dal protocollo di immunizzazione. Ti-picamente, la malattia è indotta in specie “inbred” su-scettibili attraverso l'immunizzazione con omogenatomielinico o con singole frazioni proteiche quali MBP(“myelin basic protein”), MOG (“myelin oligodendro-cyte glycoprotein”) e PLP (“proteolipid protein”). Taliproteine sono “processate” a livello dei linfonodi regio-nali e presentate da cellule (APC) professionali, qualile cellule dendritiche, a linfociti T CD4+ reattivi versol'antigene immunogenico. Alcune di queste cellule at-tivate sono in grado di attraversare la barriera emato-encefalica (BEE) e migrare nel SNC dove riconosconole proteine mieliniche “self” presentate nell'ambito dimolecole MHC di classe II su cellule presentanti l'anti-gene quali macrofagi, microglia e forse astrociti. Il ri-lascio di citochine (IL-1, TNF-α, IL-12 ed altre anco-ra) e chemochine (per esempio IP-10, RANTES, MIP-1a) pro-infiammatorie produce una cascata di fenome-ni immunologici che inducono l'espressione di molecoledi adesione sull'endotelio dei piccoli vasi capillari al-terando perciò la permeabilità della BEE e richiaman-do una seconda ondata di linfociti T nel sito di infiam-mazione (Fig. 26.1). Nei roditori, durante le fasi inizialidella malattia, la risposta T è generalmente focalizzataverso un epitopo immunodominante dell'antigene mieli-nico ed è mediata da popolazioni linfocitarie clonali.Nelle fasi più tardive della malattia altri epitopi nell'am-bito della stessa molecola e di altre molecole risultanoesposte in seguito al danno tissutale e pertanto diven-tano il bersaglio della risposta autoimmune, un fenome-no noto come “epitope spreading”. Il ruolo patologicodi questi linfociti T antigene-specifici è stato dimostra-to dagli esperimenti di trasferimento passivo che han-no permesso di indurre la malattia grazie al trasferimen-to di cellule T da un animale malato ad un altro sanosingenico. Nell'uomo così come in altri modelli animali“outbred” la risposta cellulare periferica verso le pro-teine mieliniche è risultata tuttavia più complessa edeterogenea, non permettendo così l'utilizzo di quellestrategie terapeutiche atte colpire le popolazioni ence-falitogeniche clonali che erano state usate con succes-so nei modelli “inbred”. Almeno tre possono essere lespiegazioni di questi diversi risultati tra l'uomo e le spe-cie animali studiate. Innanzitutto le diverse caratteristi-che genetiche delle specie, “outbred” i primi, “inbred”le seconde. In secondo luogo un'iniziale restrizione

1114 Malattie del sistema nervoso
della risposta autoimmune potrebbe andare perdutanell'uomo, specie nella quale gli studi vengono esegui-ti solo dopo la diagnosi, momento probabilmente lon-tano dalla fase iniziale subclinica del processo autoim-mune. Allo stesso modo, un'eventuale restrizione dellarisposta potrebbe non essere osservabile nel sangue pe-riferico e pertanto risultare perduta per l'impossibilitàdi studiare “in vivo” la risposta immune nell'organo ber-saglio, cioè nel SNC. L'utilizzo di primati “outbred”
(per esempio il macaco ed il marmoset) ha permesso disuperare i limiti dovuti al background genetico dellespecie utilizzate fornendo un modello sperimentale piùsimile all'uomo.
Molteplici meccanismi effettori sono probabilmentealla base del danno mielinico nella EAS (Fig. 26.1). Traquesti giocano un ruolo fondamentale oltre ai linfociti Te alle cellule “Natural Killer” ad azione citotossica, an-che autoanticorpi verso le proteine mieliniche, nonchè
M∅ M∅
Linfociti T attivati CD4Linfociti T
attivati CD4
Assone
TNF-α
M∅
IL-4
RANTESIP-10
ROI
NO
Microglia
BBB
Neuron
Oligodendrocita
C C
Linfocita B
VC
AM
HLA
DR
ICA
MVLA
4
LFA
2
MMPs
ROI
TNF-α, IFN-γ
VC
AM
HLA
DR
ICA
M
CCC
Astrocita
Th1 CD4+ Th2 CD4+
CD28/B7
TCR/Ag/MHC
IL-12, IL-1
IL-12, IL-1
MIP-1αIP-10
TNF-α, IFN-γ
MMPs
IFN-γ
TNF-β
CD8+
TGF-β
IL-10
NO
MMPs
NKCellula NK
M∅ M∅
Linfociti T attivati CD4Linfociti T
attivati CD4
Assone
TNF-αTNF-α
M∅
IL-4IL-4
RANTESIP-10IP-10
ROIROI
NONO
Microglia
BBB
Neuron
Oligodendrocita
CC CC
Linfocita B
VC
AM
HLA
DR
ICA
M
VC
AM
HLA
DR
ICA
MVLA
4
LFA
2
MMPsMMPs
ROIROI
TNF-α, IFN-γTNF-α, IFN-γ
VC
AM
HLA
DR
ICA
M
VC
AM
HLA
DR
ICA
M
CCCCCC
Astrocita
Th1 CD4+ Th2 CD4+
CD28/B7
TCR/Ag/MHC
IL-12, IL-1IL-12, IL-1
IL-12, IL-1IL-12, IL-1
MIP-1αMIP-1αIP-10IP-10
TNF-α, IFN-γTNF-α, IFN-γ
MMPsMMPs
IFN-γIFN-γ
TNF-β
CD8+
TGF-βTGF-β
IL-10IL-10
NONO
MMPsMMPs
NKCellula NK
Figura 26.1 - Ipotesi patogenetica della sclerosi multiplaLinfociti T CD4+ circolanti nel sangue periferico sono attivati da un evento "trigger", verosimilmente un patogeno ubiqui-tario, che ne determina la migrazione attraverso la barriera ematoencefalica. Il passaggio della barriera è mediatodall'interazione tra recettori sulle cellule endoteliali, selectine ed integrine, ed i loro ligandi sulla superficie dei linfociti.All'interno del sistema nervoso centrale avviene il riconoscimento, da parte dei linfociti attivati, di auto-antigeni mielinicipresentati da macrofagi e microglia, attraverso un meccanismo di mimetismo molecolare tra la proteina mielinica e laproteina del patogeno. L'innesco della risposta autoimmunitaria comporta un aumento della permeabilità della barrieracausata dal rilascio di metalloproteasi con il conseguente richiamo di nuovi linfociti dalla periferia mediato da chemochinequali RANTES, MIP-1α ed IP-10. Il propagarsi dell'infiammazione determina il coinvolgimento di nuovi attori quali i linfocitiT CD8 ad azione citotossica, i linfociti B, i macrofagi, la microglia e, in minor misura, gli astrociti. L'attivazione locale dimolteplici meccanismi effettori risulterà in un danno sulla guaina mielinica, sugli oligodendrociti e, in un secondo tempoanche sui neuroni, che è mediato da citochine, complemento, anticorpi specifici per antigeni mielinici, proteasi, derivatidell'ossido nitrico e radicali liberi dell'ossigeno. Il propagarsi o l'arresto della cascata autoimmune dipenderà, almeno inparte, dal bilancio netto tra stimoli pro-infiammatori rilasciati dai linfociti T helper 1, ed in minor misura da macrofagi emicroglia, quali INFγ, TNF-α, TNF-β e stimoli anti-infiammatori quali per esempio TGF-β, IL-10, IL-4 prodotti dai linfociti Thelper 2.Abbreviazioni: CD4: linfocita T CD4; CD8: linfocita T CD8 citotossico; M∅ : macrofago; Th1: linfocita CD4 T-helper; BEE: bar-riera ematoencefalica; NK: cellule Natural Killer; NO: ossido nitrico; MMPs: metalloproteasi; ROI: radicali dell'ossigeno; C:complemento; TCR/Ag/MHC: complesso trimolecolare recettore linfocitario per l'antigene/antigene/complesso maggiore diistocompatibilità; CD28/B7: complesso di costimolazione; VCAM, ICAM: integrine; VLA4, LFA2: recettori per le integrine

1115Malattie demielinizzanti
fattori solubili quali citochine, radicali liberi dell'ossige-no e fattori del complemento. Esistono dati sperimentalinell'EAS che dimostrano che la demielinizzazione richie-de l'uso sinergico di anticorpi, e cellule T sensibilizzatecontro antigeni mielinici. Recentemente è stato dimostra-ta la presenza di anticorpi anti-MOG attaccati alla mielinain fase di degenerazione nelle lesioni di soggetti con SM.L'importanza di una risposta immunitaria mediata dailinfociti B è indicata inoltre dalla presenza di immuno-globuline IgG oligoclonali, presenti nel liquor in più del90% dei casi di SM.
Un ruolo fondamentale nella propagazione e nellamodulazione della risposta immunitaria nel SNC è poisvolto da un complesso network di molecole solubilichiamate citochine e chemochine. Benchè su quest'argo-mento nuovi dati siano continuamente segnalati dai la-boratori di tutto il mondo in base a ricerche sull'animalee sull'uomo, è probabile che il risultato delle complesseinterazioni tra diverse popolazioni cellulari a livello delSNC sia dovuto all'effetto netto di stimoli pro- e anti-in-fiammatori mediati da citochine e chemochine. Le pri-me sono prodotte da molte cellule del SNC (per esem-pio microglia ed astrociti) e praticamente da tutte le cel-lule del sistema immunitario. In base alla loro azione sidividono schematicamente in citochine pro-infiam-matorie, quali ad esempio l'interferone gamma (IFN-γ)e il tumor necrosis factor alfa (TNF-α) e citochine anti-infiammatorie, quali l'interleuchina 4 (IL-4), il IL-10 edil tumor growth factor beta (TGF-β). L'utilizzo di strate-gie sperimentali atte a bloccare l'azione pro-infiammato-ria di alcune citochine o di potenziare l'azione anti-in-fiammatoria di altre ha permesso di trattare con succes-so l'EAS. Molte di queste terapie sono ora in fase di stu-dio nell'uomo. Le maggiori difficoltà nell'utilizzo di que-ste molecole derivano dalla loro azione sistemica checomporta effetti spesso indesiderati su altri organi, e nellaloro grande ridondanza biologica per cui lo stesso effet-to viene mediato da molteplici citochine rendendo cosìscarsamente efficace l'intervento sulla singola sostanza.L'utilizzo di tecnologie, quali la terapia genica, atte a ri-lasciare la molecola desiderata direttamente nel SNCpotrebbe risolvere, almeno, in parte questi problemi. Re-centemente l'attenzione si è concentrata sulle chemochi-ne, una popolazione eterogenea di sostanze capaci di re-golare l'attivazione e le migrazione di cellule immuno-competenti. In particolare, è stato osservata la presenzadi alcune chemochine quali RANTES e IP-10 nelle plac-che di animali con EAS e di soggetti con SM. Analoga-mente, le cellule infiltranti le lesioni esprimono i recettoriper tali chemochine, rispettivamente CCR5 e CXCR5.L'utilizzo di molecole in grado di bloccare l'interazionetra chemochina ed il suo ligando potrebbe essere utiliz-zata a scopo terapeutico per impedire la migrazione dicellule patogeniche dalla periferia al SNC.
NEUROPATOLOGIA
La malattia si caratterizza per la presenza di numero-se aree di demielinizzazione nella sostanza bianca o plac-che. Al taglio dell'encefalo le placche di antica data han-no un'aumentata consistenza, bordi ben delimitati e uncolorito grigiastro, mentre le placche più recenti hannouna diminuita consistenza e un colorito più roseo del tes-suto circostante. Le placche sono preferenzialmente di-stribuite intorno ai ventricoli laterali, al pavimento del-l'acquedotto e al quarto ventricolo, anche se talvolta pos-sono avere sede sottocorticale o sconfinare in parte nel-la sostanza grigia. Il corpo calloso, i nervi ottici, il tron-co cerebrale sono costantemente interessati, così come ilmidollo spinale, specie nel suo tratto cervicale, con areedi demielinizzazione distribuite nelle colonne dorsali,nelle regioni subpiali e intorno al solco anteriore. Isto-logicamente le placche sono rappresentate da aree didemielinizzazione con un variabile grado di infiltrazio-ne cellulare, perdita di oligodendrociti, astrogliosi reat-tiva, variabile nella sua intensità a seconda della fase dievoluzione (Fig. 26.2).
Le lesioni precoci sono caratterizzate da un infiltratoinfiammatorio perivascolare, formato da linfociti T e B,plasmacellule cui segue la comparsa di macrofagi cheiniziano ad aggredire la guaina mielinica. Gli oligoden-drociti, nelle fasi iniziali, possono proliferare, verosimil-
Fig. 26.2 - Placca di grosse dimensioni che circonda il cor-no occipitale del ventricolo laterale (Colorazione Spielmeyer).

1116 Malattie del sistema nervoso
mente nel tentativo di ricostruire la mielina e riparare lalesione, in alcuni casi con successo e parziale remieliniz-zazione (placca ombra). Con il passare del tempo, al cen-tro della placca gli oligodendrociti e la mielina scompa-iono completamente, gli astrociti proliferano ed inizia-no a formare un denso intreccio di processi fibrillaricicatriziali, i macrofagi carichi di detriti mielinici si di-spongono a sede perivascolare, mentre ai margini dellalesione si osserva un bordo attivo ipercellulare formatoda astrociti, linfociti, cellule mononucleate, macrofagi,responsabili della progressione centrifuga della lesione.Nelle fasi più tardive la placca diventa inattiva e si tra-sforma in una cicatrice nettamente demarcata dal tessu-to circostante, nell'ambito della quale si evidenzia unadensa gliosi fibrillare, una completa perdita di oligoden-drociti con associata perdita assonale. In tale fase gliinfiltrati infiammatori sono molto modesti. Il dannoassonale tuttavia non caratterizza solo le placche più vec-chie ma è presente anche nelle lesioni acute, come dimo-strato recentemente, ed è probabilmente secondario aglistessi meccanismi infiammatori che inducono demieliniz-zazione.
Dal punto di vista immunoistochimico macrofagi elinfociti predominano nelle placche in fase attiva. Cellu-le T α/β CD4+ ed in minor misura CD8+, linfociti γ/δ,B e plasmacellule sono riconoscibili negli infiltrati peri-vascolari. I macrofagi sono numericamente le cellule piùrilevanti nelle placche. Recenti studi di microscopia elet-tronica hanno permesso di visualizzare i macrofagi nel-l'atto di distruggere la mielina attraverso un meccanismoche, almeno in parte, è mediato da anticorpi specifici perproteine mieliniche quali la MOG e dalla liberazione difattori solubili quali l'ossido nitrico (NO) ed altri radica-li liberi dell'ossigeno. Grande attenzione è stata dedica-ta all'identificazione delle cellule che esprimono gliantigeni di istocompatibilità di I e II classe, poichè attra-verso tali molecole le cellule immunocompetenti presen-tano le proteine mieliniche alle cellule T e sono quindiin grado di innescare e propagare nel tempo una rispo-sta autoimmune. La microglia attivata, i macrofagi e par-ticolari cellule a disposizione perivascolare esprimonomolecole HLA-DR, mentre solo più raramente lo fannole cellule endoteliali e gli astrociti. Recenti studi hannodimostrato che in talune circostanze anche i neuroni dan-neggiati e pertanto elettricamente inattivi sono in gradodi esprimere antigeni di classe I. La presenza a livello delSNC di cellule con capacità di presentare l'antigene(APC) “professionali”,quali le cellule dendritiche, è tut-tora discusso.
Alcune varianti della SM, che si caratterizzano per lapresenza di un particolare decorso clinico e per peculia-ri alterazioni neuropatologiche, sono da molti anni co-nosciute e rappresentano varianti anatomo-cliniche. NellaSM acuta di Margburg, la sintomatologia ha un decorso
tumultuoso della durata di pochi mesi e le alterazionineuropatologiche consistono in marcati infiltrati infiam-matori con edema ed estesa distruzione assonale, espres-sione dell'acuzie e della gravità del processo infiamma-torio. Nella neuromielite ottica, descritta da Devic, le le-sioni interessano i nervi ottici e il midollo spinale. L'esor-dio è acuto e il decorso rapidamente ingravescente, an-che se è possibile in alcuni casi un recupero. Sul pianoneuropatologico la neuromielite ottica dimostra una mag-giore gravità del processo infiammatorio, che può por-tare ad una distruzione tissutale con necrosi midollare.Nella sclerosi diffusa di Schilder, che colpisce soggettiin età infantile, la demielinizzazione è estesa e interessadiffusamente la sostanza bianca degli emisferi cerebrali,del tronco, e del cervelletto, e non deve essere confusacon le leucodistrofie, e in particolare con la adrenoleu-codistrofia. Clinicamente si manifesta con segni di in-gravescente compromissione diffusa e focale del SNC.Nella sclerosi concentrica di Balò zone di demielinizza-zione si alternano a zone di normale mielinizzazione,assumendo spesso una disposizione concentrica. Talevariante della SM ha spesso, ma non costantemente, undecorso rapidamente evolutivo.
DEMIELINIZZAZIONE, DANNO ASSONALE ECONDUZIONE NERVOSA
La mielina è formata dai concentrici avvolgimentidella membrana plasmatica degli oligodendrociti lungol'assone ed ha la funzione di isolare l'impulso nervoso chesi propaga rapidamente dal corpo cellulare lungo l'asso-ne, passando da un nodo di Ranvier al successivo conmodalità saltatoria. Studi sperimentali hanno dimostra-to che la demielinizzazione ha importanti effetti sullaconduzione nervosa. Nelle fibre demielinizzate la con-duzione saltatoria non è più possibile e l'impulso nervo-so procede con più lentezza, per propagazione continuao viene cortocircuitato nelle aree denudate dell'assone.Il rallentamento della conduzione nervosa nella viapiramidale è responsabile della fatica, costantemente pre-sente nei malati di SM, e il blocco della conduzione ner-vosa della perdita di funzione. Gli assoni demielinizzatidiventano sensibili a fattori ambientali quali l'aumentodella temperatura che peggiora la conduzione nervosa,le variazioni metaboliche del “milieu” extra-cellulare egli stimoli meccanici, con comparsa di potenziali di azio-ne improvvisi dovuti, ad es., alla flessione del capo, comenel fenomeno di Lhermitte, o con spasmi tonici secon-dari al movimento. La demielinizzazione può inoltre ge-nerare impulsi ectopici, all'origine del fenomeno di tra-smissione efaptica tra assoni demielinizzati contigui. Talimeccanismi di conduzione anomala sono alla base difenomeni parossistici sensitivi o motori, quali la nevral-

1117Malattie demielinizzanti
gia trigeminale, le parestesie, le contrazioni muscolari to-niche prolungate. Nelle fasi acute ed iniziali della malattiala presenza di infiltrati infiammatori, edema e demieli-nizzazione sono responsabili di una sofferenza assonaleche trova riscontro in una perdita di funzione che puòessere parzialmente recuperata con la regressione dell'in-fiammazione, l'utilizzo di vie di conduzione alternativeed attraverso un tentativo di rimielinizzazione. A tale con-dizione fisiopatologica corrisponde probabilmente la faseclinica di malattia caratterizzata da ricadute e remissio-ni. La cronicizzazione della lesione implica un processodi tipo gliotico cicatriziale, a cui corrisponde una mar-cata riduzione della componente infiammatoria e si as-socia ad una sofferenza duratura dell'assone cui segue undanno cellulare irreversibile. Al danno assonale corri-sponde la mancata regressione dei sintomi e la stabiliz-zazione del deficit neurologico ovvero la fase cronico-progressiva della malattia.
SINTOMATOLOGIA
I sintomi iniziali sono variabili, a secondadella sede lesionale, ma alcuni sintomi ricorro-no più frequentemente, poiché le aree di demie-linizzazione si distribuiscono in sedi preferen-ziali. I disturbi si manifestano e raggiungono illoro acme in poche ore o in alcuni giorni. Nellamaggior parte dei casi il sintomo iniziale èun'ipostenia ad uno o più arti (40% dei casi),una neurite ottica (22%), un disturbo soggetti-vo della sensibilità (tipo parestesie e disestesie)(21%), diplopia, vertigine o disturbi della min-zione. Tali sintomi possono comparire isolata-mente (esordio monosintomatico) o in associa-zione (esordio polisintomatico). I disturbi ini-ziali tendono nella maggior parte dei casi a re-gredire dopo un periodo di tempo variabile edin seguito possono ripresentarsi o possono com-parire altri sintomi e segni di sofferenza focaledel SNC. Virtualmente ogni sintomo può rap-presentare l'esordio così come verificarsi suc-cessivamente nel corso della malattia. Pochemalattie sono così variabili ed imprevedibilicome la SM, e diversi per ogni singolo caso sa-ranno l'età di esordio, il sintomo iniziale, la fre-quenza delle ricadute, il decorso della malattia,la disabilità e la sua progressione. Nei casi avan-zati, tuttavia, i disturbi motori, la spasticità,
l'atassia, le turbe della sensibilità, i deficit visi-vi e le turbe sfinteriche sono pressoché costan-temente presenti.
Disturbi piramidali. - L'interessamento delsistema piramidale con conseguente ipostenia espasticità, localizzata ad uno o più arti, è unevento costante e rappresenta una importantecausa di disabilità. Al suo esordio la compro-missione piramidale si può manifestare con va-rie modalità, talvolta in maniera insidiosa, conuna ipostenia agli arti inferiori, molto spessosolo dopo affaticamento o lunghe camminate, etendenza al recupero dopo un periodo di ripo-so, altre volte in maniera subacuta con unaparaparesi o emiparesi. L'ipostenia colpisce piùspesso gli arti inferiori, in genere in manieraasimmetrica, o un solo arto inferiore o un emi-lato corporeo; meno frequente è la compromis-sione isolata di un arto superiore e rara quellaisolata di ambedue gli arti superiori. L'obietti-vità neurologica evidenzia oltre all'iposteniaanche la presenza di riflessi osteotindinei viva-ci o policinetici, riflessi patologici quali l'Hoff-mann e il fenomeno di Babinski, clono del pie-de e più raramente della rotula; i riflessi addo-minali sono diminuiti o aboliti, spesso precoce-mente. In alcuni casi la spasticità agli arti infe-riori può essere utilie per mantenere la stazio-ne eretta e per camminare, ma in altri casi, spe-cie quando l'arto inferiore è iperesteso e il pie-de flesso plantarmente, può al contrario impe-dire la marcia. In rari casi, quando la lesioneinteressa l'ingresso delle radici posteriori o an-che la bianca adiacente le corna posteriori oanteriori, i riflessi profondi possono essere di-minuiti o aboliti e possono comparire segni diatrofia muscolare.
La fatica. - La fatica è un sintomo molto co-mune (circa l'80% dei soggetti) e la maggiorparte dei pazienti si lamenta di un eccessivo af-faticamento che compare anche dopo limitate ebanali attività: tale disturbo, di notevole impor-tanza pratica, correla con la presenza di una

1118 Malattie del sistema nervoso
compromissione della via piramidale, come di-mostrato dagli studi del tempo di conduzionecentrale. La fatica deve essere distinta dai piùfrequenti sintomi associati alla depressione.
Disturbi della sensibilità. - Sono spesso il sin-tomo di esordio e la loro comparsa lungo il corsodella malattia è costante. Sono dovuti a lesioni deicordoni posteriori, delle vie spino-talamiche odelle zone d'ingresso delle radici posteriori, e ven-gono descritti dai pazienti come sensazione di “in-torpidimento”, di “carne morta”, di formicolio, difasciatura, di gonfiore, ecc. La sede dei disturbisensitivi è la più varia: spesso iniziano ad un pie-de, per poi interessare l'altro piede e propagarsi aisettori prossimali degli arti inferiori, al perineo eall'addome; talvolta sono interessate una o ambe-due le mani, con una sensazione come “avere iguanti”; in altri casi ancora una lesione al midol-lo sacrale è responsabile di una ipo-anestesiaperineale con associata perdita della sensazionedel passaggio di urine e feci o, in caso di lesione alivello del midollo toracico, di una sensazione di“cintura” o di “corsetto che stringe” al torace.
Il dolore somatico non è un sintomo raro espesso è erroneamente sottovalutato. Il doloreneuropatico ha spesso un andamento parossi-stico come, ad esempio, nel caso della nevral-gia trigeminale e degli spasmi tonici dolorosinotturni. In altri casi il dolore, per esempio lom-bare, è dovuto ad una abnorme postura o adipertonia della muscolatura dorso-lombare. Sen-sazioni dolorose di stiramento agli arti inferio-ri, sono più frequenti durante le ore notturne esi associano con la spasticità.
All'esame obiettivo, specie nelle fasi inizia-li, non si evidenziano marcate alterazioni, ma ilpaziente può avvertire lo stimolo tattile o termi-co o dolorifico come “diverso” e “lontano” o fa-stidioso. Nelle fasi più avanzate residuano spes-so parestesie e disestesie alle dita delle mani edei piedi, ipoestesia distale agli arti inferiori emolto frequentemente si evidenzia una compro-missione delle sensibilità profonde, e in parti-colare della pallestesia agli arti inferiori.
Disturbi cerebellari. - Non sono un sintomofrequente all'esordio, ma diventano comuni suc-cessivamente. I sintomi ed i segni cerebellarihanno una scarsa tendenza alla regressione equando aprono la malattia hanno un cattivo si-gnificato prognostico. Il tremore intenzionale ela dismetria agli arti sono particolarmenteinvalidanti, sono scarsamente sensibili alle te-rapie sintomatiche, e causano grave disabilità eimportante limitazione dell'autonomia. La disar-tria cerebellare, poco frequente all'inizio dellamalattia, non è rara nelle fasi più tardive cosìcome l'andatura atasso-spastica che è un sinto-mo pressochè costante nel corso della malattia.Il nistagmo può essere espressione di lesionedelle vie cerebellari o delle vie internucleari neltronco encefalico.
Nervo ottico. - Il nervo ottico è particolar-mente vulnerabile: una neurite ottica è il sinto-mo d'esordio nel 22% dei casi. Il rischio che unaneurite ottica evolva in SM clinicamente defi-nita aumenta progressivamente con il passaredegli anni fino a diventare circa il 50% a 10anni. Il riscontro al momento della diagnosi diuna RM encefalica positiva, aumenta la possi-bilità di conversione in malattia conclamata.Nella maggior parte dei casi di SM di lungadurata una compromissione del nervo ottico,anche clinicamente non manifesta, si può evi-denziare con l'allungamento di latenza dei po-tenziali evocati visivi (PEV). La neurite ottica,in genere unilaterale, raramente bilaterale, èspesso associata o preceduta da dolore sopraor-bitario o del globo oculare, aggravato dai mo-vimenti oculari. Il calo della acuità visiva èspesso rapido ed i pazienti riferiscono di averela vista “annebbiata” o di vedere come attraver-so un vetro “appannato” o “smerigliato”. Il calodella vista è variabile, da pochi decimi fino, ra-ramente, alla completa cecità, e non è correg-gibile con lenti. L'esame del fundus oculare puòevidenziare una papilla ottica normale nellaneurite ottica retrobulbare, o una sfumatura deimargini papillari (papillite) quando è colpita la

1119Malattie demielinizzanti
porzione anteriore del nervo ottico. Successiva-mente la papilla ottica diventa pallida, specie neimargini temporali, anche quando il recupero ècompleto. Una papilla ottica pallida e atroficaè di comune riscontro nei casi avanzati di SM.L'esame del campo visivo evidenzia in genereuno scotoma centrale, poiché è colpita la partecentrale del nervo ottico ove decorrono le fibredi origine maculare; talvolta il deficit campime-trico può essere più ampio e presentare difettiparziali. Il fenomeno di Uhthoff si caratterizzaper una diminuzione dell'acuità visiva con l'au-mento della temperatura corporea, che segue unesercizio fisico o la comparsa di febbre, ed espri-me il peggioramento della conduzione nervosanelle fibre demielinizzate, secondario all'eleva-zione della temperatura corporea. Il recupero,dopo una neurite ottica, è lento e variabile e ta-lora permangono esiti, come una diminuita sen-sibilità ai colori. Il chiasma e le vie ottiche sonofrequenti sedi di demielinizzazione, ma deficitcampimetrici quali emianopsie sono rari.
Nervi oculomotori. - Una lesione del VI, delIII o più raramente del IV nervo cranico puòessere responsabile dell'insorgenza di diplopia,spesso a decorso favorevole. Più frequente è laparalisi internucleare, che si manifesta con undeficit della adduzione e nistagmo orizzontalenell'occhio abdotto, dovuta a una lesione del fa-scicolo longitudinale mediale. La paralisi inter-nucleare è caratteristica, anche se non patogno-monica, della SM.
Altri nervi cranici. - Una nevralgia trige-minale non è evento infrequente e quando insor-ge in un individuo di età inferiore ai 50 anni,soprattutto se associata ad un disturbo obietti-vo delle sensibilità, deve comunque far pensa-re alla possibilità di un origine demielinizzante.È causata da una lesione della zona di ingressodel nervo trigeminale nel ponte, ma anche daplacche interessanti i tratti trigeminali discen-denti o il nucleo del trigemino. Ipoestesia edisestesie nel territorio trigeminale possono es-
sere associate. Una paralisi periferica del faccia-le e talvolta miochimie in tale territorio sonopossibili e indicano l'esistenza di una lesioneintratroncale. Vertigini soggettive possono com-parire nel 30% dei casi così come un'ipoacusia,espressione di un interessamento delle viecocleari del nervo acustico, spesso con scarsatendenza alla guarigione. Il nistagmo è frequen-te, tende a persistere e può essere pendolare,fasico o tonico e spesso è associato a sintomicerebellari. Nei soggetti con estese lesioni tron-cali e nelle fasi tardive della malattia riso e pian-to spastico possono essere presenti.
Disturbi affettivi e cognitivi. - La depressio-ne è un sintomo frequente. In effetti la profon-da e totale modificazione della vita, la consape-volezza di essere affetto da una malattia pro-gressivamente invalidante, l'assenza di terapiecapaci di modificare in modo significativo ildecorso, l'incertezza del futuro, il timore di do-vere dipendere da altri in un periodo di temponon lontano, rendono ampiamente conto dellafrequenza di disturbi depressivi. La depressio-ne è quindi, nella maggior parte dei casi, di tipo“secondario”. Aree di demielinizzazione a sedeperiventricolare e localizzate nella sostanza gri-gia possono essere responsabili di veri e propridisturbi cognitivi, recentemente evidenziati evalorizzati: la memoria, l'attenzione visuo-spa-ziale, la capacità di calcolo, possono esserecompromesse; un vero deterioramento menta-le non è frequente, salvo in alcuni casi di ma-lattia in fase avanzata. In certi casi può esserepresente un' euforia inadeguata, espressione diuna compromissione psicorganica. Episodipsicotici maniacali, anche al di fuori della tera-pia steroidea, o turbe dissociative, sono statidescritti, e possono in alcuni rari casi essere ilsintomo d'esordio della malattia.
Disturbi sfinterici e sessuali. - Molto fre-quenti sono i disturbi sfinterici: la minzione im-periosa è spesso sintomo precoce e talora si as-socia ad incontinenza; in altri casi è presente

1120 Malattie del sistema nervoso
difficoltà ad iniziare il mitto con incapacità asvuotare la vescica e conseguente residuo urina-rio da cui derivano frequenti infezioni delle vieurinarie. Nei casi avanzati i disturbi della min-zione sono di tipo misto. Un rallentato transitointestinale è comune ed è scarsamente sensibi-le alle usuali terapie.
Frequente è il rilievo di turbe sessuali, condiminuizione della libido o vera e propria im-potenza, dovute a lesioni dei centri spinali, an-che se aspetti psicologici e depressivi sonoun'importante concausa.
Altri sintomi. - Molto caratteristici, anche senon particolarmente frequenti, sono i "sintomiparossistici", che si manifestano con un esordioimprovviso, ripetizione del sintomo con le stes-se caratteristiche a breve distanza di tempo,spesso scatenati da movimenti o da stimolisensitivi. Fra questi sono: il “segno di Lhermit-te” consistente nell'improvvisa comparsa di unasensazione a tipo “scossa elettrica” per flessioneo più raramente estensione del capo, con irra-diazione lungo la schiena fino agli arti inferiorio più raramente agli arti superiori; la “nevral-gia trigeminale”, con caratteristiche simili aquella idiopatica, ma ad insorgenza più preco-ce; le crisi di disartria e atassia parossistica, bre-vi attacchi della durata di 20-30 secondi, ripe-tentisi ogni 30-60 minuti, di disartria o di turbedella coordinazione, in genere localizzate agliarti inferiori; contrazioni toniche, con assun-zioni di una postura in flessione all'arto superio-re e in estensione all'arto inferiore, spesso pre-cedute da sintomi sensitivi, scatenate dai movi-menti, della durata di 1 o 2 minuti; dolori im-provvisi o parestesie interessanti il volto ouno degli arti. Tali sintomi parossistici sono ingenere l'espressione di una lesione troncale o,come nel caso del “segno di Lhermitte”, del mi-dollo cervicale, durano per un periodo di tem-po variabile, da alcuni giorni ad alcuni mesi, etendono a regredire spontaneamente o con tera-pia sintomatica. In generale si può dire che, pro-prio per le caratteristiche di disseminazione
spaziale delle placche, numerosi altri sintomineurologici si possono riscontrare quali: sinto-mi tipo “narcolessia”, per lesioni a livello ipo-talamico o della sostanza reticolare; movimen-ti involontari patologici, quali tremore e coreo-atetosi; cefalea, in genere priva di caratteristi-che particolari; crisi convulsive generalizzate oparziali, per altro ben controllate dalla terapia.
Raro è l'interessamento del sistema nervosoperiferico. L'assenza di compromissione perife-rica in una malattia in cui il bersaglio è la guai-na mielinica, presente nel SNC e SNP, può sor-prendere. È probabile che le differenze strutturalinella composizione proteolipidica della mielinaperiferica e centrale giochi un ruolo nella diver-sa suscettibilià al processo autoimmune. Rari casiin cui coesistono segni di sofferenza centrale eperiferica sono stati segnalati, ma non è chiaro sesi tratti o meno di una concomitanza casuale. Èben noto infine il rapporto tra SM ed altre ma-lattie di origine autoimmune, quali le tiroiditi, laspondilite anchilosante, il diabete mellito di tipoI, anche se tali associazioni non sono per la veri-tà più numerose di quelle prevedibili. L'associa-zione con l'uveite è stata raramente segnalata, mai possibili motivi della presenza contemporaneadi uveite e SM sono ancora oscuri.
ESAMI COMPLEMENTARI
La diagnosi si basa sui dati clinici e in parti-colare sulla disseminazione temporale e spazia-le dei disturbi e dei segni neurologici. La neces-sità di una diagnosi certa ha assunto una rilevan-za particolare in questi ultimi anni di fronte allapossibilità di utilizzare precocemente terapiecapaci di modificare il decorso naturale dellamalattia. Tuttavia, in assenza di caratteristichecliniche patognomoniche o di un esame di la-boratorio definitivo, la SM rimane in ultimaanalisi una diagnosi di esclusione basata su in-dagini paracliniche.
Analisi del liquor cerebro-spinale. - Benchèin molti paesi sia considerata una indagine non

1121Malattie demielinizzanti
più necessaria a fronte di una documentazioneneuroradiologica probativa, l'analisi del liquora nostroavviso fornisce a tutt'oggi dati indispen-sabili alla diagnosi. Sebbene vi possa essere unmodesta pleiocitosi fino ad un massimo di 50cellule/mm3, nella maggior parte dei casi il nu-mero di cellule, prevalentemente mononucleati,è normale (1-5 cellule/mm3). Il reperto più fre-quente è l'aumento delle immunoglobulineliquorali che testimonia una sintesi intratecaledi IgG e in minor misura di IgA e IgM. Talereperto risulta da un aumento dell'indice diLink, un semplice rapporto matematico che mi-sura le immunoglobuline rispetto all'albuminadel liquor. La prova più sicura per evidenziareuna risposta umorale nel SNC è tuttavia il ri-scontro, mediante isoelectrofocusing o immu-noblot, di bande oligoclonali di IgG su liquor,dimostrabile in circa il 95% di pazienti con SM.Tali bande rappresentano verosimilmente unarisposta cellulare B aspecifica diretta contro unampio pannello di antigeni, espressione di unagenerica attivazione immunitaria. La presenzadi bande oligoclonali non è tuttavia specificadella SM, osservandosi anche in altre malattieinfiammatorie a carico del SNC, quali, ad esem-pio, l'infezione da HIV o la malattia di Lyme.
Potenziali evocati - Il rallentamento dellaconduzione nervosa, dovuto alla perdita dellaguaina mielinica, può essere dimostrato dai po-tenziali evocati, ottenuti dalla stimolazione dellevie visive, uditive, somatosensoriali o della viamotoria centrale. Il difetto di conduzione si e-videnzia come un aumento della latenza men-tre l'ampiezza del potenziale è in genere inva-riata. I potenziali evocati permettono il ricono-scimento di una lesione subclinica, in sedi di-verse da quelle clinicamente evidenti, e posso-no dimostrare quindi la disseminazione spazialedella malattia. In un paziente con una para-paresi, ad esempio, il rallentamento della con-duzione nelle vie visive dimostra l'esistenza diuna compromissione di almeno due sistemi, equindi almeno due sedi lesionali. I potenziali
evocati visivi (PEV) sono alterati circa nel 90%dei pazienti con una storia clinica di neurite ot-tica e in più dei 2/3 dei casi definiti di SM, an-che in assenza di una precisa storia clinica diturbe visive. I potenziali evocati somestesici(PES), espressione della conduzione nei cordoniposteriori del midollo, sono rallentati circa nel70-90% dei casi. I potenziali evocati uditivi han-no una minor percentuale di positività, pari acirca il 50% dei casi. Il tempo di conduzionecentrale della via motoria esprime il grado diintegrità del fascio piramidale. I potenziali evo-cati hanno inoltre una loro particolare importan-za nell'evidenziare una compromissione di vienervose in casi dubbi, in cui l'organicità o lafunzionalità dei disturbi appare incerta.
Neuroimmagini. - La TC è stata diffusamenteutilizzata negli anni ’70, sia per escludere altrepossibili cause di malattia, sia perché può evi-denziare ipodensità parenchimali, specie neicasi di lesioni estese e confluenti. In seguito èstata completamente soppiantata dall'uso dellarisonanza magnetica nucleare (RM). La RM ele tecniche correlate rappresentano la metodicapiù sensibile, benchè scarsamente specifica, perdefinire la diagnosi e monitorare l'evoluzionedella malattia. La capacità della RM di visua-lizzare lesioni clinicamente asintomatiche hacondotto all'uso di questa tecnica per documen-tare la disseminazione spaziale delle lesioni inpazienti con sintomi altamente suggestivi diSM. Nelle sequenze in densità protonica/T2 sievidenzia tipicamente la presenza di aree iperin-tense nella sostanza bianca degli emisferi cere-brali. Nelle immagini in DP/T2, alcuni criteriaumentano sensibilmente l'accuratezza delladiagnosi. Tra questi la presenza di aree peri-ventricolari, di lesioni in fossa cranica posterio-re e nella corona radiata sopra il corpo calloso,di lesioni di diametro > 6mm. In T1 è possibi-le riscontrare aree ipointense che identificanoplacche croniche; al contrario, lesioni acute vi-sibili in DP/T2 spesso non sono visualizzabiliin T1. Le lesioni visualizzate in T1 appaiono
1122 Malattie del sistema nervoso
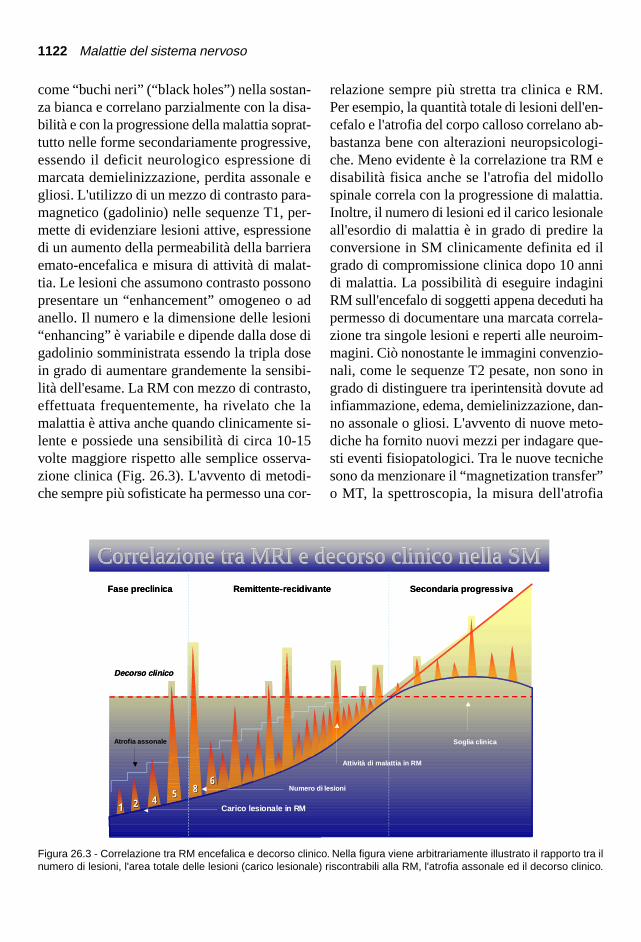
come “buchi neri” (“black holes”) nella sostan-za bianca e correlano parzialmente con la disa-bilità e con la progressione della malattia soprat-tutto nelle forme secondariamente progressive,essendo il deficit neurologico espressione dimarcata demielinizzazione, perdita assonale egliosi. L'utilizzo di un mezzo di contrasto para-magnetico (gadolinio) nelle sequenze T1, per-mette di evidenziare lesioni attive, espressionedi un aumento della permeabilità della barrieraemato-encefalica e misura di attività di malat-tia. Le lesioni che assumono contrasto possonopresentare un “enhancement” omogeneo o adanello. Il numero e la dimensione delle lesioni“enhancing” è variabile e dipende dalla dose digadolinio somministrata essendo la tripla dosein grado di aumentare grandemente la sensibi-lità dell'esame. La RM con mezzo di contrasto,effettuata frequentemente, ha rivelato che lamalattia è attiva anche quando clinicamente si-lente e possiede una sensibilità di circa 10-15volte maggiore rispetto alle semplice osserva-zione clinica (Fig. 26.3). L'avvento di metodi-che sempre più sofisticate ha permesso una cor-
relazione sempre più stretta tra clinica e RM.Per esempio, la quantità totale di lesioni dell'en-cefalo e l'atrofia del corpo calloso correlano ab-bastanza bene con alterazioni neuropsicologi-che. Meno evidente è la correlazione tra RM edisabilità fisica anche se l'atrofia del midollospinale correla con la progressione di malattia.Inoltre, il numero di lesioni ed il carico lesionaleall'esordio di malattia è in grado di predire laconversione in SM clinicamente definita ed ilgrado di compromissione clinica dopo 10 annidi malattia. La possibilità di eseguire indaginiRM sull'encefalo di soggetti appena deceduti hapermesso di documentare una marcata correla-zione tra singole lesioni e reperti alle neuroim-magini. Ciò nonostante le immagini convenzio-nali, come le sequenze T2 pesate, non sono ingrado di distinguere tra iperintensità dovute adinfiammazione, edema, demielinizzazione, dan-no assonale o gliosi. L'avvento di nuove meto-diche ha fornito nuovi mezzi per indagare que-sti eventi fisiopatologici. Tra le nuove tecnichesono da menzionare il “magnetization transfer”o MT, la spettroscopia, la misura dell'atrofia
Fase preclinica
Correlazione tra MRI e decorso clinicoCorrelazione tra MRI e decorso clinicoCorrelazione tra MRI e decorso clinico nellanellanella SMSMSMRemittente-recidivante Secondaria progressiva
Decorso clinico
Carico lesionale in RM
Numero di lesioni
11 22 4455 88
Atrofia assonale
Attività di malattia in RM
Soglia clinica
66
Fase preclinica
Correlazione tra MRI e decorso clinicoCorrelazione tra MRI e decorso clinicoCorrelazione tra MRI e decorso clinico nellanellanella SMSMSMRemittente-recidivante Secondaria progressiva
Decorso clinico
Carico lesionale in RM
Numero di lesioni
11 22 4455 88
Atrofia assonale
Attività di malattia in RM
Soglia clinica
66
Figura 26.3 - Correlazione tra RM encefalica e decorso clinico. Nella figura viene arbitrariamente illustrato il rapporto tra ilnumero di lesioni, l'area totale delle lesioni (carico lesionale) riscontrabili alla RM, l'atrofia assonale ed il decorso clinico.

1123Malattie demielinizzanti
cerebrale e più recentemente la tecnica di “dif-fusion” e la risonanza magnetica funzionale. Il'MT misura lo scambio di magnetizzazione traprotoni legati alle macromolecole e protoni li-beri. Fattori che influenzano l'equilibrio tra i duepool di protoni modificano il MT e pertanto in-fluenzano il rapporto (MTR o “magnetizationtransfer ratio”) tra l'intensità di segnale di im-magini ottenute con o senza un impulso di ra-diofrequenze. L'edema e l'infiammazione chedeterminano un aumento del pool di protoni li-beri, causano una riduzione del 3-5% del'MTR.La demielinizzazione, il danno assonale e, piùin generale, le lesioni che recano danno in modosostanziale alla struttura del parenchima, ridu-cono in modo più marcato il'MTR. Il'MTR èpertanto in grado di documentare l'eterogenei-tà delle lesioni e addirittura di evidenziare un'al-terazione di segnale nella sostanza bianca appa-rentemente normale. La spettroscopia in RMNmisura i metaboliti tissutali e quindi le le modi-ficazioni biochimiche del tessuto patologico enormale. È possibile identificare quattro princi-pali picchi di risonanza: quello del N-acetil-aspartato (NAA) presente esclusivamente neineuroni, quello della colina che misura i fosfo-lipidi di membrana, ed i picchi della creatina edel lattato. Una riduzione del NAA è indicativadi danno assonale e correla con la disabilità cli-nica mentre un aumento della colina suggerisceuna danno della guaina mielinica. Sulla basedelle metodiche più tradizionali schematica-mente si può immaginare che l'evoluzione diuna singola lesione inizi con l'alterazione dellabarriera emato-encefalica, che si associa a pre-sa di contrasto, seguita dalla comparsa di unalesione infiammatoria visibile in DP/T2 e nelleimmagini T1 senza contrasto. La risoluzionedella componente infiammatoria esita in un'ipe-rintensità nelle immagini DP/T2 talora associataalla presenza di “buchi neri” in T1. Tali lesio-ni, inizialmente stabili, possono poi aumentaredi volume, presentando aspetti confluenti edeventualmente assumere nuovamente contrastocome segno di riattivazione infiammatoria.
L'utilizzo e l'elaborazione di tecniche semprepiù sofisticate permetterà una più accurata do-cumentazione del danno.
DIAGNOSI
Si basa sulla presenza anamnestica di almenodue episodi di sofferenza focale del SNC, sul ri-scontro obiettivo di almeno due diverse sedi dicompromissione della sostanza bianca o delle vielunghe di connessione, su un decorso a ricaduteo cronico progressivo da almeno 6 mesi, sull'etàdi esordio, in genere fra i 15 e i 50 anni, e sullaesclusione di altre possibili cause di malattia neu-rologica. I dati clinici sono essenziali e spessosufficienti per una diagnosi di certezza, ma gliesami complementari, quali i potenziali evocatie la RM, capaci di evidenziare sedi lesionali cli-nicamente silenti, e l'esame del liquor, che dimo-stra una sintesi di IgG oligoclonali, sono neces-sari per convalidare il giudizio clinico. Nella ta-bella 26.1 sono riportati i criteri diagnostici cli-nici e di laboratorio proposti da Poser.
Nella diagnosi differenziale sono da considerareinnanzitutto le malattie che possono causare unasofferenza multifocale del SNC con un decorsoremittente, quali ad esempio, le vasculiti sistemi-che o primitivamente cerebrali, il lupus eritema-toso sistemico, la malattia di Behçet, la malattiadi Sjogren e la sarcoidosi. La compromissioneviscerale e l'alterazione degli esami ematochimicipossono essere indicativi di una malattia auto-immune sistemica. Malattie infettive, quali laneurosifilide, l'AIDS, la malattia di Lyme posso-no manifestarsi con una compromissione multifo-cale del SNC, con bande oligoclonali e sintesiintratecale di IgG, ma gli esami di laboratorio spe-cifici permettono, in genere, di risolvere il proble-ma. Nelle aree endemiche per la malattia di Lymeuna discreta percentuale della popolazione gene-rale ha una positività sierologica, e può quindi ca-pitare di osservare pazienti con una storia clinicatipica di SM e con anticorpi anti-Borrelia nel sie-ro. La compromissione del SNC in corso di ma-lattia di Lyme è estremamente rara, e pertanto il

1124 Malattie del sistema nervoso
sospetto di neuroborreliosi deve essere sempreconfermato dalla positività dei tests diagnostici suliquor.
L' adrenoleucodistrofia si può presentare nel-l'età adulta con una paraparesi spastica progres-siva. La malattia è legata al sesso, ma casi lievisono stati descritti anche nelle femmine porta-trici. La diagnosi può essere correttamente sta-bilita dal dosaggio ematico degli acidi grassi acatena lunga. La malattia di Leber, dovuta amutazioni interessanti il DNA mitocondriale,deve essere tenuta in considerazione quando siverifichi una perdita acuta della vista con atrofiaottica bilaterale in adolescenti o giovani adulti.La familiarità e l'esame oftalmologico sono utiliper indirizzare la diagnosi. Particolarmente dif-ficile può essere la diagnosi differenziale conl'arteriopatia cerebrale autosomica dominantecon infarti sottocorticali e leucoencefalopatia(CADASIL). L'ereditarietà, la frequente asso-ciazione con l'emicrania, il quadro RM rapida-mente evolutivo, l'assenza di alterazioni liquo-rali e l'assenza di interessamento del midollosono elementi di differenza tra le malattie.
L'esordio ictale della sintomatologia può in-dirizzare verso una malattia cerebrovascolare,mentre un decorso progressivo impone l'esclu-sione di una lesione tumorale. Nei casi con unaparaparesi ingravescente le indagini di labora-torio e strumentali devono escludere mielopatiespondilogenetiche, malformazioni della cernie-ra atlo-occipitale, lesioni compressive intra oextramidollari, malattie carenziali. Alcune mal-formazioni vascolari intramidollari possonomimare la SM, presentandosi con un decorsosubacuto e con remissione, e alcune mielopatieinfettive, come la paraparesi spastica tropicaledovuta ad una infezione da virus HTLV1, ecce-zionale nel nostro paese, ma diffusa nei Caraibi,nel Sud America e nel Giappone, hanno un de-corso molto simile alle forme midollari croni-co progressive di SM. Va infine ricordato chespesso la diagnosi differenziale più complessaè con un disturbo psichiatrico di conversione. Idati clinici, associati alla RM e all'esame del li-quor permettono nella maggior parte dei casiuna diagnosi certa. L'errore attualmente più fre-quente è quello di porre una diagnosi sulla base
Tabella 26.1 - Criteri diagnostici della sclerosi multipla.
A. Sclerosi multipla clinicamente definita.
A1 Due episodi ed evidenza clinica di due diverse e separate sedi lesionali
A2 Due episodi, evidenza clinica di una sede lesionale ed evidenza paraclinica di un’altra separata lesione
B. Sclerosi multipla definita da esami di laboratorio.
B1 Due episodi; evidenza clinica o paraclinica di una sede lesionale; presenza di bande oligoclonali o di una aumen-
tata sintesi intratecale di IgG all’esame del liquor
B2 Un episodio; evidenza clinica di due diverse sedi lesionali; presenza di bande oligoclonali o di una aumentata
sintesi intratecale di IgG all’esame del liquor
B3 Un episodio; evidenza clinica di una sede lesionale e paraclinica di un’altra separata lesione; presenza di bande
oligoclonali o di una aumentata sintesi intratecale di IgG all’esame del liquor
C. Sclerosi multipla clinicamente probabile.
C1 Due episodi ed evidenza clinica di una sede lesionale
C2 Un episodio ed evidenza clinica di due sedi lesionali separate
C3 Un episodio, evidenza clinica di una sede lesionale ed evidenza paraclinica di un’altra separata lesione
D. Sclerosi multipla probabile da esami di laboratorio.
D1 Due episodi e presenza di bande oligoclonali o di una aumentata sintesi intratecale di IgG all’esame del liquor
Nota: l’evidenza paraclinica di sedi lesionali può essere dimostrata con lo studio dei potenziali evocati, la TC, la RM o
con le prove urodinamiche.
1125Malattie demielinizzanti
della RM, esame di alta sensibilità ma non spe-cifico, in soggetti in cui la storia e l'obiettivitàclinica non siano adeguate e manchi un esamedel liquor cerebrospinale.
DECORSO E PROGNOSI
Il decorso della malattia è variabile e nel sin-golo caso è spesso imprevedibile anche se, ingenerale, le modalità di decorso più comuni efrequenti sono ben note. Nell'80% circa dei casila malattia si presenta e procede per ricadute,con comparsa acuta e subacuta di un sintomoclinico che raggiunge il suo acme in giorni osettimane, per poi regredire parzialmente ocompletamente in circa 1 o 2 mesi. La minimadurata di una ricaduta è convenzionalmente sta-bilita in 24 ore e fluttuazioni di sintomi preesi-stenti o la comparsa di nuovi sintomi con du-rata inferiore non hanno rilevanza clinica. Ilsecondo episodio di deficit neurologico si puòverificare entro un periodo di tempo molto va-riabile che generalmente si aggira intorno ai 2anni ma che in un 10-15% dei casi può durare
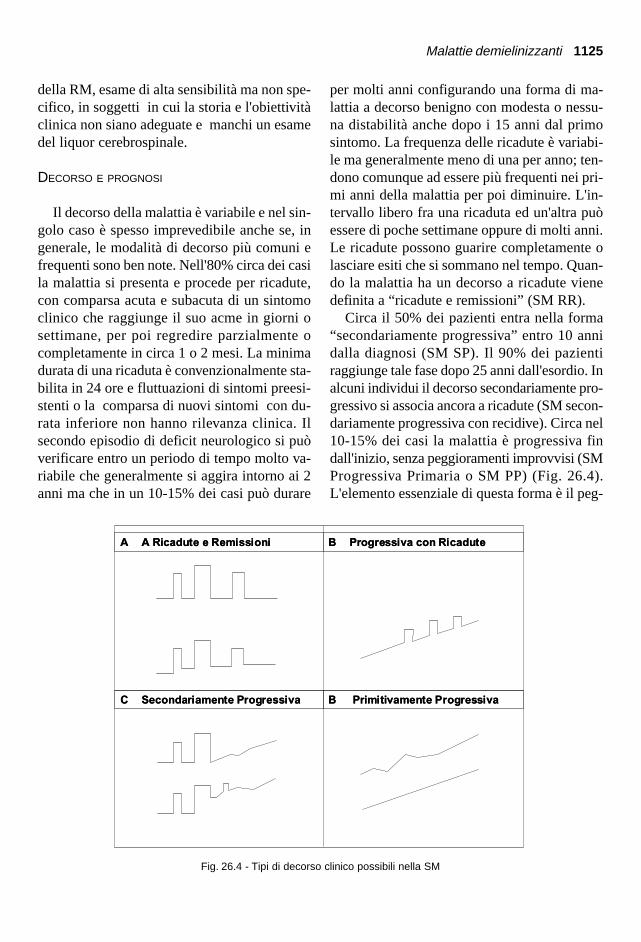
per molti anni configurando una forma di ma-lattia a decorso benigno con modesta o nessu-na distabilità anche dopo i 15 anni dal primosintomo. La frequenza delle ricadute è variabi-le ma generalmente meno di una per anno; ten-dono comunque ad essere più frequenti nei pri-mi anni della malattia per poi diminuire. L'in-tervallo libero fra una ricaduta ed un'altra puòessere di poche settimane oppure di molti anni.Le ricadute possono guarire completamente olasciare esiti che si sommano nel tempo. Quan-do la malattia ha un decorso a ricadute vienedefinita a “ricadute e remissioni” (SM RR).
Circa il 50% dei pazienti entra nella forma“secondariamente progressiva” entro 10 annidalla diagnosi (SM SP). Il 90% dei pazientiraggiunge tale fase dopo 25 anni dall'esordio. Inalcuni individui il decorso secondariamente pro-gressivo si associa ancora a ricadute (SM secon-dariamente progressiva con recidive). Circa nel10-15% dei casi la malattia è progressiva findall'inizio, senza peggioramenti improvvisi (SMProgressiva Primaria o SM PP) (Fig. 26.4).L'elemento essenziale di questa forma è il peg-
Fig. 26.4 - Tipi di decorso clinico possibili nella SM
� ���������������� �� � �� ��������� ��������
� ������������� �������� �� ����������� �������
� ���������������� �� � �� ��������� ��������
� ������������� �������� �� ����������� �������

1126 Malattie del sistema nervoso
gioramento graduale talora con modeste fluttua-zioni ma senza precedenti recidive. Se l'esordioè in età giovanile, il decorso è in genere a rica-dute e remissioni, mentre se la malattia iniziapiù tardivamente, intorno ai 45-50 anni, si os-serva spesso una lenta progressione dei distur-bi, prevalentemente a carico del midollo spina-le. Nel 5% dei casi il decorso è maligno fin dal-l'inizio, con rapida comparsa di gravi segni neu-rologici e compromissione dell'autonomia.
Anche se è particolarmente difficile, in unamalattia così imprevedibile, formulare una pro-gnosi corretta, alcuni fattori possono essere pre-si in considerazione: il decorso appare più favo-revole nel sesso femminile rispetto al sesso ma-schile, l'esordio precoce è associato ad una pro-gnosi migliore, mentre l'esordio tardivo è spes-so seguito da un decorso progressivo con rapidacomparsa di disabilità. Sintomi d'esordio di tiposensitivo e interessamento dei nervi cranici, qualiil nervo ottico, sono associati ad un decorso piùfavorevole rispetto ai casi in cui sintomi pira-midali o cerebellari segnano l'inizio della malat-tia. Il numero e la frequenza di ricadute nei pri-mi anni di malattia sembra determinare una piùrapida progressione verso una disabilità grave. Lacomparsa di un decorso di tipo progressivo rive-ste certamente un significato prognostico sfavo-revole e l'insorgenza di una disabilità neurologi-ca diventa probabile nel volgere di pochi anni.Circa il 50% dei casi raggiunge in 15 anni un li-vello tale in cui la deambulazione autonoma nonè più possibile, ma necessita di appoggio uni obilaterale. Nello stesso periodo, il 10% è costrettosulla sedia a rotelle. Ciò nonostante, le attualiterapie ed il miglioramento generale della assi-stenza medica hanno reso migliore la qualità del-la vita e ne hanno prolungato la durata che sem-bra essere solo lievemente ridotta rispetto alla po-polazione generale.
SCALE DI VALUTAZIONE
L'impatto sociale della malattia, nonché l'e-norme impatto economico che la SM suscita in
tutto il mondo ha imposto la creazione di crite-ri non soltanto diagnostici ma anche valutativi,comuni tra gli operatori che trattano e gestisco-no questa malattia. Di qui è nata l'esigenza diavere a disposizione, oltre che parametri para-clinici come la RM, anche scale cliniche di va-lutazione che permettano un confronto relativa-mente obiettivo di un esame neurologico tra e-saminatori diversi, e, soprattutto, il confrontodelle condizioni cliniche di uno stesso pazientea distanza di tempo. L'enorme diffusione di“trials” clinici con il coinvolgimento di Centriin tutto il mondo ha fatto sì che queste scaleavessero una vasta diffusione tra i neurologi. Inparticolare, la scala di disabilità di Kurtzke(Expanded Disability Status Scale o EDSS),benché poco sensibile, è ormai uno strumentonecessario per la valutazione routinaria di qua-lunque paziente con SM (Tabella 26.2). La scalasi basa su “Sistemi Funzionali”, per esempio“funzione piramidale”, “funzione cerebellare”etc cui viene attribuito un punteggio da 0 a 5.Sulla base dei punteggi per ciascun sistema fun-zionale viene quindi attribuito uno score totaleche va da 0, quando il paziente è asintomaticoe privo di segni patologici, a punteggi via via piùalti a seconda della gravità della malattia. Il 6 èuno dei punteggi più importanti per la suarilevanza clinica, in quanto è indicativo dellaperdita di autonomia clinica e della necessità diutilizzare un appoggio per deambulare. Un'al-tra scala di facile utilizzo è l' “AmbulationIndex” che misura sostanzialmente le capacitàdi deambulazione del paziente. La “Scripps Sca-le” e la “MS Functional Composite” sono sca-le più complesse che al momento sono utilizzatesoltanto nell'ambito di studi clinici al fine divalutare l'impatto di una terapia su parametriclinici più fini.
TERAPIA
La classica affermazione che non esiste alcu-na terapia in grado di influenzare il decorso del-la malattia è stata smentita nell'ultima decade

1127Malattie demielinizzanti
Tabella 26.2 - Kurtzke Extended Disability Status Scale (EDSS)
0.0 Esame neurologico normale [tutti i sistemi funzionali (SF) sono di grado 0].1.0 Non c’e disabilità, segni minimi in un SF (cioè, grado 1).1.5 Non c’e disabilità, segni minimi in più di un SF (più SF di grado 1).2.0 Disabilità minima in un SF (un SF di grado 2, altri di grado 0 o 1).2.5 Disabilità minima in due SF (due di grado 2, altri di grado 0 o 1).3.0 Disabilità moderate in un SF (uno di grado 3, altri di grado 0 o 1), o disabilità lieve in tre o quattro SF (tre o
quattro di grado 2, altri di grado 0 o 1), il paziente è del tutto autonomo nella deambulazione.3.5 Il paziente è del tutto autonomo nella deambulazione ma ha una disabilità moderate in un SF (di grado 3) e
uno o due SF di grado 2; oppure due SF di grado 3; oppure cinque SF di grado 2 (altri di grado 0 e 1).4.0 Il paziente è del tutto autonomo nella deambulazione senza aiuto, autosufficiente, anche per 12 ore al gior-
no nonostante una disabilità relativamente marcata consistente in un SF di grado 4 (altri di grado 0 e 1), ocombinazioni di gradi inferiori che superano i limiti precedenti; il paziente è in grado di camminare senzaaiuto o senza fermarsi per circa 500 metri.
4.5 Il paziente è del tutto autonomo nella deambulazione senza aiuto, anche per tutto il giorno; in grado di lavo-rare durante tutto il giorno, ma puo avere qualche limitazione per un’attività completa e richiedere un mini-mo di assistenza; si caratterizza per una disabilità relativamente marcata consistente in un SF di grado 4(altri di grado 0 e 1) o combinazioni di gradi inferiori che superano i punteggi precedenti; è in grado di cam-minare senza aiuto o senza fermarsi per circa 300 metri.
5.0 Il paziente è in grado di camminare senza aiuto e senza fermarsi per circa 200 metri; la disabilità è suffi-cientemente marcata da intralciare una completa attività quotidiana (per esempio lavorare per tutto il gior-no senza provvedimenti particolari). (Di solito i SF equivalenti sono uno solo di grado 5, altri di grado 0 e 1,oppure combinazioni di gradi inferiori.) II paziente deambula per circa 200 metri senza aiuto o senza fer-marsi.
5.5 Il paziente è in grado di camminare senza aiuto o senza fermarsi per circa 100 metri; la disabilità è suffi-cientemente marcata da impedire una complete attività quotidiana (di solito i SF equivalenti sono uno solodi grado 5, altri di grado 0 e 1, o combinazioni di gradi inferiori).
6.0 Il paziente necessita di assistenza saltuaria o costante da un lato (bastone, gruccia, cinghie) per cammina-re per circa 100 metri con o senza fermarsi (di solito i SF equivalenti sono combinazioni con piu di due SFdi grado 3).
6.5 Il paziente necessita di assistenza bilaterale costante (bastoni, grucce, cinghie) per camminare per circa20 metri senza fermarsi (di solito i SF equivalenti sono combinazioni con piu di due SF di grado 3).
7.0 Il paziente è incapace di camminare per oltre 5 metri anche con aiuto, ed è essenzialmente obbligato suuna sedia a rotelle, è in grado si spostarsi da solo sulla sedia a rotelle e di trasferirsi da essa ad altra sede(letto, poltrona); passa in carrozzella circa 12 ore al giorno (di solito i SF equivalenti sono combinazioni conpiù di un SF di grado 4+, raramente il SF piramidale e solo di grado 5).
7.5 Il paziente è incapace di fare piu di qualche passo, è obbligato sulla sedia a rotelle; può aver bisogno diaiuto per trasferirsi; si sposta da solo sulla carrozzella standard per un giorno intero. Può aver bisogno diuna carrozzella a motore (di solito i SF equivalenti sono combinazioni con piu di un SF di grado 4+; moltoraramente il solo SF piramidale è di grado 5).
8.0 Il paziente è essenzialmente obbligato a letto o su una sedia a rotelle o viene trasportato sulla carrozzella,ma può stare fuori dal letto per gran parte del giorno; mantiene molte funzioni di assistenza personale; hageneralmente un uso efficace degli arti superiori (di solito i SF equivalenti sono combinazioni, con piu si-stemi di grado 4).
8.5 Il paziente è essenzialmente obbligato a letto per una buona parte del giorno; ha un qualche uso efficace diuno (o di tutti e due) gli arti superiori; mantiene alcune funzioni di assistenza personale (di solito i SF equi-valenti sono combinazioni di grado 4 in piu di un SF).
9.0 Paziente obbligato a letto e dipendente; puo solo comunicare e mangiare (viene alimentato). (Di solito i SFequivalenti sono combinazioni, per la maggior parse di grado 4+.)
9.5 Paziente obbligato a letto e totalmente dipendente; incapace di comunicare efficacemente o di mangiare/deglutire (di solito i SF equivalenti sono combinazioni, quasi tutti di grado 4+).
10.0 Decesso dovuto a SM.

1128 Malattie del sistema nervoso
dall'ingresso sul mercato di farmaci in grado dimodificarne la storia naturale. Tuttavia, mentreil riconoscimento del ruolo svolto dall'attaccoautoimmunitario nella patogenesi dalla malat-tia ha determinato l'utilizzo di terapie immuno-modulanti e immunosoppressive in grado di in-fluenzare il decorso della malattia nelle sue fasiprecoci, sono tuttora carenti i farmaci in gradodi agire sulle conseguenze di questo attacco epertanto di determinare un recupero funziona-le una volta che si sia instaurata la disabilità. Lescelte terapeutiche a disposizione permettono,a seconda della fase della malattia e del suodecorso, di trattare efficacemente l'episodio acu-to, ridurre la frequenza delle ricadute e, parzial-mente, la disabilità e di migliorare alcuni sin-tomi.
Terapia dell'attacco acuto - Il trattamentodelle ricadute consiste nell'uso di steroidi, ge-neralmente per infusione endovenosa in bolo,ma talora anche per via intramuscolare o per os.Avendo tuttavia l'episodio acuto una spontaneatendenza alla regressione, qualora i disturbi si-ano lievi, è possibile non somministrare alcunaterapia in considerazione dei potenziali effetticollaterali del cortisone che, se usato ripetu-tamente, tende a perdere efficacia. Le ricadutepiù gravi vanno trattate con steroidi. Benchè inletteratura siano descritti numerosi schemi tera-peutici è ormai ampiamente accettato l'utilizzodi metilprednisolone endovena 1000 mg per 5giorni seguito da una riduzione graduale del far-maco (tapering) per os per 15 giorni. L'uso diACTH è ormai poco diffuso e scarsamente sup-portato da studi clinici. Gli steroidi esercitereb-bero la loro azione riducendo l'infiammazioneattraverso un'azione pro-apoptotica sugli ele-menti cellulari mononucleati e stabilizzando labarriera emato-encefalica, come dimostrato an-che dagli studi in RM. Il loro uso determina unapiù rapida regressione dei sintomi, ma la loroutilità, a lungo termine, e in particolare la lorocapacità di influenzare il decorso della malat-tia non sono mai state dimostrate.
Terapia diretta a prevenire le ricadute e a ral-lentare la progressione - L'ipotesi che la SM siauna malattia immunomediata è alla base deitrattamenti volti ad influenzarne il decorso at-traverso una azione immunomodulante e/oimmunosoppressiva. Il progredire delle cono-scenze sui meccanismi patogenetici della malat-tia e la possibilità di ridurre e controllare glieffetti collaterali delle immuno-terapie ne hapermesso un utilizzo sempre più precoce e mi-rato.
Trattamenti immunomodulanti - L' inter-ferone beta (IFNβ) è stato il primo farmaco adessere approvato dalla Food and Drug Admini-stration e dell'EMEA Europeo quale terapianella SM a ricadute e remissioni. Gli interferonisono una classe di glicoproteine ad azione anti-virale prodotte da cellule del sistema emato-poietico. Gli interferoni di tipo I includonol'IFNα e l'IFNβ prodotti rispettivamente daileucociti e dai fibroblasti mentre l'INFγ (tipo II)è prodotto dalle cellule NK e dai linfociti T ef-fettori. Nelle pratica commerciale sono dispo-nibili 2 formulazioni commerciali. Il primo,l'IFNβ1b è lievemente modificato rispetto al na-turale, l'INFβ1a, non essendo glicosilato e con-tenendo una serina in posizione 17 al posto diuna cisteina.
Nel 1993 sono stati pubblicati i dati del pri-mo studio che dimostrava una riduzione dellafrequenza di ricadute in pazienti affetti da SMa ricadute e remissioni trattati con INFβ1b. Idati RM evidenziavano una diminuzione delcarico lesionale ed una riduzione del numero dilesioni nuove identificabili con la RM. Da al-lora altre 2 formulazioni commerciali di IFNsono disponibili sul mercato: esse differisconoper le modalità di somministrazione (sottocutevs intramuscolare), la posologia (una volta allasettimana vs giorni alterni), la frequenza dicomparsa di anticorpi anti-IFN e la dose. Indi-pendentemente dalle formulazioni, la cui effi-cacia non è ancora stata efficacemente analiz-zata in un serio studio comparativo, è possibile

1129Malattie demielinizzanti
affermare che l'enorme mole di studi pubblica-ti negli ultimi anni ha confermato l'efficacia dell'INFβ per i pazienti con SM a ricadute e remis-sioni sulla frequenza, riduzione di circa il 30%,gravità delle ricadute nonché su quasi tutti iparametri di attività di malattia (riduzione dicirca il 70%) misurati in risonanza magnetica.Meno certi sono al momento i dati riguardantil'efficacia sulla disabilità cumulativa. Altrettantocontroverso è, sulla base della letteratura piùrecente, l'efficacia dell'INFβ sulle forme secon-dariamente progressive. Al contrario, recentis-sime evidenze sembrano suggerire l'utilitàdell'INFβ, anche a basse dosi, nel ridurre o ral-lentare la conversione in malattia conclamata dipazienti con un singolo episodio monosintoma-tico, suggestivo di malattia demielinizzante. Ilcumulo delle evidenze raccolte negli ultimi annisembra pertanto suggerire che l'INFβ sia effica-ce sia nelle forme iniziali di malattia che inquelle più avanzate, sempre che sia comunquedocumentabile, clinicamente o con la RM, at-tività di malattia.
Il glatiramer acetato (GA) è un polimero lun-go circa 40-100 aminoacidi, composto da quat-tro aminoacidi (L-acido glutamico, L-alanina, L-tirosina, L-lisina) disposti tra loro in modo casua-le ma in costante rapporto molare, in grado dilegarsi in modo specifico con molecole DR delMHC. GA è in grado di sopprimere l'EAE. L'uti-lizzo subcutaneo giornaliero è in grado di ridur-re, con risultati sovrapponibili a quelli dell'IFN,il numero di ricadute in pazienti con SM. Il GAriduce inoltre in modo significativo l'attività dimalattia misurata in RM. Recenti lavori sugge-rirebbero che il GA funzionerebbe come “anti-gene universale” la cui continua somministrazio-ne indurrebbe un “riconoscimento degenerato”da parte di linfociti T specifici per proteine mieli-niche, con conseguente induzione di una rispo-sta di tipo anti-infiammatoria Th2.
La plasmaferesi è stata utilizzata in associa-zione a farmaci immunosoppressori, in partico-lare la ciclofosfamide. Il razionale del suo usorisiede nella rimozione di anticorpi circolanti,
immunocomplessi, complemento, citochine edaltri fattori solubili dell'infiammazione. Nono-stante non manchino lavori che ne sostengonol'efficacia, la sua utilità non è stata documenta-ta con sufficiente certezza per cui viene consi-derato un trattamento di seconda scelta delle ri-cadute resistenti alla terapia cortisonica.
Due studi recenti hanno dimostrato l'effica-cia delle immunoglobuline ad alte dosi (IGEV)nel ridurre significativamente il numero di rica-dute. Dubbio è invece l'effetto sulla risonanzae sulla disabilità. Il meccanismo di azione del-le IGEV consisterebbe sia in una azione immu-nomodulante sia in un'ipotetica capacità di pro-muovere la remielinizzazione. A dispetto deimodesti effetti collaterali e dei sopra citati risul-tati, per alcuni aspetti comparabili a quelli del-l'IFNβ e del glatiramer acetato, le IGEV trova-no modesta applicazione pratica anche a causadegli altri costi.
Farmaci immunosoppressori aspecifici - L'a-zatioprina (AZA) è rapidamente metabolizzatain 6-mercaptopurina che ha un effetto inibentela sintesi degli acidi nucleici, particolarmenteevidente su cellule a rapida replicazione comei linfociti. L'AZA viene impiegata da moltotempo nella terapia della SM ma, nonostante ilsuo lungo uso, ancora incerta è la sua utilità ele opinioni dei ricercatori sono discordanti.Poco usata negli USA, l'AZA è più utilizzata inEuropa: una meta-analisi delle esperienze effet-tuate ha evidenziato che l'AZA è in grado di ri-durre la frequenza delle ricadute e di rallentarela progressione della malattia, sebbene in misu-ra modesta. L'AZA deve essere utilizzata aldosaggio di 2,5-3 mg/kg/die, considerando chela azione immunosoppressiva inizia dopo 3-6mesi. Seppure potenzialmente cancerogena,l'utilizzo cronico di AZA non è mai stato asso-ciato ad un'aumentata incidenza di neoplasie neisoggetti trattati rispetto ai controlli. Uno studiorecente suggerisce un possibile effetto anche suiparametri di RM. Per la sua scarsa tossicità, lasua efficacia, anche se modesta, e il suo bassocosto, l'AZA trova ancora oggi applicazione.

1130 Malattie del sistema nervoso
La ciclofosfamide (CY) è un agente alchi-lante il DNA ad azione citotossica ed immuno-soppressiva. La disponibilità negli ultimi anni difarmaci in grado di ridurne gli effetti collateraliquali il vomito, la nausea, l'amenorrea, la cistiteemorragica, la suscettibilità ad infezioni oppor-tunistiche e l'alopecia ha prepotentemente ripro-posto questo farmaco per il trattamento delleforme di SM più gravi. Benché in letteraturanon vi sia un univoco consenso sulle dosi da uti-lizzare, nella nostra esperienza la somministra-zione per 6 mesi di 1 grammo/m2 endovena diCY in boli mensili è in grado di ridurre la rapi-da progressione di quelle forme di malattia re-sistenti ai comuni immunomodulanti. In alcu-ne particolari situazioni di estrema gravità èpossibile utilizzare in ambiente sterile dosi fino4 grammi/m2.
Un'amino-antraciclina, il Mitoxantrone, èprobabilmente il farmaco immunosoppressivoche ha dato i risultati più incoraggianti negliultimi anni. È stata dimostrato che, alla dose di12 mg/mg per 6 mesi, può rallentare la progres-sione di malattia nelle forme secondariamenteprogressive e di ridurre l'attività di malattia mi-surata alla risonanza magnetica con singoladose di gadolinio di circa il 90%. La sua relati-va maneggevolezza, pur limitata da un'impor-tante cardiotossicità cumulativa (non è possibilesuperare la dose totale di 120 - 140 mg), lo hareso uno dei farmaci più utilizzati nelle formesecondariamente progressive dove vi siano an-cora importanti segni di attività clinica e radio-logica di malattia.
Altre terapie immunosoppressive per la scle-rosi multipla ma ormai scarsamente utilizzate oaddirittura abbandonate a causa dell'inefficaciao degli effetti collaterali, includono la linomide,la cladribina, la ciclosporina, il methotrexate, el'irradiazione linfonodale totale (TLI).
Il trapianto di cellule ematopoietiche stami-nali autologhe, procedura che comporta ancoraimportanti rischi di mortalità (5-8%), rappresen-ta al momento l'ultima frontiera per quelle for-me particolarmente gravi in cui le terapie tradi-
zionali non abbiano sortito effetto. I risultatipreliminari sembrano suggerire la soppressionecompleta dell'attività di malattia misurata in ri-sonanza ma l'utilità clinica di tale trattamento èancora dubbia.
Immunoterapia selettiva - Per immunoterapiaselettiva intendiamo un insieme di approcci concui si tenta di bloccare in modo relativamenteselettivo l'interazione tra le supposte celluleeffettrici del danno e l'organo bersaglio. Sebbe-ne un'analisi approfondita di questi trattamentiesuli dagli scopi di questo capitolo è utile men-zionare alcune terapie che sono al momento giàin fase di studio nell'uomo e il cui possibile uti-lizzo potrebbe rappresentare un ulteriore passoavanti nella cura della SM. Esistono terapie spe-rimentali indirizzate a bloccare o modificarel'azione dei linfociti T effettori: in particolare:1) gli "altered peptide ligand o APL" sono pro-teine modificate strutturalmente in grado di al-terare la risposta autoaggressiva verso le prote-ine della mielina; 2) i peptidi del recettore perl'antigene dei linfociti T (TCR) inducono unarisposta immune contro le cellule autoaggressi-ve che esprimono sulla loro superficie questostesso recettore; 3) la somministrazione dimielina orale aumenta la produzione da parte dicellule del sistema immunitario di citochine adazione anti-infiammatoria; 4) l'utilizzo di cito-chine ad azione anti-infiammatoria, come l'in-terleuchina IL-10 e l'IL-4 oppure di anticorpicontro le citochine ad azione pro-infiammato-ria quali il TNFa, IL-1, IL-12 potrebbe modu-lare la risposta autoimmunitaria; 6) il comples-so solubile costituito dalla proteina basica del-la mielina (MBP) legata al suo recettore MHCdi classe II (AG284) riduce la risposta autoim-munitaria nei confronti della stessa MBP; 7) glianticorpi contro il complesso B7/CD28 e CD40/CD40L inibiscono la costimolazione necessariaper l'attivazione delle cellule T autoaggressive.
Esistono inoltre farmaci che inibiscono l'in-gresso delle cellule autoaggressive nel SNC edhanno principalmente lo scopo di inibirne il

1131Malattie demielinizzanti
passaggio dal sangue periferico attraverso labarriera ematoencefalica. Sono oggi in fase distudio anticorpi anti-molecole d'adesione comeil VLA-4 e l'antigene CD11/CD18 ed inibitoridelle metalloproteasi, enzimi necessari alle cel-lule del sistema immunitario per attraversare labarriera ematoencefalica.
Tra le nuove frontiere della terapia per lasclerosi multipla citiamo ancora gli agenti vol-ti a promuovere la riparazione del danno e larimielinizzazione della lesione, nella speranzache ciò conduca anche al recupero della funzio-ne danneggiata. Inoltre sono in fase di studioalcuni farmaci ad azione neuroprotettiva comei fattori di crescita (IGF-1), il riluzolo, l'elipro-dil e altri (Pirfenidone) che riducono il proces-so cicatriziale gliotico secondario alla demie-linizzazione. Infine, si guarda con grande atten-zione alla possibilità in un prossimo futuro ditrapiantare direttamente nella lesione gli oligo-dendrociti, cioè le cellule che normalmente for-mano la mielina.
Infine una delle grandi sfide nella cura dellaSM è di arrivare a rilasciare le sostanze terapeu-tiche direttamente a livello del SNC invece chenel sangue periferico dove esse sono probabil-mente assai meno efficaci. A questo scopo i ri-cercatori stanno cercando di modificare geneti-camente cellule o virus in grado di trasportarenell'organo bersaglio, in modo innocuo per ilpaziente, molecole che possano spegnere lamalattia (ad esempio una citochina ad azioneanti-infiammatoria) e promuovere la riparazio-ne del danno (ad esempio un fattore di cresci-ta).
Terapia sintomatica - Con il passare del tem-po le ricadute non regrediscono completamen-te e gli esiti neurologici tendono a sommarsi.Una terapia solo sintomatica non è da sottova-lutare, se è in grado di migliorare alcuni distur-bi, in particolar modo nelle forme più avanzatedi malattia allorchè le terapie in grado di mo-dificarne il decorso naturale sono ormai ineffi-caci.
Non sempre la spasticità necessita di esseretrattata, poiché in molti casi l'aumento del tonomuscolare può essere utile per mantenere la sta-zione eretta. Il baclofen, al dosaggio di 50-75mg al dì è in grado di ridurre la spasticità, maspesso può accentuare l'ipostenia e la finestraterapeutica è piuttosto stretta. Il dantrolene (25-100 mg al dì) può essere utile ma non va dimen-ticata la sua possibile epatotossicità. Il diazepam(5-20 mg al dì) può dare sedazione, ma può es-sere utile assumerlo alla sera, prima di coricar-si, per diminuire gli spasmi notturni e il clonoindotto dai movimenti durante il sonno. Latinazidina, al dosaggio di 12-18 mg al dì, è par-ticolarmente attiva, ma per il suo effetto seda-tivo non è ben sopportata da tutti i pazienti. Neicasi avanzati, in cui coesistono gravi disturbisfinterici e la motilità agli arti superiori è mar-catamente compromessa, possono essere presiin considerazione trattamenti più aggressivi,quali l'installazione di pompe intratecali a rila-scio controllato di baclofen, la somministrazio-ne mirata di tossina botulinica a livello dei di-stretti muscolari più colpiti da spasticità. Lemanifestazioni parossistiche della malattia, qua-li la nevralgia del trigemino, gli spasmi tonicie i disturbi sensitivi parossistici, rispondonomolto bene ai farmaci antiepilettici quali lacarbamazepina (400-1200 mg/die), il gabapen-tin (300-1200 mg/die) e la lamotrigina (50-200mg/die). La stessa terapia, così come l'amitripti-lina alle dosi di 30-50 mg/die, può essere utiliz-zata per i dolori muscolari, la sensazione di sti-ramento e le parestesie. Il tremore cerebellaree l'atassia sono sintomi particolarmente invali-danti e poco sensibili alla terapia sintomatica.Di qualche utilità, peraltro modesta, possonoessere i beta-bloccanti, come il propranololo egli antidepressivi serotoninergici quali il trazo-done ed altri farmaci quali il primidone ed il bu-spirone. La sensazione di fatica che frequente-mente viene allegata dai pazienti può trarre lie-ve beneficio dall'impiego di amantadina, 4-diaminopiridina e, come recentemente descrit-to, di modafenil (200 mg/die).

1132 Malattie del sistema nervoso
I disturbi sfinterici sono particolarmente dif-ficili da trattare e richiedono spesso l'esecuzio-ne di prove urodinamiche, al fine di chiarire ilmeccanismo e l'importanza della dissinergia fradetrusore e gli sfinteri. La minzione imperiosapuò essere parzialmente controllata dagli anti-colinergici come la propantelina o l'oxibutina,od anche dall'amitriptilina. Le difficoltà a svuo-tare la vescica possono essere al contrario mi-gliorate da farmaci colinomimetici. Particolar-mente utile, quando il residuo di urina è supe-riore ai 200 ml, è la cateterizzazione intermit-tente, che il paziente può eseguire da solo, e cheha parecchi vantaggi, innanzitutto di impedireinfezioni urinarie. La stipsi, disturbo frequenteanche in casi iniziali, può essere trattata condieta ricca in scorie, emollienti, microclismi.
Il trattamento fisioterapico ha particolarerilevanza nella terapia sintomatica e di suppor-to. Il fisioterapista deve evitare di affaticare ec-cessivamente il paziente, deve combattere l'iper-tonia di determinati gruppi muscolari, consiglia-re l'uso di particolari protesi. Alcune modifi-cazioni nell'abitazione possono essere consiglia-te, per facilitare la deambulazione, la puliziapersonale, l'attività in cucina. Il calore eccessi-vo, quale quello di un bagno caldo, deve essereevitato, mentre particolarmente utile è la fisio-terapia in piscina, dove possono essere effettuatialcuni movimenti altrimenti impossibili. L'ab-bassamento della temperatura corporea, otteni-bile con bagni freddi o anche con particolariapparecchi, migliora, specie nei pazienti termo-sensibili, la conduzione nervosa e può essere unsemplice metodo utilizzato per migliorare leprestazioni motorie, per la durata di circa 1-2ore dopo il raffreddamento.
Conclusioni
Le strategie terapeutiche in una malattia cosìdifficile e imprevedibile sono in costante diveni-re, in rapporto ai progressi della ricerca di basee applicata, ed all'enorme interesse economicoche questa malattia suscita. Pur nell'incertezza e
nella mancanza di una terapia sicuramente effi-cace, esistono alcune linee guida di comporta-mento. L'episodio acuto, se lieve, non va tratta-to, mentre se di gravità moderata o marcata vacurato con terapia steroidea, ed in particolare conun bolo di metilprednisolone e.v. La disponibi-lità corrente di terapie in grado di modificare si-gnificativamente il decorso naturale della malat-tia quali l'IFNβ in primo luogo, e, in alternativa,il glatiramer acetato, l'AZA, l'IGEV suggerisco-no di trattare precocemente il paziente con SM.Qualora tali trattamenti siano inefficaci, è utiletrattare il paziente ancora clinicamente attivo conun farmaco immunosoppressore, mitoxantrone ociclofosfamide in modo tanto più aggressivoquanto più tumultuoso è il decorso della malat-tia. A questo proposito vale la pena ricordare chela maggior parte delle terapie attualmente dispo-nibili possono rallentare il decorso della malat-tia e pertanto la comparsa di un danno, che la RMha dimostrato essere spesso clinicamente silen-te. Al contrario, una volta che il deterioramentoneurologico si è stabilizzato e la disabilità è di-ventata permanente, non vi sono terapie in gra-do di far regredire e riparare il danno. In questafase bisogna effettuare soprattutto terapia sinto-matica, associata ad un prolungato e persona-lizzato trattamento fisioterapico. Purtroppo inmolti casi la malattia progredisce nonostante lepiù varie strategie terapeutiche, ma il profondoscetticismo del passato sembra svanire, per la-sciare il posto alla convinzione che sia possibileincidere sul decorso della malattia, anche se lameta è ancora lontana.
Encefalomielite acuta disseminata(EAD)
L'encefalomielite acuta disseminata è stataclassicamente descritta come una sindrome adecorso monofasico, secondaria ad una vacci-nazione (“encefalomielite post-vaccinica”) op-pure ad un'infezione(“encefalomielite post-in-fettiva”), caratterizzata da diffuse aree perive-

1133Malattie demielinizzanti
nulari di infiammazione, edema e demieliniz-zazione. In rari casi, è stata descritta una formaiperacuta, clinicamente tumultuosa, caratteriz-zata istologicamente da infiltrati perivenularinecrotico-emorragici o leucoencefalite acutaemorragica di Weston Hurst. Talora, non è pos-sibile associare una sindrome clinica di EAD adun episodio infettivo o alla somminstrazione divaccino ed allora il quadro è definito “encefa-lomielite acuta disseminata idiopatica”.
A) ENCEFALOMIELITE ACUTA DISSEMINATA POST-VACCINICA
I primi casi di EAD vennero descritti in se-guito alla somministrazione di vaccini contro larabbia e contro il vaiolo (vaccino di Semple). Inparticolare, venne dimostrato che la comparsadi encefalomielite in seguito a somministrazio-ne di vaccino anti-vaioloso, estratto da cervel-lo di coniglio, non era dovuto all'effetto citopa-tico del virus sulle cellule del SNC, ma ad unarisposta abnorme del sistema immunitario con-tro certi componenti delle guaine mieliniche.Tali studi dimostrarono, per la prima volta, lapossibilità che meccanismi immunitari potesse-ro danneggiare strutture cerebrali, e furono allabase della successiva creazione di un modelloanimale di autoimmunità del sistema nervosocentrale (encefalite autoimmune sperimentale).L'introduzione di vaccini preparati senza utiliz-zare tessuto neurale ha notevolmente diminuitol'incidenza di questa malattia. Negli ultimi anni,sono state riportate sporadiche osservazioni diEAD dopo vaccinazione anti-influenzale, anti-vaiolosa, anti-pertosse, anti-difterite, anti-rosolia,anti-rabbia e anti-morbillo. È stata inoltre segna-lata la comparsa di una sindrome tipo EAD dopola somministrazione di alcuni antibiotici.
B) ENCEFALOMIELITE ACUTA DISSEMINATA POST-INFETTIVA
L'EAD post-infettiva si associa più frequen-temente ai comuni esantemi infantili di origine
virale. Complicazioni neurologiche secondariead infezione morbillosa si verificano in circa 1su 500-1000 soggetti. Relativamente frequenteè la comparsa di un quadro di EAD dopo infe-zione con il virus della varicella (1 su 4.000-10.000); più raramente è stata descritto comecomplicazione di rosolia, parotite, influenza,mononucleosi o infezione da virus coxsackie oda mycoplasma.
EZIOPATOGENESI
Dati clinici, epidemiologici e sperimentalidepongono per una reazione autoimmunitariadiretta contro antigeni mielinici. Infatti l'ence-falite autoimmune sperimentale rappresenta ilmodello ottimale di malattia autoimmunitariaindotta da un'immunizzazione o da un'infezio-ne. A conferma di quest'ipotesi, è stata segna-lata un'aumentata risposta verso la proteinabasica della mielina (MBP) nel sangue e nelliquor di pazienti con EAD. Tale risposta potreb-be essere indotta dalla somiglianza molecolaretra proteine del virus infettante e componentimieliniche del SNC (“molecular mimicry”), op-pure dal danno cerebrale indotto dall'infezionecui seguirebbe una sensibilizzazione verso nuo-vi antigeni cerebrali.
Il rapporto di questo gruppo di malattie conla SM, benché difficilmente inquadrabile, appa-re per molti aspetti molto stretto ed è ben notala possibilità che si verifichi una cronicizza-zione della malattia il cui decorso diventa spes-so indistinguibile da quello di una tipica SM.
Neuropatologia - L'EAD è una sindrome ca-ratterizzata dalla presenza di infiltrati perivenu-lari di cellule mononucleate, prevalentementemacrofagi e linfociti, cui si associano edema emarcata demielinizzazione caratteristicamenteperivascolare.
Sintomatologia e decorso - L'esordio è spes-so improvviso, l'evoluzione rapida e la progno-si estremamente variabile. Nell'EAD post-in-

1134 Malattie del sistema nervoso
fettiva l'insorgenza dei sintomi neurologici puòessere preceduta da cefalea, febbre, sonnolen-za e talora da modesti segni meningei. Il qua-dro clinico è invariabilmente caratterizzato dasegni di sofferenza focale del SNC che evol-vono nel corso di ore e raggiungono l'acme inpochi giorni. I sintomi possono riflettere unasofferenza cortico-sottocorticale con emi-paresi, disturbi delle sensibilità, del troncoencefalico con deficit multipli dei nervi cra-nici, del cervelletto con atassia, dei gangli dellabase con movimenti involontari e del midollospinale con paraparesi, disturbi sfinterici. Fre-quente è l'insorgenza di crisi comiziali. Il mi-glioramento del quadro clinico, quando occor-re, si verifica nell'arco di giorni, ma, più spes-so, di settimane. La mortalità può raggiunge-re il 10-25% dei casi. Spesso sono presenti esitiquali crisi epilettiche o ritardo nello sviluppopsico-motorio dell'individuo. È possibile che siverifichino ricadute o addirittura una croniciz-zazione del quadro clinico.
Diagnosi - È semplice quando sia possibilestabilire un rapporto temporale tra pregressainfezione virale o somministrazione di vaccinoe la sintomatologia neurologica multifocale adesordio acuto o sub-acuto. In circa l'80% deicasi è possibile osservare una pleiocitosi liquo-rale con cellule mononucleate (fino a 200 cel-lule/mmc); talora possono essere presenti anchepolimorfonucleati, espressione di un processoacuto di tipo necrotizzante, e un modesto au-mento delle proteine (50-150 mg/dl). È statadescritta la presenza di bande oligoclonali nelliquor, per lo più, a carattere transitorio. Inda-gini elettrofisiologiche sono spesso in grado didimostrare, accanto ad una sofferenza del SNC,anche un interessamento del SNP. La RM dimo-stra, nella maggior parte dei casi, aree dissemi-nate a livello della sostanza bianca cerebrale cheassumono contrasto nelle sequenze T1 pesate,a dimostrazione di un danno di barriera.
La diagnosi differenziale tra EAD e una for-ma acuta di sclerosi multipla (SM) può essere
difficile. Sono suggestivi dell'EAD la presenzadi un simultaneo coinvolgimento dei nervi ot-tici (neurite ottica bilaterale), di mielite trasver-sa completa, segni meningei, convulsioni ecoma.
L'esame del liquor nella SM non evidenziageneralmente un aumento delle proteine liquo-rali e di polimorfonucleati; la pleiocitosi inol-tre, raggiunge raramente valori superiori a 50cellule/mmc. Infine, l'EAD ha un decorso mo-nofasico, spesso tumultuoso, e più frequente-mente colpisce l'età infantile. In casi ad esordioparticolarmente rapido e grave con prevalenteinteressamento emisferico, è necessario prende-re in considerazione l'ipotesi di un'encefalitevirale (ad esempio, da Herpes Simplex).
Terapia - Consiste nella somministrazione dimetilprednisolone e.v. ad alto dosaggio, secon-do i protocolli comunemente utilizzati nella te-rapia della SM. in fase acuta.
Leucoencefalite acuta emorragica di Wen-ston-Hurst - Questa rara forma di EAD è ge-neralmente preceduta da una comune infiamma-zione delle vie aeree superiori benchè talora nonsia possibile identificare anamnesticamente al-cun episodio infettivo. L'esame neuropatologicorivela aree perivascolari disseminate di necrosifibrinoide, infiltrati cellulari, edema e stravasiemorragici mentre non è mai presente demie-linizzazione. La malattia, più comune nei ma-schi, è caratterizzata da un esordio brusco e tu-multuoso con febbre, convulsioni e coma. Ta-lora, l'insorgenza di segni focali è mascheratadalla rapida insorgenza di disturbi dello stato dicoscienza. L'evoluzione è rapida e frequente-mente fatale. L'esame del liquor nella maggiorparte dei casi rivela un aumento della pressio-ne intrarachidea, proteinorrachia, marcata pleio-citosi con cellule mononucleate e polimor-fonucleati. La diagnosi differenziale includemalattie cerebrali focali a rapida evoluzionequali l'encefalite da herpes simplex, gli ascessie le neoplasie cerebrali a rapida crescita.

1135Malattie demielinizzanti
Riferimenti bibliografici
BROCKE, S., GAUR A., PIERCY C., GAUTAM A., GIJBELS K.,FATHMAN C.G., STEINMAN L.: Induction of relapsing pa-ralysis in experimental autoimmune encephalomyelitis bybacterial superantigen. Nature 365, 642-644, 1993.
GOLD, R., HARTUNG H.P., TOYKA K.V.: Animal models forautoimmune demyelinating disorders of the nervoussystem. Mol. Med. Today 6, 88-91, 2000.
LUBLIN, F. D. AND S. C. REINGOLD. Defining the clinicalcourse of multiple sclerosis: results of an internationalsurvey. National Multiple Sclerosis Society (USA) Advi-sory Committee on Clinical Trials of New Agents in Mul-tiple Sclerosis. Neurology 46, 907-911, 1996.
MILLER S.D., VANDERLUGT C.L., BEGOLKA W.S., PAO W.,YAUCH R.L., NEVILLE K.L., KATZ-LEVY Y., CARRIZOSA A.,KIM B.S.: Persistent infection with Theiler's virus leadsto CNS autoimmunity via epitope spreading. Nat. Med.3, 1133-1136, 1997.
OKSENBERG J.R., BARANZINI S.E., BARCELLOS L.F., HAUSER
S.L.: Multiple sclerosis: genomic rewards. J Neuroim-munol. 113, 171-184, 2001.
POLIANI P.L., BROK H., FURLAN R., RUFFINI F., BERGAMI A.,DESINA G., MARCONI P.C., ROVARIS M., UCCELLI A., GLO-RIOSO J.C., PENNA G., ADORINI L., COMI G., MARTINO G.:Delivery to the central nervous system of a nonreplicativeherpes simplex type 1 vector engineered with the inter-leukin 4 gene protects rhesus monkeys from hyperacuteautoimmune encephalomyelitis. Hum.Gene Ther. 12,905-920, 2001.
POSER C.M., PATY D.W., SCHEINBERG L., MCDONALD W.I.,DAVIS F.A., EBERS G.C., JOHNSON K.P., SIBLEY W.A.,SILBERBERG D.H., TOURTELLOTTE W.W.: New diagnosticcriteria for multiple sclerosis: guidelines for researchprotocols. Ann. Neurol. 13, 227-231, 1983.
STEINMAN L.: Escape from “horror autotoxicus”: patho-genesis and treatment of autoimmune disease. Cell 80,7-10, 1995.


















