
Dossier N.15 Le epatopatie autoimmuni - service.synlab.it · fegato e sistema immune...
28
Fegato e sistema immune Le epatopatie autoimmuni Dossier N.15
Transcript of Dossier N.15 Le epatopatie autoimmuni - service.synlab.it · fegato e sistema immune...

Fegato e sistema immune
Le epatopatieautoimmuni
Dossier N.15


3
Dossier N. 15Ottobre 2011
Parte i: Le maLattie autoimmuNi DeL fegatoIntroduzione 4Epatite autoimmune 6Cirrosi Biliare Primitiva 9Colangite sclerosante primitiva 12
Parte 2: gLi autoaNticorPi NeLLe maLattie ePaticheGli Autoanticorpi nelle malattie epatiche 14La diagnostica di laboratorio nelle epatopatie autoimmuni 16Gli autoanticorpi 18Diagnostica di laboratorio della Cirrosi Biliare Primitiva (PBC) 24Diagnostica di laboratorio della Colangite Sclerosante (PSC) 26Impressum 26Bibliografia Parte 1 e 2 26

4
iNtroDuZioNe
fegato e sistema immuneNell‘antichità il fegato ha sempre suscitato un notevole fascino. Non solo veniva considerato essenziale per la vita, in quanto da quest‘organo si pensava originasse il sangue, ma addirittura si credeva fosse sede dell‘anima, della compassione e dell‘amore. Anche nella letteratura Araba, mentre il cuore era considerato la sede dell‘intelletto, il fegato quella dell‘anima.Nelle arti divinatorie molti popoli sacrificavano animali (soprattutto il montone) e dall‘osservazione del fegato e delle vie biliari traevano auspici sul futuro e il destino.Oggi chiaramente non crediamo più che il fegato sia la sede dell‘anima o dell‘amore, ma siamo semplicemente consapevoli che sia il laboratorio centrale, dove innumerevoli reazioni biochimiche si svolgono allo scopo di mantenere in equilibrio il nostro organismo.Un‘altra peculiare caratteristica del fegato è rappresentata dal fatto che è un organo in cui il sistema immune in presenza di vari agenti esterni è relativamente poco efficace; così nelle epatiti B e C è frequente la non completa eliminazione del virus con cronicizzazione della malattia e frequente evoluzione in cirrosi. Altro esempio è la malaria dove a livello epatico si constata una quasi assenza di risposta immunologica nei confronti del parassita malarico che migra attraverso il fegato stesso e dove subisce una sostanziale parte della sua maturazione. Molteplici meccanismi sono stati invocati per spiegare i motivi per cui nel fegato la risposta immunologica è spesso deviata verso una relativa immunotolleranza. Recentemente si sta sottolineando, per spiegare questa caratteristica, il
ruolo della presentazione degli Antigeni. Nel fegato le cellule deputate a questo compito sono molteplici: le cellule dentritiche, i fagociti, le cellule di Ito, le stesse cellule epatiche, ecc. Molte di queste cellule avrebbero le caratteristiche di favorire più una immunosoppressione che una immunostimolazione (secrezione di IL10, TGFbeta1, particolari chemochine, ecc.). A questo proposito si legga una recente review di Ian Nicholas Crispe („Liver antigenpresenting cells“ J.Hepatol. 54, 357365, 2011).

5
Pertanto, essendo in circostanze normali il fegato un organo dal punto di vista immunologico relativamente tollerante, le malattie autoimmuni sono più rare rispetto a quelle interessanti altri organi o apparati (tiroiditi, artrite reumatoide, glomerulonefriti, diabete mellito di tipo 1, ecc.).Anche per questa relativa rarità le diagnosi di queste malattie sono sovente tardive o addirittura misconosciute, soprattutto in quei paesi dove le forme virali, tossiche e metaboliche che colpiscono il fegato sono assai frequenti. Non scordiamo che la seconda reale causa di cirrosi criptogenetica, dopo quella di steatoepatite non alcolica, è proprio l‘epatite autoimmune.E ciò è particolarmente grave, considerando che un trattamento adeguato è in grado di portare in remissione totale la malattia in quasi tutti i pazienti. Ne consegue l‘ovvia importanza di una diagnosi il più possibile precoce e precisa. Purtroppo spesso la clinica non aiuta, essendo la sintomatologia assai variabile, andando da forme blande e poco evolutive a epatiti fulminanti, per fortuna assai rare, per le quali l‘unico intervento utile è un urgente trapianto di fegato. La diagnostica risiede sull‘esclusione di altre cause di malattia, ma soprattutto su esami immunologici di laboratorio (positività di anticorpi antinucleo, antimuscolo liscio, anti LKM, anti SLA/LP), sulla biopsia epatica e su algoritmi clinici.Un‘altra patologia del fegato interessante le vie biliari, dove è anche coinvolto il sistema immune con tipica presenza di colestasi cronica, è la cirrosi biliare primitiva. Anche in questo caso la diagnosi è incentrata su test immunologici (positività di anticorpi antimitocondrio) ed esame istologico del fegato.Infine esistono, anche se meno numerose, delle forme intermedie (cosiddette „overlap syndromes“) dove è presente, in vario grado, sia una componente colestatica, sia una componente infiammatoria e che richiedono trattamenti misti che agiscono sulle vie biliari (acido ursodesossicolico) e sull‘attività epatica (cortisone).
Questa Brochure è rivolta ai Medici, soprattutto di Medicina generale Ospedalieri e non Ospedalieri, ai Biologi laboratoristi, che „lavorano sul campo“, ed ha lo scopo di presentare i dati essenziali riguardanti la clinica e la diagnostica delle principali malattie autoimmuni del fegato. Gli autori sono dei Clinici e Ricercatori noti in tutto il mondo, con lavori personali pubblicati sulle più qualificate Riviste Internazionali.Dal momento che fondamentali per una diagnosi precisa sono il riscontro della presenza di relativamente specifici, spesso eterogenei autoanticorpi, abbiamo dedicato un capitolo alla diagnostica. Da sottolineare che le metodiche sono varie, alcune complesse, che spesso richiedono esperienza del personale di laboratorio e del Medico che deve interpretare e correlare questi dati con la clinica. Non è certamente come valutare i livelli delle transaminasi sieriche!
Gaetano Ideo

6
ePatite autoimmuNe
Il fegato può essere aggredito da noxae patogene di varia natura, non ultima quella autoimmune. Si può affermare altresì che il fegato sia stato il “pioniere” dell’autoimmunità, essendo stato il primo organo ad essere identificato come bersaglio di tale tipologia di danno [1]. Sessant’anni fa, prima delle scoperte di Ian R. Mackay sull’epatite autoimmune, si credeva che tutte le epatiti croniche “attive” fossero il risultato di un’infezione virale persistente nel fegato [2, 3]. Ad oggi le malattie autoimmuni del fegato si possono classificare in tre grandi entità, che racchiudono al loro interno alcune varianti, così come sindromi da sovrapposizione cosiddette “overlap”. L‘Epatite Autoimmune è un’infiammazione cronica del fegato a causa sconosciuta, nella quale viene persa la tolleranza immunitaria verso l’epatocita [4]. Si caratterizza per: a) un’epatite periportale (infiammazione dello spazio portale); b) movimento di autoanticorpi e aumento delle immunoglobuline cirocolanti c) esclusione di altre patologie epatiche ed epatiti croniche (per es. da virus HCV, HBV, emocromatosi, morbo di Wilson ecc., cirrosi biliare primitiva, steatoepatite alcolica) [5]. La malattia colpisce soprattutto le donne, sia in età pediatrica che in età adulta [6]. E’ un processo autoimmune e pertanto si associa, talora, ad altri quadri autoimmuni come la colite ulcerosa [7] e la malattia di Graves [8].
classificazioneSi è soliti classificare l’epatite autoimmune sulla base del riscontro degli autoanticorpi, che sono per il tipo 1 rappresentati massimamente dagli ANA (antinucleo) e SMA (antimuscolo liscio); il tipo 2 presenta invece gli anticorpi LKM1 (liverkidney microsomial antibodies type 1) che non coesistono con i primi citati. Gli autoanticorpi antiLKM1 sono diretti contro il citocromo P450 (CYP) 2D6. L’epatite autoimmune cosiddetta di tipo 3 si caratterizza per anticorpi anti-SLA (soluble liver antigens) e anti-LP (liver-pancreas). L’11% dei pazienti con epatite autoimmune di tipo 1 ha antiSLA. Altri tipi di anticorpi riscontrati sono quelli diretti contro le asialoglicoproteine (anti-ASGPR), contro il citosol epatico di tipo 1 (anti-LC1) e anticorpi citoplasmatici antineutrofili perinucleari (pANCA). Questi ultimi sono stati riscontrati specialmente nella colangite sclerosante [911].
1

7
DiagnosiI criteri diagnostici per l’epatite autoimmune sono un rialzo di immunoglobuline > di 1,5 volte rispetto al limite superiore della norma e i titoli sierici di SMA ed ANA ed LKM1 > di 1:80. Non vi deve essere stato uso di alcool, né epatite cronica virus B e C correlata, né da virus di EpsteinBarr, né Cytomegalovirus.
Esiste un punteggio (score), vedi tabella 1, per fare diagnosi di certezza dell‘epatite autoimmune [12].
criterio PuNteggio
Sesso femminile +2Rapporto ALP:AST (o ALT)< 1.5 +21.53.0 0> 3.0 2Immunoglobuline sieriche o IgG> 2.0 +31.52.0 +21.01.5 +1< 1.0 0ANA, ASMA, anti-LKM1> 1:80 +31:80 +21:40 +1<1:40 0AMAPositivo 4Negativo 0Markers virali epatititiciPositivi 3Negativi +3Utilizzo di farmaci epatotossiciSi 4No +1Consumo di alcol<25 g/die +2>60 g/die 2IstologiaEpatite dell’interfaccia +3Infiltrato prevalentemente linfoplasmacitoide +1Rosetting liver cells +1Nessuna delle precedenti 5Alterazioni dei dotti biliari 3Altre lesioni 3Altre malattie autoimmuni +2Parametri aggiuntiviPositivita’ per altri autoanticorpi (pANCA, antiSLA, antiASGPR)
+2
HLA DR3 o DR4 +1Risposta al trattamento immunosoppressivoCompleta +2Riacutizzazione +3
Interpretazione del punteggio totale: pretrattamento: certezza della patologia se > 15, probabilità se 1015. posttrattamento: certezza della patologia se > 17, probabilità se 1217.
8
Quando la curaLa gravità dell’attività infiammatoria viene valutata sulla base degli indici biochimici di citolisi, per es. un rialzo delle AST almeno di 10 volte (10X) la norma o più, oppure AST > 5X associata ad ipergammaglobulinemia. Il quadro istologico evidenzierà ponti necrotici o necrobiosi confluente, associati ad infiltrato linfoplasmacellulare ed epatite d’interfaccia. Meritano una terapia i soggetti che presentino un rialzo delle transaminasi, delle immunoglobuline, evidenza istologica di epatite
dell’interfaccia o attività necroinfiammatoria. Il trattamento precoce sembra essere associato ad una prognosi più favorevole. Una biopsia epatica a tempo zero è utile per la diagnosi ma anche per studiare grado e stadio dell’epatopatia [16]. Sono molto pochi i pazienti che debbono essere esclusi dal trattamento e sono rappresentati da coloro che presentano una cirrosi scompensata in lista per trapianto oppure con cirrosi epatica e senza attività di malattia [4]. L’età di per sé non costituisce un’esclusione dalla terapia [17].
terapiaSi basa su cicli di cortisonici per 4 settimane iniziando con 60 mg di prednisone. Segue un periodo di décalage di 10 mg a settimana, poi cicli di mantenimento attorno ai 10 mg o meno; tuttavia è possibile una terapia di combinazione con cortisonico + azatioprina, che permette di ridurre al minimo il dosaggio di steroide durante il mantenimento [1820]. In caso questi farmaci siano controindicati o mal tollerati, oppure si assista ad una non risposta o ad una risposta solo parziale, si può ricorrere ad immunosoppressori di seconda scelta: ciclosporina [21] e micofenolato [22]. La Budesonide sembra sortire una risposta migliore rispetto a quella del prednisone, con minori effetti collaterali [23].
eziologia e patogenesi del danno epaticoL’origine della malattia è ancora oggetto di controversie [13].Sono riportate diverse associazioni con antigeni di istocompatibilità e in particolare HLA A1 e B8 [14]I meccanismi che stanno alla base del danno epatico sono rappresentati da:A) citotossicità cellulo-mediata anticorpodipendente.
Esistono autoanticorpi diretti contro le normali proteine di membrana dell’epatocita ed il complesso antigeneanticorpo sulla superficie dell’epatocita diventa bersaglio per i linfociti che hanno i recettori Fc per le molecole anticorpali (cellule Natural Killer).
B) citotossicità diretta. E’ un meccanismo mediato dai T CD8+ in cui un autoantigene malattiaspecifico viene esposto in modo anomalo sulla superficie dell’epatocita in associazione agli antigeni HLA (cioè di istocompatibilità): ne deriva che il linfocita citotossico non riconosce più come self la struttura e si attiva per distruggerla. Le linfochine che vengono liberate facilitano la comunicazione tra le cellule e promuovono l’espressione di neoantigeni HLA di classe II. Esiste dunque in entrambi i casi un incremento dell’immunoreattività cellulomediata [15].
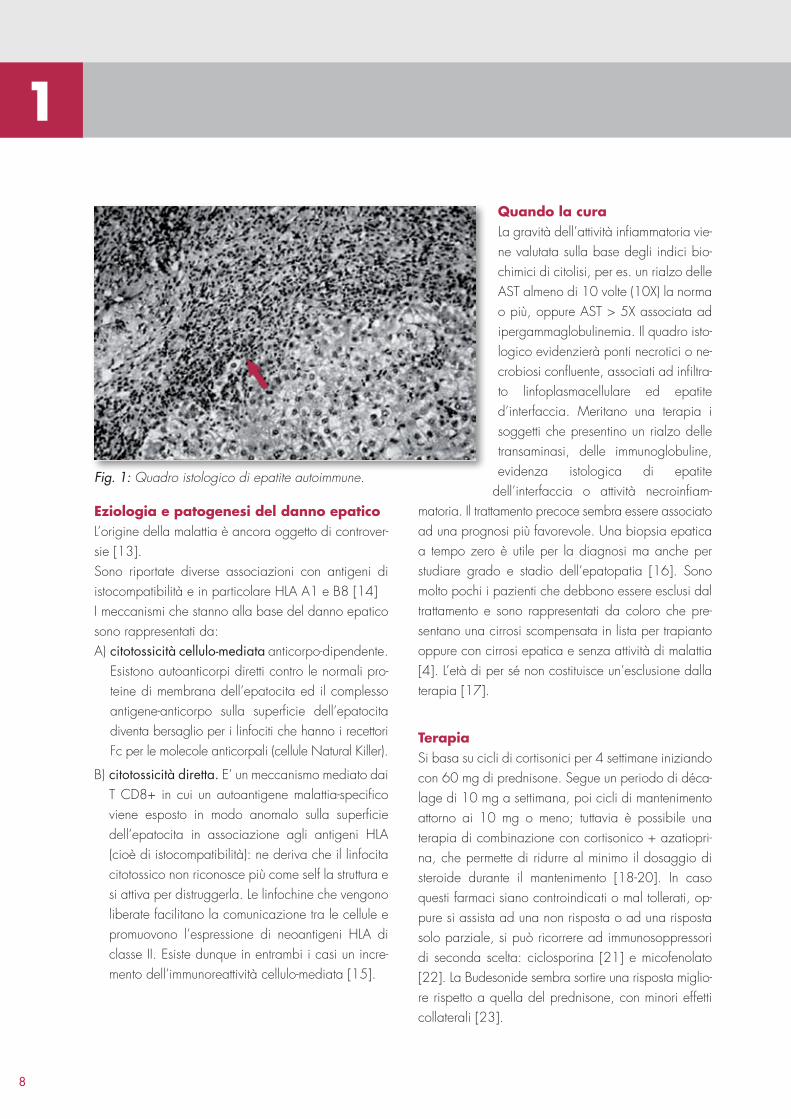
Fig. 1: Quadro istologico di epatite autoimmune.
1

La Cirrosi Biliare Primitiva (CBP) è una malattia cronica del fegato caratterizzata da innalzamento degli indici di colestasi, positività degli autoanticorpi antimitocondrio (AMA), istologia epatica peculiare (dapprima proliferazione duttulare, poi duttopenia e infine fibrosi e cirrosi; reperto caratteristico è la presenza di granulomi). Il termine “cirrosi”, applicato a questa malattia, è da considerarsi quantomeno inesatto, dal momento che la essa rappresenta solamente l’ultima fase della storia naturale, dopo 1020 anni dall’esordio, in assenza di trattamento [24].
eziologiaNonostante sia noto il coinvolgimento del sistema immunitario nella CBP, non è a tutt’oggi stato individuato alcun agente causale. Vi sono però numerose evidenze rispetto ai fattori di rischio ambientali: il fumo di sigaretta e le infezioni ricorrenti delle mucose, e in particolare del tratto urinario (specie se sostenute da E. Coli) hanno un ruolo riconosciuto. Un altro batterio, Novosphingobium Aromaticivorans, gramnegativo, aerobio, freeliving, ubiquitario nell’ambiente (acque, suolo) potrebbe avere un ruolo di iniziatore della risposta autoimmune. Il ruolo dei cosmetici, così come delle gravidanze e di terapie ormonali non è invece del tutto chiarito [25]. Oltre alle esposizioni ambientali, i fattori genetici sono critici nel determinare la suscettibilità alla malattia. Ciò risulta evidente anche per una certa quota di clustering familiare e di concordanza fra i gemelli monozigotici. Particolari varianti alleliche del MHC di classe II sembrano conferire una maggiore suscettibilità alla malattia. In generale viene riportata un’associazione con DRB1*08 ma con ampie differenze geografiche; gli alleli DRB1*11 e *13 sembrano invece avere un effetto protettivo. Gli alleli HLA non sembrano invece essere un fattore determinante particolari espressioni fenotipiche della malattia stessa, come la presenza di cirrosi o il titolo anticorpale. Numerosi componenti dell’immunità innata (C4*Q0, C4B*2, NRAMP1/SLC11A1, MBL, VDR) e adattativa (CTLA4, IL beta, TNF alpha, IL12A, IL12RB2) si sono dimostrati in grado di determinare un aumentato rischio di sviluppare CBP [26].
epidemiologiaLa malattia ha una prevalenza molto variabile a seconda dell’area geografica ed è compresa fra 6.7 e 402 per milione. E’ probabile, tuttavia, che una simile variabilità non rispecchi il dato reale, ma sia piuttosto il frutto di una diversa sensibilità alla diagnosi fra le differenti aree geografiche, oppure in una profonda differenza nelle metodiche di case finding [27]. Non esistono dati affidabili di prevalenza/incidenza in Italia perché è una patologia di interesse specialistico che spesso viene indirizzata a centri di riferimento. E’ più frequente nelle donne con un rapporto femmine:maschi di 10:1. Una simile polarizzazione di tale rapporto potrebbe essere dovuta a difetti del cromosoma X e particolarmente alla monosomia dell’X nelle cellule mononucleate del sangue periferico [28]. Il 20% dei pazienti con CBP presenta simultaneamente o consecutivamente un’altra malattia autoimmune, fra cui le più frequenti sono sclerosi sistemica, tirodite autoimmune, sindrome di Sjogren, malattia celiaca [29].
DiagnosiNella metà dei casi la PBC viene diagnosticata in modo casuale quando, per altri accertamenti o durante uno screening, vengono rilevati anomali livelli dei marcatori di patologia epatica: le transaminasi (AST e ALT) e soprattutto gli indici di colestasi (gammaGT e Fosfatasi Alcalina).
La diagnosi è data dalla presenza di almeno due dei seguenti tre criteri:
•positività della ricerca degli AMA (che si riscontra nel 95% dei casi) a titolo adeguato (1:40 all‘immu nofluorescenza indiretta);
•persistenza per oltre 6 mesi di alti valori di fosfatasi alcalina (maggiori di 1.5 volte);
•biopsia epatica compatibile.
Gli AMA sono considerati anticorpi altamente specifici e sensibili, e la loro presenza nel siero è virtualmente diagnostica. Talora si rileva anche la presenza degli ANA, soprattutto nei casi in cui siano assenti gli AMA [30].
cirrosi BiLiare PrimitiVa
9

10
La biopsia epatica dà la certezza diagnostica e mostra una „colangite cronica destruente non suppurativa“ (ovvero la presenza di granulomi che danneggiano i dotti biliari). Il danno istologico è classificato in 4 stadi, solo l‘ultimo dei quali corrisponde ad una franca cirrosi:
•Stadio 1: Infiltrato infiammatorio portale
•Stadio 2: Infiammazione e/o fibrosi periportale con proliferazione dei piccoli dotti biliari
•Stadio 3: Setti Fibrosi
•Stadio 4: Cirrosi Biliare (noduli di rigenerazione) [31]
aspetti cliniciLa CBP è una malattia cronica ad andamento, nella maggior parte dei casi, indolente. Inizialmente la sintomatologia è aspecifica e, dove presente, è dominata da astenia e prurito. Può comparire ittero, steatorrea, xantelasmi, iperpigmentazione cutanea. Oggi raramente si osserva una malattia avanzata al momento della diagnosi, con segni di cirrosi, ipertensione portale e/o insufficienza epatica. La colestasi cronica, però, produce un danno epatico che, se mantenuto efficiente, conduce alla cirrosi. Le complicanze extraepatiche della colestasi cronica comprendono il malassorbimento di vitamine liposolubili (in particolare D3) con conseguente osteoporosi anche
precoce o accelerata. Raramente può associarsi acidosi tubulare renale di tipo 1 (distale) [32]. I dati di laboratorio documentano tipicamente un quadro di colestasi anitterica con aumento della fosfatasi alcalina e delle gammaGT, del colesterolo totale solitamente con valori tra i 220 e i 290 mg/dl. L’ipercolesterolemia, tuttavia, se mantenuta entro tali valori, non sembra associarsi ad un aumentato rischio cardiovascolare e non necessiterebbe di terapia. Può associarsi un rialzo delle transaminasi, di solito modesto. Aumentati valori di IgM sono piuttosto frequenti. I casi più aggressivi sono caratterizzati da duttopenia precoce con severa colestasi itterica e da sviluppo di fibrosi e cirrosi in un tempo medio di circa 5 anni. La diagnosi differenziale si pone con la colangite sclerosante primitiva, con epatiti di diversa natura (virali, autoimmuni, tossiche, da accumulo…) che presentino aspetti di colestasi, la colelitiasi, con il Morbo di Caroli e la Fibrosi Epatica Congenita. Altre malattie che possono dare colestasi sono inoltre la sarcoidosi e il linfoma di Hodgkin.
Esistono poi sindromi cosiddette “overlap” con Epatite Autoimmune (AIH), caratterizzate da elevazione più marcata delle transaminasi, risposta allo steroide simile a quella osservata in AIH e istologicamente aspetti di epatite d’interfaccia con peacemeal necrosis e infiltrato plasmacellulare, oltre ai reperti caratteristici della colangiopatia granulomatosa. La diagnosi di Sindrome Overlap risponde ai 3 criteri seguenti: ALT > 5 volte la norma, IgG > 2 volte la norma e/o anticorpi AntiSm positivi, istologia caratterizzata da infiammazione periportale o perisettale moderata o severa [33].
complicanzeRilevante è la possibilità che alla PBC si associno altre malattie autoimmuni, per questo è consigliabile effettuare uno screening sierologico per rilevarne la presenza. Fino al 70% delle pazienti lamenta i sintomi della Sindrome di Sjögren (caratterizzata da secchezza orale ed oculare e da artralgie). Frequente è anche la tiroidite di Hashimoto, che può presentare i sintomi dell‘ipotiroidismo (stanchezza, torpore, aumento ponderale). Meno comuni sono la celiachia, l‘epatite auto
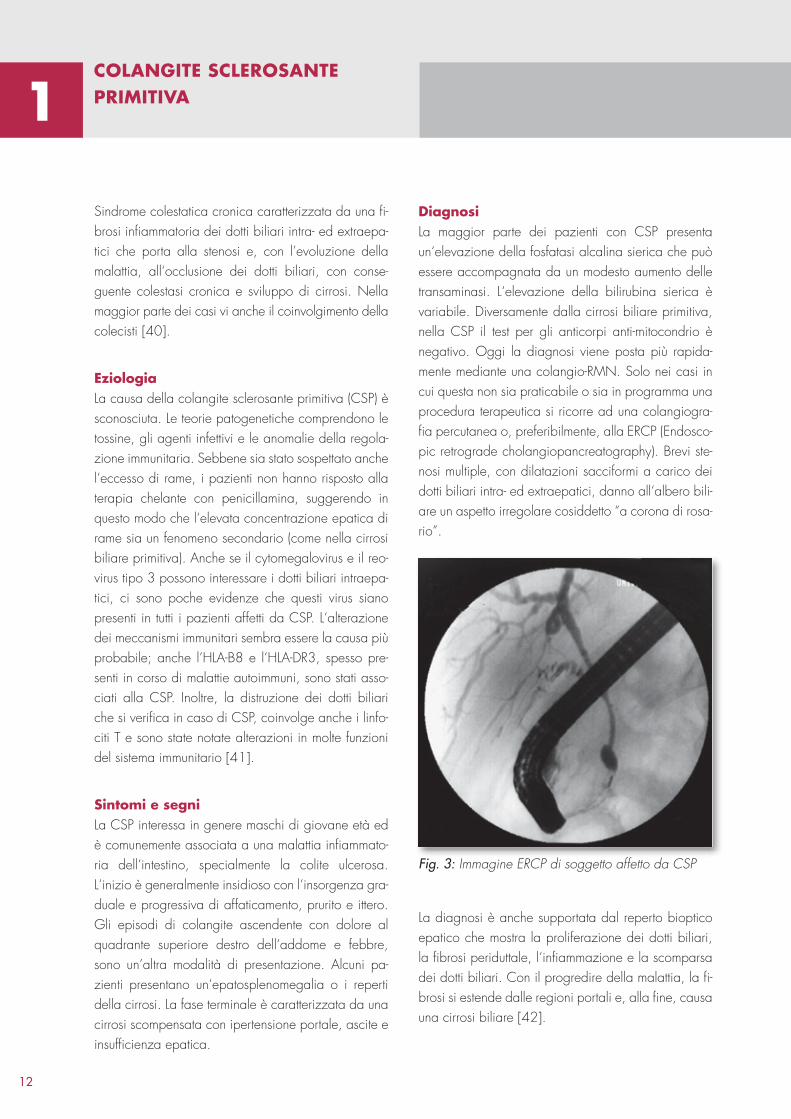
Fig. 2: Quadro istologico di cirrosi biliare primitiva
1

quella della popolazione generale. L’UDCA è globalmente un farmaco sicuro, con pochi effetti collaterali. Alcuni pazienti possono sviluppare diarrea, sintomi da reflusso, cui si può ovviare con l’assunzione del farmaco alla fine del pasto, e con l’impiego di un PPI (Inibitori di Pompa Protonica). Nei pazienti con colestasi marcata e prurito, l’introduzione di UDCA a pieno dosaggio può aumentare il prurito, sicché è buona norma in questi pazienti iniziare il trattamento con dosaggi sub ottimali, e quindi aumentare in 48 settimane. Si considera risposta ottimale una livello di bilirubina totale < 1 mg/dl, AST < 2 volte la norma e ALP < 3 volte la norma. Circa il 40% dei pazienti ha una risposta subottimale e richiede perciò terapie adiuvanti, ovvero l’arruolamento in trial clinici [37]
E’ utile altresì provvedere alla supplementazione con calcio e vitamina D per prevenire, ove possibile, la perdita di massa ossea. Un problema considerevole nella gestione dei pazienti con CBP è il trattamento del prurito, che si può giovare dell’impiego di colestiramina. Altri farmaci efficaci sono la rifampicina, gli antiistaminici, il naloxone. La plasmaferesi e la MARS (Molecular Adsorbent Recirculating System) sono provvedimenti utili nei casi gravi e resistenti ad altri trattamenti [38]. Il trapianto di fegato trova indicazione nella malattia avanzata, complicata da cirrosi scompensata e/o insufficienza epatica, come pure nei casi di prurito refrattario e severamente invalidante. La CBP ricorre posttrapianto in circa il 20% dei casi [39].
Alcuni farmaci vecchi e nuovi, promettenti, fra cui menzioniamo agonisti di FXR (recettore X dei Farnesoidi, coinvolto nella regolazione della sintesi degli acidi biliari) e farmaci biologici sono in sperimentazione per l’impiego nella CBP, nella speranza di ottenere una terapia soddisfacente anche in quella percentuale, non trascurabile, di casi refrattari ai trattamenti attuali.
11
immune (sindrome overlap), ed il diabete di tipo 1. Più rare sono l‘artrite reumatoide, la porpora trombocitopenica immune, la sclerosi sistemica, e la glomerulo nefrite membranosa. L‘osteoporosi è piuttosto frequente in quanto 1) le pazienti sono donne di mezza età e pertanto possono essere affette dall‘osteoporosi postmenopausale 2) la stasi biliare non consente un corretto assorbimento intestinale della vitamina D esogena (che è liposolubile) 3) il danno epatico determina una ridotta attivazione della vitamina D endogena [34]. Inoltre è aumentato il rischio di epatocarcinoma, per il quale è consigliabile intraprendere un programma di sorveglianza semestrale (ecografia epatica e dosaggio dell‘alfafetoproteina) [35]. Nelle fasi avanzate della malattia compaiono i segni dell‘insufficienza epatica e dell‘ipertensione portale e non vi è differenza con la cirrosi da altra causa.
PrognosiI principali fattori prognostici sono:
•la precocità della diagnosi;
•lo stadio istopatologico rivelato dalla biopsia;
•l‘entità della sintomatologia;
•la positività a specifici tipi di ANA (anticorpi antinucleo), come gli antigp210 e gli antisp100;
•la risposta alla terapia (in particolare la diminuzione della fosfatasi alcalina ed i livelli di bilirubina);
•la sovrapposizione con altre patologie autoimmuni.
La prognosi a breve termine (fino a 2 anni) può essere calcolata secondo il „Mayo Score“, un sistema di punteggio elaborato dalla Mayo Clinic di Rochester [36]
terapiaL’unica terapia attualmente approvata per la CBP è l’acido ursodesossicolico (UDCA) a dosaggio di 1315 mg/kg/die, che è in grado di abbassare fino al range di normalità gli indici di colestasi e migliorare la prognosi di questi pazienti che, nella maggior parte degli studi longterm si è dimostrata paragonabile a quella dei gruppi di controllo. La spettanza di vita dei pazienti che rispondono a UDCA è sovrapponibile a
12
coLaNgite scLerosaNte PrimitiVa
Sindrome colestatica cronica caratterizzata da una fibrosi infiammatoria dei dotti biliari intra ed extraepatici che porta alla stenosi e, con l’evoluzione della malattia, all‘occlusione dei dotti biliari, con conseguente colestasi cronica e sviluppo di cirrosi. Nella maggior parte dei casi vi anche il coinvolgimento della colecisti [40].
eziologiaLa causa della colangite sclerosante primitiva (CSP) è sconosciuta. Le teorie patogenetiche comprendono le tossine, gli agenti infettivi e le anomalie della regolazione immunitaria. Sebbene sia stato sospettato anche l‘eccesso di rame, i pazienti non hanno risposto alla terapia chelante con penicillamina, suggerendo in questo modo che l‘elevata concentrazione epatica di rame sia un fenomeno secondario (come nella cirrosi biliare primitiva). Anche se il cytomegalovirus e il reovirus tipo 3 possono interessare i dotti biliari intraepatici, ci sono poche evidenze che questi virus siano presenti in tutti i pazienti affetti da CSP. L‘alterazione dei meccanismi immunitari sembra essere la causa più probabile; anche l‘HLAB8 e l‘HLADR3, spesso presenti in corso di malattie autoimmuni, sono stati associati alla CSP. Inoltre, la distruzione dei dotti biliari che si verifica in caso di CSP, coinvolge anche i linfociti T e sono state notate alterazioni in molte funzioni del sistema immunitario [41].
sintomi e segni La CSP interessa in genere maschi di giovane età ed è comunemente associata a una malattia infiammatoria dell‘intestino, specialmente la colite ulcerosa. L‘inizio è generalmente insidioso con l‘insorgenza graduale e progressiva di affaticamento, prurito e ittero. Gli episodi di colangite ascendente con dolore al quadrante superiore destro dell‘addome e febbre, sono un’altra modalità di presentazione. Alcuni pazienti presentano un‘epatosplenomegalia o i reperti della cirrosi. La fase terminale è caratterizzata da una cirrosi scompensata con ipertensione portale, ascite e insufficienza epatica.
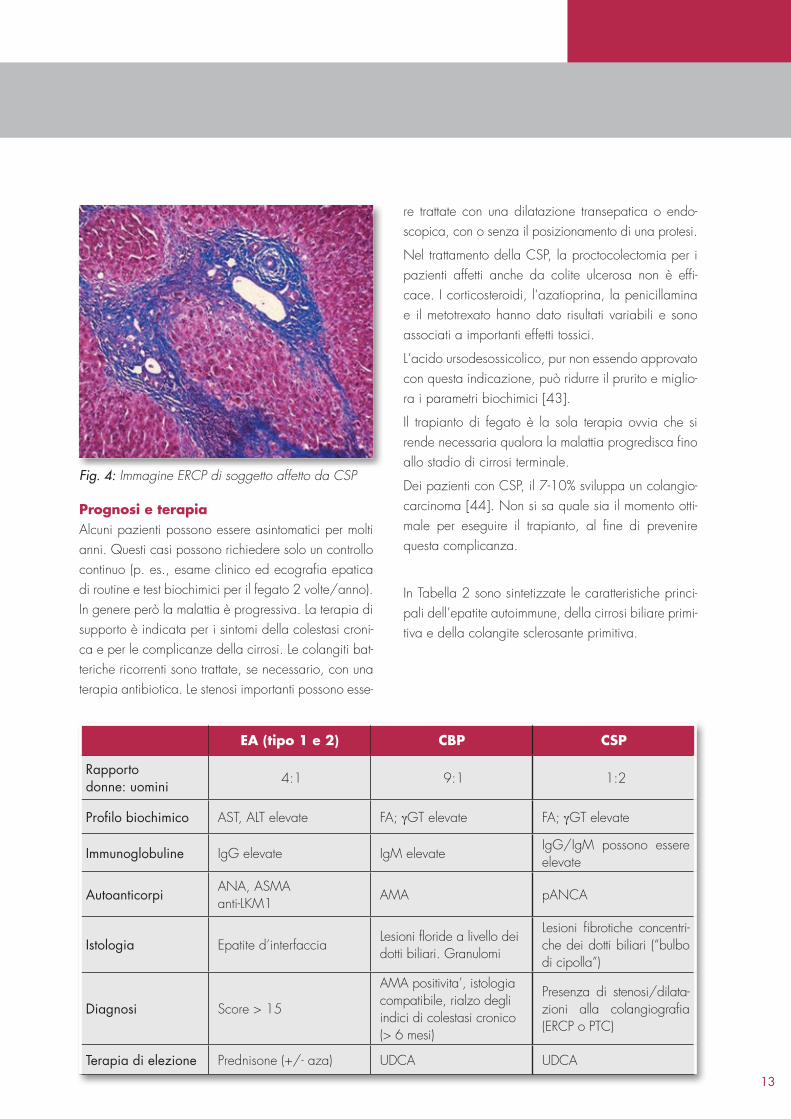
Diagnosi La maggior parte dei pazienti con CSP presenta un‘elevazione della fosfatasi alcalina sierica che può essere accompagnata da un modesto aumento delle transaminasi. L‘elevazione della bilirubina sierica è variabile. Diversamente dalla cirrosi biliare primitiva, nella CSP il test per gli anticorpi antimitocondrio è negativo. Oggi la diagnosi viene posta più rapidamente mediante una colangioRMN. Solo nei casi in cui questa non sia praticabile o sia in programma una procedura terapeutica si ricorre ad una colangiografia percutanea o, preferibilmente, alla ERCP (Endoscopic retrograde cholangiopancreatography). Brevi stenosi multiple, con dilatazioni sacciformi a carico dei dotti biliari intra ed extraepatici, danno all‘albero biliare un aspetto irregolare cosiddetto “a corona di rosario”.
La diagnosi è anche supportata dal reperto bioptico epatico che mostra la proliferazione dei dotti biliari, la fibrosi periduttale, l‘infiammazione e la scomparsa dei dotti biliari. Con il progredire della malattia, la fibrosi si estende dalle regioni portali e, alla fine, causa una cirrosi biliare [42].
Fig. 3: Immagine ERCP di soggetto affetto da CSP
1
13
Prognosi e terapia Alcuni pazienti possono essere asintomatici per molti anni. Questi casi possono richiedere solo un controllo continuo (p. es., esame clinico ed ecografia epatica di routine e test biochimici per il fegato 2 volte/anno). In genere però la malattia è progressiva. La terapia di supporto è indicata per i sintomi della colestasi cronica e per le complicanze della cirrosi. Le colangiti batteriche ricorrenti sono trattate, se necessario, con una terapia antibiotica. Le stenosi importanti possono esse
re trattate con una dilatazione transepatica o endoscopica, con o senza il posizionamento di una protesi.
Nel trattamento della CSP, la proctocolectomia per i pazienti affetti anche da colite ulcerosa non è efficace. I corticosteroidi, l‘azatioprina, la penicillamina e il metotrexato hanno dato risultati variabili e sono associati a importanti effetti tossici.
L‘acido ursodesossicolico, pur non essendo approvato con questa indicazione, può ridurre il prurito e migliora i parametri biochimici [43].
Il trapianto di fegato è la sola terapia ovvia che si rende necessaria qualora la malattia progredisca fino allo stadio di cirrosi terminale.
Dei pazienti con CSP, il 710% sviluppa un colangiocarcinoma [44]. Non si sa quale sia il momento ottimale per eseguire il trapianto, al fine di prevenire questa complicanza.
In Tabella 2 sono sintetizzate le caratteristiche principali dell’epatite autoimmune, della cirrosi biliare primitiva e della colangite sclerosante primitiva.
Fig. 4: Immagine ERCP di soggetto affetto da CSP
ea (tipo 1 e 2) cBP csP
Rapporto donne: uomini
4:1 9:1 1:2
Profilo biochimico AST, ALT elevate FA; γGT elevate FA; γGT elevate
Immunoglobuline IgG elevate IgM elevateIgG/IgM possono essere elevate
AutoanticorpiANA, ASMA antiLKM1
AMA pANCA
Istologia Epatite d’interfacciaLesioni floride a livello dei dotti biliari. Granulomi
Lesioni fibrotiche concentriche dei dotti biliari (“bulbo di cipolla”)
Diagnosi Score > 15
AMA positivita’, istologia compatibile, rialzo degli indici di colestasi cronico (> 6 mesi)
Presenza di stenosi/dilatazioni alla colangiografia (ERCP o PTC)
Terapia di elezione Prednisone (+/ aza) UDCA UDCA

Gli autoanticorpi specifici epatici si riscontrano nelle Epatopatie autoimmuni primarie, ma anche nelle forme secondarie, come le Malattie autoimmuni extraepatiche o nell’ambito di infezioni epatiche.
Le tre forme principali di epatopatie autoimmuni primarie (Malattie autoimmuni del fegato – AILD) sono:
•L’epatite autoimmune (AIH – Autoimmune Hepatitis)
•Cirrosi biliare primitiva (PBC – Primary biliary cirrhosis)
•Colangite sclerosante primitiva (PSC – Primary sclerosing cholangitis)
Gli autoanticorpi specifici sono importanti come supporto alla diagnostica, in associazione alla sintomatologia clinica, al fine di differenziare le diverse forme di epatopatie. La determinazione degli autoanticorpi diventa quindi essenziale ai fini della diagnosi.
L’epatite autoimmune viene classificata a seconda degli autoanticorpi individuati in Tipo I (test ANA e/o ASMA e/o antiSLA/LP) e Tipo II (antiLKM1)1.
Per quanto riguarda la cirrosi biliare primitiva, nella maggior parte dei casi (9095%) vengono individuati autoanticorpi antimitocondri (AMA), occasionalmente associati con specifici patterns di autoanticorpi anti nucleo ANA (nuclear dot/SP100, Nuclear pore glycoprotein210 (gp210), Lamin B receptor (LBR). In rari casi, tuttavia, non possono essere rilevati autoanticorpi. Alcuni autori definiscono la cirrosi biliare primitiva AMAnegativa come Colangite autoimmune (AIC)2. Per quanto riguarda i pazienti affetti da colangite sclerosante primitiva, fino nell’80% dei casi sono stati individuati infatti autoanticorpi anticitoplasma dei neutrofili tipo pANCA , che tuttavia non sono marcatori specifici della malattia.
In alcuni pazienti si riscontra la sovrapposizione di più malattie autoimmuni, sindrome da Overlap, come ad esempio l’associazione tra epatite autoimmune e cirrosi biliare primitiva, o tra epatite autoimmune e colangite sclerosante primitiva. L’overlap tra AIH e PBC si presenta in due forme: AIH con colangite autoimmune (PBC AMAnegativa) o AIH con cirrosi biliare primitiva. Tra i bambini e gli adolescenti si osserva invece
14
gLi autoaNticorPi NeLLe maLattie ePatiche2

più frequentemente la sindrome da overlap AIH/PSC. Per quanto riguarda l’overlap tra PSC e PBC, esistono solo “case report”. L’individuazione di una sindrome da overlap può essere complicata e vengono individuate per lo più a seguito dell’analisi dei rispettivi autoanticorpi e dell’analisi istologica.
La formazione degli autoanticorpi epatici può tuttavia avvenire in un secondo momento e questo accade soprattutto per le malattie autoimmuni extraepatiche o sistemiche che determinano la formazione di alcuni autoanticorpi. Per esempio, nel corso della vita del 2550% dei pazienti affetti da LES (Lupus Eritematoso Sistemico) si riscontrano autoanticorpi specifici epatici. Di questi pazienti, circa il 70% soddisfa i criteri diagnostici di AILD (Autoimmune Liver Disease), ma solo il 20% ha cambiamenti istologici conformi alla malattia. La prevalenza di AILD nei pazienti affetti da LES è circa del 5%3.
Le altre associazioni con patologie autoimmuni di origine extraepatica sono:
•AIH e tiroidite autoimmune
•PBC e sindrome CREST
•PSC e colite ulcerosa
Una formazione di autoanticorpi secondaria può essere osservata anche in caso di epatite virale: in presenza di infezione da HCV, in più del 50% dei casi vengono individuati autoanticorpi, per lo più sono fattori reumatoidi con crioglobulinemia. Nel 510% dei casi questi autoanticorpi sono altamente predittivi e portano spesso a sintomi clinicamente manifesti. Nel 25% circa dei pazienti con infezione da HCV si riscontrano anticorpi antiLKM1, che sono normalmente associati all’epatite autoimmune di tipo II; quindi i pazienti con anticorpi LKM1 dovrebbero in ogni caso essere sottoposti ad accertamenti per verificare la presenza di un’infezione da HCV4.
Comunque, un’infezione da HCV con la presenza di autoanticorpi anti LKM1 non pregiudica il successo della terapia con sostanze immunostimolanti come l’Interferone α.
Le malattie autoimmuni sono caratterizzate da una triade di fattori ricorrenti:
•La predominanza di sesso femminile
•La presenza di autoanticorpi
•Una buona risposta alla terapia immunosoppressiva
Per le Malattie epatiche autoimmuni queste caratteristiche sono parzialmente soddisfatte rendendo più complessa la determinazione di queste patologie.
Infatti, sebbene la Cirrosi Biliare Primitiva abbia una maggiore predominanza nel genere femminile e autoanticorpi altamente specifici, non risponde o risponde limitatamente alla terapia immunosoppressiva; mentre la Colangite Sclerosante Primitiva ha una predominanza negli uomini e non presenta autoanticorpi specifici.
15

16
La DiagNostica Di LaBoratorio NeLLe ePatoPatie autoimmuNi6,7
Diagnostica di base•Transaminasi (GOT/AST, GPT/ALT)•Parametri di colestasi (gGT, ALP)
A causa dell’eterogeneità delle epatopatie autoimmuni, ogni paziente con livelli di transaminasi (aminotransferasi) e parametri di colestasi elevati deve essere sottoposto a diagnosi differenziale per le epatopatie autoimmuni.
Diagnosi differenziale delle epatiti (Tabella 1)Per chiarire l’origine non autoimmune di un’epatopatia bisogna procedere a seconda delle probabili cause come indicato di seguito:In primo luogo si effettua una valutazione dei risultati clinici e dei test di funzionalità epatica, con particolare riguardo all’eventuale abuso di farmaci e di alcol.Quindi si dovrebbe valutare la presenza di epatite virale cronica, in primo luogo attraverso i parametri HBsAg e antiHCV.Per valutare la presenza di emocromatosi o della malattia di Wilson si esegue la determinazione di ferritina, saturazione della transferrina e ferrossidasi, concentrazione di rame nelle urine.Infine, si raccomanda la determinazione quantitativa dell’ α1antitripsina. La tipizzazione elettroforetica o genetica deve avvenire ad una concentrazione ridotta.Per determinare la steatoepatite non alcolica (NASH), che può essere conseguenza di un diabete mellito non controllato o di una iperlipidemia, si esegue la determinazione dei livelli di colesterolo, trigliceridi e HbA1c.
I risultati di elettroforesi del siero e l’aumento delle immunoglobuline sono in grado di fornire la prima evidenza della presenza di un’epatopatia autoimmune. In questo senso queste analisi dovrebbero essere incluse nella valutazione di una patologia del fegato.
2
17
elettroforesi del siero Nel caso di molte epatopatie croniche, nell’elettroforesi del siero si riscontra un’ipergammaglobulinemia. Ciò è particolarmente evidente nel caso dell’epatite autoimmune, mentre in presenza di cirrosi biliare primitiva risulta meno evidente e, nel caso della colangite sclerosante primitiva, invece, non si riscontrano particolari variazioni.
Determinazione quantitativa delle immunoglobuline•IgG: un innalzamento selettivo delle IgG, solitamente
> 1,5 volte rispetto al valore massimo di riferimento, è tipico dell’AIH.
•IgM: nel caso delle epatopatie autoimmuni colestatiche (PBC e PSC) si presenta un innalzamento selettivo delle IgM, più significativo nel caso della PBC.
•IgG ed IgM: un innalzamento combinato di IgG ed IgM è stato riscontrato nei pazienti affetti da sindrome da Overlap (AIH/PBC e AIH/PSC).
•IgA: un innalzamento selettivo delle IgA, al contrario, è indicativo di un’epatotossicità (alcol, droghe, steatoepatite non alcolica)
Determinazione human Leukocyte antigen (hLa)8
Per tutte le tre forme di epatopatie autoimmuni considerate in questo dossier sono state identificate delle associazioni HLA. Queste associazioni sono generalmente deboli, quindi non dovrebbero essere utilizzate per la valutazione della malattia epatica nella diagnostica di routine. Nelle popolazioni europee gli aplotipi HLAB8, DR3 o DR4 sono associati alla presenza di epatite autoimmune, mentre in Giappone è l’aplotipo DR4 e in Sudamerica il DR13. Le associazioni con PBC e PSC sono invece più limitate.
PatoLogia DiagNostica Di Base DiagNostica Di LaBoratorio sPecifica osserVaZioNi
Per tutte le forme Transaminasi (AST/GOT, ALT/GPT)Parametri di colestasi (ALP, γGT)
Anamnesi
Tossica (Alcolica) Rilevazione dell‘alcol, CDT nel siero
Ammoniaca, Etilglucuronide nelle urine (Metabolita dell’alcol)
Anamnesi; spesso diagnosi per esclusione
Tossica (Farmaci) Farmacogenetica Anamnesi; spesso diagnosi per esclusione
Virale Sierologia: HAV, HBV, HCV, HDV (IgG e IgM)
PCR quantitativo per HBV e HCV e, se necessaria, tipizzazione genetica
Diagnosi definitiva da laboratorio
Emocromatosi Ferritina, saturazione della transferrina
HFE (gene) Diagnosi definitiva da laboratorio
Deficit α 1-AT Elettroforesi sierica, quantitativa Determinazione quantitativa α1AT
Tipizzazione α 1AT (chimica e genetica delle proteine)
Diagnosi definitiva da laboratorio
Morbo di Wilson Ceruloplasmina, rame libero nel siero, rame nelle urine
Diagnostica genetica Diagnosi definitiva da laboratorio
NASH (epatite non alcolica)
Trigliceridi, Colesterolemia, HDL, LDL Glucosio, HbA1c
Istologia Definizione diabete, obesità, disturbi nel metabolismo dei grassi
Malattie epati-che autoimmuni (si veda tabella 2)
Per il 90% diagnosi definitiva da laboratorio; per il 10% diagnosi da esclusione in accordo con l’istologia
Quota: 1020% delle epatiti croniche
Tab. 1: Diagnosi differenziale per sospetto di epatite
in IFA, ancora meglio in associazione con ELISA o Immunoblot in particolare per l’antigene SLA). In caso non vengano individuati autoanticorpi, si deve procedere con la determinazione di anticorpi antiLC1: questi anticorpi si trovano solitamente in associazione con LKM1, e in alcuni casi, soprattutto nei bambini, possono presentarsi in presenza di epatite autoimmune di tipo II, oppure anche come singoli autoanticorpi. Gli autoanticorpi ASGPR, LMA (Liver Membrane Autoantibodies) ed LSP (Liver Specific Protein) hanno poco valore diagnostico supplementare, mentre in caso di sospetta colangite sclerosante primitiva si dovrebbe determinare il pANCA.
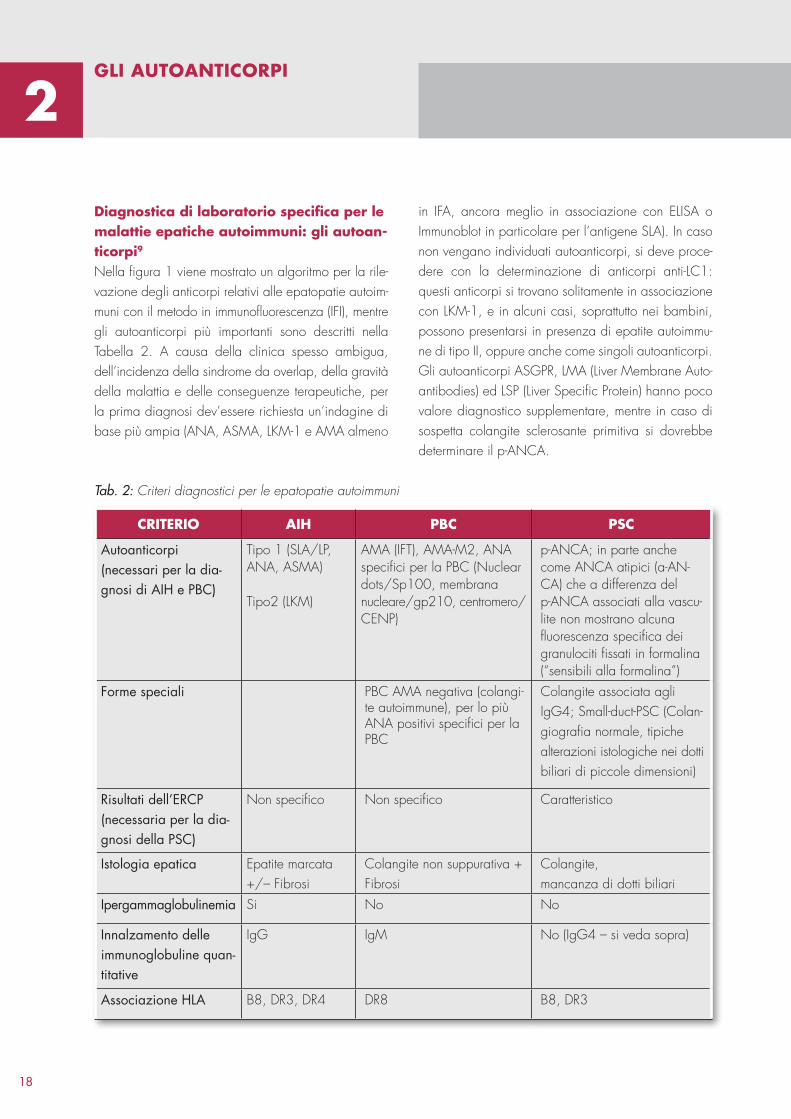
Diagnostica di laboratorio specifica per le malattie epatiche autoimmuni: gli autoanticorpi9
Nella figura 1 viene mostrato un algoritmo per la rilevazione degli anticorpi relativi alle epatopatie autoimmuni con il metodo in immunofluorescenza (IFI), mentre gli autoanticorpi più importanti sono descritti nella Tabella 2. A causa della clinica spesso ambigua, dell’incidenza della sindrome da overlap, della gravità della malattia e delle conseguenze terapeutiche, per la prima diagnosi dev’essere richiesta un’indagine di base più ampia (ANA, ASMA, LKM1 e AMA almeno
gLi autoaNticorPi
18
Tab. 2: Criteri diagnostici per le epatopatie autoimmuni
2
criterio aih PBc Psc
Autoanticorpi (necessari per la dia-gnosi di AIH e PBC)
Tipo 1 (SLA/LP, ANA, ASMA)
Tipo2 (LKM)
AMA (IFT), AMAM2, ANA specifici per la PBC (Nuclear dots/Sp100, membrana nucleare/gp210, centromero/CENP)
pANCA; in parte anche come ANCA atipici (aANCA) che a differenza del pANCA associati alla vasculite non mostrano alcuna fluorescenza specifica dei granulociti fissati in formalina (“sensibili alla formalina”)
Forme speciali PBC AMA negativa (colangite autoimmune), per lo più ANA positivi specifici per la PBC
Colangite associata agli IgG4; SmallductPSC (Colangiografia normale, tipiche alterazioni istologiche nei dotti biliari di piccole dimensioni)
Risultati dell’ERCP (necessaria per la dia-gnosi della PSC)
Non specifico Non specifico Caratteristico
Istologia epatica Epatite marcata +/– Fibrosi
Colangite non suppurativa + Fibrosi
Colangite, mancanza di dotti biliari
Ipergammaglobulinemia Si No No
Innalzamento delle immunoglobuline quan-titative
IgG IgM No (IgG4 – si veda sopra)
Associazione HLA B8, DR3, DR4 DR8 B8, DR3
19
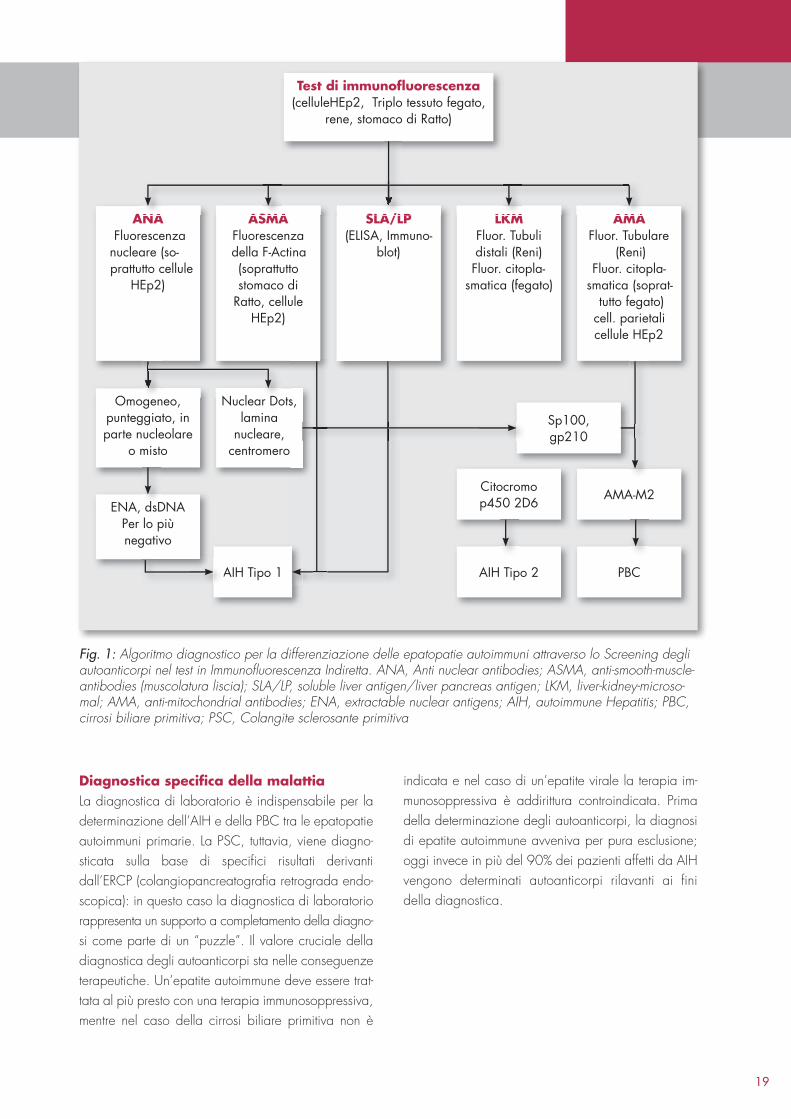
Fig. 1: Algoritmo diagnostico per la differenziazione delle epatopatie autoimmuni attraverso lo Screening degli autoanticorpi nel test in Immunofluorescenza Indiretta. ANA, Anti nuclear antibodies; ASMA, anti-smooth-muscle-antibodies (muscolatura liscia); SLA/LP, soluble liver antigen/liver pancreas antigen; LKM, liver-kidney-microso-mal; AMA, anti-mitochondrial antibodies; ENA, extractable nuclear antigens; AIH, autoimmune Hepatitis; PBC, cirrosi biliare primitiva; PSC, Colangite sclerosante primitiva
ANA Fluorescenza nucleare (so-prattutto cellule
HEp2)
Omogeneo,punteggiato, inparte nucleolare
o misto
ENA, dsDNAPer lo piùnegativo
ASMAFluorescenza della F-Actina(soprattutto stomaco di
Ratto, cellule HEp2)
Nuclear Dots,lamina
nucleare,centromero
AIH Tipo 1
SLA/LP(ELISA, Immuno-
blot)
Test di immunofluorescenza(celluleHEp2, Triplo tessuto fegato,
rene, stomaco di Ratto)
LKMFluor. Tubuli distali (Reni) Fluor. citopla-
smatica (fegato)
AIH Tipo 2
Citocromop450 2D6
Sp100,gp210
PBC
AMA-M2
AMAFluor. Tubulare
(Reni) Fluor. citopla-
smatica (soprat-tutto fegato)
cell. parietalicellule HEp2
M AMA SLA/LP LKA/LANA AS
s,
d D
A
N l D
SMASMA KMKM MAMA
Diagnostica specifica della malattiaLa diagnostica di laboratorio è indispensabile per la determinazione dell’AIH e della PBC tra le epatopatie autoimmuni primarie. La PSC, tuttavia, viene diagnosticata sulla base di specifici risultati derivanti dall’ERCP (colangiopancreatografia retrograda endoscopica): in questo caso la diagnostica di laboratorio rappresenta un supporto a completamento della diagnosi come parte di un “puzzle”. Il valore cruciale della diagnostica degli autoanticorpi sta nelle conseguenze terapeutiche. Un’epatite autoimmune deve essere trattata al più presto con una terapia immunosoppressiva, mentre nel caso della cirrosi biliare primitiva non è
indicata e nel caso di un’epatite virale la terapia immunosoppressiva è addirittura controindicata. Prima della determinazione degli autoanticorpi, la diagnosi di epatite autoimmune avveniva per pura esclusione; oggi invece in più del 90% dei pazienti affetti da AIH vengono determinati autoanticorpi rilavanti ai fini della diagnostica.
20
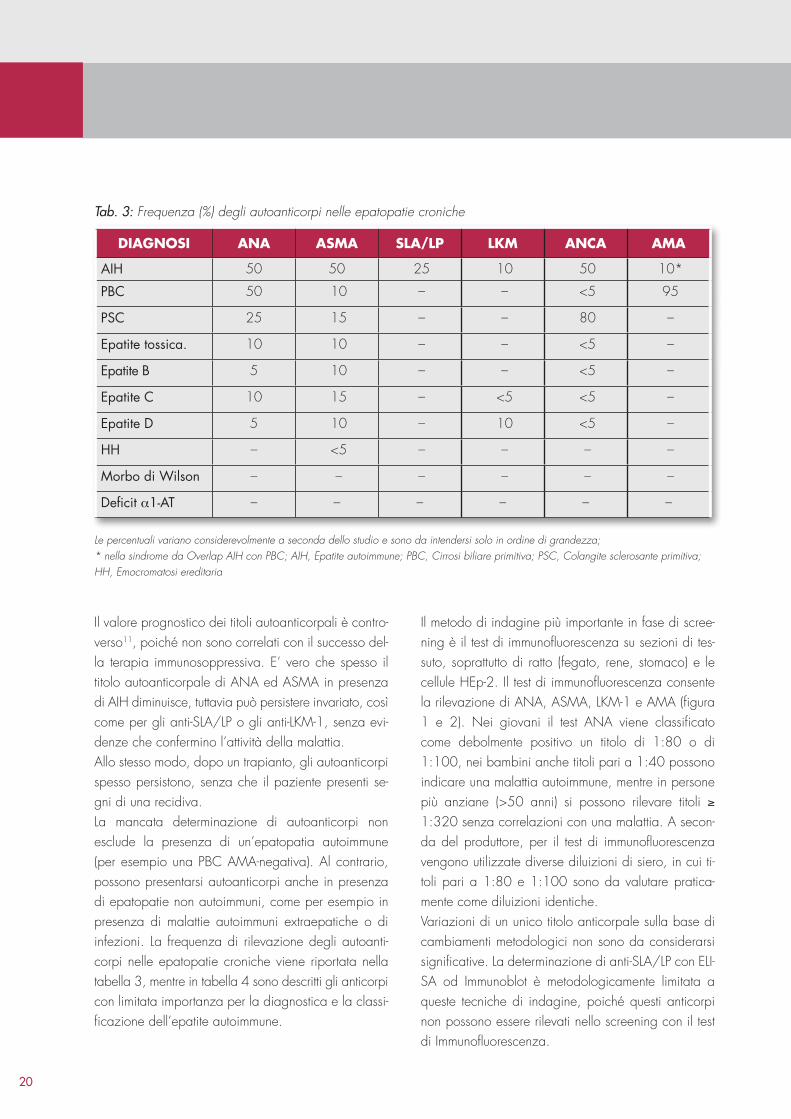
Il valore prognostico dei titoli autoanticorpali è controverso11, poiché non sono correlati con il successo della terapia immunosoppressiva. E’ vero che spesso il titolo autoanticorpale di ANA ed ASMA in presenza di AIH diminuisce, tuttavia può persistere invariato, così come per gli antiSLA/LP o gli antiLKM1, senza evidenze che confermino l’attività della malattia.Allo stesso modo, dopo un trapianto, gli autoanticorpi spesso persistono, senza che il paziente presenti segni di una recidiva.La mancata determinazione di autoanticorpi non esclude la presenza di un’epatopatia autoimmune (per esempio una PBC AMAnegativa). Al contrario, possono presentarsi autoanticorpi anche in presenza di epatopatie non autoimmuni, come per esempio in presenza di malattie autoimmuni extraepatiche o di infezioni. La frequenza di rilevazione degli autoanticorpi nelle epatopatie croniche viene riportata nella tabella 3, mentre in tabella 4 sono descritti gli anticorpi con limitata importanza per la diagnostica e la classificazione dell’epatite autoimmune.
Il metodo di indagine più importante in fase di screening è il test di immunofluorescenza su sezioni di tessuto, soprattutto di ratto (fegato, rene, stomaco) e le cellule HEp2. Il test di immunofluorescenza consente la rilevazione di ANA, ASMA, LKM1 e AMA (figura 1 e 2). Nei giovani il test ANA viene classificato come debolmente positivo un titolo di 1:80 o di 1:100, nei bambini anche titoli pari a 1:40 possono indicare una malattia autoimmune, mentre in persone più anziane (>50 anni) si possono rilevare titoli ≥ 1:320 senza correlazioni con una malattia. A seconda del produttore, per il test di immunofluorescenza vengono utilizzate diverse diluizioni di siero, in cui titoli pari a 1:80 e 1:100 sono da valutare praticamente come diluizioni identiche.Variazioni di un unico titolo anticorpale sulla base di cambiamenti metodologici non sono da considerarsi significative. La determinazione di antiSLA/LP con ELISA od Immunoblot è metodologicamente limitata a queste tecniche di indagine, poiché questi anticorpi non possono essere rilevati nello screening con il test di Immunofluorescenza.
DiagNosi aNa asma sLa/LP LKm aNca ama
AIH 50 50 25 10 50 10*
PBC 50 10 – – <5 95
PSC 25 15 – – 80 –
Epatite tossica. 10 10 – – <5 –
Epatite B 5 10 – – <5 –
Epatite C 10 15 – <5 <5 –
Epatite D 5 10 – 10 <5 –
HH – <5 – – – –
Morbo di Wilson – – – – – –
Deficit α1-AT – – – – – –
Le percentuali variano considerevolmente a seconda dello studio e sono da intendersi solo in ordine di grandezza; * nella sindrome da Overlap AIH con PBC; AIH, Epatite autoimmune; PBC, Cirrosi biliare primitiva; PSC, Colangite sclerosante primitiva; HH, Emocromatosi ereditaria
Tab. 3: Frequenza (%) degli autoanticorpi nelle epatopatie croniche
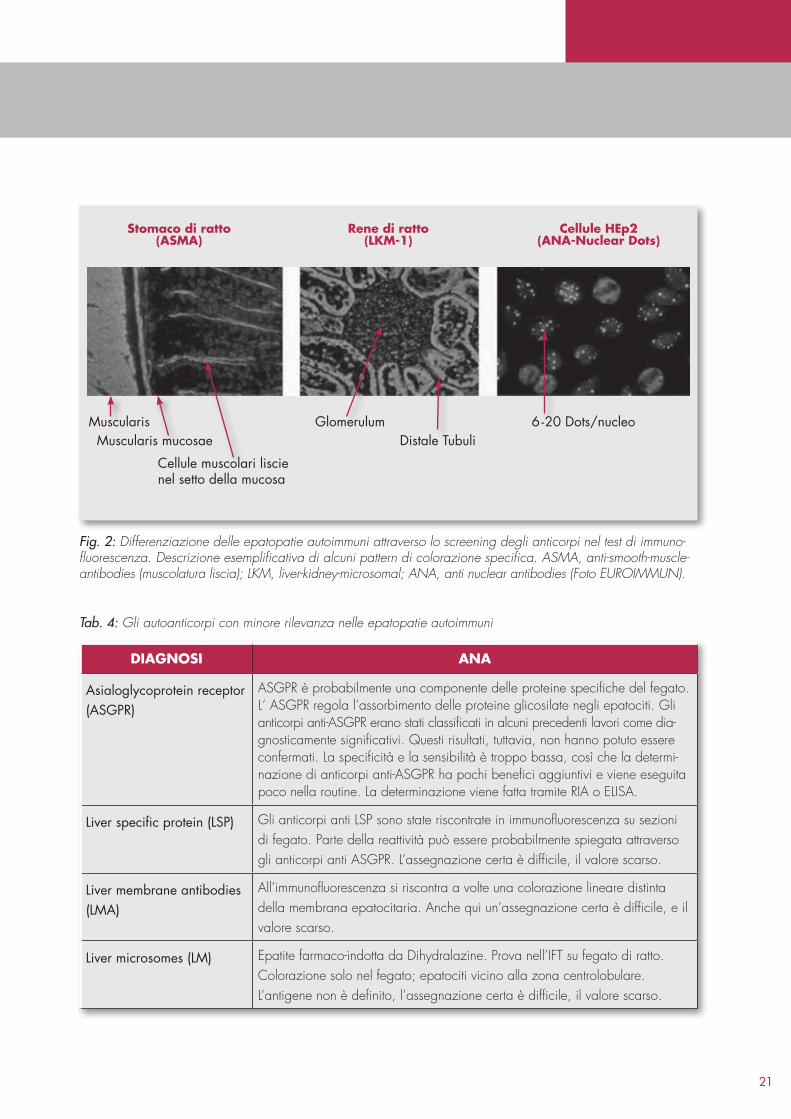
21
Muscularis Glomerulum 6-20 Dots/nucleoMuscularis mucosae Distale Tubuli
Cellule muscolari liscienel setto della mucosa
stomaco di ratto(asma)
cellule hep2(aNaNuclear Dots)
rene di ratto(LKm1)
Fig. 2: Differenziazione delle epatopatie autoimmuni attraverso lo screening degli anticorpi nel test di immuno-fluorescenza. Descrizione esemplificativa di alcuni pattern di colorazione specifica. ASMA, anti-smooth-muscle-antibodies (muscolatura liscia); LKM, liver-kidney-microsomal; ANA, anti nuclear antibodies (Foto EUROIMMUN).
DiagNosi aNa
Asialoglycoprotein receptor (ASGPR)
ASGPR è probabilmente una componente delle proteine specifiche del fegato. L‘ ASGPR regola l’assorbimento delle proteine glicosilate negli epatociti. Gli anticorpi antiASGPR erano stati classificati in alcuni precedenti lavori come dia gnosticamente significativi. Questi risultati, tuttavia, non hanno potuto essere confermati. La specificità e la sensibilità è troppo bassa, così che la determinazione di anticorpi antiASGPR ha pochi benefici aggiuntivi e viene eseguita poco nella routine. La determinazione viene fatta tramite RIA o ELISA.
Liver specific protein (LSP) Gli anticorpi anti LSP sono state riscontrate in immunofluorescenza su sezioni di fegato. Parte della reattività può essere probabilmente spiegata attraverso gli anticorpi anti ASGPR. L’assegnazione certa è difficile, il valore scarso.
Liver membrane antibodies (LMA)
All’immunofluorescenza si riscontra a volte una colorazione lineare distinta della membrana epatocitaria. Anche qui un’assegnazione certa è difficile, e il valore scarso.
Liver microsomes (LM) Epatite farmacoindotta da Dihydralazine. Prova nell’IFT su fegato di ratto. Colorazione solo nel fegato; epatociti vicino alla zona centrolobulare. L’antigene non è definito, l’assegnazione certa è difficile, il valore scarso.
Tab. 4: Gli autoanticorpi con minore rilevanza nelle epatopatie autoimmuni

22
Fig. 3: Percentuale di pazienti affetti da PBC diagnosticati attraverso autoanticorpi specifici. Attraverso una combinazione di tutti gli anticorpi e dei metodi è possibile diagnosticare quasi il 100% dei casi di PBC.
complesso, queste proteine sono molto più rare del PDH come auto antigeni.
La sensibilità può essere aumentata ulteriormente senza deterioramento della specificità quando gli antigeni ANA specifici della PBC (gp210 e SP100) sono inclusi nella diagnostica (Figura 3).Gli anticorpi SLA/LP in ELISA sono altamente specifici per AIH, ma dovrebbero essere confermati con un Immunoblot sulla base delle conseguenze terapeutiche. L’ELISA può in alcuni casi presentare dei falsi positivi, per esempio con gli anticorpi eterofili, e con anticorpi crossreattivi, che per esempio si formano nell’ambito di infezioni virali.Soprattutto in presenza di bassi titoli in ELISA, viene richiesto un controllo intermedio e un secondo test di conferma (ELISA di un altro fornitore, Blot). Diversi studi dimostrano una maggiore sensibilità dell’Immunoassay (ELISA, Blot) rispetto al test di immunofluorescenza, per cui è assolutamente giustificato, in casi dubbi, considerare una combinazione di risultati ottenuti con diversi metodi anche in presenza di IFA negativo. La figura 3 mostra la percentuale di pazienti diagnosticati con la combinazione di diversi metodi .
test di confermaIl pattern rappresentativo di fluorescenza descritto indirizza già verso quale struttura cellulare sono diretti gli autoanticorpi; quindi la fluorescenza risultante dalla presenza di anticorpi anti ASMA, LKM1 e AMA, è relativamente caratteristica e suggestiva di epatite autoimmune o di cirrosi biliare primitiva. Occa sionalmente i pattern di fluorescenza non possono essere assegnati in modo univoco (soprattutto la distin zione tra AMA e LKM1), così talvolta è necessario eseguire ulteriori test per l’antigene specifico:
Anti-LKM-1: la conferma di anticorpi antiLKM1 avviene tramite ELISA o Immunoblot, Antigene Citocromo P450 2D6.
AMA: per confermare questi anticorpi è adatto l’Immunoblot o l’ELISA con piruvato deidrogenasi (PDH)E2 come antigene (AMAM2). Per aumentare la sensibilità, in alcuni sistemi sono stati implementati anche 2 ulteriori epitopi (“branched chain alphaketoacid dehydrogenase” (BCKDH), ossoglutarato deidrogenasi (OCKDH), in parte sottoforma di proteine ricombinanti. Nel
ca. 80% ca. 90% ca. 93% ca. 98%
AM-AIFI AMA-Immunoassay(Blot, ELISA):
PDH-E2
AMA-Immunoassay(Blot, ELISA):
PDH-E2BCKD-E2OADC-E2(AMA-M2)
ANA specifici per la PBC (IFI,
Blot, ELISA):Nuclear Dots, Sp100,
gp210 lamina nucleare in IFI, LBR (inner nuclear
membrane protein lamin B receptor)

23
Fig. 4: Immunoblot per la differenziazione di epato-patie autoimmuni attraverso la determinazione di autoanticorpi tipici (EUROIMMUN). AMA-M2, anti-corpi anti-mitocondri vs l’antigene principale degli AMA, la sottounità E2 del piruvato deidrogenasi (PDH-E2); 3E BPO: proteina ricombinante con com-ponenti E2 dell’antigene “bersaglio” degli AMA-M2 (Branched Chain Keto Dehydrogenase-E2, Pyruvat-Dehydrogenase-E2, Oxo acid Dehydrogenase-E2); Sp100, antigeni specifici degli ANA anti Nuclear Dots; PML, proteina descritta nei pazienti con leuce-mia acuta promielocitica; gp210, antigene principa-le degli ANA con fluorescenza membranosa; LKM-1, Liver-kidney-microsomal antibodies; LC-1, liver cytosol antibodies; SLA/LP, soluble liver antigen / liver pan-creas antigen; Ro-52, Sub-Antigen des SS-A/Ro Antigens
AMA M2
3E (BPO)
Sp100
PML
gp210
LKM-1
LC-1
SLA/LP
Ro-52
control
classificazione dell’epatite autoimmune12
L’AIH si classifica in Tipo 1 e Tipo 2 in funzione degli autoanticorpi rilevati contro l’antigene “bersaglio”. In entrambe le tipologie predomina comunemente il sesso femminile.
Tipo 1: ANA e/o ASMA o anti-SLA/LP positivoQuesta è la tipologia più comune di AIH e colpisce prevalentemente le giovani donne. La malattia si presenta precocemente, ha un decorso severo e reagisce bene all’immunosoppressione. Circa il 20% dei pazienti presenta un’insorgenza acuta analoga a quella del virus dell’epatite.
Tipo2: anti-LKM-1 positivoLa malattia si presenta spesso già nell’infanzia, ma presenta un picco d’incidenza nella fascia 3565 anni. Il decorso della malattia è peggiore rispetto al tipo 1, nella metà dei casi si presenta una diagnosi di cirrosi epatica. Rispetto al Tipo 1, il Tipo 2 presenta più comunemente un’associazione con sindrome autoimmuni extraepatiche come la tiroidite, l’artrite, la neuropatia, l’anemia perniciosa. Secondo alcuni autori esiste anche un Tipo 3, definito dall’individuazione di autoanticorpi SLA. Tuttavia questa è l’unica differenza sostanziale rispetto al tipo 1; per questo motivo la classificazione del tipo 3 non è stata unanimemente condivisa ed è stata abbandonata.

24
DiagNostica Di LaBoratorio DeLLa cirrosi BiLiare PrimitiVa (PBc)
A partire dalla scoperta e dalla clonazione degli autoanticorpi mitocondriali, la diagnostica di laboratorio per la PBC ha fatto enormi progressi. Sulla base delle reattività nell’Immunoblot sono stati definiti nove diversi sottotipi di AMA, tra i quali, per quanto riguarda la diagnostica di laboratorio, è stato studiato soprattutto l’AMAM2, altamente specifico e quasi patognomonico. Gli anticorpi AMAM2 possono essere diretti contro tre antigeni principali: piruvato deidrogenasi (PDH); Branched chain keto acid dehydrogenase E1, alpha polipeptide (BCKDHA); ossoglutarato deidrogenasi (NADP+).Gli AMAM4, AMAM8 e ANAM9 hanno un minore valore diagnostico22 (Tabella 5).
Oltre agli AMA, anche determinati ANA possono indicare la presenza di PBC. A tal proposito valgono gli ANA anti antigene SP100, che nel test di immunofluorescenza sono evidenziati come pattern “nuclear dots”, così come gli anticorpi anti gp210, che nell’IFI determinano una positività della lamina nucleare. Questi ANA possono essere ritrovati sia come anticorpi isolati, sia in combinazione con gli AMA. Gli AMA sono rilevabili nel 9095% dei pazienti affetti da PBC quando vengono utilizzate più metodi che includano tutti gli antigeni conosciuti. Gli ANA specifici per la PBC sono rilevabili in circa il 50% dei pazienti AMA negativi, così che, in definitiva, solo nel 25% dei pazienti la PBC può non essere chiaramente diagnosticata per mezzo degli autoanticorpi (Figura 3, Tabella 6)23,24. Il quadro clinico della PBC AMAnegativa viene definita anche Colangite autoimmune (AIC).
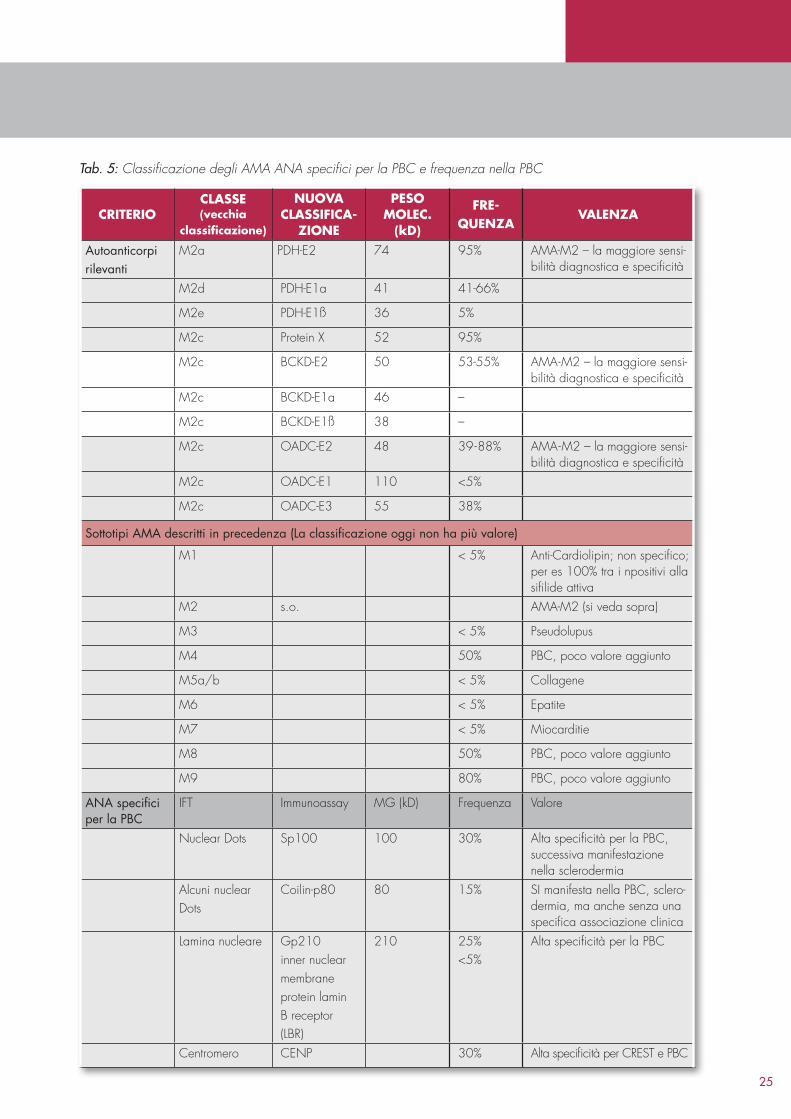
25
criteriocLasse (vecchia
classificazione)
NuoVa cLassifica
ZioNe
Peso moLec.
(kD)
freQueNZa
VaLeNZa
Autoanticorpi rilevanti
M2a PDHE2 74 95% AMAM2 – la maggiore sensibilità diagnostica e specificità
M2d PDHE1a 41 4166%
M2e PDHE1ß 36 5%
M2c Protein X 52 95%
M2c BCKDE2 50 5355% AMAM2 – la maggiore sensibilità diagnostica e specificità
M2c BCKDE1a 46 –
M2c BCKDE1ß 38 –
M2c OADCE2 48 3988% AMAM2 – la maggiore sensibilità diagnostica e specificità
M2c OADCE1 110 <5%
M2c OADCE3 55 38%
Sottotipi AMA descritti in precedenza (La classificazione oggi non ha più valore)
M1 < 5% AntiCardiolipin; non specifico; per es 100% tra i npositivi alla sifilide attiva
M2 s.o. AMAM2 (si veda sopra)
M3 < 5% Pseudolupus
M4 50% PBC, poco valore aggiunto
M5a/b < 5% Collagene
M6 < 5% Epatite
M7 < 5% Miocarditie
M8 50% PBC, poco valore aggiunto
M9 80% PBC, poco valore aggiunto
ANA specifici per la PBC
IFT Immunoassay MG (kD) Frequenza Valore
Nuclear Dots Sp100 100 30% Alta specificità per la PBC, successiva manifestazione nella sclerodermia
Alcuni nuclear Dots
Coilinp80 80 15% SI manifesta nella PBC, sclerodermia, ma anche senza una specifica associazione clinica
Lamina nucleare Gp210 inner nuclearmembraneprotein laminB receptor(LBR)
210 25%<5%
Alta specificità per la PBC
Centromero CENP 30% Alta specificità per CREST e PBC
Tab. 5: Classificazione degli AMA ANA specifici per la PBC e frequenza nella PBC

26
DiagNostica Di LaBoratorio DeLLa coLaNgite scLerosaNte (Psc)
BiBLiografiaParte 11. 1.Invernizzi, P. and I.R. Mackay, Autoimmune liver diseases. World J Gastroenterol,
2008. 14(21): p. 32901.
2. Mackay, I.R., Chronic active hepatitides. Front Gastrointest Res, 1975. 1: p. 14287.
3. Mackay, I.R., Historical reflections on autoimmune hepatitis. World J Gastroenterol,
2008. 14(21): p. 3292300.
4. Strassburg, C.P., Autoimmune hepatitis. Best Pract Res Clin Gastroenterol, 2010.
24(5): p. 66782.
5. Kochar, R. and M. Fallon, Diagnostic criteria for autoimmune hepatitis: what is the
gold standard? Hepatology, 2010. 51(1): p. 3501; author reply 351.
6. Ngu, J.H., et al., Populationbased epidemiology study of autoimmune hepatitis: a
disease of older women? J Gastroenterol Hepatol, 2010. 25(10): p. 16816.
7. Navaneethan, U. and B. Shen, Hepatopancreatobiliary manifestations and complica
tions associated with inflammatory bowel disease. Inflamm Bowel Dis, 2010. 16(9):
p. 1598619.
8. Nakamura, H., et al., Prevalence of interrelated autoantibodies in thyroid diseases
and autoimmune disorders. J Endocrinol Invest, 2008. 31(10): p. 8615.
9. Gelpi, C., E.J. Sontheimer, and J.L. RodriguezSanchez, Autoantibodies against a se
rine tRNAprotein complex implicated in cotranslational selenocysteine insertion. Proc
Natl Acad Sci U S A, 1992. 89(20): p. 973943.
10. Volkmann, M., et al., Soluble liver antigen: isolation of a 35kd recombinant protein
(SLAp35) specifically recognizing sera from patients with autoimmune hepatitis. He
patology, 2001. 33(3): p. 5916.
11. Wies, I., et al., Identification of target antigen for SLA/LP autoantibodies in autoimmu
ne hepatitis. Lancet, 2000. 355(9214): p. 15105.
12. Alvarez, F., et al., International Autoimmune Hepatitis Group Report: review of criteria
for diagnosis of autoimmune hepatitis. J Hepatol, 1999. 31(5): p. 92938.
13. Vogel, A., M.P. Manns, and C.P. Strassburg, Autoimmunity and viruses. Clin Liver Dis,
2002. 6(3): p. 73953.
14. Mackay, I.R. and P.J. Morris, Association of autoimmune active chronic hepatitis with
HLA1,8. Lancet, 1972. 2(7781): p. 7935.
15. Longhi, M.S., et al., Aetiopathogenesis of autoimmune hepatitis. J Autoimmun, 2010.
34(1): p. 714.
imPressumParte 1Le malattie autoimmuni del fegato, a cura di P. Invernizzi, L. Moroni, I. Bianchi, A. Lleo, M. PoddaDr. Pietro InvernizziDr. Luca MoroniDr.ssa Ilaria BianchiDr.ssa Ana LleoCentro per le Malattie Autoimmuni del Fegato, Clinica Medica, Dipartimento di MedicinaIRCCS Istituto Clinico Humanitas Milano
Prof. Mauro Podda Responsabile Dipartimento Medicina IRCCS Istituto Clinico Humanitas Milano
Parte 2Gli Autoanticorpi nelle malattie epatiche, a cura di R. Gruber, S. Borgmann
Allo stadio iniziale asintomatico è evidente solo un innalzamento dei valori dell’ALP e delle gGT ed eventualmente un leggero innalzamento delle amminotransferasi (Transaminasi). Nel corso della malattia l’ALP può aumentare fino a 20 volte rispetto ai valori normali, mentre le amminotransferasi possono aumentare fino a 5 volte. Al momento della diagnosi, in circa il 50% dei pazienti la bilirubina presenta un lieve innalzamento e aumenta poi continuamente presentando valori oscillanti. La concentrazione di acidi biliari è aumentata di molto rispetto ai normali valori, mentre tutte le forme della colestasi vanno definite per diagnostica differenziale.
La diagnosi specifica di PSC viene effettuata in contrapposizione a quelle dell’AIH e della PBC per mezzo del quadro fornito dalla colangiopancreatografia retrograda endoscopica e sussidiata solo marginalmente dal laboratorio. Utile alla diagnosi si è rivelata la determinazione degli anticorpi anti citoplasma dei neutrofili (ANCA) con un quadro fluoroscopico pANCA in etanolo, presente anche nell’80% dei pazienti affetti da colite ulcerosa. Questo quadro fluoroscopico, a differenza del pANCA anti –Mieloperossidasi specifico associato alla vasculite, non presenta sui granulociti fissati in formalina una colorazione granulare citoplasmatica, ma un’insolita fluorescenza filamentosa o addirittura nessuna fluorescenza. Questi ANCA vengono definiti quindi come “sensibili alla formalina” o aANCA (ANCA atipici), o, secondo alcuni autori, xANCA27

27
16. Werner, M., et al., Characteristics and longterm outcome of patients with autoimmu
ne hepatitis related to the initial treatment response. Scand J Gastroenterol, 2010.
45(4): p. 45767.
17. Strassburg, C.P. and M.P. Manns, Autoimmune hepatitis in the elderly: what is the
difference? J Hepatol, 2006. 45(4): p. 4802.
18. Kirk, A.P., et al., Late results of the Royal Free Hospital prospective controlled trial of
prednisolone therapy in hepatitis B surface antigen negative chronic active hepatitis.
Gut, 1980. 21(1): p. 7883.
19. Soloway, R.D., et al., Clinical, biochemical, and histological remission of severe
chronic active liver disease: a controlled study of treatments and early prognosis.
Gastroenterology, 1972. 63(5): p. 82033.
20. Manns, M.P. and C.P. Strassburg, Autoimmune hepatitis: clinical challenges. Gastro
enterology, 2001. 120(6): p. 150217.
21. Alvarez, F., et al., Shortterm cyclosporine induces a remission of autoimmune hepatitis
in children. J Hepatol, 1999. 30(2): p. 2227.
22. Richardson, P.D., P.D. James, and S.D. Ryder, Mycophenolate mofetil for maintenance
of remission in autoimmune hepatitis in patients resistant to or intolerant of azathiopri
ne. J Hepatol, 2000. 33(3): p. 3715.
23. Manns, M.P., et al., Budesonide induces remission more effectively than prednisone
in a controlled trial of patients with autoimmune hepatitis. Gastroenterology, 2010.
139(4): p. 1198206.
24. Nguyen, D.L., B.D. Juran, and K.N. Lazaridis, Primary biliary cirrhosis. Best Pract Res
Clin Gastroenterol, 2010. 24(5): p. 64754.
25. Selmi, C. and M.E. Gershwin, The role of environmental factors in primary biliary
cirrhosis. Trends Immunol, 2009. 30(8): p. 41520.
26. Invernizzi, P., et al., Human leukocyte antigen polymorphisms in Italian primary biliary
cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology,
2008. 48(6): p. 190612.
27. Invernizzi, P., Geoepidemiology of autoimmune liver diseases. J Autoimmun, 2010.
34(3): p. J3006.
28. Invernizzi, P., et al., Frequency of monosomy X in women with primary biliary cirrho
sis. Lancet, 2004. 363(9408): p. 5335.
29. Poupon, R., Primary biliary cirrhosis: a 2010 update. J Hepatol, 2010. 52(5): p.
74558.
30. Lindor, K.D., et al., Primary biliary cirrhosis. Hepatology, 2009. 50(1): p. 291308.
31. Bergasa, N.V., et al., Primary biliary cirrhosis: report of a focus study group. Hepato
logy, 2004. 40(4): p. 101320.
32. Talwalkar, J.A. and K.D. Lindor, Primary biliary cirrhosis. Lancet, 2003. 362(9377):
p. 5361.
33. Boberg, K.M., et al., Overlap syndromes: the International Autoimmune Hepatitis
Group (IAIHG) position statement on a controversial issue. J Hepatol, 2011. 54(2):
p. 37485.
34. Pares, A. and N. Guanabens, Osteoporosis in primary biliary cirrhosis: pathogenesis
and treatment. Clin Liver Dis, 2008. 12(2): p. 40724; x.
35. Bruix, J. and M. Sherman, Management of hepatocellular carcinoma. Hepatology,
2005. 42(5): p. 120836.
36. Jacob, D.A., et al., Mayo risk score for primary biliary cirrhosis: a useful tool for the
prediction of course after liver transplantation? Ann Transplant, 2008. 13(3): p. 3542.
37. Corpechot, C., et al., The effect of ursodeoxycholic acid therapy on the natural
course of primary biliary cirrhosis. Gastroenterology, 2005. 128(2): p. 297303.
38. Kremer, A.E., R.P. Oude Elferink, and U. Beuers, Pathophysiology and current ma
nagement of pruritus in liver disease. Gastroenterol Clin Biol, 2011.
39. MacQuillan, G.C. and J. Neuberger, Liver transplantation for primary biliary cirrhosis.
Clin Liver Dis, 2003. 7(4): p. 94156, ix.
40. Wiesner, R.H. and N.F. LaRusso, Clinicopathologic features of the syndrome of prima
ry sclerosing cholangitis. Gastroenterology, 1980. 79(2): p. 2006.
41. Karlsen, T.H., E. Schrumpf, and K.M. Boberg, Primary sclerosing cholangitis. Best
Pract Res Clin Gastroenterol, 2010. 24(5): p. 65566.
42. Chapman, R., et al., Diagnosis and management of primary sclerosing cholangitis.
Hepatology, 2010. 51(2): p. 66078.
43. Lindor, K.D., Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing
CholangitisUrsodeoxycholic Acid Study Group. N Engl J Med, 1997. 336(10): p.
6915.
44. Boberg, K.M., et al., Cholangiocarcinoma in primary sclerosing cholangitis: risk fac
tors and clinical presentation. Scand J Gastroenterol, 2002. 37(10): p. 120511.
Parte 21. Treichel U, Gerken G, Meyer zum Buschenfelde KH. 1995. Autoantikörper bei chroni
scher Hepatitis. In Labor und Diagnose, ed. L Thomas. Frankfurt: THBooks
2. Beuers U. Hepatic overlap syndromes. J Hepatol 2005; 42 Suppl: S939
3. Efe C, Purnak T, Ozaslan E. Systemic lupus erythematosus and autoimmune hepatitis.
Rheumatol Int 2011; 31: 419
4. Strassburg CP, Manns MP. Autoimmune hepatitis versus viral hepatitis C. Liver 1995;
15: 22532
5. Monti V, Aghemo A, Rumi MG, Donato MF, Del Ninno E, et al. The prevalence, clinical
features and response to antiviral therapy of patients with chronic hepatitis C who are
seropositive for liverkidney microsome type 1 antibodies. Antivir Ther 2005; 10: 71520
6. Czaja AJ. Autoimmune liver disease. Curr Opin Gastroenterol 2009; 25: 21522
7. Manns MP, Czaja AJ, Gorham JD, Krawitt EL, MieliVergani G, et al. Diagnosis and
management of autoimmune hepatitis. Hepatology 2010; 51: 2193213
8. Feld JJ, Heathcote EJ. Epidemiology of autoimmune liver disease. J Gastroenterol Hepa
tol 2003; 18: 111828
9. Krawitt EL. Discrimination of autoimmune hepatitis: autoantibody typing and beyond. J
Gastroenterol 2011; 46 Suppl 1: 3941
10. Lohse AW, Hennes E. Diagnostic criteria for autoimmune hepatitis. Hepatol Res 2007;
37 Suppl 3: S509
11. Czaja AJ. Autoantibodies as prognostic markers in autoimmune liver disease. Dig Dis
Sci 2010; 55: 214461
12. Krawitt EL. Autoimmune hepatitis. N Engl J Med 2006; 354: 5466
13. Takahashi H, Zeniya M. Acute presentation of autoimmune hepatitis: Does it exist? A
published work review. Hepatol Res 2011; 41: 498504
14. Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, et al. Simplified criteria for
the diagnosis of autoimmune hepatitis. Hepatology 2008; 48: 16976
15. Nguyen DL, Juran BD, Lazaridis KN. Primary biliary cirrhosis. Best Pract Res Clin
Gastroenterol 2010; 24: 64754
16. Tozzoli R. The diagnostic role of autoantibodies in the prediction of organspecific
autoimmune diseases. Clin Chem Lab Med 2008; 46: 57787
17. Flisiak R, Pelszynska M, Prokopowicz D, Rogalska M, Grygoruk U. High concentrati
on of antimitochondrial antibodies predicts progressive primary biliary cirrhosis.
World J Gastroenterol 2005; 11: 57069
18. Hirschfield GM, Gershwin ME. Primary biliary cirrhosis: one disease with many faces.
Isr Med Assoc J 2011; 13: 559
19. Sarkar K, Miller FW. Possible roles and determinants of microchimerism in autoimmu
ne and other disorders. Autoimmun Rev 2004; 3: 45463
20. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, et al. Primary biliary cirrhosis in
monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroen
terology 2004; 127: 48592
21. Hirschfield GM, Siminovitch KA. Toward the molecular dissection of primary biliary
cirrhosis. Hepatology 2009; 50: 134750
22. Berg PA, Binder T, Lindner H, Bannaski H, Maas D, et al. Heterogenität mitochondri
aler Antikörper. Dtsch Med Wschr 1975; 100: 11237
23. Muratori P, Muratori L, Ferrari R, Cassani F, Bianchi G, et al. Characterization and
clinical impact of antinuclear antibodies in primary biliary cirrhosis. Am J Gastroente
rol 2003; 98: 4317
24. Worman HJ, Courvalin JC. Antinuclear antibodies specific for primary biliary cirrhosis.
Autoimmun Rev 2003; 2: 2117
25. Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med 1995; 332:
92433
26. Khosroshahi A, Stone JH. A clinical overview of IgG4related systemic disease. Curr
Opin Rheumatol 2011; 23: 5766
27. Hov JR, Boberg KM, Karlsen TH. Autoantibodies in primary sclerosing cholangitis.
World J Gastroenterol 2008; 14: 378191

I10
008
_08_
Aut
oim
mun
olog
ia
synlab Italia S.r.l.Via Orzinuovi 111 25125 Brescia
Tel. 030 3514085
www.synlab.it


















