
CHIMICA - MyPoli - Homemypoliuni.weebly.com/uploads/8/1/6/8/81687434/tutto_il_corso.pdf · tra loro...
34
CHIMICA Introduzione La chimica studia le proprietà delle sostanze e le reazioni che le trasformano in altre sostanze. Si definisce elemento lo stato più semplice della materia, con proprietà fisiche e chimiche esclusive. Esso è costituito da una sola specie atomica. In totale gli elementi sono un centinaio, ordinati dai chimici in base alle loro proprietà che variano in modo periodici, sistematico e prevedibile. Sono catalogati nella tavola periodica, realizzata per la prima volta da Mendeleev. Si parla invece di composto quando si è in presenza di una materia costituita da due o più elementi chimicamente legati tra loro. La miscela è invece un gruppo di due o più sostanza che sono mescolate fisicamente e non chimicamente. Le miscele si dividono in: Miscele omogenee: in queste miscele i componenti si mescolano così bene che perdono parte delle loro proprietà e non è più possibile distinguerli separatamente. I componenti di tali miscele possono essere mescolati in moltissimi proporzioni, talvolta però con qualche limitazione. Nonostante i componenti si mescolino bene è possibile separarli cambiando il loro stato di aggregazione o sfruttando la diversa solubilità. Inoltre le proprietà di una miscela omogenea sono le stesse in ogni suo punto. Miscele eterogenee: in queste miscele i componenti mantengono le proprie caratteristiche e ciò permette di individuarli anche se sono ben mescolati. Questi componenti possono essere mescoli nelle più diverse quantità e proporzioni. Possono essere separati mantenendo immutate le loro proprietà e inoltre tali proprietà possono risultare diverse in diversi punti della miscela stessa. Le miscele eterogenee possono essere separate nei due o più componenti principali tramite: 1. Decantazione = è una tecnica di separazione che consente di separare due sostanze di un miscuglio eterogeneo per mezzo della forza di gravità. Nel caso di miscele eterogenee solido- liquido, la decantazione consiste nel lasciar depositare sul fondo di un recipiente il solido sino a quando tutto il liquido sovrastante risulti limpido. 2. Centrifugazione = è un metodo che consente di separare due sostanza di un miscuglio eterogeneo per messo della forza centrifuga che spinge le particelle di un solido sul fondo della provetta 3. Filtrazione = è un metodo di separazione che viene usato per separare un solido dal liquido in cui è disperso. Consiste nel versare il miscuglio eterogeneo delle due sostanze su un filtro di materiale poroso (in genere, carta) sovrapposto a un imbuto. Il filtro normalmente un disco di carta, viene collocato all'interno di un imbuto dopo essere stato opportunamente piegato in modo da ottenere un cono. Materia Sostanza pura Fe, O 2 , H 2 O, Au Elemento O 2 , Fe, Au Atomico Fe, Au Molecolare O 2 Composto H 2 O, NaCl Ionico NaCl Molecolare H 2 O Miscela Omogenea soluzione salina Eterogenea acqua + olio
Transcript of CHIMICA - MyPoli - Homemypoliuni.weebly.com/uploads/8/1/6/8/81687434/tutto_il_corso.pdf · tra loro...

CHIMICA
Introduzione La chimica studia le proprietà delle sostanze e le reazioni che le trasformano in altre sostanze. Si definisce elemento lo stato più semplice della materia, con proprietà fisiche e chimiche esclusive. Esso è costituito da una sola specie atomica. In totale gli elementi sono un centinaio, ordinati dai chimici in base alle loro proprietà che variano in modo periodici, sistematico e prevedibile. Sono catalogati nella tavola periodica, realizzata per la prima volta da Mendeleev. Si parla invece di composto quando si è in presenza di una materia costituita da due o più elementi chimicamente legati tra loro. La miscela è invece un gruppo di due o più sostanza che sono mescolate fisicamente e non chimicamente. Le miscele si dividono in:
Miscele omogenee: in queste miscele i componenti si mescolano così bene che perdono parte delle loro proprietà e non è più possibile distinguerli separatamente. I componenti di tali miscele possono essere mescolati in moltissimi proporzioni, talvolta però con qualche limitazione. Nonostante i componenti si mescolino bene è possibile separarli cambiando il loro stato di aggregazione o sfruttando la diversa solubilità. Inoltre le proprietà di una miscela omogenea sono le stesse in ogni suo punto.
Miscele eterogenee: in queste miscele i componenti mantengono le proprie caratteristiche e ciò permette di individuarli anche se sono ben mescolati. Questi componenti possono essere mescoli nelle più diverse quantità e proporzioni. Possono essere separati mantenendo immutate le loro proprietà e inoltre tali proprietà possono risultare diverse in diversi punti della miscela stessa. Le miscele eterogenee possono essere separate nei due o più componenti principali tramite: 1. Decantazione = è una tecnica di separazione che consente di separare due sostanze di un
miscuglio eterogeneo per mezzo della forza di gravità. Nel caso di miscele eterogenee solido-liquido, la decantazione consiste nel lasciar depositare sul fondo di un recipiente il solido sino a quando tutto il liquido sovrastante risulti limpido.
2. Centrifugazione = è un metodo che consente di separare due sostanza di un miscuglio eterogeneo per messo della forza centrifuga che spinge le particelle di un solido sul fondo della provetta
3. Filtrazione = è un metodo di separazione che viene usato per separare un solido dal liquido in cui è disperso. Consiste nel versare il miscuglio eterogeneo delle due sostanze su un filtro di materiale poroso (in genere, carta) sovrapposto a un imbuto. Il filtro normalmente un disco di carta, viene collocato all'interno di un imbuto dopo essere stato opportunamente piegato in modo da ottenere un cono.
Materia
Sostanza pura
Fe, O2, H2O, Au
Elemento
O2, Fe, Au
Atomico
Fe, Au
Molecolare
O2
Composto
H2O, NaCl
Ionico
NaCl
Molecolare
H2O
Miscela
Omogenea
soluzione salina
Eterogenea
acqua + olio

STORIA DELLA MATERIA
1) Democrito Il primo studioso ad intuire che la materia era formata da piccolissime particelle fu il filosofo greco Democrito. Egli chiamò queste particelle atomi.
2) Dalton Bisognerà aspettare fino a Dalton per la formulazione di una teoria chimica sulla base di quanto aveva già intuito Democrito. Egli affermò che la materia era costituita da atomi cioè la più piccola parte di un elemento. Gli atomi di uno stesso elemento, secondo lo studioso, sono tutti uguali e possono reagire tra loro solo in numeri interi. Egli intuì anche che la materia non si crea né si distrugge, ma si trasforma soltanto e quindi, secondo Dalton, in una reazione chimica gli atomi rimangono inalterati in numero e qualità. Tale proposizione è la diretta conseguenza della legge di Lavoisier sulla conservazione della masse (il cardine di tutta la chimica). Atomo = sferetta indivisibile di materia neutra.
3) Thomson Solo grazie a Thomson e all’esperimento dei raggi catodici si scoprì l’elettrone. L’esperimento consisteva nell’usare il tubo di Crookes, cioè un tubo di vetro resistente che viene mantenuto sotto vuoto spinto, alle estremità del quale sono applicati due elettrodi collegati rispettivamente con il polo positivo (anodo) e con il polo negativo (catodo) di un generatore di corrente. Se il tutto era sottoposto ad un campo elettrico si osservava che i raggi catodici, che si creavano nel tubo, deviavano perso il polo positivo quindi dovevano essere cariche negativamente. Dal momento in cui i raggi non dipendevano dalla natura del catodo Thomson comprese che queste particelle negative erano proprie di ogni atomo, iniziando quindi a determinarne le proprietà chimiche. Secondo gli elettroni galleggiano in una nuvola carica positivamente che bilancia le cariche negative degli elettroni assicurando così la neutralità tipica dell’atomo stesso. Elettrone = particella carica negativamente Protone = particella carica positivamente
4) Rutherford (Marie Curie) La scoperta del nucleo si deve però solamente a Rutherford. In un esperimento, che consisteva nel bombardare una lamina sottile d’oro con raggi α, egli osservò che alcuni raggi rimbalzano indietro, altri venivano deviati di angoli più o meno ampi e molti attraversavano la lamina senza subire deviazioni. Rutherford demolisce quindi l’ipotesi di Thomson affermando l’esistenza di un nucleo all’intero dell’atomo costituito da protoni. Rutherford poté eseguire questo esprimono solo dopo aver scoperto, così come Marie Curie, che elementi riscaldati emettono radiazioni. Per capire dunque le proprietà di queste radiazioni vennero usati campi magnetici ed elettrici. Le radiazioni potevano essere di tre tipi:
α = radiazioni cariche positivamente
β = radiazioni cariche negativamente
γ = radiazioni neutre
5) Chadwick Egli formulò poi un nuovo modello dopo aver scoperto il neutrone affermando che l’atomo, una struttura vuota, era costituito da un nucleo di protoni e neutroni attorno al quale ruotano gli elettroni. Neutroni = particella priva di carica

Cenni sulle onde I parametri caratteristici di un onda sono due: 1. Ampiezza = spostamento massimo dell’oscillazione dalla condizione
di assenza di perturbazione 2. Lunghezza d’onda (λ) = distanza tra due massimi o due minimi
successivi espressa in metri 3. Frequenza (v) = numero di massimi o minimi che passano per un
determinato punto in ogni secondo espresso con s-1 4. Velocità: c = λ * v
Si definiscono onde stazionarie quelle onde che rimangono sempre nella stessa porzione di spazio. Esse sono caratterizzate da nodi e la loro
lunghezza soddisfa questa equazione: 𝐿 = 𝑛𝜆
2 . In una circonferenza deve
essere contenuto un numero intero di lunghezze d’onda: 2𝜋 = 𝑛𝜆.
Meccanica quantistica: doppia natura della luce Secondo la fisica classica le onde erano un flusso continuo di energia e di conseguenza anche la luce, essendo un’onda, è un flusso di energia. In seguito all’esperimento dell’effetto fotoelettrico si arrivò però alla conclusione che la luce era costituita da pacchetti di energia detti fotoni. L’effetto fotoelettrico è un esperimento che consiste nell’inviare un fascio di luce su una lamina detta emettitore ed osservarne il comportamento: da questa lamina verrà emesso un elettrone che verrà poi attratto dal collettore che si trova a potenziale opposto (positivo) rispetto all’emettitore. Il risultato sarà una corrente misurabile tramite l’ausilio dell’amperometro. Questo esperimento mostra che, affinché l’elettrone venga espulso il fascio di luce deve avere una determinata frequenza v maggiore della frequenza detta di soglia v0. Questo comportamento venne spiegato da Einstein tramite il modello a fotoni cioè pacchetti di energia discreti: 𝐸 = ℎ ∗ 𝑣 . Gli stati energetici accessibili all’elettrone sono quantizzati, per cui solo ad un opportuno contenuto energetico il fotone riesce a strappare l’elettrone al metallo: la radiazione elettromagnetica propaga dunque energia attraverso pacchetti discreti.
Nel momento in cui si eccita un elemento chimico esso emette luce; questa emissione viene impressa in uno spettro detto:
Assorbimento: fonte luce – gas – fessura – prisma – spettro di assorbimento
Emissione: gas riscaldato – fessura – prisma – spettro di emissione
6) Bohr A partire dallo studio degli spettri dell’atomo di idrogeno Bohr ipotizzò il modello atomico quantizzato secondo cui i raggi delle orbite si trovano a una precisa distanza dal nucleo. Per staccare un elettrone in un’orbita vicino al nucleo sarà necessaria un’energia maggiore rispetto alle orbite più lontane. Questi pacchetti indivisibili di energia elettromagnetica sono detti fotoni. Bohr dunque fu in grado di interpretare correttamente lo spettro atomico dell’idrogeno monoelettronico ma non fu in grado di interpretare spettri atomi polielettronici.
Meccanica quantistica: dualismo corpuscolare Secondo la meccanica quantistica la particella ha sia un comportamento corpuscolare (oggetti macroscopici) sia un comportamento ondulatorio (oggetti microscopici). Einstein affermò che la luce assumeva questo doppio comportamento mentre De Broglie fu il primo ad affermare che la materia assumeva sia un comportamento ondulatorio che uno corpuscolare. De Broglie affermò che ogni particella di materia e ogni fotone che muove con una certa quantità di moto pari a p mostra un
comportamento ondulatorio: 𝜆 =ℎ
𝑝 .

Dove h è la costante di Planck e p è appunto la quantità di moto. Secondo dunque il principio di indeterminazione di Heisenberg non è possibile conoscere contemporaneamente la posizione e la
velocità dell’elettrone. (Δ𝑝)(Δ𝑥) ≥ℎ
4𝜋.
7) Schrodinger Il modello ondulatorio dell'elettrone consente di stabilire le zone dello spazio attorno al nucleo di un atomo ove è massima la densità della carica elettrica negativa dovuta agli elettroni dell'atomo stesso. Tale conoscenza è possibile grazie alla equazione di E. Schrodinger, che rappresenta, in tre dimensioni, l'onda stazionaria associata ad un elettrone, dalla cui risoluzione si ottengono funzioni (funzioni d'onda) rappresentabili graficamente che consentono di conoscere la distribuzione della densità di carica elettrica negativa nello spazio attorno al nucleo. Dunque, le soluzioni dell'equazione di Schrodinger, dette funzioni d'onda e indicate con la lettera Ψ (psi), permettono di conoscere lo stato di un elettrone. Anche se la funzione Ψ, in realtà, non ha significato fisico diretto, la funzione Ψ2, calcolata per una determinata porzione di spazio, fornisce la probabilità di trovare l'elettrone in essa.

STRUTTURA ATOMICA E SISTEMA PERIODICO
Atomo Dopo diversi e innumerevoli studi si è arrivati a definire l’atomo come una struttura vuota costituita da un nucleo di protoni e neutroni attorno al quale ruotano gli elettroni. Si parla i numero atomico (Z) per indicare il numero di protoni mentre di usa il termine numero di massa (A) per indicare la somma del numero di protoni e del numero di neutroni. Il numero di neutroni si può dunque calcolare come A-Z. Tutti gli atomi di uno stesso elemento hanno ugual numero di protoni ma possono avere diverso numero di neutroni dando così vita a diversi isotopi. Gli isotopi sono dunque atomi che hanno lo stesso numero atomico (e quindi appartengono allo stesso elemento), ma differiscono per il numero di massa. Z = n° protoni
A = n° protoni + n° neutroni
N° neutroni = A - Z
Massa molecolare e numero di Avogadro Dal momento in cui gli atomi hanno dimensioni molto ridotte è stato necessario introdurre una nuova unità di misura detta unità di massa atomica (uma) cioè 1/12 della massa dell’isotopo 12 del carbonio, contenente 6 neutroni nel nucleo. La massa atomici si differenzia ovviamente dalla massa molecolare. Per confrontare dunque due elementi presenti in quantità diverse non è efficiente sfruttare la massa molecolare poiché un atomo può pesare di più di un altro e quindi è stato introdotti il numero di Avogadro cioè il numero esatto di atomi contenuti un 12 grammi dell’isotopo 12 del carbonio: 6.02*1023. La quantità di materia che contiene un numero di Avogadro di particelle misura in mole (mol). In questo modo è possibile confrontare 2 o più sostanze a partire dal numero di atomi. Numero Avogadro: N0 = 6.022*1023
Numero particelle contenute in un campione: N = N0 * n(mol)
Numero moli: n = 𝑚 (𝑔)
𝑀𝑀 (𝑔
𝑚𝑜𝑙)
Orbitali atomici Il modello ondulatorio dell'elettrone consente di stabilire le zone dello spazio attorno al nucleo di un atomo ove è massima la densità della carica elettrica negativa dovuta agli elettroni dell'atomo stesso. A partire da questo modello si è arrivati a descrivere la quantizzazione degli stati degli elettroni tramite diversi numeri quantici:
Numero quantico principale n indica la dimensione dell’orbitale e
l’energia dell’elettrone
Numero quantico secondario (momento
angolare) l indica la forma dell’orbitale 0 < l < n-1
Numero quantico magnetico m indica l’orientamento dell’orbitale
nello spazio -l < m < l
Numero quantico di spin ms indica il senso dell’orientazione
dell’elettrone ±
1
2
N.B.: n = coincide con il numero dei sottolivelli nel livello in oggetto
2l+1 = numero di orbitali nel sottolivello in oggetto n2 = numero di orbitali di un livello elettronico 2n2 = numero massimo di elettroni in un livello
Ogni orbitale è dunque caratterizzato da dimensione, forma, orientamento nello spazio e contenuto energico. La forma dell’orbitale stesso è indicata dalla funzione d’onda al quadrato. Gli elettroni non

risentono totalmente della carica positiva del nucleo ma risentono della carica effettiva. Dal momento in cui in un atomo ci sono più elettroni questi risentono non solo della forza attrattiva del nucleo ma anche della forza repulsiva tra gli elettroni stessi. Elettroni più interni tendono a fare un’azione di schermo maggiore rispetto agli elettroni più esterni quindi la carica efficace che subiscono è sempre inferiore alla carica nucleare effettiva. Gli elettroni che si trovano in posizione 1s avranno un’azione di schermo maggiore di quelli che si trovano in 2s. Dal momento in cui l’elettrone nell’orbitale s spende più tempo vicino al nucleo dell’elettrone p avente lo stesso numero quantico n, 2s è più penetrante di 2p perché 2s scherma 2p. Per n = 3 3s è più penetrante. N.B.: tutti gli elettroni che hanno lo stesso n appartengono allo stesso strato. N.B.: gli orbitali sono caratterizzati al massimo da 2 elettroni con 3 numeri quantici uguali un solo numero quantico (ms) diverso.
Proprietà magnetiche A partire dal principio di Pauli derivano anche le proprietà magnetiche della materia; le cariche con diversa direzionalità possono infatti creare un campo magnetico. Dalla configurazione elettronica dipendono le proprietà fisiche magnetiche della materia. Se l’atomo possiede due elettroni spaiati allora si creeranno dei dipoli magnetici e quindi tali sostanze risulteranno essere paramagnetiche. Se invece l’atomo possiede degli elettroni accoppiati allora gli elettroni saranno debolmente respinti dal campo magnetico e quindi si avranno sostanze diamagnetiche.
Configurazione elettronica Principio di esclusione di Pauli = due elettroni in un atomo non possono avere tutti e quattro i
numeri quantici uguali: ogni orbitale può essere occupato da un massimo di 2 elettroni aventi spin opposto.
Regola di Hund = se sono disponibili più orbitali aventi la stessa energia gli elettroni si disporranno preferenzialmente in orbitali diversi e con lo stesso valore del numero quantico di spin.
Principio dell’Aufbau = le strutture elettroniche degli atomi nello stato fondamentali possono essere determinate ordinando gli orbitali secondo energie crescenti e riempendoli nel rispetto del principio di Pauli e della regola di Hund a cominciare dall’energia più basse N.B.: gli elettroni sono prima assegnati al sottolivello con valore n più basso
1s2 2s2 2p4 3s2 3p6 4s2 3d10 4p6 5s2 4d10 5p6
Classificazione degli elementi Come già accennato Mendeleev aveva intuito che gli elementi possono essere raggruppati in base al numero atomico e per questo aveva lasciato degli spazi vuoti all’interno della sua classificazione. Gli elementi si classificano quindi in periodi e gruppi. Mendeleev aveva anche capito che le proprietà fisiche di un elemento dipendono dalla configurazione esterna degli orbitali e quindi da come sono riempiti i gusci esterni.
Elementi del I° gruppo = metalli alcalini (ns1 – tranne H) che cedono un elettrone per cercare di essere stabili [1 carica]
Elementi del II° gruppo = metalli alcalino-terrosi (ns2) che cedono due elettroni per cercare di assumere la configurazione del gas nobile che li precede [1 carica doppia]
Elementi del VII° gruppo = alogeni (ns2, np5) che prendono un elettrone per assumere la configurazione del gas nobile che li segue
Elementi del VIII° gruppo = gas nobili (ns2, np6) N.B.: l’energia necessaria per strappare elettroni viene fornita quindi si avrà una reazione endotermica; l’energia che si libera quando si acquisisce elettroni darà una reazione esotermica. I metalli e il non metalli sono divisi nella tavola periodica da una linea. A cavallo di questa linea sono presenti semimetalli cioè elementi che hanno proprietà intermedie come lucentezza e conducibilità termica. I metalli invece sono tutti quegli elementi che possono presentarsi solidi o come gas molecolari e hanno tendenza a creare legami con H. I non metalli al contrario sono elementi che si formano a partire dall’unione di atomi tramite legame ionico. Le proprietà periodiche dipendono da:

1. Carica nucleare effettiva = carica di cui risente un particolare elettrone di un atomo polielettronico come conseguenza della presenza del bilanciamento delle forze attrattive del nucleo e quelle repulsive degli altri elettroni: 𝑧𝑒𝑓𝑓 = 𝑧 − 𝑠 dove z è il numero atomico e s rappresenta lo schermo
degli elettroni più interni. 2. Raggio atomico = cioè la distanza di avvicinamento tra due atomi dello stesso elemento di una
molecola (gas). Il raggio diminuisce lungo il periodo e cresce lungo il gruppo. N.B.: un catione (elemento che perde e-) ha raggio atomico inferiore rispetto all’elemento da cui deriva Un anione (elemento che acquista e-) ha raggio atomico maggiore dell’elemento da cui deriva
3. Energia di ionizzazione = minima quantità di energia necessaria per rimuovere un elettrone da un atomo nello stato fondamentale; energia necessaria per trasformare l’atomo di un elemento nel suo catione monopositivo. Cresce lungo periodo e decresce lungo il gruppo. N.B.: esiste anche l’energia di 2a o 3a ionizzazione cioè la quantità di energia necessaria per rimuovere un elettrone dallo ione monopositivo o dallo ione 2vv positivo
4. Affinità elettronica = quantità di energia che un atomo libera allorché cattura un elettrone che si trova a distanza infinita con valore 0 di energia cinetica: 𝑋 + 𝑒− → 𝑋− + 𝐸𝐴
5. Elettronegatività = misura della tendenza di un atomo ad attrarre verso di sé gli elettroni del legame covalente

LEGAMI INTRAMOLECOLARI
Introduzione La maggior parte degli elementi in natura non esistono allo stato atomico. Per legarsi tra loro a formare delle molecole, gli atomi modificano la distribuzione degli elettroni attorno al proprio nucleo. Gli elettroni coinvolti nella formazione del legame sono quelli degli strati più esterni: gusci di valenza. I tipi di legami intermolecolari sono 3: 1. Ionico che dà origine a composti ionici 2. Covalente che dà origine a composti molecolari 3. Metallico
Se due atomi possono reagire spontaneamente per formare molecole o composti se la formazione del legame porta ad uno sviluppo di energia: un sistema punta sempre ad avere il minor contenuto energetico possibile. Ogni elemento cercherà dunque di raggiungere la sua forma più stabile completando il proprio guscio di valenza seconda quella che viene definita regola dell’ottetto: quando due o più atomi si legano tra loro, ridistribuiscono gli elettroni del guscio di valenza in modo che ogni atomo abbia nel suo guscio di valenza 8 elettroni (ad eccezione dell’idrogeno). Gli atomi impegnati in un legame raggiungono l’ottetto in due diversi modi:
Trasferimento di elettroni da una atomo all’altro (legame ionico)
Compartecipazione di una o più coppie di elettroni (legame covalente)
Legame ionico Il legame ionico si realizza quando la differenza di elettronegatività fra i due elementi che intendono legarsi è superiore a 1,9. In realtà non è proprio un legame ma solo un’attrazione elettrostatica. Si verifica il trasferimento di uno o più elettroni dall'atomo meno elettronegativo (che perdendo elettroni diventa uno ione positivo = catione) all’atomo più elettronegativo (che acquistando elettroni diventa uno ione negativo = anione). Il legame ionico è la conseguenza dell'attrazione elettrostatica che si manifesta tra i due ioni di carica opposta. Il legame ionico avviene tendenzialmente tra atomi dei primi due gruppi e atomi degli ultimi gruppi poiché entrambi, cedendo o acquisendo elettroni hanno la possibilità di raggiungere l’ottetto nel guscio di valenza. Es: 𝐾 → 𝐾+ + 𝑒− 𝐹 + 𝑒− → 𝐹− 𝐾 + 𝐹 → 𝐾+ + 𝐹−
Dal punto di vista energetico l’energia di ionizzazione del potassio è positiva (processo endotermico); l’affinità elettronica del fluoro è negativa (processo esotermico) quindi la formazione del sale dovrebbe avvenire tramite un processo endotermico, ma ciò non avviene poiché i Sali che si ottengono combinando in rapporto 1:1 un metallo alcalino e un alogeno sono stabili e dunque la loro formazione avviene tramite processo esotermico.
Come già accennato il legame ionico porta alla formazione di composti ionici come ad esempio NaCl. Dal momento in cui il volume di un atomo è dato dallo spazio occupato dai suoi orbitali; e spendo che un catione è sempre più piccolo dell’atomo neutro mentre un anione sarà sempre più grande dello stesso
atomo neutro si avrà che la disposizione degli atomi di un solido ionico sarà la seguente: gli anioni saranno circondati da cationi e viceversa. Con il termine energia reticolare si intende la quantità di energia necessaria per separare una mole di cristallo nei suoi ioni allo stato gassoso. L’energia reticolare equivale all’energia di stabilizzazione elettrostatica.
Legame covalente Il legame covalente si forma tra atomi che hanno un alto valore di elettronegatività e quindi tra atomi "non metallici". In questo tipo di legame si ha la compartecipazione di coppie di elettroni: per predire la formazione di legami tra due elementi secondo Lewis era necessario andare ad osservare la configurazione esterna dell’atomo. SI definisce distanza di legame la distanza tra i centri dei 2 atomi legati. Al contrario di parla di lunghezza di legame la distanza media tra gli atomi coinvolti nel legame

stesso. Tale lunghezza del legame H-X decresce lungo il periodo, mentre la lunghezza di legame X-X cresce lungo il gruppo all’aumentare del numero quantico principale. Si definisce invece energia di legame l’energia che occorre fornire per rompere il legame portando i due atomi a distanza infinita; tale energia decresce all’aumentare del numero quantico principale. A seconda che nel legame siano coinvolte una, due o tre coppie di elettroni si parla di ordine di legame 1, ordine di legame 2 e ordine di legame 3; ad un maggior ordine di legame corrisponde una minor distanza di legame e una maggior energia di legame. Se la formazione di legami covalenti avviene tramite sovrapposizione di orbitali s con s (H-H) o p con p (F-F)la condivisione sarà paritetica; se invece la sovrapposizione avviene fra orbitali p e s come per HF la condivisione non sarà paritetica poiché i singoli elementi hanno diversa energia di ionizzazione e diversa affinità elettronica tra loro quindi sarà necessario quantificare questa disparità. Per farlo è necessario valutare prima l’energia di dissociazione del primo elemento (H-H) poi bisognerà valutare quella del secondo elemento (F-F) e infine valutare l’energia di dissociazione del legame H-F nella
molecola HF. Nell’ipotesi di perfetta condivisione tra gli elettroni si avrebbe che ∆𝐸𝐴𝐵 = √∆𝐸𝐴 ∗ ∆𝐸𝐵
, in realtà ciò non avviene poiché l’energia di dissociazione fra i due atomi appartenenti a elementi diversi non corrisponde a questa media. Quando la densità elettronica è concentrata in modo simmetrico rispetto alla linea che congiunge i nuclei si parla di legami σ, altrimenti, se non c’è la possibilità di trovare l’elettrone lungo l’asse internucleare si parla di legami π. Nello specifico esistono tre tipi di legame covalente: 1. Legame covalente puro si realizza tra atomi dello stesso elemento. 2. Legame covalente polare si realizza tra atomi di elementi diversi (ma i due atomi devono avere
una differenza di elettronegatività inferiore a 1,9). In questo tipo di legame vi è la presenza di un dipolo elettrico: la compartecipazione delle densità elettroniche porta a un parziale trasferimento della carica dall’elemento meno elettronegativo a quello più elettronegativo. Per indicare questo momento dipolare si usa una freccia che va dal δ+ al δ, dall’atomo meno elettronegativo a quello più elettronegativo.
3. Legame covalente dativo = un atomo fornisce tutti e due gli atomi da condividere (0 < ∆𝐸 < 0,4)
Legame metallico La maggior parte dei metalli è malleabile, può dunque essere ridotta in lamini sottili inoltre è duttile cioè può essere ridotta in forma di fili. Un modello che permette di schematizzare bene il legame metallico e i composti metallici è il modello del mare di elettroni: in questo modello il metallo è descritto disponendo i cationi metallici in un mare di elettroni di valenza. Gli elettroni sono legati al metallo dall’attrazione elettrostatica con i cationi e sono uniformante distribuiti sull’intera struttura. Nonostante ciò gli elettroni sono mobili. Gli orbitali metallici sono estremamente numerosi e la differenza di energia fra due orbitali è estremamente piccola. A partire dall’energia di ionizzazione si può diagnosticare il carattere metallico di un elemento. Una teoria del legame metallico, e in genere del legame nei solidi, più quantitativa e più estesa, è quella detta teoria delle bande dei solidi (v. stato solido, fisica dello). Secondo la teoria delle bande, tutti gli elettroni di un solido occupano livelli energetici permessi che sono così strettamente vicini da essere praticamente continui. Questi gruppi di livelli ravvicinati, o bande di energia, sono separati da intervalli energetici (gap) di varia lunghezza, che gli elettroni non possono occupare. Le bande che sono completamente riempite dagli elettroni non possono condurre la corrente elettrica, cosicché gli isolanti, cioè i non conduttori, hanno sempre bande completamente riempite separate da quelle vuote da ampi gap di energia. Nei metalli avviene o che una banda permessa è riempita solo parzialmente, o che una banda riempita si sovrappone a una banda vuota (gap di energia uguale a zero); in entrambi i casi si ha la possibilità di condurre la corrente elettrica. La teoria delle bande spiega in modo elegante il meccanismo di riflessione pressoché totale della luce visibile da parte di superfici metalliche ben pulite. Il fotone della luce visibile incidente eccita un elettrone dal livello più alto della sua banda energetica parzialmente riempita a un livello del continuo

tra quelli permessi e non occupati di energia più alta, nella stessa banda. Successivamente l'elettrone eccitato ricade nel più alto livello occupato della banda, provocando l'emissione di un fotone visibile, esattamente della stessa frequenza di quello incidente, e quindi dando luogo alla "riflessione" della luce.
Le leghe Metalli diversi possono essere spesso fusi insieme per dar luogo a nuovi sistemi di tipo metallico, detti leghe. Le leghe possono essere classificate come:
Soluzioni solide = miscele omogenee in cui i componenti sono dispersi in modo casuale ed uniforme. I composti intermetallici sono leghe omogenee che hanno determinate proprietà e composizioni.
Leghe sostituzionali = si formano quando due componenti metallici hanno raggi atomici e caratteristiche di legame chimico simili
Leghe interstiziali = il componenti presente nelle posizioni interstiziali tra atomi solventi deve avere un raggio atomico legante più piccolo di quello dell’atomo solvente.
Leghe eterogenee = i componenti non sono dispersi in modo uniforme
Disegnare le formule di struttura Per poter disegnare la struttura molecolare di un composto è necessario eseguire questi passi: 1. Si determina il numero di valenza di ogni elemento. Se si è in presenza di un catione si sottrae un
elettrone, al contrario se vi è un anione si aggiunge un elettrone 2. Scrivere ora tramite la notazione di Lewis gli elettroni di valenza e disegnare i legami. Ricordarsi
di disporre al centro l’elemento meno elettronegativo 3. Controllare che i legami formati completino l’ottetto nei gusci di valenza (una singola coppia di
elettroni se siamo in presenza dell’idrogeno) 4. Se vi sono elettroni che avanzano disporli generalmente attorno all’atomo centrale poiché molti
composti non seguono la regola dell’ottetto 5. Se non ci sono elettroni sufficiente per fornire all’atomo centrale un ottetto introdurre legami
multipli Si definisce carica formale la carica assegnata a un atomo presente in una molecola o in uno ione poliatomico assumendo che gli elettroni di legame siano equamente condivisi a prescindere dall’elettronegatività di ciascun atomo e si calcolano: Carica formale = n°e−di valenza −
[n° coppie e−solitarie +1
2n°e−legame
Generalmente si sceglie la struttura di Lewis nella quale egli atomi portano cariche formali più vicine a 0. N.B.: la carica di un elemento non è determinata dalla sua natura ma dalla partnership con l’altro elemento Per determinare la geometria molecolare di un composto è necessario utilizzare la teoria VSEPR secondo cui la geometria del sistema è controllata dalla repulsione che si realizza fra gli elettroni del guscio di valenza dell’atomo centrale: essi si disporranno nello spazio in modo da essere il più lontano possibili gli uni dagli altri. Per stabili tale geometria è fondamentale il numero sterico:
𝑛𝑢𝑚𝑒𝑟𝑜 𝑠𝑡𝑒𝑟𝑖𝑐𝑜 = (𝑛°𝑎𝑡𝑜𝑚𝑖 𝑙𝑒𝑔𝑎𝑡𝑖 𝑎 𝑞𝑢𝑒𝑙𝑙𝑜 𝑐𝑒𝑛𝑡𝑟𝑎𝑙𝑒) + (𝑛𝑢𝑚𝑒𝑟𝑜 𝑑𝑖 𝑐𝑜𝑝𝑝𝑖𝑒 𝑠𝑜𝑙𝑖𝑡𝑎𝑟𝑖𝑒 𝑠𝑢𝑙𝑙′𝑎𝑡𝑜𝑚𝑜 𝑐𝑒𝑛𝑡𝑟𝑎𝑙𝑒)
Geometria in base al numero sterico: SN = 2 sp lineare 180° SN = 3 sp2 trigonale planare 120° SN = 4 sp3 tetraedrica 109.5° SN = 5 sp3d bipiramidale trigonale 90° e 120° SN = 6 sp3d2 ottaedrica 90°
Eccezioni regola ottetto 1. Ottetti incompleti (es: Be, B) 2. Ottetti espansi (es: F) 3. Radicali = specie con elettroni spaiati. Tali molecole tendono a dimerarsi

Orbitali ibridi Ibridazione sp3 (singolo legame) = "mescolamento" dell'orbitale 2s con i tre orbitali 2p; tale
mescolamento è matematico, delle funzioni d'onda dell'orbitale e quindi non è un reale fenomeno fisico. Come risultato si ottengono 4 nuovi orbitali identici tra loro, di forma, energia e disposizione nello spazio del tutto diverse da quelle originarie. Questa operazione matematica prende il nome di ibridazione. I nuovi 4 orbitali ibridi, chiamati sp3, hanno per 1/4 le caratteristiche dell'orbitale s di partenza e per 3/4 le caratteristiche degli orbitali 2p. Il 3 esponente di p indica il numero di orbitali p che partecipano alla formazione dell'ibrido.
Ibridazione sp2 (doppio legame) = dal mescolamento di un orbitale s con due orbitali di tipo p si ottengono 3 orbitali ibridi detti orbitali sp2 che si dispongono su di un piano formando angoli di 120° l'uno dall'altro (geometria trigonale planare). L'orbitale p non coinvolto nell'ibridazione si dispone perpendicolarmente al piano formato dai tre orbitali ibridi sp2.
Teoria degli orbitali molecolari Il punto di partenza di questa teoria è di considerare che al legame tra atomi non concorrano solo gli elettroni di valenza, ma in generale tutti gli elettroni degli atomi costituenti la molecola. Nella molecola così concepita non esistono più gli elettroni che appartengono ai singoli atomi, ma essi sono tutti ridistribuiti nella molecola su nuovi livelli energetici, denominati orbitali molecolari. Gli orbitali molecolari sono centrati attorno a tutti i nuclei di una molecola. Lo studio dei loro livelli energetici e del modo in cui si dispongono in essi gli elettroni permette di conoscere la stabilità della molecola considerata. Traducendo in termini matematici questa affermazione, e cioè applicando l'equazione di Schrödinger ad una molecola, vale a dire ad un sistema formato da un insieme di elettroni appartenenti indifferentemente a due o più nuclei di atomi uguali o diversi, è possibile descrivere, tramite le soluzioni di questa equazione, sia l'energia sia la forma geometrica della molecola. Mentre gli orbitali atomici sono funzioni matematiche che descrivono il comportamento di un elettrone in un atomo, gli orbitali molecolari sono funzioni matematiche che descrivono il comportamento di un elettrone in una molecola. Un orbitale è una soluzione dell'equazione di Schrödinger che permette di individuare le zone di spazio in cui è possibile trovare l'elettrone con il massimo di probabilità. La probabilità di trovare un elettrone in un generico punto dello spazio (x,y,z) è direttamente legata al quadrato del valore che la funzione d'onda assume nel punto (x,y,z). Tramite il metodo L.C.A.O. (Linear combination of atomic orbitals) si possono combinare linearmente le autofunzioni d'onda associate ai legami presenti nella molecola per ottenere un orbitale molecolare. I calcoli dimostrano che la combinazione lineare di due funzioni fornisce due combinazioni relative a due orbitali molecolari: un orbitale di legame, derivante dalla sovrapposizione "in fase" delle funzioni d'onda (Ψ1 + Ψ2 + ... + Ψn) caratterizzato da una certa stabilità (notare che la sovrapposizione in fase rafforza la probabilità di trovare l'elettrone nel dominio delle funzioni d'onda) e da un orbitale di antilegame meno stabile del precedente, dovuto alla sovrapposizione fuori fase delle due funzioni d'onda. Esiste anche la possibilità di ottenere un orbitale non legante, caratterizzato dal fatto di non influenzare sostanzialmente la stabilità di una molecola, ed avente, nel caso di una generica molecola A--B un carattere che può essere puramente di A o puramente di B.



FORZE INTERMOLECOLARI
Introduzione I legami intermolecolari sono generalmente molto più deboli di quelli ionici o covalenti; è quindi richiesta una minore energia per evaporare un liquido o per far fondere un solido che per rompere i legami covalenti nelle molecole. Esistono diversi tipi di forze intermolecolari per quanto riguarda i liquidi (messe in ordine per punto di ebollizione dal più alto al più basso):
Ione – ione
Legame idrogeno = è una speciale attrazione intermolecolare tra l’atomo di idrogeno in un legame polare e una coppia elettronica non condivisa presente su un piccolo ione o atomo elettronegativi
Ione – dipolo = si formano tra uno ione e la carica parziale localizzare sull’estremità di una molecola polare (es: NaCl)
Dipolo – dipolo = si creano tra molecole polari quando l’estremità positiva di una molecola si trova vicina all’estremità negativa di un’altra. Per molecole di massa e misura circa uguali la forza delle attrazioni intermolecolari aumenta all’aumentare della polarità. Per molecole con polarità confrontabile invece quelle con volumi molecolari inferiori presentano forze attrattive più alte
Dipolo – dipolo indotto = se la molecola di una sostanza non dotta di momento dipolare si avvicina ad una sostanza dotata di momento dipolare la prima può essere soggetta a ridistribuzione istantanea della densità di carica
Dipolo indotto – dipolo indotto o forza di dispersione di London = si creano tra molecole apolari. Le forze di dispersione tendono ad aumentare la loro intensità con l’aumentare del peso molecolare
FASI CHIMICHE
Introduzione Si definisce fase chimica la parte omogenea di un sistema chimico-fisico delimitata da superfici fisiche ben definite. Esistono tre diversi fasi: liquida, gassosa e solida. Nella maggior parte dei casi una sostanza può esistere in tutte e tre le fasi in funzione di temperatura, pressione e volume. Il passaggio da una fase chimica ad un’altra è detto transizione di fase.
Proprietà della materia Gas = assume la forma ed il volume del suo contenitore, è comprimibile e fluisce velocemente
Liquido = non si espande fino al riempimento del contenitore, è virtualmente comprimibile, fluisce velocemente e la diffusione avviene lentamente
Solido = conserva la sua forma e il suo volume, è incomprimibile e non fluisce. Lo stato di una qualsiasi sostanza dipende dal bilancio tra le energie cinetiche delle particelle e le energie d’attrazione tra le particelle stesse. L’energia cinetica dipende dalla temperatura tende a mantenere le particelle separate e in movimento al contrario le interazioni intermolecolari (i legami chimici) tendono a legare le particelle insieme. Come è facilmente intuibile sostanze gassose a temperatura ambiente presentano interazioni intermolecolari più deboli di quelle liquide a loro volta le sostanze liquidi hanno forze intermolecolari più deboli di quelle solide.

STATO GASSOSO
Proprietà dei gas Un gas si espande spontaneamente e riempire il suo contenitore, di conseguenza il volume di un gas è uguale a quello del suo contenitore. I gas inoltre sono molto comprimibili: quando sono compresso il loro volume diminuisce infine i gas formano miscele omogenee a dispetto della loro identità e proporzioni relative dei componenti gassosi. La fase gassosa è caratterizzata da grandezze estensive come il volume e grandezze intensive come pressione e temperatura. Si definisce pressione l’effetto che il gas esercita sulle pareti del contenitore.
Legge di Boyle: tale legge lega la pressione al volume: il volume di una certa quantità di gas
mantenuta a temperatura costante è inversamente proporzionale alla pressione. 𝑃 ∗ 𝑉 = 𝐶𝐵 𝑃1𝑉1 = 𝑃2𝑉2 (t ed n costanti) 𝑃1 = 𝑃0 + 𝑃0𝑐𝑇 𝑃0
𝑇0 =
𝑃1
𝑇1 (V ed n costanti) 𝑐 =
1
273 𝑇 = 𝑡1 − 𝑡2
Legge di Charles: tale legge instaura una relazione tra temperatura e volume: a pressioni sufficientemente basse il volume di un gas varia linearmente con la temperatura. 𝑉
𝑇= 𝐶𝐶 𝑉1 = 𝑉0 + 𝑉0𝑐𝑇
𝑉0
𝑇0 =
𝑉1
𝑇1
Legge di Avogadro: volumi uguali di gas diversi misurati nelle stesse condizioni di T e P, contengono lo stesso numero di particelle cioè di moli di gas. Il volume di un gas mantenuto a T e P costante è direttamente proporzionale al numero di moli del gas.
Legge dei gas ideali: 𝑃𝑉 = 𝑛𝑅𝑇 𝑃1𝑉1
𝑇1 =
𝑃2𝑉2
𝑇2
Legge di Dalton: la pressione totale di una miscela di gas è la somma delle
pressioni parziali dei singoli gas. Con l’espressione pressione parziali si indica la pressione che ogni gas di una miscela eserciterebbe sulle pareti se fosse d solo nel recipiente.
𝑋𝑎 =𝑛𝑎
𝑛𝑡𝑜𝑡 𝑝𝑎 = 𝑋𝑎 𝑃𝑡𝑜𝑡 𝑣𝑎 = 𝑋𝑎 𝑉𝑡𝑜𝑡
Teoria cinetica dei gas La teoria cinetica dei gas identifica le ragioni molecolari che stanno alla base delle legge dei gas ideali: 1. Un gas è costituito da un grandissimo numero di particelle (atomi, molecole) il cui volume è molto
minore (nullo) rispetto al volume disponibile al gas. 2. Le particelle sono in moto costante, rettilineo, casuale e con una certa distribuzione di velocità 3. Le particelle collidono tra loro e con le pareti tramite urti elastici (senza perdita di energia). Con
gli urti le singolo particelle possono acquistare o cedere energia, ma l’energia totale resta costante.
4. Le particelle (tranne che durante gli urti, che avvengono in tempi estremamente piccoli) non interagiscono tra loro né con le pareti del contenitore in ci il gas è confinato.
Le particelle si urtano e rimbalzano: la pressione risulta essere un effetto netto negli urti. Maggiore è il numero di particelle, maggiore è il numero di urti e quindi maggiore sarà la pressione esercitata sulle pareti del contenitore.
1 atm = 760 mmHg = 760 Torr = 1,01 * 105 Pa
Condizioni normali: 0°C 1 atm Condizioni standard: 25°C 1 atm

STATO LIQUIDO
Proprietà dei liquidi Densità: le molecole dei liquidi sono in contatto reciproco, anche se possono scorrere le une sulle
altre, mentre le molecole dei gas sono separate tra loro. Per questo i liquidi hanno in genere densità maggiore rispetto ai gas (la densità, , è definita come il rapporto tra la massa e il volume del fluido).
Viscosità: grandezza fisica che misura la resistenza che le particelle di un fluido incontrano nello scorrere le une sulle altre. La viscosità dipende dalla temperatura: nei gas aumenta con la temperatura, poiché aumenta il moto termico tra le particelle del gas, mentre nei liquidi temperatura e viscosità sono inversamente proporzionali, perché aumentando la temperatura diminuisce la coesione tra le molecole.
Tensione superficiale: la forza di coesione che si esercita fra le molecole superficiali di un liquido. La tensione superficiale diminuisce all'aumento della temperatura.
Soluzioni Si definiscono soluzioni tutte quelle fasi liquidi nelle quali sono presenti diverse sostanze. Le soluzioni sono sistemi omogenei che contengono dunque due o più sostanze: o due liquidi o due solidi o una gas e un solido. Nelle soluzioni liquide il componente presente in maggiore percentuale è detto solvente mentre il componente presente in percentuale minore è il soluto. Una soluzione si forma spontaneamente se il processo porta a una diminuzione del contenuto energetico del sistema. La formazione di soluzioni è dunque favorita dall’aumento di entropia che accompagna il mescolamento [ulteriori nozioni sulle soluzioni in: equilibrio chimico].
Proprietà colligative Abbassamento della pressione di vapore = aggiungendo un soluto non volatile ad un solvente si
ha sempre un abbassamento della pressione di vapore:
𝑃𝑠𝑜𝑙𝑢𝑧𝑖𝑜𝑛𝑒 = 𝑋𝑠𝑜𝑙𝑣𝑒𝑛𝑡𝑒 ∗ 𝑃𝑠𝑜𝑙𝑣𝑒𝑛𝑡𝑒0
Innalzamento ebullioscopico = il punto di ebollizione di una soluzione è più alto di quello del liquido puro ∆𝑇𝑏 = 𝑘𝑏 ∗ 𝑚 (𝑚𝑜𝑙𝑎𝑙𝑖𝑡à) ∗ 𝑣(𝑐𝑜𝑒𝑓𝑓𝑖𝑐𝑖𝑒𝑛𝑡𝑒 𝑉𝑎𝑛′𝑡𝐻𝑜𝑓𝑓)
Abbassamento crioscopico = il punto di congelamento di una soluzione è più basso di quello del liquido puro ∆𝑇𝑐 = 𝑘𝑐 ∗ 𝑚 (𝑚𝑜𝑙𝑎𝑙𝑖𝑡à) ∗ 𝑣(𝑐𝑜𝑒𝑓𝑓𝑖𝑐𝑖𝑒𝑛𝑡𝑒 𝑉𝑎𝑛′𝑡𝐻𝑜𝑓𝑓)
Pressione osmotica = movimento del solvente dalla soluzione meno concentrata a quella più concentrata 𝜋𝑉 = 𝑛𝑅𝑇 Se due soluzioni di identica pressione osmotica sono separate da una membrana semipermeabile non si ha alcuna osmosi. Le due soluzioni sono isotoniche. Se una soluzione ha una pressione osmotica inferiore, è ipotonica rispetto a quella più concentrata. La soluzione più concentrata è ipertonica rispetto alla soluzione diluita.
Il coefficiente di van 't Hoff (o fattore di dissociazione) è un coefficiente correttivo che viene
introdotto nel calcolo delle proprietà colligative nel caso in cui la soluzione contenga elettroliti. Esso
è un fattore di correzione adimensionale che esprime la quantità di particelle o ioni che
effettivamente si producono dalla dissoluzione di una mole di soluto (inizialmente allo stato solido) in
un solvente.

STATO SOLIDO
Introduzione solidi I solidi possono essere di due principali tipi:
Solido cristallino = caratterizzato da una forma propria ed è incomprimibile
Solido amorfo = caratterizzato da una forma impropria ed è incomprimibile
Solido cristallino Il fenomeno che porta alla formazione di un solido cristallino è detta cristallizzazione; tale fenomeno è spontaneo in quanto tende a diminuire il contenuto di energia libera. Il mattino che costituisce il reticolo cristallino è detta cella elementare. Tali solidi sono caratterizzati da una disposizione degli atomi e degli ioni ben definita nello spazio. Si definisce numero di coordinazione il numero di particelle immediatamente circostanti ad una particella della struttura cristallina. I solidi cristallini hanno un valore del punto di fusione ben definito. In presenza di un solvente molto polare (come l'acqua) danno luogo a soluzioni in cui gli ioni esistono solvatati in fase liquida. Sia tali soluzioni sia i solidi ionici fusi sono in grado di condurre la corrente elettrica, mentre allo stato solido ciò non è possibile perché gli elettroni sono implicati rigidamente nel legame ionico così come lo sono gli ioni. I solidi cristallini possono essere ulteriormente classificati in base alla disposizione spaziale assunta dalle particelle che li compongono, ossia al loro reticolo cristallino e all'essere formati da un singolo cristallo (monocristalli) o da più cristalli (policristalli) aggregati tra loro.
Solido amorfo I solidi amorfi, al contrario non sono caratterizzati da una struttura ben definita ed organizzata. Come conseguenza di ciò, sono caratterizzati da maggior contenuto entropico rispetto ai solidi ionici e non hanno punto di fusione ben definito (i legami tra le particelle non hanno tutti la stessa forza) e costante nel tempo.

TERMODINAMICA
Introduzione Lo studio dell’energia e delle sue trasformazioni prende il nome di termodinamica. La termodinamica nasce con la rivoluzione industriale quando gli scienziati e gli ingegneri ebbero modo di studiare le relazioni che sussistevano tra calore, lavoro e contenuto energetico dei combustibili nel tentativo di massimizzare il funzionamento delle macchine a vapore. Definiamo:
Lavoro = energia utilizzata per spostare un oggetto, dotato di massa, da un punto ad un altro
Calore = energia utilizzata per determinare l’aumento di temperatura di un oggetto
Energia = capacità di compiere un lavoro o di trasferire del calore. L’unità di misura dell’energia è comunemente la caloria definita come la quantità di energia richiesta per aumentare la temperatura di 1 g di acqua da 14,5°C a 15,5°C.
Tipi di energia Esistono vari tipi di energia come: 1. Energia cinetica: cioè l’energia collegata al movimento 2. Energia potenziale: che è correlata alla posizione di un oggetto rispetto agli altri e che dipende
dalla forza impressa sull’oggetto stesso. Un tipo di energia potenziale è l’energia potenziale elettrostatica che deriva dall’interazione tra le particelle cariche.
3. Energia interna di un sistema è la somma di tutte le energie cinetiche e potenziali di tutti i componenti del sistema. Generalmente non è possibile conoscere il valore assoluto di U, ma si può sperare conoscere ΔE cioè la differenza tra Efinale e Einiziale. ΔE = Efinale - Einiziale
Sistema e ambiente In termodinamica sono fondamentali due concetti: sistema e ambiente, cioè tutto ciò che circonda il sistema stesso. Si definisce sistema una porzione di universo, reale o immaginaria, separato dal restante universo tramite confini fisici (sistema reale) o condizioni matematiche (sistema immaginario). Il sistema può essere:
Aperto = un sistema può scambiare sia energia che materia con l’ambiente
Chiuso = un sistema può scambiare energia, ma non materia con l’ambiente
Isolato = un sistema non può scambiare né energia né materia con l’ambiente
Adiabatico = un sistema non può scambiare né energia né materia con l’ambiente, ma può invece scambiare calore
Da un punto di vista termodinamico, il sistema possiede delle proprietà che possono essere definite estensive o intensive. Le proprietà intensive sono quelle proprietà il cui valore non dipende dalla quantità di materia o dalle dimensioni del campione, ma soltanto dalla sua natura e dalle condizioni nelle quali si trova. Al contrario una proprietà si dice estensiva se il suo valore dipende dalle dimensioni del corpo a cui ci si riferisce. Si definisce stato termodinamico una condizione macroscopica per cui le proprietà che lo descrivo non variano nel tempo e sono determinate in modo univoco. Viceversa il processo termodinamico consiste nella modificazione dello stato termodinamico di n sistema per cui il sistema stesso passa da uno stato ad un altro; lo stato termodinamico non dipende dal processo termodinamico con cui lo si raggiunge. Un processo può essere:
Reversibile = passaggio da uno stato all’altro per cambiamento infinitesimi
Irreversibile = passaggio da uno stato ad un altro per cambiamento repentino Si definiscono funzioni di stato tutte quelle proprietà determinate univocamente dallo stato termodinamico del sistema; tali proprietà permetto di descrivere il sistema prescindendo dalla sua storia pregressa e sono di importanza primaria per comprendere e manipolare le variazioni dei sistemi stessi.
Trasferimento di energia Il trasferimento di energia da un sistema al suo ambiente (o viceversa) si verifica sotto forma di:
Lavoro (w) cioè l’energia trasferita ad un oggetto quando viene messo in movimento da una forza in opposizione ad un’altra. 𝑤 = 𝐹 ∗ 𝑑
1 𝑐𝑎𝑙 = 4,184 𝐽

Quando il lavoro avviene a pressione costante il lavoro può essere calcolato come 𝑤 = −𝑃 ∗ ∆𝑉
Calore cioè l’energia che viene trasferita da un corpo più caldo a uno più freddo. ˗ Può essere calcolato conoscendo capacità termica e sapendo se la variazione di temperatura
avviene a V costante 𝑞𝑝 = 𝑛 𝑐𝑝 ∆𝑇
˗ Può essere calcolato conoscendo capacità termica e sapendo se la variazione di temperatura avviene a P costante 𝑞𝑣 = 𝑛 𝑐𝑣 ∆𝑇
˗ Può essere calcolato conoscendo calore specifico 𝑞 = 𝑚 𝑐𝑠 ∗ ∆𝑇
Calorimetria Capacità termica = quantità di calore necessaria per aumentare di 1 °K la sua temperatura La
capacità termica di un oggetto è elevata, tanto maggiore è il calore richiesto per ottenere un determinato incremento di temperatura.
Capacità termica molare = quantità di calore necessaria per aumentare di 1 °K la temperatura di una mole di sostanza. Essa cambia a seconda che la variazione di temperatura avvenga a V costante o a P costante.
Calore specifico = quantità di calore richiesta perché un grammo-massa della sostanza aumenti
all’aumentare della temperatura di 1 °C 𝑐𝑠 = 𝑞
𝑚 ∆𝑇
I° principio della termodinamica Il primo principio della termodinamica (enunciato da R. Clausius nel 1865) afferma che: l'energia può essere convertita da una forma in un'altra ma non può essere né creata né distrutta ΔE = 𝑞 + 𝑤 In altri termini afferma che l’energia dell’universo rimane sempre costante.
ΔE𝑢𝑛𝑖𝑣 = ΔE𝑠𝑖𝑠 + ΔE𝑎𝑚𝑏 = 0 Tale formula permette di calcolare la variazioni di energia interna del sistema misurando lo scambio di lavoro e di calore tra sistema ed ambiente stesso. q e w singolarmente non sono funzioni di stato poiché dipendono dal processo che porta il sistema da uno stato iniziale A a uno stato finale B, ma la loro somma è funzione di stato poiché indipendente da tale processo. Si deve ricordare che qualunque variazione nell’energia del sistema è accompagnata da una corrispondente variazioni opposta nell’ambiente. In una reazione chimica lo stato iniziale del sistema è identificato dai reagenti e lo stato finale viene identificato dai prodotti. N.B.: ΔE > 0 energia ceduta dall’ambiente al sistema (processo endotermico)
ΔE < 0 energia ceduta dal sistema all’ambiente (processo esotermico)
Positivo (> 0) Negativo (< 0)
q Il sistema guadagna calore Il sistema perde calore
w Il lavoro è svolto dall’ambiente sul sistema
Il lavoro è svolto dal sistema sull’ambiente
ΔE Guadagno netto di energia da parte del sistema
Perdita netta di energia da parte del sistema
Entalpia È una funzione termodinamica che corrisponde all’energia interna del sistema più il prodotto tra pressione e volume del sistema 𝐻 = 𝐸 + 𝑃𝑉 È una funzione di stato poiché l’energia interna, la pressione e il volume sono tutte funzioni di stato; è infatti stabilita univocamente dallo stato iniziale e dallo stato finale indipendentemente dal precorso seguito per passare dall’uno all’altro. L’entalpia è una proprietà estensiva. In un processo a pressione costante la variazioni di entalpia ΔH è calcolabile come: Δ𝐻 = Δ𝐸 + 𝑃Δ𝑉 = 𝑞𝑝
La variazioni di entalpia equivale quindi al calore guadagnato o perso a pressione costante. N.B.: ΔH > 0 calore ceduto dall’ambiente al sistema (processo endotermico + non spontanea)
ΔH < 0 calore ceduto dal sistema all’ambiente (processo esotermico + reazione spontanea) Lo stato standard di una sostanza si identifica con la sua forma pura a pressione atmosferica (1 atm) e con una temperatura pari a 298 °K. La variazione di entalpia standard di una reazione è dunque definita come la variazione di entalpia che si ottiene quando tutti i reagenti e tutti i prodotti si trovano nel loro stato standard. Si chiama entalpia di formazione (∆H°f) di una sostanza pura la variazione di entalpia associata alla formazione di una mole di tale sostanza a partire dagli elementi componenti, in

condizioni standard (t = 25°C; P = 1 atm). Per definizione l’entalpia standard di formazione di ciascun elemento nella sua forma più stabile è zero. La variazione di entalpia associata ad una reazione chimica è detta entalpia di reazione o semplicemente calore di reazione e si indica con ∆H°reaz. La variazione di entalpia di una reazione, che dipende dallo stato dei reagenti e dei prodotti, è uguale in valore assoluto ed opposto al ∆H della corrispondente reazione inversa. L’entalpia di legame di un composto è la variazione di entalpia, espressa solitamente in kJ/mol, che accompagna il processo di formazione del composto, partendo dagli atomi isolati.
Legge di Hess Afferma che se una reazione è fatta avvenire attraverso una serie di passaggi, il ∆H della reazione globale sarà uguale alla somma delle variazioni di entalpia corrispondenti ai singoli passaggi. Dato un particolare set di reagenti e di prodotti il valore del ∆H è lo stesso sia che la reazione avvenga in un singolo passaggio, sia che avvenga con una serie di passaggi.
Termochimica Ci sono tre tipi di trasformazione fisiche particolarmente importanti, governate da altrettante leggi termodinamiche: 1. Trasformazioni isoterme: avvengono a temperatura costante, mentre variano la pressione e il
volume. Dato che T è costante e, dal momento in cui E dipende da T si ha che ΔE = 0.
∆𝐸 = 0
∆𝐻 = 0
𝑤 = − ∫ 𝑃 𝑑𝑉𝑣2
𝑣1
= −𝑛𝑅𝑇 ∫1
𝑉 𝑑𝑉
𝑣2
𝑣1
= −𝑛𝑅𝑇 𝑙𝑛𝑣2
𝑣1
𝑞 = −𝑤 = 𝑛𝑅𝑇 𝑙𝑛𝑣2
𝑣1
2. Trasformazioni isobare: avvengono a pressione costante, mentre variano la temperatura e il
volume. ∆𝐸 = 𝑞 + 𝑤 = 𝑛∆𝑇(𝑐𝑣 − 𝑅) = 𝑛∆𝑇𝑐𝑝
∆𝐻 = 𝑞𝑝
3. Trasformazioni isocore: avvengono a volume costante, mentre variano la temperatura e la
pressione. 𝑤 = 0
∆𝐸 = 𝑞𝑣 = 𝑛∆𝑇𝑐𝑣
∆𝐻 = ∆𝑇𝑛(𝑐𝑣 + 𝑅)
4. Processi adiabatici: processo o trasformazione fisica delle variabili macroscopiche di un sistema
termodinamico (pressione, temperatura, volume) da uno stato fisico ad un altro senza scambi di calore con l'ambiente circostante al sistema.
∆𝐸 = 𝑤 = 𝑛∆𝑇𝑐𝑣
∆𝐻 = 𝑛∆𝑇𝑐𝑝
II° principio della termodinamica Il secondo principio della termodinamica, noto anche come enunciato di Clausius, afferma che: il calore non può spontaneamente fluire da un corpo freddo a uno più caldo; in altri termini afferma che lo stato inziale (ambiente + sistema) spontaneamente evolve nello stato finale (ambiente + sistema)

se e solo se lo stato finale è più probabile cioè ha maggior disordine di materia e/o energia di quello iniziale. Ciò può verificarsi in due modi: 1. Reazione disordinante: aumentando il disordine delle particelle dispersione materia 2. Reazione ordinante: disperdendo l’energia su un numero maggiore di particelle dispersione
energia N.B.: Se una reazione chimica è disordinante ed esotermica allora è sempre spontanea
Se una reazione chimica è ordinante ed endotermica allora la reazione non avviene mai Se si verifica una sola condizione (orinante/esotermica/disordinante/endotermica) allora è necessario analizzare la temperatura: a T basse prevale la dispersione di energia mentre a T alta prevale la dispersione di materia.
Un sistema può essere considerato un macrostato divisibile in microstati cioè le configurazioni (i modi) che si possono realizzare per avere una determinata disposizione (combinazione), ovvero un macrostato. Il numero di microstati che compongono un macrostato vengono chiamati probabilità termodinamica W. Si chiama dunque entropia, S, la grandezza termodinamica che esprime lo stato di disordine di un sistema dato. L’entropia si lega alla probabilità e quindi ai microstati tramite la relazione di Boltzman: 𝑆 = 𝑘 𝑙𝑛𝑊 N.B.: ΔSsistema > 0 processo disordinante
ΔSsistema < 0 processo ordinante
L’entropia, che è una funzione di stato, si può calcolare come variazione ∆𝑆 = 𝑞
𝑇.
Inoltre si può afferma che l’entropia dell’universo è pari alla somma dell’entropia del sistema più quella dell’ambiente: ∆𝑆𝑢𝑛𝑖𝑣𝑒𝑟𝑠𝑜 = ∆𝑆𝑠𝑖𝑠𝑡𝑒𝑚𝑎 + ∆𝑆𝑎𝑚𝑏𝑖𝑒𝑛𝑡𝑒 . L’entropia S aumenta all’aumentare di T mentre la variazione di entropia ΔS diminuisce all’aumentare di T. L’entropia di qualunque sostante nel suo stato di equilibrio raggiunge il valore 0 alla temperatura dello zero assoluto.
III° principio della termodinamica Il terzo principio della termodinamica, noto come principio di Nernst, afferma che allo zero assoluto, l'entropia di un cristallo puro è uguale a zero 𝑆 = 0 quando T = 0°K Il terzo principio consente di determinare i valori assoluti dell'entropia. Poiché conosciamo il punto in cui l'entropia ha valore zero, si può determinare la reale quantità di entropia che una sostanza possiede a una temperatura superiore a 0 K. Se l'entropia di una mole di una sostanza è determinata alla temperatura di 298 K (25 °C) e alla pressione di 1 atm, è chiamata entropia standard (S°) espressa in cal/K o joule/K. Una volta note le entropie di diverse sostanze possiamo calcolare la variazione di entropia standard (Δ S°), per le reazioni chimiche
∆𝑆° = ∑ 𝑆𝑝𝑟𝑜𝑑𝑜𝑡𝑡𝑖0 − ∑ 𝑆𝑟𝑒𝑎𝑔𝑒𝑛𝑡𝑖
0 dove S° per ciascuna specie è moltiplicato per il coefficiente
stechiometrico di reazione. Il Δ S°f (entropia standard di formazione di un composto) si ricava dai valori di S°.
Energia libera di Gibbs Tra il 1875 e il 1876, lo statunitense J. W. Gibbs dimostrò inequivocabilmente che l'unico criterio per stabilire la spontaneità di una reazione è la sua capacità di produrre lavoro utile. Ciò significa che se a temperatura e pressione costante una reazione può produrre lavoro utile essa è termodinamicamente consentita, cioè spontanea. Si definisce dunque l’energia libera di Gibbs come: 𝐺 = 𝐻 − 𝑇𝑆 da cui deriva ∆𝐺 = ∆𝐻 − 𝑇∆𝑆 N.B.: ΔGsistema > 0 processo non spontaneo
ΔGsistema = 0 processo reversibile ΔGsistema < 0 processo spontaneo
Schema generale
ΔH < 0 ΔS > 0 ΔG < 0 Reazione sempre spontanea
ΔH < 0 ΔS < 0 / Reazione spontanea a basse temperature
ΔH > 0 ΔS > 0 / Reazione spontanea ad alte temperature
ΔH > 0 ΔS < 0 ΔG > 0 Reazione mai spontanea

EQUILIBRI
Soluzioni Una soluzione è un miscuglio omogeneo di due o più componenti. Essa può essere:
Satura = è una soluzione che esiste in equilibrio dinamico con il soluto presente come corpo di fondo. Si parla di equilibrio dinamico in quanto molecole di soluto presenti come corpo di fondo si sciolgono mentre molecole di soluto presenti in soluzione precipitano continuamente: le molecole del solvente solvatano molecole del soluto. Per solvatazione in chimica si intende l'interazione tra soluto e solvente che porta le singole molecole di soluto disciolto a circondarsi di molecole di solvente.
Ideale = è una soluzione caratterizzata da 3 proprietà: 1. Le molecole del soluto non devono interagire fra loro 2. Ad alte concentrazioni la soluzione devia dal comportamento ideale 3. L’aggiunta di un solvente a una soluzione satura fa diminuire la concentrazione delle sostanze
disciolte. Il sistema tende allora a reagire per neutralizzare la modificazione: altro solido passa in soluzione.
Dissoluzione e precipitazione I processi di dissoluzione e di precipitazione non si portano istantaneamente all’equilibrio, ma possono richiede anche parecchio tempo di agitazione della sospensione del soluto in solvente prima di arrivare all’equilibrio, cioè alla soluzione satura. Nel momento in cui una soluzione diventa soprassatura, cioè quando nel solvente è presente una quantità di soluto maggiore di quella presente all’equilibrio, può a sua volta passare molto tempo prima che si verifichi la precipitazione.
Solubilità Si definisce solubilità la massima quantità, in grammi o moli, di una certa sostanza che può essere sciolta in un certo solvente in condizioni di equilibrio, ad una certa temperatura. La solubilità di una sostanza non dipende solo dalla temperatura, ma anche dalla presenza di altri soluti (la presenza di un acido, ad esempio, può avere grande influenza sulla solubilità di una sostanza). Nello specifico, in un composto ionico i fattori che influenzano la solubilità sono: 1. Presenza di ioni comune = la solubilità di un sale poco solubile decresce in presenza di un secondo
soluto che fornisce uno ione comune 2. Il pH della soluzione = la solubilità di Sali poco solubili che contengo anioni basici aumenta
all’aumentare di [H+], cioè al diminuire del Ph.
Equilibri di solubilità Quasi tutti i sali sono degli elettroliti forti quindi anche i sali poco solubili si dissociano completamente. Immaginando di avere una soluzione acquosa satura di un generico sale poco solubile si avrà in soluzione anche una certa quantità di ioni derivanti dallo stesso sale. Tra il sale disciolto e i suoi ioni in soluzione si instaura immediatamente un equilibrio noto come equilibrio di solubilità. Poiché questa è una reazione di equilibrio sarà necessariamente presente una costante, nota come prodotto di solubilità e indicata con il simbolo kps, che mostra quanto il solido stesso sia solubile in acqua. Il prodotto di solubilità è uguale al prodotto delle concentrazioni degli ioni coinvolti nell’equilibrio, ciascuno elevata ad un esponente pari al proprio coefficiente stechiometrico nell’equazione di equilibrio. Il raggiungimento dell’equilibrio può avvenire sia a partire dai reagenti puri, sia a partire dai prodotti; analizzando il prodotto delle concentrazioni iniziali (Q) e il kps è possibile intuire quando la precipitazione del solido inizia o finisce:
Se Q (prodotto delle concentrazioni iniziali) > kps la precipitazione inizia quando Q = kps
Se Q = kps il sistema è all’equilibrio (soluzione satura)
Se Q < kps si ha dissoluzione del solido fino a che Q = kps
Solubilità Sali
Solubili > 10 gL-1 Parzialmente solubili da 0,1 a 10 gL-1 Insolubili < 0,1 gL-1

Equilibrio di fase L'equilibrio liquido-vapore è la condizione in cui due fasi, quella liquida e quella vapore, stanno in equilibrio termodinamico tra loro. L'equilibrio che si viene a creare è di tipo dinamico, ovvero la velocità di evaporazione del liquido eguaglia la velocità di condensazione del vapore. Infatti l'interfaccia liquido-vapore è interessata da continui scambi di materia tra le due fasi. All’equilibrio inoltre la tensione di vapore dell’acqua e il suo volume non cambiano nel tempo. Dal punto di vista macroscopico si assiste a una situazione statica, mentre nel microscopico è di tipo dinamico.
Equilibrio chimico Analizzando una qualsiasi reazione chimica del tipo 𝑎𝐴 + 𝑏𝐵 ⇄ 𝑐𝐶 + 𝑑𝐷 si può affermare di essere in equilibrio chimico nel momento in cui non tutte le molecole dei reagenti si trasformano completamente in molecole di prodotti. In questi tipi di reazioni infatti solo alcune molecole di reagenti si trasformano in molecole di prodotti e viceversa. Si parla dunque di equilibrio chimico quando reazioni opposte procedono alla stessa velocità: la velocità di formazione dei prodotti è uguale alla velocità con cui i reagenti sono formati dai prodotti. Una volta che l’equilibrio è raggiunto le concentrazioni rimangono costanti. All’equilibrio inoltre il rapporto di specifici termini espressi in funzione delle concentrazioni è uguale a una costante. La composizione del sistema all’equilibrio dipende esclusivamente dalla temperatura, non dalla composizione di partenza. I sistemi in equilibrio: 1. Non mostrano a livello macroscopico nessuna variazioni misurabile 2. Sono raggiunti attraverso variazioni spontanee 3. Mostrano un bilanciamento dinamico tra processo diretto e inverso che avvengono con la stessa
velocità 4. L’equilibrio è raggiuto si partendo dai reagenti che partendo dai prodotti.
Per poter esprime il rapporto fra le concentrazioni dei reagenti e dei prodotti presenti all’equilibrio fu postulata nel 1864 da Guldberg e Waage la legge di azione di massa secondo cui lo stato di equilibrio è caratterizzato da una costante nota come kc. L’espressione di tale costante dipende, dal punto di vista matematico, dalla stechiometria della reazione, mentre dal punto di vista chimico-fisico dalla temperatura. Quando nell’espressione della costante di equilibrio si usano le pressioni parziali espresse in atmosfere è possibile denotare la costante di equilibrio come kp
𝑘𝑐 = [𝐷]𝑑[𝐶]𝑐
[𝐴]𝑎[𝐵]𝑏
𝑘𝑝 = [𝑃𝐷]𝑑[𝑃𝐶]𝑐
[𝑃𝐴]𝑎[𝑃𝐵]𝑏
𝑘𝑝 = 𝑘𝑐 (𝑅𝑇)∆𝑛
Interpretazione delle costanti di equilibrio Generalmente, per qualsiasi costante di equilibrio k, è possibile affermare:
Se k >> 1 l’equilibrio è spostato a destra (i prodotti dominano)
Se k << 1 l’equilibrio è spostato a sinistra (i reagenti dominano) È inoltre possibile affermare che l’espressione della costante di equilibrio per una reazione scritta in un senso è il reciproco di quella della reazione scritta nel senso inverso. La costante di equilibrio di una reazione che è stata moltiplicata per un dato numero è la costante di equilibrio elevata ad un esponente uguale a quel numero. Infine la costante di equilibrio per una reazione netta composta da due o più stadi è il prodotto delle costanti di equilibrio dei singoli stadi.
Direzione di evoluzione di un sistema Con la lettera Q si indica anche il quoziente di reazione cioè un numero ottenuto sostituendo le concentrazioni o le pressioni parziali iniziali dei prodotti e dei reagenti dell’espressione della costante di equilibrio.

(𝑃𝐷)𝑑(𝑃𝐶)𝑐
(𝑃𝐴)𝑎(𝑃𝐵)𝑏 = 𝐾(𝑇)
pressioni dei componenti all’equilibrio
𝑃𝐷
𝑑𝑃𝐶𝑐
𝑃𝐴𝑎𝑃𝐵
𝑏 = 𝑄
pressioni dei componenti istantanee A partire dall’analisi di Q e di k è possibile stabile la direzione verso cui procederà la reazione per realizzare l’equilibrio:
Q > k la reazione si sposta da destar verso sinistra perché la concentrazione dei prodotti è troppo grande e quella dei reagenti troppo piccola (la reazione raggiunge l’equilibrio formando più reagenti)
Q = K il sistema è all’equilibrio
Q < k la reazione si sposta da sinistra verso destra poiché la concentrazione dei prodotti è troppo piccola e quella dei reagenti è troppo grande (la reazione raggiunge l’equilibrio formando più prodotti)
Equilibri eterogeni Si definiscono equilibri eterogenei tutti quegli equilibri che si instaurano tra sostanze facenti parte di fasi diverse. A partire da questi tipi di equilibri è possibile generalizzare la legge dell’azione di massa: 1. I gas entrano nelle espressioni dell’equilibrio come pressioni parziali 2. Le specie disciolte entrano nelle espressioni dell’equilibrio come concentrazioni 3. I liquidi e i solidi non compaiono nelle espressioni dell’equilibrio, né vi compare il solvente quando
si opera con una soluzione (a meno che non sia particolarmente concentrata) 4. Le pressioni parziali e le concentrazioni delle specie che compaiono al numeratore e al
denominatore di k sono elevate ciascuna all’esponente pari al suo coefficiente stechiometrico nell’equazione bilanciata.
Principio di Le Chatelier Secondo il principio di Le Chatelier quando un sistema in equilibrio è soggetto a una modifica dall’esterno, reagisce per controbilanciare tale modifica. Ciò vale per tutti i parametri fisici e chimici del sistema stesso. In altre parole se un sistema all’equilibrio viene disturbato da una variazione di temperatura, pressione, o concentrazione di uno dei componenti, il sistema sposterà la relativa posizione di equilibrio in modo da neutralizzare l’effetto della variazione. Di conseguenza, se un sistema chimico è all’equilibrio e si aggiunge una sostanza, la reazione si sposterà in modo da ristabilire l’equilibrio consumando parte della sostanza aggiunta. Per contro, la rimozione di una sostanza indurrà la reazione a spostarsi nel senso che forma quella sostanza in quantità maggiore. A temperatura costante, la diminuzione del volume di una miscela gassosa di equilibrio induce il sistema a spostarsi nel senso che riduce il numero di moli di gas. Quando aumentiamo la temperatura di un sistema all’equilibrio, è come se avessimo aggiunto un reagente ad una reazione endotermica o un prodotto ad una reazione esotermica. L’equilibrio si sposta nel senso che consuma il reagente o il prodotto eccedente, vale a dire nominalmente il calore.
Processo endotermico: l’aumento di T provoca un aumento di K. L’equilibrio si sposta da sinistra a destra: verso i prodotti per controbilanciare la perturbazione
Processo esotermico: l’aumento di T provoca una diminuzione di K. L’equilibrio si sposta da destra a sinistra: verso i reagenti.

ACIDI E BASI
Arrhenius Negli anni 80 del 1800 il chimico svedese Arrhenius correlò il comportamento acido con la presenza di ioni H+ e il comportamento basico con la presenza i ioni OH-in soluzioni acquose. Egli definì quindi gli acidi come quelle sostanze che quando sono disciolte in acqua aumentano la concentrazione degli ioni H+, mentre le basi sono quelle sostanze che disciolte in acqua aumentano la concentrazione di ioni OH-
La definizione di Arrhenius fa riferimento esclusivamente all’acqua come solvente. Il concetto di acidi e basi espresso da Arrhenius è quindi limitante poiché ristretto alle soluzioni acquose.
Bronsted-Lowry Bronsted-Lowry proposero una definizioni di acidi e basi a partire dalla loro abilità di trasferire protoni: essi definirono un acido come una sostanza che può donare un protone ad un’altra sostanza. Analogamente, una base è una sostanza che può accettare un protone. Entrambi questi scienziati posero dunque l’accento sul trasferimento di protoni; ciò permise di studiare anche le reazioni che avvengono in qualsiasi solvente. In altre parole una sostanza può funzione come un acido sole so un’altra sostanza si comporta simultaneamente come una base. N.B.: alcune sostanze possono agire da acido in una reazione e da base in un’altra. Tali sostanze che possono agire sia da acido che da base sono dette anfiprotiche.
Lewis Lewis definì invece gli acidi come sostanza in grado di accettare una o più coppie di elettroni liberi, mentre parlò di basi come sostanze in grado di donare una o più coppie di elettroni liberi.
Reazioni acido-base In ogni equilibrio acido-base ogni acido possiede la corrispettiva base coniugata e viceversa. Si definisce base coniugata l’acido che è stato privato di un protone. Si parla invece di acido coniugato quando si ha una base più un protone. Più forte è un acido, più debole è la sua base coniugata; più forte è la base, più debole è il suo acido coniugato. In ogni reazione acido-base è favorito il trasferimento del protone verso la base più forte. In tal modo, la reazione di un acido di una base forte tende a formare un acido o una base debole. Si definiscono acidi monoprotici gli acidi che liberano un unico ione H+ per molecola, mentre si definiscono poliprotici tutti gli acidi che hanno più di un idrogeno ionizzabile. Tendenzialmente è sempre più facile rimuovere il primo protone di un acido poliprotico rispetto al secondo. Quando una soluzione di un acido è mescolata con la soluzione di una base ne consegue una reazione di neutralizzazione.
Autoionizzazione dell’acqua Nell'acqua sono presenti degli ioni, anche se in concentrazione molto limitata, che si formano secondo la seguente reazione: Si tratta di un normale equilibrio acido-base secondo il quale una molecola d'acqua si comporta da acido e un'altra molecola di acqua si comporta da base. Questa reazione è detta di autoionizzazione o di autoprotolisi. Questo equilibrio è estremamente spostato a sinistra poiché le molecole d’acqua implicate in tale processo di ionizzazione sono pochissime. In acqua pura sono dunque presenti, anche se in quantità minime, ioni OH- e H3O+. La costante di equilibrio per la reazione di autoionizzazione dell’acqua viene detta prodotto ionico dell’acqua ed indicata dal simbolo Kw: 𝑘𝑤 = [𝐻3𝑂+][𝑂𝐻−] = [𝐻+][𝑂𝐻−] = 1,0 ∗ 10−14 . Tale equazione è considerata valida per ogni soluzione diluita e può dunque essere usata per calcolare la concentrazione degli H3O+, se nota quella degli OH- e viceversa. Una soluzione in cui la concentrazione degli ioni H+ è pari a quella degli ioni OH- è detta neutra. Tendenzialmente però, in una soluzione, le concentrazioni di OH- e H+ non sono uguali; se la concentrazione di uno di questi due ioni aumenta, la concentrazione dell’altra diminuisce, in modo tale che il loro prodotto rimanga sempre una costante.

N.B: Soluzione acida [H+] > [OH-] Soluzione basica [H+] < [OH-]
La scala del pH La concentrazione molare di H+ in una soluzione acquosa è normalmente molto bassa. Per convenienza quindi si usa esprimere [H+] in termini di pH cioè meno il logaritmo in base 10 di [H+]. Esiste anche il pOH definito come meno il logaritmo in base 10 di [OH-]. 𝑝𝐻 = − log10 [𝐻3𝑂+] 𝑝𝑂𝐻 = − log10 [𝑂𝐻−] 𝑝𝐻 + 𝑝𝑂𝐻 = 14 Il pH di una soluzione può essere misurato rapidamente e accuratamente con un pH-metro. La sonda per pH è generalmente un elettrodo a vetro che misura la differenza di potenziale elettrico su due lati di una sottile membrana di vetro posta all'estremità dell'elettrodo, tale differenza di potenziale è legata alla differenza tra le concentrazioni degli ioni idrogeno all'interno e all'esterno della membrana. Un altro modo per conoscere il pH di una sostanza è quello di usare degli indicatori acido-base, come, ad esempio, una cartina tornasole; sebbene siano meno precisi permettono di capire l’acidità o la basicità di una determinata sostanza. Un indicatore altro non è che un acido o una base molto debole e che quindi in soluzione acquosa è coinvolto in un equilibrio di dissociazione. Molti coloranti naturali sono indicatori di pH, come ad esempio la cianidina. La cianidina è un pigmento organico naturale appartenente alla classe delle antocianidine. Ha un caratteristico colore rosso-arancio, anche se questo può cambiare con il pH, le soluzioni del composto sono di colore rosso a pH <3, viola a pH 7-8, e blu a pH >11.
Forti Gli acidi e le basi elettroliti forti, cioè che si dissociano completamente in soluzione, sono chiamati acidi forti e basi forti. Per un acido forte la quantità di ioni H+ presenti in soluzione è praticamente equivalente alla quantità di molecole dell’acido stesso aggiunte alla soluzione e quindi il pH si calcola: 𝑝𝐻 = − log10 [𝑎𝑐𝑖𝑑𝑜]. L’equilibrio è spostato quantitativamente verso destra.
Deboli e costante di ionizzazione Quelli che invece si disciolgono parzialmente si chiamano acidi deboli e basi deboli. Gli acidi e le basi deboli sono dunque parzialmente ionizzati in soluzione acquosa. Per esprimere quanto un acido o una base debole sia ionizzato possiamo utilizzare la costante di equilibrio per la reazione di dissociazione. Tale costante prende il nome di costante di dissociazione acida o basico e si indica con i simboli ka e kb. L’ordine di grandezza di ka indica la tendenza dell’acido a ionizzarsi in acqua: maggiore è il valore di ka, più forte è l’acido. Stessa cosa per kb.
𝐻𝐴 + 𝐻2𝑂 ⇌ 𝐻3𝑂+ + 𝐴− 𝐻𝐴 ⇌ 𝐻+ + 𝐴− 𝑘𝑎 = [𝐻3𝑂+][𝐴−]
[𝐻𝐴]=
𝑘𝑤
𝑘𝑏
𝐵𝑂𝐻 + 𝐻2𝑂 ⇌ 𝐵+ + 𝑂𝐻− 𝐵𝑂𝐻 ⇌ 𝐵+ + 𝑂𝐻− 𝑘𝑏 = [𝐵+][𝑂𝐻−]
[𝐵𝑂𝐻]=
𝑘𝑤
𝑘𝑎
𝑘𝑤 = 𝑘𝑎 ∗ 𝑘𝑏 𝑝𝑘𝑎 + 𝑝𝑘𝑏 = 𝑝𝑘𝑤 = 14,00 (a 25°C)
Per acido: [𝐻3𝑂+] ≃ √𝑘𝑎[𝐻𝐴] 𝑝𝐻 ≃ −log10 √𝑘𝑎[𝐻𝐴]
Per base: [𝑂𝐻−] ≃ √𝑘𝑏[𝐵𝑂𝐻] 𝑝𝑂𝐻 ≃ −log10 √𝑘𝑏[𝐵𝑂𝐻]
Soluzione acida
[H+] > 10-7 pH < 7 [OH-] < 10-7
Soluzione neutra
[H+] = 10-7 pH = 7 [OH-] = 10-7
Soluzione basica
[H+] < 10-7 pH > 7 [OH-] > 10-7
Acidi forti Basi forti
Cloridrico, HCl Idrossido di sodio, NaOH Bromidrico, HBr Idrossido di litio, LiOH Iodidrico, HI Idrossido di potassio, KOH Clorico, HClO3 Diidrossido di bario, Ba(OH)2 Perclorico, HClO4 Diidrossido di calcio, Ca(OH)2 Nitrico, HNO3 Solforico, H2SO4

Acidità e struttura dei composti Prendendo il considerazione un composto X-O-H è possibile intuire se si comporterà da acido o da base ragionando sull’elettronegatività di X e di H:
Elettronegatività X > elettronegatività di H: il composto è acido poiché O attrae elettroni che lo legano ad H e verrà dunque liberato lo ione H+
Elettronegatività X < elettronegatività di H: il composto è basico poiché O attrae gli elettroni che lo legano a X e verrà dunque liberato lo ione OH-
Proprietà acido-basiche delle soluzioni saline Quasi tutti i sali sono elettroliti forti; di conseguenza le proprietà acido-basiche dei sali sono dovute al comportamento dei cationi e anioni che li costituiscono. Molti degli ioni liberati sono in grado di reagire con l’acqua a formare H+ e OH-: questo tipo di reazioni viene detta idrolisi. È inoltre possibile prevedere qualitativamente il pH di una soluzione acquosa salina considerando gli ioni che formano il sale:
Acido forte + base forte = soluzione neutra (es: HCl + NaOH)
Acido forte + base debole = idrolisi acida (es: HCl + NH3). Il pH dipende dal valore della ka del catione
Acido debole + base forte = idrolisi basica (es: CH3COOH + NaOH). Il pH dipende dal valore della kb dell’anione
Acido debole + base debole = sale il cui catione è l’acido coniugato della base debole e in cui l’anione è la base coniugata dell’acido debole. Il pH della soluzione dipende dai relativi valori della ka e della kb
Soluzioni tampone Si dicono soluzioni tampone quelle soluzioni in cui sono presenti un acido debole e la sua base coniugata (oppure una base debole e il suo acido coniugato) in un rapporto fra le concentrazioni stechiometriche compresa tra 0,1 e 10. In altre parole le soluzioni tampone sono soluzioni contenenti miscele di soluti che impediscono significative variazioni di pH per aggiunta di moderate quantità di acidi e di basi forti. Le proprietà di queste soluzioni sono: 1. Il pH non varia con la diluizione poiché in queste soluzione il pH dipende esclusivamente dal
rapporto tra le concentrazioni stechiometriche e dalla ka (o dalla kb) 2. Il pH tende a rimanere costante per piccole aggiunte di acidi e basi forti 3. Il potere tamponante di una soluzione nei confronti di piccole aggiunte di ioni H+ e OH- è massimo
quando [𝐻𝐴]
[𝐴−]= 1
Si definisce effetto dello ione comune l’entità della dissociazione di un elettrolita debole che decresce per aggiunta alla soluzione di un elettrolita forte che abbia uno ione in comune con l’elettrolita debole (cioè è l’effetto provocato su un equilibrio per aggiunta di uno ione “in comune” ad uno già presente nell’equilibrio stesso). Considerando ora un sale che si dissocia completamente
𝐻𝑋 + 𝐻2𝑂 ⇄ 𝑋− + 𝐻3𝑂+ 𝑘𝑎 = [𝐻3𝑂+][𝑋−]
[𝐻𝑋]
𝑀𝑋 → 𝑀+ + 𝑋− tutto il sale si dissocia La dissociazioni del sale fa ulteriormente spostare l’equilibrio della dissociazioni dell’acido verso sinistra. Quindi possiamo approssimare la concentrazione della forma indissociata HX con [HX] cioè la concentrazione iniziale dell’acido e quella dell’anione X- con quella del sale [X-].
Per acido: [𝐻3𝑂+] = 𝑘𝑎 ∗ [𝐻𝑋]
[𝑋−] 𝑝𝐻 = 𝑝𝑘𝑎 + log10
[𝑋−]
[𝐻𝑋]
Per base: [𝑂𝐻−] = 𝑘𝑏 ∗ [𝑋𝑂𝐻]
[𝑋+] 𝑝𝑂𝐻 = 𝑝𝑘𝑏 + log10
[𝑋+]
[𝑋𝑂𝐻]
Titolazione acido-base La titolazione è una tecnica di analisi chimica utilizzata per determinare la concentrazione incognita di un acido o di una base in una soluzione (analita) facendola reagire con una soluzione a concentrazione (titolo) nota rispettivamente di una base o un acido (titolante). Durante ‘aggiunta della soluzione basica
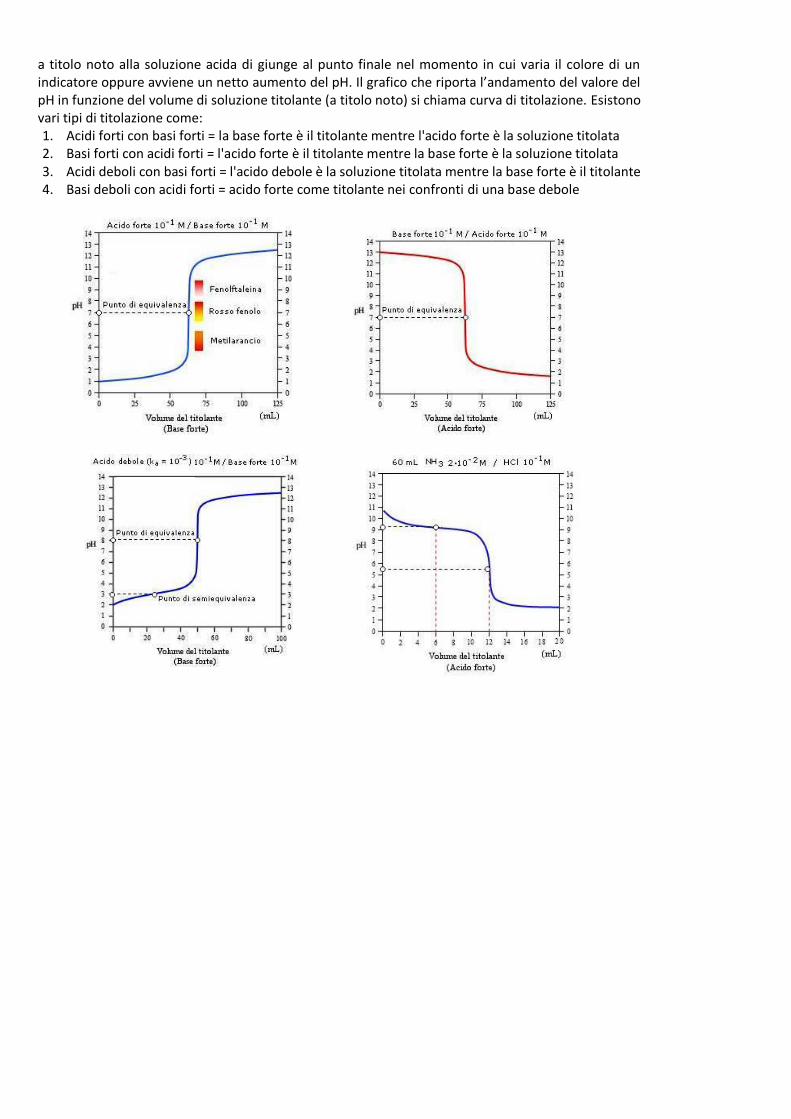
a titolo noto alla soluzione acida di giunge al punto finale nel momento in cui varia il colore di un indicatore oppure avviene un netto aumento del pH. Il grafico che riporta l’andamento del valore del pH in funzione del volume di soluzione titolante (a titolo noto) si chiama curva di titolazione. Esistono vari tipi di titolazione come: 1. Acidi forti con basi forti = la base forte è il titolante mentre l'acido forte è la soluzione titolata 2. Basi forti con acidi forti = l'acido forte è il titolante mentre la base forte è la soluzione titolata 3. Acidi deboli con basi forti = l'acido debole è la soluzione titolata mentre la base forte è il titolante 4. Basi deboli con acidi forti = acido forte come titolante nei confronti di una base debole

ELETTROCHIMICA
Celle galvaniche L’energia rilasciata n una redox spontanea può essere utilizzata per ottenere del lavoro elettrico: proprio su questo principio si basano le celle galvaniche o voltaiche cioè le comuni pile. Una pila è costituita da due elettrodi collegati tra loro tramite un filo metallico, attraverso il quale avviene il passaggio di elettroni. Questo filo metallico a sua volta è connesso a un amperometro che permette di misurare il passaggio di corrente. Ciascuno dei due componenti di una cella voltaica è chiamata semicella. La semicella in cui avviene la reazione di ossidazione (OX) prende il nome di anodo mentre la semicella dove avviene la riduzione (RED) prende il nome di catodo. Affinché la cella voltaica possa funzionare correttamente deve avere un ΔG < 0: le soluzioni delle due semicelle devono dunque rimanere elettricamente neutre e perciò è necessario un ponte salino che permette una migrazione di ioni in modo da mantenere la neutralità elettrica delle soluzioni stesse. In ogni cella voltaica gli elettroni fluiscono dall’anodo al catodo attraverso il circuito esterno a causa della differenza di energia potenziale. L’energia potenziale degli elettroni è più alta nell’anodo e dunque gli elettroni fluiscono spontaneamente attraverso il circuito esterno al catodo. Poiché gli elettroni carichi negativamente fluiscono dall’anodo al catodo, l’anodo in una cella voltaica è indicato con un segno negativo e il catodo con un segno positivo. Tale energia potenziale è misurata in Volt ed è detta forza elettromotrice (fem) poiché rappresenta la froza guida che spinge l’elettrone attraverso il circuito esterno. La fem di una cella è spesso indicata con Ecell. Nelle reazioni di cella che procedono spentamente, come quelle di una cella voltaica, il potenziale di cella è positivo. La fem di una particolare cella voltaica dipende dalle reazioni specifiche che avvengono al catodo e all’anodo, dalla concentrazione dei reagenti, dei prodotti e dalla temperatura (se non è specificata la assumiamo pari a 25 °C).
Potenziale di riduzione standard In condizioni standard la fem è detta fem standard o potenziale standard. Il potenziale di cella E°cell è dato dal potenziale di riduzione standard della reazione catodica meno il potenziale di riduzione
standard della reazione anodica: 𝐸𝑐𝑒𝑙𝑙° = 𝐸𝑐𝑎𝑡𝑜𝑑𝑜
° − 𝐸𝑎𝑛𝑜𝑑𝑜°
Per definire in una coppia redox chi è anodo e chi è catodo è necessario confrontare i potenziali di riduzione delle due specie metalliche: gli elettroni fluiranno dalla specie che ha potenziale di riduzione minore (anodo - ossida) a quello maggiore (catodo - riduce). Se assegniamo un potenziale di riduzione standard ad una certa semi-reazione di riferimento, possiamo determinare il potenziale di riduzione standard di altre semi-reazioni, relativamente a questo riferimento. La semi-reazione di riferimento utilizzata è:
2𝐻+ + 2𝑒− → 𝐻2 𝐸𝑟𝑖𝑑° = 0 𝑉
Se la specie chimica ha un potenziale di riduzione negativo sarà riducente forte, se invece il suo potenziale di riduzione è positivo sarà ossidante forte. Il comportamento di tutte le altre specie che hanno potenziale di riduzione medio dipenderò dalla coppia redox a cui sono legate. N.B.: E > 0 indica un processo spontaneo E < 0 indica un processo non spontaneo
Disproporzione Una specie che può sia trasformarsi nella sua specie ossidata che in quella ridotta si chiama disproporzione o dismutazione (es: Cu).

Fem: condizioni standard La variazione dell’energia libera di Gibbs è la misura della spontaneità di un processo che avviene a temperatura e pressione costante. Dal momento che la forza elettromotrice, E, di una reazione redox indica se la reazione è spontanea o meno è possibile instaurare una relazione tra queste due entità:
∆𝐺 = −𝑛𝐹𝐸 dove F è la costante di Faraday 1 𝐹 = 96485 𝐶
𝑚𝑜𝑙= 96485
𝐽
𝑉𝑚𝑜𝑙 . Da questa formula
derivata la corrispettiva ∆𝐺° = −𝑛𝐹∆𝐸° = −𝑅𝑇𝑙𝑛𝐾 dove K rappresenta la costante di equilibrio.
Fem: condizioni non standard (equazione Nernst) La fem generata in condizioni non standard può essere calcolare utilizzando l’equazione determinata da Nernst, un chimico tedesco che stabilì molto leggi fondamentali dell’elettrochimica. ∆𝐺 = ∆𝐺° + 𝑅𝑇𝑙𝑛𝑄
∆𝐺 = −𝑛𝐹∆𝐸
∆𝐺° = −𝑛𝐹∆𝐸°
−𝑛𝐹∆𝐸 = −𝑛𝐹∆𝐸° + 𝑅𝑇𝑙𝑛𝑄
∆𝐸 = ∆𝐸° −𝑅𝑇
𝑛𝐹𝑙𝑛𝑄
Celle a concentrazione Le pile a concentrazioni sono pile in grado di erogare corrente elettrica sfruttando due semicelle galvaniche contenenti la medesima specie chimica a due concentrazioni diverse. In altre parole le pile a concentrazione sono pile formate da due elettrodi dello stesso metallo immersi in due soluzioni di un suo sale a diversa concentrazione. Le due soluzioni sono separate anch’esse dal un ponte salino.
Elettrolisi L’elettrolisi è un processo che utilizza energia elettrica per fare avvenire reazioni redox naturalmente spontanee in senso contrario. Nelle celle elettrolitiche avviene la trasformazione dell’energia elettrica in energia chimica. Come nelle celle voltaiche l’elettrodo nel qual avviene la riduzione è chiamato catodo e l’elettrodo al quale avviene l’ossidazione è detto anodo. L’elettrodeposizione utilizza ad esempio l’elettrolisi per depositare un sottile strato di metallo su un altro metallo in modo da migliorarne l’aspetto o la resistenza alla corrosione.
Cella galvanica Cella elettrolitica
Il flusso di elettroni avviene spontaneamente
Un potenziale esterno forza gli elettroni in un flusso opposto a quello spontaneo
Trasforma energia chimica in energia elettrica.
Trasforma energia elettrica in energia chimica
Tipi di elettrodi Esistono due tipi di elettrodi: 1. Inerti = non danno reazioni, ma fungono solo da superficie ove avvengono le reazioni di
ossidazione e riduzione 2. Attivi = partecipano al processo di elettrolisi
Tipi di pile Esistono due tipi di pile:
1. Pile primarie = non sono ricaricabili. Prendendo ad esempio la batteria al piombo si ha l’anodo di Pb e il catodo di PbO2. Se lasciati per molto tempo in condizione di scarica i cristalli piccoli di PbSO4 si trasformano in pochi cristalli grossi rendendo poco efficiente per la ricarica.
2. Pile secondarie = sono ricaricabili. Collegando la batteria a un generatore esterno le reazioni si invertono.

Corrosione La corrosione è l'ossidazione dei metalli. Il metallo che si trova nel suo stato fondamentale perde degli elettroni e passa alla sua forma ossidata. Se c'è qualcosa che si ossida c'è anche qualcosa che si riduce. La reazione di corrosione è anche termodinamicamente favorita. L’ossigeno nell’aria è l’ossidante ma l’acqua è la maggior responsabile della corrosione. La presenza infatti dell'acqua permette di veicolare gli ioni e rende quindi più veloce la reazione di ossidazione. Quando si è in condizioni di elevate umidità (sopra il 60%) si viene a creare un velo sopra il metallo che consente all’ossigeno disciolto si ossidare il
ferro o qualsiasi metallo. Anche il pH e la presenza di catalizzatori possono influire sulla reazione di corrosione. La ruggine non è altro che ossido di ferro variamente idrato. Questa reazione che termodinamicamente favorita ha un altro vantaggio cioè che l'ossido di ferro è scarsamente solubile in acqua. La reazione di formazione della ruggine è anche elettrochimicamente favorita e
spostata verso i prodotti, non c'è dunque un minimo di equilibrio. Sempre dal punto di vista termodinamico nel caso in cui ci fosse una diminuzione del pH e quindi si ha un aumento di ioni H+ la riduzione dell'ossigeno è favorita così come è favorita l'ossidazione del ferro. Se lavoriamo a pH molto basici invece la reazione di corrosione del ferro viene meno perché non c'è niente che può ridursi e niente che può ossidarsi. Una superficie metallica perfettamente asciutta non può essere aggredita dall'ossigeno perché la reazione e cineticamente molto lenta. Se l'acqua contiene inquinanti acidi, il potere ossidante di O2 aumenta e quindi aumenta la corrosione (fattore termodinamico). Anche la presenza di sali aumenta la corrosione, perché aumenta la conducibilità dell'acqua (fattore cinetico).
Motivi che causano la corrosione Corrosione del ferro può venire per diversi motivi:
1. Deformazione meccaniche = se il pezzo di ferro viene deformato si creano punti di tensione nel ferro. Si formano così zone in cui il ferro si ossida più facilmente
2. Corrosione galvanica = impurezza di metalli più nobili. Se il blocco di ferro e impuro di un metallo con potenziale di riduzione maggiore di quello dal ferro stesso vi è un’accelerazione della corrosione poiché il metallo impuro agisce da catalizzatore.
3. Aerazione differenziale = zone a differenti concentrazione di ossigeno. Vi sono zone ad elevata concentrazione di ossigeno mentre altre sono a bassa concentrazione di ossigeno. La zona a maggiore concentrazione di O2 diventa positiva richiamando elettroni dalla zona a minor concentrazione di ossigeno dove quindi il ferro si ossida. I danni per la corrosione da aereazione differenziale sono maggiori in acque stagnanti che in acque correnti: in quest’ultime è infatti minore la probabilità che si creino zone con forti differenze di concentrazione di ossigeno. La corrosione per aereazione differenziale si verifica anche su strutture di ferro verniciate: se viene scrostata parte della vernice che protegge il erro, la superficie esposta all’aria è più ricca di ossigeno mentre le zone sotto la vernice sono più povere di ossigeno stesso. Nella zona sotto la vernice si formano quindi incavi non visibili che portano alla perdita di forza strutturale di travi e altri supporti. Il danno più serio non è dunque la ruggine visibile ma il danno al di sotto delle superfici verniciate.
Protezione conto la corrosione Rivestimento con impermeabile all’acqua Rivestimento con un altro metallo che abbia potenziale di riduzione maggiore di quello di ferro
(es: Ag) É possibile anche rivestire il ferro mediante processi di elettrolisi con metalli che hanno potenziali
di riduzione più basso di quello del ferro come zincatura, cromatura. In questo caso il ferro accelererà l'ossidazione del metallo che lo riveste che quindi ha un potenziale di riduzione maggiore del suo. Queste reazioni di ossidazione molto veloci si chiamano passivazione. Si vengono così a formare degli ossidi che proteggono la barra di ferro e di zinco.
Inibitori o protezione catodica

CINETICA CHIMICA
Introduzione La cinetica chimica è una branca della chimica che si occupa di esaminare quali sono i fattori che influiscono sul tempo affinché una data reazione giunga a completezza. Quando si parla di cinetica si parla dunque della velocità con cui la concentrazione dei reagenti varia nel tempo.
Parametri “macroscopici” La velocità di una reazione chimica dipende principalmente da tre fattori: 1. Concentrazione dei reagenti = maggiore è la concentrazione maggiore è la velocità di reazione
questo perché aumentando la concentrazione aumenta la frequenza con cui le molecole dei reagenti si scontrano tra loro e ciò comporta un incremento della velocità.
2. Temperatura della reazione = maggiore è la temperatura, maggiore è la velocità di reazione poiché aumentando la temperatura aumenta anche l’energia cinetica delle molecole e quindi aumenta la frequenza e l’energia con la quale si scontrano tra loro
3. Superficie dei reagenti = maggiore è a superficie, maggiore è la velocità di reazione 4. Lo stato fisico dei reagenti = i reagenti per reagire devono essere in contatto tra loro. Più
velocemente si urtano facilmente reagiscono. Quando i reagenti sono presenti in fasi differenti la reazione è limitata dalla zona in cui avviene il contatto; quindi reazioni che coinvolgono i solidi avvengono più velocemente se l’area del solido è più ampia
5. Presenza di un catalizzatore = i catalizzatori sono sostanze che aumentano la velocità della reazione senza essere consumate e quindi senza partecipare attivamente alla reazione
Parametri “microscopici” – meccanismo di reazione
A livello molecolare la velocità di una reazione dipende dalla frequenza degli urti fra le molecole. Più è alta la frequenza degli urti maggiore sarà la velocità di reazione. Affinché un urto conduca a reazione deve tuttavia avvenire a un’energia sufficiente per allungare i legami ad una lunghezza critica e con l’orientamento adeguato per consentire la formazione dei nuovi legami nelle posizioni appropriate. Dunque solo gli urti la cui energia di collisione supera un certo valore critico, sono efficaci per la formazione di molecole di prodotti. Questa energia minima richiesta è detta energia di attivazione (Ea) e varia al variare della reazione. Quando la temperatura è bassa solo una piccola frazione di molecole raggiunte questo valore critico. Dunque la velocità di molte reazioni aumenta rapidamente all’aumentare della temperatura. Arrhenius propose una formula per conoscere la velocità di reazione k:
𝑘 = 𝐴𝑒−𝐸𝑎/𝑅𝑇. In questa formula A è il fattore di frequenza, costante o quasi al variare della temperatura, con cui avvengono gli scontri fra molecole. All’aumentare di Ea, k diminuisce quindi anche la velocità della reazione diminuisce all’aumentare di Ea.
Velocità di reazione La velocità di reazione è definita come la variazione della concertazione dei reagenti o dei prodotti nell’unità di tempo. Si definisce velocità media il rapporto tra la variazione della concentrazione dei reagenti e dei prodotti fratto il tempo trascorso. Prendendo in considerazione una qualsiasi reazione: 𝑎𝐴 + 𝑏𝐵 → 𝑐𝐶 + 𝑑𝐷
Si definisce velocita istantanea = − 1
𝑎
∆[𝐴]
∆𝑡= −
1
𝑏
∆[𝐵]
∆𝑡= −
1
𝑐
∆[𝐶]
∆𝑡= −
1
𝑑
∆[𝐷]
∆𝑡 = k [𝐴]𝑚 [𝐵]𝑛
dove k è detta costante di velocità e varia con la temperatura, non dipende dalla concentrazione. Gli esponenti m ed n (non prevedibili dalla forma dell’equazioni chimica, ma solo sperimentalmente) in una legge di velocità come quella sopracitata sono detti ordini di reazione. L’ordine globale di reazione è dato dalla somma degli ordini rispetto ad ogni reagenti che figura nella legge di velocità.

Reazione di ordine zero La legge cinetica delle reazioni di ordine zero è data dalla
seguente relazione matematica: 𝑘[𝑅]0 = −∆[𝑅]
∆𝑡 da cui risulta che
la velocità di reazione dipende dalla concentrazione dei reagenti. È possibile anche conoscere il tempo necessario affinché la
concentrazione iniziale dimezzi: 𝑡1/2 = [𝑅]0
2𝑘
Riportando in un sistema di assi cartesiani la [A] in funzione del tempo t, si ottiene una retta con pendenza -k.
Reazione del primo ordine Nella reazione di primo ordine la velocità dipende dalla concentrazione di un singolo reagente elevato ad uno. Per una reazione del primo ordine la legge di velocità può essere
espressa come: 𝑣 = −∆[𝐴]
∆𝑡= 𝑘[𝐴]. Integrando questa
espressione possiamo ottenere una relazione che mette in relazione la concentrazione di A all’inizio della reazione alla
relativa contrazione ad un generico tempo t: 𝑙𝑛[𝐴]𝑡
[𝐴]0= −𝑘𝑡.
Il tempo necessario affinché la concentrazione dei reagenti
si dimezzi si calcola: 𝑡1/2 = 𝑙𝑛2
𝑘=
0,6931
𝑘
Reazione del secondo ordine Nella reazione di primo ordine la velocità dipende dalla concentrazione di un singolo reagente elevato alla seconda. Per una reazione del primo ordine la legge di velocità può essere espressa come: 𝑣 = 𝑘[𝐴]2. Integrando questa espressione possiamo ottenere una relazione che mette in relazione la concentrazione di A all’inizio della reazione alla
relativa contrazione ad un generico tempo t: 1
[𝐴𝑡]−
1
[𝐴0]= 𝑘𝑡.
Il tempo necessario affinché la concentrazione dei reagenti si
dimezzi si calcola: 𝑡1/2 = 1
𝑘[𝐴0]
Catalisi e catalizzatori I catalizzatori sono sostanze che, aggiunte in piccole quantità ad una reazione chimica, modificano il valore della velocità di reazione senza venir consumati durante la reazione stessa, non compaiono nelle equazioni globali di reazione e non provocano variazioni del valore della costante di equilibrio. I catalizzatori possono essere distinti in due categorie:
Catalizzatori eterogenei = il catalizzatore si trova in una fase diversa da quella dei reagenti e dei prodotti
Catalizzatori omogenei = il catalizzatore si trova nella stessa fase dei reagenti e dei prodotti La catalizzazione abbassa Ea e varia le formazioni degli intermedi di reazione cioè. Gli enzimi sono esempi di catalizzatori.

Reazione a catena Una qualunque reazione procede attraverso una serie di stadi elementari, alcuni dei quali si ripetono più volte. Gli stadi principali sono ter: 1. Inizio catena = si formano uno o più intermedi di reazione 2. Propagazione catena = si formano i prodotti e si generano gli intermedi 3. Termine catena = gli intermedi si combinano formando un prodotto stabile Le reazioni a catena in cui i numeri degli intermedi di reazione, formati in uno o più stadi di propagazione, aumenta, sono dette reazioni a catena ramificata. Molte reazioni industriali di sintesi di polimeri sono reazioni a catena.


















