
MITOCONDRI E DEMENZA - univacalabria.it · Miopatia mitocondriale:(a) ... • Caratterizzata da...
143
Dott. Michelangelo Mancuso Dipartimento di Neuroscienze AOUP Pisa MITOCONDRI E DEMENZA
Transcript of MITOCONDRI E DEMENZA - univacalabria.it · Miopatia mitocondriale:(a) ... • Caratterizzata da...

Dott. Michelangelo Mancuso
Dipartimento di Neuroscienze
AOUP Pisa
MITOCONDRI E DEMENZA

• MITOCONDRIO E STRESS OSSIDATIVO
• MITOCONDRIO COME CAUSA DI
DEMENZA
• MITOCHONDRIAL CASCADE
HYPOTHESIS IN ALZHEIMER DISEASE

• Un mitocondrio è un organulo cellulare
presente in tutti gli eucarioti.
• I mitocondri sono presenti nel citoplasma
di tutte le cellule animali a metabolismo
aerobico.
• Questo organulo può essere considerato
come la centrale energetica della
cellula: infatti esso ricava energia dai
substrati organici per produrre un
gradiente ionico che viene sfruttato per
produrre adenosintrifosfato (ATP).

EVIDENZE:
• presenza di molecole di cardiolipina ed assenza di colesterolo nella membrana
interna
• la presenza di un DNA circolare a doppia elica
• presenza di ribosomi propri e di una doppia membrana
• assenza di istoni
• ribosomi sensibili ad alcuni antibiotici (come il cloramfenicolo).
• replicazione, per scissione binaria, autonoma rispetto alla cellula.
La teoria endosimbiotica afferma che i
mitocondri deriverebbero da ancestrali
batteri, dotati di metabolismo ossidativo,
inglobati dalle cellule eucariote con
conseguente mutuo beneficio.
I mitocondri sarebbero, quindi, cellule
simbiontiche ottimizzate per produrre molta
energia e consumarne pochissima.
E' stato calcolato che questa simbiosi doni
un vantaggio energetico alle cellule
eucariote da 3 a 4 ordini di grandezza in
più. Salvador Dalì, Simbiosi donna-animale (1928)
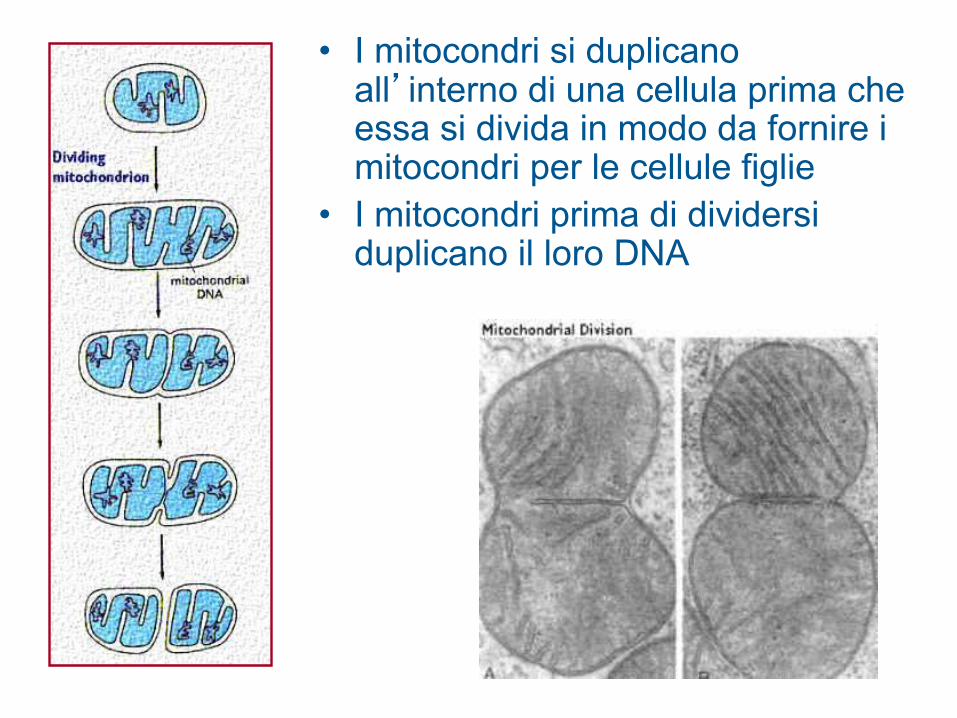
• I mitocondri si duplicano all’interno di una cellula prima che essa si divida in modo da fornire i mitocondri per le cellule figlie
• I mitocondri prima di dividersi duplicano il loro DNA
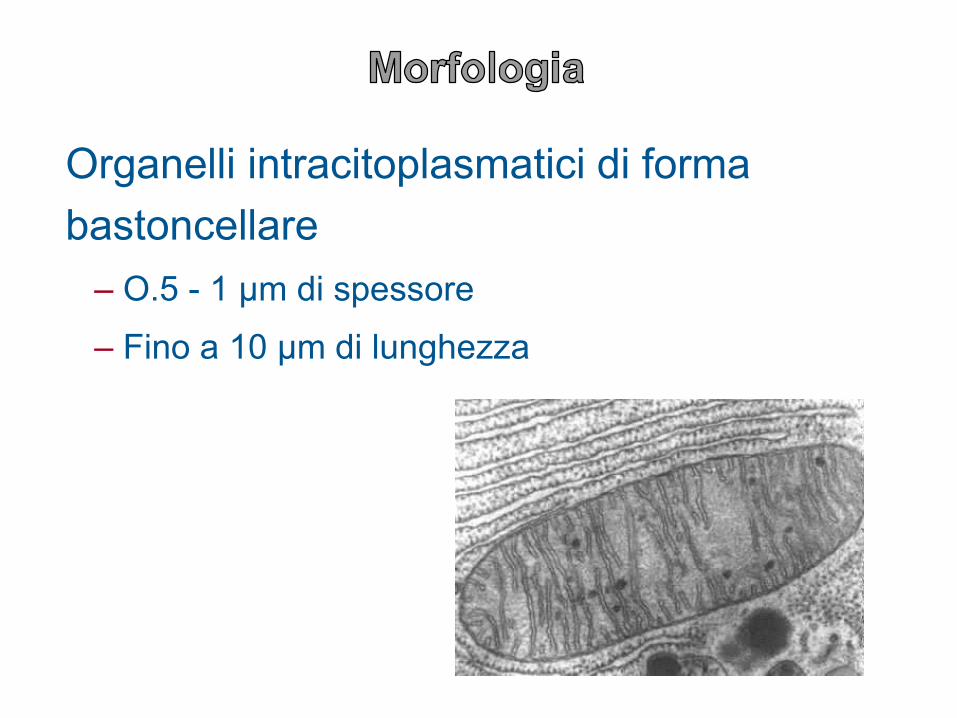
Organelli intracitoplasmatici di forma
bastoncellare
– O.5 - 1 µm di spessore
– Fino a 10 µm di lunghezza
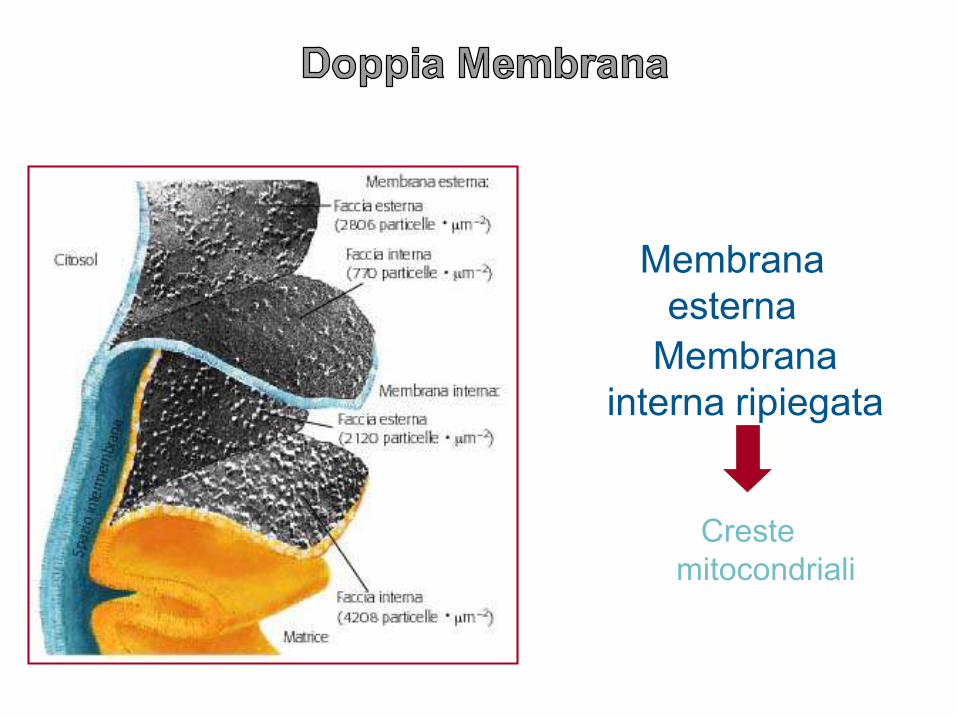
Membrana
esterna
Membrana
interna ripiegata
Creste
mitocondriali
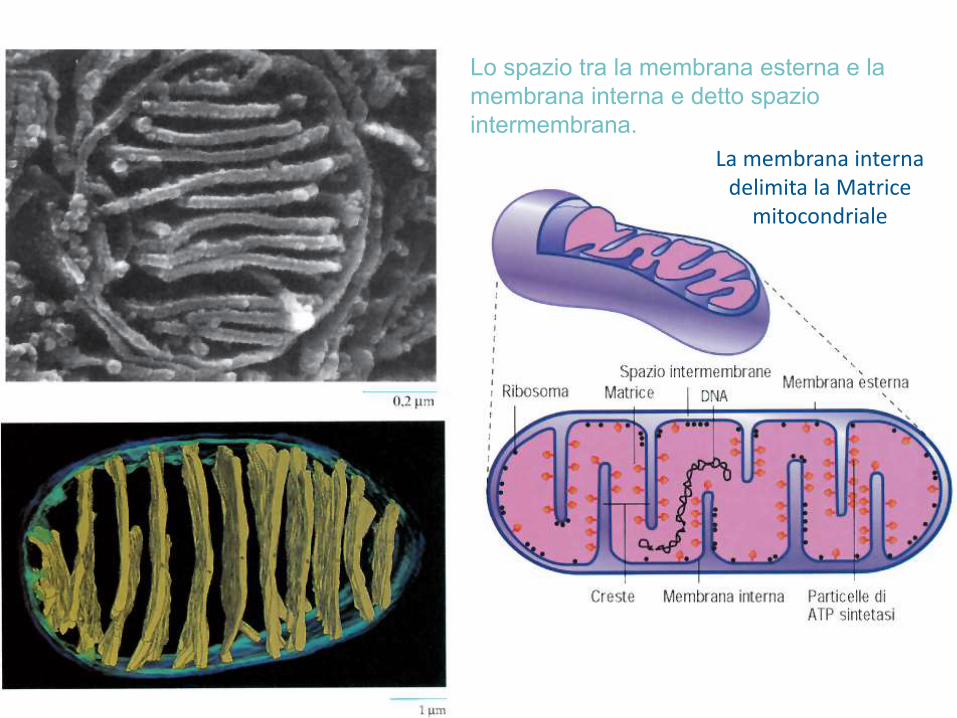
Lo spazio tra la membrana esterna e la
membrana interna e detto spazio
intermembrana.
La membrana interna delimita la Matrice
mitocondriale
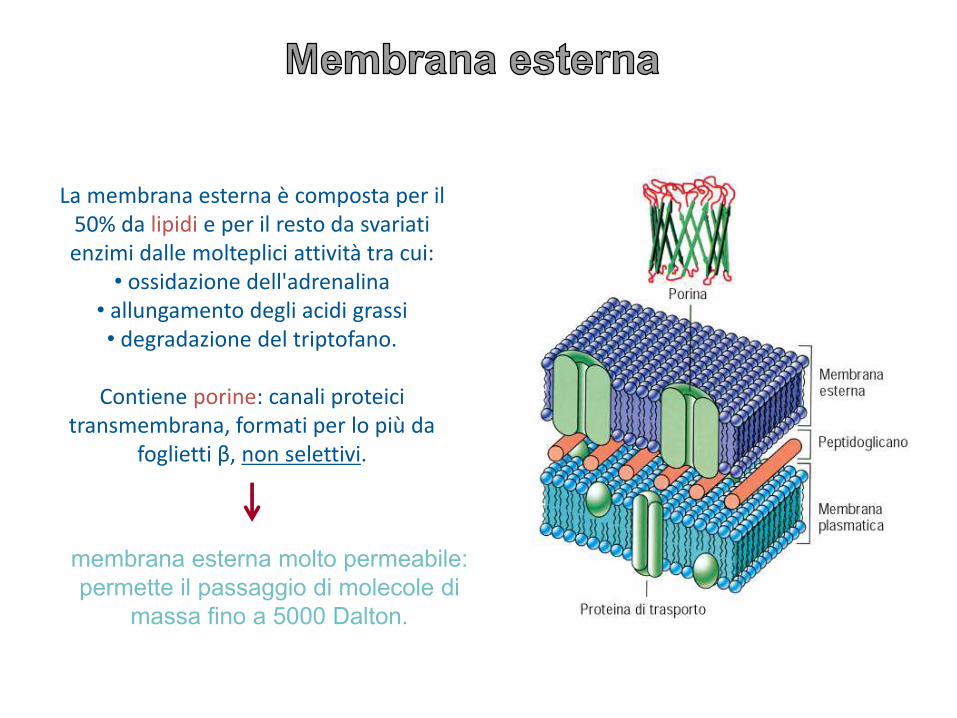
La membrana esterna è composta per il 50% da lipidi e per il resto da svariati enzimi dalle molteplici attività tra cui:
• ossidazione dell'adrenalina • allungamento degli acidi grassi • degradazione del triptofano.
Contiene porine: canali proteici transmembrana, formati per lo più da
foglietti β, non selettivi.
membrana esterna molto permeabile:
permette il passaggio di molecole di
massa fino a 5000 Dalton.

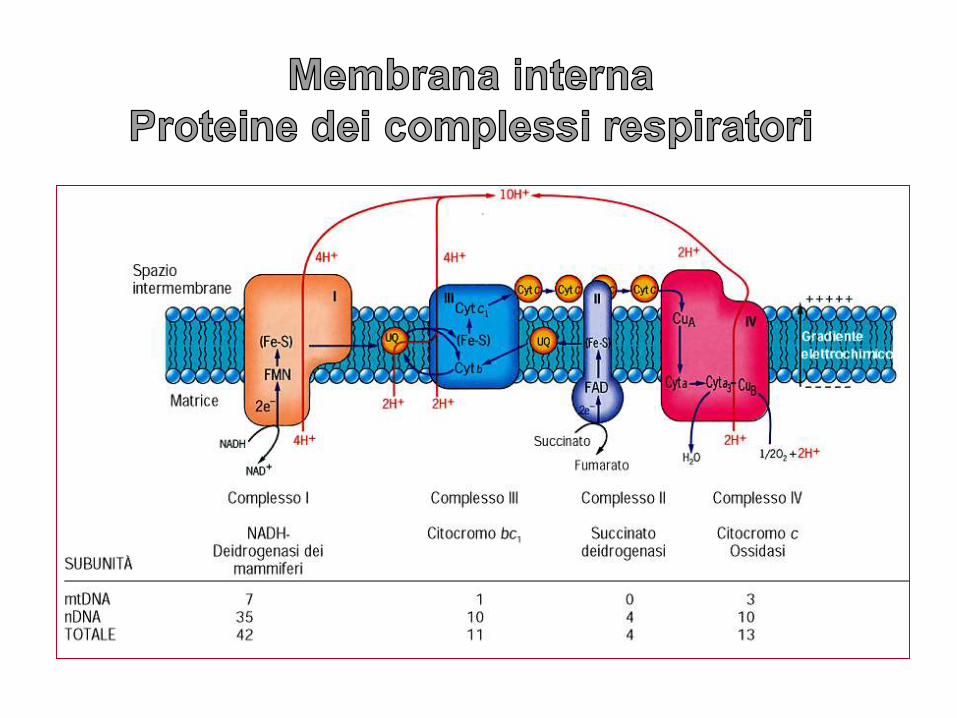
La membrana interna ha un rapporto proteine/lipidi che si aggira intorno a 3:1 e contiene più di 100 molecole polipeptidiche.
La membrana interna, contrariamente a quella esterna, è selettivamente permeabile, priva di porine, ma con trasportatori transmembrana altamente selettivi per ogni molecola o ione.
Si distinguono,
quindi due facce
della membrana
interna
versante della matrice - o versante N in ragione del
potenziale di membrana neutro -
versante citosolico (in quanto viene facilmente raggiunto dalle piccole molecole del citosol cellulare) – o versante P in ragione del potenziale di membrana positivo nello spazio intermembranoso interno -

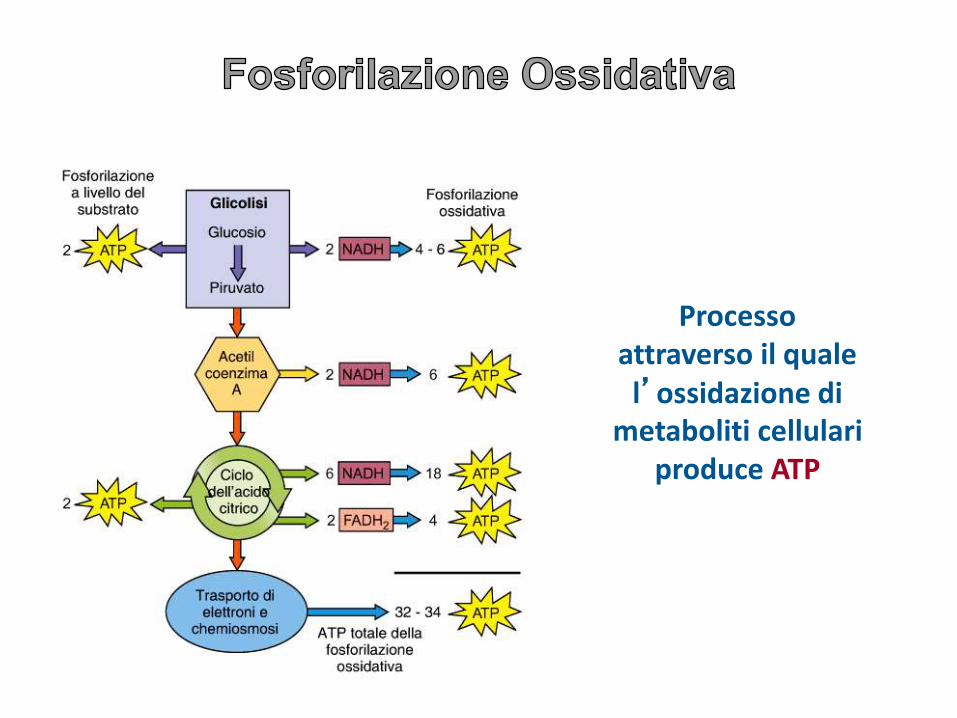
Processo attraverso il quale l’ossidazione di
metaboliti cellulari produce ATP
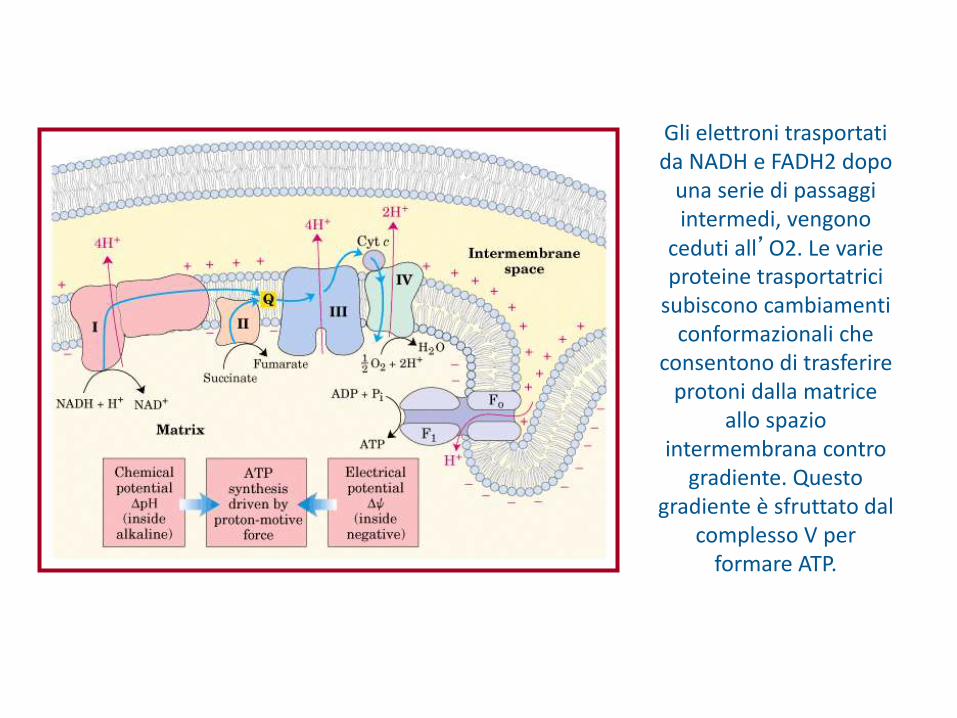
Gli elettroni trasportati da NADH e FADH2 dopo
una serie di passaggi intermedi, vengono
ceduti all’O2. Le varie proteine trasportatrici
subiscono cambiamenti conformazionali che
consentono di trasferire protoni dalla matrice
allo spazio intermembrana contro
gradiente. Questo gradiente è sfruttato dal
complesso V per formare ATP.


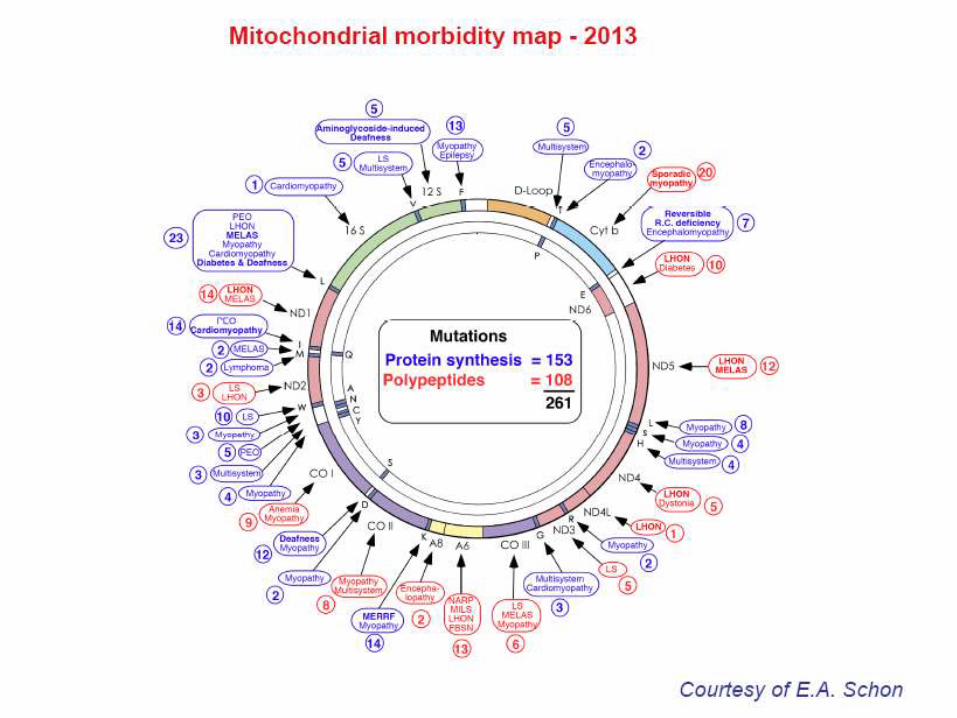
I mitocondri possiedono un proprio DNA costituito da un
cromosoma circolare nudo, superavvolto a doppia elica,
16.569 pb codificanti, 3% del DNA totale.
In ogni mitocondrio sono presenti da 2 a 10 copie di mtDNA.
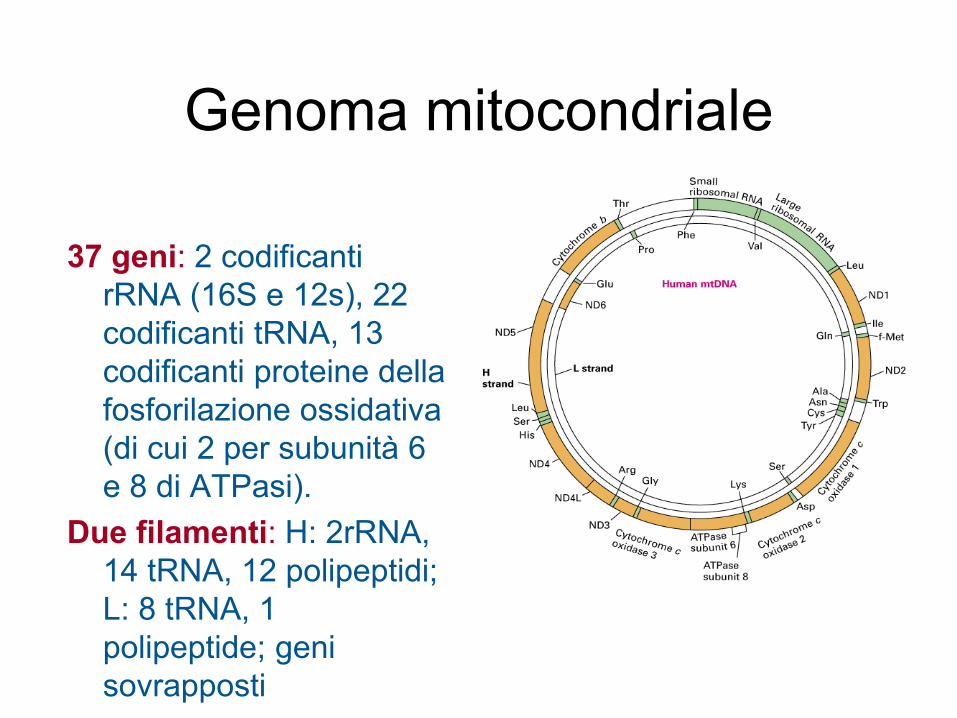
Genoma mitocondriale
37 geni: 2 codificanti
rRNA (16S e 12s), 22
codificanti tRNA, 13
codificanti proteine della
fosforilazione ossidativa
(di cui 2 per subunità 6
e 8 di ATPasi).
Due filamenti: H: 2rRNA,
14 tRNA, 12 polipeptidi;
L: 8 tRNA, 1
polipeptide; geni
sovrapposti
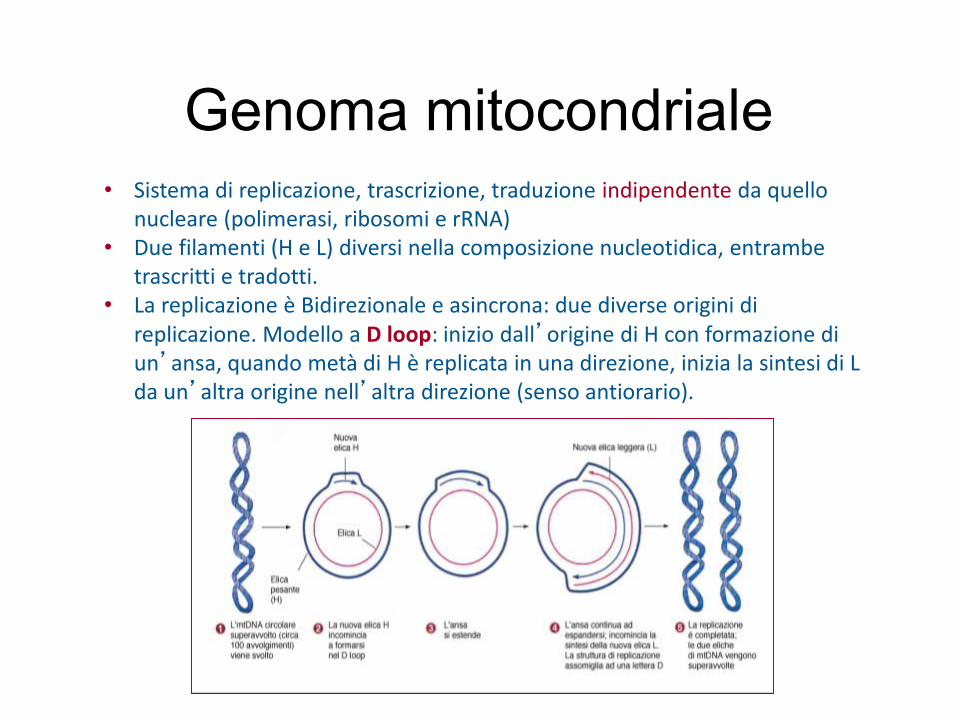
Genoma mitocondriale• Sistema di replicazione, trascrizione, traduzione indipendente da quello
nucleare (polimerasi, ribosomi e rRNA)• Due filamenti (H e L) diversi nella composizione nucleotidica, entrambe
trascritti e tradotti.• La replicazione è Bidirezionale e asincrona: due diverse origini di
replicazione. Modello a D loop: inizio dall’origine di H con formazione di un’ansa, quando metà di H è replicata in una direzione, inizia la sintesi di L da un’altra origine nell’altra direzione (senso antiorario).

• Non esistono introni• Codice genetico diverso da quello nucleare.
• Non esiste sistema di riparo delle mutazioni (tasso di mutazione è 10-20 volte superiore a quello nucleare)
Genoma mitocondriale
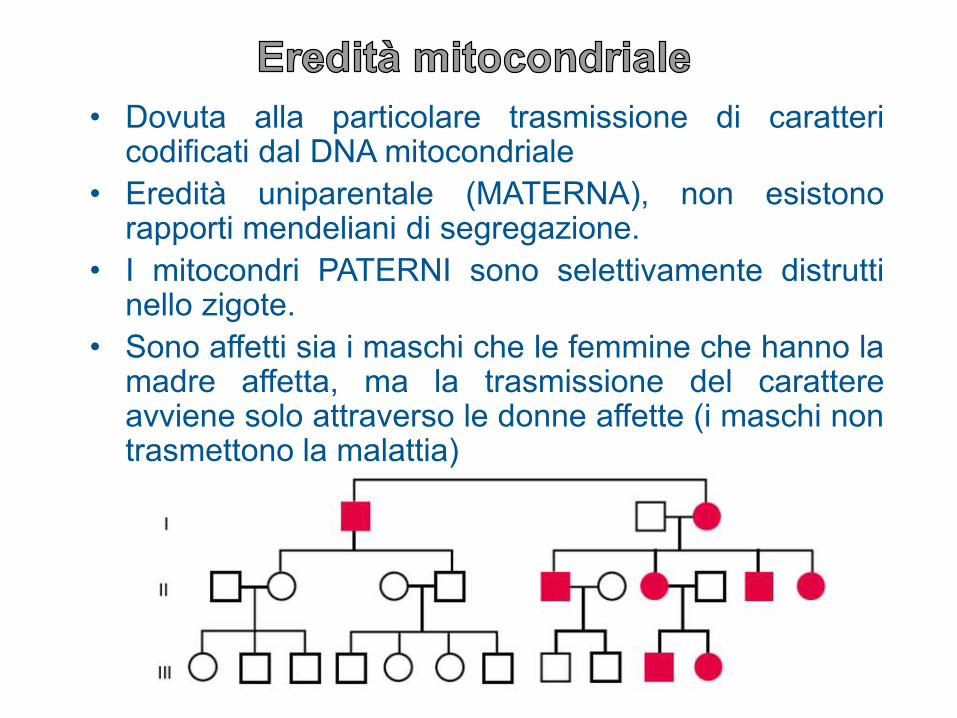
• Dovuta alla particolare trasmissione di carattericodificati dal DNA mitocondriale
• Eredità uniparentale (MATERNA), non esistonorapporti mendeliani di segregazione.
• I mitocondri PATERNI sono selettivamente distruttinello zigote.
• Sono affetti sia i maschi che le femmine che hanno lamadre affetta, ma la trasmissione del carattereavviene solo attraverso le donne affette (i maschi nontrasmettono la malattia)

• Ciascuna cellula contiene migliaia di molecole di mtDNA(poliplasmia)
• Una mutazione nel mtDNA viene trasmessa ai mitocondri inmaniera casuale perchè durante la divisione cellulare imitocondri sono distribuiti alle cellule figlie in modo casuale
• Una cellula può ricevere dalla cellula madre una popolazionedi mtDNA omogenea, sana o mutante (omoplasmia)
• Alternativamente può ricevere una popolazione mista dimolecole normali e mutate (eteroplasmia)
• Se le cellule eteroplasmiche sono capostipiti di vari tessuti,l’espressione fenotipica sarà multisistemica.
Omoplasmia
(genoma normale)
Omoplasmia
(genoma mutato)Eteroplasmia
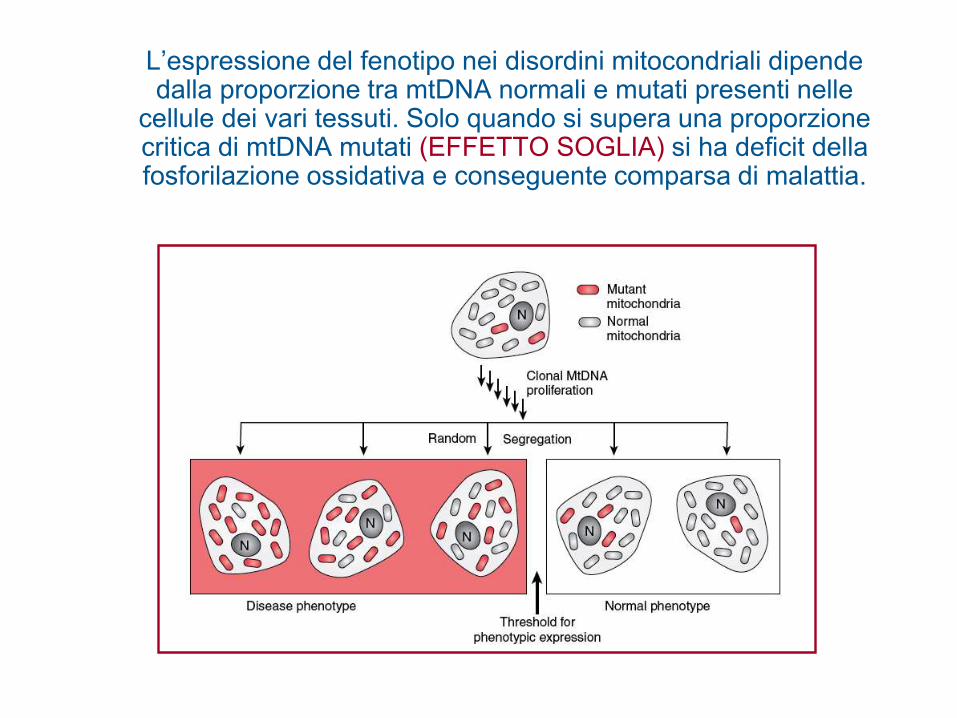
L’espressione del fenotipo nei disordini mitocondriali dipende dalla proporzione tra mtDNA normali e mutati presenti nelle
cellule dei vari tessuti. Solo quando si supera una proporzione critica di mtDNA mutati (EFFETTO SOGLIA) si ha deficit della fosforilazione ossidativa e conseguente comparsa di malattia.

Variabilità fenotipica: è espressa dal rapporto mtDNA mutante/mtDNa normale nella cellula. La severità del difetto
biochimico e la rilevanza clinica delle manifestazioni patologiche sono variabili e dipendono quindi da due fattori:
la percentuale di mtDNA mutante presente nella cellula; la dipendenza funzionale dei diversi tessuti dal metabolismo
ossidativo. Per tale motivo il sistema nervoso centrale, il muscolo striato
scheletrico, il cuore, il rene ed il fegato sono gli organi più frequentemente coinvolti.
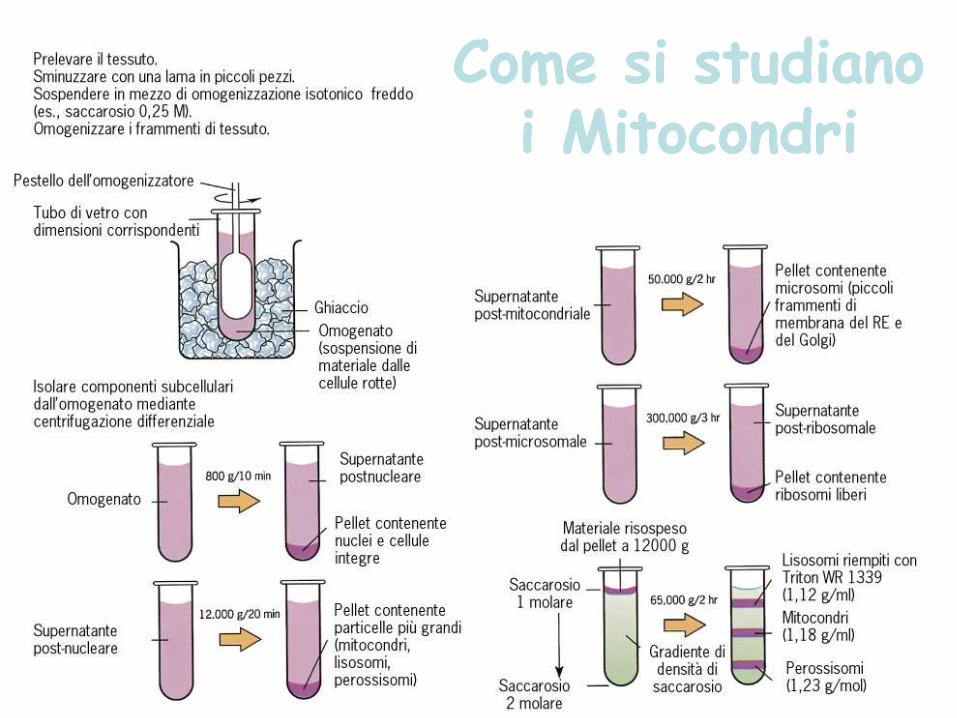
Come si studiano i Mitocondri
Come si studiano i Mitocondri
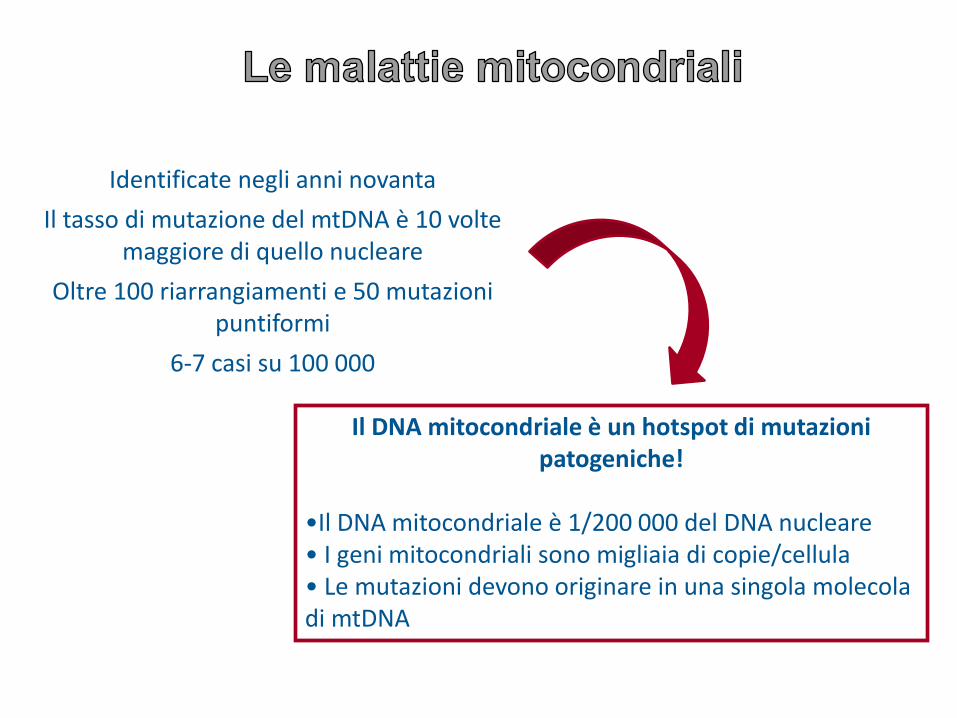
Identificate negli anni novanta
Il tasso di mutazione del mtDNA è 10 volte maggiore di quello nucleare
Oltre 100 riarrangiamenti e 50 mutazioni puntiformi
6-7 casi su 100 000
Il DNA mitocondriale è un hotspot di mutazioni patogeniche!
•Il DNA mitocondriale è 1/200 000 del DNA nucleare• I geni mitocondriali sono migliaia di copie/cellula• Le mutazioni devono originare in una singola molecola di mtDNA
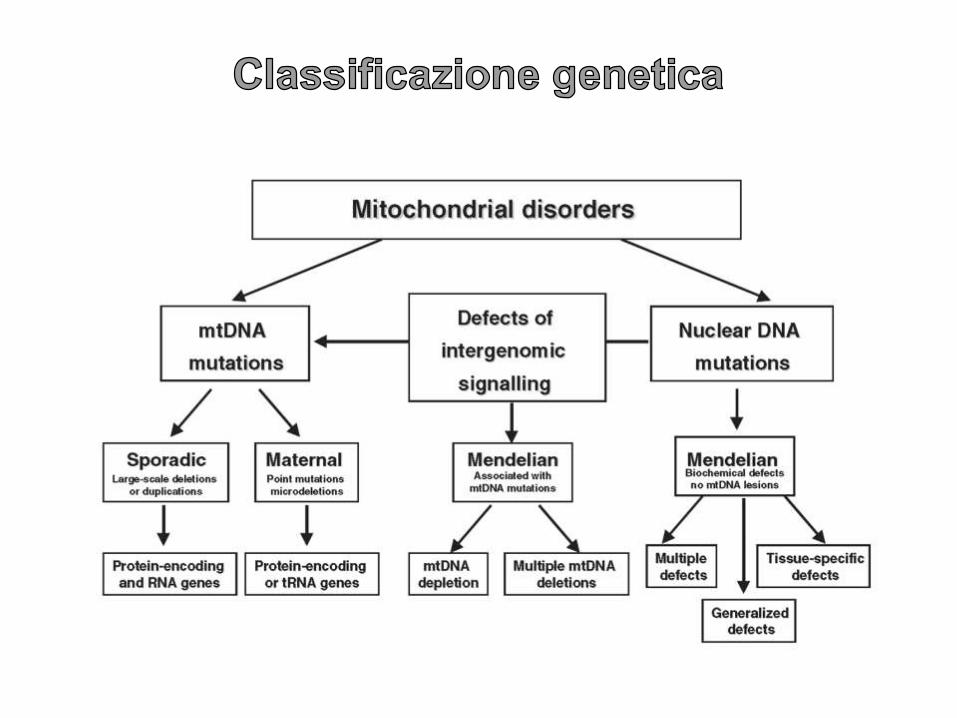

Le mutazioni possono
essere MISSENSO,
INSERZIONI,
DELEZIONI e
DEPLEZIONI
Mutazioni a carico del
mtDNA riducono la
produzione di ATP
danneggiando i diversi
organi a secondo del
loro fabbisogno
energetico

•Ptosi palpebrale
•Oftalmoplegia esterna
•Intolleranza all’esercizio fisico
•Debolezza muscolare
•Crampi, mialgie
•Atassia
•Degenerazione retinica
•Ipoacusia neurosensoriale
•Ritardo o deterioramento mentale
•Atrofia ottica
•Crisi epilettiche e mioclonie
•Stroke
•Cefalea emicranica
•Parkinsonismo
•Neuropatia
•Blocchi di branca
•Cardiomiopatia
•Tubulopatia renale
•Diabete mellito
•Ipotiroidismo
•Ipoparatiroidismo
•Bassa statura
•Lipomatosi multipla
•Insufficienza epatica

danno ossidativo
ROS-mediato
ALTERAZIONI DEL mtDNA
DIFETTO ENZIMATICO DELLA CATENA RESPIRATORIA
MITOCONDRIALE
produzione
cellulare di ATP

DNA
proteine
lipidi di
membrana
DANNO CELLULARE
IRREVERSIBILE
MORTE
CELLULARE
Radicali liberi
Ruolo patogenetico
dello stress ossidativo sui tessuti
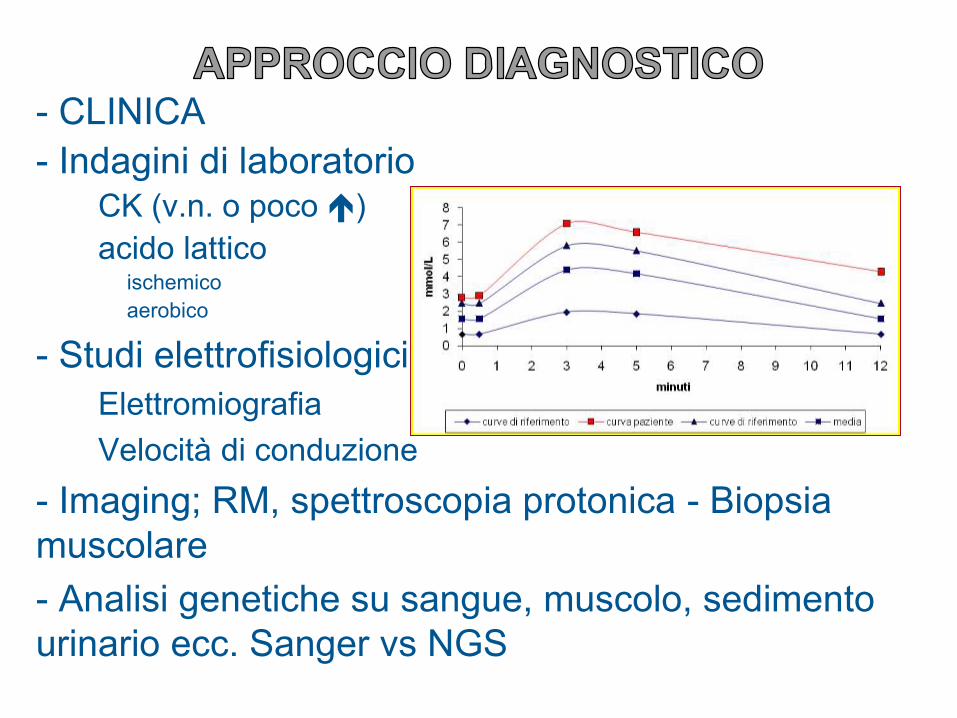
- CLINICA
- Indagini di laboratorioCK (v.n. o poco )
acido latticoischemico
aerobico
- Studi elettrofisiologici
Elettromiografia
Velocità di conduzione
- Imaging; RM, spettroscopia protonica - Biopsia
muscolare
- Analisi genetiche su sangue, muscolo, sedimento
urinario ecc. Sanger vs NGS

• Microscopia ottica
(e.e., tricromica di
Gomori)
• Indagini
istochimiche (COX,
SDH)
• Studi biochimici
• Indagini genetiche:
mtDNA
• Microscopia
elettronica
(raramente richiesta)

Miopatia mitocondriale:(a) fibre con il caratteristico aspetto“ragged red”, dovuto alla proliferazione mitocondriale. (b) Conla reazione istoenzimatica per la succinico-deidrogenasi (SDH)le fibre sono intensamente marcate (*). (c) fibre COXiporeattive-negative
(Tricromica di Gomori, ingrandimento originale
40x). Fibra muscolare con aspetto tipo "ragged
red". Fibre muscolari contenenti un numero
eccessivo di mitocondri a struttura anomala,
espressione di alterata proliferazione mitocondriale
A
B
C
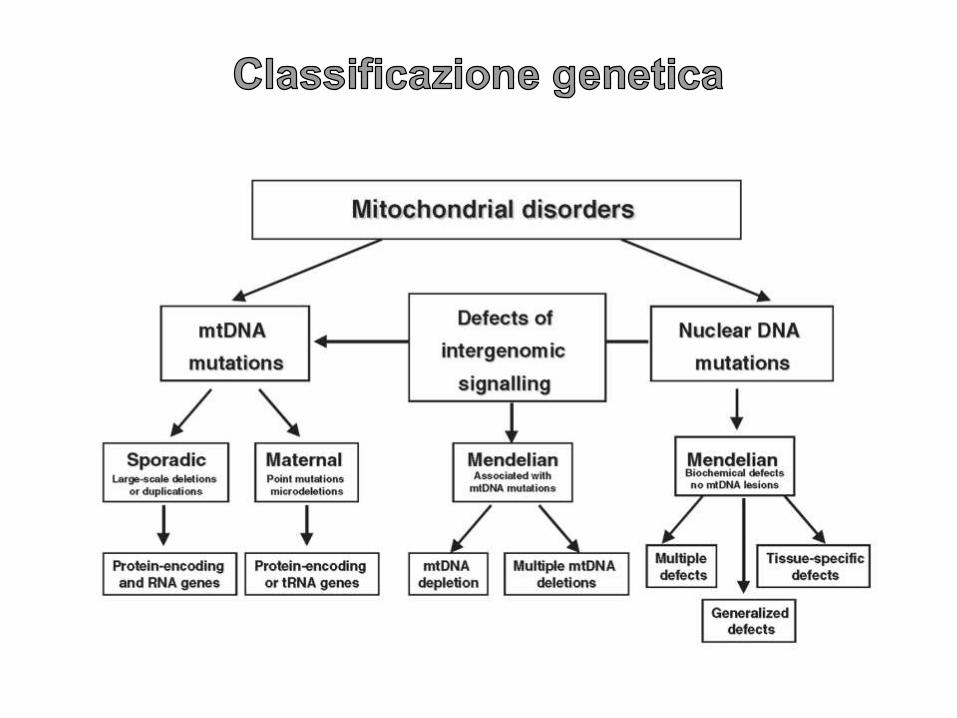

FENOTIPO ETEROGENEO
INTERESSAMENTO COMUNE DEL SNC
SINTOMATOLOGIA CENTRALE MULTISISTEMICA

Telethon-UILDM Clinical Grant 2009 GUP09004
“NETWORK Nazionale Malattie Mitocondriali”
La nostra esperienza…..

Steering CommitteeG Siciliano& M Mancuso Pisa
G Uziel, Milano
E Bertini, Roma
GP Comi, Milano
T Mongini,Torino
A Toscano, Messina
V Carelli, Bologna
C Angelini, Padova
S Servidei, Roma
P Tonin, Verona
C Minetti, Genova Mitocon

Italian Network of Mitochondrial Diseases
• Corrado Angelini
• Luca Bello
• Enrico Bertini
• Claudio Bruno
• Elena Caldarazzo Ienco
• Valerio Carelli
• Michela Catteruccia
• Giacomo Pietro Comi
• Maria Alice Donati
• Maria Teresa Dotti
• Antonio Federico
• Massimiliano Filosto
• Costanza Lamperti
• Michelangelo Mancuso
• Carlo Minetti
• MITOCON
• Maurizio Moggio
• Tiziana Mongini
• Isabella Moroni
• Olimpia Musumeci
• Daniele Orsucci
• Elena Pegoraro
• Dario Ronchi
• Filippo Maria Santorelli
• Mauro Scarpelli
• Monica Sciacco
• Serenella Servidei
• Gabriele Siciliano
• Paola Tonin
• Antonio Toscano
• Graziella Uziel
• Maria Lucia Valentino
• Liliana Vercelli
• Massimo Zeviani

Oversee Committee
G Logroscino, Bari
S. DiMauro, Columbia University, New York
J. Montoya, Zaragoza, Spain
RW Taylor, Newcastle

Siena
Cagliari

The network has reached the following goals:
1. Establishment of an Italian network of clinical
centers with expertise on MM
2. Creation of a validated web based database,
harmonized with other European Databases and
Networks
3. Characterization of a big cohort of MM cases
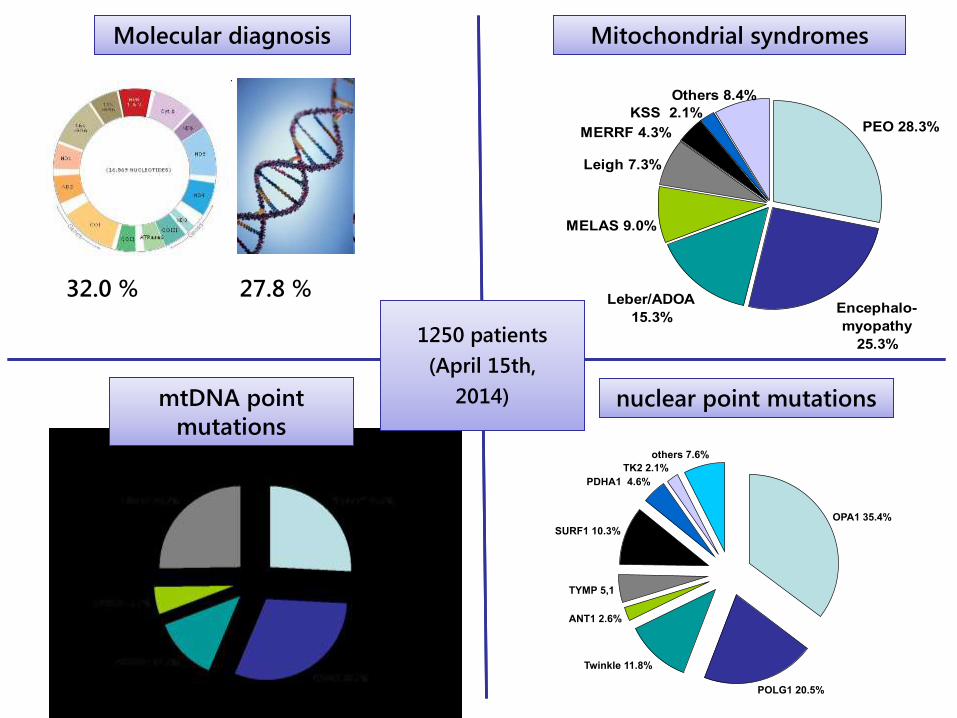
32.0 % 27.8 %
Molecular diagnosisMolecular diagnosis
PEO 28.3%
Encephalo-
myopathy
25.3%
Leber/ADOA
15.3%
MELAS 9.0%
Leigh 7.3%
KSS 2.1%
MERRF 4.3%
Others 8.4%
Mitochondrial syndromesMitochondrial syndromes
mtDNA point
mutations
mtDNA point
mutations
nuclear point mutationsnuclear point mutations
OPA1 35.4%
POLG1 20.5%
Twinkle 11.8%
ANT1 2.6%
TYMP 5,1
SURF1 10.3%
PDHA1 4.6%
TK2 2.1%
others 7.6%
1250 patients
(April 15th,
2014)
1250 patients
(April 15th,
2014)


3 CLASSICI FENOTIPI
PEO
KEARN SAYRE SYNDROME
PEARSON SYNDROME



QUADRO CLINICO
• Crisi miocloniche generalizzate
• Atassia/demenza/sordità neurosensoriale/ atrofia ottica/debolezza
mucolare con atrofia
• Deficit stenico prevale ai cingoli
• Polineuropatia sensorimotoria diffusa con piede cavo
• Esordio precoce, cardiomiopatia con blocco di conduzione e
insufficienza respiratoria
DATI DI LABORATORIO
• CK: normale/elevata
• Lattato sierico: aumentato
• TC/RMN encefalo: atrofia cerebrale e cerebellare
• EEG: rallentamento diffuso con scoppi di punte e onde lente

QUADRO CLINICO
• Esordio nel I anno di vita 10% entro i 15 anni 60-80%
• Caratterizzata da encefalomiopatia mitocondrale
• Episodi ricorrenti simil-ictus con cefalea tipo emicranico, nausea,
vomito, emiparesi, emianopsia o cecità corticale. Provocati da
infezioni o sforzi fisici.
• Deficit forza muscolare prossimale e intolleranza allo sforzo
DATI DI LABORATORIO
• CK: normale/elevata
• Lattato sierico e nel LCS: aumentato
• TC/RMN encefalo: atrofia corticale / depositi di Ca⁺⁺ nei gangli della
base
• EEG: attività di punte e onde lente
• EMG/VdC: nella norma
• BIOPSIA muscolare: indistinguibile da altre miopatie mitocondrali.

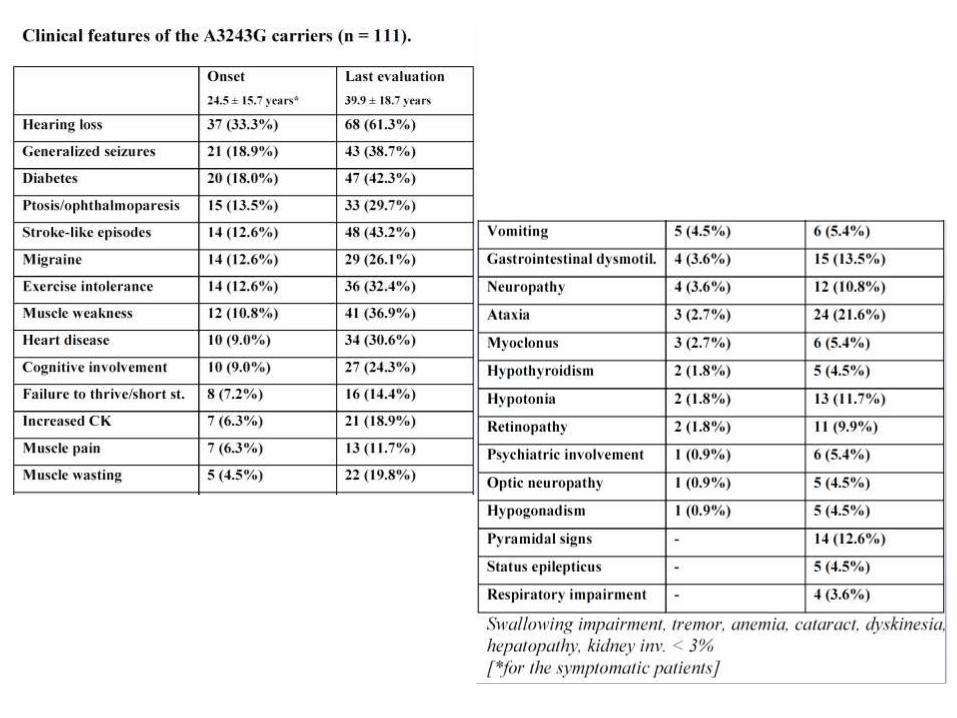
Decadimento cognitivo e Mitocondriopatie


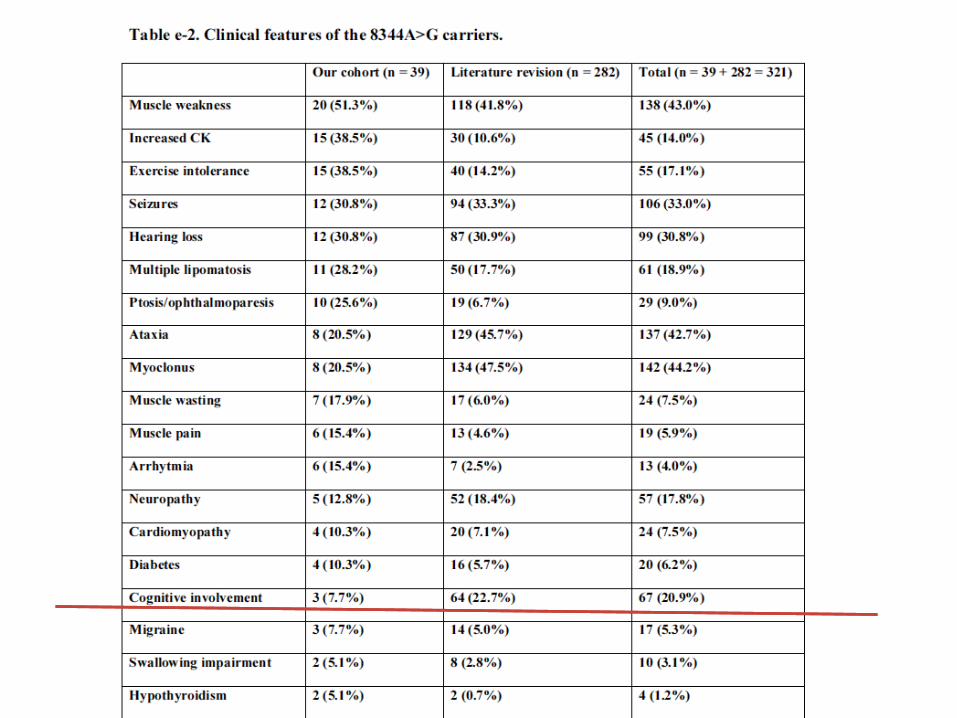
RESULTS: the m.3243A>G mutation
• 118 patients harboring the A8344G mutation have
been identified among the 1150 mitochondrial patients
(10.9%) present in our database (April 15th, 2013).
• They represented the 34.2% (118/345) of all mtDNA
point mutations.

MRI
DWI

MENDELIAN MD

Temple Lee Parker Mitochondria 3 B
MITOCHONDRIAL MEDICINE: ADVANCES IN UNDERSTANDING PATHOLOGIES AND
THERAPEUTICAL STRATEGIES, Pisa 2006

ONCE UPON A TIME…..
POLG

Nat Genetics 2001

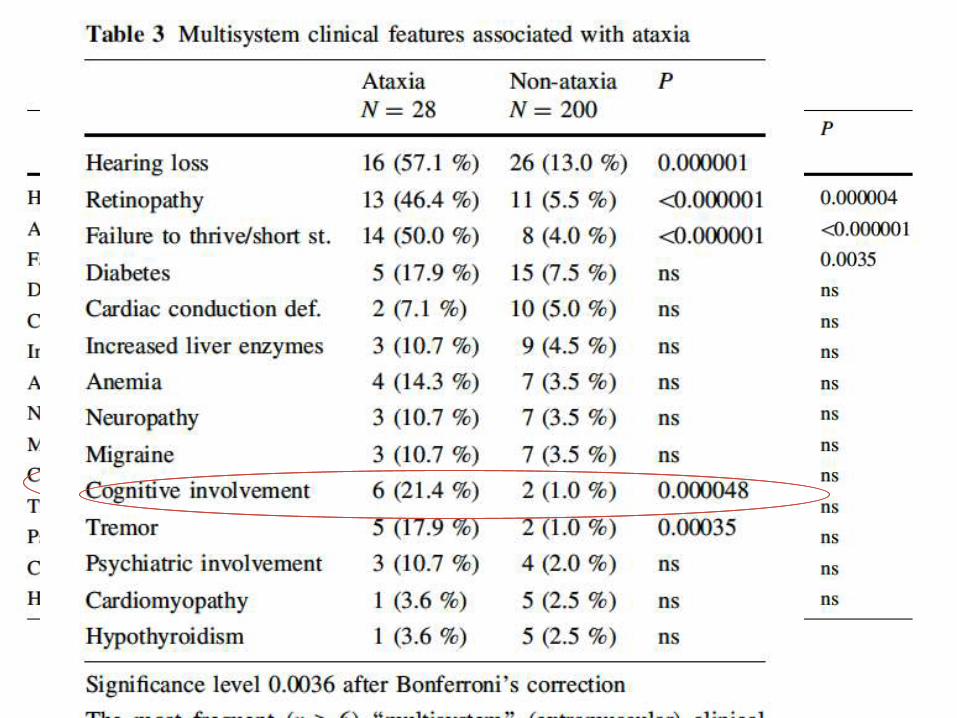
Mitochondrial ataxias
• mitochondrial recessive ataxia syndrome
(MIRAS)
• sensory ataxia neuropathy dysarthria and
ophthalmoplegia (SANDO)

Mitochondrial ataxias – SANDO
• Caused by mutations in mitochondrial POLG gene
• POLG encodes the catalytic subunit of mitochondrial DNA
polymerase
• Adult-onset disease (typically between 16–40 years of age)
• Typical clinical symptoms include:
– Sensory ataxic neuropathy
– Dysarthria
– Chronic progressive external ophthalmoplegia
SANDO, sensory ataxic neuropathy, dysarthria and
ophthalmoparesis Mancuso et al. 2004

Mitochondrial ataxias – MIRAS
• Caused by homozygous or compound heterozygous mutations
in the POLG1 gene
• Results in decreased complex I, II & III of the electron transport
chain
• Cerebellar ataxia
• Commonly associated with:
– Mild cognitive impairment
– Psychiatric abnormalities
– Involuntary movements
– Seizures
– PNP
• Patients may be prone to adverse reactions to valproate
manifesting as acute liver failure
MIRAS, mitochondrial recessive ataxia syndrome

Caso Clinico
• Donna 75 anni
• In anamnesi: ipertrofia ventricolo sinistro, tireopatia
• Anamnesi familiare:cardiopatia e morte improvvisa, ptosi
palpebrale (madre, nonna materna, zia materna)
• Astenia muscolare, facile affaticabilità, ptosi palpebrale
• DA 2 AA CIRCA DIST MEMORIA A BREVE TERMINE,
MMSE 21
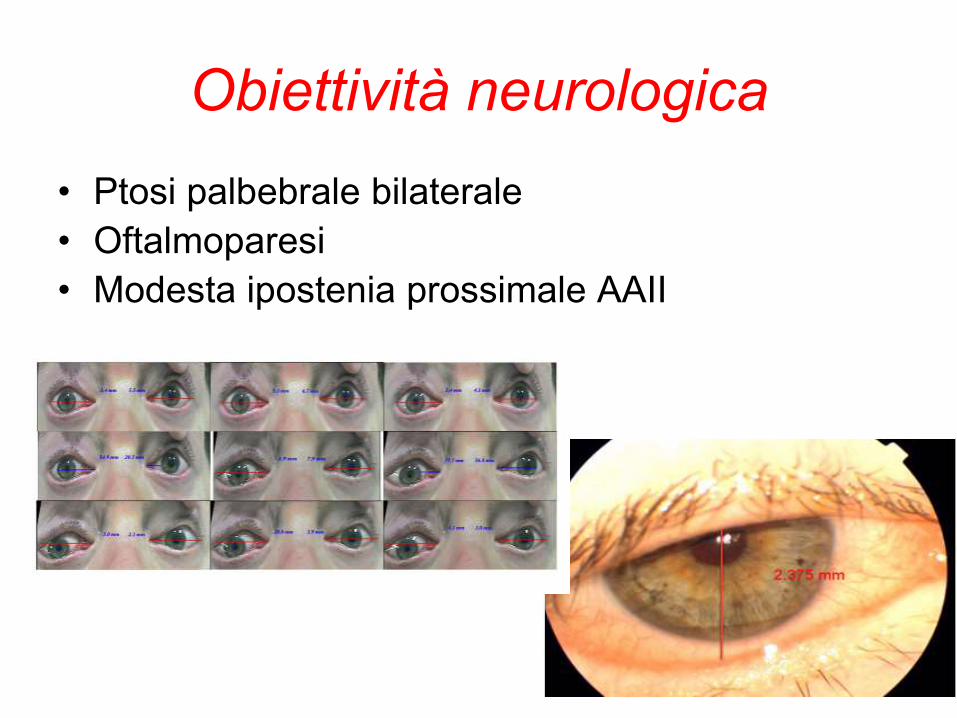
Obiettività neurologica
• Ptosi palbebrale bilaterale
• Oftalmoparesi
• Modesta ipostenia prossimale AAII
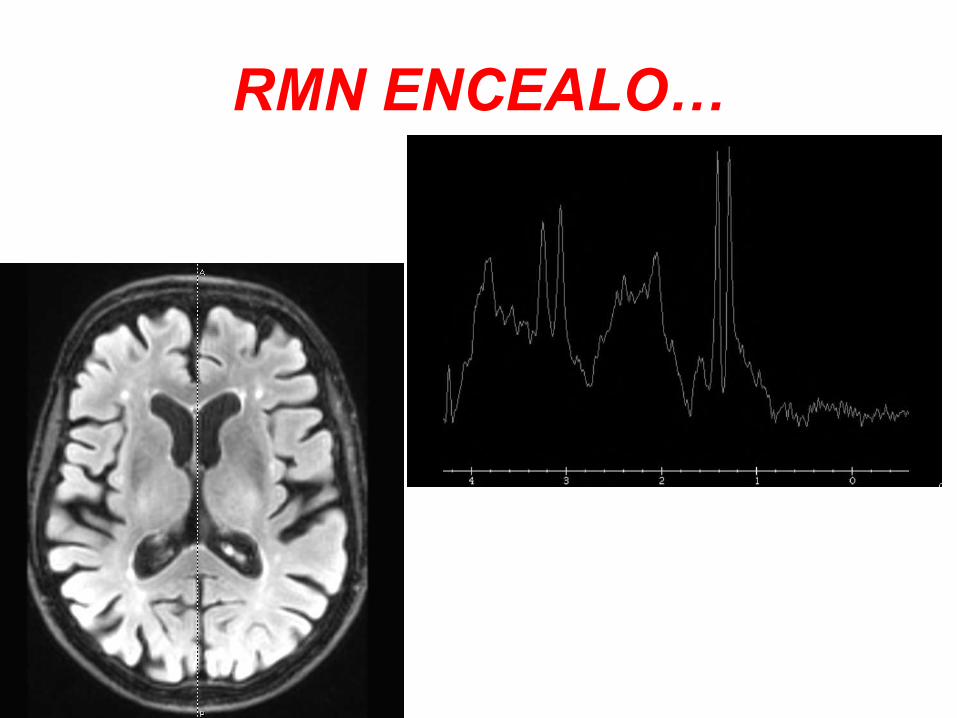
RMN ENCEALO…
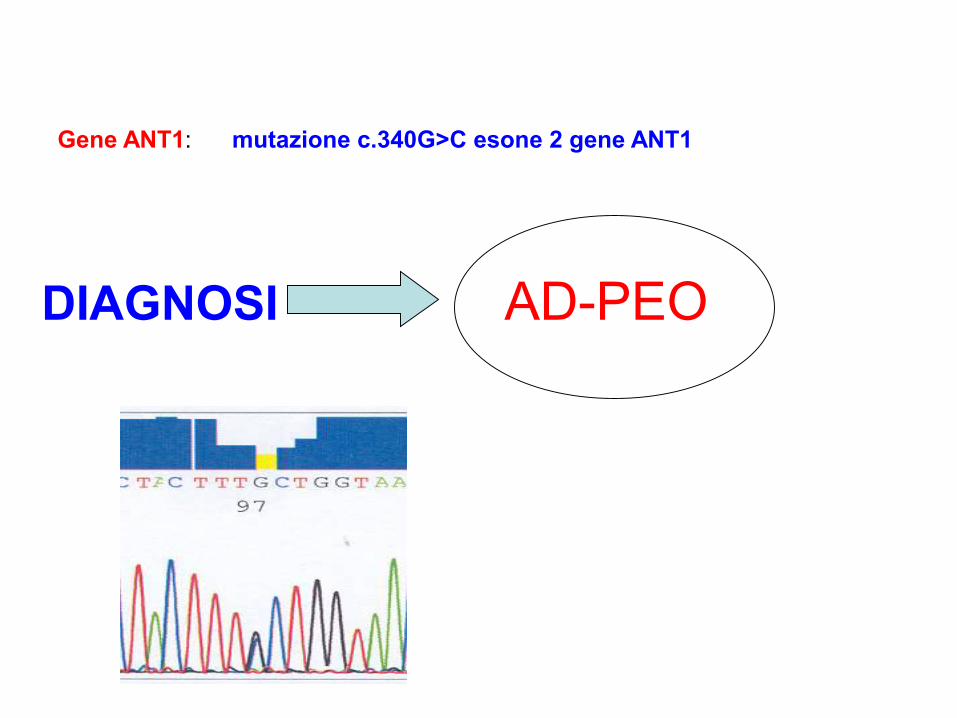
Gene ANT1: mutazione c.340G>C esone 2 gene ANT1
DIAGNOSI AD-PEO

Work in progress…

Le premesse….


Obiettivo dello studioEffettuare una valutazione neuropsicologica e neurometabolica di
screening in una popolazione adulta di entrambi i sessi, affetta da
Malattia Mitocondriale
Pazienti•Pazienti adulti di entrambi i sessi affetti da malattia mitocondriale
Età compresa tra 18 e 75 anni
In grado di comprendere ed aderire a quanto richiesto dal protocollo dello
studio
Consenso informato scritto

Disegno dello studio e Metodi
• Studio osservazionale, monocentrico, prospettico/retrospettivo
• Visita basale per verificare i criteri di inclusione/esclusione dallo studio
• Somministrazione di una batteria di test neuropsicologici
Procedura dello studio e valutazione
• 40 pazienti reclutati affetti da malattia mitocondriale
• Acqusizione consenso informato scritto
• Anamnesi accurata
• Esclusione di esami di laboratori alterati (emocromo, funzionalità epatica e renale, profilo lipidico e glicemico, Vitamina B12, folati e omocisteina)
• RMN encefalo
• Batteria neuropsicologica
• PET cerebrale con FDG


Allo stato attuale….
• 21 pazienti arruolati
• 12 con batteria neuropsicologica alterata
• PET data in progress

1. CARATTERI GENERALI
2. NELLE MALATTIE NEURODEGENERATIVE
LO STRESS OSSIDATIVO
(OS)

1. OS: caratteri generaliStress chimico indotto dalla presenza, in un organismo vivente, di una eccessiva
produzione di Specie Chimiche Reattive (SCR) secondario ad un’aumentata produzione
delle stesse e/o a una ridotta efficacia dei sistemi fisiologici di difesa antiossidanti.

Le Specie Chimiche Reattive
radicaliche
(radicali liberi)
non radicaliche
reazioni a catena che aumentano il danno ossidativo
specie reattive dell’ ossigeno (ROS)
specie reattive dell’ azoto (RNS)
specie reattive del carbonio (RCS)
Specie chimica Formula Natura
Anione
superossido·O2ˉ R
Ossigeno
singoletto1O2* R (?)
Perossido di
idrogenoH2O2 NR
Idrossile HO· R
Alcossile RO· R
(Alchil)idroperossi
leROOH NR
Ossido nitrico NO· R
Diossido nitrico NO2· R
Acido nirtoso HNO2 NR
Perossinitrito ONOO- NR
(Alchil)tiile (da R-
SH)RS· R

I radicali liberiGli atomi sono chimicamente stabili quandociascun elettrone dell ’ orbitale più esterno èassociato ad un secondo elettrone che ruota indirezione opposta (appaiato) .
Processi endogeni ed esogeni possono“stappare” un elettrone dall’orbitale esternolasciando un elettrone spaiato; gli atomicaratterizzati dalla presenza di un elettronespaiato sono definiti radicali liberi
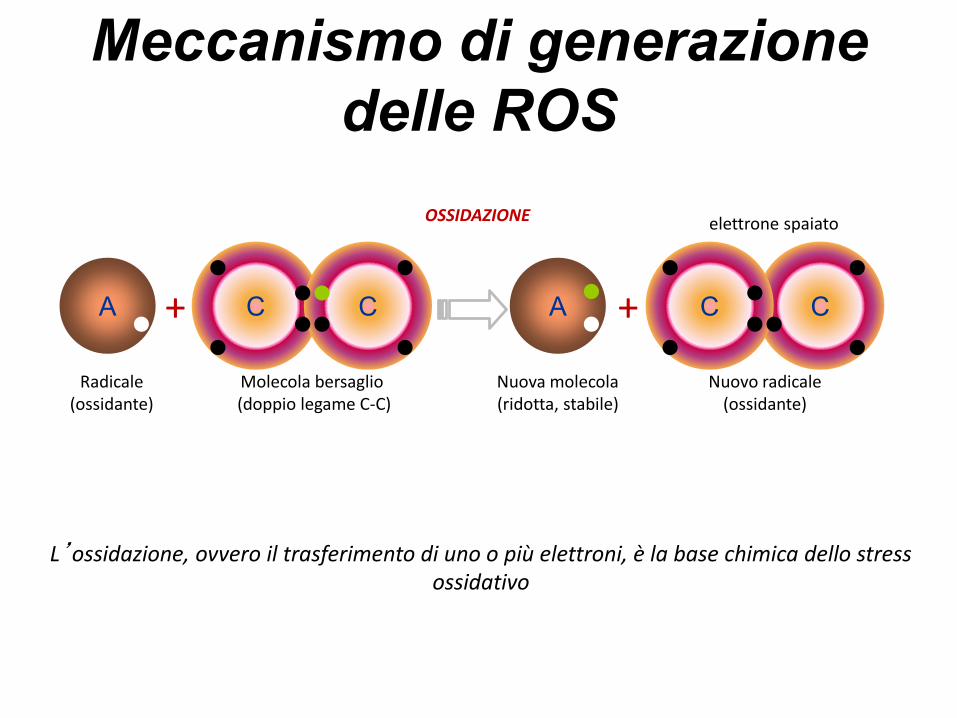
Meccanismo di generazione
delle ROS
++
Radicale (ossidante)
A A
Nuova molecola (ridotta, stabile)
Molecola bersaglio(doppio legame C-C)
C C
Nuovo radicale(ossidante)
CC
elettrone spaiatoOSSIDAZIONE
L’ossidazione, ovvero il trasferimento di uno o più elettroni, è la base chimica dello stress ossidativo

A concentrazioni moderate le ROS partecipano attivamente ad una varietà di processi
biologici complessi, implicati nella normale crescita cellulare quali la trasduzione del
segnale, il controllo dell’espressione genica, la senescenza cellulare, l’apoptosi. Sono in
grado di bloccare l’azione patogena di diversi microrganismi.
A concentrazioni elevate le ROS sono dannosi per l’organismo in quanto attaccano i
maggiori costituenti della cellula (proteine, acidi nucleici, lipidi) partecipando così a
processi complessi quali l’invecchiamento e ad un gran numero di patologie.
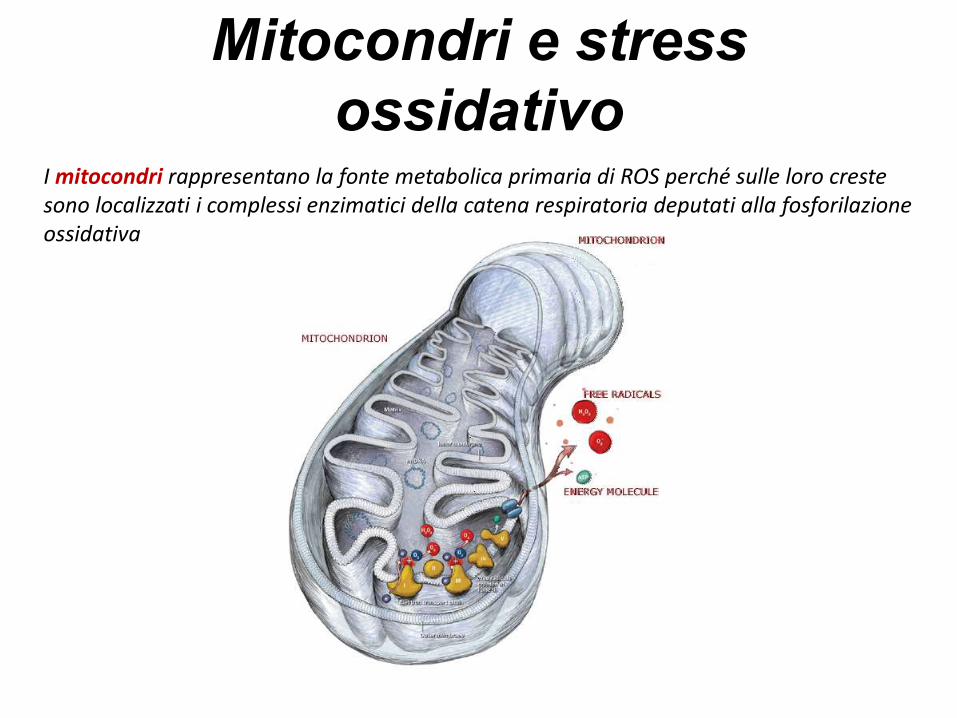
Mitocondri e stress
ossidativoI mitocondri rappresentano la fonte metabolica primaria di ROS perché sulle loro creste sono localizzati i complessi enzimatici della catena respiratoria deputati alla fosforilazione ossidativa

La Catena di Trasporto degli
Elettroni
·O2-
O2
H2O2
SOD
Durante i processi di fosforilazione ossidativa, il 4-5% dell ’ ossigeno non vienecompletamente ridotto ad H2O, ma forma prodotti intermedi dell’O2 altamente reattivi
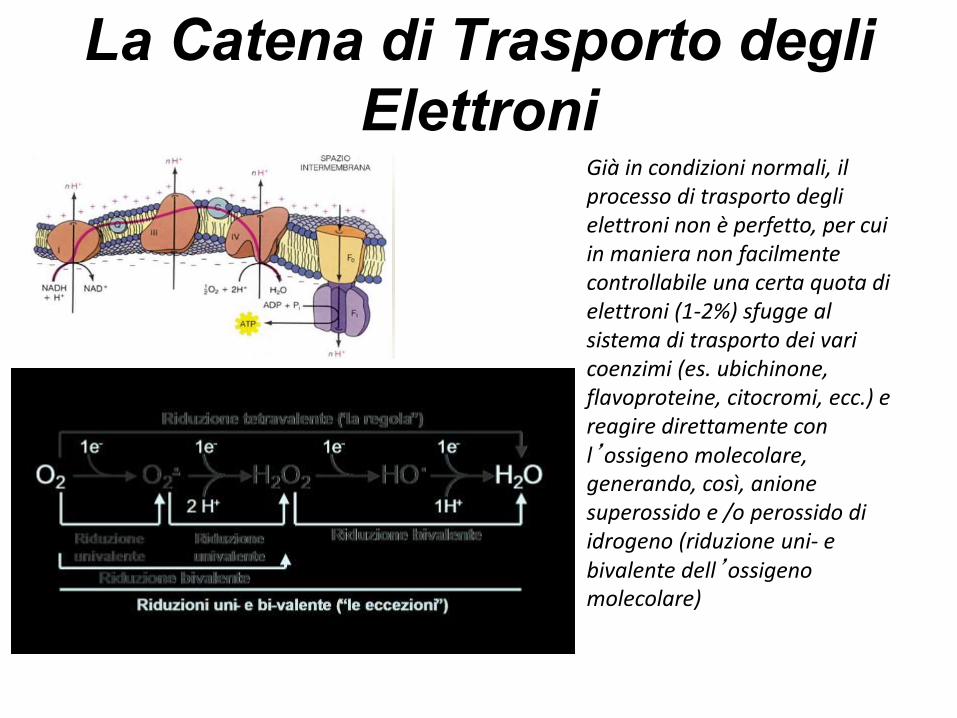
Già in condizioni normali, il processo di trasporto degli elettroni non è perfetto, per cui in maniera non facilmente controllabile una certa quota di elettroni (1-2%) sfugge al sistema di trasporto dei vari coenzimi (es. ubichinone, flavoproteine, citocromi, ecc.) e reagire direttamente con l’ossigeno molecolare, generando, così, anione superossido e /o perossido di idrogeno (riduzione uni- e bivalente dell’ossigeno molecolare)
La Catena di Trasporto degli
Elettroni

Effetti dello stress ossidativo sulla
struttura e sulle funzioni cellulari
BIOMARCATORI DI DANNO OS
a. Danno ai lipidi
Perossidazione lipidica:
Sostanze Reattive all’acido
Tiobarbiturico (TBARS)
Malondialdeide (MDA)
F2-isoprostano
4-idrossi nonenale (4-HNE)
b. Danno alle proteine
proteine nitrosilate
carbonil proteine
c. Danno al DNA
8-idrossi desossi guanosina
(8-OHdG)
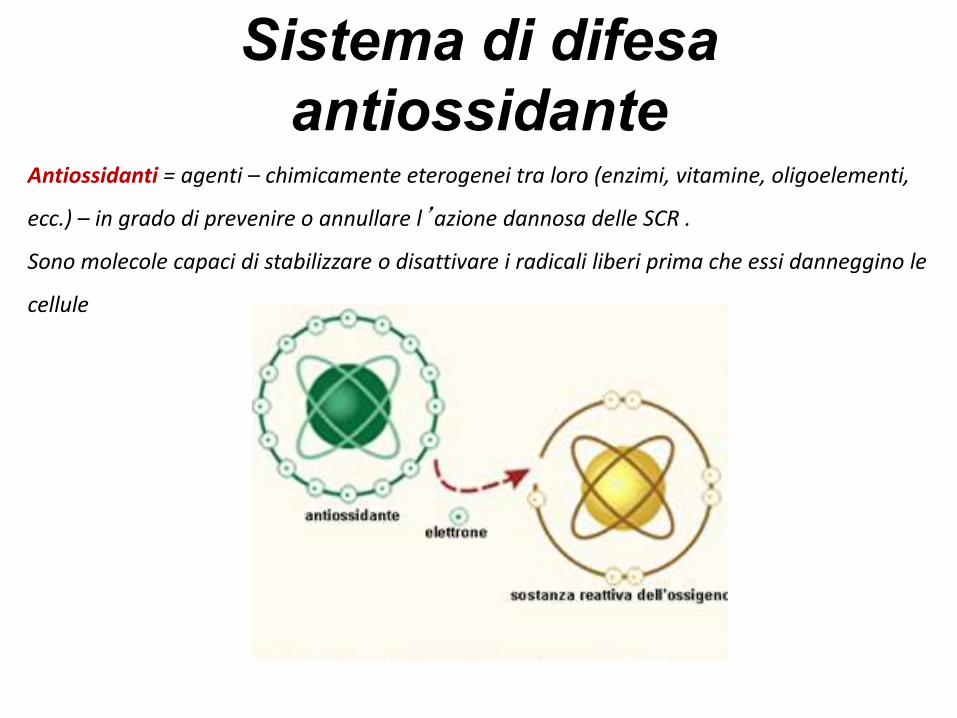
Sistema di difesa
antiossidanteAntiossidanti = agenti – chimicamente eterogenei tra loro (enzimi, vitamine, oligoelementi,
ecc.) – in grado di prevenire o annullare l’azione dannosa delle SCR .
Sono molecole capaci di stabilizzare o disattivare i radicali liberi prima che essi danneggino le
cellule

Classificazione degli
antiossidanti
Gli antiossidanti possono essere classificati secondo diversi criteri:
sulla base dell’origine: endogeni ed esogeni,
sulla base della struttura chimica: enzimatici e non enzimatici,
sulla base della solubilità: in liposolubili e idrosolubili.
Considerando, invece, il meccanismo d’azione prevalente, risulta molto utile sotto il profilo
fisiopatologico suddividere gli antiossidanti in 3 gruppi principali:
preventivi,
scavenger
di riparo.
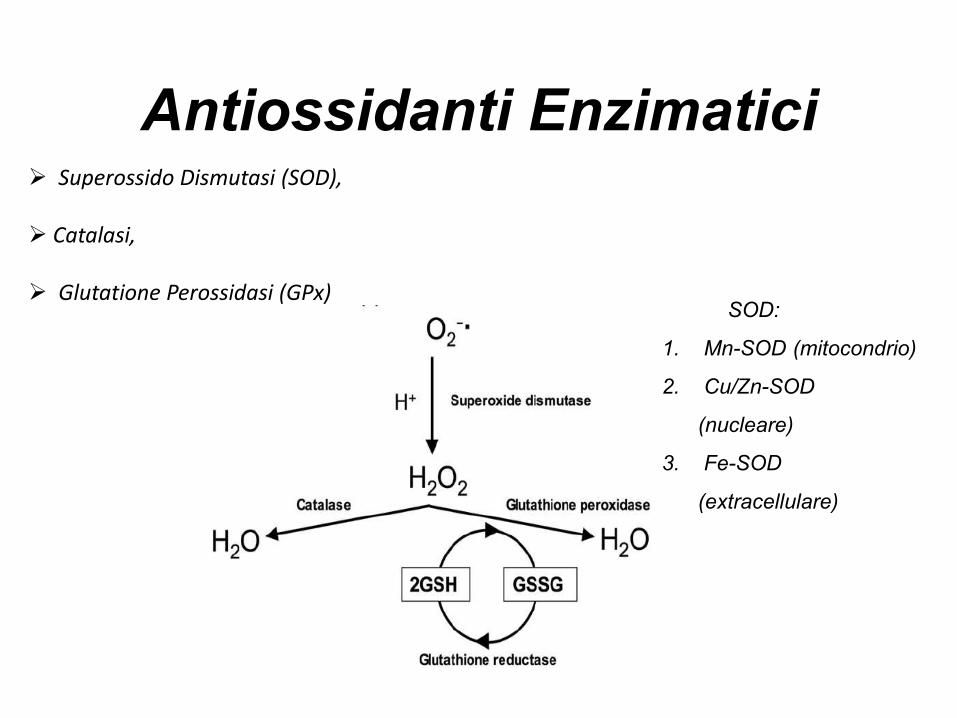
Antiossidanti Enzimatici Superossido Dismutasi (SOD),
Catalasi,
Glutatione Perossidasi (GPx)SOD:
1. Mn-SOD (mitocondrio)
2. Cu/Zn-SOD
(nucleare)
3. Fe-SOD
(extracellulare)

Antiossidanti non enzimatici
vitamina E;
vitamina C;
molecole tioliche (-SH):
- tioredoxina (TRX),
- acido α-lipoico (ALA),
- glutatione (GSH)
altre molecole
- melatonina,
- carotenoidi,
- flavonoidi
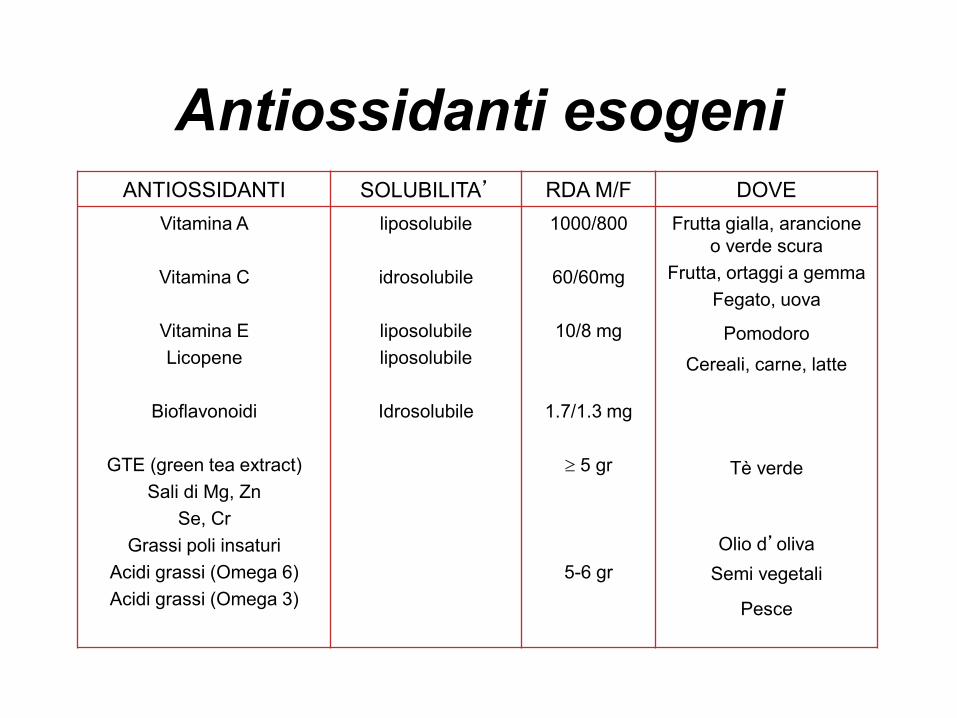
Antiossidanti esogeniANTIOSSIDANTI SOLUBILITA’ RDA M/F DOVE
Vitamina A
Vitamina C
Vitamina E
Licopene
Bioflavonoidi
GTE (green tea extract)
Sali di Mg, Zn
Se, Cr
Grassi poli insaturi
Acidi grassi (Omega 6)
Acidi grassi (Omega 3)
liposolubile
idrosolubile
liposolubile
liposolubile
Idrosolubile
1000/800
60/60mg
10/8 mg
1.7/1.3 mg
5 gr
5-6 gr
Frutta gialla, arancione
o verde scura
Frutta, ortaggi a gemma
Fegato, uova
Pomodoro
Cereali, carne, latte
Tè verde
Olio d’oliva
Semi vegetali
Pesce

Meccanismo d’azione degli
antiossidanti
Antiossidanti preventivi
Bloccano la formazione dei radicali
Antiossidanti scavenger
Bloccano la reazione di inizio
Bloccano la reazione di propagazione
Riparano il danno e ricostituiscono le membrane
Antiossidanti di riparo e de novo
Inquinanti, fumo, radiazioni, metalli pesanti,farmaci, alcool, ischemia-riperfusione, …
Specie Chimiche Reattive
Attacco di biomolecole(glicidi, lipidi, proteine, ecc)
Reazioni radicaliche a catena
Danno ossidativo
malattie
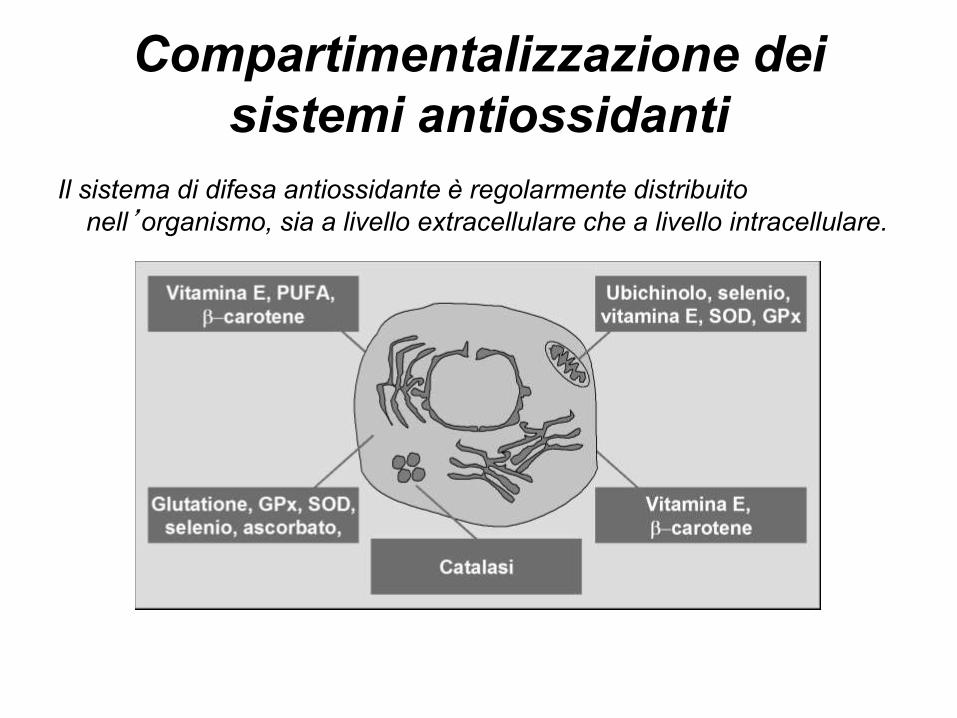
Compartimentalizzazione dei
sistemi antiossidanti
Il sistema di difesa antiossidante è regolarmente distribuito
nell’organismo, sia a livello extracellulare che a livello intracellulare.
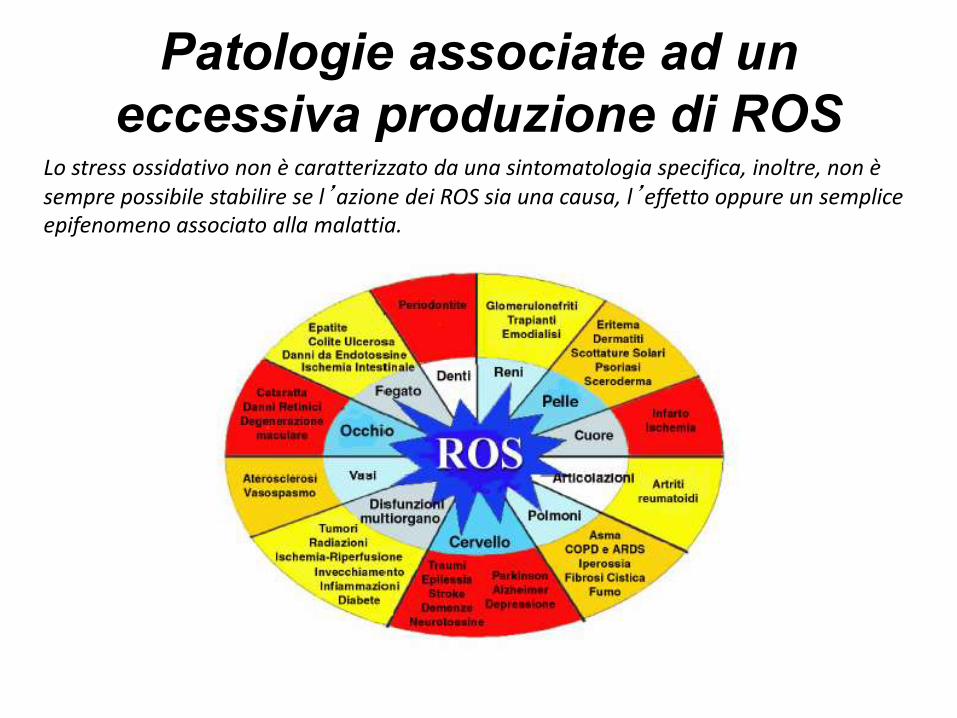
Patologie associate ad un
eccessiva produzione di ROSLo stress ossidativo non è caratterizzato da una sintomatologia specifica, inoltre, non è sempre possibile stabilire se l’azione dei ROS sia una causa, l’effetto oppure un semplice epifenomeno associato alla malattia.

2. Lo stress ossidativo nelle malattie
neurodegenerative
È noto un coinvolgimento delle specie radicaliche nel processo di degenerazione
neuronale e un’ associazione tra questo e la patogenesi delle malattie neurodegenerative.
Il SNC è altamente vulnerabile al danno ossidativo a causa di:
elevato consumo di ossigeno,
elevato contenuto lipidico (PUFA),
bassa concentrazione di antiossidanti
Un’eccessiva produzione di prossidanti e/o la caduta dei meccanismi antiossidanti di difesa,
potrebbero far superare facilmente il valore soglia oltre il quale il danno neurodegenerativo
diventerebbe importante
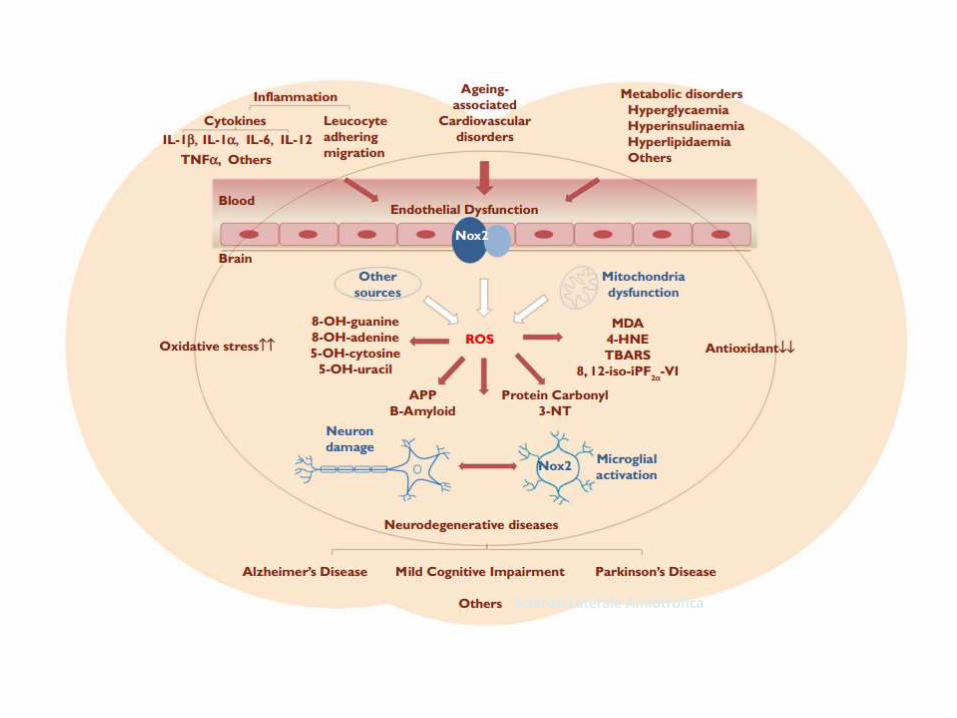
Sclerosi Laterale Amiotrofica

OS nella Malattia di Alzheimer
Neurosci Bull March, 2014
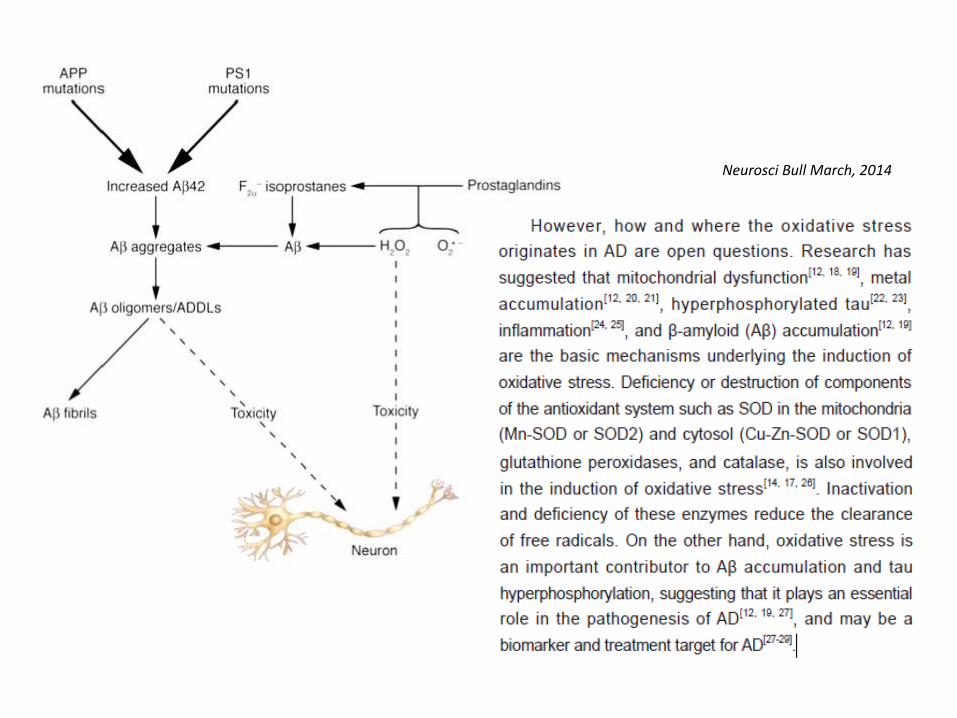
Neurosci Bull March, 2014
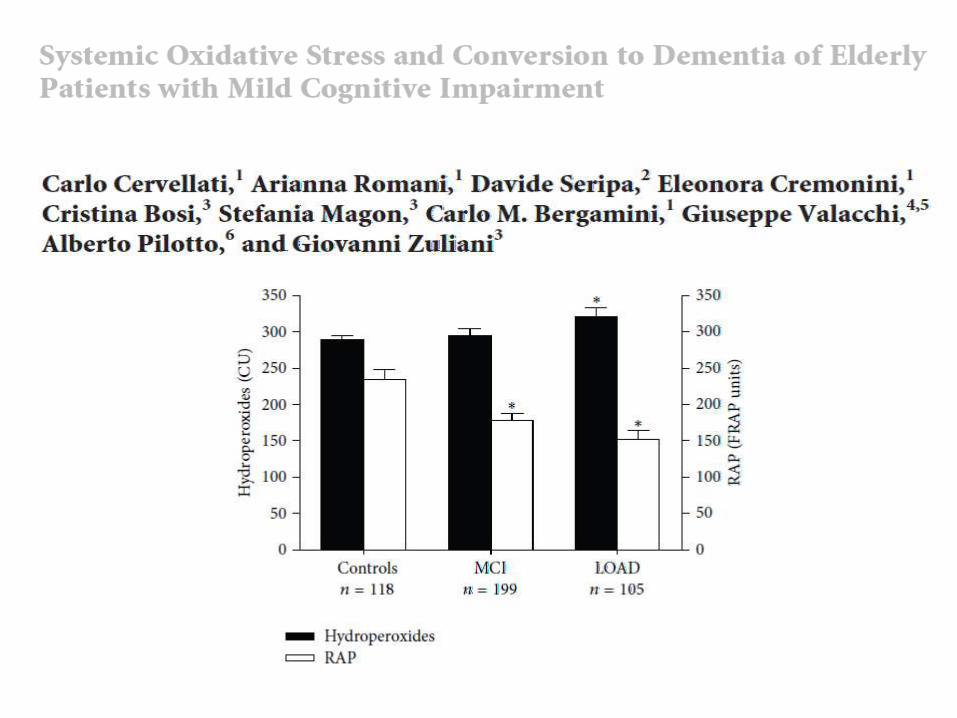

[Mayo Clin Proc. 2011; 86(9):876-884]
J. Eric Ahlskog, PhD, MD; Yonas E. Geda, MD, MSc; Neill R. Graff-Radford, MBBCh, FRCP; and Ronald C. Petersen,
Stili di vita e stress ossidativo
Modelli murini SOD1-G93A sottoposti a training (motor or swimming-ALS mice)
il nuoto ritarda in maniera significativa la morte dei motoneuroni
spinali
Severine Deforges, Julien Branchu, Olivier Biondi, Clement Grondard, Claude Pariset, Sylvie Lecolle, Philippe Lopes, Pierre-Paul Vidal, Christophe Chanoine and Frederic Charbonnier ´
[J Physiol 587.14 (2009) pp 3561–3571]
Studi prospettici di meta-analisi documentano una diminuzione
significativa del rischio di sviluppare demenza nei
soggetti che praticano una moderata attività fisica di tipo
aerobica

…..Decadimento cognitivo, mitocondri e stress
ossidativo….

Sempre maggiori evidenze considerano la compromissione del metabolismo e il danno
ossidativo coinvolti nella patogenesi delle malattie neurodegenerative
I mitocondri sono la maggior fonte cellulare di energia e ROS
MITOCONDRI E NEURODEGENERAZIONE
1980: “mitochondrial-free-radical hypothesis”: alla base del fisiologico
invecchiamento cellulare e tissutale vi è il progressivo accumulo di
alterazioni irreversibili indotte da radicali liberi dell’ossigeno (reactive
oxygen species-ROS)
(Harman, 1992)
I mitocondri hanno un ruolo chiave nella
neurodegenerazione?

Mitocondrio, Stress ossidativo e
Malattia di AlzheimerDanno cellulare
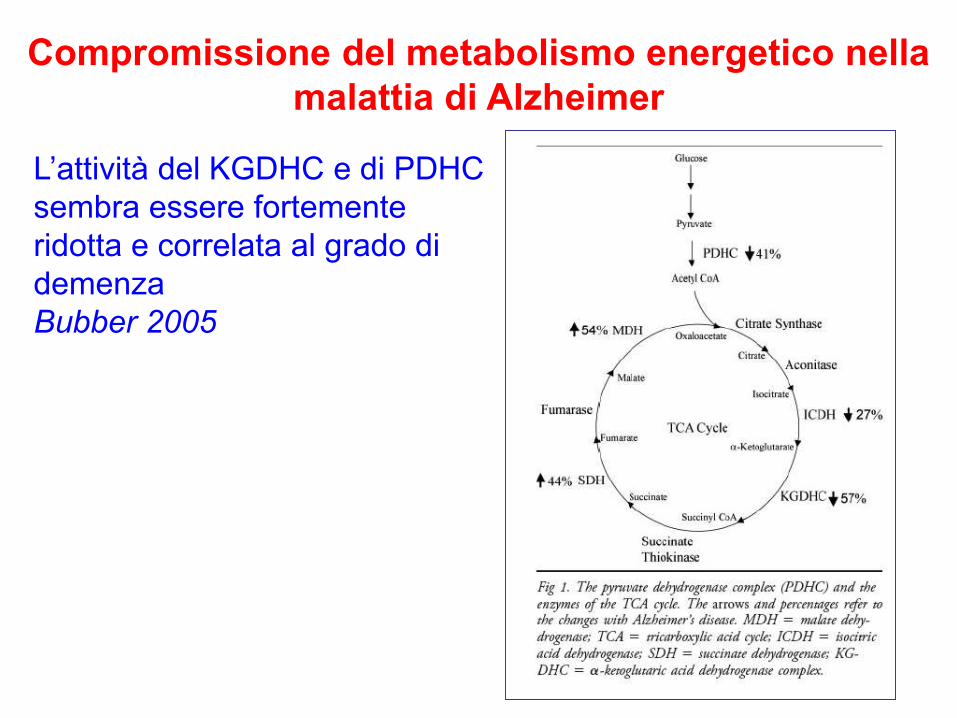
Compromissione del metabolismo energetico nella
malattia di Alzheimer
L’attività del KGDHC e di PDHC
sembra essere fortemente
ridotta e correlata al grado di
demenza
Bubber 2005
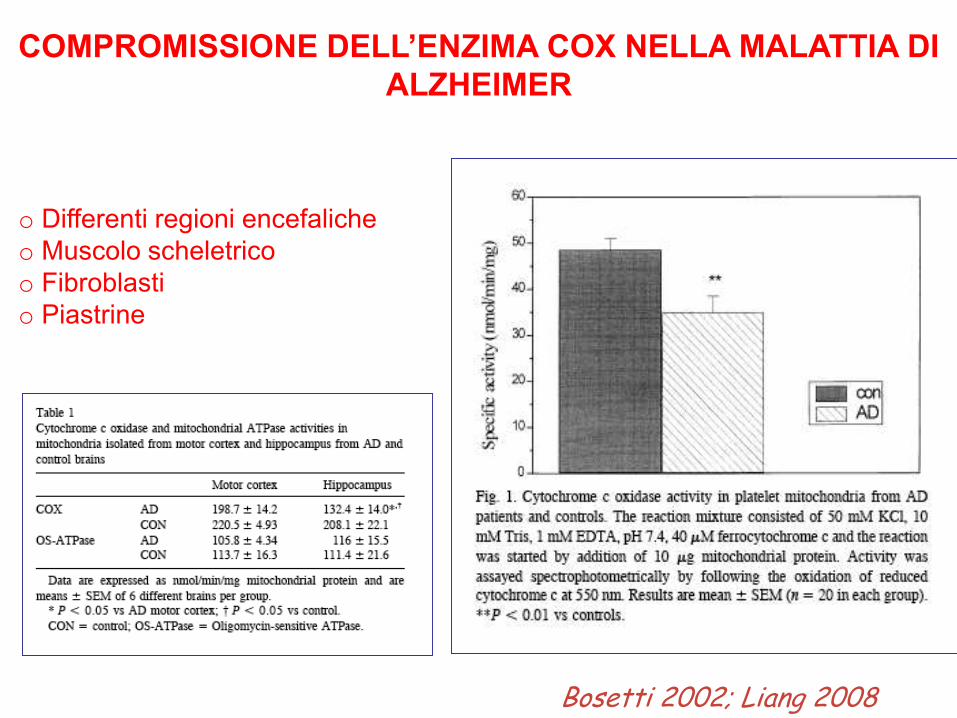
COMPROMISSIONE DELL’ENZIMA COX NELLA MALATTIA DI
ALZHEIMER
o Differenti regioni encefaliche
o Muscolo scheletrico
o Fibroblasti
o Piastrine
Bosetti 2002; Liang 2008
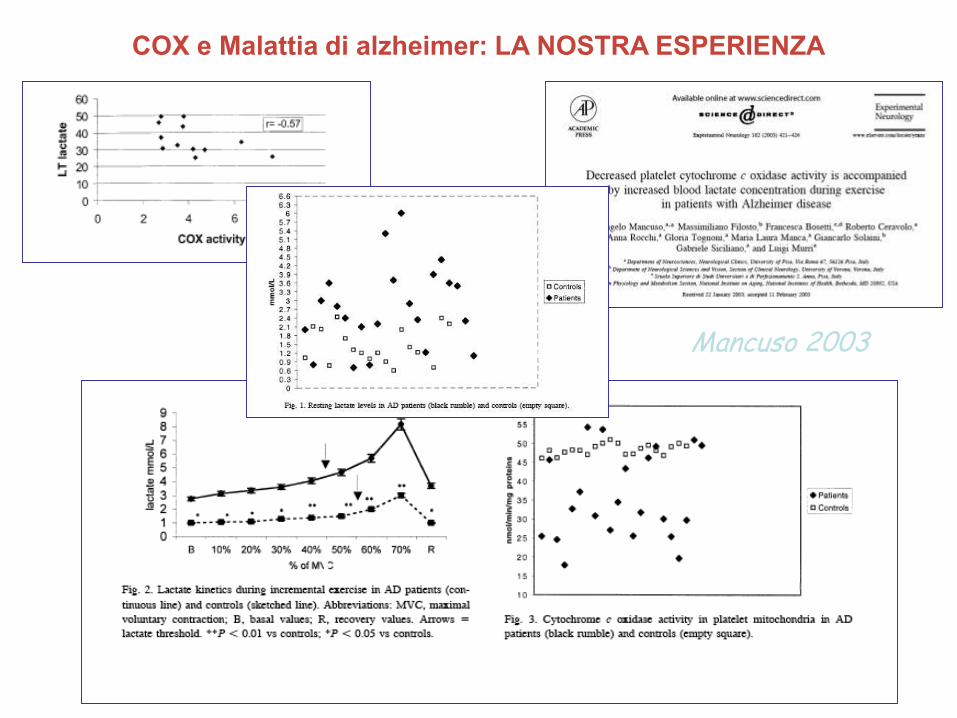
COX e Malattia di alzheimer: LA NOSTRA ESPERIENZA
Mancuso 2003

I MODELLI CIBRIDI DELLA MALATTIA DI
ALZHEIMER
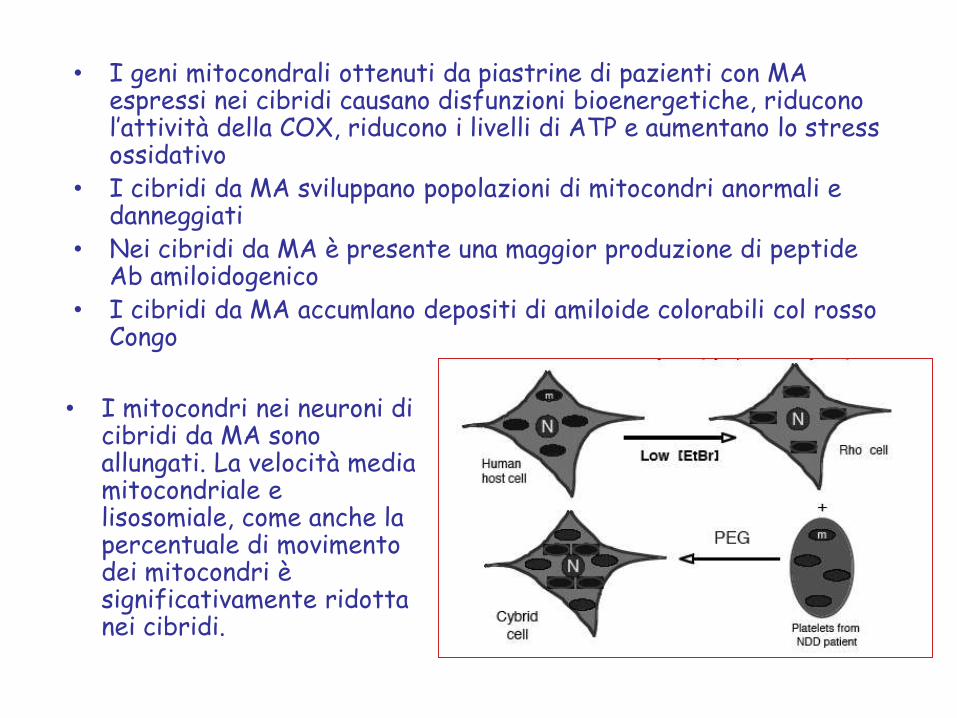
• I geni mitocondrali ottenuti da piastrine di pazienti con MA espressi nei cibridi causano disfunzioni bioenergetiche, riducono l’attività della COX, riducono i livelli di ATP e aumentano lo stress ossidativo
• I cibridi da MA sviluppano popolazioni di mitocondri anormali e danneggiati
• Nei cibridi da MA è presente una maggior produzione di peptide Ab amiloidogenico
• I cibridi da MA accumlano depositi di amiloide colorabili col rosso Congo
• I mitocondri nei neuroni di cibridi da MA sono allungati. La velocità media mitocondriale e lisosomiale, come anche la percentuale di movimento dei mitocondri è significativamente ridotta nei cibridi.
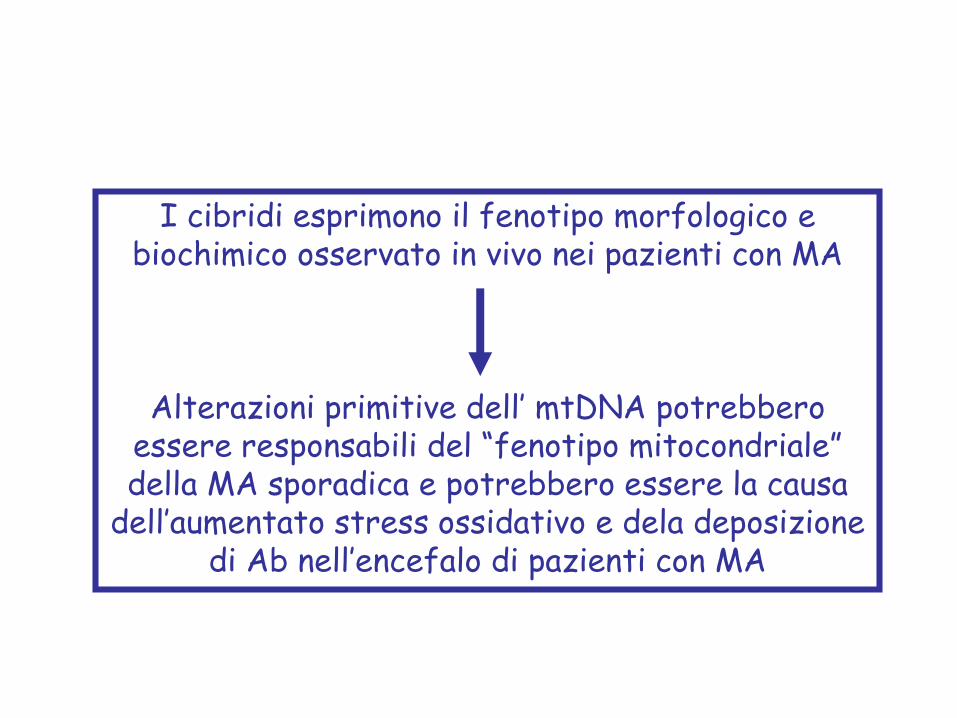
I cibridi esprimono il fenotipo morfologico e biochimico osservato in vivo nei pazienti con MA
Alterazioni primitive dell’ mtDNA potrebbero essere responsabili del “fenotipo mitocondriale” della MA sporadica e potrebbero essere la causa
dell’aumentato stress ossidativo e dela deposizione di Ab nell’encefalo di pazienti con MA

GENETICA MITOCONDRIALE E MA

GENETICA MITOCONDRIALE E MA
• Nel tessuto cerebrale di pazienti MA aumento di circa 15 volte della
“common deletion” Corral-Debrinski 1994
• Sostituzione A4336G più frequente in pazienti MA Shoffner 1993
• Due cambiamenti eteroplasmici nella regione di controllo CR sono
specifici del tessuto cerebrale dei soggetti con demenza di Alzheimer:
T414G e T477C Wallace and Beal
AD OGGI NESSUNA MUTAZIONE NEL DNA MITOCONDRIALE PATOGENETICA
PER AD

Caso Clinico• Uomo di 64 anni
• Da 2 anni deficit di memoria ingravescente
• Obiettività neurologica: nella norma. Adl
COMPROMESSE
Valutazione neuropsicologica:
Compromesse le prove per la memoria immediata
verbale, visiva e per l'apprendimento. Compromesse
le prove per l'attenzione. Compromessa la prova per
le gnosie visuo-percettive. Compromesse le prove
per le capacità astrattive. Compromesso il Mini
Mental State. Compromesse le prove per la prassia
costruttiva. Destrimane, disturbo del linguaggio
inquadrabile in una afasia di tipo transorticale
sensoriale di grado modesto.
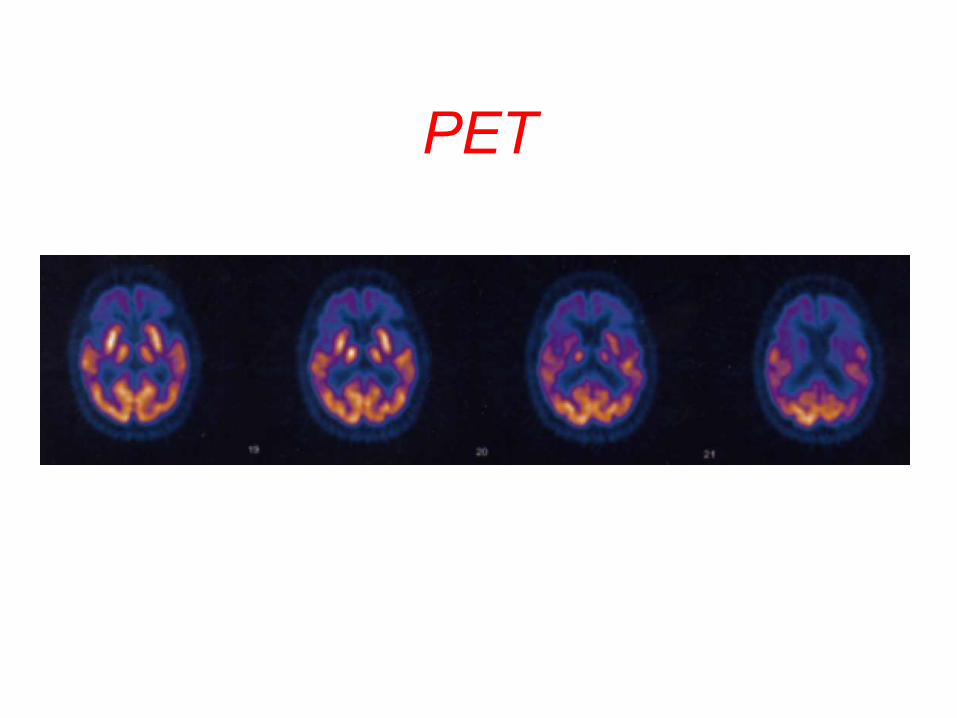
PET
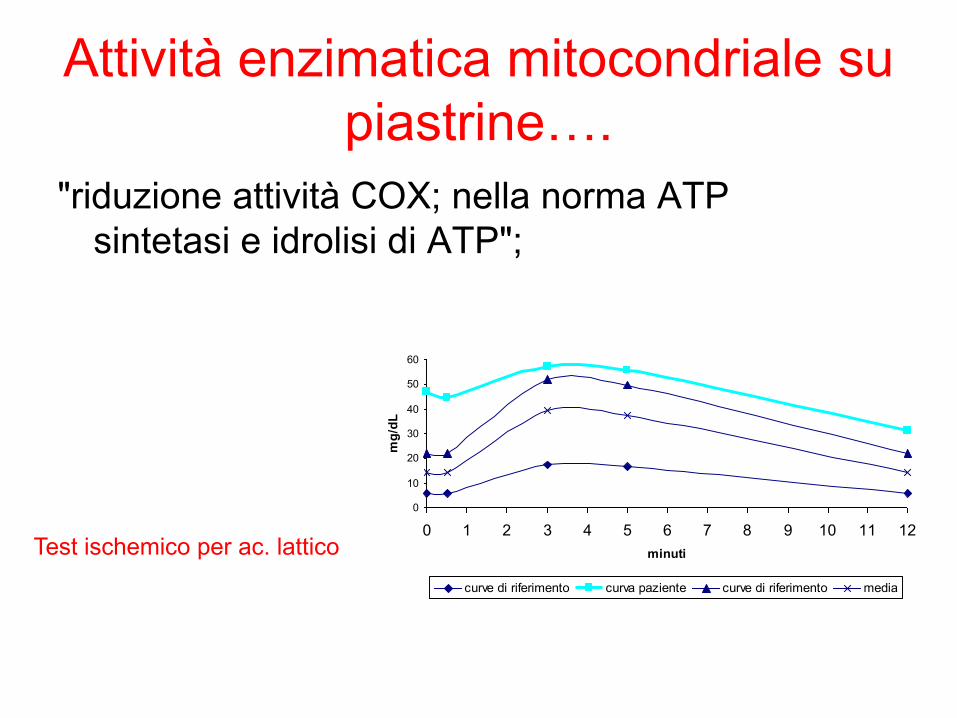
Attività enzimatica mitocondriale su
piastrine….
"riduzione attività COX; nella norma ATP
sintetasi e idrolisi di ATP";
0
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10 11 12
minuti
mg
/dL
curve di riferimento curva paziente curve di riferimento media
Test ischemico per ac. lattico
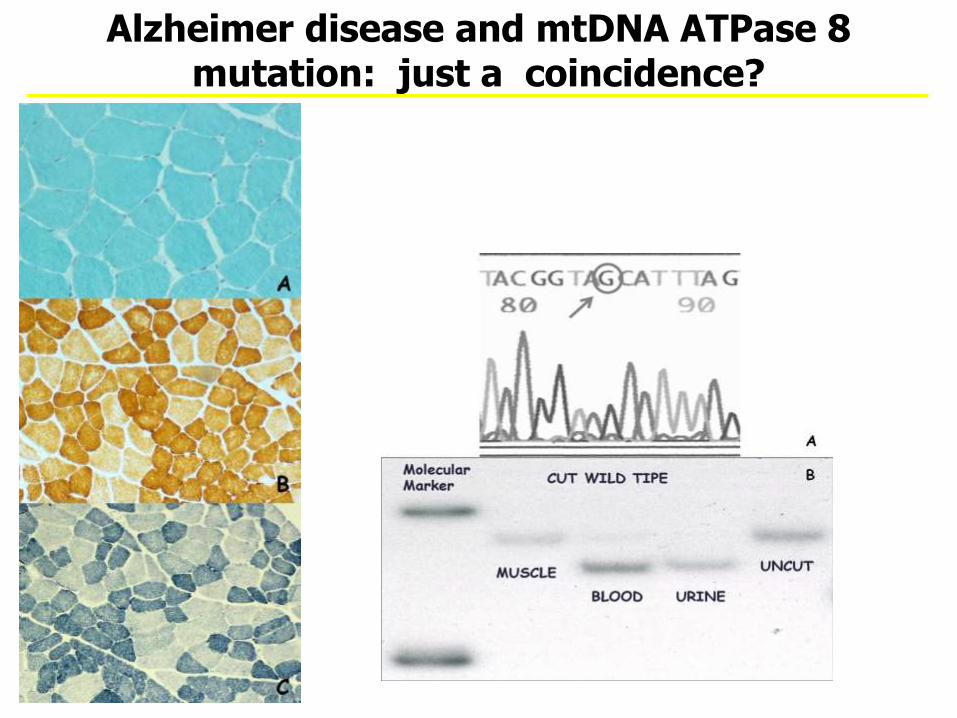
A 64-year-old man with AD and increased blood lactate. Complex IV and V activities were mildly reduced in muscle. Analysis of the mtDNA revealed an m.8381A>G mutation in ATPase 8 (ThrAla)
Alzheimer disease and mtDNA ATPase 8 mutation: just a coincidence?

ANOMALIE MITOCONDRIALI A TUTTI I LIVELLI (INCLUSI I
CIBRIDI)
ASSENZA MUTAZIONI PATOGENE mtDNA
???

Motilità degli spermatozoi
Capacità di adattamento a fredde condizioni climatiche
Leber
Multiplesclerosis Parkinson
ALS
Friedreich
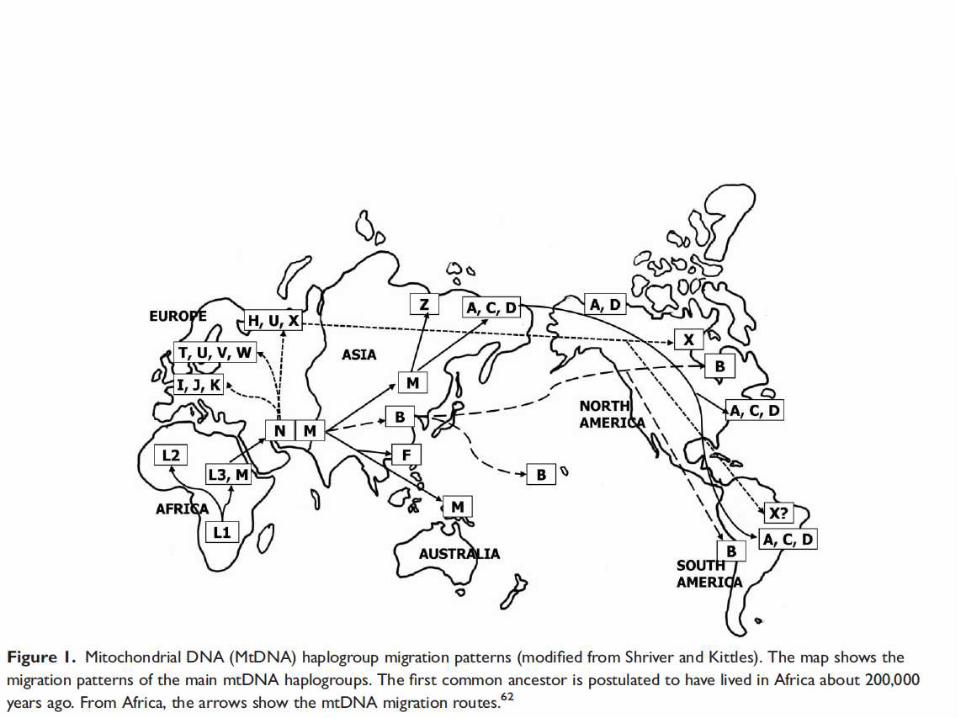
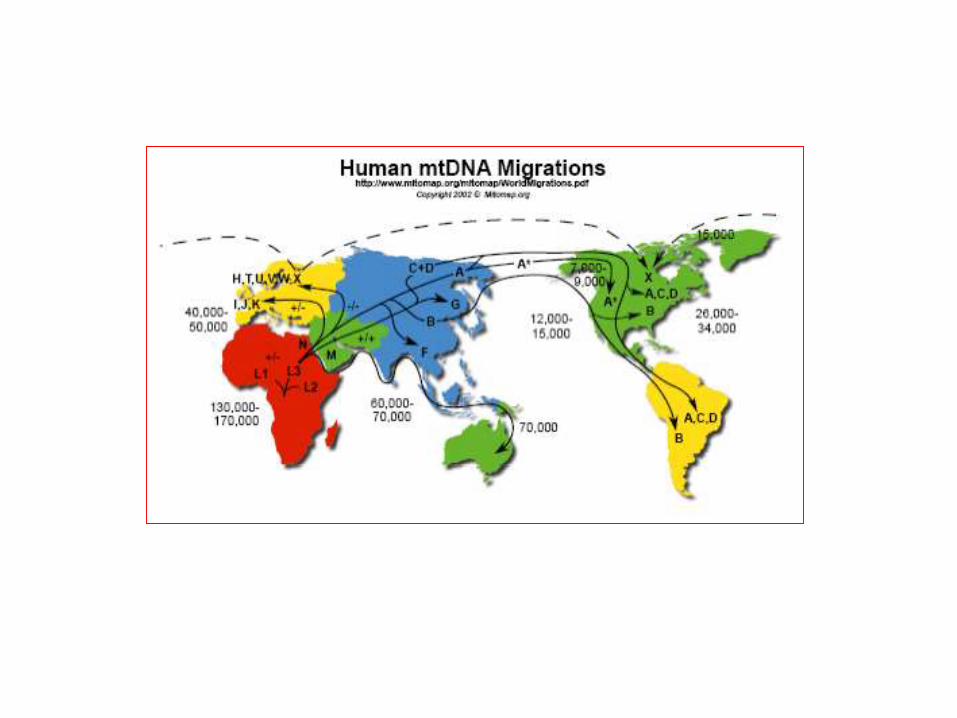
MITOCHONDRIAL HAPLOGROUPS
aging
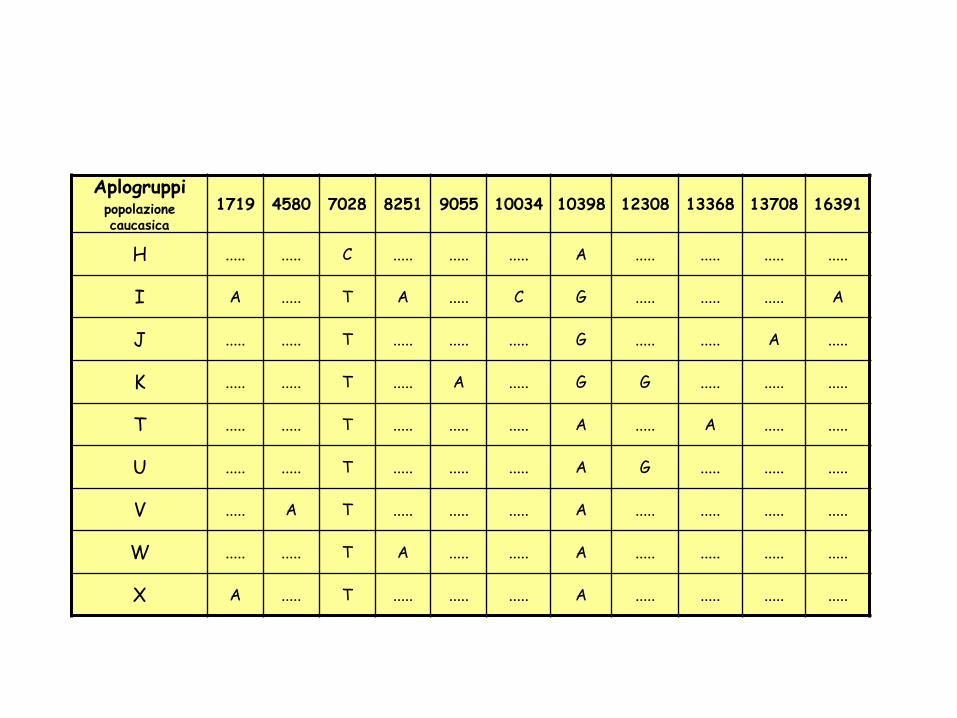
Aplogruppipopolazione caucasica
1719 4580 7028 8251 9055 10034 10398 12308 13368 13708 16391
H ..... ..... C ..... ..... ..... A ..... ..... ..... .....
I A ..... T A ..... C G ..... ..... ..... A
J ..... ..... T ..... ..... ..... G ..... ..... A .....
K ..... ..... T ..... A ..... G G ..... ..... .....
T ..... ..... T ..... ..... ..... A ..... A ..... .....
U ..... ..... T ..... ..... ..... A G ..... ..... .....
V ..... A T ..... ..... ..... A ..... ..... ..... .....
W ..... ..... T A ..... ..... A ..... ..... ..... .....
X A ..... T ..... ..... ..... A ..... ..... ..... .....

MITOCHONDRIAL HAPLOGROUPS IN AD (1)
o Haplogroup T under-represented in AD; haplogroup J over-represented. Chagnon 1999
o Haplogroups K and U present at a lower frequency in AD ApoE4 carriers than in non-carriers. K and U may act by neutralising the effect of this major AD risk factor. Carrieri 2001
o Males with haplogroup U had increased risk of AD, while females demonstrated a decreased risk with the same U haplogroup. Van der Walt 2004

o Two studies (including only neuropathologically proven cases of AD patients of European descent) indicated that mtDNA haplogroups were not associated with AD. Chinnery 2000, Elson 2006
MITOCHONDRIAL HAPLOGROUPS IN AD (2)


HAPLOGROUPS IN AD: OUR EXPERIENCE (1)
o 209 unrelated sporadic AD subjects (79♂ 130♀ mean age: 74.0±7.6 years, range 50–94)
o 191 healthy, age-matched controls (93♂, 98♀, mean age: 69.1±6.6, range 55–92)
o To minimize the risk of false associations between gene markers and disease, we enrolled in the study only patients and controls of clear Tuscan origin (at least three generations)
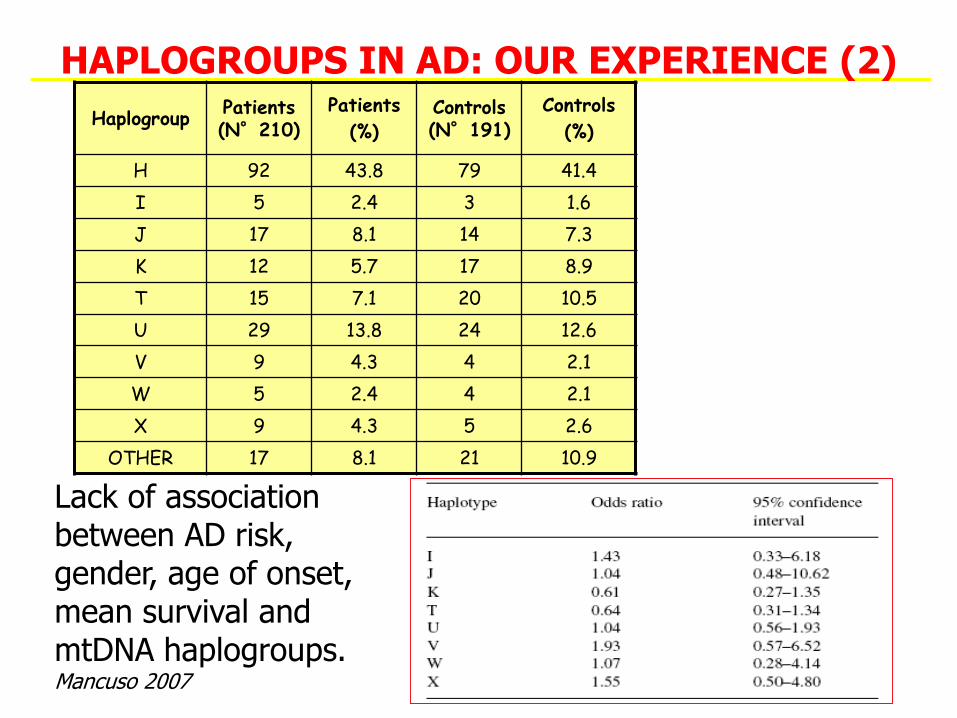
HAPLOGROUPS IN AD: OUR EXPERIENCE (2)
Lack of association between AD risk, gender, age of onset, mean survival and mtDNA haplogroups. Mancuso 2007
HaplogroupPatients (N°210)
Patients
(%)Controls (N°191)
Controls
(%)
H 92 43.8 79 41.4
I 5 2.4 3 1.6
J 17 8.1 14 7.3
K 12 5.7 17 8.9
T 15 7.1 20 10.5
U 29 13.8 24 12.6
V 9 4.3 4 2.1
W 5 2.4 4 2.1
X 9 4.3 5 2.6
OTHER 17 8.1 21 10.9

Patients4
without 4
Haplogroups 4(%) without 4(%) OR vs H (95% CI)
H 47 50.5
I 3 1.5 3.45 (0.30-39.71)
J 13 8 1.72 (0.58-5.08)
K 5 5 1.03 (0.23-4.65)
T 9 6 1.72 (0.51-5.84)
U 11 18 0.67 (0.25-1.80)
V 1.5 5 0.34 (0.04-3.10)
W 1.5 3.5 0.57 (0.06-5.78)
X 8 3.5 2.87 (0.64-12.91)
No relationship
HAPLOGROUPS IN AD: OUR EXPERIENCE (3)

HAPLOGROUPS AND AD: CONCLUSION
o Although it has been suggested that inherited haplogroups K and U may influence AD risk in Caucasians, this is still an unresolved question. To date, mtDNA haplogroups do not seem to play a major role in AD.
o The mtDNA alterations that cybrid models induce to hypothesize might be due to somatic factors, i.e. ROS.
o APP, Aβ-induced mitochondrial toxicity, oxidative stress, somatic mtDNA damage, mitochondrial dysfunction and apoptosis seem to be interconnected in the cascade leading to neurodegeneration and dementia.

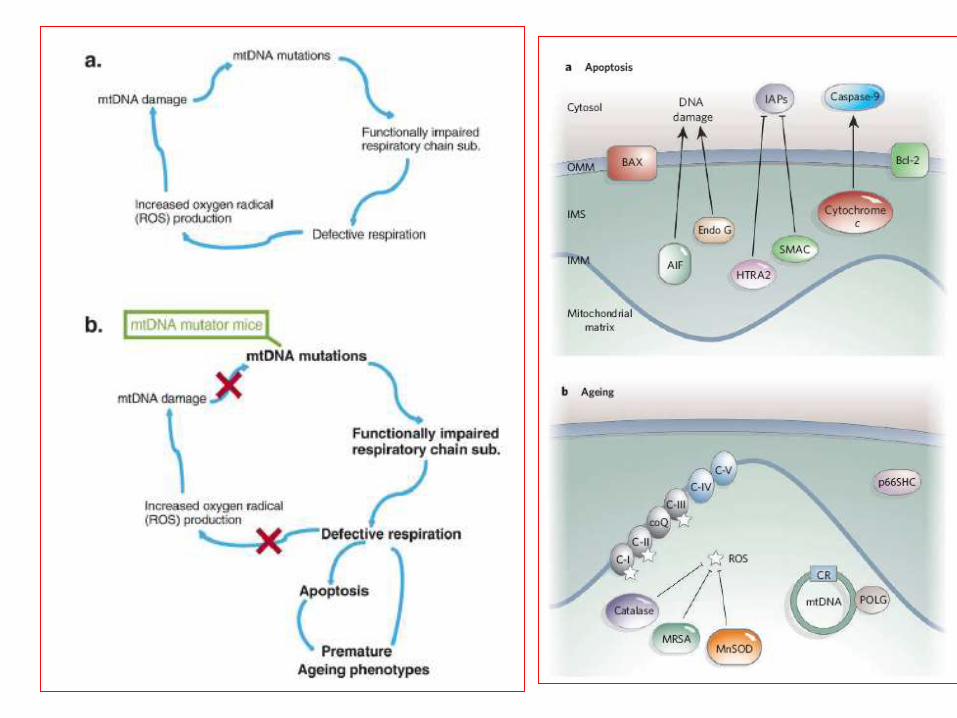
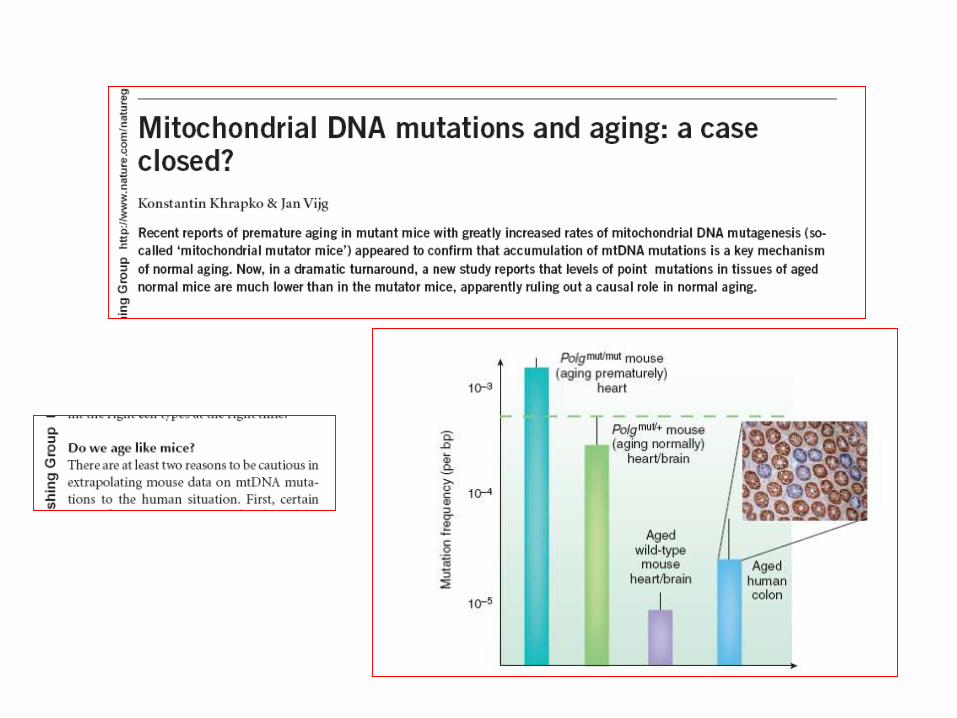
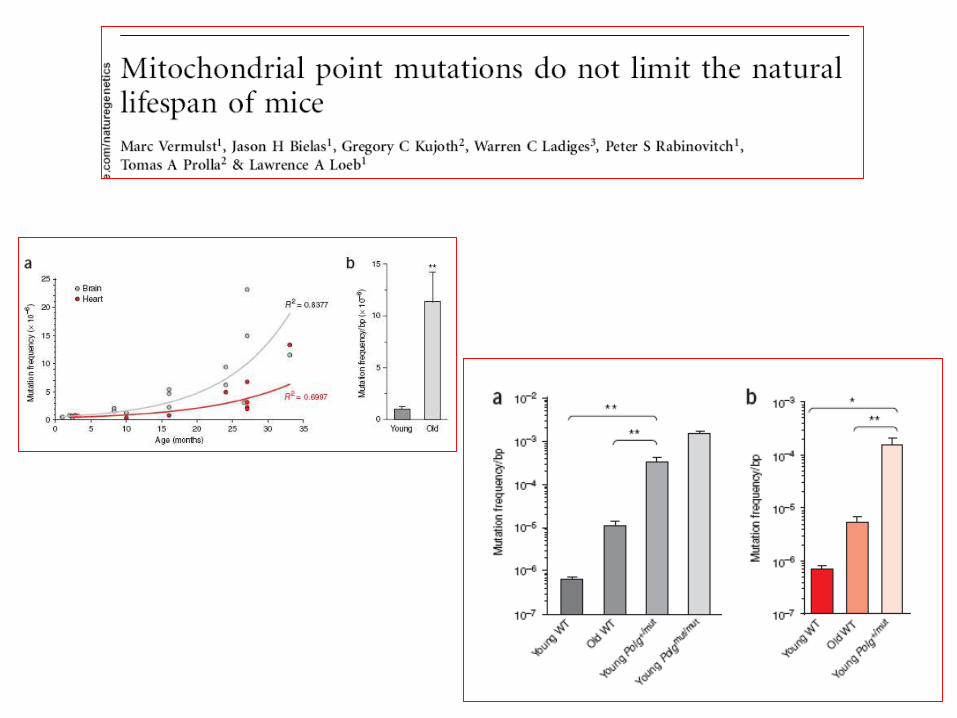
Lin&Beal Nature 2006Trifunovic 2006
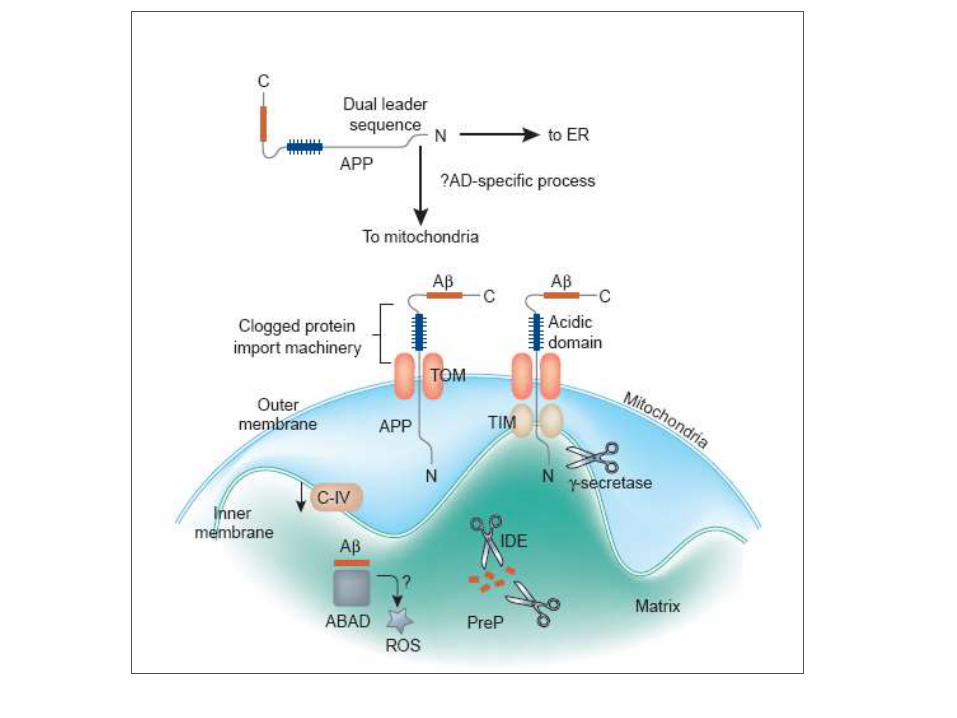
Devi J Neurosci 2006

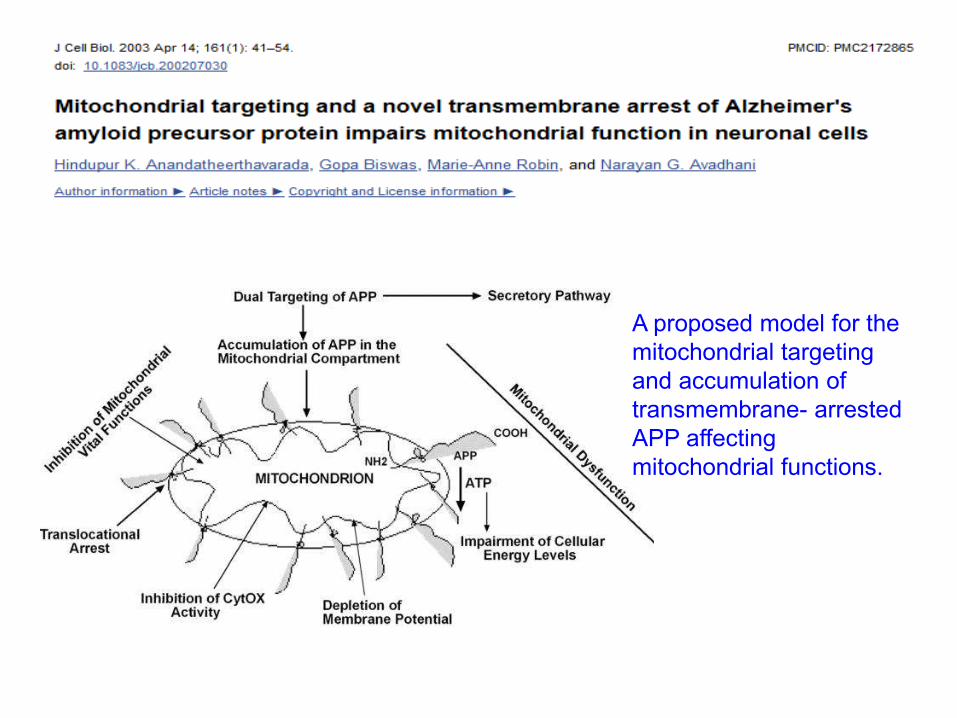
A proposed model for the
mitochondrial targeting
and accumulation of
transmembrane- arrested
APP affecting
mitochondrial functions.
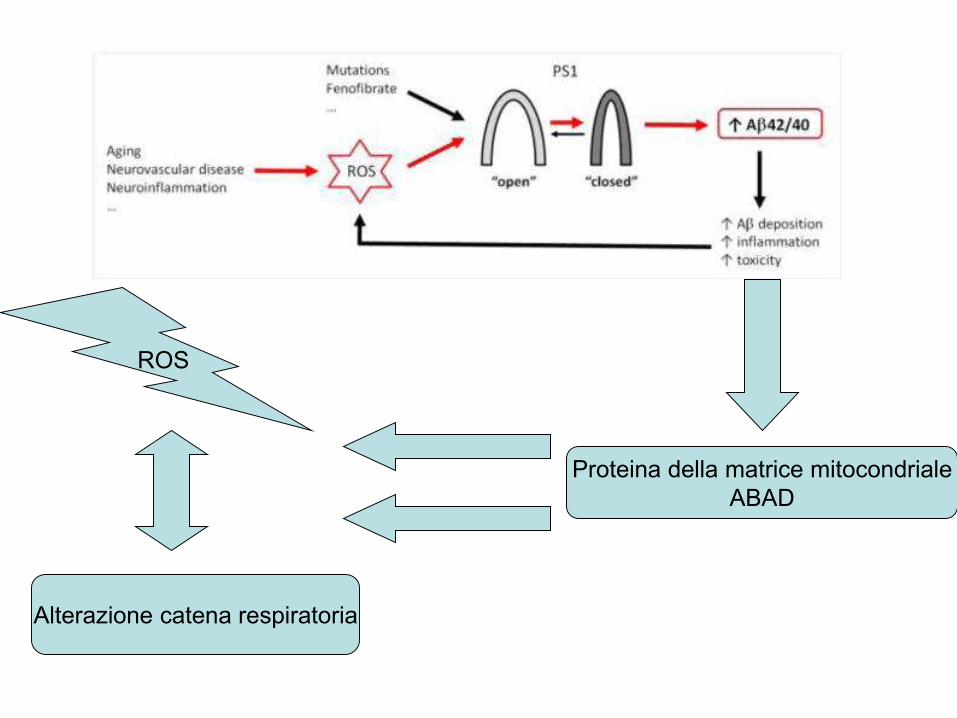
Proteina della matrice mitocondriale
ABAD
ROS
Alterazione catena respiratoria
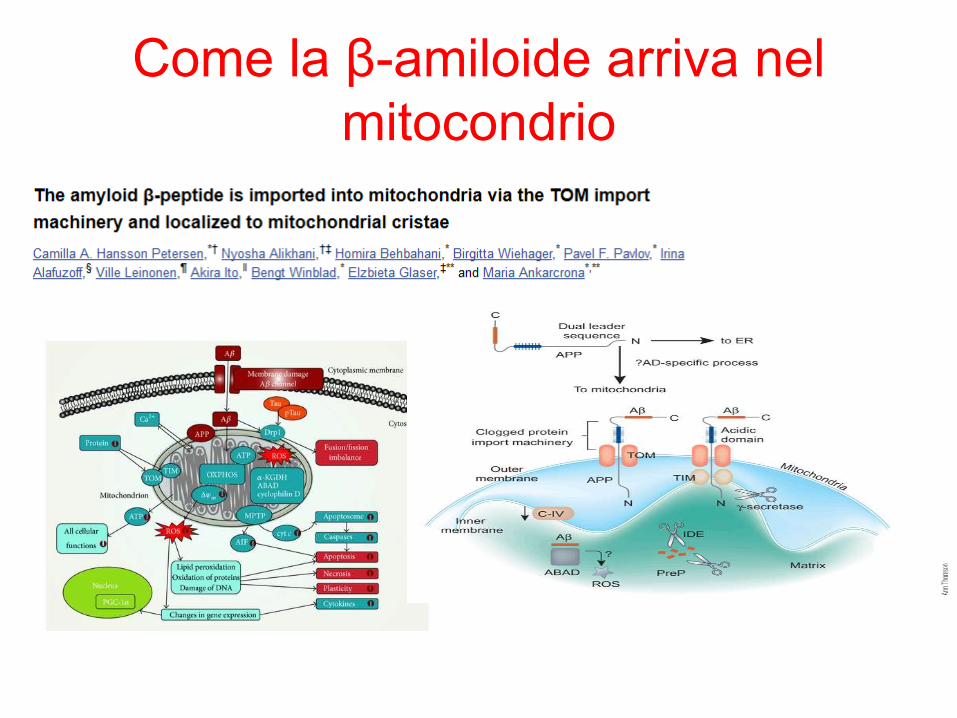
Come la β-amiloide arriva nel
mitocondrio
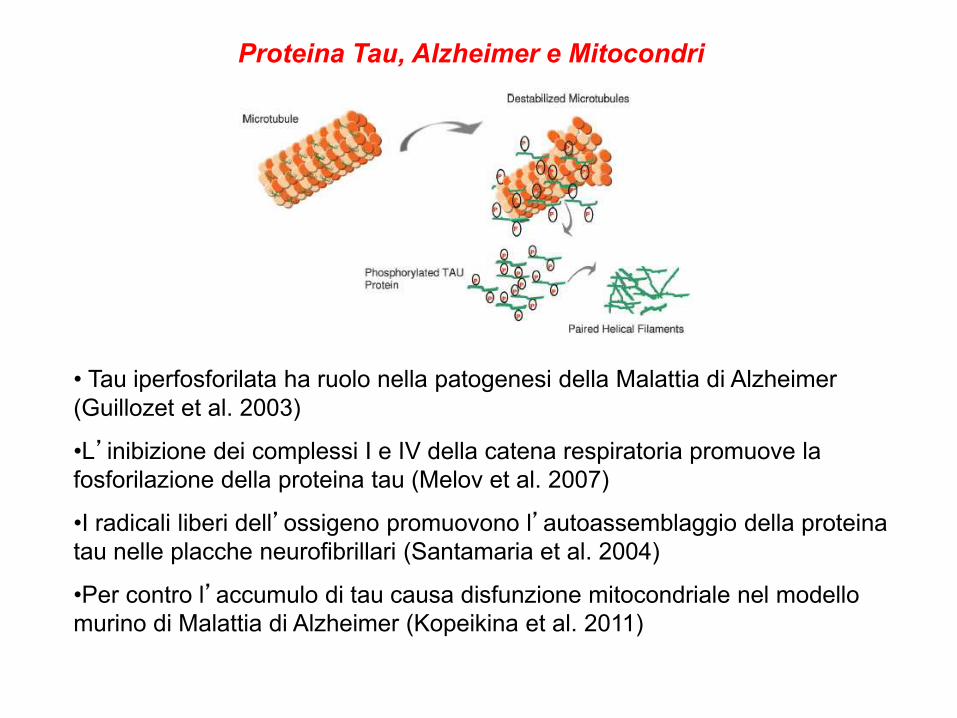
Proteina Tau, Alzheimer e Mitocondri
• Tau iperfosforilata ha ruolo nella patogenesi della Malattia di Alzheimer
(Guillozet et al. 2003)
•L’inibizione dei complessi I e IV della catena respiratoria promuove la
fosforilazione della proteina tau (Melov et al. 2007)
•I radicali liberi dell’ossigeno promuovono l’autoassemblaggio della proteina
tau nelle placche neurofibrillari (Santamaria et al. 2004)
•Per contro l’accumulo di tau causa disfunzione mitocondriale nel modello
murino di Malattia di Alzheimer (Kopeikina et al. 2011)
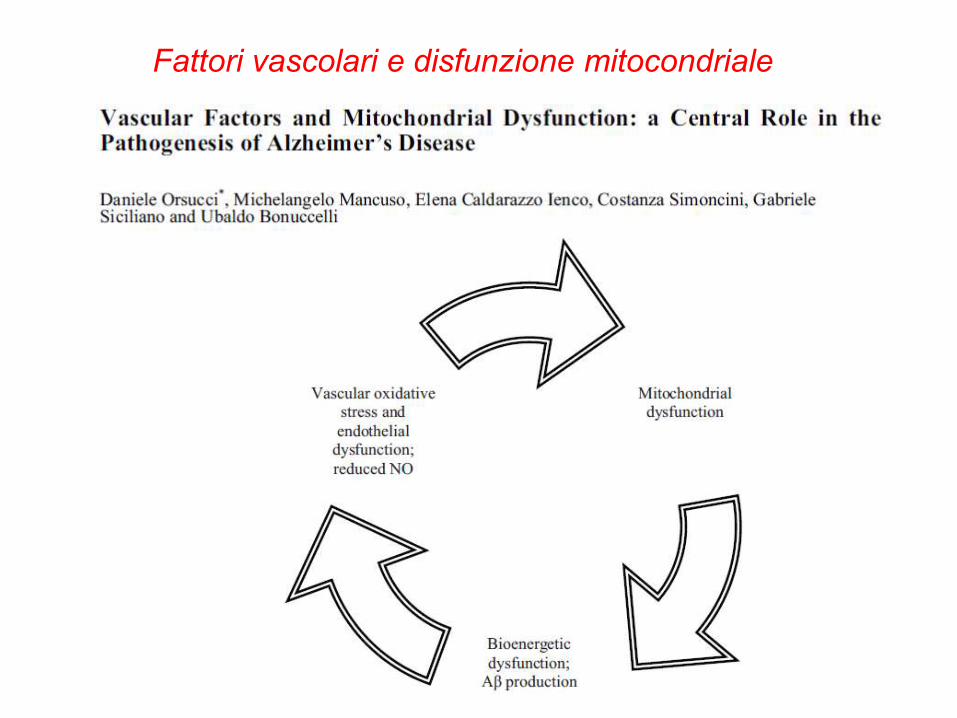
Fattori vascolari e disfunzione mitocondriale

CONCLUSIONI I
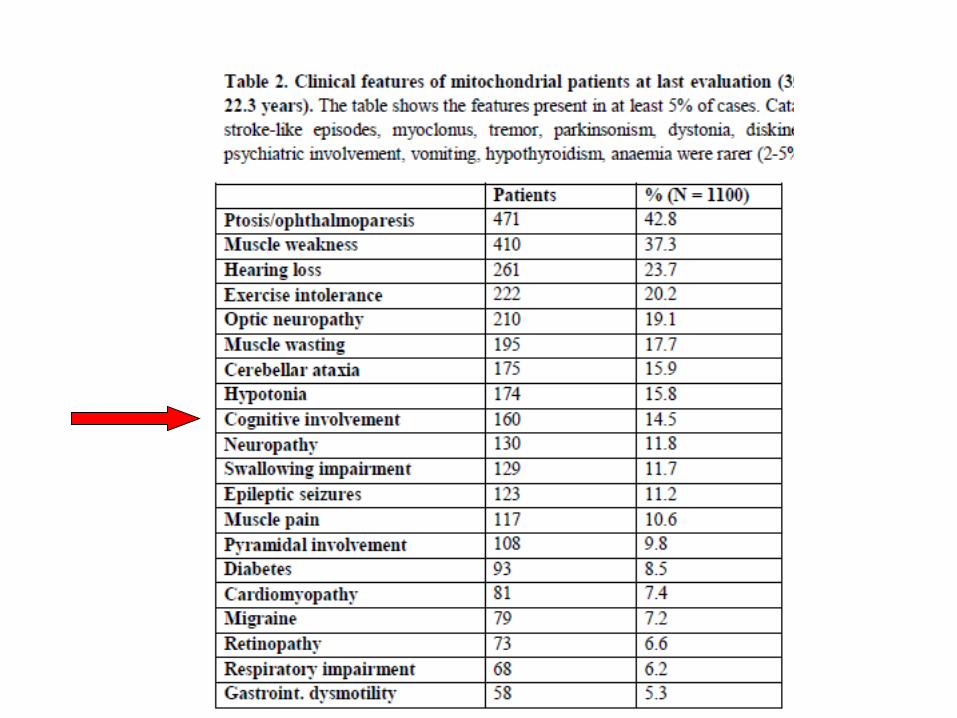
COGNITIVE IMPAIRMENT AS A COMMON FINDING IN MD
PLEASE PAY ATTENTION TO THE FOLLOWING THE RED FLAGS
-Central Nervous System: seizures, ataxia, myoclonus,
sensoryneural hearing loss, cognitive decline, psychomotor
retardation or regression, acute focal signs, migraine, tremor
and parkinsonism.
-Muscles and nerves: exercise intolerance, weakness,
dysphagia, respiratory failure, ptosis, ophtalmoparesis, cramps,
muscle wasting and paresthesia
-Eye: retinopathy, cataract, optic atrophy
-Endocrine: diabetes, short status, dysthyroidism
-Heart: cardiomyopathy, arrhythmia, sudden death
-Gastrointestinal: dysmotility, cachexia, pseudo-obstruction
-Kidney and liver impairment

CONCLUSIONI II
LA MALATTIA DI ALZHEIMER NON E’CAUSATA DA
ALTERAZIONI DNA MT
CONVERGENZA FISIOPATOLOGICA TRA AMYLOID
AND MITOCHONDRIAL CASCADE HYPOTHESIS

MY Professor in Calabria…

Grazie


















