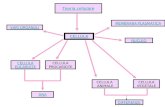
Cellula tumorale : perdita delle caratteristiche sociali della cellula normale controllo della...
47
Cellula tumorale : perdita delle caratteristiche “sociali ” della cellula normale • controllo della proliferazione • risposta e dipendenza dai segnali da altre cellule, dallo stroma, da fattori circolanti • differenziamento • funzioni specifiche • monitoraggio di danni e invecchiamento • morte cellulare programmata
-
Upload
fina-riccio -
Category
Documents
-
view
219 -
download
1
Transcript of Cellula tumorale : perdita delle caratteristiche sociali della cellula normale controllo della...

Cellula tumorale :perdita delle caratteristiche “sociali ” della cellula normale
• controllo della proliferazione• risposta e dipendenza dai segnali
da altre cellule, dallo stroma, da fattori circolanti• differenziamento• funzioni specifiche• monitoraggio di danni e invecchiamento• morte cellulare programmata

Identificati attraverso lo studio dei “tumori ereditari”Rb1 retinoblastomaNF 1 e 2 neurofibromatosi 1 e 2APC poliposi adenomatosa famigliareTSC I e II sclerosi tuberosa tipo 1 e 2VHL sindrome di Von Hippel-LindauCDKN2A (p16) melanoma famigliareMEN1 neoplasie endocrine multiple 2SDH B-C-D paragangliomi e feocromocitomi famigliariFH leiomiomatosi e tumori renalipTEN sindrome di CowdenSMAD4 poliposi giovanile del colonBRCA1 e 2 ca. famigliare della mammella e dell’ovaioMSH2 MLH1 MSH6 PMS1 PMS2 ca. famigliare del colon
Identificati attraverso lo studio dei “tumori sporadici”
WT1 tumore di Wilms’p53 multi tumor suppressor genecadE tumore gastrico diffusoMET RET KIT ca. renale papillifero, MEN2, GISTMYH poliposi recessiva del colon
Geni di suscettibilità al cancro

GENIONCO - SOPPRESSORI
Proto -ONCOGENI
PROLIFERAZIONE APOPTOSI CELLULARE
SORVEGLIANZA dell’INTEGRITA’ del GENOMA
DIFFERENZIAMENTO
GENI del RIPAROdel DNA: “care takers”

Geni di predispozione AD…ma a livello cellulare :X
Recessivi Parzialmente recessivi Dominanti
XX
mutazioni somatiche casualmente acquisiteda una o più cellule
Tumori spessomultipli e bilaterali
Modello Rb
espansione del compartoproliferitivo delle cripteintestinali
XX
mutazioni somatiche acquisite dalle celluleproliferanti
Adenomi multipli
Modello APC
X
stimolo proliferativocon IPERPLASIA dellecellule che esprimonol’oncogene
Tumori spessomultipli e bilaterali
Modello RET

Geni guardiani dellacorretta proliferazione cellulare
Rischio del 90-100 %
Geni di organizzazione di un tessutocomunicazione tra stroma e cellule
Rischio del 20-40 %
Geni di sorveglianza dell’integrità del genoma
Rischio del 40-80 %
Geni di suscettibilità al cancro

neurotecheomanevi
nevi
Astrocitoma anaplastico multifocale
Chirurgiapolichemioterapia
radioterapiaanalisi genetica TP53 e p16 (INK4)
TP53 R273H INK4del (p16, p14, p15)
glioblastoma multiforme a 15 aa 5 melanomi al capo a 12 aa

D9S
974
D9S
736
D9S
958
hM
p1α
I1E
x 2E
x 3
Ex 1α
D9S
942
D9S
1748E
x 1βD
9S1604
D9S
1752E
x 2
Ex 1
D9S
975
9p tel.9p cent.
D9S
1749
22 Kb
H HH
11 Kb
p16 – CDKN2A
p14
p15 – CDKN2B
Locus INK4 , 9p21
R RLOH LOH LOHNI NI NINILOH

p16
p14
CDK4CiclinaD

Servizi di consulenza geneticain campo oncologico
forme di predisposizione geneticamentedeterminate allo sviluppo di tumori
• riconoscerle• diagnosticarle mediante analisi genetiche• studiare le manifestazioni cliniche• gestire il rischio oncologico
difetti genetici molto rariresponsabili di circa il 5 % di tutti i casi di tumore

05/27/2003
dec 23 aa Ca. colon Ca. colon Ca. colon Senilità Ca. colon Linfoma
npl vescica npl laringe Ca. colon Ca. Ovaio
sano
2
Informazione: errataerrata correttacorretta mancantemancante
Validazione dei datianamnestici…..

05/27/2003
dec 23 aa BrCa BrCa 74 npl laringe BrCa 43 BrCa 44 Prostata 71 Linfoma
npl vescica sano Pancreas 42 Ca. Ovaio
npl laringe
2
Validazione dei datianamnestici…..….un problema legale, etico, organizzativo.

Predisposizioni SINDROMICHE
caratteristiche fenotipiche, lesioni amartomatose
benigne e pre-neoplastiche multiple
diagnosi CLINICA
• neurofibromatosi 1 e 2
• poliposi adenomatosa fam. (FAP)
• poliposi giovanile
• sindrome di Peutz-Jeghers
• sindrome di Cowden
• neoplasie endocrine multiple 1 e 2
• sindrome di Von Hippel - Lindau
• sindrome di Carney
• sindrome di Denys-Drash, WAGR
* riconoscerle* considerarle “ malattie ereditarie ”

24/03/2004
12PNST
16 macchie
48 macchie
Tumori in NF1(PNST, gliomi, astrocitomi, feocromocitomi)forma di predisposizionead espressività variabile

Neurofibromatosineoplasie benigne ad origine dalle cellule di Schwann
delle guaine dei nervi periferici
• Neurofibromatosi tipo I– 1 su 3.000 - 4.000 individui
– gene NF1 in 17q11.2 (60 esoni)
• Neurofibromatosi tipo II– 1 su 40.000 individui
– gene NF2 in 22q12.2 (16 esoni)

• più di 6 macchie caffè-latte• lentigginosi delle pieghe• neurofibromi cutanei e delle radici
dei nervi• noduli di Lisch (amartomi
dell’iride)• neurofibromi plessiformi• anomalie ossee e scoliosi• glioma delle vie ottiche• ipertensione e vasculopatia• tumori (schwannomi maligni,
neoplasie cerebrali, leucemia)• difficoltà di apprendimento
• rare macchie• rari schwannomi cutanei• schwannomi dei nervi spinali
o cranici• schwannoma bilaterale della
branca vestibolare del nervo acustico
• meningiomi• ependimomi, astrocitomi• opacità sub-capsulare
posteriore del cristallino
Neurofibromatosi :
NF1 NF2

Neurofibromatosi tipo 1criteri diagnostici NIH (1987) presenza di 2 o più dei seguenti :
• sei o più macchie caffè-latte (95%)> 5 mm in età pre-pubere, > 15 mm in età post-pubere
• due o più neurofibromi o uno plessiforme (20%)
• lentigginosi in regione ascellare o inguinale (90%)
• glioma ottico (tumore della via ottica)
• due o più noduli di Lisch (amartomi dell’iride)
• una tipica lesione ossea displasia dello sfenoide displasia o ispessimento della corticale delle ossa lunghe
con o senza pseudo-artrosi
• un parente di primo grado con diagnosi di NF1





Segni e sintomi Età comparsa
manifestazioni ossee congenite
macchie caffè latte alla nascita o nei primi anni di vita
neurofibromi plessiformi
della faccia e collo :
in altre sedi :
primo anno di vita
entro l’adolescenza
lentiggini 3-5 anni
glioma ottico primi 6 anni di vita
scoliosi displastica tra i 6 e i 10 anni
MPNST adolescenti e adulti
neurofibromi cutanei e sottocutanei
dopo la pubertà(aumentano in gravidanza)
ipertensione qualsiasi età

Gene NF1 17q11.2
60 esoni – mRNA di 9022 basi – 2818 aa

NF1: neurofibrominaGTPase activating protein

50% dei casi sono dovuti a nuove mutazioniMutazioni del gene NF1 possono essere identificate in più del 95% dei casi
Neurofibromatosi tipo 1
missense o in frame del/ins : 10%

Neurofibromatosi tipo 1 : PTT
- estrazione dell’RNA- retrotrascrizione dell’mRNA di NF1 (poliT)- amplificazione di frammenti sovrapposti- traduzione in vitro (reticolociti di coniglio)- separazione dei frammenti proteici in gel
di poliacrilamide- ricerca di frammenti più corti
Frammento a
Frammento b
Frammento c
Frammento d Frammento f
Frammento e
AAAAAAAAAATTTTTTTTTT
cDNA
STOP

11/01/2005
40 schwannoma vestibolare (dx) 39schwannoma vestibolare (sx) 39meningioma (rocca petrosa) 39
macchie cutanee (4 cl)
18
76 schwannoma vestibolare (dx) 69schwannoma vestibolare (sx) 69
meningioma (etmoide) 69meningioma (frontale sx) 69
69 67Vescica 47
Vie urinarie 62
42
14
38
67 40Tumori in NF2(schwannomi, meningiomi, astrocitomi, gliomi, ependimomi)

Neurofibromatosi tipo 2
Sintomi di esordio• sordità monolaterale (35%)
• debolezza focale (12%)
• ronzio, acufeni (10%)
• sordità bilaterale (9%)
• difetto dell’equilibrio (8%)
• apoplessia (8%)
• perdita di sensibilità focale (6%)
• cecità (1%)
• diagnosi per famigliarità (11%)
Forma grave< 25 anni
presenza di meningiomi
e schwannomi in altre sedi
Forma lieve> 25 anni
schwannoma bilaterale del nervo acustico
Forma intermediaaltri fenotipi

Neurofibromatosi tipo 2
gene NF2 in 22q12.216 esoni (+ 1 splicing alternativo)
proteine di 595 e 590 aa
50% dei casi sono dovuti a nuove mutazioni25-30% dei nuovi casi sono mosaici somatici
mutazioni del gene NF2 vengono identificate nel 65% dei casi circa- 10% : delezioni parziali o complete del gene- 10% : mutazioni missenso o delezioni in frame- 80% : mutazioni troncanti (non senso, frameshift, splicing)

NF2: merlinacoordinamento dei segnali tra fattori
di crescita e adesione cellulare

familial clustering
diagnosi PROBABILISTICA
• neoplasie RARE
- sindrome di Li-Fraumeni
- ca. midollare della tiroide ecc.
- retinoblastoma
- tumore gastrico diffuso
- sindrome di Purtillo-Duncan
• neoplasie FREQUENTI- ca. famigliare mammella - ovaio
- HNPCC
- melanoma famigliare
probabilità pre-testalta
probabilità pre-test da definireper ogni caso
Predisposizioni NON sindromiche

TP53IVS 3 -11 C>G (delezione in frame: aa 33-125)

Sindrome di Li - Fraumeni
• 1 caso di sarcoma < 45 anni• 1 parente di 1° grado con un “cancro” < 45 anni• 1 parente di 2° grado con un “cancro” < 45 anni
o un sarcoma a qualsiasi età
Li-Fraumeni “like”: nuclei famigliari senza sarcomi
Li-Fraumeni “variant”: tre tumori primitivi nello stesso soggetto (I°< 45 aa)
npl. <12 aa o npl. Li-F <45aa + parente con npl. Li-F any age + parente any cancer < 60aa
Li-Fraumeni “incompleta”: 2 criteri su 3

Fattori predittivi di mutazione TP53
• rabdomiosarcoma embrionale < 4 anni
• sarcomi < 20 anni
• carcinoma della mammella < age 40
• presenza di : npl cerebrali (bambini o giovani adulti)ca. giovanile cortico-surrene
• assenza di : leucemie e linfomi come primi tumori
Prevalenza: 1:50.000 ???Ereditarietà: autosomica dominantePenetranza: del 20% nell’infanzia, del 50% a 40 aa,
del 90% a 60 aa
Sindrome di Li - Fraumeni

Sindrome di Li - Fraumeni
• 25% carcinoma della mammella : età media 36,2 anni
• 22% sarcomi
11% osteosarcomi : età media 16 aa
11% sa. tessuti molli : RMSE 5-6 aa
IFM 25 aa
LMS 45 aa
• 14% tumori cerebrali : infanzia, 30-40 aa
74%: astrocitomi e glioblastomi
15%: medulloblastomi e PNET
9%: papillomi dei plessi corioidei
2%: ependimomi, schwannomi• 4,2% LLA e HD• 4% ca. gastrico, ca polmonare 46 aa• 3,6% ca. corteccia surrenalica < 16 aa• 2,5% ca. colon• 1.3% ca. prostata e ca. pancreas• 0.8-0.6% ca. vescica, epatoca., Wilms, epatoblast.• 0.4-0.2% ca. nasofaringe e laringe, melanoma, tum. germinali, neuroblastoma

• 15 % sviluppo di 2° tumore (latenza: 1-27 anni)
RA 57 % (64 % se 1° tum. STS)
RR 83 se 1° tumore < 19 aa
RR 9.7 se 1° tumore 20 - 44 aa
RR 1.5 se 1° tumore < 45 aa• 4 % sviluppo di un 3° tumore RA 38 %• 2 % sviluppo di un 4° tumore
tra i casi di 2° tumore : il 66 % dei pazienti irradiati (35-70 Gy)
sviluppano un tumore solido nel campo di RT
Sindrome di Li - Fraumeni

39Paraganglioma 35
+
32Paraganglioma 13Paraganglioma 29
+
21 9 6
62Polmone 62
+
62Paraganglioma 29
+
69
39 36 33
58 68 colon 67
62
2
11
2
60
SDH-BW47X

Controllo della sintesi e degradazione diHIF1

05/14/2003
MTC
MTC
MTC
MTC
MTC
MTC
MTC
MTC
MTC
MTC
MTC
MTC
micro MTC
CCH
RET: Cys618Ser

familial clustering
diagnosi PROBABILISTICA
• neoplasie RARE
- sindrome di Li-Fraumeni
- ca. midollare della tiroide ecc.
- retinoblastoma
- tumore gastrico diffuso
- sindrome di Purtillo-Duncan
• neoplasie FREQUENTI- ca. famigliare mammella - ovaio
- HNPCC
- melanoma famigliare
probabilità pre-testalta
probabilità pre-test da definireper ogni caso
Predisposizioni NON sindromiche



Diagnosi di predisposizioneallo sviluppo di tumori
Tumore sporadicofrequenza attesa nella popolazione generale
Tumori Famigliari• aggregazione casuale• comuni fattori di rischio• varianti genetiche a bassa penetranza
Tumori Ereditaripredisposizioni geneticamente determinate ad alta penetranza
predisposizioni sindromiche

TUMORI FAMIGLIARI TUMORI EREDITARI
utilità pratica :“sorveglianza clinica ottimale”
Criteri per la definizione dei programmi di “prevenzione”:
Aggregazioni famigliari di tumori(mammella, ovaio, colon, utero, melanomi)
- entità del rischio (basso, medio, alto)- distribuzione del rischio per età (età di inizio dei controlli)- rapidità di sviluppo della neoplasia (periodicità)- prognosi della neoplasia (tipo di strategia preventiva)

TUMORI FAMIGLIARI TUMORI EREDITARI
strumenti dignostici :
probabilità di mutazione vs. aggregazione casuale
test geneticogeni maggiori di suscettibilità
varianti genetiche a bassa penetranza ??
Aggregazioni famigliari di tumori(mammella, ovaio, colon, utero, melanomi)

Aggregazioni famigliari di tumoripercorso diagnostico
• ricostruzione della storia famigliare
• stima della probabilità di mutazionein base a:
- n° collaterali affetti- pattern di ereditarietà- età di insorgenza della malattia- eventuale presenza di tumori multipli- n° ed età dei collaterali sani
• ricorso al test genetico

Stime di PROBABILITA’ pre - testdi identificare un difetto genetico
Regressione logistica
caratteristiche qualificantidei nuclei famigliari a
mutazione nota
probabilità di mutazionein geni noti
Modello Bayesiano
probabilità diaggregazione casuale
<> difetto genetico
• frequenza allelica• penetranza• frequenza della npl nella
polazione generale

Probability to carry a BRCA mutationpersonal : familial :