
Bonezzi BUonoCoRe - paviapainschool.it di fisiopatologia.pdf · spetti di siopatoloia e terapia del...
68
Cesare BONEZZI Michelangelo BUONOCORE ASPETTI DI FISIOPATOLOGIA E TERAPIA DEL DOLORE
Transcript of Bonezzi BUonoCoRe - paviapainschool.it di fisiopatologia.pdf · spetti di siopatoloia e terapia del...

Cesare Bonezzi
Michelangelo BUonoCoRe
CO
D. 0
1820
022
ASPETTI DI FISIOPATOLOGIA
E TERAPIA DEL DOLORE


Michelangelo BUONOCORE
Cesare BONEzzi
ASPETTI DI FISIOPATOLOGIA
E TERAPIA DEL DOLORECORSO
ECM-FADProgetto di Formazione a DistanzaResponsabile ScientificoCesare BonezziDirettore Unità di Ricerca in Fisiopatologia e terapia del dolore - Fondazione Salvatore Maugeri IRCCS Pavia
TutorMichelangelo BuonocoreServizio di Neurofisiopatologia del Dolore IRCCS Fondazione Maugeri Pavia
Per partecipare alla FAD collegarsi al sito: ...-fad.itdall’01/09/2013 al 31/12/2013
Corso ECM/FAD realizzato con il contributo non condizionato di
ASPETTI DI FISIOPATOLOGIA
E TERAPIA DEL DOLORE
Michelangelo BUONOCORE
Cesare BONEzzi
Michelangelo BUONOCORE
Cesare B

Copyright © 2013 Momento Medico S.r.l.
11ED2305-04/13
Tutti i diritti di traduzione, riproduzione, adattamento parziale o totale con qualsiasi mezzo (compresi microfilms, copie fotostatiche o xerografiche) sono riservati

IndIce
1. Il dolore: aspetti generali e classificazioni 5
2. Meccanismi della sensibilizzazione periferica 12
3. Genesi ectopica degli impulsi 21
4. Meccanismi della sensibilizzazione centrale 31
5. La terapia combinata nel trattamento
del dolore cronico 43


1 Il dolore: AspettI GenerAlI e clAssIfIcAzIonICesare Bonezzi
L’Associazione Internazionale per lo Studio del Dolore (IASP) definisce il dolore come “un’esperienza spiacevole, sensoriale ed emozionale, correlata con un danno tissutale o descritta in tali termini” (Merskey 1994). Nella definizione vi sono due parole che noi riteniamo essenziali: “l’esperienza” come ultimo atto della nocicezio-ne, e il “danno” come primo fattore responsabile. Nella lingua greca antica la parola esperienza era indicata con ε′μπειρι′α (empeirìa), composta da ε′υ, η′
∼υ (in, all’inter-
no) e πει∼ρα (prova) volendo significare che con l’esperienza il soggetto è in grado di “saggiare” all’interno la realtà. Ma nella filosofia della scienza l’esperienza è il fondamento delle osservazioni scientifiche basate sulle “sensate esperienze” e sulle “dimostrazioni necessarie”. Il dolore provato è una esperienza e costruisce l’esperienza per l’interpretazione di ogni altro dolore provato successivamente.
Il danno sta ad indicare che il dolore ha una sua origine in una lesione del nostro corpo che, a sua volta, è in grado di generare meccanismi patogenetici. Nella pratica clinica quotidiana moltissime malattie sono accompagnate da dolore, sia come sintomo marginale del quadro clinico, sia come elemento dominante. In questa confusa quantità, come è possibile costruire una classificazione del dolore se non partendo dal danno?
Quando ci si riferisce ad una possibile classificazione del dolore, acuto e cronico che sia, si pensa ad un elenco di patologie di varia eziologia e appartenenza (presenti nelle varie discipline medico-chiurgiche), che ovviamente sono caratterizzate dalla presenza di dolore. La mancanza di una classificazione del dolore come malattia a sé stante costituisce sicuramente un freno al progredire del sapere clinico ed epidemio-logico in ambito sanitario.
A tale proposito J.J. Bonica (1990) ha commentato la situazione della scienza che si occupa di dolore con l’espressione “una moderna torre di Babele”.
In effetti manca ancor oggi un linguaggio condiviso e una classificazione del dolore come malattia che tenga conto della sua eziologia, della sua patogenesi ed ovviamente del quadro clinico che determina e caratterizza. Nella letteratura troviamo molti tentativi di classificazione del dolore in base alle discipline mediche (neurologia, reumatologia, ortopedia ecc.), alla malattia di base (neoplasia ecc.) o alla diversa sede della malattia stessa, al tessuto interessato (articolazioni, muscoli ecc.), alla durata del dolore. Un interessante sforzo si sta osservando in questi ultimi tempi con il tentativo di una clas-sificazione basata sul meccanismo patogenetico che sottende al dolore dichiarato da un paziente. In particolare riteniamo importante chiarire alcune di queste classificazioni e di capire quali implicazioni possano avere nella pratica clinica.
1. In base alla durata del doloreMolto spesso si sente definire (e diagnosticare) il dolore di un paziente con il
termine “cronico” non solo perché l’anamnesi dimostra la sua lunga durata (cronico deriva dal greco Kronos, che significa “di lunga durata”) ma per attribuirgli un valore

6Aspetti di fisiopatologia e terapia del dolore
fisiopatologico. Arricchito di questo termine, il dolore diviene una sindrome clinica ovvero una realtà complessa e difficilmente curabile. Vediamo in particolare cosa si intende per dolore acuto e dolore cronico.
Dolore acuto. Si divide in fisiologico e patologico. Il dolore acuto fisiologico è sempre evocato, ovvero causato da uno stimolo che deve essere “sovra-soglia” ovve-ro di intensità sufficiente a generare nei nocicettori tissutali un potenziale d’azione, senza provocare un danno tissutale. La stimolazione dei recettori è transitoria. Ne è un esempio lo stimolo termico caldo. Ha la funzione di allerta (scopo preventivo) e genera sempre una risposta riflessa che ha lo scopo di impedire il verificarsi di un danno tissutale. Dura in genere pochi secondi ed è di intensità proporzionale alla causa che lo ha generato. Il dolore acuto patologico è invece causato da un danno tissutale che si mantiene per un tempo breve (ore o giorni). Il dolore acuto può essere spontaneo ed evocato da uno stimolo non necessariamente doloroso. Scompare con la guarigione del danno tissutale ed ha uno scopo protettivo in quanto avverte il paziente della presenza del danno e induce ad accertamenti medici. Si pensi ad una ferita, ad una ustione, ad un ascesso dentale.
Dolore cronico (sempre patologico). Il dolore che continua per giorni o settimane possiamo dire che è un “dolore cronico”. Se analizziamo la letteratura scientifica tro-viamo infatti varie misure del tempo di persistenza del dolore. Alcuni autori parlano di tre mesi, altri di sei e altri ancora di dodici. La IASP, nel tentativo di risolvere la questione, sottolinea che è cronico quel dolore che persiste al di lá del tempo ragionevole di un normale decorso di una malattia (Turk 2001, Main 2001, Thienhous 2001). La presenza di una malattia cronica può certamente spiegare la presenza di dolore e una certa insopportazione da parte del paziente. Il termine “cronico” viene però utilizzato anche per definire il dolore da un punto di vista fisiopatologico nel senso che sottin-tende la presenza di meccanismi patogenetici propri in grado di mantenere il dolore nel tempo, scatenati dalla persistenza stessa del dolore. Ma nessuno ha mai dimostrato che il persistere del dolore genera dolore cronico. Si è invece osservato che esistono meccanismi patogenetici del dolore che, vuoi perché ancora poco conosciuti o vuoi perché non esistono terapie efficaci, causano un dolore continuo e persistente nel tempo.
Ci riferiamo alle sindromi da deafferentazione come l’avulsione del plesso brachiale o ai casi di apoptosi del primo neurone sensitivo. Con il termine di “dolore cronico” possiamo anche definire quei casi in cui, dopo un evento lesivo o malattia iniziale, si instaurano modificazioni biologiche, psicologiche e sociali che portano il quadro clinico in una condizione di complessità in cui è difficile ritrovare la causa iniziale e i meccanismi del dolore sono molteplici e sovrapposti.
Possiamo quindi parlare di cronico quando il dolore continua nel tempo perché è causato dalla presenza di una malattia cronica (è cronica la malattia), o quando si è instaurato un meccanismo patogenetico cronico proprio del dolore o infine quando il paziente sviluppa una vera e propria “malattia” per l’instaurarsi di un quadro clinico che comprende manifestazioni patologiche che appartengono alla sfera fisica, a quella psicologica e a quella sociale (Bonezzi 2012).
Dolore persistente (sempre patologico). Il termine “persistente” viene in genere utilizzato per definire un dolore che si mantiene nel tempo. Questo termine viene comunemente associato al dolore postoperatorio che perdura nel tempo. Ne sono un esempio il dolore post-mastectomia, post-toracotomia, post-amputazione, post-erniotomia inguinale o dopo interventi sulle articolazioni. In un interessante articolo Cousins avvicina i due termini di cronico e persistente in un unico quadro clinico caratterizzato da fattori bio-psico-sociali (Cousins 2007).

7 Il dolore: aspetti generali e classificazioni
2. In base alla conoscenza dell’eziologia il dolore può essere suddiviso in: idiopatico, nocicettivo, neuropatico
Dolore “idiopatico”. Questo termine indica quelle forme cliniche in cui non sembra esistere una causa evidente in grado di spiegare la presenza del dolore. Tra le più importanti vengono riportate la nevralgia essenziale del trigemino, la bocca che brucia, le Complez Regional Pain Syndrome, la fibromialgia. Alcuni Autori (Lipowsky 1990) sottolineano la concomitanza di meccanismi fisiopatologici periferici e di fattori psicologici.
Un’importante classificazione divide il dolore in base al tessuto interessato dalla lesione e all’origine dell’impulso doloroso. Si distingue il dolore nocicettivo somato-viscerale, in cui il dolore nasce da una patologia interessante i tessuti del corpo e dalla stimolazione dei nocicettori tissutali (siti normotopici), dal dolore neuropatico periferico e centrale, in cui la patologia interessa la via somatosensoriale che conduce il dolore ed in cui l’impulso nasce lungo la stessa via (in siti cosiddetti “ectopici”).
Il dolore “nocicettivo” è definito come “il dolore che nasce da un danno attuale o potenziale ai tessuti (con esclusione del sistema nervoso) e che è dovuto alla at-tivazione dei nocicettori (Merskey 1994, IASP: http://www.iasp-pain.org). Questo gruppo comprende tutte le sindromi in cui sono coinvolti i tessuti somatici (ossa e articolazioni, fasce, tendini e muscoli, rivestimenti cutanei e mucosi, sierose) ed i tessuti viscerali del corpo.
Il dolore nasce dai nocicettori tissutali ed è condotto dalle vie afferenti al midollo spinale. Di fondamentale importanza è l’integrità del sistema somatosensoriale depu-tato alla conduzione degli impulsi nocicettivi. In genere le sindromi viscerali vengono classificate in base alla sede del viscere d’origine (dolore addominale, dolore pelvico-perineale, dolore toracico ecc.) e al viscere coinvolto.
Diversamente le sindromi somatiche vengono raccolte in base al tessuto (sindromi miofasciali, sindromi articolari ecc) o alle sedi dove il dolore è più frequente (dolore lombare, cervicale ecc). Gli studi di fisiopatologia hanno poi spostato l’attenzione sui meccanismi che sottendono a questo dolore (“nocicettivo”), sulla ipersensibilità dei nocicettori da parte di processi infiammatori e di sostanze algogene di varia natura, nonché sulla ipersensibilità dei neuroni spinali che determina un incremento dell’in-tensità, una allargamento del territorio in cui il dolore viene percepito e la comparsa di un segno clinico importante come l’allodinia meccanica dinamica nelle aree sane circostanti il danno.
Non sempre l’infiammazione è all’origine del dolore in quanto le modificazioni strutturali, come quelle che possono avvenire in una articolazione colpita da processi degenerativi, sono in grado di causare il dolore in quanto responsabili dell’insorgenza di stimoli di intensità elevata (dolore nocicettivo meccanico strutturale).
Un aspetto clinico importante di queste sindromi, per quanto riguarda l’esame clinico del paziente, è il cosiddetto “dolore riferito”. Nelle patologie viscerali, miofa-sciali e articolari il dolore viene localizzato dal paziente in aree del corpo più o meno estese che non hanno nulla a che vedere con la zona e il tessuto sofferente. Si pensi al dolore all’arto superiore sinistro nell’angina cardiaca, al dolore nell’arto inferiore da sacroileite che mima una estensione neurologica radicolare. Alla sua origine si ritie-ne siano presenti i fenomeni della convergenza e della sensibilizzazione spinale. Ai neuroni spinali giungono fibre afferenti provenienti non solo dal tessuto danneggiato ma anche da altri visceri e da altre strutture somatiche.
Si crea così la situazione che il paziente localizza il dolore in altri visceri ed in altre sedi del corpo che hanno lo stesso segmento spinale. Questo dolore riferito è in genere percepito dal paziente come profondo e si può accompagnare ad iperalgesia e ad allodinia.

8Aspetti di fisiopatologia e terapia del dolore
Le sIndroMI cLInIche deL doLore neuropatIco perIferIco e centraLe
Nella seconda edizione della “Classification of chronic pain” disponibile online sul sito della International Association Study of Pain (IASP: http://www.iasp-pain.org) vengono riportate le definizioni principali riguardanti il tema “dolore”.
Il dolore “neuropatico” è definito come “il dolore causato da una lesione o da una malattia del sistema nervoso somatosensoriale”. È scritto inoltre che il “dolore neuropatico” è un termine clinico che richiede una lesione dimostrabile o una malat-tia che soddisfi i criteri diagnostici neurologici. Il termine “lesione” è comunemente utilizzato quando gli strumenti diagnostici (radiologici, neurofisiologici, bioptici o di laboratorio) mostrano una anormalità o quando vi è stato un trauma evidente. Il termine “malattia” è utilizzato quando è nota la causa della lesione (ischemia, vasculite, diabete e altro). Viene introdotta per la prima volta la definizione di sistema somatosensoriale per identificare il sistema sensitivo afferente che porta le informazioni provenienti da tutto il corpo, sia dagli organi e tessuti del corpo sia dall’esterno (vista, udito e olfatto). In base alla sede della lesione o malattia, interessante la parte periferica o centrale del sistema somatosensoriale, si distingue un dolore neuropatico periferico e un dolore neuropatico centrale. La sola presenza di sintomi o segni (come il dolore evocato da uno stimolo tattile) non giustifica l’uso del termine “neuropatico”. Nella nota della IASP si sottolinea inoltre che in alcuni casi, come la nevralgia essenziale del trigemino, dove non sono rilevabili dati oggettivi di lesione o malattia, sia importante l’aspetto clinico, così come nella neuropatia post-erpetica è rilevante la storia. Poiché è frequente che le indagini non siano in grado di portare ad una definizione certa di dolore neuropatico, sempre nella nota, si ritiene importante il giudizio clinico per poter giungere ad una diagnosi. Le principali sindromi cliniche neuropatiche secondo alcuni Autori (Jensen 2001) sono riportate nella tabella 1 e sono suddivise in base alla sede della lesione neurologica nel sistema nervoso periferico, spinale ed encefalico.
Nell’ambito del dolore neuropatico troviamo altre classificazioni come quella di Baron (2010) che riportiamo perché molto completa e dettagliata. In essa troviamo:1) Neuropatie periferiche dolorose: arto fantasma, dolore del moncone, dolore da lesione parziale o totale del nervo, dolore da neuroma postraumatico o postchirurgico, sindrome da intrappolamento, da mastectomia, da toracotomia, nevralgia di Morton, cicatrici dolorose, herpes zoster e neuropatia posterpetica, mononeuropatia diabetica, amiotrofia diabetica, neuropatie ischemiche, borrelliosi, connettivopatie (vasculiti), amiotrofia nevralgica, neoplasie nervose periferiche, plessopatie attiniche, plessopatie, nevralgie trigeminali e glossofaringeo.
Tabella 1. classificazione del dolore neuropatico (Jensen 2001)
Periferico Spinale Encefalico
neuropatieherpes zosterLesioni nervose traumaticheamputazioniplessopatieradicolopatieavulsionineoplasienevralgia trigeminale
sclerosi multiplaLesioni spinali traumatichearacnoiditeneoplasiesiringomieliaInfarto spinale
Infartosclerosi multiplaneoplasiesiringomieliaparkinsonepilessie

9 Il dolore: aspetti generali e classificazioni
2) Le polineuropatie distinte in:• Metaboliche o nutrizionali: diabetiche, alcoliche, da amiloidosi, ipotiroidismo• Da farmaci: antiretrovirali, cisplatino, oxaliplatino, disulfiram, etambutolo, iso-
niazide, nitrofurantoina, talidomide, metiltiouracile, vincristina, cloramfenicolo, metronidazolo, taxoidi, oro.
• Da sostanze tossiche: acrilamide, arsenico, clioquinolo, dinitrofenolo, ossido di etilene, pentaclorofenolo, tallio.
• Ereditarie: neuropatie da amiloidosi, malatti di Fabry, Charcot-Marie-Tooth, tipo 2B, neuropatie sensitive e autonomiche ereditarie
• Neoplastiche: neuropatie periferiche paraneoplastiche, mieloma • Infettive o postinfettive, immunitarie: poliradicoloneuropatie infiammatorie (sin-
drome di Guillain-Barré), borrelliosi, HIV• Altre polineuropatie: eritromelalgia idiopatica, neuropatia small-fibre. 3) Sindrome da dolore centrale• Lesioni ischemiche in particolare del tronco e del talamo o mieliche (infarto, emor-
ragie, malformazioni vascolari)• Sclerosi multipla• Lesioni traumatiche mieliche o encefaliche• Siringomielia e siringobulbia• Tumori• Ascessi• Epilessia• Morbo di Parkinson4) Sindromi neuropatiche dolorose complesse Complex regional pain syndromes tipo I e II (altrimenti definite come distrofie sim-patico riflesse, causalgia)5) Mixed pain syndromesDolore lombare cronico con radicolopatia, dolore da cancro, invasione neoplastica del plesso, complex regional pain syndromes.Possiamo concludere con quanto riportato recentemente (2010) dalla IASP che pro-pone alcune possibili soluzioni classificatorie. Il dolore neuropatico potrebbe essere distinto (IASP update 2010):1) In base alla sede: periferica (nervo, plesso, ganglio della radice dorsale, radice) e centrale (spinale, tronco, talamo, corteccia)2) In base al fattore eziologico: trauma, ischemia o emorragia, infiammazione, neuro-tossicità, neurodegenerazione, paraneoplastica, metabolica, da deficienza vitaminica, neoplastica3) In base ai sintomi e segni: qualità del dolore, perdite sensoriali, ipersensibilità4) In base al meccanismo patogenetico: scariche ectopiche, perdita del sistema ini-bitorio, sensibilizzazione periferica, sensibilizzazione centrale.
Tra queste possibilità l’ultima, a nostro avviso, è la più importante, non solo per quanto riguarda il dolore neuropatico ma anche il dolore nocicettivo, perché definisce le vere cause del dolore offrendo indicazioni precise al trattamento. In altre parole, possiamo identificare tre gruppi patologici: le sindromi di dolore nocicettivo, le sindromi di dolore neuropatico ed infine il dolore malattia. In quest’ultimo gruppo sono raccolti quei casi in cui, accanto ai meccanismi patogenetici propri del dolore nocicettivo o neuropatico, si sono sviluppati meccanismi connessi a comportamenti reattivi del paziente interessanti la sfera psico-sociale (Bonezzi 2012).
Se al meccanismo è possibile associare, mediante opportune indagini cliniche e strumentali, il tessuto colpito (articolazione, viscere, muscolo, tendine, nervo) è altresì possibile individuare tecniche antalgiche mirate ed efficaci. Di fronte ad un dolore lom-

10Aspetti di fisiopatologia e terapia del dolore
bare di tipo nocicettivo infiammatorio è possibile stabilire un trattamento farmacologico ma anche, una volta identificata l’eventuale faccetta articolare coinvolta, procedere a blocchi selettivi radioguidati o anche a denervazione delle afferenze sensitive.
La classificazione basata sul meccanismo patogenetico: le sindromi del dolore nocicettivo, quelle del dolore neuropatico e del dolore malattia (Tabella 2)
Questa classificazione nasce dal presupposto che attraverso una valutazione clinica e strumentale si possano raccogliere sintomi e segni appartenenti a diversi meccanismi patogenetici, permettendo una diagnosi dettagliata e utile ai fini terapeutici.
La valutazione dell’area di dolore e la presenza di allodinia statica primaria (evo-cata mediante stimoli pressori o termici caldi, dal movimento attivo e passivo) per-mette di identificare l’esistenza di una ipersensibilità dei nocicettori tissutali come fondamentale meccanismo del dolore nocicettivo somatico o viscerale (Gold 2010, Koltzenburg 1995, Woolf 2007).
Un dolore evocato, a volte intenso e disabilitante, da un movimento di una arti-colazione deformata (si pensi alla coxartrosi) che si attenua progressivamente per-mettendo al paziente di muoversi, potrebbe non dipendere da una sensibilizzazione dei nocicettori tissutali ma avere un meccanismo patogenetico legato alla deformità stessa e ad un eccessivo stimolo di nocicettori non sensibilizzati. Questo dolore viene chiamato meccanico-strutturale.
La presenza di una perdita delle sensibilità in un’area di dolore lascia supporre che il dolore percepito in quell’area nasca da una lesione delle fibre afferenti e ad una ipersensibilità della fibra con origine ectopica degli impulsi afferenti. In questi casi parliamo di dolore neuropatico periferico.
Il dolore avvertito dal paziente in un’area estesa completamente priva di innerva-zione per grave lesione delle vie neurologiche a monte del primo neurone induce ad ipotizzare una ipersensibilità dei neuroni centrali da deafferentazione e un dolore neuropatico centrale.
Inoltre la presenza di un dolore più o meno intenso allo sfioramento della cute priva di danno o di deficit sensitivi (allodinia dinamica meccanica) è dovuta al coin-volgimento dei neuroni spinali e alla cosiddetta ipersensibilità dei neuroni spinali. A questo meccanismo concorrono gli impulsi nocicettivi che arrivano dal nocicettore o dal sito ectopico.
Una valutazione clinica che comprende una accurata indagine psicologica del paziente permette di identificare quei casi in cui, accanto al dolore, i comportamenti reattivi, la fragilità della persona, e altre manifestazioni della sfera psico-sociale, in-serendoli in una gruppo a sé stante che definiamo “dolore malattia” (Bonezzi 2012). Le sindromi cliniche che noi osserviamo possono presentare questi meccanismi, in forma singola o complessa, e la loro individuazione è utile, come vedremo, alla scelta del trattamento.
Tabella 2. una proposta di classificazione patogenetica
Sindromi del dolore nocicettivo somato-viscerale
Sindromi del dolore neuropatico periferico e centrale
Sindromi del dolore malattia
Ipersensibilità del nocicettore e del neurone spinaleIpersensibilità spinale da input di fibre amieliniche
Ipersensibilità della fibra con genesi ectopica degli impulsiIpersensibilità spinale da input di fibre amieliniche e da deafferentazione
tutti i meccanismi prima citati a cui si associano meccanismi generati da comportamenti reattivi di tipo bio-psico-sociale

11 Il dolore: aspetti generali e classificazioni
La “torre dI BaBeLe”
Per sottolineare la confusione che è presente nel mondo scientifico e nel “real world” riportiamo un esempio. Nel lavoro recente (2011) di Tesfaye e Expert Pannel sulle neuropatie diabetiche compare una tabella che elenca i meccanismi del dolore neuropatico. Nella parte della tabella riguardante i meccanismi periferici del dolore neuropatico si riporta la “peripheral sensitization”.
Questo meccanismo è proprio del dolore nocicettivo, ma viene qui inserito perché, secondo molti altri Autori, le modificazioni della sensibilità del nocicettore tissutale sono una forma di “neuro-patia”. In altre parole tutto il dolore diviene così neuropati-co. Per meglio comprendere questa confusione si riporta in lingua originale lo scritto di un altro Autore (Tolle 2010): “Two of the mechanisms that can cause neuropathic pain conditions are central and peripheral sensitization. Central sensitization occurs as a result of increased responsiveness of spinal cord pain transmission neurons, while peripheral sensitization is produced by the lowering of nociceptor activation thresholds following exposure to inflammatory mediators, such as nerve growth factor or bradykinin, released at the site of tissue injury”.
La sensibilizzazione dei nocicettori tissutali da parte dei mediatori infiammatori è alla base del dolore nocicettivo così come la sensibilizzazione centrale, generata e sostenuta da afferenze nocicettive condotte da fibre amieliniche, presente sia nel dolore nocicettivo che neuropatico.
BIBLIoGrafIa
Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment Lancet Neurol 2010; 9: 807-19.
Bonezzi C, Demartini L, Buonocore M. Chronic pain: not only a matter of time. Minerva Anestesiol. 2012 Jun; 78 (6): 704-11.
Bonica JJ. The management of pain. Lea & Febiger 1990 Second edition.
Cousins MJ. Persistent Pain: A Disease Entity. Journal of Pain and Symptom Management Vol. 33 No. 2S February 2007.
Gold MS and Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nature Medicine 2010; 11: 1248-1257.
Jensen TS, Gottrup H, Sindrup SH, Bach FW. The clinical picture of neuropathic pain. European Journal of Pharmacology 429 (2001); 1-11.
IASP Clinical Update Vol. XVIII, Issue 7 September 2010.
Lipowsky ZJ. Chronic idiopathic pain syndrome. Ann Med 1990; 22: 213-7.
Main CJ, Spanswick CC. Pain management: an interdisciplinary approach. Elsevier. 2001 pp. 93.
Merskey H, Bogduk N. Classification of chronic pain: descriptions of chronic pain syndromes and defini-tions of pain terms. Seattle, WA: IASP Press; 1994.
Tesfaye S, Vileikyte L, Rayman G, Sindrup SH, Per-kins BA, Baconja M, Vinik AI, A. J. Boulton M, on behalf of The Toronto Expert Panel on Diabetic Neuropathy. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assess-ment and management. Diabetes Metab Res Rev 2011; 27: 629-638.
Thienhaus O, Cole BE. “Classification of pain”. In Weiner, RS. Pain management: A practical guide for clinicians (6 ed.). American Academy of Pain Management 2002.
Tolle ThR. Challenges with current treatment of neuro-pathic pain. European Journal of Pain Supplements 4 (2010) 161-165.
Turk, DC, Okifuji A. Pain terms and taxonomies. In Loeser D, Butler SH, Chapman JJ, et al.. Bonica’s management of pain (3 ed.). Lippincott Williams & Wilkins. 2001; pp. 18-25.
Koltzenburg M. Stability and plasticity of nociceptor function and their relationship to provoked and ongoing pain. The Neurosciences, Vol 7, 1995: pp 199-210.
Woolf CJ, and Ma Q: Nociceptors—Noxious Stimulus Detectors Neuron 2007: August 2; 353-364.

2 MeccAnIsMI dellA sensIbIlIzzAzIone perIferIcAMichelangelo Buonocore
GeneraLItà suL doLore
Se si esclude il dolore fisiologico, che generalmente non si accompagna ad una lesione ma evita che essa si determini, ed una forma molto rara di dolore patologico puramente meccanico, nella maggior parte dei casi il dolore clinico è il frutto dello sviluppo di una ipersensibilità agli stimoli. Con questo termine si intende lo spostamento a sinistra della curva stimolo-risposta riferita al dolore fisiologico (Figura 1). In termini pratici, tutte le volte che si sviluppa una condizione di ipersensibilità agli stimoli il dolore viene avvertito per intensità di stimoli che normalmente non evocano la sensazione dolorosa (allodinia) oppure esso viene avvertito di intensità più elevata in seguito a stimoli che anche normalmente sono avvertiti come dolorosi (iperalgesia). Quando l’ipersensibilità diventa molto marcata, essa può portare ad una condizione per cui il dolore viene avvertito indipendentemente da qualsiasi tipo di stimolazione portata. È il dolore spontaneo che, per quanto appena espresso, almeno dal punto di vista fisiopatologico, è un dolore più grave di quello evocato. I punti cruciali per lo sviluppo di ipersensibilità agli stimoli dolorosi sono localizzabili a livello dei tessuti lesionati, lungo le vie del sistema somatosensoriale oppure a livello del sistema nervoso centrale, midollo spinale incluso. Il complesso di fenomeni algogeni che si sviluppa nei tessuti periferici lesionali va usualmente sotto il nome di sensibilizzazione periferica, ad indicare che il dolore può essere giustificato in toto da fenomeni che avvengono nel tessuto periferico lesionato. Col termine di “sen-sibilizzazione” centrale si intende invece quell’insieme di fenomeni che si sviluppa nel
Figura 1. La figura mostra due esemplificative curve stimolo-risposta. La prima a destra è rappresentata dal dolore fisiologico. La curva di sinistra è il risultato dello sviluppo di una lieve condizione di ipersensibilità agli stimoli dolorosi, con comparsa delle condizioni note come allodinia ed iperalgesia (per le definizioni vedere testo).
0
Intensità dello stimolo100%
100%Intensità
del dolore
allodinia
Iperalgesia
dolore fisiologico

13 Meccanismi della sensibilizzazione periferica
sistema nervoso centrale in seguito ad una lesione algogena periferica e che porta ad un’amplificazione del dolore con tipica estensione dello stesso in area extralesionale. Infine, un’altra importante sede di ipersensibilità agli stimoli è rappresentata dal sistema nervoso stesso dove gli impulsi nervosi che generano la sensazione dolorifica si autoge-nerano senza che siano coinvolte le terminazioni nervose, cioè quelle strutture recettoriali che sono fisiologicamente deputate alla trasduzione degli stimoli da un tipo di energia (meccanica, termica, chimica) ad energia elettrica (potenziale d’azione). Questo tipo di sensibilizzazione è alla base del cosiddetto dolore neuropatico. Sintetizzando, ogni dolore che origina dai recettori del dolore “sensibilizzati” viene definito come dolore nocicettivo, mentre ogni dolore che origina direttamente dalle fibre nervose viene definito come dolore neuropatico. In questo capitolo saranno illustrati i meccanismi alla base della sensibilizzazione periferica (dolore nocicettivo), nei prossimi due saranno affrontati i meccanismi della genesi ectopica degli impulsi nel sistema nervoso (dolore neuropatico) e della sensibilizzazione centrale (fenomeno comune ad entrambi i suddetti processi).
IpersensIBILItà perIferIca, concettI GeneraLI
A differenza del dolore fisiologico, che è basato su un sistema abbastanza rigido e stereotipato, il dolore patologico è basato su un sistema molto plastico e variabile. Una delle parti più dinamiche in tal senso è rappresentata dai tessuti periferici. Gran parte del dolore che si incontra in patologia è infatti il risultato di fenomeni di sensibilizzazione che occorrono in seguito a lesioni interessanti i tessuti extra-nervosi. È il cosiddetto dolore nocicettivo. Il primo fenomeno che si osserva, una volta che la lesione si è verificata, è rappresentato dallo sviluppo della sensibilizzazione periferica. In ambito algologico, il termine di sensibilizzazione periferica si riferisce ad un insieme di fenomeni che porta le terminazioni libere delle fibre nocicettive ad abbassare la loro soglia di scarica, fino ad arrivare, nei casi di maggiore intensità, alla scarica spontanea. La sensibilizzazione periferica è in genere la conseguenza dello sviluppo di fenomeni infiammatori nei tes-suti dove sono localizzate le terminazioni nervose in grado di trasdurre impulsi nocivi o potenzialmente nocivi per l’organismo. Tali terminazioni sono rappresentate dalle cosiddette terminazioni libere, cioè non connesse ad un particolare tipo di recettore. Esse possono essere considerate come assoni nudi su cui ci sono specifici canali in grado di modificare il flusso ionico attraverso le membrane neuronali. Ogni tipo di dolore che si genera dai recettori specifici del dolore (terminazioni libere) viene comunemente definito dolore nocicettivo. Le conseguenti modifiche del potenziale della membrana delle ter-minazioni libere possono creare differenze di potenziale locali che, se raggiungono una certa intensità, sono in grado di generare il potenziale d’azione nervoso. In altri termini, le modificazioni dei potenziali di membrana abbassano la soglia di scarica delle termi-nazioni nervose che incominciano a scaricare per stimoli di intensità più bassa rispetto a quella necessaria per evocare dolore in condizioni fisiologiche. Quando i fenomeni di ipersensibilità periferica sono particolarmente intensi, la soglia di attivazione delle terminazioni si abbassa parecchio fino alla scomparsa di una vera e propria soglia. Allora la scarica avviene indipendentemente dagli stimoli e si configura quello che clinicamente viene definito come dolore spontaneo. Comunque esso venga generato, come è noto, il potenziale d’azione, una volta insorto, si propaga lungo la fibra fino alla prima sinapsi che incontra, dove, inducendo la liberazione di neurotrasmettitori, si esaurisce. La liberazione di neurotrasmettitori è il meccanismo mediante il quale l’impulso si propaga dal sistema nervoso periferico al sistema nervoso centrale. Come avviene a livello del recettore periferico, anche a livello delle sinapsi si creano dei potenziali locali che, se sufficiente-mente intensi, innescano la ripartenza degli impulsi, mediante la generazione di nuovi potenziali d’azione che corrono lungo le fibre nervose dei secondi neuroni nocicettivi.

14Aspetti di fisiopatologia e terapia del dolore
GLI stIMoLI nocIcettIvI
Perché si avverta dolore e si possano generare fenomeni riferibili alla sensibiliz-zazione periferica è indispensabile che gli stimoli siano di elevata intensità. Quando i fenomeni di ipersensibilità si sono instaurati, allora anche stimoli di intensità medio-bassa diventano in grado di evocare il dolore. La natura degli stimoli nocicettivi è variabile ma tre sono i tipi di “energia” che, una volta trasdotti, possono generare dolore. Il primo tipo è quello chimico. È ben noto come il rilascio di alcune sostanze nei tessuti sia in grado di generare quelle condizioni che portano ad avvertire dolore. Tra queste vanno sicuramente ricordati alcuni ioni positivi come gli ioni idrogeno H+ o gli ioni potassio K+, ma anche sostanze quali la bradichinina, la serotonina, alcune prostaglandine ed anche l’ATP. Un ruolo particolare sembra svolto da i recettori TRPV1, come dimostrato sperimentalmente dal fatto che le sostanze che li attivano, come ad esempio la capsaicina, inducono i classici segni dell’infiammazione (dolore, eritema, edema e calore). Un altro tipo di stimolazione in grado di creare dolore per attivazione delle terminazioni nervose intra tissutali è la stimolazione termica, soprattutto per stimoli caldi, ma anche per stimoli freddi. L’esempio classico è quello delle ustioni che si accompagnano al tipico dolore che, all’inizio, è sempre continuo e spontaneo. Infine, non meno importanti, appaiono gli stimoli meccanici che spesso attraverso la liberazione di sostanze proinfiammatorie ed algogene, cioè attraverso stimoli chimici, sono a loro volta in grado di creare condizioni di ipersensibilità nei tessuti lesi.
IL ruoLo deLL’InfIaMMazIone
L’insorgenza di una sensibilizzazione periferica agli stimoli dolorosi appare in gran parte legata allo sviluppo di fenomeni infiammatori. Come è storicamente ben noto, fin dai tempi di Celso (I secolo dopo Cristo) il dolore è uno degli elementi fondamentali dell’infiammazione (calor, rubor, tumor e dolor). È ben noto infatti come l’infiam-mazione possegga meccanismi algogeni specifici, spesso bersaglio delle più diffuse terapie antidolorifiche. Vale la pena di ricordare che l’infiammazione non è di per sé un fenomeno negativo in quanto essa rappresenta un elemento di difesa naturale e innato nei confronti degli attacchi che l’organismo subisce dall’ambiente circostante, siano essi microrganismi, traumi o neoplasie. Essa viene considerata una vera e propria barriera, come lo sono le difese strutturali anatomiche, le risposte difensive fisiologiche e quelle immunitarie ancestrali come ad esempio la fagocitosi (Tabella 1). Anche se l’idea più diffusa dell’infiammazione si rifà ad una reazione ad un evento ben localizzabile e cir-coscrivibile, il realtà l’infiammazione è il processo finale comune di numerosi processi patologici che vanno dai traumi, alle reazioni allergiche, all’ischemia, alle risposte autoimmuni, ai dismetabolismi (Tabella 2).
È questo il motivo per cui, soprattutto nei danni persistenti e non autolimitanti, anche la somministrazione dei più importanti antinfiammatori, i corticosteroidei, non è in grado di eliminare completamente la lesione e quindi anche il dolore ad essa correlato. Esistono al-meno 5 diversi tipi di infiammazione: non specifica (es. l’infiammazione post-traumatica), allergica (es. l’infiammazione che si accompagna all’orticaria), da immunocomplessi (es. l’infiammazione delle vasculiti o del LES), da anticorpi citotossici (es quella dell’anemia emolitica), cellulo-mediata (es. quella della tubercolosi). In generale si può dire che l’in-fiammazione sia strettamente legata alle risposte immunitarie dell’organismo, anche se non tutte le volte che si verifica una risposta immunitaria, questa viene accompagnata da una reazione infiammatoria. L’osservazione poi che non sempre la reazione infiammatoria si accompagna ad un danno tissutale ha fatto trarre la considerazione che, un po’ come il dolore, l’infiammazione può essere fisiologica (quando previene l’insorgenza del danno) oppure patologica (quando rappresenta una risposta al danno che si è già verificato).

15 Meccanismi della sensibilizzazione periferica
Considerato che i mediatori sono essenzialmente gli stessi, secondo alcuni Autori è possibile cogliere la differenza tra risposta infiammatoria fisiologica e patologica nel fatto che la prima è di entità minore rispetto alla seconda. Ma non tutti i ricercatori sono d’accordo su questa affermazione. A proposito di mediatori, un gran numero di sostanze è stato identificato negli ultimi decenni, anche se il peso di ciascun elemento nella com-plessa cascata di eventi che portano all’infiammazione non è facilmente calcolabile. Un contributo importante sembra comunque certo per alcune “famiglie” di sostanze che sono state identificate nei tessuti infiammati. Il ruolo delle chinine per esempio non è più in discussione, vista l’enorme messe di dati accumulata negli anni. Anche perché tale gruppo di sostanze, oltre a svolgere un’azione pro-infiammatoria diretta, rappresenta l’innesco per la liberazione di numerosi altri mediatori dell’infiammazione quali le citochine. Sotto
Tabella 1. risposte immunitarie innate e adattative (in accordo con de Leo e Yezierski, 2001)
Immunità innata o aspecifica (barriere difensive)
anatomiafisiologiafagocitosiInfiammazione
Immunità adattativa o specifica (proprietà) specificitàdiversitàMemoriariconoscimento del self / non self
Tabella 2. tipi diversi di infiammazioni che si riscontrano nella pratica clinica (in accordo con ali et al. 1997)
Tipo di immunità Riconoscimento Cellule Mediatori Meccanismi Malattie non specifica via alternativa
del complementoneutrofili e macrofagi
complemento Liberazione di mediatori citotossici da neutrofili e macrofagi attivati da sostanze derivate dal complemento
trauma, sepsi da gram-negativi
allergica(immediata)
Ige Mast cellule, basofili, eosinofili
Istamina e leucotrieni
attivazione di mast cellule, basofili ed eosinofili da parte del legame antigene-Ige
orticaria, rinite, asma, anafilassi
da immuno-complessi
IgG, IgM neutrofili e macrofagi
complemento attivazione di neutrofili e macrofagi da immunocomplessi fissati dal complemento
Malattie reumatiche, glomerulonefriti, vasculiti, lupus eritematoso sistemico
da anticorpi citotossici
IgG, IgM neutrofili e macrofagi
complemento Lisi o fagocitosi di antigeni circolanti
Malattie autoimmuni, anemia emolitica
cellulo-mediata(ritardata)
Linfociti t Linfociti e macrofagi
citochine attivazione di macrofagi e rilascio di mediatori citotossici da parte di citochine rilasciate da cellule t-helper
tubercolosi, polimiosite, sarcoidosi

16Aspetti di fisiopatologia e terapia del dolore
questo nome si identifica un gruppo di proteine e glicoproteine che può essere liberato da diverse cellule dell’organismo e che interviene sicuramente nelle reazioni infiammatorie collegate allo sviluppo di ipersensibilità agli stimoli dolorosi. Vi è ampia dimostrazione in letteratura che alcune citochine quali il TNF (Tumor Necrosis Factor) e diverse in-terleuchine aumentano la scarica delle fibre nocicettive in corso di infiammazione. Le citochine facilitano lo sviluppo di reazioni infiammatorie in vario modo. Oltre all’azione sulla liberazione di ossido nitrico, comune a molti percorsi connessi all’infiammazione, attraverso l’induzione dell’ossido nitrico sintetasi, le citochine facilitano la liberazione di sostanza P dalle terminazioni nervose e attivano enzimi coinvolti nell’infiammazione quale la ciclossigenasi 2, meglio nota come COX-2. Per quanto riguarda in modo spe-cifico il TNF, attualmente forse la citochina legata all’infiammazione più studiata, esso favorisce la reazione infiammatoria acuta e attiva le cellule immunitarie. È ben nota la sua produzione dai macrofagi, ma non solo da questi. Anche i leucociti CD4+, i linfociti NK (Natural Killer) e i neuroni stessi sono stati visti essere in grado di liberare TNF. Ritornando a concetti più generali, quello che non è ancora molto chiaro è come mai in alcuni casi l’infiammazione abbia un’azione benefica (infiammazione fisiologica) ed altre volte sia essa stessa causa di persistenza di malattia (infiammazione patologica). A questo proposito, come già accennato in precedenza, c’è discussione sul fatto che sia solo una questione di quantità dei fenomeni infiammatori. Altri Autori hanno considerato come possibile fattore scatenante la durata dei fenomeni infiammatori. In altri termini, dopo l’insulto ricevuto l’organismo reagirebbe con diverse modalità, tra cui l’infiammazione, nel tentativo di prevenire e/o riparare i danni. Se però questo processo non avviene in un determinato tempo, l’organismo perderebbe il controllo sulla catena di eventi che si accompagnano all’infiammazione acuta e questa diventerebbe cronica, sostenendo il perdurare e il non guarire di alcune malattie croniche infiammatorie. Ovviamente anche le ben note alterate risposte immunitarie di riconoscimento/non riconoscimento dei tessuti dell’organismo giocano un ruolo molto importante in questi processi.
stress ossIdatIvo, superossIdodIsMutasI (sod) e InfIaMMazIone
Nella fisiologica attività tissutale i sistemi di ossidazione/antiossidazione sono ten-denzialmente in equilibrio tra di loro. Quando però un tessuto viene interessato da un processo patologico spesso si verifica uno sbilanciamento a favore dei fenomeni ossida-tivi. Tale condizione è nota col termine di stress ossidativo. Trattasi di un accumulo di specie reattive derivate dall’ossigeno denominate con l’acronimo ROS (Reacting Oxygen Species) e appartenenti alla specie chimica definita radicali liberi. La formazione delle ROS rappresenta uno strumento di difesa, ma se esse persistono a lungo in sede lesionale possono innescare pericolosi meccanismi fisiopatologici fino ad indurre ulteriori lesioni dei tessuti, con interessamento anche dei tessuti sani circostanti. Uno dei meccanismi che gli organismi possiedono per combattere lo stress ossidativo è quello di attivare un sistema di difesa cellulare endogena che prevede l’utilizzo della superossidodismutasi (SOD), un enzima che appartiene alla classe delle ossidoreduttasi. La SOD, la cui azione enzimatica è nota fin dal 1969, è considerato uno dei più importanti enzimi antiossidanti presenti negli organismi viventi. La sua attività “difensiva” è basata sulla capacità di trasformare, grazie all’utilizzo di ioni idrogeno (H+), il superossido (l’anione O
2-) in ossigeno (O
2)
e perossido di idrogeno (H2O
2). In altri termini, la SOD catalizza la seguente reazione:
2 O2– + 2 H+ O
2 +H
2O
2
Attualmente sono state identificate 3 forme di SOD: la SOD1, presente nel citosol, la SOD2, presente nei mitocondri e la SOD3, presente sulle superfici extracellulari. Poiché però il perossido di idrogeno è anch’esso un ossidante, per una completa detossificazione

17 Meccanismi della sensibilizzazione periferica
è necessario che il perossido di idrogeno venga trasformato in acqua. Tale operazione è compiuta dall’enzima glutatione perossidasi, un’altra ossidoreduttasi che catalizza la seguente reazione:
2 glutatione + H2O
2 glutatione disolfuro + 2 H
2O
Numerose condizioni fisiopatologiche si accompagnano a stress ossidativo. Tra di esse spiccano quelle sostenute dallo sviluppo di fenomeni infiammatori. Alla formazio-ne di radicali liberi durante l’infiammazione concorrono diversi fattori. Una parte dello stress ossidativo osservato durante l’infiammazione, è sicuramente legata ai neutrofili, leucociti che spesso si accumulano in sede di lesione infiammatoria. È infatti noto che quando i neutrofili vengono attivati, iniziano a secernere le ROS che rappresentano, tra le altre cose, uno strumento per indurre la morte cellulare di eventuali batteri presenti nella sede dell’infiammazione. In tali condizioni la SOD può contribuire, neutralizzando il superossido, a riequilibrare il bilancio ossidazione/antiossidazione esplicando pertanto un’azione antinfiammatoria. Un altro meccanismo antinfiammatorio posseduto dalla SOD è rappresentato da una sua azione diretta sui neutrofili, di cui è in grado di indurre l’apoptosi (morte cellulare programmata). È ben noto come l’accumulo di tali cellule rappresenti uno dei fattori associati allo sviluppo di infiammazione. I neutrofili sono infatti tra le prime cellule a giungere nei siti lesionali, attratti dalle chemiochine, sostanze liberate dalle cellule endoteliali dei vasi presenti in sede lesionale. Una volta giunti nella sede dell’infiammazione, i neutrofili, oltre a mettere in atto le loro capacità di fagocitosi e a liberare le ROS, secernono anche alcuni tipi di proteasi, nonché una notevole quantità di chemiochine. Queste ultime amplificano la risposta infiammatoria attirando nuovi neu-trofili, mentre la liberazione di ROS e proteasi è potenzialmente dannosa per i tessuti in cui vengono liberati. Considerato quindi che la rimozione dei neutrofili mediante apoptosi rappresenta un meccanismo cruciale per l’inattivazione dei fenomeni infiammatori, l’a-poptosi dei neutrofili è considerata un possibile bersaglio per il controllo terapeutico delle lesioni tissutali mediate dai neutrofili. Ne consegue che la SOD, grazie alla sua potente azione inducente l’apoptosi dei neutrofili, può rappresentare un importante strumento per ridurre l’entità dei fenomeni infiammatori in sede lesionale. Il meccanismo attraverso cui la SOD induce l’apoptosi non è completamente noto e numerose ipotesi sono state formulate. Tra le altre si segnala quella che prevede un ruolo significativo del perossido di idrogeno. Poiché questo si libera dalla reazione di trasformazione del superossido mediato dalla SOD (vedi sopra), il ruolo positivo di quest’ultima, in caso di infiammazione, non si limiterebbe alla neutralizzazione del superossido, ma risiederebbe anche nella produzione di perossido di idrogeno con conseguente apoptosi dei neutrofili. In altre parole, è possibile che i neutrofili liberino ROS nella fase infiammatoria acuta nel tentativo di distruggere quanti più batteri possibili, ma così facendo programmino la loro morte cellulare visto che l’intervento della SOD, mediante la neutralizzazione delle ROS, genera quel peros-sido d’idrogeno che appare in grado di indurre la loro apoptosi. Ciò spiegherebbe anche perché in condizioni cliniche caratterizzate da una ridotta produzione di ROS, come ad esempio l’ipossia, si osservi una ridotta apoptosi dei neutrofili. L’azione della SOD sui neutrofili risulta ancora più importante se si considera che i glucocorticoidi, giustamente considerati come i più potenti antinfiammatori, inducono l’apoptosi dei T-linfociti e degli eosinofili, ma addirittura inibiscono l’apoptosi dei neutrofili.
L’InfIaMMazIone neuroGena
Uno dei meccanismi più noti dello sviluppo di ipersensibilità periferica è rappresentato dall’infiammazione neurogena. Con tale termine si intende quell’insieme di fenomeni che porta alla liberazione di sostanze pro-infiammatorie e potenzialmente algogene, in

18Aspetti di fisiopatologia e terapia del dolore
seguito all’attivazione antidromica di fibre nervose. In pratica, tutte le volte che un po-tenziale d’azione viaggia antidromicamente, cioè dalla fibra verso il recettore, una volta arrivato alla fine della corsa, a livello del recettore, esso è in grado di liberare sostanze pro-infiammatorie e potenzialmente algogene. Questo fenomeno non si riscontra durante l’attivazione di tutti i tipi di fibre, ma solo quando ad essere attivate sono le fibre amielini-che afferenti (dette anche C afferenti o C sensitive o fibre C delle radici dorsali). Diverse sostanze vengono liberate in corso di attivazione antidromica delle fibre C, ma quelle più note e studiate sono la sostanza P ed il CGRP (Calcitonin Gene Related Peptide). Appare interessante sottolineare come in condizioni fisiologiche la liberazione di sostanza P e CGRP avvenga solo da determinate fibre nervose, mentre in corso di infiammazione tali sostanze vengono liberate anche da fibre che normalmente non sono in grado di liberarle. Tutto questo contribuisce al mantenimento e all’amplificazione dei fenomeni infiammatori e pro-nocicettivi. Uno dei meccanismi attraverso cui sostanza P e CGRP potenziano i fenomeni infiammatori è rappresentato dalla loro capacità di richiamare e far accumulare neutrofili nell’interstizio (Figura 2). Ciò avviene per modificazione indotta soprattutto dall’attivazione di cellule endoteliali e dal richiamo ed attivazione di macrofagi. Come si inserisce questo discorso nel meccanismo della sensibilizzazione periferica? Se si pensa alla morfologia delle terminazioni libere si capisce come queste terminazioni finali che si diramano dallo stesso assone rappresentino una vera e propria unità che non è solo anatomica, ma anche funzionale. Quello che si verifica in caso di attivazione costante delle terminazioni libere, come avviene per esempio in corso di infiammazione, è sostanzialmente questo: ogniqualvolta, in seguito ad uno stimolo, si genera un potenziale d’azione a partenza da una terminazione, questo impulso oltre che viaggiare verso il midollo spinale, torna anche indietro, attraverso un’altra termi-nazione che in quel momento non sta trasmettendo impulsi (Figura 3). Questo tornare indietro altro non è che un’attivazione antidromica di una fibra amielinica e quindi libera sostanze proinfiammatorie e potenzialmente algogene dalla terminazione in tal modo attivata. Questo meccanismo è anche alla base di quel fenomeno che porta allo sviluppo di segni di infiammazione intorno all’aria lesionale, in cui si possono, tra l’altro, regi-
Figura 2. Meccanismi attraverso i quali la liberazione di sostanza p e cGrp può attirare neutrofili a livello interstiziale, favorendo l’infiammazione neurogena.
sostanza pcGrp
espressione di recettori per i peptidi
liberazione molecole di adesione cellulare
citochine
accumulo interstiziale di neutrofili
sostanza pcGrp
endotelio
macrofago

19 Meccanismi della sensibilizzazione periferica
strare fenomeni di ipersensibilità agli stimoli meccanici, noti come allodinia/iperalgesia secondaria (così chiamata per distinguerla da quella che si sviluppa all’interno dell’area lesionale, denominata allodinia/iperalgesia primaria). In tutto questo appare chiaro il ruolo del sistema nervoso periferico e delle fibre nervose amieliniche in particolare. La dimostrazione dell’importante ruolo giocata da tale sistema è data dall’osservazione, sia sperimentale che clinica, che le lesioni nervose che fanno degenerare gli assoni di piccolo calibro riducono nettamente, e a volte aboliscono completamente, tutti i fenomeni tipici dell’infiammazione neurogena.
IL ruoLo deI GanGLI sensItIvI neLL’IpersensIBILItà perIferIca
Come descritto precedentemente, l’impulso generato in periferia viaggia senza osta-coli verso la prima sinapsi posta nel corno posteriore del midollo. Lungo questo tragitto esso può essere modificato, ed in particolare amplificato, a livello dei gangli sensitivi delle radici posteriori. È stato infatti dimostrato che, in caso di ipersensibilità sviluppata in un tessuto da fenomeni infiammatori, l’informazione di amplificazione dei segnali generati in periferia si avvale anche di un meccanismo che si verifica nei gangli sensitivi delle radici dorsali. In tali condizioni nuovi canali del sodio si evidenziano nei gangli sensitivi generando una sorta di ipersensibilità da passaggio di impulsi: ogni impulso che proviene dalla periferia, sia esso fisiologicamente o patologicamente generato, arrivato a livello gangliare subisce un’amplificazione che è in grado di incrementare la sensazione dolorosa.
IL ruoLo deLL’IscheMIa
Un ruolo particolare viene giocato dall’ischemia. Quando i tessuti vanno incontro ad ischemia essi sviluppano condizioni di ipersensibilità. Un esempio clinico molto noto è rappresentato dalla claudicatio intermittens che si osserva nei pazienti con insufficienza vascolare periferica. Quando un muscolo viene attivato, come si verifica normalmente in caso di esercizio muscolare, le richieste energetiche e metaboliche aumentano e quindi tutto quello che ostacola un corretto adattamento da parte dell’organismo finisce per creare un condizione di sofferenza tissutale. Nelle fasi iniziali della vasculopatia periferica obliterante le arterie, il compenso a riposo è soddisfacente e il paziente non avverte alcun sintomo quando è a riposo. Se però inizia a contrarre i muscoli, dopo una certa quantità di sforzo muscolare, che varia con la gravità dell’arteriopatia, il paziente avverte dolore. Tale sensazione spiacevole è legata al fatto che uno stimolo meccanico
assone
terminazioni libere
stimolo nocicettivo
Figura 3. Illustrazione schematica di come un impulso generato a livello di una terminazione nervosa possa, attraverso un riflesso assonale, ritornare indietro e liberare sostanze pro-infiammatorie nel tessuto interessato dalla lesione.

20Aspetti di fisiopatologia e terapia del dolore
(contrazione muscolare) che normalmente non attiva i nocicettori, in condizioni di ischemia lo fa. In altre parole, l’ischemia abbassa la soglia del dolore muscolare, che si riduce sempre più al progredire dell’arteriopatia. È per questo motivo che il paziente riferisce che la distanza che riesce a percorrere senza avvertire dolore si riduce sempre di più. Spesso il paziente è in grado di stabilire con una certa precisione quanti metri (o quanti passi) riesce a percorrere senza dolore. Il fenomeno della claudicatio che si osserva nei pazienti con vasculopatie periferiche è pertanto il frutto di una progressiva sensibilizzazione delle terminazioni nervose nocicettive che sono presenti nei muscoli utilizzati per la deambulazione. Ma qual è il meccanismo intrinseco che porta a tale ipersensibilità periferica? Il fenomeno è complesso ma un ruolo importante viene si-curamente svolto dai recettori vanilloidi TRPV1 che, oltre ad attivarsi per progressivi incrementi della temperatura fino a livelli francamente nocicettivi, si attivano anche in condizioni di ischemia perché sensibili agli idrogeno ioni. Come è ben noto, l’attivazione dei TRPV1 si accompagna ad una sensazione urente, ad un abbassamento della soglia al dolore a numerosi stimoli e, se persiste sufficientemente a lungo, allo sviluppo di tutti i segni dell’infiammazione, inclusi allodinia, iperalgesia, e dolore spontaneo. Almeno per il dolore che insorge in corso di contrazioni effettuate in condizioni di ischemia, anche un altro meccanismo è stato dimostrato nell’animale: l’attivazione di nocicettori silenti. In sintesi, quando il muscolo si contrae in condizioni di ischemia, si osserva la scarica di nocicettori che non si attivavamo, anche per stimolazioni molto intense, in condizioni non-ischemiche. L’attivazione dei nocicettori muscolari silenti induce una sommazione spazio-temporale degli impulsi nocicettivi provenienti dalla periferia. Ciò è sufficiente ad aumentare la frequenza di scarica dei nocicettori spinali, condizione necessaria, sebbene non sufficiente, per avvertire le sensazioni dolorose.
BIBLIoGrafIa
Ali H, Haribabu B, Richardson RM, Snyderman R. Mecha-nisms of inflammation and leukocyte activation. Med Clin North Am. 1997 Jan; 81 (1): 1-28.
Buonocore M. Meccanismi patogenetici del dolore da infiammazione. Contro il dolore, Excerpta Medica, Milano, 2002: 3-17.
Buonocore M, Bonezzi C. Fisiopatologia del dolore. Mo-mento Medico, Salerno, 2005.
Buonocore M. Meccanismi del dolore periferico. In: Merca-dante. S. «Il dolore. Valutazione diagnosi e trattamento». Masson, Milano, 2006: 87-96.
Calixto JB, Cabrini DA, Ferreira J, Campos MM. Kinins in pain and inflammation. Pain. 2000 Jul; 87 (1): 1-5.
Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001 Oct 19; 429 (1-3): 161-76.
Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Anti-oxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001 Mar; 53 (1) :135-59.
De Leo JA, Yezierski RP. The role of neuroinfammation and neuroimmune activation in persistent pain. Pain 2001 Feb 1; 90 (1-2): 1-6
Henriksson KG Hypersensitivity in Muscle Pain Syn-dromes Curr Pain Headache Rep. 2003, 7: 426-432.
Koltzenburg M. Neural mechanisms of cutaneous nocicep-tive pain. The Clin J Pain 2000, 16, S131-S138.
Mense S. The pathogenesis of muscle pain. Curr Pain Headache Rep. 2003 Dec; 7 (6): 419-25.
Merskey H, Bogduk N. Classification of chronic pain: descriptions of chronic pain syndromes and definitions of pain terms. 2nd ed. Seattle: IASP Press; 1994.
Portenoy RK Basic mechanisms. In: Portenoy RK e Kanner RM (Eds): Pain Management: Theory and Practice. F.A. Davis Company, Philadelphia, 1996: 19-39.
Savill J. Apoptosis in resolution of inflammation. J Leukoc Biol. 1997 Apr; 61 (4): 375-80.
Schlereth T, Birklein F. Mast cells: source of inflammation in complex regional pain syndrome? Anesthesiology. 2012 Apr; 116 (4): 756-7.
Sommer C. The role of cytockines in pain In: Josè Castro-Lopes (Ed) Current Topics in Pain- 12th World Congress on Pain IASOP-Press, Seattle, 2008: 95-113.
Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD. Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol Sci. 2001 Mar; 22 (3): 146-52.
Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004 Jun; 309 (3): 869-78. Epub 2004 Feb 26.
Woolf CJ, Decosterd I. Implications of recent advances in the understanding of pain pathophysiology for the assessment of pain in patients. Pain, 1999, Supplement 6: S141-S147.
Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000 Jun 9; 288 (5472): 1765-9.

3 GenesI ectopIcA deGlI IMpulsI Michelangelo Buonocore
doLore neuropatIco
Come già anticipato nel precedente capitolo, non sempre il dolore origina dalle terminazioni nervose delle fibre di piccolo calibro, fisiologicamente deputate alla trasmissione di impulsi nocicettivi. In alcuni casi esso origina da impulsi generati direttamente a livello delle fibre nervose. Un dolore che presenta tale caratteristica fisiopatologica è definito dolore neuropatico, la cui definizione ufficiale attuale è quella adottata dallo Special Interest Group della IASP (NeupSIG) e cioè “dolore che origina come diretta conseguenza di una lesione o di una malattia interessante il sistema nervoso somatosensoriale”.
Il tipo di fibra nervosa da cui originano gli impulsi che contribuiscono all’insor-genza di un dolore neuropatico non sembra importante, purché appartenente al sistema somatosensoriale. Nella sua patogenesi possono infatti essere coinvolte sia fibre di piccolo calibro, quelle che fisiologicamente veicolano il dolore, sia fibre di grande calibro, quelle che fisiologicamente veicolano sensazioni non dolorose, quali quelle tattili. In altre parole, se nel dolore nocicettivo le fibre coinvolte sono quelle del dolore fisiologico, nel dolore neuropatico anche fibre che fisiologicamente non conducono il dolore sono in grado di generare quelle sensazioni spiacevoli che noi chiamiamo dolore. Come è allora possibile che una fibra che è normalmente deputata a trasmettere sensazioni non dolorose diventa in grado di farlo quando lesionata? La risposta non è facile perché i meccanismi del dolore neuropatico non sono a tutt’oggi completamen-te noti, ma è molto probabile che un ruolo venga giocato dalla frequenza di scarica della fibra. È infatti noto che i potenziali che viaggiano lungo le fibre nervose sono generati dai recettori, cioè da quelle strutture che trasducono gli stimoli di altra natura (soprattutto meccanici, termici, chimici) in stimoli elettrici. Le capacità intrinseche dei recettori condizionano pertanto la frequenza di scarica delle fibre nervose. Se invece che dal recettore, la fibra viene attivata direttamente, per una sorta di cortocircuito che si viene a creare in un punto preciso della fibra, allora la sua frequenza di scarica può essere completamente diversa rispetto a quella normalmente generata dall’attivazione del recettore (Figura 1).
Esistono evidenze sperimentali che, anche nell’uomo, hanno evidenziato come, quando un impulso attraversa un sito lesionale esso può essere bloccato oppure può propagarsi a distanza. Nel primo caso si ha una riduzione della sensibilità veicolata dalla fibra lesionata, ma nel secondo caso si possono generare due situazioni anomale che possono ingannare il sistema nervoso centrale. La prima è dovuta al fatto che i potenziali passano da una frequenza di scarica regolare ad una irregolare e caotica. La seconda situazione anomala che si può creare è legata al fatto che le frequenze di scarica diventano più elevate di quelle fisiologiche (Figura 2). Poiché è ben noto che il sistema nervoso funziona a modulazione di frequenza, entrambi i suddetti fe-
3
22Aspetti di fisiopatologia e terapia del dolore
nomeni sono in grado di attivare in modo anomalo il sistema nervoso, ingannandolo, e rendendo possibile lo sviluppo di sensazioni spiacevoli per l’individuo. Inoltre, in queste particolari condizioni fisiopatologiche gli impulsi possono passare (ed essere
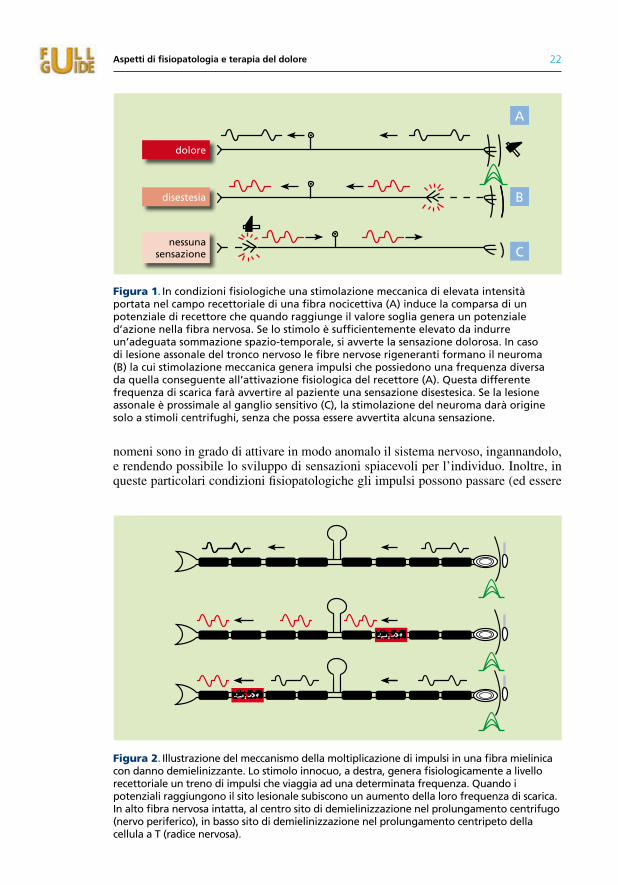
Figura 1. In condizioni fisiologiche una stimolazione meccanica di elevata intensità portata nel campo recettoriale di una fibra nocicettiva (a) induce la comparsa di un potenziale di recettore che quando raggiunge il valore soglia genera un potenziale d’azione nella fibra nervosa. se lo stimolo è sufficientemente elevato da indurre un’adeguata sommazione spazio-temporale, si avverte la sensazione dolorosa. In caso di lesione assonale del tronco nervoso le fibre nervose rigeneranti formano il neuroma (B) la cui stimolazione meccanica genera impulsi che possiedono una frequenza diversa da quella conseguente all’attivazione fisiologica del recettore (a). Questa differente frequenza di scarica farà avvertire al paziente una sensazione disestesica. se la lesione assonale è prossimale al ganglio sensitivo (c), la stimolazione del neuroma darà origine solo a stimoli centrifughi, senza che possa essere avvertita alcuna sensazione.
a
B
c
dolore
disestesia
nessuna sensazione
Figura 2. Illustrazione del meccanismo della moltiplicazione di impulsi in una fibra mielinica con danno demielinizzante. Lo stimolo innocuo, a destra, genera fisiologicamente a livello recettoriale un treno di impulsi che viaggia ad una determinata frequenza. Quando i potenziali raggiungono il sito lesionale subiscono un aumento della loro frequenza di scarica. In alto fibra nervosa intatta, al centro sito di demielinizzazione nel prolungamento centrifugo (nervo periferico), in basso sito di demielinizzazione nel prolungamento centripeto della cellula a t (radice nervosa).

23 Genesi ectopica degli impulsi
riferiti) a fibre che non sono state per niente stimolate in periferia (Figura 3). In ogni caso, indipendentemente dal meccanismo specifico di generazione, il dolore neuro-patico è sempre caratterizzato dalla legge di proiezione specifica. Questa legge della fisiologia afferma che, tutte le volte che una fibra nervosa viene stimolata ed attivata, direttamente (bypassando il recettore) o indirettamente (attraverso il recettore), in un suo punto (qualsiasi) la sensazione evocata dalla stimolazione viene sempre riferita nel punto dove ci sono (o ci dovrebbero essere, in caso di danno assonale) i recettori. La dimostrazione è data dalla stimolazione elettrica (che crea stimoli ectopici nei punti di stimolazione): ovunque venga portata la stimolazione, la sensazione di scossa elettrica viene avvertita sempre nel territorio del nervo, del plesso, della radice, della parte di midollo o encefalo stimolati. Un altro esempio della veridicità di tale legge è rappresentata dalla tipica disestesia evocata nel territorio del nervo ulnare tutte le volte che, inavvertitamente, il nervo ulnare viene meccanicamente stimolato a livello del gomito, nel punto di passaggio nella doccia olecranica.
La GenesI ectopIca deGLI IMpuLsI
Come illustrato precedentemente il dolore neuropatico è caratterizzato dal fatto che, in questa forma di dolore, gli impulsi si generano direttamente dalle fibre nervose e non dalle terminazioni delle fibre stesse (recettori). Il termine ectopico (fuori luogo) sta proprio a significare che gli impulsi generatori del dolore non si generano nel posto giusto (quello fisiologicamente deputato allo scopo). A testimonianza della differenza tra stimoli fisiologici e stimoli ectopici vi è tra l’altro il fatto che i primi si generano in un punto specifico (recettore) e viaggiano solo in direzione ortodromica (verso il sistema nervoso centrale) mentre i secondi si generano in un punto qualsiasi della fibra e si propagano sia in direzione centripeta (ortodromica) che centrifuga (antidromica). Appare inoltre importante sottolineare come potenziali ectopici si possono generare su qualsiasi tipo di fibra, indipendentemente dal loro calibro e velocità di conduzione. Come per il dolore nocicettivo (a genesi normotopica) anche per il dolore neuropatico (a genesi ectopica) si può distinguere un meccanismo di scarica spontanea (indipendente cioè da qualsiasi stimolo) da un meccanismo di scarica evocata (stimolo-dipendente). La possibilità di evocare un dolore neuropatico in seguito ad una stimolazione meccanica viene sfruttato in semeiotica in quel famoso segno che viene definito segno di Tinel, dal nome del suo primo descrittore. Trattasi della possibilità di evocare con stimoli meccanici lievi una sensazione parestesico-disestesico-dolorosa quando si stimola il
Figura 3. disegno schematico illustrante il meccanismo dell’efapsi tra fibre mieliniche con danno demielinizzante. L’attivazione mediante stimoli innocui della fibra nervosa illustrata in alto induce potenziali d’azione che viaggiano fisiologicamente fino al sito di demielinizzazione. Qui, a causa della perdita dell’isolamento rappresentato dalla mielina, essi subiscono un aumento della frequenza e, soprattutto, passano nella fibra limitrofa illustrata in basso.

24Aspetti di fisiopatologia e terapia del dolore
moncone prossimale di un nervo interrotto o il sito patologico di un nervo leso ma non interrotto. In altre situazioni, ad esempio in alcune forme di polineuropatia, gli impulsi ectopici sono generati spontaneamente nelle fibre sensitive degeneranti generando un complesso di disestesie e dolore che non dipendono da stimolazioni particolari. Per quanto riguarda gli aspetti neuropatologici, appare importante sottolineare che non esiste un quadro specifico di danno nervoso che più di altri è stato associato alla genesi di impulsi ectopici. In particolare, sia danni assonali che danni demielinizzanti sono in grado di generare stimoli ectopici.
doLore da LesIone assonaLe senza deafferentazIone
Un’altra differenziazione importante è quella che prevede possibili differenze fisiopatologiche algogene a seconda che la lesione sia assonale o demielinizzante. La presenza di un danno assonale, cioè di una degenerazione della parte distale di una fibra sensitiva e mantenimento di un moncone prossimale integro fa pensare subito alla condizione di ipereccitabilità in cui si viene a trovare il moncone prossimale dopo poco tempo dall’interruzione assonale. È ben noto infatti che nel momento in cui un danno assonale si verifica, immediatamente nel corpo cellulare si attivano i processi riparativi che preparano il moncone prossimale per la ricrescita. Nel corpo cellulare inizia la produzione di elementi strutturali e funzionali, quali gli elementi citoschele-trici, i canali ionici, i recettori, i neurotrasmettitori, che vengono trasportati verso la periferia, dove si trova l’interruzione assonale, mediante i ben noti flussi assonali. Il risultato finale di tutto questo fervore metabolico è rappresentato dal fatto che sulle fibre rigeneranti si creano dei bottoni germinativi altamente sensibili a stimoli meccanici, chimici e termici. Questa ipersensibilità, che si genera ogni qualvolta esiste un siffatto danno assonale, non si accompagna però necessariamente ad un dolore clinicamente significativo. L’esempio di questo appare lampante nel fatto che nella gran parte delle lesioni nervose, anche gravi, non si sviluppano dolori attribuibili alla lesione nervosa. Così come non è controvertibile il fatto che durante alcune operazioni chirurgiche il chirurgo taglia deliberatamente un nervo per evitare che questi resti intrappolato (e generi dolore) nell’anatomia sovvertita dall’intervento. Un esempio di tale asporta-zione deliberata ed indolore di rami nevosi è rappresentata dall’intervento per ernia inguinale dove spesso viene sezionato ed asportato un pezzo del nervo ilio-inguinale. Altro esempio del genere sono gli interventi di innesto nervoso, quando, per riparare un nervo leso, si preleva un pezzo di un nervo sano, frequentemente il nervo surale e lo si giustappone tra i monconi del nervo lesionato. Se tale asportazione generasse un dolore, nessuno più utilizzerebbe questa tecnica che, invece, viene ancora attualmente utilizzata e non crea nuovi dolori nel paziente operato.
È anche vero, però, che in alcuni casi l’ipersensibilità che si sviluppa sulle fibre ner-vose rigeneranti rappresenta una importante causa di dolore. È ben noto che quando le fibre nervose rigeneranti non trovano più la strada da percorrere perché il connettivo del nervo è stato interrotto da un trauma o perché una lesione ha occluso i tubi endoneurali si forma un vero e proprio groviglio di fibre nervose rigeneranti in ogni direzione che va sotto il nome di neuroma. Come già detto precedentemente l’ipersensibilità delle fibre nervose rigeneranti che si organizzano a formare il neuroma non genera neces-sariamente dolore. In altre parole il neuroma si forma ogni qualvolta il connettivo di un tronco nervoso viene interrotto o lesionato, ma solo raramente il neuroma diventa algogeno. Quando ciò si verifica? Il neuroma genera dolore solo quando le fibre nervose rigeneranti che lo costituiscono vengono stimolate. Questa stimolazione può essere meccanica, termica o semplicemente chimica, ma essa è condizione indispensabile per la genesi di potenziali nervosi ectopici algogeni. Una causa relativamente frequente

25 Genesi ectopica degli impulsi
di neuroma doloroso è rappresentato dalla formazione del neuroma in stretto rapporto con una fascia muscolare o un tendine. In tale condizione tutte le volte che un muscolo viene contratto, il neuroma viene stimolato ed il paziente avverte dolore.
Uno dei motivi più importanti e più studiati della genesi ectopica di impulsi è dato da modificazioni che avvengono a carico dei canali del sodio. Questi canali (ce ne sono di diversi tipi) sono stati ripetutamente correlati alla genesi di potenziali ec-topici nelle fibre nervose. Quando si verifica un danno assonale, il numero di canali del sodio presenti sulle membrane del moncone prossimale aumenta (Figura 4). Tale
incremento si accompagna ad una maggiore eccitabilità della fibra nervosa in quan-to i flussi di ioni sodio dall’esterno all’interno della cellula sono facilitati e questo aumenta le probabilità che si generi un potenziale d’azione perché l’entrata di ioni positivi all’interno della cellula nervosa la rende più facilmente eccitabile. Vale la pena sottolineare come, in caso di danno assonale, l’espressione sulle membrane di nuovi canali del sodio non avvenga solo nel sito lesionale, ma in numerose altre parti del neurone, incluso il corpo cellulare.
L’aumento dei canali del sodio sulle membrane dipende in modo inversamente proporzionale alla velocità del flusso assonale. Infatti, quando si verificano alcune condizioni che sono note essere in grado di rallentare il flusso assonale (esposizione al freddo, uso di farmaci neurotossici) il numero di canali del sodio che appaiono sulle membrane neuronali è maggiore.
doLore da LesIone assonaLe con deafferentazIone
In realtà il termine di dolore neuropatico è un po’ limitativo in quanto i meccanismi di insorgenza possono essere veramente molto diversi tra di loro. Se è indubbio che una ipereccitabilità/scarica spontanea di una fibra nervosa periferica lesa è in grado di creare una ipersensibilità neuronale sufficientemente intensa per lo sviluppo di una
Figura 4. Illustrazione schematica dello sviluppo di nuovi canali del sodio in caso di lesione assonale di un nervo periferico. si noti come i nuovi canali si formino anche a distanza dal sito lesionale e come la presenza di nuovi canali del sodio si accompagni a modificazioni della membrana che portano ad una diversa frequenza di attivazione della fibra lesa.
na+
assone integro
canale del sodio assone leso

26Aspetti di fisiopatologia e terapia del dolore
sensazione dolorosa, è altrettanto vero che esistono situazioni completamente opposte dove è proprio la distruzione completa delle fibre nervose a creare il dolore.
L’esempio più importante in tal senso è rappresentato dal cosiddetto dolore da deafferentazione. Con tale termine si intende un dolore che insorge a causa della scarica spontanea di un neurone nocicettivo che abbia perso tutte le connessioni con il neurone nocicettivo che lo precede lungo la via spino-talamica. Per quanto appena detto appare molto chiaro che tale genesi ectopica di impulsi non può verificarsi nel I neurone nocicettivo perché, essendo il primo della serie, non esiste un neurone che lo precede lungo la via. Quindi, il primo neurone che può incominciare a scaricare a causa dello sviluppo di fenomeni di deafferentazione è il II neurone nocicettivo, quello che è localizzato nel corno posteriore del midollo spinale. Tale neurone viene a trovarsi in una condizione di deafferentazione nel momento in cui gravi lesioni interessanti il sistema nervoso periferico lo disconnettono dalla periferia. Ciò si verifica esclusiva-mente per lesioni interessanti il ganglio sensitivo e/o il suo prolungamento centripeto.
È importante sottolineare come, in quest’ultimo caso, gran parte del neurone sopravvive alla lesione, ma l’attivazione patologica delle fibre nervose non produce alcuna sensazione dolorosa (Figura 1).
La tipica lesione gangliare che può deafferentare il II neurone nocicettivo è la riattivazione del virus della varicella, dormiente nel ganglio sensitivo. In alcuni casi la reinfezione (herpes zoster) porta alla degenerazione del I neurone sensitivo, con conseguente sviluppo di quel quadro clinico doloroso che viene denominato nevralgia post-herpetica (Figura 5).
Un classico esempio della seconda, tipica lesione nervosa periferica deafferentante è rappresentata dall’avulsione post-traumatica del plesso brachiale. In questi casi, la lesione, irreversibile, avviene a monte del ganglio sensitivo dove i prolungamenti cen-trifughi delle cellule a T vengono letteralmente strappati dai loro siti di connessione a livello del corno posteriore del midollo spinale (Figura 5). Cosa hanno di particolare le suddette lesioni rispetto agli altri esempi di possibile lesione gangliare o radicolare?
Figura 5. esempi di deafferentazione del secondo neurone nocicettivo (midollare) da degenerazione del moncone prossimale delle cellule a t in seguito a lesione radicolare (in alto) o dell’intero neurone periferico in seguito a lesione gangliare (in basso).

27 Genesi ectopica degli impulsi
Le caratteristiche delle lesioni in grado di deafferentare i secondi neuroni posti nel corno posteriore del midollo sono essenzialmente due.
La prima è rappresentata dal fatto che la lesione deve essere grave, portando a degenerazione tutte le fibre nervose presenti nelle strutture lese.
La seconda è rappresentata dal fatto che più radici limitrofe devono essere con-temporaneamente interessate. In particolare, per quanto riguarda quest’ultimo aspetto, è molto importante ricordare come la connessione tra radici e livelli corrispondenti midollari non sia biunivoca. Infatti un segmento midollare riceve fibre da due o più radici limitrofe e non solo da quella corrispondente. Così come le fibre che decorrono in un’unica radice posteriore non si connettono solo con il segmento midollare (corno posteriore) corrispondente, ma anche, almeno, con quello immediatamente superiore ed inferiore. Questo complesso sistema di interconnessioni fa sì che anche una lesione completa di una sola radice o di un ganglio non siano in grado di indurre deafferenta-zione, per indurre la quale c’è quindi bisogno di lesioni che interessino diversi livelli segmentali (Figura 6).
Come già descritto in precedenza, nella genesi del dolore neuropatico è frequente il riscontro di alterazioni a carico dei canali del sodio che, se iperespressi, sono in grado di creare quelle condizioni di ipereccitabilità in grado di generare e sostenere una scarica ectopica di impulsi. Anche in caso di dolore da deafferentazione è stata considerata l’ipotesi di una disregolazione di tali canali come possibile meccanismo algico. In casi di dolore riferibili al meccanismo della deafferentazione è stata infatti segnalata una iperespressione del canale denominato Nav 1.3 sia nei corpi cellulari dei neuroni del corno posteriore del midollo (secondi neuroni della via), sia nei neuroni talamici (terzi neuroni della via).
Figura 6. schema illustrante come, a causa delle connessioni multiple tra primo e secondo neurone sensitivo, la deafferentazione del secondo neurone si verifichi solo in caso di degenerazione contemporanea di numerosi neuroni periferici limitrofi. In alto condizione di normalità; al centro, lieve danno assonale; in basso grave danno assonale con deafferentazione.
normale
lieve danno assonale(I neurone sensitivo)
grave danno assonale(I neurone sensitivo)deafferentazione del II neurone sensitivo

28Aspetti di fisiopatologia e terapia del dolore
doLore da LesIone nervosa senza danno assonaLe
Altro ragionamento va fatto per le lesioni che non si accompagnano a degenera-zione assonale. Le più frequenti e conosciute sono quelle demielinizzanti. Appare subito apparentemente contraddittorio il fatto che lesioni demielinizzanti interessanti le fibre sensitive possano dare sia sintomi positivi (parestesie, disestesie, dolori) che negativi (ipoestesie). La risposta a tale osservazione non è chiara, ma sulla base delle esperienze acquisite in anestesia loco-regionale, è ipotizzabile che il blocco della con-duzione avvenga solo quando un tratto sufficiente lungo e continuo di fibra nervosa venga interessato dalla lesione. In altre parole se le lesioni sono corte e/o discontinue esse possono sostenere sintomi positivi in quanto gli impulsi ivi generati possono propagarsi a distanza (Figura 2).
Se, al contrario le lesioni interessano lunghi tratti di fibra nervosa, anche nell’e-ventualità che la genesi ectopica avvenga, essa non può propagarsi prossimalmente fino ad arrivare a livello cerebrale dove le sensazioni vengono avvertite. Nelle fibre amieliniche la differenziazione suddetta è meno applicabile perché, a differenza delle fibre mieliniche che hanno una conduzione nervosa saltatoria da un nodo di Ranvier e l’altro, le fibre amieliniche hanno una trasmissione punto a punto. Ciò rende altamente improbabile una genesi ectopica senza contemporaneo blocco della trasmissione. A parità di lunghezza lesionale infatti, i nodi di Ranvier bloccati sono sempre maggiori nelle fibre amieliniche rispetto a quelle mielinizzate.
Così come, sempre a parità di lesione, il numero di nodi di Ranvier bloccati nelle fibre scarsamente mielinizzate (A-delta) sarà maggiore rispetto a quelli bloccati nelle fibre molto mielinizzate (A-beta). Anche nelle lesioni demielinizzanti gioca un ruolo l’aumento dei canali del sodio. È infatti noto che, lì dove l’assone perde la sua guaina mielinica è presente un numero maggior di canali del sodio, anche se non vi è stato alcun danno a carico dell’assone stesso. L’importanza dei canali del sodio nella genesi del dolore ectopico è chiaramente indicato anche da due fenomeni tra loro comple-tamente opposti. È infatti noto come due forme di dolore neuropatico molto grave, l’eritromelalgia ereditaria e il disturbo doloroso estremo parossistico, sono sostenute da una mutazione genica interessante i geni che codificano un particolare tipo di canali del sodio (Na 1.7). Al contrario l’assenza congenita del canale Nav 1.7 si accompagna ad insensibilità al dolore con mantenimento delle altre modalità sensitive.
La scarsa aBItuazIone e La refrattarIetà
Uno degli aspetti più significativi che differenzia il dolore ectopico (neuropatico) da quello normotopico (nocicettivo) è il diverso comportamento nei confronti dell’abi-tuazione. Nel dolore ectopico è stato osservato come i fenomeni abituativi sono molto più inefficienti rispetto al dolore nocicettivo.
I motivi di questa scarsa abituazione degli impulsi generati direttamente dalle fibre nervose sono molti e non completamente noti. Uno di questi è rappresentato dal fatto che ogni impulso nervoso che viene generato da un recettore subisce una sorta di filtro rappresentato dal potenziale di recettore. È questo quel piccolo potenziale che si crea a livello delle parti più estreme delle fibre nervose sensitive in seguito agli stimoli adeguati per quel tipo di recettore.
Come è ben noto non tutti i potenziali di recettore fanno partire degli impulsi ner-vosi lungo la fibra (i potenziali d’azione), ma solo quelli che raggiungono una certa differenza di potenziale che permette al potenziale d’azione di raggiungere la soglia critica e cioè quella differenza di potenziale tra l’interno e l’esterno della cellula in grado di scatenare quel fenomeno tutto o nulla che genera il potenziale d’azione. Quan-do lo stimolo (ectopico) si forma direttamente a livello delle fibre nervose il “filtro”

29 Genesi ectopica degli impulsi
rappresentato dal recettore non c’è e pertanto anche i fenomeni di abituazione, che portano ad una riduzione delle scariche neuronali fino al loro esaurimento, risultano più deboli. In alcuni casi di dolore neuropatico però si osserva un fenomeno opposto: dopo che si è generata la scarica di impulsi nervosi che ha fatto avvertire al paziente la sensazione dolorosa, per un certo periodo di tempo non è più possibile far attivare le fibre nervose.
In altre parole, l’attivazione ripetuta di uno stesso sito di genesi ectopica di impulsi rende quel sito non eccitabile per un intervallo di tempo più o meno lungo. Tale ca-ratteristica è stata evidenziata negli animali, ma è presente in alcuni pazienti affetti da nevralgia trigeminale. Questi pazienti dopo aver avvertito la tipica scossa di dolore in seguito alla stimolazione di un punto grilletto (trigger point doloroso) possono stimolare lo stesso punto senza più avvertire dolore. I pazienti con nevralgia trigeminale che hanno un dolore scatenato dall’apertura della bocca, arrivano a sfruttare tale caratteristica del dolore per potersi alimentare: si provocano volontariamente il dolore stimolando adeguatamente il trigger point dopodiché si alimentano sfruttando l’impossibilità ad evocare il dolore nel momento di refrattarietà nervosa.
prIncIpI terapeutIcI per IL doLore neuropatIco
La terapia del dolore sarà ampiamente trattata in un capitolo successivo. Qui di seguito verranno brevemente trattati alcuni principi terapeutici strettamente correlati alla fisiopatologia del dolore neuropatico. Anzitutto va premesso che il dolore a genesi ectopica è un dolore resistente alle comuni terapie. Attualmente le probabilità di un buon controllo del dolore in un paziente affetto da vero dolore neuropatico sono molto basse e si aggirano intorno al 30-40%.
La caratteristica patogenetica che lo contraddistingue sposta infatti i bersagli te-rapeutici dai recettori e dai canali alle membrane neuronali. In pratica, tutte le terapie in grado di inibire l’eccitabilità della membrana neuronale posseggono teoricamente un’azione antalgica nelle forme cliniche di dolore neuropatico. Non è un caso che molti dei farmaci che mostrano un qualche effetto sul dolore neuropatico sono far-maci appartenenti alla categoria degli antiepilettici. Sono questi in genere farmaci che riducono l’ipereccitabilità delle membrane neuronali, fenomeno che è alla base sia dell’epilessia che del dolore neuropatico. Molti di questi farmaci posseggono la caratteristica di bloccare la trasmissione degli impulsi che viaggiano a frequenze ele-vate, mentre non modificano la trasmissione degli impulsi a bassa o media frequenza. Tale azione farmacologica viene svolta, il più delle volte, prolungando il periodo di inattivazione dei canali del sodio, condizione in grado di per sé di bloccare le scariche ad alta frequenza.
L’esempio più noto di farmaco inibitorio della trasmissione neuronale, utilizzato sia in epilessia che in terapia antalgica, è sicuramente quello della carbamazepina, farmaco tutt’ora insuperato, in termini di efficacia, nel trattamento della nevralgia trigeminale. Il meccanismo più importante della carbamazepina è il blocco dei canali del sodio. Come visto precedentemente, questi canali sono normali costituenti delle membrane neuronali, ma in alcune condizioni patologiche essi risultano iperespressi e contribuiscono significativamente al mantenimento di ipereccitabilità delle membrane neuronali, fenomeno che quando intenso porta alla genesi ectopica di potenziali d’a-zione. Purtroppo l’utilizzo clinico dei farmaci stabilizzanti di membrana appare molto limitato dall’insorgenza di effetti indesiderati sul sistema nervoso centrale, caratteristica questa che appare chiaramente legata al fatto che tutti i suddetti farmaci sono lipofilici e pertanto in grado di passare la barriera ematoencefalica.

30Aspetti di fisiopatologia e terapia del dolore
BIBLIoGrafIa
Baron R. Peripheral Neuropathic Pain: From Mecha-nisms to Symptoms The Clinical Journal of Pain 2000, 16: S12-S20.
Basbaum A, Bushnell MC. Pain: Basic Mechanisms. In: Giamberardino MA (Ed): Pain 2002 - Un Update Review: Refresher Course Syllabus. IASP Press, Seattle, 2002: 3-7.
Buonocore M. Meccanismi fisiopatologici del dolore neurogeno In: M.Buonocore, C. Bonezzi Il dolore neurogeno: dalla definizione alla terapia. I libri della Fondazione Maugeri, PI-ME Press, Pavia, 1999, 25-34.
Buonocore M. Rehabilitation of chronic neuropathic pain syndromes. Directions in Rehabilitation Coun-seling 2002 (b); 13: 117-25.
Buonocore M, Bonezzi C. Fisiopatologia del dolore. Momento Medico, Salerno, 2005.
Calvo M, Dawes JM, Bennett DL. The role of the im-mune system in the generation of neuropathic pain. Lancet Neurol. 2012 Jul; 11 (7) :629-42.
Campbell JN. Nerve lesions and the generation of pain. Muscle Nerve. 2001 Oct; 24 (10): 1261-73.
Campero M, Serra J, Marchettini P, Ochoa JL. Ectopic impulse generation and autoexcitation in single myelinated afferent fibers in patients with peripheral neuropathy and positive sensory symptoms. Muscle Nerve, 1998, 21: 1661-1667.
Devor M. Neuropathic pain and injured nerve: pe-ripheral mechanisms. British Medical Bullettin, 1991, 47: 619-630.
Devor M. Sodium channels and mechanisms of neuro-pathic pain. J Pain. 2006 Jan; 7 (1 Suppl 1): S3-S12.
Dubový P. Wallerian degeneration and peripheral nerve conditions for both axonal regeneration and neuropathic pain induction. Ann Anat. 2011 Jul; 193 (4): 267-75.
Dyck PJ, Windebank AJ. Diabetic and nondiabetic lumbosacral radiculoplexus neuropathies: new in-sights into pathophysiology and treatment. Muscle Nerve. 2002 Apr; 25 (4): 477-91.
Gorson KC, Herrmann DN, Thiagarajan R, Brannagan TH, Chin RL, Kinsella LJ, Ropper AH. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry. 2008 Feb; 79 (2): 163-9.
Portenoy RK Basic mechanisms. In: Portenoy RK e Kanner RM (Eds): Pain Management: Theory and Practice. F.A. Davis Company, Philadelphia, 1996: 19-39.
Serra J, Ochoa J, Campero M. Human Studies of Primary Nociceptors in Neuropathic Pain. In: Hansson T, Fields HL, Hill RG, Marchettini P (Eds) Neuropathic Pain: Pathophysiology and treatment. Progress in Pain Research and Management, Vol. 21 IASP Press, Seattle, 2001: 63-83.
Stemkowski PL, Smith PA. Sensory neurons, ion channels, inflammation and the onset of neuropathic pain. Can J Neurol Sci. 2012 Jul; 39 (4): 416-35.
Sunderland S The painful sequelae of injuries to pe-ripheral nerves. Nerves and Nerve Injuries, chapter 34. Churchill Livingston, 1978: 377-420.
Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, Hansson P, Hughes R, Nurmikko T, Serra J. Neuropathic pain: redefini-tion and a grading system for clinical and research purposes. Neurology. 2008 Apr 29; 70 (18): 1630-5. Epub 2007 Nov 14.
Wall PD, Gutnick M. Properties of afferent nerve im-pulses originating from a neuroma. Nature 1974a, 248: 740-743.
Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. The Lancet, 1999, 353: 1959-64.

MeccAnIsMI dellA sensIbIlIzzAzIone centrAleMichelangelo Buonocore
4
Buona parte delle nuove conoscenze sul dolore che si sono sviluppate nelle ultime decadi sono dovute a studi effettuati a livello del sistema nervoso centrale e del mi-dollo spinale in particolare. Ciò perché il midollo spinale, oltre ad essere una stazione importante per la trasmissione degli impulsi nervosi, rappresenta anche un importante luogo di modulazione degli stessi (Figura 1). Non fanno eccezione gli impulsi noci-cettivi. Una volta infatti che gli impulsi provenienti da fibre nocicettive raggiungono il sistema nervoso centrale, che ha il suo “avamposto” a livello del midollo spinale,
Figura 1. La trasmissione midollare di impulsi nocicettivi attraverso la sinapsi midollare dipende dall’equilibrio tra neurotrasmettitori eccitatori, il cui capostipite è il glutammato (GLu), ed inibitori, il cui capostipite è l’acido gamma-amino-butirrico (GaBa). In condizioni di prevalenza di glutammato (al centro) la trasmissione di impulsi viene facilitata, in prevalenza di GaBa inibita (in basso). Quando i due tipi di neurotrasmettitori sono in equilibrio (in alto) la trasmissione degli impulsi non subisce significative modificazioni a livello sinaptico.
GLu GaBa
GLu GaBa
GLu GaBa

32Aspetti di fisiopatologia e terapia del dolore
essi possono creare ulteriori fenomeni di amplificazione, frequentemente definiti col termine di sensibilizzazione centrale.
MeccanIsMI GeneraLI deLLa sensIBILIzzazIone centraLe
Perché i suddetti processi si possano attivare è necessario che gli stimoli che arri-vano dalla periferia siano costanti. Stimoli ripetuti di breve entità, per quanto intensi, che però non generino un dolore spontaneo persistente non sono in grado di indurre la sensibilizzazione centrale. Non tutti i tessuti possiedono le stesse potenzialità di generare fenomeni di sensibilizzazione spinale. Vi sono per esempio alcuni studi su-gli animali che hanno confrontato lesioni muscolari e cutanee ed hanno chiaramente dimostrato come le lesioni muscolari possiedano una maggiore capacità di produrre una sensibilizzazione spinale. Numerosi meccanismi sono stati tirati in ballo per giusti-ficare l’insorgenza dei fenomeni che vanno sotto il nome di sensibilizzazione centrale. Considerato che il corno posteriore del midollo è la sede della prima sinapsi nocicet-tiva, una semplice classificazione divide i meccanismi fisiopatologici in presinaptici e postsinaptici. Va però ricordato come la stragrande maggioranza dei meccanismi di sensibilizzazione spinale sia legata al fatto che il secondo neurone nocicettivo, sito nel corno posteriore, da cui parte la via spino-talamica, diventa ipersensibile agli stimoli. Così come vale la pena di segnalare come una ipersensibilità che raggiunga elevati livelli possa alla fine portare alla scarica spontanea dei neuroni post-sinaptici, diven-tando pertanto completamente indipendente rispetto agli eventuali stimoli, intensi o lievi, provenienti dalla periferia mediante i primi neuroni nocicettivi.
Tra i meccanismi presinaptici vanno sicuramente ricordati l’insieme di modifica-zioni fenotipiche che si verificano ogniqualvolta in un tessuto si verifica una lesione. La plasticità del sistema in questo contesto appare molto elevata e gran parte delle numerosissime modificazioni osservate nell’ambito della biologia molecolare del dolore appartengono a questo tipo di modificazioni. Un altro fenomeno presinaptico frequentemente descritto è rappresentato dal fatto che gli stimoli nocivi o potenzial-mente nocivi inducono il rilascio di peptidi che non vengono liberati da stimoli di lieve entità. Il rilascio di questi peptidi innesca poi altri meccanismi che rendono il sistema ipersensibile a stimoli successivi. Un vero e proprio rimaneggiamento anatomico avviene quando si verifica una degenerazione massiva dei neuroni di piccolo calibro che arrivano al corno posteriore del midollo. In tali condizioni, negli animali, è stato dimostrato un proliferare (sprouting) di fibre nervose non nocicettive, di grande calibro, che dalle lamine più profonde del corno posteriore vanno a connettersi, in modo non fisiologico, con i secondi neuroni nocicettivi presenti nelle lamine più esterne del corno posteriore, creando in tal modo un corto circuito tra impulsi non nocicettivi, mediati dalle fibre nervose di grande calibro e i circuiti spinali della nocicezione.
I meccanismi postsinaptici sono molto diversi tra loro a tal punto da essere divi-sibili in due grandi categorie: i meccanismi eccitatori e quelli inibitori. Tra i primi si annoverano, tra gli altri, l’attivazione di kinasi, la fosforilazione dei recettori NMDA (N-Methil-D-Aspartato), l’attivazione di NOS (Nitric Oxide Synthase) e di COX2 (Cyclooxygenase 2). Tra i meccanismi inibitori vanno annoverati la ridotta espressio-ne di sostanze inibitorie spinali (es GABA, acido gamma-amino-butirrico), la ridotta espressione di recettori inibitori, la morte di neuroni inibitori.
MeccanIsMI specIfIcI correLatI aLLa sensIBILIzzazIone spInaLe
Come detto precedentemente, è il neurone presente nel corno posteriore del midollo che sviluppa quei fenomeni di ipersensibilità agli stimoli che sono alla base del più complesso fenomeno che viene denominato sensibilizzazione centrale. Ma quali sono

33 Meccanismi della sensibilizzazione centrale
i meccanismi neurofisiopatologici che sostengono la sensibilizzazione centrale? Se ne conoscono diversi e i più importanti saranno trattati essenzialmente qui di seguito. Il primo è rappresentato dalla riduzione della soglia di scarica. Tale fenomeno avviene sia per i neuroni nocicettivi specifici che per quelli ad ampio spettro dinamico, molto noti anche come neuroni WDR (dall’inglese Wide Dynamic Range) o come neuroni convergenti. In condizione fisiologiche, i neuroni nocicettivi specifici, localizzati negli strati più esterni del corno posteriore del midollo, hanno caratteristicamente un’alta soglia di scarica in quanto producono potenziali d’azione che viaggiano nelle vie spino-talamiche, direttamente a frequenze nocicettive (alte frequenze), solo in seguito a stimoli di elevata intensità (Figura 2).
Quando si sviluppano i fenomeni di sensibilizzazione centrale la soglia di scarica di tali neuroni si abbassa (Figura 3). In altre parole, essi si attivano producendo poten-ziali che viaggiano a frequenze elevate anche quando le stimolazioni in periferia sono state di lieve entità. Un fenomeno simile avviene anche per i neuroni ad alto spettro dinamico, localizzati negli strati più profondi del corno posteriore del midollo. Questi neuroni, che fisiologicamente rispondono a stimoli lievi con basse frequenze di scarica, in condizioni di ipereccitabilità rispondono ad alta frequenza anche per stimolazioni periferiche di lieve entità. In tal modo anch’essi riducono la loro soglia di scarica a frequenze nocicettive.
Come già accennato in precedenza, la riduzione della soglia di scarica dei neu-roni nocicettivi spinali si può verificare anche per attivazione dei neuroni nocicettivi specifici da parte di abnormi connessioni dei loro corpi cellulari con le fibre nervose periferiche di grande calibro che veicolano, in condizioni fisiologiche, le sensazioni non dolorose quali tatto e propriocezione. È stato infatti dimostrato, negli animali, che lesioni prossimali e selettive di fibre periferiche nocicettive (di piccolo calibro)
Figura 2. nella figura viene illustrata la risposta in frequenza agli stimoli dei neuroni nocicettivi presenti nel corno posteriore del midollo. si noti come il neurone nocicettivo specifico (ns) risponda solo a stimolazioni di elevata intensità e quello ad ampio spettro dinamico (Wdr) a stimoli di intensità sia bassa che elevata.
120
90
60
30
0
120
90
60
30
0
tocco pressione stretta
tocco pressione stretta
imp
/sec
imp
/sec
ns
Wdr

34Aspetti di fisiopatologia e terapia del dolore
Figura 3. nella figura viene illustrato come in caso di ipersensibilità dei neuroni convergenti (Wdr) (in basso), si verifichi un aumento della frequenza di scarica neuronale rispetto alle condizioni fisiologiche (in alto). ne consegue che gli stimoli innocui acquisiscono la capacità di indurre risposte in frequenza simili a quelle che lo stesso neurone aveva in condizioni fisiologiche in conseguenza di stimolazioni nocicettive. pertanto, in caso di ipersensibilità dei Wdr, uno stimolo non nocivo può essere interpretato, a livello del sistema nervoso centrale, come stimolo nocivo e dare origine alla sensazione dolorosa.
120
90
60
30
0
120
90
60
30
0
tocco pressione stretta
tocco pressione stretta
imp
/sec
imp
/sec
neurone ad ampio spettro dinamico(Wdr)
condizioni fisiologiche
Ipersensibilità
possono indurre l’attivazione di fenomeni di gemmazione (sprouting) di rami nervose collaterali che partendo dalle lamine interne del corno posteriore, dove sono localizzate le terminazioni delle fibre nervose di grande calibro, si spostano verso le lamine più superficiali, dove sono localizzati i neuroni nocicettivi specifici (Figura 4). Grazie a tali connessioni stimoli periferici di bassa intensità, che fisiologicamente attivano potenziali nervosi nelle fibre di grande calibro, una volta arrivati a livello midollare, grazie alle suddette neo-connessioni, attivano i corpi cellulari dei neuroni nocicettivi, usualmente attivati solo da stimoli di elevata intensità. Il risultato finale di questo nuovo circuito è che i nocicettori specifici vengono attivati da stimoli di lieve entità (riduzione della soglia di scarica nocicettiva).
Un secondo meccanismo correlato alla sensibilizzazione centrale è dato dall’au-mento della frequenza di scarica. Nei neuroni ad ampio spettro dinamico esiste una proporzionalità diretta tra intensità della stimolazione periferica e frequenza di scarica neuronale. In parole semplici, stimoli di lieve entità danno risposte in bassa frequen-za, mentre stimoli di elevata intensità danno stimoli di elevata frequenza. In caso di sviluppo di ipersensibilità spinale anche stimoli di lieve intensità sono in grado di far scaricare ad alta frequenza il neurone ad ampio spettro dinamico. Tra i meccanismi meglio studiati in questo ambito vi è il fenomeno del wind-up (Figura 5).
Trattasi di una sensibilizzazione spinale che interessa le fibre nocicettive le quali, se stimolate ad alta frequenza (superiore a 0.3 Hz e cioè uno stimolo ogni 3 secondi) mostrano un aumento esponenziale della frequenza di scarica del neurone nocicettivo spinale senza cioè che tra uno stimolo e l’altro il sistema ritorni alle condizioni basali. Questo fenomeno, molto studiato negli anni ’70 ed ’80 del secolo scorso, ha in realtà
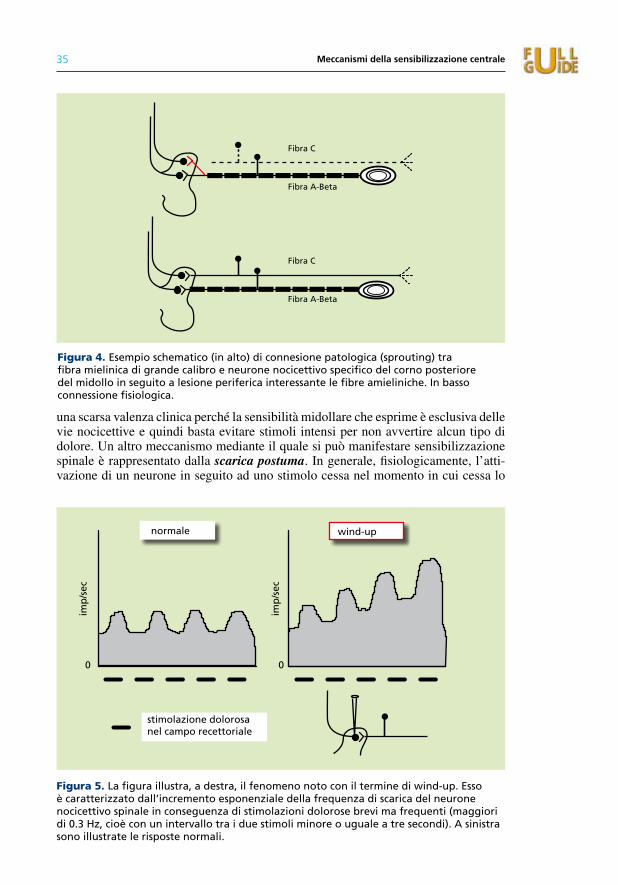
35 Meccanismi della sensibilizzazione centrale
Figura 4. esempio schematico (in alto) di connesione patologica (sprouting) tra fibra mielinica di grande calibro e neurone nocicettivo specifico del corno posteriore del midollo in seguito a lesione periferica interessante le fibre amieliniche. In basso connessione fisiologica.
fibra c
fibra a-Beta
fibra c
fibra a-Beta
Figura 5. La figura illustra, a destra, il fenomeno noto con il termine di wind-up. esso è caratterizzato dall’incremento esponenziale della frequenza di scarica del neurone nocicettivo spinale in conseguenza di stimolazioni dolorose brevi ma frequenti (maggiori di 0.3 hz, cioè con un intervallo tra i due stimoli minore o uguale a tre secondi). a sinistra sono illustrate le risposte normali.
0
imp
/sec
0
imp
/sec
stimolazione dolorosanel campo recettoriale
normale wind-up
una scarsa valenza clinica perché la sensibilità midollare che esprime è esclusiva delle vie nocicettive e quindi basta evitare stimoli intensi per non avvertire alcun tipo di dolore. Un altro meccanismo mediante il quale si può manifestare sensibilizzazione spinale è rappresentato dalla scarica postuma. In generale, fisiologicamente, l’atti-vazione di un neurone in seguito ad uno stimolo cessa nel momento in cui cessa lo

36Aspetti di fisiopatologia e terapia del dolore
stimolo. In caso di sviluppo di fenomeni di ipersensibilità, la scarica può continuare ben oltre il tempo dello stimolo (scarica postuma). Questo porta a due conseguenze.
La prima è rappresentata dal fatto che se lo stimolo che ha iniziato la scarica è stato sufficiente a generare il dolore, con la persistenza della scarica neuronale persiste anche la sensazione dolorosa.
La seconda è relativa ad un fenomeno di sommazione. Se il primo stimolo ha at-tivato il neurone che ha continuato a scaricare anche dopo la fine della stimolazione, se il neurone viene attivato durante questa scarica postuma la sua soglia di attivazione è più bassa e la frequenza di scarica sarà più alta di quella ottenuta dai due singoli stimoli, e quindi il dolore sarà più intenso.
Forse però, tra tutti i meccanismi tipici della sensibilizzazione spinale, quello più caratteristico, anche perché più visibile clinicamente, è l’ingrandimento del campo recettoriale. In condizioni fisiologiche sappiamo che ogni neurone spinale possiede un suo campo recettoriale che viene facilmente identificato stimolando i tessuti periferici e vedendo se tale stimolazione induce delle risposte a livello del neurone spinale da cui viene registrata l’attività. Sappiamo anche che i campi recettoriali hanno diverse caratteristiche nei diversi neuroni nocicettivi: i neuroni nocicettivi ad ampio spettro dinamico possiedono campi recettoriali ben più grandi dei neuroni nocicettivi specifici (Figura 6). Ma entrambi i neuroni nocicettivi possiedono la capacità di espandere il loro campo recettoriale, sebbene questa caratteristica sia nettamente più importante per i neuroni ad ampio spettro dinamico. Il meccanismo dell’allargamento del campo recettoriale è attualmente spiegato dal fatto che i dendriti del neurone nocicettivo
Figura 6. nella figura vengono illustrati i campi recettoriali dei neuroni nocicettivi presenti nel corno posteriore del midollo. In alto il neurone nocicettivo specifico (ns) in basso il neurone ad ampio spettro dinamico (Wdr). si noti come il primo abbia un piccolo campo recettoriale al contrario del secondo che possiede un campo recettoriale molto più vasto.
neurone nocicettivospecifico(ns)
neurone ad ampio spettro dinamico(Wdr)

37 Meccanismi della sensibilizzazione centrale
spinale hanno numerose connessioni con le fibre nervose periferiche e che alcune di queste connessioni sono fisiologicamente inattive. È solo in caso di sensibilizzazione del neurone nocicettivo spinale che si attivano tutte le connessioni possibili, incluse quelle silenti, permettendo così al neurone spinale di ricevere informazioni da un ter-ritorio periferico più vasto rispetto a quello originale. Vale la pena di ricordare come, secondo alcuni autori, la sensibilizzazione spinale può essere considerata responsabile anche di un abbassamento della soglia del dolore nell’area lesionale, ma non tutti gli esperti sono d’accordo.
In ogni caso, considerato l’ampio sviluppo di fenomeni di ipersensibilità in sede di lesione tissutale, non è improbabile che sia la sensibilizzazione centrale che quella periferica siano coinvolti. Ma la manifestazione più importante della sensibilizzazione spinale è rappresentata dalla scarica spontanea (e continua) dei nocicettori spinali, che si accompagna al dolore spontaneo, quello cioè presente indipendentemente da qualsiasi stimolazione. È questo un fenomeno che si osserva solo quando le cause della sensibilizzazione sono importanti e/o intense e/o persistenti per cui il neurone non riesce in nessun modo ad attuare quei meccanismi di autoinibizione che pur pos-siede. Come già accennato in precedenza la presenza di un’attivazione costante del neurone nocicettivo spinale lo rende anche più sensibile agli stimoli, giustificando in tal modo il frequente riscontro clinico di un’allodinia/iperalgesia nelle stesse aree dove il paziente riferisce il dolore spontaneo.
sensIBILIzzazIone centraLe da auMentate afferenze perIferIche (concettI GeneraLI)
Sicuramente la più importante causa di sensibilizzazione spinale è rappresentata da un aumento delle afferenze al midollo spinale. In particolare, studi effettuati su animali hanno chiaramente dimostrato che non tutte le fibre giocano uno stesso ruolo. Le fibre nervose sensitive in grado di creare la sensibilizzazione spinale sono esclu-sivamente le fibre C (fibre amieliniche). Come già accennato in precedenza stimoli brevi per quanto intensi, non sono considerati in grado di creare le condizioni per lo sviluppo di una sensibilizzazione centrale. Quello che appare cruciale è la costanza della scarica neuronale periferica che arriva al midollo. Studi su animali hanno cal-colato che i primi fenomeni di sensibilizzazione centrale si colgono già dopo circa un minuto di stimolazione periferica. Perché proprio le fibre C? La motivazione risiede nel fatto che solo tali fibre sono in grado di liberare alcune sostanze coinvolte nella trasmissione nocicettiva e pertanto legate, direttamente o indirettamente, ai fenomeni di modulazione del dolore.
Il neurotrasmettitore che più di ogni altro sembra avere un ruolo nella induzione dei fenomeni di sensibilizzazione centrale è la sostanza P, la cui liberazione sinaptica è strettamente legata alla scarica delle fibre amieliniche (C). Secondo alcuni autori la sostanza P svolge tale ruolo sensibilizzante perché si lega al recettore NK1 presente sulla membrana post-sinaptica. Quando questo recettore viene attivato, sulla membrana post-sinaptica (del nocicettore spinale) si attivano una serie di processi che, attraver-so l’entrata del calcio nella cellula, riducono il potenziale di membrana rendendo il neurone ipereccitabile (Figura 7). È interessante ricordare che le fibre amieliniche C liberino glutammato per stimoli di lieve intensità e glutammato e sostanza P quando attivate ad alta intensità. Ciò giustifica come mai non sempre l’attivazione delle fibre C afferenti evoca dolore, ma contrasta con alcuni studi che hanno segnalato come la scarica periferica afferente che induce la sensibilizzazione centrale non deve essere necessariamente dolorosa. È noto da tempo che un altro tipo di recettori che gioca un ruolo cruciale è rappresentato dal recettore NMDA (N-Metil-D-Aspartato). È stato

38Aspetti di fisiopatologia e terapia del dolore
infatti più volte dimostrato che il loro blocco riduce l’allodinia che si sviluppa in corso di dolore neuropatico. Anche i recettori NMDA, che sono recettori per il glutammato, possono essere attivati dalla sostanza P. E quando questo avviene, si genera un poten-ziale post-sinaptico eccitatorio che persiste nel tempo, rendendo pertanto ipereccitabile la membrana post-sinaptica (quella del neurone nocicettivo spinale).
sensIBILIzzazIone centraLe da LesIone tIssutaLe
Non tutti i tessuti periferici giocano lo stesso ruolo nello sviluppo di fenomeni di sensibilizzazione centrale. Esistono numerose evidenze che dimostrano l’importanza del tipo di tessuto periferico lesionato, quello cioè da cui partono gli stimoli che arrivati al midollo creano la sensibilizzazione centrale. È per esempio ben noto come le lesioni tissutali non nervose si accompagnino sistematicamente ad uno sviluppo di ipersen-sibilità midollare. Gran parte di queste lesioni sono di tipo periferico e rientrano tra quelle già descritte nel capitolo della sensibilizzazione periferica. Come detto in altra parte, non tutti i tipi di tessuto hanno la stessa capacità di generare sensibilizzazione centrale. È per esempio noto che a parità di intensità lesionale, le lesioni muscolari si accompagnano a fenomeni di ipersensibilità centrale più marcati, rispetto a quelle cutanee.
Un caso molto particolare è rappresentato dalla cosiddetta ipersensibilità tattile progressiva. Come accennato precedentemente, è la liberazione di sostanza P da parte delle fibre amieliniche che innesca i fenomeni di sensibilizzazione centrale. Ebbene, in alcuni studi animali è stato dimostrato che l’infiammazione persistente di un tessuto periferico può indurre una trasformazione fenotipica delle fibre mielinizzate di grande calibro che acquisiscono anch’esse la capacità di liberare sostanza P, cosa che non avviene in condizioni fisiologiche. Ne consegue che stimoli tattili, che attivano fibre nervose periferiche di grande calibro, quando sono portati su un tessuto infiammato, possono anch’essi liberare sostanza P a livello midollare, contribuendo al mantenimento ed eventualmente all’aumento dei fenomeni di ipersensibilità midollare.
Figura 7. Illustrazione schematica dei meccanismi di sensibilizzazione del neurone ad ampio spettro dinamico in conseguenza di una lesione tissutale. L’attivazione spontanea della fibra amielinica (fibra c) da parte della lesione tissutale libera elevate quantità di sostanza p nello spazio sinaptico midollare. In tal modo si rendono attivi alcuni recettori “silenti” sulla membrana post-sinaptica (nMda, kainate/aMpa, metabotrobico) che, una volta eccitati, ne inducono la depolarizzazione attraverso l’entrata di calcio nella cellula. ne risulta una ipereccitabilità del secondo neurone nocicettivo.
p
fibra c
fibra a-BetanMda
KaInate/aMpa
MetaBotroBIco
Wdr
p = sostanza p
ca++
ca++

39 Meccanismi della sensibilizzazione centrale
sensIBILIzzazIone centraLe da LesIone nervosa perIferIca
Non solo le lesioni tissutali ma anche quelle nervose periferiche possono innescare fenomeni di ipersensibilità midollare. In tali condizioni si sviluppa una condizione di ipersensibilità da lesione nervosa periferica. È ben noto come una lesione parziale di un nervo possa generare scariche ectopiche a livello delle fibre amieliniche lesionate. Come visto nel precedente capitolo, la genesi ectopica degli impulsi genera impulsi che si propagano sia prossimalmente che distalmente. Gli impulsi così generati, che si propagano prossimalmente lungo le fibre amieliniche, inducono una liberazione di neurotrasmettitori nelle sinapsi spinali, esattamente come avrebbero fatto impulsi generati normotopicamente (dalle terminazioni libere) in caso di lesione tissutale. In caso di scariche intense e prolungate, esattamente come avviene per stimoli nocicettivi dovuti a lesioni tessutali non nervose, si innescano tutti i processi che portano alla sensibilizzazione centrale. Per completezza di informazione vanno ricordati due im-portanti aspetti che possono diversificare le manifestazioni cliniche di quanto appena illustrato. Il primo è rappresentato dall’osservazione che, in caso di lesione nervosa periferica, gran parte delle scariche ectopiche sperimentalmente registrate, sono state evidenziate nelle fibre nervose di grande calibro, che fisiologicamente veicolano le sensibilità tattile e propriocettiva, e non nelle fibre nervose di piccolo calibro, che fisiologicamente veicolano le sensibilità termiche e dolorifiche. Ne consegue che non è obbligatorio che una lesione nervosa periferica induca una sensibilizzazione spinale. Il secondo aspetto, non meno importante, è rappresentato dalla possibilità che anche in caso di lesione nervosa periferica, così come in seguito a lesione tissutale infiammatorie, le fibre nervose periferiche di grande calibro possono modificare il loro fenotipo e incominciare a liberare sostanze pronocicettive dalle sinapsi spinali, rendendo così possibile una sensibilizzazione centrale generata e sostenuta da fibre nervose di grande calibro. Quest’ultimo meccanismo contribuisce, insieme agli altri meccanismi illustrati prima, all’estrema variabilità di espressione clinica del dolore che consegue ad una lesione del sistema nervoso periferico.
sensIBILIzzazIone spInaLe da auMentata facILItazIone/ dIMInuIta InIBIzIone dIscendente
Ormai da molti anni è ben noto come quello che si verifica a livello della trasmis-sione sinaptica sensitiva midollare sia influenzato da sistemi costituiti da fibre nervose discendenti a partenza cerebrale e proiettanti direttamente sulle terminazioni sensitive presinaptiche. Tale influenza può essere sia eccitatoria che inibitoria, con un equili-brio oscillante a favore dell’una o dell’altra azione. Impulsi eccitatori provenienti dal troncoencefalo possono quindi facilitare la trasmissione degli impulsi nocicettivi a livello midollare e creare condizioni di ipereccitabilità intra ed extralesionali, tipiche della sensibilizzazione centrale. Ma il meccanismo forse più importante in tale ambito è quello dell’inibizione. Questa si può attuare attraverso l’azione su neuroni inibitori midollari oppure, sull’attivazione diretta di fibre discendenti inibitorie. L’inibizione sovrasegmentaria è un meccanismo subcontinuo che serve a ridurre gli impulsi no-cicettivi che viaggiano in direzione centripeta. Esso rappresenta un vero e proprio servo-meccanismo che fa sì che molti degli impulsi di lieve entità, e quindi non po-tenzialmente nocivi, arrivino fino al cervello e si trasformino in sensazioni spiacevoli. I sistemi inibitori discendenti appaiono molto importanti anche per la possibilità di modulazione farmacologica in quanto essi sono rappresentati prevalentemente da vie serotoninergiche e noradrenergiche. Ogni azione che aumenti la disponibilità di sero-tonina e noradrenalina è pertanto in grado, teoricamente, di avere un’azione antalgica a livello midollare.

40Aspetti di fisiopatologia e terapia del dolore
Effetti clinici della sensibilizzazione spinale in area extralesionale L’effetto più evidente ed importante della sensibilizzazione centrale è rappresentato dalla comparsa del dolore in zone extralesionali. Le manifestazioni cliniche della sensibilizzazione centrale sono diverse potendo il dolore essere spontaneo od evocato e la manifestazione extralesionale essere vicina o lontana. Numerosi sono gli esempi di sensibilizzazione centrale. Se per esempio duole il dente ed il dolore persiste per qualche ora in modo intenso, continuo o subcontinuo, il dolore diffonde e diventa spesso difficile identi-ficare, sulla sola descrizione del paziente, da quale dente sia partito il tutto. Un altro esempio molto noto di sensibilizzazione centrale è il dolore dell’infarto miocardico che viene avvertito al lato mediale del braccio.
Altrettanto nota è l’irradiazione alla spalla destra del dolore in caso di interessa-mento della parte superiore (capsula) del fegato. Un altro dolore riferito si riscontra nel caso che nei muscoli si sviluppino quei punti dolorosi muscolari denominati trig-ger points. Questi, a differenza dei tender points che non diffondono, hanno proprio la caratteristica di irradiare a distanza la sensazione dolorosa. Tutti questi fenomeni, apparentemente poco comprensibili, in realtà avvengono per fenomeni di convergenza delle fibre sensitive periferiche su uno stesso pool neuronale, generalmente localiz-zato a livello del corno posteriore del midollo. In altri termini l’attivazione di fibre nocicettive periferiche finisce per interessare non solo le fibre nocicettive centrali cor-rispondenti, quelle cioè poste sulla stessa via nervosa, ma anche una parte di neuroni nocicettivi centrali posti lì vicino. La convergenza di impulsi infatti non è in grado da sola di spiegare il fenomeno. Se si trattasse infatti di una “semplice” convergenza di 2 vie periferiche su una stessa via centrale, allora tutte le volte che una delle due vie periferiche viene attivata si dovrebbe avere una sensazione di dolore nella parte con la lesione reale e nella zona corporea innervata dalle altre fibre convergenti. Invece il fenomeno si verifica solo quando l’attivazione di una via è sufficientemente prolungata da creare stabili fenomeni di ipersensibilità. Uno degli aspetti della sensibilizzazione centrale è rappresentato dal dolore riferito. Come già detto, è nozione comune il fatto che un dolore cardiaco possa essere avvertito anche nel braccio sinistro, oppure che il dolore dell’appendicite tenda ad irradiarsi verso la radice dell’arto inferiore destro. L’avvertire il dolore a distanza dal sito anatomico con la lesione viene denominato dolore riferito. Tale fenomeno è interpretato come il risultato di una convergenza di vie periferiche che veicolano la sensibilità dolorifica periferica su comuni neuroni nocicettivi spinali. Sicuramente la convergenza esiste, ma perché essa si esprima in termini clinici è necessario che i neuroni convergenti presenti nel corno posteriore del midollo siano in qualche modo sensibilizzati. Ed essi sono effettivamente sensibilizzati dalle scariche nocicettive provenienti dalla lesione. Quello che però appare sempre più evidente è che la convergenza non è frutto di connessioni rigide perché altrimenti il dolore riferito sarebbe presente ogniqualvolta una via nervosa viene attivata. E non è così. Il meccanismo più verosimile è che l’attivazione e la sensibilizzazione del secondo neurone nocicettivo attivano neuroni di connessione normalmente silenti che permettono al fenomeno clinico di presentarsi. In altre parole esisterebbero delle vie separate che si attivano per singoli stimoli nocicettivi provenienti dai due tessuti tra loro lontani e che non interferiscono tra loro fino al momento della sensibilizzazione di uno dei due neuroni centrali. È solo allora che si creerebbero le connessioni tra i neuroni centrali, presenti nello stesso pool neuronale dello stesso corno posteriore, in grado di ingannare il cervello sul reale sito lesionale. Come già accennato in pre-cedenza, in riferimento alla possibilità di sensibilizzare i neuroni centrali e quindi di generare dolori riferiti, non tutti i tessuti si comportano allo stesso modo. Oltre alla già citata differenza tra cute e muscolo, è abbastanza accettato il fatto che i dolori primitivi viscerali tendono a riferire alla cute (Figura 8), mentre i dolori primitivi dei muscoli

41 Meccanismi della sensibilizzazione centrale
no, mantenendo la loro proiezione su tessuti profondi quali tendini, legamenti ed arti-colazioni. Per esempio, il dolore sperimentale evocato da sostanze algogene iniettate nel tibiale anteriore si irradia tipicamente alla caviglia (Figura 9). Secondo alcuni autori lo schema di diffusione del dolore muscolare segue più quello dello sclerotoma che del dermatoma. Vale la pena infine di sottolineare come il termine dolore riferito si riferisca ad un dolore spontaneo, ma tale tipo di dolore a distanza non è il solo risultato della sensibilizzazione spinale. Esiste infatti anche la comparsa a distanza di aree di dolore evocato (allodinia e/o iperalgesia) che, come il dolore riferito (spontaneo) si distribuiscono nel corpo seguendo rigidi principi di metameria.
Figura 8. esempio di convergenza di due fibre nervose periferiche provenienti da tessuti diversi sullo stesso neurone midollare ad ampio spettro dinamico.
Figura 9. Localizzazione del dolore sperimentale locale (dL) e riferito (dr) in seguito all’iniezione di sostanze algogene nel muscolo tibiale anteriore. si noti come il dolore locale insorga nella sede dell’iniezione e quello riferito a livello della caviglia omolaterale. In queste condizioni sperimentali viene chiaramente rispettata la regola che prevede che la sede del dolore riferito sia sempre più distale rispetto al dolore locale.
dr
dL

42Aspetti di fisiopatologia e terapia del dolore
BIBLIoGrafIa
Arendt-Nielsen L, Svensson P Referred Muscle Pain: Basic and Clinical Findings The Clinical Journal of Pain. 2001, 17: 11-19.
Buonocore M. Meccanismi di centralizzazione del do-lore In: “Contro il dolore. Stato dell’arte”. Volume 1. Excerpta Medica, 2002. Pag. 3-16.
Buonocore M, Bonezzi C. Fisiopatologia del dolore. Momento Medico, Salerno, 2005.
Basbaum Spinal Mechanisms of Acute and Persistent Pain. Regional Anesthesia and Pain Medicine 1999, 24: 59-67.
Capra NF, Ro JW Human and animal experimental models of acute and chronic muscle pain: intra-muscular algesic injection. Pain, 2004, 110: 3-7.
Curatolo M. Diagnosis of altered central pain process-ing. Spine (Phila Pa 1976). 2011 Dec 1; 36 (25 Suppl): S200-4.
Graven-Nielsen T, Arendt-Nielsen L. Induction and assessment of muscle pain, referred pain, and muscular hyperalgesia. Curr Pain Headache Rep. 2003 Dec; 7 (6): 443-51.
Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that pos-sess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002 Oct 15; 22 (20): 9086-98.
Le Bars D, Dickenson AH, Besson JM. Diffuse nox-ious inhibitory controls (DNIC). I. Effects on dorsal horn convergent neurones in the rat. Pain. 1979 Jun; 6 (3): 283-304.
Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999, 79: 75-82.
Ma QP, Woolf CJ. Progressive tactile hypersensitivity: an inflammation-induced incremental increase in
the excitability of the spinal cord. Pain. 1996 Sep; 67 (1): 97-106.
Mannion RJ, Woolf CJ. Pain mechanisms and manage-ment: a central perspective. The Clinical Journal of Pain 2000, 16, S144-S156.
Mease PJ, Hanna S, Frakes EP, Altman RD. Pain mechanisms in osteoarthritis: understanding the role of central pain and current approaches to its treatment. J Rheumatol. 2011 Aug; 38 (8): 1546-51.
Melzack R, Wall PD. Pain mechanisms: a new theory. Science 1965, 150: 971-979.
Ossipov MH, Lai J, Malan TP, Vanderah TW, Porreca F. Tonic descending facilitation as a mechanism of neuropathic pain. In: Hanson PT, Fields HL, Hill RG, Marchettini P. (Eds) Neuropathic Pain: Pathophysiology and Teatment. Seattle, IASP Press, 2001: 107-124.
Terman GW, Bonica JJ Meccanismi spinali e loro mod-ulazione. In: Loeser JD (Ed) Bonica’s - Trattamento del dolore. Delfino Editore, Roma, 2002: 73-152.
Traub RJ. Spinal modulation of the induction of central sensitization. Brain Res. 1997 Dec 5; 778 (1): 34-42.
Wall PD, Woolf CJ. Muscle but not cutaneous C-afferent input produces prolonged increases in the excitability of the flexion reflex in the rat. J Physiol. 1984 Nov; 356: 443-58.
Wall P.D. Neurophysiological mechanisms of referred pain and hyperalgesia. In: Vecchiet L, Albe-Fessard D, Lindblom U., Giamberardino MA (Eds) New Trends in Referred Pain and Hiperalgesia. Elsevier, 1993: 3-12.
Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000 Jun 9; 288 (5472): 1765-9.

lA “terApIA coMbInAtA” nel trAttAMento del dolore cronIcoCesare Bonezzi e Laura Demartini
5IntroduzIone: I ModeLLI dI trattaMento
Nell’ambito del trattamento del dolore (acuto o cronico) le scelte terapeutiche seguono modelli differenti tra loro in quanto l’approccio diagnostico al dolore e a chi lo prova non è affatto omogeneo e condiviso. I farmaci sono sempre gli stessi ma la loro prescrizione e soprattutto il piano terapeutico variano da scuola a scuola.
a) La scala analgesica della WhoIl modello più diffuso è quello introdotto dalla Organizzazione Mondiale della
Sanità (WHO’ pain ladder), che consiglia di modulare la scelta e quindi la prescrizione dei diversi farmaci in base alla intensità del dolore. Applicata soprattutto nel dolore causato dalla malattia oncologica, la cosiddetta “scala” è stata progressivamente introdotta nella cura di qualsiasi tipo di dolore, sia acuto che cronico. I farmaci, an-tinfiammatori o oppioidi, ad esempio, sono prescritti, secondo le indicazioni previste dalla normativa nazionale, seguendo il criterio dell’intensità del dolore dichiarato dal paziente. Alcuni oppioidi sono prescrivibili in presenza di dolore “lieve-moderato” (primo e secondo gradino della scala), altri di dolore “severo” (terzo gradino). Sempre nel modello intensità-dipendente si fa riferimento ad un quarto gradino dove trovano posto le procedure antalgiche invasive come la somministrazione di oppioidi nel canale vertebrale (perdurale o subaracnoideo), le neuro-lesioni o la neuro-stimolazione. A questo gradino si accede dopo l’insuccesso delle terapia farmacologiche adeguata-mente prescritte e assunte. Non vengono affatto citati gli interventi antalgici minori (blocchi, infiltrazioni e altro) che possono essere associati alle terapie farmacologiche per ottenere un miglior risultato antalgico. In base a quanto fin qui esposto, una volta definita la patologia di base (neoplasia, osteoartrosi, neuropatia diabetica ecc.) non è necessario esaminare il dolore e i meccanismi che lo generano ma è sufficiente conoscere la sua intensità per decidere il trattamento. Il criterio dell’intensità si basa sul principio che gli impulsi afferenti al midollo spinale, provenienti dalla periferia invasa dalla neoplasia o dall’infiammazione, sono condotti dalle vie nocicettive e che possono essere più o meno elevati in base alla gravità ed estensione della lesione. Più stimoli arrivano, più dolore viene percepito, più potente deve essere il farmaco utilizzato o più dose deve essere prescritta. Sempre nella fatidica scala è consigliata la contemporanea somministrazione dei cosiddetti farmaci adiuvanti (antinfiammatori, antiepilettici, antidepressivi ecc.), prima di superare ogni gradino. Il criterio che ne regola la prescrizione e il “tentativo terapeutico” e non la relazione tra loro meccanismo d’azione e il meccanismo patogenetico del dolore percepito.

44Aspetti di fisiopatologia e terapia del dolore
b) Le sindromi cliniche e la scelta della terapiaUn secondo modello, che si è diffuso nel campo del dolore neuropatico, si basa sulla
prescrizione di quei farmaci di cui gli studi clinici, randomizzati e controllati, hanno dimostrato l’efficacia in alcune sindromi algiche, come la neuropatia post-erpetica, la neuropatia diabetica e la nevralgia essenziale del trigemino, indipendentemente dai meccanismi patogenetici, dalle differenze riscontrate nei sintomi e nei segni clinici osservati. Questo modello prescrittivo è quello che segue le cosiddette “linee guida” delle diverse società scientifiche o di boards scientifici (O’Connor 2009, Attal 2010, Backonia 2006). Una volta dimostrata l’efficacia di un dato farmaco in una sindrome clinica si presume che lo debba essere in tutte le altre che appartengono alla stessa tipologia. L’esperienza clinica ha dimostrato che le risposte sono incostanti e variabili da caso a caso anche all’interno dello stesso quadro clinico e della stessa diagnosi. Le stesse sindromi utilizzate come standard clinici nella ricerca farmacologica sono sindromi complesse, che spesso si manifestano con caratteristiche diverse da paziente a paziente ed i cui il dolore è sostenuto da meccanismi patogenetici differenti a volte contemporaneamente presenti.
c) I sintomi come “guida” alla terapiaUn terzo modello è rappresentato dalla correlazione tra un tipo di farmaco e la
caratteristica del dolore spontaneo od evocato lamentato dal paziente. Ciò vale so-prattutto nel trattamento del dolore neuropatico. Un esempio è dato dall’indicazione all’impiego del la carbamazepina in presenza di un dolore che il paziente descrive come “ lancinante”. È ampiamente risaputo nella pratica clinica che un dolore “lanci-nante” può essere causato da meccanismi patogenetici differenti interessanti il nervo o le strutture miofasciali. Appare evidente che non è pensabile impostare un piano terapeutico basandosi sui valori numerici dell’intensità del dolore o sulla descrizione che il paziente fa della suo dolore (Rull 1969).
d) I meccanismi patogenetici Le conoscenze della fisiopatologia del dolore e dei meccanismi patogenetici ci per-
mettono di trattare il dolore con un approccio più specialistico che, dopo un momento diagnostico fondamentale, permette di definire un piano di cura basato su scelte farma-cologiche, di blocco e invasive sempre più efficaci (Basbaum 2001,Max 2000, Woolf 1998, 1999, Clifford 2004). Benché già sottolineato nelle parti precedenti, è importante ricordare che nella pratica clinica quotidiana esistono fondamentalmente due tipi di dolore, il dolore nocicettivo e quello neuropatico, e una via nervosa somato-sensoriale che conduce gli impulsi ai centri superiori. Nel dolore nocicettivo la via è integra e funge da conduttore degli impulsi nati a livello dei nocicettori tissutali, nel dolore neuropatico, la via è interessata da processi patologici e genera essa stessa gli impulsi a livello dei cosiddetti siti ectopici. Il dolore subisce importanti modificazioni a livello delle sinapsi spinali sia inibendo (in condizioni normali) sia amplificando gli impulsi afferenti (quando le afferenze provenienti lungo le fibre amieliniche sono continue e intense). Ne consegue che il dolore diviene più intenso, più esteso e scatenato da stimoli non necessariamente dolorosi (allodinia dinamica meccanica) in un vasto territorio. In breve possiamo af-fermare che il dolore può originare dai nocicettori tissutali (dolore nocicettivo), dai siti ectopici (dolore neuropatico periferico) e che i neuroni spinali possono amplificare gli impulsi ma possono anche generarli essi stessi. Questo accade nel dolore cosiddetto da deafferentazione ovvero nelle lesioni gravi delle vie afferenti. Il neurone che non riceve più impulsi dalla periferia, genera dolore (dolore neuropatico centrale). Possiamo quindi definire che i punti in cui concentrare l’azione dei farmaci sono nel tessuti periferici dove i nocicettori sono coinvolti, lungo le fibre nervose dove si sviluppano i siti ectopici, a

45 La “Terapia combinata” nel trattamento del dolore cronico
livello spinale dove l’impulso attraversa le sinapsi verso i neuroni di secondo ordine. Se questi sono le sedi di origine degli impulsi, i meccanismi patogenetici sui cui i farmaci devono agire sono la ipersensibilità del nocicettore, l’ipersensibilità delle fibre con facilitata genesi ectopica di impulsi, l’ipersensibilità del neurone post-sinaptico con fa-cilitata trasmissione trans-sinaptica o scarica spontanea da deafferentazione. Poiché alla ipersensibilità del neurone spinale concorrono più fattori come la persistenza di impulsi nocicettivi afferenti, la riduzione dei sistemi inibitori, l’aumento dei sistemi eccitatori, si comprende come più scelte terapeutiche sono possibili. Una volta che la diagnosi clinica e strumentale ci permette di definire i meccanismi patogenetici coinvolti, conoscendo il meccanismo d’azione dei farmaci è possibili definire un piano terapeutico efficace. Più farmaci possono essere associati tra loro (subito o in una strategia add-on) in modo da agire sui diversi meccanismi contemporaneamente presenti (complementarietà), in modo da sfruttare eventuali sinergie (potenziamento reciproco) o capacità di antagonizzare gli effetti collaterali. Si attua così una “terapia combinata”. Nella pratica clinica incontria-mo sindromi algiche che appaiono complesse per la contemporanea presenza di segni e sintomi appartenenti sia al dolore nocicettivo, sia a quello neuropatico periferico e centrale. Gli eventi traumatici, la lesione erpetica e l’invasione neoplastica, primitiva o metastatica, sono i principali responsabili di queste forme. La complessità deriva dalla presenza di segni e sintomi di diversa natura, spiegabili solo per il contemporaneo sovrap-porsi di più meccanismi patogenetici, e dalla variabilità che si riscontra nell’evoluzione della malattia. Per definire queste sindromi algiche viene spesso utilizzato il termine di “dolore misto”. Misto non è il dolore, che è o nocicettivo o neuropatico (non esiste un altro tipo di dolore), ma bensì il quadro clinico che li vede contemporaneamente presenti e talvolta interagente tra loro.
La terapIa Basata suL MeccanIsMo patoGenetIco
1) farmaci che agiscono sull’ipersensibilità del nocicettoreIn questo gruppo raccogliamo i farmaci che, a livello del processo infiammatorio,
agiscono riducendo le sostanze algogene o direttamente l’ipersensibilità del nocicet-tore. Questi farmaci agiscono sul dolore spontaneo ed evocato modificando la soglia del nocicettore e riportandola ai valori normali. In un certo senso sono farmaci anti-allodinici perché riducono fino a togliere il dolore evocato da stimoli non dolorosi (o allodinia primaria).
Figura 1. La via nocicettiva periferica e i meccanismi patogenetici.
nocIcettore tIssutaLe
IpersensIBILItà deL nocIcettore
I neurone sensItIvo
IpersensIBILItà dI fIBra con orIGIne ectopIca
deGLI IMpuLsI
IpersensIBILItà deL neurone spInaLe da eccesso d’IMpuLsI
o da deafferentazIone
sIsteMa GLIaLesIsteMa
InIBItorIo
II neurone
WDRN

46Aspetti di fisiopatologia e terapia del dolore
farmaci che agiscono sulle sostanze algogene
Tra questi farmaci compaiono gli antinfiammatori non steroidei (FANS) (Attal 2000), i corticosteroidi (Abram 2000), gli inibitori specifici della ciclossigenasi-2 (COX 2).
a) Farmaci antinfiammatori non steroidei o FANS
È noto che i FANS sono gravati da effetti collaterali importanti a livello della fun-zionalità renale (con esclusione dell’acido acetilsalicilico) a livello dell’aggregazione piastrinica e del tratto digerente. (Morrison 2001, Girlon 2003). È altrettanto noto che l’azione gastrointestinale può essere modulata con la contemporanea associazione di inibitori di pompa (terapia combinata a scopo di ridurre gli effetti collaterali). Questo effetto protettivo può ridursi in presenza di paracetamolo (Rahme 2008) come dimo-strato da uno studio compiuto su quasi 650.000 pazienti anziani a cui era stato prescritto solo un FANS tradizionale con o senza inibitori di pompa e una associazione di FANS e paracetamolo con o senza inibitori di pompa. L’associazione di FANS e paraceta-molo aumentava il rischio di sanguinamento gastrico nonostante l’inibitori di pompa.
I FANS presentano inoltre due aspetti fondamentali che devono essere tenuti in considerazione: il primo è rappresentato dalla risposta individuale ai FANS ed il secondo dalla presenza di un affetto antalgico “tetto” per cui all’aumento della dose non corrisponde una aumento dell’efficacia (Jacox 1994).
Un altro aspetto interessante è dato dalla presenza di prostaglandina E2 (PGE
2)
nel sistema nervoso spinale e sovra-spinale in presenza di un processo infiammatorio tissutale periferico (Samad 2001). A tale presenza è connessa una riduzione dei siste-mi inibitori spinali. Alcuni FANS come il Diclofenac e soprattutto i COX-2 inibitori hanno una contemporanea azione antalgica “centrale” (azione su due meccanismi patogenetici). Omoigui nel 2007 sostiene che il processo patologico fondamentale nel dolore cronico è l’infiammazione e che esiste per ciascun caso uno specifico “profile” dei mediatori infiammatori. Nell’artrosi, ad esempio, sono coinvolte l’interleukina 1 β, l’interleukina -6, il Tumor Necrosis Factor TNF-α, nelle patologie erniarie cer-vicali e lombari le prostaglandine, il Tumor Necrosis Factor TNF-α, l’interleukina 1 β, nella fibromialgia la sostanza P, l’interleukina 1 β, l’interleukina -6, e così nel dolore neuropatico la sostanza P. Le prostaglandine, il glutammato, l’interleukina 1 β, l’interleukina -6, il Tumor Necrosis Factor (TNF-α). Questa diversa presenza di mediatori della flogosi spiega anche la ridotta efficacia dei farmaci antinfiammatori tradizionali in alcuni casi di flogosi e spinge la ricerca a individuare nuove molecole attive sui diversi mediatori.
Tabella 1. considerazioni sui farmaci antinfiammatori.
Considerazioni sul trattamento con FANS
I farmaci antinfiammatori non steroidei sono utili quando il meccanismo patogenetico è costituito dalla ipersensibilità dei nocicettori tissutali ad opera di mediatori della flogosi ma:a) vi è una risposta individuale ai vari fansb) hanno un effetto tettoc) I fans e i coX-2 sono gravati da effetti collaterali indesideratid) non sono certo efficaci per tutti i mediatori della flogosie) alcuni fans ed i coX-2 hanno un’azione periferica ed una spinale
possono essere “combinati” con:a) farmaci gastroprotettivi (associazione protettiva)b) farmaci che agiscono a livello sinapticoc) farmaci che agiscono a livello di siti ectopici

47 La “Terapia combinata” nel trattamento del dolore cronico
b) I farmaci antinfiammatori steroidei, i corticosteroidi
Gli steroidi orali sono spesso utilizzati nel trattamento del dolore cronico quando il meccanismo patogenetico è quello della flogosi tissutale. Vengono ampiamente prescritti nelle malattie reumatiche, nelle vasculiti, nelle artropatie in genere ma anche nelle patologie infiammatorie neurologiche sia radicolari che periferiche (come nella sindrome del tunnel carpale – Chang 2002). Si deve tener conto dei pazienti immuno-depressi. Per quanto riguarda la somministrazione di steroidi nell’Herpes acuto non esistono controindicazioni perché la riattivazione dell’infezione erpetica è mediata dalle IgG e non dalle IgM e quindi non inibita dagli steroidi (Toliver 1997 e Pardo 1997). Il metilprednisolone, il triamcinolone, il betametasone ed il desametasone ven-gono iniettati per via intra-articolare o peri-articolare (Benzon 2007). I corticosteroidi (metilprednisone e traimcinolone) sono indicati nella somministrazione peridurale nel trattamento delle radicolopatie. Sono insorte controversie sulla somministrazione di metilprednisone peridurale per il pericolo di aracnoiditi o altre lesioni neurologiche (Bernat 1976). La tossicità presunta è stata ridimensionata dallo studio di Kotani del 2000 in cui non si sono osservati danni neurologici sono somministrazione intratecale di metilprednisone a dosi elevate (80 mg) in pazienti con neuropatia post-erpetica.
Gli anti-NGF, anti-IL-1 and anti-TNFa
Il Nerve Grow Factor NGF è rilasciato nei traumi tissutali e nell’infiammazione da un certo numero di mastcellule, macrofagi, linfociti, fibroblasti e keratinociti, ed è considerato un fattore algogeno importante soprattutto quando l’infiammazione costituisce il processo patogenetico principale. Il NGF sembra agire anche diretta-mente sul nocicettore aumentandone l’eccitabilità (Rukwied 2010 e Radtke 2010). L’azione è mediata dai recettori tirosin-kinasi o TrKA (Zampieri 2006). Il Tanezumab è un anticorpo monoclonale di origine umana in fase utilizzato nelle osteoartrosi di ginocchio, nel low back pain, nella cisitite interstiziale, nel dolore osseo da cancro, nelle endometriosi e in alcune sindromi neuropatiche.
Senza entrare nei particolari si sottolinea il ruolo terapeutico nelle patologie reuma-tiche come l’artrite reumatoide e la spondilite anchilosante dei farmaci che agiscono contro l’Interleukina 1 e il TNFa. La loro azione non si esplica solo a livello periferico ma anche a livello spinale (Schaible 2006).
farmaci che agiscono direttamente sul nocicettore
A questo gruppo appartiene la Capsaicina somministrata mediante cerotto ad alta concentrazione (8%) e gli anestetici locali (Lidocaina patch) somministrati per via transcutanea. Anche gli oppioidi agiscono a livello nocicettoriale quando vengono somministrati nei tessuti coinvolti (Stein 1999, Abram 2000). La loro azione sembra ridurre la liberazione di mediatori della flogosi.
La capsaicina, o 6-nonenamide, N-[(4-idrossi-3-metossifenil) metil]-8-metile, (6E), è un agonista altamente selettivo del recettore vanilloide 1 TRPV1 (transient receptor potential vanilloid 1). L’effetto iniziale della capsaicina è l’attivazione dei nocicettori cutanei che esprimono il TRPV1, che causa sensazione di puntura ed eritema dovuti al rilascio di neuropeptidi vasoattivi. In seguito all’esposizione alla capsaicina, i nocicettori cutanei sembrano diventare meno sensibili a stimoli di tipo chimico (pH) e termico (>45°C). Questi effetti tardivi della capsaicina sono denomi-nati spesso “desensibilizzazione” e si ritiene siano alla base del miglioramento della sintomatologia dolorosa. Poiché sono coinvolti solo i recettori TRPV1 e solo nervi cutanei che esprimono il TRPV1, rimane inalterata la capacità di avvertire stimoli termici, meccanici e vibratori. Le alterazioni indotte dalla capsaicina nei nocicettori

48Aspetti di fisiopatologia e terapia del dolore
cutanei sono reversibili ed è stato segnalato ed osservato che la normale funzionalità viene ripristinata nel giro di settimane nei volontari sani. L’indicazione principale è quella della neuropatia post-erpetica ma anche quella delle neuropatie periferiche. L’esatto meccanismo d’azione non è ancora completamente noto ma si suppone che l’azione sui recettori TRPV1 sia in grado di impedire che sostanze nocicettive liberate nei tessuti periferici da fibre danneggiate non possano sensibilizzare i nocicettori sani rimasti presenti (Irving 2011, Anand 2011).
Tra gli anestetici locali rientrano EMLA (eutectic mixture of local anesthetic) e la lidocaina, le cui formulazioni topiche (gel e patch al 5%) sono state dimostrate efficaci in pazienti con neuropatia post-erpetica e in alcuni altri tipi di poli e mononeuropatie (Rowbotham 1995, 1996). Le formulazioni in commercio in Italia sono Luan pomata al 2,5% e Xilocaina pomata al 5%. I livelli serici raggiunti dopo l’applicazione topica sono di ordini di grandezza non significativi per effetto cardiaco. La lidocaina patch al 5% (cerotto di 10x14 cm contenente 700 mg di lidocaina) applicata sulla zona di dolore utilizzando fino a tre patch per 12 ore. Il maccanismo d’azione è legato al blocco dei canali del sodio locali e presenti sui nocicettori o sulle fibre (siti ectopici) presenti nell’epidermide e nel derma. Ne è stata dimostrata l’efficacia nel trattamento della neuropatia post-erpetica (Galer 1999) e nelle neuropatie periferiche (Meier 2003). Il patch di lidocaina viene considerato come farmaco di prima scelta nella cura delle sindromi algiche neuropatiche (Dworkin 2003).
2) I farmaci che agiscono a livello della sinapsi spinale riducendo l’ipersensibilità del secondo neurone
In questo numeroso gruppo dobbiamo distinguere i farmaci in base alla loro azione sulla sinapsi spinale. I farmaci che modulano la trasmissione sinaptica agendo sull’elemento presinaptico:
A: gli oppioidi
Gli oppioidi agiscono a livello spinale e sopraspinale. Le sedi spinali sono rappre-sentate, a livello presinaptico (75%), dal terminale dei nocicettori delle fibre afferenti C dove riducono la liberazione di peptidi e glutammato (a livello spinale prevalgono i recettori di tipo mu e delta), riducendo il meccanismo eccitatorio esercitato dal mes-saggio afferente, e, a livello postsinaptico, dal neurone spinale, inibendo l’invio delle
Figura 2. I farmaci che agiscono sulla ipersensibilità del nocicettore tissutale.
nocIcettore tIssutaLe
IpersensIBILItà deL nocIcettorefarmaci antinfiammatori steroidei e non steroidei
capsaicina e Lidocaina patchanti tnf e nGf
I neurone sensItIvo
sIsteMa GLIaLesIsteMa
InIBItorIo
II neurone
WDRN

49 La “Terapia combinata” nel trattamento del dolore cronico
informazioni ai neuroni superiori. Altrettanto noto è il meccanismo d’azione sulla nocicezione degli oppioidi a livello sopraspinale dove sono presenti recettori di tipo mu, delta e kappa. Possono agire sulle vie discendenti noradrenergiche e serotoniner-giche, che intervengono nella modulazione della nocicezione (Dickenson 1999). Alla norepinefrina, rilasciata dalle vie discendenti, che agisce sui recettori adrenergici a
2, si
associano altri elementi inibitori come l’adenosina e l’acido y-aminobutirrico (GABA) che concorrono a modulare le vie nocicettive a livello spinale. A livello presinaptico l’attivazione dei recettori oppioidi porta alla chiusura dei canali del calcio voltaggio-dipendenti limitando la liberazione di neurotrasmettitori come la norepinefrina, il glutammato, la serotonina, la sostanza-P e l’aceticolina (Kaneko 1994, Piros 1995). A livello postsinaptico gli oppioidi iperpolarizzano la cellula (neuroni di proiezione e interneuroni), attivando i canali del potassio (Yoshimura 1983, Dickenson 2005).
L’impiego degli oppioidi nel dolore cronico inizia dopo gli anni ’80, gravato dalla disputa sulla sua efficacia o inefficacia nel dolore neuropatico. Tralasciando i particolari di questa disputa e ogni rigida suddivisione tra dolore nocicettivo e neuropatico (ancor oggi controversa) si è visto che gli oppioidi agiscono nel dolore cronico a livello dei meccanismi patogenetici del dolore coinvolti nell’origine e trasmissione del messaggio nocicettivo lungo le fibre C.
L’instaurarsi del meccanismo patogenetico costituito dall’ipersensibilità dei neuroni spinali rende necessario un incremento della dose d’oppioidi, in quanto è richiesta una azione inibitoria maggiore per controllare l’aumentata eccitabilità (Dickenson 2005), come d’altronde è dimostrato dalla maggior efficacia della morfina nei modelli animali dopo somministrazione di antagonisti dei recettori NMDA (Dickenson 2001). Mannion (2000) consiglia la via intratecale per favorire l’azione postsinaptica degli oppioidi.
Prima del 2003 gli unici dati, riguardanti la somministrazione nel dolore non on-cologico, provenienti da studi controllati, si riferiscono al loro impiego nella nevralgia post-erpetica (Watson 1998). In questo studio l’ossicodone a rilascio prolungato si è dimostrato più efficace del placebo nel controllo del dolore continuo, di quello acces-sionale e dell’allodinia, in 38 pazienti (trial crossover). Altrettanto interessanti sono gli studi di Rowbotham (1991), che dimostrano l’efficacia di una somministrazione endovenosa lenta di morfina (0,3 mg/Kg/ora) nei confronti del placebo nel controllare il dolore da nevralgia post-erpetica, e di Dellemijn (1997), che, utilizzando l’infusione di fentanyl (5 mg/Kg/h per 5 ore) ed un placebo attivo, dimostra lefficacia degli oppioidi nel dolore neuropatico. Lo stesso Dellemijn in una successiva pubblicazione (1999) sottolinea la possibilità di utilizzo degli oppiacei nel dolore nocicettivo e neuropatico. In uno studio randomizzato, doppio cieco, crossover, dove la morfina a lento rilascio ed il metadone sono stati confrontati con due triciclici (nortriptilina e amitriptilina), si è dimostrato che la morfina a lento rilascio è significativamente migliore della nor-triptilina e che il metadone è paragonabile sia all’amitriptilina che alla nortriptilina (Raja 2002). Questi dati sottolineano che gli oppioidi possono essere somministrati in alternativa agli antidepressivi triciclici nella neuropatia post-erpetica tenendo presente gli effetti collaterali talvolta intollerati (Argoff 2004). Va però considerato che da un punto di vista patogenetico il dolore nella neuropatia post-erpetica può avere più origini. Vi sono casi in cui è presente una sindrome da deafferentazione con dolore centrale e casi in cui è presente una condizione flogistica tissutale che stimola i neuroni sani a cui contribuisce una anormale liberazione di neurotrofine come il Nerve Grow Factor (NGF), il fattore neurotrofico (glial cell line-derived neurotrophic factor o GDNF), o citochine pro-infiammatorie, che non vengono controllate dai FANS (Anand 2011).
In un articolo (2003) Mendell e Sahenk esaminano le neuropatie dolorose sensitive (tra cui elencano le neuropatie da piccole fibre, le neuropatie diabetiche periferiche, le neu-ropatie da connettivopatie, le neuropatie paraneoplastiche e amiloidotiche, le neuropatie

50Aspetti di fisiopatologia e terapia del dolore
autonomiche ereditarie, le tossiche arseniche e da HIV), elencano i farmaci utilizzabili e concludono che il gabapentin può essere considerato il farmaco di prima scelta, che gli antidepressivi triciclici seppur utili sono gravati da effetti indesiderati, che il tramadolo è utilissimo in associazione al gabapentin o in sua sostituzione, che l’ossicodone e la morfina a lento rilascio sono indicati seppur senza aspettarsi una completa risoluzione del dolore. In uno studio double-blind e controllato su pazienti con neuropatia diabetica è stata dimostrata l’efficacia dell’ossicodone a lento rilascio a dosaggi compresi tra 10 e 99 mg/die. L’efficacia del Tramadolo è stata dimostrata in due studi controllati di cui il primo si riferiva al trattamento della neuropatia diabetica (Harati 1998) e il secondo al dolore neuropatico di diversa tipologia (Sindrup 1999). Il tramadolo si è rivelato superiore al placebo in entrambe gli studi con un NNT di 3.1 e 4.3. Nel secondo studio si è osservato che oltre al dolore spontaneo si riduceva il dolore evocato dal tatto e l’al-lodinia meccanica indotta sperimentalmente. In tutti questi studi viene registrata una elevata percentuale di abbandono dello studio per evidenti effetti collaterali. Il Trama-dolo è un farmaco analgesico che agisce sia con meccanismo oppioide che con quello monoaminergico (come gli antidepressivi triciclici). Va sottolineato che la tolleranza e la dipendenza sono evenienze rare anche dopo lunga somministrazione di tramadolo. Alla luce di quanto abbiamo fin qui esposto appare evidente che gli oppioidi possono agire in numerosi quadri algici dove esiste un impulso nocicettivo (indipendente dal tessuto d’origine) e dove esiste un’integrità della sinapsi tra primo e secondo neurone. Appare altresì evidente come sia opportuno instaurare una terapia combinata. Il passaggio dalla somministrazione sistemica a quella epidurale e a quella intratecale mediante impianto di sistemi di infusione permette il raggiungimento di risultati migliori sia in casi in cui l’oppiaceo ha determinato l’insorgenza di effetti collaterali importanti, sia in quei casi in cui la somministrazione sistemica non ha dato una analgesia soddisfacente. Nella pratica clinica quotidiana vengono utilizzati i diversi oppioidi a disposizione, dalla morfina al fentanil, dall’oxicodone all’idromorfone e al tapentadolo. Nello stesso tempo sono state introdotte alcune associazioni (terapia conbinata) che aumentano l’efficacia come nel caso dell’oxicodone e del paracetamolo, oppure altre associazioni (terapia preventiva) in cui all’oppioide (oxicodone) è stato unito il naloxone, per antagonizzare l’effetto del farmaco sui recettori intestinali e combattere la stipsi. Un farmaco oppioide recente, il tapentadolo, presenta un doppio meccanismo nel senso che agisce direttamente sui recet-tori spinali, sia sui sistemi noradrenergici potenziando l’azione inibitoria. Questo doppio meccanismo ne potenzia l’effetto antalgico e riduce la comparse di effetti collaterali.
B: il Paracetamolo
Poniamo qui il paracetamolo anche se la sua azione si ritiene si esplichi sia sui meccanismi inibitori discendenti sia a livello postsinaptico spinale. Secondo alcuni
Tabella 2. considerazioni sui farmaci oppioidi.
Considerazioni sull’impiego degli oppioidi:I farmaci oppioidi agiscono modulando l’impulso nocicettivo a livello della sinapsi spinale, sul meccanismo patogenetico della ipersensibilità dei neuroni spinali ad opera di impulsi nocicettivi afferenti.a) vi è una risposta individuale ai vari oppioidib) hanno un effetto tettoc) sono gravati da effetti collaterali indesiderati nel breve e nel lungo periodod) non sono efficaci nella deafferentazione del neurone spinalepossono essere “combinati” con:a) farmaci contro la stitichezza e la nausea (azione protettiva)b) farmaci ce agiscono sui sistemi inibitori ed eccitatori spinali

51 La “Terapia combinata” nel trattamento del dolore cronico
Autori il paracetamolo attiverebbe a livello centrale le vie discendenti serotoninergiche di modulazione inibitoria sulle afferenze primarie nocicettive (azione indiretta sui recettori 5HT
3) (Alhaider 1991, Sandrini 2003). Sono stati recentemente individuati a
livello del sistema nervoso centrale i recettori COX-3 (variante dei COX-1), sensibili al paracetamolo, che sembrano giocare un ruolo chiave nella biosintesi dei prostanoidi, mediatori importanti del dolore e della febbre (Shawab 2003).
La prostaglandina E2 (PGE
2) è uno dei maggiori mediatori della flogosi che sensi-
bilizza i nocicettori ed inoltre causa la comparsa di allodinia e di iperalgesia quando è somministrata per via intratecale. L’azione della PG E
2 è mediata dai recettori G-
protein-coupled prostanoid (EP) di cui sono presenti quattro tipi a livello del midollo spinale, indicando un effetto postsinaptico della PG E
2. L’attivazione del recettore EP1
comporta l’entrata di calcio nella cellula mentre la stimolazione dei recettori EP2 e EP4 aumenta l’AMP ciclico intracellulare e quella del recettori EP4 riduce la concen-trazione dell’AMP intracellulare (Ebersberger 2002). Pertanto l’inibizione dei COX-3 potrebbe modificare l’eccitabilità del neurone postsinaptico. Il paracetamolo così come alcuni antinfiammatori (tra cui il diclofenac) sembrerebbero agire attraverso questo meccanismo inibitorio. Al dosaggio terapeutico singolo di 1 grammo si raggiunge il picco ematico e quindi liquorale che corrisponde all’effetto analgesico massimale, mentre a dosi superiori si ha solo un effetto a “plateau”.
C: i Farmaci che agiscono sui canali del calcio
I canali del calcio voltaggio-dipendenti (VDCC) sono importanti nella trasmissione sensitiva. In risposta alla depolarizzazione di membrana essi mediano l’entrata del Ca++ in numerosi tipi di cellule e pertanto intervengono nella trasmissione sinaptica delle informazioni sensitive provenienti dalla periferia attraverso il controllo dei complessi eventi che vedono coinvolte la depolarizzazione, la liberazione di neurotrasmettitori, l’eccitabilità neuronale, le modificazioni intracellulari (Dickenson 2004).
I neuroni sensitivi esprimono varie classi di VDCC (L, N, P, Q, R e T) distinte per profili elettrofisiologici e farmacologici. Le subunità a
2-d determinano il tipo di canale
e contengono il poro che permette l’entrata del calcio nel neurone o nel terminale, quando viene depolarizzata. Alcuni canali (N, P/Q & R) sono presenti a livello presi-naptico sui terminali e influenzano la liberazione di trasmettitori allorché il potenziale d’azione arriva dalla periferia (esponendo i neuroni spinali ad un maggiore presenza di trasmettitori). Altri tipi di canali del calcio (N, T & L) sono invece presenti a livel-lo postsinaptico e contribuiscono all’eccitabilità del neurone. Recentemente i canali del calcio sono stati suddivisi in tre famiglie (Ca 1, 2 e 3) in base alle caratteristiche anatomiche e funzionali (Ertel 2000).
D: Alfa 2 delta ligandi (Gabapentin e Pregabalin)
Questi farmaci si legano alle subunità a2-d dei canali del calcio a livello sinaptico
sui terminali nocicettivi delle fibre C e agiscono limitando l’entrata del calcio e quindi la liberazione di neurotrasmettitori nella sinapsi (Dahl 2004).
Secondo Tremont-Lukats (2002) il gabapentin agirebbe non solo sui canali del calcio di tipo a
2-d ma anche attraverso i siti glicinici dei recettori complessi NMDA e
aumentando la presenza di GABA senza però agire direttamente sul sistema gabaergico. Queste ipotesi farebbero rientrare il gabapentin tra i farmaci che abbiamo già citato con azione sui sistemi inibitori centrali (Backonja 1998). Secondo alcuni Autori (Pan 1999) il gabapentin agirebbe anche sui canali del sodio per azione diretta o indiretta, senza modificare la conduzione nervosa. Il gabapentin è stato ampiamente studiato in passato nella terapia delle neuropatie e nel dolore oncologico (Caraceni 1999, Bennet

52Aspetti di fisiopatologia e terapia del dolore
2000), partendo dagli studi controllati sulla sua efficacia nel dolore da neuropatia dia-betica (NNT di 3.7 (2.4±8.3) Backonja et al., 1998). e nella neuropatia postherpetica (NNT di 3.2 (2.4±5.0) Rowbotham 1998). La scarsità di effetti collaterali (confusione, nausea, astenia) e la flessibilità del dosaggio l’hanno fatto indicare come farmaco di prima scelta in numerose neuropatie. Questa azione si è rivelata particolarmente utile nel dolore causato dalla malattia neoplastica dove il gabapentin ha dimostrato un’a-zione sinergica con gli oppioidi (Caraceni 1999, Bennett 2004). Questo farmaco in passato (Dworkin 2003) ed ora il pregabalin (Freynhagen 2005) sono entrati a far parte dell’”evidente-based approach” del dolore cronico insieme agli oppioidi, agli antide-pressivi triciclici, alla lidocaina. Il gabapentin ed ora il pregabalin hanno dimostrato una chiara efficacia nei modelli animali (Field 1999), nelle sindromi di dolore cronico neuropatico e particolarmente nella neuropatia diabetica, in quella post-erpetica, ma anche nei casi di fibromialgia. I positivi risultati osservati e la modesta incidenza di effetti collaterali hanno fatto si che questi due farmaci siano considerati come farmaci di prima scelta nel trattamento del dolore neuropatico (Freynhagen 2005). Il pregabalin, contrariamente al gabapentin, viene utilizzato a bassi dosaggi con ridotta incidenza di effetti collaterali. Il basso dosaggio è dovuto alla maggior biodisponibilitò e al rapido assorbimento. Inoltre, diversamente dal gabapentin, la concentrazione plasmatica di pregabalin aumenta in modo lineare con l’aumento della dose (Wesche 2005).
3) I farmaci che agiscono sui sistemi inibitori discendenti e spinaliI neuroni spinali sono sottoposti ad una modulazione (eccitatoria ed inibitoria),
tonicamente attiva, da parte di fibre discendenti e di interneuroni locali. Differenti trasmettitori e recettori si sviluppano nelle corna dorsali (Mannion 2000) e tra questi i recettori GABA, i recettori glicinici, e quelli degli oppioidi.
I farmaci antidepressivi
Il loro utilizzo è stato inizialmente proposto per la componente depressiva che accompagnava la sintomatologia dolorosa in pazienti con neuropatia post-erpetica (Woodforde 1965). In seguito venne proposta l’associazione di un antidepressivo triciclico (amitriptilina) con un neurolettico (flufenazina) ritenendo quest’ultimo il principio attivo ed utilizzando l’antidepressivo per combatterne gli effetti collaterali (Taub 1985). Sono ormai molto numerosi gli studi controllati che dimostrano l’efficacia dei triciclici (amitriptilina, nortriptilina) in diversi tipi di neuropatie (Gomez-Peres 1985, Watson 1982). L’impressione clinica è che questi farmaci agiscano più sulla componente urente continua, sul dolore sordo, e meno sul dolore lancinante più sen-sibile agli antiepilettici. Gli studi clinici dimostrano che l’efficacia degli antidepressivi non ha alcuna relazione con la tipologia del dolore (McQuay 1996).
L’effetto degli antidepressivi sul dolore è mediato sia dall’inibizione del reuptake delle monoamine (noradrenalina e serotonina) a livello del sistema nervoso centrale con conseguente aumento dell’attivazione delle vie discendenti di modulazione del dolore, sia dalla modulazione dei canali del sodio (per cui sono citati anche nella parte riguardante i farmaci ad azione sui canali del sodio). È noto che sono presenti vie discendenti che modulano la trasmissione nocicettiva tra cui i sistemi inibitori endo-geni serotoninergici che originano a livello del grigio periaqueduttale e, scendendo, raggiungono le corna dorsali. Dal locus ceruleus sorge la vie inibitoria noradrenergica anch’essa diretta alle corna posteriori. Gli effetti inibitori di queste due vie utilizzano anche gli interneuroni GABAergici. La loro azione inibitoria fa si che questi farmaci trovino indicazione nel trattamento del dolore cronico a diversa eziologia. Nel dolore cronico nocicettivo essi si dimostrano utili in sinergia con gli analgesici periferici ed oppioidi potenziandone l’azione e riducendone la richiesta in termini di dosaggio.

53 La “Terapia combinata” nel trattamento del dolore cronico
Da numerosi autori i farmaci antidepressivi, in particolar modo i triciclici, sono considerati i farmaci di prima scelta nel trattamento delle neuropatie periferiche e centrali, ad eccezione della nevralgia trigeminale (Galer 1995, Warson 1995). Questi farmaci possono esercitare un’azione importante sia nella genesi ectopica periferica, sia sui neuroni centrali deafferentati. Tra gli antidepressivi troviamo i cosiddetti triciclici, gli inibitori selettivi del reuptake della serotonina (SSRI) e gli inibitori del reuptake della noradrenalina (SNRI). Il meccanismo con cui questi farmaci agiscono sulla noci-cezione è quello del reuptake delle amie biogene come la serotonina e la noradrenalina.
Gli antidepressivi triciclici comprendono le amine terziarie (amitriptilina e clomi-pramina) che inibiscono il reuptake della serotonina e della noradrenalina, e le amine secondarie (nortriptilina e disipramina) relativamente selettive per il reuptake della no-radrenalina. Tra i cosiddetti SNRI ricordiamo la venlafaxina. Vi è la controindicazione all’uso degli antidepressivi triciclici in pazienti con glaucoma, ipertrofia prostatica benigna, infarto miocardico. Alcuni effetti collaterali abbastanza fastidiosi ricorrono frequentemente a seguito di terapia con antidepressivi triciclici: secchezza delle fauci, confusione, stipsi, aumento ponderale, difficoltà alla minzione, perdita di memoria, ipotensione ortostatica, alterazioni della conduzione cardiaca. I triciclici si sono rivelati efficaci in pazienti depressi e non depressi con una azione analgesica che è indipen-dente dalle alterazioni dell’umore e che diviene significativa solo dopo 1 o 2 settimane di assunzione. L’efficacia è stata dimostrata sia nella neuropatia diabetica (Max 1987 - 1992, McQuay 1996)), sia in altre forme di dolore neuropatico (Warson 1992, Vrethem 1997). I dosaggi indicati sono all’inizio limitati (10-25 mg) e in somministrazione unica prima di coricarsi, aumentano progressivamente di 10-25 mg ogni 1 o 2 settimane fino a 100-150 mg/die. Le amine secondarie, come la nortriptilina o la desipramina) tendono ad avere meno effetti sedativi e meno azione anticolinergica (Wolfe 2004). Gli inibitori selettivi del reuptake della serotonina (SSRI) si sono dimostrati meno efficaci degli antidepressivi triciclici nel trattamento del dolore neuropatico (Syndrup 1990, 1992). La venlafaxina è un antidepressivo che inibisce il reuptake sia della serotonina, sia della noradrenalina ed ha una minima attività muscarinica e istaminergica rispetto ai triciclici (Syndrup 2003, 2000). I risultati ottenuti nel trattamento della neuropatia post-erpetica ha indotto alcuni autori a valutare una possibile azione profilattica somministrando anti-depressivi triciclici e oppioidi nella fase acuta dell’eruzione (Winnie 1993, Kanazi 2000, Dworkin 2000). Lo studio controllato, doppio cieco, di Bowsher (1997) ha dimostrato l’efficacia della somministrazione di amitriptilina nel prevenire la temuta neuropatia. Tra gli inibitori del reuptake della serotonina e della noradrenalina è presente la Duloxetina che si è dimostrata efficace nel dolore neuropatico ed in particolare in pazienti con sin-drome depressiva (Goldstein 2005).
clonidina
L’alfa 2 agonista clonidina, è stata dimostrata essere efficace, per applicazione transdermica, in una percentuale di pazienti con dolore correlato a neuropatia diabetica e a neuropatia post-erpetica (Byas-Smith 1995, Abadir 1996). Gli sprouts periferici, in sede di lesione di un nervo, dimostrano una aumentata sensibilità alla noradrenalina, la cui liberazione dai terminali simpatici viene inibita dalla clonidina.
Lo stesso farmaco è noto per avere effetto analgesico quando somministrato per via spinale, ma il risultato sembra mediato da attività sui recettori a
2 presinaptici che
intervengono nella liberazione di neurotrasmettitori (Mannion 2000). L’effetto antal-gico è stato dimostrato a seguito di somministrazione orale (singola dose di 0,2 mg) ma la maggior parte delle pubblicazioni riguardanti l’uso di clonidina nel trattamento del dolore neuropatico si riferiscono a somministrazioni peridurali o subaracnoidee (Siddall 2000, Bennet 2000). In somministrazione subaracnoidea continua, il farmaco

54Aspetti di fisiopatologia e terapia del dolore
viene spesso associato a morfina o ad altri oppioidi (Hassenbusch 2000). La sommi-nistrazione sistemica per via orale, pur ottenendo l’effetto antipertensivo, non sembra avere una significativa efficacia sul dolore.
Baclofen
Il baclofen è stato introdotto in commercio per terapia della spasticità di origine centrale. La sua azione si espleta tramite l’attivazione dei recettori GABA-B presinap-tici spinali. Nella terapia del dolore è considerato un farmaco di seconda scelta nella nevralgia trigeminale ed è stato studiato su diverse patologie con dolore neuropatico con risultati discordanti. Nella nostra esperienza, la terapia con baclofen per via orale si è rivelata efficace in caso di dolore radicolare e/o spinale accompagnato da crampi e/o ipertono muscolare (Herman 1992). La somministrazione di baclofen spinale tramite pompa d’infusione completamente impiantabile è ormai ampiamente utilizzata per il controllo della spasticità in pazienti con lesioni spinali o cerebrali: alcune di queste casistiche riportano anche controllo del dolore in una parte dei pazienti.
clonazepam
Anche sull’impiego del clonazepam sono pochi i lavori controllati che ne dimo-strino l’efficacia nel dolore neuropatico. Il clonazepam è una benzodiazepina con meccanismo d’azione leggermente diverso rispetto agli altri antiepilettici; la sua azione è infatti mediata dai recettori GABA-A con aumento della conduttanza al clo-ro che crea iperpolarizzazione cellulare e quindi riduzione dell’eccitabilità. Benchè i lavori pubblicati presentano casi aneddotici ed utilizzo soprattutto in caso di dolore lancinante, a scarica (Bartusch 1996), nella nostra pratica clinica il clonazepam si è rivelato efficace, con scarsissimi effetti collaterali, in pazienti con dolore continuo, urente, di origine centrale o periferica. La risposta si ottiene solitamente a dosaggio molto basso, 1-1,5 mg/die anche se a distanza di uno/due mesi l’efficacia antalgica risulta attenuarsi e può rendersi necessario un aumento del dosaggio.
topiramato
Il topiramato è un antiepilettico con la capacità di bloccare i canali del Na+, pro-muovere l’attività GABA interagendo con un sito diverso dalle benzodiazepine sul recettore GABA A e bloccare selettivamente i recettori AMPA/kainato glutammato
Figura 3. I farmaci che agiscono sulla ipersensibilità del neurone spinale.
nocIcettore tIssutaLe
IpersensIBILItà deL neurone spInaLeoppioidi e paracetamolo
alfa2 delta ligandiantidepressivi
clonidina, Baclofen, clonazepam, topiramato
I neurone sensItIvo
sIsteMa GLIaLesIsteMa
InIBItorIo
II neurone
WDRN

55 La “Terapia combinata” nel trattamento del dolore cronico
(Schneiderman 1998). Non sono numerosi gli studi controllati sull’uso del topiramato in pazienti con dolore neuropatico ma i primi risultati sono incoraggianti (Bajwa 1999, Edwards 2000). I dosaggi utilizzati nella pratica clinica vanno da 50 mg/die iniziali sino a 400 mg/die.
4) I farmaci che agiscono sulla fibre riducendo l’ipersensibilità a livello dei siti ectopici
In conseguenza di una lesione le fibre nervose manifestano una attività spontanea ed un aumento della sensibilità a vari stimoli termici, chimici e meccanici. Questa abnor-me attività non si sviluppa solo lungo il nervo periferico (foci ectopici), ma coinvolge anche le cellule (neuroni attivi) del ganglio della radice posteriore (DRG) (Wall 1974, Chabal 1989 e 1992, Michaelis 1997). Questa attività si accompagna all’espressione di canali ionici ed in particolar modo di canali del sodio e di recettori. L’accumulo di canali del sodio e di recettori a livello della sede di lesione e quindi di generazione ectopica d’impulsi può essere responsabile della riduzione della soglia del potenziale d’azione e dell’origine di una attività spontanea nell’afferente primario danneggiato (Devor 1999, Julius 2001, England 1996). Si consideri inoltre che il sistema nervoso simpatico interviene a livello della lesione e a livello dei corpi cellulari (Suzuki 2000).
L’attività spontanea dei siti ectopici porta alla genesi di un dolore spontaneo con ca-ratteristiche diverse in base alle fibre coinvolte. L’interessamento delle fibre nocicettivo C e Adelta suscita un dolore di varia natura, da sordo e trafittivo, o una scarica elettrica. Le ectopie che interessano le fibre mieliniche Abeta danno una sensazione disestesica più o meno intollerabile. Nelle lesioni periferiche acute sono probabilmente coinvolte (a causa dell’esteso processo flogistico) tutte le fibre ed i nerva nervorum con dolore sia spontaneo, sia evocato. Nelle forme croniche (quando recede il processo flogistico e rimane la lesione) prevale il dolore evocato a partenza dai siti ectopici lungo le fibre mieliniche. I nerva nervorum (da cui origina un dolore nocicettivo) sono coinvolti sia nelle lesioni nervose periferiche, sia in alcune polineuropatie causando un dolore in genere sordo, profondo, accompagnato da sintomi di una sensibilizzazione dei neuroni spinali (Otto 2003). I canali del sodio sono canali ionici voltaggio-dipendenti e sono ampiamente distribuiti in tutti i neuroni. L’accumulo nel punto di lesione altera le proprietà elettriche della membrana assonale e porta alla scarica ectopica ed all’ab-bassamento della soglia di depolarizzazione (Matzner 1994).
I canali del sodio comprendono una larga sub-unità alfa e a sub-unità beta. Queste ultime giocano un ruolo di regolazione importante determinando i diversi livelli di espressione e sono in grado di alterare la cinetica di inattivazione del canale. Allo stato attuale sono noti otto tipi di canali del sodio presenti nel sistema nervoso dei mammiferi. Si suddividono in due tipi: i canali tetrodotossina sensibili e tetrodotossina resistenti. Entrambe sono presenti e giocano un ruolo importante nella fisiologia della nocicezione (Block 1998). Dopo una lesione nervosa periferica sembra prevalere l’espressione di canali del sodio tetrodotossina resistenti (Novakovic 1998).
Alcuni farmaci, che fanno parte del gruppo degli antiepilettici, di quello degli an-tidepressivi e degli anestetici locali, modulano i canali del sodio. Si ritiene che questo meccanismo d’azione sia responsabile della soppressione delle scariche ectopiche che originano lungo le vie nervose danneggiate e a livello del corrispondente ganglio della radice posteriore. Comunque i farmaci attualmente disponibili non sono selettivi per i canali del sodio tetrodotossina-resistenti e questa non specificità può esitare in un blocco di molteplici altri canali comportante il rischio di tossicità ad alte concentrazioni del farmaco. I dati a disposizione sostengono che i farmaci che agiscono sui canali del sodio sono più efficaci nel dolore neuropatico da lesione delle vie nervose periferiche (Galer 1993, Edwards 1999, Bhattacharya 2009).

56Aspetti di fisiopatologia e terapia del dolore
5) farmaci che agiscono sui canali del sodio
carbamazepina
Questa molecola, derivata dall’iminostilbene, e strutturalmente vicino agli anti-depressivi triciclici, induce l’inattivazione dei canali del sodio voltaggio dipendenti riducendo le scariche ripetute ad alta frequenza dei potenziali d’azione. L’inattivazione è voltaggio dipendente con limitazione delle scariche aumentate dopo la depolarizzazione e ridotte dopo l’iperpolarizzazione (Macdonald 1995). Questo meccanismo, che porta i canali del sodio in una condizione di inattività, ritardandone il recupero, è responsabile della riduzione delle scariche spontanee registrate nei preparati sperimentali di neuroma (Burchiel 1988). L’effetto frequenza dipendente spiega perché la carbamazepina è in grado di ridurre le scariche toniche senza modificare la conduzione normale del nervo.
La carbamazepina diminuisce inoltre la liberazione di neurotrasmettitori eccitatori (probabilmente sempre per l’azione sui canali del sodio), modula i canali del calcio L-tipo ad alta soglia, coinvolti nel meccanismo della sensibilizzazione centrale, ed infine aumenta la liberazione di serotonina e accresce la trasmissione dopaminergica (Beydoun 2003). La carbamazepina rimane il farmaco di riferimento per il trattamento delle nevralgie facciali e particolarmente per le forme essenziali delle nevralgie trigeminale e glossofaringea. Il dosaggio ottimale, solitamente compreso tra 600 e 900 mg, viene raggiunto gradualmente per evitare effetti indesiderati come la sonnolenza o capogiri. Nel trattamento prolungato l’incidenza di effetti collaterali è minima e di modesta rilevanza clinica. Lo studio di McQuay (1995) sull’impiego della carbamazepina nel trattamento del dolore neuropatico ha escluso per rilevanti carenze metodologiche bel 17 sudi su 24 presi in considerazione. Nello studio di Rull (1968) viene indicato l’impiego efficace della carbamazepina nella neuropatia diabetica, così come negli studi di Gerson (1977) e di Keczkes (1980) viene utlizzata con successo nella nevralgia erpetica. McQuay (1995) riporta valori di NNT per la carbamazepina nella neuropatia diabetica di 3.3 (2±9.4) e nella nevralgia trigeminale di 2.6 (2.2±3.3) sostenendo l’indiscussa efficacia.
oxcarbazepina
Il farmaco è un keto analogo della carbamazepina rispetto alla quale possiede una migliore tollerabilità e sicurezza, ed un migliore profilo farmacocinetico. Ciò è dovuto all’assenza del 10,11-eposside, metabolita attivo della carbamazepina, responsabile degli effetti collaterali e del rash cutaneo. La carbazepina agisce come la carbamazepina sui canali del sodio voltaggio dipendenti e inibisce la liberazione di neurotrasmettitori eccita-tori (McLean 1994). Il farmaco comporta una inibizione delle scariche ad alta frequenza nelle fibre afferenti cutanee dopo ripetuta stimolazione senza alterare la conduzione degli impulsi (Ichikawa 2001). È stata inoltre dimostrata un’azione inibitoria anche sui canali del calcio N-tipo rendendo il farmaco indicato nel meccanismo della sensibilizzazione dei neuroni centrali (Stefani 1995). La oxcarbazepina ha le stesse indicazioni della carbamazepina, dimostrandosi più efficace del placebo nel trattamento della nevralgia trigeminale (Zakrzewska 1989). La dose iniziale raccomandata nel trattamento della nevralgia trigeminale è di 600 mg/die (300 mg x 2/die) con incrementi di dosaggio a intervalli di qualche giorno di 150-300 mg (Carrazana 2003). I risultati di uno studio aperto prospettico indicano che la oxcarbazepina riduce significativamente il dolore nei pazienti con neuropatia diabetica (Carrazana 2003), nella radicolopatia dolorosa (Ward 2002), nelle sindromi regionali complesse (Carrazana 2003).
Lamotrigina
La lamotrigina è un derivato feniltriazinico strutturalmente non correlato agli altri farmaci antiepilettici. Essa stabilizza i canali del sodio e inibisce le scariche ripetute del

57 La “Terapia combinata” nel trattamento del dolore cronico
potenziale d’azione in condizioni di elevata depolarizzazione neuronale (Xie 1995). Lo stesso meccanismo può spiegare la capacità di inibire la liberazione di neurotrasmet-titori eccitatori (glutammato). Più recentemente è stata dimostrata l’azione modulante sui canali del calcio N-tipo ad alta soglia facendo sorgere la possibilità di una duplice azione modulante, periferico e centrale (Grunze 1998).
Molti studi randomizzati, verso placebo, e doppio cieco, hanno utilizzato la lamo-trigina in varie neuropatie dolorose.
Di questi studi il primo condotto su 100 pazienti ad un dosaggio giornaliero di 200 mg non riporta un risultato statisticamente superiore al placebo (McCleane 1999), mentre altri due studi eseguiti in pazienti con neuropatia da HIV (Simpson 2003) e con neuropatia diabetica (Eisemberg 2001), ne documentano l’efficacia. Nell’ultimo studio la lamotrigina viene somministrata alla dose gironaliera di 200-400 mg e non si sono registrati effetti collaterali probabilmente per la lenta titration utilizzata. La lamotrigina si è dimostrata efficace anche nel trattamento di forme refrattarie di ne-vralgia trigeminale (Zakrzewska 1997).
Altri Autori (Canavero 1996) ne sostengono l’efficacia nel dolore neuropatico centrale. L’effetto centrale della lamotrigina sembra essere correlato al blocco, diretto o indiretto, dei recettori NMDA. Si raccomanda infatti di utilizzare la lamotrigina iniziando al dosaggio di 25 mg alla sera per due settimane, aumentando settimanal-mente la dose di 25-50 mg fino a raggiungere la dose massima di 400 mg/die. Gli effetti collaterali, rash cutanei, sonnolenza, diplopia, sindrome di Stevens-Johnson, possono essere modesti o gravi.
Lidocaina
La lidocaina è un anestetico locale amidico che stabilizza la membrana neuronale modulando i canali del sodio voltaggio-dipendenti. È possibile ottenere un blocco completo dell’attività ectopica con dosaggi del farmaco 2-3 volti inferiori a quelli necessari per il blocco della conduzione nervosa in fibre normali. La possibilità di un trattamento efficace con bassi dosaggi di farmaco garantisce un ridotto rischio di effetti collaterali sistemici (Rowbotham 2000).
La lidocaina è somministrata per via endovenosa ad un dosaggio massimo di 5 mg/kg di peso del paziente (2,5-5 mg/kg), ma spesso anche a dosaggi inferiori, in bolo o in infusione di breve durata (30-60 min). L’effetto inizia rapidamente e si manifesta in genere per 1-2 ore. Al dosaggio indicato l’analgesia non si associa a blocco della conduzione delle fibre nervose sane e pertanto non si osserva alcuna area di anestesia locale (Devor 1992). Durante il trattamento possono insorgere effetti collaterali tra cui cefalea, fotofobia, alterazioni del ritmo cardiaco, tremori e disturbi gastroenterici (Attal 2000).
antidepressivi triciclici
Uno dei meccanismi antalgici degli antidepressivi è la loro capacità di bloccare i canali del sodio. Numerosi studi compiuti in animali hanno dimostrato che l’amitriptilina ha un potente effetto anestetico di lunga durata sia in somministrazioni periferiche lungo il decorso dello sciatico, sia per somministrazione intratecale (Sudoh 2003). Si è inoltre visto che tale effetto è simile a quello della bupivacaina ma con una maggior durata (Chen 2004).
oppioidi
Alcuni oppioidi agiscono anche sui canali del sodio. Tra questi al primo posto vi è la meperidina seguita dalla buprenorfina, che si comporta come anestetico locale sui canali del sodio voltaggio dipendenti, dal fentanyl e dal tramadolo (Smith 2012).

58Aspetti di fisiopatologia e terapia del dolore
6) farmaci ad azione sul primo neurone agendo sui fattori di flogosi e sul trofismo tissutale
Accanto ai farmaci che agiscono sui canali del sodio possiamo aggiungere:1. Farmaci che agiscono sui fattori della flogosi intorno al nervo leso (i corticosteroidi)2. Farmaci che agiscono direttamente sul trofismo tissutale (l’acido alfa lipoico)
I corticosteroidi
È noto che dopo una lesione nervosa periferica si sviluppano nella sede di lesione mediatori della flogosi e citochine pro-infiammatorie che contribuiscono alla ipersen-sibilità delle fibre a livello dei siti ectopici. La somministrazione di metilprednisolone a livello della lesione sembra interferire con questo meccanismo (Eker 2012).
L’acido alfa Lipoico
Dagli studi compiuti nei pazienti diabetici con polineuropatie dolorose agli arti si è constatata l’efficacia dell’acido alfa lipoico (Ziegler 2008, Ruessman 2009) e l’uti-lità della sua prescrizione nel controllo dei sintomi neuropatici di tali pazienti (Tang 2007). L’efficacia, nonché la sua sicurezza, è stata ribadita in altri studi controllati, randomizzati e a doppio cieco (Ziegler 1995 e 1997, Rejanovic 1999, Ruhnau 1999, Ametov 2003). Da questi studi emerge anche un dato interessante che è rappresentato dal miglioramento significativo dei segni negativi connessi alla polineuropatia. Si è infatti osservato un miglioramento della sensibilità allo stimolo puntiforme e alla pres-sione, ma anche dei riflessi muscolo-tendinei. In questi studi inoltre si è registrato un miglioramento già durante la seconda settimana di trattamento. L’ipotesi riguardante il meccanismo d’azione faceva riferimento ad una azione sulla microcircolazione e quindi sulla perfusione per effetto antiossidante posseduto dalla sostanza (Haak 2000). In altre parole il meccanismo con cui l’acido alfa lipoico migliora i sintomi neuropatici può essere riferito al miglioramento del flusso ematico mediato dall’azione antiossidante. L’acido alfa lipoico inoltre riduce nel plasma i livelli di interleukina 6 e dell’attiva-tore 1 plasminogeno (PAI-1) e quindi possiede un meccanismo antinfiammatorio e antitrombotico (Han 2012). In uno studio compiuto su pazienti con polineuropatia diabetica e dolore in cui si è valutato il passaggio da acido alfa lipoico a gabapentina si è costatata una efficacia antalgica del primo farmaco a fronte di un insuccesso del secondo e soprattutto la necessità, per il mantenimento dell’analgesia, di continuare il trattamento per lungo tempo (Ruessman 2009).
Figura 4. I farmaci che agiscono sulla ipersensibilità di fibra e sui siti ectopici.
nocIcettore tIssutaLe
IpersensIBILItà dI fIBra e svILuppo dI sItI ectopIcIcarbamazepina e oxcarbazepina
Lidocainaantidepressivi triciclici, oppioidi
I neurone sensItIvo
sIsteMa GLIaLesIsteMa
InIBItorIo
II neurone
WDRN

59 La “Terapia combinata” nel trattamento del dolore cronico
Nella recente review (Han 2012) riguardante specificatamente l’acido alfa lipoico nel trattamento della neuropatia diabetica si sottolinea l’evidenza che il trattamento con il farmaco alla dose di 300-600 mg/die per 2-4 settimane è in grado di migliorare significativamente la conduzione nervosa e i sintomi neuropatici dei pazienti.
Di Geronimo (2009) consiglia la somministrazione di acido alfa lipoico (in associa-zione all’acido gamma linolenico) nel trattamento della sindrome del tunnel carpale e Ranieri (2010) nel trattamento del dolore nelle lesioni traumatiche nervose. Per quanto riguarda altre indicazioni dell’acido alfa lipoico nel trattamento del dolore neuropatico va sottolineato il lavoro di Zakrzewska (2005) che riporta l’utilità nei pazienti con la sindrome della bocca che brucia recentemente attribuita ad una patologia delle piccole fibre. In un recente lavoro (Gurvits 2013) l’acido alfa lipoico viene indicato come parte di una associazione di farmaci che comprende il clonazepam, gli antidepressivi e la capsaicina nel trattamento della bocca che brucia.
Recenti studi hanno messo in evidenza le proprietà neurotrofiche di un polifenolo estratto dalla corteccia di magnolia, l’Honokiolo, che esplica la sua azione neurotrofica inducendo la biosintesi di componenti della membrana cellulare, con un meccanismo che è stato definito simile a quello del Nerve-Growth Factor (NGF).
L’Honokiolo è un polifenolo estratto dalla corteccia di magnolia. In letteratura sono state documentate numerose azioni farmacologiche quali quella antinfiammatoria e quella antiossidante. Honokiolo ha mostrato attività neurotrofica in colture di neuroni corticali di ratto. L’Honokiolo ha dimostrato di avere anche un effetto neuroprotettivo mediato dall’azione di inibizione sulla apoptosi neuronale. Honokiolo attiva la Fosfolipasi C, che libera dalla membrana cellulare inositolo trifosfato (IP3). Il conseguente aumento di Ca2+ intracellulare attiva a cascata una serie di fattori di trascrizione (CAM – MEK – ERK) che, una volta entrati nel nucleo, attivano la trascrizione di geni che codificano per proteine che inducono proliferazione cellulare e dunque con azione neurotrofica. In estrema sintesi, Honokiolo induce la biosintesi di componenti della membrana cellulare, con un meccanismo che è stato definito simile a quello del Nerve-Growth Factor (NGF). Honokiolo ha dimostrato di possedere inoltre una attività antinfiammatoria, in quanto inibisce il fattore di trascrizione nucleare NF-kB, responsabile dell’attivazione della trascrizione di importanti citochine infiammatorie come IL-1, IL-6 e TNF-α. L’effetto di Honokiolo sui mediatori dell’infiammazione è correlato anche alla sua capacità di legarsi con i recettori dell’acido γ-amino butirrico (GABA) favorendone l’attivazione. I recettori periferici GABA sono presenti nelle cellule del sistema immunitario ed inibi-
Figura 5. I farmaci che agiscono sui fattori tissutali e sulla rigenerazione del nervo.
nocIcettore tIssutaLe
danno deL prIMo neuronecorticosteroidi
acido alfa Lipoico
I neurone sensItIvo
sIsteMa GLIaLesIsteMa
InIBItorIo
II neurone
WDRN

60Aspetti di fisiopatologia e terapia del dolore
scono l’attivazione linfocitaria nelle malattie infiammatorie. L’azione antinfiammatoria dell’Honokiolo è stata osservata anche in modelli sperimentali di artrite reumatoide; in questi studi Honokiolo ha dimostrato di proteggere le cartilagini articolari e di ridurre i livelli di citokine pro-infiammatorie e metalloproteasi sia a livello ematico che a livello articolare. Da quanto fin qui esposto si può concludere che, in una strategia terapeutica basata sulla associazione di più farmaci basata sui meccanismi patogenetici, anche far-maci che non hanno un chiaro riconoscimento nella categoria degli “analgesici” possono trovare una precisa collocazione. Il loro impiego, sicuramente protratto nel tempo, può avere anche il significato di favorire una progressiva riduzione dei farmaci analgesici per il miglioramento del danno neurologico.
La terapia combinata: dalla diagnosi alla scelta dei farmaciAlla base di una corretta scelta terapeutica si deve porre una approfondita valu-
tazione diagnostica clinica e strumentale che identifichi le co-morbilità mediche e psicologiche ma soprattutto i meccanismi patogenetici del dolore. Nella pratica clinica quotidiana la maggior parte dei quadri clinici di dolore mostrano la presenza di più meccanismi e pertanto la terapia dovrebbe essere “combinata”. Nello stesso tempo è importante conoscere l’azione di un farmaco nei confronti di uno o più meccanismi, gli effetti collaterali e le interazioni con gli altri farmaci eventualmente scelti nella terapia combinata. Le associazioni di farmaci possono seguire vari scopi:1) Efficacia terapeutica in quanto:a. Si agisce su tutti i meccanismi patogenetici coinvolti (corticosteroidi e oppioidi ad
esempio)b. Si sfrutta l’eventuale potenziamento reciproco (paracetamolo e tramadolo ad
esempio)c. Si unisce un’azione sui meccanismi patogenetici del dolore ed una sugli elementi
che influiscono sul danno neurologico (acido alfa lipoico)2) Protezione sugli effetti collaterali:a. Si ottiene una riduzione del dosaggio e indirettamente una riduzione degli effetti
collateralib. Si riduce l’incidenza e l’entità degli effetti collaterali (antiemetici, gastroprotettori,
antistipsi). Pertanto è possibile consigliare l’associazione di “farmaci ad azione sul nocicettore”
insieme a “farmaci che agiscono a livello spinale” che in pratica si traduce nella pre-scrizione di Antinfiammatori e di paracetamolo o oppioidi, a cui si possono aggiungere farmaci che riducono l’eccitabilità spinale come i gabapentinoidi o gli antidepressivi triciclici. È di fondamentale importanza non associare farmaci che hanno lo stesso meccanismo d’azione e che interagiscono tra loro a vari livelli. Già nel 2002 Curatolo sosteneva che alcune combinazioni si erano dimostrate utili. Nel trattamento del dolore postoperatorio, ad esempio, l’associazione di farmaci antinfiammatori non steroidei o di paracetamolo alla somministrazione endovenosa di morfina si rivelava efficace. Così anche l’associazione di farmaci antinfiammatori al paracetamolo dava un risultato antalgico migliore dei singoli farmaci separati. Ma anche nel dolore da cancro l’asso-ciazione di antinfiammatori agli oppioidi comporta un maggior successo terapeutico. Lo stesso Autore conclude lo studio proponendo un metodo matematico per individuare le possibili combinazioni efficaci in studi clinici controllati e randomizzati. Un altro modello è quello fin qui esposto e si basa sulla diagnosi dei meccanismi patogenetici coinvolti e la conoscenza del meccanismo d’azione dei farmaci. Chen (2011) sottoli-nea l’importanza della terapia combinata nel trattamento del dolore neuropatico. Egli riporta i diversi meccanismi patogenetici che intervengono nella genesi del dolore e sostiene l’importanza di una terapia mirata con diversi farmaci. Tra questi annovera

61 La “Terapia combinata” nel trattamento del dolore cronico
l’associazione di gabapentina e nortriptilina, di gabapentina e oxicodone, di prega-balin e oxicodone e di pregabalin e di COX-2, ma anche la possibilità di associare a terapia farmacologiche altre modalità terapeutiche come le terapie comportamentali e riabilitative e altre ancora. Nello stesso tempo rimane da stabilire se sia meglio iniziare subito con una terapia combinata con i farmaci ritenuti utili sulla base dei meccanismi patogenetici coinvolti o procedere con un farmaco per volta, aggiungendo successiva-mente (add-on) gli altri farmaci indicati. Solo gli studi controllati ci potranno fornire una risposta anche se nella pratica clinica quotidiana possiamo sostenere che la scelta sarà anche dettata da molti fattori quali l’età, i trattamenti già in corso e le co-morbilità presenti, la complessità del quadro clinico (Chaparro 2012). Si consiglia di iniziare con dosi ridotte e di aumentare le dosi lentamente, soprattutto nel paziente anziano. La lenta titration permette di svilupparsi una tolleranza agli effetti collaterali. È importante la valutazione continua del risultato, il monitoraggio degli eventuali effetti collaterali e la valutazione delle attività della vita quotidiana. Il farmaco inefficace va rapidamente sostituito. Partendo dalla classificazione presentata nel capitolo introduttivo i termini “nocicettivo”, “neuropatico” e “malattia” ci permettono di identificare tre gruppi di pazienti caratterizzati dalla presenza di meccanismi patogenetici a loro volta respon-sabili di segni e sintomi che l’esame clinico e strumentale dovrebbero identificare. Per ciascuno di questi gruppi è possibile definire un preciso piano diagnostico-terapeutico, farmacologico e invasivo, che tenga conto delle variabili cliniche possibili e delle comorbilità presenti. La tassonomia tradizionale perde quindi il ruolo che ha sempre avuto di indicare il trattamento ideale. Ogni sindrome deve essere rivista alla luce di un nuovo approccio al dolore e a chi lo prova.
BIBLIoGrafIa
Abadir AR, Kraynack BJ, Maida J 2nd, Gintautas J. Postherpetic neuralgia: response to topical clonidine. Proc. West Pharmacol Soc. 39: 47-48, 1996.
Abram SE. Neural blockade for neuropathic pain. The Clin J of Pain 2000,16: S56-S61.
Alhaider AA et al. ” Spinal 5HT3 receptor mediated antinocicep-tion: possible release of GABA.” The J of Neuroscience 1991, 11C7: 11881-88.
Ametov A, Barinov A, Dyck PJ et al. and the SYDNEY Trial Study Group. The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care 2003; 26: 770-6.
Anand P. and Bley K. Topical capsaicin for pain management: therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8% patch. British Journal of Anaesthesia 2011; 17: 1-13.
Argoff CE, Katz N, Backonja M: Treatment of postherpetic nevral-gia: a review of therapeutic options. J Pain Symtom Manage 2004; 28: 396-411.
Attal N, Cruccu G, Baron R, Haanpa M, Hansson P, Jensen TS and Nurmikko T. EFNS guidelines on the pharmacological treat-ment of neuropathic pain: 2010 revision. European Journal of Neurology 2010, 17: 1113-1123.
Attal N, Gaude V, Brasseur L, Dupuy M, Guirim, F, Parker F, Bouhassira D: Intravenous lidocaine in central pain: a double-blind, placebo-controlled, psychophysical study. Neurology 54: 564-574, 2000.
Attal N. Chronic neuropathic pain: mechanisms and treatment. The Clin J of Pain 2000, 16: S118-S130.
Backonja M, et al. “Gabapentin in the symptomatic treatment of painful neuropathy in patients with diabetes mellitus; a rand-omized controlled trial” JAMA 1998.
Backonja MM, Irving G, and Argoff Ch. Rational Multidrug Therapy in the Treatment of Neuropathic Pain Current Pain and Headache Reports 2006, 10:34-38.
Bajwa Z, et al. Topiramate relevies refractory intercostal nevralgia. Neurology 1999; 52: 1917.
Bartusch SL, Sanders BJ, D’Alessio JC, Jernigan JR. Clonazepam for the treatment of lancinating phantom limb pain. Clin J Pain 12 (1): 59-62, 1996 Mar.
Basbaum JD, AJ: Molecular mechanisms of nociception. Nature 2001; 413: 203-210.
Bennet G, Deer T, Du Pen S, Rauck R, Yaksh T, Hassenbusch SJ. Future directions in the management of pain by intraspinal drug delivery. J Pain Symptom Manage 2000, 20: S44-50.
Bennett MI, Simpson KH. “Gabapentin in the treatment of neuro-pathic pain”. Palliat Med. 2004 Jan; 18 (1): 5-11.
Benzon H, Teng-Leong C, McCarthy R, Benzon H. Comparison of the particulate sizes of different steroids and effect of dilution: a review of the relative neurotoxicities of the steroids. Anesthesiol-ogy 2007; 106: 331-8.
Bernat J, Sandowsky C, Vincent F, Nordgren R, Margolis G. Sclerosing spinal pachymeningitis: a complication of intrathe-cal administration of Depo-Medrol_R for multiple sclerosis. J Neurol Neurosurg Psychiatry. 1976; 39: 1124-8.
Bertolotto F, Massone A. Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Dugs RD 2012; 12:1-6.
Beydoun A, Kobets SA, Carrazana EJ: Efficacy of Oxcarbazepine in the treatment of painful diabetic neuropathy. Clin J Pain 2004; 20: 174-178.
Bhattacharya A, Wickenden AD, Chaplan SR: Sodium Channel Blockers for the Treatment of Neuropathic Pain- Vol. 6, 663-678, October 2009 © The American Society for Experimental NeuroTherapeutics, Inc.
Biewenga GP, Haene GR, Bast A. The pharmacology of the antioxi-dant lipoic acid. Gen Pharmacol 1997; 29:315-31
Block JA, McLachlan EM, Belmonte C: Tetrodotoxin-resistent impulses in single nociceptor nerve terminals in guinea-pig cornea. J. Physiol 1998; 512: 211-217.
Bowsher D.: The effects of pre-emptive treatment of postherpetic nevralgia with amitriptyline: a randomized, double-blind, place-bo-controlled trial. J Pain Symtom Manage 1997; 13: 327-331.
Burchiel KJ: Carbamazepine inhibits spontaneous activity in experi-mental neuromas. Exp Neurol 1988; 102: 249-253.
Byas-Smith MG, Max MB, Muir J, Kingman A. Transdermal clonidine compared to placebo in painful diabetic neuropathy using a two-stage “enriched enrollment” design. Pain 60 (3): 267-274, 1995 Mar.

62Aspetti di fisiopatologia e terapia del dolore
Caraceni A. et al. “Gabapentin as an adjuvant to opioid analgesia for neuropathic cancer pain” J Pain Symptom Manag. 1999.
Carrazana E, Mikoshiba J: Rationale and evidente for the use of oxcarbazepine in neuropathic pain. J Pain Symptom Manage 2003; 25: 531-535.
Chabal C, Jacobson L, Russel LC, Burchiel KJ: “Pain responses to perineuromal injection of normal saline, gallamine end lidocaine in humans”. Pain 1989: 36; 321.
Chabal C, Jacobson L, Russel LC, Burchiel KJ:”Pain response to perineuromal injection of normal saline, epinephrine, lidocaine in humans” Pain 1992: 49; 9-12.
Chang M, Ger L, Hsieh P, Huang S. A randomised clinical trial of oral steroids in the treatment of carpal tunnel syndrome: a long term follow up. J Neurol Neurosurg Psychiatry. 2002; 73: 710-4.
Chaparro LE, Wiffen PJ, Moore RA, Gilron I: Combination phar-macotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev. 2012 Jul 11; 7.
Chen Y-W, Huang K-L, et al. Intrathecal tri-cyclic antidepressant produce spinal anestesia. Pain 2004; 112: 106-112
Cho YS, Lee J, Lee TH, Lee EY, Lee KU, Park JY, Moon HB. α-Lipoic acid inhibits airway infl ammation and hyperre-sponsiveness
Clifford J. Woolf: Dissecting out mechanisms responsible for peripheral neuropathic pain: Implications for diagnosis and therapy- Life Sciences 74 (2004) 2605-2610.
Curatolo M: Drug combinations in pain treatment: a review of the published evidence and method for finding the optimal com-bination. Best Practice & Researche Clinical Anaesthesiology 2002; 4: 507-519.
Cuzzocrea S et al. Antioxidant Therapy: A New Pharmacological Approach in Shock, Infl ammation, and Ischemia/Reperfusion Injury. Pharmacol Rev 2001; 53:135-59.
Dahl JB, Mathiesen O, Moniche S. Protective premedication: an op-tion with gabapentin and related drugs? A review of gabapentin and pregabalin in the treatment of post-operative pain. Acta Anaesthesiol Scand 2004; 48: 1130-36.
Dellemijn P.:”Are opioids effective in releiving neuropathic pain?” Pain 8 (1999), 453-462.
Devor M, Seltzer Z: Pathophysiology of damaged nerves in relation to chronic pain. In Wall PD, Melzack R. (Eds), Textbook of Pain, Edimburg; Churchill Livingstone, 1999, pp 128-164.
Devor M, Wall P, Catalan N: Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain 1992; 48: 261-268.
Di Geronimo G, Fonzone Caccese A, Caruso I, Soldati A, Passaretti U. Treatment of carpal tunnel syndrome with alpha-lipoic acid. European Review for Medical and Pharmacological Sciences 2009; 13: 133-139.
Dickenson AH, Matthews E, SuzukiR: Central nervous system mechanisms of pain in peripheral neuropathy. In Neuropathic Pain Pathophysiology and treatment. In Hanson PT, Fields HL, Hill RG, Marchettini P, Eds, Progress in pain research and management, Vol 21, IASP Press, 2001, p. 106-185.
Dickenson AH, Sozuki K: Function and dysfunction of opioid receptors in spinal cord. In Kalso E, McQuay H, Wiesenfeld-Hallin Z (Eds) Opioid Sensitività of Chronic Noncancer Pain, Progress in Pain Reasearch and Management. Vol 14, Seattle, IASP Press, 1999, pp 17-44
Dickenson AH, Suzuki R, Matthews EA et al. Balancing excitation and inhibitions in spinal circuits. In The Pain System in Normal and Pathological States; a primer for clincians. Progress in Pain Researche and Management. Vol 31, Edited by L. Villanueva, A. Dickenson and H. Ollat, IASP Press, Seattle, 2004, p. 79-105.
Dickenson AH, Suzuki R: Opioids in neuropathic pain: clues from animal studies. European J Pain 2005; 9: 113-116.
Dworkin RH, Backonja M, Rowbotham MC, Allen RR, Argoff CR, Bennett GJ, Bushnell MC, Farrar JT, Galer BS, Haythornthwaite JA, Hewitt DJ, Loeser JD, Max MB, Saltarelli M, Schmader KE, Stein C, Thompson D, Turk DC, Wallace MS, Watkins LR, Weinstein SM: Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Arch Neurol 60: 1524-1534, 2003.
Dworkin RH, et al “Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations”. Arch Neurol. 2003 Nov; 60 (11): 1520.
Dworkin RH, Perkins RM, Nagasake EM: Prospects for the preven-tion of postherpetic nevralgia in herpes zoster patients. Clin J Pain 2000; 16: S90-S100.
Ebersberger A, et al. Differential role of spinal prostaglandin receprots subtype EP1-EP4 in rats with normal and inflamed knee joints. Progress in Pain Research and Management 2, IASP Press 2002.
Edwards AD. The role of systemic lidocaine in neuropathic pain management. J Intraven Nurs 1999; 22: 273-279.
Edwards K, Glantz M, Button J et al. Efficacy and safety of topiramate in the treatment of painful diabetic neuropathy. Neurologo 2000; 54: 81.
Eisemberg E, Lurie Y, Braker et al. Lamotrigine reduces painful diabetic neuropathy: a randomized, controlle study. Neurology 2001; 57: 505-509.
Eker HE, Cok OY, Aribogan A, and Arslan G. Management of Neuropathic Pain with Methylprednisolone at the Site of Nerve Injury Pain Medicine 2012; 13: 443-452.
England JD, Happel LT, Kline DG, et al. Sodium channel accu-mulation in humans with painful neuromas. Neurology 1996; 47: 272-276.
Ertel EA, Campbell KP, Harpold MM, et al. Nomenclature of voltage-gated calcium channels. Neurol 2000; 25: 253-280.
Field MJ, McCleary S, Hughes S, Singh L: Gabapentin and pre-gabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by sterptozocin in the rat. Pain 1999; 80: 391-398.
Freynhagen R, Strojek K, Griesing T, et al. Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo-controlled trial of flexibleand fixed-dose regimens. Pain 2005; 115 (3): 254-63.
Galer BS, Miller KV, Rowbotham MC. Response to intravenous lidocaine infusion differs based on clinical diagnosis and site of nervous system injury. Neurologo 1993; 43: 1233-1235.
Galer BS, Rowbotham MC, Perander J,Friedman E: Topical lido-caine patch relieves postherpetic neuralgia more effectively than a vehicle topical path: results of an enriched enrollment study. Pain 80:533-538, 1999.
Galer BS. Neuropathic pain of peripheral origin: Advances in pharmacologic treatment. Neurology 45 (suppl 9): 17-25, 1995.
Gerson G, Jones R, Luscombe O: Studies on concomitant use of carbamazepine and clomipramine for relief of posthereptic nevralgia. Postgrad Med J 1977; 53: 104-109.
Gilron I, Milne B, Hong M. Cyclooxygenase-2 inhibitors in postop-erative pain management; current evidence and future directions. Anesthesiology 2003;99:1198-2008.
Goldstein D, Lu Y, Detke MJ, Lee TC, Iyengar S: Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain 116 (2005) 109-118.
Gomez-Perez FJ, Rull JA, Dies H, et al. Nortriptyline and fluphen-azine in the symptomatic treatment of diabetic neuropathy: a double-blind cross-over study. Pain 23: 395-400, 1985.
Group. Treatment of symptomatic diabetic polyneuropathy with the antioxidant α-lipoic acid. A 7-month multicenter randomized controlled trial (ALADIN III Study). Diabetes Care 1999; 22:1296-301.
Grunze H, von Wegerer J, Green RW, et al. Modulation of calcium and potassium currents by lamotrigine. Neuropsychobiology 1995; 38: 131-138.
Gurvits GE, Tan A: Burning mouth syndrome. World J Gastroen-terol. 2013 Feb 7;19(5):665-72.
Haak E, Usadel KH, Kusterer K, Amini P, Frommeyer R, Tritschler HJ et al. Effects of alpha-lipoic acid on microcirculation in pa-tients with peripheral diabetic neuropathy. Exp Clin Endocrinol Diabetes 2000; 108: 168–174.9: 123-125
Han T, Bai J, Wei Liu and Hu Y: A systematic review and meta-analysis of a-lipoic acid in the treatment of diabetic peripheral neuropathy. European Journal of Endocrinology (2012); 167: 465-471.
Harati Y, Gooch L, Swenson M, et al. Double blind randomized trial of tramadol for the treatment of diabetic neuropathy. Neurologo 1998; 50: 1842-1846.
Hassenbusch, Portenoy RK. Current practices in intraspinal therapy – a survey of clinical trends and decision making. J of Pain and Symptom Manage 2000, 20: S4-S11.
Herman RM, D’Luzansky SC, Ippolito R. Intrathecal baclofen suppresses central pain in patients with spinal lesions. A pilot study. Clin J Pain 8: 338-345, 1992.
Hounsom L, Horrobin DF, Tritschler H, Corder R, Tomlinson DR. A lipoic acid-gamma linolenic acid conjugate is effective against multiple indices of experimental diabetic neuropathy. Diabetologia 1998; 41: 839-43

63 La “Terapia combinata” nel trattamento del dolore cronico
Ichikava K, Koyama N, Kiguchi S, et al: Inhibitory effect of oxcar-bazepine on high-frequency firing in peripheral nerve fibers. Eur J Pharmacol 2001; 420: 119-122.
Irving GA, Backonja M, Rauck R, Lynn R. Webster LR et al.NGX-4010, a Capsaicin 8% Dermal Patch, Administered Alone or in Combination With Systemic Neuropathic Pain Medications, Reduces Pain in Patients With Postherpetic Neu-ralgia Clin J Pain 2011.
Jacox A, Carr DB, Payne R, et al. Management of cancer pain. Clini-cal Practice Guideline No. 9. AHCPR Publication No. 94-0592. Rockville, MD: Agency for Health Care Policy and Research, U.S. Department of Health and Human Services, Public Health Service; 1994, p. 257.
Jamal GA, Carmichael H. The Effect of Gamma-Linolenic Acid on Human Diabetic Peripheral Neuropathy: a Double-Blind Placebo-Controlled Trial. Diabet Med 1990; 7:319-23.
Jameel NM, Shekhar MA, Vishwanath BS. α-Lipoic acid: an inhibi-tor of secretory phospholipase A2 with antiinfl ammatoryactivity. Life Science 2006; 80:146-53.
Kalso E, Allan L, Dellermijn PL, Faura CC, et al. Recomanda-tion for using opioids in chronic non-cancer pain. Eur J Pain 2003; 7: 381-386.
Kanazi GE, Johnson RW, Dworkin RH: Treatment of postherpetic nevralgia: an update. Drugs 2000; 59: 1113-1126.
Kaneko S, Fukuda K, Yada N, et al. Ca++ channel inhibition by kappa opioid receptors ecpressed in Xenopus oocytes. Neurore-port 1994; 5: 2506-2558.
Keczkes K, Basheer A: Do corticostroids prevent postherpetic nevralgia? Brit J Derm 1980; 102: 551-555.
Kim HY, Chung JM et al. Increased production of mitochondrial superoxide in the spinal cord induces pain behaviours in mice: the effect of mitochondrial electron transport complex inhibitors. Neurosciences Letters 2008; 447:87-91.
Kotani N, Kushikata T et al. Intratecal methylprednisolone for intractable postherpetic neuralgia. NEJM 2000; 343: 1514-1519.
Kupers E, Gybels J: The consumption of fentanyl is increased in rats with nociceptive but not with neuropathic pain: a patient-controlled analgesia outcome. Anesthesia 1992; 47: 757-767.
Macdonald RL, Kelly KM: Antiepileptic drug mechanisms of action. Epilepsia 1995; 36(suppl 2): S2-S12.
Maier CM, Chan PH. Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist 2002; 8(4):323-34.
Mannion RJ, Woolf CJ: Pain mechanisms and management: a central perspective. The Clinical J of Pain 2000; 16: S144-S156.
Matzner O, Devor M: Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J. Neurophysiol 1994; 72: 349-359.
Max MB, Cullane M, Shaker SC et al. Amitriptyline relieves diabetic neuropathy pain in patient with normal or depressed mood. Neurology 1987; 37: 589-596.
Max MB, Lynch SA, Mair J, et al. Effects of disipramine, amitrip-tyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med 326: 1250-1256, 1992.
Max MG: Is Mechanism-Based Pain Treatment Attainable? Clini-cal Trial Issues. The Journal of Pain, Vol 1, No. 3 (Fall), Suppl 1, 2000: pp 2-9.
McCleane G: 200 mg daily of lamotrigine has no analgesic effect in neuropathic pain: a randomized, double.blind, placebo controlled trial. Pain 1999; 83: 105-107.
McLean MJ, Scmutz M, Wamil AW: Oxcarbazepine mechanisms of action. Epilepsia 1994; 35(suppl 3): 55-59.
McQuay H, Carrol D, Jadad A: Anticonvulsant drugs for manage-ment of pain. BMJ 1995; 311: 1047-1052.
McQuay HJ, Tramer M, Nye BA, et al. A systematic review of antidepressant in neuropathic pain. Pain 1996; 68: 217-227.
Memeo A, Loriero M. Acido tioctico e acetil-L-carnitina nel trat-tamento del dolore sciatico causato da ernia del disco. Clin Drug Invest 2008; 28:495-500.
Mendell JR, Sahenk Z: Painful sensory neuropathy. N Engl J of Med 2003; 348: 3243-53.
Michaelis M, Vogel C, Blenk KH, Janig W: Algesic excite axot-omised afferent nerve fibres within the first hours following nerve transection in rats. Pain 1997; 72: 347-354.
Milesi MA. Effect of an oral supplementation with a proprietary melon juice concentrate (Extramel®) on stress and fatigue in healthy people: a pilot, double-blind, placebo-controlled clinical trial. Nutrition Journal 2009; 8:40.
Morrison R, Carney M, Manfredi P. Pain management. In: Rosenthal RA, Zenilman ME, Katlic MR, editors, Chapter 12 in Principles and practice of geriatric surgery. New York, NY: Springer; 2001:160-171.
Naik AK, Tandan SK, Dudhgaonkar SP, Jadhav SH, Kataria M, Prakash VR, Kumar D. Role of oxidative stress in pathophysi-ology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur J Pain 2006; 10:573-9.
Nakajima S et al. Oral supplementation with melon superoxide dismutase extract promotes antioxidant defences in the brain and prevents stress-induced impairment of spatial memory. Behavioural Brain Research 2009; 200:15-21
Novakovic SD, Tzoumaka E, McGivern JG et al. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurone in normal and neuropathic conditions. J Neurosi 1998; 18: 2174-2187.
O’Connor AB, Dworkin EH. Treatment of Neuropathic Pain: An Overview of Recent Guidelines. The American Journal of Medicine 2009: 122; S22-S32.
Omoigui S. The biochemical origin of pain – Proposing a new law of pain: The origin of all pain is inflammation and the inflam-matory response. Part 1 of 3 – A unifying law of pain Medical Hypotheses (2007) 69, 70-82.
Otto M, Back S, Bach FW, Jensen TS, Sindrup SH: Pain phenomena and possible mechanisms in patients with painful polyneuropa-thy. Pain 2003;101: 187-192.
Pardo ES, Berger JM, Toliver KT. Post herpetic neuralgia. Semin Anesth. 1997; 16 (2): 132-135.
Radtke C, Vogt PM, Devor M, Kocsis JD. Keratinocytes acting on injured afferents induce extreme neuronal hyperexcitability and chronic pain. Pain. 2010; 148 (1): 94-102.
Rahme E, Barkam A, Nedjar H, et al. Hospitalizations for upper and lower GI events associated with traditional NSAIDs and acetaminophen among elderly in Quebec, Canada. Am J Gas-troenterol. 2008; 103 (4): 872-82.
Raja SN, Haythornthwaite Ja, Pappagallo M et al: Opioids versus antide-pressant in posthereptic nevralgia. Neurology 2002; 59: 1015-1021.
Ranieri M, Sciuscio M, Cortese A, Stasi M, Panza F, Megna M, Fiore P, Santamato A. Possible role of alpha-lipoic acid in the treatment of peripheral nerve injuries. J Brachial Plex Peripher Nerve Inj. 2010 Aug 31; 5:15.
Ranieri M, Sciuscio M, Cortese AM, Santamato A, Di Teo L, Ianieri G, Bellomo RG, Stasi M, Megna M The use of alphalipoic acid (ALA), gamma linolenic acid (GLA) and rehabilitation in the treatment of back pain: effect on health-related quality of life. Int J Immunopathol Pharmacol 2009; 22(S3):45-50.
Reljanovic M, Reichel G, Rett K et al. Treatment of diabetic pe-ripheral neuropathy with the antioxidant thioctic acid (a-lipoic acid). A two-year multicenter randomized double blind placebo-controlled trial (ALADIN II). Free Radic Res 1999; 31: 171-9.
Rowbotham M, et al.”Gabapentin for the treatment of post-herpetic neuralgia: a randomized controlled trial” JAMA 1998.
Rowbotham M, Petersen K, Davies P et al. recent developments in the treatment of neuropathic pain. In Proc. 9th WCP, Vol 16 (ed.: Devor et al) Seattle: IASP, p. 831-855, 2000.
Rowbotham MC, Davies PJ, Verkempinck CM, Galer BS. Lidocaine patch: double-blind controlled study of a new treatment method for portherpetic neuralgia. Pain 65: 39-45, 1996.
Rowbotham MC, Davies PS, Fields HL. Topical lidocaine gel relieves postherpetic neuralgia. Ann Neurol 37: 246-253, 1995.
Rowbotham MC, Reisner KL, Fields HL: Both intravenous lidocaine and morphine reduce the pain of postherpetic nevralgia. Neurol-ogy 1991; 41: 1024-1028.
Ruessmann HJ Switching from pathogenetic treatment with alpha-lipoic acid to gabapentin and other analgesics in painful diabetic neuropathy: a real-world study in outpatients. J Diabetes Complications. 2009 May-Jun; 23 (3): 174-7.
Ruhnau KJ, Meissner HP, Finn JR et al. Effect of a 3-week oral treat-ment with the antioxidant thioctic acid (a-lipoic acid) in symp-tomatic diabetic polyneuropathy. Diabet Med 1999; 16: 1040-3.
Rukwied R, Mayer A, Kluschina O, Obreja O, Schley M, Schmelz M. NGF induces non-inflammatory localized and lasting mechanical and thermal hypersensitivity in human skin. Pain. 2010; 148(3):407–413.
Rull J, Quilibreva R, et al: Symptomatic treatment of peripheral neuropathy with CDZ. Diabetologia 1969; 5: 215-218.
Samad T, Moore K, Sapirstein A, et al. Interleukin-1[beta]-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001; 410 (6827): 471-75.

64Aspetti di fisiopatologia e terapia del dolore
Sandrini M, Pini LA, Vitale G: Differential involvement of central 5-HT1B and 5-HT3 receptor subtypes in the antinociceptive effect of paracetamol. Inflamm. res 2003; 52: 347-352.
Schaible HG,Schmelz M, Tegeder I:Pathophysiology and treatment of pain in joint disease. Advanced Drug Delivery Reviews 58 (2006) 323-342.
Schawab JM, Schluesener HJ, Meyermann R, Serhan CN: COX-3 the anzyme and the concept: steps towards highly specialized pathways and precision therapeutics? Prostaglandins Leukot Essent Fatty Acids 2003; 68: 339-43.
Schneiderman JH: Topiramate: pharmacokinesics and pharmaco-dynamics. Can J Neurol Sci 1998; 25: 53-55.
Schwartz ES, Kim HY et al. Persistent pain is dependent on spinal mitochondrial antioxidant levels. J Neurosci 2009 Jan; 29(1):159-68.
Siddal PJ, Molloy AR, Walker S, Mather LE, Rutkowski SB, Cousins MJ. The efficacy of intrathecal morphine and clonidine in the treatment of pain after spinal cord injury. Anesth Analg 2000, 91: 1493-1498.
Simpson DM, McArthur JC, Olney R, et al. Lamotrigine for HIV-associated painful sensory neuropathies: a placebo-controlled trial. Neurologo 2003; 60: 1284-1289.
Smith HS: Opioids and Neuropathic Pain. Pain Physician 2012; 15:ES93-ES110.
Stefani A, Pisani A, De Murtas M, et al: Action of GP 47779, the active metabolite of oxcarbazepine on the corticostriatal system II. Modulation of high-voltage-activated calciom currents. Epilepsia 1995; 36: 997-1002.
Stein A, Yassouridis A, Szopka C, Helmke K, Stein C. Intraarticular morphine versus dexamethasone in chronic arthritis. Pain 1999, 83: 525-532.
Sudoh Y, Cahoon EE, Gerner P, Wang GK: “Tryciclic antidepres-sants as long-acting local anesthetics” Pain 2003: 103: 49-55.
Suzuki R, Dckenson AH: Neuropathic pain: nerve bursting with excitement. Neuroreport 2000; 11: R17-21.
Syndrup SH, Andersen S, Madsen C et al. Tramadol relieves pain and allodynia in polyneuropathy. Pain 1999; 83: 85-90.
Syndrup SH, Bach FW, Madsen C. et al. Venlafaxine versus imipra-mine in painful polyneuropathy: a randomized controlled trial. Neurology 2003; 60: 1284-1289.
Syndrup SH, Gram LF, Brosen et al. The selective serotonin reup-take inhibitor paroxetine is effective in the treatment of diabetic neuropathy symptoms. Pain 1990; 42: 135-144.
Syndrup SH, Jensen TS: Pharmacologic treatment of pain in po-lineuropathy. Neurology 2000; 55: 915-920.
Tan E et al. The Oxidative Response in the Chronic Constriction Injury Model of Neuropathic Pain. J Surg Res 2009;152:84-8.
Tang J, Wingerchuk D.M, Crum, B.A. Rubin D.I, Demaerschalk B.M. Alpha-Lipoic Acid May Improve SymptomaticDiabetic Polyneuropathy The Neurologist 2007; 13: 164-167.
Taub A. Relief of postherpetic neuralgia with psychotropic drugs. J Neurosurg 39: 235-239, 1973.
Tesfaye S, Vileikyte L, Rayman G, Sindrup SH, Perkins BA, Baconja M, Vinik AI, Boulton AJM, on behalf of The Toronto Expert Panel on Diabetic Neuropathy. Painful diabetic peripheral neu-ropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metab Res Rev 2011; 27: 629-638.
Thacker MA et al. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analog 2007; 105:838-47.
Toliver, KT, Berger JM, Pardo ES. Review of Herpes Zoster. Semin Anesth. 1997;16 (2): 127-131.
Tremont-Lukats IW, Megeff C, Bachonia M. “Anticonvulsants for neuropathic pain syndromes: mechanisms of action and place in therapy” Drug 2002.
Vetro A, Mantia F, Mantia R. terapia dei confl itti disco-radicolari. Comparazione clinica tra ossigeno-ozono terapia paravertebrale e trattamento combinato ossigeno-ozono terapia e un prodotto a base di acido α-lipoici (ALAnerv®). Minerva Ortop Traumatol 2006; 57:57-63.
Viggiano E, Monda M et al. Persistent facial pain increases superox-ide anion production in the spinal trigeminal nucleus.
Vrethem M, Boivie J, Arnquist H, Holmgren H, Lindstron T, Thorell L-H: A comparison of amithriptyline and naprotiline in the treat-ment of painful polyneuropathy in diabetics and nondiabetics. 1997; 13: 313-323.
Wagner R, Heckman HM, Myers RR. Wallerian degeneration and hyperalgesia after peripheral nerve injury are glutathione- de-pendent. Pain 1998; 77:173-9.
Wall PD, Gutnick M: Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Exp Neurol 1974; 43: 580-593.
Ward S, Royal MA, Jenson M: An open label trial of oxcarbazepine in patients with radiculopathy refractory to gabapentin. J Pain 2002; 3: 42.
Watson CP, Evans RJ, Reed K, Merskey H, Goldsmith L, Warsh J. Amitriptyline versus placebo in postherpetic neuralgia. Neurol-ogy 32: 671-673, 1982.
Watson CPN, Babul N: Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic nevralgia. Neurology 1998; 50: 1837-1841.
Watson CPN, Chipman M, Reed K, Evans RJ, Birkett N. Am-itrptyline versus maprotiline in postherpetic neuralgia: a randomized, double-blind, crossover trial. Pain 48: 29-36, 1992.
Watson CPN. The treatment of postherpetic neuralgia. Neurology 45 (suppl 8): S58-S60, 1995.
Wesche D, Bockbrader H. A pharmacokinetic comparison of prega-balin and gabapentin. J Pain 2005; 6 (3): S29-S29.
WHO’s Pain Ladder. www.who.int/cancer/palliative/painladder/en/Winnie AP, Hartwell PAW: Relationship between time of treatment
of acute herpes zoster with sympathetic blockade and prevention of postherpetic nevralgia: clinical support for a new theory of the mechanism by which sympathetic blockade provides theraputic benefit. Re Anesth 1993; 18: 177-182.
Wolfe GI, Trivedi JR: Painful peripheral neuropathy and its nonsur-gical treatment. Muscle Nerve 2004; 30: 3-19.
Woodforde JM, Dwyer B, McEwen BW et al. The treatment of postherpetic neuralgia. Med J Aust 2: 869-872, 1965.
Woolf CJ, Bennett GJ, Doherty M, et al: Towards a mechanism-based classification of pain? Pain 77:227-229, 1998.
Woolf CJ, Decostered I: Implications of recent advances in the understanding of pain pathophysiology for the assessment of pain in patients. Pain 6:141-147, 1999 (suppl).
Xie X, Lancaster B, Peakman T et al. Interaction of the antiepilectic drug lamotrigine with recombinant rat brain type IIA Na+ chan-nels and with native Na+ channels in rat hippocampal neurones. Pflugers Arch 1995; 430: 437-446.
Yoshimura M, North RA. Substantia gelatinosa neurones hyper-polarized in vitro by enkefalin. Nature 1983; 305: 529-530.
Zakrzewska J, Patsalos P: Oxcarbazepine: a new drug in the man-agement of intractable trigeminal nevralgia. J Neurol Neurosurg Psychiat 1989; 52: 472-476.
Zakrzewska JM, Chaudhry Z, Nurmikko TJ, et al. Lamotrigine (Lamictal) in refractory trigeminal nevralgia: results from a doubl-blind placebo controlled crossover trial. Pain 1997; 73: 223-230.
Zakrzewska JM, Forssell H, Glenny AM. Interventions for the treat-ment of burning mouth syndrome. Cochrane Database Syst Rev. 2005 Jan 25; (1): CD002779.
Zampieri N, Chao MV. Mechanisms of neurotrophin receptor signal-ling. Biochem Soc Trans. 2006; 34 (pt 4): 607-611.
Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, Munzel U, Yakhno N, Raz I, Novosadova M, Maus J, Samigul-lin R. Oral treatment with α-lipoic acid imroves symptomatic diabetic polyneuropathy. The Sydney 2 trial. Diabetes Care 2006; 29:2365-70.
Ziegler D, Hanefeld M, Ruhnau KJ et al. and the ALADIN Study Group. Treatment of symptomatic diabetic peripheral neuropathy with the antioxidant a-lipoic acid. Diabetologia 1995; 38: 1425-33.
Ziegler D, Hanefeld M, Ruhnau K-J, Hasche H, Lobisch M, Schutte K, Kerum G, Malessa R, The ALADIN III Study
Ziegler D, Schatz H, Conrad F, Gries FA, Ulrich H, Reichel G. Ef-fect of treatment with the antioxidant a-lipoic acid on cardiac autonomic neuropathy in NIDDM patients. A 4-month rand-omized, controlled multicenter trial (DEKAN Study). Diabetes Care 1997; 20: 369-73.
Ziegler D, Sohr CG, Nourooz-Zadeh J. Oxidative stress and antioxidant defence in relation to the seveity of diabetic poly-neuropathy and cardiovascular autonomic neuropathy. Diabetes Care 2004; 27:2178-83.
Ziegler D. Painful Diabetic Neuropathy. Diabetes Care 2009; Suppl 2(32):S414-S419.
Ziegler D. Treatment of Diabetic Neuropathy and Neuropathic Pain. How far have we come? Diabetes Care 2008, 31 (Suppl. 2): S255-S261.
23 Studies into the safety and efficacy of tanezumab in pain patients. Clinicaltrials.gov Web Site. http://www.clinicaltrial.gov/ct2/results?term=tanezumab. Accessed March 23, 2010.


Cesare Bonezzi
Michelangelo BUonoCoRe
CO
D. 0
1820
022
ASPETTI DI FISIOPATOLOGIA
E TERAPIA DEL DOLORE


















