
CORSO INTEGRATO DI GENETICA -...
18
1 CORSO INTEGRATO DI GENETICA a.a.2010-2011 Prof. Pier Franco Pignatti 21.10.2010 Lezioni N. 17 e 18 Malattie da espansione di triplette (Neri-Genuardi cap. 15) Mutazioni dinamiche, premutazioni, anticipazione Le sequenze di DNA ripetuto sono spesso coinvolte in malattie Crossing-over diseguale Scambio diseguale fra cromatidi fratelli Conversione genica Duplicazione Delezione Inserzione Inversione Espansione di ripetizioni instabili
Transcript of CORSO INTEGRATO DI GENETICA -...

1
CORSO INTEGRATO DI GENETICA
a.a.2010-2011Prof. Pier Franco Pignatti
21.10.2010Lezioni N. 17 e 18
Malattie da espansione di triplette(Neri-Genuardi cap. 15)
Mutazioni dinamiche, premutazioni, anticipazione
Le sequenze di DNA ripetuto sono spesso coinvolte in malattie�Crossing-over diseguale�Scambio diseguale fra cromatidi fratelli�Conversione genica�Duplicazione�Delezione�Inserzione�Inversione�Espansione di ripetizioni instabili

2
NEJM 332:1499, 1995
Espansione di triplette
Meccanismo di espansione
3
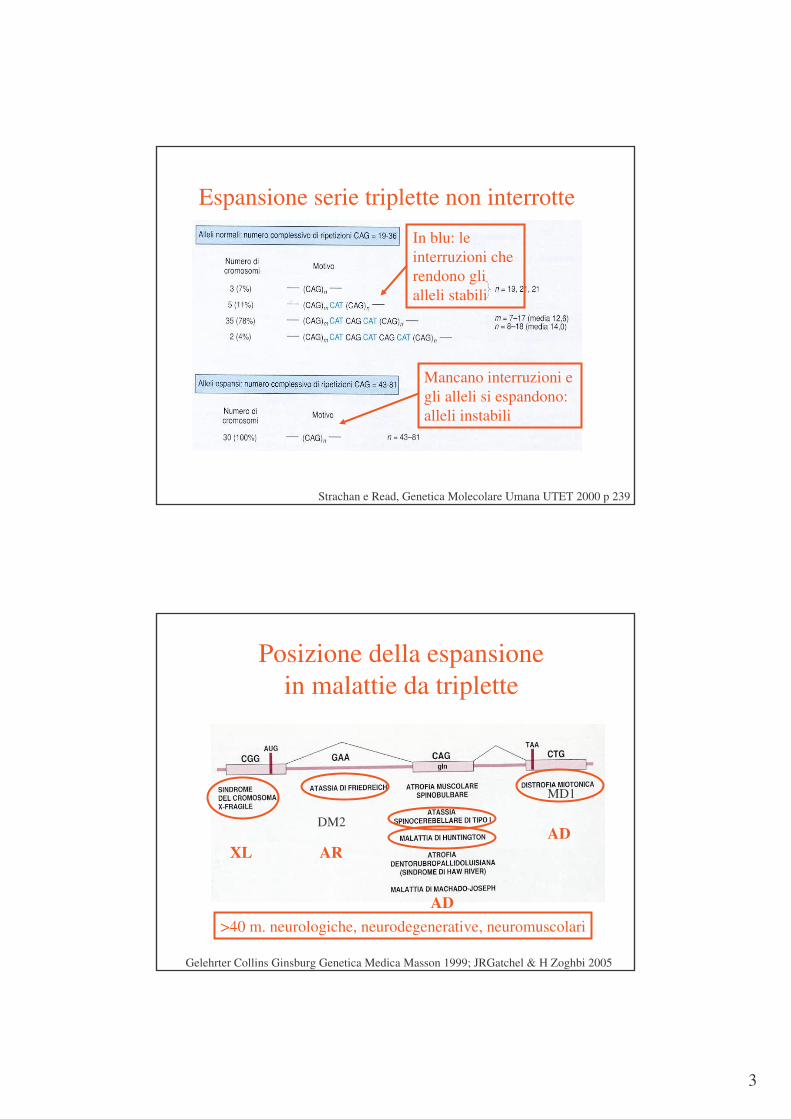
Espansione serie triplette non interrotte
Strachan e Read, Genetica Molecolare Umana UTET 2000 p 239
In blu: le interruzioni che rendono gli alleli stabili
Mancano interruzioni e gli alleli si espandono: alleli instabili
Posizione della espansione in malattie da triplette
Gelehrter Collins Ginsburg Genetica Medica Masson 1999; JRGatchel & H Zoghbi 2005
XL ARAD
AD>40 m. neurologiche, neurodegenerative, neuromuscolari
MD1
DM2

4
SINDROME DEL CROMOSOMA X FRAGILE
1
SINDROME DELL’X FRAGILE (FXS)
La forma più comune di ritardo mentale ereditario (FMR) 1:4.000 maschi, 1:6.000 femmine (S. di Martin e Bell,1943)
Sito fragile Xq27.3 (FRAX-A) in terreno privo di folato (Lubs 1969)

5
ESPANSIONE in FXS
Gelehrter Collins Ginsburg Genetica Medica Masson 1999
0-54 triplette
55-200 triplette
200-1300triplette
1991:identificazione gene FMR-1, la prima mutazione dinamica, che si modifica nel passaggio da una generazione all’altra
Espansione di triplette gene FMR1
Griffiths et al., Genetica, Zanichelli 1996, Fig.18.12
Paradosso di Sherman: aumento del numero di casi (penetranza) col passare delle generazioni
Trasmettitore sano (NTM)
6
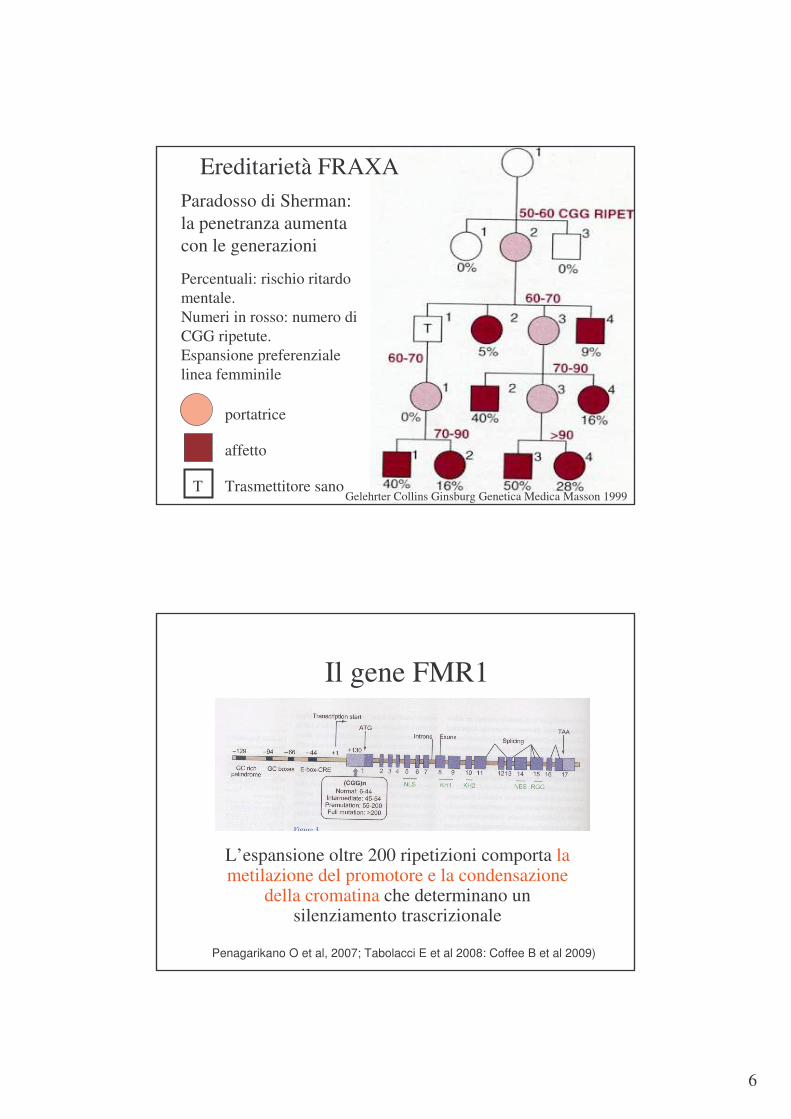
Ereditarietà FRAXA
affetto
portatrice
T Trasmettitore sano
Paradosso di Sherman: la penetranza aumenta con le generazioni
Percentuali: rischio ritardo mentale.Numeri in rosso: numero di CGG ripetute. Espansione preferenziale linea femminile
Gelehrter Collins Ginsburg Genetica Medica Masson 1999
Il gene FMR1
L’espansione oltre 200 ripetizioni comporta la metilazione del promotore e la condensazione
della cromatina che determinano un silenziamento trascrizionale
Penagarikano O et al, 2007; Tabolacci E et al 2008: Coffee B et al 2009)

7
Inattivazione promotore e repressione trascrizione in FXS
NA Di Prospero e KH Fischbeck 2005
MECP2: Metil CpGbinding protein 2 (S. Rett)MBD: Metil CpGbinding domainproteinHDAC: Histonedeacetilasi che reprimono ulteriormente la trascrizione
Funzione del gene FMR1
Topi con gene Fmr1 inattivato hanno la perdita della proteina FMRP e alterata maturazione della trasmissione glutamatergica durante il periodo critico perinatale.
Conclusione: FMRP è necessaria per il normale progresso della maturazione sinaptica nello sviluppo dell’organismo
(Harlow EG et al, 2010)

8
AJHG 74:805, 2004
premutazione (55-200)
L’aumentodel numero di ripetizioni diminuisce l’efficienza di traduzione in un sistema di espressione in cellule neurali o epiteliali
ESPANSIONE FMR1 e TRASCRIZIONE
PREMUTAZIONE FMR1
La premutazione (55-200 CGG) può dare :• Sindrome Tremore-Atassia associata a X
fragile (FXTAS) • Lievi sintomi psichiatrici • Insufficenza ovarica e menopausa precoci
(POF)• Atassia isolata adulti
PJ Hagerman e RJ Hagerman, 2004

9
ATASSIA DI FRIEDREICH
2
ATASSIA DI FRIEDREICH (FRDA)
La sindrome atassica ereditaria più frequente nelle popolazioni caucasoidi (3-4:100.000, AR)
Insorgenza pubertà, perdita progressiva coordinazione muovimenti ed equilibrio
Deficit di energia: anche cardiomiopatia ipertrofica che può condurre a morte precoce
Gene FXN (frataxina) mappato cr. 9q13Triplette GAA nel primo introne: da 35-40 normale a
70-1000 espansione causano silenziamento genico

10
FrataxinaProteina mitocondriale che mantiene
omeostasi Fe mitocondriale ed ha anche ruolo protettivo del danno nucleare
FRDA: la funzione mitocondriale è alterata con sovraccarico di Fe, difetto produzione ATP, difetto funzionamento neuroni e cardiomiociti; instabilità cromosomica, aumento sensibilità mutageni chimici e specie reattive dell’ossigeno (ROS)
Tentativi terapeutici di rimozione Femitocondri mediante un chelante e di aumento espressione con eritropoietina e inibitori della deacetilasi istonica
POLIGLUTAMINOPATIE
3

11
POLIGLUTAMINOPATIE
Una famiglia di 9 malattie neurodegenerativeIl tratto poliglutaminico conferisce un
guadagno di funzione tossico alle proteine che porta a inizio tardivo della malattia e perdita progressiva di neuroni in regioni specifiche del SNC.
Possibile contributo di modificazioni post-traduzionali della proteina
NEJM 340:1974, 1999
SCA
POLIGLUTAMINOPATIE4
SBMAHD
DRPLA

12
MALATTIA DI HUNTINGTON (HD)George Huntington (1872) descrive la malattia caratterizzata da disabilitàmotoria progressiva e deterioramento mentale, prevalenza 3-7:10.000, AD
Read e Donnai, Genetica Clinica, Zanichelli 2007
Gelehrter Collins Ginsburg Genetica Medica Masson 1999
Famiglia con HD
Famiglia venezuelana che ha permesso la localizzazione del gene sul cromosoma 4 nel 1983 (Gusella et al Science225:1320,1984)

13
Le ripetizioni CAG (Gln, Q) in HD
“Uno studio mondiale della mutazione HD”Conclusioni: la lunghezza delle ripetizioni è un marcatore sensibile e specifico della ereditarietàdella mutazione
NEJM 330:1401,1994
40-121triplette: HD11-36 triplette: N
1993: identificazione del gene IT15
Amplificazione del gene IT15
S: DNA da SpermaL: DNA da Linfociti
50 copie
20 copie
Nature Genet 1993;387
Si nota particolare instabilità delle triplette ripetute nella meiosi maschile. Infatti si può avere insorgenza precoce della malattia trasmessa dal padre. L’età di esordio è inversamente correlata alla lunghezza della espansione, anche nel tessuto cerebrale

14
Possibile meccanismo patogenetico HD
NEJM 340:1975,1999
Formazione di aggregati proteici che impediscono la degradazione proteasomalee inducono morte cellulare. Una proteina striato specifica lega la poliQ-Htt
DISTROFIA MIOTONICA
4

15
DISTROFIA MIOTONICA (MD)Malattia di Steinert (1909) : miotonia, debolezza e
difficoltà rilassamento muscolare, deperimento, anomalie conduzione cardiaca, …
La più frequente fra le distrofie muscolari a ereditarietà autosomica (1:8.000)
Ptosi, debolezza faccia, iposviluppo muscolo mandibola e sternomastoideo, amimia faccia e aspetto scavato volto
Numero di triplette ed età di esordio MD
Gelehrter Collins Ginsburg Genetica Medica Masson 1999 p.33
C: congenitaG: giovanileA: adultaSC: subclinica(nessun segno clinico tranne elettromiogrammaanomalo o altro)

16
Famiglia con MD
Numero di triplette:- in nero allele N (5-37 triplette)- in rosso alleleespanso (50-1.000 triplette)
Gelehrter Collins Ginsburg Genetica Medica Masson 1999, p.33
L’età di esordio diminuisce con il passare delle generazioni mentre aumenta la gravità delle manifestazioni cliniche: il fenomeno della anticipazione
AJHG 74:793, 2004
Espansione di sequenza CTG (MD1) o CCTG (MD2), non tradotte
MODELLO di GUADAGNO DI FUNZIONE dell’RNA per MD1 e MD2

17
Neutralizzazione dell’RNA tossico in MD1
Cooper TA 2009
Meccanismi patogenetici
FXS
MD1
HD
Strachan T, Read A. Human Molecular Genetics, 4°ed. 2011 pg 425

18
Effetti della espansione
Gelehrter Collins Ginsburg Genetica Medica Masson 1999; JRGatchel & H Zoghbi 2005
Perdita funzione Guadagno funz
XL AR AD
AD
Guadagno funzione RNA
>40 m. neurologiche, neurodegenerative, neuromuscolari
MD1


















