
Manuale di Gestione della Qualità Manuale...ASP – Palermo Laboratorio Medico di Sanità Pubblica...
58
ASP – Palermo Laboratorio Medico di Sanità Pubblica MANUALE DI GESTIONE DELLA QUALITA’ Pagina 1 di 57 Manuale di Gestione della Qualità Edizione: 7 Revisione: 0 Data Emissione: 14/09/18 Motivo della revisione : Revisione totale del documento Redatto da: Responsabile della Qualità Dr.ssa Maria Angela Rampulla (firmato in originale) Firma: dott.ssa Rampulla M.A. Data: 17/09/2018 Verifica/Approvazione: Responsabile Laboratorio Dr.ssa Teresa Barone (firmato in originale) Firma: Dr.ssa Barone T. Data: 17/09/18
Transcript of Manuale di Gestione della Qualità Manuale...ASP – Palermo Laboratorio Medico di Sanità Pubblica...

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 1 di 57
Manuale di Gestione della Qualità
Edizione: 7 Revisione: 0 Data Emissione: 14/09/18
Motivo della revisione : Revisione totale del documento
Redatto da: Responsabile della Qualità Dr.ssa Maria Angela Rampulla (firmato in originale)
Firma: dott.ssa Rampulla M.A. Data: 17/09/2018
Verifica/Approvazione: Responsabile Laboratorio Dr.ssa Teresa Barone (firmato in originale)
Firma: Dr.ssa Barone T. Data: 17/09/18
ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 2 di 57
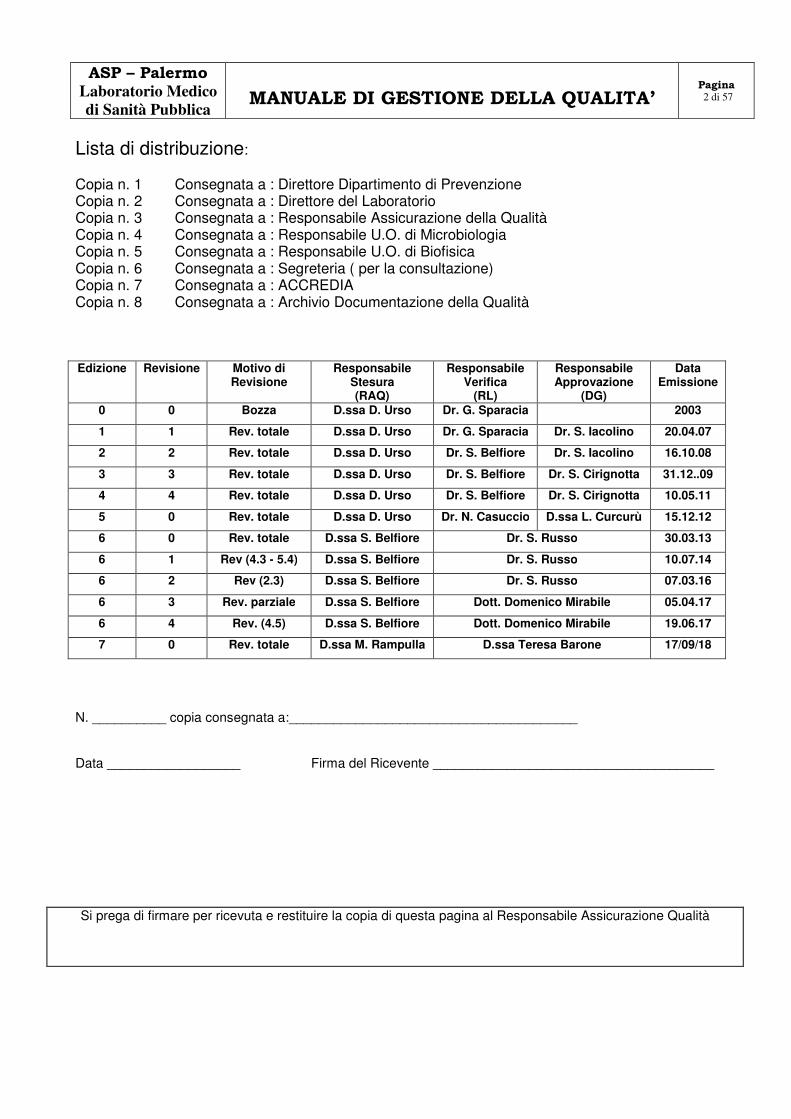
Lista di distribuzione: Copia n. 1 Consegnata a : Direttore Dipartimento di Prevenzione Copia n. 2 Consegnata a : Direttore del Laboratorio Copia n. 3 Consegnata a : Responsabile Assicurazione della Qualità Copia n. 4 Consegnata a : Responsabile U.O. di Microbiologia Copia n. 5 Consegnata a : Responsabile U.O. di Biofisica Copia n. 6 Consegnata a : Segreteria ( per la consultazione) Copia n. 7 Consegnata a : ACCREDIA Copia n. 8 Consegnata a : Archivio Documentazione della Qualità
Edizione Revisione Motivo di Revisione
Responsabile Stesura (RAQ)
Responsabile Verifica
(RL)
Responsabile Approvazione
(DG)
Data Emissione
0 0 Bozza D.ssa D. Urso Dr. G. Sparacia 2003
1 1 Rev. totale D.ssa D. Urso Dr. G. Sparacia Dr. S. Iacolino 20.04.07
2 2 Rev. totale D.ssa D. Urso Dr. S. Belfiore Dr. S. Iacolino 16.10.08
3 3 Rev. totale D.ssa D. Urso Dr. S. Belfiore Dr. S. Cirignotta 31.12..09
4 4 Rev. totale D.ssa D. Urso Dr. S. Belfiore Dr. S. Cirignotta 10.05.11
5 0 Rev. totale D.ssa D. Urso Dr. N. Casuccio D.ssa L. Curcurù 15.12.12
6 0 Rev. totale D.ssa S. Belfiore Dr. S. Russo 30.03.13
6 1 Rev (4.3 - 5.4) D.ssa S. Belfiore Dr. S. Russo 10.07.14
6 2 Rev (2.3) D.ssa S. Belfiore Dr. S. Russo 07.03.16
6 3 Rev. parziale D.ssa S. Belfiore Dott. Domenico Mirabile 05.04.17
6 4 Rev. (4.5) D.ssa S. Belfiore Dott. Domenico Mirabile 19.06.17
7 0 Rev. totale D.ssa M. Rampulla D.ssa Teresa Barone 17/09/18
N. __________ copia consegnata a:_______________________________________
Data __________________ Firma del Ricevente ______________________________________
Si prega di firmare per ricevuta e restituire la copia di questa pagina al Responsabile Assicurazione Qualità

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 3 di 57
Generalità
La struttura del presente Manuale è articolata in sezioni e capitoli. Le prime tre sezioni espongono i riferimenti normativi, le definizioni adottate e le abbreviazioni utilizzate, mentre le sezioni 4 e 5 descrivono il sistema di gestione della qualità adottato dal laboratorio in applicazione dei punti della Norma UNI CEI EN ISO/ IEC 17025:05. Ciascun capitolo e sotto capitolo riporta lo stesso riferimento numerico della Norma.
INDICE
SEZIONE CAPITOLO
TITOLO RIFERIMENTO
REV PAGG
UNI CEI EN ISO IEC 17025:2005
REG. RT-08 ACCREDIA
1.1 Compiti istituzionali
0 6
1.2 Attività istituzionali del LSP
0 7
1.3 Articolazione organizzativa
0 7
2.1 Scopo e campo di applicazione
0 9
2.2 Controllo e responsabilità del Manuale di Gestione della Qualità
0 9
2.3 Riferimenti Normativi
0 10
3.1 Definizioni
0 12
3.2 Abbreviazioni
0 13
4.1 Organizzazione 4.1.1 -4.1.2 -4.1.3- 4.1.4-
4.1.5
4.1.1 -4.1.2 -4.1.3- 4.1.4-
4.1.5 0 14
Imparzialità, indipendenza e integrità del personale
4.1.5 4.1.5 0 15
Riservatezza 4.1.5 4.1.5
0 15
Comunicazione interna 4.1.6 4.1.6
0 16
4.2 Sistema di gestione 4.2.1 4.2.1
0 16
Politica della qualità 4.2.2-4.2.3-
4.2.4-4.2.5-4.2.6 4.2.2-4.2.3-
4.2.4-4.2.5-4.2.6 0 17
4.3 Tenuta sotto controllo della documentazione 4.3.1 4.3.1
0 18
Sistema documentale della qualità 4.3.1 4.3.1
0 18
Documentazione Interna 4.3.2 - 4.3.3 4.3.2 – 4.3.3
0 18
Gestione della documentazione esterna 4.3.2 4.3.2
0 20

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 4 di 57
SEZIONE CAPITOLO
TITOLO RIFERIMENTO
A NORME REV PAGG.
UNI CEI EN ISO IEC 17025:2005
REG. RT-08 ACCREDIA
4.4 Riesame delle richieste, delle offerte e dei contratti 4.4.1 - 4.4.2 - 4.4.3 - 4.4.4 -
4.4.5
4.4.1 - 4.4.2 - 4.4.3 - 4.4.4 - 4.4.5 0 21
4.5 Subappalto delle prove e delle tarature 4.5.1 – 4.5.2 – 4.5.3 – 4.5.4
4.5.1 – 4.5.2 – 4.5.3 – 4.5.4 0 22
4.6 Approvvigionamento di servizi e di forniture 4.6.1 – 4.6.2 –
4.6.3 4.6.1 – 4.6.2 –
4.6.3 0 22
Qualifica dei fornitori 4.6.4 4.6.4
0 23
4.7 Servizi al cliente 4.7.1 – 4.7.2 4.7.1 – 4.7.2
0 23
4.8 Gestione dei reclami 4.8 4.8
0 24
4.9 Tenuta sotto controllo delle attività di prova e taratura non conformi
0 24
Gestione delle non conformità e azioni correttive
4.9.1 – 4.9.2 – 4.11.1 – 4.11.2 – 4.11.3 – 4.11.4
4.9.1 – 4.9.2 – 4.11.1 – 4.11.2 – 4.11.3 – 4.11.4 –
0 24
4.10 Miglioramento e gestione dei cambiamenti 4.10 4.10
0 25
4.11 Azioni correttive 4.11.1 -4.11.2 -
4.11.3 – 4.11.4 – 4.11.5
4.11.1 -4.11.2 -4.11.3 – 4.11.4-
4.11.5 0 25
4.12 Azioni preventive 4.12.1 – 4.12.2 4.12.1 – 4.12.2
0 26
4.13 Tenuta sotto controllo delle registrazioni 4.13.1.1-4.13.1.2-4.13.1.3-4.13.1.4
4.13.1.1-4.13.1.2-4.13.1.3-4.13.1.4 0 26
4.14 Audit interni 4.14.1 – 4.14.2 4.14.3 – 4.14.4
4.14.1 – 4.14.2 4.14.3 – 4.14.4 0 29
4.15 Riesame della Direzione 4.15.1 -4.15.2 4.15.1 -4.15.2
0 29
5.1 Requisiti tecnici 5.1.1 – 5.1.2 5.1.1 – 5.1.2
0 31
5.2 Personale 5.2.1 – 5.2.2 -
5.2.3 5.2.4 – 5.2.5
5.2.1 – 5.2.2 - 5.2.3 5.2.4 – 5.2.5
0 31
5.3 Luogo di lavoro e condizioni ambientali 5.3.1 – 5.3.2 –
5.3.3 5.3.4 – 5.3.5 5.3.1 – 5.3.2 –
5.3.3 5.3.4 – 5.3.5 0 37
Planimetria e identificazione dei locali 5.3.3 5.3.3
0 38
Monitoraggio ambientale 5.3.2 5.3.2
0 38
5.4 Metodi di prova e taratura e validazione dei metodi
0 38
Identificazione dei metodi di prova 5.4.1 4.4.1
0 38
Scelta dei metodi di prova 5.4.2 – 5.4.3 –
5.4.4 5.4.2 – 5.4.3 –
5.4.4 0 39
ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 5 di 57
SEZIONE CAPITOLO
TITOLO RIFERIMENTO
A NORME REV PAGG.
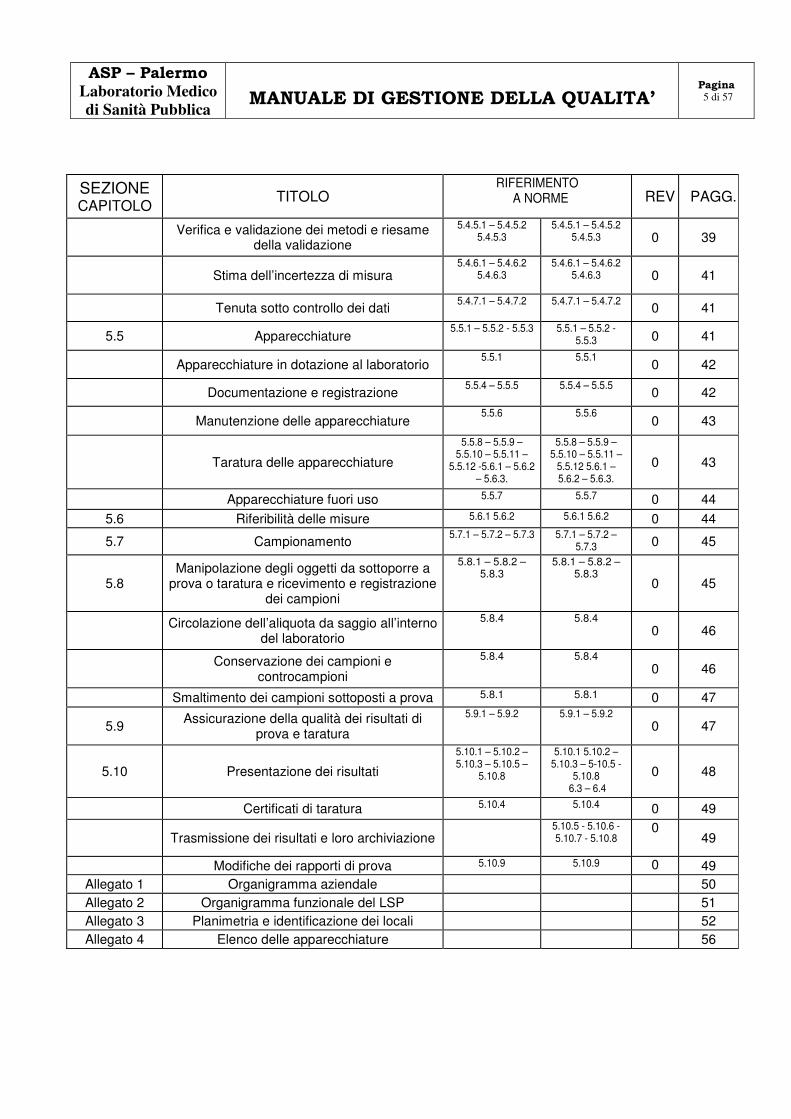
Verifica e validazione dei metodi e riesame della validazione
5.4.5.1 – 5.4.5.2 5.4.5.3
5.4.5.1 – 5.4.5.2 5.4.5.3 0 39
Stima dell’incertezza di misura 5.4.6.1 – 5.4.6.2
5.4.6.3 5.4.6.1 – 5.4.6.2
5.4.6.3 0 41
Tenuta sotto controllo dei dati 5.4.7.1 – 5.4.7.2 5.4.7.1 – 5.4.7.2
0 41
5.5 Apparecchiature 5.5.1 – 5.5.2 - 5.5.3 5.5.1 – 5.5.2 -
5.5.3 0 41
Apparecchiature in dotazione al laboratorio 5.5.1 5.5.1
0 42
Documentazione e registrazione 5.5.4 – 5.5.5 5.5.4 – 5.5.5
0 42
Manutenzione delle apparecchiature 5.5.6 5.5.6
0 43
Taratura delle apparecchiature
5.5.8 – 5.5.9 – 5.5.10 – 5.5.11 –
5.5.12 -5.6.1 – 5.6.2 – 5.6.3.
5.5.8 – 5.5.9 – 5.5.10 – 5.5.11 –
5.5.12 5.6.1 – 5.6.2 – 5.6.3.
0 43
Apparecchiature fuori uso 5.5.7 5.5.7 0 44
5.6 Riferibilità delle misure 5.6.1 5.6.2 5.6.1 5.6.2 0 44
5.7 Campionamento 5.7.1 – 5.7.2 – 5.7.3 5.7.1 – 5.7.2 –
5.7.3 0 45
5.8 Manipolazione degli oggetti da sottoporre a
prova o taratura e ricevimento e registrazione dei campioni
5.8.1 – 5.8.2 – 5.8.3
5.8.1 – 5.8.2 – 5.8.3
0 45
Circolazione dell’aliquota da saggio all’interno del laboratorio
5.8.4 5.8.4 0 46
Conservazione dei campioni e controcampioni
5.8.4 5.8.4 0 46
Smaltimento dei campioni sottoposti a prova 5.8.1 5.8.1 0 47
5.9 Assicurazione della qualità dei risultati di prova e taratura
5.9.1 – 5.9.2 5.9.1 – 5.9.2 0 47
5.10 Presentazione dei risultati
5.10.1 – 5.10.2 – 5.10.3 – 5.10.5 –
5.10.8
5.10.1 5.10.2 – 5.10.3 – 5-10.5 -
5.10.8 6.3 – 6.4
0 48
Certificati di taratura 5.10.4 5.10.4 0 49
Trasmissione dei risultati e loro archiviazione 5.10.5 - 5.10.6 -
5.10.7 - 5.10.8 0
49
Modifiche dei rapporti di prova 5.10.9 5.10.9 0 49 Allegato 1 Organigramma aziendale 50 Allegato 2 Organigramma funzionale del LSP 51 Allegato 3 Planimetria e identificazione dei locali 52 Allegato 4 Elenco delle apparecchiature 56

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 6 di 57
SEZIONE: 1 EDIZ. 7 – REV. 0
INDICE Cap. Rev. Titolo
1.1 0 Compiti istituzionali del laboratorio
1.2 0 Attività del Laboratorio
1.3 0 Articolazione organizzativa
1.1 COMPITI ISTITUZIONALI DEL LABORATORIO
Il Laboratorio Medico di Sanità Pubblica (di seguito identificato in maniera semplificativa: Laboratorio, Laboratorio di Sanità Pubblica o LSP) dell’ASP di Palermo, ha sede unica in Via Carmelo Onorato N. 6. (Tel. 0917033505 - Fax 0917033514 e-mail [email protected] – pec: [email protected].).
Dal 10/07/1995, con la legge istitutiva delle Aziende Unità Sanitarie Locali, il Laboratorio diventa l’organo tecnico analitico del Settore di Igiene e Sanità Pubblica.
Con la Legge Regionale n° 5 del 14 aprile 2009 “Norme per il riordino del Servizio sanitario regionale”, il Laboratorio di Sanità Pubblica è mantenuto all’interno del Dipartimento di Prevenzione dell’”ASP di Palermo” come previsto dal DA 6 agosto 2004: “Organizzazione del Laboratorio di Sanità Pubblica dei Dipartimenti di Prevenzione delle Aziende Unità Sanitarie Locali”.
Con successivo Atto Aziendale Delibera n. 198 del 17/03/2016, adeguato alle prescrizioni di cui al DA 210/2016 del 12/02/2016, il LSP è organizzato in due unità operative: U.O.S Microbiologia, parassitologia e virologia e U.O.S Biofisica negli ambienti di vita e di lavoro. In modo transmurale con la U.O.S. Tossicologia e biochimica, afferente al Dipartimento di Diagnostica di Laboratorio, condivide obiettivi di tutela della salute dei cittadini in base ai piani di intervento emanati dall’Assessorato Regionale alla Salute, che risultano funzionali alle attività del LSP per il controllo chimico di alimenti e acque.
Nell’ambito della tutela della salute collettiva, dai rischi legati a fattori fisici e microbiologici presenti negli alimenti, nelle acque destinate al consumo umano, negli ambienti di vita e di lavoro, il LSP svolge attività analitica e di consulenza a supporto, delle competenze del Dipartimento di Prevenzione, e di tutte le Autorità Sanitarie Competenti.
In data 19 Giugno 2009, il LSP è stato riconosciuto dal ISS-ORL quale Laboratorio conforme alla Norma Europea UNI CEI EN ISO/IEC 17025:05 e alle altre disposizioni vigenti in materia di sicurezza alimentare, registrato nell’elenco dei Laboratori riconosciuti con il numero identificativo NR 0070. In data 21.12.2010, ha ottenuto l’accreditamento da parte dell’Ente Unico ACCREDIA con numero di identificazione 1101, operando in conformità alla Norma UNI CEI EN ISO/IEC 17025:05 e al Regolamento RT-08. L’accreditamento è disciplinato da giusta convenzione, e rappresenta il riconoscimento della competenza tecnica del laboratorio per l’effettuazione di specifiche prove volte al controllo ufficiale. L’elenco aggiornato delle prove accreditate è consultabile sul sito http://www.accredia.it

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 7 di 57
SEZIONE: 1 EDIZ. 7 – REV. 0
1.2 ATTIVITÀ ISTITUZIONALI DEL LSP
Le attività individuate nel D.A. del 06.08.2004 e s.m.i. sono: ▪ profilassi delle malattie infettive, cronico, degenerative e professionali; ▪ controllo degli alimenti e delle bevande; ▪ controllo bromatologico alimenti e bevande; ▪ controllo delle acque destinate al consumo umano; ▪ controllo delle acque minerali e termali; ▪ controllo delle acque di balneazione; ▪ controllo delle acque destinate a scopo ludico-ricreativo; ▪ controllo delle acque destinate alla molluschicoltura ed alla acquicoltura; ▪ controlli indoor in ambienti di vita e di lavoro; ▪ attività analitiche per la ricerca delle fibre di amianto ▪ attività di riconoscimento e analitiche sui funghi
In base alle competenze istituzionali attribuite, il laboratorio svolge attività di controllo ufficiale nell’ambito della sicurezza alimentare, secondo quanto stabilito nel Regolamento CE 882/04.
Proprio in garanzia della sicurezza alimentare, applica i piani di controllo individuati nel documento programmatico redatto dalla Regione in base ai piani nazionali, e garantisce il supporto analitico sull’intero territorio regionale, secondo le matrici e le rispettive prove. Per i dettagli si rimanda al documento PRIS e successive integrazioni.
1.3 ARTICOLAZIONE ORGANIZZATIVA
Il LSP è Unità Operativa Complessa articolata in tre Unità Operative Semplici (UOS) individuate dal D.A.
06.08.2004 e recepite nell’Atto Aziendale dell’ASP:
• UOS Microbiologia Parassitologia e Virologia
• UOS Biofisica negli Ambienti di Vita e di Lavoro
UOS Microbiologia, Parassitologia e
Virologia
Campo di Attività
Laboratorio Microbiologia
degli Alimenti
Analisi microbiologiche di alimenti, acque in distribuzione, acque confezionate (acque da bere - acque minerali) acque superficiali, acque profonde, acque ludico ricreative (piscina), acque di balneazione, sedimenti marini
Laboratorio di preparazione terreni Preparazione dei terreni e reagenti necessari alle prove
Laboratorio di diagnostica microbiol. Identificazione biochimica e test di conforma microbiol.
Laboratorio Microbiologia
Ambientale
Ricerca di Legionella Pneumophila, nelle acque destinate al
consumo umano e negli ambienti confinati

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 8 di 57
SEZIONE: 1 EDIZ. 7 – REV. 0
UOS Biofisica Negli Ambienti di Vita e
di Lavoro
Campo di Attività
Centro di riferimento regionale per
l'amianto
Ricerca delle fibre aerodisperse di amianto (ambienti indoor),
analisi di massa per la ricerca ed identificazione delle fibre di
amianto, analisi per certificati di restituibilità
Laboratorio (SEM -Spettrometria IR) Analisi in microscopia elettronica, diffrattometria, in
spettrofotometria IR
Laboratorio preparazione campioni Preparazione dei campioni da sottoporre a prova
Laboratorio ambientale Accertamenti analitici per la valutazione della salubrità degli
ambienti confinati.
Ispettorato Micologico Ispettorato Micologico, riconoscimento funghi, accertamenti analitici
e merceologici su funghi freschi e secchi

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 9 di 57
SEZIONE: 2 EDIZ. 7 – REV. 0
INDICE:
Cap. Rev. Titolo
2.1 0 Scopo e campo di applicazione del MQ
2.2 0 Controllo e responsabilità del MQ
2.3 0 Riferimenti normativi
2.1 SCOPO E CAMPO D’APPLICAZIONE ( UNI CEI EN ISO/IEC 17025:2005 - 4.2.2. / RT-08 – 4.2.2 - 6.1)
Il presente Manuale di Gestione della Qualità (MQ) è stato redatto con lo scopo di creare un
documento che descriva il Sistema di Qualità sviluppato dal Laboratorio Medico di Sanità Pubblica dell’Azienda Sanitaria Provinciale di Palermo (ASP), in conformità alla normativa di riferimento UNI CEI EN ISO/IEC 17025:2005 e al Regolamento ACCREDIA RT- 08.
È rivolto a tutte le attività svolte dal Laboratorio per la gestione delle prove.
Oltre a descrivere le attività e le modalità di applicazione, elenca le procedure di riferimento, permettendo al personale, anche a quello di nuova assunzione, di prendere visione dell’organizzazione gestionale e tecnico-analitica del Laboratorio.
La sua struttura, dettagliatamente descritta nella PG/002 “Modalità per la stesura, verifica, approvazione e revisione del Manuale della Qualità”, è articolata in sezioni e capitoli. Le prime tre sezioni espongono i riferimenti normativi, le definizioni adottate e le abbreviazioni utilizzate, mentre le sezioni 4 e 5 descrivono il sistema di gestione della qualità adottato dal laboratorio in applicazione dei punti della Norma UNI CEI EN ISO/ IEC 17025:2005.
Ciascun capitolo e sottocapitolo riporta lo stesso riferimento numerico della Norma.
2.2 CONTROLLO E RESPONSABILITÀ DEL MANUALE DI GESTIONE DELLA QUALITÀ ( UNI CEI EN ISO/IEC 17025:2005 - 4.2.2./RT-08 – 4.2.2)
Il MQ è il documento più importante del Sistema di Qualità, in quanto racchiude e sintetizza il sistema stesso.
Viene gestito conformemente agli altri documenti, in maniera controllata, mettendo in evidenza le figure
coinvolte nella stesura, verifica, approvazione revisione, distribuzione ed archiviazione, secondo quanto descritto nelle PG/002 e PG/003. La tabella sintetizza le responsabilità assegnate durante le varie fasi di realizzazione e gestione del documento:

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 10 di 57
SEZIONE: 2 EDIZ. 7 – REV. 0
Responsabilità
Stesura/Revisione Responsabile della Qualità
Verifica Responsabile del Laboratorio
Approvazione Responsabile del Laboratorio
Distribuzione Responsabile della Qualità
Archiviazione Responsabile della Qualità
2.3 RIFERIMENTI NORMATIVI ( UNI CEI EN ISO/IEC 17025:2005 - 4.1.2./ RT-08 – 4.1.2 )
Il Sistema di Qualità e le Procedure descritte nel MQ trovano riferimento nei seguenti documenti:
• UNI CEI EN ISO/IEC 17025 :2005 « Requisiti generali per la competenza dei laboratori di prova e taratura »
• UNI EN ISO 19011 :2012 « Linee guida per audit di sistemi di gestione »
• UNI CEI EN ISO/IEC 17000 :2005 « Valutazione delle conformità – vocabolario e principi generali »
• Rapporti Istisan 13/41 : « Guida Eurachem – Terminologia per le misurazioni analitiche – Introduzione VIM 3° edizione »
• VIM 3° edizione « UNI CEI 70099 : Vocabolario internazionale di metrologia » consultabile on line : (http://www.ceiweb.it/it/lavori-normativi-it/vim/vim-contenuti.html)
• Regolamento ACCREDIA RT-08 « Prescrizioni per l’accreditamento dei laboratori di prova »
• Regolamento ACCREDIA RT-24 « Prove valutative »
• Regolamento ACCREDIA RG-09 « Regolamento per l’utilizzo del marchio ACCREDIA »
• Regolamento ACCREDIA RG-02 « Regolamento per l’accreditamento dei Laboratori di Prova per la sicurezza degli alimenti e dei Laboratori medici »
• DT-0002 REV 1 : « Guida per la valutazione e l’espressione dell’incertezza nelle misurazioni »
• DT-05-DT REV 1 : « Introduzione ai criteri di valutazione dell’incertezza di misura nelle tarature »
• Euramet/cgt-18/v04 : « Guidelines on the calibration of non automatic nweighing instruments »
• EA-4/02 : « Expression of the uncertainty of measurement in calibration »
• EURACHEM – AML 2013 : « Accreditation for microbiological Laboratories »
• EA-4/16 : « EA guidelines on the expression of uncertainty in quantitative testing »
• UNI EN ISO 9000 :2015 « Sistemi di gestione della qualità. Fondamenti e vocabolario»
• ISO/TS 19036 :2006 - Amd 1 :2009 « Microbiology of food and animal feeding stuff- Guidelines for the estimation of measurement uncertainty for quantitative determinations»
• UNI EN ISO 7218 :2007 – Amd. 1 :2013 « Microbiologia di alimenti e mangimi per animali, requisiti generali e guida per le analisi microbiologiche »
• Decreto Ministeriale 06.09.1994 «Normative e metodologie tecniche di applicazione,...., relativa alla cessazione dell’impiego dell’amianto»

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 11 di 57
SEZIONE: 2 EDIZ. 7 – REV. 0
• DPR 08.08.1994 : « Linee guida protezione amianto»
• UNI EN ISO 9001/2015 :Quality management Systems - Requirements
• DM 14.05.1996 : « Requisiti minimi dei laboratori pubblici e privati che intendono effettuare attività analitiche sull’amianto»
• UNI EN ISO 10012 :2004 « Sistemi di gestione della misurazione - Requisiti per i processi e le apparecchiature di misurazione»
Ulteriori documenti di riferimento, sono elencati nel documento ACCREDIA LS-04 (vedi sito)
• Reg CE n. 882/04 : « relativo ai controlli ufficiali intesi a verificare le conformità alla normativa in materia di mangimi e di alimenti e alle norme sulla salute e benessere degli alimenti »
• Reg CE n. 2073/2005 (s.m.i) « sui criteri microbiologici applicabili ai prodotti alimentari »
• Decisione Commissione del 12.08.2002 : attuazione direttiva 96/23/CE e 2002/657/CE «relativa al rendimento dei metodi analitici e all’interpretazione dei risultati»
• Reg. CE 401/2006
• Direttiva 414/1991
• Reg. CE 178/2002
• DL 114/2006
• DL 31/2001 : «Attuazione direttiva 98/83/CE relativa alla qualità delle acque destinate al consumo umano»
• Reg CE 1829 e 1830/2003
• DA 210/2016
REGOLAMENTI AZIENDALI :
1. Atto Aziendale n. 198 del 17/03/2016 2. Disposizioni in materia di disciplina del personale della Dirigenza sanitaria, professionale, tecnica e
amministrativa Delibera 509 del 28 giugno 2017 3. Disposizioni in materia di disciplina del personale della Dirigenza Medica e Veterinaria 4. Disposizioni in materia di disciplina del personale 5. Regolamento per l’acquisizione in economia di beni e servizi (del 280/13.3.2014) 6. Approvazione aziendale (nota 13040 del 07.06.13) del DPR n. 62 del 16.04.13 « Codice di
comportamento dei dipendenti pubblici, a norma dell’articolo 54 del decreto legisletivo 30.03.2001 n. 165 »
7. Delibera 623 del 23/12/2013 “Sistema gestione segnalazioni reclami”, 8. PTPC 2018 Piano anticorruzione ASP Palermo 9. Codice di comportamento dei dipendenti dell’a.s.p. Palermo Allegato 3 al P.T.P.C. ASP Palermo 10. Documento Valutazione dei Rischi 11. Regolamento Anticorruzione 12. DL. 81/2008 13. Regolamento Aziendale Privacy DL 196/2003
Tutti i regolamenti aziendali aggiornati sono visionabili sul sito www.asppalermo.org

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 12 di 57
SEZIONE: 3 EDIZ. 7 – REV. 0
INDICE:
Cap. Rev. Titolo
3.1 0 Definizioni
3.2 0 Abbreviazioni
3.1 DEFINIZIONI
In applicazione al Regolamento RT-08 punto 3.1, il Laboratorio ha adottato alcune delle definizioni riportate dalla UNI CEI EN ISO/IEC 17000:2005, dalla ISO 9000:2005 e dal UNI CEI 70099:2008, Vocabolario Internazionale di Metrologia (VIM) Di seguito vengono riportati i termini, di carattere generale, ritenuti più importanti:
1. Accreditamento: Attestazione di terza parte relativa ad un organismo di valutazione della conformità che comporta la dimostrazione formale della sua competenza ad eseguire compiti specifici di valutazione della conformità
2. Qualità: grado in cui un insieme di caratteristiche intrinseche soddisfa i requisiti.
3. Requisito: esigenza o aspettativa che può essere espressa generalmente implicita o cogente
4. Assicurazione della Qualità: Parte della gestione della qualità mirata a dare fiducia che i requisiti per la qualità siano soddisfatti
5. Cliente/Richiedente: Organizzazione o persona che riceve un prodotto. Soggetto per il quale il laboratorio effettua le prove accreditate.
6. Certificato di un materiale di riferimento: Un documento che certifica che uno o più valori di una proprietà per un dato materiale di riferimento sono stati ottenuti mediante le procedure idonee ad ottenere valori validi di tali proprietà.
7. Certificazione: Procedimento per stabilire, mediante operazioni tecnicamente valide, il valore misurato di una o più proprietà di un materiale o sostanza. Il procedimento porta all’emissione di un certificato.
8. Controllo di Qualità: Insieme delle tecniche e delle attività a carattere operativo messe in atto per soddisfare i requisiti di qualità.
9. Laboratorio di prova : Laboratorio che esegue le prove.
10. Materiale di riferimento: Materiale o sostanza, per la quale una o più proprietà sono sufficientemente ben stabilite, da essere utilizzata per la taratura di un’apparecchiatura, per la valutazione di un metodo di misura o per l’assegnazione di valori ai materiali.
11. Materiale di riferimento certificato: Un materiale di riferimento in cui il valore di una o più proprietà è certificato mediante una procedura tecnicamente valida, accompagnata o riferibile a un certificato emesso da un organismo di certificazione. Un materiale di riferimento può essere certificato individualmente o mediante esame di campioni rappresentativi prelevati da un lotto.
12. Metodo di prova: Procedura tecnica specificata per eseguire una prova.
13. Metodo di prova ufficiale: metodo di prova riportato o richiamato in documenti normativi cogenti e/o pubblicato su gazzetta ufficiale italiana o dell’unione europea, o comunque richiamato o riportato in un documento emesso da una autorità quale regione, provincia, ecc.. la qualifica di ufficiale è una proprietà trasversale indipendente dal grado di esaustività dei contenuti

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 13 di 57
SEZIONE: 3 EDIZ. 7 – REV. 0
14. Metodo di prova normalizzato: metodo approvato da organismi di normazione nazionali, europei o internazionali (ad es. UNI, CEI, CEN, ISO, UNICHIM, ASTM, AOAC, ecc) o da organismi pubblici autorevoli (es. USDA, FDA, EPA,NIOSH, UPAC, APHA, OIV, OIE, WHO, APAT,CNR,IRSA, ISPRA, NMKL,ecc.).
15. Metodo di prova non normalizzato: metodo emesso da organizzazioni tecniche nazionali o internazionali e metodo sviluppato da laboratori/centri di riferimento nazionali o comunitari o da centri di referenza nazionali accreditati. Elemento discriminante è che la responsabilità dei dati forniti è riferita non all’organizzazione che lo ha emesso, ma ai singoli autori.
16. Metodo di prova interno: metodo di prova messo a punto o adottato da un laboratorio sulla base di conoscenze desunte dalla letteratura scientifica e/o da esperienza pratica.
17. Organismo di accreditamento: Organismo autorevole che rilascia l’accreditamento (esso stesso non è un organismo di valutazione)
18. Organismo di valutazione della conformità: organismo che fornisce servizi di valutazione della conformità
19. Prova: determinazione di una o più caratteristiche di un oggetto di valutazione della conformità secondo una procedura
20. Procedura: modo specificato per svolgere un’attività o un processo
21. Rapporto di prova: Documento che riporta i risultati di una prova e altre informazioni ad essa relative. Per quanto riguarda le altre definizioni omesse nel presente MQ si rimanda alla UNI CEI EN ISO/IEC 17000:2005.
3.2 ABBREVIAZIONI
Le sigle adoperate dal Laboratorio, riportate nel MQ e negli altri documenti, sono le seguenti:
AP Apparecchiatura ASP Azienda Sanitaria Provinciale SIAN Servizio Igiene degli alimenti e nutrizione DG Direttore Generale DP Dipartimento di Prevenzione DDP Direttore del Dipartimento di Prevenzione DL Dipartimento Diagnostica di Laboratorio DDL Direttore del Dipartimento Diagnostica di Laboratorio LSP Laboratorio Medico di Sanità Pubblica MP Metodi di prova MQ Manuale della Qualità PG Procedura Generale PI Procedura di prova integrativa PO Procedura Operativa PT Personale tecnico PA Personale Accettazione Campioni di prova RAQ Responsabile di Assicurazione della Qualità RL Responsabile del Laboratorio RP Rapporto di prova RT Responsabile Tecnico RUO Responsabile di Unità Operativa SQ Sistema di Qualità UAQ Unità di Assicurazione della Qualità

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 14 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
INDICE:
Cap. Rev. Titolo
4.1 0 Organizzazione
4.2 0 Sistema di gestione
4.3 0 Tenuta sotto controllo della documentazione
4.4 0 Riesame delle richieste offerte e contratti
4.5 0 Sub appalto delle prove e tarature
4.6 0 Approvvigionamento di servizi e forniture
4.7 0 Servizi al cliente
4.8 0 Gestione dei reclami
4.9 0 Tenuta sotto controllo delle attività di prova e taratura non conformi
4.10 0 Miglioramento e gestione dei cambiamenti
4.11 0 Azioni correttive
4.12 0 Azioni preventive
4.13 0 Tenuta sotto controllo delle registrazioni
4.14 0 Audit interni
4.15 0 Riesame della Direzione
4.1 ORGANIZZAZIONE
( UNI CEI EN ISO/IEC 17025:2005 – 4.1.1. – 4.1.2 – 4.1.3- 4.1.4/RT 08 4.1.1-4.1.2- 4.1.4 note )
Come già espresso nella sezione 1, il Laboratorio Medico di Sanità Pubblica, è una struttura complessa con sede unica, dell’Azienda Sanitaria Provinciale di Palermo. Il Decreto Assessoriale che ne ha definito l’istituzione (GURS 35 del 20.08.2004), conformemente ai punti 4.1.3. e 4.1.4 della Norma, ha così fissato alcuni criteri organizzativi: art. 1: per l’esercizio delle funzioni di natura tecnico scientifica ed analitica, dei Dipartimenti di Prevenzione delle Aziende Unità Sanitarie Locali della Regione, degli enti locali, degli enti pubblici e dell’ARPA, è istituito il Laboratorio di Sanità Pubblica (LSP), che si identifica con gli attuali Reparti medico-micrografici dei Laboratori d’igiene e profilassi con la relativa dotazione dei beni mobili, immobili, strumentali e il personale di pertinenza. art. 2 : il LSP, servizio interareale, è una unità operativa complessa, dotato di autonomia tecnica-funzionale, organizzativa e gestionale ed opera sotto la direzione di un responsabile tecnico-organizzativo. art. 3 : il Laboratorio si articola in 3 Unità Operative Semplici (successivamente modificato in 2 Unità Operative Semplici con successivo Atto Aziendale Delibera n. 198 del 17/03/2016) art 4: il LSP, attraverso le sue UOS, assolve alle attribuzioni istituzionali di natura tecnico-analitica nello svolgimento delle materie di competenza sanitaria, già svolte e identificate con la circolare n. 1045/2001.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 15 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
Il personale dirigente e tecnico, dal RL al RAQ ai RUO, secondo le competenze individuate,
sovrintende e vigila sul personale, affinché siano messe in opera le disposizioni aziendali, predispone l’aggiornamento e la formazione, in garanzia della qualità delle prove, e provvede a formulare, agli organi preposti, quanto necessario per la corretta esecuzione delle stesse; assicura un efficace sistema di comunicazione interna in modo da consentire la partecipazione attiva di tutto il personale, per il raggiungimento degli obiettivi.
Le responsabilità individuate per ogni figura professionale e le deleghe di funzione, sono dettagliate nella sezione 5.
IMPARZIALITA’, INDIPENDENZA E INTEGRITA’ DEL PERSONALE
(UNI CEI EN ISO/IEC 17025:2005 4.1.5/ RT 08 4.1.5)
L’ASP di Palermo opera attivamente al fine di garantire l’integrità del proprio personale nell’espletamento delle proprie funzioni avendo emanato un proprio regolamento inerente il “Piano Triennale di Prevenzione Della Corruzione2018-2019-2020” cui si rimanda per maggiori chiarimenti. Tutto il personale in servizio a qualunque titolo deve adeguarsi alle misure di prevenzione e contrasto alla corruzione e di trasparenza adottate dall’ASP Palermo, la loro violazione costituisce illecito disciplinare (art. 1, comma 14, l. n. 190/2012).
Il Piano Triennale prevede inoltre uno specifico codice di comportamento che armonizza le proprie previsioni con le misure di trasparenza e di prevenzione della corruzione contenute nel Piano Nazionale Anticorruzione (PNA) e nei Piani Triennali per la prevenzione della corruzione (PTPCT) aziendali. Tutti i regolamenti aziendali sono disponibili, per la consultazione, nella pagina web dell’azienda (www.asppalermo).
Conformemente a quanto previsto al punto 4.1.4 (nota 2) e 4.1.5, il Laboratorio, svolgendo attività di controllo ufficiale, non effettua prestazioni che potrebbero determinare conflitti di interesse.
RISERVATEZZA (UNI CEI EN ISO/IEC 17025:2005 4.1.5/ RT 08 4.1.5)
In conformità al DL 196/03, L’azienda Sanitaria, con delibera n. 0496 del 21.07.2010, ha pubblicato “il Regolamento della Privacy Aziendale”, che descrive le linee guida per il trattamento dei dati personali e sensibili, identificando le funzioni e le figure che devono sovrintendere al rispetto del DL 196 sia in garanzia degli utenti che dei dipendenti.
Tutto il personale, ed in particolare quello adibito alle attività di prova, deve mantenere la segretezza
sulle informazioni di cui viene a conoscenza, nello svolgimento delle attività di laboratorio. In generale, all’interno del laboratorio devono circolare, quando possibile, campioni di prova contraddistinti dal solo numero di codifica;
• i RP devono essere trasmessi unicamente ai destinatari specificati nella lista di distribuzione • nessun documento, o anche parte di esso, può essere riprodotto o divulgato senza l’autorizzazione scritta del RL • l’accesso ai laboratori è, di norma, vietato agli estranei. Anche il cliente per accedere, deve essere autorizzato dal RUO.
Copia integrale del Manuale della Privacy è stata consegnata a ciascun RUO. Il Laboratorio periodicamente aggiorna l’elenco degli incaricati alla gestione della Privacy, comunicandolo all’Ufficio relazioni con il Pubblico.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 16 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
COMUNICAZIONE INTERNA ( UNI CEI EN ISO/IEC 17025:2005 – 4.1.6 RT-08 – 4.1.6) La direzione si rapporta con il personale, attraverso comunicazione scritta (ordini di servizio,
disposizioni, circolari) e incontri di lavoro, dove vengono discusse le tematiche riguardanti la gestione del laboratorio.
Relativamente all’attuazione del sistema qualità, si avvale di documenti e moduli pertinenti a
procedure, programmi, obiettivi, istruzioni, che devono essere condivisi dal personale, sia per la comprensione del sistema stesso, che per l’applicazione di quanto di propria competenza.
Gli incontri di lavoro possono avere carattere di periodicità in occasione di specifici eventi quali ad
esempio gli incontri annuali conseguenti al riscontro del riesame della direzione successivo ad Audit esterni o interni, ma più frequentemente scaturiscono da esigenze tecniche o gestionali su imput di specifiche disposizioni aziendali e/o di adeguamento a specifiche norme.
4.2 SISTEMA DI GESTIONE
L’assetto istituzionale della ASP di Palermo è stabilito con Atto Aziendale adottato ai sensi del DLGs 502/92 e s.m.i. e della LR n.5 del 14/04/2009 tenendo conto delle linee guida regionali di cui ai DA 0736/10 e DA 1360 del 03/08/2015.
RAPPRESENTANTE LEGALE
Il Direttore Generale, nominato dalla Giunta Regionale, rappresenta legalmente l’ASP di Palermo, di cui il Laboratorio Medico di Sanità Pubblica fa parte.
Esercita tutti i poteri di gestione di ordinaria e straordinaria amministrazione, predispone e adotta i
programmi di attività, i bilanci annuali e triennali, i conti consuntivi, la struttura operativa, la dotazione organica; sovrintende all’attuazione dei programmi e ne assicura la gestione, determina articolazioni e modalità organizzative della struttura operativa centrale e periferica; determina, secondo il programma annuale, la distribuzione delle risorse ai Dipartimenti, in rapporto agli obiettivi assegnati.
Il DG, è coadiuvato dal Direttore Sanitario e dal Direttore Amministrativo. Le tre Direzioni costituiscono la struttura operativa centrale, a sua volta articolata in Dipartimenti,
Servizi, Distretti e quanto previsto dall’Atto Aziendale. La struttura organizzativa e le linee di dipendenza gerarchica e di collegamento funzionale, sono
riportate nello schema dell’allegato 1. Nell’ambito dell’accreditamento, il Direttore Generale, o suo delegato, sottoscrive la convenzione con
l’Ente Unico di Riconoscimento, assumendo l’impegno attraverso la direzione del laboratorio, a garantire il miglioramento continuo.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 17 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
POLITICA DELLA QUALITA’
( UNI CEI EN ISO/IEC 17025:2005 4.2.1-4.2.2-4.2.3-4.2.4-4.2.5-4.2.6/ RT08 4.2.1-4.2.2-4.2.3-4.2.4-4.2.5-4.2.6)
Il Laboratorio, secondo i principi generali enunciati, nella Norma UNI CEI EN ISO/IEC 17025:2005, ha manifestato la necessità di sviluppare uno specifico SQ, attraverso la pianificazione, stesura e messa in atto di procedure, riconoscendo che un sistema di qualità è necessario non solo per il mantenimento dello standard qualitativo, ma anche per promuovere un piano continuo di miglioramento, in modo da garantire, nell’ambito delle proprie competenze e dei controlli ufficiali, la qualità delle prove.
Il Direttore Generale, con delibera n. 283 del 02/05/2017, ha emanato l’Atto Aziendale che all’articolo
4 stabilisce l’applicazione sistematica della metodologia e degli strumenti di un sistema di gestione della qualità orientato al miglioramento continuo dell’offerta sanitaria. Tali obiettivi vengono sviluppati anche negli articoli successivi in prospettiva di un miglioramento continuo della qualità dei servizi offerti, identificando di volta in volta le figure professionali coinvolte nell’assicurazione della qualità a livello aziendale. Il Sistema di Gestione della Qualità del Laboratorio pertanto si integra con l’Atto Aziendale e ne esplicita gli aspetti organizzativi e funzionali, aderendo ai principi dettati dalla UNI EN ISO 17025:2005.
Con l’approvazione del Manuale della Qualità, il RL esprime l’impegno a perseguire una politica della
Qualità, atta a garantire la conformità alla Norma UNI CEI EN ISO/IEC 17025:05 e al Regolamento ACCREDIA RT-08.
DICHIARAZIONE DELLA POLITICA PER LA QUALITA’
“Il Direttore del Laboratorio, di concerto con la Direzione Aziendale dell’ASP di Palermo, nella consapevolezza
della necessità di una visione globale dell’organizzazione aziendale, nella quale l’aspetto “qualità” rappresenta
l’obiettivo primario, ritiene necessario formulare in maniera dettagliata gli obiettivi e le strategie per garantire al
Laboratorio Medico di Sanità Pubblica di operare in conformità alla norma UNI CEI EN ISO/IEC 17025 e al
Regolamento ACCREDIA RT-08, e pertanto dichiara:
Gli Obiettivi, della politica per la qualità, identificati sono:
- Ottemperanza ai requisiti richiesti (cogenti e non) per l’erogazione di servizi
- Soddisfazione delle parti interessate con riferimento ai contratti/ convenzioni stipulate con Assessorati, Enti
pubblici, Università, altre Aziende Sanitarie, con privati nell’ambito dei compiti istituzionali
- Soddisfazione delle aspettative implicite ed esplicite delle parti interessate
- Soddisfazione dei requisiti di sicurezza d’igiene e tutela ambientale previsti dalle leggi in vigore
- Conoscenza delle tecniche e tecnologie per l’effettuazione delle prove e tarature
- Conoscenza delle leggi cogenti e delle norme di riferimento
- Identificazione dei percorsi per i contatti e le comunicazioni
- Efficace gestione del sistema di qualità attraverso l’applicazione di quanto prescritto nel manuale e nelle
procedure adottate
- Ottimizzazione dell’organizzazione interna
- Ampliamento dei servizi in linea con le strategie aziendali nel rispetto dell’integrità del sistema di qualità
implementato.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 18 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
In base agli obiettivi prefissati, La Direzione s’impegna ad attuare le seguenti strategie:
- Mantenere integro il sistema di Gestione per la Qualità implementato dal Laboratorio, secondo UNI CEI EN
ISO/IEC 17025 e Regolamento ACCREDIA RT-08, qualora vengano pianificati e/o attuati dei cambiamenti al
sistema stesso
- Arricchire la professionalità degli operatori attraverso specifica formazione
- Ottemperare a punto 4.1.5 della norma, identificando percorsi, responsabilità e competenze, al fine di
motivare e responsabilizzare il personale che opera nell’ambito del SQ
- Nominare un Responsabile del Sistema di Qualità attribuendo autonomia gestionale e operativa
- Garantire l’affidabilità dei fornitori attraverso un sistema di valutazione di “ditta /prodotto”
- Verificare, attraverso il Riesame della Direzione, effettuato con cadenza annuale, il grado di
raggiungimento/scostamento dagli obiettivi prefissati: reclami, non conformità, qualifica dei fornitori,
miglioramento, ecc..”
La Direzione
(documento firmato in originale)
4.3 TENUTA SOTTO CONTROLLO DELLA DOCUMENTAZIONE
SISTEMA DOCUMENTALE DELLA QUALITÀ (UNI CEI EN ISO/IEC 17025:2005 4.3.1 RT-08 4.3.1 )
Il SQ del Laboratorio viene gestito attraverso un sistema documentale, sia esso cartaceo o informatico, avente lo scopo di assicurare che ogni attività sia supportata da adeguata documentazione che ne descriva i campi e modalità di applicazione, le responsabilità, che sia disponibile alle persone interessate, e accessibile ai percorsi di audit e al riesame.
La documentazione del SQ può essere divisa in documentazione interna e documentazione prodotta da terzi, ma che, per le proprietà intrinseche, diventa parte integrante del SQ.
Documenti interni Documenti esterni
MQ Norme, linee guida Leggi e decreti e Regolamenti aziendali
PG Richieste e corrispondenza relative all’attività
PO Certificati di qualità, Schede di sicurezza, Certificati di taratura, Listini
MP Attestati di corsi di formazione
Modulistica Modulistica aziendale
Registrazioni della qualità (vedi MQ punto 4.13)
DOCUMENTAZIONE INTERNA:
APPROVAZIONE E DIFFUSIONE E MODIFICHE DEI DOCUMENTI: ( UNI CEI EN ISO/IEC 17025:2005 – 4.3.2- 4.3.3/RT-08 4.3.2-4.3.3)
STESURA, APPROVAZIONE, EMISSIONE, REVISIONE E DISTRIBUZIONE
Il RL ha stabilito le modalità per la gestione della documentazione del sistema qualità, fornendo ai responsabili, tutti gli strumenti necessari per la stesura, verifica, approvazione, emissione e revisione dei documenti stessi. Ciascun documento, secondo la tipologia, è identificabile per : codifica, di tipo alfa numerico, stesura, verifica, stato di revisione e data di entrata in vigore, coincidente con la data di emissione, sottoscritta dal RL.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 19 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0 Tale documentazione è sottoposta a verifica nel corso di Audit interni ed esterni, qualora dovesse
ravvisarsi la necessità di aggiornamento, o per risoluzioni di “non conformità”. Sul documento revisionato, viene riportato, oltre al nuovo indice di revisione, il motivo della revisione: se trattasi di revisione parziale o totale del documento. Nel caso di revisioni parziali, le parti revisionate o aggiunte sono evidenziate in grassetto.
Correzioni o integrazioni manuali, sono consentite, in attesa dell’emissione della nuova revisione, purché controfirmate dal RAQ, e notificate per iscritto a tutti i possessori di copia del documento. La revisione aggiornata viene quindi distribuita, dopo la convalida di emissione da parte del RL, con il ritiro contemporaneo della precedente stesura. La revisione totale del documento non porta all’interno parti significative in quanto sostituisce in toto il documento stesso. La assegnazione della documentazione avviene attraverso una lista di distribuzione, riportata nel frontespizio. Nel caso avvenga in formato pdf, il destinatario sottoscrive la presa in carico della copia.
Prima della distribuzione, ciascun documento viene verificato dall’UAQ, che ne accerta la completezza. Il RAQ predispone inoltre, una lista della documentazione del SQ, in modo che ciascuna UOS e servizi interessati, disponga di una visione aggiornata del sistema documentale della Qualità. Il “Manuale di Gestione della Qualità” prevede due livelli d’indicizzazione, uno, identificato dall’edizione, e l’altro dalla revisione: l’edizione si riferisce all’ultima revisione totale del documento, mentre la revisione interessa la modifica delle sezioni e capitoli interni. Ciascuna sezione può andare in revisione, ed essere sostituita, mantenendo invariato il numero dell’edizione. Le parti revisionate sono identificabili perché in grassetto.
La tabella sottostante, riportata nella PG/003, sintetizza le fasi di gestione della documentazione e le rispettive responsabilità:
TABELLA RIEPILOGATIVA
Tipo di documento
identificazione stesura verifica approvazione
destinatari archiviazione
MQ
- Nome – Edizione – Revisione - Data
RAQ
RL
RL
DG-DP-RL-RAQ-RUO-SEG-ACCREDIA ARCHIVIO
DISTRIBUZ. PDF
RAQ
PG
- Nome – Codice – revisione – data
RUO
RAQ
RL
RL-RAQ-RUO-SEG- ARCHIVIO
DISTRIBUZ. PDF
RAQ
PO
- Nome – Codice – revisione – data
RUO
RAQ
RL
RL-RAQ-RUO-SEG- ARCHIVIO
DISTRIBUZ. PDF
RAQ
MP
- Nome – Codice – revisione – data
RUO
RAQ
RL
RL-RAQ-RUO-UOS- ARCHIVIO
DISTRIBUZ. PDF
RAQ
REGISTRAZIONI DELLA QUALITA’
- titolo- riferimento alla PG con
revisione - data
RUO
RAQ
RL
SECONDO L’ATTIVITA’
RAQ
MODULISTICA
- Nome- Revisione- riferimento a
PG/PO (allegati)
RUO
RAQ
RL
-ATTIVITA’ DI PERTINENZA
- SEGRETERIA
RAQ
REGISTRAZIONI TECNICHE
- Nome- Revisione- riferimento a PG/PO (allegati)
RUO
RAQ
RL
-ATTIVITA’ DI PERTINENZA
RAQ
Registro codifica campioni
-Nome - Anno- Codifica (da….a..) RUO (format)
RAQ (format)
RL (format)
PA
PA
Quaderni di lavoro
-Numero progressivo- operatore, - U.O.
RUO (format)
RAQ (format)
RL (format)
DIR/PT
RUO
NORME-GUIDE TECNICHE
-Titolo-rev/edizione-anno -ATTIVITA’ DI PERTINENZA
RAQ
LEGGI REGOLAMENTI
-Titolo-rev/edizione-anno -ATTIVITA’ DI PERTINENZA - RICHIEDENTE
RAQ
Cataloghi, listini
Titolo A DISPOSIZIONE RAQ
Attestati corsi di formazione
Ente di formazio
ne
RAQ Segreteria
Certificazioni qualità,
Riferimento prodotto- lotto RAQ
RAQ
SCHEDE DI SICUREZZA PT
Riferimento prodotto UU.OO.SS.
UU.OO.SS.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 20 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
GESTIONE DELLA DOCUMENTAZIONE ESTERNA
“MODULISTICA AZIENDALE, CORRISPONDENZA”
INDICIZZAZIONE, DISTRIBUZIONE, EMISSIONE, ARCHIVIAZIONE
( UNI CEI EN ISO/IEC 17025:2005 – 4.3.2.1 RT-08- 4.3.2.1 )
Tutta la documentazione che afferisce al LSP, o che viene prodotta, è gestita in modo da assicurare riconoscimento univoco, rintracciabilità dall’ingresso alla emissione, fino alla archiviazione. La documentazione in ingresso e/o prodotta, può essere così riassunta:
1. Circolari e Regolamenti aziendali
2. Delibere riguardanti:
• Gare di fornitura ( allegati alle
aggiudicazione)
• Autorizzazioni e/o disposizioni di servizio
• Incarichi
• Adozioni di procedimenti
3. Corrispondenza per compiti di istituto
4. Piani di lavoro, obiettivi aziendali
5. Verbali di riunioni
6. Richieste in relazione alla attività
7. Moduli – schede- emessi ed adottati
dall’Azienda
8. Verbali di campionamento
9. Leggi, Norme e decreti
La documentazione in ingresso può pervenire attraverso: consegna diretta, FAX, servizio postale, e-mail, consultazione siti ufficiali. L’Ufficio segreteria procede alla registrazione, fanno eccezione i verbali di campionamento che vengono gestiti dal servizio accettazione per la codifica (PG/015).
La registrazione avviene in un protocollo generale, dove al documento viene apposto il numero di protocollo, la data di ricevimento, e vengono annotati gli estremi identificativi (richiedente, protocollo di provenienza, oggetto).
La posta viene quindi consegnata al RL, in apposita carpetta custodita dalla segreteria. Il RL, prende visione della documentazione già protocollata, e predispone la lista di distribuzione. La corrispondenza in busta chiusa, viene visionata dal RL e consegnata al personale di segreteria per la registrazione, portando già l’annotazione del destinatario. Per la documentazione prodotta per l’esterno, il numero di protocollo viene assegnato nel momento dell’emissione, dopo l’approvazione da parte del RL o suo sostituto. Lista di distribuzione
Il personale di segreteria, presa visione di quanto disposto dal RL, compila la lista di distribuzione, annotando: numero di protocollo del documento da distribuire, l’oggetto del documento, il nominativo del destinatario, firma del compilatore.
Il documento viene consegnato al destinatario che firma per ricevuta, apponendo la data di ritiro del documento in oggetto. La copia del documento e la lista di distribuzione vengono quindi archiviati negli appositi armadi, custoditi dalla segreteria. Non viene prodotta la lista di distribuzione per quei documenti divulgativi o di cui basta prenderne visione, in tal caso è sufficiente l’indicazione da parte del RL del destinatario. I documenti d’interesse generale, vengono affissi in bacheca o notificati dal RL attraverso comunicazione scritta, dando la possibilità di prendere visione dell’intera documentazione presso la segreteria.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 21 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
Aggiornamento della documentazione esterna soggetta a revisione
Le figure addette alla verifica e revisione degli aggiornamenti sono identificati nella tabella sottostante:
Documentazione Responsabilità Documentazione aziendale Segreteria Documentazione relativa alle attività (Norme – metodi – regolamenti - disposizioni)
RAQ/RUO con cadenza trimestrale
L’aggiornamento delle Norme viene controllato con cadenza trimestrale dai RUO e riportato nelle
relazioni trimestrali che vengono redatte da ciascuna UOS al fine di formalizzare la richiesta di acquisto ove necessaria. Tutte le informazioni al riguardo sono definite nelle Procedure Gestionali.
• PG/003: “Modalità di gestione della documentazione e dei dati informatici” che viene inviata all’URP per la pubblicazione sul sito Aziendale
• PG/001: “Modalità per la stesura, identificazione, approvazione, emissione, revisione e distribuzione delle PG e PO”
• PG/002: “Modalità per la stesura, approvazione, autorizzazione e revisione del Manuale della Qualità”. • PG/013: “Modalità per la stesura emissione e revisione dei metodi di prova”
4.4. RIESAME DELLE RICHIESTE, DELLE OFFERTE E DEI CONTRATTI ( UNI CEI EN ISO/IEC 17025:2005 / RT-08– 4.4.1-4.4.2 - 4.4.3 - 4.4.4 - 4.4.5/ RT-08 – 4.4.1-4.4.2 - 4.4.3 - 4.4.4 - 4.4.5 -6.2 )
Il LSP effettua attività di prova, sia per l’Azienda di appartenenza, che per i “clienti” che si avvalgono dell’intervento del laboratorio. I clienti più frequenti sono:
1. Aziende Sanitarie Provinciali
2. Assessorato Regionale per la salute ( Ispettorato Sanitario)
3. Organismi di controllo istituzionali ( NAS- NOE- altro)
4. Procura
5. Privati
Le attività di prova svolte per l’Azienda sono essenzialmente quelle previste dal D.A. 06.08.2004 e sono quando possibile, anticipate da documento programmatico dagli organi competenti:
- controllo ufficiale dei prodotti alimentari e bevande - acqua per il consumo umano - acque per la balneazione - indagini epidemiologiche (Legionella) - controlli per la ricerca di amianto
Mentre per i restanti clienti, le attività sono definite nelle singole richieste o in specifici contratti stipulati ad hoc con la ASP di Palermo tenuto conto delle decisioni adottate da parte del Dipartimento Risorse Umane Sviluppo Organizzativo e Affari Generali.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 22 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
Il Laboratorio ha stabilito pertanto, i criteri per il riesame delle richieste, delle offerte e dei contratti, in modo da poter assicurare al contraente: 1. che i metodi utilizzati siano adeguati alla richiesta, documentati, rispondenti a norme e metodi ufficiali 2. che il laboratorio abbia la capacità e le risorse professionali e strumentali per eseguire le prove 3. che i metodi di prova siano appropriati a soddisfare la richiesta del cliente 4. che i tempi di consegna dei RDP siano congrui alle richieste in relazione alla loro complessità e/o tempistica di esecuzione degli esami analitici richiesti 5. che siano applicati gli importi registrati nel Tariffario Regionale ( G.U.R.S. 18.06.2004 n. 26 e/o eventuali aggiornamenti) 6. che il cliente venga avvertito, su possibili scostamenti da quanto concordato, anche prevedendo la riformulazione del contratto in caso di aggiornamenti di norme e/o disposizioni subentranti.
4.5 SUBAPPALTO DELLE PROVE E DELLE TARATURE (UNI CEI EN ISO/IEC 17025:2005 – RT-08 – 4.5.1 – 4.5.2 – 4.5.3 – 4.5.4)
Il Laboratorio esegue in generale tutte le prove analitiche richieste e non sub-appalta di regola alcuna attività, pertanto non si applicano i punti da 4.5.1 a 4.5.4 della Norma. Nel caso in cui il cliente dovesse chiedere l’effettuazione di prove che il laboratorio non è in grado di eseguire, lo stesso, procede ad assistere il cliente, unicamente per venire incontro alle esigenze, collaborando nell’individuazione della struttura idonea, in grado di assicurare i requisiti richiesti per la specifica attività. Nell’ambito dell’applicazione dei piani regionali per la sicurezza alimentare, potrebbe verificarsi la necessità di conferire campioni ad altro laboratorio competente, in questo caso però non si tratta di subappalto, ma di attività assegnata per decreto o disposizione assessoriale ad altro laboratorio competente, che emette rapporto di prova e risponde direttamente dell’attività analitica.
4.6 APPROVVIGIONAMENTO DI SERVIZI E FORNITURE (UNI CEI EN ISO/IEC 17025:2005 – RT-08 - 4.6.1 – 4.6.2 – 4.6.3 )
Gli approvvigionamenti di materiali specifici, di consumo, e di apparecchiature, vengono effettuati secondo quanto stabilito e deliberato dal Direttore Generale, con il decentramento delle funzioni amministrative articolate all’interno dei Dipartimenti: Regolamento per l’acquisizione in economia di beni e servizi (del 280/13.3.2014)
Per quanto riguarda i reagenti e i prodotti consumabili per analisi, il Laboratorio comunica il fabbisogno annuale al Dipartimento di Diagnostica di Laboratorio, detentore del budget specifico, mentre si rivolge al Dipartimento Appalti e Forniture per l’acquisto di strumentazioni o di beni in economia, previa autorizzazione del DP (per dipendenza gestionale) e del DDL in quanto responsabile degli approvvigionamenti. In base alle richieste inoltrate, i Dipartimenti espletano gare pubbliche o trattative private. Le richieste inoltrate dal Laboratorio, precisano quantità e caratteristiche dei prodotti in base alle previsioni di analisi. Il Dipartimento di Diagnostica di Laboratorio, acquisita l’autorizzazione e dopo la dovuta registrazione, inoltra le richieste al servizio economale, che attiva le indagini di mercato, in base all’elenco fornitori qualificati predisposto dal laboratorio. Per i dettagli si rimanda alla PO/044 “Modalità per l’approvvigionamento di beni e servizi”.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 23 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0 Per assicurare il mantenimento della qualità delle prove e tarature, il LSP ha sviluppato una
procedura per il controllo dei materiali critici e materiali di riferimento:
- al ricevimento della fornitura: vengono verificati, conformità della bolla di accompagnamento alla richiesta, quantità (acconto/saldo), temperatura di trasporto. - all’apertura delle forniture, all’interno delle singole UOS, vengono verificati conformità dei singoli prodotti, lotto/scadenza, certificati di qualità, schede di sicurezza , ove previste, stato dei materiali.
Nel caso di non conformità alla consegna, i prodotti vengono restituiti alla farmacia territoriale con eventuale nota di reclamo, nel caso di non conformità dopo immagazzinamento, la stessa viene gestita attraverso la registrazione, l’analisi delle cause e l’eventuale correzione/azione correttiva. Per i dettagli si rimanda alla PG/011.
QUALIFICA DEI FORNITORI ( UNI CEI EN ISO/IEC 17025:2005 – 4.6.4/RT-08 – 4.6.4)
Per l’espletamento delle gare e delle trattative private, l’Azienda ha elaborato un albo fornitori, al quale attinge di volta in volta secondo necessità. Il Laboratorio, a sua volta, ha proceduto a qualificare i propri fornitori, secondo i criteri stabiliti nella procedura PG/010 “Qualifica fornitori” dove sono riportati i requisiti minimi ritenuti indispensabili e i punteggi assegnati. Utilizzando tale strumento, il Laboratorio ha prodotto un elenco che mette in relazione: fornitori, prodotti e/o strumenti e punteggi ottenuti.
L’elenco dei fornitori qualificati viene aggiornato con cadenza annuale, in base alla qualità dei servizi
ricevuti, e serve anche come riferimento per l’elenco dei fornitori aziendali. Per i dettagli si rimanda alla PG/010.
4.7 SERVIZI AL CLIENTE ( UNI CEI EN ISO/IEC 17025:2005- RT-08 – 4.7.1 – 4.7.2 /RT-08– 4.7.1 – 4.7.2)
Il Laboratorio e ogni UOS si interfaccia con clienti diversi. La loro tipologia si può suddividere in: clienti interni (esempio SIAN - SISPE - SIAV, Personale di vigilanza sanitaria etc.), clienti esterni sia istituzionali (esempio NAS, autorità giudiziaria, etc.) che privati richiedenti prove di laboratorio ad esclusivo uso personale purché non in contrasto con i compiti istituzionali.
Le UOS durante l’orario di lavoro, forniscono assistenza ai clienti sia direttamente che tramite via
telefonica. Il personale di vigilanza deputato al campionamento, viene ricevuto dall’Ufficio Accettazione
Campioni così come descritto nella Sezione 5 capitolo 6.1. Solo per le attività ambulatoriali come l’ispettorato micologico afferente al LSP, sono previsti giorni e fasce orarie di ricevimento.
Per quanto riguarda i privati che si avvalgono del diritto di presenziare alla esecuzione delle prove su invito a garanzia del diritto alla difesa, è cura del RUO o suo incaricato, di informarli sulle procedure di sicurezza adottate, sui metodi di prova utilizzati, garantendo contestualmente, il rispetto della riservatezza dei dati propri e delle altre attività in corso.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 24 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0 Il Personale della Accettazione ha il compito di fornire al cliente, gli idonei mezzi di protezione
individuale, e di accompagnarlo presso il laboratorio dove si svolgeranno le prove, dopo la consegna di un pass registrando contestualmente nominativo e motivazione dell’accesso ai locali del Laboratorio, trattenendo un documento identificativo, che viene restituito alla fine della visita. Per valutare l’efficacia del servizio prestato, il laboratorio sottopone al cliente un questionario sul grado di soddisfazione.
4.8 GESTIONE DEI RECLAMI ( UNI CEI EN ISO/IEC 17025:2005 – 4.8/ RT-08 4.8)
La ASP di Palermo ha regolamentato la gestione dei reclami al suo interno con la Delibera 623 del 23/12/2013 “Sistema gestione segnalazioni reclami” che regola la gestione dei reclami con gli utenti esterni tramite Ufficio Relazione col Pubblico Aziendale.
Per quanto riguarda la gestione di reclami o disservizi con le altre Unità Operative aziendali, per procedere alla risoluzione delle problematiche che possono comunque occorrere, il Laboratorio ha sviluppato una specifica procedura gestionale cui si rimanda PG/006.
4.9 TENUTA SOTTO CONTROLLO DELLE ATTIVITA’ DI PROVA E/O DI TARATURA NON CONFORMI Gestione delle non conformità e azioni correttive ( UNI CEI EN ISO/IEC 17025:2005 – 4.9.1-4.9.2-4.11.1-4.11.2-4.11.3-4.11.4/ RT-08– 4.9.1-4.9.2-4.11.1-4.11.2-4.11.3-4.11.4-6.5 )
Il laboratorio attua una politica e delle procedure, per ridurre quanto più possibile il verificarsi di non conformità durante l’attività di prova e/o taratura: ciascun operatore che rileva “non conformità”, ha l’obbligo di compilare l’apposita scheda e segnalare al RUO competente e al RAQ, tutte le informazioni che possono essere utili per individuare gli inconvenienti rilevati, per avviare quelle azioni atte a correggere e prevenire successive non conformità. La politica e le procedure devono assicurare che: 1. siano individuate le responsabilità e le autorità per la gestione delle attività non conformi e per il trattamento correttivo della non conformità 2. sia effettuata l’analisi delle cause e una valutazione dell’importanza della non conformità, attraverso la verifica delle attività coinvolte, delle prove che possono essere state influenzate e dei RP emessi che riportano informazioni al cliente potenzialmente errate. Secondo la gravità della non conformità, può essere prevista l’interruzione dell’attività coinvolta, in tal caso si procede ad informare il cliente, qualora la non conformità abbia influenzato prove da lui richieste, ritirando, ove necessario, i relativi rapporti di prova. Inoltre si procede alla comunicazione alle autorità competenti in merito alla prova oggetto di interruzione, nonché all’Ente di Accreditamento Accredia se fosse necessario sospendere le attività di prova accreditate. 3. siano adottate tempestive azioni correttive, o che sia accettata la non conformità, in quanto non pregiudizievole per le prove e/o tarature 4. vengano emessi i rapporti di prova sostitutivi, nel caso di non conformità che possono pregiudicare l’utilizzo degli stessi o l’interpretazione dei risultati. 5. sia identificata la responsabilità di chi autorizza la ripresa o la continuità delle attività coinvolte dalla non conformità 6. siano attivati eventuali monitoraggi delle azioni correttive 7. sia valutata l’efficacia dell’azione correttiva

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 25 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
Per quanto riguarda le non conformità rilevate in corso di Audit interni o esterni, è cura del RL redigere le schede di non conformità in riferimento ai rilievi. Le modalità sono descritte nella PG/005 “Criteri generali per la verifica del sistema qualità e la gestione delle Non Conformità ed azioni correttive”.
4.10 MIGLIORAMENTO E GESTIONE DEI CAMBIAMENTI ( UNI CEI EN ISO/IEC 17025:2005 – 4.10 RT-08 -4.10)
Il sistema di gestione della qualità, è progettato in modo da garantire un miglioramento continuo, tenendo conto delle esigenze di tutte le parti coinvolte. In genere, durante il riesame della Direzione, in corso di audit interni ed esterni, vengono formulate necessità di miglioramento del sistema nella sua totalità o di attività specifiche. Il miglioramento, che rappresenta l’obiettivo permanente del laboratorio, deve tenere conto di alcuni aspetti fondamentali, quali: le esigenze del cliente, la soddisfazione di ritorno (qualità percepita), il coinvolgimento del personale, la gestione attraverso procedure, la valutazione delle risorse, la formulazione degli obiettivi attraverso l’analisi delle attività, l’identificazione dei criteri per stabilire l’efficacia del miglioramento, l’identificazione delle responsabilità e dei tempi di realizzazione. Per quanto riguarda la gestione dei cambiamenti, il Laboratorio procede alla valutazione e pianificazione del cambiamento da concretizzare, garantendo nel contempo l’integrità del sistema di qualità e orientando le scelte e le modifiche in modo da produrre un miglioramento delle attività/servizi erogati. Per i dettagli si rimanda alla PG/004 “Il Miglioramento continuo del sistema di qualità e la gestione dei cambiamenti, dei punti critici e le azioni preventive”.
4.11 AZIONI CORRETTIVE ( UNI CEI EN ISO/IEC 17025:2005 4.11.1- 4.11.2 – 4.11.3 – 4.11.4 – 4.11.5/ RT 08 05 4.11.1- 4.11.2 – 4.11.3 – 4.11.4 – 4.11.5)
Il laboratorio dispone di specifica procedura (PG/005) per la gestione delle non conformità e azioni correttive. Queste vengono attuate, successivamente ad una attenta analisi delle cause, al fine di eliminare la possibilità che la medesima non conformità si ripresenti. La ricerca delle cause che hanno determinato la non conformità risulta a volte complessa e non sempre determinabile, sia perché possono presentarsi più fattori causali, sia perché le cause possono non essere imputabili al laboratorio stesso. La procedura adottata dal laboratorio identifica le responsabilità, in base alla fase applicativa, le figure deputate all’attuazione dell’azione correttiva, l’attivazione di eventuali monitoraggi o di Audit suppletivi, e valutazioni dell’efficacia di risoluzione. Le registrazioni delle non conformità e delle azioni correttive costituiscono oggetto di riesame della Direzione.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 26 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
4.12 AZIONI PREVENTIVE ( UNI CEI EN ISO/IEC 17025:2005 – 4.12.1-4.12.2 RT-08 – 4.12.1-4.12.2 )
Nel corso di Audit interni o esterni, durante il riesame della direzione, o nel corso dell’attività, possono essere messi in luce dei punti deboli del SQ che potrebbero essere potenziali cause di non conformità. Il SQ prevede in questi casi, l’analisi dei punti critici e delle potenziali cause di non conformità, pianificando le azioni preventive atte ad anticipare l’insorgenza di tali eventi: • Tipo di azione • Identificazione del responsabile dell’attuazione • Inizio dell’azione e previsione del tempo di realizzazione • Monitoraggio • Responsabile del monitoraggio • Valutazione dell’efficacia/efficienza dell’azione preventiva • Approvazione e chiusura dell’azione preventiva, o scelta di altra azione preventiva in sostituzione di azione non efficace. Le azioni preventive rappresentano un miglioramento del sistema di qualità. Vedi PG/004 “Il Miglioramento continuo del sistema di qualità e la gestione dei cambiamenti, dei punti critici e le azioni preventive”
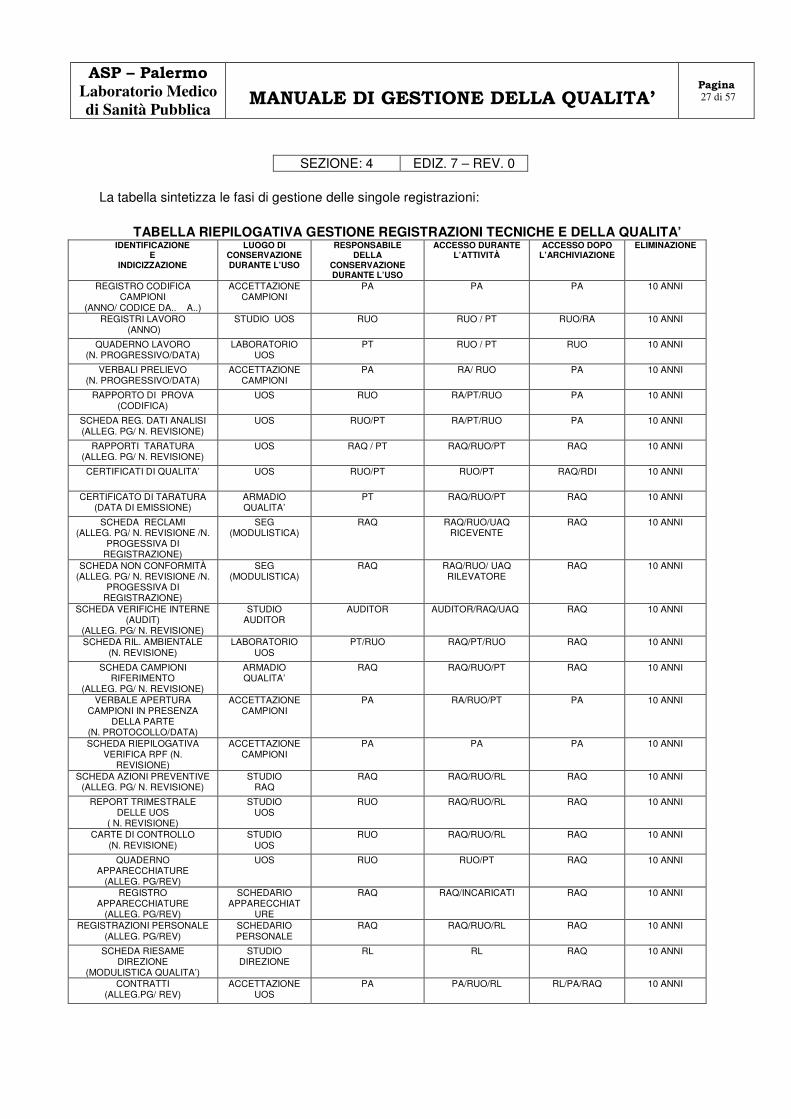
4.13 TENUTA SOTTO CONTROLLO DELLE REGISTRAZIONI ( UNI CEI EN ISO/IEC 17025:2005 /RT-08: 4.13.1.1 - 4.13.1.2 - 4.13.1.3 - .4.13.1.4.- 4.13.4 – 4.13.2.1. – 4.13.2.2. – 4.13.2.3 )
Il laboratorio ha elaborato delle procedure per il controllo delle registrazioni tecniche e relative alla qualità, individuando un sistema d’identificazione, di raccolta, d’indicizzazione, di accesso, di archiviazione e di eliminazione.
Tutte le registrazioni sono catalogate in maniera univoca, permettendo un facile reperimento delle
stesse, custodite in locali, idonei ad impedire il deterioramento dei documenti, e dotati di controllo di accesso. Tutte le registrazioni relativa alla qualità, compreso i rapporti di prova e i certificati di taratura e tutta la documentazione correlata: fogli di lavoro, verbali di campionamento, corrispondenze, sono custodite secondo prontuario di scarto delle aziende sanitarie (in genere 10 anni), e accessibili ai percorsi di Audit. Secondo il tipo di registrazione, la stessa viene gestita in copia cartacea o informatica. Gli errori inerenti le registrazioni su copia cartacea, non vengono eliminati ma barrati, e controfirmati dal responsabile della correzione. Il RUO convalida in genere la correzione apportata. Dove possibile va annotato il motivo della correzione.
ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 27 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
La tabella sintetizza le fasi di gestione delle singole registrazioni:
TABELLA RIEPILOGATIVA GESTIONE REGISTRAZIONI TECNICHE E DELLA QUALITA’ IDENTIFICAZIONE
E INDICIZZAZIONE
LUOGO DI CONSERVAZIONE DURANTE L’USO
RESPONSABILE DELLA
CONSERVAZIONE DURANTE L’USO
ACCESSO DURANTE L’ATTIVITÀ
ACCESSO DOPO L’ARCHIVIAZIONE
ELIMINAZIONE
REGISTRO CODIFICA CAMPIONI
(ANNO/ CODICE DA.. A..)
ACCETTAZIONE CAMPIONI
PA PA PA 10 ANNI
REGISTRI LAVORO (ANNO)
STUDIO UOS RUO RUO / PT RUO/RA 10 ANNI
QUADERNO LAVORO (N. PROGRESSIVO/DATA)
LABORATORIO UOS
PT RUO / PT RUO 10 ANNI
VERBALI PRELIEVO (N. PROGRESSIVO/DATA)
ACCETTAZIONE CAMPIONI
PA RA/ RUO PA 10 ANNI
RAPPORTO DI PROVA (CODIFICA)
UOS RUO RA/PT/RUO PA 10 ANNI
SCHEDA REG. DATI ANALISI (ALLEG. PG/ N. REVISIONE)
UOS RUO/PT RA/PT/RUO PA 10 ANNI
RAPPORTI TARATURA (ALLEG. PG/ N. REVISIONE)
UOS RAQ / PT RAQ/RUO/PT RAQ 10 ANNI
CERTIFICATI DI QUALITA’ UOS RUO/PT RUO/PT RAQ/RDI 10 ANNI
CERTIFICATO DI TARATURA (DATA DI EMISSIONE)
ARMADIO QUALITA’
PT RAQ/RUO/PT RAQ 10 ANNI
SCHEDA RECLAMI (ALLEG. PG/ N. REVISIONE /N.
PROGESSIVA DI REGISTRAZIONE)
SEG (MODULISTICA)
RAQ RAQ/RUO/UAQ RICEVENTE
RAQ 10 ANNI
SCHEDA NON CONFORMITÀ (ALLEG. PG/ N. REVISIONE /N.
PROGESSIVA DI REGISTRAZIONE)
SEG (MODULISTICA)
RAQ RAQ/RUO/ UAQ RILEVATORE
RAQ 10 ANNI
SCHEDA VERIFICHE INTERNE (AUDIT)
(ALLEG. PG/ N. REVISIONE)
STUDIO AUDITOR
AUDITOR AUDITOR/RAQ/UAQ RAQ 10 ANNI
SCHEDA RIL. AMBIENTALE (N. REVISIONE)
LABORATORIO UOS
PT/RUO RAQ/PT/RUO RAQ 10 ANNI
SCHEDA CAMPIONI RIFERIMENTO
(ALLEG. PG/ N. REVISIONE)
ARMADIO QUALITA’
RAQ RAQ/RUO/PT RAQ 10 ANNI
VERBALE APERTURA CAMPIONI IN PRESENZA
DELLA PARTE (N. PROTOCOLLO/DATA)
ACCETTAZIONE CAMPIONI
PA RA/RUO/PT PA 10 ANNI
SCHEDA RIEPILOGATIVA VERIFICA RPF (N.
REVISIONE)
ACCETTAZIONE CAMPIONI
PA PA PA 10 ANNI
SCHEDA AZIONI PREVENTIVE (ALLEG. PG/ N. REVISIONE)
STUDIO RAQ
RAQ RAQ/RUO/RL RAQ 10 ANNI
REPORT TRIMESTRALE DELLE UOS
( N. REVISIONE)
STUDIO UOS
RUO RAQ/RUO/RL RAQ 10 ANNI
CARTE DI CONTROLLO (N. REVISIONE)
STUDIO UOS
RUO RAQ/RUO/RL RAQ 10 ANNI
QUADERNO APPARECCHIATURE
(ALLEG. PG/REV)
UOS RUO RUO/PT RAQ 10 ANNI
REGISTRO APPARECCHIATURE
(ALLEG. PG/REV)
SCHEDARIO APPARECCHIAT
URE
RAQ RAQ/INCARICATI RAQ 10 ANNI
REGISTRAZIONI PERSONALE (ALLEG. PG/REV)
SCHEDARIO PERSONALE
RAQ RAQ/RUO/RL RAQ 10 ANNI
SCHEDA RIESAME DIREZIONE
(MODULISTICA QUALITA’)
STUDIO DIREZIONE
RL RL RAQ 10 ANNI
CONTRATTI (ALLEG.PG/ REV)
ACCETTAZIONE UOS
PA PA/RUO/RL RL/PA/RAQ 10 ANNI

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 28 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
Ciascuna attività organizzativa (UOS – servizi in staff alla Direzione) che gestisce registrazioni tecniche, è dotata di terminali che coadiuvano l’esercizio delle funzioni attribuite. Non disponendo di un sistema unico di collegamento (intranet), ciascuna attività, utilizza specifici file per facilitare la gestione delle proprie competenze.
Mentre all’interno delle singole UOS vengono aggiornati i dati prodotti dalle attività di reparto, salvati
dopo l’immagazzinamento nel supporto magnetico fisso, e prodotti in formato cartaceo, secondo il tipo di documento, il Responsabile gestione dati informatici, in garanzia della tracciabilità dei dati, provvede con cadenze definite, al salvataggio progressivo dei file gestiti dalle UOS su apposito supporto magnetico. La tabella sottostante sintetizza responsabilità e tempi di intervento.
TIPOLOGIA DI
REGISTRAZIONE
ATTIVITÀ
COINVOLTA
RESPONSABILE SALVATAGGIO DATI
DA PARTE DEL RESPONSABILE
(RDI)
REGISTRI CAMPIONI UOS RUO TRIMESTRALE MAGAZZINO DI CARICO E
SCARICO UOS PT TRIMESTRALE
FOGLI DI CALCOLO – CARTE DI CONTROLLO (*)
UOS RUO/RAQ CARTACEO
TRIMESTRALE
RENDICONTAZIONE PER L’AZIENDA (*)
(PRESENZA/ASSENZA-INDENNITÀ ACCESSORIE)
SEGRETERIA PS TRIMESTRALE
RENDICONTAZIONE PER L’AZIENDA (*)
(RENDICONTAZIONE MENSILE- ATTIVITÀ
PRODUTTIVE)
STAFF - RL UOS TRIMESTRALE
FLUSSI INFORMATIVI (MINISTERO-
ASSESSORATO)
UOS RESPONSABILE FLUSSI UOS
TRIMESTRALE
DOCUMENTI DEL SQ : MQ (*) STAFF-RL RAQ SECONDO REVISIONE DOCUMENTI DEL SQ :
MP-PG-PO-ALLEGATI (*) STAFF-RL RUO SECONDO REVISIONE
DOCUMENTI DEL SQ : MODULISTICA (*)
STAFF-RL RAQ/RUO CARTACEO
SECONDO REVISIONE
DOCUMENTI DEL SQ : SCHEDE PERSONALE (*)
STAFF-RL RAQ CARTACEO
CERTIFICATI DI QUALITÀ PRODOTTI
UOS PT TRIMESTRALE
ELENCO FORNITORI (*) STAFF-RL RAQ SECONDO REVISIONE RAPPORTI DI PROVA (*) UOS RUO/DIR TRIMESTRALE
ELENCHI (PG-PO-MP) TUTTE RAQ SECONDO REVISIONE REPORT STRUMENTALI UOS PT TRIMESTRALE
Per i dettagli si rimanda alla PG/007 “Gestione delle registrazioni tecniche e della qualità” e PG/003
“Gestione della documentazione e dei dati informatici”.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 29 di 57
SEZIONE: 4 EDIZ. 7 – REV. 0
4.14 AUDIT INTERNI (UNI CEI EN ISO/IEC 17025:2005 / RT-08 – 4.14.1-4.14.2-4.13.3-4.14.4-4.14.5 )
Per garantire un controllo continuo del SQ, il RL, indipendentemente alle verifiche esterne, con cadenza almeno annuale, pianifica le verifiche interne, commissionando al RAQ e all’Auditor l’elaborazione del programma.
Le verifiche, predisposte dal gruppo di audit, previo avviso all’attività coinvolta, devono accertare che
gli elementi del sistema, presi di volta in volta in esame, rispondano ai punti della Norma UNI CEI EN ISO/IEC 17025:2005 e al Regolamento RT-08 di Accredia, tenendo conto delle carenze evidenziate negli audit precedenti.
Il gruppo di audit è formato da personale del Laboratorio che si occupa, ove possibile, di attività diverse
da quelle sottoposte alla verifica stessa, e ha ricevuto dal RAQ adeguata formazione sulla norma UNI EN ISO /IEC 17025:2005. Il gruppo di Audit coadiuva e supporta l’Auditor interno che è in possesso di attestato di formazione specifica come da norma UNI EN ISO 19011:2012.
Le risultanze dell’Audit vengono registrate e comunicate al committente (RL) e le eventuali non
conformità vengono annotate sugli appositi moduli, per attivare l’analisi delle cause e le eventuali azioni correttive.
In base alle risultanze, possono essere commissionati Audit supplementari. Il RAQ, assicura che le
verifiche si svolgano secondo il programma stabilito, in accordo alla procedura PG/005 “Criteri generali per la verifica del sistema qualità e la gestione delle Non Conformità ed azioni correttive”.
4.15 RIESAME DELLA DIREZIONE ( UNI CEI EN ISO/IEC 17025:05 – RT-08 – 4.15.1 – 4.15.2 )
Il Riesame della Direzione, condotto con cadenza annuale, rappresenta il momento di sintesi del Sistema di Qualità, permette la valutazione di quanto implementato nell’anno in corso, e pone le basi per la formulazione degli obiettivi di miglioramento. Esso scaturisce dall’esame delle singole attività o da riesami parziali, e permette di evidenziare i punti deboli del sistema, di introdurre eventuali azioni correttive e preventive, ed elaborare il nuovo piano di miglioramento. Riesami suppletivi sono condotti dal RL sia a seguito della valutazione esterna annuale, che in seguito ad Audit interno, trattando adeguatamente i punti oggetto di osservazione/non conformità. Il riesame annuale deve tenere conto di:
� Adeguatezza della politica di qualità � Idoneità delle procedure � Relazione del RAQ � Rapporti o relazioni dei responsabili di
funzioni � Esiti audit interni � Valutazioni esterne
� Azioni correttive e azioni preventive � Risultati delle prove comparative
interlaboratorio e intralaboratorio � Reclami � Informazione di ritorno da parte del cliente � Variazioni carichi di lavoro � Variazioni di tipologia di lavoro

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 30 di 57
� Piani di miglioramento � Addestramento e formazione del personale
� Risorse del laboratorio (umane – strumentali - approvvigionamenti).
SEZIONE: 4 EDIZ. 7 – REV. 0
Ad analisi ultimata, il RL riassume quanto sottoposto a riesame, compilando una scheda riepilogativa, che mostra in maniera sintetica, i punti di forza del laboratorio e permette di evidenziare i punti critici rilevati.
Dopo la stesura del report, il RL, notifica per conoscenza al Dipartimento di Prevenzione quanto rilevato, e
congiuntamente riunisce il personale, per la verbalizzazione del riesame al fine di esplicitare gli scopi, gli obiettivi e i piani di azione per la gestione del sistema qualità per l’anno successivo. Durante tale incontro vengono analizzati: • quanto evidenziato dall’analisi dei documenti sottoposti a valutazione. • i punti di debolezza messi in evidenza • i miglioramenti raggiunti e i punti di forza verificati
Inoltre vengono definiti i criteri di realizzazione, i mezzi necessari, le responsabilità, i tempi di attuazione e gli indicatori di efficacia o efficienza. Per quanto riguarda la gestione delle non conformità, delle azioni preventive e del miglioramento, si rimanda alle procedure PG/005, PG/004.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 31 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0
INDICE :
Cap. Rev. Titolo
5.1 0 Requisiti tecnici
5.2 0 Personale
5.3 0 Luogo di lavoro e condizioni ambientali
5.4 0 Metodi di prova e taratura e validazione dei metodi
5.5 0 Apparecchiature
5.6 0 Riferibilità delle misure
5.7 0 Campionamento
5.8 0 Manipolazione degli oggetti da sottoporre a prova o taratura
5.9 0 Assicurazione della qualità dei risultati di prova e taratura
5.10 0 Presentazione dei risultati
5.1 REQUISITI TECNICI (UNI CEI EN ISO/IEC 17025:2005- RT-08 – 5.1.1 – 5.1.2 )
La correttezza delle prove e tarature è influenzata da numerosi fattori, i quali pur se in misura differente, apportano un proprio contributo di “imprecisione”. La somma di tali fattori, determina l’incertezza estesa del misurato. Nella sezione 5 del presente Manuale, vengono analizzati i contributi più rilevanti, a partire da quello umano, fino a quello strumentale e ambientale. La conoscenza dei fattori che influenzano le prove e le tarature è indispensabile per la validazione dei metodi di prova e per una corretta taratura strumentale: le procedure adottate dal laboratorio consentono di tenere sotto controllo tali fattori e di quantificare singolarmente o globalmente, il grado di influenza sulle prove.
5.2 PERSONALE ( UNI CEI EN ISO/IEC 17025:2005 – RT-08 -5.2.1 – 4.1.5) Il Laboratorio dispone di un organigramma funzionale di dipendenza gerarchica come riportato
nell’allegato n.2. E’ presente inoltre di un organigramma nominale in cui è specificata l’appartenenza del personale alle aree di pertinenza del Laboratorio.
QUALIFICA DEL PERSONALE (UNI CEI EN ISO/IEC 17025:2005-RT-08 5.2.1 – 5.2.5)
FIGURE PROFESSIONALI: COMPITI, RESPONSABILITÀ
� IL RESPONSABILE DEL LABORATORIO (RL):
Il RL viene scelto dal DG dell’azienda, in base ai criteri stabiliti in conformità al DPR 484/97. � è responsabile della gestione del Laboratorio, dell’organizzazione e funzionamento in conformità ai criteri

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 32 di 57
generali stabiliti dalla Norma UNI CEI EN ISO/IEC 17025:2005, e dal regolamento ACCREDIA RT-08, è inoltre responsabile, per quanto di competenza, della conformità a quanto richiesto dal Reg. CE 882/04 e delle disposizioni ad esso correlate
SEZIONE: 5 EDIZ. 7 – REV. 0 � garantisce l’integrità del sistema di qualità, e promuove il miglioramento continuo attraverso obiettivi specifici � garantisce, in base alle risorse disponibili, gli obiettivi di budget assegnati dall’azienda, sia quali obiettivi annuali,
correlati al sistema premiante, sia triennali � garantisce la gestione delle non conformità e dei reclami e ne approva le risoluzioni � verifica e approva il MQ, le PG e PO emesse dal Laboratorio; garantendone l’idoneità e l’aggiornamento � assicura l’esistenza di un sistema di gestione, dalla registrazione all’archiviazione dei dati analitici e della
documentazione � attua la politica della Qualità stabilita dal DG dell’Azienda; � nomina il RAQ; � individua le responsabilità di chi esegue le prove e tarature, chi pianifica le prove e tarature e valuta i risultati,
chi esprime interpretazioni dei dati analitici, chi propone, valida e sviluppa modifiche ai metodi di prova. � Formalizza, con disposizione di servizio, a ciascun operatore, le proprie mansioni e responsabilità � attesta la validità delle prove, verificandone annualmente il mantenimento � garantisce la formazione e l’aggiornamento del personale, attraverso programmi di formazione interni. Notifica inoltre, con cadenza annuale, le necessità formative al Dipartimento di Prevenzione � abilita il personale ad effettuare prove e tarature e ad utilizzare le specifiche attrezzature, garantendo l’idoneità di quanto necessario � apponendo la propria firma, è responsabile di tutti i documenti del SQ � commissiona gli audit interni e riesamina, annualmente, il SQ � formalizza i contratti col cliente e riesamina annualmente le richieste e le offerte � garantisce l’incolumità dei lavoratori e la sicurezza dei luoghi di lavoro (per le funzioni delegate dal Responsabile della Sicurezza aziendale) � nomina la persona che in sua assenza è delegata a svolgere specifiche funzioni
� IL RESPONSABILE ASSICURAZIONE DELLA QUALITÀ (RAQ):
viene individuato dal RL, deve essere in possesso di titolo di studi in materie scientifiche e avere effettuato specifica formazione sulla Norma UNI CEI EN ISO/IEC 17025:2005, integrata dai regolamenti ACCREDIA. � collabora con il RL, dal quale dipende direttamente, nel predisporre il SQ � ha il compito della stesura e della distribuzione controllata del MQ e della verifica e distribuzione delle PG e PO e MP � collabora con il RL e RUO nella gestione delle non conformità e azioni correttive e nella gestione dei reclami � convalida il piano di formazione del personale; e provvede alla formazione interna del SQ � mantiene copia di tutti i documenti relativi al SQ. � verifica con il RUO i requisiti del personale per l’abilitazione alle prove e all’uso degli strumenti � collabora con il RL e i RUO per il programma di controllo di qualità � verifica con il RUO la validazione dei metodi e il mantenimento dei requisiti di validazione � partecipa insieme all’UAQ alla verifica periodica del SQ, programmando con l’Auditor i percorsi di audit � aggiorna l’elenco dei documenti del SQ � redige e aggiorna il programma di manutenzione e taratura degli strumenti � gestisce la documentazione del personale relativa al SQ

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 33 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0
� IL SOSTITUTO DEL RL
Viene individuato fra i dirigenti del Laboratorio, e provvede: � all’ordinaria gestione del laboratorio, continuando ad espletare i compiti della propria funzione � al controllo della regolare attuazione del SQ � ai rapporti con terzi che riguardano l’ordinaria amministrazione � a riferire al rientro del RL, le attività svolte e quanto in corso di attuazione
� IL SOSTITUTO DEL RAQ
Viene individuato dal RAQ fra i componenti dell’UAQ, deve avere specifica formazione sulla Norma UNI CEI EN ISO/IEC 17025:2005 e deve affiancare il RAQ per un periodo di addestramento. In assenza del titolare si occupa della gestione ordinaria della Qualità, mantenendo compiti e funzioni preesistenti.
� IL RESPONSABILE DI UNITÀ OPERATIVA SEMPLICE (RUO)
E’ nominato dal DG con incarico a tempo determinato scelto da una rosa di idonei selezionati tramite concorso interno. Il RUO deve assicurare: � programmazione ed organizzazione delle attività dell’UO � la supervisione del personale tecnico assegnato; � l’efficienza delle apparecchiature assegnate; � la scelta dei metodi di prova, la verifica dell’applicabilità, la stesura dei metodi di prova interni e delle procedure
di prova integrativa; e delle PO inerenti le specifiche attività � la validazione dei metodi di prova e il mantenimento dei requisiti di validazione � l’addestramento del personale, qualificazione e aggiornamento � verifica con il RAQ i requisiti del personale per l’abilitazione alle prove e all’uso degli strumenti � la stesura delle PG su incarico del RL � attuazione delle prescrizioni contenute nel MQ, PG, PO � rispetto delle norme vigenti in materia di salute e sicurezza sul lavoro � collabora con il RL e il RAQ per il programma di controllo di qualità � la pianificazione degli acquisti � la relazione trimestrale delle attività e delle esigenze � la verifica e l’eventuale aggiornamento della documentazione relativa all’attività (Norme ISO, Regolamenti,
Metodi Ufficiali, etc.) e sua registrazione nella relazione trimestrale � la verifica dei dati grezzi, degli eventuali calcoli e di quanto registrato nei quaderni di lavoro � l’esecuzione di prove per le quali è abilitato, riguardanti lo specifico settore, in mancanza del personale addetto
o per particolari situazioni � assistenza al cliente che sovrintende alle prove, informandolo delle procedure di sicurezza, dei metodi di prova adottati, garantendo al tempo stesso la riservatezza � elabora le offerte e i contratti rispetto alle richieste inoltrate dal cliente � predispone la rendicontazione mensile delle attività � la stesura e la firma dei RP su delega del Responsabile del Laboratorio
� IL SOSTITUTO DEL RUO:
Viene individuato fra i DIR, e oltre ad espletare i compiti inerenti la propria funzione, si occupa dell’ordinaria gestione dell’UO assicurando quanto definito per il RUO.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 34 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0 � IL DIRIGENTE:
Viene assegnato alle UO secondo il titolo di studio, l’esperienza maturata e le competenze individuate. Il DIR con incarico di funzione è nominato dal DG su richiesta del RL. Il DIR deve assicurare: � applicazione delle PG e PO � lo studio e la proposta dei metodi di prova, di metodi interni e delle procedure di prova integrativa � la conformità dei dati grezzi ai risultati riportati sulla scheda di lavoro � l’emissione delle richieste di acquisto dei materiali e delle apparecchiature � lo svolgimento delle prove assegnate e la supervisione dell’attività svolta dal PT � stesura e firma dei RP, in assenza del RUO � eventuali altri incarichi conferiti con ordine di servizio dal RL e RUO Nelle UO dove non c’è assegnato un secondo Dirigente, le responsabilità che dovrebbero ricadere su questa figura professionale, vengono assunte dal RUO. � IL PERSONALE TECNICO (Tecnici di laboratorio biomedico, Periti Chimici, Tecnici della prevenzione)
Deve essere in possesso dello specifico diploma di laurea o diploma equipollente o dello specifico diploma di scuola media superiore con abilitazione (perito chimico), secondo l’indirizzo. Deve: � essere formato sul SQ e deve applicare quanto stabilito nel MQ- PG-PO � eseguire le prove di laboratorio per il quale è abilitato, garantendo la tracciabilità delle fasi analitiche e applicando i calcoli statistici correlati ai risultati di prova � garantire il buon funzionamento delle apparecchiature della U.O. di appartenenza, effettuando manutenzione ordinaria e controllo di taratura, secondo quanto stabilito dalle procedure specifiche � gestire le scorte dei materiali e reagenti necessari alle analisi, con relativa registrazione � gestire secondo le specifiche procedure, i materiali critici e i materiali di riferimento � garantire la preparazione e relativa registrazione dei reagenti, materiale e terreni � garantire la decontaminazione di materiali infetti o tossici � garantire il corretto conferimento dei rifiuti � custodire la documentazione loro affidata � conservare i contro-campioni secondo la tipologia di matrice � custodire i contro-campioni non conformi per i quali è stata richiesta revisione � smaltire secondo indicazione del RUO, i controcampioni dopo 60 giorni dalla trasmissione del RP
� IL PERSONALE DELL’ACCETTAZIONE DEI CAMPIONI (PA)
Il PA viene designato dal RL, ed è alle sue dirette dipendenze, 1 ha la responsabilità dell’accettazione, codifica, registrazione, campioni di prova. Il PA, secondo quanto definito nella PG/015, deve: � controllare il campione consegnato e la relativa documentazione e rilasciare al cliente copia della ricevuta di conformità � compilare il registro di codifica e assegnare il numero di codice al campione � predisporre la scheda identificativa dell’aliquota da saggio � predisporre il verbale di apertura campione in presenza della parte � consegnare l’aliquota da saggio e relativa scheda al RUO di competenza � predisporre il fascicolo del campione di prova � verificare la corretta compilazione del RP

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 35 di 57
� conferire alla segreteria i RP verificati, per la trasmissione al RL � consegnare al cliente il RP (solo per i RP che andranno ritirati direttamente al LSP)
SEZIONE: 5 EDIZ. 7 – REV. 0
� archiviare, a lavoro ultimato, il fascicolo relativo al campione
� IL PERSONALE AUSILIARIO SOCIO SANITARIO
Il personale Ausiliario garantisce: � La pulizia giornaliera della vetreria e degli accessori di laboratorio � La pulizia giornaliera delle superfici di lavoro � La pulizia delle superfici esterne ed interne delle apparecchiature come stabilito nel programma della
PO/021 � Il ritiro del materiale dal magazzino economale e/o proveniente dalla farmacia territoriale � Il ritiro dalle UO dei contenitori dei rifiuti speciali e il conferimento nei locali di deposito temporaneo � Il conferimento dei rifiuti alla ditta incaricata dello smaltimento � La collaborazione con il personale tecnico per le attività di reparto.
� IL PERSONALE DI SEGRETERIA
Il personale di segreteria svolge funzione amministrativa e coadiuva il RL nella gestione delle attività. Rientrano fra i compiti specifici: � gestione del protocollo � ricezione e trasmissione della documentazione � gestione della documentazione relativa al personale � archiviazione della documentazione (ad eccezione della documentazione del SQ) � richieste fornitura del materiale di cancelleria e casermaggio � rendicontazione relativa al personale � gestione dei buoni pasto e dei buoni benzina � compilazione delle liste di distribuzione della documentazione secondo quanto espresso dal RL � gestione della modulistica aziendale e della modulistica di sistema (reclami/ non conformità) � collaborazione con il RL e il RAQ per le comunicazioni interne
� UAQ
L’Unità Assicurazione della Qualità è costituita da un gruppo di operatori (DIR- PT) e ha il compito di coadiuvare il RAQ e l’Auditor in specifiche applicazioni del SQ. L’UAQ deve: � partecipare ai percorsi di audit � controllare la documentazione del SQ prima dell’emissione � coadiuvare il RAQ nella gestione dei quaderni e registri delle apparecchiature � presentare nel corso di tavoli tecnici, le informazioni raccolte e necessità espresse dalle UOS
� REFERENTI TECNICI
Individuati fra i DIR e PT delle UOS, coadiuvano il RAQ nella gestione delle registrazioni tecniche e delle carte di controllo. Rispondono al Team Ispettivo circa i dati di validazione delle prove, e delle verifiche di mantenimento.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 36 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0
DOCUMENTAZIONE RELATIVA AL PERSONALE ( UNI CEI EN ISO/IEC 17025:2005 –RT-08 – 5.2.4 – 5.2.5 )
La documentazione relativa al personale, comprende: � assegnazione di compiti (ordini di servizio), nella quale vengono precisate per ogni dipendente, l’UOS di appartenenza, la posizione gerarchica all’interno di essa, le competenze assegnate e le responsabilità. � registrazione dei dati anagrafici e del titolo di studio posseduto. Alla scheda viene allegato un curriculum professionale, aggiornato periodicamente. � Registrazione dei dati sulla formazione, addestramento, abilitazione e partecipazione a corsi, congressi, seminari, ecc.. da aggiornare di volta in volta. La documentazione riguardante il personale viene archiviata, a cura del RAQ, in modo riservato, ma accessibile ai percorsi di audit. Le modalità di gestione della documentazione, relativa al personale, è riportata nella PG/008 “Modalità di gestione della documentazione relativa al personale”.
DELEGA DI RESPONSABILITÀ (UNI CEI EN ISO/IEC 17025:2005 – RT-08 - 4.1.5 ) Sono previste deleghe di responsabilità per la sostituzione dei titolari assenti, per le seguenti funzioni: � RL ( Responsabile del Laboratorio); � RUO ( Responsabile di Unità Operativa); � RAQ ( Responsabile di Assicurazione della Qualità).
FORMAZIONE E ADDESTRAMENTO PROFESSIONALE DEL PERSONALE (UNI CEI EN ISO/IEC 17025:2005 –RT-08 - 5.2.2 – 5.2.3)
Il RL, in accordo con il RAQ, e in collaborazione con i RUO, tenuto conto del piano di formazione aziendale e del contratto di lavoro dei dirigenti e del comparto, predispone ogni anno un piano di formazione e addestramento del personale, che prevede corsi e seminari interni o esterni al laboratorio, partecipazione a convegni, giornate di studio, etc. Tenuto conto della formazione dell’anno precedente, delle esigenze aziendali, delle eventuali nuove commissioni attribuite al laboratorio, viene redatto con cadenza annuale, un piano che esprime le necessità di formazione e approfondimento, interna e/o esterna. Tale piano oltre che a programmare la formazione su specifiche attività istituzionali (balneazione, amianto, sostanza d’abuso, etc.),prevede un aggiornamento continuo sul sistema di qualità (Norme di Riferimento: UNI CEI EN ISO/IEC 17025:2005 e Regolamenti ACCREDIA) e su prove e tarature (aggiornamento metodi, validazione, calcolo dell’incertezza di misura, controllo delle tarature). Il programma di formazione predisposto, viene inviato ad inizio anno al Dipartimento di Prevenzione e all’Alta Direzione per la valutazione. Dopo l’approvazione del piano, la richiesta di partecipazione agli specifici corsi, convegni, ecc, viene convalidata dal RL, in base all’organizzazione del lavoro all’interno dell’UO di appartenenza, alle commissioni attribuite all’operatore, previo parere del RUO di competenza. Tale richiesta viene quindi inoltrata al Dipartimento di Prevenzione per l’autorizzazione e completamento dell’iter burocratico. Per quanto riguarda il personale neoassunto o assegnato a nuovo incarico, il RUO di pertinenza emette un piano di formazione/addestramento specifico, secondo le commissioni previste o attribuite, che comprende l’addestramento su diverse prove e uso di apparecchiature per l’acquisizione dei requisiti specifici e la verifica dell’efficacia. La formazione sul sistema di qualità e la valutazione della formazione raggiunta è a carico del RAQ.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 37 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0
La valutazione tecnica viene effettuata in base ai criteri definiti nelle specifiche procedure, tenendo conto della: � Capacità di riprodurre un dato analitico, sullo stesso campione con la medesima tecnica in tempi diversi ma
ravvicinati (ripetibilità) � Capacità di effettuare prove in doppio, dove la differenza dei valori, risulti uguale o inferiore al limite di
ripetibilità, adottato in fase di abilitazione al metodo � Capacità di ottenere dati proporzionali effettuando semine di differenti diluizioni (indice di proporzionalità) � Capacità di produrre un dato statisticamente correlato con il dato prodotto da un altro operatore, sul medesimo
campione, applicando lo stesso metodo (riproducibilità intralaboratorio) � Capacità di produrre un dato statisticamente correlato, con il dato prodotto da un altro operatore, di diverso
laboratorio, utilizzando altra strumentazione, sul medesimo campione, applicando un metodo uguale e/o diverso (Z-score)
� Capacità di applicare metodi qualitativi e di evidenziare i veri positivi e i veri negativi (sensibilità e specificità). I criteri vengono adottati sia in fase di abilitazione che per la verifica annuale di mantenimento (PG/009, alla PG/017).
Anche per l’abilitazione all’esecuzione delle tarature e per la verifica di mantenimento sono stato definiti dei criteri di valutazione:
� Per l’abilitazione del personale (almeno due operatori), dopo aver partecipato a specifico corso formativo di metrologia, si procede ad un confronto delle varianze (F-test) calcolate in fase di misura dello scarto tipo di ripetibilità. I dati sono elaborati con l’utilizzo del modulo 32 rev. del 30/12/01 ARPAT. I risultati vengono registrati nell’allegato 1 della PG/018
� Per la verifica del mantenimento dell’abilitazione all’esecuzione delle tarature, con frequenza semestrale gli operatori abilitati alle tarature eseguono delle misurazioni in doppio: la differenza deve essere inferiore o uguale al limite di ripetibilità calcolato in fase di abilitazione. I dati sono registrati nell’allegato 2 della PG/018.
5.3 LUOGHI DI LAVORO E CONDIZIONI AMBIENTALI ( UNI CEI EN ISO/IEC 17025:2005 – RT-08- 5.3.1 – 5.3.2 – 5.3.3 – 5.3.4 – 5.3.5 )
Il Laboratorio dispone di locali ampi, ben arieggiati e ben illuminati dalla luce solare. Gli ambienti sono ben distinti in modo da: prevenire le contaminazioni crociate, differenziare le attività secondo un criterio di funzionalità e facilitare il controllo degli accessi.
I locali destinati all’esecuzione delle prove sono dotati di: � energia elettrica � gas (ove necessario) � acqua potabile � impianto di climatizzazione � di superfici e pareti lavabili e disinfettabili I locali sono attrezzati con adeguata strumentazione e dotati di strutture di supporto (cappe a flusso laminare, cappe chimiche, armadi di sicurezza dispositivi di protezione individuale, ecc..). Tutti i laboratori sono contrassegnati da apposito segnale di rischio secondo la destinazione d’uso. Anche i locali destinati al deposito temporaneo dei i rifiuti, sono ben identificati, dotati di serratura per il controllo degli accessi e contrassegnati da apposito segnale di rischio.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 38 di 57
I locali adibiti ad ufficio sono confortevoli e dotati di arredi idonei per la sicurezza dei dati personali e sensibili. I corridoi sono ampi, luminosi, dotati di estintori e cassette di pronto soccorso.
SEZIONE: 5 EDIZ. 7 – REV. 0
PLANIMETRIA E IDENTIFICAZIONE DEI LOCALI (UNI CEI EN ISO/IEC 17025:2005- RT-08 – 5.3.3 )
Il Laboratorio di Sanità Pubblica, si articola in 2 Unità Operative, ciascuna dotata di propri locali dove tutte le attività tecniche si svolgono in maniera indipendente.
L’accesso ai locali, destinati alle prove è controllato: un elenco affisso riporta i nominativi dei soggetti autorizzati. Il cliente può accedere dopo la consegna dei dispositivi necessari, e in presenza del personale autorizzato.
Al 1° piano, nella parte centrale della struttura, si trovano la Direzione, lo studio del RAQ, la segreteria e l’Accettazione campioni di prova. Di seguito si articola l’UOS di Microbiologia (vedi sez. 1 pag 11).
La UOS di Biofisica negli ambienti di vita e di lavoro, che è anche “Centro di Riferimento Regionale per l’amianto” ha sede nei locali del piano terra.
La planimetria dei locali e la loro disposizione sono accluse quale allegato N. 3 del MQ.
MONITORAGGIO AMBIENTALE (UNI CEI EN ISO/IEC 17025:2005 – RT-08– 5.3.2 )
Per evitare che condizioni ambientali sfavorevoli possano avere influenza sulle prove e tarature, il Laboratorio ha predisposto una procedura operativa ( PO/021) che descrive, diversificando per le singole UO, le attività di pulizia, disinfezione, e monitoraggio, da eseguire, secondo calendario, con cadenza giornaliera, settimanale, mensile e semestrale. Per quanto riguarda l’UOS di Biofisica, la sanificazione delle superfici di lavoro e il controllo della contaminazione ambientale da fibra di amianto, avviene con specifica procedura identificata PO/027/3.
5.4 METODI DI PROVA E TARATURA E VALIDAZIONE DEI METODI
IDENTIFICAZIONE DEI METODI DI PROVA (UNI CEI EN ISO/IEC 17025:2005 – 5.4.1/RT-08 -– 5.4.1 - 6.8 )
Il Laboratorio deve garantire l’uso di metodi appropriati secondo il campo di attività, e l’adeguatezza della strumentazione necessaria. Pertanto, deve acquisire i metodi, identificarli in maniera univoca, renderli disponibili al personale, predisporre le eventuali procedure integrative, garantire l’eventuale aggiornamento.
I metodi di prova adottati dal LSP, siano essi ufficiali, editi, normati e interni, sono catalogati con un codice
identificativo composto da: MP – XXX – 1) 2) secondo l’UOS di pertinenza, dove con MP si intende Metodo di Prova, con XXX, il numero progressivo a tre cifre a partire da 001 – e con 1) UOS di Microbiologia, 2) UOS di Biofisica. Ogni gruppo di caratteri è separato da un trattino.
Nel caso in cui, i metodi pubblicati non forniscano sufficienti dettagli, vengono messi a punto metodi di
prova integrativa, che puntualizzano e spiegano dettagliatamente le prescrizioni contenute nel metodo. Le procedure di prova integrativa, vengono autorizzate dal RL, insieme all’autorizzazione del metodo
ufficiale, del quale sono parte integrante. Le procedure di prova integrativa al metodo, sono codificate con: PI-XXX-1) 2), dove PI, indica Procedura di
prova Integrativa, XXX indica il numero progressivo a tre cifre del MP del quale è parte integrante, 1) 2) identificano l’UOS di pertinenza. Ogni gruppo di caratteri è separato da un trattino.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 39 di 57
L’operatore tecnico che esegue la prova, ha disponibile copia controllata del metodo di prova.
SEZIONE: 5 EDIZ. 7 – REV. 0
SCELTA DEI METODI DI PROVA (UNI CEI EN ISO/IEC 17025:2005 RT-08 - 5.4.2-5.4.3-5.4.4)
Il LSP, utilizza, per le prove sui campioni da analizzare: � Metodi ufficiali di analisi (riportati su documenti normativi cogenti, GURI e GUCE) � Metodi normalizzati da Organizzazioni nazionali o internazionali (UNI, CEI, ISO,etc.) � Metodi non normalizzati, editi da Enti Nazionali e Internazionali (ISTISAN, IRSA) � Metodi interni Ad eccezione dei metodi riportati in direttive (es: Reg CE) che vanno applicati in quanto ritenuti cogenti, la scelta del metodo di prova, la verifica dell’applicabilità e della fattibilità, compete al RUO, il quale deve prediligere come prima scelta l’uso di metodi di prova normalizzati, e in assenza di questi, l’uso di metodi non normalizzati o interni (PG/013). Il metodo viene progettato sull’apposito allegato e dopo l’approvazione della scelta e progettazione, viene validato. Il metodo di prova, è in ultimo autorizzato dal RL.
VERIFICA, VALIDAZIONE DEI METODI E RIESAME DELLA VALIDAZIONE
(UNI CEI EN ISO/IEC 17025:2005 RT-08 5.4.5)
Il punto 5.4.5 della Norma 17025:2005 prescrive che i laboratori di prova, prima della messa in uso di un metodo di prova, procedano alla validazione o verifica dello stesso, al fine di evidenziare che i requisiti richiesti siano soddisfatti. Pertanto il laboratorio ha provveduto a pianificare tali processi nelle procedure gestionali riportate in calce al capitolo. Le fasi e le responsabilità individuate sono così riassunte:
ATTIVITA’
RESPONSABILITA’
Scelta del metodo RUO Pianificazione della validazione RUO/RAQ
VALIDAZIONE PRIMARIA:
Verifica con matrici certificate /ceppi ATCC RUO Elaborazione statistica dei dati RUO/RAQ
Valutazione dei dati ottenuti RL Archiviazione dei dati RAQ
Autorizzazione del metodo RL VALIDAZIONE SECONDARIA (VERIFICA):
Verifica con matrici certificate o ring test RUO Elaborazione dei dati RUO/RAQ
Valutazione dei dati ottenuti RL Archiviazione dei dati RAQ
Autorizzazione RL Riesame della Validazione RUO/RAQ/RL
Rivalidazione RUO/RAQ/RL
Il LSP adotta prevalentemente metodi di prova ufficiali o normalizzati, pertanto deve verificare la capacità di applicare correttamente gli stessi.
Secondo la tipologia di prove, la verifica avviene attraverso la stima di parametri valutativi diversi: per

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 40 di 57
le prove qualitative, la stima si effettua prendendo in esame i parametri “ Sensibilità e Specificità”, mentre per le prove quantitative, vengono presi in considerazione parametri che esprimono la precisione: “ripetibilità e riproducibilità”.
SEZIONE: 5 EDIZ. 7 – REV. 0 Nel caso di metodi non normalizzati o interni, o nel caso di campi di applicazione diversi, il laboratorio procede alla validazione, secondo quanto richiesto dal punto 5.4.4. della Norma, utilizzando o circuiti di prova conformi alla UNI CEI EN ISO/IEC 17043:2010 secondo quanto prescritto dal regolamento Accredia RT-24, o matrici certificate. La validazione dei metodi non comprende il campionamento e il trasporto del campione, in quanto non effettuato dal laboratorio. Dopo la validazione, il RL sottoscrive la dichiarazione di validazione, e autorizza la messa in uso del metodo. Con cadenza annuale, i metodi vengono sottoposti a riesame, per verificare il mantenimento della validazione, partecipando, ove possibile a circuiti di prova o in assenza, con l’uso di materiali di riferimento. L’incertezza di misura, qualora fosse necessario, viene ricalcolata. Il riesame di validazione dei metodi di prova microbiologici, prevede come parametri di verifica, precisione ed esattezza, attraverso la valutazione:
Metodo Parametro Valutazione Accettabilità
Quantitativo Precisione Riproducibilità intralaboratorio Omogeneità con dati validazione
Esattezza Z- Score ± 2 (critico ± 3)
Qualitativo Precisione Sensibilità ≥ a quella calcolata in fase di validazione
Precisione Specificità ≥ a quella calcolata in fase di validazione
Il riesame periodico della validazione dei restanti metodi è distinto invece su due livelli, uno annuale e uno quadriennale.
Il riesame annuale del mantenimento della validazione applicabile sia ai metodi di prova validati sia a quelli
interni o non validati, prevede la verifica dei seguenti criteri di rendimento: • Precisione • Recupero • Esattezza
La precisione viene valutata attraverso una prova in doppio: la differenza deve essere inferiore o uguale al limite di ripetibilità adottato in fase di validazione del metodo. Il recupero, quando applicabile, viene valutato fortificando un campione a concentrazione nota con una quantità nota di standard. Il limite di accettabilità è +/- 3 volte la deviazione standard del recupero medio, studiato in fase di validazione del metodo.
L’esattezza è valutata tramite la partecipazione a circuiti interlaboratorio o quando non disponibili, con matrice di riferimento certificato.
Il riesame quadriennale per i metodi interni prevede la verifica di tutti i criteri di rendimento previsti dalla
procedura, mentre per i metodi normati è sufficiente la verifica del rapporto Sr sperimentale σr indicato dalla norma o prefissato dal laboratorio. La rivalidazione dei restanti metodi adottati dal laboratorio, viene programmata in base ai dati disponibili, nel caso di variazioni sostanziali dei metodi o in corso di riesame della direzione. Per maggiori dettagli si rimanda alla procedura PG/017

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 41 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0
STIMA DELL’INCERTEZZA DI MISURA (UNI CEI EN ISO/IEC 17025:2005 RT-08 -5.4.6)
Dai dati di validazione, il laboratorio ricava l’incertezza di misura da associare ai risultati di prova e taratura, accertando che la stessa, sia stata stimata a livelli significativi rispetto ai limiti di legge. L’approccio per la valutazione dell’incertezza deve tenere conto di tutti i fattori che possono influenzarla: il laboratorio, quando possibile, cerca di identificare, per ciascun componente, l’influenza che lo stesso ha su uno specifico metodo di prova (approccio metrologico). Il livello di rigore adottato dipende dai requisiti del metodo di prova, dai requisiti del cliente, dalla ristrettezza dei limiti su cui sono basate le decisione di conformità ad una specifica. Quando il metodo di prova lo consente, il laboratorio procede a stimare l’incertezza attraverso la valutazione della riproducibilità. Tali parametri di precisione vengono verificati,di regola, con cadenza annuale. Il risultato di una prova, viene espresso associando il valore ottenuto con la propria incertezza di misura, solo nel caso in cui il valore è significativamente inferiore al limite di legge, può essere omessa l’espressione dell’incertezza di misura, calcolata in sede di validazione. Per i dettagli si rimanda alla PO/016/1.
TENUTA SOTTO CONTROLLO DEI DATI (UNI CEI EN ISO/IEC 17025:2005 RT-08 -5.4.7)
L’operatore tecnico che esegue la prova, ha la responsabilità di riportare ogni dato, compreso l’esecuzione dei calcoli, nel proprio quaderno di laboratorio e di trascrivere i risultati sulla scheda di registrazione dati, che viene consegnata al RUO per la compilazione del Rapporto di Prova. Nel caso in cui venissero utilizzati calcoli con l’ausilio di fogli excel, gli stessi devono essere preventivamente validati e messi in sicurezza attraverso blocco con password (PG/003) Il RUO, prima della stesura del RP, ha il compito di valutare i dati analitici trascritti sulla scheda, con le informazioni riguardanti il campione in suo possesso. Il RUO inoltre, registra i dati relativi al campione e effettua salvataggio su supporto magnetico, secondo quanto stabilito nel regolamento aziendale della privacy e nella PG/007.
5.5 APPARECCHIATURE (UNI CEI EN ISO/IEC 17025:2005 – RT 08: 5.5.1-5.5.2-5.5.3)
Il LSP dispone di dotazione strumentale adeguata alle prove. Tale strumentazione viene tenuta sotto controllo, al fine di garantirne il buon funzionamento, attraverso manutenzioni ordinarie e programmate. La strumentazione soggetta a taratura, viene verificata secondo un programma di controllo di taratura stabilito dal RUO. Il RAQ riassume in un unico schema il programma di taratura e di manutenzione per i singoli strumenti. Il personale che utilizza la strumentazione in dotazione viene abilitato e autorizzato all’uso, dopo comprovata capacità di utilizzo, almeno per le strumentazioni sofisticate. Restano a disposizione dell’operatore, manuali e guide per la corretta manutenzione e istruzione, nonché le specifiche PO.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 42 di 57
SEZIONE: 5 EDIZ. 7 – REV. 0
APPARECCHIATURE IN DOTAZIONE AL LABORATORIO (UNI CEI EN ISO/IEC 17025:2005 – RT 08: 5.5.4-5.5.5 )
Le apparecchiature utilizzate dalle UUOOSS del LSP sono riportate nell’elenco allegato al presente MQ. Nell’elenco viene riportato: � La denominazione dell’apparecchiatura � L’indicazione se l’apparecchiatura è soggetta o meno a taratura. Tutte le notizie riguardanti le apparecchiature, sono registrate in una scheda d’identificazione, conservata nel fascicolo dello strumento, una copia di essa si trova nel quaderno dell’apparecchiatura, custodito nell’apposito schedario, gestito dal RAQ.
DOCUMENTAZIONE E REGISTRAZIONE (UNI CEI EN ISO/IEC 17025:2005 5.5.4-5.5.5)
Come descritto nella PG/012, per ogni singola apparecchiatura del laboratorio, sono state approntate: 1. Scheda d’identificazione 2. Scheda d’identificazione dei componenti 3. Scheda di manutenzione preventiva 4. Scheda di manutenzione straordinaria 5. Scheda di taratura 6. Rapporto di taratura 7. Etichetta di apparecchiatura sottoposta a taratura 8. Etichetta di apparecchiatura fuori servizio Questi documenti (ad eccezione del rapporto di taratura), le eventuali carte di controllo dell’apparecchio e la corrispondente PO, vengono raccolti nel quaderno dell’apparecchiatura, in prossimità della stessa. Viene predisposto anche un registro dell’apparecchiatura, custodito dal RAQ in apposito schedario, che raccoglie tutti i documenti riguardanti l’apparecchio. La scheda d’identificazione riporta le seguenti informazioni: � Denominazione del Laboratorio; � Numero della scheda; � Codice dell’apparecchiatura; � Nome dell’apparecchiatura; � Classe dell’apparecchiatura; � Numero di matricola, modello/tipo, numero di serie; � Elenco degli eventuali componenti; � Data di ricevimento, di collaudo, d’inizio servizio, e numero di inventario; � Nome del produttore e nome del distributore; � Collocazione (n° di stanza e piano); � Tipo di assistenza tecnica (interna o esterna) e ditta; � Elenco delle persone abilitate all’uso; � Taratura (si, no) e sua frequenza; � Controllo (si, no) e sua frequenza; � Collocazione del Manuale;

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 43 di 57
� Responsabile della taratura; � Responsabile dell’apparecchio; � Riferimento di procedura di taratura e procedura di manutenzione e uso;
SEZIONE: 5 EDIZ. 7 – REV. 0 La scheda identificativa dei componenti, riporta le stesse informazioni di quella delle apparecchiature.
MANUTENZIONE DELLE APPARECCHIATURE (UNI CEI EN ISO/IEC 17025:2005 5.5.6-5.5.7)
La manutenzione e la taratura delle apparecchiature di prova, sono operazioni indispensabili non solo per assicurare la qualità del risultato analitico, ma per garantire l’affidabilità nel tempo delle apparecchiature stesse. La manutenzione si divide in ordinaria, preventiva e straordinaria. La manutenzione ordinaria prevede tutte quelle operazioni che vanno effettuate giornalmente, o prima dell’uso dell’apparecchiatura, al fine di assicurare il buon funzionamento. La manutenzione preventiva comprende gli interventi effettuati a tempi prefissati, per evitare il decadimento dell’apparecchio. Sono affidati in convenzione ad una ditta specializzata. La manutenzione straordinaria comprende gli interventi effettuati dopo il verificarsi di guasti o malfunzionamenti; vengono effettuati, dopo richiesta del LSP, da personale specializzato, o se espressamente indicato nel manuale, da personale della ditta costruttrice. In tal caso il laboratorio provvede a segnalare il fermo dello strumento, apponendo il cartello “Fuori servizio”. Sono state predisposte per ogni apparecchiatura delle schede di manutenzione, collocate nel quaderno dell’apparecchio, dove vengono annotati tipo di inconvenienti e interventi cronologici effettuati. Le schede, vengono archiviate periodicamente nel registro dell’apparecchiatura. In sostituzione della scheda può essere archiviato il rapporto rilasciato dalla ditta.
TARATURA DELLE APPARECCHIATURE (UNI CEI EN ISO/IEC 17025:2005- RT 08: 5.5.8-5.5.9-5.5.10-5.5.11-5.6.1-5.6.2-5.6.3)
Il LSP ha in dotazione apparecchiature che per la loro diversità necessitano di tarature differenti, pertanto oltre la descrizione riportata nella PG/012, ogni strumento riporta nella propria PO la descrizione, se necessaria, della specifica taratura. Il personale abilitato all’uso degli strumenti di prova e taratura, deve possedere i requisiti per effettuare le verifiche di taratura necessarie, e deve essere in grado di gestire materiali e campioni di riferimento certificati. Come stabilito dalla PG/018 “requisiti del personale che esegue tarature e controllo di tarature”, Il RUO individua fra gli operatori tecnici abilitati all’uso dell’apparecchiatura, quelli idonei a verificare le tarature. La scheda di taratura è riportata in allegato alla PG/012, e conservata nel quaderno dell’apparecchio, in prossimità dello stesso; una volta completata, viene archiviata nel registro dell’apparecchio a cura del RAQ. Le carte di controllo, vengono conservate nel quaderno dell’apparecchio, in prossimità dello stesso, e archiviate, una volta completate, nel registro dell’apparecchio, a cura del RAQ. Per la verifica di taratura dei termometri, delle bilance e degli apparecchi a temperatura controllata, il LSP ha elaborato specifiche PO, che definiscono i limiti di accettabilità relativi a scostamento e incertezze delle tarature interne ed esterne: • PO/022 Taratura e verifica di taratura dei termometri: calcoli statistici • PO/026. Taratura e controlli di taratura delle apparecchiature a temperatura controllata • PO/015 Manutenzione, modalità d’uso e taratura delle bilance.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 44 di 57
Per le apparecchiature di prova, il fornitore deve assicurare e il Laboratorio verificare che l’incertezza di misura fornita dallo strumento sia tale da non condizionare l’incertezza totale dello stesso metodo di prova.
SEZIONE: 5 EDIZ. 7 – REV. 0
APPARECCHIATURA FUORI SERVIZIO (UNI CEI EN ISO/IEC 17025:2005 5.5.7)
Tutte le apparecchiature o componenti di esse, che hanno mostrato durante il controllo giornaliero un malfunzionamento, o che non sono state sottoposte a taratura o a manutenzione nei tempi stabiliti, non devono essere utilizzate. Su questi strumenti, il responsabile, deve apporre il cartello che ne vieta l’uso. Il cartello riporta: � La dicitura FUORI SERVIZIO;
� L’avvertenza NON UTILIZZARE ;
� La data d’inizio del fuori servizio;
� Il motivo del fuori servizio.
Se si è evidenziato un malfunzionamento, deve essere attivato un intervento di manutenzione straordinaria.
Al fine di garantire il cliente e la qualità delle prove, il Laboratorio, dopo aver aperto l’eventuale non conformità,
esamina la durata della stessa e l’effetto sulle prove, procedendo, qualora si ravvedesse la necessità, a ritirare i
relativi rapporti di prova, dando comunicazione al cliente, ed eventualmente ad ACCREDIA.
5.6 RIFERIBILITA’ DELLE MISURE (UNI CEI EN ISO/IEC 17025:2005- RT 08: 5.6.1-5.6.2-5.6.3) Tutte le apparecchiature soggette a taratura, in dotazione al laboratorio, utilizzate per le prove, sono tenute sotto controllo attraverso un programma di taratura. Per effettuare i controlli di taratura, il LSP dispone di campioni e materiali di riferimento certificati, riferibili a campioni o materiali primari.
I campioni utilizzati per la taratura delle apparecchiature di prova e misura, sono certificati da Centri LAT o in mutuo riconoscimento, accreditati per le grandezze di interesse, per campi di misura ed incertezze appropriati.
Il Laboratorio dispone di campioni certificati con incertezza dichiarata, idonea a soddisfare le tarature
interne (taratura termosonde di seconda linea in conformità alla ISO 7218/2007 Amd. 1:2013, masse classe E per taratura di bilance analitiche e tecniche). Le PO/022 e PO/015 definiscono i criteri di accettabilità adottati dal laboratorio.
In base alla frequenza d’uso dei campioni certificati, e l’adeguata conservazione, la validità dei certificati di
taratura è stabilita di regola in: • 3 anni per le sonde termometriche • 5 anni per le masse
Allo scadere del tempo prefissato, il RAQ provvede al rinnovo della certificazione inviando i campioni di riferimento ai Centri autorizzati secondo i requisiti sopra descritti. È responsabilità del RAQ verificare l’andamento dell’incertezza associata al campione attraverso l’analisi delle differenti certificazioni e i controlli intermedi effettuati dal laboratorio con cadenza semestrale.

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 45 di 57
Qualora il campione di riferimento non sia più in grado di garantire i criteri di accettabilità stabiliti dal
Laboratorio, viene declassato limitandone il campo di applicazione.
SEZIONE: 5 EDIZ. 7 – REV. 0 Nel caso in cui i criteri vengono costantemente soddisfatti, i rinnovi di certificazione possono essere
prolungati. Per i materiali di riferimento certificati, è lo stesso produttore che dichiara il periodo di validità, con la
scadenza del prodotto, in base alle modalità di conservazione. Tale periodo può essere esteso qualora il laboratorio sia in grado di attestare l’efficacia attraverso verifiche di laboratorio (PG/011).
Solo i Responsabili delle tarature possono prelevare i campioni di riferimento custoditi dal RAQ. Le apparecchiature dopo la taratura vengono tenute sotto controllo attraverso l’uso di campioni o materiali di seconda linea.
5.7 CAMPIONAMENTO (UNI CEI EN ISO/IEC 17025:2005 - RT 08: 5.7.1-5.7.2-5.7.3)
I campionamenti per il controllo ufficiale vengono eseguiti, di regola, da personale sanitario delle Aziende Sanitarie Provinciali (Dirigenti e Tecnici della prevenzione), o da organismi di polizia giudiziaria abilitati a controlli sanitari secondo quanto previsto dalle vigenti disposizioni legislative nazionali e dai regolamenti comunitari. Le registrazioni vengono effettuate in appositi verbali approvati dagli organi competenti in materia di applicazione di controlli ufficiali (Assessorato per la Salute, Ministero della Salute, etc.). I campi da compilare descritti nel verbale soddisfano tutte le esigenze per una corretta attività di prova, e forniscono le informazioni che vanno trasmesse tramite i “flussi informativi” agli organi competenti. All’atto del campionamento, qualsiasi scostamento dalla corretta applicazione della normativa di riferimento, deve essere registrata nel verbale, con relativa motivazione. Quantunque il Laboratorio non effettui di norma campionamenti, sono state messe a punto distinte PO secondo il campo di applicazione disponibili per la consultazione: • PO/024 che descrive in maniera dettagliata le modalità da seguire per il prelevamento di prodotti alimentari sfusi o confezionati, non ricompresi nei regolamenti • PO/030/3, che descrive il campionamento di aria e di massa per la ricerca delle fibre di amianto. Per i campioni effettuati dal cliente, ove possibile, vengono date indicazioni riguardo le modalità di campionamento, conservazione e trasporto fino al conferimento al reparto accettazione del laboratorio .
5.8 MANIPOLAZIONE DEGLI OGGETTI DA SOTTOPORRE A PROVA RICEVIMENTO E REGISTRAZIONE DEI CAMPIONI (UNI CEI EN ISO/IEC 17025:2005 5.8.1-5.8.2-5.8.3)
Di seguito vengono descritte, sinteticamente, le modalità di gestione per i campioni da sottoporre a prova: Il RA, in presenza del consegnante, procede al controllo della conformità del campione, e compila una ricevuta in doppia copia, firmata da entrambi. La ricevuta prevede la valutazione dell’integrità del campione e del sigillo, della temperatura di trasporto, il numero di aliquota e unità campionarie, come indicato nella PG/015. Una copia viene rilasciata al consegnante, l’altra viene archiviata nel fascicolo del campione. Nel caso

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 46 di 57
d’informazioni complete, che permettono la chiara identificazione del campione e la richiesta di analisi, il RA compila il registro di codifica/protocollo, riportando tutte le informazioni del verbale di prelevamento, e assegna ad ogni campione un codice di identificazione di tipo alfanumerico, codice che accompagna il materiale da saggio in tutto il percorso, dalla registrazione alla emissione del Rapporto di prova, così composto: 00/XXXXX, dove:
SEZIONE: 5 EDIZ. 7 – REV. 0 � 00 indica le ultime due cifre dell’anno in corso � XXXXX indica il numero progressivo da 00001 a 99999. I due caratteri sono separati da una barra. Sul materiale da saggio, viene collocata una targhetta adesiva o viene trascritto, il codice d’identificazione, come da registro campioni. Nel caso d’informazioni incomplete (impossibilità d’identificazione del campione, carenze documentali, richieste analitiche non chiare o non pertinenti) il RA, rimette il campione al mittente, ad eccezione di quei campioni che possono essere custoditi in apposita area, distinta da quella destinata ai campioni codificati, nell’attesa del completamento delle informazioni necessarie per l’esaustiva identificazione e quindi per la codifica. Dopo la codifica, il RA, predispone il fascicolo dei documenti, recante lo stesso numero di codice del materiale da saggio, che contiene tutta la documentazione di supporto al campione e, ad analisi ultimate, anche il rapporto di prova. Il RA, dopo la trasmissione dei risultati, procede all’archiviazione del fascicolo.
CIRCOLAZIONE DELL’ALIQUOTA DA SAGGIO E DEL DOCUMENTO DI ACCOMPAGNAMENTO, ALL’INTERNO DEL LABORATORIO (UNI CEI EN ISO/IEC 17025:2005 5.8.4)
Dopo la registrazione, l’addetto all’accettazione provvede a custodire l’aliquota da saggio, secondo la natura della stessa, all’idonea temperatura di conservazione, e a consegnare al RUO la corrispondente scheda di accompagnamento. Il RUO, viste le informazioni registrate nella scheda di accompagnamento, compila e firma la scheda di trasmissione dati, trascrivendo: • le informazioni relative al materiale da saggio: codice – parametri di prova • la comunicazione di invito alla parte • il nome dell’operatore tecnico a cui viene affidata l’aliquota da saggio per le prove • la data di inizio delle analisi. La scheda di trasmissione dati viene riportata in accettazione, per essere consegnata al laboratorio competente, con l’aliquota da saggio e i rispettivi contro campioni di prova. L’operatore tecnico, preleva, dall’aliquota da saggio, la quantità necessaria all’espletamento delle prove, riponendo in frigorifero la rimanente parte fino alla conclusione delle analisi. Per i dettagli si rimanda alla procedura PG/015.
CONSERVAZIONE DEI CAMPIONI E CONTRO CAMPIONI (UNI CEI EN ISO/IEC 17025:2005 5.8.4)
Dopo la consegna dei campioni di prova, il PT si occupa della corretta conservazione SIA del materiale da saggio residuo che dei contro campioni:

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 47 di 57
1. prodotto stabile: in armadi o scaffalature 2. prodotto deteriorabile: un’aliquota in frigorifero a + 3°C ±2°C per la ripetizione di eventuali parametri non conformi, un’aliquota in congelatore a -15/-18°C, a disposizione dell’autorità giudiziaria. 3. acque minerali: acque di sorgente o della linea d’imbottigliamento, in frigorifero a +3/+5 °C.
SEZIONE: 5 EDIZ. 7 – REV. 0
4. prodotto surgelato: in congelatore a -15/-18°C. 5. campioni di amianto: conferimento all’UOS di Biofisica I campioni biologici vengono conservati, negli appositi locali resi in sicurezza, sotto la responsabilità del Dirigente incaricato.
SMALTIMENTO DEI CAMPIONI SOTTOPOSTI A PROVA (UNI CEI EN ISO/IEC 17025:2005 5.8.1)
Dopo l’espletamento delle analisi, il residuo dell’aliquota da saggio (alimenti) viene smaltito dagli operatori secondo la conformità, come rifiuto urbano o speciale. Nel caso di residuo di campioni di acqua, gli stessi vengono eliminati attraverso la rete fognaria. I contro campioni vanno conservati per la durata di sessanta giorni a decorrere dalla data di trasmissione dei Rapporti di prova, e poi smaltiti come: • rifiuto speciale ospedaliero, se le analisi hanno rilevato non conformità • rifiuto solido urbano, se le analisi sono risultate conformi.
I tempi di conservazione e di smaltimento del contro campione, vengono registrati dal RUO, nella scheda di accompagnamento dell’aliquota da saggio, allegata al Rapporto di Prova. La procedura PG/015 “Modalità di gestione dei campioni di prova - Criteri generali” definisce dettagliatamente ogni operazione. Per i dettagli sullo smaltimento dei rifiuti si rimanda alla PO/025.
5.9 ASSICURAZIONE DELLA QUALITÀ DEI RISULTATI DI PROVA E TARATURA
(UNI CEI EN ISO/IEC 17025:2005 5.9.1)
Il laboratorio assicura la qualità dei risultati di prova e taratura, attraverso controlli e verifiche che vengono effettuati periodicamente secondo quanto descritto nella PG/016. Gli strumenti utilizzati per assicurare la qualità delle prove e tarature sono così riassunti: 1. prove intra laboratorio utilizzando materiali di riferimento certificati (PG/011) 2. partecipazione a circuiti di prova che operano in conformità alla ISO/IEC 17043:2010, secondo i requisiti minimi adottati dal laboratorio, in conformità a quanto richiesto dal Regolamento ACCREDIA (RT-24):
• Almeno un’attività analitica per ciascun tipologia di prova, prima della valutazione per l’accreditamento. Nel caso in cui risulti che non siano disponibili circuiti inerenti il settore di competenza, il laboratorio fornirà evidenza dellʼindagine effettuata e delle altre attività di assicurazione della qualità eseguite (es. controllo di qualità interno) • Almeno un’attività analitica relativa a ciascuna delle sub-discipline maggiori dopo l’accreditamento e prima della valutazione per il rinnovo dell’accreditamento. • verifica del mantenimento del limite di ripetibilità/operatore I risultati delle prove, permettono di ricavare le linee di tendenza, rappresentate mediante carte di controllo. Per quanto riguarda gli esiti negativi, il laboratorio procede secondo quanto richiesto dalla RT24, alla verifica e ripetizione su campione supplementare, e alla sospensione del marchio con comunicazione all’ente Accredia, nel caso di due esiti negativi.
La qualità delle prove viene inoltre assicurata attraverso: • controllo dei materiali e campioni di seconda linea e confronto col rispettivo materiale/campione certificato

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 48 di 57
• controllo dei materiali critici in ingresso (PG/011) • qualifica dei fornitori (PG/010) • formazione e aggiornamento del personale (PG/009) • requisiti del personale che esegue controlli di taratura (PG/018)
SEZIONE: 5 EDIZ. 7 – REV. 0
5.10 PRESENTAZIONE DEI RISULTATI (UNI CEI EN ISO/IEC 17025:2005 5.10.1- 5.10.2-5.10.3-5.10.5/RT-08 -5.10.1- 5.10.2-5.10.3-5.10.5- 6.3-6.4)
MODULI IMPIEGATI
Il laboratorio, per comunicare i risultati relativi alle analisi condotte su campioni di prova, utilizza un modulo che riporta quanto specificato nella PG/014 “Rapporto di prova - Criteri generali”. Il Rapporto di prova è strutturato in modo da garantire il punto 5.10 dell’UNI CEI EN ISO/IEC 17025:2005 e quanto prescritto da ACCREDIA nel RT-08, comprendendo le seguenti informazioni: • I dati identificativi del Laboratorio e dell’Unità Operativa responsabile della prova; • Il titolo “RAPPORTO DI PROVA” • L’identificazione univoca del documento e la numerazione delle pagine • L’identificazione del cliente (Lista di distribuzione) • L’identificazione del campione sottoposto a prova:
1. Natura del campione (prodotto, confezione, lotto, scadenza) 2. Numero di codifica e data
• L’identificazione dell’organismo che ha operato il prelievo, della data, del numero di verbale e di tutte le informazioni relative al campione riferite nel verbale, compreso la motivazione del prelievo • Numero dell’aliquota del campione sottoposta ad analisi • Data d’inizio e di conclusione delle analisi • Data di eliminazione dei contro campioni • I caratteri organolettici (ove previsti) • La preparazione del campione (ove prevista) La registrazione delle prove specifica: 1. Prove eseguite 2. Metodo di prova 3. Risultato, con unità di misura e relativa incertezza di misura 4. Valore limite espresso nel riferimento legislativo • L’eventuale valutazione del risultato analitico rispetto il limite di legge, espresso in base ai criteri di conformità tenendo conto dell’incertezza di misura associata ai risultati, definiti nella PO/016-1 (specificando nell’apposito capitolo del Rapporto di prova: Pareri e interpretazioni – non oggetto dell’accreditamento ACCREDIA) • Il Rapporto di Prova riporta la firma del Responsabile di Unità Operativa delegato dal Responsabile del Laboratorio alla sua emissione o in sua assenza, di altro dirigente appositamente individuato.
Il Rapporto di prova può essere articolato in maniera differente dalle distinte UUOOSS secondo la tipologia delle prove, purché vengano rispettati tutti i requisiti richiesti dalla norma e specificati nella PG/014. I rapporti di prova riportanti prove accreditate, secondo il Regolamento RG-09, sono identificabili dal marchio ACCREDIA e le prove non accreditate eventualmente ricomprese sono ben individuabili perché contrassegnate con il simbolo: *. Non viene utilizzato rapporto di prova con marchio ACCREDIA, quando lo stesso non prevede

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 49 di 57
prove accreditate.
SEZIONE: 5 EDIZ. 7 – REV. 0
CERTIFICATI DI TARATURA (UNI CEI EN ISO/IEC 17025:2005 5.10.4)
Il LSP è inserito fra i laboratori di prova, e le tarature che effettua sono esclusivamente per uso interno. I report di taratura soddisfano le parti di pertinenza del punto 5.10.4 della Norma, prevedendo: - la registrazione della temperatura ambientale, ove prevista, -l’incertezza di misura -la riferibilità delle misure secondo campo di applicazione
TRASMISSIONE DEI RISULTATI E LORO ARCHIVIAZIONE (UNI CEI EN ISO/IEC 17025:2005 5.10.5-5.10.6-5.10.7-5.10.8)
Dopo avere completata la stesura del RP, il RUO, lo consegna, insieme alla scheda di accompagnamento dell’aliquota da saggio, al PA che allega tutta la documentazione riguardante il campione da lui custodita, e dopo aver proceduto alla verifica della concordanza dei dati registrati, predispone la scheda di avvenuto controllo del RP. Invia quindi la totalità del fascicolo alla firma del RL. Il RL appone la firma di convalida finale. Il RP, passa quindi alla segreteria che ha la responsabilità della trasmissione agli organi competenti e di riconsegnare il fascicolo al RA. Il PA, controlla i dati della trasmissione e le relative ricevute, quindi procede all’archiviazione delle pratiche. In caso di richiesta di duplicato, l’originale archiviato viene copiato e ritrasmesso con apposto il timbro “copia conforme” e firma del Responsabile. Il laboratorio conserva la documentazione relativa ai RP per 10 anni (PG /007)
MODIFICHE AI RAPPORTI DI PROVA (UNI CEI EN ISO/IEC 17025:2005 5.10.9) Come previsto nel documento RT-08, il laboratorio provvede a riemettere RP nel caso si verificassero: • Uso improprio del marchio Accredia (o riferimento all’accreditamento) • Errori dei risultati di prova • Carenze o errori che potrebbero comportare un uso improprio del documento
In tali casi, si procede all’emissione di un nuovo rapporto di prova, che annulla e sostituisce il precedente, il quale, pur non avendo più validità, rimarrà in ogni caso agli atti, congiuntamente a quello di nuova emissione. Il documento riformulato viene identificato univocamente dal numero di codice attribuito al campione associato al simbolo distintivo: #. Nella legenda dei RP viene specificato che la presenza di tale simbolo annulla il documento precedentemente emesso, che pertanto non ha più alcuna validità. E’ consentita soltanto la correzione manuale di un RP, quando interessa il campo descrittivo del campione o nel caso di piccoli aggiustamenti (recapito telefonico, etc.). Non è possibile apportare correzioni manuali nello specchietto relativo ai risultati di prova. In base all’errore rilevato, la remissione di un RP impone la verifica contemporanea dei RP emessi in

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 50 di 57
Dipartimento Diagnostica di laboratorio
contemporanea. Per i dettagli si rimanda alla PG/005
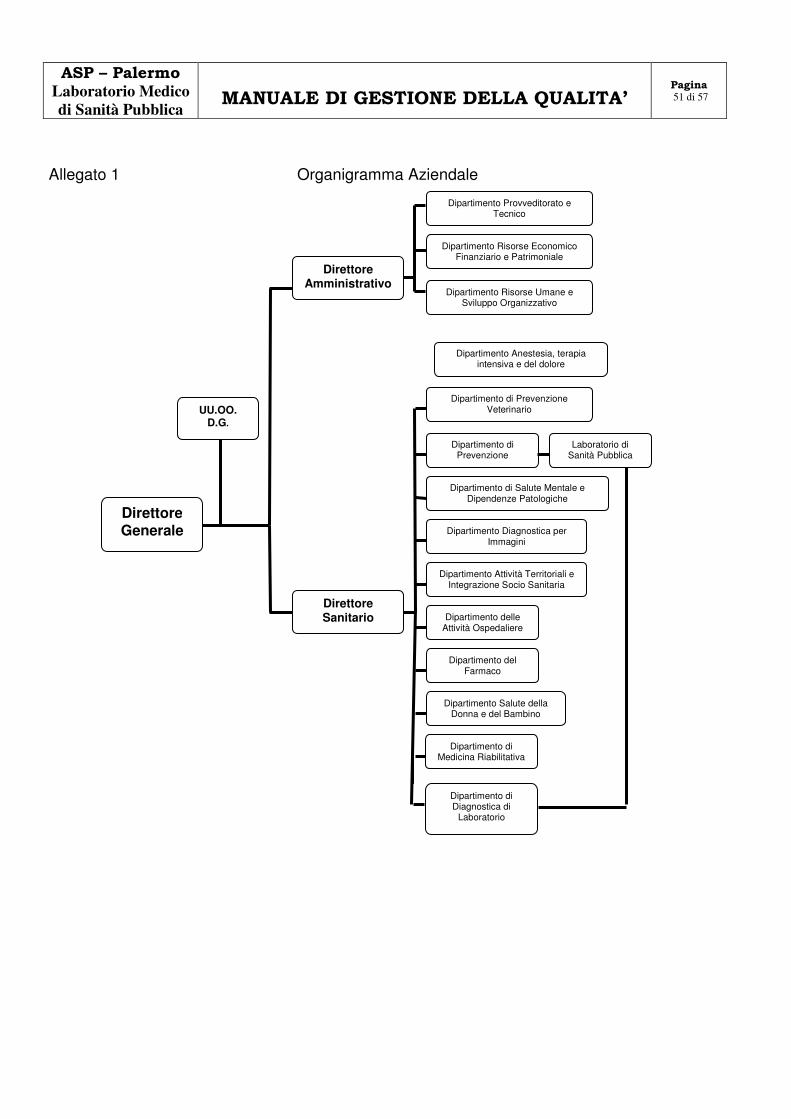
ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 51 di 57
Direttore Generale
UU.OO. D.G.
Direttore Amministrativo
Dipartimento Provveditorato e Tecnico
Dipartimento Risorse Economico Finanziario e Patrimoniale
Dipartimento Risorse Umane e Sviluppo Organizzativo
Dipartimento di Prevenzione
Dipartimento di Salute Mentale e Dipendenze Patologiche
Direttore Sanitario
Dipartimento Salute della Donna e del Bambino
Dipartimento del Farmaco
Dipartimento delle Attività Ospedaliere
Dipartimento Attività Territoriali e Integrazione Socio Sanitaria
Dipartimento Diagnostica per Immagini
Laboratorio di Sanità Pubblica
Dipartimento di Prevenzione Veterinario
Dipartimento di Medicina Riabilitativa
Dipartimento Anestesia, terapia intensiva e del dolore
Dipartimento di Diagnostica di
Laboratorio
Allegato 1 Organigramma Aziendale
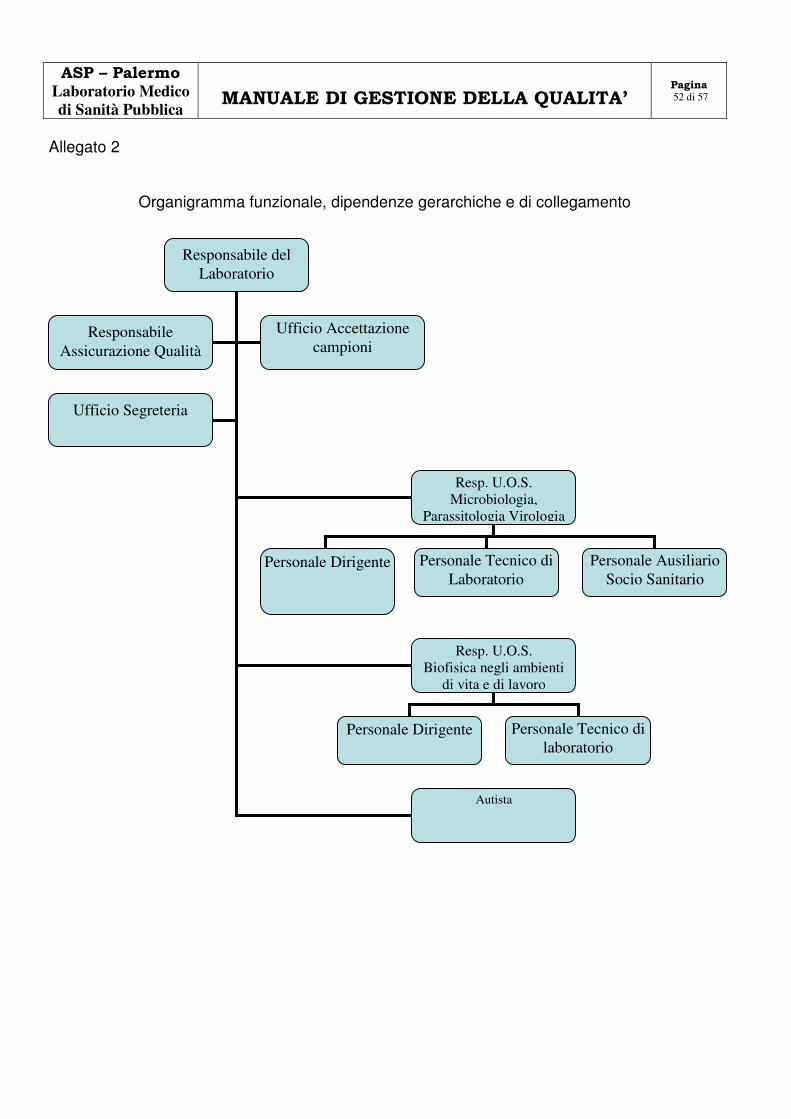
ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 52 di 57
Allegato 2
Organigramma funzionale, dipendenze gerarchiche e di collegamento
Responsabile del
Laboratorio
Resp. U.O.S.
Microbiologia,
Parassitologia Virologia
Resp. U.O.S.
Biofisica negli ambienti
di vita e di lavoro
Responsabile
Assicurazione Qualità
Ufficio Accettazione
campioni
Ufficio Segreteria
Personale Dirigente
Autista
Personale Ausiliario
Socio Sanitario
Personale Tecnico di
Laboratorio
Personale Dirigente Personale Tecnico di
laboratorio

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 53 di 57
Allegato 3
Planimetria e identificazione dei locali:
N. 1 Studio
N. 2 Direzione
N. 3 Accettazione campioni di prova
N. 4 Segreteria
N. 5 Studio RAQ/RUO
N. 6 Studio
N. 7 Studio
N. 8 Lab. Microbiologia alimenti e acque
N. 9 Lab. Microbiologia alimenti e acque
N. 10 Conservazione terreni di coltura
N. 11 Lab. Microbiologia Speciale (Legionella pn.)
N. 12 Servizi igienici
N. 13 Studio tecnici
N. 14 Magazzino
N. 15 Lab. Preparazione terreni di coltura
N. 16 Sterilizzazione
N. 17 Lavaggio vetreria
N. 18 EX Lab. PCR real time (OGM – biologia molecolare)
N. 19 EX Lab. PCR real time (OGM – biologia molecolare)
N. 20 Lab. di diagnostica microbiologica
N. 21 Archivio documenti
N. 22 Studio
N. 23 Laboratorio chimico
N. 24 Laboratorio chimico
N. 24 Laboratorio chimico
N. 25 Laboratorio chimico
N. 26 Laboratorio chimico
N. 27 Laboratorio chimico

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 54 di 57
N. 28 Laboratorio chimico
N. 29 Magazzino
N. 30 Studio
N. 31 Servizi igienici
N. 32 Laboratorio chimico
N. 33 Locale apparecchiature
N. 34 Laboratorio chimico
N. 35 Laboratorio chimico
N. 36 Studio
N. 37 Ingresso Laboratori Amianto (piano terra)
N. 38 Laboratorio Microscopia Elettronica
N. 39 Servizio Igienico
N. 40 Servizio Igienico
N. 41 Laboratorio preparazione campioni
N. 42 Laboratorio Diffrattometria a raggi X
N. 43 Laboratorio Spettrofotometria IR
N. 44 Sala attesa UO Biofisica
N. 45 Studio
N. 46 Laboratorio amianto
N. 47 Laboratorio
N. 48 Servizio Igienico
N. 49 Laboratorio analisi amianto ( aria)
N. 50 Laboratorio analisi amianto ( massa)
N. 51 Studio
N. 52 Studio
N. 53 Servizi Igienici

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 55 di 57

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 56 di 57

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 57 di 57
Allegato 4
ELENCO APPARECCHIATURE IN DOTAZIONE ALLA UO DI MICROBIOLOGIA
Denominazione
Taratura (si-no)
Incubatore refrigerato si Incubatore refrigerato si
Incubatore si Cappa a flusso laminare si
Frigocongelatore si Frigorifero a due ante si
Frigorifero si Frigorifero a tre ante si
Aspiratore no Bilancia si
Stomacher 400 no Stomacher 400 no
Stomacher 3500 no Incubatore si
Misuratore di acqua libera si pH-metro si
Bagno termostatico digitale si Armadio termostatico si
Cabina T3 no Stufa a secco si
Agitatore magnetico a piastra no Agitatore magnetico a piastra no
Piastra riscaldante no Bilancia si
Cappa aspirante no Incubatore si Frigorifero si
Microscopio no Lavavetreria no Lavavetreria no
Lavavetreria a ultrasuoni no Sistema API si Congelatore si Congelatore si
Fotometro per ELISA si Incubatore si

ASP – Palermo Laboratorio Medico
di Sanità Pubblica
MANUALE DI GESTIONE DELLA QUALITA’ Pagina 58 di 57
Denominazione
Taratura (si-no) Sistema di filtrazione no Sistema di filtrazione no
Pompa da vuoto no Pompa da vuoto no
Autoclave si Autoclave si Autoclave si
Frigorifero a tre ante si Cappa a Flusso Laminare si
Elettroforesi no Transilluminatore UV no Heavy Duty Blender no
Termomixer si Ultracentrifuga no
ELENCO APPARECCHIATURE IN DOTAZIONE ALLA UO
DI BIOFISICA
Denominazione
Calibrazione e /o
Taratura (si-no)
Microscopio ottico a contrasto di fase Si Cappa a flusso laminare II Classe Si
Cappa a flusso laminare di III classe Si Microscopio elettronico Si Diffrattometro a raggi X Si
Spettrofotometro IR Si Pompe aspiranti Si
Mulino macina campioni No


















