
CICLO XXVII COORDINATORE Prof. Silvano Capitani - Unifeeprints.unife.it/1012/1/Tesi John Charles...
95
Università degli Studi di Ferrara DOTTORATO DI RICERCA IN SCIENZE BIOMEDICHE CICLO XXVII COORDINATORE Prof. Silvano Capitani Mechanism of progression in cervical intraepithelial neoplasia Settore Scientifico Disciplinare BIO/13 Dottorando Tutore Dott. John Charles Rotondo Prof. Mauro Tognon Anni 2012/2014
Transcript of CICLO XXVII COORDINATORE Prof. Silvano Capitani - Unifeeprints.unife.it/1012/1/Tesi John Charles...

Università degli Studi di Ferrara
DOTTORATO DI RICERCA IN SCIENZE BIOMEDICHE
CICLO XXVII
COORDINATORE Prof. Silvano Capitani
Mechanism of progression in cervical intraepithelial neoplasia
Settore Scientifico Disciplinare BIO/13
Dottorando TutoreDott. John Charles Rotondo Prof. Mauro Tognon
Anni 2012/2014

INDEX
INTRODUCTION 3
OBJECTIVE AND AIMS OF THE STUDY 25
MATERIALS AND METHODS 26
RESULTS 36
DISCUSSION 58
REFERENCES 68
2

INTRODUCTION
CERVICAL INTRAEPITHELIAL NEOPLASIA
Classification of the cervical intraepithelial neoplasia
The cervical intraepithelial neoplasia (CIN) is a pre-cancerous lesion of uterine
cervical squamous cell carcinoma (SCC) and is characterized by potentially premalignant
transformation and abnormal growth, named dysplasia, of cervical keratinocytes. The
histological classification system provides three CIN grades referred to as CIN1 (mild
dysplasia), CIN2 (moderate dysplasia) and CIN3 (severe dysplasia) lesions. Each CIN
grade remains confined to the cervical epithelium (Figure 1, A,
www.lookfordiagnosis.com) and has a variable potential evolution towards the cancer.
CIN1 lesions are characterized by keratinocytes with abnormal cell growth, perinuclear
cytoplasmic vacuolation, named koilocytosis, and increase volume of the nucleus. This
histologic change is confined to the basal third of the cervical epithelium (Figure 1, B).
CIN2 lesions, compared to CIN1, are characterized by higher cytologic atypia and cellular
disorganization, while koilocytosis is lower or absent. The histological abnormalities are
confined to the basal two thirds of the cervical epithelium (Figure 1, C). CIN2 cervical
cells show two main features: (i) atypical mitotic figures induced by aneuploidy; (ii)
abnormal nucleus/cytoplasm ratio. CIN3 lesions are characterized by keratinocytes
endowed with high proliferative index, immaturity and vertical orientation. Abnormal
keratinocytes exceed the two thirds of the epithelium or expand throughout the thickness of
the epithelium, altering the whole cyto-architecture of the tissue (Figure 1, D).
Figure 1. Histological representation of normal and CIN tissue. (A) Normal cervical
epithelium; (B) CIN1, disorganization of the lower third of the epithelium; (C) CIN2,
3

disorganization of the lower half of the epithelium with viral infection; (D) CIN3, the
disorganization of the epithelium is evident in more than 2/3 lower.
Epidemiology of the CIN lesions
The incidence of CIN lesions in a population depends on several factors such as
etiopathogenetic factors, the efficiency of prevention programs, the immunologic status,
and the age of the female. The World Health Organization (WHO) has estimated that the
annual incidence of CIN lesions among women aged 25-65 years who undergo cervical
SCC screening for the first time was 3-10% for CIN 1 and 1-5% for CIN2 and CIN3. CIN2
and CIN3 lesions are generally diagnosed in women between 25 and 35 years of age
(Kumar et al., 2007). On the contrary, cervical SCC is usually detected in women over 40
years of age, typically 8 to 13 years after a diagnosis of CIN3. Women can develop CIN
lesions at any age?. The prevalence of CIN lesions varies from 1/10,000 in women aged
15-19 years to 1/1,000 in women aged 25-29 and then it reduces to 8/10,000 in women
aged 25-29 years (WHO 2010). In developing Countries, like Nigeria, the mean age for
CIN lesion is about 37 years. The prevalence is 3.6% for CIN1, 0.8% for CIN2 and 0.4%
for CIN3. Such a result is due to the lack of screening methods and it mirrors the typical
trend of developing Nations (Oguntayo and Samaila, 2012).
Clinical progression of CIN lesions
The temporal progression CIN1> CIN2> CIN3 represents the initial steps in
tumorigenesis of cervical SCC. However, CIN lesions do not always progress towards
cancer. Indeed, the histological and molecular phenotype of CIN reflects a fine balance
between the factors that promote/accelerate or reduce/slow down disease progression
(Melsheimer et al, 2001). About 90% low-grade CIN lesions tend to regress spontaneously
while 10% women progress to high grade CIN2 and CIN3 lesions. Typically, the risk of
CIN progression to cancer is related to the severity of CIN lesion. Indeed, the probability
that a CIN3 lesion progresses towards cervical SCC is significantly higher than CIN1. The
percentage of regression and progression of different grades of CIN lesions are shown in
Table 1:
4
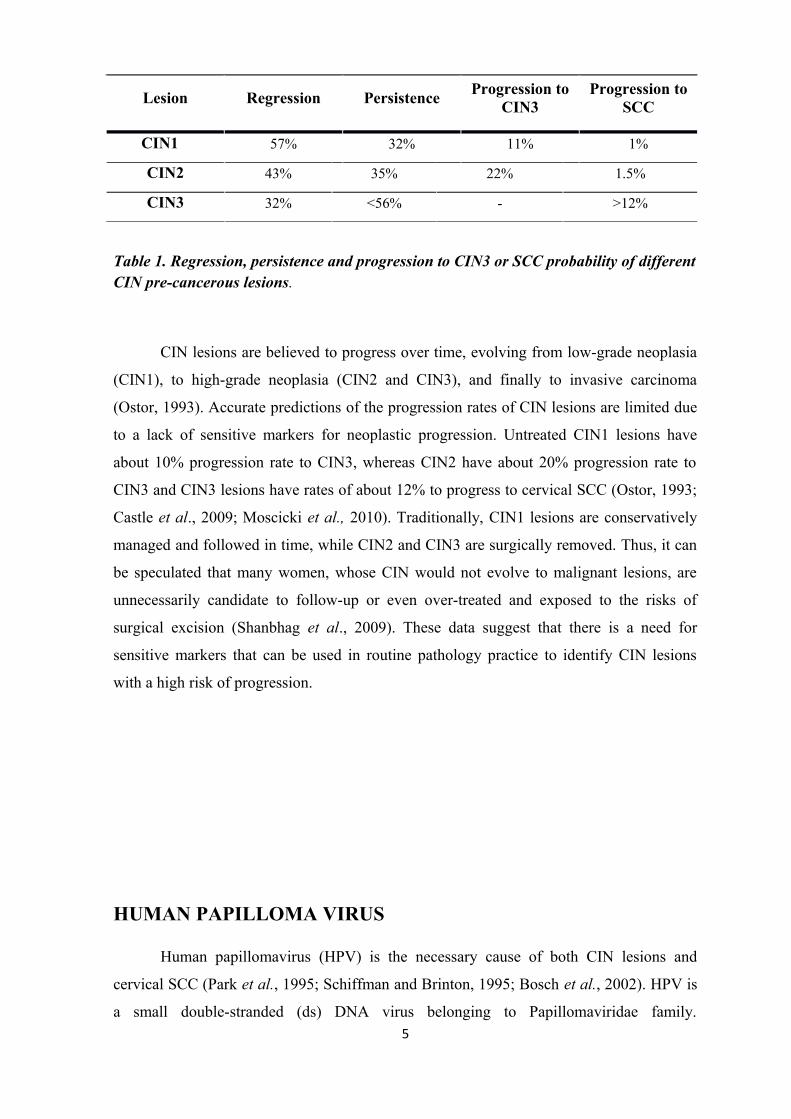
Lesion Regression PersistenceProgression to
CIN3Progression to
SCC
CIN1 57% 32% 11% 1%
CIN2 43% 35% 22% 1.5%
CIN3 32% <56% - >12%
Table 1. Regression, persistence and progression to CIN3 or SCC probability of different CIN pre-cancerous lesions.
CIN lesions are believed to progress over time, evolving from low-grade neoplasia
(CIN1), to high-grade neoplasia (CIN2 and CIN3), and finally to invasive carcinoma
(Ostor, 1993). Accurate predictions of the progression rates of CIN lesions are limited due
to a lack of sensitive markers for neoplastic progression. Untreated CIN1 lesions have
about 10% progression rate to CIN3, whereas CIN2 have about 20% progression rate to
CIN3 and CIN3 lesions have rates of about 12% to progress to cervical SCC (Ostor, 1993;
Castle et al., 2009; Moscicki et al., 2010). Traditionally, CIN1 lesions are conservatively
managed and followed in time, while CIN2 and CIN3 are surgically removed. Thus, it can
be speculated that many women, whose CIN would not evolve to malignant lesions, are
unnecessarily candidate to follow-up or even over-treated and exposed to the risks of
surgical excision (Shanbhag et al., 2009). These data suggest that there is a need for
sensitive markers that can be used in routine pathology practice to identify CIN lesions
with a high risk of progression.
HUMAN PAPILLOMA VIRUS
Human papillomavirus (HPV) is the necessary cause of both CIN lesions and
cervical SCC (Park et al., 1995; Schiffman and Brinton, 1995; Bosch et al., 2002). HPV is
a small double-stranded (ds) DNA virus belonging to Papillomaviridae family. 5

Papillomaviridae family is divided into 16 genres defined phylogenetically by the letters of
the greek alphabet (De Villiers et al., 2004). Five of them, Alpha, Beta, Gamma Mu and
Nu, belong to the human papillomavirus (Figure 2). The α-Papillomavirus are
predominantly mucosal, while other genres are predominantly cutaneous. Approximately
40 HPV types show ano-genital tropism and are transmitted by sexual activity. Among
these, some HPV cause genital warts, while others produce persistent infections that may
evolve in CIN lesions and cervical SCC (Schiffman et al., 2003). For these reasons, HPVs
are divided into high- and low- oncogenic risk due to their ability to induce benign genital
warts or CIN lesions and cervical SCC (Table 2). High-risk HPVs are detected in 90% of
cervical cancers (Clifford et al., 2003; Muñoz et al., 2003). High-risk HPV16 and HPV18
are detected in approximately in 50% and in 10-20% of all cervical cancers, respectively
(Muñoz et al., 2003). Low-risk HPVs are associated with 90% of the warts, benign
squamous papillomas and CIN1 lesions, that in most cases resolve spontaneously (Muñoz
et al., 2003).
HPV Group HPV type
High Risk 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, 82
Low Risk 6, 11, 40, 42, 43, 44, 54, 61, 70, 72, 81, CP6108
Probably High-Risk 26, 53, 66, 68, 73, 82
Table 2. Epidemiologic classification of High-, Low- and Probably High-risk HPVs
6
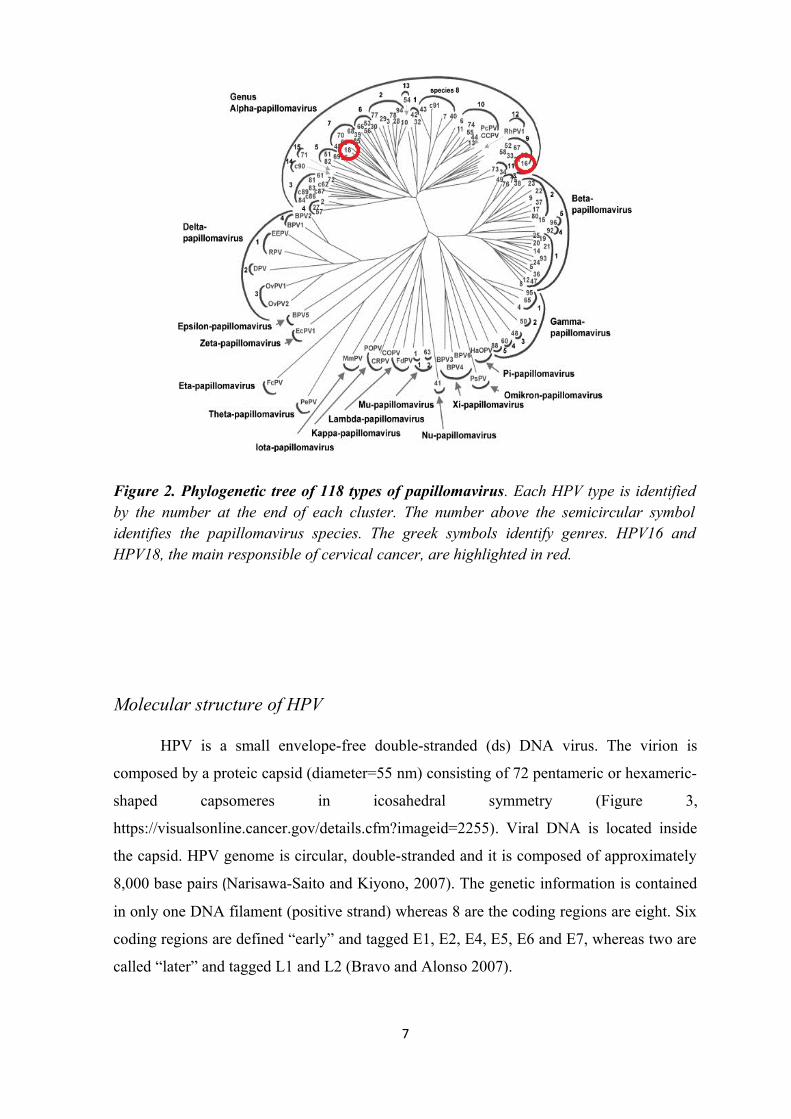
Figure 2. Phylogenetic tree of 118 types of papillomavirus. Each HPV type is identified by the number at the end of each cluster. The number above the semicircular symbol identifies the papillomavirus species. The greek symbols identify genres. HPV16 and HPV18, the main responsible of cervical cancer, are highlighted in red.
Molecular structure of HPV
HPV is a small envelope-free double-stranded (ds) DNA virus. The virion is
composed by a proteic capsid (diameter=55 nm) consisting of 72 pentameric or hexameric-
shaped capsomeres in icosahedral symmetry (Figure 3,
https://visualsonline.cancer.gov/details.cfm?imageid=2255). Viral DNA is located inside
the capsid. HPV genome is circular, double-stranded and it is composed of approximately
8,000 base pairs (Narisawa-Saito and Kiyono, 2007). The genetic information is contained
in only one DNA filament (positive strand) whereas 8 are the coding regions are eight. Six
coding regions are defined “early” and tagged E1, E2, E4, E5, E6 and E7, whereas two are
called “later” and tagged L1 and L2 (Bravo and Alonso 2007).
7

Figure 3. Electron micrograph of a HPV 16.
HPV genome can be divided into three regions: (i) Long Control Region (LCR), or
upstream regulatory region (URR), which is a non-coding region required for viral
replication and viral DNA encapsidation; (ii) Early region (E), that represents 45% of the
viral DNA and contains the six E genes expressed early in the HPV life cycle; (iii) Late
Region (L), corresponds to 40% of the viral DNA and encodes for the structural proteins of
capsid, L1 and L2 (Muñoz et al., 2006). The “E” and “L” regions are referred to as ORF
(Open Reading Frame) and are separated by the LCR region (Muñoz et al., 2006).
The viral genes are transcribed in a clockwise direction, but the transcription is not
temporally sequential. The encoded proteins have the same nomenclature of genes: six
nonstructural regulatory proteins, E1, E2, E4, E5, E6 and E7 that interact with the host
genome and proteins to replicate viral DNA, and two structural proteins L1 and L2,
expressed after replication of viral DNA. Each viral protein has a specific function in the
viral replication cycle. E1 and E2 are specifically involved in DNA replication, while E4,
8

E5, E6, and E7 contribute to the initial destabilization of proliferation and differentiation of
the host cell (Bravo and Alonso, 2007). E1 plays a major role in the initial phase of
replication of the viral genome as it presents the ATP-dependent helicase activity that
allows recognition and beginning of viral DNA replication. E2 gene encodes several
proteins that regulate early viral gene transcription and viral DNA replication, thanks to a
structural sequence that binds specific DNA regions (Wilson et al., 2002). At low levels,
E2 binds specific recognition sequences and activates early viral promoters, while at high
concentrations it represses viral transcription by blocking the binding to transcription
factors. E2 plays a key role in the direct regulation of the levels of E6 and E7 oncoproteins.
Indeed, loss of E2 expression is the first stage of neoplastic transformation. Despite its
name, E4 is expressed during the final phases of the viral replication cycle: it seems to
interact with the keratin intermediate filaments, making them mechanically unstable and
thus facilitating the release of mature virions from the keratinocytes. E5 is a highly
hydrophobic protein made up of 83 amino acids that participates to viral DNA replication
along with E1, E2 and E4 proteins. His Its expression induces several cellular changes.
Indeed, E5 enhances the signal of growth factors (Crusius et al., 1998) activates MAPK
pathway and down-regulates MHC class I and class II (Ashrafi et al., 2005). Specifically,
E5: (i) activates the EGF receptor to promote cell proliferation; (ii) enhances the activity of
transcription factors, such as c-jun and c-fos thereby favoring cellular mitosis; (iii)
inactivates p21; (iv) prevents apoptosis resulting in DNA damage: (v) prevents transport of
the major histocompatibility complex (MHC) class I and class II. Recent works proved that
E5 contributes to hyperplasia, regulates growth and invasion in cervical cancer cell lines
and promotes cervical carcinogenesis in conjunction with E6 and E7 (Genther et al., 2005;
Maufort et al., 2010). A study of Kabsch conducted, in 2002, indicates that E5 could
inhibit apoptosis, forcing the cell to remain in a continuous proliferative state.
E6 and E7 are the two oncoproteins of High Risk HPVs. These two viral proteins
are essential to induce and maintain cell transformation because they interfere with the
normal controls of cell cycle and apoptosis. E6 is one of the first proteins to be expressed
during HPV infection. This protein is formed by 150 amino acids with a molecular weight
of 18 kD. Typically, E6 protein of High Risk HPVs is located in the nucleus and has no
intrinsic enzymatic activity. E6 is involved in different cellular pathways, which involve a
large number of different proteins: transcription factors, pro-apoptotic proteins, proteins
involved in the formation and maintenance of cellular architecture, polarity and adhesion,
and factors involved in DNA replication and repair. (Zur Hausen, 2002). E6 protein binds
9

to tumor suppressor protein p53, its principal target, thereby leading to p53 degradation
(Scheffner et al., 1990; Werness et al., 1990). p53 plays a key role in apoptosis and cell
cycle regulation, through several mechanisms: (i) it induces apoptosis in the case of
irreparably damaged DNA; (ii) it activates p21 (an inhibitor of cyclin/cyclin-dependent
kinase complex, cdk) consequently holding cell cycle at the G1/S regulation point on DNA
damage recognition and finally blocking cell growth (Vogelstein et al., 2000). Therefore,
functional inactivation of p53 by E6 results in G1/S and G2/M deregulation of cell cycle
control leading to abnormalities in DNA replication and genomic structure. The carboxyl
(C)-terminal of E6 protein carries a short sequence that interacts with a specific set of PDZ
domains included in several human proteins (Scott and Klingelhutz, 2014). PDZ domains
(an acronym from the initial of the proteins PSD95, DLG, and ZO1 on which they have
been identified) are small domains that bind to peptide ligands through a consensus
sequence XX(S/T/Y)X(V/L/M) located on target proteins. The PDZ protein family present
a conserved domain that is often found in proteins located in the areas of contact between
cells, such as tight junctions between epithelial cells or neural cell synaptic junctions.
Furthermore, PDZ proteins are implicated in signal transduction and polarity. The
members of PDZ protein family (MUPP-1, and hDlg, hSCRIB, MAGI-1, -2 and -3, GIPC,
PATJ, PTPN3 and PSD95) bind the C-terminus of the protein E6 of oncogenic HPVs and
are subsequently degraded in a proteasome dependent manner (Gardiol et al., 1999;
Glaunsinger et al., 2000; Nakagawa and Huibregtse, 2000; Massimi et al., 2004). As a
consequence of the degradation of PDZ family proteins, cellular contact mediated by
adherence junctions is lost with a consequent lack of cell polarity. These alterations were
observed in High Risk HPV-associated transformed cells.
E6 protein is also able to induce cell immortalization via telomerase activation and
specifically activating the expression of the catalytic subunit of telomerase, i.e. telomerase
reverse transcriptase TERT. Telomerases are RNA-dependent polymerases that add
telomere repeats to the ends of chromosomes (Klingelhutz et al., 1996; Kiyono et al.,
1998; McMurray and McCance, 2003; Howie et al., 2009). They are ribonucleoproteins
made up by RNA and a protein component, which is the reverse transcriptase subunit
TERT. This subunit catalyzes the addition of deoxynucleotides in a TTAGGG sequence to
the ends of telomeres (Shampay and Blackburn, 1988). Thereby preventing the
degradation of the chromosomal ends during DNA replication. Both proliferative stem
cells and cancer cells express human TERT (hTERT). The expression of hTERT allows
telomerase activity and suppression of cell senescence in germ cells. The E6 of some HPV
10

types is able to activate the TERT promoter and this mechanism is strongly associated with
cancer (Van Doorslaer and Burk, 2012). Even though the exact molecular mechanism of
promoter activation is still unclear, it probably involves E6AP binding (Gewin and
Galloway, 2001). Furthermore, E6 activates the transcription of hTERT through the
transcription factor c-myc (Veldman et al., 2001) that displaces the USF transcriptional
repressor from the E box in the TERT promoter (McMurray and McCance, 2003).
According to other models, E6 and E6AP bind to a repressor of TERT transcription called
NFX1-91, inducing its degradation. The interaction between E6-E6AP and NFX1-91
allows myc to bind to the hTERT promoter and activates it (Gewin et al., 2004). As a
consequence of the interaction with E6-E6AP, NFXI-91 is ubiquitinated and degradated,
thereby removing transcriptional repression at the TERT promoter. Conversely, a splice
variant of NFX1 referred to as NFX1-123 is unable to interact with E6 and hence
transcriptional repression is kept despite HPV infection (Katzenellenbogen et al., 2007).
Furthermore, NFX1-123 stabilizes TERT transcripts in HPV-16 E6 expressing cells by
binding to poly-(A) binding proteins (Katzenellenbogen et al., 2007; Katzenellenbogen et
al., 2009). Interestingly, E6 is able to bind directly TERT proteins, (Liu et al., 2009) even
though the biological consequences of this interaction are still unknown (Liu et al., 2009).
HPV E7 is a phosphoprotein made up of 100 amino acid residues that is not
encoded by all papillomaviruses. E7 is composed by three domains: conserved region (CR)
1, CR2 and CR3. The E7 amino terminus region contains two regions corresponding to the
small portion of CR1 and nearly the entire CR2, whose sequence is similar to the
adenovirus (Ad) E1A (Phelps et al., 1988). The CR1 and CR2 domains are separated by a
non-conserved sequence of variable size and amino-acidic composition. The CR3 is
located at the carboxyl terminal that contains a zinc-binding site important for
dimerization. Such a site is made up by two CXXC domains separated by 29-30 amino
acid residues (Barbosa et al., 1989; McIntyre et al., 1993). When one or both the cystein
residues of one zinc-binding site is mutated, the virus is no longer able to immortalize
human keratinocytes in the dermis (HFK) and transform rodent cells (Dyson et al., 1989).
E7 could presents post-translational modifications. Indeed, E7 contains a consensus
phosphorylation site for: (i) protein kinase C (PKC) on threonine 7 in the CR1 homology
domain (Liang et al., 2008); (ii) casein kinase II (CKII) in the CR2 domain (Firzlaff et al.,
1989; Barbosa et al., 1990); (iii) unknown kinase in CR3 domain (Massimi and Banks,
2000). E7 can be either carboxyl terminally polyaminated or amino (N)-terminally
ubiquitinated and subsequently degradated via proteasome (Reinstein et al., 2000; Jeon et
11

al., 2003). This oncoprotein is usually located in the nucleus, but can be potentially
shuttled between the two cellular compartments, thanks to nuclear export sequences
(Knapp et al., 2009). E7 interacts with several cellular proteins: transcription factors and
proteins that remodel chromatin, negative regulators of the cell cycle, and components of
the innate immune response. As previously reported, E7 has oncogenic activity as it
efficiently immortalizes human keratinocytes via the combined action with E6 (Felsani et
al., 2006). In addition to that, E7 binds pRb and other members of Rb family: p107 and
p130 (Dyson et al., 1989; Davies et al., 1993). E7 associates to pRB through the conserved
LXCXE sequence motif within CR2 domain (Dyson et al., 1989). pRb is a nuclear tumor
suppressor phosphoprotein belonging to the pocket protein family, whose members have a
interacting region with various proteins (Korenjak and Brehm, 2005). pRb prevents
excessive cell growth by inhibiting cell cycle progression and can be considered a
“molecular brake” of the transition from G1 to S phase. pRb regulates cell cycle
progression through the interaction with E2F/DP, a dimer of E2F protein and a
dimerization partner (DP) protein (Wu et al., 1995) that pushes the cell cycle into S phase
(Funk et al., 1997). The active form of pRB normally binds and inactivates E2F/DP,
blocking cell cycle in G1 phase. Furthermore, the complex pRb-E2F/DP interacts with
several chromatin remodeling enzymes such as acetylases and methylases, thereby
silencing numerous promoter of genes involved in the progression from G1 to S phase. On
the contrary, when the cell is ready to divide, Rb is inactivated through phosphorylation,
allowing the progression from G1 to S phase. The other two members of retinoblastoma
family that interact with E7 (p107 and p130) are involved in the control of the different
phases of the cell cycle. Specifically, p130 has a regulatory function during the G0/G1
phase, while p107 is active in the G1/S transition. E7 is able to bind p107 and p130,
allowing cell progression from G1 to S phase and enhancing cellular proliferative
characteristics (Dyson et al., 1989; Howie et al., 2009). E7 is able to interact with the
cyclin-dependent kinase, CDK2 dependent cyclin A and E that normally regulate cell cycle
(Arroyo et al., 1993). Furthermore, it associates to inhibitors of cyclin/cdk complex as p21
and p27, thereby blocking their inhibitory action and fostering the activity of the complex
cyclin/cdk (Funk et al., 1997). p16INK4a is an inhibitor of cyclin/cdk complex, whose
overexpression normally induces cycle arrest. E7 counteracts the inhibitory role of
p16INK4a through mechanisms that have not been completely clarified yet. For this
reason, p16INK4a was considered as a molecular marker for the CIN pre-cancerous lesions
(Razmpoosh et al., 2014). Furthermore, E7 is able to bind histone deacetylases (HDACs), a
12

class of enzymes that remove acetyl groups (O=C-CH3) from the ε-N-acetyl lysine amino
acid on the histones and increase their ability to bind to DNA, thereby enhancing genetic
transcription. The interaction between E7 and HDACs causes chromatin packing and
consequently blocks transcription. Since pRb normally binds and recruits HDAC at level
of E2F inducible promoters, E7 can prevent histone deacetylation also in an indirect way,
i.e. through the inhibition of pRb (Brehm et al., 1999). Furthermore, in the case of
persistent HPV infection, HDACs recruitment allows E7 to silence specific genes, such as
interferon regulatory factor 1 (IRF1), whose expression is important in immune response
(Park et al., 2000). Since the viral genome is unable to remain permanently in episomal
form when a mutation in the E7-HDAC binding site of E7 is induced (Longworth and
Laimins, 2004), it has been deducted that the association with HDAC allows E7 to
maintain the viral DNA in episomal form. The molecular mechanism of such an interaction
is still unclear, but it has been proposed that HDAC directly deacetylates of E2F, causing
loss of function. Another important action of E7 is the transactivation of the enzyme
phosphatase Cdc25, which allows the dephosphorylation and activation of complex
cyclin/cdk and is required for cell cycle progression (Jinno et al., 1994). Another capacity
of E7 is the ability to induce genomic instability. In fact, several HPV-positive cancer cells
contain different aneuploidies, indicating that changes in the number of chromosomes are
important events in tumor progression. The presence of E7 is sufficient to induce an
abnormal increase in the number of chromosomes in primary human keratinocytes
(Duensing et al., 2000). Mutated E7 proteins that do not bind or degrade pRb, but are
associated with p107, induce centrosome abnormalities during cell division (Duensing and
Munger, 2003). These anomalies are also observed in cells lacking p53 and pRb and in
embryonic fibroblasts of knock-out mice for pRb, p130 and p107. It has been speculated
that a combination of family members of pRb or other factors can induce centrosome
abnormalities during HPV infection (Duensing and Munger, 2003).
The late proteins L1 and L2 have a structural function. L1 is the major viral capsid protein
common to all the HPV protein. L1 self-assemble into 72 pentamers and is the most
exposed region of the capsid and the target of the majority of neutralizing antibodies. L2
protein is highly variable among different types of HPV. This viral protein plays mainly
regulatory and structural. In fact, it presents a nuclear localization signal and it is involved
into the selective viral DNA encapsidation.
13

The life cycle of HPVs in uterine cervix
HPV infections are mainly common in keratinocytes of the skin or mucous
membranes presenting pluristratified epithelium, the only tissue in which they can
replicate. In particular, HPV infection starts with the initial contact of the virus with micro-
abrasions of the cutaneous or mucous membranes that expose segments of the basement
region of the pluri-stratified epithelia, composed by stem elements (figure 1, A; Figure 4).
Following the same process, genital HPV attack the uterine cervix, whose covering surface
is thin and hence particularly susceptible to suffer from micro-abrasions, making it even
more vulnerable to infection. As far as we know, the virus enters in the basal cells through
endocytosis, even though the exact mechanism (classical endocytosis or receptor-mediated
endocytosis) is still unclear. Presumably the heparan sulfate proteoglycans, located at the
cell surface and in the extracellular matrix, mediate the initial phase of cell infection (Joyce
et al., 1999; Patterson et al., 2005). As for other viruses, (Chung et al., 1998; Summerford
et al., 1999) it seems that secondary receptors as efficient as the integrin receptor α4β6 are
needed for HPV infection (Evander et al., 1997; Bossis et al., 2005). After receptor
recognition, the process of endocytosis occurs through clathrin-coated vesicles. After the
entrance of the virus into the cytoplasmic compartment (Culp and Christensen, 2004), the
capsid protein is disassembled in the lysosomes and the viral DNA is transferred into the
nucleus through the minor capsid protein L2 (Day et al., 2003). The viral DNA remains in
episomal form in the nucleus in the number of 10-100 copies/cell equivalent and this
process does not usually cause cytological abnormalities (Schiller et al., 2010). HPV early
genes E1/E2 are expressed at low levels in the nucleus and trigger the replication of the
viral genome, which is almost completely mediated by the replicative machinery of the
host keratinocyte (Figure 4). The life cycle of HPV is intimately associated with the
proliferative activity and differentiating status of the host keratinocyte. In fact, quick
cellular replication is necessary for an increased viral progeny. E2 protein activates and
controls the expression of E6 and E7 that stimulate the host cell to proliferate, thereby
stimulating cell growth.
14

Figure 4. HPV life cycle. Graphical representation of the key events in HPV life cycle: from the first interaction with the basal layer by the virions to the DNA replication, viral gene expression and viral release.
Furthermore, E6 and E7 proteins induce a block of cell cycle progression keeping
the cell in an undifferentiated state. The final effect is an increased proliferation of
undifferentiated and infected keratinocytes (Doorbar et al., 2012). The viral cycle follows
the natural maturation of the epithelium, where basal keratinocytes progressively
differentiate while moving towards the most superficial layers. In the intermediate cell
layers, viral DNA replication stops and the expression of E4 and E5 early genes starts
(Figure 4, http://www.clinsci.org/cs/). The proteins transcript of E4 activate the expression
of the late genes L1 and L2, while and proteins transcript of E5 encapsidates the viral DNA
within the virion (Doorbar et al., 2012). The virus is completely assembled in the
squamous cells of the upper layer of the pluri-stratified epithelium, where the keratin is
destroyed and virions are released by dead cells (Figure 4). The infectious process is slow
as it takes about 12–24 hours for the initiation of transcription and the final passage of life
cycle is the exfoliation of superficial keratinocytes and subsequent release of HPV virions
into the surrounding cervix environment (Doorbar et al., 2012). The complete life cycle of
HPV from the first interaction with the basal layer to the viral release, requires about 6-12
weeks.
15

HPV and cell proliferation
During the full replicative cycle of low-risk HPV the genome remains in episomal
form and the production of mature viral progeny occurs. On the contrary, during high-risk
HPV infections viral DNA is integrated into the DNA of the host cell. This molecular
mechanism has a double effect: it decreases viral progeny on the one hand and it increases
proliferative capacity in the host cell on the other hand. Indeed, the integration occurs at
the level of the E2 ORF, blocking the repressive action of E2 on viral oncoproteins E6 and
E7 and leading to uncontrolled expression. As recently reported, E6 and E7 stimulate cell
cycle progression by inhibiting the activity of important regulatory proteins of the cycle,
p53 and pRb, with well known molecular mechanisms (Munger et al., 1989; Sheffner et
al., 1990). E5 seems to be involved in the stimulation of cell proliferation. In fact, it can
inhibit apoptosis and lead the cell to a high proliferative status.
HPV and tumorigenesis
A crucial aspect of the replicative cycle of cervical high-risk HPVs is the possibility
to induce persistent infections. As previously reported, the integration of viral DNA into
the DNA of the host cell causes the disruption of E2 gene sequence and leads to the
uncontrolled and continuous production of viral oncoproteins E6 and E7. Consequently,
the epithelial cells that express E6 and E7 have a growth advantage compared to those that
contain the viral DNA in episomal form. This is the first mechanism in the multistep
process of cervical neoplastic transformation. E6, E7 and E5 are critical for the
tumorigenic process, as they stimulate cell proliferation, induce cell survival and modulate
keratinocytes differentiation, as confirmed by experimental models. In fact, when the
expression of E6 and E7 is inhibited in cultured transformed cells the malignant phenotype
is reverted, while the activation of E2 in in vitro experiments blocks cell proliferation in
cervical SCC cell lines (Goodwin et al., 1998; Jiang and Milner, 2005). During high-risk
HPVs infection, the tumorigenic process changes the histological characteristics of the
cervical squamous pluri-stratified epithelium. However, the viral DNA is present only in
the episomal form and the expression levels of oncoproteins E6 and E7 are relatively low
(Schiller et al., 2010). This phase is generally referred to as CIN1 lesion. Conversely, when
the viral DNA begins to integrate in shuffle mode and becomes equally available in the
16

episomal and integrated form, the cervical pre-neoplastic lesion acquires tumorigenic
potential (CIN2). Finally, cervical keratinocytes of CIN3 lesions are no longer able to
regulate their own proliferative and differentiation capacity, due to the uncontrolled
expression of E6 and E7. Epidemiological and experimental data indicate that the presence
of High-risk HPVs in cervical keratinocytes is necessary yet not sufficient to induce
cervical SCC. Indeed, other genetic and epigenetic events would be implicated in the
multifactorial process of neoplastic transformation (Zur Hausen, 2002), justifying the
presence of several functional and structural abnormalities of both oncogenes and tumor
suppressor genes as well as epigenetic modifications in cervical SCCs. As to the proto-
oncogenes, cell mutations and/or gene amplification have been identified in the genes
encoding for the subunits of PIK3CA, RAS, MYC, ErbB2 and cIAP1 (Zhang et al., 2002;
Imoto et al., 2002; Bertelsen et al., 2006). As to the tumor suppressor genes, loss of PTEN,
CADM1 and NOTCH seems to be associated with tumor progression (Cheung et al., 2004;
Overmeer et al., 2008; Vande and Klingelhutz, 2013). The epigenetic deregulation and its
role in cervical pre-neoplastic CIN progression or in SCC will be dealt in detail in the
following chapter.
EPIGENETIC AND CANCER
It is well known that epigenetic alterations, i.e. improper DNA methylation and
histone modification, are strongly involved, as other molecular phenomena, like genetic
mutations or gene expression changes, in a cell’s transformation to cancer (Novak, 2004).
Furthermore, the DNA methylation and histone modifications defects could be operate
alone or in concert with several other molecular alterations involved in cancer
development.
Epigenetics can be defined as the study of all heritable chemical modifications that
affect the phenotype without altering the genotype. Indeed, it is defined as ‘‘the study of
mitotically and/or meiotically heritable changes in gene function that cannot be explained
by changes in DNA sequence’’ (Russo et al., 1996). Furthermore, epigenetics modification
may have short- or long-term and even trans-generational effect. Epigenetics modification
are well-established and common phenomena in all cells and are involved in the regulation 17

of gene expression. At the basis of the epigenetics process there are the regulation of
chromatin remodeling and regulation of gene expression that are accomplished through
two related mechanisms: DNA methylation and posttranslational histone modifications.
From a biological standpoint, epigenetics modifications are phenomenon that plays a key
role in a diversity of processes, such as embryonic development, immune system response,
infertility and cancer development. However, aberrant epigenetic modifications may have
the same negative effect of a gene mutation, because the expression of the DNA is altered.
Consequently, aberrant epigenetic modifications may be associated, or induces, disease.
While the genetic code is considered static, i.e. it is the same in each cell for the entire life
of the organism, the epigenetics phenomena are dynamic, tissue-specific and provides to
change the phenotype because of various factors such as environmental factors (Li et al.,
2006). This dynamic state may suggest the possibility of reversibility of epigenetic
changes, and then a possible therapeutic target for a certain number of diseases. (Egger et
al., 2009)
Aberrant CpGs methylations, hypo- and hypermethylation, have long been
associated with diseases and cancer. DNA methylation is a biochemical process based to
the addition of a methyl group to the S-adenosyl-methionine to the 5’ position of cytosine
belonging to CpG DNA dinucleotide (Biermann and Steger 2007). This activity is
mediated by DNA methyltransferases (DNMTs) family enzymes. Specifically, the family
of enzimes DNMT1 is involved in the maintenance of methylation patterns (Bestor et al.,
1988); whereas, enzymes DNMTs 3a and 3b (Okano et al., 1999) are required for de novo
methylation activity (Lei et al., 1996; Okano et al., 1999). Another protein, DNMT3L, is
homologous to DNMT3s, but has no catalytic activity and assists the DNMTs by
increasing their ability to bind DNA and stimulating their activity (Chedin et al., 2002;
Suetake et al., 2004). Takai and Jones formulated the accepted definition of a CpG island
as a 500, or higher, base pair segment of DNA with a G+C equal to, or greater than, 55%
and with a CpG frequency of at least 0.65 of the statistically expected value (Takai and
Jones, 2002). In mammals 70% to 80% of CpG cytosines are methylated (Jabbari and
Bernardi 2004). CpG islands are located preferentially in high concentrations within
promoters genes (Bird et al., 1995) with a frequency about 40% (Fatemi et al., 2005),
indicating that they are involved in the regulation of gene expression (Deaton and Bird,
2011). Indeed, DNA hypomethylation in CpG islands correlates with gene expression
(Biermann and Steger, 2007), whereas DNA methylation in CpG islands is associated with
trascriptional repression (Deaton and Bird, 2011). Hypermethylation in CpG islands
18

located at gene promoter region cause gene suppression because: (i) DNA methylation
itself prevents binding of transcriptional factors (Choy et al., 2010); (ii) methylated DNA
is bound by methyl-CpG-binding domain proteins (MBDs) (Nan et al., 1993), which,
subsequently recruit other factors involved in gene expression inhibition (Illingworth and
Bird, 2009; Thomson et al., 2010);
In general, these epigenetic modifications, causes defective gene expression,
improper condensation and chromosomal instability (Attila and Lorincs, 2014). In cancer
cells, both of these alterations could coexist in concert or alone. As previously reported,
CpG islands are typically located within, or close to, gene promoter and are involved in
gene expression regulation. Whereas CpG islands preceding tumor suppressor gene
promoters are generally unmethylated, in cancer cells it is possible that an abnormal
increase of methylation level at this region induce gene silencing. Indeed,
hypermethylation of tumor suppressor gene promoters is often associated with the
silencing of those genes. The tumorigenic process is induced when genes that regulate the
cell cycle are silenced, allowing cells to increase their proliferative capacity and reproduce
uncontrollably (Esteller, 2007). Two important genes that acquire hypermethylation were
two cell-cycle inhibitor referred to as p16 and p15 in different types of cancer (Viet et al.,
2007). Various genes involved in DNA repair, such as O-6-methylguanine-DNA
methyltransferase (MGMT), undergo defective methylation in many types of carcinomas
(Esteller et al., 1999; Weller et al., 2010). Furthermore, another DNA-repair gene found to
be hypermethylated is MLH1. This gene was detected whit this defective epigenetics
aberration in gastric cancer (Li et al., 2014). Others suppressor genes known to be
hypermethylated during cancer progression were: RB, Cyclin-dependent kinase inhibitor;
the Von Hippel–Lindau tumor suppressor gene VHL; and E-cadherin, a calcium-dependent
cell-cell adhesion glycoprotein (Greger et al.,1989; Santini et al., 2001). Typically, during
cancer development, while hypermetilation generally occurs in a single molecular target,
DNA hypomethylation is a general phenomenon. Genomic instability is the principal
consequence, but sometime transcriptional activation of oncogenes may occur. Indeed,
aberrant hypomethylation of CpG islands located at proto-oncogene promoters could lead
to an increase of expression of these genes. In normal cell, repetitive genomic sequence,
such as LINE, SINE, IAP and Alu elements are highly methylated. These genomic
sequences ensure genomic integrity/stability and improper hypometylation phenomena
could induce undesired mitotic recombination. Indeed, loss of DNA methylation in SAT2
(juxtacentromeric satellite 2) and SATα (centromeric satellite α) was detected in breast
19

cancer and may contribute to progression of ovarian cancer (Widshwendter et al., 2004).
DNA loss of methylation of individual gene is rather in common. Indeed, frequently, most
of promoter regulatory region are under this epigenetic control, belong to tissue specific
genes. For instance, a cancer/testis (CT) antigens, expressed in normal condition in adult
male testis, are epigenetically and aberrantly activated in various types of human cancers
(Caballero and Chen, 2009). Furthermore, in colorectal cancer and melanoma, the CT
antigens MAGE are reactivated by promoter hypomethylation (Weber et al., 1994).
High-risk HPVs and epigenetics deregulation in cancer
The multifactorial process of cervical SCCs high-risk HPVs induced, includes,
among the various aspects, epigenetic changes in the host genome. The apoptotic pathway
presents numerous genes, which epigenetic aberrations are involved with the onset of
cervical SCCs. The gene coding two members of tumor necrosis factor receptor
superfamily, referred to as decoy receptors (DcR1/DcR2) can be the target for abnormal
methylation that leads to their silencing in cervical SCCs, suggesting that cervical cancer
cells may obtain a growth advantage, probably due to the down-regulation of decoy
receptor DcR1/DcR2 (Van Noesel et al., 2002; Shivapurkar et al., 2004). Interestingly,
despite cervical SCCs show down-regulation of hTERT mRNA, a study has found a
correlation between reduced expression, and catalytic subunit activity, with hTERT gene
promoter demethylation (Guilleret and Benhattar, 2003). p73, a member of the p53 family,
involved in cellular response to DNA damage induced by radiation and chemotherapeutic
agents, presents two independent promoters that have opposite activities. One of this two
promoters presents within the exon 1, is rich in CpG dinucleotides and its transcriptional
silencing through hypermethylation represents a mechanism for inactivation of this gene in
cervical SCCs (Liu et al., 2004). As previously reported, a lot of number of cell cycle-
related genes is deregulated during cancer development. Aberrant methylation of the p16
gene promoter occurs in situ as well as in invasive tumors with a frequency of ranged
between 10-100% (Nakashima et al., 1999). Furthermore, p16 hypermethylation is
progressively most frequent during CIN pre-cancerous lesions progression. Indeed is
present in 17.6% of CIN I, 42.1% of CIN II, 55.0% of CIN III, and 65.0% of invasive
20

cancers (Huang et al., 2011). Two other genes cycle-related genes epigenetically involved
in cervical SCCs onset and progression are CCNA1 and the fragile histidine triad (FHIT).
The CCNA1 gene encodes Cyclin A1, which regulates the cell cycle. CCNA1
downregulation due to promoter hypermethylation was detected in 60% and 93.3% of
microinvasive cancers, and invasive cancers, respectively (Yang et al., 2010). FHIT is
another protein involved in cell cycle regulation and apoptosis. Epigenetic silencing of this
gene by promoter hypermethylation is common in cervical cancer (Ki et al., 2008).
Furthermore, the signal transduction pathway Wnt, named for its most upstream ligands,
the Wnts, is epigenetically deregulated during cervical SCC development. In this pathway
were detected promoter aberrant methylations in PTEN (Cheung et al., 2004), E-cadherin
(Widschwendter et al., 2004) and APC (Virmani et al., 2001) in various cervical SCCs
cases. Two genes belonging to the Fanconi anemia (FA)-BRAC pathway referred to as
BRCA and FANCF present aberrant methylation in their promoter in cervical SCCs
(Marsit et al., 2004; Narayan et al., 2003). Furthermore, in a study conducted in patients
with SCC, BRCA1 promoter hypermethylated cases present a frequency of 6.1%, in
invasive SCCs, whereas FANCF hypermethylation rate was 30%. Thus, hypermentilation
of these genes was mutually exclusive in the analyzed cases, suggesting the important role
of epigenetics aberrations in this pathway for cancer. (Narayan et al., 2004). Other
molecular pathways that have genes, which promoter carries epigenetics deregulations
include: mismatch repair, metastasis/cell death, cell differentiation and DNA repair
(Narayan et al., 2003; Virmani et al., 2001).
21

MOLECULAR STUDIES OF CIN PROGRESSION
The molecular bases involved in the pre-cancerous CIN lesions progression to
cervical SCC were previously investigated in different studies. These studies were carried
out at both tissue and cellular level using microarray analysis or HPV DNA transfection
assays, respectively.
In order to compare gene expression signature on biopsies from cervical SCC with
normal tissues, different studies were performed using microarray technique. (Cheng et al.,
2002; Chen et al., 2003; Wong et al., 2003; Rosty et al., 2005; Wong et al., 2006; Chao et
al., 2006). However, in all these studies have not been investigated the molecular
mechanisms involved in the progression of CIN lesions to the cervical SCC. These
mechanism were subsequently investigated in other works when were used, in addition to
cervical SCC and normal tissues, CIN biopsies as well (Gius et al., 2007; Arvantis and
Spandidos, 2008; Song et al., 2008; Rajcumar et al., 2011). A work conducted by Gius and
colleagues shows that proproliferative/immunosuppressive genes, such as p16INK4a,
KIF23 and CENPF are up regulated in CIN1 lesions, probably due to the epithelial
response to human papillomavirus infection, while proangiogenic stromal/epithelial
interaction genes, such as HINT1, TAGLN and TBX19 and proinvasive genes, such as
DSG3, MMP3 are mainly up-regulated in CIN2 and CIN3 lesions, respectively. These
results suggest a cooperative signaling interaction between stroma and tumor cells. Finally,
the signature pattern detected in CIN3 and SCC probably represents epithelial tumor cell
overcrowding (Gius et al., 2007).
Microarray studies were performed on biopsies of tumor and normal tissue. This
study model may affect the actual gene expression profile of the type of cell under study,
as keratinocytes, in this case. Such a problem has to be ascribed to tissue samples - in
particular cervical CIN or normal tissue samples - that contain a significant number of
stromal cells and contaminants derived from the host immune cells, such as monocytes,
dendritic cells and lymphocytes.
In vitro HPV-transfected human skin keratinocytes represent good models to mimic
the molecular and morphological characteristics of cancerous cells (Pirisi et al., 1987;;
Zyzak et al., 1994; Creek et al., 1995; Borger et al., 2000; Akerman et al., 2001; Chang
and Laimins, 2000; Nees et al., 2001; Oh et al., 2001). In vitro models of HPV-transfected
cells showed loss of differentiation, overexpression of EGF receptor and resistance to
22

TGF-beta-induced growth inhibition (Pirisi et al., 1988). Several studies have applied
microarray technology to this model with the aim of identifying the molecular processes
involved in HPV-induced transformation and tumor development (Ruutu et al., 2002;
Duffy et al., 2003; Kravchenco-Balasha et al., 2009). However, HPV-transfected
keratinocytes may not mirror the actual in vivo situation because they do not derive from
the uterine cervix, but from other parts of the body. Moreover, keratinocytes were not
naturally infected by high-risk HPVs. Consequently, the molecular pathways detected in
this model could be different from those involved in HPV induced progression to cervical
SCC.
Only few gene-expression studies have been performed on cervical keratinocytes
that were naturally infected with HPV, i.e. in vitro neoplastic HPV-keratinocytes derived
from CIN lesions (Gray et al., 2010). In those studies, the gene expression profile was
investigated in HPV16-infected keratinocytes derived from low-grade CIN1 lesions (Gray
et al., 2010). A work of Nees and colleagues used, as a study model, primary cultures of
ectocervical keratinocytes obtained from cervical tissue from hysterectomies. The cells
used were infected with retroviruses expressing E6 and E7 genes of HPV16 (Nees et al.,
2001). In 1989 Stanley and colleagues set up a human cervical keratinocyte cell line,
referred to as W12, from a low-grade cervical lesion histologically diagnosed as CIN1.
This keratinocyte cell line represented a good model to study the natural history of cervical
neoplasia. In fact, the same model was used to identify groups of genes that carried
expression changes due to HPV-16 integration (Alazawi et al., 2002). Santin and
colleagues analyzed gene expression on in vitro cultures of cervical keratinocytes using
microarray assay. Even though cultured keratinocytes did not derive from CIN pre-
cancerous lesions, the study was conducted on 15 primary cervical cell lines: 11 HPV-16
or HPV-18 positive cervical SCC primary cultures and 4 cell lines of normal cervical
keratinocytes (Santin et al., 2005).
23

OBJECTIVE AND AIMS
OF THE STUDY
Overall, the main objective of my study was to investigate the molecular
mechanisms occurring in neoplastic progression of CIN2 and CIN3 cervical pre-cancerous
lesions. To this purpose, HPV16-CIN2 and HPV16-CIN3 keratinocytes derived from high-
grade CIN2 and CIN3 pre-cancerous lesions were investigated by microarray analysis and
DNA methylation of gene promoters. Aims of this study were:
I
To set up a cell culture protocol able to derive pre-neoplastic and normal cervical
keratinocytes from small tissue fragments of naturally high-risk HPV-infected CIN2 and
CIN3 lesions and normal uterine cervix, respectively. Cultures of CIN and normal cervical
keratinocytes were stained with immunofluorescence technique in order to investigate
expression of epithelial and cervical markers.
II
To investigate the gene expression profile of HPV16-CIN2 and HPV16-CIN3
keratinocytes. To this purpose, HPV16-CIN2 and HPV16-CIN3 keratinocytes and the
corresponding normal cervical keratinocytes were subjected to microarray analysis, Real-
Time Quantitative RT- PCR, and Immunohistochemistry analysis.
III
24

To analyze the mRNA expression levels and DNA methylation status of target
genes in HPV16-CIN2 and HPV16-CIN3 keratinocytes. To this purpose, HPV16-CIN2
and HPV16-CIN3 keratinocytes and the corresponding normal cervical keratinocytes were
subjected to Real-Time Quantitative RT- PCR, and bisulfite sequencing analysis.
MATERIALS AND METHODS
CERVICAL UTERINE SPECIMENS
Small tissue fragments (2-3 mm3) were taken from CIN biopsies, CIN2 or CIN3
pre-cancerous lesions, after surgery excision. The corresponding surrounding normal
tissues were also provided. The patients had undergone electrosurgical conisation under
colposcopic examination using 5% acetic acid and Lugol’s iodine solution, which stains
the pathologic tissue with a white color, whereas stains normal tissue brown (Stafl and
Wilbanks, 1991). CIN and normal specimens were selected and divided by the
gynecologist during surgery and CIN specimens were classified by pathologists according
to international criteria (Horvat et al., 2008). Informed written consent was obtained from
all patients in accordance with our institutional guidelines.
CIN2, CIN3 AND NORMAL TISSUE PREPARATION
Each tissue fragment was transferred into a 50 ml centrifuge tube and submerged in
an ice-bath containing 20 ml of DMEM:F12 transporting medium (with L-glutamine, 15
mM HEPES and 3.151 g/L glucose; Lonza, Milan, Italy) with 200 U ml-1
Penicillin/Streptomycin (10,000 U ml-1 penicillin, 10,000 U ml-1 streptomycin; Lonza,
Milan, Italy), 0.25 μg ml-1 Amphotericin B (250 μg ml-1; Lonza, Milan, Italy). Under a
25

sterile hood, the transporting medium was removed and the tissue fragments were rinsed
with 10 ml of sterile PBS 1X (without calcium or magnesium; Lonza, Milan, Italy,) and
mixed manually 3-4 times. The samples were centrifuged at 400 g at RT for 10 minutes
and the PBS 1X was discarded. The samples were washed again twice and afterwards the
PBS 1X was discarded. The tissue fragments were lifted with tweezers and placed in a 10
cm Petri-dish. Afterwards the tissue fragments were finely cut with a disposable blade.
During cutting, it was followed the direction of the blade edge, and the tissue was not
dragged laterally; to cut the tissue in all directions, the Petri-dish was turned around
periodically. Then, DMEM:F12 medium (with L-glutamine, 15 mM HEPES and 3.151 g/L
glucose; Lonza, Milan, Italy), 4.5 ml, was deposited on the Petri-dish and the minced
specimens were aspirated and transferred into a T25 flask (Corning, Pero, Italy); 500 µl of
2000 u/ml Type II collagenase enzyme solution (final concentration 200 u/ml, Sigma-
Aldrich, Milan, Italy) were added and tissue digestion was performed at 37 °C, with 5%
CO2, for 24 hrs.
CIN2, CIN3 AND NORMAL CULTURE SETUP
After digestion, cell suspension was gently mixed to optimally disaggregate all
tissue fragment residues, and then transferred the into a 10 ml conical tube and centrifuged
at 400g at RT, for 10 minutes. The supernatant was discarded and the cell pellet was
washed twice with 2 ml of PBS 1X. The cell pellet was suspended in 5 ml of DMEM:F12
complete medium (with L-glutamine, 15 mM HEPES and 3.151 g/L glucose; Lonza,
Milan, Italy) with 200 U ml-1 Penicillin/Streptomycin (10,000 U ml-1 penicillin, 10,000 U
ml-1 streptomycin; Lonza, Milan, Italy), 0.25 μg ml-1 Amphotericin B (250 μg ml -1; Lonza,
Milan, Italy) and 10% v/v of Fetal Bovine Serum (FBS, Lonza, Milan, Italy) and seeded in
a T25 flask. Cell cultures were incubated at 37 °C, with 5% CO2. After two days, the cell
suspension was transferred into a 10 ml conical tube whereas the attached cells were
washed twice with 5 ml of PBS 1X; then, 5 ml of fresh DMEM:F12 complete medium was
added. Cell suspension was centrifuged at 400g at RT for 10 minutes. The cell pellet was
26

washed twice with 2 ml of PBS 1X, then suspended in 5 ml of DMEM:F12 complete
medium and transferred into a new T25 flask (T2 flask). The cell suspension in the T2
flask was incubated at 37 °C, with 5% CO2, and leaved to attach for 2 days. After two
days, the cell suspension of the T2 flask was discarded, the attached cells were washed
twice with PBS1X and 5 ml of fresh DMEM:F12 complete medium was added.
CIN2, CIN3 AND NORMAL PRIMARY COLONY EXPANSION
Colonies grown in T1 and T2 flask were analysed with Inverted Nikon TE2000E
microscope; the larger colonies, and made up of small cells, were selected for expansion.
DMEM:F12 complete medium was removed and cells were washed twice with 5 ml of
PBS 1X. The upper surface of the T1 and T2 flasks was opened at the top with a red-hot
sterile disposable blade. The flasks were carefully opened in order to avoid dropping
plastic fragments onto the layer where the cells were located. After discarding of the
PBS1X, glass cylinders (height 10 mm, external diameter 9 mm, internal diameter 7 mm;
Elettrofor s.a.s., Borsea, Italy) were used to isolate the single colonies. Glass cylinders
were sealed with a silicone rubber and placed around the colonies; after pressing lightly
down, so that the cylinders adhered well to the bottom of the T25 flasks, cells were
detached with 50 µl of 0.05% w/v Trypsin (from bovine pancreas; Sigma-Aldrich, Milan,
Italy)/0.01% w/v Ethylenediaminetetraacetic acid (EDTA; Sigma-Aldrich, Milan, Italy) in
a PBS 1X solution, at 37°C for 5 minutes. After incubation, most of the cells from different
colonies were completely detached. However, cells from some colonies were difficult to
detach and further 3-5 minutes of incubation at 37°C were needed. Then, cell suspension
from each colony was recovered and seeded in a well of 6 well culture plate (Corning,
Pero, Italy) with 2 ml of DMEM:F12 complete medium. Cells were left to attach overnight
at 37 °C, with 5% CO2. After incubation, the DMEM:F12 complete medium was changed
with 2 ml of DMEM:F12 complete medium/defined Keratinocyte Serum Free Medium
(dKSFM, Invitrogen, Monza, Italy) (ratio 1:1) (DMEM:F12/dKSFM) medium. Cells were
incubated at 37 °C, with 5% CO2 and the DMEM:F12/dKSFM medium was changed every
27

3 days. Cell cultures became 80% confluent typically in 4 days for CIN2 and CIN3
keratinocyte colonies and in 7 days for normal keratinocyte colonies. In order to obtain
huge amounts of normal and neoplastic keratinocytes, the subconfluent cells were detached
from each well with 500 µl Trypsin/EDTA solution at 37 °C for 5 minutes, and then
transferred into a new T25 flasks with 5 ml DMEM:F12/dKSFM medium. Cell cultures
were incubated at 37 °C, with 5% CO2 and the medium was changed every 3 days. CIN2
and CIN3 keratinocyte colonies grew more quickly than normal keratinocyte colonies.
Therefore, 10 days were sufficient to reach sub-confluence for CIN2 and CIN3
keratinocytes and 15 days or more were needed in order for sub-confluence to be reached
by normal keratinocytes.
IMMUNOFLUORESCENCE ASSAYS
Single CIN3 and normal colonies were isolated with cloning rings, keratinocytes
were subdivided onto different coverslips and cells were grown on microscope slides.
Keratinocytes were fixed by immersing the slides in jars filled with paraformaldehyde
solution (4% formaldehyde and 0.5% Triton X-100 in PBS 1X) and incubated at 37°C for
20 min. Keratinocytes were then blocked with 10% goat serum in PBS 1X at 37°C for 1h.
Subsequently keratinocytes were incubated with different mouse anti-human monoclonal
antibodies (mabs). To determine the epithelial and cervical markers, immunofluorescence
staining with K5, K14, K17, and K19 keratins and with p63 (Dako SpA, Milan, Italy) was
performed, as previously described (Quade et al., 2001; Radu et al., 2002; Martens et al.,
2004; Harper et al., 2007; Tudor et al., 2007). The substitution of primary antibodies with
PBS 1X served as a negative control. Digital images from a Nikon TE2000E microscope
were captured using the ACT- 1 software for the DXM1200F digital camera (Nikon,
Florence, Italy). The percentage of cells expressing different keratin markers in the
colonies was quantified by counting 1,000 cells in four randomly selected fields/colony.
28

DNA ISOLATION
DNA was isolated from a small fraction of CIN2, CIN3, and normal specimens and
from corresponding CIN2, CIN3, and normal keratinocytes according to standard
procedures, as described (Barrandon and Green, 1987; Bononi et al., 2012; Rotondo et al.
2012). All DNA was stored at -80°C until the time of analysis. Furthermore, before
molecular analysis, all DNA was first quantified using the Nanodrop spectrophotometer
(ND-1000, NanoDrop Technologies, Wilmington, DE USA). To test the suitability of the
extracted DNA for PCR analysis, isolated DNA was PCR amplified with ß-globin primers
(Pancaldi et al., 2009).
HPV DETECTION AND GENOTYPING
Purified DNA from CIN2, CIN3, and normal specimens and from corresponding
cultured CIN2, CIN3 and normal keratinocytes was amplified for HPV sequences with the
general primers GP5-GP6, which enable detection of HPV -6b, -11, -16, -18, -31, -33
genotypes. PCR analysis was carried out with 500 ng human genomic DNA (Barrandon
and Green, 1987; Bononi et al., 2012; Martini et al., 2004). HPV PCR product sizes were
139 bp for HPV -6b, -11 and -33 genotypes, 142 bp for HPV -16 and -31 genotypes, and
145 bp for HPV -18 genotype. PCR products were electrophoretically separated on 2.5%
agarose gel. To further assess PCR product specificity, a restriction endonuclease analysis
of HPV sequences was performed with RsaI digestion (Barrandon and Green, 1987;
Bononi et al., 2012). DNA digestion was performed at 37°C for 2 h. The digested DNA
products were electrophoretically separated on 20% acrylamide gel, and DNA fragment
size GP5-GP6 amplified DNA of CIN and HPV PCR (positive control) products were
compared.
29

RNA ISOLATION
Total RNA was extracted from CIN2, CIN3, and normal keratinocytes by use of a
RNAzol kit (Life Technologies, Milan, Italy), according to the manufacturer’s instructions.
All RNA was stored at -80°C until the time of analysis. Furthermore, before molecular
analysis, all RNA was first quantified using the Nanodrop spectrophotometer (ND-1000,
NanoDrop Technologies, Wilmington, DE USA).
WHOLE HUMAN GENOME EXPRESSION DETECTION BY
OLIGO MICROARRAY
RNA from CIN2, CIN3 and normal keratinocytes was hybridized on Agilent whole
human genome oligo microarray (Agilent Technologies, Palo Alto, CA). This microarray
consists of 60-mer DNA probes which have been synthesized in situ and represent 41,000
unique human transcripts. One-colour gene expression was performed according to the
manufacturer’s procedure. Briefly, RNA quality was assessed with Agilent 2100
Bioanalyzer (Agilent Technologies). Low quality RNA (RNA integrity number below 7)
was 6 excluded from microarray analyses. Labelled cRNA was synthesized from 500 ng of
total RNA using the Low RNA Input Linear Amplification Kit (Agilent Technologies) in
the presence of cyanine 3-CTP (Perkin-Elmer Life Sciences, Boston, MA). Hybridizations
were performed at 65°C for 17 hours in a rotating oven. Images at 5 μm resolution were
generated by the Agilent scanner, and Feature Extraction 10.5 software (Agilent
Technologies) was used to obtain the microarray raw data.
MICROARRAY DATA ANALYSIS
30

Microarray raw data were analyzed by use of GeneSpring GX 10 software (Agilent
Technologies). Data transformation was applied to set all the negative raw values at 1.0,
followed by normalization at the 75th percentile. A filter on low gene expression was used
to keep only the probes expressed in at least one sample (flagged as Marginal or Present).
Then, samples were grouped according to their differentiation status and compared.
Differentially expressed genes were selected as having a 1.5-fold expression difference
between their geometrical mean in two or more groups of interest, and a statistically
significant p-value (<0.05) according to ANOVA (analysis of variance) and Benjamini and
Hoechberg correction for reduction of false-positive values. Differentially expressed genes
were employed for sample cluster analysis by use of the Pearson correlation as a measure
of similarity. The microarray raw data have been deposited at ArrayExpress
(http://www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-2019.
REVERSE TRANSCRIPTION QUANTITATIVE REAL-TIME
PCR (RT-QPCR)
The differential expression of selected genes in CIN2, CIN3 and normal
keratinocytes was validated by Reverse Transcription quantitative real-time PCR (RT-
qPCR). Briefly, 300 ng total RNA was reverse transcribed with a random hexamer primer
using High Capacity RNA-to-cDNA Kit (Roche Applied Science, Milan, Italy), according
to the manufacturer’s instructions. qPCR monitoring was performed with the ABI 7500
Fast Real Time PCR system (Roche Applied Science) and Power SYBR Green PCR
Master Mix (Roche Applied Science). The following eight genes were investigated: RARB
(Bohlken et al., 2009), IRF6 (Restivo et al., 2011), TIMP3 (Bernot et al., 2010), APOC1
(Oue et al., 2004), MSX1 (Chetcuti et al., 2011), PHGDH (Liu et al., 2013), C-JUN (De-
Castro et al., 2004) and p63 (Yalcin-Ozuysal1 et al., 2010). The glyceraldehyde-3-
phosphate dehydrogenase gene (GAPDH) was used as an internal control (Chetcuti et al.,
31

2011). Each assay was performed in triplicate. Data analysis was performed with the 2-
⊿⊿Ct
method.
IMMUNOHISTOCHEMICAL (IHC) ANALYSIS
Immunohistochemical (IHC) analysis was performed on paraffin-embedded
specimens from 30 patients with CIN lesions (10 CIN1, 10 CIN2 and 10 CIN3), from 4
with invasive squamous cell cervical cancer, and from 10 with normal cervical tissue. The
IHC staining was performed by use of the Multimeric Detection Kit (Universal DAB
Detection Kit Ultraview, Roche Tissue Diagnostics (CH), on a BenchMark XT
immunostainer (Roche T.D.). Paraffin-embedded tissue sections (4 μm thick) were stained
with mouse monoclonal 3PGDH antibody sc-100317 (Santa Cruz Biotechnology, Santa
Cruz, CA) (dilution, 1:50). HeLa cells, processed as tissues, ie. pelleted, fixed and paraffin-
embedded, were used as positive controls, as recommended by the manufacturer. Staining
intensity and the distribution of staining were assessed by two pathologists. Staining was
graded as negative (no staining) and as weak, moderate, or strong intensity.
SODIUM BISULFITE TREATMENT OF DNA
DNA from CIN2, CIN3 and normal keratinocytes was treated with sodium bisulfite
using the Epitect Bisulfite kit (Qiagen, Milan, Italy) as previously described (Rotondo et
al ., 2013). Sodium bisulfite treatment induces the conversion of unmethylated cytosines of
DNA to uracil, while leaving the 5-methylcytosines unchanged. Samples were then
purified using DNA purification columns (Epitect Bisulfite kit, Qiagen).
32

RARB METHYLATION PCR PRIMERS DESIGN
RARB methylation PCR primers were designed using the MethPrimer informatics
software (Li and Dahiya, 2002). Briefly, MethPrimer is a software for methylation
designing PCR primers for methylation investigation. In a first step, this software is able to
search for all CpGs in a limit input sequence of 5 Mbp. In a second step the software can
design primers within the imput sequence through general parameters changes, like
product Size, primer Tm, etc. (Li and Dahiya, 2002).
BISULFITE TREATED DNA PCR OF RARB AND IRF6
PROMOTER REGION
The methylation assay was performed at the promoter region of RARB and IRF6
genes. 150 ng of sodium bisulfite-treated CIN2, CIN3 and normal keratinocytes DNA was
amplified at the RARB and IRF6 (Botti et al., 2011) loci by Bisulfite treated DNA PCR.
The RARB promoter region studied contained 14 CpG islands whereas the IRF6 promoter
region studied contained 25 CpGs.
DNA CLONING AND SEQUENCING
Amplified products were purified with the QIAquick PCR Purification Kit (Qiagen)
and then cloned with the TOPO TA cloning kit (Invitrogen), using the Turbo Competent E.
coli bacteria strain (EuroClone) and the pCR 2.1-TOPO vector (Invitrogen), according to
manufacturer’s instructions. Selection of bacterial clones containing the fragment of
interest was performed using selective LB growth medium with ampicillin (100 μg/ml,
33

Sigma-Aldrich). For each DNA sample, 10 positive clones were selected for sequencing
analysis. Single clones were sequenced using automated ABI Prism Genetic (Analyzer
Applied Biosystems).
STATISTICAL ANALYSIS
The observed RARB and IRF6 epigenotype frequencies (i.e. methylated CpGs)
were compared between groups using the chi-square trend test with Yates' correction. All
statistical analyses were carried out using Graph Pad Prism version 5.0 for Windows
(Graph Pad, La Jolla, CA, USA). P-values < 0.05 were considered statistically significant.
34

RESULTS
HPV-DNA ANALYSIS OF CIN2, CIN3 AND NORMAL
SPECIMENS AND KERATINOCYTES
In a first step of our experiments, high-risk HPV16 presence in CIN2 and CIN3
specimen was investigated. To this purpose, CIN and normal specimens, were screened by
PCR for HPV DNA sequences. All normal samples were negative for HPV sequences
(Figure 5, A). CIN2 and CIN3 keratinocyte specimens tested positive for HPV16
sequences were selected for the present study (Figure 5, A, B). The DNA fragment sizes
for HPV types are shown in the table 3. Subsequently, in a second step, in order to confirm
HPV detection and genotyping, the same DNA analysis was performed in CIN2 and CIN3
keratinocytes. Previous CIN specimens data were confirmed.
35

HPV type Total length (bp) Length of RsaI restrictionfragments (bp)
30HPV-6 139 42
67
30HPV-11 139 109
30HPV-16 142 42
70
30HPV-18 145 38
77
30HPV-31 142 112
30HPV-33 139 39
70
Table 3. DNA fragment sizes for HPV types spanned by GP5/GP6 primer set and fragment lengths generated by RsaI restriction enzyme digestion.
36

Figure 5. HPV PCR and HPV genotyping. A: the agarose gel shows HPV PCR results obtained from the CIN2, CIN3, and normal keratinocyte DNA specimens. HPV PCR products are only visible in the CIN2 and CIN3 samples (lanes 1 and 2). MW: molecular weight markers are 100 bp (left); HPV-16: PCR positive control; C-: negative control of the PCR reaction without DNA template. B: polyacrylamide gel shows HPV genotyping of HPV PCR products from the CIN2 and CIN3 specimens. The CIN2 (lanes 1 and 2) specimens and CIN3 (lanes 1 and 2) are positive for HPV-16. Fragment lengths are reported in table 3. M.W.: molecular weight markers are 100 bp (M.W. I) and 50 bp (M.W. II) ladder. HPV-6b, -11, -16, -18, -31 and -33: HPV controls.
37

CIN2, CIN3 AND NORMAL KERATINOCYTES CULTURES
SETUP
The protocol is developed in two main steps: (i) CIN and normal keratinocyte
primary culture preparation, (ii) CIN and normal keratinocyte primary colony expansion.
CIN tissue fragments are obtained from CIN patients during the excision of a cone-shaped
portion of preneoplastic tissue under colposcopic examination. Since excided CIN tissue
specimens include surrounding normal tissue, a small fragment of normal cervical tissue
can be taken from each corresponding CIN tissue specimen. The isolation of all the total
cells from the small CIN and normal cervical tissue relies on a collagenase digestion step.
Then, separated cells are washed, counted and seeded in T25 flasks (T1) with DMEM:F12
medium and 10% foetal bovine serum (FBS). Approximately, 1x104-2x105 cells are
isolated from each CIN and normal tissue fragment. After seeding, cells are left to attach
for 48 h; then, any unattached cell suspension is recovered, washed and reseeded for a
further 48 h in new T25 flasks (T2). This re-seeding procedure allows keratinocytes
endowed with slow attachment capability to be rescued later and therefore enables total
primary colony numbers per specimen to be increased. Representative primary
keratinocyte colonies from a CIN3 specimen are shown in figure 6. CIN2 and CIN3 tissues
give the best primary cultures, producing approximately 200-400 colonies/tissue. Normal
cervical tissues give rise to a lower number of colonies, ranging from 50 to 80
colonies/tissue. Frequently, T2 flasks develop many more colonies than T1 flasks. The
duration of the procedure is 3-4 weeks; however, CIN2 and CIN3 keratinocyte colonies
can be well visible in 2-3 weeks. Keratinocytes and fibroblasts are well distinguishable in
primary cell cultures. Indeed, keratinocytes grow forming colonies whereas fibroblasts
proliferate sparsely (Figure 6, A), or in disordered cell clusters or in parallel bundle groups
(Figure 6, C). Keratinocyte colonies grow surrounded by fibroblasts (Figure 6, B, C) or
isolated (Figure 6, A). CIN2, CIN3 and normal primary cultures develop three different
types of colonies, which are classified on their cell content and morphology: type I
colonies contain cells which are irregularly sized, flattened or spindle-shaped and are
loosely spaced (endowed with a low proliferation rate) (Figure 6, A); type II colonies
consist of small, compact and uniform sized cells (endowed with a high proliferation rate)
(Figure 6, B); type III colonies contain smaller, more compact and more uniform size cells
than those of type II (endowed with a very high proliferative rate) (Figure 6, C). CIN2 and
CIN3 primary cultures develop approximately 70% type II/type III colonies and 30% type
I colonies, whereas normal primary cultures 50% type II and 50% type I colonies. Type II
38

and III colonies can be expanded efficiently since they are endowed with a high
proliferative capability.
Figure 6. Keratinocyte primary colonies of a CIN3 culture. (A) A type I colony. Arrow indicates a fibroblast; (B) a type II colony. Arrow indicates a fibroblast; (C) a type III colony. Arrows indicate parallel bundle groups of fibroblasts.
IMMUNOFLUORESCENCE CHARACTERIZATION
Immunofluorescence characterization on round coverslip was assessed in CIN3
(Figure 7) and normal primary and expanded colonies. All CIN3 and normal colonies were
analyzed for K14, K17, and K19 keratins and p63 expression. The CIN3 primary colonies
react strongly for K14 (Figure 7, A) , K17 (Figure 7, B) , K19 (Figure 7, C) as well as for
p63 in all cells (Figure 7, D). In some normal primary colonies K14 and K19 stained
strongly in all cells whereas K19 reacted with a moderate signal in only 50% of cells.
Furthermore, staining for p63 was negative. The CIN3 and normal colonies expanded with
the new mixture DMEM-F12/dKSFM (1:1 ratio) cell culture medium stained strongly for
K14, K17, K19, and p63 (Figure 7, A-D, insert), and for K14 and K19. Expanded colonies
maintain the same staminal and epithelial markers as the primary colonies from which they
originated as shown in figure 7.
39

Figure 7. Immunofluorescence staining. Panels A-D: immunofluorescence staining of type III colonies from a CIN3 culture. Cells react strongly to K14 (A), K17 (B) and K19 (C) keratins (markers of cervical keratinocytes; cytoplasmic signal) and p63 (D) (marker of cervical staminal keratinocytes; nuclear signal). Keratinocytes from type III colonies expanded in the new medium mixture maintain high expression of K14 (panel A, inset), K17 (panel B, inset), K19 (panel C, inset) and p63 (panel D, inset).
IDENTIFICATION OF DIFFERENTIALLY EXPRESSED GENES
IN CIN KERATINOCYTES
To identify genes associated with progression of CIN lesions, it was examined the
gene expression profiles in two CIN2, two CIN3, and two normal keratinocytes obtained
40

by tissue cultures from four CIN patients. The genes modulated during the progression
from normal to CIN2 keratinocytes and from CIN2 to CIN3 keratinocytes were
investigated. One hundred and thirteen genes were significantly down-regulated in CIN2
keratinocytes compared with normal keratinocytes, and 211 genes were down-regulated in
CIN3 compared with CIN2 keratinocytes (P<0.05 and fold-change >2). A consistent
down-regulation from normal to CIN2 keratinocytes, and from CIN2 to CIN3
keratinocytes, was observed for the following 23 genes: INHBB, SLC38A11, IRX2, RARB,
TIMP3, ALDH1A3, ABCB4, EFNB2, C6orf168, ATP2A3, FMO3, FMO4, NCAM2, TLR2,
IRF6, SYNM, HIST1H2AC, FRMPD4, LIMS3, C1orf96, FBXO32, HIST2H2BE and IFIT2
(Table 4 and Figure 8, A). One hundred seventy-five genes were significantly up-regulated
in CIN2 keratinocytes compared with normal keratinocytes, and 94 were up-regulated in
CIN3 keratinocytes compared with CIN2 keratinocytes (P<0.05 and fold-change >2). A
consistent up-regulation from normal to CIN2 keratinocytes and from CIN2 to CIN3
keratinocytes was detected for 14 genes: FOXD2, SCARA5, OLFM1, LPAR1, SFRP2,
MSX1, APOC1, KLF2, TAGLN, SFXN2, TMEM54, PHGDH, SPOCD1, and ARNTL2
(Table 4 and Figure 8, B). Genes consistently up- or down-regulated during transition from
CIN2 to CIN3 were used to perform a hierarchical clustering analysis (Figure 9). Normal,
CIN2 and CIN3 keratinocytes grouped in three different clusters and showed a distinct
gene expression pattern (Figure 9).
41

Figure 8. Genes differentially expressed revealed by microarray analysis..A: Intersection between the 113 genes down-regulated (p<0.05 and FC<0.5) in CIN2 keratinocytes (CIN2) compared with normal keratinocytes and the 221 genes down-regulated (p<0.05 and FC<0.5) in CIN3 keratinocytes (CIN3) compared with CIN2 keratinocytes (CIN2). Twenty-three genes are in common. B: Intersection between the 175 genes up-regulated (p<0.05 and FC>2) in CIN2 keratinocytes (CIN2) compared with normal keratinocytes, and the 94 genes up-regulated (p<0.05 and FC>2) in CIN3 keratinocytes (CIN3) compared with CIN2 keratinocytes (CIN2). Fourteen genes are in common.
_________________________________________________________________________Gene symbol Gene Bank acc. no. Function
42
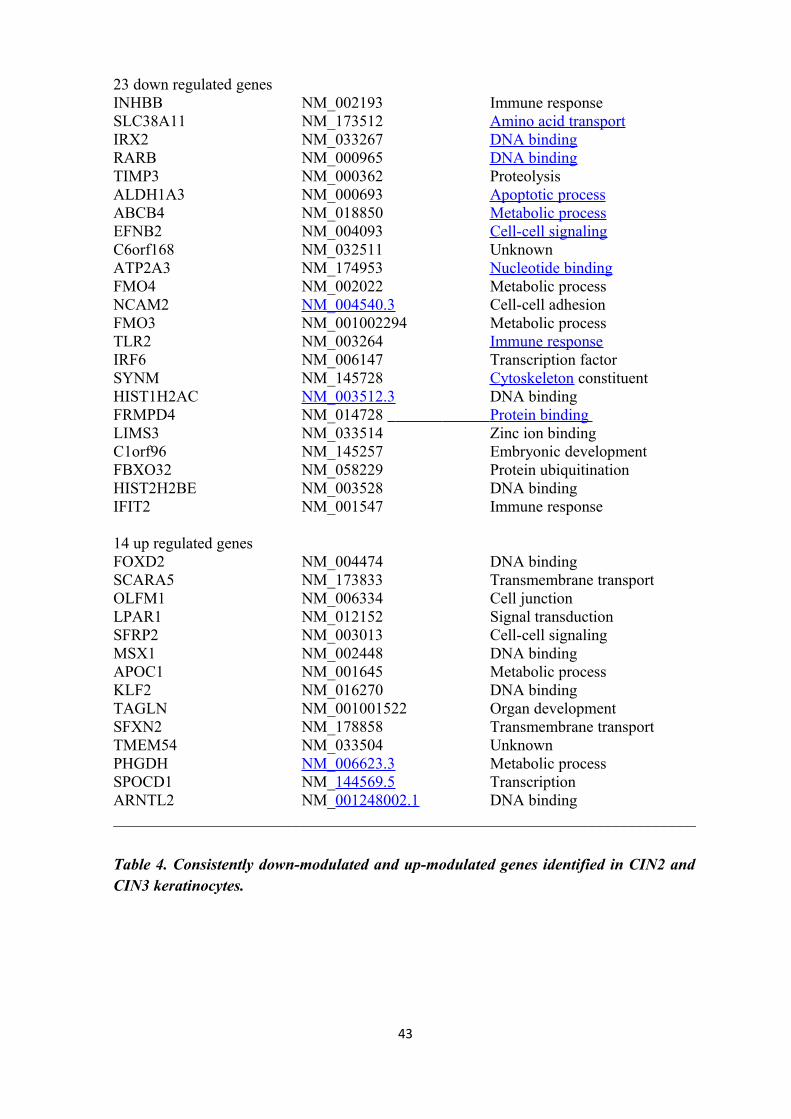
23 down regulated genesINHBB NM_002193 Immune responseSLC38A11 NM_173512 Amino acid transportIRX2 NM_033267 DNA bindingRARB NM_000965 DNA bindingTIMP3 NM_000362 ProteolysisALDH1A3 NM_000693 Apoptotic processABCB4 NM_018850 Metabolic processEFNB2 NM_004093 Cell-cell signalingC6orf168 NM_032511 UnknownATP2A3 NM_174953 Nucleotide bindingFMO4 NM_002022 Metabolic processNCAM2 NM_004540.3 Cell-cell adhesion FMO3 NM_001002294 Metabolic processTLR2 NM_003264 Immune responseIRF6 NM_006147 Transcription factor SYNM NM_145728 Cytoskeleton constituentHIST1H2AC NM_003512.3 DNA bindingFRMPD4 NM_014728 Protein binding LIMS3 NM_033514 Zinc ion bindingC1orf96 NM_145257 Embryonic development FBXO32 NM_058229 Protein ubiquitinationHIST2H2BE NM_003528 DNA bindingIFIT2 NM_001547 Immune response
14 up regulated genesFOXD2 NM_004474 DNA bindingSCARA5 NM_173833 Transmembrane transportOLFM1 NM_006334 Cell junctionLPAR1 NM_012152 Signal transductionSFRP2 NM_003013 Cell-cell signaling MSX1 NM_002448 DNA bindingAPOC1 NM_001645 Metabolic processKLF2 NM_016270 DNA binding TAGLN NM_001001522 Organ developmentSFXN2 NM_178858 Transmembrane transportTMEM54 NM_033504 UnknownPHGDH NM_006623.3 Metabolic processSPOCD1 NM_144569.5 TranscriptionARNTL2 NM_001248002.1 DNA binding_________________________________________________________________________
Table 4. Consistently down-modulated and up-modulated genes identified in CIN2 and CIN3 keratinocytes.
43

Figure 9. Cluster analysis of normal keratinocytes, CIN2 and CIN3 keratinocytes performed in accordance to the expression of commonly modulated genes, both annotated (with gene symbol) and not annotated (N/A). Genes are in rows, samples in columns. The colors of the genes represented on the heat map correspond to the values normalized on miRNA average expression across all samples (see color bar); up-regulated miRNAs are in red, down-regulated miRNAs in green.
VALIDATION OF MICRARRAY DATA BY RT-QPCR
44

To confirm the gene expression data identified by microarray analysis, total RNA
isolated from the two CIN2, two CIN3 and two normal keratinocyte specimens was
subjected to QRT-PCR analysis. Three consistently down-regulated genes from the 23
candidate genes (RARB, IRF6, and TIMP3) and three consistently up-regulated genes from
the 14 candidate genes (APOC1, MSX1, and PHGDH) were chosen. Representative
amplification plot is showed in figure. In accordance with microarray analysis, the mRNA
expression level of RARB, IRF6, and TIMP3 was significantly decreased from that of
normal and CIN2 keratinocytes and from that of CIN2 and CIN3 keratinocytes (Figure 11).
Specifically, the RARB gene was significantly down-regulated by 8.9-fold and 29.2-fold in
CIN2 and CIN3 keratinocytes, respectively, compared to normal keratinocytes (Figure 11).
As for RARB, IRF6 also resulted down-expressed: 3.6-fold and 7.2-fold in CIN2 and CIN3
keratinocytes, respectively, compared to normal keratinocytes (Figure 11). Similarly,
TIMP3 gene resulted significantly down-regulated by 5.6-fold and 13.7-fold in CIN2 and
CIN3 keratinocytes, respectively, compared to normal keratinocytes (Figure 11).
Differences in RARB, IRF6 and TIMP3 mRNA expression levels between CIN2 and CIN3
keratinocytes were statistically significant (p<0.0001).
45
Figure 10. Reappresentative real-time quantitative RT-PCR amplification plot of: (A) RARB, IRF6, TIMP3, and the housekeeping gene GAPDH and (B) APOC1, MSX1, PHGDH and GAPDH.
46
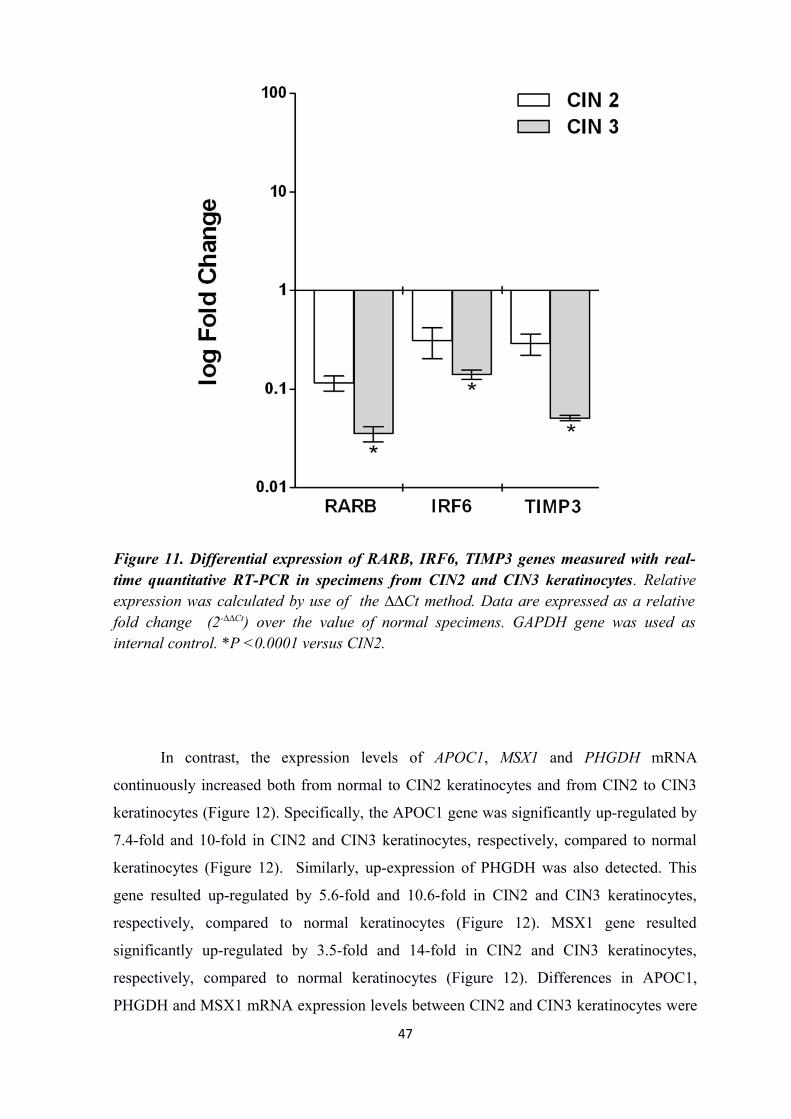
Figure 11. Differential expression of RARB, IRF6, TIMP3 genes measured with real-time quantitative RT-PCR in specimens from CIN2 and CIN3 keratinocytes. Relative expression was calculated by use of the ∆∆Ct method. Data are expressed as a relative fold change (2-∆∆Ct) over the value of normal specimens. GAPDH gene was used as internal control. *P <0.0001 versus CIN2.
In contrast, the expression levels of APOC1, MSX1 and PHGDH mRNA
continuously increased both from normal to CIN2 keratinocytes and from CIN2 to CIN3
keratinocytes (Figure 12). Specifically, the APOC1 gene was significantly up-regulated by
7.4-fold and 10-fold in CIN2 and CIN3 keratinocytes, respectively, compared to normal
keratinocytes (Figure 12). Similarly, up-expression of PHGDH was also detected. This
gene resulted up-regulated by 5.6-fold and 10.6-fold in CIN2 and CIN3 keratinocytes,
respectively, compared to normal keratinocytes (Figure 12). MSX1 gene resulted
significantly up-regulated by 3.5-fold and 14-fold in CIN2 and CIN3 keratinocytes,
respectively, compared to normal keratinocytes (Figure 12). Differences in APOC1,
PHGDH and MSX1 mRNA expression levels between CIN2 and CIN3 keratinocytes were
47

statistically significant (p<0.0001).These findings indicate that the data obtained from the
microarray analysis are reliable and that these 37 genes may be significant contributors in
the progression of high-grade HPV16 CIN lesions.
Figure12. Differential expression of APOC1, MSX1, PHGDH genes measured with real-time quantitative RT-PCR in specimens from CIN2 and CIN3 keratinocytes. Relative expression was calculated by use of the ∆∆Ct method. Data are expressed as a relative fold change (2-∆∆Ct) over the value of normal specimens. GAPDH gene was used as internal control. *P <0.0001 versus CIN2.
IHC ANALYSES
The IHC staining of CIN and normal cervical tissue samples validated PHGDH
protein expression. PHGDH staining was cytoplasmatic. As shown in figure 13, in normal
48

tissues, PHGDH was weakly expressed and only in the proliferative compartment of the
epithelium. In cervical cancer tissue, strong staining was present in all tissue sections.
Strong staining of dysplastic cells was present in 9/10 CIN3, 7/10 CIN2 and 10/10 CIN1
lesions; one out of ten CIN3 and 3/10 CIN2 lesions showed moderate staining of dysplastic
cells (data not shown). PHGDH staining extended to the superficial layers in CIN3 cases,
to the mid layer in CIN2 lesions, and to the lower third of the epithelium in CIN1 lesions,
corresponding to the extension of the dysplastic proliferative compartment in the different
types of lesions. Nondysplastic cells in CIN cases were stained only weakly or not at all.
The specificity of IHC staining for PHGDH protein in CINs and cervical cancers indicates
that PHGDH is likely associated with tumorigenesis.
49

Figure 13. Immunohistochemical analysis of PHGDH in CIN, cervical cancer, and normal tissues. Immunohistochemical analysis with anti-PHGDH polyclonal antibody confirmed the elevated expression of this protein in dysplastic cells from CIN1 (B), CIN2 (C), CIN3 (D) lesions, in tumor cells from invasive carcinoma (E), and in HeLa cells (F) compared with the expression in proliferative cells from normal cervical tissue (A).
RARB, C-JUN, IRF6 AND P63 GENE EXPRESSION ANALYSIS50

In order to explore the expression trend of RARB, c-Jun, IRF6 and p63 mRNAs, 10
CIN2, 10 CIN3, positive for HPV16, and 10 normal keratinocyte specimens was subjected
to QRT-PCR analysis. As reported previously in this work (page 47), the mRNA
expression level of RARB and IRF6, resulted significantly decreased from that of normal
and CIN2 keratinocytes and from that of CIN2 and CIN3 keratinocytes (Figure 14, B)
(P<0.0001). Specifically, RARB was significantly down-regulated by 9.3-fold and by
35.8-fold in CIN2 and CIN3 keratinocytes, respectively, compared to normal keratinocytes
(figure 3, B). Differences in RARB expression levels between CIN2 and CIN3
keratinocytes were statistically significant (p>0.0001). In contrast, the amount of c-Jun
mRNA was found to correlate with the degree of pre-neoplastic CIN lesion: the lowest in
CIN2 (4.3-fold up-regulated compared to normal) and the highest in CIN3 (22 up-
regulated compared to normal) (Figure 14, B) (P<0.0001). Indeed, differences in c-Jun
expression levels between CIN2 and CIN3 keratinocytes were statistically significant
(p>0.0001). As for RARB, IRF6 mRNA was Similarly down-regulated by 2.9-fold and
6.7-fold in CIN2 and CIN3 group, respectively, compared to normal keratinocytes (figure
15, B). Contrariwise, p63 was significantly high expressed by 3.6-fold and by 15-fold in
CIN2 and CIN3 keratinocytes, respectively, compared to normal keratinocytes (figure 15,
B). Differences in IRF6 and p63 expression levels between CIN2 and CIN3 keratinocytes
were statistically significant (p>0.0001).
RARB AND IRF6 PROMOTER METHYLATION ANALYSIS
The methylation status of the RARB and IRF6 loci was investigated by sequencing
analysis of the cloned PCR products. A total of 120 clones were investigated for the DNA
methylation status of RARB and IRF6 loci in bisulfite-treated DNA samples from CIN
keratinocytes. RARB and IRF6 clones derived from two HPV16-CIN2, two HPV16-CIN3
and two normal RARB and IRF6 PCR products (10 from each sample) were randomly
selected for DNA sequencing. The overall distribution of the different clones within CIN
keratinocytes, within CIN2 and normal as well as within CIN3 and normal keratinocytes
was evaluated. Only RARB or IRF6 hypermethylated clones i.e., clones showing 50% (or
more than 50%) methylated CpG islands, were taken into account in this analysis. RARB 51

hypermethylation was detected in 9/11 (45%) of the clones from the CIN2 keratinocytes
and in 16/20 (80%) of the clones from the CIN3 keratinocytes. Whereas, in normal
keratinocytes, RARB hypermethylation was never detected. The difference in RARB
hypermethylation between CIN2 and CIN3 keratinocytes was statistically significant
(P<0.05, Figure 14, A, Figure 15) as well as difference between CIN2 and normal
keratinocytes (p<0.01, Figure 14, A, Figure 15) and CIN3 and normal keratinocytes
(p<0.0001, Figure 14, A, Figure 15). IRF6 hypermethylation was detected in 6/20 (40%) of
the clones from the CIN2 keratinocytes and in 11/20 (55%) of the clones from the CIN3
keratinocytes. Whereas, as for RARB, in normal keratinocytes, it was never detected
(Figure 14, A, Figure 15). No significant differences in IRF6 hypermethylation frequencies
were evaluated between CIN2 and CIN3 keratinocytes (p>0.05, Figure 16, A, Figure 17).
However, the difference between CIN2 and normal keratinocytes (p<0.05, Figure 16, A,
Figure 17) as well as CIN3 and normal keratinocytes (p<0.0001, fig. 3. A) resulted
statistically significant.
52

Figure 14. RARB promoter methylation pattern and RARB and c-Jun differential expression. (A) Bisulfite-PCR sequencing for (A) RARB gene promoter region in one representative Normal, CIN2 and CIN3 cultured keratinocytes. Filled-in and clear circles represent methylated and unmethylated CpG islands, respectively. The CpG islands within the RARB locus is numbered on the upper side of the circles. Each line represents one clone. For the analysis, were considered ten clones per sample. (B) RARB and c-Jun differential expression measured by real-time quantitative RT-PCR in specimens from CIN2 and CIN3 keratinocytes. Relative expression was calculated using the ∆∆Ct method. Data are expressed as a relative fold change (2-∆∆Ct) over the value of normal specimens. *P <0.0001 versus CIN2.
53
Figure 15. Frequency of RARB hypermethylation clones in normal, CIN2 and CIN3 keratinocytes. Only clones showing 50% or more than 50% methylated CpG islands were taken into account in this analysis
54
Figure 16. IRF6 promoter methylation pattern and IRF6 and p63 differential expression. (A) Bisulfite-PCR sequencing for (A) IRF6 gene promoter region in one representative Normal, CIN2 and CIN3 cultured keratinocytes. Filled-in and clear circles represent methylated and unmethylated CpG islands, respectively. The CpG islands within the IRF6 locus is numbered on the upper side of the circles. Each line represents one clone. For the analysis, were considered ten clones per sample. (B) IRF6 and p63 differential expression measured by real-time quantitative RT-PCR in specimens from CIN2 and CIN3 keratinocytes. Relative expression was calculated using the ∆∆Ct method. Data are expressed as a relative fold change (2-∆∆Ct) over the value of normal specimens. *P <0.0001 versus CIN2.
Figure 17. Frequency of IRF6 hypermethylation clones in normal, CIN2 and CIN3 keratinocytes. Only clones showing 50% or more than 50% methylated CpG islands were taken into account in this analysis
55

DISCUSSION
CIN AND NORMAL KERATINOCYTES CULTURES
Recognition of the role of Human Papilloma Virus (HPV) as the etiologic agent in
cervical cancer has focused attention on the mechanisms inducing transformation in
cervical keratinocytes (Zur Hausen, 1996; Togtema et al., 2012; Lindel et al., 2012; Herfs
et al., 2012; Kaczkowski et al., 2012; Tan et al., 2012; Malinowski, 2005). Due to
difficulties in propagating HPV in culture (Angeletti, 2005; Pyeon et al., 2005. McLaughli
and Meyers ,2005). Molecular characterization of naturally infected-HPV keratinocytes
derived from cervical preneoplastic lesions is a promising cell study model for elucidating
HPV-induced cancerogenesis in the human cervix. The HPV transformation process occurs
56

in multiphase steps which are clinically diagnosed as cervical intraepithelial neoplasia
(CIN) -1, 2 and 3 (Mitchell et al., 1996; Schiffman et al., 2007). Two published methods
have been previously described for culturing keratinocytes derived from CIN1-3 lesions as
well as from normal cervical tissues (Freshney and Freshney, 2002; Green, et al., 1977).
With one method, keratinocytes are grown in co-culture with mouse fibroblasts which are
inactivated with mitomycin C or gamma rays (feeder cells) and submerged in culture
medium (Freshney and Freshney, 2002). The alternative approach regards the employment
of defined keratinocyte-specific medium which allows keratinocyte expansion without the
use of fibroblast feeder layers (Green, et al., 1977). However, there are some limitations to
culturing CIN and normal cervical keratinocytes by these methods. Firstly, a large number
of pure CIN or normal keratinocytes, i.e. free from cervical fibroblasts, is needed to start
primary cultures. Whilst an adequate number of normal cervical keratinocytes can be
isolated from cervical epithelium obtained from hysterectomy specimens (Narisawa-Saito
and Kiyono, 2007) CIN tissue specimens are usually very small and do not allow a
sufficient number of pure keratinocytes to be isolated for culturing purposes. Secondly, the
procedure for isolating pure keratinocytes requires several steps, which increase the risk of
cell culture contamination.
Therefore, the development of a rapid and easy method of enabling cultures of CIN
and normal cervical keratinocytes to be grown using small amounts of starting material
was considered useful. The search for a new culturing method began with the observation
that seeding the total number of cells isolated from a complete enzymatic digestion of CIN
or normal cervical tissues, a typical range of morphologically differing keratinocyte
colonies, based on their cell content and general morphology, could be obtained.
Additional studies from our laboratories demonstrated that by sufficiently diluting cells
during primary propagation, CIN and normal keratinocyte colonies develop clonally.
Furthermore, different CIN and normal keratinocyte primary clonal colonies consisted of
small, compact and uniform size cells, indicating that cultured keratinocytes were endowed
with high proliferative potential (Barrandon and Green, 1985, 1987). On the basis of these
observations, a cell culturing method which employs small CIN and normal cervical tissue
fragments to derive primary CIN and normal keratinocyte clonal colonies capable of
further expansion was set up.
Although two different methods for CIN and normal cervical keratinocyte cultures
have been reported, a very few studies regarding cellular and molecular characterization of
naturally infected HPV CIN1-3 or normal cervical keratinocytes, have been published to
57

date. These studies have been carried out in W12 and CIN 612 cells, two naturally HPV16-
infected keratinocyte lines derived from CIN1 lesions (Rader et al., 1990; Bedell et al.,
1991; Gray et al., 2010; Dall et al., 2008). The reason for lacking of researchers on CIN
keratinocytes is that previous culturing methods work well when large pieces of CIN or
normal cervical tissues are employed, but these are rarely obtained from gynaecological
surgery. By using murine fibroblasts as feeder layers, the number of CIN or normal
keratinocytes seeded must maintain a 1:2 density ratio with the murine fibroblasts co-
cultured in vitro (for an example, 1x104/cm2:2x104/cm2) (Rader et al., 1990; Freshney and
Freshney, 2002). Alternatively, the number of CIN or normal keratinocytes seeded must be
at a density of 2x105/cm2 (Barrandon and Green, 1987), using the specific keratinocyte
defined medium without murine fibroblasts. The main procedure for isolating pure
keratinocytes includes dermal sheet elimination, by dispase digestion or tissue scraping,
and two consecutive digestions of cervical epithelial tissues by trypsin-EDTA. Due to the
small size of CIN tissues, CIN keratinocytes are also isolated following migration from
micro-dissected CIN explants. However, the major problem in this latter procedure is that
CIN keratinocytes are rescued at a low number and are frequently highly contaminated by
cervical fibroblasts, which do not allow primary cultures to be started successfully.
The protocol described in this work is very simple compared to previous CIN and
normal tissue culture methods. CIN and normal cervical keratinocyte primary colonies can
be obtained in a single technical passage, i.e. after tissue digestion. Consequently, the time
required is highly reduced. Moreover, our culture method enables CIN and normal
keratinocytes to be grown at a clonal rate by seeding the cells at a high dilution during
primary propagation. This is an important aspect since CIN lesions are clonally
heterogeneous, due to the multiphase steps of HPV-induced carcinogenesis; therefore,
clonal CIN keratinocyte cultures allow different CIN keratinocyte clones of the
preneoplastic specimen to be faithfully reproduced in vitro. Finally, primary CIN and
normal keratinocyte clones can be expanded at highly proliferative rates over different
passages with no sign of differentiation. When taken altogether, these results may provide
important findings on the mechanisms induced by HPV during onset/progression of
cervical neoplasia. CIN2, CIN3 and normal cervical primary culture preparation provides
the onset of a series of problems due mainly to the presence of endogenous microbiological
contaminants, bacteria and fungi, which are naturally present in the cervical region. The
contamination of primary cultures is relevant due to this aspect. Notably, the higher the
cervical preneoplastic grade, the higher the endogen microbial contamination and therefore
58

the risk of culture contamination. CIN2 and CIN3 tissues give the best primary culture
results as the higher the preneoplastic grade of keratinocytes, the higher the ability to
attach and proliferate clonally. On the contrary, keratinocytes from normal tissues attach
slowly and differentiate rapidly and much fewer primary colonies can be obtained than
with high grade CIN2-CIN3 tissues. Nevertheless, the successful rate of normal primary
keratinocyte cultures is approximately 5-6 out of 10 and nearly 10 out of 10 of CIN2-CIN3
primary keratinocyte cultures. This protocol is based on a cellular culture method, which
was used in a recent publication of my lab (Bononi et al., 2012) and employs the total
number of cells, i.e. fibroblasts and keratinocytes, isolated from small tissue fragments of
CIN or normal cervical tissue samples. The isolation of all the cells overcomes the issues
arising from isolating pure populations of keratinocytes including low cell numbers and
exogenous microbial contamination, and avoids keratinocyte loss from starting tissues.
Although the presence of live fibroblasts is considered detrimental to keratinocyte cultures,
due to overgrowth being caused (Freshney and Freshney 2002), this problem is avoided by
seeding the all cells at a very high dilution, i.e. in large culture surfaces. In this way,
primary keratinocyte colonies can be obtained before fibroblasts colonize the cultures. On
the other hand, high cell dilution allows keratinocytes to reach optimal density for clonal
growth (Barrandon and Green, 1985, 1987). Furthermore, were formulated a new medium
mixture which permits CIN and normal keratinocyte colonies to be expanded singly at a
highly proliferative rate for different passages without signs of differentiation.
Immunofluorescence staining showed that CIN and normal keratinocyte primary colonies,
as well as expanded colonies, expressed K14, K17, and K19 indicating their common
origin from basal and parabasal layers of the cervical epithelia (Smedts et al., 1990,
Martens et al., 2004; Akgűl et al., 2007). P63 stained strongly, with nuclear localization, in
CIN3 keratinocytes suggesting that those keratinocytes present stemness characteristics.
Indeed, p63 has previously been detected in stem cells from CIN and normal ectocervical
epithelia (Quade et al., 2001; Martens et al., 2004). Therefore, naturally HPV infected
keratinocyte clones have the potential to provide valuable insights into the mechanisms
induced by HPV during cervical cancerogenesis. Since cultured cells represent a useful
model for investigating the onset/progression of the carcinogenesis processes (Goldstein et
al., 2011; Karst and Drapkin, 2012) and cellular and molecular studies on naturally HPV-
infected keratinocytes have been poorly reported, our protocol may provide opportunities
for exploring mechanisms which occur during CIN transition, as well as gene expression
59

changes in progression toward cervical cancer in vitro, which to date is far from having
been elucidated (Woodman et al., 2007; Galloway, 2009)
GENE EXPRESSION CHANGES AND CIN PROGRESSION
In this study, gene expression changes were investigated, for the first time, in
naturally HPV16-infected CIN2 and CIN3 keratinocytes by microarray analysis. Since the
majority of high-grade HPV16-CIN lesions are the result of 2-dimensional intra-mucosal
extension of a single monoclonal cell population infected by the oncogenic HPV (Ueda et
al., 2003), two CIN keratinocyte colonies, representative of all neoplastic keratinocytes
comprising the CIN lesion, were chosen from each CIN tissue culture for the analysis of
microarray. Moreover, in order to avoid the risk of a selection bias inherent in any long
term in vitro growth, were collected CIN and normal keratinocyte colonies from primary
cultures. Were used DNA microarray technology in order to compare changes of gene
expression, which characterize two key stages of progression of HPV16-cervical lesions in
vivo. It was encouraging that the hierarchical clustering analysis of the data neatly grouped
CIN keratinocytes together according to their status: CIN2 or CIN3. Moreover, the finding
that our dataset contained many expected gene expression changes (such as those involving
cell proliferation and differentiation) affirmed validity of the dataset. Therefore, the results
obtained in CIN2 and CIN3 keratinocytes by microarray analysis likely parallel the
molecular changes that underlie progression to high-grade HPV16 CIN. Notably, the
number of genes down-regulated in CIN3 keratinocytes compared with the number in
CIN2 keratinocytes (n=211) was almost double that in normal keratinocytes compared
with CIN2 keratinocytes (n=113). In contrast, 175 genes were increased significantly in
CIN2 compared with normal keratinocytes, and 94 genes were increased in CIN3
compared with CIN2 keratinocytes. These findings suggest that the majority of genes
down-regulated and up-regulated during the progression of cervical neoplasia affect the
transition from CIN2 to CIN3 lesions and from normal to CIN2 lesions, respectively. 60

Twenty-three under-expressed and 14 over-expressed genes, which may play an important
role in CIN progression, were selected. These genes are associated with HPV-mediated
transformation, including the down-regulation of antiviral immune response-inducible
genes and up-regulation of proliferation-related genes. Indeed, INHBB, TLtlrR2, TIMP3,
RARB, IFIT2 and EFNB2 were consistently under-expressed, whereas APOC1 and
PHGDH were consistently overexpressed in CIN2 and CIN3 keratinocytes. Notably, most
of the down-regulated genes, such as INHBB, TLR2, TIMP3 and RARB, are the target of
promoter methylation in various tumours (De Oliveira et al., 2011; Gu et al., 2008;
Karpinski et al., 2011; Wang et al., 2005), including HPV-related cancers (Stephen et al.,
2010), high-grade CIN lesions (Terra et al., 2007), and HPV cervical cancers (Sun et al.,
2011), suggesting an important role of these genes in tumour development. Therefore, it is
possible that the consistent decrease in mRNA expression in CIN2 and CIN3 keratinocytes
accounts for increasing CpG-island methylations at the gene promoter level. In line with
this, HPV16 can induce cellular gene promoter hypermethylation (Leonard et al., 2012).
These findings this findings leaded me to explore explore, in the next step of my
investigation, the putative link between gene expression, at mRNA levels, and gene
promoter methylation status in high-grade CIN keratinocytes. A major feature of CIN2 and
CIN3 keratinocytes is the consistent overexpression of genes that play a role in tumour
invasiveness. Indeed, consistent up-regulation of LPAR1, a pro-angiogenesis gene
(Hayashi et al., 2012), and ARNTL2 (Mazzoccoli et al., 2012), MSX1 (Chetcuti et al.,
2011), and TAGLN (Rho et al., 2009), which are pro-invasive genes, was present in
progression from CIN2 to CIN3 keratinocytes. These findings are in agreement with
results of a recent microarray study in which transition to CIN2 stage coincided with the
activation of pro-angiogenesis pathways, whereas the transition to CIN3 and then to
invasive cancer was characterized by a pro-invasive gene expression (Gius et al., 2007).
Therefore, these up-regulated genes detected in CIN2 and CIN3 keratinocytes likely are
associated with cervical cancer and may be prognostic markers for CIN progression. An
interesting characteristic of CIN2 and CIN3 keratinocytes is the disruption of cell-
differentiation programs. Indeed, a series of differentiation-induced genes, such as
ALDH1A3 (Muzio et al., 2012), IRX2 (Lewis et al., 1999), IRF6 (Botti et al., 2011),
SYNM (De Souza Martins et al., 2011), and ATP2A3 (Korosec et al., 2009), were down-
regulated in late-stage CIN keratinocytes. On the other hand, stemness-related genes, such
as KLF2 (Gillich et al., 2012), were increased in the transition of CIN keratinocytes from
CIN2 to CIN3. These data are in accordance with the oncogenic activities of HPV16 E6-
61

E7 viral oncoproteins that dysregulate the cell cycle through differentiation program
disruption (Rosty et al., 2005). Finally, two up-regulated genes, SCARA5 and SFRP2
change in opposite directions in HPV-related tumors or other cancers (Huang et al., 2010).
Further investigations are needed to clarify the role of these genes in CIN progression. I
focused on one of the up-regulated genes, PHGDH, which encodes an enzyme that controls
the flux from glycolysis into the serine biosynthesis pathway, although a novel non-
metabolic role of PHGDH also has been reported. Various studies have found increasing
PHGDH expression in human astrocytomas according to increasing tumour grade (Liu et
al., 2013), and PHGDH recently was found overexpressed in cervical cancer (Locasale et
al., 2011). These observations prompted me to investigate PHGDH expression in CINs and
invasive cancer in order to explore its contribution to cervical carcinogenesis. Expression
of PHGDH mRNA was continuously increased during conversion from normal to CIN2
keratinocytes and from CIN2 to CIN3 keratinocytes. Consistently, a strong staining of
PHGDH was observed in dysplastic cells from almost all CIN lesions and in tumour cells
from all cervical cancer tissues, whereas expression was less and only in the proliferative
compartment of the epithelium from normal tissues, a finding which suggests that the
protein expression of this gene is enhanced according to the degree of malignant
transformation. In conclusion, this microarray study revealed 37 down-expressed or over-
expressed genes which may contribute to progression of CIN. mRNA expression of one of
the up-regulated genes, PHGDH, was significantly greater in CIN2 keratinocytes than in
normal keratinocytes and in CIN3 keratinocytes than in CIN2 keratinocytes. In addition,
protein expression of PHGDH increased from CIN1 to cancer according to the degree of
malignant transformation. Thus, PHGDH likely plays an important role in the initiation
and progression of cervical tumorigenesis and may be a prognostic marker for progression
of CIN to invasive cancer.
GENES PROMOTER METHYLATION AND CIN PROGRESSION
Together with genetics changes, epigenetic alteration, such as improper DNA
promoter methylation, have an important role in cancer onset and progression. Indeed, the
CpGs methylation status of some gene promoter regions is a significant prognostic variable 62

for various tumor types, including cervical SCC (Van Noesel et al., 2002; Shivapurkar et
al., 2004). Particularly, aberrant promoter methylation play a important role in
downregulation of tumor suppressor genes in SCC. This phenomenon could lead to cell
transformation or cervical SCC formation. As previously reported, a certain number of
tumor suppressor genes such as p16 (Nakashima et al., 1999), CCNA1 ( Yang et al., 2010)
PTEN (Cheung et al., 2004), E-cadherin (Widschwendter et al., 2004) and APC (Virmani
et al., 2001) present improper hypermethylation in cervical carcinogenesis. Furthermore,
p16 hypermethylation was detected progressively higher in CIN pre-cancerous lesions
progression (Huang et al., 2011). Using DNA cloning and bisulfite sequencing, it was
investigated the epigenetic status of RARB and IRF6 promoter region in neoplastic
progression of CIN pre-cancerous lesions. These genes have been suggested to be
implicated as actual or potentitumour suppressor genes, and are aberrantly methylations of
their promotereds were detected in variousdifferent cancers (Piperi et al., 2010; Botti et
al., 2011). The RARB gene encodes for retinoic acid receptor beta, a member of the
thyroid-steroid hormone receptor superfamily of nuclear transcriptional regulators, which
binds the retinoic acid. Interaction of retinoic acid with RARB receptor induces cell growth
and differentiation as well as embryonic morphogenesis (Soprano et al., 2004). RARB
improper methylation was detected in several tumors, like breast (Park et al., 2012),
prostate (Serenaite et al., 2015) and ovarian cancer (Flanagan et al., 2013) and
oropharyngeal SCC (Van Kempen et al., 2014). Moreover, methylation defects in RARB
promoter region were detected in CIN3 biopsies (Vasiljević et al., 2014) and in primary
human foreskin keratinocytes transfected with episomial form of HPV16 (Leonard et al.,
2013).
This study shows,for the first time, that an extensive methylation defects, i.e.
hypermethylation, occur at the RARB tumor suppressor gene promoter in naturally
HPV16-infected CIN2 and CIN3 keratinocytes. Similar data were obtained data were
showed in HPV16-positive exfoliated cervical CIN2 and CIN3 samples (Chang et al.,
2011), and in cervical SCC samples (Narayan et al., 2003). In the present study,
hypermethylation of RARB gene promoter was significantly higher in CIN23 keratinocytes
compared to CIN23 keratinocytes (p<0.05). ThereforeFurthermore, these results indicate
that methylation defects at the RARB locus frequently occur in CIN keratinocytes,
suggesting that increased methylation of the gene promoter is predictive ofwith CIN2
progression towards >CIN3 progression. It can be hypothesized that the presence of
HPV16 could be involved in this improper promoter methylation, as previously reported
63

(Leonard et al., 2012). In my study, in CIN2 and CIN3 keratinocytes wereas found a
progressive down-regulation of RARB and a progressive up-regulation of the proto-
oncogene c-Jun transcript, respectively (p<0.0001). This negative correlation between
RARB and c-JUN was previously reported in Cervical Carcinoma HeLa Cells (De-Castro
et al., 2004). It is conceivable that the progressive RARB hypermethylation may cause a
consequent RARB mRNA down-regulation and this reduction of expression could enhance
c-JUN expression (De-Castro et al., 2004). As previously mentioned, c-JUN expression is
increased in in SV40 immortalized human epidermal keratinocytes, transfected with HPV-
16 E5 coding sequence (Chen et al., 1996). Moreover, it is know that HPV-16 E5
suppresses the expression of tumor suppressor gene p21 by c-JUN activation inducing
human keratinocytes immortalization. (Tsao et al., 1996). This might be one of the
mechanisms by which HPV-16 stimulates cell proliferation by c-Jun activation and p21
inactivation. It is conceivable that the same molecular effect could occur in naturally HPV-
16-positive CIN keratinocytes used in my study.
As for RARB, this study shows that extensive methylation defects, i.e.
hypermethylation, occur also at the IRF6 gene promoter in HPV16-positive CIN2 and
CIN3 keratinocytes. This gene encodes for a member of the interferon regulatory
transcription factor (IRF) family. The encoded IRF6 protein regulates craniofacial
development and epidermal proliferation and may be a transcriptional activator.
ReFurthermore, recently findings suggest have suppose that IRF6 exhibits tumor
suppressor activity IRF6 (Botti et al., 2011). Despite the epigenetics data of the present
study show extensive DNA hypermethylation in IRF6 gene promoter region in CIN
keratinocytes compared to normal, this trend was not in progression between CIN2 and
CIN3 keratinocytes. However, it is appreciable a methylation increase, even though not
statistically significant (p>0.05), between these two groups. These epigenetics data are
consistent with a work conducted by Botti and colleagues, where IRF6 gene promoter
region was aberrantly methylated in primary SCCs cultures and SCC cell lines (Botti et al.,
2011). Furthermore, in the present study was observed a reduced IRF6 expression in the
same cell cultures in association with hypermethylation of whose its IRF6 promoter region
is hypermethylated. Accordingly, in this work, In my work, was detected a similar IRF6
expression was detected to be reduced trend in HPV16-positive CIN2 and CIN3
keratinocytes were IRF6 promoter resulted hypermethylated. Specifically, was
evaluatedIndeed, it was observed a progressively down-regulation of IRF6 transcript from
normal to CIN2 as well as from CIN2 to CIN3 (p<0.0001) in HPV16-positive
64

keratinocytes whit progressive CIN2 to CIN3 IRF6 promoter region hypermethylation.
Therefore, it is conceivable that the hypermethylation of IRF6 promoter region may cause
a consequent IRF6 transcript reduction in CIN keratinocytes. It is known that IRF6 is
involved in squamous differentiation (Biggs et al., 2012). This biological effect is limited
by p63, a protein present in squamous epithelia that promotes renewal of basal
keratinocytes (Okuyama et al., 2007). Furthermore, IRF6 is a p63 transcriptional target that
limits keratinocyte proliferation by inducing p63 proteasome-mediated degradation
(Moretti et al., 2010). In the present work an increased of p63 expression was detected an
increased p63 expression in HPV16-positive CIN2 and CIN3 keratinocytes compared to
normal. This expression was confirmedevaluated also by immunofluorescence, where p63
resulted highly expressed in CIN3 keratinocytes with nuclear localization.
MoreoverFurthermore, p63 transcript up-regulation resulted in progression from CIN2 to
CIN3 and in progressive negative correlation with IRF6 expression (p<0.0001). It is
conceivable that the deregulation of this molecular loop, altering the critical balance
between differentiation and proliferation during CIN progression, as previously reported in
Neck Squamous Cell carcinoma (Nicolas Stransky et al., 2011), ectodermal dysplasias
(Moretti et al., 2010) and cleft palate (Thomason et al., 2010). These findings led us to
hypothesize that RARB and IRF6 gene promoter hypermethylation could be a potential
prognostic epigenetic marker for HPV16-positive CIN progression. Such changes might
therefore be used as markers of cervical neoplasia, either alone or in conjunction with other
molecular prognostic markers. Furthermore, RARB, c-Jun, IRF6 and p63 deregulation may
contribute to progression of CIN pre-cancerous lesions.
CONCLUSIONS
In conclusion, in my study the molecular mechanisms occurring in CIN lesions
progression were investigated. In particular, it was investigated gene expression profiles
and methylation status of gene promoters in a novel study model, i.e. primary colonies of
CIN2 and CIN3 keratinocytes derived from HPV16-CIN2 and CIN3 lesions. Gene
expression analysis revealed 37 down-expressed or over-expressed genes which may
contribute to CIN progression. One of these genes, the phosphoglycerate dehydrogenase,
which resulted over-expressed at both mRNA and protein level in CIN2 and CIN3
65

keratinocytes and in CIN2, CIN3 and cancer tissues, respectively, is likely to be associated
with tumorigenesis and may be a potential prognostic marker for CIN progression.
Aberrant promoter hypermethylation of RARB and IRF6 genes also may be a potential
epigenetic prognostic marker for CIN progression.
REFERENCES
Akgűl B, Ghali L, Davies D, Pfister H, Leigh IM, Storey A. HPV8 early genes modulate differantiation and cell cycle of primary human adult keratinocytes. Exp Dermatol. 2007. 16:590–9.
Akerman GS, Tolleson WH, Brown KL, Zyzak LL, Mourateva E, Engin TS, Basaraba A, Coker AL, Creek KE, Pirisi L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signalling shortens the life span of normal human keratinocytes. Cancer Res. 2001; 61: 3837–43.
Alazawi W, Pett M, Arch B, Scott L, Freeman T, Stanley MA, Coleman N. Changes in Cervical Keratinocyte Gene Expression Associated with Integration of Human Papillomavirus 16. Cancer Res. 2002; 62: 6959–65.
Angeletti PC. Replication and encapsidation of papillomaviruses in Saccharomyces cerevisiae. Methods Mol Med. 2005; 119, 247-60.
Arroyo M, Bagchi S, Raychaudhuri P. Association of the human papillomavirus type 16 E7 protein with the S-phase specific E2F-cyclin A complex. Mol Cell Biol.1993;13:6537–46.
66

Arvantis DA, Spandidos DA. Deregulation of the G1/S phase transition in cancer and squamous intraepithelial lesions of the uterine cervix: A case control study. Oncol Rep. 2008; 20: 751-60.
Ashrafi GH, Haghshenas MR, Marchetti B, O’Brien PM, Campo MS. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer. 2005; 113: 276-83.
Attila T. The Promise and the Problems of Epigenetics Biomarkers in Cancer. Expert Opin Med Diagn. 2011; 5: 375–9
Barbosa MS, Lowy DR, Schiller JT. Papillomavirus polypeptides E6 and E7 are zinc-binding proteins. J Virol. 1989; 63:1404-7.
Barbosa MS, Edmonds C, Fisher C, Schiller JT, Lowy DR, Vousden KH. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 1990; 9:153–160.
Barrandon Y, Green H. Cell-Size as a Determinant of the Clone-Forming Ability of Human Keratinocytes. Proc Natl Acad Sci U S A. 1985; 82:5390-4.
Barrandon Y, Green H. Three clonal types of keratinocytes with different capacities for
Multiplication. Proc Natl Acad Sci USA. 1987; 84:2302–6.
Bedell MA, Hudson JB, Golub TR, Turyk ME, Hosken M, Wilbanks GD, Laimins LA. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J Virol. 1991; 65, 2254-60
Bernot D, Barruet E, Poggi M, Bonardo B, Alessi MC, Peiretti F. Down-regulation of Tissue Inhibitor of Metalloproteinase-3 (TIMP-3) Expression Is Necessary for Adipocyte Differentiation. J Biol Chem. 2010; 285:6508-14.
Bertelsen BI, Steine SJ, Sandvei R, Molven A, Laerum OD. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int J Cancer. 2006; 118:1877-83.
67

Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol. 1988; 20;203:971-83.
Biermann K, Steger K. Epigenetics in male germ cells. J Androl. 2007; 28:466-80.
Biggs LC1, Rhea L, Schutte BC, Dunnwald M. Interferon regulatory factor 6 is necessary, but not sufficient, for keratinocyte differentiation. J Invest Dermatol. 2012; Jan;132:50-8.
Bird A, Tate P, Nan X, Campoy J, Meehan R, Cross S, Tweedie S, Charlton J, Macleod D. Studies of DNA methylation in animals. J Cell Sci Suppl. 1995;19:37-9.
Bohlken A, Cheung BB, Bell JL, Koach J, Smith S, Sekyere E, Thomas W, Norris M, Haber M, Lovejoy DB, Richardson DR, Marshall GM. ATP7A is a novel target of retinoic acid receptor beta2 in neuroblastoma cells. Br J Cancer. 2009;100:96-105.
Bononi I, Bosi S, Bonaccorsi G, Marci R, Patella A, Ferretti S, Tognon M, Garutti P, Martini F. Establishment of keratinocyte colonies from small-sized cervical intraepithelial neoplasia specimens. J Cell Physiol. 2012; 227:3787-95.
Borger DR, Mi Y, Geslani G, Zyzak LL, Batova A, Engin TS, Pirisi L, Creek KE. Retinoic acid resistance at late stages of human papillomavirus type 16-mediated transformation of human keratinocytes arises despite intact retinoid signaling and is due to a loss of sensitivity to transforming growth factor-β. Virology. 2000; 270: 397–407.
Bosch FX, de Sanjosé S. Human papillomavirus in cervical cancer. Curr Oncol Rep. 2002; 4:175-83.
Bossis I, Roden RB, Gambhira R, Yang R, Tagaya M, Howley PM, Meneses PI. Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J. Virol. 2005; 79, 6723–31.
68

Botti E, Spallone G, Moretti F, Marinari B, Pinetti V, Galanti S, De Meo PD, De Nicola F, Ganci F, Castrignanò T, Pesole G, Chimenti S, Guerrini L, Fanciulli M, Blandino G, Karin M, Costanzo A. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc Natl Acad Sci U S A. 2011;
Bravo IG, Alonso A. Phylogeny and evolution of papillomaviruses based on the E1 and E2 proteins. Virus Genes. 2007; 60:249-62.
Brehm A, Nielsen SJ, Miska EA, Mccance DJ, Reid JL, BannisterAJ, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999; 18:2449–2458.
Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014-21.
Castle PE, Schiffman M, Wheeler CM, Solomon D. Evidence for frequent regression of cervical intraepithelial neoplasia-grade 2. Obstet Gynecol. 2009;113(1):18-25.
Chang YE, Laimins LA. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol. 2000; 74:4174–82.
Chang S, Reimers LL, and Burka RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol. 2011 Apr; 121(1): 59–63.
Chao A, Wang TH, Lee YS, Hsueh S, Chao AS, Chang TC, Kung WH, Huang SL, Chao FY, Wei ML, Lai CH. Molecular characterization of adenocarcinoma and squamous carcinoma of the uterine cervix using microarray analysis of gene expression. Int J Cancer. 2006; 119:91–8.
Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002; 99:16916-21.
69

Chen Y, Miller C, Mosher R, Zhao X, Deeds J, Morrissey M, Bryant B, Yang D, Meyer R, Cronin F, Gostout BS, Smith-McCune K, Schlegel R. Identification of Cervical Cancer Markers by cDNA and Tissue Microarrays. Cancer research. 2003; 63: 1927–1935.
Cheng Q, Lau WM, Tay SK, Chew SH, Ho TH, Hui KM. Identification and characterization of genes involved in the carcinogenesis of human squamous cell cervical carcinoma. Int. J. Cancer. 2002; 98, 419–426.
Chen SL, Tsai TZ, Han CP, Tsao YP. Mutational analysis of human papillomavirus type 11 E5a oncoprotein. J Virol. 1996 Jun;70(6):3502–8.
Chetcuti A, Aktas S, Mackie N, Ulger C, Toruner G, Alkan M, Catchpoole D.. Expression profiling reveals MSX1 and EphB2 expression correlates with the invasion capacity of Wilms tumors. Ped blood & cancer. 2011; 57:950-957.
Cheung TH, Lo KW, Yim SF, Chan LK, Heung MS, Chan CS, Cheung AY, Chung TK, Wong YF. Epigenetic and genetic alternation of PTEN in cervical neoplasm. Gynecol Oncol. 2004;93:621-7.
Choy MK, Movassagh M, Goh HG, Bennett MR, Down TA, Foo RS. Genome-wide conserved consensus transcription factor binding motifs are hyper-methylated. BMC Genomics. 2010; 11:519.
Chung CS, Hsiao JC, Chang YS, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol. 1998; 72:1577–85.
Creek KE, Geslani G, Batova A, Pirisi L. Progressive loss of sensitivity to growth control by retinoic acid and transforming growth factor-β at late stages of human papillomavirus type 16-initiated transformation of human keratinocytes. Adv Exp Med Biol. 1995; 375: 117–35.
70

Crusius K, Auvinen E, Steuer B, Gaissert H, Alonso A. The human papillomavirus type 16 E5-protein modulates ligand-depedent activation of the EGF receptor family in the human epithelial cell line HaCaT. Exp Cell Res. 1998; 241: 76-83.
Culp TD, Christensen ND. Kinetics of in vitro adsorption and entry of papillomavirus virions. Virology. 2004; 319:152-61.
Dall KL, Scarpini CG, Roberts I, Winder DM, Stanley MA, Muralidhar B, Herdman MT, Pett MR, Coleman N. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res. 2008; 68:8249-59.
Davies R, Hicks R, Crook T, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993; 67:2521-8.
Day PM, Lowy DR, Schiller JT. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology. 2003; 307:1–11.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011 May 15; 25:1010-22
De-Castro Arce J, Soto U, Van Riggelen J, Schwarz E, zur Hausen H, Rösl F. Ectopic expression of nonliganded retinoic acid receptor beta abrogates AP-1 activity by selective degradation of c-Jun in cervical carcinoma cells. J Biol Chem. 2004; 29;279:45408-16.
De Oliveira NF, Andia DC, Planello AC, Pasetto S, Marques MR, Nociti FH, Jr., Line SR, De Souza AP. TLR2 and TLR4 gene promoter methylation status during chronic periodontitis. J Clin Periodontol. 2011; 38:975-83.
De Souza Martins SC, Agbulut O, Diguet N, Larcher JC, Paulsen BS, Rehen SK, Moura-Neto V, Paulin D, Li Z, Xue Z. Dynamic expression of synemin isoforms in mouse embryonic stem cells and neural derivatives. BMC cell biol. 2011; 12:51.
71

De Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004; 324:17-27
De Villiers EM, Gunst K, Stein H, Scherübl H. Esophageal squamous cell cancer in patients with head and neck cancer: Prevalence of human papillomavirus DNA sequences. Int J Cancer. 2004;109:253-8.
Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. The biology and life-cycle of human papillomaviruses. Vaccine. 2012; 20:30.
Duffy CL, Phillips SL, Klingelhutz AJ. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology. 2003; 314:196–205.
Dyson N, Buchkovich K, Whyte P, Harlow E. Cellular proteins that are targetted by DNA tumor viruses for transformation. Princess Takamatsu Symp. 1989; 20:191-8.
Dyson N, Howley PM, Münger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989; 17;243:934-7.
Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle.Proc. Natl. Acad. Sci. USA, 2000; 97: 10002-7.
Duensing S, Münger K. Centrosome abnormalities and genomic instability induced by human papillomavirus oncoproteins. Prog Cell Cycle Res. 2003; 5:383-91.
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004; 429:457-63.
72

Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 200;16 Spec No 1:R50-9.
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999; 59:793-7.
Evander M, Frazer IH, Payne E, Mei Qi Y, Hengst K and McMillan NAJ. Identification of the α6 integrin as a candidate receptor for papillomaviruses. J Virol. 1997; 71: 2449–2456.
Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ, Jones PA. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005; 33:e176.
Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006; 25:5277-85
Firzlaff JM, Galloway DA, Eisenman RN, Luscher B. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989;1:44–53.
Flanagan JM, Wilhelm-Benartzi CS, Metcalf M, Kaye SB, Brown R. Association of somatic DNA methylation variability with progression-free survival and toxicity in ovarian cancer patients. Ann Oncol. 2013; 24:2813-8.
Freshney RI, Freshney MG. Culture of epithelial cells (Wiley-Liss, New York, 2002).
Funk JO, Waga S, Harry JB, Espling E, Stillman B, Galloway DA. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997; 11:2090-100.
Galloway DA. Human papillomaviruses: a growing field. Genes Dev. 2009; 23:138-42.
73

Gardiol D, Kühne C, Glaunsinger B, Lee SS, Javier R, Banks L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene. 1999 Sep 30;18:5487-96.
Genther Williams SM, Disbrow GL, Schlegel R, Lee D, Threadgill DW, Lambert PF. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 2005; 65:6534-42.
Gewin L, Galloway DA. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J Virol. 2001; 75:7198-201.
Gewin L, Myers H, Kiyono T, Galloway DA. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004 Sep 15;18:2269-82.
Gillich A, Bao S, Grabole N, Hayashi K, Trotter MW, Pasque V, Magnusdottir E, Surani MA. Epiblast stem cell-based system reveals reprogramming synergy of germline factors. Cell stem cell. 2012; 10:425-439.
Gius D1, Funk MC, Chuang EY, Feng S, Huettner PC, Nguyen L, Bradbury CM, Mishra M, Gao S, Buttin BM, Cohn DE, Powell MA, Horowitz NS, Whitcomb BP, Rader JS. Profiling Microdissected Epithelium and Stroma to Model Genomic Signatures for Cervical Carcinogenesis Accommodating for Covariates. Cancer Res. 2007; 67: 7113-23.
Goldstein AS, Drake JM, Burnes DL, Finley DS, Zhang H, Reiter RE, Huang J, Witte ON. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat Protoc. 2011; 6:656-67.
Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene. 2000;19:5270-80.
74

Goodwin EC, Naeger LK, Breiding DE, Androphy EJ, DiMaio D. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J Virol. 1998; 72:3925-34.
Gray RH1, Serwadda D, Kong X, Makumbi F, Kigozi G, Gravitt PE, Watya S, Nalugoda F, Ssempijja V, Tobian AA, Kiwanuka N, Moulton LH, Sewankambo NK, Reynolds SJ, Quinn TC, Iga B, Laeyendecker O, Oliver AE, Wawer MJ. Male circumcision decreases acquisition and increases clearance of high-risk human papillomavirus in HIV-negative men: a randomized trial in Rakai, Uganda. J Infect Dis. 2010; 201:1455–62.
Gray E1, Pett MR, Ward D, Winder DM, Stanley MA, Roberts I, Scarpini CG, Coleman N. In vitro progression of human papillomavirus 16 episome-associated cervical neoplasia displays fundamental similarities to integrant-associated carcinogenesis. Cancer Res. 2010; 70:4081-91.
Green H, Rheinwald JG, Sun TT. Properties of an epithelial cell type in culture: the epidermal keratinocyte and its dependence on products of the fibroblast. Prog Clin Biol Res. 1977 17:493-500.
Greger V1, Passarge E, Höpping W, Messmer E, Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989; 83:155-8.
Gu P, Xing X, Taenzer M, Roecken C, Weichert W, Ivanauskas A, Pross M, Peitz U, Malfertheiner P, Schmid RM, Ebert M. Frequent loss of TIMP-3 expression in progression of esophageal and gastric adenocarcinomas. Neoplasia. 2008;10:563-72.
Guilleret I, Benhattar J. Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp Cell Res. 2003; 289:326-34.
Hayashi M, Okabe K, Kato K, Okumura M, Fukui R, Fukushima N, Tsujiuchi T. Differential function of lysophosphatidic acid receptors in cell proliferation and migration of neuroblastoma cells. Cancer letters. 2012; 316:91-96
75

Herfs M, Yamamoto Y, Laury A, Wang X, Nucci MR, McLaughlin-Drubin ME, Münger K, Feldman S, McKeon FD, Xian W, Crum CP. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc Natl Acad Sci U S A. 2012; 109:10516-21.
Horvat R, Herbert A, Jordan J, Bulten J, Wiener HG. Techniques and quality assurance guidelines for histopathology. In: Arbyn M, Anttila A, Jordan J, Ronco G, Schenck U, Segnan N,Wiener HG, Herbert A, Daniel J, von Karsa L, editors. European guidelines for quality assurance in cervical cancer screening, 2nd edition. 2008. Belgium: International Agency for Research on Cancer. European Communities. pp 171–189
Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384:324-34.
Huang LW, Pan HS, Lin YH, Seow KM, Chen HJ, Hwang JL. P16 methylation is an early event in cervical carcinogenesis. Int J Gynecol Cancer. 2011; 21:452-6.
Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, Wang YP, Xiao HS, Han ZG.Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Inves. 2010; 120:223-241.
Illingworth RS, Bird AP. CpG islands--'a rough guide'. FEBS Lett. 2009; 583:1713-20.
Imoto I, Tsuda H, Hirasawa A. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 2002; 62: 4860-66.
Jabbari K, Bernardi G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene. 2004; 333:143-9.
76

Jiang M, Milner J. Selective silencing of viral gene E6 and E7 expression in HPV-positive human cervical carcinoma cells using small interfering RNAs. Methods Mol Biol. 2005; 292: 401-20.
Jeon JH, Choi KH, Cho SY, Kim CW, Shin DM, Kwon JC, Song KY, Park SC, Kim IG. Transglutaminase 2 inhibits Rb binding of human papilloma-virus E7 by incorporating polyamine. EMBO J. 2003; 22:5273–82.
Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994; 13:1549-1556.
Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem. 1999; 274:5810–22.
Kabsch K, Alonso A. The human papillomavirus type 16 (HPV-16) E5 protein sensitizes human keratinocytes to apoptosis induced by osmotic stress. Oncogene. 2002; 21:947-53.
Kaczkowski B, Morevati M, Rossing M, Cilius F, Norrild B. A Decade of Global mRNA and miRNA Profiling of HPV-Positive Cell Lines and Clinical Specimens. Open Virol J. 2012 ;6:216-31.
Karpinski P, Myszka A, Ramsey D, Kielan W, Sasiadek MM. Detection of viral DNA sequences in sporadic colorectal cancers in relation to CpG island methylation and methylator phenotype. Tumour biology. 2011; 32:653-9.
Karst AM, Drapkin R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat Protoc. 2012; 7, 1755-64.
Katzenellenbogen RA, Egelkrout EM, Vliet-Gregg P, Gewin LC, Gafken PR, Galloway DA. NFX1-123 and poly(A) binding proteins synergistically augment
77

activation of telomerase in human papillomavirus type 16 E6-expressing cells. J Virol. 2007;81:3786-96.
Katzenellenbogen RA, Vliet-Gregg P, Xu M, Galloway DA. NFX1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J Virol. 2009;83:6446-56.
Ki KD, Lee SK, Tong SY, Lee JM, Song DH, Chi SG. Role of 5'-CpG island hypermethylation of the FHIT gene in cervical carcinoma. J Gynecol Oncol. 2008;19:117-22.
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998; 396:84-8.
Klingelhutz AJ, Foster SA, McDougall JK.Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996; 380:79-82.
Knapp AA, McManus PM, Bockstall K, Moroianu J. Identification of the nuclear localization and export signals of high risk HPV16 E7 oncoprotein. Virology. 2009; 383:60–68.
Korenjak M1, Brehm A. E2F-Rb complexes regulating transcription of genes important for differentiation and development. Curr Opin Genet Dev. 2005; 15:520-7.
Korosec B, Glavac D, Volavsek M, Ravnik-Glavac M. ATP2A3 gene is involved in cancer susceptibility. Cancer Genet Cytogenet. 2009; 188:88-94.
Kravchenco-Balasha N, Mizrachy-Schwartz S, Klein S, Levitzki A. Shift from Apoptotic to Necrotic Cell Death during Human Papillomavirus-induced Transformation of Keratinocytes. J Biol Chem. 2009; 284:17717-27.
78

Lei H, Oh SP, Okano M, Jüttermann R, Goss KA, Jaenisch R, Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195-205.
Leonard SM, Wei W, Collins SI, Pereira M, Diyaf A, Constandinou-Williams C, Young LS, Roberts S, Woodman CB. Oncogenic human papillomavirus imposes an instructive pattern of DNA methylation changes which parallel the natural history of cervical HPV infection in young women. Carcinogenesis. 2012; 33:1286-93
Lewis MT, Ross S, Strickland PA, Snyder CJ, Daniel CW. Regulated expression patterns of IRX-2, an Iroquois-class homeobox gene, in the human breast. Cell Tissue Res. 1999; 296:49-554.
Liang YJ, Chang HS, Wang CY, Yu WC. DYRK1A stabilizes HPV16E7 oncoprotein through phosphorylation of the threonine 5 and threonine 7 residues. Int. J. Biochem. Cell. Biol. 2008; 40:2431–41.
Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18:1427-31.
Li Y, Yang Y, Lu Y, Herman JG, Brock MV, Zhao P, Guo M. Predictive value of CHFR and MLH1 methylation in human gastric cancer. Gastric Cancer. 2014; Apr 21. [Epub ahead of print]
Li ZX, Ma X, Wang ZH. A differentially methylated region of the DAZ1 gene in spermatic and somatic cells. Asian J Androl. 2006; 8:61-7.
Lindel K, Rieken S, Daffinger S, Weber KJ, de Villiers EM, Debus J. The transcriptional regulator gene E2 of the Human Papillomavirus (HPV) 16 influences the radiosensitivity of cervical keratinocytes. Radiat Oncol. 2012; 7:187.
Liu X, Dakic A, Zhang Y, Dai Y, Chen R, Schlegel R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc. Natl. Acad. Sci. USA, 2009; 106:18780–5
79

Liu J, Guo S, Li Q, Yang L, Xia Z, Zhang L, Huang Z, Zhang N. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. Journal neurooncol. 2013; 111:245-55.
Liu SS, Leung RC, Chan KY, Chiu PM, Cheung AN, Tam KF, Ng TY, Wong LC, Ngan HY. p73 expression is associated with the cellular radiosensitivity in cervical cancer after radiotherapy. Clin Cancer Res. 2004;10:3309-16.
Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Heiden MG. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature genetics. 2011; 43:869-74.
Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol. 2004; 78:3533-41.
Malinowski DP. Molecular diagnostic assays for cervical neoplasia: emerging markers for the detection of high-grade cervical disease. Biotechniques. 2005; Suppl:17-23.
Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2004; 24:1000-4.
Martens JE, Arends J, Van der Linden PJ, De Boer BA, Helmerhorst TJ. Cytokeratin 17 and p63 are markers of the HPV target cells, the cervical stem cells. Anticancer Res. 2004; 24:771–4.
Martini F, Iaccheri L, Martinelli M, Martinello R, Grandi E, Mollica G, Tognon M. Papilloma and polyoma DNA tumor virus sequences in female genital tumors. Cancer Invest. 2004; 22:697-705.
Massimi P, Gammoh N, Thomas M, Banks L. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation. Oncogene. 2004; 23:8033-9.
80

Massimi P, Banks L. Differential phosphorylation of the HPV-16 E7 oncoprotein during the cell cycle. Virology. 2000; 276:388–94.
Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 2010; 70:2924-31.
Mazzoccoli G, Pazienza V, Panza A, Valvano MR, Benegiamo G, Vinciguerra M, Andriulli A, Piepoli A. ARNTL2 and SERPINE1: potential biomarkers for tumor aggressiveness in colorectal cancer. J Cancer Res Clin Oncol. 2012; 138:501-11.
McIntyre MC, Frattini MG, Grossman SR, Laimins LA. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J Virol. 1993; 67:3142-50.
McLaughlin-Drubin ME, Meyers C. Propagation of infectious, high-risk HPV in organotypic "raft" culture. Methods Mol Med. 2005; 119:171-86.
McMurray HR, McCance DJ. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J Virol. 2003; 77:9852-61.
Melsheimer P, Klaes R, Doeberitz MV, Bastert G. Prospective clinical study comparing DNA flow cytometry and HPV typing as predictive tests for persistence and progression of CIN I/II. Cytometry. 2001; 46:166-71.
Mitchell MF, Tortolero-Luna G, Wright T, Sarkar A, Richards-Kortum R, Hong WK, Schottenfeld D. Cervical human papillomavirus infection and intraepithelial neoplasia: a review. J Natl Cancer Inst Monogr. 1996; 21:17-25.
Moretti F, Marinari B, Lo Iacono N, Botti E, Giunta A, Spallone G, Garaffo G, Vernersson-Lindahl E, Merlo G, Mills AA, Ballarò C, Alemà S, Chimenti S, Guerrini L, Costanzo A. A regulatory feedback loop involving p63 and IRF6 links
81

the pathogenesis of 2 genetically different human ectodermal dysplasias. J Clin Invest. 2010; 120:1570-7.
Moscicki AB, Ma Y, Wibbelsman C, Darragh TM, Powers A, Farhat S, Shiboski S. Rate of and risks for regression of cervical intraepithelial neoplasia 2 in adolescents and young women. Obstet Gynecol. 2010; 116:1373-80.
Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003; Feb 6;348:518-27.
Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006; 24 Suppl 3:S3/1-10.
Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989; 8:4099-105.
Muzio G, Maggiora M, Paiuzzi E, Oraldi M, Canuto RA. Aldehyde dehydrogenases and cell proliferation. Free rad biol & med. 2012; 52:735-746.
Nakashima R, Fujita M, Enomoto T, Haba T, Yoshino K, Wada H, Kurachi H, Sasaki M, Wakasa K, Inoue M, Buzard G, Murata Y. Alteration of p16 and p15 genes in human uterine tumours. Br J Cancer. 1999; 80:458-67.
Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993; 21:4886-92.
Nakagawa S, Huibregtse JM. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol Cell Biol. 2000; 20:8244-53.
82

Narayan G, Arias-Pulido H, Koul S, Vargas H, Zhang FF, Villella J, Schneider A, Terry MB, Mansukhani M, Murty VV. Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: its relationship to clinical outcome. 2003; 13:2-24.
Narayan G, Arias-Pulido H, Nandula SV, Basso K, Sugirtharaj DD, Vargas H, Mansukhani M, Villella J, Meyer L, Schneider A, Gissmann L, Dürst M, Pothuri B,Murty VV. Promoter hypermethylation of FANCF: disruption of Fanconi Anemia-BRCA pathway in cervical cancer. Cancer Res. 2004; 64:2994-7.
Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007; 98:1505-11.
Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation associated and NF-κB-responsive genes in cervical keratinocytes. J Virol 2001; 75:4283–96.
Novak K. Epigenetics changes in cancer cells. Med Gen Med. 2004; 6:17.
Oguntayo, Samaila. Cervical Intraepithelial Neoplasia (CIN) (Squamous Dysplasia). Intraepithelial Neoplasia. 2012; ISBN: 978-953-307-987-5
Oh ST, Kyo S, Laimins LA. Telomerase Activation by Human Papillomavirus Type 16 E6 Protein: Induction of Human Telomerase Reverse Transcriptase Expression through Myc and GC-Rich Sp1 Binding Sites. J virol. 2001; 75:559–66.
Okano M, Xie S, Li E. Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Res. 1998; 26:2536-40.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247-57.
83

Okuyama R, Ogawa E, Nagoshi H, Yabuki M, Kurihara A, Terui T, Aiba S, Obinata M, Tagami H, Ikawa S. p53 homologue, p51/p63, maintains the immaturity of keratinocyte stem cells by inhibiting Notch1 activity. Oncogene. 2007; 26:4478-88.
Ostor AG. Natural history of cervical intraepithelial neoplasia: a critical review. Int J Gynecol Pathol. 1993; 12:186-92.
Overmeer RM, Henken FE, Snijders PJ, Claassen-Kramer D, Berkhof J, Helmerhorst TJ, Heideman DA, Wilting SM, Murakami Y, Ito A, Meijer CJ, Steenbergen RD. Association between dense CADM1 promoter methylation and reduced protein expression in high-grade CIN and cervical SCC. J Pathol. 2008; 215:388-97.
Oue N, Hamai Y, Mitani Y, Matsumura S, Oshimo Y, Aung PP, Kuraoka K, Nakayama H, Yasui W. Gene expression profile of gastric carcinoma: identification of genes and tags potentially involved in invasion, metastasis, and carcinogenesis by serial analysis of gene expression. Cancer res. 2004; 64:2397-405.
Pancaldi C, Balatti V, Guaschino R, Vaniglia F, Corallini A, Martini F, Mutti L, Tognon M. Simian virus 40 sequences in blood specimens from healthy individuals of Casale Monferrato, an industrial town with a history of asbestos pollution. J Infect. 2009; 58:53-60.
Park TW, Fujiwara H, Wright TC. Molecular biology of cervical cancer and its precursors. Cancer. 1995; 76(10 Suppl):1902-13.
Park EJ, Kim HJ, Kwon ES, Hwang SE, Namkoong SJ. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000; 275:6679–764.
Park SY, Kwon HJ, Choi Y, Lee HE, Kim SW, Kim JH, Kim IA, Jung N, Cho NY, Kang GH. Distinct patterns of promoter CpG island methylation of breast cancer subtypes are associated with stem cell phenotypes. Mod Pathol. 2012; 25:185-96.
84

Patterson NA, Smith JL, Ozbun MA. Human papillomavirus type 31b infection of human keratinocytes does not require heparan sulfate. J Virol. 2005; 79: 6838–6847.
Phelps WC, Yee CL, Münger K, Howley PM. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell. 1988; 53:539-47.
Piperi C, Themistocleous MS, Papavassiliou GA, Farmaki E, Levidou G, Korkolopoulou P, Adamopoulos C, Papavassiliou AG. High incidence of MGMT and RARbeta promoter methylation in primary glioblastomas: association with histopathological characteristics, inflammatory mediators and clinical outcome. Mol Med. 2010; 16:1-9.
Pirisi L, Yasumoto S, Feller M, Doniger J, DiPaolo JA. Transformation of human fibroblasts and keratinocytes with human papillomavirus type 16 DNA. J Virol. 1987; 61:1061–6.
Pirisi L, Creek KE, Doniger J, DiPaolo JA. Continuous cell lines with altered growth and differentiation properties originate after transfection of human keratinocytes with human papillomavirus type 16 DNA. Carcinogenesis. 1988; 9:1573–9.
Pyeon D, Lambert PF, Ahlquist P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc Natl Acad Sci U S A. 2005; 102:9311-6.
Quade BJ, Yang A, Wang Y, Sun D, Park J, Sheets EE, Cviko A, Federschneider JM, Peters R, McKeon FD, Crum CP. Expression of the p53 homologue p63 in early cervical neoplasia. Gynecol Oncol. 2001; 80:24–29
Rader JS, Golub TR, Hudson JB, Patel D, Bedell MA, Laimins LA. In vitro differentiation of epithelial cells from cervical neoplasias resembles in vivo lesions. Oncogene. 1990; 5:571-6.
Radu E, Simionescu O, Regalia T, Dumitrescu D, Popescu LM. Stem cells (p63þ) in keratinocyte cultures from human adult skin. J Cell Mol Med. 2002; 6:593–598.
85

Rajcumar T, Sabitha K, Vijayalakshmi N, Shirley S, Bose MV, gopal G, Selvaluxmy G. Identification and validation of genes involved in cervical tumorigenesis. BMC Cancer. 2011; 11:80.
Razmpoosh M, Sansregret A, Oligny LL, Patey N, Dormoy-Raclet V, Ducruet T, Bouron-Dal Soglio D. Assessment of correlation between p16INK4a staining, specific subtype of human papillomavirus, and progression of LSIL/CIN1 lesions: first comparative study. Am J Clin Pathol. 2014;142:104-10.
Reinstein E, Scheffner M, Oren M, Ciechanover A, Schwartz A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000;19:5944–50.
Restivo G, Nguyen BC, Dziunycz P, Ristorcelli E, Ryan RJH, Ozuysal OY, Di Piazza M, Radtke F, Dixon MJ, Hofbauer GFL, Lefort K, Dotto GP. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. Embo J. 2011; 30:4571-85.
Rho JH, Roehrl MH, Wang JY. Tissue proteomics reveals differential and compartment-specific expression of the homologs transgelin and transgelin-2 in lung adenocarcinoma and its stroma. J Proteome Res. 2009; 8:5610-18.
Rosty C, Sheffer M, Tsafrir D, Stransky N, Tsafrir I, Peter M, de Crémoux P, de La Rochefordière A, Salmon R, Dorval T, Thiery JP, Couturier J. Identification of a proliferation gene cluster associated with HPV E6/E7 expression level and viral DNA load in invasive cervical carcinoma. Oncogene. 2005; 24:7094–104.
Rotondo JC, Bosi S, Bazzan E, Di Domenico M, De Mattei M, Selvatici R, Patella A, Marci R, Tognon M, Martini F. Methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples of infertile couples correlates with recurrent spontaneous abortion. Hum Reprod. 2012; 27:3632-8.
Rotondo JC, Selvatici R, Di Domenico M, Marci R, Vesce F, Tognon M, Martini F. Methylation loss at H19 imprinted gene correlates with methylenetetrahydrofolate
86

reductase gene promoter hypermethylation in semen samples from infertile males. Epigenetics. 2013; 8:990-7. doi: 10.4161/epi.25798. Epub 2013 Jul 24.
Russo VEA, Martienssen RA, Riggs AD. Epigenetic mechanisms of gene regulation. Cold Spring Harbor Monograph. 1996; Volume 32.
Ruutu M, Peitsaro P, Johansson B, Syrjanen S. Transcriptional profiling of a human papillomavirus 33-positive squamous epithelial cell line which acquired a selective growth advantage after viral integration. Int J Cancer. 2002; 100:318–26.
Santin AD, Zhan F, Bignotti E, Siegel ER, Cané S, Bellone S, Palmieri M, Anfossi S, Thomas M, Burnett A, KayHH, Roman JJ, O’Brien TJ, Tian E, Cannon MJ, Shaughnessy Jr. J, Pecorelli S. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology. 2005; 331:269-91.
Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann Intern Med. 2001; 134:573-86.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990; 63:1129-36.
Schiffman M, Castle PE. Human papillomavirus: epidemiology and public health. Arch Pathol Lab Med. 2003; 127:930-4.
Schiffman MH, Brinton LA. The epidemiology of cervical carcinogenesis. Cancer. 1995; 76:1888-901.
Schiller JT, Day PM, Kines RC. Current understanding of the mechanism of HPV infection. Gynecol Oncol. 2010; 118:12-17.
87

Scott B, Vande P, Aloysius JK. Papillomavirus E6 oncoproteins. Virology. 2013; 445:115–37.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990; 63:1129-36.
Shampay J, Blackburn EH. Generation of telomere-length heterogeneity in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1988; 85:534-8.
Shanbhag S, Clark H, Timmaraju V, Bhattacharya S, Cruickshank M. Pregnancy outcome after treatment for cervical intraepithelial neoplasia. Obstet Gynecol. 2009; 114:727-35.
Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Rodriguez CS. Human papillomavirus and cervical cancer. Lancet. 2007; 370, 890-907.
Shivapurkar N, Toyooka S, Toyooka KO, Reddy J, Miyajima K, Suzuki M, Shigematsu H, Takahashi T, Parikh G, Pass HI, Chaudhary PM, Gazdar AF. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. Int J Cancer. 2004; 109:786-92.
Serenaite I, Daniunaite K, Jankevicius F, Laurinavicius A, Petroska D, Lazutka JR, Jarmalaite S. Heterogeneity of DNA methylation in multifocal prostate cancer. Virchows Arch. 2015;466(1):53-9.
Smedts F, Ramaekers F, Robben H, Pruszczynski M, van Muijen G, Lane B, Leigh I, Vooijs P. Changing patterns of keratin expression during progression of cervical intraepithelial neoplasia. Am J Pathol. 1990; 136:657–68.
Song JY, Lee JK, Lee NW, Jung HH, Kim SH, Lee KW. Microarray analysis of normal cervix, carcinoma in situ, and invasive cervical cancer: identification of candidate genes in pathogenesis of invasion in cervical cancer. Int J Gynecol Cancer. 2008; 18:1051-9.
88

Soprano DR, Qin P, Soprano KJ. Retinoic acid receptors and cancers. Annu Rev Nutr. 2004; 24:201-21.
Stafl A, Wilbanks GD. An international terminology of colposcopy: report of the Nomenclature Committee of the International Federation of Cervical Pathology and Colposcopy. Obstet Gynecol. 1991; 77:313-4.
Stanley MA, Browne H. M, Appleby M, and Minson AC. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int J Cancer. 1989; 43: 672–6.
Stephen JK, Chen KM, Shah V, Schweitzer VG, Gardner G, Benninger MS, Worsham MJ. Consistent DNA hypermethylation patterns in laryngeal papillomas. Int J Head Neck Surg. 2010; 1:69-77.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, Grandis JR. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157-60.
Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem. 2004; 279:27816-23.
Summerford C, Bartlett JS, Samulski RJ. αVβ5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat Med. 1999; 5:78–82.
Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynec oncol. 2011; 121:59-63.
Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol. 2003; 3:235-40.
89

Tan CL, Gunaratne J, Lai D, Carthagena L, Wang Q, Xue YZ, Quek LS, Doorbar J, Bachelerie F, Thierry F, Bellanger S. HPV-18 E2^E4 chimera: 2 new spliced transcripts and proteins induced by keratinocyte differentiation. Virology. 2012; 429:47-56.
Terra AP, Murta EF, Maluf PJ, Caballero OL, Brait M, Adad SJ. Aberrant promoter methylation can be useful as a marker of recurrent disease in patients with cervical intraepithelial neoplasia grade III. Tumori 2007; 93:572-79.
Thomason HA, Zhou H, Kouwenhoven EN, Dotto GP, Restivo G, Nguyen BC, Little H, Dixon MJ, van Bokhoven H, Dixon J. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J Clin Invest. 2010;120:1561-9.
Togtema M, Pichardo S, Jackson R, Lambert PF, Curiel L, Zehbe I. Sonoporation delivery of monoclonal antibodies against human papillomavirus 16 E6 restores p53 expression in transformed cervical keratinocytes. PLoS One. 2012; 7:e50730.
Y P Tsao, L Y Li, T C Tsai, and S L Chen. Human papillomavirus type 11 and 16 E5 represses p21(WafI/SdiI/CipI) gene expression in fibroblasts and keratinocytes. J Virol. 1996; 70:7535–9.
Tudor D, Chaudry F, Harper L, Mackenzie CI. The in vitro behavior and patterns of
colony formation of murine epithelial stem cells. Cell Prolif. 2007; 40:706–20.
Ueda Y, Enomoto T, Miyatake T, Ozaki K, Yoshizaki T, Kanao H, Ueno Y, Nakashima R, Shroyer KR, Murata Y. Monoclonal expansion with integration of high-risk type human papillomaviruses is an initial step for cervical carcinogenesis: association of clonal status and human papillomavirus infection with clinical outcome in cervical intraepithelial neoplasia. J techl meth pathol. 2003; 83:1517-1527.
Van Noesel MM, van Bezouw S, Salomons GS, Voûte PA, Pieters R, Baylin SB, Herman JG, Versteeg R. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Res. 2002; 62:2157-61.
90

Van Doorslaer K, Burk RD. Association between hTERT activation by HPV E6 proteins and oncogenic risk.Virology. 2010; 433:216-9.
van Kempen PM, van Bockel L, Braunius WW, Moelans CB, van Olst M, de Jong R, Stegeman I, van Diest PJ, Grolman W, Willems SM. HPV-positive oropharyngeal squamous cell carcinoma is associated with TIMP3 and CADM1 promoter hypermethylation. Cancer Med. 2014; 3:1185-96.
Vande Pol SB1, Klingelhutz AJ. Papillomavirus E6 oncoproteins. Virology. 2013; 445:115-37
Veldman T, Horikawa I, Barrett JC, Schlegel R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol. 2001; 75:4467-72.
Vasiljević N, Scibior-Bentkowska D, Brentnall AR, Cuzick J, Lorincz AT. Credentialing of DNA methylation assays for human genes as diagnostic biomarkers of cervical intraepithelial neoplasia in high-risk HPV positive women. Gynecol Oncol. 2014; 132:709-14.
Viet CT, Jordan RC, Schmidt BL. DNA promoter hypermethylation in saliva for the early diagnosis of oral cancer. J Calif Dent Assoc. 2007; 35:844-9.
Vinay K, Abul K, Nelson F, Richard M. Robbins Basic Pathology. 2007; 8th Edition
Virmani AK, Muller C, Rathi A, Zoechbauer-Mueller S, Mathis M, Gazdar AF. Aberrant methylation during cervical carcinogenesis. Clin Cancer Res. 2001; 7:584-9.
Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408:307-10.
91

Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, Treisman J, Rosenberg SA. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-28-deoxycytidine. Cancer Res. 1994; 54:1766-71.
Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990; 248:76-9.
Widschwendter M, Jiang G, Woods C, Müller HM, Fiegl H, Goebel G, Marth C, Müller-Holzner E, Zeimet AG, Laird PW, Ehrlich M. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004; 64:4472-80.
Wilson VG, West M, Woytek K, Rangasamy D. Papillomavirus E1 proteins: form, function, and features. Virus Genes. 2002; 24: 275-90.
Wang YP, Yu QJ, Cho AH, Rondeau G, Welsh J, Adamson E, Mercola D, McClelland M. Survey of differentially methylated promoters in prostate cancer cell lines. Neoplasia. 2005; 7:748-60.
WHO - Integrating Health Care for Sexual and Reproductive Health and Chronic Diseases. Comprehensive cervical cancer control – a guide to essential practice, chapter 2. 2006.
Wong YF, Selvanayagam ZE, Wei N, Porter J, Vittal R, Hu R, Lin Y, Liao J, Shih JW, Cheung TH, Lo KWK, Yim SF, Yip SK, Ngong DT, Siu N, Chan LKY, Chan CS, Kong T, Kutlina E, McKinnon RD, Denhardt DT, Chin K-V, Chung TKH. Expression Genomics of Cervical Cancer: Molecular Classification and Prediction of Radiotherapy Response by DNA Microarray. Clin Can Res. 2003; 9:5486–92.
Wong YF, Cheung TH, Tsao GS, Lo KW, Yim SF, Wang VW, Heung MM, Chan SC, Chan LK, Ho TW, Wong KW, Li C. Genome-wide gene expression profiling of cervical cancer in Hong Kong women by oligonucleotide microarray. Int J Cancer. 2006; 118:2461–9.
Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007; 7:11-22.
92

Wu CL1, Zukerberg LR, Ngwu C, Harlow E, Lees JA. In vivo association of E2F and DP family proteins. Mol Cell Biol. 1995;15:2536-46.
Yalcin-Ozuysal O, Fiche M, Guitierrez M, Wagner KU, Raffoul W, Brisken C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010; 17:1600-12.
Yang N, Nijhuis ER, Volders HH, Eijsink JJ, Lendvai A, Zhang B, Hollema H, Schuuring E, Wisman GB, van der Zee AG. Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cell Oncol. 2010; 32:131-43.
Zhang A, Maner S, Betz R. Genetic alterations in cervical carcinomas: frequent low-level amplifications of oncogenes are associated with human papillomavirus infection. Int J Cancer. 2002; 101:427-33.
Zyzak LL, MacDonald LM, Batova A, Forand R, Creek KE, Pirisi L. Increased levels and constitutive tyrosine phosphorylation of the epidermal growth factor receptor contribute to autonomous growth of human papillomavirus type 16 immortalized human keratinocytes. Cell Growth Differ. 1994; 5:537–47.
Zur Hausen H. Papillomavirus infections a major cause of human cancers. Biochim Biophys Acta. 1996; 1288:55-78.
Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002; 2:342-50.
https://visualsonline.cancer.gov/details.cfm?imageid=2255
http://www.clinsci.org/cs/
93

During my doctoral period, conducted at the laboratories of Professors Mauro
Tognon and Fernanda Martini, in addition to the work presented in this thesis, I have
published or contributed significantly to the following publications:
94

Rotondo JC, Bosi S, Bassi C, Ferracin M, Lanza G, Gafà R, Magri E, Selvatici R, Torresani S, Marci R, Garutti P, Negrini M, Tognon M, Martini F. Gene Expression Changes in Progression of Cervical Neoplasia Revealed by Microarray Analysis of Cervical Neoplastic Keratinocytes. Journal of Cellular Physiology. April 2015 Sep. I.F.= 3.87
Mazzoni E, Pietrobon S, Masini I, Rotondo JC, Gentile M, Fainardi E, Casetta I, Castellazzi M, Granieri E, Caniati ML, Tola MR, Guerra G, Martini F, Tognon M. Significant low prevalence of antibodies reacting with simian virus 40 mimotopes in serum samples from patients affected by inflammatory neurologic diseases, including multiple sclerosis. PLoS One. Nov 2014. I.F.= 3.53
Mazzoni E, Martini F, Corallini A, Taronna A, Barbanti-Brodano G, Querzoli P, Magri E, Rotondo JC, Dolcetti R, Vaccher E, Tognon M. Serologic investigation on undifferentiated nasopharyngeal carcinoma and Simian Virus 40 infection. Head Neck. Sep 2014. I.F.= 3
Mazzoni E, Gerosa M, Lupidi F, Corallini A, Taronna AP, D'Agostino A, Bovenzi M, Ruggeri G, Casali F, Rotondo JC, Rezza G, Barbanti-Brodano G, Tognon M, Martini F. Significant prevalence of antibodies reacting with simian virus 40 mimotopes in sera from patients affected by glioblastoma multiforme. Neuro-Oncology. 2013 Dec. I.F. = 5.28
Rotondo JC, Selvatici R, Di Domenico M, Marci R, Vesce F, Tognon M, Martini F. Methylation loss at H19 imprinted gene correlates with methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples from infertile males. Epigenetics. 2013 Jul 24;8(9). I.F. = 5.1
Rotondo JC, Bosi S, Bazzan E, Di Domenico M, De Mattei M, Selvatici R, Patella A, Marci R, Tognon M, Martini. Methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples of infertile couples correlates with recurrent spontaneous abortion. Human Reproduction. 2012 Dec;27(12):3632-8. I.F. = 4.58
95


















