
Dispensa di Citofluorimetria - spaziotlb.com · Università degli Studi di Brescia . ... capacità...
74
APPLICAZIONI DELLA CITOFLUORIMETRIA IN ANATOMIA PATOLOGICA: PLOIDIA E IMMUNOFENOTIPIZZAZIONE TUMORALE Moris Cadei Sezione di Anatomia Patologica Università degli Studi di Brescia
Transcript of Dispensa di Citofluorimetria - spaziotlb.com · Università degli Studi di Brescia . ... capacità...

APPLICAZIONI DELLA CITOFLUORIMETRIA IN ANATOMIA
PATOLOGICA: PLOIDIA E IMMUNOFENOTIPIZZAZIONE TUMORALE
Moris Cadei
Sezione di Anatomia Patologica Università degli Studi di Brescia

INTRODUZIONE
E’ opinione comune nei laboratori di Anatomia Patologica che la classificazione istopatologica di per sè non è sufficiente a prevedere l’aggressività biologica di una neoplasia. Lo studio di un tumore solido e delle sue caratteristiche fenotipiche (rapida crescita, invasione locale, capacità di produrre metastasi a distanza) oltre che biologiche (proliferazione, contenuto di DNA, contenuto proteico) è irrinunciabile qualora si voglia avere una visione il più possibile completa dello stadio patologico e della potenziale evolutività clinica della malattia. Da qui, l’interesse verso tutta una serie di metodiche che possono fornire informazioni utili al patologo riguardo il grado di instabilità genetica di una neoplasia e di conseguenza l’acquisizione, da parte del tumore, di un “fenotipo” ad alta aggressività biologica e quindi ad elevato rischio di progressione. Tra le numerose tecniche proposte, la Citometria a Flusso è l’unica che consente un’analisi multiparametrica che può fornire nell’arco di poco tempo due o più informazioni contemporaneamente sullo stesso tessuto.

PRINCIPI DI CITOFLUORIMETRIA
La citometria a flusso è una tecnologia che consente, in completa
automazione, la determinazione di diverse caratteristiche individuali fisiche e/o chimiche, delle cellule, mentre scorrono una ad una in un mezzo liquido grazie ad uno o più sensori ottici od elettronici (Shapiro HM. 1988). Una popolazione cellulare può quindi essere studiata prendendo in esame contemporaneamente parametri fisici (volume ed organizzazione cellulare) biochimici (come attività enzimatica, ph, ioni Ca++) molecolari ( pattern antigenico e genetico) e funzionali (stato di attivazione e proliferazione cellulare). Si tratta pertanto di un’analisi multiparametrica caratterizzata da rapidità, sensibilità, accuratezza e da alta significatività statistica, che consente una valutazione sia quantitativa (percentuale di espressione), che qualitativa (misura dell’intensità ed omogeneità di espressione) dei diversi parametri. II Citofluorimetro
Ogni citofluorimetro è costituito essenzialmente da quattro parti: 1- un sistema idropneumatico che, nella cella di misura, provvede allo
scorrimento del campione ed alla focalizzazione idrodinamica delle cellule in esso contenute. E’ costituito da un circuito idraulico e da un gas (azoto od aria sotto pressione) ai quali spetta il trasporto e l’allineamento delle singole cellule nel punto di misura. Il principio universale accettato è quello della focalizzazione idrodinamica in regime di flusso laminare, che consente di far fluire le cellule nel punto esatto di misura.
Agendo quindi sul sistema pneumatico, che controlla la differenza di pressione tra core flow (flusso interno contenente la cellula) e sheat flow (asse ideale di flusso), si regola la velocità di efflusso delle cellule (flow rate) normalmente valutata in numero di cellule al secondo (Geido E. 1988).
La sospensione cellulare, monodispersa e non contenente aggregati di grosse dimensioni, viene spinta verso il punto di misura, attraverso una camera di flusso nella quale avvengono la progressiva diluizione ed allineamento in singola fila delle cellule. Nello strumento circola una soluzione salina isotonica che “inguaina” laminarmente la sospensione cellulare mantenendola allineata al centro della camera di flusso.
Essendo la camera di dimensioni piccole (75-250 Φm) è importante filtrare i campioni al fine di evitare intoppi della fluidica.

La pressione di spinta e la progressiva diluizione del campione determinano una velocità di conta più o meno costante (Rate), che può essere regolata in base alle necessità. Esistono inoltre numerose varianti nel tipo e nella geometria, tra cui ad esempio le semplici camere coniche con un piccolo orifizio (Nozzle), le camere a cuvetta di quarzo e quelle capillari.
2- Un sistema ottico che provvede all’illuminazione delle cellule nel punto di misura ed alla raccolta dei segnali luminosi (Scatter e Fluorescenza) generati dalla interazione delle cellule con il fascio luminoso. E’ questo uno degli aspetti più critici della citofluorimetria: una corretta focalizzazione della luce di eccitazione sulle cellule è il requisito base per ottenere misurazioni attendibili. Ciò deriva dalla necessità che tutte le cellule ricevono la stessa intensità di eccitazione. Le caratteristiche delle sorgenti di luce devono garantire una adeguata eccitazione delle molecole della sostanza fluorescente (fluorocromo) legate alle cellule. Inoltre la sorgente di luce deve fornire un’illuminazione spazialmente uniforme nella zona di interazione con le cellule per garantire un’adeguata proporzionalità tra contenuto di sostanza ed intensità di fluoresceza.
Esistono due tipi di sorgenti luminose: laser ad Argon e lampade a vapori di mercurio o allo xenon (vedi schema del citofluorimetro). I vantaggi principali dell’impiego del laser risiedono nell’intensità della luce ed eccitazione ottenibili e nella monocromaticità delle righe emesse, mentre le lampade sono più versatili perché hanno una maggiore estensione delle lunghezze d’onda selezionabili.
3- Un sistema di trasduzione ottico-elettronico. I segnali provenienti dai sensori (fotomoltiplicatori) del citofluorimetro
sono impulsi elettrici proporzionali all’entità luminosa del parametro misurato. Questi segnali vengono prima amplificati in modo lineare o logaritmico, poi trasformati da un convertitore analogico-digitale, che campiona le misure registrate in vari momenti fornendo i relativi valori numerici. Questi singoli valori vengono quindi a costituire un “quanto” citometrico, a cui viene dato il nome di “EVENTO”.
4- Un elaboratore di dati che consenta in una successiva fase di analisi di poter estrarre dalla distribuzione i parametri rilevanti dal punto di vista biologico. Lo sviluppo degli strumenti di misura è stato fin dall’inizio accompagnato dall’utilizzo di modelli matematici non specifici di analisi delle distribuzioni (fasi del ciclo, CV). E’ quindi necessario fare ricorso a modelli statistici per i diversi tipi di misure (contenuto di DNA, misure di immunofluorescenza).
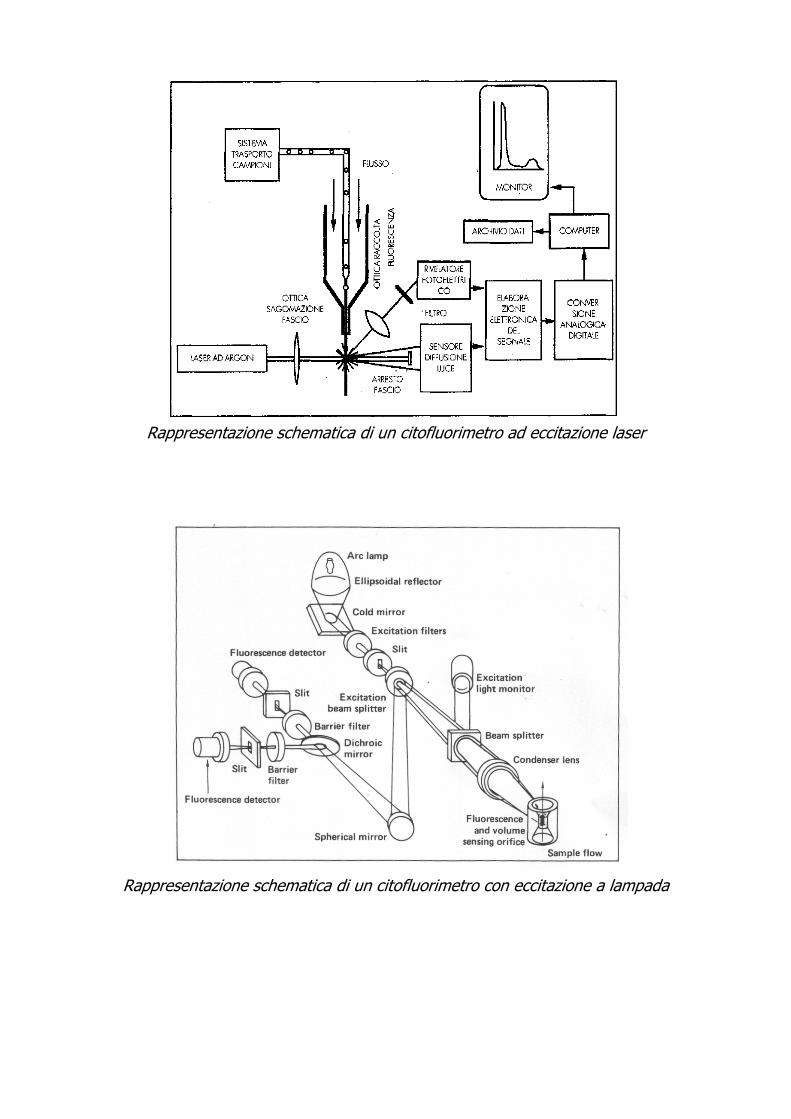
Rappresentazione schematica di un citofluorimetro ad eccitazione laser
Rappresentazione schematica di un citofluorimetro con eccitazione a lampada

ANALISI DEL CONTENUTO DI DNA (PLOIDIA)
Il complesso degli studi effettuati in questi ultimi trent’anni sulla genesi dei tumori ha portato alla conclusione che esiste un comune denominatore delle loro differenti forme che è rappresentato dalla loro base genetica. In particolare appare sempre più chiaro che un tumore si produce quando una serie di alterazioni del genoma (mutazioni) si accumula in una singola cellula somatica causandovi la perdita del controllo della crescita cellulare. L’approccio genetico allo studio dei tumori è divenuto di conseguenza sempre più essenziale e ciò non solo per rispondere a problematiche scientifiche inerenti la loro genesi e la loro evoluzione, ma anche sotto il profilo pratico in ordine alla possibilità di diagnosi sempre più precoce, al monitoraggio terapeutico ed ai fini prognostici. Si tratta in ogni caso di un approccio piuttosto complesso; la trasformazione neoplastica è infatti un processo “multifattoriale “ con alterazioni del genoma che possono riguardare i geni (mutazioni geniche) ad azione di tipo dominante o di tipo recessivo, sia il cariotipo a livello dei singoli cromosomi (mutazioni cromosomiche o anomalie strutturali) oppure dell’assetto cromosomico complessivo (mutazioni genomiche o anomalie cromosomiche numeriche). Per affrontare lo studio del fenomeno tumorale è necessario pertanto utilizzare il complesso delle tecnologie genetiche oggi disponibili ed in particolare di quelle che puntano al genoma in modo diretto. I procedimenti e le metodologie di analisi diretta del genoma attualmente disponibili sono numerose ma nel complesso possono essere distinte in tre grandi categorie: a) tecniche di “genetica molecolare” che analizzano il genoma a livello del
DNA e quindi dell’informazione stessa e dei geni in particolare, con procedure di tipo biochimico-molecolare, come quella del DNA – ricombinante;
b) tecnche di “citogenetica” che, diversamente dalle prime, mantengono intatto il contenuto morfostrutturale delle cellule e si rivolgono ai più alti livelli di organizzazione del genoma quali sono, come è noto, i cromosomi ed al loro assetto complessivo o cariotipo;
c) tecniche di “citometria a flusso” che, pur non consentendo di accertare le più fini alterazioni cromosomiche, permettono di identificare i casi in cui l’alterazione del patrimonio genetico, comporta una variazione del cariotipo pari al 4% circa (Forabosco A, 1992).

Il contenuto di DNA cellulare misurato mediante le tecniche di
citometria a flusso ha indubbiamente giocato un ruolo importante nello sviluppo delle conoscenze sulla genesi e la natura dei tumori. Divenuto ormai una delle caratteristiche biologiche più diffuse nello studio di diverse neoplasie non solo a livello di ricerca di base ma anche in ambito clinico– analitico, in particolare esso consente di valutare, in una patologia tumorale, due aspetti importanti che, in aggiunta ai tradizionali parametri clinici ed istologici, sono assai utili nel precisare il quadro diagnostico e spesso nel definire una terapia: il grado di alterazione rispetto al contenuto diploide delle cellule normali (ploidia del DNA) e lo stato proliferativo della popolazione cellulare.
Si è potuto determinare che un contenuto anomalo di DNA è il marker più ampiamente condiviso di malignità apprezzabile nel 75% dei tumori solidi umani (Barlogie B,1983): il grado di anomalia varia in funzione del tipo di neoplasia ed oscilla da valori ipodiploidi a valori iperottaploidi. Non è raro incontrare cloni multipli di cellule tumorali con diverso contenuto di DNA all’interno della stessa neoplasia (Teodori L, 1983; Mauro F, 1986).
Malgrado la presenza di numerosi e complessi meccanismi biochimici alla base della riproduzione cellulare e del suo controllo, oltre alla determinazione della ploidia, l’analisi citofluorimetrica del DNA riflette lo stato proliferativo della popolazione tumorale con particolare riferimento alla identificazione di “momenti diversi” del ciclo cellulare(Baserga R, 1976; Baserga R, 1985). Infatti la determinazione della cinetica cellulare permettendo la valutazione simultanea del genotipo e di alcuni fenotipi tumorali, aumenta il potere discriminativo dell’analisi citofluorimetrica nel caso di cellule aventi lo stesso contenuto cellulare di DNA.
I progressi nella comprensione dei meccanismi molecolari della proliferazione cellulare unitamente a quelli metodologico – strumentali nel campo dell’analisi cellulare automatizzata, costituiscono un esempio di come la tendenza attuale ad una ricerca oncologica finalizzata alla clinica, abbia portato allo sviluppo di metodologie sofisticate ma al tempo stesso sufficientemente semplici da poter essere applicate a studi in ambito clinico.
La citologia analitica (sviluppatasi in parallelo alla evoluzione tecnologica della microscopia), si pone l’obiettivo di quantificare vari componenti cellulari nonché di caratterizzare la morfologia di cellule e tessuti anche con l’aiuto di modelli matematici e di analisi statistiche che permettono di elaborare in modo completo i dati ottenuti.
La citometria a flusso in particolare è potenzialmente in grado di ricondurre a numeri caratteristiche biologiche diverse fra loro che sarebbero valutate più empiricamente con osservazioni soggettive semi-quantitative.
Il contenuto di DNA nucleare è certamente il parametro più significativo sotto questo aspetto ed il fatto di essere divenuto praticamente il primo

“target” della citometria ha contribuito a farne uno dei parametri biologici più diffusamente studiati in molti tipi di neoplasie. Dai laboratori di ricerca dove è iniziata e si è sviluppata questa metodica, i dati ricavati dall’analisi sono progressivamente passati alla utilizzazione in ambito clinico.A questa transizione hanno contribuito in misura variabile almeno due fattori: il primo legato allo sviluppo di strumentazioni sempre più orientate verso un impiego clinico-analitico, il secondo legato al sempre più rapido accumularsi di studi clinici volti a dimostrare il valore di fattori prognostici nella maggior parte delle neoplasie solide.
I risultati non sono stati peraltro univoci, soprattutto a causa di problemi insiti nella strutturazione di molti di questi studi e dalla mancanza, sino a poco tempo fa, di una standardizzazione tecnico-metodologica che renda uniformi (e quindi comparabili tra loro) i risultati provenienti da istituzioni diverse.
Va detto che oggi, un’accurata diagnosi e classificazione delle neoplasie, richiede sempre più spesso un approccio multiparametrico che includa: lo studio della morfologia, l’immunofenotipizzazione, lo studio citofluorimetrico del DNA, lo citogenetica classica e la genetica molecolare.
Come per tutte le altre branche applicative della citofluorimetria a flusso, un requisito essenziale per un’interpretazione corretta di ognuno di questi studi è ovviamente un campione tecnicamente ottimale (Danova M, 1994).
Per la natura stessa dei campioni da analizzare è possibile ottenere con facilità sospensioni cellulari da campioni freschi (Meyer JS, 1988), congelati (Clark GM, 1989; Dressler, 1988) o inclusi in paraffina (Hedley DW, 1983; Kallioniemi OP, 1987).
I vantaggi principali della metodica di estrazione dei nuclei da paraffina sono da un lato la necessità di effettuare analisi retrospettive su campioni d’archivio di pazienti di cui si conosce il follow-up, dall’altro la possibilità di effettuare l’analisi del contenuto di DNA anche quando non si disponga di un prelievo congelato della parte tumorale, come ad esempio nelle lesioni minime della mammella.
In letteratura, tuttavia, vengono riportati risultati discordanti sull’attendibilità della metodica di estrazione dei nuclei da paraffina; in particolari alcuni Autori riportano la perdita di intere linee cellulari a contenuto di DNA anomalo (Frierson HF, 1988; Jacobsen AB,1988; Krause JR, 1992; Mervak T, 1986; Zalupski MM, 1993).

Tecniche per l’ottenimento di sospensioni monocellulari
Un passaggio critico nell’analisi del contenuto cellulare di DNA nei tumori solidi umani è la preparazione di una sospensione monocellulare del campione tumorale(Darzynkiewicz Z, 1990).
Sono stati condotti a questo scopo molti studi in cui le tecniche sono state ottimizzate per i diversi tipi di tessuto (Thornthwaite JT, 1980; Wooley RC, 1979).
In generale, il grado di difficoltà per l’ottenimento di tali sospensioni è strettamente dipendente dal tipo di neoplasia; in particolare, poiché la natura del materiale coesivo intercellulare (tessuto connettivo, proteine e glicoproteine) varia a seconda del tessuto (Kleinmann HK, 1981) l’efficacia delle tecniche di dissociazione è influenzata dalla natura di questi costituenti.
E’ noto che il metodo di disaggregazione del tessuto può alterare le proprietà delle cellule in sospensione. Queste alterazioni possono essere morfologiche e visibili al microscopio, oppure dare origine alla perdita di costituenti cellulari come antigeni di superficie (Milas L, 1973), proteine, o alla degradazione dei polisomi (Hosick HL, 1971).
Le tecniche di disaggregazione più comunemente usate in citometria a flusso, anche in associazione tra loro, variano a seconda che il campione sia costituito da materiale fresco, congelato o incluso in paraffina.
Disaggregazione maccanica del tessuto fresco o congelato
Il trattamento di tipo meccanico con bisturi è effettuato sia su campioni a fresco che dopo congelamento (-20°C e –80°C). Nel primo caso mediante un accurato “scraping” con bisturi è possibile ottenere una sospensione cellulare che, in una elevata frequenza, mantiene l’integrità e le principali caratteristiche morfologiche della singola cellula (Cornacchiari A, 1995).
Nel secondo caso il campione opportunamente scongelato viene trattato con bisturi e successivamente con tamponi dedicati (TRIS-EDTA) fino ad ottenere una sospensione di nuclei.

Metodo di estrazione del DNA da tessuto fresco o congelato per la
valutazione della ploidia
Sospensione monocellulare da scraping della superficie neoplastica o tessuto
congelato in azoto liquido e conservato a -80 °C:
- disaggregare il frammento neoplastico con bisturi in PBS pH 7,4;
- filtrare la sospensione con filtri di nylon da 50 micron in provette immerse in
ghiaccio;
- centrifugare a 2000 rpm per 10';
- risospendere il pellet nella soluzione per isolare i nuclei TRIS-HCl-EDTA (vedi
ricetta) ;
- colorare in rapporto 1:1 con miscela di colorazione DAPI/Sr101 o IP/Fitc;
- leggere al citofluorimetro;
TRIS-HCl-EDTA
(sciogliere in 100 ml di acqua distillata: 0,097 g di TRIZMA-base, 0,661 g di TRIS-HCl, 0,29 g di NaCl, 0,04 g di EDTA (tetraNa), 0,5 ml di Nonidet P-40
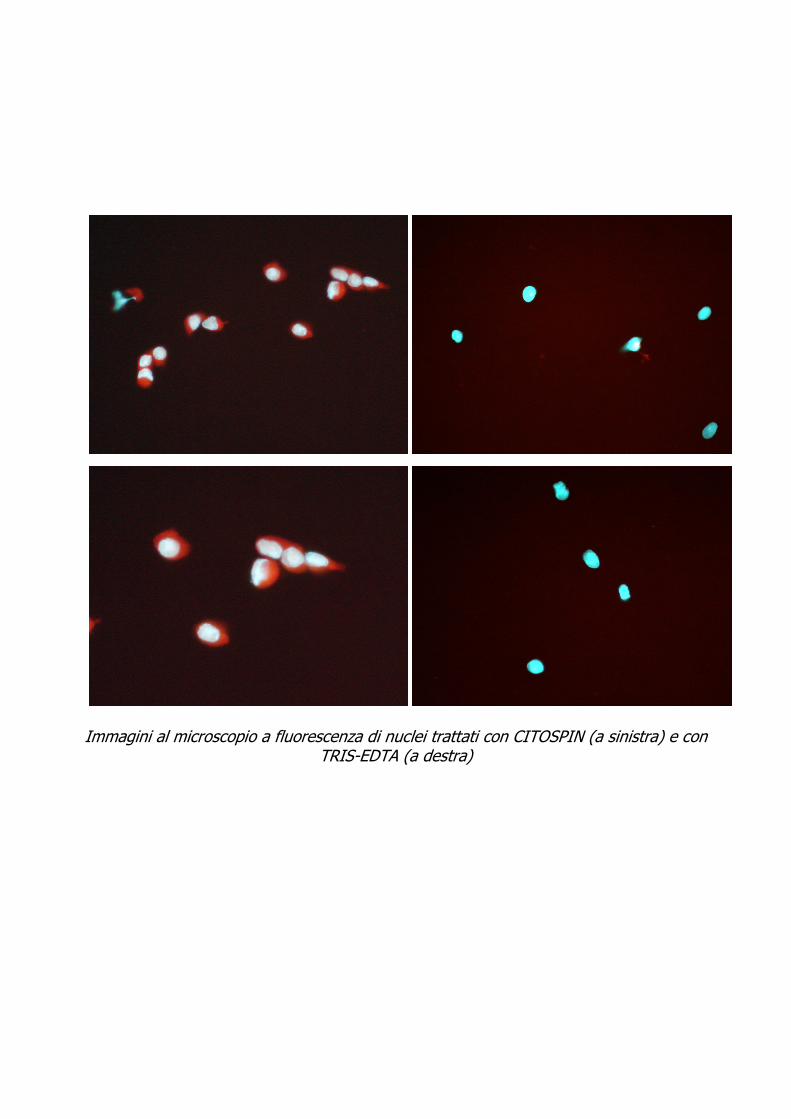
Immagini al microscopio a fluorescenza di nuclei trattati con CITOSPIN (a sinistra) e con
TRIS-EDTA (a destra)

Disaggregazione del materiale fissato in formalina ed incluso in paraffina La metodica per campioni fissati in formalina e inclusi in paraffina
sviluppata e proposta da Hedley DW (1983; 1989) offre il vantaggio di operare studi retrospettici su pazienti con decorso clinico noto e di superare il limite dovuto all’esiguità del numero dei casi negli studi prospettici.
Numerosi autori hanno confrontato questo metodo con quello inizialmente basato sull’esame di sospensioni nucleari ottenute da materiale fresco o congelato. Alcuni di essi per confrontare la qualità e la riproducibilità degli istogrammi ottenuti dai due diversi metodi, hanno notato alcuni svantaggi relativi a questo tipo di approccio, evidenziando in genere la perdita di intere linee cellulari a contenuto anomalo di DNA (Kallioniemi OP, 1987; De Vita R, 1990). La minore qualità della misura sia in termini di coefficiente di variazione, sia per la presenza di detriti cellulari determinano inoltre una sottostima della frequenza di aneuploidia.
I risultati di questi lavori sono stati spesso contrastanti e non conclusivi e questo ci ha stimolato a verificare la validità della metodica di estrazione dei nuclei da materiale incluso in paraffina. INSERIRE IL NOSTRO LAVORO SULLA PARAFFINA!!!
In conclusione, la misura del DNA da materiale paraffinato, se da una
parte consente di poter conoscere morfologicamente la popolazione cellulare su cui effettuare l’analisi del DNA, dall’altra presenta l’inconveniente di una minor riuscita della misurazione (che può dipendere dallo stato di fissazione del tessuto e dal tempo di conservazione del tessuto incluso), conseguente ad una più facile distruzione dei nuclei in frammenti che possono anche precludere la lettura dei risultati ( De Vita R, 1992; Di Vinci A, 1992).

Metodo di estrazione del DNA da tessuto paraffinato
per la valutazione della ploidia
Sezioni da 40 micron sparaffinate e idratate attaverso la scala decrescente degli alcooli:
- xilolo per una notte;
- alcool e xilolo 50% per 10’;
- alcool assoluto per 10’;
- alcool 95% per 10’;
- alcool 70% per 10’;
- alcool 50% per 10’;
- passaggio in pepsina allo 0.25% in HCl 0,1 N ph 1,5 per 60’ a 37°C;
- centrifugare per 5-10’ a 1500 rpm;
- passaggio in pepsina alla stessa concentrazione per 30’ a 37°C;
- centrifugare per 5-10’ a 1500 rpm;
- bloccare l’attivita’ della pepsina con Tris pH 8,6 centrifugando a 1500 rpm per 10’;
- sostituzione del Tris pH 8,6 con PBS pH 7,4;
- filtrare con filtri di nylon con pori del diametro di 50 micron;
- colorare con miscela d’incubazione DAPI/SR101 o IP/FITC;
- leggere al citofluorimetro.
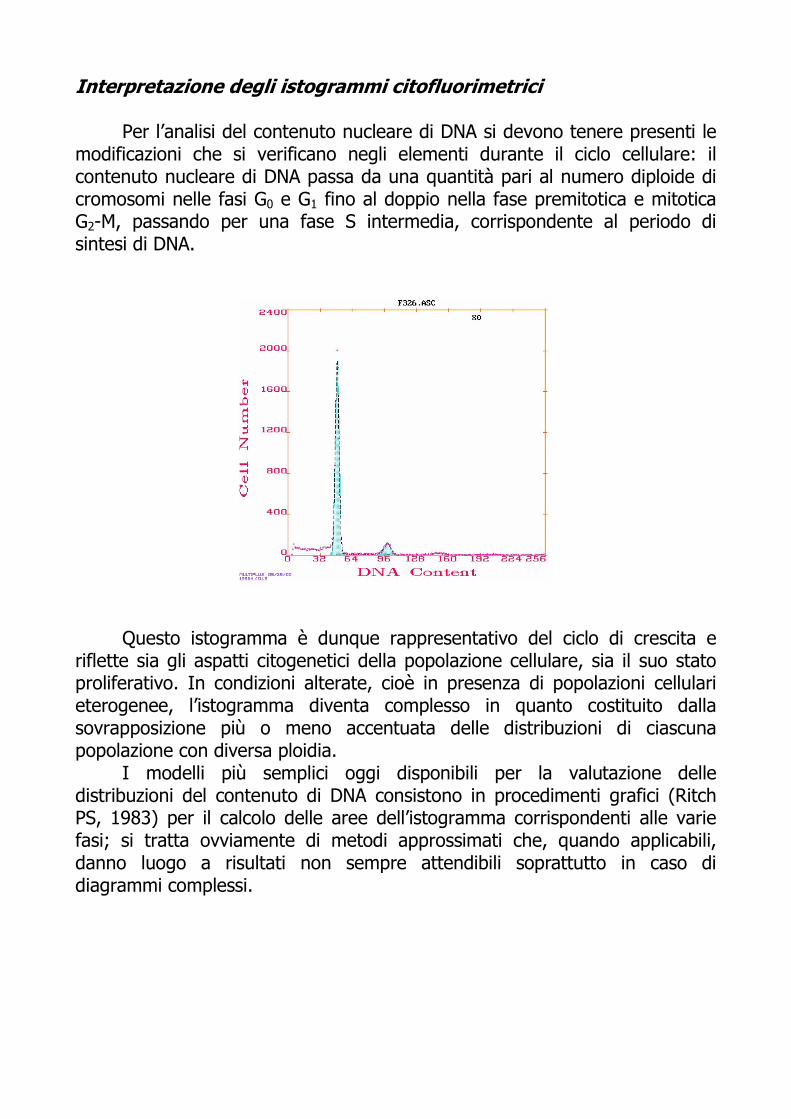
Interpretazione degli istogrammi citofluorimetrici Per l’analisi del contenuto nucleare di DNA si devono tenere presenti le
modificazioni che si verificano negli elementi durante il ciclo cellulare: il contenuto nucleare di DNA passa da una quantità pari al numero diploide di cromosomi nelle fasi G0 e G1 fino al doppio nella fase premitotica e mitotica G2-M, passando per una fase S intermedia, corrispondente al periodo di sintesi di DNA.
Questo istogramma è dunque rappresentativo del ciclo di crescita e riflette sia gli aspatti citogenetici della popolazione cellulare, sia il suo stato proliferativo. In condizioni alterate, cioè in presenza di popolazioni cellulari eterogenee, l’istogramma diventa complesso in quanto costituito dalla sovrapposizione più o meno accentuata delle distribuzioni di ciascuna popolazione con diversa ploidia.
I modelli più semplici oggi disponibili per la valutazione delle distribuzioni del contenuto di DNA consistono in procedimenti grafici (Ritch PS, 1983) per il calcolo delle aree dell’istogramma corrispondenti alle varie fasi; si tratta ovviamente di metodi approssimati che, quando applicabili, danno luogo a risultati non sempre attendibili soprattutto in caso di diagrammi complessi.

In alternativa sono stati proposti metodi analitici (Dressler LG, 1987;
Lampariello F, 1988), basati su opportune rappresentazioni della distribuzione del contenuto di DNA e dei fenomeni legati al procedimento di misura (modelli matematici), che si avvalgono dell’uso di tecniche di adattamento ottimo ai dati sperimentali per la stima dei parametri.
Questi metodi, implemantati su calcolatore, forniscono stime dei parametri biologici di interesse anche in situazioni “difficili” in cui la qualità della misura è relativamente modesta o in cui ha notevole sovrapposizione tra le varie componenti dell’istogramma. In particolare, si tratta innanzitutto di considerare il noto effetto della “dispersione di misura”: le cellule di una sottopopolazione omogenea, ad esempio quelle in fase G0-G1, pur avendo lo stesso contenuto di DNA non compaiono in corrispondenza della stessa intensità di fluorescenza ma si accumulano intorno ad un valore medio di questa formando un “picco” più o meno largo. Questa dispersione di fluorescenza è dovuta all’inevitabile imprecisione del sistema di misura, e in particolare ad imperfezioni nel processo di colorazione delle cellule e ad instabilità di funzionamento delle varie unità strumentali (fluidica, ottica ed elettronica).
In base alla larghezza del picco G0-G1 si può quindi valutare la qualità di una misura: a tale scopo viene usato il Coefficiente di Variazione (CV) dato dalla Deviazione Standard del picco (che si suppone rappresentato da una funzione gaussiana) rapportata al suo valore medio. Esso esprime in pratica la ristrettezza della curva gaussiana del picco. I valori comunemente riscontrabili oscillano tra il 2% ed il 5% e sono più bassi nelle analisi ottenute da materiale fresco o congelato. Con tali valori di CV bisogna tenere presente che una popolazioni con indice di DNA molto ravvicinato potrebbe dare luogo a due picchi tra loro no facilmente distinguibili come entità separate.
E’ quindi importante tenere conto della forma del picco: se esso è perfettamente simmetrico, è assai probabile che esso rappresenti una singola popolazione cellulare, mentre se presenta delle asimmetrie bisogna considerare la possibilità di una sovrapposizione di due diverse popolazioni cellulari che va oltre la capacità risolutiva dello strumento. Perciò, più il picco risulta stretto, minori sono state le cause di dispersione e quindi migliore è la qualità della misura.
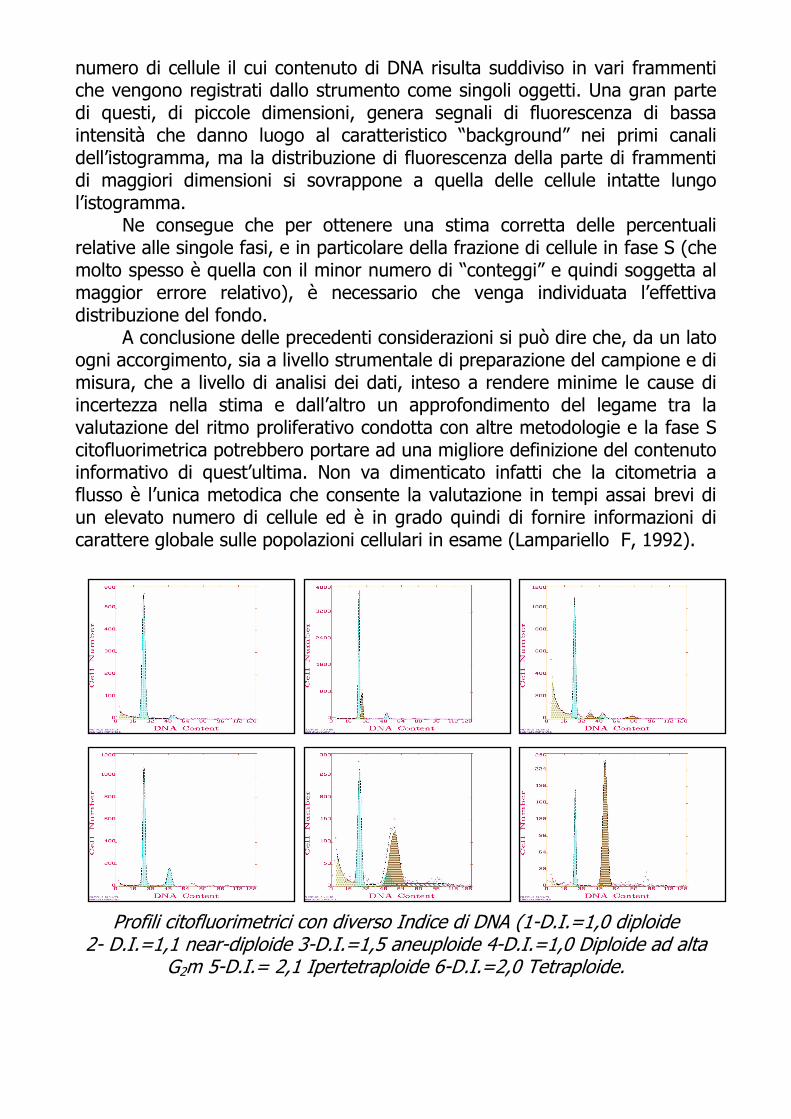
Il fenomeno della dispersione è di ostacolo alla precisa identificazione del grado di ploidia, cioè del grado di alterazione del contenuto di DNA; occorre infatti individuare dall'istogramma le intensità di fluorescenza corrispondenti al contenuto di DNA delle cellule in fase G0-G1 della popolazione tumorale e di quella diploide normale in cui il rapporto, detto indice di DNA o DNA INDEX (D.I.), è una misura del grado di ploidia.
Per convenzione picchi con un valore di D.I. compreso tra 1.85 e 2.15 vengono considerati nella regione G2-M, per cui , se la percentuale di eventi

del picco è inferiore al 20% degli eventi presenti nel ciclo cellulare il caso è considerato diploide, mentre se la percentuale supera il 20% il caso è classificato tetraploide (Kallioniemi OP, 1988). Ora, anche in condizioni strumentali ottimali, soprattutto nel caso di popolazioni cellulari anomale, il picco può risultare piuttosto largo ed anche alquanto asimmetrico (ad un elevato CV si può riferire infatti ad una variabilità intrinseca del campione cellulare intesa anche come presenza di sottopopolazioni con modeste anomalie nel contenuto di DNA) rendendo più problematica una corretta determinazione dell’indice di DNA.
Un’ulteriore causa di errore nella valutazione dell’indice di DNA è dovuta all’eventualità di un’imperfetta taratura strumentale con lo spostamento dello zero nel convertitore analogico-digitale del citometro (Givan AL, 1988) da cui deriva una non esatta proporzionalità tra intensità di fluorescenza e contenuto di DNA.
Pertanto, l’introduzione di accorgimenti atti a correggere quest’ultimo effetto e l’uso di un appropriato modello matematico che tenga conto della dispersione di fluorescenza consentono di rendere più attendibile l’interpretazione del dato citometrico. L’impiego di un modello della dispersione è invece essenziale per una corretta distinzione delle tre fasi del ciclo: infatti per effetto di essa si ha una sovrapposizione tra il picco G0-G1 e la distribuzione iniziale delle cellule in fase di sintesi e tra la parte finale di questa e il picco G2-M.
Negli istogrammi del DNA, viene frequentemente osservato che il picco G2-M non compare esattamente al doppio dell’intensità di fluorescenza corrispondente al picco G0-G1, pur essendo il contenuto di DNA delle cellule nelle due fasi l’uno il doppio dell’altro. Questo fenomeno è legato a fattori strumentali: in particolare esso può essere attribuito in parte ad un’imperfetta taratura strumentale, eventualità a cui si è già accennato ed in parte ad un effetto di non linearità del legame tra contenuto di DNA e intensità di fluorescenza (effetto di saturazione). Nella rappresentazione matematica della distribuzione dell’intensità di fluorescenza occorre ovviamente tener conto anche di questo fenomeno.
Rimane infine da considerare il problema della corretta valutazione del fondo, causato dalla presenza di frammenti cellulari, negli istogrammi relativi a campioni provenienti da tumori solidi. Questo problema costituisce tutt’ora una rilevante incognita ai fini della determinazione della percentuale di cellule in fase S a partire da istogrammi citofluorimetrici del DNA, per cui non tutti gli studiosi sono concordi sull’attendibilità di questa variabile come indice dell’attività proliferativa.
La necessità di disporre di una sospensione cellulare monodispersa, comporta l’applicazione di una procedura di disaggregazione meccanica o enzimatica per eliminare la struttura intercellulare. Questo procedimento di preparazione provoca inevitabilmente anche la frammentazione di un certo
numero di cellule il cui contenuto di DNA risulta suddiviso in vari frammenti che vengono registrati dallo strumento come singoli oggetti. Una gran parte di questi, di piccole dimensioni, genera segnali di fluorescenza di bassa intensità che danno luogo al caratteristico “background” nei primi canali dell’istogramma, ma la distribuzione di fluorescenza della parte di frammenti di maggiori dimensioni si sovrappone a quella delle cellule intatte lungo l’istogramma.
Ne consegue che per ottenere una stima corretta delle percentuali relative alle singole fasi, e in particolare della frazione di cellule in fase S (che molto spesso è quella con il minor numero di “conteggi” e quindi soggetta al maggior errore relativo), è necessario che venga individuata l’effettiva distribuzione del fondo.
A conclusione delle precedenti considerazioni si può dire che, da un lato ogni accorgimento, sia a livello strumentale di preparazione del campione e di misura, che a livello di analisi dei dati, inteso a rendere minime le cause di incertezza nella stima e dall’altro un approfondimento del legame tra la valutazione del ritmo proliferativo condotta con altre metodologie e la fase S citofluorimetrica potrebbero portare ad una migliore definizione del contenuto informativo di quest’ultima. Non va dimenticato infatti che la citometria a flusso è l’unica metodica che consente la valutazione in tempi assai brevi di un elevato numero di cellule ed è in grado quindi di fornire informazioni di carattere globale sulle popolazioni cellulari in esame (Lampariello F, 1992).
Profili citofluorimetrici con diverso Indice di DNA (1-D.I.=1,0 diploide 2- D.I.=1,1 near-diploide 3-D.I.=1,5 aneuploide 4-D.I.=1,0 Diploide ad alta
G2m 5-D.I.= 2,1 Ipertetraploide 6-D.I.=2,0 Tetraploide.

ANALISI BIPARAMETRICA DNA/PROTEINE Il recente sviluppo delle conoscenze sulle proteine quali fattori di controllo della moltiplicazione cellulare ha permesso di identificare proteine specifiche, che sarebbero responsabili dell’attivazione dei geni controllanti la divisione cellulare. L’intervento di queste proteine attivanti la divisione cellulare si svolgerebbe in corrispondenza della fase G0-G1, cioè della fase di transizione in cui le cellule “prendono la decisione per la divisione”. Le proteine totali rappresentano, oltre al DNA, il componente cellulare più importante; per questa ragione esse sono ampiamente considerate non solo per lo studio del metabolismo cellulare, ma anche e soprattutto per monitorare la dinamica della proliferazione cellulare e della trasformazione neoplastica. L’importanza delle proteine nella crescita tumorale è indicata da tempo dal ruolo di alcune proteine specifiche, come i recettori ormonali la cui presenza può permettere di prevedere la risposta di un tumore alla terapia ormonale. Parlando di proteine si intendono tutte le differenti componenti proteiche della cellula, sia citoplasmatiche, che nucleari, siano esse strutturali o facenti parte del metabolismo cellulare. E’ noto tuttavia che, per quanto riguarda la dinamica della proliferazione cellulare, le proteine strutturali sono poco coinvolte, mentre lo sono molto di più le nucleoproteine (Mazzini G.). Il ruolo delle proteine nucleari nel controllo della espressione genica e della struttura cromosomica è stata oggetto di numerosi studi, saggi e monografie per oltre dieci anni. Generalmente le proteine nucleari possono essere divise in due gruppi: gli istoni e le proteine non istoniche. Le proteine istoniche Gli istoni furono isolati per la prima volta da Albrecht Kossel nel 1884, non molto tempo dopo la scoperta del DNA (il DNA fu scoperto da F.Miescher nel 1871). Gli istoni si distinguono dalla maggior parte delle altre proteine cellulari per la loro abbondanza, la piccola dimensione e la forte carica positiva. Gli istoni si possono facilmente isolare mediante trattamento della cromatina con acidi o soluzioni ad alta concentrazione salina. La loro rimozione mediante questi metodi dimostra che essi sono associati alla cromatina mediante attrazioni elettrostatiche piuttosto che mediante legami covalenti.

La forte carica positiva degli istoni è determinata dalla presenza di residui di lisina e arginina, che costituiscono una parte preponderante della componente aminoacidica di queste proteine, il cui peso molecolare varia tra 11.000 e 15.000 dalton, ad eccezione di H1 che ha un peso molecolare compreso tra 21.000 e 28.000 dalton negli eucarioti superiori (vedi tabella).
ISTONE PESO MOLECOLARE
NUMERO DI AMMINOACIDI
CONTENUTO DI AMMINOACIDI BASICI
H1 H2A H2B H3 H4
17.000-28.000 13.900 13.800 15.300 11.300
200-265 129-155 121-148
135 102
27% lisina, 2% arginina 11% lisina, 9% arginina 16% lisina, 6% arginina 10% lisina, 15% arginina 11% lisina, 4% arginina
Caratteristiche chimiche degli istoni
Gli istoni di molti eucarioti sono stati completamente sequenziati, cosa che ha permesso di comprendere le strutture fondamentali dei diversi tipi di istoni; in tutti i tipi, infatti, H1, H2A, H2B, H3 e H4, gli aminoacidi basici e apolari sono organizzati in domini separati quando gli istoni sono in soluzione. I domini apolari di ogni istone formano una singola regione globulare. I domini basici si estendono come braccia distese da una o entrambe le estremità della regione globulare. H2A, H2B, H3 e H4 sono chiamati istoni del core, in quanto formano il nucleo dalla struttura a forma di perla attorno alla quale si avvolge il DNA a formare un nucleosoma. H1 è chiamato istone di giunzione in quanto si pensa si combini in prossimità di un segmento di DNA che funziona da congiunzione tra un nucleosoma e il successivo. Il confronto tra le sequenze degli istoni di specie diverse ha rivelato che, in particolare quelli degli eucarioti superiori, sono tra le proteine più conservate. Il grado di conservazione è approssimativamente inversamente proporzionale al sistema di numerazione degli istoni; così H3 e H4 sono i più conservati, H2A e H2B sono mediamente conservati e H1 è il meno conservato. Per ogni tipo di istone, tranne che per H4, esistono comunemente diverse varianti, che presentano una sequenza leggermente diversa anche all’ interno della stessa specie. Il numero di variazioni, come osservato per la conservazione della sequenza, è approssimativamente inversamente proporzionale all’ordine di numerazione. La maggior parte delle specie di vertebrati presentano due o tre varianti, che differiscono tra loro per una o due posizioni aminoacidiche. H1 è

di gran lunga il più variabile; per quanto riguarda gli istoni del core, le varianti vengono solitamente identificate dal tipo dell’istone seguito da un punto e da un numero. L’esistenza di diverse varianti anche in individui appartenenti alla stessa specie indica che la maggior parte degli istoni è codificata da più di un gene. Tale ipotesi è confermata da studi di sequenza, che rivelano che gli istoni di tutte le specie eucariotiche sono codificati da geni presenti in copie multiple, da decine a centinaia, con diverse duplicazioni dei geni che codificano ciascuna variante. Gli specifici cambiamenti che avvengono in relazione allo sviluppo e alla divisione cellulare indicano che le varianti non rappresentano dei cambiamenti casuali; non funzionali delle proteine istoniche. Piuttosto, esse sono necessarie per la modificazione della struttura della cromatina, così critica per il normale procedere della cellula verso il differenziamento e la duplicazione. I cinque tipi di istoni sono ampiamente modificati dall’aggiunta di gruppi chimici a singoli residui aminoacidici. Semplici modificazioni includono l’aggiunta di gruppi acetilici, metilici o fosfato ad aminoacidi localizzati nelle braccia cariche positivamente degli istoni. Altre modificazioni coinvolgono l’aggiunta di strutture chimiche relativamente complesse: una proteina chiamata ubiquitina, e una complessa unità nucleotidica il gruppo ADP ribosio. Le varie modificazioni degli istoni sono correlate ad importanti meccanismi cellulari come la regolazione genica, la replicazione del DNA, l’assemblaggio e la condensazione della cromatina, la divisione cellulare (Stephen L. Wolfe, 1994).

Le proteine non istoniche Col nome di proteine non istoniche si indicano tutte quelle proteine, esclusi gli istoni, che sono associate al DNA nella cromatina. La maggior parte delle proteine di questo vasto gruppo sono cariche negativamente o neutre a pH fisiologico. In questo gruppo si trovano proteine che regolano l’attività dei singoli geni. Esse sono, quindi, deputate al controllo dell’attività cellulare e del differenziamento. Il numero totale dei diversi tipi di proteine non istoniche si è dimostrato difficile da determinare; poiché la maggior parte di esse, a differenza degli istoni, presentano proprietà chimiche simili a quelle di altre proteine cellulari, è spesso difficile determinare quali polipeptidi di estratti siano proteine cromosomiche non istoniche e quali siano contaminanti provenienti da altre fonti cellulari. La distinzione fra proteine intere e frammenti polipeptidici presenta anch’essa delle difficoltà, in quanto molte proteine non istoniche si degradano facilmente originando frammenti che possono sembrare appartenenti a diverse proteine. Inoltre, molte proteine non istoniche sono presenti nel nucleo in quantità così piccole da essere difficili da individuare. Queste difficoltà hanno prodotto un’ampia varietà nel numero e nei tipi di polipeptidi identificati come proteine non istoniche. Generalmente le proteine non istoniche vengono classificante in: • proteine che regolano la trascrizione genica; • enzimi e fattori attivi nella trascrizione, replicazione, ricombinazione, riparazione e modificazione del DNA e delle proteine cromosomiche; • proteine associate all’RNA; • proteine che contribuiscono al mantenimento della struttura della cromatina o alla conversione della cromatina dallo stato decondensato allo stato altamente compattato. Molto interesse oggi ha suscitato lo studio delle modalità di interazione delle proteine non istoniche col DNA. Non c’è dubbio che queste proteine possano riconoscere e legare specificamente sequenze e molecole di DNA completamente avvolte a doppia elica. Le proteine di regolazione evidentemente si legano a specifiche sequenze di DNA nella doppia elica principalmente leggendo i contorni delle coppie di basi esposti a livello del solco maggiore. Tale processo comporta la formazione di legami idrogeno tra gli atomi delle coppie di basi e i gruppi laterali degli aminoacidi della proteina di regolazione (Stephen L. Wolfe, 1994). Proteine nucleari e patologia neoplastica Il lavoro più recente presente in letteratura su questo tipo di analisi appartiene al gruppo della Teodori, la quale, in uno studio del 1998

concernente l’Esofago di Barrett, sottolinea l’importanza e l’utilità dell’analisi biparametrica DNA/Proteine. La metaplasia intestinale identificata col nome di Esofago di Barrett appare frequentemente associata ad un aumentato rischio di adenocarcinoma esofageo e la displasia compare solamente come uno stadio intermedio della progressione da metaplasia a neoplasia. La determinazione simultanea di DNA e proteine, realizzata con una combinazione di DAPI e sulforodamina, insieme ad una attenta analisi istologica sono state applicate a 362 campioni provenienti da 30 pazienti affetti da tale displasia e sottoposti a follow-up dal 1985. Nove casi sono risultati aneuploidi, cinque dei quali (IV gruppo) con un profilo inequivocabile, mentre, negli altri quattro casi (III gruppo) l’aneuploidia è stata determinata solo in seguito ad analisi biparametrica. Ventuno pazienti sono stati classificati, invece, come diploidi; dodici di questi (II gruppo) erano caratterizzati da un’elevata fase G1 ovvero da un contenuto proteico comparabile con quello del primo gruppo costituito da nove pazienti con profilo diploide, ma con un basso-moderato contenuto proteico. A tre pazienti del quarto gruppo è stato diagnosticato un adenocarcinoma in situ dopo 5, 6 e 10 anni rispettivamente. In due pazienti del terzo gruppo, sono stati osservati, invece, un basso ed un alto grado displastico (frequentemente associato ad adenocarcinoma) al terzo a al sesto anno di follow-up, rispettivamente. Solamente un caso del primo gruppo mostrava inizialmente un alto contenuto proteico, quindi un assetto aneuploide e infine la comparsa dell’adenocarcinoma (dopo 12 anni). Nessuno dei pazienti diploidi è progredito verso la displasia o il cancro, comparso invece, in sei dei tredici pazienti aneuploidi. In conclusione, è possibile affermare che l’analisi citofluorimetrica DNA/Proteine è un importante fattore predittivo di rischio neoplastico, nei casi di displasia, nonchè marker di malignità e come tale può essere impiegato per identificare pazienti a più elevato rischio e per monitorare i diversi stadi della proliferazione maligna in studi di follow-up a lungo termine.
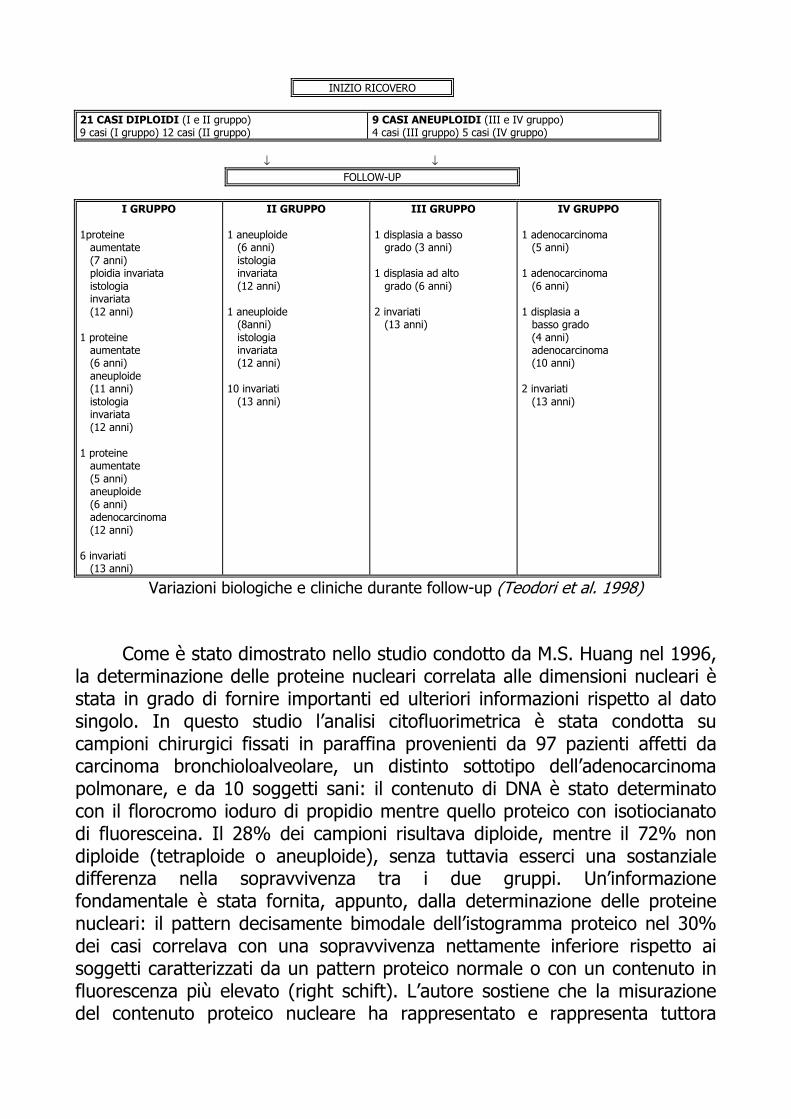
INIZIO RICOVERO
21 CASI DIPLOIDI (I e II gruppo) 9 casi (I gruppo) 12 casi (II gruppo)
9 CASI ANEUPLOIDI (III e IV gruppo) 4 casi (III gruppo) 5 casi (IV gruppo)
↓ ↓
FOLLOW-UP
I GRUPPO 1proteine aumentate (7 anni) ploidia invariata istologia invariata (12 anni) 1 proteine aumentate (6 anni) aneuploide (11 anni) istologia invariata (12 anni) 1 proteine aumentate (5 anni) aneuploide (6 anni) adenocarcinoma (12 anni) 6 invariati (13 anni)
II GRUPPO
1 aneuploide (6 anni) istologia invariata (12 anni) 1 aneuploide (8anni) istologia invariata (12 anni) 10 invariati (13 anni)
III GRUPPO 1 displasia a basso grado (3 anni) 1 displasia ad alto grado (6 anni) 2 invariati (13 anni)
IV GRUPPO
1 adenocarcinoma (5 anni) 1 adenocarcinoma (6 anni) 1 displasia a basso grado (4 anni) adenocarcinoma (10 anni) 2 invariati (13 anni)
Variazioni biologiche e cliniche durante follow-up (Teodori et al. 1998)
Come è stato dimostrato nello studio condotto da M.S. Huang nel 1996, la determinazione delle proteine nucleari correlata alle dimensioni nucleari è stata in grado di fornire importanti ed ulteriori informazioni rispetto al dato singolo. In questo studio l’analisi citofluorimetrica è stata condotta su campioni chirurgici fissati in paraffina provenienti da 97 pazienti affetti da carcinoma bronchioloalveolare, un distinto sottotipo dell’adenocarcinoma polmonare, e da 10 soggetti sani: il contenuto di DNA è stato determinato con il florocromo ioduro di propidio mentre quello proteico con isotiocianato di fluoresceina. Il 28% dei campioni risultava diploide, mentre il 72% non diploide (tetraploide o aneuploide), senza tuttavia esserci una sostanziale differenza nella sopravvivenza tra i due gruppi. Un’informazione fondamentale è stata fornita, appunto, dalla determinazione delle proteine nucleari: il pattern decisamente bimodale dell’istogramma proteico nel 30% dei casi correlava con una sopravvivenza nettamente inferiore rispetto ai soggetti caratterizzati da un pattern proteico normale o con un contenuto in fluorescenza più elevato (right schift). L’autore sostiene che la misurazione del contenuto proteico nucleare ha rappresentato e rappresenta tuttora

un’importante e indipendente indicatore prognostico quando associato alle dimensioni nucleari e alla determinazione del contenuto di DNA (Huang Ming S. et al. 1996).
Distribuzione gaussiana del contenuto proteico
Già alcuni anni prima G. Ciancio (1993) anticipava il metodo citato precedentemente introducendo l’utilizzo di ribonucleasi (RNasi). Le sospensioni nucleari ottenute dalle sezioni incluse in paraffina successivamente a idratazione, incubazione in una soluzione di pepsina allo 0,5%, lavaggio e risospensione in tampone contenente detergente non ionico, sono state colorate con isotiocianato di fluoresceina e ioduro di propidio in presenza di RNasi. Numerosi tipi di tessuti tumorali sono stati analizzati, inclusi mammella, colon, timo e rene: i migliori risultati sono stati ottenuti con un trattamento con pepsina iniziale di 1,5 ore anzichè 0,5 ottenendo la riduzione del coefficiente di variazione di entrambi i parametri DNA e proteine nucleari, senza tuttavia ridurre il livello di proteine nucleari o determinare una significativa disgregazione dei nuclei. Poichè i nuclei tumorali aneuploidi hanno tipicamente un livello più alto di proteine rispetto ai nuclei diploidi, la tecnica permette la miglior definizione di popolazioni near-diploidi (Ciancio G.et al. 1993). Nello studio condotto nel 1992 da L. Teodori et al. allo scopo di distinguere sottopopolazioni cellulari neoplastiche con differente significato prognostico e diagnostico, misurazioni biparametriche DNA/Proteine sono state condotte simultaneamente su campioni di tessuto polmonare provenienti da 110 pazienti affetti da malattie neoplastiche e non. L’analisi biparametrica ha dimostrato che le cellule caratterizzate da un’elevata fluorescenza rossa (che è indicativa del contenuto proteico), associabile ad una crescita incontrollata, sono state più frequentemente riscontrate in tumori maligni, che in campioni di tessuto normale.

Inoltre, l’analisi biparametrica ha permesso il riconoscimento di linee cellulari tumorali addizionali indicanti che la frequenza dei tumori diploidi determinati citrometricamente era più bassa di quella descritta precedentemente con la sola analisi monoparametrica. L’identificazione di sottopopolazioni aneuploidi attraverso l’analisi biparametrica in campioni definiti diploidi dall’analisi monoparametrica e negativi dal punto di vista clinico e istologico, assume certamente un importante valore diagnostico. I risultati hanno, inoltre, mostrato la presenza di multiple sottopopolazioni proteiche in cloni caratterizzati dalla stessa ploidia, il che sta ad indicare un più alto livello di eterogeneità cellulare di quella dimostrata dalla sola analisi citofluorimetrica del DNA (Teodori L. et al. 1992). Parlando di analisi biparametrica DNA/Proteine non si deve dimenticare un’altra importantissima tecnica di colorazione introdotta da Mazzini et al. nel 1987 e basata sull’utilizzo della coppia di coloranti DACM e ioduro di propidio. Si tratta di una metodica alternativa alla precedente (PI/FITC), che utilizza un fluorocromo N-(7-dimetilamino-4-metil-cumarinil) maleimide (DACM) avente affinità per i gruppi funzionali -SH (tioli), coinvolti nella struttura dei nucleoli e attivamente sintetizzati nel ciclo cellulare e di cui le proteine nucleari (segnatamente la componente non-istonica) sono particolarmente ricche. La colorazione dei nuclei isolati con DACM consente una più selettiva marcatura delle proteine nucleari. Le caratteristiche spettrofluorimetriche di questo colorante consentono inoltre, una sua associazione allo ioduro di propidio, potendo essere entrambi eccitati in ultravioletto. Per quanto concerne l’emissione, la fluorescenza blu di DACM è ben separabile da quella rossa del propidio, il che rende la metodica di impego ideale con strumentazione a lampada dove l’eccitazione della FITC può non essere ottimale (Mazzini G. et al. 1987). Come dimostrato da Ffrench et al. nel 1985, la determinazione simultanea di Dna e proteine può fornire preziose informazioni anche riguardo la proliferazione e il metabolismo cellulari. Il contenuto proteico cellulare correlato con parametri nucleari geometrici può essere sfruttato, infatti, per valutare la maturazione e la differenziazione di una cellula. Nell’esperimento sono stati analizzati, attraverso citometria a flusso, i leucoblasti provenienti da 50 casi di leucemia mieloide acuta contemporaneamente ad un esame morfometrico semiautomatico eseguito su aspirati midollari. L’istogramma biparametrico ottenuto colorando il DNA con ioduro di propidio e le proteine con isotiocianato di fluoresceina ha permesso di correlare un basso contenuto proteico con una sottopopolazione cellulare non proliferante (frazione LPC) e con la fase di sintesi (fase S). La più accentuata dispersione dei valori proteici,rispetto a quelli della fase S, suggerisce inoltre, la possibilità di individuare più chiaramente eventuali

differenze tra popolazioni cellulari. Questa ipotesi è stata verificata studiando il significato prognostico del ciclo cellulare; i pazienti seguiti sono stati divisi in due gruppi in base al conseguimento di una completa remissione o meno. Trenta casi hanno presentato un netto miglioramento entro due mesi, gli altri venti casi sono deceduti. A differenza dei valori della fase S, quelli della fase LPC erano significativamente diversi nei due gruppi e più precisamente risultavano maggiori nel gruppo in cui si era ottenuta una completa guarigione. Il valore prognostico della frazione LPC può essere quindi definito come nella Tabella che segue:
LPC (%) REMISSIONE NON COMPLETA
REMISSIONE COMPLETA
>20% 7 20
<20% 13 10
Valore prognostico della frazione LPC
Nel presente studio è stato inoltre dimostrato come la differenziazione e la maturazione cellulare siano in grado di modificare le dimensioni del nucleo; la combinazione dei risultati ottenuti permatte di ottenere, in ultima analisi, utili informazioni per la classificazione citologica (Ffrench M. et al. 1985). La rapida procedura citofluorimetrica sviluppata da Pollack nel 1984 per la determinazione simultanea di DNA e proteine a base di isotiocianato di fluorescina e ioduro di propidio impiegati su nuclei isolati non presupponeva la centrifugazione dei campioni ed era facilmente utilizzabile per la colorazione di linfociti umani di sangue periferico stimolati con fitoemoagglutinina, di sospensioni di cellule linfoidi tumorali murine EL4 e di campioni di adenocarcinomi prostatici di ratto R3327-G. Gli istogrammi derivati dall’analisi dei linfociti umani di sangue periferico non stimolati con fitoemoagglutinina e di quelli trattati con actinomicina D, idrossiurea, 3H-TdR (timidina tritiata), colcemide, o con una combinazione di idrossiurea e colcemide hanno mostrato che: • le cellule quiescenti della fase G1Q sono distinguibili da quelle della fase
“tardiva” G1 (G1AB); • le cellule della fase “precoce” G2 (G2A) sono distinguibili da quelle della
fase “tardiva”G2 (G2B); • le cellule mitotiche sono distinguibili da quelle della fase G2. Mentre il trattamento con idrossiurea ha determinato un aumento del numero di cellule diploidi aventi un elevato contenuto proteico nucleare (G1AB), l’incubazione con 3H-TdR ha causato l’incremento del numero di quelle ad elevato contenuto proteico, ma con assetto tetraploide (G2B). L’incubazione con colcemide ha, invece, bloccato le cellule mitotiche, che si

sono così rese riconoscibili per il basso contenuto proteico nucleare (minore rispetto a quello dei nuclei della fase G2A), e per l’assetto tetraploide. Gli istogrammi DNA/Proteine delle cellule linfoidi tumorali murine EL4 trattate e non trattate con colcemide hanno confermato un risultato sovrapponibile all’esperimento precedente. Il metodo è stato inoltre utilizzato per valutare la risposta dei tumori prostatici di ratto R3327-G androgeno-sensibili alla privazione di androgeno successiva alla castrazione(Pollack A. 1984). Come si è potuto facilmente apprendere dalla letteratura finora analizzata, le analisi biparametriche DNA/Proteine forniscono maggiori dettagli rispetto alle sole misurazioni monoparametriche; in particolare, mentre l’analisi monoparametrica del DNA offre informazioni circa la distribuzione delle cellule nelle fasi G0-G1, S e G2-M, la determinazione simultanea di un secondo parametro, come le proteine, ha permesso l’identificazione di nuovi compartimenti all’interno del ciclo cellulare (G1A e G1B). Darzynkiewicz et al., nel 1981 usando arancio di acridina per quantificare simultaneamente singole e doppie eliche di acidi nucleici, è stato in grado di delineare i compartimenti G1Q, G1A, G1B, S, G2 ed M.
L’applicazione di questa tecnica alle analisi di tessuti neoplastici di
vescica indica che l’informazione aggiuntiva che se ne ricava è fondamentale per l’identificazione di cellule linfoidi contaminanti, così come per selezionare preventivamente cellule di tessuti benigni e maligni non identificabili esclusivamente su base cinetica. L’iniziale metodo di colorazione con arancio di acridina, tuttavia, richiedeva cellule intere e non era quindi adattabile all’analisi multiparametrica di tumori fibrosi. Successivamente Piwnicka et al. usando arancio di acridina, e Roti Roti et al. usando ioduro di propidio e isotiocianato di fluorescina hanno introdotto metodi multiparametrici

impiegando nuclei isolati, il che rende queste tecniche più adatte anche per l’analisi di tumori solidi. Nel 1980 l’analisi citofluorimetrica è stata impiegata anche per monitorare la radioattività causata dall’incorporazione di timidina tritiata (3H-TdR) da parte di linfociti umani di sangue periferico stimolati con fitoemoagglutinina e successivamente colorati con ioduro di propidio. L’esperimento, condotto da Pollack et al., prevedeva l’utilizzo di differenti concentrazioni di timidina tritiata in intervalli di tempo diversi su microcolture di linfociti. I risultati, ottenuti confrontando i due tipi di istogrammi del DNA rilevati rispettivamente su cellule incubate con 3H-TdR e cellule non incubate, hanno dimostrato che la timidina tritiata, quando somministrata in particolari condizioni (marcatura sia a breve termine continua che a impulsi) a linfociti proliferanti, determina un significativo incremento della popolazione cellulare tetraploide proporzionale alla concentrazione e al tempo di marcatura con timidina tritiata e che sembra rappresentare un blocco in fase G2. Infine, è stata confermata la possibilità che la marcatura a breve termine continua rappresenti un modo per saggiare sostanze che interferiscono con la proliferazione linfocitaria e con la stima dei tempi del ciclo cellulare (A.Pollack et al. 1980). Dallo studio condotto nel 1979 da Blair e Roti Roti in cui sono stati verificati attraverso l’analisi citofluorimetrica gli effetti dell’ipertermia sul contenuto proteico di cellule HeLa, è emerso che il contenuto in FITC è direttamente proporzionale al contenuto proteico nucleare e che la distribuzione nucleare della FITC è da attribuire al legame con le proteine non istoniche della cromatina. Sfruttando metodi citofluorimetrici e biochimici, infatti, è stato possibile osservare un aumento del contenuto proteico dei nuclei isolati, fissati e colorati con isotiocianato di fluorescina (FITC) di cellule HeLa “scaldate” per 7.5, 15 o 30 minuti a 45°C. La fluorescenza emessa dai nuclei così trattati è risultata rispettivamente 1.46 ± 0.05, 1.88 ± 0.14 e 2.02 ± 0.14 rispetto alle cellule di controllo. Inoltre, l’incremento del contenuto proteico è risultato maggiore proporzionalmente ad un minore tempo di riscaldamento (inferiore a 15 minuti). Per contro, l’aumento proteico è stato determinato anche biochimicamente ed è risultato 1.18 ± 0.04, 1.30 ± 0.06 e 1.39 ± 0.05 nei rispettivi tempi di trattamento. I risultati ottenuti concordano perfettamente, sebbene non lo dimostrino direttamente, con la convinzione che la componente non-cromatinica non aumenta significativamente successivamente a riscaldamento. Pertanto, l’incremento proteico potrebbe essere dovuto a proteine nucleari solubili e proteine cromosomiali debolmente legate, che, normalmente perdute durante il processo di isolamento dei nuclei, rafforzano il loro legame con la cromatina, quando sottoposte a trattamento ipertermico (Owen C. Blair, Roti Roti et al. 1979).

L’importanza biologica della determinazione del contenuto di DNA e proteine, sia nelle cellule che nei cromosomi singoli, ha determinato un grande sviluppo delle tecniche di colorazione e di misura di queste sostanze. Oggi, infatti, è disponibile una vasta serie di coloranti fluorescenti con meccanismi di legame e caretteristiche spettrali assai diverse. Otto diverse combinazioni di coloranti fluorescenti per la determinazione simultanea di DNA e proteine erano state elaborate e valutate già nel 1978 da Stohr et al.: ioduro di propidio (PI) con isotiocianato di fluoresceina (FITC), fluorescamina (FC) e dansilclorito (DANS); diamidinofenilindolo (DAPI) con sulforodamina (SR101), isotiocianato di tetrametilrodamina (RITC) e nitrobenzodiazolo (NBD); acriflavina (AF) con isotiocianato sulfonico stilbene (SITS) e DAPI. Gli studi, condotti su tre diverse linee cellulari tumorali, sono stati di due tipi: l’analisi biparametrica eseguita con il citofluorimetro HEIFAS e l’analisi spettroscopica dei due gruppi di fluorocromi basata sulla loro massima eccitazione. DANS e NBD, a differenza dei restanti fluorocromi impiegati nelle diverse combinazioni, non hanno fornito un risultato comparabile con quello ottenuto dalla distribuzione delle proteine con citometria a flusso. Relativamente ai tempi di preparazione dei campioni e ai passaggi in centrifuga previsti dai protocolli di colorazione e alla stabilità dei fluorocromi utilizzati, il metodo con DAPI/SR101 è risultato essere il più veloce nonchè il più semplice per lo studio simultaneo di DNA/Proteine nell’arco di 30 min (M. Stohr et al. 1978).
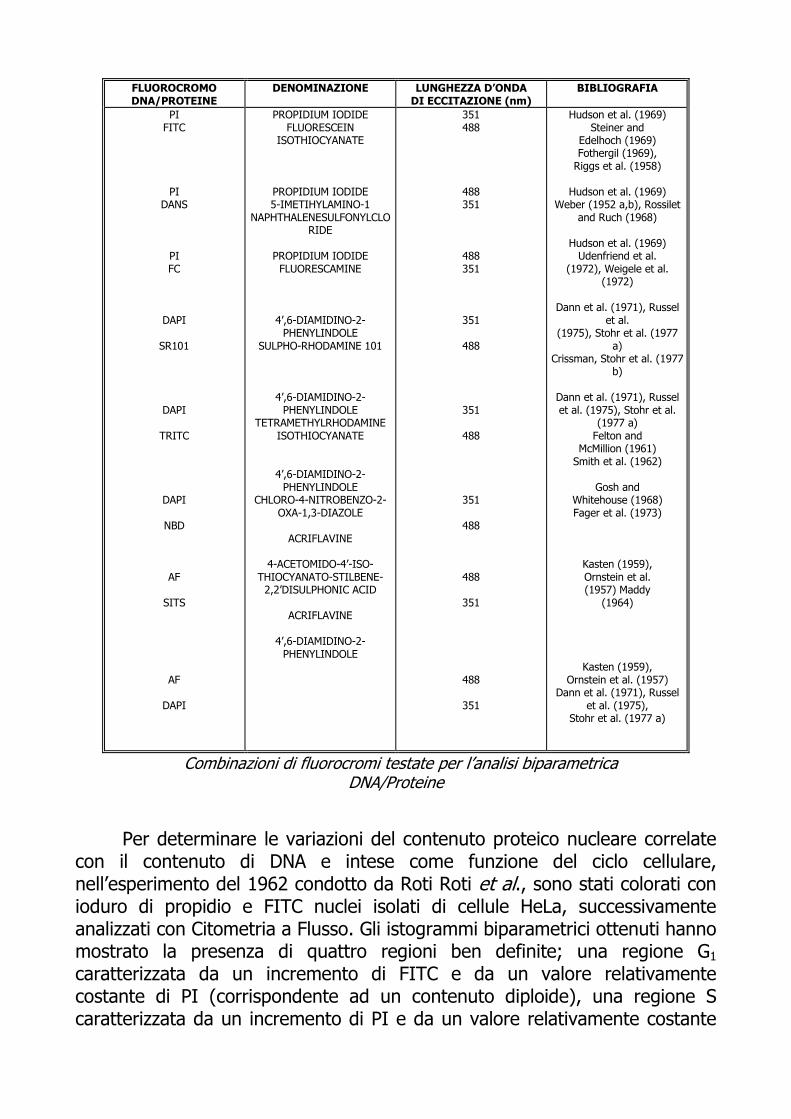
FLUOROCROMO DNA/PROTEINE
DENOMINAZIONE LUNGHEZZA D’ONDA DI ECCITAZIONE (nm)
BIBLIOGRAFIA
PI FITC
PI DANS
PI FC
DAPI
SR101
DAPI
TRITC
DAPI
NBD
AF
SITS
AF
DAPI
PROPIDIUM IODIDE FLUORESCEIN
ISOTHIOCYANATE
PROPIDIUM IODIDE 5-IMETIHYLAMINO-1
NAPHTHALENESULFONYLCLORIDE
PROPIDIUM IODIDE
FLUORESCAMINE
4’,6-DIAMIDINO-2-PHENYLINDOLE
SULPHO-RHODAMINE 101
4’,6-DIAMIDINO-2-PHENYLINDOLE
TETRAMETHYLRHODAMINE ISOTHIOCYANATE
4’,6-DIAMIDINO-2-PHENYLINDOLE
CHLORO-4-NITROBENZO-2-OXA-1,3-DIAZOLE
ACRIFLAVINE
4-ACETOMIDO-4’-ISO-
THIOCYANATO-STILBENE-2,2’DISULPHONIC ACID
ACRIFLAVINE
4’,6-DIAMIDINO-2-
PHENYLINDOLE
351 488
488 351
488 351
351
488
351
488
351
488
488
351
488
351
Hudson et al. (1969) Steiner and
Edelhoch (1969) Fothergil (1969),
Riggs et al. (1958)
Hudson et al. (1969) Weber (1952 a,b), Rossilet
and Ruch (1968)
Hudson et al. (1969) Udenfriend et al.
(1972), Weigele et al. (1972)
Dann et al. (1971), Russel
et al. (1975), Stohr et al. (1977
a) Crissman, Stohr et al. (1977
b)
Dann et al. (1971), Russel et al. (1975), Stohr et al.
(1977 a) Felton and
McMillion (1961) Smith et al. (1962)
Gosh and
Whitehouse (1968) Fager et al. (1973)
Kasten (1959), Ornstein et al. (1957) Maddy
(1964)
Kasten (1959), Ornstein et al. (1957)
Dann et al. (1971), Russel et al. (1975),
Stohr et al. (1977 a)
Combinazioni di fluorocromi testate per l’analisi biparametrica DNA/Proteine
Per determinare le variazioni del contenuto proteico nucleare correlate con il contenuto di DNA e intese come funzione del ciclo cellulare, nell’esperimento del 1962 condotto da Roti Roti et al., sono stati colorati con ioduro di propidio e FITC nuclei isolati di cellule HeLa, successivamente analizzati con Citometria a Flusso. Gli istogrammi biparametrici ottenuti hanno mostrato la presenza di quattro regioni ben definite; una regione G1 caratterizzata da un incremento di FITC e da un valore relativamente costante di PI (corrispondente ad un contenuto diploide), una regione S caratterizzata da un incremento di PI e da un valore relativamente costante

di FITC, una regione G2 caratterizzata da un incremento di FITC e da un valore costante di PI (corrispondente ad un contenuto tetraploide) e infine una regione “intermedia” caratterizzata cioè da un livello di FITC e PI leggermente inferiori a quelli osservati nella popolazione tetraploide. La correlazione tra queste regioni e il ciclo cellulare è stata confermata da due studi in particolare; il primo riguarda la distribuzione dei nuclei nell’istogramma dopo marcatura “a impulsi” con (14C)TdR, il secondo l’uscita delle cellule dalla fase G1 che è stata osservata dopo aggiunta di colcemide alle colture cellulari di HeLa. Il contenuto proteico nucleare non è aumentato uniformemente durante il ciclo cellulare, ha anzi, raggiunto il picco massimo nel corso della fase G1. Questo conferma che il contenuto di proteine nucleari, misurato in termini di intensità di fluorescenza verde emessa dalla FITC, può essere considerato come un secondo parametro ciclo cellulare-dipendente. La conclusione trova conferma anche nell’osservazione che la sintesi di proteine cromosomiali non-istoniche, verificandosi prima dell’inizio della fase S, potrebbe rappresentare un evento critico e fondamentale per la sintesi di DNA (J.L.Roti Roti et al. 1962). Metodo di analisi per la quantificazione del contenuto proteico
Sulla base delle considerazioni sovraesposte è facile capire l’importanza di focalizzare la determinazione prevalentemente alla componente proteica nucleare, quando si intende studiare la proliferazione cellulare. Un metodo semplice per raggiungere tale scopo è quello di utilizzare nuclei isolati, anziché cellule, in questo modo viene eliminato il contributo delle proteine citoplasmatiche, che è di solito considerevole. Più recentemente si è cercato di raggiungere lo stesso scopo per altra via, ovvero cercando di utilizzare colorazioni citochimiche specificamente sviluppate per la messa in evidenza di particolari componenti proteiche. Anzichè considerare il gruppo funzionale aminico comune a tutte le proteine, si considera il radicale sulfidrilico (SH); esso non è ugualmente rappresentato in tutte le componenti proteiche cellulari, ma è maggiormente contenuto nelle nucleoproteine. Il N-(7-dimetilamino-4-metil-cumarinil) maleimide è un colorante specifico per i gruppi tiolici, da tempo impiegato per determinazioni biochimiche. Le sue caratteristiche chimico fisiche sono inoltre particolarmente adatte ad un suo impiego in Citometria a Flusso. Importante è la sua capacità di attraversare la membrana cellulare integra e quindi la possibilità di colorare cellule non fissate. In queste condizioni di impiego si ottiene la massima intensità di fluorescenza ad una determinata concentrazione di colorante. Cellule fissate in etanolo mostrano intensità di fluorescenza più bassa in ragione del fatto che il fissativo provoca la

formazione di ponti disolfuro fra radicali sulfidrilici vicini e quindi conseguente diminuzione di quelli disponibili al colorante. La determinazione delle proteine nucleari non correlata ad altri parametri cellulari è pochissimo utilizzata per lo scarso significato del dato singolo; numerose sono invece le applicazioni basate sulla determinazione contemporanea di proteine e altri componenti cellulari, primo fra tutti il DNA. Analisi spettrofluorimetriche effettuate sulla coppia di coloranti DACM-ioduro di propidio hanno consentito di verificare la fattibilità della colorazione simultanea di DNA e proteine nucleari. Utilizzando una singola eccitazione a 366 nm è possibile eccitare sufficientemente entrambi i coloranti misurando la fluorescenza blu del DACM fra 450 e 530 nm e quella rossa del propidio oltre 620 nm. Queste condizioni di misura sono state sperimentate su colture di cellule in crescita esponenziale, fissate e suddivise in aliquote, colorate sia con i coloranti singoli, che con una miscela di DACM e ioduro di propidio. E’ stato così verificato che l’interferenza reciproca dei due fluorocromi è minima in queste condizioni sperimentali. Attualmente questa metodologia è applicata allo studio delle caratteristiche proliferative e della ploidia nel carcinoma della mammella. I risultati preliminari ottenuti, analizzando oltre cento campioni, hanno consentito di stabilire che la metodica è affidabile e che è particolarmente utilizzabile con sistemi di misura provvisti di eccitazione mediante lampada a mercurio. Generalmente il contenuto di nucleoproteine è basso in campioni di tessuto normale di controllo e in casi con con diagnosi benigna rispetto a casi di carcinoma a vari stadi e con presenza di aneuploidia (Mazzini G.). La misura del contenuto proteico cellulare utilizza la classica reazione tra il gruppo -N=C-S- dell’isotiocianato di fluorescina (FITC) o di rodamina (RITC) e il gruppo NH2 delle proteine. Il legame tra FITC e proteine è di tipo covalente e quindi assai stabile ed anche la fluorescenza del complesso FITC-proteine è stabile, diversamente dalle soluzioni acquose di FITC. Ciò consente di usare questa colorazione per misure quantitative del contenuto proteico cellulare. Va tuttavia ricordato che le procedure di colorazione richiedono particolare cura: ciò deriva dalla cinetica di colorazione relativamente lenta, dalla suddetta instabilità delle soluzioni FITC e dalla minore efficienza quantica del colorante legato rispetto a quello libero. La colorazione delle proteine con FITC, inoltre, consente l’uso simultaneo dello ioduro di propidio (IP) come colorante del DNA in quanto, mentre gli spettri di eccitazione dei due fluorocromi sono sufficientemente sovrapposti, le emissioni risultano ben separate (Starace G.). Una metodica alternativa alla precedente, originariamente proposta da Stohr prevede l’utilizzo della miscela DAPI/SR101 (sulforodamina) e la lettura dei preparati con citometri a flusso provvisti di eccitazione con lampada a vapori di mercurio (Stohr M. et al., 1977).

IP/DACM IP/FITC DAPI/SR101
Eccitazione(nm) 488 366 488 488 350 590
Emissione (nm) 623 450-530 623 530 470 620
Lunghezze d’onda di eccitazione e di emissione dei fluorocromi più utilizzati
Campioni fissati in formalina e inclusi in paraffina Da una sezione di 3 micron colorata con Ematossilina-Eosina è stata selezionata un’area con prevalente componente tumorale da sottoporre all’analisi citofluorimetrica. Il metodo utilizzato per l’analisi del contenuto di DNA prevede il recupero dell’area prescelta dal blocchetto in paraffina e la processazione secondo il metodo di Hedley (Hedley at al. 1983) parzialmente modificato.

Metodo di estrazione del DNA da tessuto fissato e incluso in
paraffina per l’analisi biparametrica DNA/Proteine
Sezioni da 50 micron sparaffinate e idratate:
- xilolo per una notte;
- alcool e xilolo 50% per 10’;
- alcool assoluto per 10’;
- alcool 95% per 10’;
- alcool 70% per 10’;
- alcool 50% per 10’;
- pepsina allo 0.25% in HCl 0,1 N ph 1,5 per 60’ a 37°C;
- centrifugare per 5-10’ a 1500 rpm;
- pepsina alla stessa concentrazione per 30’ a 37°C;
- centrifugare per 5-10’ a 1500 rpm;
- bloccare l’attivita’ della pepsina con Tris pH 8,6 centrifugando a 1500 rpm
per 10’;
- sostituire il Tris pH 8,6 con PBS pH 7,4;
- filtrare con filtri di nylon con pori del diametro di 50 micron;
- colorare con miscela d’incubazione IP/FITC;
- leggere al citofluorimetro.
L’analisi del contenuto proteico effettuata sulle stesse sospensioni monocellulari consiste nel colorare i nuclei in due passaggi successivi con una “miscela d’incubazione” proposta da Ming S. Huang e composta da FITC 3 µg/ml in NaHCO3 a pH 8,0 e IODURO DI PROPIDIO al 50 µg/ml in PBS. La scelta della miscela colorante è stata effettuata dopo numerose prove su tessuto normale (linfonodi e mammella) e neoplastico in base alla sorgente di eccitazione del citofluorimetro e all’intensità della fluorescenza emessa. La lettura dei preparati è stata effettuata al Citofluorimetro COULTER PROFILE II (COULTER) e il controllo ottico della fluorescenza al Microscopio a Fluorescenza JENA.

I parametri selezionati per l’analisi erano i seguenti: istogramma 1: LFS/LSS; istogramma 2: COUNT/FL3; istogramma 3: COUNT/FL1; istogramma 4: FL1/FL3 (FL1=FITC, FL3=IP, LSF=Light Forward Scatter, LSS= Light Side Scatter). Campioni congelati Di ogni tumore giunto in reparto per l’esame estemporaneo, un frammento di tessuto neoplastico viene congelato in isopentano pre-raffredato in azoto liquido e conservato in freezer a -80°C. La preparazione dei campioni per l’analisi citofluorimetrica DNA/Proteine prevede la disgregazione meccanica con una lama da bisturi in tampone fosfato (PBS) del tessuto congelato, la filtratura della sospensione attraverso filtri di nylon dello spessore di 50 micron e l’immediata colorazione con una miscela fluorescente composta da 0,016 mg/ml di DAPI e 0,01 mg/ml di Sr101.
Metodo di estrazione del DNA da tessuto fresco o congelato per la
valutazione del contenuto proteico
Sospensione monocellulare da scraping della superficie neoplastica o tessuto congelato
in azoto liquido e conservato a -80 °C:
- disaggregare il frammento neoplastico con bisturi in PBS pH 7,4;
- filtrare la sospensione con filtri di nylon da 50 micron in provette immerse in
ghiaccio;
- centrifugare a 2000 rpm per 10';
- risospendere il pellet nella soluzione per isolare i nuclei TRIS-HCl-EDTA (vedi
ricetta) ;
- colorare in rapporto 1:1 con miscela di colorazione DAPI/Sr101 o IP/Fitc;
- leggere al citofluorimetro;

Le sospensioni monodisperse, colorate con una miscela DAPI-Sr101 (inserire preparazione!!!) sono state successivamente lette al citofluorimetro PARTEC CCA (Dako Spa) provvisto di lampada ad arco a vapori di mercurio. Anche in questo caso, di ogni preparato 50 microlitri di soluzione venivano strisciati su un vetrino e controllati al Microscopio a fluorescenza. I parametri selezionati per l’analisi erano: istogramma 1: FL1/FL2; istogramma 2: COUNT/FL1; istogramma 3: COUNT/FL2 (FL1=DAPI, FL2=SR101).

STUDIO CITOFLUORIMETRICO DEL CICLO CELLULARE
Le conoscenze sul ciclo cellulare, i suoi marcatori ed i fattori di controllo che ne regolano l’attività hanno avuto un recente sviluppo soprattutto per quanto riguarda la comprensione di quei segnali che causano “l'immissione nel ciclo" di cellule che prima erano quiescenti o in fase G0. Enorme interesse
suscita inoltre la conoscenza di quei meccanismi che inducono le cellule tumorali ad entrare in fase di sintesi (fase S) ed a proliferare.
Grazie a metodiche in grado di evidenziare in modo preciso le diverse fasi in cui una singola cellula si trova (G0, G1, S e G2/M), è possibile
completare il quadro diagnostico di patologie tumorali sempre più complesse. La determinazione delle cellule in mitosi (indice mitotico) è la metodica
più antica e tradizionale, basata sulla valutazione quantitativa al microscopio ottico di questo evento morfologico del ciclo, nelle sue fasi: profase, metafase, anafase e telofase.
Il vantaggio di questa osservazione è la sua fattibilità su sezioni fissate e colorate secondo le metodiche utilizzate comunemente in anatomia patologica per scopi diagnostici. La limitazione è rappresentata dal fatto che la mitosi occupa un intervallo molto limitato del ciclo, per cui rappresenta un evento scarsamente presente in tumori caratterizzati da una bassa attività proliferativa.
Inoltre, in un processo di proliferazione anomala, come quella tumorale, la mitosi può, ed è stato dimostrato, essere una fase finale della vita della cellula per l’impossibilità delle cellule con anomalie cromosomiche di procedere nel ciclo proliferativo.
A partire dagli anni 50 studi biochimici condotti su linee cellulari sincronizzate con l’impiego di precursori degli acidi nucleici e delle proteine marcati radioattivamente, hanno dimostrato che il periodo che intercorre tra una fase mitotica e quella successiva, denominato intercinetico od erroneamente “a riposo”, è in realtà un periodo di intensa attività metabolica dell’acido ribonucleico e delle proteine in preparazione (G1) o a completamento (G2) della fase S, durante il quale, accanto alla sintesi di RNA e proteine si verifica la duplicazione del materiale genetico e di DNA (Howard A. 1953).
La conoscenza dell’esistenza di precise fasi non morfologicamente riconoscibili ma identificabili su precisi eventi metabolici ha portato alla proposizione e messa a punto di nuove metodologie volte essenzialmente alla caratterizzazione delle fasi del ciclo cellulare.
L’utilizzo di anticorpi monoclonali e policlonali (PCNA, MIB1 e cicline) che riconoscono il loro antigene in campioni fissati in formalina ed inclusi in paraffina è già da alcuni anni una realtà quotidiana del laboratorio di

immunistochimica. Il livello di standardizzazione tecnica raggiunto attraverso l’applicazione del trattamento con forno a micronde alle sezioni, si contrappone al criterio di quantificazione della positività che risulta in molti casi indaginoso (analizzatore di immagini), lungo e a volte soggettivo.
Interessante sarebbe invece l’utilizzo di questi marcatori in citofluorimetria sfruttando anticorpi marcati con traccianti fluorescenti.
In questa linea di indagini si colloca il nostro studio, dedicato all’allestimento di un metodo, come descritto in letteratura prevalentemente applicato a linee cellulari stabilizzate, da applicare a materiale congelato ed incluso in paraffina proveniente da tumori solidi umani.
Meccanismi di controllo delle fasi del ciclo La cinetica di una cellula attivamente proliferante è regolata da due tipi di geni:
- I proto-oncogeni, che codificano per delle proteine (recettori) in grado di trasmettere all’interno della cellula i segnali, come i fattori di crescita, mediante fosforilazione e attivazione di altre proteine note come trasduttori. Gli oncogeni sono versioni alterate di questi proto-oncogeni che codificano per delle oncoproteine in grado di trasferire continuamente il segnale ai trasduttori anche in assenza del ligando (fattore di crescita).
- I geni soppressori hanno la funzione di regolare la proliferazione producendo delle anti-oncoproteine in grado di legarsi al DNA bloccandone la trascrizione. Le cellule tumorali hanno delle tipiche alterazioni di questi geni che perdono così di funzionalità e non sono più in grado di limitare la proliferazione (Tab. 1).
Tipo di Tumore
Oncogeni e anti-oncogeni
Attivati/inattivati
Colon
Mammella Polmone Pancreas Tiroide Gliomi AML
Ras, p53, DCC
p53, RB, c-myc, Cerb-B2 p53, RB, C-, N-, L-myc
ras, p53 ras, ret, trk, gsp Cerb-B2, p53, RB
ras, fms
Tumori umani con oncogeni ed anti-oncogeni attivati/inattivati

Il gene p53, localizzato sul braccio corto del cromosoma 17, codifica per una anti-oncoproteina fosforilata di 393 aminoacidi caratterizzata da una brevissima emivita (30 minuti) e presente principalmente nel nucleo ed occasionalmente nel citoplasma perinucleare.
La p53 è un fattore di trascrizione che legandosi a livello di particolari sequenze del DNA (promotori o enhancers) accelera o ritarda la sintesi dei trascritti della RNA-polimerasi II. Le forme mutate, generalmente dovute ad una perdita di eterozigosi sul 17p per mutazione di un allele normale, sono molto più stabili da un punto di vista metabolico con conseguente accumulo cellulare a causa dell’aumentata emivita della proteina (Ulrich SJ. 1992).
Un altro gene il cui prodotto è legato all’attività proliferativa cellulare è il bcl-2 (B-cell leukaemia-limphoma), la sua sovra-espressione è stata originariamente scoperta sui linfomi follicolari umani come conseguenza di una traslocazione cromosomica t(14;18) (Ngan B-Y. 1988). Questa traslocazione porta il gene bcl-2 in prossimità di loci per l’Ig causando così la over-espressione della proteina BCL-2. Questo gene codifica per una proteina integrale di membrana di circa 25 Kda che si trova preferibilmente all’interno della cellula piuttosto che sulla superficie cellulare.
La proteina BCL-2 è localizzata sulla membrana interna e su quella più esterna dei mitocondri, all’esterno del nucleo e sul reticolo endoplasmatico (Akao Y. 1994).
Normalmente questa proteina è espressa in tessuti che sono in continuo rinnovamento come quelli fetali o della cute (Lebrun DP 1993). Una sovraespressione della proteina BCL-2 è riportata in numerosi tumori maligni in cui si verifica perciò una aumentata sopravvivenza cellulare come nei cancri gastrici, colon - rettali e della prostata oltre a linfomi e leucemie (Pezzella F. 1990).
Questi risultati suggeriscono un possibile ruolo patologico del bcl-2 nella carcinogenesi. Per quanto riguarda i tumori mammari questa proteina è presente nel 70% dei casi con una eterogenea correlazione nei confronti dei tipici parametri prognostici di questa neoplasia (Gee JMW 1994).
ll passaggio delle cellule da uno stadio all'altro del ciclo è regolato da una serie di fattori che ne assicurano la progressione in modo ordinato. Negli ultimi dieci anni sono stati condotti studi approfonditi sulle basi molecolari che regolano la cinetica del ciclo nel passaggio da una all'altra fase. Tramite l'utilizzo di mutanti del ciclo ottenuti grazie alla genetica e l'aggiunta di proteine specifiche alle colture in esame è stato possibile dimostrare che la regolazione delle varie fasi del ciclo dipende molto dalle modificazioni finemente regolate dell'attività di un complesso sistema polipeptidico denominato MPF (maturation-promoting factor) che, contenuto all'interno del citoplasma (Murray AW.1988, Gould KL. 1989) è il principale regolatore del passaggio della cellula in due punti del ciclo cellulare: lo start in cui la cellula in G1 replica o meno il DNA, costringendosi

ad entrare in fase S in caso di sintesi; un punto di transizione in cui la cellula decide di entrare in mitosi.
L'MPF è una fosfoproteina dall'attività fluttuante garantita da una duplice configurazione ciclo-correlata: 1) pre-MPF non appare in interfase poiché vi si trova in una forma fosforilata inattiva; 2) MPF in forma attiva che è riconoscibile nella fase M delle cellule mitotiche e degli oociti (Cyert MS and Kirschner MW, 1988).Nella frazione maggiormente purificata della sua molecola si possono riconoscere due principali polipeptidi con diversa massa molecolare (32 KDa e 45 KDa). Lohka e collaboratori, basandosi su studi fatti sul lievito Saccharomyces C. che dimostravano come il gene cdc 2 fosse implicato nella regolazione della mitosi, identificarono il prodotto di questo gene: la proteina p34 uno dei polipeptidi che costituiscono l'MPF.
Studi fatti successivamente da Nurse valorizzarono ulteriormente la teoria sulla p34 e resero possibile la sua identificazione come una protein-chinasi cioè un enzima che trasferisce gruppi fosfato dall'ATP alle proteine (Nurse P. 1990). Il fatto che la proteina CDC2 è una componente dell'MPF non spiegava perché questo fattore fosse attivo durante la mitosi ma non durante l'interfase, visto che la concentrazione di p34 rimaneva costante per tutto il ciclo, si pensò che forse era la seconda componente dell'MPF ad attivare o meno la CDC2 e quindi a regolare l'attività dell'MPF stesso.
Questa seconda porzione dell'MPF fu identificata all'inizio degli anni '80, venne chiamata ciclina per le sue modificazioni ciclo correlate, infatti pur essendo prodotta costantemente come la p34 lungo il ciclo, si accumula in interfase e scompare nell'anafase mitotica a causa di una rapida degradazione in mitosi. Si scoprirà successivamente che è la ciclina B1, codificata dal gene cdc13 l'effettiva subunità regolatrice dell'MPF e che di conseguenza partecipa all'attivazione della proteina p34 (Murray AW.1989). Questa attivazione, sulla quale è basato un meccanismo di controllo che regola l'entrata di tutte le cellule eucariotiche in mitosi, avviene secondo
Nurse grazie a due eventi chiave: la defosforilazione della p34cdc-2 a livello del sito di legame dell'ATP e l'associazione con la ciclina. Come già accennato, il livello di p34 rimane invariato lungo tutto il ciclo delle cellule proliferanti, ma la sua attività catalitica si fa sentire nella fase M, ciò dimostra che la sua è una regolazione post-trascrizionale.
Nell’interfase la ciclina B1 si associa al prodotto del gene cdc2 formando il complesso pre-MPF che essendo inattivo (Kirschner M.1992) si accumula durante questa fase. Il complesso cdc2/cyc B1 è stabilizzato dalla fosforilazione della subunità p34-catalitica a livello di un residuo di treonina (Thr 161), fosforilazione poi necessaria all’attivazione stessa. Visto che la semplice unione delle cicline con la proteina cdc2 non è sufficiente ad attivare il complesso chinasico dell’MPF, dovranno esserci delle
reazioni in grado di modificare la p34cdc2 e la ciclina, prima che l’MPF diventi
funzionale. Il controllo di queste modificazioni, secondo Russel e Nurse

(Russel P. 1987) è da imputare a due geni, scoperti in Schizosaccharomyces pombe, i cui messaggeri presentano interessanti variazioni funzionalmente correlate con l’entrata in mitosi:
- il gene wee1 che inibisce la mitosi poichè codifica per l’enzima tirosin-
fosfoprotein-chinasi che fosforilando la p34cdc-2 su un residuo di treonina (Thr 14) e su uno di tirosina (Tyr 15) sul sito di legame per l’ATP, rende inattivo il complesso MPF.
- il gene cdc25+ induce invece la mitosi poichè codifica per l’enzima tirosin-fosfoprotein-fosfatasi in grado di rimuovere i fosfati inibitori attivando
la p34cdc-2 e di conseguenza l’MPF. Negli eucariotici più evoluti le protein-chinasi che inibiscono l’MPF sono codificate non solo dal gene wee1 ma anche dal gene mik1 che può fosforilare anche un residuo di treonina (Thr 161) della p34 (Pines J. 1991). Questo ha indotto a ipotizzare che fosse l’attività antagonista di questi due gruppi di geni a regolare l’attivazione del complesso cdc2/cycB1 (pre-MPF/ MPF) e l’entrata in mitosi delle cellule eucariotiche.
L’MPF attivo poi non controlla solo i processi che riguardano la divisione fisica del nucleo ma attiva anche una ciclin-proteasi cioè un enzima in grado di degradare le cicline mitotiche, dato che le cellule non sono in grado di terminare la divisione fino a quando queste cicline non sono praticamente scomparse. Cicline
Sono state individuate varie cicline il cui accumulo in diversi punti del ciclo permette l’attivazione di particolari passaggi.
Generalmente le cicline sono ubiquitarie negli eucarioti e presentano in tutte le specie un 30% di identità di sequenza per circa 200 residui aminoacidi a livello di una regione centrale denominata “cyclin box” (Hunt T. 1989). Piccole variazioni all’interno di questa regione distinguono le cicline in più classi caratterizzate da differenti cinetiche di accumulo durante il ciclo ed in grado di azionare eventi diversi. Le cicline G2:
La ciclina B1 è una molecola chiave che controlla l’entrata delle cellule in mitosi essendo l’unità regolatrice della subunità catalitica dell’MPF. Si accumula in G2 ed è bruscamente degradata, attraverso una via ubiquitino-
mediata, alla fine della mitosi. La ciclina A, scoperta negli invertebrati marini mostra un pattern
d’espressione simile a quello della B1 con la differenza che questa ciclina compare già a livello della transizione G1/S (Swenson K. 1986).
La ciclina F, la più grande della famiglia (87kDa) è stata recentemente scoperta in lieviti cdc4 mutanti. Il suo mRNA attraversa l'intero ciclo cellulare

raggiungendo un picco d’espressione in G2 e decresce prima del declino
dell’mRNA della ciclina B1. La localizzazione cellulare della proteina è all’interno del nucleo in prossimità della membrana nucleare.
La ciclina G partecipa all’inibizione delle cdcks (Okamoto K. 1994). La ciclina H si lega alla protein chinasi cdk7 attivando la fosforilazione
della treonina 161 necessaria per consolidare il legame in G2 tra la P34 e la ciclina B. Le cicline G1:
Ciclina C: i suoi livelli oscillano lievemente durante tutto il ciclo cellulare.
Ciclina E: ha un'espressione massima nel passaggio tra G1 ad S (Koff
A. 1991). La ciclina D è espressa ad un livello piuttosto costante nelle cellule che
proliferano continuamente, si presume perciò che le cicline D siano indotte nella risposta a fattori di crescita ed a differenza delle cicline E, A e B1
periodicamente espresse, queste possono non essere coinvolte nella macchina replicativa del ciclo cellulare.
Valutazione della proliferzione cellulare nei tumori solidi
La prognosi di una neoplasia viene classicamente definita dalle caratteristiche morfologiche, dall’istotipo, dal grado di differenziazione, dalle sue dimensioni, dalla sua estensione locale e dallo stato linfonodale. Negli ultimi anni si è cercato di associare a questi parametri morfologici informazioni che descrivessero le caratteristiche biologiche del tumore. A questo proposito primariamente ci si è rivolti allo studio della proliferazione cellulare dato che nel tumore, come in qualsiasi tessuto non neoplastico, le dimensioni dipendono dal rapporto esistente tra l’indice di proliferazione delle cellule e la quantità di cellule che vanno incontro a morte.
Nelle neoplasie, tuttavia, le cellule hanno un ritmo proliferativo più elevato se proporzionato al tessuto normale circostante e di conseguenza, una crescita maggiore (Mangili F. 1997).
Tra i parametri proposti per la valutazione biologica delle neoplasie umane, lo studio dell'attività proliferativa è senza dubbio quello di maggior interesse.
Infatti esso può fornire informazioni sia sull’evoluzione della patologia tumorale, sia nella ricerca applicata dove la conoscenza di tali meccanismi può portare anche alla comprensione dei processi di morte cellulare programmata (apoptosi).
Esistono varie metodiche utilizzate per lo studio non solo qualitativo ma anche quantitativo dell'attività proliferativa cellulare (Tab.2):

- morfologiche - biochimiche: Timidina tritiata Timidina chinasi BrdU - citofluorimetriche: DNA ploidia e fase S - immunistochimiche: Ki-67 PCNA
- citochimiche e istochimiche: AgNORs Valutazione morfologica
La determinazione dell'indice mitotico è la metodica più antica per identificare le cellule in divisione mediante rivelazione al microscopio ottico.
I conteggi mitotici danno una buona indicazione dell’attività proliferativa mediante visualizzazione su sezioni fissate e colorate anche se ha il difetto di essere un metodo troppo laborioso se condotto correttamente infatti il numero di cellule da contare deve essere elevato ed inoltre la mitosi, in campioni tumorali, è spesso un evento finale della vita delle cellule neoplastiche che a causa delle loro aberrazioni cromosomiche spesso non riescono a progredire nel ciclo proliferativo. Determinazione biochimica Mediante l’utilizzo di precursori del DNA marcati, che al momento della sintesi vengono incorporati all’interno del DNA di neo-produzione, è possibile ottenere una marcatura delle cellule in fase S e rilevarla mediante autoradiografia. Si può ottenere questo mediante l’impiego di timidina tritiata (3H-Tdr). L’indice delle cellule in fase S può essere ottenuto poi mediante conteggio al microscopio oppure in modo automatico con l’uso di sistemi di analisi di immagine computerizzata. I problemi di questa tecnica sono dovuti all’uso di un isotopo radioattivo, laboriose tecniche di autoradiografia e l’obbligo di utilizzare materiale fresco perché le cellule devono essere in grado di incorporare la timidina tritiata.

L’uso della timidina kinasi è un enzima di recupero dei nucleotidi pirimidinici che esiste in due forme: TK1 è una forma citosolica che mostra una maggiore attività nelle cellule in divisione ma assente nelle resting cells (cellule in G0). TK2 è invece di origine mitocondriale e rimane relativamente costante in tutto il ciclo.
Anche la bromodeossiuridina (BrdU) viene utilizzata per marcare il DNA in fase S. La BrdU è una base pirimidinica alogenata analoga della timidina che viene aggiunta al terreno di coltura delle cellule e da loro incorporata nel DNA di neo-sintesi. Attualmente l’avvenuta incorporazione di BrdU viene routinariamente rilevata con tecniche immunoistochimiche che utilizzano specifici anticorpi anti-BrdU marcati con fluorocromi o con perossidasi, è così possibile calcolare un indice di marcatura detto labelling Index paragonabile all’indice ottenuto con la timidina tritiata.
Il principale vantaggio è la rapidità di questa tecnica che però necessita come la precedente di materiale fresco e tecniche da associare per il recupero dell’antigenicità come un pretrattamento con HCl che denatura il DNA al fine di poter fare esporre la BrdU espressa su catene di DNA a singola elica. Determinazione citofluorimetrica
La quantificazione della percentuale di cellule in fase S può essere fatta anche mediante il dosaggio del contenuto di DNA in cui si utilizzano tecniche citofotometriche (analisi di immagine) grazia alla reazione di Feulgen e citofluorimentriche a flusso, previa reazione con opportuni fluorocromi fenantridinici (Ioduro di Propidio e Bromuro di Etidio) ed indolici (DAPI e DIPI);
Questi processi citochimici ormai assodati hanno avuto, soprattutto nel periodo iniziale, dei problemi di tipo distribuzionale ovvero dalla differente distribuzione della cromatina nel nucleo e di conseguenza del colorante nell’area da misurare.
Il risultato dell’analisi quantitativa del DNA è tradotto in un istogramma di frequenza dei valori ottenuti per ogni singola cellula; in base all’intensità dell’emissione del fluorocromo (quantità di DNA) è possibile collocare la cellula all’interno delle fasi del ciclo e quantificarne la percentuale (Vedi figura). Di conseguenza in base alla distribuzione della popolazione nelle diverse fasi del ciclo sarà possibile definire la ploidia basandosi sulla precedente lettura di un controllo diploide (Indice di DNA o DNA Index).
L’accuratezza dell’analisi è rappresentata dal coefficiente di variazione (CV) che è in funzione dell’ampiezza del picco della fase G0-G1. Un’accessiva ampiezza del CV può significare una analisi poco sensibile, condizionare la
capacità di riconoscere eventuali aneuploidie ed alterare l’accuratezza della valutazione della fase S. Questo valore è influenzato dal tipo di tessuto in analisi, dalla sua fissazione e dalla metodica utilizzata per ottenere la sua disaggregazione.
Determinazione immunoistochimica
I metodi immunoistochimici maggiormente usati per lo studio del ciclo cellulare sono quelli che utilizzano anticorpi specifici contro gli antigeni PCNA e KI-67. PCNA
E’ una proteina di 36 Kda che funziona come un fattore ausiliario della DNA polimerasi-delta ed ha una funzione enzimatica essenziale nella replicazione del DNA. In passato per la rivelazione dell’Antigene Nucleare di Proliferazione Cellulare (PCNA) venivano utilizzati anticorpi provenienti da sieri di alcuni pazienti affetti da leucemia mieloide cronica (Takasaki Y. 1981). Tuttavia era difficile trovare sieri che non contenessero anticorpi in grado di riconoscere anche altri antigeni nucleari. Attualmente vengono preferiti anticorpi monoclonali specifici anti-PCNA prodotti e immessi sul mercato dalle principali aziende commerciali.
Grazie all’impiego di questo nuovo strumento è stato possibile ottenere nuove informazioni sull’attività proliferativa di tessuti normali e neoplastici e la dimostrazione della correlazione tra percentuale di positività alla PCNA e grado di malignità in tumori della mammella, polmone e sistema nervoso centrale (Dawson AE. 1990).
La PCNA è correlata direttamente con lo stato proliferativo della cellula ed il suo andamento all’interno del ciclo prevede la comparsa in tarda fase G1,

raggiungendo la sua massima espressione nella fase S e infine diminuendo durante la G2/M (Celis JE.1984).
Quando la cellula si trova in mitosi ha un contenuto molto basso di PCNA a causa della rottura dell’involucro nucleare con conseguente dispersione del materiale nel citoplasma e dalla degradazione della PCNA stessa. Sono state così classificati due tipi di PCNA: il primo con localizzazione citoplasmatica e conseguentemente più solubile del secondo che essendo strettamente legata ai siti di replicazione del DNA all’interno del nucleo risulta insolubile.
La presenza di cellule con positività intracitoplasmatica avvalora l’ipotesi che la PCNA, come altre proteine ciclo-correlate, prima si accumuli nel citoplasma e poi venga traslocata all’interno del nucleo dove andrà a svolgere la sua funzione regolatrice (Pines J. 1991). Ki-67
Oltre alla già citata PCNA abbiamo un altro anticorpo che viene largamente utilizzato, ormai routinariamente in anatomia patologica per lo studio immunocitochimico della percentuale di cellule ciclanti: è l’anticorpo anti Ki-67.
Questo anticorpo monoclonale come il suo equivalente per il materiale incluso in paraffina MIB-1, è in grado di riconoscere una proteina a localizzazione nucleare (codificata da un complesso genico ubicato sul cromosoma 10), costituita da due subunità di peso molecolare pari rispettivamente a 395 Kdalton e 345 Kdalton. Questo antigene è espresso sia nelle cellule normali che in quelle tumorali ma in quantità diverse. La sua espressione inizia nella fase G1, subisce un progressivo incremento quantitativo nel corso delle fasi S e G2 ed infine raggiunge il suo picco massimo nella fase M. Determinazione citochimica ed istochimica Ag-NORs
Questa metodica che sfrutta una semplice impregnazione argentica è utile per evidenziare una certa attività proliferativa collegata direttamente alla presenza di proteine nucleari. I geni che codificano per queste proteine sono presenti sulle braccia corte dei cromosomi acrocentrici 13, 14, 15, 21, 22 a livello delle costrizioni secondarie, note come Regioni Organizzatrici Nucleolari (NOR).
Questi geni sono trascritti velocemente dalla RNA Polimerasi I e i prodotti di questa trascrizione, le subunità 45S e 5S, vengono inglobate in un complesso citoplasmatico all’interno del quale avviene l’assemblaggio dei
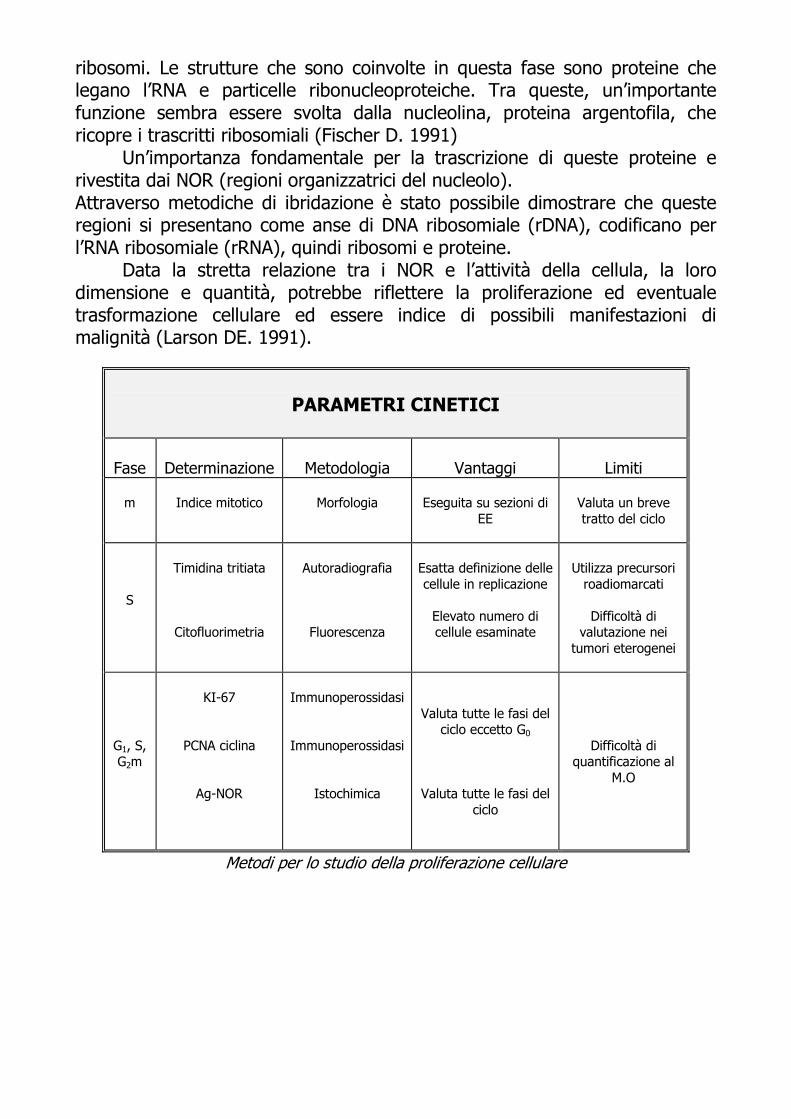
ribosomi. Le strutture che sono coinvolte in questa fase sono proteine che legano l’RNA e particelle ribonucleoproteiche. Tra queste, un’importante funzione sembra essere svolta dalla nucleolina, proteina argentofila, che ricopre i trascritti ribosomiali (Fischer D. 1991)
Un’importanza fondamentale per la trascrizione di queste proteine e rivestita dai NOR (regioni organizzatrici del nucleolo). Attraverso metodiche di ibridazione è stato possibile dimostrare che queste regioni si presentano come anse di DNA ribosomiale (rDNA), codificano per l’RNA ribosomiale (rRNA), quindi ribosomi e proteine.
Data la stretta relazione tra i NOR e l’attività della cellula, la loro dimensione e quantità, potrebbe riflettere la proliferazione ed eventuale trasformazione cellulare ed essere indice di possibili manifestazioni di malignità (Larson DE. 1991).
PARAMETRI CINETICI
Fase
Determinazione
Metodologia
Vantaggi
Limiti
m
Indice mitotico
Morfologia
Eseguita su sezioni di
EE
Valuta un breve tratto del ciclo
S
Timidina tritiata
Citofluorimetria
Autoradiografia
Fluorescenza
Esatta definizione delle cellule in replicazione
Elevato numero di cellule esaminate
Utilizza precursori
roadiomarcati
Difficoltà di valutazione nei
tumori eterogenei
G1, S, G2m
KI-67
PCNA ciclina
Ag-NOR
Immunoperossidasi
Immunoperossidasi
Istochimica
Valuta tutte le fasi del ciclo eccetto G0
Valuta tutte le fasi del ciclo
Difficoltà di quantificazione al
M.O
Metodi per lo studio della proliferazione cellulare

Allestimento delle sospensioni nucleari da cellule in coltura Le colture cellulari di tumori solidi cresciute in piastra vengono private del terreno di coltura e lavate con PBS. Mediante l’uso di tripsina viene facilitato il distacco delle cellule adese alle pareti della piastra che così possono essere raccolte e successivamente centrifugate per 10’ a 1500 RPM in modo da ottenere un pellet. A questo punto tolto il PBS vengono fissate con alcool 70° a temperatura ambiente, goccia a goccia, sotto agitazione e poi messe in frigorifero a -4°C per 30 minuti. da scraping a fresco La superficie tumorale “a fresco” viene grattata (scraping) attraverso la lama di un bisturi e risospesa in PBS. Successivamente la sospensione viene filtrata attraverso un filtro di nylon con una porosità di 50 micron di diametro che consente il passaggio dei soli nuclei. Dopo centrifuga a 3400 RPM per 10’ si toglie il surnatante (PBS) e si fissa con etanolo 70° freddo (-20°C) goccia a goccia sotto agitazione. Il campione viene poi lasciato in frigorifero a -4°C per almeno 30’. da tessuto congelato Dal materiale congelato preventivamente selezionato ne vengono tagliate delle sezioni poi disaggregate in PBS mediante l’uso di bisturi. La soluzione viene quindi filtrata con filtri di nylon con porosità di 50 micron e poi centrifugata per 10’ a 3400 RPM. Dopo aver tolto il surnatante la sospensione viene fissata con etanolo 70° freddo (-20°C) goccia a goccia sotto agitazione e lasciata in frigorifero a -4°C per almeno 30’.

Metodo citofluorimetrico di rivelazione dell’antigene nucleare Ki-67 Indiretto (ab secondario coniugato con FITC) per sospensioni monocellulari di
cellule in coltura
- Dopo la fissazione le cellule vengono centrifugate a 3400 RPM e viene tolto il
surnatante
- Lavaggio in soluzione permeabilizzante composta da TRITON allo 0.01% in PBS.
- Mettere subito i campioni in centrifuga a 3400 RPM per 10’.
- Togliere il surnatante e risospendere in una soluzione di Albumina -Bovina 1% in PBS
per 10’.
- Centrifugare le cellule per 10’ a 3400 RPM togliere il surnatante.
- Incubare con l’anticorpo monoclonale MIB-1 (IMMUNOTECH, IgG1 mouse anti-
human, cod. IM0505).
- Per ogni caso si allestisce in questa fase il controllo negativo omettendo l’anticorpo.
- Aggiungere PBS e centrifugare per 10’ a 3400 RPM
- Incubare per un’ora con anticorpo secondario coniugato con FITC (Bouty, Goat
F(ab)2 anti-mouse Ig’s) diluito 1:50 in PBS.
- Risospendere in PBS
- Con una piccola aliquota di ogni campione contrastata con DAPI diluito 1:10 viene
preparato un vetrino per la valutazione preventiva al microscopio ottico a
fluorescenza
- Si esegue la lettura al citofluorimetro.
Indiretto (ab secondario coniugato con FITC) per sospensioni nucleari a fresco e
da tessuto congelato
- Dopo la fissazione le cellule vengono centrifugate a 3400 RPM e viene tolto il
surnatante
- Lavaggio in soluzione permeabilizzante composta da TRITON allo 0.01% in PBS.
- Mettere subito i campioni in centrifuga a 3400 RPM per 10’.
- Togliere il surnatante e risospendere in una soluzione di Albumina -Bovina 1% in PBS
per 10’.
- Centrifugare le cellule per 10’ a 3400 RPM togliere il surnatante.
- Incubare per un’ora con l’anticorpo monoclonale MIB-1 (IMMUNOTECH, IgG1 mouse
anti-human, cod. IM0505)
- Per ogni caso si allestisce in questa fase il controllo negativo omettendo l’anticorpo.
- Aggiungere PBS e centrifugare per 10’ a 3400 RPM
- Incubare per un’ora con l’anticorpo secondario coniugato con FITC (Bouty, Goat
F(ab’)2 anti-mouse Ig’s) diluito 1:50 in PBS
- Lavare con PBS per 10’ e centrifugare per 10’ a 3400 RPM
- Risospendere in PBS
- Con una piccola aliquota di ogni campione contrastata con DAPI diluito 1:10 viene
preparato un vetrino per la valutazione preventiva al microscopio ottico a

fluorescenza
- Si esegue la lettura al citofluorimetro.
Indiretto (ab secondario biotinilato e terziario streptavidinato) per sospensioni
nucleari da tessuto congelato
- Dopo la fissazione le cellule vengono centrifugate a 3400 RPM e viene tolto il
surnatante
- Lavaggio in soluzione permeabilizzante composta da TRITON allo 0.01% in PBS.
- Mettere subito i campioni in centrifuga a 3400 RPM per 10’.
- Togliere il surnatante e risospendere in una soluzione di Albumina -Bovina 1% in PBS
per 10’.
- Centrifugare le cellule per 10’ a 3400 RPM togliere il surnatante.
- Incubare per un’ora con l’anticorpo monoclonale MIB-1 (IMMUNOTECH, IgG1 mouse
anti-human, cod. IM0505)
- Per ogni caso si allestisce in questa fase il controllo negativo omettendo l’anticorpo.
- Aggiungere PBS e centrifugare per 10’ a 3400 RPM
- Togliere il surnatante ed incubare per un’ora con l’anticorpo secondario biotinilato
(LAB VISION, IgG Goat Anti-mouse e Anti-rabbit IgG cod. TP-125-HL).
- Incubare anche i controlli negativi.
- Lavare con PBS per 10’ e centrifugare per 10’ a 3400 RPM
- Togliere il Surnatante ed incubare per un’ora con Streptavidina coniugata con FITC
(DAKO, cod. F 0422) diluito 1:50 in PBS
- Incubare anche i controlli negativi
- Aggiungere PBS per lavare e centrifugare a 3400 RPM per 10’
- Con una piccola aliquota di ogni campione contrastata con DAPI diluito 1:10 viene
preparato un vetrino per la valutazione preventiva al microscopio ottico a
fluorescenza
- Si esegue la lettura al citofluorimetro.

LA IMMUNOFENOTIPIZZAZIONE CITOFLUORIMETRICA DEI TUMORI SOLIDI
Tipizzazione del tessuto linfoide Il sistema immunitario si è sviluppato per proteggere gli organismi dalle infezioni.
La risposta immunitaria è basata su tre momenti distinti: 1. il riconoscimento di ciò che è estraneo all’individuo, cioè la
capacità di distinguere il self dal not self; 2. la capacità di approntare una reazione appropriata, rivolta ad
eliminare l’antigene stesso; 3. l’acquisizione di una memoria immunitaria; l’esistenza della
memoria viene dedotta dal fatto che un successivo incontro con una sostanza che in precedenza è stata riconosciuta come estranea evoca una risposta di grado maggiore e molto più veloce rispetto a quella osservabile in occasione del primo contatto.
Poiché questo meccanismo è cruciale per la sopravvivenza dell’individuo il tessuto linfoide, in particolare i linfonodi, sono presenti in tutto il corpo, più numerosi nelle aree che drenano organi in contatto con l’ambiente esterno. In questo modo l’antigene penetrato nell’organismo raggiunge gli organi deputati al suo riconoscimento: i linfonodi regionali per gli antigeni penetrati nei tessuti e veicolati dalla linfa, la milza per gli antigeni trasportati dal circolo ematico. Gli apparati respiratorio e digerente hanno inoltre strutture che assomigliano a linfonodi direttamente appena al di sotto del rivestimento epiteliale, il cosiddetto tessuto linfoide mucosa associato (MALT). L’intero sistema che drena i differenti gruppi di linfonodi (mediastinici, mesenterici, inguinali superficiali e profondi, ascellari, cervicali, ecc.) converge in un unico vaso linfatico, il dotto toracico, che si apre nel circolo venoso, creando un efficace sistema di filtrazione.
Gli antigeni estranei penetrati nell’organismo, dopo essere stati fagocitati da macrofagi o da cellule dendritiche, cosiddette “cellule presentanti l’antigene”, vengono trasportati ai linfonodi regionali dove, tramite interazioni cellulo-cellulari o liberazione di citochine, attivano cloni linfoidi che porteranno all’eliminazione degli organismi estranei. Poiché ciascun linfocita riconosce un solo antigene, la capacità di rispondere ad una enorme varietà di antigeni dipende

dallo sviluppo di numerose popolazioni linfocitarie con differente specificità del recettore (immunoglobuline per le cellule B e recettore T cellulare per i linfociti T).
In seguito si assiste ad una ulteriore differenziazione delle cellule rispondenti, con coinvolgimento ed attivazione di tutto il sistema effettore (produzione di anticorpi, attivazione di macrofagi, generazione di cellule citotossiche, etc.), con lo scopo di eliminare l’antigene e risolvere il danno tissutale secondario all’infezione.
Una volta che l’antigene è stato eliminato, alcuni segnali di controllo riportano in stato di riposo il sistema immunitario. Il contatto con un antigene estraneo porta alla formazione di cellule memoria, in grado cioè di riconoscere in maniera specifica quel determinato antigene.
La popolazione linfoide viene distinta in base a differenti antigeni presenti sulla membrana cellulare in linfociti B e linfociti T. Ciascuna popolazione ha funzioni proprie. I linfociti B sono deputati alla cosiddetta “immunità umorale”, basata sulla produzione di immunoglobuline che hanno la capacità di legare in modo specifico i determinanti antigenici: le tossine batteriche possono essere neutralizzate, si può provocare la lisi cellulare mediata dal complemento, può essere favorita la fagocitosi attraverso l’ opsonizzazione specifica e può essere prevenuta la penetrazione dei virus in certe cellule. Le attività biologiche dei linfociti T (cosiddetta “immunità cellulo-mediata”) sono rappresentate dalla capacità di lisare direttamente cellule il cui corredo antigenico sia stato modificato da un’infezione virale e certe cellule tumorali, e di produrre sostanze solubili non antigene specifiche (“linfochine”) in grado di attivare i macrofagi e potenziare o inibire le classi di linfociti (Vedi figura).

Ontogenesi linfoide B-cellulare (midollo osseo)
e T-cellulare (timo)
L’architettura degli organi linfoidi è distinta in aree B-dipendenti
ed aree T-dipendenti. La conoscenza dei normali processi di differenziazione linfoide e la distribuzione nei vari compartimenti costituisce una premessa indispensabile per la comprensione dei processi neoplastici delle cellule da essi derivati (linfomi). I linfomi rappresentano infatti proliferazioni neoplastiche di cellule arrestate (“congelate”) in un particolare stadio maturativo e come tali mostrano caratteri citologici e fenotipici identici o molto simili a quelli delle cellule normali da cui derivano.
Nel 1994 la classificazione R.E.A.L., poi ripresa dalla classificazione dell’Organizzazione Mondiale della Sanità (2001) [tab. 1], ha codificato per ciascun linfoma specifici caratteri citologici, morfologici, fenotipici e di biologia molecolare.
In breve, i passi che ci portano ad una diagnosi di linfoma sono i seguenti:
a. riconoscere la natura linfoide di una popolazione cellulare;
b. dimostrare la monoclonalità (segno di malignità) di una proliferazione linfoide;
c. classificare i linfomi. Nella pratica quotidiana la diagnosi dei differenti istotipi di
linfoma è basata sulla morfologia e sul fenotipo; più raramente si rendono necessari studi di biologia molecolare.
Per immunofenotipizzazione si intende la caratterizzazione del fenotipo cellulare, cioè l’identificazione degli antigeni espressi da

quella cellula linfoide, mediante reazioni immunologiche (reazioni antigene-anticorpo).
Gli approcci metodologici all’analisi del fenotipo cellulare sono sostanzialmente tre:
- lo studio immunoistochimico su sezioni di tessuto fissato ed incluso in paraffina o sezioni criostatiche;
- immunocitochimico di strisci cellulari o citocentrifugati ottenuti da sospensioni cellulari;
- lo studio dei marcatori di superficie mediante citometria a flusso (CTF) su sospensioni cellulari.
Ognuno dei tre approcci possiede vantaggi e svantaggi. Il tessuto fissato ed incluso può causare la perdita di alcuni antigeni nel caso di una fissazione eccessivamente prolungata, solo parte degli anticorpi funzionano su tessuto fissato e ha tempi di processazione relativamente lunghi. Il materiale congelato offre tempi di trattamento relativamente più brevi ed è disponibile un ampio spettro di anticorpi, ma non consente una adeguata valutazione citologica della popolazione linfoide in conseguenza degli artefatti da congelamento.
L’esame citofluorimetrico consente di ottenere informazioni anche su materiali esigui come la citologia ottenuta mediante agoaspirazione con ago sottile (tecnica poco invasiva), ma necessita che la sospensione cellulare sia trattata in tempi rapidi in quanto il campione è soggetto a rapido deterioramento.
L’analisi delle sospensioni cellulari viene eseguita in citofluorimetria, tecnica che permette la rivelazione di antigeni cellulari di membrana mediante l’utilizzo di anticorpi coniugati a fluorocromi (molecole che colpite da un raggio laser di una determinata lunghezza d’onda, emettono fluorescenza che può essere quantificata).
La citofluorimetria quindi è un sistema rapido, versatile ed altamente riproducibile che permette di stabilire la presenza o l’assenza di un determinato antigene di membrana su un elevato numero di cellule; è inoltre l’unica tecnica che permette di valutare la contemporanea espressione di due o tre antigeni da parte della stessa cellula e di quantificare il numero di molecole di uno specifico antigene.
La doppia marcatura degli anticorpi con FITC (Isotiocianato di Fluoresceina) e PE (Ficoeritrina), è la tecnica più usata ed accettata nei laboratori di analisi.
Quando un marcatore di superficie che definisce una specifica funzione delle sottoclassi cellulari è coespresso in altre

sottopopolazioni, l’uso di fluorescenze multiple è l’unico metodo per ottenere una definizione corretta di un determinato tipo cellulare.
Gli anticorpi utilizzati nella diagnostica dei processi linfoproliferativi sono identificati dalla sigla CD (Cluster of Differentiation) seguita da un numero (es. CD19, CD20, etc), secondo la Nomenclatura raccomandata dall’Organizzazione Mondiale della Sanità per gli antigeni di differenziazione dei Leucociti umani (Shapiro,1998).
La possibilità di vedere dalla singola cellula l’espressione contemporanea di antigeni diversi, consente di ricavare informazioni superiori a quelle che si otterrebbero con la tecnica della singola marcatura (immunoistochimica tradizionale).
La selezione delle popolazioni che si vogliono studiare, viene fatta attraverso una operazione elettronica detta ”gating”, che consente di analizzare solo le cellule che hanno le caratteristiche fisiche o di fluorescenza volute. I dati ottenuti in questo modo sono estremamente più significativi rispetto ad una analisi in toto.
La citofluorimetria, entrata nei laboratori di Anatomia Patologica come supporto alle tecniche di analisi convenzionali, si prospetta come un tecnica affidabile e rapida per la determinazione del fenotipo delle proliferazioni linfomatose.

LINFOMA DI HODGKIN
Classico:
Sclerosi nodulare Cellularità mista Deplezione linfocitaria Ricco in linfociti
Predominanza linfocitica
LINFOMI NON HODGKIN
Linfomi B
Linfomi T
Linfociti precursori
Leucemia/Linfoma linfoblastico
Linfociti maturi
Leucemia/Linfoma linfocitico a piccoli linfociti Leucemia prolinfocitica Linfoma linfoplasmacitico Linfoma marginale splenico Leucemia a cellule capellute Discrasia plasmacellulare (mieloma, plasmacitoma solitario dell’osso, plasmacitoma extraosseo) Linfoma della zona marginale extranodale (MALT) Linfoma della zona marginale nodale Linfoma follicolare (GI, GII, GIII) Linfoma mantellare Linfoma diffuso a grandi cellule Linfoma a grandi cellule primitivo del medistino Linfoma a grandi cellule intravascolare Linfoma primitivo delle sierose (PEL) Linfoma di Burkitt
Linfociti precursori
Leucemia/Linfoma linfoblastico Linfoma blastico a cellule NK
Linfociti maturi
Leucemia prolinfocitica Leucemia a grandi linfociti granulari Leucemia a cellule Natural Killer, aggressiva Linfoma/leucemia a cellule T dell’adulto Linfoma a cellule T/NK di tipo nasale Linfoma a cellule T di tipo eteropatico Linfoma a cellule T epato-splenico (gamma/delta) Linfoma a cellule T del sottocute, simil-panniculitico Micosi fungoide Sindrome di Sezary Linfoma a grandi cellule anaplastiche primitivo cutaneo Linfoma a cellule T periferico, non specificato Linfoma a cellule T angioimmunoblastico Linfoma a grandi cellule anaplastiche
Classificazione dei linfomi secondo l’Organizzazione Mondiale della Sanità (2001) Tumors of haematopoietic and lymphoid tissues. World Health Organization
IARC PRESS, 2001.
Tipizzazione dei tumori epiteliali
Nel citoplasma delle cellule animali è presente una rete tridimensionale di filamenti proteici, detta citoscheletro, costituita da più complessi molecolari interagenti tra di loro che partecipano alla funzione di mantenimento della forma cellulare e al movimento intracitoplasmatico di organelli e di vescicole.
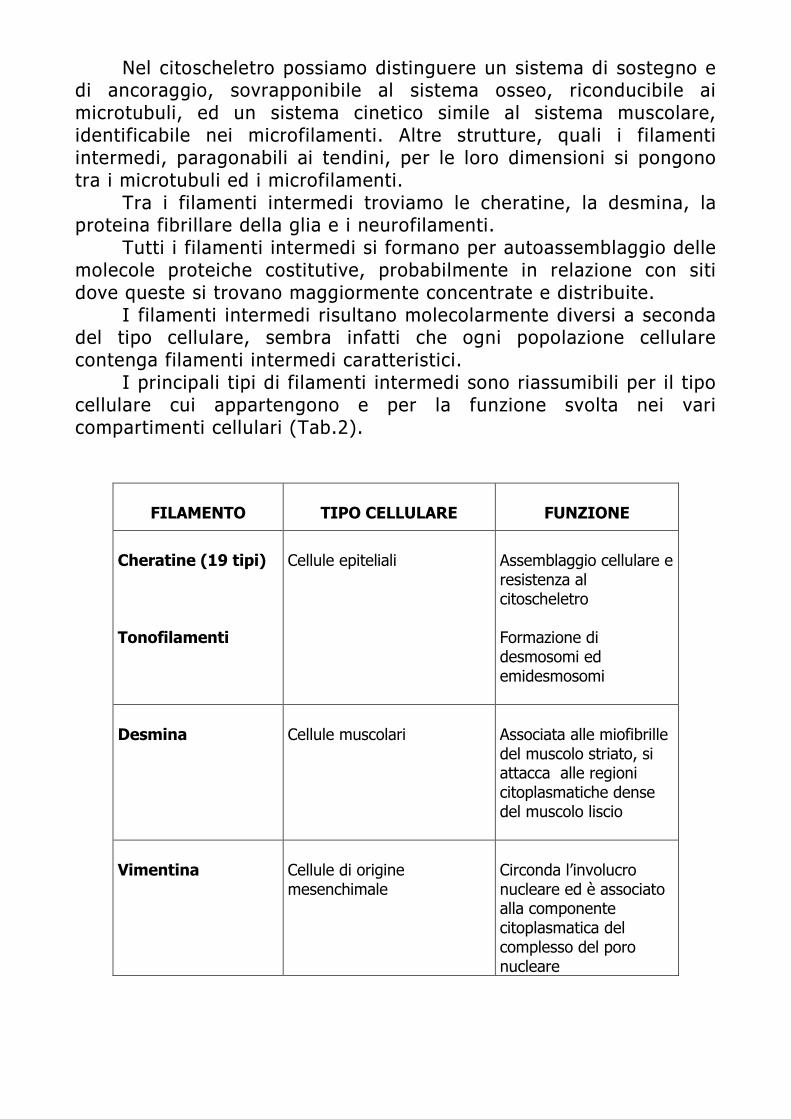
Nel citoscheletro possiamo distinguere un sistema di sostegno e di ancoraggio, sovrapponibile al sistema osseo, riconducibile ai microtubuli, ed un sistema cinetico simile al sistema muscolare, identificabile nei microfilamenti. Altre strutture, quali i filamenti intermedi, paragonabili ai tendini, per le loro dimensioni si pongono tra i microtubuli ed i microfilamenti.
Tra i filamenti intermedi troviamo le cheratine, la desmina, la proteina fibrillare della glia e i neurofilamenti.
Tutti i filamenti intermedi si formano per autoassemblaggio delle molecole proteiche costitutive, probabilmente in relazione con siti dove queste si trovano maggiormente concentrate e distribuite.
I filamenti intermedi risultano molecolarmente diversi a seconda del tipo cellulare, sembra infatti che ogni popolazione cellulare contenga filamenti intermedi caratteristici.
I principali tipi di filamenti intermedi sono riassumibili per il tipo cellulare cui appartengono e per la funzione svolta nei vari compartimenti cellulari (Tab.2).
FILAMENTO
TIPO CELLULARE
FUNZIONE
Cheratine (19 tipi) Tonofilamenti
Cellule epiteliali
Assemblaggio cellulare e resistenza al citoscheletro Formazione di desmosomi ed emidesmosomi
Desmina
Cellule muscolari
Associata alle miofibrille del muscolo striato, si attacca alle regioni citoplasmatiche dense del muscolo liscio
Vimentina
Cellule di origine mesenchimale
Circonda l’involucro nucleare ed è associato alla componente citoplasmatica del complesso del poro nucleare

Proteina fibrillare acida (GFAP)
Astrociti, cellule di Schwann, oligodendrociti
Partecipa all’impalcatura strutturale delle cellule gliali
Neurofilamenti
Neuroni
Forma il citoscheletro di assoni e dendriti, partecipa alla formazione dello stato di gel del citoplasma
Classi di filamenti intermedi
Le citocheratine interagiscono sia con la membrana nucleare che citoplasmatica e possono costituire dal 30% al 60% delle proteine citoplasmatiche totali. Le citocheratine sono proteine costituite da differenti subunità polipeptidiche. In rapporto al peso molecolare vengono distinte 19 tipi di citocheratine (classificazione di Moll-1987): differenti epiteli contengono differenti citocheratine.
I singoli tipi di citocheratine vengono riconosciute da specifici anticorpi monoclonali. Tali anticorpi sono utilizzati nella pratica diagnostica quotidiana sia per identificare una proliferazione tumorale come di natura epiteliale, sia riconoscere i singoli istotipi tumorali (soprattutto identificare l’epitelio e quindi l’organo di origine di un carcinoma) in rapporto alla differente distribuzione dei tipi di citocheratine. Possono ancora essere utilizzate per identificare e distinguere le cellule luminali dalle cellule basali e mioepiteliali.
La possibilità di risalire al tipo originario di tumore è sicuramente utile per il trattamento terapeutico (es. terapia ormonale in neoplasie riconosciute come di origine mammaria), ma anche per la possibilità di risalire alla neoplasia primitiva nel caso di primo riscontro di localizzazione metastatica. Va tuttavia ricordato che differenti tipi di citocheratine possono essere espressi da diversi epiteli e le neoplasie epiteliali maligne possono perdere la capacità di sintetizzare citocheratine presenti nell’epitelio di origine.
Gli anticorpi possono risconoscere un singolo tipo di citocheratina e l’anticorpo viene identificato con il numero corrispondente alla classificazione di Moll (es. CK7, CK8, CK9, CK18, CK19, CK20) o cocktails di citocheratine, in tale caso l’anticorpo viene identificato con una sigla commerciale o con i tipi di citocheratine riconosciute (es. CK5/6, CAM 5.2, KL1, CK903, ecc.).

Le citocheratine che più frequentemente vengono utilizzate per identificare l’epitelio di origine in carcinomi poco differenziati o nelle localizzazioni metastatiche sono la CK7 e la CK20: si può risalire con discreta approssimazione all’organo di origine di una neoplasia in base alla differente espressione immunoistochimiche di queste due citocheratine.
La CK 20 è in grado inoltre di identificare le cellule di Merkel (cellule neuroendocrine dell’epidermide) e le neoplasie da esse derivate (carcinoma a cellule di Merkel), dove caratteristicamente si presenta con una colorazione globulare paranucleare.
L’anticorpo anti-CK5/6 è utile nella diagnosi differenziale tra carcinoma metastatico pleurico (negativo), ed il mesotelioma epiteliale (positivo).
La CK19 è in grado di riconoscere i carcinomi papillari della tiroide, anche ad architettura follicolare, facilitando la diagnosi differenziale con le neoplasie tiroidee follicolari.
Le citocheratine riconosciute dall’anticorpo CK903 sono presenti nelle cellule basali e non nelle cellule epiteliali secernenti della ghiandola prostatica e mammaria, risultando pertanto particolarmente utile nella diagnosi differenziale tra lesioni in situ (persistenza delle cellule basali) e carcinomi infiltranti (assenza di cellule basali) (Monfair-1999).
Le ricerche compiute sull’identificazione delle cellule tumorali nel carcinoma della mammella e del tratto genitale femminile, hanno riportato l’espressione delle citocheratine 8, 18, 7 e 17, nel carcinoma duttale della ghiandola mammaria e nel tumore dell’endometrio (Nagle,1988, Kimming,2001, Stimpfl,1999, Moll,1982, Dabbs,2002).
Il carcinoma renale è la terza neoplasia più comune del sistema genito-urinario. L’istotipo più frequente è il “carcinoma a cellule chiare”. Esistono però altri istotipi: carcinoma cromofobo, carcinoma papillare, carcinoma di Bellini (carcinoma dei dotti collettori) e l’oncocitoma (Wu-2002). Queste neoplasie possono essere distinte, oltre che in base a criteri cito-morfologici, anche in base a differente espressione di citocheratine.
Carcinoma a cellule
chiare
Carcinoma cromofobo
Carcinoma papillare
Carcinoma Di Bellini
Oncocitoma
CK18 + Non
studiata
+
+ Non
Studiata
AE1/
AE3
+ + + +
+

CK7 - + +
Non
studiata
+
CK20 - - - Non
studiata
Non
Studiata
Differente espressione delle citocheratine nei carcinomi renali
E’ da ricordare che l’espressione delle citocheratine non è ristretta solo ai tumori di origine epiteliale: sono stati descritti rari casi di neoplasie non epiteliali con espressione di citocheratine: melanomi, linfomi e sarcomi. In tali casi l’espressione di anticorpi specifici consente di porre la diagnosi corretta.
Nella maggior parte dei laboratori di anatomia patologica, la determinazione della citocheratina tissutale avviene attraverso l’analisi immunoistochimica. Solo negli ultimi anni, tale analisi è stata resa possibile anche attraverso metodiche citofluorimetriche.
Nei casi di tumore epiteliale è possibile applicare questo tipo di determinazione per dimostrare, simultaneamente all’analisi del DNA, la presenza di positività alla citocheratina.
Come dimostrano alcuni lavori in letteratura (Leonardi,2000, Kimming,2001, Dabbs,2002, Glocova,1996.) negli istogrammi citofluorimetrici l’utilizzo degli anticorpi specifici coniugati (CK/FITC) permette di selezionare la popolazione neoplastica che esprime maggiormente la componente epiteliale, escludendo la contaminazione dovuta alla presenza di cellule linfoidi ed infiammatorie.
Tipizzazione della popolazione linfoide.
Per testare le metodiche citofluorimetriche su tessuto linfoide abbiamo richiesto l’invio di sangue intero di pazienti sani dal laboratorio di analisi chimico-cliniche e dal reparto di otorinolaringoiatria pediatrica di tonsille o adenoidi asportate per tonsillite o adenoidite recidivanti.
Per ciò che riguarda il sangue intero il nostro interesse è stato rivolto alla tipizzazione di un tessuto che rispondeva alle nostre aspettative per ciò che riguardava la distribuzione dimensionale (Fig.3) e al controllo della funzionalità degli anticorpi disponibili presso il nostro laboratorio, mentre per il tessuto solido abbiamo avuto l’opportunità di esaminare 7 casi di tonsille e 1 caso di adenoidi. L’età dei pazienti è compresa tra i 2 e i 13 anni, con una

media di 7 anni. Per ciascun caso la diagnosi istologica è stata di: tonsillite/adenoidite immunoreattiva priva di caratteri di specificità.
Citogramma di distribuzione del sangue normale.
Il pezzo operatorio asportato veniva trasportato a fresco
(ghiaccio) in una garza imbevuta di soluzione fisiologica presso il II° servizio di Anatomia Patologia.
Il lasso di tempo intercorso tra il momento dell’asportazione e la processazione del campione è risultato inferiore a 30 minuti in sei casi, mentre in due casi è risultato compreso tra 30 e 120 minuti. Per ciascun pezzo operatorio è stato prelevato un microfrustolo conservato in PBS a 4°C e sono state eseguite manovre agoaspirative con ago di 25 gauge per ottenere materiale citologico con metodica sostanzialmente sovrapponibile a quanto viene effettuato in vivo (citologia agoaspirativa). Il frustolo è stato trattato con metodica identica a quanto verrà descritto per l’espressione di citocheratine. Il materiale citologico è stato conservato sempre in PBS a 4°C1. Solo in 2 pazienti è stato esaminato il materiale prelevato con tecnica agoaspirativa, mentre in 7 pazienti l’esame citofluorimetrico è stato eseguito su frustolo.
In corso di citologia diagnostica 5 pazienti affetti da linfoma con sospetta recidiva di malattia o evoluzione in un linfoma più aggressivo, seguiti presso il day hospital di Ematologia – 3° servizio di Medicina Interna degli Spedali Civili di Brescia - il materiale citologico è stato prelevato tramite agoaspirazione con ago sottile (25 gauge); per ciascun paziente sono state eseguite 4-5 manovre agoaspirative. Il materiale ottenuto da una singola manovra agoaspirativa o il lavaggio dei singoli aghi utilizzati dopo preparazione di un singolo striscio citologico è stato sospeso in PBS mantenuto a 4°C.
1 Il mezzo di conservazione più uti lizzato f ino al momento dell’analisi è il tampone fosfato
(PBS) preferibilmente a 4°C, medium che consente una migliore conservazione della qualità citologica ed evita il fenomeno dell’aggregazione cellulare.
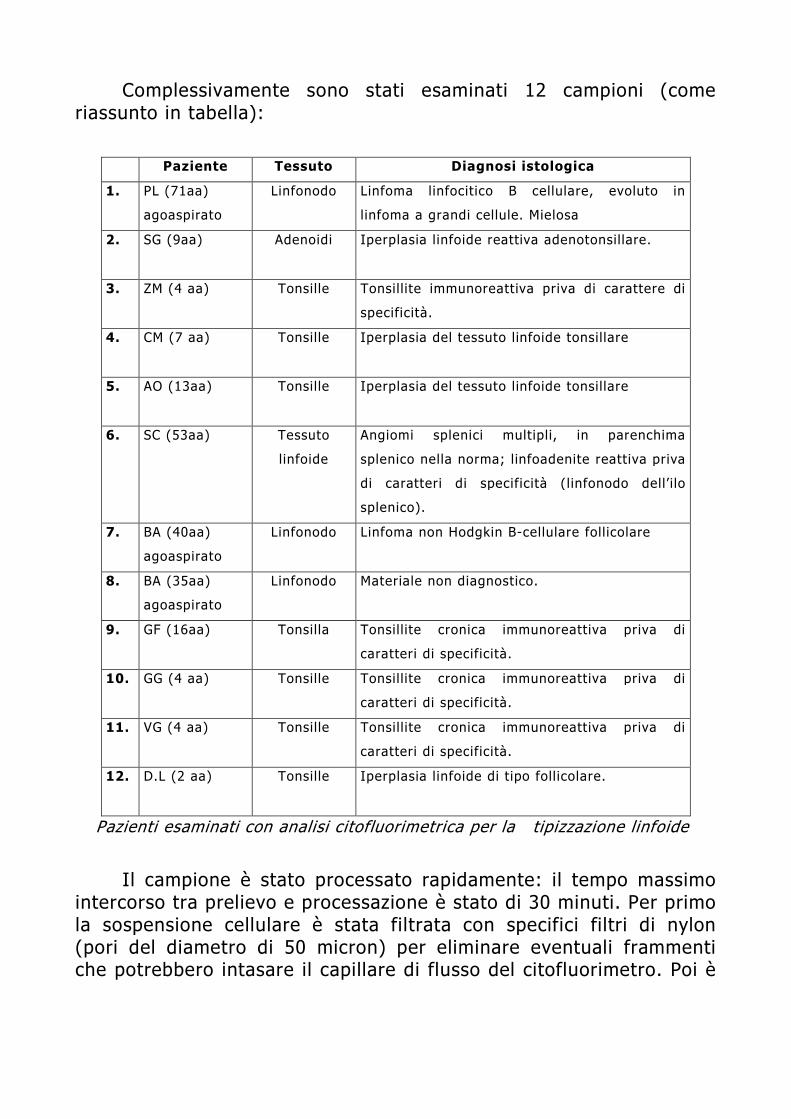
Complessivamente sono stati esaminati 12 campioni (come
riassunto in tabella):
Paziente Tessuto Diagnosi istologica
1. PL (71aa)
agoaspirato
Linfonodo Linfoma linfocitico B cellulare, evoluto in
linfoma a grandi cellule. Mielosa
2. SG (9aa)
Adenoidi Iperplasia linfoide reattiva adenotonsillare.
3. ZM (4 aa)
Tonsille Tonsillite immunoreattiva priva di carattere di
specificità.
4. CM (7 aa)
Tonsille Iperplasia del tessuto linfoide tonsillare
5. AO (13aa)
Tonsille Iperplasia del tessuto linfoide tonsillare
6. SC (53aa)
Tessuto
linfoide
Angiomi splenici multipli, in parenchima
splenico nella norma; linfoadenite reattiva priva
di caratteri di specificità (linfonodo dell’ i lo
splenico).
7. BA (40aa)
agoaspirato
Linfonodo Linfoma non Hodgkin B-cellulare follicolare
8. BA (35aa)
agoaspirato
Linfonodo Materiale non diagnostico.
9. GF (16aa)
Tonsilla Tonsillite cronica immunoreattiva priva di
caratteri di specificità.
10. GG (4 aa)
Tonsille Tonsillite cronica immunoreattiva priva di
caratteri di specificità.
11. VG (4 aa) Tonsille Tonsillite cronica immunoreattiva priva di
caratteri di specificità.
12. D.L (2 aa) Tonsille
Iperplasia linfoide di tipo follicolare.
Pazienti esaminati con analisi citofluorimetrica per la tipizzazione linfoide
Il campione è stato processato rapidamente: il tempo massimo
intercorso tra prelievo e processazione è stato di 30 minuti. Per primo la sospensione cellulare è stata filtrata con specifici filtri di nylon (pori del diametro di 50 micron) per eliminare eventuali frammenti che potrebbero intasare il capillare di flusso del citofluorimetro. Poi è

stata valutata la cellularità del preparato e la vitalità cellulare in camera di Burker2, tramite colorante sopravitale (Trypan Blue 3). A questo punto si è eseguita la metodica di determinazione antigenica attraverso l’incubazione della sospensione cellulare con anticorpi specifici. Metodica di determinazione antigenica:
1. Centrifugazione a 1800 rpm per 10 min. 2. Eliminazione del surnatante. 3. Risospensione del pellet in un ml di PBS. 4. Incubazione di 100 :l di campione con 10 :l di anticorpo
coniugati al fluorocromo e conservazione al buio a 4° C per 45 minuti4.
5. Lisi dei globuli rossi e stabilizzazione del campione con la soluzione C (fissativo per le membrane cellulari a base di parafolmaldeide) proposta dalla ditta Coulter5.
6. Incubazione al buio a 4°C per 15 min e lavaggio con 500 :l di PBS6. 7. Valutazione al citofluorimetro impostato con un protocollo specifico
per l’analisi della doppia fluorescenza. Anticorpi testati:
2 La camera di Burker o contaglobuli è uno strumento analitico utilizzato nei laboratori per la
determinazione della conta cellulare. La vitalità cellulare in genere riflette il tempo intercorso tra il prelievo e l’analisi. I leucociti conservati in PBS entrano in apoptosi dopo poche ore, per cui non è consigliato utilizzare campioni rimasti troppo a lungo all’aria e con vitalità inferiore al 90%, in quanto le cellule morte adsorbono in maniera aspecif ica gli anticorpi coniugati aumentando il background della f luorescenza. 3 La sospensione cellulare è colorata con trypan blu e successivamente si effettua una conta
con la camera di Burker. La percentuale di cellule vitali si ottiene considerando il numero di cellule non colorate (vitali) rispetto a quelle colorate (non vitali). La conta deve essere effettuata entro i primi 5-15 minuti dall ’aggiunta del colorante. 4 Il protocollo della tipizzazione prevede un tempo di incubazione con anticorpi monoclonali di
30-45 minuti a 4°C in quanto la bassa temperatura riduce i fenomi di capping e d’internalizzazione del complesso antigene/anticorpo. Il tempo d’incubazione con gli anticorpi non può essere inferiore perché porterebbe a sottostimare gli antigeni riconosciuti da anticorpi con bassa aff inità di legame. Il rapporto cellule/anticorpo è uno dei fattori che più influenzano la qualità dell’analisi. Ogni anticorpo ha infatti un range di concentrazione ottimale, è necessario pertanto procedere alla titolazione dei vari anticorpi nel caso in cui non sia già stabilita la concentrazione che da la migliore discriminazione tra le cellule positive e quelle negative. 5 La lisi delle emazie si può ottenere con una soluzione di cloruro di ammonio o con reagenti
commerciali che dovrebbero quindi essere utilizzati dopo la marcatura con l’anticorpo per evitare alterazioni dell’antigene. 6 I lavaggi dopo la marcatura e la lisi delle emazie, allontanano la quota di anticorpo non
legato, ma comportano una piccola perdita cellulare.

Indagine citofluorimetrica con anticorpi monoclonali coniugati ad isotiocianato di fluoresceina (FITC) e ficoeritrina (PE) fornito dalle ditte Coulter e Bio-D:
- CD3FITC-CD19PE per linfociti T e B. - CD3FITC-CD56PE per linfociti NK. - CD45FITC-CD14PE per la popolazione leucocitaria comune. - Controlli isotipici (IgG1FITC, IgG2aFITC) per verificare il livello di fluorescenza di base7.
Di ogni caso è stata eseguita l’analisi immunoistochimica di anticorpi in
grado di riconoscere le medesime popolazioni cellulari linfoidi su sezioni fissate in formalina e incluse in paraffina secondo il protocollo classico in uso presso il nostro laboratorio onde poter confrontare il risultato citofluorimetrico con quello immunoistochimico. Per i campioni ottenuti con citologia agoaspirativa sono state eseguite colorazioni immunocitochimiche. Gli anticorpi utilizzati sono i seguenti:
- CD3p linfociti T (pan-T) - CD20 linfociti B (pan-B) - CD15 forme segmentate mieloidi e monociti - CD45 (antigene leucocitario comune) - CD56 linfociti natural killer
Acquisizione ed analisi citofluorimetrica:
L’analisi citofluorimetrica è stata effettuata dopo l’impostazione di un adeguato protocollo di acquisizione in cui sono stati selezionati i seguenti parametri:
- istogramma 1: FS vs SS - istogramma 2: LFL2 vs LFL1 - istogramma 3: LFL2 - istogramma 4: LFL1 Nei casi in cui dot plot degli scatter è risultato particolarmente
complesso per l’identificazione delle diverse popolazioni cellulari è stato impostato un parametro fisico, l’SSC, combinato con la fluorescenza del CD45 che riconosce la popolazione leucocitaria totale.
7 Ogni lettura deve infine includere un controllo non reattivo (controllo isotipico) coniugato con lo stesso
fluorocromo degli anticorpi utilizzati per la valutazione del livello dell’autofluorescenza e l’individuazione del cut-off di positività .

La prima fase prevede la verifica del settaggio dello strumento, l’impostazione del numero di eventi da acquisire e la definizione di un gate per l’individuazione della popolazione di interesse8.
I segnali relativi alle fluorescenze (LFL1 e LFL2) sono stati impostati in scala logaritmica mentre per i parametri di scatter la scala era lineare9.
La metodica effettuata sullo strumento prevede la lettura del controllo isotipico ed in rapida successione dei campioni coniugati con gli anticorpi marcati.
Per ogni provetta sono stati letti almeno 10.000 eventi ed i dati sono stati conservati su floppy-disk.
La valutazione della reazione immunofluorescente avviene in fase di post-acquisizione attraverso l’analisi degli istogrammi che riproducono la quantificazione percentuale della positività espressa dalla popolazione selezionata.
I parametri utilizzati nella valutazione qualitativa e quantitativa della metodica sono:
-tipo di fissativo ideale -percentuale di vitalità cellulare -percentuale di popolazione linfoide selezionata nel gate -percentuale di legame all’anticorpo primario
Tipizzazione della popolazione epiteliale.
Sono stati studiati 9 casi di tessuto solido di origine epiteliale (2 carcinomi dell’utero, 2 carcinomi della mammella, 1 cisti ovarica, 3 carcinomi renali, 1 metastasi linfonodale), giunti presso il nostro laboratorio per la diagnosi istopatologica (come riportato in tabella):
N Pazienti Tessuto Diagnosi istologica
1. LP (54 aa)
Utero Carcinoma sieroso papillare
2. CG (74 aa)
Utero Carcinoma mucinoso
3. CS (33 aa) Linfonodo Metastasi di carcinoma mammario
8 La sensibilità, la linearità e la stabil ità dello strumento devono essere controllate
regolarmente. Esistono in commercio microsfere, legate a diversi f luorocromi, che consentono di verif icare l’eff icienza dei singoli fotomoltiplicatori. Ciò è anche possibile grazie all’util izzo di standard noti. 9 Può essere necessaria l’amplificazione logaritmica degli scatter quando sono presenti cellule di grandi
dimensioni che possono finire fuori scala. Si definisce inoltre una soglia del segnale di forward scatter per discriminare le cellule nucleate dai detriti.
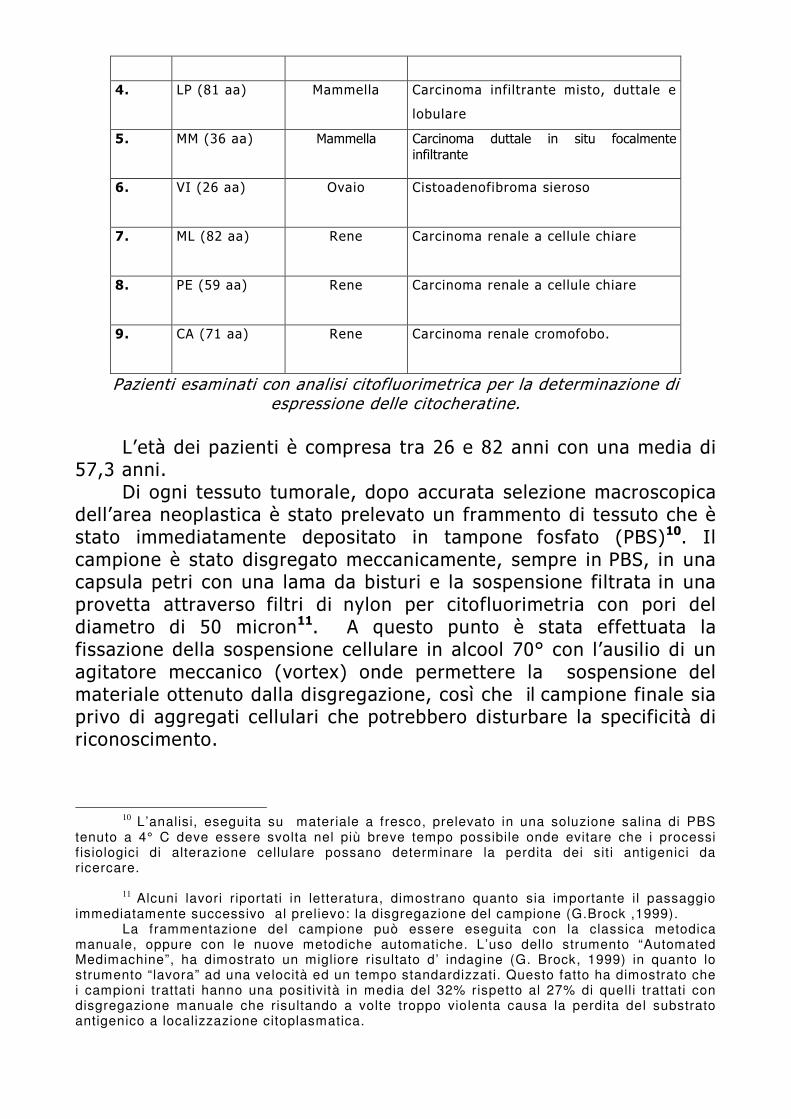
4. LP (81 aa)
Mammella Carcinoma infiltrante misto, duttale e
lobulare
5. MM (36 aa)
Mammella Carcinoma duttale in situ focalmente infiltrante
6. VI (26 aa)
Ovaio Cistoadenofibroma sieroso
7. ML (82 aa)
Rene Carcinoma renale a cellule chiare
8. PE (59 aa)
Rene Carcinoma renale a cellule chiare
9. CA (71 aa)
Rene Carcinoma renale cromofobo.
Pazienti esaminati con analisi citofluorimetrica per la determinazione di espressione delle citocheratine.
L’età dei pazienti è compresa tra 26 e 82 anni con una media di
57,3 anni. Di ogni tessuto tumorale, dopo accurata selezione macroscopica
dell’area neoplastica è stato prelevato un frammento di tessuto che è stato immediatamente depositato in tampone fosfato (PBS)10. Il campione è stato disgregato meccanicamente, sempre in PBS, in una capsula petri con una lama da bisturi e la sospensione filtrata in una provetta attraverso filtri di nylon per citofluorimetria con pori del diametro di 50 micron11. A questo punto è stata effettuata la fissazione della sospensione cellulare in alcool 70° con l’ausilio di un
agitatore meccanico (vortex) onde permettere la sospensione del materiale ottenuto dalla disgregazione, così che il campione finale sia privo di aggregati cellulari che potrebbero disturbare la specificità di riconoscimento.
10 L’analisi, eseguita su materiale a fresco, prelevato in una soluzione salina di PBS
tenuto a 4° C deve essere svolta nel più breve tempo possibile onde evitare che i processi f isiologici di alterazione cellulare possano determinare la perdita dei sit i antigenici da ricercare.
11 Alcuni lavori riportati in letteratura, dimostrano quanto sia importante il passaggio
immediatamente successivo al prelievo: la disgregazione del campione (G.Brock ,1999). La frammentazione del campione può essere eseguita con la classica metodica
manuale, oppure con le nuove metodiche automatiche. L’uso dello strumento “Automated Medimachine”, ha dimostrato un migliore risultato d’ indagine (G. Brock, 1999) in quanto lo strumento “lavora” ad una velocità ed un tempo standardizzati. Questo fatto ha dimostrato che i campioni trattati hanno una positività in media del 32% rispetto al 27% di quell i trattati con disgregazione manuale che risultando a volte troppo violenta causa la perdita del substrato antigenico a localizzazione citoplasmatica.

Prima di procedere alla processazione della sospensione monocellulare per la determinazione antigenica, si è effettuata la conta cellulare12. Metodica di determinazione antigenica:
1. Centrifugazione a 1800 rpm per 10 minuti. 2. Eliminazione del surnatante e lavaggio con soluzione salina di
Normal Goat Serum e PBS (rapporto 1:5 )13. 3. Centrifugazione a 1800 rpm per 10 minuti. 4. Secondo lavaggio con soluzione PBS/NGS. 5. Centrifugazione a 1800 rpm per 10 minuti. 6. Incubazione di 100 :l di sospensione cellulare con 20 :l di anticorpo
CK8/18 (l’anticorpo riconosce le citocheratine a peso molecolare 8 e 18 sec. Moll-1987) coniugato ad Isotiocianato di Fluoresceina (FITC) fornito dalla ditta Ylem al buio a 4°C per 2 ore.
7. Si procede a questo punto anche all’allestimento del controllo negativo (stessa procedura ma con omissione dell’anticorpo primario).
8. Successivamente si effettua il lavaggio dei campioni con la soluzione PBS/NGS per eliminare l’eccesso di anticorpo non coniugato.
9. Si centrifuga a 1800 rpm per 10 minuti. 10. Si elimina il surnatante e si effettua la controcolorazione con
ioduro di propidio 50 mg/ml per la lettura citofluorimetrica e 0,5 mg/ml per la valutazione citomorfologica al microscopio a fluorescenza.
Di ogni caso è stata eseguita l’analisi immunoistochimica con due anticorpi che riconoscono specificatamente la CK8 e la CK18, su sezioni fissate in formalina e incluse in paraffina secondo il protocollo classico in uso presso il nostro laboratorio onde poter confrontare il risultato citofluorimetrico con quello immunoistochimico.
12 La quantif icazione della popolazione cellulare da tipizzare è necessaria in quanto le reazioni di riconoscimento antigene-anticorpo richiedono rapporti specif ici di legame. In questa fase ci si avvale di due colorazioni: con Ioduro di Propidio oppure con Trypan Blue.
Lo Ioduro di Propidio permette di analizzare, mediante quantif icazione citofluorimetrica, le cellule morte che adsorbono maggiormente il colorante e danno una fluorescenza più elevata delle cellule vive. La colorazione con Trypan Blue permette di valutare, tramite la conta con la camera di Burker, la percentuale di cellule vitali che risulteranno non permeabili al colorante.
13
Il protocollo classico prevede un trattamento di lavaggi del campione per precludere la possibil ità di eventuali legami con siti aspecif ici. Questi lavaggi sono effettuati anche successivamente all’incubazione, in modo da eliminare l’eccesso di anticorpo non coniugato. In questi passaggi pre- e post-incubazione, deve essere seguito attentamente il momento della centrifugazione del materiale, in quanto i diversi cicli in centrifuga risultano una delle cause di perdita del campione.

Acquisizione ed analisi citofluorimetrica:
L’indagine citofluorimetrica dei campioni processati è stata eseguita adottando un protocollo di analisi che comprende i seguenti parametri:
- istogramma 1 : FL3P vs FL3 - istogramma 2 : FLS vs FL3 - istogramma 3 : FL3 - istogramma 4 : FL1 vs FL3
Allo stesso modo della tecnica di determinazione della popolazione
linfoide, la metodica per l’identificazione della popolazione epiteliale, necessita di una prima fase di verifica del settaggio dello strumento, l’impostazione del numero di eventi da acquisire e la definizione di un gate per l’individuazione della popolazione di interesse.
Prima dell’analisi del campione coniugato con l’anticorpo si effettua l’impostazione del cut-off di positività attraverso l’utilizzo del controllo negativo (campione in cui è stato omesso l’anticorpo primario e colorato con Ioduro di Propidio). Successivamente viene effettuata la lettura del campione coniugato con l’anticorpo fluoresceinato mantenendo la conta cellulare sui diecimila eventi e conservando i dati acquisiti su floppy-disc.
Terminata la fase di acquisizione, si valuta la positività della reazione immunofluorescente, analizzando le percentuali di positività della popolazione d’interesse quantificata dallo strumento.
I parametri utilizzati nella valutazione qualitativa e quantitativa della metodica sono:
-percentuale di vitalità cellulare -tempo di incubazione con l’anticorpo primario -percentuale di positività citofluorimetrica alla CK -valutazione cito-morfologica al microscopio della sospensione fluorescente
Conclusioni L’approccio classico alla tipizzazione immunofenotipica è rivolto
all’esame citofluorimetrico su microfrustoli di tessuto linfoide disgregati meccanicamente e trasformati in sospensione cellulare.

Il nostro interesse primario è invece quello di utilizzare l’analisi citofluorimetrica su sospensioni monocellulari ottenute con tecnica citologica agoaspirativa.
Prima di introdurre una nuova metodica con utilità diagnostica nel laboratorio, dove peraltro la citometria a flusso viene sfruttata routinariamente per la valutazione della ploidia nelle cellule epiteliali maligne, ci siamo documentati sulla metodica standard impiegata in altri laboratori ed abbiamo scelto anticorpi idonei tra quelli proposti dalle differenti ditte.
Durante l’applicazione della metodica scelta ci siamo trovati via via a dover risolvere problematiche principalmente di tipo tecnico, sia testando differenti possibili soluzioni da noi individuate, sia confrontandoci con l’esperienza di altri laboratori.
I maggiori problemi sono stati riscontrati nel tipo di soluzione utilizzata per conservare le cellule da esaminare. Il Citospin e l’EDTA, sostanza utilizzata come anticoagulante per il sangue intero, non sono risultati idonei in quanto hanno dimostrato di non preservare l’antigenicità cellulare. Il miglior fissativo è risultato il tampone fosfato (PBS) utilizzato a 4°C, che rallenta i fenomeni di autolisi cellulare, evitando inoltre fenomeni di abnorme espressione antigenica, come è risultato nel caso di G.F. (sospensione cellulare fissata in Citospin), in cui la popolazione linfoide mostrava innaturale coespressione di CD3 e CD19 nel 25% delle cellule testate.
Un altro passaggio indispensabile per la conservazione del campione è risultato il lasso di tempo che intercorre tra il prelievo e l’esame del campione.
Periodi superiori a 30-60 minuti influiscono negativamente sulla vitalità cellulare, come evidenziato attraverso la conta cellulare con la camera di Burker. Un altro suggerimento che abbiamo estratto dalle prove effettuate è che i migliori risultati si ottengono utilizzando un “gate” di analisi quantitativa cellulare impostato su una percentuale media di popolazione linfoide del 75% (tabella).

Pazienti Conta cellulare
(n°cell per ml)
Gate
%
1 B.A. 700.000 80
2 B.A. 250.000 73
3 P.L. 1.000.000 87
4 S.C. 900.000 61
Valore medio 75
Conta cellulare con camera di Burker
In tutti i casi vi è stata corrispondenza nel fenotipo della
popolazione linfoide prevalente: in 6 casi B-cellulare ed in 1 caso T-cellulare anche se il rapporto tra linfociti T e linfociti B ottenuto con tecnica immunoistochimica è stato decisamente superiore a quanto osservato in citofluorimetria.
La discrepanza tra le percentuali di linfociti T e B ottenute con citofluorimetria ed immunoistochimica su tessuto potrebbe essere secondaria al fatto che il materiale utilizzato per lo studio è stato prelevato da tonsille o adenoidi caratterizzate da marcata iperplasia follicolare. In tale situazione le cellule B sono immerse in una fitta rete di cellule follicolari dendritiche (si tratta di elementi indispensabili alla maturazione delle cellule centrofollicolari) che mantengono coesi gli elementi linfoidi in aggregati definiti linfo-istiocitici. Tali aggregati potrebbero essere risultati troppo voluminosi ed eliminati durante il processo di filtrazione della sospensione cellulare. Utilizzando filtri di 50 micron tutti gli aggregati di cellule centrofollicolari comprendenti più di 4-5 elementi sono stati eliminati; già di per sé le cellule B centrofollicolari, sia i centrociti ma soprattutto i centroblasti sono più voluminosi dei linfociti T.
Molto probabilmente i linfociti B che abbiamo esaminato con tecnica citofluorimetrica sono derivati dal mantello dei follicoli secondari (tali cellule hanno dimensioni sostanzialmente sovrapponibili ai piccoli linfociti T).
A conferma di tale ipotesi sarebbe stato necessario testare le sospensioni cellulare con CD10 (marcatore delle cellule centrofollicolari). Nell’unico campione valutabile ottenuto da agoaspirato diagnostico, durante il quale vengono aspirate in prevalenza singole cellule, vi è stata sostanziale corrispondenza tra il dato ottenuto con tecnica citofluorimetrica e immunocitochimica.

Tra i casi pervenuti presso il nostro Servizio, abbiamo avuto l’opportunità di testare 3 campioni ottenuti in corso di citologia agoaspirativa diagnostica da 3 differenti pazienti affetti da linfoma con sospetta recidiva di malattia o evoluzione in un linfoma più aggressivo.
In due pazienti non è stato possibile eseguire la determinazione antigenica con citofluorimetria in quanto la popolazione linfoide era quantitativamente troppo esigua. In uno di questi il materiale è risultato insufficiente per la diagnosi anche alla valutazione degli strisci citologici.
In un caso (P.L.) la positività del 36% per l’anticorpo CD19 ha indirizzato la diagnosi verso una linfoproliferazione di tipo B, confermata successivamente dalla diagnosi citologica di Linfoma non Hodgkin linfocitico/Leucemia Linfocitica Cronica B cellulare.
Anche nel caso S.C. (come riportato nella tabella corrispondente) il dato citofluorimetrico è risultato sostanzialmente paragonabile a quanto ottenuto con la valutazione immunocitochimica.
Per quanto riguarda il tipo di sospensione cellulare su cui sono state effettuate le prove, c’è da evidenziare che i campioni ottenuti dal prelievo tramite citologia agoaspirativa non hanno dato sostanziali differenze rispetto ai campioni ottenuti dalla disgregazione meccanica del tessuto solido.
A conferma della buona sovrapposizione dei dati ottenuti con le due tecniche, la preparazione del caso D.L. mostrava una cellularità sufficiente ed una buona morfologia della popolazione analizzata, con un dato di positività di legame del 30% all’anticorpo CD3 nell’ agoaspirato, ed una positività del 24% nel tessuto solido. Questi risultati ci portano ad affermare che la citologia agoaspirativa, tecnica rapida e poco invasiva, può essere un metodo proponibile per questo tipo di analisi.
I risultati ottenuti nelle ultime prove con tecnica standardizzata (soluzione di PBS a 4°C, esame eseguito entro 30 min. dal prelievo citologico, gate di cellule esaminate >25%) hanno mostrato come le modifiche introdotte alla metodica scelta inizialmente ne abbiano migliorato significativamente la qualità, come anche evidenziato dalla corrispondenza dei dati ottenuti in citometria a flusso e con metodica immunoisto-citochimica.
Durante le prove di tipizzazione sul tessuto epiteliale, il nostro studio è stato impostato sull’analisi di frammenti tumorali di origine epiteliale disgregati in sospensione cellulare.

Il tipo di metodica impiegata in questa analisi non ha richiesto significative modifiche rispetto a quanto pubblicato in letteratura (Leonardi, 2000).
Nella tipizzazione del tessuto epiteliale tumorale si ricercano
principalmente cellule positive all’anticorpo anti-citocheratina. Tali anticorpi sono utilizzati per identificare sia una
proliferazione tumorale di natura epiteliale, sia i singoli istotipi tumorali (soprattutto identificare l’epitelio e quindi l’organo di origine di un carcinoma) in rapporto alla differente distribuzione dei tipi di citocheratine.
Nei casi di tumore epiteliale da noi analizzati è stato possibile applicare questo tipo di determinazione per dimostrare, simultaneamente all’analisi del DNA (ploidia), la presenza di positività alla citocheratina, infatti abbiamo potuto dimostrare negli istogrammi citofluorimetrici che l’utilizzo degli anticorpi specifici coniugati (CK/FITC) permette di selezionare la popolazione neoplastica che esprime maggiormente la componente epiteliale, escludendo la contaminazione dovuta alla presenza di cellule linfoidi ed infiammatorie.
Abbiamo valutato che i risultati ottenuti con la tecnica citofluorimetrica sono stati influenzati nei passaggi di trattamento del campione che precedono la fase di tipizzazione. Infatti, in un campione di carcinoma della mammella (L.P.) ed in un caso di carcinoma renale (C.A.), non abbiamo raggiunto un risultato citofluorimetricamente valutabile a causa dell’esiguità del materiale.
Successivamente in due pazienti con carcinoma renale (P.E. e M.L.) abbiamo ottenuto percentuali di positività relativamente basse (rispettivamente 10% ed 11%), anche queste due determinazioni potrebbero essere state influenzate dal trattamento preanalitico, infatti le preparazioni citologiche della sospensione cellulare mostravano nella morfologia una perdita del citoplasma e quindi delle strutture citocheratiniche di circa il 40%.
In seguito all’ottimizzazione della metodica utilizzata, siamo riusciti a quantificare positività all’anticorpo specifico superiori al 50%, come dimostrato nel caso di carcinoma uterino (C.G.) e nel caso di cistoadenofibroma sieroso (V.I.).
Dopo aver effettuato e valutato le prove di colorazione immunoistochimica sui medesimi campioni, possiamo affermare che esiste una buona corrispondenza nei due risultati, anche se il numero di cellule marcate con tecnica immunoistochimica è risultato superiore di quanto riscontrato con tecnica citofluorimetrica. Tale discrepanza potrebbe essere secondaria al fatto che durante la disgregazione

meccanica del frustolo tissutale una parte delle cellule sia stata danneggiata, in particolare abbia perso in parte o completamente il citoplasma e pertanto il citoscheletro risultando negativa alla citofluorimetria.
Tale risultato tuttavia non mette in dubbio la validità diagnostica della metodica, in quanto ciò che è importante è evidenziare la natura epiteliale della popolazione neoplastica (positività alla citocheratina) anche in una quota minoritaria degli elementi tumorali.
Su questi è comunque possibile testare i differenti tipi di
citocheratine per individuare l’organo di origine del carcinoma. In conclusione lo scopo che ci eravamo prefissi: introdurre nel
nostro laboratorio nuove metodiche, ci pare sia stato sostanzialmente raggiunto. La fenotipizzazione della popolazione linfoide con tecnica citofluorimetrica, ottimizzata su tonsille ed adenoidi, ha mostrato la sua utilità anche su materiale citologico ottenuto in corso di citologia agoaspirativa diagnostica. Pur se in un unico campione è stato dimostrato fenotipo B-cellulare nella maggioranza della popolazione linfoide aspirata, suggerendo un linfoma non Hodgkin, confermato all’esame citologico e immunocitologico dei preparati strisciati.
Anche per l’espressione delle citocheratine la tecnica citofluorimetrica ha mostrato capacità di riconoscimento di antigeni espressi nel citoplasma cellulare.
Lo studio fenotipico di popolazioni cellulari su sospensioni con tecnica citofluorimetrica ci consente di avere i risultati entro brevissimo tempo (circa due ore, invece di 24 ore per l’immunocitochimica ed almeno 72 ore per l’immunoistochimica) rendendo più rapida la comunicazione del referto al clinico di riferimento per quel determinato paziente.
Inoltre la valutazione del fenotipo linfoide da materiale recuperato in corso di citologia agoaspirativa ci consente di eseguire un minor numero di passaggi agoaspirativi (è possibile recuperare il materiale da esaminare “risciacquando” in PBS gli aghi utilizzati per l’esame citologico), con vantaggio per il paziente (manovra più breve e meno dolorosa) e con minor rischio di necrosi della neoformazione agoaspirata (la necrosi ischemica post-agoaspirazione è una conseguenza possibile di tale manovra, che talora impedisce o limita fortemente la diagnosi istologica sulla neoformazione completamente asportata).
La possibilità di avere a disposizione i risultati del fenotipo della popolazione linfoide aspirata ci permette infine di comunicare in giornata al medico curante il risultato dell’esame, riducendo i tempi di ulteriori indagini clinico-strumentali o terapeutiche.

Questa dispensa è stata realizzata grazie ai contenuti ed alle immagini delle tesi degli studenti del Corso di Laurea in Tecniche di Laboratorio Biomedico dell’Università degli Studi si Brescia che hanno frequentato il Laboratorio di Anatomia Patologica per il tirocinio tecnico-pratico e che ho seguito personalmente in qualità di Tutor didattico. Il materiale scientifico presentato è ad esclusivo uso personale e non può essere riprodotto. Per informazioni potete contattarmi direttamente all’indirizzo mail: [email protected] oppure visitando il gruppo Facebook TLB-Tecnici di Laboratorio Bresciani o la pagina SpazioTlb.


















