
Azienda Ospedaliera San Camillo-Forlanini · ( emoglobinopatie strutturali con diversa stabilità o...
17
Azienda Ospedaliera San Camillo-Forlanini Centro di Riferimento Regionale per la Diagnosi e Terapia delle Anemie Ereditarie ( Codice Centro 12090102 ) Medico responsabile: RONDINELLI MARIA BEATRICE- tel. 06587034733–3558 Circonvallazione Gianicolense 87- 00152 Roma [email protected] Azienda Policlinico Umberto I Centro Malattie Rare Ematologiche-Ematologia (Codice Centro 12090602) Medici responsabili: FIORINA GIONA - tel.06/49974739 Ematologia, Via Benevento n° 6 –00161 Roma [email protected] PELLEGRINA PUGLIESE - tel. 0649976550 - UOC Immunoematologia e Medicina Trasfusionale, Via Chieti 7 – 00161 Roma [email protected] SMACCHIA MARIA PAOLA- tel. 06 49979259 - DAI Pediatria e NPI - Pediatria Edificio A Viale Regina Elena n. 324 – 00161 Roma m.smacchia@policlinicoumberto1 IRCCS Ospedale Pediatrico Bambino Gesù Centro di Riferimento Regionale per la Diagnosi e Terapia delle Anemie Ereditarie (Codice Centro: 12090406) Medico responsabile: LUISA STROCCHIO - tel. 06/68592679 - [email protected] Piazza S. Onofrio, 4 - 00165 Roma (Padiglione Giovanni Paolo II, piano 3)
-
Upload
truongcong -
Category
Documents
-
view
248 -
download
0
Transcript of Azienda Ospedaliera San Camillo-Forlanini · ( emoglobinopatie strutturali con diversa stabilità o...

Azienda Ospedaliera San Camillo-Forlanini
Centro di Riferimento Regionale per la Diagnosi e Terapia delle Anemie Ereditarie
( Codice Centro 12090102 )
Medico responsabile: RONDINELLI MARIA BEATRICE- tel. 06587034733–3558
Circonvallazione Gianicolense 87- 00152 Roma
Azienda Policlinico Umberto I
Centro Malattie Rare Ematologiche-Ematologia
(Codice Centro 12090602)
Medici responsabili: FIORINA GIONA - tel.06/49974739 Ematologia, Via Benevento n° 6 –00161 Roma
PELLEGRINA PUGLIESE - tel. 0649976550 - UOC Immunoematologia e Medicina Trasfusionale,
Via Chieti 7 – 00161 Roma [email protected]
SMACCHIA MARIA PAOLA- tel. 06 49979259 - DAI Pediatria e NPI - Pediatria Edificio A
Viale Regina Elena n. 324 – 00161 Roma m.smacchia@policlinicoumberto1
IRCCS Ospedale Pediatrico Bambino Gesù
Centro di Riferimento Regionale per la Diagnosi e Terapia delle Anemie Ereditarie
(Codice Centro: 12090406)
Medico responsabile: LUISA STROCCHIO - tel. 06/68592679 - [email protected]
Piazza S. Onofrio, 4 - 00165 Roma (Padiglione Giovanni Paolo II, piano 3)

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
2
Fondazione Policlinico Universitario A Gemelli
Largo A. Gemelli 8 00168 Roma
Centro di Riferimento Regionale per la Diagnosi e Terapia delle Anemie Ereditarie
(Codice Centro 12090503)
Coordinatore VALERIO DE STEFANO Tel 063015 4968 o 4206 [email protected]
Medici Responsabili: BIANCA MARIA RICERCA (Ematologia) Tel 0630154968 [email protected]
ANTONIO RUGGIERO (Pediatria) Tel 063015 5134 [email protected]
OSPEDALE S. EUGENIO
Piazzale dell’Umanesimo 10- 00144 Roma
UO Talassemici – Malattie Rare del Globulo Rosso
Medico responsabile : SORRENTINO FRANCESCO – tel 0651002560 - [email protected]
ANEMIE EREDITARIE (RDG010)
PERCORSO DIAGNOSTICO TERAPEUTICO ASSISTENZIALE (elaborato nel mese di Ottobre 2016 - a cura dei a cura dei Centri di Riferimento –AO San Camillo-Forlanini/ Azienda
Policlinico Umberto I-Università Sapienza / Ospedale Pediatrico Bambino Gesù / Fondazione Policlinico Gemell i- Roma
1. Inquadramento della malattia ……………….…………………………………………………………...……3
2. Strumenti per la diagnosi …………………………..…………………………………………….……………6
3. Terapia ……………………………………………………………………………………..……………………...8
4. Controlli di salute..………………………………………..…………………..……….…………….………....11
5. Modalità di accesso al Centro e servizi offerti ………………………………………………..……….….13
6. Collaborazioni con altri Centri nazionali e internazionali…………………………………………..…..16
7. Rapporti con le Associazioni……………………………………………………..……….……...……...…..16

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
3
1. Inquadramento della malattia
Le anemie ereditarie sono un gruppo eterogeneo di malattie del sangue caratterizzate da una ridotta concentrazione di emoglobina, dovute a difetti che riguardano la sintesi dell’emoglobina, la membrana eritrocitaria, gli enzimi eritrocitari oppure a insufficiente produzione da difetto dell’eritropoiesi midollare.
EMOGLOBINOPATIE Talassemie
Drepanocitosi
Altre emoglobinopatie clinicamente rilevanti
PATOLOGIE DELLA MEMBRANA ERITROCITARIA Sferocitosi
Ellissocitosi
Stomatocitosi
PATOLOGIE DA DIFETTO ENZIMI ERITROCITARI Deficit di glucosio-6-fosfato deidrogenasi (G6PD)
Deficit di piruvato-chinasi (PK)
Altre enzimopenie clinicamente rilevanti
ANEMIE CONGENITE DA PREVALENTE DIFETTO MIDOLLARE
Anemia di Fanconi
Anemia di Blackfan-Diamond
Anemie diseritropoietiche congenite
Anemie sideroblastiche congenite
1.1. EMOGLOBINOPATIE Sono le anemie ereditarie più frequenti e l’Italia è uno dei paesi con maggiore incidenza. La malattia, inizialmente endemica in Sardegna, in Sicilia , nell’Italia del sud e nel Delta del Po, si è diffusa in seguito ai fenomeni migratori spec ie nel periodo post bellico .Ad oggi si contano circa 7000 pazienti e oltre 2,5 milioni di portatori sani. In considerazione inoltre dei flussi migratori, attualmente l’8% della popolazione italiana è costituita da migranti provenienti sopratutto da zone ad alta incidenza di emoglobinopatie, per cui il numero dei pazienti e portatori verosimilmente tenderà ad aumentare. In teoria oggi non esiste un paese del mondo in cui le emoglobinopatie non siano presenti e fin dal 2006 l’Executive Board dell’Organizzazione Mondiale della Sanità indicava le Talassemie e l’Anemia falciforme come problematiche maggiori che richiedono azioni immediate da parte dei governi. Le Anemie legate ad emoglobinopatie sono dovute a difetti quantitativi di sintesi globinica ( Talassemie ) o qualitative ( emoglobinopatie strutturali con diversa stabilità o solubilità o affinità per l’ossigeno , ad es. l’ Anemia Falciforme o Drepanocitosi ).
Le sindromi talassemiche sono malattie genetiche a trasmissione autosomica recessiva caratterizzate da ridotta o assente sintesi delle catene emoglobiniche. L’alta incidenza nei paesi rivieraschi del Mediterraneo e la prima descrizione della malattia da parte di Cooley e Lee nel 1925 in bambini originari di tale area giustificano il sinonimo di Anemia Mediterranea. Il difetto genetico può interessare le diverse catene globiniche. Ad oggi sono state individuate circa 400 mutazioni: le più significative sono a carico delle catene β , α e δ e pertanto si distinguono, beta-talassemie, alfa-talassemie e delta-beta talassemie. Ne fanno parte anche un gruppo di disordini definiti Persistenza di Emoglobina Fetale (HbF) ereditaria. Nel complesso le talassemie costituiscono la patologia paradigmatica dei disordini della aggregazione proteica. Nella Beta Talassemia (la forma più frequente nel nostro paese) le α catene in eccesso si aggregano tra loro e insieme agli emi formano gli emicromi che precipitano e danneggiano la membrana cellulare. L’eccesso di emi inoltre innesca la produzione di specie reattive dell’ossigeno (ROS) che reagiscono con il Ferro non altrimenti utilizzabile. Ne deriva una tossicità ferro-mediata con apoptosi dei precursori eritroidi, riduzione dei processi di differenziazione midollare e riduzione degli eritrociti circolanti con ridotta sopravvivenza per emolisi e quindi anemia. Questi eventi fisiopatologici determinano l’espansione midollare con caratteristiche anomalie ossee, eritropoiesi ectopica con epatomegalia, splenomegalia, il re-ciclyng del ferro e la soppressione della produzione della epcidina ( sostanza chiave del metabolismo del ferro) determinano l’emosiderosi e fibrosi di organi vitali.

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
4
Le talassemie sono malattie genetiche a carattere autosomico recessivo, per cui si distinguono le condizioni di: - portatore sano (eterozigote) - soggetto affetto (omozigote o eterozigote composto)
A queste condizioni corrispondono i seguenti quadri clinici - forma di gravità severa (talassemia maior) - forma intermedia (talassemia intermedia) - forma silente (condizione di eterozigosi o portatore di talassemia)
La Drepanocitosi o Anemia a Cellule falciformi con tali termini si definiscono un gruppo di disordini del globulo rosso ad ereditarietà autosomica recessiva, associati alla predominante presenza di emoglobina S ( HbS) , una variante patogenica della catena beta globinica . Tale variante può essere presente in omozigosi (HbS/S) o in eterozigosi mista con altre varianti beta globiniche : Hb C, β÷ Tal e β°Tal ( Microdrepanocitosi o Talasso-drepanocitosi ) o, più raramente, con le varianti D-Punjab, O-Arab e E. E’ la prima malattia molecolare identificata: la prima descrizione risale al 1910 (Herrick ) . Nel 1949 vennero dimostrate la presenza della emoglobinopatia (Pauling) e la trasmissione mendelliana del difetto ( Neel ).La condizione è causata da una mutazione puntiforme del gene beta globinico. La sostituzione di una base di adenina con una di timina determina la conversione della tripletta GAG in GTG che codifica per l’aminoacido Valina al posto dell’Acido Glutammico in posizione 6 della catena globinica. Tale mutazioni determina particolari caratteristiche di solubilità e stabilità della globina. In determinati condizioni infatti la HbS polimerizza (sickling) aggregandosi per formare una struttura filamentosa elicoidale (tactoide) che determina la caratteristica deformazione a falce dell’eritrocita. Inizialmente si forma un nucleo costituito solo da pochi tetrameri con HbS e il sickling è reversibile. Successivamente il processo diventa rapidamente esplosivo e irreversibile. La quantità e velocità di polimerizzazione è dovuta principalmente alla concentrazione intracellulare di HbS (fattore determinante). Altri fattori favorenti sono la ridotta pO2, l’elevato pH, la ridotta Temperatura. Rallentano e/o contrastano la polimerizzazione la concentrazione di 2-3 difosfoglicerato, la presenza di Hb Fetale e la presenza di concomitanti difetti delle alfa catene globiniche. I globuli rossi così trasformati non sono in grado di procedere attraverso il lume capillare e aderiscono all’endotelio vasale, coinvolgendo leucociti e piastrine, innescano il processo infiammatorio e attivano la cascata coagulativa e tendono ad emolizzare, Il paziente portatore del difetto ( HbS < 50%) è prevalentemente asintomatico. Per contro l’espressione fenotipica della malattia è costituita da eventi vaso-occlusivi intermittenti e dalla anemia emolitica cronica che possono essere contemporaneamente presenti nel paziente con predominanza dell’uno o dell’altro quadro clinico. Gli eventi vaso-occlusivi determinano lesioni ischemiche con sintomatologica acuta e cronica dolorosa che possono interessare qualsiasi distretto del corpo, possono essere potenzialmente fatali e richiedono intervento medico immediato, con modalità di urgenza o emergenza non differibile. Lo stato emolitico cronico determina segni e sintomi riconducibili allo stato anemico e formazione di colelitiasi. I pazienti che dimostrano un fenotipo prevalentemente emolitico possono essere relativamente protetti dalle sindromi vaso-occlusive ma risultano particolarmente predisposti a sviluppare ipertensione polmonare, priapismo e ulcere malleolari. 1.2. PATOLOGIE DELLA MEMBRANA ERITROCITARIA I difetti di membrana del globulo rosso danno origine ad un gruppo di malattie definite dal particolare aspetto che assumono le emazie dei soggetti affetti La Sferocitosi ereditaria è trasmessa come carattere autosomico dominante a penetranza variabile. Esiste una anomalia della sintesi o della struttura delle proteine di membrana eritrocitaria (spectrina, anchirina e proteina 3) che, di conseguenza, risulta permeabile al sodio. Le emazie presentano una ridotta resistenza osmotica che rende necessario, un aumentato dispendio energetico aumentato per impedire un eccessivo ingresso di sodio ed acqua. .Ad esaurimento di tale meccanismo difensivo la cellula, rigonfia e meno elastica, rimane intrappolata e sequestrata dal sistema reticolo-endoteliale della milza. L’emolisi è extra-vascolare. Simile alla Sferocitosi è la Elissocitosi il cui difetto di membrana è costituito da una anomalia della spectrina che impedisce all’emazia di riassumere la sua forma a disco biconcavo dopo passaggi nei capillari periferici. Altre forme rarissime di patologia di membrana sono le Stomatocitosi caratterizzate dalla particolare forma delle emazie che presenta nella zona centrale una fessura a forma di bocca . Il termine definisce un gruppo di disordini a trasmissione autosomica dominante, caratterizzata da alterata permeabilità della membrana e che determina emolisi delle emazie. Differiscono dalle sferocitosi ereditarie in quanto il difetto di membrana determina alterazione prevalentemente degli scambi elettrolitici nei globuli rossi. La loro individuazione è importante per i risvolti terapeutici.

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
5
1.3. PATOLOGIE DA DIFETTO ENZIMI ERITROCITARI Le anomalie ereditarie degli enzimi eritrocitari costituiscono un gruppo distinto di disordini genetici, tutti, tranne uno (il deficit di glucosio-6-fosfato deidrogenasi), rari o rarissimi. La maggior parte degli enzimi coinvolti sono “housekeeping”, cioè presenti in tutte le cellule; tuttavia, gli effetti della loro carenza finiscono per evidenziarsi soprattutto nei globuli rossi. 1) Il Deficit di glucosio-6-fosfato deidrogenasi (G6PD), noto come favismo, è una malattia genetica ereditaria a trasmissione X-linked dovuta a deficit dell'enzima G6PD, essenziale per la vitalità degli eritrociti e in particolare per i processi ossidoriduttivi che in essi si svolgono. La carenza di questo enzima provoca un'improvvisa distruzione dei globuli rossi (emolisi) con la comparsa di crisi emolitiche acute dopo assunzione di fave, bibite contenenti chinino (Chinotto, Shweps) o addizionati con vitamina C, alcuni farmaci (ad esempio sulfamidici, salicilici, chinidina, ecc.) (elenco presente nel sito http://www.g6pd.org) o in seguito a episodi infettivi (epatite virale, polmonite e febbre tifoide), che agiscono da "fattori scatenanti". Esistono più di 400 varianti di deficit di G6PD, distinte per caratteristiche biochimiche e funzionali: una forma precoce, più severa, quasi esclusiva dei maschi, che si manifesta già alla nascita con ittero neonatale grave in cui non è identificabile nessun fattore scatenante; una forma da deficit enzimatico severo (forma mediterranea); una forma da deficit moderato e una forma asintomatica. La gran parte degli individui affetti sono asintomatici. I pazienti sintomatici sono quasi esclusivamente maschi, per via dell'ereditarietà correlata al cromosoma X di questa malattia; le portatrici di sesso femminile potrebbero comunque manifestare clinicamente la malattia. È una malattia ubiquitaria, la cui incidenza è particolarmente elevata in alcune aree geografiche (Africa, Medio Oriente…). 2) Il Deficit di piruvato-chinasi (PK) è una eritroenzimopatia a trasmissione autosomica recessiva. Il deficit di PK comporta una minore disponibilità di ATP che determina un ridotto funzionamento della pompa sodio-potassio, con danno di membrana, diminuita plasticità della stessa e prematura distruzione degli eritrociti nel fegato e nella milza. Il quadro clinico è quello di un'anemia emolitica cronica di variabile severità, che può manifestarsi come anemia trasfusione-dipendente o come emolisi compensata senza anemia. Ittero e splenomegalia sono sempre presenti. La colelitiasi può svilupparsi sopratutto in età adulta. La frequenza stimata nella popolazione Caucasica è di 1/20.000. 3) Altri deficit enzimatici, clinicamente rilevanti, sono rarissimi: il deficit di glucosio fosfato isomerasi (GPI), di fosfofruttochinasi (PFK), di triosofosfato isomerasi (TPI), di esochinasi (HK), di adenilatochinasi (AK) e di pirimidin-5’-nucleotidasi (P5’N). Nei casi in cui l’enzima compromesso è espresso anche in altri tessuti, alle manifestazioni ematologiche si possono associare quadri sintomatologici più complessi, quali miopatia nel deficit di PFK, suscettibilità alle infezioni ed anormalità neuromuscolari nella carenza di TPI, ritardo mentale ed altre anormalità neurologiche nei deficit di fosfogliceratochinasi (PGK) e di aldolasi. Le manifestazioni cliniche spesso insorgono in età pediatrica o neonatale, e nei casi più gravi si può verificare anche la morte intrauterina. 1.4. ANEMIE EREDITARIE DA PREVALENTE DIFETTO MIDOLLARE Le anemie da difetto dell’eritropoiesi midollare sono un gruppo eterogeneo di condizioni determinate da una ridotta/assente produzione midollare che può coinvolgere selettivamente la serie eritroide o più linee emopoietiche (come l’anemia di Blackfan-Diamond e l’anemia di Fanconi) o da eritropoiesi inefficace (come l’anemia sideroblastica). L’Anemia di Fanconi è un disordine caratterizzato dalla variabile presenza di progressiva insufficienza midollare che può interessare le tre filiere emopoietiche (con anemia, piastrinopenia e/o neutropenia), multiple anomalie somatiche congenite e predisposizione allo sviluppo di leucemie e altre forme di neoplasie. A livello cellulare, caratteristiche della patologia sono la fragilità cromosomica e l'estrema sensibilità al trattamento con agenti che inducono danno al DNA. Alla base dell’anemia di Fanconi sono state attualmente individuate mutazioni in almeno 19 geni denominati FANC, coinvolti nei meccanismi di riparazione del danno al DNA, trasmesse con ereditarietà di tipo autosomico recessivo o, in una minoranza di casi, X-linked recessivo. L’Anemia di Blackfan-Diamond è una forma di insufficienza midollare costituzionale che interessa la serie eritroide, caratterizzata da grave anemia presente dalla nascita o dai primi mesi di vita, malformazioni congenite e aumentato rischio di neoplasie. Alla base della patologia, vi sono mutazioni dei geni coinvolti nella sintesi delle strutture ribosomiali, che possono essere sporadicheo trasmesse come carattere autosomico dominante. Le anemie diseritropoietiche congenite (CDA) sono un raro gruppo di disordini ereditari caratterizzati da anemia iporigenerativa, associata a caratteristici reperti midollari ed emocromatosi secondaria. La classificazione attuale riconosce 5 tipi: CDA tipo I, CDA tipo II, CDA tipo III familiare, CDA tipo III sporadica, forme “varianti”. Dal punto di vista clinico le varie forme sono molto simili: l’anemia, spesso evidente già presente in epoca neonatale, è lieve-moderata, per cui generalmente i pazienti non sono trasfusione-dipendenti. Il quadro clinico è spesso caratterizzato da ittero e splenomegalia; in una minoranza di casi inoltre, si associano anomalie scheletriche degli arti inferiori (CDA tipo I).

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
6
Le anemie sideroblastiche congenite rappresentano un gruppo di disordini ematologici, rari in età pediatrica, caratterizzati da un difetto della sintesi dell’eme, in cui il mancato utilizzo di ferro da parte dei precursori eritroidi determina un’eritropoiesi inefficace con sviluppo di anemia. Sono accomunate dalla presenza, allo striscio di sangue midollare, di eritroblasti contenenti accumuli di ferro in regione perinucleare (cosiddetti sideroblasti ad anello), evidenziabili con specifiche colorazioni (colorazione di Pearls). Le forme congenite (X-linked, autosomiche recessive o a ereditarietà materna, come nella sindrome di Pearson) possono essere suddivise in forme sindromiche e non-sindromiche (a seconda della presenza o meno di anomalie associate). L’età di insorgenza è variabile (dalla prima infanzia all’età adulta), così come il grado di anemia (da lieve a severo), con reticolocitopenia.
2. Strumenti per la diagnosi L’iter per la diagnosi delle emoglobinopatie comprende l’esecuzione di esami di I e II livello. Gli esami di I livello forniscono le informazioni utili per l’approfondimento diagnostico di II livello. 2.1 EMOGLOBINOPATIE L’iter per la diagnosi delle emoglobinopatie comprende l’esecuzione di esami di I e II livello. Gli esami di I livello forniscono le informazioni utili per individuare lo stato di paziente affetto o di portatore e prevenire patologie determinate geneticamente che nella condizione di eterozigosi sono asintomatiche. Oltre 400 difetti talassemici sono una sfida per il laboratorio considerando la loro elevata eterogeneità fenotipica. La richiesta può avvenire in diversi ambiti e prodotta da diversi specialisti. È indispensabile che lo specialista riporti nella richiesta il quesito diagnostico specifico ESAMI di I LIVELLO - Esame emocromocitometrico completo e reticolociti - Striscio di sangue periferico per l’esame della morfologia eritrocitaria - Glicemia, azotemia,creatinina - AST, ALT, GGT, bilirubina totale/diretta, aptoglobina, LDH - Sideremia, ferritina, transferrina, percentuale di saturazione della transferrina - Test di Coombs diretto e indiretto - High Performance Liquid Chromatography (HPLC) delle frazioni emoglobiniche, per la determinazione
quantitativa di HbS, HbA2 e HbF - Test di falcizzazione con metabisolfitodisodio al 2%. - In diagnosi prenatale, tecniche di genetica molecolare per la ricerca delle mutazioni sul DNA fetale estratto da
cellule del liquido amniotico o dei villicoriali. ESAMI DI II LIVELLO
Tali esami servono a caratterizzare i difetti molecolari e permettere la diagnosi differenziale tra /α/δTalassemia.
Sono obbligatori in caso di rischio di coppia in portatori accertati o sospetti, sospetta α talassemia in entrambi i partner - sintesi delle catene globiniche - analisi del DNA
ESAMI DIAGNOSTICI DI COMPLETAMENTO DEL QUADRO CLINICO: Ecografia del fegato,colecisti e vie biliari,milza Ecocardiogramma Doppler trans-cranico ( per pazienti in età pediatrica affetti da Anemia Falciforme) Valutazione clinico-funzionale dell’apparato respiratorio Valutazione della crescita (nei bambini Eventuale valutazione del SNC con tecniche di imaging (nella drepanocitosi) Valutazione di alcune funzioni cognitive (nei bambini affetti da drepanocitosi)
2.2 PATOLOGIE DELLA MEMBRANA ERITROCITARIA Storia familiare e anamnesi ESAMI di I LIVELLO - Esame emocromocitometrico completo e reticolociti - Striscio di sangue periferico per l’esame della morfologia eritrocitaria - indici di emolisi - bilancio marziale

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
7
- resistenze osmotiche eritrocitarie (ROE) - SDS-PAGE delle proteine di membrana e loro quantificazione percentuale - Test di lisi al glicerolo a PH 7.4 e a PH 6.85 - Resistenza osmotica a fresco e dopo incubazione a 37 C° - Pink test - EMA binding test - Test dell'autoemolisi (senza additivi, + glucosio, + ATP) - Ricerca di corpi inclusi eritrocitari, delle emoglobine instabili, screening delle emoglobine (HPLC) 2.3 PATOLOGIE DA DIFETTO ENZIMI ERITROCITARI Storia familiare e anamnesi ESAMI I LIVELLO - Esame emocromocitometrico completo e reticolociti, - morfologia eritrocitaria, - bilirubina, aptoglobina, LDH, - stato del ferro (sideremia, ferritina, transferrina, indice di saturazione della transferrina) ESAMI DI II LIVELLO STUDIO DEL METABOLOSMO ERITROCITARIO Glicolisi: PK, HK, GPI, GAPD, PGK, PFK, TPI, LDH, Aldolasi, Enolasi, DPGM, MPGM. Enzimi dello shunt: G6PD, 6PGD, GR, GSH-Px. Altri enzimi eritrocitari: AK, PYR, 5'N, NADH diaf, PGM, catalasi ESAMI DI III LIVELLO Caratterizzazione molecolare del gene coinvolto o, nel caso dei difetti di membrana dei polimorfismi satelliti 2.4 ANEMIE EREDITARIE DA PREVALENTE DIFETTO MIDOLLARE ESAMI di I LIVELLO - Esame emocromocitometrico completo con MCV e conta reticolocitaria; - Striscio di sangue venoso periferico; - Glicemia, azotemia, creatininemia; - GOT, GPT, GGT, bilirubina totale/diretta, aptoglobina,LDH; - Dosaggio amilasi/lipasi sieriche, elastasi fecale, valutazione equilibrio acido-base; - agoaspirato midollare per valutazione morfologica, colorazioni per il ferro, analisi citogenetica convenzionale ed
eventuale FISH; - biopsia osteomidollare; - High Performance Liquid Chromatography (HPLC) delle frazioni emoglobiniche, per la determinazione di HbF; - dosaggio di alfa-fetoproteina sierica; - Test di fragilità cromosomica al diepossibutano (DEB test); - Dosaggio adenosina deaminasi eritrocitaria (ADA) ESAMI di APPROFONDIMENTO/DIAGNOSTICA DIFFERENZIALE - Analisi citofluorimetrica su sangue midollare; - ricerca di cloni PNH mediante citofluorimetria multiparametrica; - dosaggio eritropoietina sierica; - test clonogenici per la valutazione della crescita in vitro dei precursori eritroidi; - Dosaggio di vitamina B12 e acido folico; - Valutazione dello stato marziale; - Fibrinogeno e ferritina; - studio della lunghezza dei telomeri; - sierologia o ricerca del genoma virale di Parvovirus B19, EBV, CMV, virus epatitici; - Screening per autoanticorpi. ESAMI di II LIVELLO Anemia di Fanconi Nella maggior parte dei casi la diagnosi può essere posta con la combinazione di obiettività clinica e positività del DEB test. Esame diagnostico di più recente applicazione è rappresentato dall’analisi del ciclo cellulare mediantecitometria a flusso di linfociti da sangue periferico che permette di documentare un accumulo di cellule in fase G2/M 6. Una volta posta la diagnosi, analisi mirate a una completa definizione diagnostica del paziente sono: - l’analisi di complementazione (eseguita in un numero limitato di centri di ricerca);

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
8
- l’analisi genetica per la ricerca di mutazioni dei geni FANC: non generalmente indicata tra le indagini diagnostiche di prima linea; possibile eccezione è rappresentata da famiglie con mutazioni note di un gene FANC in cui tale analisi permette: 1) la diagnostica prenatale e il counseling genetico; 2) l’estensione dell’indagine al nucleo familiare al fine di identificare eventuali soggetti affetti ancora fenotipicamente silenti; 3) la accurata genotipizzazione di fratelli/sorelle al fine di escludere eventuali soggetti omozigoti dai potenziali donatori di cellule staminali emopoietiche ai fini trapiantologici7.
Anemia di Blackfan-Diamond - analisi genetica per la ricerca di mutazioni dei geni coinvolti nella sintesi delle strutture ribosomiali: tale analisi, oltre a permettere una completa definizione diagnostica del paziente affetto, riveste un ruolo di rilievo relativamente a: 1) diagnostica prenatale e counseling genetico nei casi di mutazioni non sporadiche; 2) estensione dell’indagine al nucleo familiare al fine di identificare eventuali soggetti affetti ancora fenotipicamente silenti; 3) accurata genotipizzazione di fratelli/sorelle al fine di escludere eventuali soggetti affetti dai potenziali donatori di cellule staminali emopoietiche ai fini trapiantologici. Anemie diseritropoietiche congenite (CDA)La diagnosi è fortemente indirizzata dall’esame dello striscio di sangue midollare che mostra caratteristiche anomalie degli eritroblasti, quali ponti di cromatina (CDA tipo I), eritroblasti binucleati (CDA tipo I a II), eritroblasti giganti multi nucleati (CDA tipo III) ed è confermata da: - analisi geneticaper la ricerca di mutazioni dei geni CDAN1 e C15ORF41 (CDA tipo I), SEC23B (CDA tipo II), KIF23 (CDA tipo III), KLF1 e GATA-1 (forme “varianti”). Anemie sideroblastiche - analisi genetica per la ricerca delle mutazioni note (le più frequenti coinvolgenti il gene ALAS2, responsabile
della forma X-linked, XLSA) 11. ESAMI DI COMPLETAMENTO DEL QUADRO CLINICO:
- studio radiografico di eventuali distretti sospetti per anomalie scheletriche congenite; - studio ecografico di addome, reni e vie urinarie; - valutazione urologica con attenzione alla possibile presenza di reflusso vescico-ureterale, infezioni delle vie
urinarie e malformazioni genito-urinarie; - valutazioneauxo-endocrinologica, comprensiva di valutazione dell’accrescimento staturo-ponderale, della
funzionalità tiroidea, della tolleranza glucidica, dello stato lipidico e della densità minerale ossea; - valutazione otorinolaringoiatrica mirata ad indagare eventuali deficit dell’udito o anomalie strutturali dell’orecchio e
all’identificazione precoce di neoplasie del distretto testa-collo; - valutazione oftalmologica, in caso di indicazione clinica; - valutazione cardiologica con ecocardiogramma; - valutazione ginecologica, specie in caso di riscontro di anomalie renali.
3. Terapia
3.1 EMOGLOBINOPATIE TALASSEMIE Terapia Trasfusionale. Il regime trasfusionale, ormai standardizzato, prevede il mantenimento di un valore dell’emoglobina pre-trasfusionale tra 9,0-10,0 gr/dl. Una adeguata terapia trasfusionale assicura al paziente una crescita ed uno sviluppo regolare ed una buona qualità di vita. La migliore strategia trasfusionale prevede il mantenimento del target con il minor numero di accessi ospedalieri possibili. Il trattamento raccomandato comprende regolari trasfusioni di sangue ogni 2-4 settimane.. La trasfusione si effettua con concentrato eritrocitario, filtrato e leucodepleto, che in casi selezionati è sottoposto a lavaggio. La quantità di GR da trasfondere in ogni seduta è stabilita in base a: - aumento di Hb necessario
- peso del paziente - ematocrito (Ht) dell’unità da trasfondere
Inevitabilmente tale trattamento comporta un sovraccarico di ferro in quanto ogni unità di sangue trasfuso contiene 200 -250 mg di ferro ( circa 1,08 mg/ml), che con il tempo si accumula nei vari organi. Inoltre questo trattamento si associa alla possibile trasmissione di infezioni virali Terapia ferrochelante. Il regime ipertrasfusionale nella Talassemia Major e l’aumentato assorbimento del ferro intestinale nella Talassemia Intermedia determinano uno stato di emosiderosi che può causare danni a molti tessuti ed organi e può essere letale se non viene eliminato con la terapia chelante.Tre sono gli obiettivi della terapia ferrochelante:

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
9
- Mantenere un corretto bilancio del ferro con valori tissutali ottimali - Mantenere un effetto di chelazione continua per ottenere una rimozione del pool di ferro labile intracellulare (LIC) ed extracellulare (Ferro non legato alla transferrina o NTBI che genera ferro labile plasmatico o LPI ) - Mantenere un ampio margine terapeutico di sicurezza La terapia ferrochelante dovrà essere iniziata quando si evidenzia un iniziale sovraccarico di ferro trasfusionale dopo circa 10-12 trasfusioni di emazie e/o quando si rileva un valore di ferritina circa 1000 ng/ml, anche se il solo valore di ferritina non rispecchia la reale entità della siderosi corporea. La possibilità di utilizzare altre metodiche per la valutazione della emosiderosi quali la determinazione della liver iron concentration ( LIC), la RMN T2* cardiaca ed epatica, lo Squid epatico, forniscono insieme alla ferritina ed alla sideruria una stima più precisa del grado di accumulo di ferro negli organi. Attualmente i farmaci chelanti in distribuzione sono: Desferioxaminache si somministra in infusione sottocutanea per non meno di 10 ore per un minimo di 6 notti a settimana, alla dose di circa 50 mg/Kg peso corporeo. Deferiproneè un chelante orale in compresse, che viene somministrato alla dose di 75-100mg/Kg/die suddivisa in tre dosi giornaliere Deferasirox è un chelante in sospensione orale somministrato alla dose di 10/40 mg/Kg/die in una unica somministrazione giornaliera. DREPANOCITOSI La drepanocitosi è una malattia ereditaria cronica dove la prevenzione delle temibili manifestazioni cliniche e stimolare una corretta autogesione da parte del paziente rivestono un ruolo di primaria importanza, ma pur non disponendo di farmaci che impediscano la gelificazione delle catene emoglobiniche, le sue manifestazioni possono essere adeguatamente trattate e prevenute. I consigli di prevenzione nell’autogestione della malattia rivestono un ruolo fondamentale per evitare situazioni che portano ad una minore disponibilità di ossigeno o ne aumentano il fabbisogno e che porterebbero quindi al fenomeno della falcizzazione. Ogni distretto vascolarizzato può essere interessato da tale evento, ma maggiormente gli organi a flusso ematico più lento (osso, milza, midollare renale, fegato) o quelli ad attività metabolica più elevata. Le crisi dolorose variano per gravità e per durata.
Nella fase acuta della crisi falcemica:
- analgesia da iniziare prima possibile
- idratazione e.v.
- se febbre:antipiretico
- se segni di infezione iniziare antibioticoterapia a largo spettro
- eventuale ossigenoterapia
- eventuale trasfusione di emazie o scambio eritrocitario manuale ( diluizione della percentuale di HbS) o eritrocitoaferesi ( sostituzione della maggior quota di HbS). In ogni caso il trattamento trasfusionale deve rispettare valori di Hb post non > 10 g/dl e di Ematocrito non > 30 % al fine di evitare pericolosi aumenti di viscosità ematica.
- Per questi pazienti è, vista l’alta incidenza di alloimmunizzazioni è necessario la corretta tipizzazione per AB0, Rh e Kell. Per pazienti che iniziano il trattamento rasfusionale in età giovanile o adulta e risultano portatori di almeno un allo-anticorpo, è indicata la tipizzazione per i sistemi Duffy e MNSs
Per la gestione degli eventi acuti utile l’algoritmo pubblicato nel sito www.site-italia.org della Società Italiana di Talassemia ed emoglobinopatie L’unico approccio terapeutico efficace e poco tossico di largo impiego è la IDROSSIUREA (HU). E’ un farmaco citotossico che determina incremento della HbF con meccanismi ancora non del tutto chiari. E’ un potente fattore che contrasta la polimerizzazione della HbS attraverso la riduzione della percentuale della emoglobina mutata e la formazione di ibridi HbF/HbS. Il trattamento con HU è correlato a una riduzione delle frequenza delle crisi vaso-occlusive, degli episodi di sindrome toracica acuta, di stroke, di priapismo , dei sintomi legati all’anemia e della mortalità in genere. Indipendentemente dall’induzione alla produzione di Hb F, determina benefici perché determina una miglior idratazione cellulare, inibisce le proteine di membrana coinvolte nei processi di adesione cellulare e funge da donatore di ossido nitrico, responsabile della vasocostrizione . E’ fortemente raccomandato anche in paziento di età pediatrica . , induce importanti variazioni, anche a livello cellulare sulla produzione delle citochine e dell’attività endoteliale.Similmente ai pazienti affetti da talassemia , tutti i soggetti sottoposti a regolari trasfusioni di emazie , con

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
10
carico trasfusionale cumulativo di almeno 20 trasfusioni e ferritina > 1000 ng/ml, necessitano della valutazione dell’emosiderosi ed a eventuale trattamento chelante con le modalità esposte nel capitolo riservato alle talassemie.
TRAPIANTO DI CELLULE STAMINALI EMATOPOIETICHE (TCSE) NELLA TALASSEMIA MAJOR E NELLA DREPANOCITOSI
I significativi miglioramenti raggiunti negli ultimi 30 anni in termini di terapia di supporto hanno condotto a un notevole miglioramento dell’aspettativa e della qualità di vita dei pazienti con talassemia major e drepanocitosi, portando tuttavia alla luce nuove necessità mediche/assistenziali legate al progressivo sviluppo di complicanze a lungo termine correlate sia alla condizione di cronicità della patologia sia al trattamento della stessa. Al momento attuale, il trapianto allogenico di cellule staminali ematopoietiche (TCSE) rimane l’unica strategia consolidata potenzialmente in grado di curare in modo definitivo questi pazienti. Tuttavia, la possibilità di offrire la procedura trapiantologica in patologie in cui l’aspettativa di vita può raggiungere attualmente la terza-quarta decade di vita va accuratamente ponderata in relazione al rischio di morbidità e mortalità trapianto-correlate. I notevoli progressi ottenuti in campo trapiantologico hanno permesso di raggiungere, nelle emoglobinopatie, probabilità di sopravvivenza globale e libera da malattia dopo TCSE da fratello/sorella HLA-identico superiori al 90% e 80%, rispettivamente. Le attuali raccomandazioni relative al TCSE nelle emoglobinopatie (EBMT Paediatric Diseases Working Party and Inborn Error Working Party; AIEOP) suggeriscono le seguenti indicazioni:
Talassemia: l’esperienza trapiantologica acquisita negli ultimi 30 anni ha permesso di identificare parametri che influenzano positivamente la probabilità di successo del TCSE, tra cui l’assenza di epatomegalia o fibrosi epatica, la regolarità della ferrochelazione e la giovane età del paziente. In considerazione di tali evidenze, la possibilità della scelta trapiantologica andrebbe offerta ai pazienti che dispongono di un donatore HLA-compatibile, in una fase precoce di malattia, prima dello sviluppo di complicanze e/o danno d’organo correlati con il sovraccarico marziale.
Drepanocitosi: le indicazioni attualmente suggerite dal Gruppo di Lavoro “Patologia del globulo rosso” dell’AIEOP includono 1) stroke; 2) sindrome toracica acuta ricorrente non responsiva al trattamento con idrossiurea; 3) crisi dolorose ricorrenti non responsive al trattamento con idrossiurea; 4) retinopatia proliferativa bilaterale o deficit visivo maggiore; 5) osteonecrosi multiple; 6) altra complicanza d’organo potenzialmente evolutiva e/o invalidante; 7) alloimmunizzazione alle trasfusioni eritrocitarie; 8) impossibilità ad aderire ai trattamenti medici proposti. Sono inoltre stati suggeriti altri fattori di rischio da prendere in considerazione nella valutazione della scelta trapiantologica dei pazienti con drepanocitosi, tra cui l’elevata velocità di flusso vascolare determinata mediante doppler transcranico (TCD), episodi ricorrenti di sequestro splenico, ipertensione polmonare, stroke silente con ritardo cognitivo, episodi ricorrenti di priapismo, nefropatia.
La possibilità di TCSE da donatore/fonte alternativi di cellule staminali ematopoietiche deve essere presa in considerazione e valutata sulla base dello status di malattia e della storia clinica del singolo paziente. In considerazione delle peculiarità di tale procedura, in particolare nelle emoglobinopatie, tale approccio dovrebbe essere riservato a Centri Trapianto con consolidata esperienza in tal senso.
3.2 PATOLOGIE DELLA MEMBRANA ERITROCITARIA Nella Sferocitosi ereditaria il trattamento si basa sulla somministrazione periodica di acido folico e di vitamina B 12 per sostenere l’iperproduzione di eritrociti. Si ricorre alla splenectomia se l’organomagalia è tale da determinare ingombro meccanico con dislocazione dei visceri e/o se si istaura un ipersplenismo. Inoltre L’emolisi cronica può determinare la formazione di colelitiasi e conseguente necessità di ricorrere alla colecistectomia. 3.3 PATOLOGIE DA DIFETTO ENZIMI ERITROCITARI Deficit di G6PD È indispensabile, al momento della diagnosi, fornire al paziente l’elenco dei farmaci e delle sostanze ossidanti da evitare. La crisi emolitica acuta da deficit di G6PD può richiede ricovero immediato per la valutazione della gravità dell’emolisi (clinica e di laboratorio). Il trattamento comporta una idratazione adeguata al fine di prevenire il danno renale conseguente all'emoglobinuria; in caso di emolisi massiva è necessario il supporto trasfusionale. Utile anche il supporto vitaminico con acido folico e vitamina B12. Deficit di PK e degli altri enzimi clinicamente rilevanti Non esistono trattamenti specifici per le anemie emolitiche croniche da difetti del metabolismo eritrocitario, e la terapia è pertanto essenzialmente di supporto con acido folico per compensare l'aumentato fabbisogno di tale vitamina. Il supporto trasfusionale può essere necessario nei casi di emolisi severa, in particolare nei bambini, nelle esacerbazioni occasionali dell’anemia dovute a infezioni o gravidanza, o durante le crisi aplastiche. La splenectomia non arresta

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
11
l'emolisi ma è spesso in grado di stabilizzare o migliorare i livelli di emoglobina, o comunque di ridurre il fabbisogno trasfusionale nei casi con anemia severa. 3.4 ANEMIE EREDITARIE DA PREVALENTE DIFETTO MIDOLLARE ANEMIA DI FANCONI In assenza di manifestazioni ematologiche o di anomalie clonali è sufficiente un monitoraggio periodico dell’esame emocromocitometrico e dell’aspirato midollare (vedi paragrafo “Controlli di salute”). In tutti i casi, è raccomandato eseguire la tipizzazione HLA di paziente e nucleo familiare per valutare l’eventuale disponibilità di potenziali donatori (fratelli/sorelle) HLA-identici nel caso si rendesse necessario un trapianto di cellule staminali ematopoietiche. Se la tipizzazione non consentisse di identificare un donatore HLA-identico nel contesto familiare, è indicata l’attivazione della ricerca nell’ambito dei registri dei donatori volontari di midollo osseo. In presenza di manifestazioni ematologiche e/o anomalie clonali, oltre all’incremento della frequenza del monitoraggio ematologico e delle valutazioni midollari (vedi paragrafo “Controlli di salute”), andranno presi in considerazione:
- terapia di supporto, mirata al trattamento delle manifestazioni legate all’insufficienza midollare con supporto trasfusionale eritrocitario e piastrinico (al fine di mantenere i valori di emoglobina entro livelli accettabili e di ridurre il rischio emorragico), profilassi/trattamento delle infezioni nei pazienti che sviluppano neutropenia (con eventuale ausilio di fattori di crescita emopoietici).
- trapianto allogenico di cellule staminali ematopoietiche, che attualmenterappresenta l’unica consolidata opzione terapeutica in grado di correggere definitivamente le manifestazioni ematologiche nei pazienti con anemia di Fanconi.
ANEMIA DI BLACKFAN-DIAMOND
- La terapia con corticosteroidi può permettere, in una buona percentuale di pazienti, di ottenere una risposta ematologica e, in alcuni casi, lunghi periodi di remissione anche dopo la sospensione del trattamento.
- Nei pazienti non responsivi alla terapia steroidea e in coloro costretti a sospendere il trattamento per importanti effetti collaterali, si rende necessaria una terapia trasfusionale associata a trattamento ferrochelante.
- In tali pazienti o in caso di mancato ottenimento di remissione ematologica, va presa in considerazione l’opzione del trapianto allogenico di cellule staminali ematopoietiche nei pazienti che dispongono di un donatore HLA-compatibile.
ANEMIE DISERITROPOIETICHE CONGENITE (CDA)Dal punto di vista terapeutico, oltre alla terapia trasfusionale laddove necessaria (specialmente nel periodo neonatale, in corso di infezioni o durante la gravidanza), alcuni pazienti
affetti da CDA tipo I rispondono alla somministrazione di interferone ; il ruolo della splenectomia non è chiaro, anche se i pazienti affetti da CDA tipo II sembrano beneficiare della procedura. In caso di riscontro di patologico accumulo di ferro andrà instaurata un’appropriata terapia ferrochelante. Infine, sono stati riportati casi di trapianto allogenico di cellule staminali ematopoietiche, in pazienti trasfusione-dipendenti, coronati da successo.
ANEMIE SIDEROBLASTICHE: Il trattamento si basa sulla somministrazione di cofattori dell’emopoiesi (piridossina) e, nelle forme non responsive, sulla terapia trasfusionale nei casi di anemia sintomatica. In considerazione della costante presenza di accumulo di ferro, vi è indicazione all’impiego di salassoterapia nelle forme di anemia lieve, e di terapia ferrochelante in quelle moderate-severe e trasfusione-dipendenti. Il trapianto allogenico di cellule staminali ematopoietiche rappresenta l’unica terapia potenzialmente curativa, da considerare nei pazienti non responsivi alle
terapie conservative e che dispongono di un donatore HLA-compatibile.
4. Controlli di salute 4.1 4.1 EMOGLOBINOPATIE
Monitoraggio esami di laboratorio e controlli strumentali A ogni trasfusione
- Esame emocromocitometrico - Visita medica breve
Ogni mese - Nei pazienti in trattamento chelante con Exjade dosaggio transaminasi, creatinina, ferritina

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
12
- Nei pazienti in trattamento chelante con Ferriprox la valutazione della granulocitopenia viene effettuata ad ogni trasfusione
- Per i pazienti affetti da Anemia falciforme in trattamento con Idrossiurea: esame emocromocitometrico con reticolociti, valutazione delle frazioni emoglobiniche in HPLC e degli indici di funzionalità renale ed epatica
Ogni 3 mesi - Esami ematochimici completi. Dosaggio ferritina
Ogni 6 mesi - Esame obiettivo completo, bilancio del ferro, coagulazione, funzionalità ghiandole endocrine, proteine totali ed
elettroforesi proteica, esame delle urine Ogni 12 mesi
- Esami strumentali e visite specialistiche, ECG, ecocardiogramma, visita cardiologia, esame audiometrico, visita oculistica + fundus oculi, ecografia addominale, MOC vertebrale e femorale, visita endocrinologia (con eventuale curva da carico orale di glucosio), visita ginecologica, bilancio del ferro (calcolo ferro introdotto/eliminato)
- Almeno annualmente per i pazienti in trattamento trasfusionale necessario il monitoraggio sierologico per virus - Per i pazienti affetti da anemia falciforme in età pediatrica: Doppler Transcranico
Ogni 24 mesi -SQUID o RMN T2* cardiaca - Angio RMN cranio per i pazienti affetti da Anemia Falciforme
ASSEMIE 4.2 PATOLOGIE DELLA MEMBRANA ERITROCITARIA
Ogni 6 mesi
Esame obiettivo completo, bilancio del ferro, coagulazione, indici di emolisi, LDH , aptoglobina , Bilirubina Totale e frazionata , proteine totali ed elettroforesi proteica, esame delle urine
Ogni 12 mesi Esami specialistici e consulenze se indicate
4.3 PATOLOGIE DA DIFETTO ENZIMI ERITROCITARI Deficit di G6PD I pazienti adeguatamente informati circa lo stile di vita da adottare eseguono almeno un controllo annuale degli esami ematochimici. Deficit di PK e degli altri enzimi clinicamente rilevanti Controlliematochimici e radiologici strumentali per monitorare l'organomegalia; la frequenza dei controlli è legata al livello di emolisi cronica del paziente.
4.4 ANEMIE EREDITARIE DA PREVALENTE DIFETTO MIDOLLARE ANEMIA DI FANCONI Monitoraggio ematologico8: 1) assenza di manifestazioni ematologiche o lieve citopenia con conte periferiche stabili in assenza di anomalie clonali:
- esame emocromocitometrico con reticolociti ogni 3-4 mesi; - valutazione annuale dell’aspirato midollare con valutazione citogenetica e biopsia osteomidollare.
2) assenza di manifestazioni ematologiche o lieve citopenia con conte periferiche stabili + evidenza di anomalie clonali: - esame emocromocitometrico con reticolociti ogni 1-2 mesi; - valutazione dell’aspirato midollare con valutazione citogenetica e biopsia osteomidollare più ravvicinata (1-6
mesi, sulla base dell’andamento clinico) 3) in caso di conte periferiche non stabili: valutazione ematologica e midollare rapida per valutare il quadro di
insufficienza midollare e la presenza di eventuale evoluzione clonale. Monitoraggio delle possibili alterazioni non ematologiche associate alla patologia: - valutazione auxo-endocrinologica annuale (valutazione dell’accrescimento staturo-ponderale, funzionalità tiroidea,
tolleranza glucidica, stato lipidico, densità minerale ossea); - valutazione otorinolaringoiatrica periodica, in particolare dopo i 10 anni di vita, prevalentemente mirata

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
13
all’identificazione precoce di neoplasie del distretto testa-collo; - il monitoraggio di altre possibili alterazioni non ematologiche associate alla patologia andrà personalizzato sulla base
del quadro clinico di ogni singolo paziente.
ANEMIA DI BLACKFAN-DIAMOND La frequenza del monitoraggio dell’esame emocromocitometrico dipende dalle condizioni cliniche del paziente, dal trattamento in atto (regime trasfusionale regolare o terapia steroidea) e dal grado di risposta al trattamento. Nei pazienti trasfusione-dipendenti, una valutazione dell’esame emocromocitometrico è indicata ogni 3-5 settimane, sulla base del programma trasfusionale; in tali pazienti è raccomandato un monitoraggio del sovraccarico marziale analogo a quello richiesto nei pazienti affetti da Thalassemia major. Nei pazienti in trattamento steroideo, il controllo delle conte periferiche dovrà essere eseguito almeno ogni 3-4 mesi. Non vi è indicazione a valutazioni midollari periodiche; l’esecuzione di agoaspirato midollare e biopsia osteomidollare è tuttavia raccomandata in caso di modifica delle conte periferiche/comparsa di citopenia bi- o trilineare. In tutti i pazienti sono raccomandati: - valutazione auxo-endocrinologica annuale (valutazione dell’accrescimento staturo-ponderale, funzionalità tiroidea,
tolleranza glucidica, densità minerale ossea); - valutazione oftalmologica periodica, in particolare nei pazienti in terapia steroidea; - il monitoraggio di altre possibili alterazioni non ematologiche associate alla patologia andrà personalizzato sulla base
del quadro clinico di ogni singolo paziente.
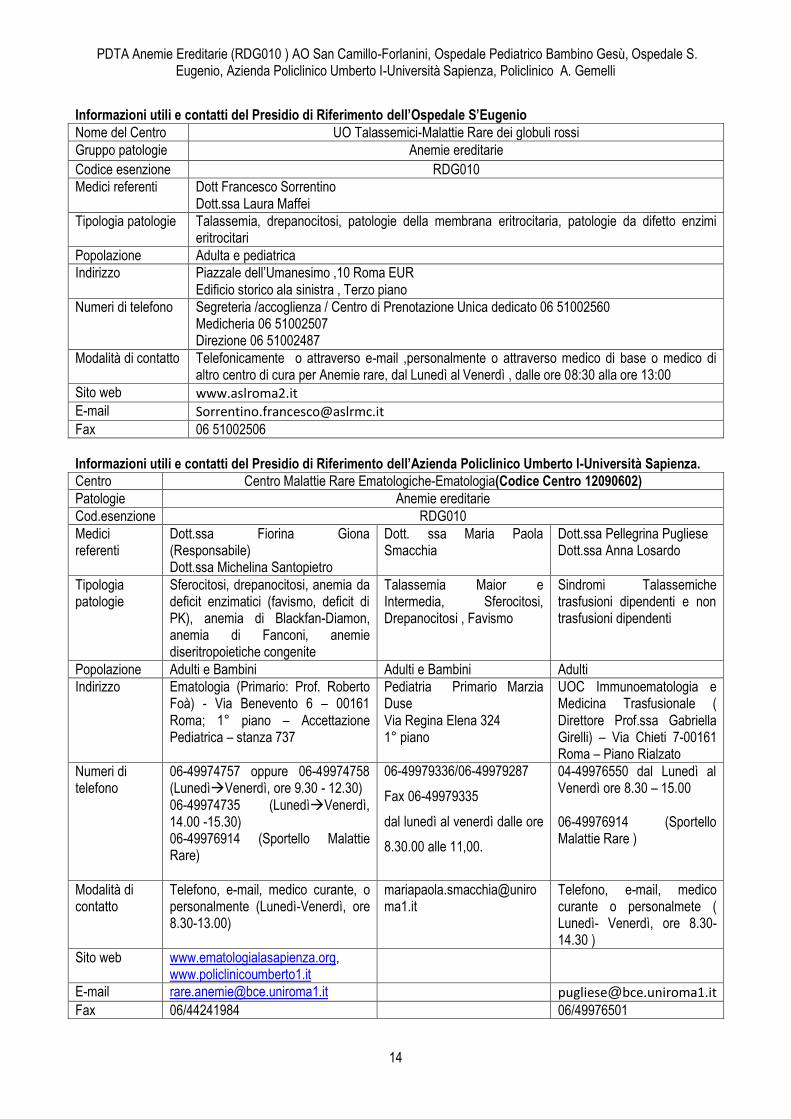
5. Modalità di accesso al Centro e servizi offerti Informazioni utili e contatti del Presidio di Riferimento dell’AO San Camillo-Forlanini.
Nome del Centro Dipartimento dei Prodotti Intermedi – Responsabile di Area Prof.ssa Paola Grammatico UOC Servizio di Immunoematologia e Medicina Trasfusionale – Padiglione Cesalpino –Direttore: Prof. Luca Pierelli
Gruppo patologie Anemie ereditarie
Codice esenzione RDG010
Medici referenti Dott.ssa Maria Beatrice Rondinelli Dott.ssa Filomena Terlizzi Dott.ssa Antonella Di Bartolomei
Tipologia patologie Talassemia, drepanocitosi, patologie della membrana eritrocitaria, patologie da difetto enzimi eritrocitari
Popolazione Adulti
Indirizzo Padiglione Cesalpino- Istituto di Ematoterapia Circonvallazione Gianicolense n 87 00152-Roma
Numeri di telefono 06/68592364 - 06/68592129 – 06/68592679
Modalità di contatto Telefono, e-mail, medico curante o personalmente , sportello delle malattie rare
Sito web http://www.scamilloforlanini.rm.it, [email protected]; [email protected], [email protected]
PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
14
Informazioni utili e contatti del Presidio di Riferimento dell’Ospedale S’Eugenio
Nome del Centro UO Talassemici-Malattie Rare dei globuli rossi
Gruppo patologie Anemie ereditarie
Codice esenzione RDG010
Medici referenti Dott Francesco Sorrentino Dott.ssa Laura Maffei
Tipologia patologie Talassemia, drepanocitosi, patologie della membrana eritrocitaria, patologie da difetto enzimi eritrocitari
Popolazione Adulta e pediatrica
Indirizzo Piazzale dell’Umanesimo ,10 Roma EUR Edificio storico ala sinistra , Terzo piano
Numeri di telefono Segreteria /accoglienza / Centro di Prenotazione Unica dedicato 06 51002560 Medicheria 06 51002507 Direzione 06 51002487
Modalità di contatto Telefonicamente o attraverso e-mail ,personalmente o attraverso medico di base o medico di altro centro di cura per Anemie rare, dal Lunedì al Venerdì , dalle ore 08:30 alla ore 13:00
Sito web www.aslroma2.it E-mail [email protected] Fax 06 51002506
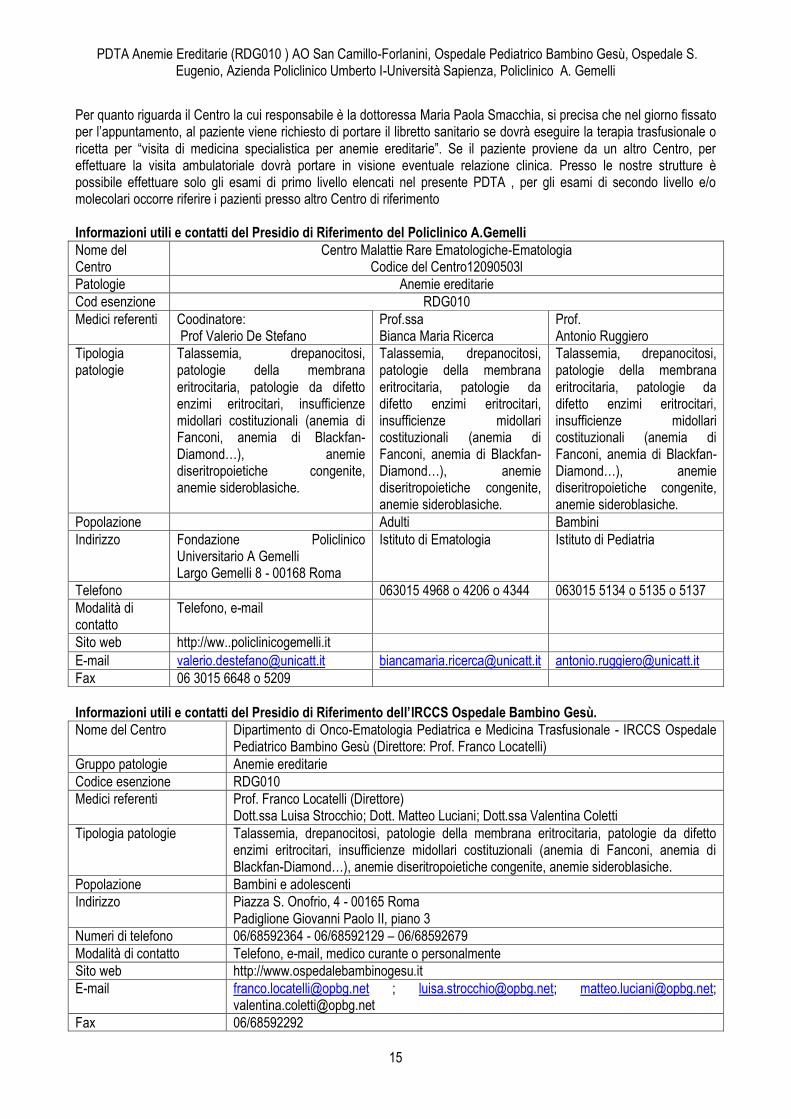
Informazioni utili e contatti del Presidio di Riferimento dell’Azienda Policlinico Umberto I-Università Sapienza.
Centro Centro Malattie Rare Ematologiche-Ematologia(Codice Centro 12090602)
Patologie Anemie ereditarie
Cod.esenzione RDG010
Medici referenti
Dott.ssa Fiorina Giona (Responsabile) Dott.ssa Michelina Santopietro
Dott. ssa Maria Paola Smacchia
Dott.ssa Pellegrina Pugliese Dott.ssa Anna Losardo
Tipologia patologie
Sferocitosi, drepanocitosi, anemia da deficit enzimatici (favismo, deficit di PK), anemia di Blackfan-Diamon, anemia di Fanconi, anemie diseritropoietiche congenite
Talassemia Maior e Intermedia, Sferocitosi, Drepanocitosi , Favismo
Sindromi Talassemiche trasfusioni dipendenti e non trasfusioni dipendenti
Popolazione Adulti e Bambini Adulti e Bambini Adulti
Indirizzo Ematologia (Primario: Prof. Roberto Foà) - Via Benevento 6 – 00161 Roma; 1° piano – Accettazione Pediatrica – stanza 737
Pediatria Primario Marzia Duse Via Regina Elena 324 1° piano
UOC Immunoematologia e Medicina Trasfusionale ( Direttore Prof.ssa Gabriella Girelli) – Via Chieti 7-00161 Roma – Piano Rialzato
Numeri di telefono
06-49974757 oppure 06-49974758 (LunedìVenerdì, ore 9.30 - 12.30) 06-49974735 (LunedìVenerdì, 14.00 -15.30) 06-49976914 (Sportello Malattie Rare)
06-49979336/06-49979287
Fax 06-49979335
dal lunedì al venerdì dalle ore
8.30.00 alle 11,00.
04-49976550 dal Lunedì al Venerdì ore 8.30 – 15.00 06-49976914 (Sportello Malattie Rare )
Modalità di contatto
Telefono, e-mail, medico curante, o personalmente (Lunedì-Venerdì, ore 8.30-13.00)
Telefono, e-mail, medico curante o personalmete ( Lunedì- Venerdì, ore 8.30- 14.30 )
Sito web www.ematologialasapienza.org, www.policlinicoumberto1.it
E-mail [email protected] [email protected] Fax 06/44241984 06/49976501
PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
15
Per quanto riguarda il Centro la cui responsabile è la dottoressa Maria Paola Smacchia, si precisa che nel giorno fissato per l’appuntamento, al paziente viene richiesto di portare il libretto sanitario se dovrà eseguire la terapia trasfusionale o ricetta per “visita di medicina specialistica per anemie ereditarie”. Se il paziente proviene da un altro Centro, per effettuare la visita ambulatoriale dovrà portare in visione eventuale relazione clinica. Presso le nostre strutture è possibile effettuare solo gli esami di primo livello elencati nel presente PDTA , per gli esami di secondo livello e/o molecolari occorre riferire i pazienti presso altro Centro di riferimento Informazioni utili e contatti del Presidio di Riferimento del Policlinico A.Gemelli
Nome del Centro
Centro Malattie Rare Ematologiche-Ematologia Codice del Centro12090503l
Patologie Anemie ereditarie
Cod esenzione RDG010
Medici referenti Coodinatore: Prof Valerio De Stefano
Prof.ssa Bianca Maria Ricerca
Prof. Antonio Ruggiero
Tipologia patologie
Talassemia, drepanocitosi, patologie della membrana eritrocitaria, patologie da difetto enzimi eritrocitari, insufficienze midollari costituzionali (anemia di Fanconi, anemia di Blackfan-Diamond…), anemie diseritropoietiche congenite, anemie sideroblasiche.
Talassemia, drepanocitosi, patologie della membrana eritrocitaria, patologie da difetto enzimi eritrocitari, insufficienze midollari costituzionali (anemia di Fanconi, anemia di Blackfan-Diamond…), anemie diseritropoietiche congenite, anemie sideroblasiche.
Talassemia, drepanocitosi, patologie della membrana eritrocitaria, patologie da difetto enzimi eritrocitari, insufficienze midollari costituzionali (anemia di Fanconi, anemia di Blackfan-Diamond…), anemie diseritropoietiche congenite, anemie sideroblasiche.
Popolazione Adulti Bambini
Indirizzo Fondazione Policlinico Universitario A Gemelli Largo Gemelli 8 - 00168 Roma
Istituto di Ematologia
Istituto di Pediatria
Telefono 063015 4968 o 4206 o 4344 063015 5134 o 5135 o 5137
Modalità di contatto
Telefono, e-mail
Sito web http://ww..policlinicogemelli.it E-mail [email protected] [email protected] [email protected] Fax 06 3015 6648 o 5209
Informazioni utili e contatti del Presidio di Riferimento dell’IRCCS Ospedale Bambino Gesù.
Nome del Centro Dipartimento di Onco-Ematologia Pediatrica e Medicina Trasfusionale - IRCCS Ospedale Pediatrico Bambino Gesù (Direttore: Prof. Franco Locatelli)
Gruppo patologie Anemie ereditarie
Codice esenzione RDG010
Medici referenti Prof. Franco Locatelli (Direttore) Dott.ssa Luisa Strocchio; Dott. Matteo Luciani; Dott.ssa Valentina Coletti
Tipologia patologie Talassemia, drepanocitosi, patologie della membrana eritrocitaria, patologie da difetto enzimi eritrocitari, insufficienze midollari costituzionali (anemia di Fanconi, anemia di Blackfan-Diamond…), anemie diseritropoietiche congenite, anemie sideroblasiche.
Popolazione Bambini e adolescenti
Indirizzo Piazza S. Onofrio, 4 - 00165 Roma Padiglione Giovanni Paolo II, piano 3
Numeri di telefono 06/68592364 - 06/68592129 – 06/68592679
Modalità di contatto Telefono, e-mail, medico curante o personalmente
Sito web http://www.ospedalebambinogesu.it
E-mail [email protected] ; [email protected]; [email protected]; [email protected]
Fax 06/68592292

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
16
6. Collaborazioni del Centro con altri centri nazionali ed internazionali IME I medici referenti fanno parte di un gruppo di studio denominato “gruppo cooperativo laziale” che vede la collaborazione dei principali centri di talassemia laziali e nazionali, impegnati nell’organizzazione di partecipazione a convegni e nell’organizzazione degli stessi. Il Centro Malattie Rare Ematologiche-Ematologia dell’Azienda Policlinico Umberto I-Università Sapienza coopera attivamente con le società scientifiche ematologiche nazionali (GdL patologie del globulo rosso dell' AIEOP- Associazione Italiana Ematologia ed Oncologia Pediatrica e SITE- Società Italiana di Talassemie ed Emoglobinopatie) e con i maggiori centri italiani esperti di anemie emolitiche congenite a livello nazionale (Azienda Ospedaliera Maggiore Policlinico-Fondazione IRCCS Cà Grande, Milano; Ente ospedaliero Ospedali Galliera, Genova; Istituto Giannina Gaslini, Genova; Azienda Ospedaliera Universitaria Federico II, Napoli)
Il Dipartimento di Onco-Ematologia Pediatrica dell’IRCCS Ospedale Pediatrico Bambino Gesù coopera attivamente con l’Associazione Italiana Ematologia ed Oncologia Pediatrica (AIEOP).
Per l’esecuzione di esami strumentali come SQUID e RMNT2* i pazienti sono riferiti all’Azienda Ospedaliero-Universitaria San Luigi Gonzaga di Orbassano (TO) o all’Ente Ospedaliero Ospedali Galliera di Genova e alcuni dei Centri partecipano allo Studio Nazionale Extension Myocardial Iron Overload in Thalassemia (eMIOT) del Consiglio Nazionale delle Ricerche (CNR) di Pisa per la valutazione dei depositi di ferro nei pazienti affetti da emoglobinopatie.
MEDIC 7. Rapporti con le Associazioni I medici I medici referenti sono iscritti alla SITE ed attraverso tale associazione partecipano a convegni regionali e nazionali ed incontri multidisciplinari per tematiche di comune interesse in questo ambito .
Altre associazioni con le quali hanno rapporti anche i pazienti edelle quali sono sostenitori sono l’AD SPEM, Gocce di vita, UNIAMO . Il Dipartimento di Onco-Ematologia Pediatrica dell’IRCCS Ospedale Pediatrico Bambino Gesù collabora con la Fondazione Europea per l’anemia di Diamond-Blackfan AnemiaOnlus - Gruppo di Sostegno DBA Italia Onlus (www.diamondblackfanitalia.org) e con l’Associazione Italiana per la Ricerca sull'Anemia di Fanconi (AIRFA) (www.airfa.it). BIBLIOGRAFIA
Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014 May; 99(5): 811–820.
Associazione Italiana Ematologia Oncologia Pediatrica. Linee-guida per la gestione della malattia drepanocitica in età pediatrica in Italia. 2012.
Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transplant. 2016 Apr;51(4):536-41.
Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749-2756.
Borgna Pignatti C. The life of patients with thalassemia maior.Haematologica 2010; 95:345-8.
Bottomley SS, Fleming MD.Sideroblastic anemia: diagnosis and management. HematolOncolClin North Am. 2014 Aug;28(4):653-70.
Cappellini MD, Cohen A, Eleftheriou A, et al. Guidelines for the Clinical Management of Thalassaemia, 2nd Revisededition. Nicosia (CY): Thalassaemia International Federation; 2008.

PDTA Anemie Ereditarie (RDG010 ) AO San Camillo-Forlanini, Ospedale Pediatrico Bambino Gesù, Ospedale S. Eugenio, Azienda Policlinico Umberto I-Università Sapienza, Policlinico A. Gemelli
17
Dossier talassemia 2009. Sfida alla Talassemia nella regione Lazio “a cura del Gruppo Cooperativo Laziale della Talassemia” con il patrocinio del Consiglio regionale del Lazio.
Hays L. Fanconi Anemia: Guidelines for Diagnosis and Management - Fourth Edition; 2014.
Iolascon A, Heimpel H, Wahlin A, TamaryH.Congenitaldyserythropoieticanemias: molecularinsights and diagnosticapproach. Blood. 2013 Sep 26;122(13):2162-6.
Ivaldi G, Leone D, Viaggi C, Pascotto D. Variabilità delle frazioni emoglobiniche dalla nascita all’età adulta in condizioni fisiologiche e patologiche. Biochimica clinica 2007, vol. 31, n. 4.
Koralkova P, van Solinge WW, van Wijk R. Rare hereditary red blood cell enzymopathies associated with hemolytic anemia - pathophysiology, clinical aspects, and laboratory diagnosis. Int J Lab Hematol. 2014 Jun;36(3):388-97.
Locatelli F, Kabbara N, Ruggeri A, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122(6):1072-1078.
MacMillan ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia - when and how? British Journal of Haematology. 2010;149(1):14-21.
Maggio A, Caronia F,Russo G. ClinicaeTerapiadellaTalassemia. Ed. SEE Firenze2000.
Mehta P, Locatelli F, Stary J, Smith FO. Bone Marrow Transplantation for Inherited Bone Marrow Failure Syndromes. Pediatric Clinics of North America. 2010;57(1):147-170.
OldJM, Traeger-SynodinosJ, Galanello R, et al. Prevention of Thalassaemias and other Haemoglobin Disorders. T.I.F. Publication,2005.
Paleari R, Cannata M,Leto F, et al. Analytical evaluation of Tosh HCL-723 automated HPCL analyzer for hemoglobin A2 and F determination “ Clinical Biochemistry 2005; 38: 159-65.
PennelDJ,Porter JB, CappelliniMD,et al “Efficacy of deferasirox in reduncing end preventing cardiac iron overload in beta-thalassemia”. Blood2010;115:2364-71.
Rund D, Rachmilewitz E. β-Thalassemia N Engl J Med 2005 ;353 : 1135-46.
Società Italiana Talassemie ed Emoglobinopatie. Raccomandazioni per la diagnostica di primo livello delle emoglobinopatie. 2012.
Società Italiana Talassemie ed Emoglobinopatie. Raccomandazioni per la gestione del paziente adulto affetto da anemia falciforme. 2014.
Società Italiana Talassemie ed Emoglobinopatie. Raccomandazioni per le strategie trasfusionali nelle emoglobinopatie. 2014.
Società per lo studio delle talassemie ed emoglobinopatie - Fondazione Italiana Leonardo Giambrone. Linee Guida per la β-Talassemia Intermedia. Ed. Edelson-Gnocchi 2005.
vanWijk R, van SolingeWW.The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood. 2005 ;106:4034-42.
Vlachos A, Muir E. How I treat Diamond-Blackfan anemia.Blood. 2010 Nov 11; 116(19):3715-23.
WoodJC“Cardiaccomplicationsinthalassemiamaior“.Hemoglobin2009;33:381-86.
Zanella A. Inherited disorders of red cell metabolism. Baillière's best practice & research. Clinical haematology. 2000, Vol 13 num.1, 1-150.
Zanella A, Bianchi P, Fermo E, Valentini G. Hereditarypyrimidine 5'-nucleotidase deficiency: from genetics to clinical manifestations.Br J Haematol. 2006;133:113-23.
Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvatekinasedeficiency: the genotypephenotypeassociation. Blood Rev. 2007;21:217-31.


















