
BfV 1! Bergamasco - ats-bg.it FV 3_784... · BollettinoFarmacovigilanzaBergamasco(2!...
17
Bollettino Farmacovigilanza Bergamasco 1 BOLLETTINO DI FARMACOVIGILANZA DELLA RETE BERGAMASCA Numero 3/2012 Comitato di Redazione: Barbaglio Giorgio, Cavallazzi Mario, Chiappa Laura, Gambera Marco, Gilberti Lavinia, Gonella Giancarlo, Lanzeni Felice, Lorini Monia M.B., Nisic Andrea, Rocchi Giovanni, Spinetti Anna, Spoldi Laura, Strippoli Antonio, Taveggia Giovanni, Tumiati Michele, Zanzottera Bruno, Zenoni Stefano. Lo sapevate ? Dal 21 luglio è attiva la nuova scheda di segnalazione di sospetta ADR! Indice terzo numero La nuova normativa di Farmacovigilanza pag 3 Case report pag 4 Andamento delle ADRs nel territorio bergamasco pag 5 Recenti Alert di farmacovigilanza pag 12 Resoconto dal convegno pag.15 Scheda ADR pag 16 BfVBergamasco Segreteria Comitato redazione Dott.ssa Laura Spoldi Servizio Farmaceutico Territoriale ASL BG Tel 035/2270755 Fax 035/270035 Email: [email protected] Casa di Cura SANDONATO OSPEDALE SANT’ISIDORO TRESCORE BALNEARIO
Transcript of BfV 1! Bergamasco - ats-bg.it FV 3_784... · BollettinoFarmacovigilanzaBergamasco(2!...

Bollettino Farmacovigilanza Bergamasco
1
BOLLETTINO DI FARMACOVIGILANZA DELLA RETE BERGAMASCA
Numero 3/2012 Comitato di Redazione: Barbaglio Giorgio, Cavallazzi Mario, Chiappa Laura, Gambera Marco, Gilberti Lavinia, Gonella Giancarlo, Lanzeni Felice, Lorini Monia M.B., Nisic Andrea, Rocchi Giovanni, Spinetti Anna, Spoldi Laura, Strippoli Antonio, Taveggia Giovanni, Tumiati Michele, Zanzottera Bruno, Zenoni Stefano.
Lo sapevate ?
Dal 21 luglio è attiva la nuova scheda di
segnalazione di sospetta ADR!
Indice terzo numero La nuova normativa di Farmacovigilanza pag 3 Case report pag 4 Andamento delle ADRs nel territorio bergamasco pag 5 Recenti Alert di farmacovigilanza pag 12 Resoconto dal convegno pag.15 Scheda ADR pag 16
BfVBergamasco
Segreteria Comitato redazione Dott.ssa Laura Spoldi Servizio Farmaceutico Territoriale ASL BG Tel 035/2270755 Fax 035/270035 Email: [email protected]
Casa di Cura
SANDONATO
OSPEDALE SANT’ISIDORO TRESCORE BALNEARIO

Bollettino Farmacovigilanza Bergamasco
2
La nuova normativa di Farmacovigilanza Laura Spoldi L’applicazione della nuova normativa europea di farmacovigilanza entrata in vigore nel luglio 2012 ha lo scopo di rafforzare il sistema di farmacovigilanza, consentendo in particolare una circolazione più rapida ed efficace delle informazioni sulle ADRs tra gli stati membri. Le due nuove disposizioni a cui ci si deve attenere sono:
• Regolamento UE 1235/2010 che modifica il regolamento CE n.726/2004
• Direttiva 2010/84/UE che modifica la direttiva n.2001/83/CE
Nuova definizione di reazione avversa La nuova definizione di reazione avversa entrata in vigore il 2 luglio di quest’anno modifica, ampliandola quella precedentemente in essere. Attualmente si definisce reazione avversa ad un farmaco l’effetto nocivo e non voluto conseguente:
• all’uso di un medicinale conformemente alle indicazioni contenute nell’autorizzazione all’immissione in commercio (AIC)
• agli errori terapeutici • agli usi non conformi alle indicazioni
contenute nell’ AIC incluso: il sovradosaggio l’uso improprio l’abuso
• all’ esposizione per motivi professionali
Non devono essere segnalati come reazioni avverse i motivi di assunzione di farmaci quali gli errori terapeutici, l’abuso anche a scopo suicidario o l’uso off label, in assenza di una conseguente reazione avversa; dovranno invece venire segnalate le eventuali reazioni avverse a farmaci insorte a seguito dei suddetti motivi di assunzione. Anche la mancata efficacia si configura come ADR ed in alcuni casi è assimilabile ad una reazione
grave, per esempio nel caso di farmaci salvavita, vaccini e contraccettivi. La progressione della malattia non è una reazione avversa, quindi non dovrebbe essere indicata come mancata efficacia del farmaco in uso. Quando segnalare Per tutti i medicinali si invitano espressamente pazienti, medici, farmacisti ed altri operatori sanitari a segnalare sospetti effetti collaterali negativi alle autorità nazionali competenti. Viene tolta la limitazione qualitativa sulle reazioni da segnalare. Scheda di segnalazione Cambia la scheda di segnalazione; saranno disponibili due nuovi formati, uno per il cittadino ed uno per l’operatore sanitario. La vecchia scheda non è stata abrogata per cui può continuare ad essere utilizzata. La nuova scheda è compilabile sia previa stampa sia direttamente in formato elettronico; una volta compilata on line la scheda deve essere firmata ed invita al responsabile locale di farmacovigilanza; l’invio elettronico non sarà possibile prima del 2015. Sarà cura del Responsabile di FV criptare tutti i dati sensibili relativi al paziente e al segnalatore qualora dovesse essere inserita in rete una relazione clinica sull’evento avverso. Important medical event (IME) LIST Per ciò che riguarda la gravità di una reazione, non è consentito il DOWNGRADE ovvero la modifica da grave a non grave; è invece consentito fare l’upgrade. Sul sito dell’ EMA (http://eudravigilance.ema.europa.eu/human/textforIME.asp) è pubblicata una list di

Bollettino Farmacovigilanza Bergamasco
3
important medical event (IME list) in cui sono riportati due gruppi di eventi: CORE LIST. Categoria di eventi da considerare sempre gravi EXPANDED LIST: categorie di eventi che a seconda delle circostanze possono essere considerati gravi Gli eventi di tre SOC sono sempre da considerare gravi:
- congenital, familial and genetic disorders
- neoplam benign, malignant and unspecified
- Infections and infestations Monitoraggio addizionale I farmaci per i quali servirà monitorare con particolare attenzione la sicurezza, verranno inseriti dall’EMA in un elenco definito di monitoraggio addizionale che riguarderà:
• Medicinali autorizzati nell’ UE che contengano una nuova sostanza attiva la quale non era contenuta in nessun medicinale autorizzato prima del 1/01/2011
• Medicinali biologici autorizzati dopo il 1/01/2011-‐
• Medicinali sottoposti ad autorizzazione condizionata
• Medicinali autorizzati in circostanze particolari
• Medicinali per i quali è richiesto un PASS (Post Autorisatio Studies)
• Medicinali individuati dal PRAC (Pharmacovigilance and Risk Assessment Committed)
Tali farmaci restano in elenco per cinque anni o fino alla cassazioni delle condizioni per cui sono stati messi in commercio in modo condizionato. L’elenco verrà reso pubblico entro la fine dell’anno e tutti i farmaci inseriti dovranno riportare la dicitura in scheda tecnica “farmaco sottoposto a monitoraggio addizionale” ed un simbolo nero, ancora in
fase di definizione( da decidere entro il 2/06/2013). Fino alla pubblicazione di tale elenco resterà attivo quello dei farmaci sottoposti a monitoraggio intensivo stilato dall’ AIFA. Sia il regolamento che la direttiva sono riportati sul portale web dell’ AIFA (www.agenziafarmaco.gov.it) in una specifica sezione denominata “Nuova legislazione in Farmacovigilanza”, si è ritenuto opportuno richiamare i cambiamenti normativi più rilevanti per l’attività degli operatori sanitari.

Bollettino Farmacovigilanza Bergamasco
4
Case Report Antonio Strippoli (A.O. Bologni – Seriate) Piastrinopenia ed emorragie multiple da Diclofenac Sodico: un caso ad esito fatale
La paziente. La paziente è una donna, 49 anni, etnia caucasica, in terapia da anni con beta-‐bloccanti e ranitidina. Da dieci giorni lamenta algie alla spalla, per cui ha assunto 6 fiale IM di Diclofenac (una fiala/die) negli ultimi 6 giorni. Evento. Il giorno 20/03/2012 la signora si presenta in PS per vomito incoercibile con striature di sangue ed addominalgie. L’addome si presenta trattabile e dolente alla palpazione; la paziente lamenta anche la comparsa, da un paio di giorni, di petecchie agli arti inferiori. La visita ginecologica esclude patologie di competenza. Esegue inoltre ECG (normale), Rx torace e addome (normali), eco addome in toto (aumento ecogenicità corticale come da nefropatia medica) ed esami ematochimici. Tra i valori rilevanti si segnalano: plt 12.000, Ht 33.6, Hb 11.3, creatinina 1.9. Il tempo di protrombina, di tromboplastina ed il fibrinogeno rientrano nel range di normalità. Sviluppo clinico e trattamento. Poiché la grave piastrinopenia è subito messa in relazione con l’utilizzo del Diclofenac, viene contattato il Centro Antiveleni OORRR BG che conferma la frequenza di trombocitopenie legate all’uso di Diclofenac. La paziente inizia a presentare enterorragia ed ematuria e viene trasferita in Rianimazione dove si inizia l’infusione di concentrato piastrinico a bassa velocità. Dopo 30 minuti dall’inizio dell’infusione la paziente perde conoscenza con improvvisa respirazione in gasping, repentino calo della saturazione, medomidriasi non fotoreagente, polso arterioso femorale filiforme. Si iniziano le manovre rianimatorie con ventilazione e in seguito intubazione, somministrazione di adrenalina nel tubo ot ed EV, somministrazione di Voluven. Si verificano due episodi di FV trattati efficacemente. Si nota scomparsa del polso femorale e si inizia massaggio cardiaco con somministrazione di 15 fiale di adrenalina. Al monitor persiste ritmo sinusale PEA con progressiva bradicardia e sottoslivellamento ST.
Esito. Dopo due ore dal ricovero si sospendono le manovre rianimatorie per assenza persistente di ritmo spontaneo e persistenza di acinesia di tutto il cuore. Si constata il decesso. La diagnosi posta è “grave piastrinopenia presumibilmente indotta da utilizzo di Diclofenac, responsabile di enterorragia, ematemesi ed ematuria”. Conclusioni. La letteratura scientifica conferma la frequenza di trombocitopenie legate all’uso di Diclofenac. Da una ricerca nel database nazionale della Rete di Farmacovigilanza sono state rintracciate 33 reazioni avverse, riferibili a trombocitopenia, di cui 26 gravi e tre mortali tutte ascrivibili all’utilizzo di FANS. Nella stessa ricerca riferita a Diclofenac sono state riportate 9 trombocitopenie, tutte gravi, con 2 decessi. Discussione. I farmaci possono indurre trombocitopenia attraverso due meccanismi principali: Ridotta produzione piastrinica (da farmaci
citotossici a livello del midollo) Incremento della distruzione delle piastrine,
immuno-‐mediata o non immuno-‐mediata con vari meccanismi eziopatogenetici proposti.
Nel segnalare che in tutti gli studi epidemiologici le classi di farmaci più frequentemente coinvolti includono sempre i FANS, oltre che anticonvulsivanti, diuretici, derivati degli alcaloidi della cincona, penicillamine e sali d’oro, si conclude che la trombocitopenia è comunque una condizione patologica multicausale e che i farmaci sono solo una delle possibili cause di tale patologia. L’incidenza della trombocitopenia immune farmaco-‐indotta è stata stimata in circa 10 casi per milione di abitanti per anno, che può aumentare in popolazioni speciali (per es. nei pazienti ospedalizzati). Questo dato contrasta quanto da noi rilevato nel database della Rete Nazionale di FV supportando la tesi secondo cui il tasso di segnalazione in Italia sia non congruo con l’effettivo numero di reazioni avverse da farmaci che si verificano.

5
Bollettino Farmacovigilanza Bergamasco
Le sospette ADRs pervenute presso l’ASL di Bergamo Laura Spoldi, Andrea Nisic
Numero di segnalazioni Nel secondo trimestre 2012 sono pervenute ed inserite nella rete nazionale di FV 38 sospette ADRs; si raggiungo così 75 segnalazioni totali nei primi sei mesi dell’anno. Sedici segnalazioni sono state inviate all’U.O. di Farmacologia dell’A.O. L. Sacco per l’inserimento nel database VIGER -‐ progetto di FV attiva – nella popolazione anziana in politerapia. Analisi dei principi attivi.
Segnalazione per ATC
3
1 1 12 2
11
65 5
1
0
2
4
6
8
10
12
A B C D G J Jv L M N R
Il maggior numero di segnalazioni continua ad essere riferito all’uso di farmaci della classe ATC J con 13 segnalazioni (11 Jv riferite a vaccini e due agli antimicrobici 2). Delle 11 reazioni ricondotte a vaccino 10 sono attribuite alla vaccinazione HPV, emerse dalla revisione delle schede di arruolamento delle pazienti nel progetto di FV attiva SAF-‐HPV; le reazioni più frequentemente segnalate sono: reazione in sede di vaccinazione, febbre, cefalea e dolori muscolari. Seguono le razioni ai farmaci della classe ATC L, farmaci antineoplastici e immunomodulatori. Analisi della gravità delle reazioni. Le sospette reazioni avverse segnalate come gravi sono 6, mentre il sesso maggiormente colpito è stato quello femminile con 28 reazioni. Le reazioni gravi sono:
• Anemizzazione da Cardioaspirin; • Agitazione e confusione da Oramorph;
• Eritema generalizzato da Mengevax ACWY ;
• Emorragia orale e mucosite da Amoxicillina e Ac. Clavulanico.
Le ulteriori due reazioni gravi hanno riguardato la classe ATC L: è stato segnalata sospetta ischemia cerebrale da Alimta mentre cianosi, desaturazione ed ipotensione sono insorti in paziente in terapia con Eloxatin. Analisi degli esiti degli eventi. Il 77% delle ADRs segnalate si è risolta completante mentre nel il 16% dei casi l’esito non è disponibile. Non sono state segnalate razioni avverse che hanno esitato in decesso.
Analisi dei segnalatori. I maggiori segnalatori risultano essere farmacisti con 19 segnalazioni. I medici di Assistenza Primaria hanno contribuito con 6 segnalazioni. Tre ADRs provengono da RSA due delle quali, gravi, sono segnalate dal personale infermieristico.

6
Bollettino Farmacovigilanza Bergamasco
Le sospette ADRs pervenute presso A.O. Ospedali Riuniti di Bergamo nel secondo trimestre 2012 Monia Lorini Numero di segnalazioni Nel secondo trimestre del 2012, sono pervenute al Responsabile di Farmacovigilanza 121 schede di segnalazione di sospetta reazione avversa a farmaco (ADRs). Rispetto allo scorso anno, il numero delle schede ricevute nello stesso periodo, sono notevolmente aumentate (nel 2011 sono state 28). Questa importante crescita è data soprattutto dall’elevato numero di reazioni avverse raccolte nell’ambito del progetto di farmacovigilanza regionale FARMAMICO (65%). Questo si riflette sia sulla classe dei medicinali maggiormente coinvolti (anticoagulanti orali), sulla tipologia delle reazioni, (variazioni INR) e sull’età dei soggetti coinvolti. Gli obiettivi principali di questo progetto di farmacovigilanza sono : 1. di verificare l’insorgenza di possibili interazioni della terapia anticoagulante orale con altre terapie (inclusi i fitoterapici e le medicine alternative), con le malattie intercorrenti e le variazioni delle abitudini di vita-‐ eventualmente non ancora segnalate in letteratura; 2.l’implementazione di interventi formativi mirati alla luce delle eventuali nuove acquisizioni. Rimangono invece numericamente costanti le segnalazioni spontanee provenienti da operatori sanitari che hanno rilevato la sospetta reazione avversa a farmaco durante la loro normale attività lavorativa. Analisi per tipologia di reazione Proprio per l’elevata percentuale di segnalazioni provenienti dal progetto FARMAMICO, delle 121 schede di segnalazioni di ADRs pervenute, il 52% (63 schede) hanno riguardato reazioni avverse che hanno interessato variazioni dell’INR (test utilizzato per valutare l'efficienza della coagulazione) e il 12% il sistema vascolare e più precisamente sanguinamenti, ematomi etc… Seguono per il 7%, reazioni avverse che hanno coinvolto la cute e il tessuto sottocutaneo
(eruzioni cutanee più o meno estese, prurito, orticaria etc..) e le reazione che coinvolgono il sistema nervoso (confusione, etc..),. Due reazioni gravi quale shock settico in un paziente con polmonite da Pseudomonas aeruginosa e insufficienza respiratoria in sepsi da Cryptococcus neoformans hanno determinato il decesso dei soggetti coinvolti. Analisi dei farmaci sospetti e della gravità I principi attivi dei farmaci indicati dal segnalatore come sospetti delle ADRs, sono 43. Suddividendoli secondo la classificazione ATC (Anatomico Terapeutico Chimico), come è prevedibile aspettarci , i medicinali maggiormente rappresentati sono quelli del gruppo B (sangue e organi ematopoietici) e più precisamente il warfarin che è coinvolto in ben 76 segnalazioni rispetto alle 121. Seguono gli antineoplastici e immunosoppressori (L), i farmaci antimicrobici (J), e quelli del sistema nervoso (N). Nel grafico seguente viene dettagliata la distribuzione.
I farmaci coinvolti nelle sospette reazioni avverse e che fanno parte del gruppo
4
87
51
1216
510
2 3
0
10
20
30
40
50
60
70
80
90
100
A B C G J L M N R S
n°
Classi farmacolog iche per ATC

7
Bollettino Farmacovigilanza Bergamasco
"speciale" dei medicinali che sono sottoposti a monitoraggio intensivo sono*: ROACTEMRA (tocilizumab) 20 mg/ml concentrato per soluzione per infusione uso ev 1 fl. 10 ml (L). La segnalazione ha riguardato una paziente di 56 anni, in trattamento con Roactemra per artrite reumatoide. Durante il terzo ciclo di somministrazione, la paziente ha manifestato shock anafilattico seguito da arresto cardiaco. Questa grave reazione, che ha messo in pericolo la vita della paziente, si è risolta grazie al pronto intervento degli operatori sanitari. REPAGLINIDE (Repaglinide) 2 mg compressa (A) Oltre a questo medicinale, la paziente assumeva anche Metformina (anch’esso classificato dal segnalatore come medicinale sospetto). La paziente in politerapia assumeva inoltre anche Norvasc, Nitroglicerina in cerotti e Corlentor (classificati come farmaci concomitanti) La reazione (astenia e ipoglicemia) ha portato all’ospedalizzazione della paziente. Se si valuta la gravità delle reazioni indicate nelle 121 schede di ADRs, le segnalazioni di reazioni avverse non gravi sono state l’82 % pari a 99 schede. Il rimanente 18% (22 schede), hanno riguardato reazioni gravi di cui:
• 9 segnalazioni di ospedalizzazione o un prolungamento dell'ospedalizzazione del paziente;
• 6 segnalazioni di pericolo di vita del paziente,
• 3 segnalazioni di invalidità grave o permanente,
• 2 segnalazioni di altra grave condizione
• 2 segnalazioni di decesso del paziente. In quest'ultimo caso, i farmaci coinvolti sono stati:
1) ARACYTIN (citarabina), ETOPOSIDE TEVA ( etoposide), ALKERAN (melfalan) e CARMUSTINA (carmustina) somministrati ad un paziente di 54 anni con linfoma non Hodgkin a cellule B follicolari sottoposto ad autotrapianto per consolidamento a seguito di persistenza di malattia dopo chemioterapia di prima linea. Durante la citopenia, dopo il trapianto autologo e previo condizionamento BEAM, il paziente ha sviluppato febbre. Per tale motivo fu intrapresa terapia antibiotica. Dopo un'iniziale miglioramento, il quadro clinico del paziente peggiorò ulteriormente, infatti manifestò insufficienza respiratoria e shock settico. L'esame colturale da bronco-‐lavaggio alveolare portò all'isolamento di Pseudomonas aeruginosa multiresistente (sensibile solo a Colistina). Nonostante le terapie instaurate per la patologia in corso, il paziente è deceduto. Utilizzando l’algoritmo di Naranjo per definire il nesso di causalità, c’è una ragionevole possibilità che il farmaco abbia causato tale evento. 2) VELCADE (bortezomib) e DESAMETASONE sol. iniettabile utilizzati nel trattamento di un paziente con mieloma multiplo. A seguito di recidiva di malattia , il paziente iniziò il trattamento chemioterapico di terza linea secondo schema Velcade-‐Desametasone. Per il progressivo decadimento delle condizioni generali e per la comparsa di febbre, il paziente fu sottoposto a prelievo per emoculture da sangue periferico, risultate positive per Cryptococcus neoformans. Nonostante la terapia in atto, le condizioni generali sono andate progressivamente deteriorandosi con peggioramento del quadro e aggravamento dell’ insufficienza respiratoria che hanno portato al decesso del paziente. Secondo l’algoritmo di Naranjo, il farmaco può aver contribuito all’evento.

8
Bollettino Farmacovigilanza Bergamasco
Analisi per sesso e per età Le segnalazioni pervenute sono equamente suddivise tra i due sessi, infatti sono state 60 per i soggetti di sesso maschile e 61 per quello femminile. Il numero più alto di segnalazioni si è concentrato nella fascia di età over 65 anni con 81 casi di reazione avversa pari al (67%), di queste il 42% (34 ADR) hanno riguardato soggetti di età superiore agli 80 anni. Questa particolarità è data dall’elevato numero di schede provenienti dal progetto FARMAMICO che coinvolge soprattutto pazienti anziani. Fa seguito la fascia dei pazienti di età compresa tra i 18-‐64 anni con 40 segnalazioni pari al 33%. Si evidenzia dalla tabella a fianco, che le ADRs gravi sono equamente distribuite tra le due fascie di età, mentre non sono pervenute segnalazioni nell’età pediatrica. Conclusioni: Nonostante i numeri elevati di schede che sono pervenute in questo trimestre, il numero delle schede che provengono da
operatori sanitari che hanno rilevato la sospetta reazione avversa a farmaco durante la loro normale attività lavorativa, risulta essere ancora contenuto.
Spesso, si è contrari a segnalare in quanto, si pensa che la segnalazione debba avvenire solo se c'è un evidente nesso di causalità, mentre il semplice sospetto è di per sé una ragione sufficiente. Voglio inoltre ricordare che con l’entrata in vigore della normativa europea, cambia la definizione di reazione avversa, definita come “effetto nocivo e non voluto conseguente all’uso di un medicinale, indipendentemente dal tipo di uso del medicinale”. Pertanto sono oggetto di segnalazione le reazioni avverse, incluse anche quelle derivanti da errore terapeutico, abuso, misuso, uso off label, sovradosaggio ed esposizione professionale.
* (Elenco istituito da AIFA ai sensi del D.M. 21/11/2003 -‐Aggiornamento n. 15 di agosto 2011),
Fascia di eta' N° ADR GRAVI
NON GRAVI
< 1 mese 1 mese-‐2 anni 2-‐14 anni 12-‐17 anni 18-‐64 anni 40 12 28 Over 65 anni 81 11 69
9
Bollettino Farmacovigilanza Bergamasco
Le sospette ADRs rilevate presso A.O. Bolognini Seriate
Antonio Strippoli Numero di segnalazioni Durante il secondo trimestre 2012 sono state inserite nella rete nazionale di farmacovigilanza 252 segnalazioni, in linea con lo stesso periodo dell’anno precedente. Analisi dei principi attivi più segnalati.
Principi attivi più segnalati
35
24
18
107 6 6 6 6 6 5 5 5 4
0
5
10
15
20
25
30
35
40
Aci
do A
cetil
salic
ilico
Am
oxic
illin
a/C
lavu
lana
to
War
farin
Vac
cini
Insu
line
Bis
opro
lolo
Furo
sem
ide
Ibup
rofe
ne
Isos
orbi
de d
initr
ato
Par
acet
amol
o
Am
oxic
illin
a
Cip
roflo
xaci
na
Val
sarta
n/id
rocl
orot
iazi
de
Levo
tirox
ina
sodi
ca
N°
AD
R
Il maggior numero di segnalazioni continua ad essere riferito all’uso di acido acetilsalicilico; seguono per numerosità le segnalazioni di reazioni avverse, per lo più riferite al distretto dermatologico, ascrivibili all’associazione amoxicillina/acido clavulanico e le emorragie da warfarin.
0
10
20
30
40
50
60
70
80
A B C D G H J L M N P R S V
Incidenza ADR per ATC
In base alla classificazione ATC, nel secondo trimestre prevalgono le reazioni da farmaci classificati in “C”, cioè dei farmaci cardiovascolari, rappresentate in particolare da bradicardie, sincopi, ipotensioni e squilibri elettrolitici.
Analisi della gravità delle reazioni. Le sospette reazioni avverse segnalate sono state ritenute gravi nel 28.3% dei casi.
Gravità delle ADRGrave28,3%
Non grave71,7%
Analisi degli esiti degli eventi. In questo trimestre non si sono registrati casi ad esito fatale; la maggior parte dei casi si è evoluta in miglioramento del quadro clinico (circa 60%) mentre circa un 20% di pazienti sono stati dimessi ma non ancora completamente guariti.
Esiti delle ADRNon
disponibile8,1%Non ancora
guarito18,4%
Miglioramento59,8%
Risoluzione completa
13,7%
Analisi dei segnalatori. Il maggior numero di segnalazioni continua a provenire dai Pronto Soccorso dell’Azienda Ospedaliera (Seriate, Alzano L.do, Piario e Lovere) e sono redatte con l’ausilio della UO Farmacia Ospedaliera. La percentuale di segnalazioni redatte autonomamente dai medici ospedalieri è in leggera crescita.
SegnalatoreMedico
ospedaliero8,7%
Infermiere0,3%
Biologo 2,8%
Farmacista 88,2%

10
Bollettino Farmacovigilanza Bergamasco
FARMAMONITOFARMAONCO
MEREAFAPS
0
10
20
30
40
19 20
35
n ̂s
egna
lazio
niLe sospette ADRs pervenute presso A.O. Treviglio Lavinia Gilberti -‐ Mariella Dimatteo Numero delle Segnalazioni Le segnalazioni di reazioni avverse inserite dall'A.O. Treviglio nella Reta Nazionale di Farmacovigilanza relative al periodo gennaio-‐giugno 2012 sono state 74. Le seguenti schede sono così distribuite tra i diversi progetti regionali di Farmacovigilanza attivi all'interno dell'Azienda: MEREAFaPS : 35 FARMAMONITO: 19 FARMAONCO : 20
Analisi dei principi attivi. Ogni scheda di segnalazione può includere uno o più farmaci quali responsabili dell'ADR. Anche nel secondo trimestre la classe ATC maggiormente coinvolta è quella appartenente agli “Antimicrobici generali per uso sistemico J”. Ciò ha contribuito ad avere, a partire dal mese di gennaio fino a giugno ben 31 segnalazioni inerenti a questa ATC. I principi attivi sospettati di aver determinato queste ADR sono rappresentati da amoxicillina/ac. clavulanico, levofloxacina, ciprofloxacina. e cotrimossazolo. Il grafico seguente illustra la distribuzione per classe farmacologica ATC:
Analisi per tipologie di reazione Dall'analisi delle 74 reazioni è emerso che il 41% delle reazioni rientra nel SOC delle “patologie della cute e del tessuto sottocutaneo” che comprendono eruzioni cutanee generalizzate, prurito e pomfi. Il 20% è relativo al SOC delle “patologie gastrointestinali” (epigastralgia e dolori addominali). Mentre, come si evince dal grafico sottostante, gli altri gruppi oscillano tra il 4-‐9%.
Analisi della gravità delle reazioni. Per quanto riguarda la gravità delle reazioni, 58 segnalazioni, pari al 85%, si riferivano a reazioni avverse non gravi e 10, pari al 15%, sono risultate gravi, mettendo in pericolo la vita del paziente, determinandone l’ospedalizzazione o un prolungamento della stessa. Analisi degli esiti degli eventi. Durante il periodo di osservazione, 27 segnalazioni (40%) hanno comportato una risoluzione completa e 37 segnalazioni (54%) hanno determinato un miglioramento.
4%4%4%
4%
5%8%
9%
20%
41%Disturbi del metabolismo e della nutrizionePatologie del sistema emolinf opoieticoPatologie v ascolariPatologie renali e urinarie
11
Bollettino Farmacovigilanza Bergamasco
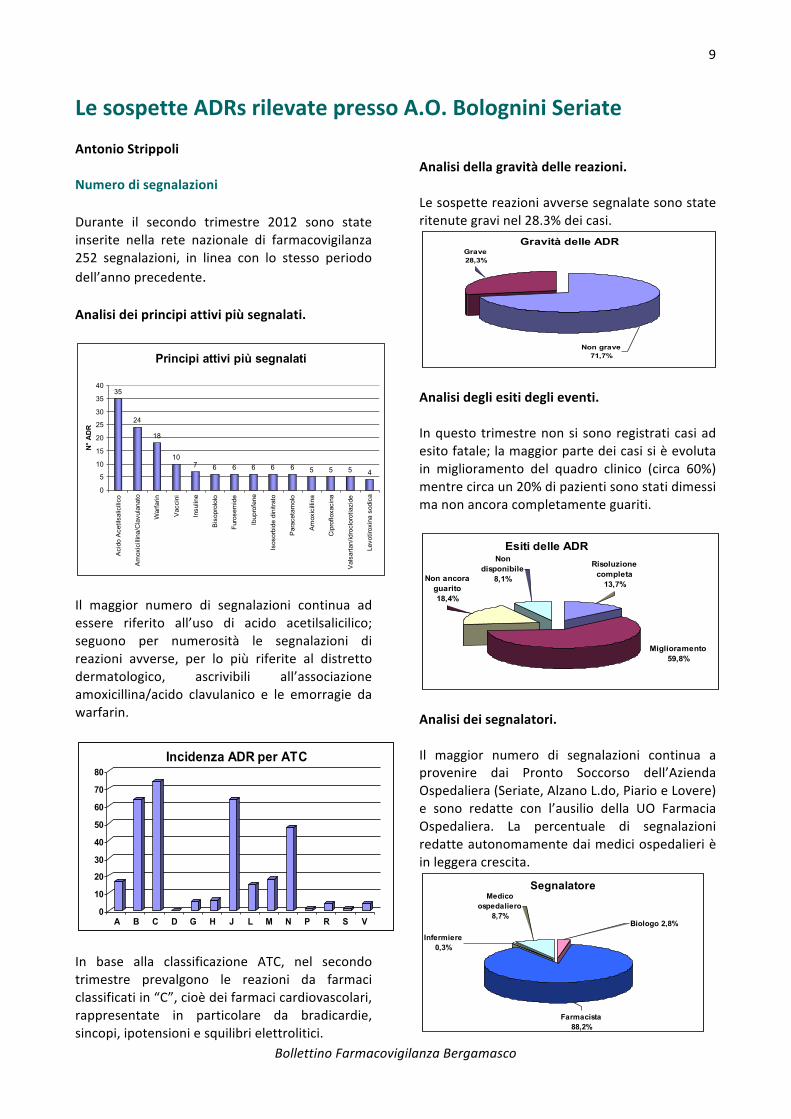
L' inserimento di nuove segnalazioni di ADR riferite al periodo aprile – giugno ha visto predominare il sesso maschile con 36 segnalazioni, rispetto al primo trimestre 2012, in cui il numero di segnalazioni ha interessato il sesso femminile.
Analisi dei segnalatori. Come si evince dal grafico sottostante le segnalazioni sono state individuate e segnalate per il 93% dai medici ospedalieri.

12
Bollettino Farmacovigilanza Bergamasco
Recenti Alert di Farmacovigilanza Andrea Nisic Avvelenamento accidentale da paracetamolo AustralianPrescriber N.35 aug 2, Number 4 Gli effetti epatotossici da overdose intenzionale da paracetamolo sono ben noti; tuttavia l’epatotossicità può manifestarsi anche a dosi terapeutiche. In molti pazienti il paracetamolo, assunto a dosi terapeutiche ha determinato epatotossicità. In uno studio di Larson AM at al. compiuto su 662 pazienti che hanno sviluppato grave epatotossicità da paracetamolo è emerso che il 48% dei pazienti hanno assunto una dose di paracetamolo non superiore ai 4 g al giorno. I fattori di rischio per lo sviluppo dell’epatotossicità iatrogena includono il digiuno, il consumo continuativo ed eccessivo di alcool e l’utilizzo concomitante di xenobiotici induttori del citocromo P450 (CYP 2E1) es. etanolo. Ulteriori fattori di rischio sono età avanzata , insufficienza renale e/o cadiopolmonare e preesistenti patologie epatiche. Il paracetamolo è normalmente metabolizzato tramite coniugazione nel fegato ed escreto nelle urine. Una piccola percentuale viene convertita dagli enzimi del CYP 2E1 e 3A4 al composto N-‐acetil-‐p-‐benzochinonimmina (NAPQI), che è poi coniugato con glutatione ed escreto. Il digiuno prolungato esaurisce i substrati necessari per la coniugazione, tra cui il glutatione, portando ad un accumulo di NAPQI. Ulteriori fattori di rischio sono l’overdose accidentale e la concomitante somministrazione di paracetamolo per via orale e per via endovenosa.
Gravi effetti collaterali da codeina in età pediatrica Food and Drug Administartion 16/08/2012 Recentemente, sono stati riportati in letteratura medica, tre casi di morte e un caso di grave depressione respiratoria avvenuti in bambini che hanno ricevuto codeina dopo interventi di tonsillectomia e / o adenoidectomia per la sindrome delle apnee ostruttive nel sonno. La codeina viene convertita in morfina nel fegato da un enzima del citocromo P450 2D6 (CYP2D6). Alcune persone hanno variazioni del DNA, per anomalie genetiche ereditarie, che rendono questo enzima più attivo, la codeina viene convertita in morfina più velocemente ed in quantità potenzialmente pericolose per la vita nei bambini di età compresa fra due e
cinque anni, è stata riscontata questa anomalia genetica ereditaria (metabolizzatori "ultra-‐rapidi" del citocromo P450 2D6). Le dosi di codeina somministrate erano all'interno del normale range terapeutico. I segni di tossicità da morfina si sono sviluppati dopo uno o due giorni
di terapia. Le concentrazioni di morfina riscontrate post-‐mortem erano più elevate dei valori normali. La FDA sta conducendo una revisione per determinare se vi sono ulteriori casi di sovradosaggio accidentale o di morte nei bambini che assumono codeina, e se questi eventi avversi si sono verificati durante il trattamento di altri tipi di dolore come il dolore post-‐operatorio in seguito altri tipi di interventi chirurgici o procedure. FDA aggiornerà il pubblico quando saranno disponibili maggiori informazioni. Dalfampiridone e rischio di convulsioni Food and Drug Administartion 27/07/2012

13
Bollettino Farmacovigilanza Bergamasco
La Food and Drug Administration (FDA) ha pubblicato una comunicazione riguardante la sicurezza del farmaco Ampyra® (dalfampridine, Acorda Therapeutics), in cui si segnala il rischio di convulsioni nelle persone con SM che stanno iniziando il trattamento con Ampyra. In America il farmaco è stato approvato nel gennaio 2010 allo scopo di migliorare la deambulazione nelle persone con SM. Le crisi epilettiche (convulsioni) erano già state segnalate come effetti collaterali. In base alle segnalazioni ricevute nel postmarketing la FDA ha osservato che la maggior parte delle crisi epilettiche si è verificata entro pochi giorni o settimane dopo l'inizio della dose raccomandata e si sono verificate anche in persone che non avevano avuto precedenti episodi di epilessia o convulsioni. Inoltre la maggior parte di coloro che hanno avuto crisi, avevano almeno 50 anni ed erano a rischio di lieve deficit della funzione renale correlato all'età. L’utilizzo del farmaco in soggetti noti per convulsioni o con alterazioni moderate o severe della funzionalità renale è controindicato. La funzionalità renale, stimata tramite il dosaggio della cretinina clearence, deve essere controllata prima di iniziare il trattamento e va monitorata almeno una volta all'anno, perché una sua alterazione può causare un aumento delle concentrazioni ematiche del farmaco che viene eliminato tramite questa via. Simvastatina: aggiornamento delle interazioni e delle controindicazioni MHRA Drug Safety Update Aug 2012 Vol.6 Issue 1
Da recente elaborazione di dati raccolti da studi clinici e report spontanei di reazioni avverse, sono emerse alcune informazioni supplementari sulla simvastatina. L’assunzione contemporanea con alcuni farmaci determina l’aumento delle concentrazioni plasmatiche della stessa accrescendo così il rischio di miopatiae e/o rabdiomiolisi. I key points sono:
• Simvastatina è ora controindicata con ciclosporina, danazolo e gemfibrozil.
• La dose massima raccomandata quando assunta in associazione con amlodipina o diltiazem è ora di 20mg al giorno.
Dal link è possibile consultare la nuova tabella che descrive e riporta le nuove interazioni descritte in letteratura. Copidogrel ed interazione con inibitori di pompa BMJ 2012; 345e:4388
Lo studio, caso-‐controllo e self-‐controlled case series, ha valutato l’associazione fra gli inibitori ed outcames negativi in pazienti in terapia con copidogrel e aspirina. Sono stati valutati 24471 soggetti effettuando un record-‐linkage tra il database United Kingdom General Practice e i dati del Myocardial Ischaemia National Audit Project (MINAP) e dell'Office for National Statistics (ricerca delle malattie cardiovascolari che connette studi personalizzati a record elettronici [CALIBER]).Gli outcome primari erano rappresentati da morte o infarto del miocardio (IM) incidente. Gli outcome secondari erano morte, IM incidente e morte per cause vascolari e non vascolari. Sono stati effettuati confronti tra utilizzo e non utilizzo di PPI. La mancanza di una associazione specifica e la discrepanza tra i risultati delle analisi finalizzate ad escludere le differenze inter-‐individuali ggeriscono che l'interazione tra PPI e clopidogrel sia clinicamente irrilevante. FDA Drug Safety Comunication: rari casi di gravi ustioni con l’utilizzo di antitolorificito topici OTC. Food and Drug Administartion 13/09/2012 FDA avverte i consumatori che alcuni prodotti over-‐the-‐counter (OTC) che vengono applicati sulla pelle per alleviare lievi dolori articolari e muscolari sono stati segnalati come causa di rari casi di lesioni cutanee

14
Bollettino Farmacovigilanza Bergamasco
gravi, ustioni chimiche che vanno dal primo al terzo grado, a livello della zona di applicazione dei prodotti; in alcuni casi rari hanno richiesto l'ospedalizzazione per ustioni gravi. I sintomi prevalenti dopo l'applicazione cutanea sono sensazioni locale di calore o di freddo. Questi farmaci sono disponibili come prodotti singoli o in combinazione e contengono mentolo, salicilato di metile e capsaicina. Le varie formulazioni in commercio includono creme, lozioni, unguenti e cerotti medicati. Nota Informativa AIFA: Ketoprofene per uso topico e rischio fotosensibilizzazione AIFA 24/09/2012
Nota Informativa Importante su ketoprofene per uso topico e rischio di reazioni di fotosensibilizzazione (I Comunicazione aprile
2010; II Comunicazione agosto 2011). Il Comitato per i Medicinali per Uso Umano (CHMP) dell’Agenzia Europea dei Medicinali (EMA) ha condotto nel 2010 una revisione scientifica dei dati di sicurezza e di efficacia dei medicinali contenenti ketoprofene per uso topico, a seguito delle segnalazioni di reazioni avverse di fotosensibilizzazione e di co-‐sensibilizzazione con l’octocrilene (filtro UV).Il CHMP ha evidenziato che i casi di fotosensibilità da ketoprofene per uso topico si verificano in seguito alla fotodegradazione del ketoprofene stesso alla luce solare, anche in caso di cielo coperto. Tale reazione avversa, anche se rara, è stata grave nella maggior parte dei casi, richiedendo l’ospedalizzazione, l’interruzione del lavoro e una permanente immunizzazione a causa del meccanismo immunologico della fotoallergia.

15
Bollettino Farmacovigilanza Bergamasco
Resoconto dal convegno: “Esperienze di Farmacovigilanza in provincia di Bergamo” Luciana Gandolfi Grande partecipazione giovedì 20 settembre in Sala Lombardia per l’evento della Farmacovigilanza. Il tema trattato nel convegno è un problema di grande attualità. Viviamo in una società vecchia: per età media siamo forse secondi al mondo dopo il Giappone. Alla nostra longenità concorrono molti fattori, in primis lo stile di vita e la genetica, si aggiunga la bassa natività e l’aumento della sopravvivenza infantile, ma certamente all’allungamento della durata di vita di uomini e donne ha dato un fondamentale contributo la scienza farmaceutica. Le patologie croniche sono controllate da adeguate terapie farmacologiche (si pensi all’ Ipertensione, al Diabete ecc.). Tali terapie devono essere protratte per tutta la vita dell’assistito e ad esse, in tempi diversi, si possono aggiungere altre terapie per medicare altri eventi acuti o altrettanto cronici. Si parla del fenomeno della “politerapia”. Il farmaco può determinare eventi avversi conosciuti e perciò attesi, ma spesso, solo con l’uso protratto e su vasta scala, può evidenziare effetti indesiderati inaspettati e sconosciuti al momento della sua immissione in commercio, che non devono essere ignorati e sottovalutati.
La sorveglianza, la segnalazione e la valutazione di effetti indesiderati da prodotti farmaceutici utilizzati per le terapie mediche sono gli obiettivi della Farmacovigilanza, una scienza clinica. La principale fonte di nuove informazioni è costituita dalla segnalazione spontanea di tali effetti. L’ASL contribuisce coinvolgendo tutti gli operatori sanitari, con la collaborazione di tutte le Strutture di ricovero e cura bergamasche, sia A.O. pubbliche che le Strutture private, tra i quali gli specialisti ospedalieri e i Medici di Assistenza Primaria e farmacisti. Questi operatori rappresentano gli attori privilegiati nel campo della farmacovigilanza, avendo maggiori contatti con il paziente cronico e potendolo osservare nel tempo e nella globalità delle sue necessità mediche. In questo quadro di lavoro collaborativo in rete tra le Aziende Sanitarie della provincia, i Responsabili aziendali di FV hanno avuto l’opportunità di documentare lo stato di avanzamento dei vari progetti di Farmacovigilanza in corso e le prospettive future in una giornata congressuale con il contributo di emeriti ospiti dalla Regione Lombardia la Dr.ssa Stefania Scotto e dall’Università di Messina il Professor Achille Caputi. Sul sito ASL sono disponibili le presentazioni degli argomenti trattati dai relatori del convegno.

16
Bollettino Farmacovigilanza Bergamasco
SCHEDA UNICA DI SEGNALAZIONE DI SOSPETTA REAZIONE AVVERSA (ADR) A cura dei medici e degli altri operatori sanitari. Inviare al responsabile di farmacovigilanza della struttura di appartenenza
(gli indirizzi dei responsabili possono essere recuperati nel sito dell’AIFA: www.agenziafarmaco.it/it/responsabili) 1. INIZIALI PAZIENTE Nome – Cognome
2. DATA di NASCITA o ETÀ
3. SESSO M" "F"
4. DATA INSORGENZA REAZIONE
5. ORIGINE ETNICA
CODICE SEGNALAZIONE
1.a. PESO (kg) 1.b. ALTEZZA (cm) 1.c. DATA ULTIMA MESTRUAZIONE
1.d. GRAVIDANZA sconosciuta
1° trimestre 2° trimestre 3° trimestre
1.e. ALLATTAMENTO
SI NO
6. DESCRIZIONE DELLA REAZIONE ED EVENTUALE DIAGNOSI (*se il segnalatore è un medico)
7. INDICARE SE LA REAZIONE OSSERVATA DERIVA DA: INTERAZIONE ERRORE TERAPEUTICO
ABUSO MISUSO
OFF LABEL OVERDOSE
ESPOSIZIONE PROFESSIONALE
8. GRAVITA' DELLA REAZIONE: GRAVE
DECESSO OSPEDALIZZAZIONE O PROLUNGAMENTO INVALIDITA' GRAVE O PERMANENTE HA MESSO IN PERICOLO DI VITA ANOMALIE CONGENITE/DEFICIT NEL NEONATO ALTRA CONDIZIONE CLINICAMENTE RILEVANTE
NON GRAVE
9. EVENTUALI ESAMI DI LABORATORIO RILEVANTI PER ADR (riportare risultati e date in cui gli accertamenti sono stati eseguiti):
10. ESITO DATA:
RISOLUZIONE COMPLETA ADR RISOLUZIONE CON POSTUMI MIGLIORAMENTO REAZIONE INVARIATA O PEGGIORATA DECESSO
dovuto alla reazione avversa il farmaco può avere contribuito non dovuto al farmaco causa sconosciuta
NON DISPONIBILE
11. AZIONI INTRAPRESE (specificare):
In caso di sospensione compilare i campi da 17 a 20
INFORMAZIONI SUI FARMACI
12. FARMACO/I SOSPETTO/I (indicare il nome della specialità medicinale o del generico*). Riportare il numero di lotto per vaccini e medicinali biologici A)
13. LOTTO
14. DOSAGGIO/FREQUENZA (specificare)
15. VIA DI SOMMINISTRAZIONE
16. DURATA DELL'USO: DAL
AL
17. IL FARMACO E’ STATO SOSPESO? SI NO 18. LA REAZIONE E' MIGLIORATA DOPO LA SOSPENSIONE? SI NO 19. IL FARMACO E’ STATO RIPRESO? SI NO 20. SONO RICOMPARSI I SINTOMI DOPO LA RISOMMINISTRAZIONE? SI NO
B)
13. LOTTO
14. DOSAGGIO/FREQUENZA (specificare)
15. VIA DI SOMMINISTRAZIONE
16. DURATA DELL'USO: DAL
AL
17. IL FARMACO E’ STATO SOSPESO? SI NO 18. LA REAZIONE E' MIGLIORATA DOPO LA SOSPENSIONE? SI NO 19. IL FARMACO E’ STATO RIPRESO? SI NO 20. SONO RICOMPARSI I SINTOMI DOPO LA RISOMMINISTRAZIONE? SI NO
C)
13. LOTTO
14. DOSAGGIO/FREQUENZA (specificare)
15. VIA DI SOMMINISTRAZIONE
16. DURATA DELL'USO: DAL
AL
17. IL FARMACO E’ STATO SOSPESO? SI NO 18. LA REAZIONE E' MIGLIORATA DOPO LA SOSPENSIONE? SI NO 19. IL FARMACO E’ STATO RIPRESO? SI NO 20. SONO RICOMPARSI I SINTOMI DOPO LA RISOMMINISTRAZIONE? SI NO
* Nel caso di vaccini specificare anche il numero di dosi e/o di richiamo, l’ora e il sito della somministrazione
Prego, girare il foglio

17
Bollettino Farmacovigilanza Bergamasco
21. INDICAZIONI O ALTRO MOTIVO PER CUI IL FARMACO È STATO USATO (le lettere fanno riferimento ai farmaci indicati precedentemente):
A:
B:
C:
22. FARMACO/I CONCOMITANTE/I (indicare il nome della specialità medicinale o del generico*). Riportare il numero di lotto per vaccini e medicinali biologici A)
23. LOTTO
24. DOSAGGIO/FREQUENZA (specificare)
25. VIA DI SOMMINISTRAZIONE
26. DURATA DELL'USO: DAL
AL
27. IL FARMACO E’ STATO SOSPESO? SI NO 28. LA REAZIONE E' MIGLIORATA DOPO LA SOSPENSIONE? SI NO 29. IL FARMACO E’ STATO RIPRESO? SI NO 30. SONO RICOMPARSI I SINTOMI DOPO LA RISOMMINISTRAZIONE? SI NO B)
23. LOTTO
24. DOSAGGIO/FREQUENZA (specificare)
25. VIA DI SOMMINISTRAZIONE
26. DURATA DELL'USO: DAL
AL
27. IL FARMACO E’ STATO SOSPESO? SI NO 28. LA REAZIONE E' MIGLIORATA DOPO LA SOSPENSIONE? SI NO 29. IL FARMACO E’ STATO RIPRESO? SI NO 30. SONO RICOMPARSI I SINTOMI DOPO LA RISOMMINISTRAZIONE? SI NO
* Nel caso di vaccini specificare anche il numero di dosi e/o di richiamo, l’ora e il sito della somministrazione
31. INDICAZIONI O ALTRO MOTIVO PER CUI IL FARMACO È STATO USATO (le lettere fanno riferimento ai farmaci indicati qui sopra):
A:
B:
32. USO CONCOMITANTE DI ALTRI PRODOTTI A BASE DI PIANTE OFFICINALI, INTEGRATORI ALIMENTARI, ecc. (specificare):
33. CONDIZIONI PREDISPONENTI e/o CONCOMITANTI (se il farmaco sospetto è un vaccino riportare l'anamnesi ed eventuali vaccini somministrati nelle 4 settimane precedenti alla somministrazione)
34. ALTRE INFORMAZIONI
INFORMAZIONI SULLA SEGNALAZIONE E SUL SEGNALATORE
35. INDICARE SE LA REAZIONE E' STATA OSSERVATA NELL'AMBITO DI: Progetto di Farmacovigilanza Attiva Registro Farmaci
Studio Osservazionale, specificare: titolo studio
tipologia
numero
36. QUALIFICA DEL SEGNALATORE □ MEDICO OSPEDALIERO 37. DATI DEL SEGNALATORE (i dati del segnalatore sono trattati in modo confidenziale)
NOME E COGNOME:
INDIRIZZO:
TEL E FAX:
E-MAIL:
MEDICO MEDICINA GENERALE PEDIATRA LIBERA SCELTA
SPECIALISTA MEDICO DISTRETTO
FARMACISTA INFERMIERE
CAV ALTRO (specificare):
38. ASL DI APPARTENENZA:
39. REGIONE:
40. DATA DI COMPILAZIONE:
41. FIRMA DEL SEGNALATORE


















