
APPROCCI DI IMMUNOTERAPIA ATTIVA E PASSIVA BAS ATI...
126
UNIVERSITÀ DEGLI STUDI DI PADOVA FACOLTÀ DI MEDICINA E CHIRURGIA DIPARTIMENTO DI SCIENZE ONCOLOGICHE E CHIRURGICHE SEZIONE DI ONCOLOGIA DOTTORATO DI RICERCA IN ONCOLOGIA E ONCOLOGIA CHIRURGICA XX CICLO APPROCCI DI IMMUNOTERAPIA ATTIVA E PASSIVA BASATI SULL'ANTIGENE TELOMERASI IN MODELLI DI CARCINOGENESI MURINA COORDINATORE: CHIAR.MO PROF. PAOLA ZANOVELLO SUPERVISORE: DOTT. VINCENZO BRONTE DOTTORANDO: DOTT. STEFANO UGEL 31 GENNAIO 2008
Transcript of APPROCCI DI IMMUNOTERAPIA ATTIVA E PASSIVA BAS ATI...

UNIVERSITÀ DEGLI STUDI DI PADOVAFACOLTÀ DI MEDICINA E CHIRURGIA
DIPARTIMENTO DI SCIENZE ONCOLOGICHEECHIRURGICHE
SEZIONEDI ONCOLOGIA
DOTTORATODI RICERCA INONCOLOGIAE ONCOLOGIACHIRURGICA
XX CICLO
APPROCCIDI IMMU NOTERAPIAATTIVA E PASSIVA
BASATI SULL'ANTIGENE TELOMERASIIN MODELLI DI CARCINOGENESI MURINA
COORDINATORE: CHIAR.MO PROF. PAOLA ZANOVELLO
SUPERVISORE:DOTT. VINCENZO BRONTE
DOTTORANDO: DOTT. STEFANOUGEL
31GENNAIO 2008


"Coloro che sognano di giorno sanno molte cose che sfuggono a chi sogna soltanto di notte."
EdgarAllan Poe


INDICE
SUMMARY ............................................................................................................ 1RIASSUNTO.......................................................................................................... 3INTRODUZIONE................................................................................................... 5
"CancerImmunoediting":unanuovavisionedel rapporto trasistemaimmunitario e tumore.......................................................................................... 5Immunoterapiae tumori.................................................................................... 15Immunoterapiaattiva: la sfidadellavaccinazioneaDNA................................ 18Immunoterapiapassiva:il successo del trasferimentoadottivo degli effettoriimmunitari CD8+...............................................................................................25Telomerasicome antigenetumoraleuniversale, candidatoidealeperl'immunoterapia anti-tumorale.......................................................................... 31Il topoTRAMP: unmodellodi studiodellacarcinogenesi prostatica.............. 33
SCOPODELLA TESI.......................................................................................... 41MATERIALI & METODI .................................................................................... 45
TOPI.................................................................................................................. 45Screeningtopi TRAMP..................................................................................... 45TERRENI DI COLTURA................................................................................. 46
Colturecellulari............................................................................................. 46Coltureutilizzateperi saggi......................................................................... 47
LINEECELLULARI ........................................................................................ 47PEPTIDI ............................................................................................................ 48ALLESTIMENTO DI COLTURELEUCOCITARIE...................................... 49
Allestimento di ColtureLeucocitarieMiste (MLC)...................................... 49Allestimento dellecoltureleucocitariestimolatedapeptide(MLPC).......... 49
ANALISI CITOFLUORIMETRICA................................................................ 50SAGGI FUNZIONALI ..................................................................................... 50
TestELISPOT( Enzyme-Linked Immunonoassobent SpotsAssays).......... 50TestE.L.I.S.A.(Enzyme-Linked ImmunosorbentAssay)............................ 50Valutazionedella rispostacitotossicatramitesaggio di rilascio di 51Cr.......51
COSTRUTTI PLASMIDICI ............................................................................. 52ANALISI SPECTRATYPING E CLONOTIPICA DEL TCR......................... 53VIRUS............................................................................................................... 54VACCINAZIONE A DNA PLASMIDICO...................................................... 54VACCINAZIONE TRAMITE VETTOREADENOVIRALE ......................... 54INOCULO DEL TUMORE.............................................................................. 54TRASFERIMENTO ADOTTIVO DI CTL ...................................................... 55
Modellodi melanomametastaticopolmonare.............................................. 55Modellodi tumoresottocutaneo................................................................... 55
ANALISI ISTOLOGICAE IMMUNOISTOCHIMICA .................................. 55ANALISI STATISTICHE ................................................................................ 56
RISULTATI .......................................................................................................... 57Comparazionetradueprotocolli di immunizzazioneattivabasatisuvaccini aDNA codificanti l'antigenetelomerasimurina(m-TERT). .............................. 57Valutazionedell'effettoterapeuticodel trasferimentoadottivo dellapopolazionepoliclonaledi linfociti T citotossici (CTL) specific i perl'epitopom-TERT198-205
in animaliportatori di tumore........................................................................... 73Identificazioneecaratterizzazionefunzionaledi uncloneCTL adalta affinitàspecificoperl'epitopom-TERT198-205 ..............................................................81

Valutazionedell'efficaciaterapeuticadelleduestrategie vaccinali aDNA versol'antigeneTERT in unmodellomurinodi carcinogenesi prostaticaspontanea............................................................................................................................87
DISCUSSIONE...................................................................................................101BIBLIOGRAFIA .................................................................................................105ABBREVIAZIONI ..............................................................................................115PUBBLICAZIONI ..............................................................................................117Ringraziamenti .......................................................................................................119

1
SUMMARY
Aim of this work was to verify the therapeutic efficacy of active and passive
vaccinationagainst m-TERT in mice bearing transplantable tumors and in an
autochthonousmousemodel of prostatecancer: the TRAMP model.To choose
the best vaccination protocol we developed DNA-based antigen-specific
expression systems employing eukaryotic plasmid expression vectors and
recombinantadenoviruses. We usedsplenocytesfrom vaccinated mice to set up
mixed leukocyte peptide cultures (MLPC) with the mouse (m)-TERT198-205
peptide. This sequenceof mouse TERT was identified as the immunodominant
epitope by screeningoverlapping peptides covering the whole protein. MLPC
were tested in IFNγ-specific E.L.IS.A. assay againstpeptide-pulsed cells and
unpulsed tumor cell lines of different histotypes, but with the same H-2b
haplotype. A persistently high level of recognition was noted against peptide-
pulsed cell lines, whereas repeated in vitro stimulation with γ-irradiated
syngeneicsplenocytespulsedwith a low doseof m-TERT198-205 peptide(0.1µM)
every week led to an increased recognition of unpulsed tumor cell lines. This
enhancedrecognition was observed only in MLPC from mice vaccinated with
DNA plasmid vector and suggesteda preferential proliferation of high-avidity
CTL clonesunderthesecultureconditions.The augment in high-avidity CTLs is
supported by TCR analysis by spectratyping,that proveda positive selection of
Vβ-11 chain of TCR during in vitro passagestogether with TCR clonotype
enrichment. We demonstratedan important therapeutic effect by m-TERT198-205
polyclonal CTLs transferin a metastaticmelanomamodel, wheremice treated
with CTLs transfer presenteda significant reduction of the lung metastases
comparedwith untreated controls, and in trasplantable tumor models, where
TERT198-205 CTLs induceda statistically significantdelayin tumor growth and a
survival increasein bothmelanoma- andprostate carcinoma-bearingmice.
To identify and isolate high affinity CTL clones specific for m-TERT198-205
epitopewe usedthelimit ing dilution techniquestarting from thepolyclonal CTLs.
We obtaineda largespectrumof clones,amongwhich cloneCTL-7 wasthemost
intriguing.In fact it wasableto recognizetumorcell lines of differenthistotypes(
melanoma,prostate carcinoma,coloncarcinomaand sarcoma) as well aspeptide-
pulsed cell lines. Transferof CTL-7 clone exerted a better therapeutic effect on
lungmetastasesgeneratedby melanomacells.

2
We also investigated the possibility to elicit a protective antitumor response
throughan active immunizationin TRAMP mice. We performeda cycle of two
biweekly i.m. injectionsof a plasmidencoding m-TERT repeated every 10 weeks
for the entire life of the mouse. Mice vaccinated against m-TERT showeda
reduction of tumorprogressionat week24th anda survival prolongationcompared
to mockvector-treatedgroups.
Thesedatarepresent a goodplatform to translate moreeffective vaccines for the
therapyof humancancers.

3
RIASSUNTO
Lo scopodi questo lavoro è statoverificarel'efficacia terapeutica di interventi di
vaccinazione attiva e passiva antitumorale, sfruttandol'antigenem-TERT, sia in
modelli di crescita tumoraleindottadall'inoculodi cellule tumorigeniche sia in un
modello murino transgenicodi carcinogenesi prostatica spontanea: il modello
murino TRAMP. Abbiamo confrontato due modalità di vaccinazione basate
sull 'impiegodi vettori plasmidicio adenovirali codificanti l'antigenedi interesse,
con lo scopo di scegliere la più efficace. Abbiamo utilizzato gli splenociti
derivantidagli animali vaccinatiper allestire colture leucocitarie stimolatecon il
peptide (MLPC) utilizzando il peptide m-TERT198-205. Questopeptide è stato
identificato comeepitopo immunodominante della ribonucleoproteina telomerasi
murinatramite screeningdi un pannellocompleto di peptidi checoprivanol'intera
sequenzadella proteina. Le MLPC sono state testate funzionalmente in saggi
E.L.IS.A. perrilasciodi IFN-γ dopococolturaconbersaglicellulari pulsaticon il
peptide m-TERT198-205 o concelluletumoralidi diversa istologiamadi medesimo
aplotipo H-2b. Abbiamo dimostrato un riconoscimento persistente delle cellule
pulsate con il peptide m-TERT198-205 da parte sia delle MLPC derivatedagli
animali vaccinati con solo DNA plasmidico che da quelle derivate dagli animali
vaccinati con il regime a "prime-boost". Solo dopo ripetuti passaggiin vitro,
duranti i quali le MLPC venivanoristimolate utilizzandosplenociti singeniciγ-
irradiati e pulsati con dosi basse di peptidem-TERT198-205 (0.1 µM), abbiamo
notato un incremento nel riconoscimento di bersagli cellulari non pulsati daparte
unicamentedelle MLPC derivatedagli animali vaccinati con DNA plamidico,
suggerendocila possibilità di averindotto la proliferazionedi linfociti a maggior
affinità per l'antigene. L'aumentodi linfociti T citotossici (CTL) a maggior
affinità è stata dimostratatramite l'analisi spectratyping del TCR, che ha rilevato
una selezionepositiva della catenaVβ-11 durantei passaggi in vitro. Abbiamo
quindi dimostratoun importanteeffetto terapeutico indotto dal trasferimento dei
CTL policlonali m-TERT specifici sia in un modello di melanomametastatico a
livello polmonare in cui gli animali trattati tramite trasferimento dei CTL m-
TERT specifici hannopresentatounariduzionesignificativa delle metastasi, sia in
due modelli tumorali trapiantabili di diversa istologia in cui il trattamento ha
indottoun significativo aumentodellasopravvivenza. Successivamente tramite la
tecnica "li miting dilution" abbiamoottenuto un ampio spettro di cloni CTL m-

4
TERT specifici, dei quali il clone CTL-7 è risultato il più interessante. Infatti il
clone CTL-7 in vitro è statocapacedi riconoscere cellule tumorali di differente
istologia (melanoma,carcinomaprostatico,carcinoma del colon e sarcoma) allo
stessomododelle cellulepulsatecon il peptide sia in saggiE.L.I.S.A. per rilascio
di IFN-γ sia in saggi di citotossicità. Inoltre il clone CTL-7 ha evidenziato un
miglior effetto terapeuticoin vivo rispetto alla coltura policlonale, infatti gli
animali trattati tramite trasferimento del cloneCTL-7 eranoefficientemente curati
dallemetastasipolmonariindottedallecelluledi melanoma.
Abbiamo inoltre studiato la possibilità di impiegare la vaccinazione attiva per
indurre unarispostaprotettivaantitumorale nel modello TRAMP. Pertanto è stato
sviluppato un protocollo di vaccinazione basato su un doppio inoculo
intramuscolo (i.m.) di DNA plasmidicoad intervalli di 15 giorni e successivi
richiami ogni 10 settimaneper tutta la vita dell'animale. Gli animali vaccinati
contro m-TERT hannoevidenziato,tramite analisi istologica, unariduzionenella
progressionedel tumoregià alla ventiquattresimasettimanae un aumento della
sopravvivenzadi circa11 settimanerispetto agli animali vaccinati con il plasmide
vuoto.
Questi dati rappresentano,quindi, un buon trampolino di lancio per traslare
l'utilizzodi vaccini nellaterapiaanti-tumoralenell 'uomo.

Introduzione
5
INTRODUZIONE
"Cancer Immunoediting": una nuova visione del rapporto tra
sistema immunitario e tumore
Nel 1909PaulErlich per primo ipotizzò che il sistema immunitario proteggesse
l’organismodallo sviluppo di tumori, che altrimenti sarebbero stati molto più
frequenti. Le prove dell’esistenzadi una relazione tra sistema immunitario e
crescita tumorale si ebbero inizialmente, nella metà del ventesimo secolo, in
seguito agli studi sul rigetto allo-genico che chiarirono il ruolo del sistema
immunitario nel riconoscimentoe nell’eliminazione di tumori trapiantabili tra
ceppi murini outbred.Questi lavori gettarono le basi per la definizione degli
alloantigenima,allo stessotempo,miseroin dubbiol’esistenzadi un meccanismo
di rigettotumore-specificoper le neoplasie. L’impi egodi ceppi inbredpermisein
seguito di dimostrare come si potesseroimmunizzare topi contro trapianti
singenicidi tumori indotti da agentichimici e virus, comprovandola presenzadi
antigeni tumore-specifici(Old andBoyse1964;Klein 1966).
I continui progressi in immunobiologiae immunogenetica, ed in particolare le
prove iniziali dell’esistenza di antigeni associati al tumore (TAA), permisero
quindi a Burnet e Thomas di formulare la cosiddetta “ ipotesi
dell’immunosorveglianza”.Secondoquesta teoria il sistema immunitario sarebbe
stato in gradodi sorvegliare costantementei tessuti dell’ospite dall’emergeredi
cellule trasformate. I neo-antigeni specifici delle cellule tumorali avrebbero
provocatounareazioneimmunologicacapacedi eliminare la neoplasiaemergente,
una necessità evolutiva per la sopravvivenza, come la protezione contro le
infezioni (Burnet1964; Burnet1970). SecondoThomas, inoltre, gli organismi più
evoluti avrebberodovuto possederedei meccanismi di protezioneversoi tumori,
simili a quelli che medianoil rigetto allogenico, mantenendoin questo modo
l’omeostasi tissutalenegli organismimulticellulari complessi.
Una serie di esperimentivennerocondotti negli anni seguenti per confermare
questa ipotesi. Inizialmente vennero util izzati diversi modelli sperimentali di
immunosoppressione indotta, tra cui la timectomia neonatale (Grant and Miller
1965; Vandeputte and De Somer1965; Burstein and Law 1971), l’impiego di
siero anti-linfocitario eterologo e metodi farmacologici (Stutman 1975), allo
scopo di indagare se lo sviluppo tumorale potesse essere influenzato dalla

Introduzione
6
soppressionedel sistemaimmunedell’ospite.Si ottennero risultati discordanti che
dimostraronosolo comegli animali immunocompromessiavessero una maggior
suscettibili tà versogli agenti infettivi e, di conseguenza, versotumori di origine
virale e verso le neoplasie linfoproliferative causate da una stimolazione
linfocitaria cronica, ma non verso tumori spontanei o indotti da agenti chimici.
Anche la sperimentazionesu topi nudi atimici, portatori di un’alterazione
immunologica a basegenetica,sembrò escludere che tali animali fosseropiù
suscettibili rispetto ai controlli allo sviluppodi tumori spontaneio chimicamente
indotti (Stutman1974;Stutman1979).
I risultati cosìottenuti,con l’uso di modelli di immunodeficienzaimperfetti, in cui
si ignoraval’azionedi altri effettori del sistema immunitario,quali,adesempio, le
cellule naturalkiller (NK) timo-indipendenti(allora sconosciute)e le cellule T γδ,
che in parte possonosvilupparsi al di fuori del timo, non provavanol’esisteza
dell’immunosorveglianzae questoportò ad abbandonare prematuramente tale
teoriaversola finedegli anniSettanta.
Successivamentevenneroeffettuati studi di tumorigenesi in topi SCID (severe-
combined immunodeficiency), che mancanodi una subunità funzionalmente
attiva di una protein chinasiDNA-dipendente (DNA-PK) necessaria al corretto
riarrangiamentodei recettori linfocitari per l’antigene. Questi topi sono perciò
incapacidi svilupparerisposteimmunitarie antigene-specificheed, in effetti, essi
mostravanomaggiore suscettibilitàai tumori indotti. Tuttavia, va tenuto presente
che DNA-PK è un enzima espressoin tutte le cellule dell’organismo ed è
coinvolto nella riparazionedi dannial DNA, per cui unasuadeficienza potrebbe
di per séfavorire lo svilupponeoplastico. I dati ottenuti, quindi, nonpermiserodi
concludere in manieradefinitiva che l’aumentata incidenzatumorale in questi
animali fossedovutaall’immunodepressione(Schuler, Weiler et al. 1986;Engel,
Svaneet al. 1997).
Tra il 1994e il 1998unaserie di scoperterinnovarono l’int eresseversol’ipotesi
dell’immunosorveglianza.In primo luogo, studi sullo sviluppo tumorale in
modelli murini deficienti per la trasduzionedel segnale della citochinainterferone
(IFN)-γ (Dighe, Richardset al. 1994), insiemeall’uso di anticorpi monoclonali
neutralizzanti l’azione della stessacitochina (Kaplan, Shankaran et al. 1998),
hanno permessodi evidenziareun ruolo protettivo dell’IFN-γ, endogenamente
prodotto, nella formazionedi neoplasietrapiantate, spontanee o chimicamente

Introduzione
7
indotte. Questacitochina sembraagire sia a livello del sistema immunitario,
promuovendonele funzioni effettrici antitumorali mediante la generazione di
linfociti T helper (Th)1 CD4+ e citotossici (CTL) CD8+ tumore-specifici e
l’attivazione dei macrofagi, sia a livello delle cellule tumorali, aumentandone
l’i mmunogenicità attraversol’induzione di molecoledel complessomaggioredi
istocompatibilità di classeI (MHC I) coinvolte nella presentazione antigenica
(Bach,Aguetet al. 1997).
Inoltre, esperimenti su topi con delezione genetica della perforina (perforina-/-),
risultavanomaggiormentesensibili all’induzione di tumori con metilcolantrene
(MCA), secomparaticonla contropartenormaleo “wild-type”. La perforinaè una
molecolarilasciata dai granuli dei CTL e cellule NK, importante per l’uccisione
delle cellule bersaglio anchedi origine tumorale. Questi studi hannosottolineato,
quindi, l’i mportanzadell’attività citolitica di diverse componenti del sistema
immune nel controllo dello sviluppo tumorale (Street, Cretney et al. 2001;
Takeda,Smyth et al. 2002).
La conferma principale a supporto dell ’esistenza del processo di
immunosorveglianzanel cancro,dipendentedai linfociti , è venutadall’uso di topi
deficientiRAG-1-/- eRAG-2-/-, i geni checodificanopergli enzimicoinvolti nella
ricombinazionedel recettoredei linfociti T (TCR) e B (BCR) e nella riparazione
di dannial DNA, espressiesclusivamentenel compartolinfoide (adifferenzadella
DNA-PK dei topi SCID presenteubiquitariamente). L’assenza di linfociti B, T e
cellule NKT rendequesti animali più suscettibili dei controlli all'insorgenza di
tumori spontaneie/oindotti (Shankaran, Ikedaet al. 2001).
Da quel momento, la disponibilitàdi ceppimurini inbredcon delezioni miratea
componenti critiche del sistema immunitario ha permesso di confermare
l’i mportanzasia del compartoinnatochedi quello adattativo nel controllo della
trasformazioneneoplastica, in particolare di cellule NKT, dei linfociti T conTCR
γδ e αβ, delle cellule NK e di citochinecomeIFN-γ e interleuchina 12 (IL-12),
chehannofunzioni almenoparzialmentesovrapponibili. L’im munosorveglianzasi
configura, quindi, comeun processoeterogeneoche richiedel’azione di diversi
effettori immuni, in manieradipendentedal tipo cellulare tumorale di origine,dal
meccanismo di trasformazione,dalla localizzazione anatomica del tumore e dal
riconoscimento immunologicoprevalente(Dunn,Bruceet al. 2002).

Introduzione
8
L’ipotesi dell’esistenzadel processo di immunosorveglianza, studiata in modelli
murini, trova conferme anche per l’uomo. Studi di follow-up su pazienti
trapiantati sottoposti a trattamenti immunosoppressivi o affetti da
immunodeficienze primarie, indicano che questi individui hanno un maggiore
rischio relativo di sviluppare tumore rispetto ad individui immunocompetenti.
Questorischio aumentatoderiva,in parte,dalla scarsaprotezioneversogli agenti
infettivi, checomporta unamaggiorincidenza di tumori indotti davirus; tuttavia,
anche un ampio spettro di sottotipi tumorali, di eziologia non virale, viene
riscontratocon maggiore frequenza in questipazienti (Bui and Schreiber 2007).
Dopoil trapianto d’organo, peresempio,è stato riportatoun incrementodi quattro
volte l’incidenzadi melanomispontanei(Sheil1986).
Ad ulteriore supportodi questi dati epidemiologici, si sono oggi accumulate
evidenzesostanziali sulla correlazionepositiva tra presenzadi linfociti infilt ranti
il tumore (TIL) e sopravvivenzadel paziente, in neoplasie di diversa origine
(Lipponen,Eskelinen et al. 1992;Mihm, Clemente et al. 1996;Naito, Saito et al.
1998; Dunn,Bruceet al. 2002).
Studi recenti dimostrano come il concetto dell’immunosorveglianza, inteso
inizialmente come un processo di protezione dell’ospite, agentesolo nelle fasi
precoci della comparsa del tumore,sia in realtà riduttivo. Il sistemaimmunitario
sembra, infatti, avere un ruolo più complesso,sia di contrastodello sviluppo
tumoralechedi rimodellamentodella neoplasianel corsodella suaprogressione,
selezionando le varianti tumorali più adatte a sopravvivere in un ambiente
immunologicamente intatto, in manierasimile a quanto avvienecon virus,batteri
e parassiti. Il passaggio ripetuto di tumori trapiantabili in ospiti
immunocompetenti generava varianti tumorali con ridotta immunogenicità
(Uyttenhove, Pilotte et al. 2003). Esperimenti con tumori trapiantabili in topi
parentali (“wil d-type”) o RAG-2-/-, hannosuccessivamentemostratochei tumori
sviluppatisi in assenzadi un sistemaimmunitario intatto sonopiù immunogenici
di tumori cresciuti in ospiti immunocompetenti (Shankaran, Ikeda et al. 2001).
Linfomi derivati da topi perforina-/- crescevanorapidamentesetrapiantatisu altri
topi perforina-/-, ma venivano invece rigettati se trapiantati su topi wild-type
(Street,Cretneyet al. 2001). L’ambienteimmunologico,quindi, selezionavarianti
tumorali che hanno maggiori possibili tà di sopravvivere in un ospite
immunocompetente, per la ridotta immunogenicità o perchè hanno acquisito

Introduzione
9
meccanismi di evasioneo soppressionedel sistemaimmunitario. Questoprocesso
è favorito dall’instabilità geneticaintrinsecadelle cellule tumorali e la selezione
interessasoprattutto i geni codificanti per antigeni tumorali, per componenti del
complesso maggiore di istocompatibilità o per componenti della via di
segnalazionedel recettoreperl’I FN-γ (Dunn,Koebel et al. 2006).
Verosimilmente il rimodellamento immunologico del tumore avviene
continuamente,anchese gli effetti maggiori di quest’azione si hannonelle fasi
precoci di crescita, quandoil tumore è istologicamente, ma non clinicamente
rilevabile.L’i mmunogenicitàdei tumori chevengonodiagnosticati, quindi, risulta
già modificata dall’interazione con il sistema immunitario, il quale non solo
proteggel’indivi duo dallo sviluppodei tumori, ma agisceanchesottoponendo il
tumoreadunapressioneselettivachenealterale caratteristicheprimarie, talvolta
generandotumori più aggressivi.Allo scopodi descrivere tale comportamento è
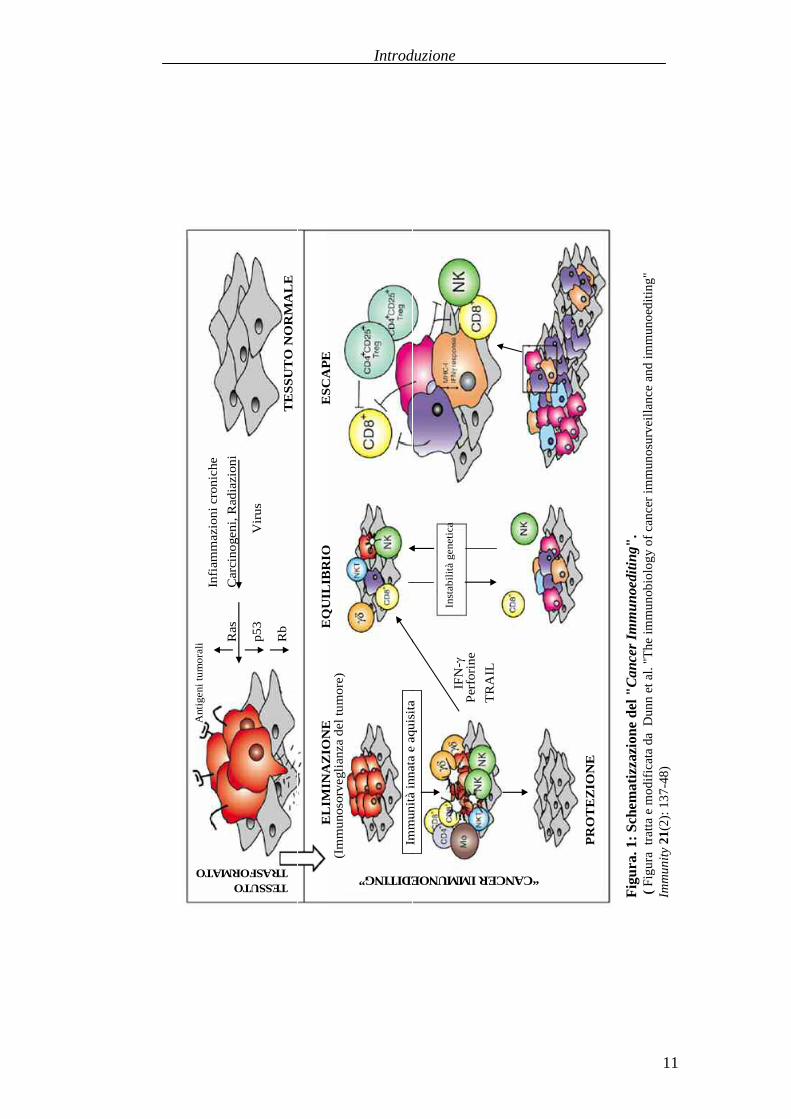
stata, perciò propostala nuova definizione di “Cancer Immunoediting”, che
comprendetre fasiprincipali: eliminazione, chepuòdare completa distruzionedel
tumore e risoluzione del processosenza avanzamento verso le fasi successive;
equilibrio, in cui cellule tumorali geneticamente instabili sopravvissutesono
sottopostead un prolungatoprocesso di selezione da linfociti ed IFN-γ per una
maggiore resistenzaall’attaccoimmune;“escape”, in cui le varianti che hanno
acquisito insensibilità all’eliminazione iniziano ad espandersi in modo
incontrollato, originando tumori clinicamente osservabili. Il grado di
rimodellamento del tumoreprobabilmentedipendedal tipo di cellule operative
durantela fasedi equilibrio, suggerendo chegli eventipossonoesseredifferenti
per ogni tessuto di origine (Dunn, Bruce et al. 2002; Dunn, Old et al. 2004)
(Figura 1). L’evasione è una diretta conseguenza delle alterazioni delle cellule
tumorali, che riguardano soprattutto i meccanismi di processazione e
presentazioneantigenicao la via di segnalazionedel recettore per IFN-γ. Queste
alterazioni riducono, da una parte, il riconoscimento e l’eliminazione degli
antigeni tumorali (“tumor ignorance”) ed attivano, dall’altra, la capacità delle
cellule neoplastiche di ostacolarele funzioni protettive del sistema immunitario.
Quest’ultimo processo può derivaredalla sovrapproduzioneda partedelle cellule
trasformatedi citochine inibitorie, quali TGF-β o IL-10 (Khong and Restifo
2002), o altri inibitori della risposta dei linfociti T, come la galectina-1
(Rubinstein, Alvarez et al. 2004) e IDO (indoleamina 2,3-diossigenasi)

Introduzione
10
(Uyttenhove, Pilotte et al. 2003), oppureda modificazioni nel reclutamento e nel
differenziamentodi diverse popolazioni cellulari. Il sistema immunitario ha
evoluto parecchi meccanismi e popolazioni cellulari per controllare la sua
attivazione e indurre tolleranzaverso le cellule T autoreattive. Il tumore può
impiegarea suovantaggioquestistessi meccanismi: ad esempio, può indurreun
differenziamentoalteratodelle cellule dendritiche (DC) in fenotipi più immaturi
non funzionali (iDC). In pazientiaffetti da tipi diversi di tumore,ad esempio, è
statariscontrata unanotevolediminuzionedelle DC ed unaprevalenza, tra queste,
di fenotipi immaturi cheesprimonolivelli bassio nulli di molecole costimolatorie
(CD80, CD86).Un numeroridottodi cellule presentanti l’antigene(APC) rende la
stimolazioneimmunepocoefficacee la presenzadi iDC, chenon forniscono un
adeguatosegnalecostimolatorioalle cellule T, puòprovocareanergia dei linfociti
T. È statopropostochediversi fattori derivanti dal tumore, tra cui VEGF, IL-10,
IL-6 e M-CSF, attivando costitutivamente il fattore trascrizionale STAT3 ed
inibendo NF-kB (nuclear factor kB), siano responsabili di questo blocco
maturativo delle DC che contribuisce alla soppressione di risposte immuni
tumore-specifiche(Gabrilovich2004).
Sono principalmente tre le popolazionicheagisconoda regolatrici della risposta
immunitaria: le T regolatorie (Treg) ovvero cellule T CD4+CD25+FOXP3+, le
cellule NKT (cellule T Natural Kill er) regolatorie e le cellule mieloidi
soppressorie (MDSC). Queste diverse popolazioni agiscono impiegando
meccanismi che non prevedonoil riconoscimento antigenico e, attualmente, sta
emergendo come un'interazionetra queste diverse popolazioni cellulari sia
un'importanteevento della regolazionenegativa della rispostaimmune. Le cellule
Treg sono state inizialmenteriscontratein malattie autoimmuni, dove sembrano
agire sopprimendo la risposta autoreattiva mediata da linfociti T CD4+. Più
recentementesi è scopertochequestecellule, costituenti circa il 10%dei linfociti
T CD4+ circolanti in animali sani,sopprimonola risposta immunitaria, mediatasia
da linfociti T CD4+ che daCD8+, anchein rispostaa patogenie tumori. Le cellule
Treg comprendonoin realtà,una composita famiglia di cellule regolatorie. Tra
questeparticolarmentestudiatein campodell'immunologiadei tumori sono le
CD4+CD25+FOXP3+: la deplezionedi questecellule in vivo, mediantel’im piego
di anticorpi anti-CD25 (PC61), prima del challengecol tumore aumenta, infatti,
l’immunosorveglianzamediatadaCTL tumore-specifici , CD4+ ecellule NK,
Introduzione
11
TESSUTOTRASFORMATO “CANCERIMMUNOEDITING”
EL
IMIN
AZ
ION
EE
QU
ILIB
RIO
ES
CA
PE
TE
SS
UT
ON
OR
MA
LE
Car
cino
geni
,Rad
iazi
oni
Infi
amm
azio
nicr
onic
he
Vir
usp5
3
Rb
Ras
Ant
igen
itu
mor
ali
(Im
mun
osor
vegl
ianz
ade
ltu
mor
e)
Imm
unit
àin
nata
eaq
uisi
ta
IFN
-γP
erfo
rine
TR
AIL
PR
OT
EZ
ION
E
Inst
abilità
gene
tica
TESSUTOTRASFORMATO “CANCERIMMUNOEDITING”
EL
IMIN
AZ
ION
EE
QU
ILIB
RIO
ES
CA
PE
TE
SS
UT
ON
OR
MA
LE
Car
cino
geni
,Rad
iazi
oni
Infi
amm
azio
nicr
onic
he
Vir
usp5
3
Rb
Ras
Ant
igen
itu
mor
ali
(Im
mun
osor
vegl
ianz
ade
ltu
mor
e)
Imm
unit
àin
nata
eaq
uisi
ta
IFN
-γP
erfo
rine
TR
AIL
PR
OT
EZ
ION
E
Inst
abilità
gene
tica
Fig
ura.
1:Sc
hem
atiz
zazi
one
del"
Can
cer
Imm
unoe
ditin
g".
(Fi
gura
trat
tae
mod
ific
ata
daD
unn
etal
."T
heim
mun
obio
logy
ofca
ncer
imm
unos
urve
illan
cean
dim
mun
oedi
ting"
Imm
unit
y21
(2):
137-
48)

Introduzione
12

Introduzione
13
induceil rigetto di diversi tumori immunogenici in vari ceppi murini ed aumenta
l’effi caciadi vaccini anti-tumorali (Onizuka, Tawaraet al. 1999). La presenzadi
queste cellule è stata riscontrataanche nell’uomo, con effetti soppressori su
linfociti T CD4+ e CD8+; in diversi pazientiaffetti da tumoreè stato riportato un
aumentodella frazionedi questa popolazione nel sangueperiferico e, in tumori
gastrointestinali, la percentuale di CD4+ CD25+FOXP3+ sembra correlare
inversamenteconla prognosi (Sasada,Kimura et al. 2003;Wolf, Wolf et al. 2003;
TerabeandBerzofsky2004).
Le cellule NKT sono una sottopopolazione di linfociti T implicata nella
regolazione della risposta immunitaria associata con malattie autoimmuni,
malattie infettive e cancro. Riconoscono antigeni glicolipidici presentati da
molecoleCD1d,attraversoun recettoreTCR checomprendeunacatenainvariante
α. Inoltre, possonoregolarele risposteimmunitariesiapromuovendolasecrezione
di citochineTh1 e Th2 tra cui IFN-γ, IL-4 ed IL-13, o reclutandoaltre cellule
immunosoppressive. Il meccanismoimmunosoppressivo sembra mediato dalla
produzione di IL-13, che in topi con tumore è aumentata rispetto agli animali
naïve e che, agendoattraversola via di IL-4Rα-STAT6, sopprimel’azione di
rigetto del tumore mediata dai CTL tumore-specific i. Probabilmente l’IL -13,
anziché agire direttamentesui CTL, induce le cellule mieloidi CD11b+Gr-1+ a
produrre TGF-β, unacitochinacheè in gradodi sopprimerela funzionedei CTL
CD8+ (Terabe, Matsui et al. 2000; Terabe and Berzofsky 2004). L'attivi tà
regolatoriadelle cellule mieloidi CD11b+Gr-1+ è dipendentedall'interazionedi
due enzimi inducibili: arginasi 1 (ARG1) e ossido nitrico sintetasi 2 (NOS2)
(Bronte andZanovello2005). Le MDSC utilizzanoARG1 e NOS2per inibire la
rispostadei linfociti T verso gli antigeni, separatamente o in combinazione, a
seconda dei segnalidettati da citochine o da molecole di membranacoinvolte
nell’interazionetra le MDSC e i linfociti T attivati. Il fenomenoprincipalelegato
all'induzionedell'enzimaARG1 è la deregolazionedella catenaCD3 ζ, elemento
principaledella trasduzionedel segnaledel TCR, con unaconseguente riduzione
della proliferazionein linfociti T attivati. In particolare, sembra che negli stadi
iniziali della crescita tumoralela downregolazione della catena ζ e l’i nibizione
della funzionalità linfocitaria siano eventiristretti ai linfociti infi ltranti il tumore,
mentrenegli stadi avanzatii fenomenisi osservino anchein cellule T circolanti in
periferia (Baniyash2004). L’ossido nitrico (NO) può regolarenegativamente le

Introduzione
14
proteine coinvolte nella segnalazioneintracellulare sia direttamente, tramite S-
nitrosilazione di residui cisteinici cruciali, sia indirettamente, attivando la
guanilato ciclasisolubile e la proteinchinasidipendente dacGMP.Nei linfociti T
il NO agisce bloccandola fosforilazione, e quindi l’attivazione, di proteine
coinvolte nelle tre principali vie di segnalazione del recettore per l’ IL-2
(JAK/STAT, Ras/MAPK, fosfoinositolo 3-chinasi/Akt), quali JAK1, JAK3,
STAT5, ERK a AKT (Mazzoni,Bronteet al. 2002). Inoltre, la deplezionedi L-
arginina citosolica da partedell’ARG1 delle MDSC modifica l’attività di NOS2,
favorendo la produzione di anione superossido (O2-) dal dominio reduttasico
dell'enzima(Xia andZweier1997;Xia, Romanet al. 1998;Bronte, Serafini et al.
2003). L’O2- si combina velocementecon altre molecole generandointermedi
reattivi dell’azoto, noti anchecomeRNOS (reactive nitrogen-oxide species), tra
cui i più noti sonoi perossinitriti, e specie reattive dell’ossigeno,i ROS(reactive
oxygenspecies), tra cui il perossidodi idrogenoH2O2. Entrambequeste classi di
composti sononote per la loro capacitàdi inibire la proliferazionedei linfociti T:
a) fungendoda messaggeriintra- e intercellulari in grado di indurre modifiche
posttraduzionali, mediantela nitrazione di residui tirosinici delle proteine che
influenzano diverse attività biologiche, come l’attivazione/inattivazione
enzimatica,il differenziamentoe la proliferazionecellulare(Schopfer, Bakeret al.
2003; Radi 2004); b) inducendol’apoptosi in linfociti T attivati dall’antigene,
mediante la deregolazionedei livelli intracellulari della proteina anti-apoptotica
BCL-2 e l’aumentodei livelli di FasLattraversosegnalazionemediatada NF-kB
(Hildeman,Mitchell et al. 2003).
In sintesi, il concetto del " Cancer Immunoediting" ci fornisce una visione
dinamicae, in un certosenso, darwinianadell' interazionetra sistema immunitario
e tumore, tale per cui le cellule tumorali sottoposte alla pressioneselettiva del
sistema immunitario tendono a selezionare patrimoni genetici codificanti
caratteristiche fenotipiche via via semprepiù aggressive,capaci alla fine di
sfuggire al controllo del sistemastesso.Questacaratteristica mobile del rapporto
tra sistamaimmunitarioe tumoredeveessersempre tenutapresente nell'iniziare
qualsiasi approccio immunoterapico e soprattutto nel giudicare l'efficacia
dell'approcciostesso.

Introduzione
15
Immunoterapia e tumori
I successiottenuti nell’ambito della vaccinoterapia anti-infettiva hanno di fatto
accesole speranzechequest’approcciopossasortire effetti altrettanto promettenti
anche nell’ambito della terapiaoncologica. Si tratta, tuttavia, di un’applicazione
chedifferiscesensibilmenteperpremesseedintenti rispetto all’im piegodi vaccini
in patologieinfettive. Utilizzatacomeprofilassidi infezioni battericheo virali , la
vaccinoterapiapredispone l’organismo a contrastare efficacemente eventuali
patogeni, mediantel’induzionedi unarisposta immunologicaefficacee duraturaa
seguito dell’i ncontro con lo stesso patogeno reso innocuo e somministrato,
talvolta, in presenza di peculiari stimoli adiuvanti. La vaccinoterapia come la
profilassi delle malattie infettive si proponequindi di sensibilizzare l’organismo
ospite per un eventuale futuro incontro con l’antigene, rappresentato nella
maggior partedei casi da proteinenon-self fortemente immunogeniche. Diversa,
invece, la situazioneperciò cheattieneagli approcci vaccinali antineoplastici che,
esclusealcune eccezioni, si propongono in primis di generare una risposta
immunitaria terapeutica, ovveroin presenza di malattia conclamata, nei confronti
di antigeni self verso cui verosimilmente esiste uno stato di tolleranza
immunologica. I limiti di quest’ultimoapproccio sonomolteplici. Esistetalora,
come detto, una forte tolleranzacentrale e periferica nei confronti di antigeni
tumoralichealtro nonsonocheproteineself, talvolta mutate, ma spessonormali o
semplicementeiper-espresse. Parlaredi tolleranzasignifica far luce su uno dei
capitoli forse più complessidell’immunologia, che comprendeeventi precoci
timici di delezionedi precursori linfocitari potenzialmente autoreattivi ed eventi
invece più tardivi, che vanno sotto il nome di tolleranza periferica, e che
comprendono fenomeni di anergizzazione mediata spessoda altri effettori del
sistema immune che proteggono l’organismo da potenziali aggressioni
autoimmuni. Recenti acquisizionihannoaltresì dimostrato, sulla scorta di sempre
più efficaci tecniche di biologia molecolare e proteomica, unadiscreta varietà di
nuovi antigeni tumore-associati riconosciuti dai linfociti T umani e murini che,
mutuandounavecchiaclassificazione, possonoesserdistinti in:
• prodotti di mutazioni geniche (ad esempio, antigene Caspase-8 del
carcinomadella testae del collo la cui mutazionenel codonedi stop causa
la lettura di una porzione non codificante in 3’; oncogenep21ras con
mutazioni puntiformi in posizione12): si tratta di forme alterate di geni

Introduzione
16
cellulari normali che controllano la proliferazione e la differenzzazione
cellulare.I proto-oncogeni, comespessovengonochiamati, possonoessere
mutati per mutazioni puntiformi, delezionio traslocazioni cromosomiche
indottedacarcinogeni,cosìdaformareoncogenii cui prodotti posseggono
attività trasformante.
• prodotti di geni silenti, di norma non espressisulla maggior parte dei
tessuti (ad esempioMAGE-1, BAGE, GAGE (Kobayashi, Higashi et al.
2000)): trattasi di geni che non sono normalmente espressinei tessuti
normali (con l’eccezionedel testicoloe della placenta), o lo sono durante
le fasi precocidello sviluppo embrionale, prima che i meccanismi della
tolleranzaversoil self sianooperativi.
• prodotti di geni virali in tumori maligni associati ad infezioni virali (ad
esempio,antigene T del SV40, prodotto del gene EBNA-1 del virus di
Epstein-Barr (Kanegane, Nomuraet al. 2002)): alcuni tumori presentano
di solito il genoma proviraleintegratonel patrimonio genetico della cellula
ospite, conseguentemente le cellule neoplastiche esprimono spesso
antigeni codificati dal genoma virale; le relative proteine vengono
sintetizzate endogenamente,processate, complessate con le molecole
MHC di classeI e quindi espresse in membrana. In tal modo le cellule
neoplastichesonoin gradodi esserericonosciutedalinfociti T specifici .
• prodotti di genidi differenziazioneespressidacelluledell’istotipo dacui il
tumorederiva (ad esempio, tirosinasi, gp100(Stennett,Riker et al. 2004),
MART-1 (Fetsch, Marincola et al. 1999)): sono proteine espresseda
cellule normali, caratteristichedi un particolare tipo di tessuto in un
determinato stadio del suo normale differenziamento; nel caso del
melanoma/melanocitigli antigenidi questogrupposonoquasitutti enzimi
del ciclo biosintetico dellamelanina.
Dato che è la stessacellula tumoralea sintetizzarela proteinaantigenica, questa
vieneprocessata al livello del citosol dal proteosoma,edi peptidi chenederivano
sono trasportati dal complesso TAP (Transporter associated with Antigen) nel
reticolo endoplasmaticodove si legano alle molecole MHC di classe I. Il
complesso così costituito, espostoa livello della membrana plasmatica, potrà
esserericonosciutodal TCR specifico.

Introduzione
17
E’ dasfatare, quindi, il vecchioassunto in baseal quale il tumoreeraconsiderato
poco antigenico; una nuova definizione di scarsamente immunogenico sembra,
alla luce delle attuali conoscenze,più appropriatae corretta. La possibili tà che a
condizionarela scarsaefficaciadella rispostanon sia l’assenza di antigenema la
circostanzaambientalee temporalenella quale il sistema immunitario viene a
contatto con il tumore apreper la vaccinoterapiaantitumorale promettenti spazi
d’intervento. Qualesia l’ antigenedascegliere,la strategia adiuvante dautilizzare,
chetipo di rispostaimmunedebbaessere generata e comemantenernel’ efficacia
a lungo termine sono solo alcuni degli interrogativi che ad oggi risultano solo
parzialmentedelucidati.
Volendodescrivere, sia pur sinteticamente,quali sianoi principali tipi d’interventi
immunologici, in corso o futuribili , é necessario ricorrere inevitabilmente a
schematismi, chepossono non semprerendercontodella complessitàdel sistema
su cui deve agire l’ approccio d’immunoterapia. Esistono, infatti, approcci
classificabili come interventi d’ immunoterapia passiva ed altri noti come
immunoterapiaattiva. Nel primo casoci si riferiscea strategie basate sull’impiego
di effettori prodotti in vitro o ex-vivo, quali anticorpi o linfociti attivati, e trasferiti
nell’ospite portatore di tumore;nel secondocaso, si fa riferimento ad interventi
chehannolo scopodi indurrein vivo, nell’ospite stessounarispostaimmunitaria
antitumoralespecifica.
Nell’ambito dell’i mmunoterapiaattiva, tre sono fondamentalmente le strategie
che possono essereassociate all’immunizzazione con un antigene tumorale per
evocaree amplificarela rispostaimmunitaria:
• aumentodi efficaciadellapresentazioneantigenica: l’im piegodi mediatori
(comeadesempio GM-CSF,Flt3 ligando,CD40L) in gradodi mobilizzare
e reclutare cellule APC (Koya, Kasahara et al. 2003), il ricorso a
ingegnerizzazione delle stesse mediante l’uso di vettori virali o ligandi
chimerici e l’im piego come immunogenodi cellule tumorali indotte ad
esprimere citochine e/o molecolecostimolatorie sono solo alcunedelle
strategieimpiegatea tal scopo(Larin, Georgiev et al. 2004).
• interferenzacon i meccanismidi tolleranzaal self: diversi sonoad oggi i
meccanismi noti che regolano quella che viene definita tolleranza
periferica e che consistenel silenziamento di eventuali risposteverso
antigeni self. Esistono, infatti, cellule regolatrici sia del compartolinfoide

Introduzione
18
(CD4+CD25+FOXP3+) (Feunou,Poulin et al. 2003;ColomboandPiconese
2007) che mieloide (CD11b/Gr1)(Bronte, Apolloni et al. 2000; Bronte,
Serafiniet al. 2003;BronteandZanovello2005)in gradodi interferire con
il priming e la fase effettrice di risposte immuni verso antigeni self.
L’utilizzo tra le strategieadiuvanti di approccicapaci di alterarel’attività
inibitoria di queste popolazioni cellulari si é dimostrato in grado di
implementaree rendereefficaceunarisposta immuneverso antigeni self,
rompendoin tal modo la tolleranzaimmunologica.
• induzione di un homing preferenzialeal sito tumorale: la possibili tà di
veicolarelinfociti antigene-specificinel sito tumoraleresta ad oggi forsela
strategiaadoggi menoesplorataedi maggiorinteresse.
Immunoterapia attiva: la sfida della vaccinazione a DNA
L’immunoterapia attiva consistenell’attivazione ed espansionedel repertorio
immunitario endogenoal fine di indurre l’im munità dell’ospite versogli antigeni
tumorali. Gli interventivaccinalisi basano,conseguentemente,nel somministrare
l'antigenein un individuo in modo da attivare gli effettori immunitari. Ad oggi
molti interventi di vaccinazionesono stati studiati nell'ambito dell'oncologia. I
vaccini di primagenerazione,ancoroggi utilizzati in trials clinici (Vil ella,Benitez
et al. 2003), sfruttano la somministrazionenel paziente delle cellule tumorali
uccise o dei loro lisati. Il vantaggiodi questi vaccini è che non richiedonola
caratterizzazionecompletadegli antigeni tumorali. D’altro canto, le principali
limitazioni ai fini dell’approvazioneda parte delle autorità regolatorie sono la
parziale caratterizzazionedi tali preparazioni (cellule intereo lisati) ed il rischio
che questi vaccini inducano risposte immuni contro numerosi antigeni self
espressi anche dalle cellulenormali.Perciò,nel contesto di un'incalzante esigenza
di standardizzazione e regolamentazione,molti sforzi puntano oggi alla
definizione di vaccini molecolari diretti contro antigeni ben caratterizzati. In
quest'otticasi inquadrano i vaccini di secondagenerazione che comprendono
l'utilizzo delle DC, di peptidi e dei vaccini genetici. Il razionale della
vaccinazionea DC si basasulle caratteristiche funzionali di questecellule che
esprimono livelli di molecoleMHC circa50 volte più elevati dei macrofagi edalti
livelli di molecoledi adesionee di costimolazionerichieste per l’attivazionedelle
cellule T naïve, rappresentandoconseguentemente la più efficacecellula APC

Introduzione
19
disponibile. I protocolli adoggi utilizzati sfruttanoDC, isolate o indotte mediante
il fattore stimolante colonie di monociti -granulociti (GM-CSF) ed IL-4 da
precursorimidollari o damonociticircolanti (Hsu,Benikeet al. 1996;Thornburg,
Boczkowski et al. 2000), maturate con lisati di cellule tumorali, caricate con i
peptidi antigenici o transfettatecon le sequenze codificanti l'antigenedi interesse.
Lavori recenti hanno dimostratoche l’espressione sostenutadell'antigene nelle
DC combina la stimolazioneantigene-specifica con una potente costimolazione;
inoltre, attraverso la selezione di epitopi ad hoc, estende le potenzialità
dell’immunizzazioneoltre le restrizioni impostedai fenotipi HLA individuali dei
pazienti (Nagorsen,Panelli et al. 2003).
I più clinicamente traslabili per un approccio vaccinale sonoi peptidi antigenici,
frammentiamminoacidiciidentificati comeepitopi immunodominanti riconosciuti
dai TIL. Questa tipologia di vaccini presenta la possibilità di migliorare le
caratteristiche antigeniche dei peptidi costituenti il vaccino, attraverso tecniche
biochimichedi sostituzioneo modificazione amminoacidiche (ligandi peptidici
alterati, APL) e di evocarerisposte puntuali verso uno o più antigeni tumorali,
limitando la comparsadi reazioni autoimmunitarie o effetti immunosoppressivi
spessoassociati all’impiego di antigeni più complessi, come cellule tumorali
intere(Romero,Cerottiniet al. 2004).
La vaccinazionegenica rappresentauna modali tà innovativa per esprimere
antigeni in vivo in modo da stimolareuna risposta immunitaria sia umorale che
cellulare. L’idea dell’utilizzo di vaccini a DNA nell’ambito dell’im munoterapia
nasce dall’osservazioneche l’inoculo di un plasmide codificante per la β-
galattosidasidi E. coli, a livello del tessutomuscolare di topo, era in gradodi
determinare la produzioneda parte di tali cellule eucariotiche della proteina
batterica(Wolff, Maloneet al. 1990). Successivamentevennedimostrato chetali
plasmidi eranocapacidi attivareeffettivamente il sistemaimmunitario contro la
proteinacodificatadallasequenzagenicainoculata. Infatti, animali inoculati conil
plasmidecodificanteper il genedella nucleoproteina del virus dell’influenza A
erano in grado di sviluppare una risposta immunitaria specifica, sia di tipo
umorale che cellulare, capacedi proteggerli dal successivo incontro con il
patogeno(Ulmer,Donnellyet al. 1993).
La costruzione di un vaccino a DNA è un processopiuttosto semplice, basato
essenzialmentenel trasferimentodella sequenzagenica codificante l’antigene

Introduzione
20
d’i nteresseall’i nterno di un vettore plasmidico dotato di quattro caratteristiche
fondamentali: un’origine di replicazione procariotica, una resistenza ad un
antibiotico, un potentepromotoreeucariotico ed unasequenzadi poliadenilazione.
Successivamente, taleplasmidevieneintrodotto in batteri competenti, amplifi cato
e quindi estratto per essere inoculato nell’ospite, una volta disciolto in una
soluzione salina. La sicurezzadi tale strategia è garantita dal fatto che, se pur
l’immunizzazionemimai normaliprocessiinfettivi , tali vettori nonsonoin grado
di integrarsinel genomadell’ospite e non possonooriginare forme patogene
pericolosecomepotrebbeaccadereconl’ut ili zzodi vettori virali o batterici.
La vaccinazione a DNA presenta due aspetti interessanti per le implicazioni
nell’attivazionedel sistemaimmunitario.In primo luogoil DNA fungedaveicolo
per indurrela produzionedellaproteinaantigenica. In questomodosonole stesse
cellule dell’ospite a sintetizzare e processare la proteina antigenica che,
analogamente alle proteineendogene, vienedigerita a livello del proteosoma ed i
relativi peptidi montati su molecoleMHC di classeI presentati sulla superficie
cellulare per esserericonosciuti dai linfociti T con TCR specifico. Inoltre, la
proteina viene ancherilasciata dalle cellule transfettate, catturata e processata
dalle cellule APC,cui spettail compitodi attivare i linfociti T e B vergini, tramite
“cross-presentation” (Doe, Selby et al. 1996) (Figura 2). Il vettore plasmidico a
DNA non possiedela solafunzionedi codificare la proteina antigenica, ma viene
altresì sfruttato in virtù dellapresenza,sulle porzioni noncodificanti, di sequenze
ripetute, proprie del genoma procariotico, in grado di attivare effettori
dell’immunità innata dell’ospite(Krieg 2002). Tali proprietà sonocontenute nelle
sequenzeipometilatedi dinucleotidicitosina–fosfato-guanina (CpG),presenti con
frequenzadi circa 20 volte maggiorenel genomaprocariotico rispetto a quello
eucariotico.
A fronte della facilità con cui tali vaccini sono costruiti, la dose,la sedee la
modalità con cui essi vengono somministrati, sono parametri fondamentali
nell’infl uenzarequalitativamentee quantitativamente la rispostaimmunitaria. Le
strategie più utilizzate sono due: la metodica “gene-gun” e l’inoculazione
intramuscolaredi DNA in soluzionesalina. L’approccio del “gene-gun” consiste
nel far adsorbireil DNA su microparticelled’oro che successivamente verranno
“sparate” nel derma dell’animale tramite la propulsione fornita da elio
pressurizzato. Il grandevantaggiodi questastrategiaè la modestaquantità di
Introduzione
21
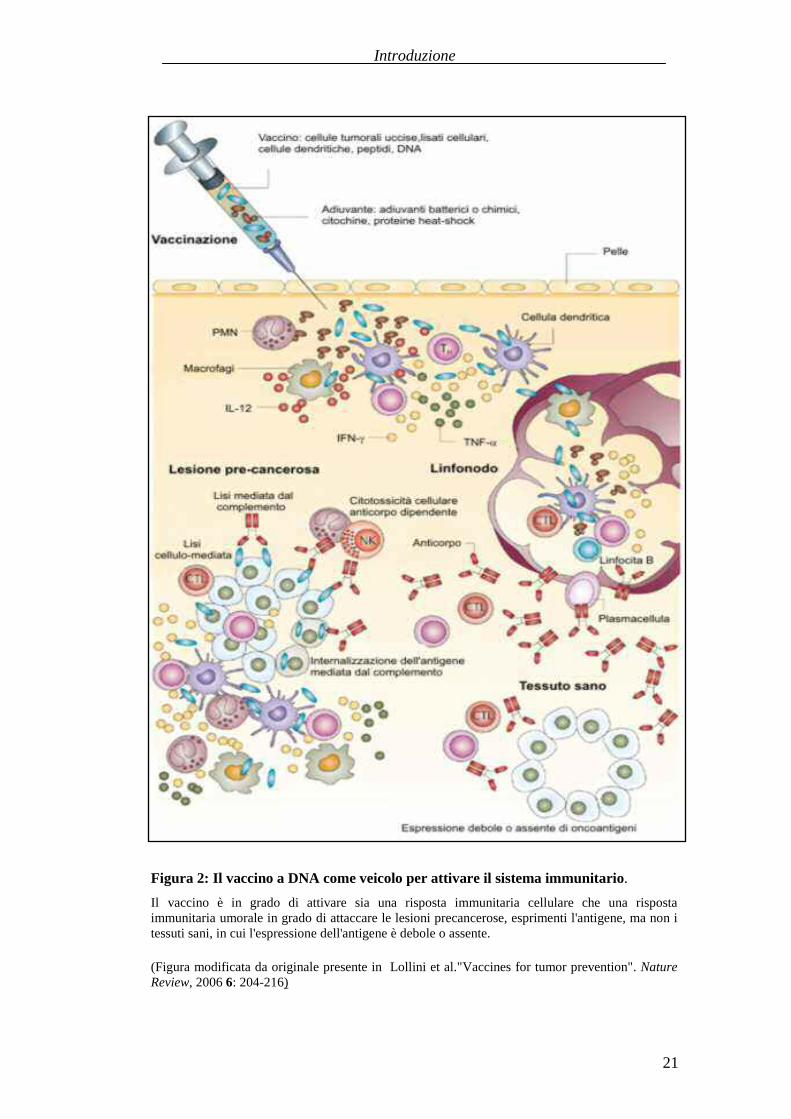
Figura 2: Il vaccino a DNA come veicolo per attivare il sistema immunitario.
Il vaccino è in grado di attivare sia una risposta immunitaria cellulare che una rispostaimmunitaria umorale in gradodi attaccarele lesioniprecancerose,esprimenti l'antigene,manon itessutisani,in cui l'espressionedell'antigene è deboleo assente.
(Figuramodificata da originale presente in Lollin i et al."Vaccinesfor tumor prevention". NatureReview, 2006 6: 204-216)

Introduzione
22

Introduzione
23
DNA necessario a stimolarela rispostaimmunitaria che difatti è dell’ordine di
grandezzadei nanogrammi(ng). L’inoculo intramuscolare di DNA in soluzione
salina, al contrario, richiede quantità di DNA dell’ordine di grandezza dei
microgrammi (µg) per indurreunapienaattivazionedegli effettori immunologici.
Utile in quest’ultimo approccio l’impiego di sostanze citocide quale la
cardiotossina, che inoculataa livello del muscolo, usuale sededi vaccinazione,
alcuni giorni prima della vaccinazioneè in gradodi migliorare la transfettabilità
dei miociti in rigenerazionea seguitodella necrosi indotta dalla tossina (Bronte,
Cingarlini et al. 2003). Altre proceduresfruttano, al contrario, veicoli quali
colloidi o liposomi per aumentarel’effi cacia del trasferimento del vaccino
all’interno della cellula, potenziandodi conseguenza la capacità della cellula di
sintetizzare la proteinaantigenicaattivandouna migliore rispostaimmunitaria.
Tale potenziamento può essere altresì ottenuto mediante elettroporazione della
sedein cui viene somministratoil vaccino. Infatti, l’applicazione di un impulso
elettrico, determinando un transienteaumento della permeabilità delle cellule,
favoriscel’ingresssoin sedeintracellularedi unamaggior quotadi DNA destinato
alla trascrizioneesuccessiva traduzione(Widera,Austinet al. 2000).
L’efficacia di questa modalità vaccinale è dimostrata dall’ini bizione della
cancerogenesi in topi transgenici Balb-neuT che overesprimonol’oncogene
trasformante rHer-2/neu nell’epitelio delle ghiandole mammarie e sviluppano
spontaneamentecarcinomadella mammella (Quaglino, Iezzi et al. 2004). Per
potenziare le vaccinazioni a DNA sono state inoltre sperimentate numerose
strategie adiuvanti, alcune delle quali prevedonoil coinoculo, unitamente al
vaccino, di sequenzegeniche codificanti per citochine (Calarota and Weiner
2004), la somministrazione esogena delle stesse citochine o molecole
costimolatrici, ed ancora, l'abbinamentodel vaccino con anticorpi depletanti o
attivanti pathway cellulari differenti. Tra gli esempi forse più meritevoli di
menzione, chehanno ancheavutoed avrannoimportanti riscontri clinici, è senza
dubbio da ricordare l’uso di citochine quali GM-CSF, IL-2 ed interferoni
(Pavlenko, Roos et al. 2004). La scelta della sede d’inoculo riveste altresì
un’importanzafondamentalenell’innescare ed indirizzare la rispostaimmunitaria.
Infatti, come riportato in letteratura, l’inoculazione intramuscolo induce
preferenzialmente risposteTh1 mentrela somministrazione intradermica, attiva
una rispostaTh2 (Pertmer,Eisenbraunet al. 1995).

Introduzione
24
L’immunoterapia antitumoralebasatasulla vaccinazione a DNA si propone,
quindi, di rompere la tolleranza immunitaria contro i TAA, che sono
frequentemente antigeni self. La tolleranzaverso tali antigeni si esprime talora
nell’impossibili tà di generare risposte immuni di lunga durata, in altri termini
nell’incapacità di generarerispostememoria. Una possibilità per superarequesto
limite può consistere nel mantenerel’attivazione linfocitaria con richiami di
vaccinazioneripetuti. A questo scopo,sonostate sperimentate diversetempistiche
di vaccinazione. Una delle strategieimpiegate consistenell’i noculo di DNA
codificanteper una proteinatumore-associata, seguito dalla somministrazione di
un vettorevirale codificanteper lo stessoantigene. Il razionale di tale approccio
risiede nella possibilità di esporre l’ospite ad una ripetuta dose di antigene,
ovviando ai limiti dell'immunizzazione con i soli vettori virali. L’in evitabile
immunitàumorale direttacontro proteinedel capsidevirale, chesi generagià dal
primo incontro con le stesse, ne preclude di fatto un impiego ripetuto. Questa
strategiaha portato ad incoraggiantirisultati nella prevenzione della malaria, che
potrebberoauspicabilmenteesser traslati in clinica (Moore and Hill 2004). Altri
protocolli al contrario sfruttano l'inoculo del vaccino a DNA seguito dalla
somministrazione di cellule ingegnerizzate per l'espressionedi citochine o
interferoni (Quaglino, Rolla et al. 2004), o scheduledi vaccinazioni basate sulla
somministrazioneripetutadel vaccinoaDNA (Quaglino, Iezzi et al. 2004).
A fronte di questovasto scenariodi protocolli vaccinali, tuttavia le risposte
cliniche oggettive dell'immunizzazionecon i vaccini di seconda generazione
restano rare ed inefficaci per l’eradicazione della malattia. Il problema più
ricorrente negli approcci clinici che sfruttano questi approcci vaccinali per la
generazionedi celluleT è la limitata disponibilità di DC autologhe. A causadella
malattia o del trattamentoal quale il paziente è sottoposto,le DC sono spesso
poco vitali o funzionalmentedanneggiate e, di conseguenza, clinicamente non
util izzabili (Grover, Kim et al. 2006). L'impossibilità di generare una risposta
immune efficace è collegata inoltre al microambiente tumorale, che come
descritto in precedenzaè affollato da cellule di origine mieloide o linfoide che
limitanola proliferazionedegli effettori immunitari. In particolare, la presenza di
due citochine (IL-10 e TGF-β) è stata associataall'inibizione della risposta
immunitaria nei confronti del tumore.IL-10 è stata descritta capacedi inibire la
fasedi priming dei linfociti T dapartedelle DC in vitro (Steinbrink,Jonuleit et al.

Introduzione
25
1999); inoltre, la suapresenzanel siero dei pazienti di certi tipi di tumoreè stata
correlataadunaprognosipiù sfavorevole (Wittke, Hoffmannet al. 1999). TGF-β
è stato descritto esserassociato alla generazione della popolazione linfocitaria
regolatoriaCD4+CD25+FOXP3+ (Peng, Laouaret al. 2004). Un datointeressante,
a conferma dell'importanzadi queste duefattori solubili, è stata un'indaginegene-
array di biopsie di pazientiaffetti di melanoma nella quale è stato evidenziato
comei trascritti di IL-10 eTGF-β risultino up-regolati (Harlin, Kunaet al. 2006)
Un dato che può in parte spiegarela mancata efficacia della vaccionterapia
antitumoraleè il fatto chei pazientiarruolati nei diversi trials clinici sonosoggetti
che presentano una malattia conclamatae di età, nella maggior parte dei casi,
avanzata. Infatti, i pazienti sottoposti agli interventivaccinali presentanounaetà
media di 65-80 anni,un arcotemporalein cui la produzionedi linfociti T naïve da
parte del timo è arrestata da tempo. Conseguentemente la rispostaal vaccino
dipenderàdal repertoiredei linfociti T memory selezionato durantela vita del
soggetto, e questo limita fortementel'innescarsi di una risposta efficace. La
presenzadi un tumoreavanzatoed in alcuni casi metastatico, inoltre, esacerba
quei fenomeni soppressivi descritti in precedenza limitando la possibilità di
ottenereunarispostaterapeuticaefficace(Finn2003).
Immunoterapia passiva: il successo del trasferimento adottivo degli
effettori immunitari CD8+
Secondo la definizione di Steven Rosenberg, l’im munoterapia cellulare è il
trasferimentoin un organismo ospite di cellule immunicon lo scopodi introdurre
nuove funzioni o di correggerequelle difettive. L’entusiasmoriposto in questo
approccioderiva dalla possibilità di manipolare,ex vivo, vie biologichecellulari e
molecolari in un modo che non sarebbe altrimenti possibile nell’ospite “ intatto”
(Dudley,Wunderlich et al. 2002).
Nell’uomo i primi approccidi trasferimentoadottivo si basavanonel trasferimento
di cellule killer attivate da linfochine (LAK) caratterizzate da un'attività
citotossicanonHLA-ristretta(Rosenberg,Mule et al. 1985)derivata in granparte
dalle cellule NK circolanti. L'utilizzo delle cellule LAK , che hanno dato ottimi
risultati nei modelli murini, ha generatonell'uomo risultati molto discutibil i. Gli
studi successivisi sono quindi indirizzati verso il trasferimento adottivo di
linfociti T CD8+ caratterizzati da un'attivitàcitotossica antigenespecifica, HLA-

Introduzione
26
ristretta. Recenti osservazionihanno dimostrato che l'effi cacia terapeutica del
trasferimentoadottivo cellulare (ACT), sia nei modelli murini che nei trials
clinici, è influenzatodallo stadio maturativo del linfocita T CD8+ (Gattinoni,
Klebanoff et al. 2005). La popolazione dei linfociti T viene suddivisa
classicamente in quattro stadi maturativi: CD8+ naïve, CD8+ effettori precoci,
CD8+ effettori intermedi e CD8+ effettori tardivi. Tale suddivisone si basa
sull'analisi fenotipica di alcuni clusterdi differenziazione (CD), sulla lunghezza
dei telomerie sull'espressione genica(Appay, Dunbar et al. 2002;Lanzavecchia
and Sallusto 2002). Un aspetto indubbiamente interessante è legato
all'osservazione che i linfociti T CD8+ effettori tardivi, se pur presentano
un'ottima attività citotossicain vitro, risultanoavere un debole impatto terapeutico
in vivo, di circa 100 volte inferiore se paragonato a quello dei linfociti T CD8+
effettori intermedi (Sussman,Pariharet al. 2004;Wang,Huang et al. 2004). Una
possibilespiegazionedi questadiversafunzionalità può esser dovutaalla perdita
del CD62L,una glicoproteinadi membranaessenziale per l'homing del linfocitaal
linfonodo.L'incapacitàdel linfocita T di migrare nei linfonodi secondari correla
con l'inefficacia terapeuticadegli effettori immunitari ed evidenzia come il
microambiente tumorale non sia grado di attivare da solo i linfociti T CD8+
(Kagamu,Touhalisky et al. 1996; Speiser,Mirandaet al. 1997). Tuttavia, l'ACT
basatasull'impiego di linfociti T effettori tardivi CD62L-CCR7- ha portato a
ottimi risultati terapeutici(Dudley,Wunderlich et al. 2002).
La ricerca sull'ACT si è oggigiorno indirizzata verso i TIL isolati dalla massa
tumoraleo dai linfonodi limitrofi alla massatumorale, in quanto nella maggior
parte dei casi essi rappresentanouna popolazione costituita da linfociti T CD8+
effettori intermedi e priva di NK (Rosenberg, Yannelli et al. 1994). I TIL possono
essereespansiin vitro utilizzandoanticorpi specifici per il CD3 e il CD28 o alte
dosi di IL-2, sottoposti a selezioneper generarelinfociti con elevata avidità di
riconoscimento dell'antigene tumorale e reinfusi nello stessopaziente in un
contestocompletamente autologo(Kawakami, Nishimuraet al. 1993). Sebbene i
protocolli perla generazionedei TIL sianotecnicamentecomplessie costosienon
sianoapplicabili a tutti i pazienti,recentirisultati hannorinnovatol'interesseverso
questaformadi terapiatumorale.
Per incrementarel'efficacia terapeuticadel'ACT sono state sperimentate varie
strategie tra le quali l'utilizzo di adiuvanti per promuoverela funzionalità dei

Introduzione
27
linfociti CD4+ Th1 endogeni, essenzialipermantenerel'attivazionedegli effettori
trasfusi (De, Mil ler-Grazianoet al. 2005), la co-infusione di TIL e linfociti T
CD4+ Th1 (Hu, Winter et al. 2000), la somministrazione di citochine IL-2, IL-7,
IL-12, IL-15 (Rosenberg,Yannelli et al. 1994; Klebanoff, Khong et al. 2005),
molecoleantagonistequali per esempiobloccanti di TGF-β (Suzuki, Kapooret al.
2004), anticorpi anti-CTLA-4 (Sutmuller, van Duivenvoorde et al. 2001), ed
anticorpi attivanti la funzionalità dei linfociti T (Grabert,Cousenset al. 2006).
Una menzionespecialeva fatta per la metodica della mielo-linfo-deplezionedel
paziente tramite radioterapia o chemioterpia, utilizzando farmaci quali
Fludarabinao Ciclofosfamide,primadel trasferimento adottivo dei TIL (Cheever,
Greenberget al. 1980; Dudley, Wunderlich et al. 2002). Il razionale della mielo-
linfo-deplezioneconsiste,in primo luogo,nell'eliminarele popolazioni linfocitarie
endogenechecompetono perle citochineomeostatichedel pool linfocitario (IL-7,
IL-15 e IL-2) e che,quindi, potrebberoostacolare la proliferazionedi linfociti T
trasferiti (Muranski, Boni et al. 2006). In secondoluogo l'eliminazione delle
popolazioni immuno-regolatoriesia del comparto linfoide (comei linfociti Treg
CD4+CD25+FOXP3+) che mieloide (come le MDSC) potrebbefavorire l'azione
effettricedelle celluleT trasferite. Inoltre, la mielo-linfo-deplezioneè in grado di
indurredelle modificazioni fenotipicheimportanti nei linfociti T trasferiti . È stato
descritto,infatti, comei TIL, chenormalmentepresentanounabassaespressione
delle proteinedi superficie CD28 e CD27, dopo trasferimento in pazienti mielo-
linfo-depletati, esprimanoentro breve tempo, alti li velli delle due proteine di
membrana (Hendriks, Gravestein et al. 2000; Acuto and Michel 2003).
L'espressione del CD28 e del CD27, oltre a migliorare l'efficacia citotossica,
determina soprattutto un incremento della sopravvivenza dei TIL trasferiti
(Powell, Dudleyet al. 2005). Similmenteancheil la catenaα del recettoreper IL-
7 (IL-7Rα), normalmentepocoespresso a livello della membranaplasmatica dei
TIL, viene iperespressadopo trasferimento nei pazienti sottoposti a un regime
mielo-linfo-depletivo (Kaech,Tanet al. 2003;Powell, Dudleyet al. 2005).
Gli agenti chimici e ionizzanti, utilizzati per indurre la mielo-linfo-deplezione,
sono in grado,inoltre, di modularel'immunogenicità delle cellule tumorali stesse,
determinando unamaggiorespressionedi molecole MHC di classeI e proteine di
membranaquali FAS e MUC-1, e questo, sianei modelli murini chenei pazienti
(Gelbard,Garnettet al. 2006). Ancora, recentemente è stato dimostrato che la

Introduzione
28
radiazione ionizzantenon solo determina nelle cellule tumorali un aumentodel
numero di peptidi antigenici presentatiin associazione alle molecole MHC di
classeI, ma soprattutto, la sintesidi nuoveedunichesequenzepeptidiche, capaci
di renderepiù immunogenicoil tumorestesso(Reits, Hodgeet al. 2006). Infine,
l'effetto citotossicodella radiazioneionizzante determinando un locale danno
tessutale ed uno stato di infiammazione, promuovela proliferazione delle DC
autologhe che stimolano l'attivazionee l'espansione dei TIL trasferiti (Kieper,
Troy et al. 2005). L' utilizzo della mielo-linfo-deplezione ha consentito di
potenziare efficacementel'ACT ottenendorisposte cliniche oggettive in circa il
50% dei pazienti con melanomametastatico (Gattinoni, Powell et al. 2006)
(Figura3-A).
Accantoa procedure per potenziarel'efficacia terapeuticadell'approccio ACT e a
traslarlanel contesto di tumori di diversaistologia, comead esempio il trapianto
delle cellule staminali ematopoietiche(HSC) in regime di mielo-linfo-deplezione
primadell'ACT (Wrzesinski, Pauloset al. 2007), la grandesfidachealimenta gli
studi in questosettoreè soprattuttoil superamento del contestoautologodi questa
procedura per ottenere una terapia "Off-the-self". In altre parole l'obiettivo è
quello di poter trasferireeffettori immunitari non più provenienti solamentedal
pazientestessoma ancheda altri donatori, quali ad esempio pazienti che hanno
risposto ottimamenteall'ACT autologa o donatorisani. In questo modo sarebbe
possibilecostruireuna libreria comunedi cloni CTL specific i per vari antigeni
tumorali, facilmente utilizzabile: i CTL potrebbero venir rapidamente testati
contro le cellule tumoralidi un pazientee unavolta identificati quelli responsivi,
amplificati e trasferiti nel malato. I primi trials clinici che sfruttano l'ACT
allogenica sono risultati molto incoraggianti (Comoli, De Palma et al. 2004)
(Figura3-B). Più innovativi sonogli studidi terapiagenicachemirano,invece,a
ricostruiredirettamentein vitro la reattività antigenica del linfocita che poi verrà
trasferito nel paziente (Kershaw, Teng et al. 2005) Questi studi si basano
sull'utili zzo di vettori lentivirali o retrovirali per indurre l'espressionedi un TCR
ad alta affinità verso l'antigene tumorale in linfociti T naïve ottenuti da un
donatoresano.In questomodoè possibile condensarele caratteristichefunzionali
di un linfocita T CD8+ effettore tardivo (TCR ad alta affinità) con quelle
fenotipiche di un linfocita T CD8+ naïve (CD62LhiCCR7hiCD27hiCD28hi),
ottenendo un potente effettore immunitario (Morgan, Dudley et al. 2006).
Introduzione
29
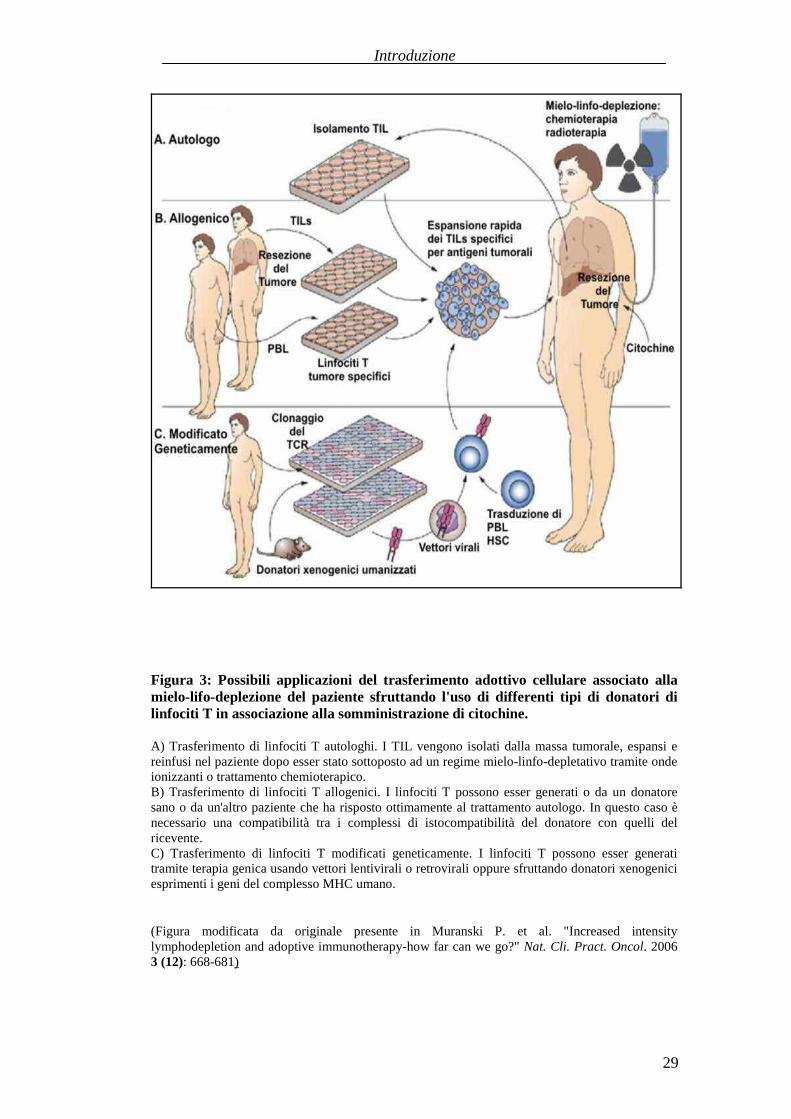
Figura 3: Possibili applicazioni del trasferimento adottivo cellulare associato allamielo-lifo-deplezione del paziente sfruttando l'uso di differenti tipi di donatori dilinfociti T in associazione alla somministrazione di citochine.
A) Trasferimentodi linfociti T autologhi. I TIL vengono isolati dalla massatumorale,espansiereinfusi nel pazientedopoesser stato sottopostoadun regimemielo-linfo-depletativotramiteondeionizzantio trattamentochemioterapico.B) Trasferimento di linfociti T allogenici. I linfociti T possonoessergenerati o da un donatoresano o da un'altro paziente cheha risposto ottimamente al trattamento autologo. In questo caso ènecessario una compatibilità tra i complessi di istocompatibilità del donatore con quelli delricevente.C) Trasferimento di linfociti T modificati geneticamente. I linfociti T possonoesser generatitramiteterapia genicausandovettori lentivirali o retrovirali oppuresfruttandodonatori xenogeniciesprimentii genidel complessoMHC umano.
(Figura modificata da originale presente in Muranski P. et al. "Increased intensitylymphodepletion andadoptive immunotherapy-how far canwe go?" Nat. Cli. Pract. Oncol. 20063 (12): 668-681)

Introduzione
30

Introduzione
31
Futuribili ma molto interessanti,infine, sonogli studi cheprevedonol'util izzo di
linfociti T da donatori xenogeniciquali, ad esempio, i topi trasgenici HLA-
A* 0201 (HHD-mice). Questianimali sonoknock-out per le molecole H2d/b del
complesso MHC murino e presentanola fusione delle molecole α-1 e α-2 del
complesso HLA-A2 umanocon la molecola α-3 del complessoH2 murino. Il
sistemaimmunitario dei topi HHD risulta quindi "educato" esclusivamente dal
complesso MHC umano.La potenzialità dell'utilizzo di donatori xenogenici è
dovuta al fatto che il loro repertoriodi linfociti T non è soggetto alla selezione
negativa verso i peptidi self umani; pertanto, si potrebbero facilmente generare
linfociti T con TCR ad alta affinità verso gli antigeni tumorali umani (Cohen,
Zhenget al. 2005;Kuball, Schmitzet al. 2005)(Figura3-C).
Telomerasi come antigene tumorale universale, candidato ideale per
l'immunoterapia anti-tumorale.
I telomeri sono sequenzeoligonucleotidiche non codificanti che chiudono le
estremità dei cromosomi, costituite generalmente dalla ripetizione del motivo
TTAGGG. Ad ogni ciclo cellularesi assistead un graduale accorciamento dei
telomeri, e una volta raggiunta una lunghezza critica, la cellula attiva un
programmagenico di morteprogrammata (HahnandMeyerson2001). Alcuni tipi
cellulari (cellule staminali, cellule germinali, linfociti attivati) risolvono questo
problema attraversol'attività dell'enzimatelomerasi. La telomerasi è costituita da
una componente di RNA, lungacirca 150 nucleotidi cheserveda stampo per la
sintesidel telomero; e daunacomponenteproteica ad attività enzimatica (TERT)
chefungedatrascrittasiinversa, in gradodi sintetizzaresolounsegmento di DNA
che è complementare alla sequenzadi RNA (Cong, Wright et al. 2002).
L'attivazione della telomerasi,intervenedo nel fenomeno di immortalizzazione
cellulare, è stata descritta anchenelle cellule tumorali (Hastie, Dempster et al.
1990; Kim, Piatyszek et al. 1994; Meyerson,Counter et al. 1997; Shay and
Wright 2002). In particolareessaè risultata esserespressain circa l'85% delle
cellule tumorali indipendentementedalla loro origine o istologia e risulta essere
pertantoun importanteantigenetumorale universale (Vonderheide, Hahn et al.
1999). Anche se la ricerca di inibitori farmacologici dell'attivi tà di telomerasi
rappresentauna promettentestrategiaper una futuribile terapia anti-tumorale, le
proprietà immunologichedi TERT suggeriscono che questa proteina sia un

Introduzione
32
potenziale bersaglio per nuovi approccidi immunoterapia contro il cancro. A tal
proposito sono da riferire le strategie immunoterapiche nei modelli murini:
l'induzione di una rispostaimmunecontro la proteina tramite vaccinazione con
DC trasfettatecon RNA codificantetelomerasiè stata descritta da Gilboa (Nair,
Heiseret al. 2000;Thornburg,Boczkowskiet al. 2000). A seguitodell'intervento
vaccinale, gli animali risultavano parzialmente protetti dall'inoculo di linee
tumorali telomerasi-positive. Inoltre, assai interessantedal puntodi vista clinico è
statal'osservazionechegli animali vaccinati risultavano sanie non sofferentiper
l'insorgenzadi patologie autoimmunirivolte contro le cellule ematopoietiche o i
tessuti normali esprimentitelomerasi(Nair, Heiseret al. 2000). Infatti, i linfonodi,
le placche del Peyer, la milza, il timo ed il fegato degli animali vaccinati
risultavanocompletamentenormali ad un'indagine istologica, anchea distanzadi
un anno dalla vaccinazione; inoltre, i li velli di IgM e IgG risultavano
sostanzialmente normali così comeil numero di granulociti , linfociti , eritrociti e
piastrinecircolantinel sangue.Questi risultati possonoesserspiegati dal fatto che
i livelli di attività di telomerasinelle cellule esprimenti fisiologicamente la
ribonucleoproteina risultanoessercircadi 100 volte più bassirispetto alle cellule
tumorali, e questoimplicherebbeun numero di complessi MHC/peptidi-TERT
specifici presentia livello di membranaplasmatica insufficienti nell'attivare una
risposta immunitaria (Gross, Graff-Dubois et al. 2004). Questeosservazioni
avvalorano il significato dell'antigenetelomerasi come antigene tumorale in
quanto la sua diversa espressione permetterebbe al sistema immune di
discriminaretra tessutonormalee tessuto neoplastico. Al tro motivo di interesseè
il fatto chemolteplici risultati scientifici suggerisconochei peptidi derivanti dalla
processazionefisiologica della proteina TERT da parte delle cellule tumorali,
vengonopresentati in associazionecon le molecole MHC di classeI e sono
riconosciuti come bersaglio da linfociti T (Vonderheide, Hahn et al. 1999).
Oggigiorno sonostati descritti numerosipotenziali peptidi di telomerasi umana
presentatida almenocinquemolecoleHLA di classeI (A2, A1, A3, A24 e B7).
Data la complessità del sistemaHLA, la maggior partedei trials sperimentali si
sono focalizzati esclusivamenteverso l'HLA-A2 essendoquesto l'istotipo più
frequente nella popolazione caucasica(Cortez-Gonzalez and Zanetti 2007).
L'impiegodi diversetecnicheper generareex vivo CTL specifici anti-telomerasi
da pazientiportatoridi tumoredimostranoche la tolleranzacentrale o periferica

Introduzione
33
verso quest'antigene non rappresentaun ostacolo importante per indurre
l'espansionedi linfociti T CD8+, anchese studi riguardantil'affinità di queste
cellule ad oggi non sono stati ancora condotti (Cortez-Gonzalez and Zanetti
2007). Interessanti risultanoessereanchele osservazioni sul numerodi linfociti T
TERT-specifici circolanti: infatti, mentrenegli individui sani il numerodi CTL
TERT-specifici è molto basso, nei pazienti oncologici sottoposti a terapie
classichesonostati trovati alti livelli di CD8+ TERT specifici (Filaci, Fravegaet
al. 2006). Inoltre,è statoriportatochei pazienti di portatori di certi tipi di tumore,
comead esempio l'epatocarcinoma,presentano nel siero alti li velli di anticorpi
reattivi contro h-TERT (Masutomi,Kanekoet al. 2002). Sullabasedi quest'intima
relazionetra la presenzadell'antigenee l'instaurarsidi unarispostaimmunitaria,
molti trials clinici di fase I di immunoterapia sono stati condotti in pazienti
portatori di tumori avanzati.Anchese sonostate utilizzate varie formulazioni di
vaccini come DC caricate con peptidi (Vonderheide, Domchek et al. 2004),
peptidi in associazione alla somministrazione di interluchine (Mavroudis,
Bolonakis et al. 2006), cellule B caricate con peptidi (Cortez-Gonzalez and
Zanetti 2007) e DC transfettate con m-RNA codificante per TERT umano(h-
TERT) (Su, Dannull et al. 2005), in vari contesti tumorali come carcinoma
mammario (Vonderheide,Domchek et al. 2004), carcinoma prostatico (Su,
Dannull et al. 2005; Cortez-Gonzalezand Zanetti 2007), melanoma (Parkhurst,
Riley et al. 2004) e carcinomapancreatico (Bernhardt, Gjertsen et al. 2006),
nessunarispostaclinica oggettivaè stata ancora descritta. Tuttavia questi trials
hanno prodotto dei dati interessanti; infatti è stata osservata l'espansionedi
linfociti T CD8+ TERT-specifici in 19 pazienti immunizzati tramite DC
transfettate con l'm-RNA codificanteh-TERT su 20 (Su, Dannull et al. 2005).
Questeosservazioni,uniti agli studi sui modelli murini, piattaforma ideale per
perfezionareprotocolli vaccinalidapotertraslare nell'uomo, rappresentanole basi
perunulterioresviluppodi vaccinibasatisuh-TERT.
Il topo TRAMP: un modello di studio della carcinogenesi prostatica.
In campo oncologico, lo sviluppo di modelli murini in grado di mimare la
patologia umanaè di importanzafondamentale per caratterizzaregli stadi della
progressione tumorale e la capacità del sistema immunitario di montare
un’opportuna rispostaantitumoralenonché per verificare l’effi cacia di nuove

Introduzione
34
terapie. Storicamente, i primi studi compiuti sui modelli animali si riferisconoa
sarcomicutaneiautoctoni indotti medianteirradiazioneo esposizione a sostanze
cancerogene, come il MCA. Altri modelli sonobasati sul trapianto sottocutedi
lineecellulari neoplastichedi origine umana in topi nudi, in mododa mimare la
crescitaneoplastica, o in alternativa, l’impianto diretto del tumore nell ’organo
bersaglio.Questomodello ortotopico permette di ottenere rispostebiologichee
terapeutichepiù simili a quantovisto nei tumori umani. Anche se tutti questi
modelli continuanoadesser utilizzati, rappresentando un’effi cace opportunità per
la ricerca ed un interessante banco di prova per sperimentare nuove terapie
antitumorali, è innegabileche essi tuttavia presentano dei limiti intrinsechialla
loro naturapseudofisiologica.I tumori trapiantabili inoltre, essendo indotti da
cancerogenichimici o fisici a dosi fortemente e acutamente genotossiche,
esprimono antigeni chesebbenesianotaloraforti antigenidi rigetto nonsi trovano
nella controparte neoplasticaa genesi spontanea. Al tro limite dei modelli di
tumore trapiantabili risiede nel fatto che la neoplasiaprogrediscein un contesto
tessutale non propriamentefisiologico, svincolato dal microambiente che si
generadurante le fasi della progressione neoplastica. Infatti, i tumori spontanei
d’origine epiteliale, comeil carcinomaprostatico, sonocompostida almenodue
insiemi cellulari che interagisconotra di loro: le cellule tumorali (componente
parenchimale) e le cellule stromali con la matrice extracellulare (componente
stromale).Le cellule normali nello stromatumorale, quali le cellule endoteliali, i
fibroblasti, i macrofagi e le mastcellule possonoesserestimolate in maniera
abnormedanumerosi fattori di crescita diffusibili , rilasciati dalle cellule tumorali,
o possono direttamenteprodurrecitochine e peptidi che, per effetto paracrino,
stimolano la crescita tumorale.Risulta, quindi, evidenteche le due componenti
sono biologicamente attive e concorronoentrambe a creare un microambiente
favorevole alla crescita e alla progressionedella neoplasia, come è evidente
nell’abnormestimolazionedella neovascolarizzazioneindotta dal tumoree, cioè,
lo sviluppo di nuovi vasi sanguignicheoriginanoda vasi preesistenti del tessuto
di origine. Tale processo, noto con il termine di neoangiogenesiè tipico della
progressionetumoraleal puntoche nessuntumorepuòcrescereoltre 1 mm3 senza
un adeguato supporto sanguigno.L’elevata cinetica replicativa delle neoplasie
trapiantabili preclude, inoltre, la possibilità che si vengano a creare quei

Introduzione
35
fisiologici e complessi meccanismi di interazione tra il sistema immunitario
dell’ospite e la neoplasia spontaneachepossonofortemente influenzarel’esito di
eventuali approcciimmunoterapici.
In questo contestosi inserisce il modello trasgenico murino del carcinoma della
prostata (TRAMP), ingegnerizzato tramite inserimento della sequenza
dell’antigeneT (Tag)di SV40 a valle del promotoredel genedella probasina(PB)
(Greenberg, DeMayoet al. 1995). L’espressionedel genePB, costituito da sette
esonie da sei introni, localizzatoa livello del braccio lungo del cromosoma X
(Xq22),è prostata-specifica, interessandoi sei lobi della ghiandola,e coincidecon
la pubertà dell’animale. L’attivazione del promotore del gene PB determina
conseguentemente la trascrizione e la traduzione della sequenza codificante
l’oncoproteina Tag, promuovendo in questo modo l’ini zio del processo
neoplastico.La proteinaTagè in gradodi interferire con la funzione di Rb e p53,
due tra le principali proteine cellulari oncosoppressorie (Ludlow 1993).
L’espressionedel trasgenePB-Tag è riscontrabile attorno alla 4a settimanad’età
dell’animale, è regolatadagli androgenied è localizzata a livello delle cellule
epiteliali della prostata, in particolarea livello dei lobi dorsolaterale e ventrale.
Successivamente, attorno alla 12a settimana d’età, il trasgene è espresso
indistintamenteda tutte le regioni della prostata e anchea livello delle vescicole
seminali; in questoperiodo, inoltre, i topi sviluppanomutazioni spontaneea
livello dei recettori per gli androgeni determinando lo sviluppo di uno stato
tumorale androgeno-indipendente, similmente a quanto accade nell’uomo. La
penetranzadi tale processoé completae l’animale muoreentro la 40a settimana
d’età.
Nel modello TRAMP la progressionedella neoplasia verso una crescita
indipendente dagli androgeniè stataampiamente dimostrata dal fatto chesei topi
vengono castrati alla 6a settimana di vita solo il 50% di essi svilupperàuna
malattia androgeno-indipendente(Eng, Charles et al. 1999); al contrario, se la
castrazione viene effettuatadopo la 12a settimana d’età, la percentuale degli
animali che svilupperranouno stato patologico ormone-indipendentesale al 70-
80% (Gingrich, Barrios et al. 1996). L’evoluzione della neoplasiain questo
modello è caratterizzata da fasi ben distinte: il tumore esordisce sottoforma
d’iperplasiaintraepiteliale(PIN), per poi passaread uno stato di carcinoma ben
differenziato e culminana in un carcinoma poco differenziato. L’iperplasia

Introduzione
36
intraepiteliale è tipica degli animali di 8-10 settimane d’età ed è caratterizzata
istologicamenteda un epitelio a stuttura cribrosa, con nuclei ipercromati e
stratificati; a questaseguela fased’ iperplasia ben differenziata tipica di animali di
12 settimane, in cui si notaun elevatonumerodi mitosi ed eventi apoptotici. La
fase d’ iperplasia poco differenziata tipica degli animali di 24-30 settimane
rappresentala fase progredita della neoplasia ed è caratterizzata da lesioni
necroticheedemorragiche(Figura4).
La tendenzasuccessivadellaprogressioneè la metastatizzazione, siaematica che
linfatica, dapprima a livello dei distretti limitrofi alla sede prostatica, quali i
linfonodiperirettali ed il collo dellavescica, poi a livello del rene, del fegato, dei
polmoni e delle ghiandole salivari. Negli animali castrati, l’i ncidenza della
metastatizzazionerisulta essereraddoppiata se paragonata agli animali non
castrati. Questo evidenziail fatto che le cellule metastatiche sono in grado di
acquisire l’indipendenza androgenica mentre le lesioni primarie rimangono
ormone-dipendenti(Greenberg, DeMayo et al. 1995). I topi TRAMP sonostati
inoltre utilizzati come risorsa per sviluppare nuove linee tumorali , che hanno
avuto un ruolo importanteper gli studi in vitro. Sonostate, infatti, isolate dalla
prostata tumorale del topo tre linee stabilizzate (C1, C2, C3) che mantengono
l’espressionedi molecoledi superficie tipiche dell’epi telio prostatico tumorale e
dispongonodi attività tumorigenica,eccetto la lineaC3, se inoculate in animali
singenicial topoTRAMP (Foster,Gingrich et al. 1997).
I carcinomiprostatici TRAMP, quantole lineederivate (Yang, Holt et al. 2001),
esprimono alcuni antigeni prostata-associati (PAA) tra cui Prostate Stem Cell
Antigen (PSCA),Six TransmembraneEpithelial Antigen of Prostate (STEAP) e
ProstateSpecific MembraneAntigen (PSMA), che presentano elevata omologia
con le rispettive isoforme umane.Il gene codificante l’antigenePSCA mappa
nell’uomo a livello del cromosoma 8 (8q.24.2) mentre nel topo a livello del
cromosoma 5, codifica per una proteina altamente glicosilata ancorata alla
superficie delle cellule stromali della prostata tramite il meccanismo del
glicosilfosfatidilinositolo (GPI). L’omologia tra l’i soforma umana con quella
murina è dell’65%. Entrambele proteinesono soggette ad internalizzazione, a
processazionee presentazionevia complesso MHC di classeI (Reiter, Gu et al.
1998). Il genecodificantel’antigeneSTEAP è localizzato nell’uomo a livello del
Introduzione
37
Fig
.4:I
ndag
ine
isto
logi
cade
ltes
suto
pros
tati
codi
topi
TR
AM
Pa
vari
stad
idim
alat
tia.
A)
pros
tata
norm
ale.
B)
neop
lasi
ain
trae
pite
liale
.C)
carc
inom
abe
ndi
ffer
enzi
ato.
D)
carc
inom
am
oder
atam
ente
diff
eren
ziat
o.E
)ca
rcin
oma
scar
sam
ente
diff
eren
ziat
o.(F
igur
am
odif
icat
ada
orig
inal
epr
esen
tene
lsito
:ww
w.G
reen
berg
lab.
fhcr
c.or
g)

Introduzione
38

Introduzione
39
cromosoma7 (7p22.3) mentrenel topomappaa livello del cromosoma5, codifica
perunaproteinatrasmembrana“a seipassi” chesottendela funzionedi canaledi
membrana;in questocaso l’omologia delle due isoforme è del 80% (Hubert,
Vivancoet al. 1999). La mappaturagenetica del PSMA nell’uomo ha rilevato la
presenzadi due geni: uno è localizzatonella regione11p11 e codifica per il
trascritto espressoa livello della prostata, l’al tro mappa a livello della regione
11q14 ma la sua espressionenon è prostata specifica. Nel topo, il gene
codificante l’antigene PSMA mappa in un unico locus ovvero a livello del
cromosoma7. Entrambele due isoforme,murina che umana,con omologia del
86% codificano per una glicoproteinadi membranadi tipoII costituita da 750
amminoacidi chesimilmentealla proteinaPSCAè soggetta ad internalizzazionee
proccessamentovia complessoMHC di classe I (Bacich, Pinto et al. 2001). Gli
mRNA specifici per le isoformemurinee umanedi PSCAe STEAP hannouna
distribuzione tissutalelimitata al tessuto prostatico e risultano iperespressi nei
tumori prostatici primari enellemetastasidei topi TRAMP.
Similmenteil PSMA, nonostante una distribuzione tissutale più ampia nel topo,
risultaup-regolato nei tessuti neoplasticiTRAMP.
Da quantodescritto è evidentechequestomodello riveste un ruolo fondamentale
nella ricerca biomedica sul carcinoma prostatico, avendo delle peculiarità
fondamentali chepossiamoriassumerein duepunti:
1. L’ evoluzione neoplasticada carcinoma in situ a tumore francamente
invasivo è simile a quantoaccade nell’uomo; questopermette di disporre
delle diversefasi della progressione neoplastica per disegnare protocolli
sperimentalivolti alla prevenzioneo alla terapia del tumore,caratteristica
questache rendepotenzialmentetraslabilealla clinica eventuali approcci
terapeutici.
2. La condivisionedi antigeniprostataassociati (PAA) conelevataomologia
con le isoforma umane;questo permettedi predisporreun reagentario che,
qualoradimostratosi efficacein modelli murini preclinici, potrebbeesser
impiegabile nella terapiadella neoplasiaumana,comedimostrail recente
impiego di anticorpi anti-PSMA che è stato in grado di indurre la
regressione infartualedi carcinomiprostatici umani impiantati in topi nudi
(Huang,Bennett et al. 2004).

Introduzione
40
Questomodello, tuttavia,presentaanchedei limiti, il più importante dei quali è
rappresentatoda una variabilità fenotipicanella progressionedella neoplasia che
risulta potenzialmentelimitante nella valutazione di un trial sprimentale. Questa
variabilità è legataal backgroundgenetico degli animali e conferma l’ipotesi di
specifiche alterazioni di uno o più geni che sopraggiungonodurantelo sviluppo
della neoplasia. L’importanzadel backgroundgenetico di partenzanell’indiri zzare
lo sviluppo della neoplasia versoun determinatopatterndi crescita è ampiamente
dimostrato in letteratura; infatti, i tumori TRAMP di background C57BL/6
risultano esseredi naturapropriamenteinfil trante invadendo con alta frequenza
l’uretrae le vescicole seminali; al contrario le massetumorali sviluppate dai topi
F1 derivati dall’i ncrocio (C57BL/6 TRAMP x FVB) sono maggiormente
localizzateed altamentevascolarizzate(Kaplan-Lefko, Chenet al. 2003). Ad una
diversa progressione della malattia del carcinoma TRAMP si associano
caratteristiche peculiari che si discostano in parte dalla medesima patologia
nell’uomo. Infatti, il tumore TRAMP presenta una percentuale maggiore di
fenotipo fill oide, rispettoa quantoravvisabile nell’uomo, ovvero unacommistione
tra un tumore di naturaconnettivalee uno di natura epiteliale. Questadoppia
naturadel tumoreTRAMP può portarea degli esiti di patologia che non sono
riferibili alla neoplasia umana.Nella stessaottica, il tumoreTRAMP presentauna
maggior percentualedi fenotiponeuroendocrino rispetto al carcinomaprostatico
umano. L’i nsorgenzadel fenotipo neuroendocrino è un eventotuttavia stocastico,
dovuto all’acquisizione da parte delle cellule epiteliali, in seguito alla
trasformazione neoplastica, di capacità secretorie tipiche delle cellule
neuroendocrine(Kaplan-Lefko, Chenet al. 2003).
Queste ultime osservazioni, se pur ridimensionano, non cancellano certo
l’importanzadel modello comepiattaforma ideale per caratterizzare il ruolo del
sistemaimmunitario nella progressionedel tumoredella prostatae nella messaa
punto di appropriatestrategieterapeutiche anti-tumorali.

Scopo della Tesi
41
SCOPO DELLA TESI
La definizionemolecolaredegli antigenitumore-associati riconosciuti dai linfociti
T e la conoscenzadei meccanismiche regolano l’attivazione linfocitaria e dei
meccanismi di escapetumoralehannoconsentito negli ultimi anni la messaa
punto, in modelli neoplasticisperimentali, di più efficaci strategie vaccinali, che
mirano ad interferire con la tolleranza immunologica e ad attivare in modo
specifico i meccanismi effettori dell’immunità adattativa,conlo scopodi generare
una rispostaimmunetumore-specifica,protettiva e di lungadurata.
Nell'ambito dell'immunoterapiaantitumorale due sono le strade potenzialmente
perseguibili: la prima è la vaccinoterapia(detta anche immunoterapia attiva), la
secondaè l'immunoterapiacellulareadottiva (o immunoterapiapassiva).
Gli interventi di vaccinoterapiamirano ad educare il sistema immunitario del
paziente a riconoscereantigeni tumori-associati (TAA) presenti nelle cellule
tumorali con lo scopodi arrivarea contrastare la crescita tumorale attraverso la
selezione e l'espansionedelle cellule effettrici proprie dell 'individuo (Lollini,
Cavallo et al. 2006). Ad oggi un numero consistentedi vaccini antitumorali sono
stati utilizzati in clinica: vaccini basatisulle stessecellule tumorali irradiate, su
cellule dendritiche transfettateo infettatecon l'antigenedi interesseo sull'inoculo
della proteina o del peptideantigenico(Mocelli n, Mandruzzato et al. 2004). Una
menzioneparticolareva fattaperi vacciniaDNA, cherappresentanounafrontiera
emergentevista l’ampia mole di dati ottenuti in ambito preclinico (Cohen 2001).
Sebbene i nuovi vaccini anti-tumorali mostrino un’aumentata efficacia nei
confronti di tumori di piccole dimensioni in modelli preclinici, l’im munoterapia
attiva è generalmente inefficace come singolo provvedimento terapeutico una
volta che la neoplasia raggiungedimensioni consistenti o diviene metastatica.
Comeè facile intuire, i limiti di questo approccio sonomolteplici e recenti meta-
analisi di protocolli clinici antitumorali basati su diverseformulazioni di vaccini
hanno rivelato che,sebbenesia possibile indurreunarispostaimmunitaria verso
antigeni tumorali nella maggiorpartedei soggetti vaccinati, le risposte cliniche
oggettive sono piuttosto limitate e si verificano in meno del 10% dei pazienti
trattati (Choudhury,Mosolitset al. 2006). La tolleranza ad antigeni tumorali self e
l’attiva immunosoppressione mediatada cellule mieloidi (Bronte and Zanovello
2005) e/o linfoidi (ColomboandPiconese2007)sonosolo alcunedelle causein
gradodi spiegarequesta modestaefficaciaterapeutica.

Scopo della Tesi
42
A differenza della vaccinoterapia,l'immunoterapia cellulareadottiva consegue il
trasferimento,in un organismoospite, di cellule immunitarie con lo scopodi
introdurrenuovefunzioni o di correggerequelle difettive (Gattinoni, Powellet al.
2006). L’entusiasmo riposto in questo approccio deriva dalla possibilità di
manipolare, ex vivo, vie biologiche cellulari e molecolari in un modo che non
sarebbealtrimenti possibile nell’ospite intatto. Linfociti T infil tranti il tumore
(TIL) possonoesserecoltivati in vitro, espansie sottoposti a selezione per
generarelinfociti con elevataavidità di riconoscimento dell'antigene tumoraleed
esserereinfusi nel paziente donatore,in un contesto completamente autologo
(Kawakami, Nishimura et al. 1993). Per incrementarel'efficacia terapeutica del
trasferimentocellulareadottivo,pazienticon melanoma sonostati trattati tramite
radioterapiao somministrazionedi farmaci quali Fludarabinao Ciclofosfamide,
allo scopodi indurreunaparziale mielo-linfo-deplezioneprimadel trasferimento
adottivo dei TIL. Qualunquesia il meccanismo d'azione, la linfodeplezione ha
consentitodi ottenererispostecliniche oggettive in circa il 50% dei pazienti con
melanomametastatico(Muranski,Boni et al. 2006).
La sceltadell'antigeneversocui mirarela strategiaimmunoterapeutica allo scopo
di generare o ristabilire unarispostaimmunitariaspecifica rappresentaun aspetto
di primariaimportanzanell'intraprendereunqualsiasipercorsoimmunoterapico.
La ribonucleoproteinanuclearetelomerasi(TERT), la cui funzione è mantenere
l'integrità delle porzioni terminali dei cromosomi durante la duplicazione del
DNA, è normalmenteinattivanellamaggior parte delle cellule somatiche,mapuò
riattivarsi in molti tipi di neoplasiefavorendoil processodi immortalizzazione
cellulare e, conseguentemente,la natura invasiva del tumore (Zakian 1997).
TERT, infatti, è espressain circa l'85% delle cellule tumorali, indipendentemente
dalla loro origine o istologia, e risulta essere pertanto un importante antigene
tumoraleuniversale.
Su questepremessesi basaquestolavoro, il cui scopoè statovalutare l'impatto
terapeutico di interventi sia di immunoterapia attiva che passiva, in contesti di
tumorigenesimurinaindottaespontanea, sfruttandol'antigeneTERT.
L'efficacia della strategiadi immunoterapia cellulare adottiva ha richiesto una
caratterizzazionefunzionale in vitro dei linfociti T citotossici selezionati ed
espansi dall'interventovaccinalee una successiva verifica della loro capacità
terapeutica,una volta trasferiti in animali portatori di tumore,nel contrastarela

Scopo della Tesi
43
progressionedella neoplasia.Analogamente, l'efficaciadell'intervento vaccinaleè
stato valutato comeeffettiva protezionedell’ animale contro la progressionedella
neoplasia, in termini d’aumentodella sopravvivenza degli animali trattati con
vacciniaDNA rispettoadanimalitrattaticonadeguati controlli negativi .

Scopo della Tesi
44

Materiali & Metodi
45
MATERIALI & METODI
TOPI
Sonostati utilizzati topi maschi transgenici TRAMP di background C57BL/6 di
aplotipo H-2b derivati da una coppia originaria del “ Greenberg Lab” (Fred
HutchisonCancerResearchCenter,Seattle, U.S.A). Attualmente disponiamo di
una coloniain omozigosi e di unacolonia in eterozigosi in backgroundC57BL/6.
I maschipositivi per l’espressionedel trasgenePB-Tag vengonoutil izzati nei
setting sperimentali all’età di sei settimane.Sono stati impiegati anche maschi
C57BL/6J di aplotipo H-2b e maschi del ceppo Balb/c di aplotipo H-2d di età
compresatra le otto e le dodici settimanedella ditta Charles River Italia S.p.A.
(Calco, Como, Italia). I topi sono stati mantenuti in gabbie di plastica, a
temperaturacostantee dieta bilanciata nello stabulario del Dipartimento di
Scienze Oncologiche e Chirurgiche, Sezione di Oncologia, dell’Università di
Padova. Tutti gli esperimentieseguiti sugli animali si sono attenuti alle linee
guidaindicate dalle direttivenazionaliedinternazionali (Workman,Balmain et al.
1988)
Screening topi TRAMP
La presenzadel trasgenePB-Tag vienevalutatatramite PCRda DNA estratto da
PBL. Gli animali vengono preliminamente anestetizzati per inalazione di etere,
successivamentetramitepasteurdi vetrovieneprelevato un piccolo quantitavo di
sangue a livello dell’occhio. Per l’estrazione del DNA è stato utilizzato il kit
“puregeneDNA isolation kit” (Gentrasystem, Germania) ed il suo protocollo
specifico. Il DNA ottenutoè statoutilizzato cometemplato per una reazione di
PCR condotta utilizzando il thermal cycler PTC-200 peltier (MY Research
Waltham,MA, U.S.A.) cheprevedeva 35 cicli di amplifi cazione (94°C 4 minuti,
94°C 1 minuto45°C15 secondi,72°C1 minuto)seguiti daun’estensionefinale a
72°C per 5 minuti.
La reazioneconsta di unamix costituitada 30 µl di c-DNA, 5 µl di buffer 10X
(Applied Biosystems, CA, U.S.A.) 6 µl MgCl2 25mM (Applied Biosystems, CA,
U.S.A.), 1 µl di dNTPs 10mM (Invitrogen, Carlsbad, CA, U.S.A.), 2.5 µl alla
concentrazionefinale di 1µM dei primersspecific i per il transgene(PB-1F: 5’-
CCG GTC GAC CGG AAG CTT CCA CAA GTG CAT T-3’; TagR: 5’-CTC

Materiali & Metodi
46
CTT TCA AGA CCT AGA AGG TCC A-3’), 2.5µl alla concentrazionefinale di
1 µM dei primersspecificiper la β-caseinadi topo(M β-caseinaR: 5’-AGA AAC
GGA ATG TTG TGG AGT-3’; M β-caseinaF:5’-GAT GTG CTC CAG GCT
AAA GTT -3’), 0.5µl Taq-DNA polymerase(Invitrogen,Carlsbad,CA, U.S.A.)e
2,5 µl di H2Operiniettabili.
Ultimatala reazionei campionivengonofatti correre in un gel d’agarosioall’1%
contenete 0.5 µg/ml di Etidio Bromuro (EtBr), assieme ad un marcatore
molecolare (1Kb Marker,MB). Le bandeattesesonorispettivamentea 0,6Kb per
il PB-Tagea0,5Kb perla β-caseina.
TERRENI DI COLTURA
Colturecellulari
DMEM 10%: DMEM (Invitrogen, Carlsbad, CA, U.S.A.), 2 mM di L-
glutammina, 10 mM di HEPES, 150 U/ml di streptomicina, 200 U/ml di
penicillina, 20 µM di 2-β-mercaptoetanolo (2β-Me) (Sigma-Aldrich, St.Louis,
MO, U.S.A.) e 10% di siero bovino fetale (FBS, Invitrogen, CA, U.S.A.)
inattivato a56°Cper30’.
DMEM 10% con rIL-2: DMEM (Invitrogen, Carlsbad,CA, U.S.A.),2 mM di L-
glutammina, 10 mM di HEPES, 150 U/ml di streptomicina, 200 U/ml di
penicillina, 20 µM di 2-β-mercaptoetanolo (2β-Me) (Sigma-Aldrich, St.Louis,
MO, U.S.A.) e 10% di siero bovino fetale (FBS, Invitrogen, CA, U.S.A.)
inattivato a 56°C per 30’ ,addizionatocon 20 U/ml di rIL-2 (Euro Cetus-Chiron,
Milano, Italia).
TRAMP 10%: DMEM (Invitrogen, Carlsbad,CA, U.S.A.) supplementato con
2mM di L-glutammina,10mM di HEPES,20µM di 2-β-mercaptoetanolo(2β-Me)
(Sigma-Aldrich, St.Louis, MO, U.S.A.), 150U/ml di streptomicina, 200 U/ml di
penicilli na e 5% siero bovino fetale (FBS, Invitrogen,CA, U.S.A.) inattivato al
calore(56° per30’), 5% NuSerum (Collaborative Biomedical Products,Bedford,
MA, U.S.A.), 5 µg/ml di Insulina (SigmaChemical, St.Louis, Co, U.S.A.) e 0.01
nM di diidrotestosterone (Sigma-Aldrich, St.Louis,MO, U.S.A.).

Materiali & Metodi
47
Coltureutilizzateperi saggi
DMEM 10%: DMEM (Invitrogen, Carlsbad, CA, U.S.A.), 2 mM di L-
glutammina, 10 mM di HEPES, 150 U/ml di streptomicina, 200 U/ml di
penicillina, 20 µM di 2β-Me (Sigma-Aldrich, St.Louis,MO, U.S.A.) e 10% di
siero fetalebovino(FBS, Invitrogen,CA, U.S.A.) inattivato al calore.
RMPI 5%: RPMI 1640 (Euroclone,Milano, Italia), 2 mM di L-glutammina , 1
mM di sodiopiruvato,150U/ml di streptomicina,200U/ml di penicill ina, 20 µM
di 2β-Me Sigma-Aldrich, St.Louis,MO, U.S.A.)e 5%di sierofetale bovino (FBS,
Invitrogen,CA, U.S.A.) inattivatoal calore.
RPMI 3%: RPMI 1640 (Euroclone,Milano, Italia), 2 mM di L-glutammina, 1
mM di sodio piruvato,150U/ml di streptomicina,200U/ml di penicil lina e 3% di
siero fetalebovino(FBS, Invitrogen,CA, U.S.A.) inattivato al calore.
LINEE CELLULARI
MBL-2 (aplotipo H-2b), linfoma indotto da virus leucemogenomurino di
Moloney(M-MuLV), mantenutoattraversopassaggisettimanali intraperitoneoin
topi singeniciC57BL/6. Talelineaèstatamantenutain colturaconDMEM 10%.
CT26 (aplotipo H-2d), linea di coloncarcinomaindifferenziato indotto mediante
carginogenesichimica,stabilizzata dopoespianto da un topo Balb/c. Tale lineaè
statamantenutain colturaDMEM 10%.
C1 (aplotipoH-2b), lineatumoraleprostaticaderivatadal topoTRAMP. Talelinea
èstatamantenutain colturaconil terrenoTRAMP
C2(aplotipoH-2b), lineatumoraleprostaticaderivatadal topoTRAMP. Talelinea
èstatamantenutain colturaconil terrenoTRAMP
C1-B71 (aplotipo H-2b), linea tumorale prostaticaderivante dal topo TRAMP
transfettata stabilmenteper l’espressionedella molecola costimolatrice B71. Tale
linea è stata mantenuta in coltura con terreno TRAMP addizionato con
l’antibiotico geneticina (G418, GIBCO) alla concentrazione di 1mg/ml che
consentedi selezionarela popolazionetransfettata.
C2-B71 (aplotipo H-2b), linea tumorale prostaticaderivante dal topo TRAMP
transfettata stabilmenteper l’espressionedella molecola costimolatrice B71. Tale
linea è stata mantenuta in coltura con terreno TRAMP addizionato con

Materiali & Metodi
48
l’antibiotico geneticina (G418, GIBCO) alla concentrazione di 1mg/ml che
consentedi selezionarela popolazionetransfettata.
MCA 203 (aplotipo H-2b), linea cellulare di fibrosarcoma.Tale linea è stata
mantenuta in colturaconDMEM 10%.
MC 38 (aplotipo H-2b), lineadi colon carcinoma.Tale linea è stata mantenutain
colturaconDMEM 10%.
EL-4 (aplotipoH-2b), lineadi timoma.Tale lineaè statamantenuta in colturacon
DMEM 10%.
B16Lu8 (aplotipo H-2b), linea di melanomamurino che metastatizza a livello
polmonareci è statafornita dal Dr. James C. Yang (SurgeryBranch,National
Istitute of Health, Bethesda, MD). Tale linea è stata mantenuta in coltura con
DMEM 10%.
B16447(aplotipo H-2b), riferita comeB16, lineacellularedi melanomamurino,ci
è statafornita dal Dr. I. J. Fidler (CancerCenter, Houston, TX). Tale lineaè stata
mantenuta in colturaconDMEM 10%.
B16-B71 (aplotipo H-2b), linea cellulare di melanoma murino transfettata
stabilmenteper l'espressionedella molecola costimolatrice B71, gentilmente
fornitaci dal Dr. P. Dalla Bona (Istituto Nazionale Tumori, Mi lano, Italia). Tale
lineaè statamantenuta in colturacon DMEM 10% addizionatocon l'antibiotico
geneticina(G418,GIBCO) allaconcentrazionedi 1 mg/ml.
clone16.1specifico per l'antigeneβ−gal: è statoottenuto dal nostro laboratorioda
topi C57BL/6J immunizzaticon il vettore plasmidico pCMV-β−gal, attraverso
ripetutestimolazioni in vitro con splenociti singenici γ-irradiati e pulsati con il
peptide β-gal96-103 (10 µmol/L) e dopo clonaggio tramite diluizione limite. Tale
CTL vienemantenutoin colturacon il terreno DMEM 10%addizionato con rIL-
2.
PEPTIDI
Il peptide m-TERT198-205 (VGRNFTNL) ristretto per aplotipo H-2b, è stato
identificato dal gruppo della Dr.ssa Elisa Scarselli dell'Istituto di Ricerca di
Biologia Molecolare (IRBM) di Pomezia, tramite screening di un pannello
completo di peptidi che coprivano l'intera sequenza di Telomerasie successiva
analisi bioinformatica utilizzandogli algoritmi predittivi BIMAS e SYFPEITHI.
Tale peptideè stato sintetizzatodalladitta JPT(Berlino, Germania). Il peptideβ-

Materiali & Metodi
49
gal96-103 (DAPIYTNV) della proteina β-galatossidasiè stato sintetizzato dalla
Technogen(Napoli, Italia).
I peptidi presentavano all’analisi mediante HPLC unapurezzamaggioredel 95%.
Il peptideliofi lizzato è statorisospeso in DMSO (Sigma-Aldrich, St.Louis, MO,
U.S.A.) estoccatoa -80°C fino al momentodell’uso.
ALLESTIMENTO DI COLTURE LEUCOCITARIE
Allestimentodi ColtureLeucocitarieMiste (MLC)
Le milze di topi maschiBalb/c (aplotipoH-2d) di 8-12 settimanedi età,e le milze
di topi C57BL/6 e TRAMP (aplotipo H-2b) sono stateprelevatesterilmentee
disgregatemeccanicamente.Si è poi proceduto alla lisi dei globuli rossi
utilizzando 4 ml di Lysing Buffer (ACK BioWhittaker Walkersivill e, MD,
U.S.A.) per 3-4 minuti. Dopo il lavaggio la sospensionecellularecosì ottenutaè
stata passata attraverso una garza di nylon per eliminare possibili aggregati
cellulari. Le sospensionicellulari così ottenutesonostate utilizzateper allestire le
MLC. In piastreda24 pozzetti(Falcon,Becton Dickinson,NJ, U.S.A.) sonostati
coltivati 3x106 splenociti Balb/c γ-irradiati e un ugual numero di splenociti
C57BL/6 o TRAMP in un volume finale di 1 ml di terrenoDMEM 10% per
pozzetto.
Allestimentodellecoltureleucocitarie stimolatedapeptide(MLPC)
Le milze di topi TRAMP e C57BL/6 (aplotipo H-2b) sono state asportate
sterilmente e tramite disgregazionemeccanica è stata ottenuta una sospensione
cellulare priva di aggregati una volta passata su una garza di nylon. La
sospensionecellulare è stata utilizzata per allestire MLPC. In piastre da 24
(Falcon,Becton Dickinson,NJ, U.S.A.) 5x106 splenociti TRAMP e/o C57BL/6
sono stati posti in colturacon terrenoDMEM 10% addizionato con il peptide di
interessealla concentrazionefinale di 1 µM. Le colture così allestitesonostate
incubateper5 giorni a 37˚C in atmosferasatura di vaporeacqueo e contenente il
5%di CO2.

Materiali & Metodi
50
ANALISI CITOFLUORIMETRICA
0.5x106 splenociti derivanti dall’MLPC sono stati risospesi in 50 µl di FACS
buffer (0.9% NaCl, 2% BSA e 0.02% NaN3, (Sigma-Aldrich, St.Louis, MO,
U.S.A.)) a cui è stato aggiunto un anticorpo anti-mouseFc-γ receptor(clone
2.4G2, ATCC HB-197) per ridurre i siti aspecifici, tale sospensionecellulare è
stata quindi incubataper 10 minuti a temperatura ambiente. E’ stato di seguito
aggiunto il tetramero specifico per m-TERT coniugato con ficoeritrina alla
concentrazionedi 5 µg/ml (Beckman Coulter, UK), o il tetramero specifico per
TRP-2 (sintetizzato nel nostro laboratorio) coniugato con ficoeritrina alla
concentrazionedi 5 µg/ml. Successivamenteè statoeffettuato un lavaggio in PBS
a cui è seguita marcaturacon CD8 Tricolor (0,1 µg/106 cellule, clone CTCD8α
Caltag, Burlingame, CA, U.S.A.) e CD3 FITC (0,1 µg/ 106 cellule, clone 145-
2C11; Caltag, Burlingame, CA, U.S.A.) in 50 µl di FACS buffer per 30 min a
+4°C. Dopo il lavaggio il campioneè stato risospesoin 200 µl di PBS 1X e
analizzatocon il citofluorimetroFACSCalibur (BD Biosciences,Mountain View,
CA, U.S.A.). I dati sono stati analizzati usandoil software Cell Quest (BD
Biosciences,MountainView, CA, U.S.A.).
SAGGI FUNZIONALI
TestELISPOT ( Enzyme-Linked ImmunonoassobentSpotsAssays)
Il saggioè stato eseguitousandoil KIT per la rilevazionedi IFN-γ murino (BD
Pharmingen, Mountain View, CA, U.S.A.) seguendo le istruzioni allegate,
util izzando le piastre MULTIscreen HTS (Millipore, NJ, U.S.A.). Il numero di
spotsèstato valutato il lettoredi piastreKS e il relativo softwar.
TestE.L.I.S.A. (Enzyme-Linked ImmunosorbentAssay)
Il surnatante delle cocolture MLPC con bersagli cellulari allogenici/singenici,
piastratiper 24 ore a 37°C in piastreda 96 pozzetti a fondo a U (Falcon,Becton
Dickinson, NJ, U.S.A.), è statoanalizzatomediante testE.L.I.S.A per rilasciodi
IFN-γ (Endogen,Rockford, U.S.A.).
Il giornoprecedenteal test,la piastrada96 pozzetti apposita(NUNC-Immuno96
microwell-plate, NUNC, Danimarca)è stata incubataovernight, a temperatura

Materiali & Metodi
51
ambiente,con100µl anticorpodi coating perla citochinain esame,diluito in PBS
1X.
Il giornodel testla piastraè stataincubataperun’oraconassay buffer (PBSBSA
2 %, pH 7.2-7.4),persaturarei siti di legameaspecifici .
La piastraè statain seguitolavatapertre volte con200µl/pozzettodi wash buffer
(50mM Tris, 0.2%Tween20; pH 7.0-7.5), per rimuoverel’anticorpo in eccesso.
Dopo il terzolavaggio,adogni pozzettosonostati aggiunti50µl di assaybuffer e
50 µl di surnatante di coltura dei campioni. In alcuni pozzetti, al posto del
campione,all’assaybuffer sono stati aggiunti50 µl di standarddel kit, partendo
dalla concentrazionedi 17500pg/ml e con diluizioni scalari 1:2 in terrenoRPMI
10 % con 2βMe, per poter costruireuna rettadi taratura delle concentrazioni di
citochinain esame.Siai campioni chegli standard sonostati allestiti in doppiette.
La piastra è statatenutain incubazioneovernight a temperatura ambiente. Sono
stati eseguiti tre lavaggiconwashbuffer e la piastraè stataincubataper un’ora a
temperaturaambientecon 100 µl di anticorpobiotinilato di detecting,diluito in
assaybuffer. Dopo tre lavaggi in washbuffer, la piastraè stata incubata per 30
minuti, a temperatura ambiente,con 100 µl di streptavidina legataa perossidasi
(poly-HRP-StreptavidinaEndogen,a 1 mg/ml) diluita 1:6000 in assay buffer.
Sonostati quindi eseguitialtri tre lavaggiin washbuffer e sonostati aggiunti 200
µl/pozzetto di substrato OPD (o-Phenylenediamine Dihydrochloride Perossidase)
(SigmaFast, OPD Peroxidase Substrate tablet set), diluito in acquadistillata. La
piastra è stata tenutaal buio. Lo sviluppodella piastra è stato bloccatomediante
l’aggiuntadi 50 µl di HCl 3 N. La piastra è statapoi analizzata medianteSpectra
Count Packardconletturaa490nm.
Valutazionedellarispostacitotossicatramitesaggiodi rilasciodi 51Cr
L’attività liti ca delle MLC e delle MLPC è stata valutata mediante un saggio
citotossicitàa breveterminedi incubazione, utilizzandola linea MBL-2 pulsata
con i due peptidi correlato/scorrelato(5-10x106 di cellule/ml di terrenoDMEM
10% con5 µg/ml di peptide),o lineeallogeniche/singenichenonpulsate.
Tali cellule (1x106) sono state marcate, in pellet, con 100 µCi Na251CrO4
(DuPont,Boston, MA, U.S.A.) per 1 h a 37 °C. Al terminedella marcaturale
cellule sonostate opportunamentelavatee quindi risospesein terreno RPMI 5%,
alla concentrazione di 2x104/ml; 100µl di tale sospensione cellulare sono stati

Materiali & Metodi
52
seminati in una micropiastrada 96 pozzetti con fondo a U (Bibby Sterlin Ltd,
Stone, U.K.) in presenzadi diluizioni scalari di cellule effettrici, distribuite
anch’essein 100µl ed in triplicato. Le micropiastre sono state centrifugate per 5
minuti a 200xg ed incubatea 37 °C per 5 h. Infine da ogni pozzettosono stati
prelevati 30µl di surnatante e trasferiti in micropiastre contenenti uno scintillante
solido (PackardInstrumentCo,Meriden,CT, U.S.A.).
Dopo aver lasciato seccarele piastre, la radioattivi tà emessaè statamisuratacon
un contatoredi scintillazioneγ per piastre(Top Count,PackardInstruments Co,
Meriden, CT, U.S.A.). La percentualedi lisi specifica è stata calcolata da
campioni allestiti in triplicatoutilizzandolaseguente formula:
)(
)(100specificorilasciodi%
RsponRMax
RsponRsperx
−−=
nellaquale:
• Rspon. in cpm = rilascio spontaneo,quantità d’isotopo rilasciato dalle
cellule bersaglio incubatein assenzadi effettori.
• Rmax. in cpm = rilascio massimo di radioattività ottenuto dopo ripetute
operazioni di scongelamentodelle cellule radiomarcate. Il trattamento
causala completarotturadellamembranacitoplasmatica.
• Rsper in cpm = rilascio sperimentalemisurato per ciascunrapporto tra
effettore/bersaglio.
COSTRUTTI PLASMIDICI
Il cDNA codificante per mouse TERT (m-TERT) ci è stato fornito dalla Geron
Corporation (Manlo, CA, U.S.A.), inserito in pBluescripted infine clonato in
pcDNA3 (Invitrogen, Carlsbad,CA, U.S.A.) tramite doppio taglio con EcoRI e
XhoI. Ottenendocosìil plasmidepcDNA3-m-TERT..
Successivamentela regionedi interesseè stata isolatadal frammento contenente il
genenonrilevantetramiterestrizione conNotI e HindIII, quindi è stata purificata
dopoestrazionedagel d’agarosioe trasferitain VR1055(in seguito riferito come
VICAL) (Vical Inc.,SanDiego,CA, USA), plasmide ottimizzato per il clonaggio
rapido e per l’espressionein vivo, precedentementelinearizzato con la medesima
reazionedi restrizione.Si è ottenuto in questomodo il plasmide VICAL-m-
TERT.

Materiali & Metodi
53
Il costruttopVlJnsA-m-TERT è statoottenutodall'inserimentodi un frammento
sintetico corrispondentealla sequenzaottimizzatadei codoniTPA-m-TERT-LTB
ottenuta dalla GENEART (Geneart GmbH, Resensburg, Germania), tramite
clonaggio dei siti BglII e SalI del vettore pVlnsA (Montgomery, Shiver et al.
1993).
Il costrutto pCMV-β-gal è stato ottenutotramite isolamentodel geneβ-gal di
E.coli dal vettore plasmidico pC4AUGβ-gal, che ci è stato fornito dal Dr.
Macgregor, tramite restrizionecon EcoRI-XbaI. Quidi subclonato nel vettore
pUC19 e infine clonato nel vettore pCMV (Addgene, MA, U.S.A.) tramite
restrizioneconNotI.
La corretta orientazionedell’inserto è stata valutata attraverso un’analisi di
restrizione. Infine l’amplificazione del costrutto è stata eseguitacon “Endofree
plasmid mega kit” (Qiagen GmbH Hilden, Germania) seguendo il specifico
protocollo.
ANALISI SPECTRATYPING E CLONOTIPICA DEL TCR
L’estrazione dell’RNA è statarealizzata con TRIZOLTM (Gibco, CA, USA), il
qualepermettel’i solamentodacellulee datessuti dell’RNA totale,preservandolo
da contaminazionida RNasi. La concentrazione dell’acido nucleico ottenutoè
stata valutata tramite spettrofotometroDU 530 Beckman usandocome ratio il
rapporto 260/280nm, e quindi portataa 0.5mg/ml. L’int egrità dell’RNA è stata
valutatatramite corsaelettroforeticain gel all’1% d’agarosioper la presenza delle
due banderibosomiali.Si è procedutoquindi alla reazione di retrotrascrizionee
successiva amplificazionedei genispecifici per il TCR, tramite PCR(Polymerase
chain reaction). Dopo l'amplificazione si è proceduto alla valutazione della
lunghezzadella regioneCDR3 del TCR tramite la tecnica dello Spectratyping.
Brevemente,un ugual volume del templato di PCR viene combinato con un
volume di dye-loading buffer e 0.2 µl di Rox-marker(Applied Biosystems, CA,
U.S.A.),quindi la reazionevieneincubata a94°Cper2 minuti e fatta correre in un
gel fluorescentedi sequenziamentoed analizzata tramite software Genescan
(Applied Biosystems,CA, U.S.A.). L'analisi clonotipicadel TCR è stata effettuata
tramitel'util izzo di un protocollogià riportato in letteratura (De Palma, Marigo et
al. 2004).

Materiali & Metodi
54
VIRUS
I costrutti adenoviraliutilizzati sono statiottenuti tramite ricombinazioneCre-lox;
espansi utilizzandola lineacellulare293,purificati tramite gradientedi densità di
cesio e infine dializzati utilizzando protocolli standard. I vettori adenovirali
util izzati esprimono unicamentela sequenza genica di interesse,inseritasotto il
promotoredel citomegaloviruspresentenel vettore. I virus vengonostoccati a -
80° gradi fino al momentodell'utilizzo.
VACCINAZIONE A DNA PLASMIDICO
Gli animali vengonopreliminarmenteanestetizzati prima di ciascunaprocedura
vaccinale che prevedal'elettroporazione del muscolo.La miscela di anestesia
consta di un farmaco ad effetto sedativoe miorilassante (Rompum,BAYER,
Germania) associato ad un analgesicomorfino-simile (KETAVET, Farmaceutici
Gellini S.p.A., Italia). 1,75 mg/5ml di Rompum e 0,06 mg/5ml di Ketavet
vengonomiscelati in soluzionefisiologica e di tale miscela viene inoculato un
volumepari a0,1ml/10grammidi pesodel topo.
Gli animali sonostatiquindi immunizzati tramite inoculodi 100µl di unamiscela
costituita da: 50 µg di DNA e 600 µg di acido poli L-glutammico (Sigma,
Chemical, St.Louis, Co, USA) miscelati in una soluzione acquosaportata alla
concentrazionefinale di 0,9 mg/100ml di NaCl. L’iniezione intramuscolaredella
soluzionecontenenteDNA é stataseguita da elettroporazione del sito di inoculo
con due pulsesdi 25 milli -secondi a voltaggio di 120 V, erogato medianteun
calibro collegatoallo strumentoT820 Electro SquarePorator (San Diego, CA,
U.S.A.).
VACCINAZIONE TRAMITE VETTORE ADENOVIRALE
L'inoculo del costrutto adenovirale ricombinante viene effettuato a livello
intramuscolare(i.m.). Gli animalivengonoimmunizzati con5x106 PFU/topo.
INOCULO DEL TUMORE
Topi C57BL/6Jsonostati inoculatiendovena(i.v.) con105 cellule B16LU8, linea
di melanomamurino. Dopo 14 giorni dall’inoculo vengonoprelevati i polmoni e
inclusi in formalina. Su questi verrà poi stimato il numero di metastasi. Topi

Materiali & Metodi
55
C57Bl/6J sonostati inoculati sottocute(s.c.)con 1x106 cellule C2 o con 0.2x106
celluleB16447.
TRASFERIMENTO ADOTTIVO DI CTL
Modellodi melanomametastaticopolmonare
Dopo 3 giorni dall'i noculo(i.v.) della linea B16Lu8,gli animali vengonoinoculati
con 5x106 CTL a topo e trattati tramitesomministrazione di 60000IU/topo/die di
IL-2 ricombinantetramite inoculo intraperitoneale (i.p.). La somministrazionedi
IL-2 vieneeseguita anchenei duesuccessivigiorni dal trasferimentoadottivo dei
CTL. Gli animali vengono quindi sacrificati dopo 15 giorni dall'i noculo del
tumoree i loro polmoni prelevatie fissati perlasuccessivacontadelle metastasi.
Modellodi tumoresottocutaneo
Quando il tumore raggiungele dimensioni di circa 10 mm2, dopo 14 giorni
dall'inoculodel tumore,gli animalivengonosottoposti al trasferimento adottivo di
5x106 diCTL, previa linfodeplezionetramiteγ-irradiazionetotal-body (500 cGy).
Gli animali vengono poi immunizzati tramite inoculo di 5x108 PFU/topo di un
vettore adenovirale ricombinante. Gli animali sono quindi trattati tramite
somministrazionedi 60000IU/topo/diedi IL-2 ricombinantetramite inoculo (i.p.).
La somministrazione di IL-2 viene eseguita anche nei due giorni successivi al
trasferimentoadottivo dei CTL. Le dimensioni del tumorevengono monitorate
ogni duegiorni. Gli animali vengonosacrificati quando il tumoreraggiungei 100
mm2.
ANALISI ISTOLOGICA E IMMUNOISTOCHIMICA
Le prostatevengono disecatedall'alberourogenitale e fissate usandoil fissativo
PLP (Parafolmaldeide/Lisina/Periodato),preparato sciogliendo 0.085 grammi di
Sodio m-Periodato (Sigma-Aldrich, St.Louis, MO, U.S.A.) in una soluzione
costituita da 10 ml di parafolmaldeideall'8% (Sigma-Aldrich, St.Louis, MO,
U.S.A.) e 30 ml di una soluzione di Lisina. Quest'ultima soluzione è preparata
sciogliendo 16.4 grammi di L-Lisina (Sigma-Aldrich, St.Louis, MO, U.S.A.) in
450 ml di acquade-ionizzatae 450 ml di buffer Sorrensons0.1 M. Il campione

Materiali & Metodi
56
viene fissato per non più di 3 ore e successivamente viene lavato nel buffer
Sorrensons 0.1 M e quindi conservatoin una soluzione di PBS addizionato di
saccarosioal 20%. Successivamenteil campione viene fissato in paraffina e
tagliatoal criostato. Le sezionidi tessutotagliate al criostato vengonolasciate ad
asciugareall’aria per tutta la nottee poi fissate in formalina tamponataal 4% per
8 minuti e in tre passaggiin alcool99° per2 minuti complessivamente. In seguito
le sezioni vengono sottopostea tre risciacqui da 5 minuti ciascunoin TBS e a
smascheramento antigenicocon tamponecitrato a pH6 in forno a microondeper
10 minuti. I vetrini sonostatiquindi incubati prima con il Protein Block Serum-
Free(DakoCytomation, Danimarca) per10 minuti e poi conl’Ab primario rabbit
anti-mouseTelomerase ( Abcam Cod. 23699,U.K) diluito 1:100 per tutta la
notte a 4°C. Le sezioni sono state quindi risciacquate con TBS ed incubate con
l’EnVision System Labelled Polymer-HRP anti Rabbit (DakoCytomation,
Danimarca) per 30’. Dopoaverrisciacquato con PBS,i vetrini sonostati incubati
con la DAB per10’ quindi risciacquaticonacquadistill ata.Il contrasto nucleare
è stato effettuato con un breve passaggioin Ematossilina di Mayer per 10’’.
L'analisi istologica prevede una colorazione Ematossilina-Eosina effettuata
tramitetre passaggi in Biocleare successivipassaggiin alcooli decrescenti (99°,
99°, 95°,80°). Quindi coloraticonEmatossilinadi Mayer per2' e30", lavaggicon
acqua di fonte e colorazionecon Eosina 0.25%, lavaggi con acquadi fonte e
passaggiin alcooli crescenti(95°, 99°, 99°). Quindi i vetrini vengonomontati con
un balsamopermanente.
ANALISI STATISTICHE
Le analisi statistiche sono state effettuate mediante test t di Student. Si è
considerataunasoglia di significativitàpari a0.05.

Risultati
57
RISULTATI
Comparazione tra due protocolli di immunizzazione attiva basati su
vaccini a DNA codificanti l'antigene telomerasi murina (m-TERT).
Il primo passo del progetto è stato identificare il miglior protocollo di
immunizzazioneattiva capace di espandere una popolazione di linfociti T
specifici per l'epitopom-TERT198-205 della ribonucleoproteinatelomerasimurina
presentatodalle molecole MHC di classe I. Abbiamo, pertanto, paragonato
l'impiego di due diverse strategie vaccinali: la prima basata sulla doppia
somministrazionedi DNA plasmidico(protocollo#1,Figura1); la seconda basata
sull 'utilizzo di un regimea"prime-boost" eterologo(protocollo#2, Figura1).
Nel primo casotopi TRAMP sonostativaccinati a partire dalla 6a settimanadi età
tramiteinoculo di 50 µg/topo del vettoreplasmidico pVlJnsA,vuotoo codificante
l'antigenem-TERT,seguito dall'elettroporazionedella seded'inoculo,e richiamati
con la medesimametodicadopo14 giorni. Nel secondocasotopi TRAMP sono
stati vaccinati a partiredalla6a settimanadi etàtramiteinoculodi 50 µg/topodel
vettore plasmidico pVlJnsA, vuoto o codificante l'antigene m-TERT, seguito
dall'elettroporazione della seded'inoculo e richiamati dopo 14 giorni tramite
somministrazionedi un vettoreadenovirale ricombinante (5x108 PFU/topo).Agli
animali vaccinati con il plasmide vuoto è stato somministrato un vettore
adenovirale ricombinantecodificante il gene reporter della medusa Aequorea
victoria: enhanced green fluorescent protein (EGFP), mentre agli animali
vaccinati con il plasmide codificantem-TERT è stato somministrato un vettore
adenovirale esprimente la medesima sequenza genica. Come controllo
dell'efficacia della modalità vaccinale,topi TRAMP sonostati vaccinati con lo
stessoprotocollo versola proteinaimmunogenicaβ-galattosidasi (β-gal). Dopo 2
settimanedal richiamo,gli animali sonostati sacrificati per effettuare unaseriedi
analisi volte a valutare l'impatto dell'intervento vaccinale in termini di selezione
edespansionedi linfociti T funzionaliespecifici per l'antigenedi interesse.
Premessa essenzialeper queste analisi è stata l'identificazione dell'epitopo
immunodominante di telomerasimurina ristretto per l’aplotipo H-2b del ceppo
C57BL/6J (allele Kb) non ancoradescritto in letteratura.L'epitopo m-TERT198-205
(VGRNFTNL) è stato identificato dal gruppo della Dr.ssa Elisa Scarselli
dell'Istituto di Ricerca di Biologia Molecolare (IRBM) di Pomezia, tramite

Risultati
58
screeningdi un pannello completodi peptidi che coprivanol'intera sequenza di
telomerasie successiva analisi bioinformatica utilizzandogli algoritmi predittivi
BIMAS eSYFPEITHI(www.bimas.ciy.nih.gov; www.syfpeithi.de/).
La capacità della vaccinazionedi indurre linfociti T specifici per l'epitopo m-
TERT198-205 è stata valutataex-vivo tramitetestELISPOTperrilascio di IFN-γ, un
saggio in cui 1x106 di splenociti di topi TRAMP, precedentemente vaccinati con il
protocollo#1 o protocollo#2, sonostati stimolati per 24 ore con 10 U/ml di IL-2
ricombinantee in presenzadi 1µg/mldel peptide immunodominate m-TERT198-205
(p-198), del peptidesubdominantem-TERT486-493 (p-486) o del peptideβ-gal96-103
(p-βgal), usato come controllo (Figura 2). Il test ha evidenziato come la
vaccinazione a "prime-boost" riesce ad espandereun numero di linfociti T
specifici per l'epitopo m-TERT198-205 (media 347 spots) maggiorerispetto alla
vaccinazione a solo DNA plasmidico (media 25,66 spots), e tale efficacia è
risultata indipendentedall’antigene. Infatti, risultati simili sono stati ottenuti
util izzando le medesime metodichevaccinali verso l'antigeneesogenoβ−gal: il
regime a "prime-boost" eterologoriesce ad espandere un numeromaggiore di
linfociti T specifici per il peptideβ-gal96-103 (media 653 spots) rispetto alla
vaccinazionea solo DNA plasmidico(media 162spots).Dato interessante è stato
il fatto che entrambele modalitàvaccinali sonorisultate molto selettive: infatti,
come si evince dalla figura 2, animali vaccinati contro l'antigene β−gal
riconosconoesclusivamenteil peptide β-gal96-103 mentre non riconosconoi due
peptidi dell'antigene telomerasi (m-TERT198-205 e m-TERT486-493, p<0.05); al
contrario,gli animali vaccinaticontrol'antigenetelomerasipresentanolinfociti T
specifici unicamente per il peptide immunodominante m-TERT198-205 non
riconoscendo né il peptide β-gal96-103 né il peptide subdominantem-TERT486-493
(p<0.05).Quest'ultimaosservazioneconfermachel'epitopo m-TERT198-205 risulta
essereeffettivamentel'epitopoimmunodominante.
A confermarela superioritàimmunogenica del regimea "prime-boost" rispetto la
vaccinazionea solo DNA plasmidico nell'espansionedi linfociti T specifici per
l'antigene di interesse,è stata anche l'analisi citofluorimetrica con tetrameri
specifici Kb m-TERT198-205 (tet-m-TERT). Il numerodi linfociti T CD3+/CD8+/m-
TERT specifici è stato valutato in vitro dopo una cocoltura di 5 giorni degli
splenociti degli animali immunizzati con il peptide m-TERT198-205 (MLPC). Le
MLPC derivanti dagli animali immunizzati con il regime "prime-boost"

Risultati
59
presentavanoun numerodi linfociti T CD3+/CD8+/tet-TERT+ pari al 4,5% mentre
le MLPC derivantidagli animalivaccinaticonsolo DNA plasmidicopresentavano
unnumero di linfociti T CD3+/CD8+/tet-TERT+ pari soloal 0,32%(Figura 3).
Questi dati nel loro insieme evidenziano come la vaccinazione a "prime-boost"
risulta esserdi gran lungapiù efficacenell'espanderelinfociti T antigenespecifici
rispettoallavaccinazioneaDNA plasmidico.
Sorprendentemente, tuttavia, a quest'espansione non ha corrisposto la stessa
differenzanella funzionalitàdei linfociti T in vitro. Infatti, sia le MLPC derivate
dalle milze degli animalivaccinaticonil regime"prime-boost"che quelle derivate
dalle milze degli animali vaccinaticon il solo DNA plasmidico eranoin gradodi
riconoscereunicamentebersagli cellulari pulsaticon il peptidespecifico, in saggi
E.L.I.SA. per rilasciodi IFN-γ. In questistessisaggicellule tumorali, telomerasi-
positive ma non pulsatecon il peptide immunodominante, non erano affatto
riconosciute,suggerendouna bassa affinità di legamedel TCR dei linfociti T
TERT-specifici al complesso peptide-Kb presentesulla superficie delle cellule
tumorali.Perselezionarelinfociti T a maggior avidità per il complessocostituito
dalle molecoledi prima classe di aplotipoKb e l'epitopom-TERT198-205 abbiamo
deciso di stimolare ripetutamente le MLPC in vitro utilizzando splenociti
singenici γ-irradiati e pulsati con dosi basse di peptide (0.1 µM). Una quantità
limitatadi peptide in coltura consentedi stimolaresolamente i linfociti T ad alta
affinità che,proliferandoselettivamente,riesconoa superarein numero i linfociti
T a bassa affinità, come precedentedimostrato da altri ricercatori per antigeni
diversi da telomerasi(Martha Miller – JayBerzofsky). Ad ogni ristimolazione le
MLPC sonostate testatein saggiE.L.I.S.A. peril rilascio di IFN-γ contro bersagli
cellulari dello stessoaplotipo (H-2b) madi differente istologia(Figura4).
Abbiamoutilizzatocomebersaglicellulari la lineadi melanomamurino B16 e la
linea di carcinoma prostatico murino C2-TRAMP e i loro corrispettivi
transfettanti per l'espressionedella molecola costimolatoria B7.1 (CD80): B16-
B7.1 e C2-B7.1. L'utilizzo della transfezione per indurre l'espressionedella
molecolaCD80 trova il suorazionalenel fatto che il legame di questamolecola
(presentenella cellula tumorale)con il corrispettivo leganteCD28 (presentea
livello dellamembranaplasmaticadel linfocita T) riducela soglia di stimolazione
necessariaad attivare il linfocita T dopo interazione tra il complesso MHC-
peptidedella cellula tumoraleedil TCR del linfocitaT.

Risultati
60
Come controllo positivo è stata utilizzata la linea di linfoma murino MBL2
pulsata con il peptide m-TERT198-205 (MBL2-p198), mentre come controllo
negativo abbiamo utilizzato la stessalinea pulsata con il peptide β-gal96-103
(MBL2-pβgal). Abbiamo scelto questa linea cellulare perchè non viene
riconosciutain vitro dalinfociti T m-TERTspecifici .
Inaspettatamente,solo le MLPC derivate dagli animali vaccinati con DNA
plasmidico hanno presentatoun incremento d'attività funzionale indotto dalle
ristimolazioni in vitro, riuscendoa riconoscereanchei bersagli cellulari tumorali
non pulsati con il peptidespecifico. Infatti, sebbene i livelli di IFN-γ rilasciati
dalle MLPC derivatedagli animali vaccinati conDNA plasmidico dopococoltura
con i vari bersagli cellulari sianorisultati inferiori rispetto a quelli del controllo
positivo MBL2-p198 questi linfociti riconosconocellule di melanoma B16 e
cellule di carcinomaprostatico C2-TRAMP, soprattutto quando queste linee
esprimono B7.1 (Figura4). Al contrariole MLPC derivate dagli animali vaccinati
con il regimea "prime-boost"presentavano un rilascio marginaledi IFN-γ dopo
cocoltura con i bersagli cellulari non pulsati con il peptideimmunodominante.
Entrambe le MLPC mantenevano, tuttavia, la loro specificità per l'antigeneanche
dopo varie ristimolazioni, riconoscendo unicamente le cellule caricate con il
peptidespecifico m-TERT198-205 e non le cellule caricatecon il peptideaspecifico
β-gal96-103 (Figura 4). Questi dati di stimolazionecon bassedosi di peptide ci
suggerisconochela vaccinazionea DNA plasmidico, anche senon è in grado di
espandereun numeroconsistentedi linfociti T specifici per telomerasi, è in grado
di dare un "priming" di popolazioni linfocitarie a più alta affinità rispetto alla
vaccinazionea "prime-boost".
A confermarequesti dati è statal'analisi “spectratyping” della catenaVβ del TCR
delle MLPC, condotta dal Dr. Raffaele De Palma dell'Università di Napoli.
Util izzando primers specifici per la regione CDR3 del TCR è stato possibile
clonarela sequenzadellacatenaVβ del TCR adalta avidità di un clone(clone7)
in gradodi riconosceretelomerasi(descritto di seguito). Questa catenaè risultata
essereVβ-11. L’analisi “spectratyping” conprimersspecifici per la catenaVβ-11
eseguitasulle MLPC derivatedagli animali vaccinati consolo DNA plasmidico a
vari passaggiin vitro, ha evidenziatocomele ripetute ristimolazioni determinano
una progressiva espansioneclonale dei linfociti T con arricchimento di TCR
caratterizzato dalla catenaVβ-11 (Figura 5A). Questa selezioneselettiva per la

Risultati
61
catenaVβ-11 del TCR risulta più evidente dall'analisi della distribuzione del
clonotipo del clone ad alta affinità all’interno delle MLPC dopo varie
ristimolazioni: la percentualedi TCR del clone7 tra i linfociti concatenaVβ-11 è
solo del 5% al primo passaggioin vitro per poi passare al 60% dopo 8
ristimolazioni in vitro (Figura 5B). E’ verisimile, quindi, che la progressiva
acquisizione della capacitàdi riconoscere bersagli cellulari telomerasi-positivi di
diversa istologia è dovuta all'espansionedi linfociti T a maggior affinità. Se
consideriamo, quindi, l’aspetto dell’affini tà di riconoscimento dell’antigene
telomerasi, la vaccinazionecon il solo DNA plasmidico risulta più efficace della
vaccinazione "prime-boost" in quanto è in grado di selezionare linfociti T
funzionalmenteefficaci nel riconoscereil complessocostituito dalle molecole di
prima classedi aplotipo Kb e l'epitopom-TERT198-205 fisiologicamente presente
sullamembranaplasmaticadellecelluletumorali.

Risultati
62

Risultati
63
Figura 1: Schematizzazione dei protocolli di vaccinazione utilizzati in questo studio.
Protocollo#1 - Doppia somministrazione di vaccino a DNA plasmidico. Topi TRAMP sonostati vaccinati a partire dalla 6a settimana di età tramite inoculo di 50 µg/topo del vettoreplasmidico pVlJnsA vuoto o codificante m-TERT, seguito dall'elettroporazionedella sede diinoculo, e successivamenterichiamati con la medesimametodicadopo 14 giorni. Dopo duesettimanedal richiamo, alcuni animali sono stati sacrificati per le analisi volte a valutarel'eff icaciadell'intervento vaccinale.Protocollo#2 - Regime a "prime boost" eterologo. Topi TRAMP sonostati vaccinati a partiredalla 6a settimanadi età tramite inoculo di 50 µg/topo del vettore plasmidico pVlJnsA vuoto ocodificantem-TERT seguitodall'elettroporazionedella sededi inoculo. Dopo14 giorni gli animalisono stati richiamati tramite somministrazionedi un vettore adenovirale ricombinantecodificantela proteinaEGFPo m-TERT (5x108 PFU/topo). Dopo duesettimanedal richiamo,alcuni animalisonostatisacrificati perle analisi volte a valutarel'efficacia dell'intervento vaccinale.
14giorni
Protocollo2
SacrificioDNA
14giorni
VIRUS
Protocollo1
SacrificioDNA DNA
14giorni
14giorni

Risultati
64
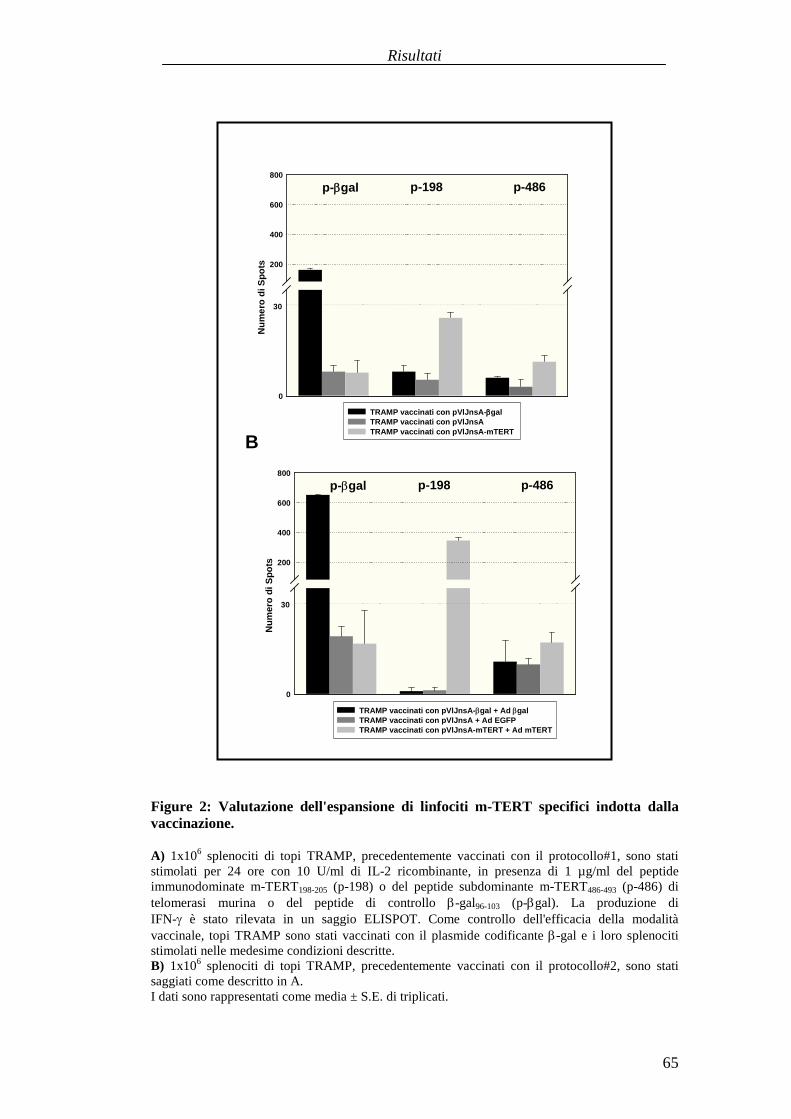
Risultati
65
Figure 2: Valutazione dell'espansione di linfociti m-TERT specifici indotta dallavaccinazione.
A) 1x106 splenociti di topi TRAMP, precedentemente vaccinati con il protocollo#1, sono statistimolati per 24 ore con 10 U/ml di IL-2 ricombinante, in presenza di 1 µg/ml del peptideimmunodominate m-TERT198-205 (p-198) o del peptide subdominantem-TERT486-493 (p-486) ditelomerasi murina o del peptide di controllo β-gal96-103 (p-βgal). La produzione diIFN-γ è stato rilevata in un saggio ELISPOT. Come controllo dell'eff icacia della modalitàvaccinale, topi TRAMP sonostati vaccinati con il plasmide codificante β-gal e i loro splenocitistimolati nelle medesimecondizioni descritte.B) 1x106 splenociti di topi TRAMP, precedentementevaccinati con il protocollo#2, sono statisaggiati comedescrittoin A.I dati sonorappresentati comemedia± S.E. di triplicati.
Nu
mer
od
iSp
ots
0
200
400
600
800
TRAMP vaccinati con pVlJnsA-βgal + Ad βgalTRAMP vaccinati con pVlJnsA + Ad EGFPTRAMP vaccinati con pVlJnsA-mTERT + Ad mTERT
p-βgal p-198 p-486
30
Nu
mer
od
iSp
ots
0
200
400
600
800
TRAMP vaccinati con pVlJnsA-βgalTRAMP vaccinati con pVlJnsATRAMP vaccinati con pVlJnsA-mTERT
30
p-βgal p-198 p-486
B

Risultati
66
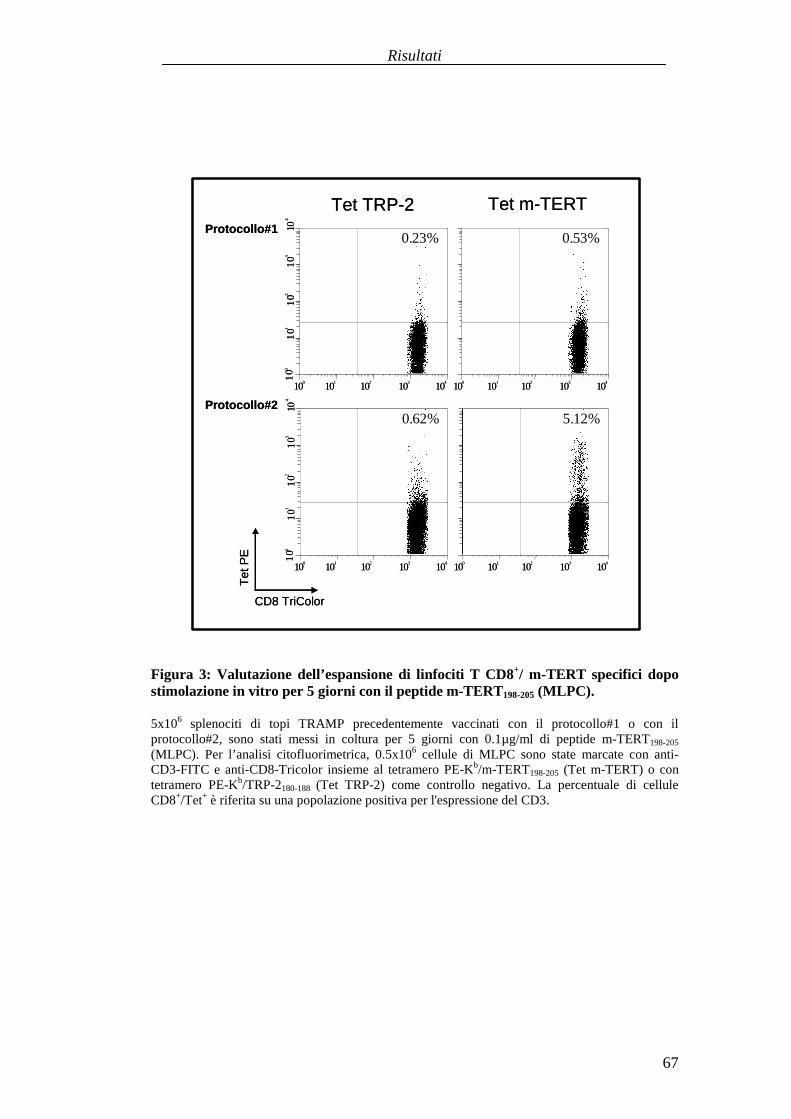
Risultati
67
Figura 3: Valutazione dell’espansione di linfociti T CD8+/ m-TERT specifici dopostimolazione in vitro per 5 giorni con il peptide m-TERT198-205 (MLPC).
5x106 splenociti di topi TRAMP precedentemente vaccinati con il protocollo#1 o con ilprotocollo#2, sono stati messi in coltura per 5 giorni con 0.1µg/ml di peptide m-TERT198-205
(MLPC). Per l’analisi citofluorimetrica, 0.5x106 cellule di MLPC sono statemarcate con anti-CD3-FITC e anti-CD8-Tricolor insiemeal tetramero PE-Kb/m-TERT198-205 (Tet m-TERT) o contetrameroPE-Kb/TRP-2180-188 (Tet TRP-2) come controllo negativo. La percentuale di celluleCD8+/Tet+ è riferita suunapopolazionepositiva perl'espressionedel CD3.
100
101
102
103
104
101
102
103
104
101
102
103
104
0.23% 0.53%
100
100
0.62% 5.12%
100
101
102
103
104
101
102
103
104
101
102
103
104
100
100
Tet m-TERTTet TRP-2Protocollo#1
Protocollo#2
CD8 TriColor
Tet
PE
100
101
102
103
104
101
102
103
104
101
102
103
104
0.23% 0.53%
100
100
0.62% 5.12%
100
101
102
103
104
101
102
103
104
101
102
103
104
100
100
Tet m-TERTTet TRP-2Protocollo#1
Protocollo#2
CD8 TriColor
Tet
PE

Risultati
68
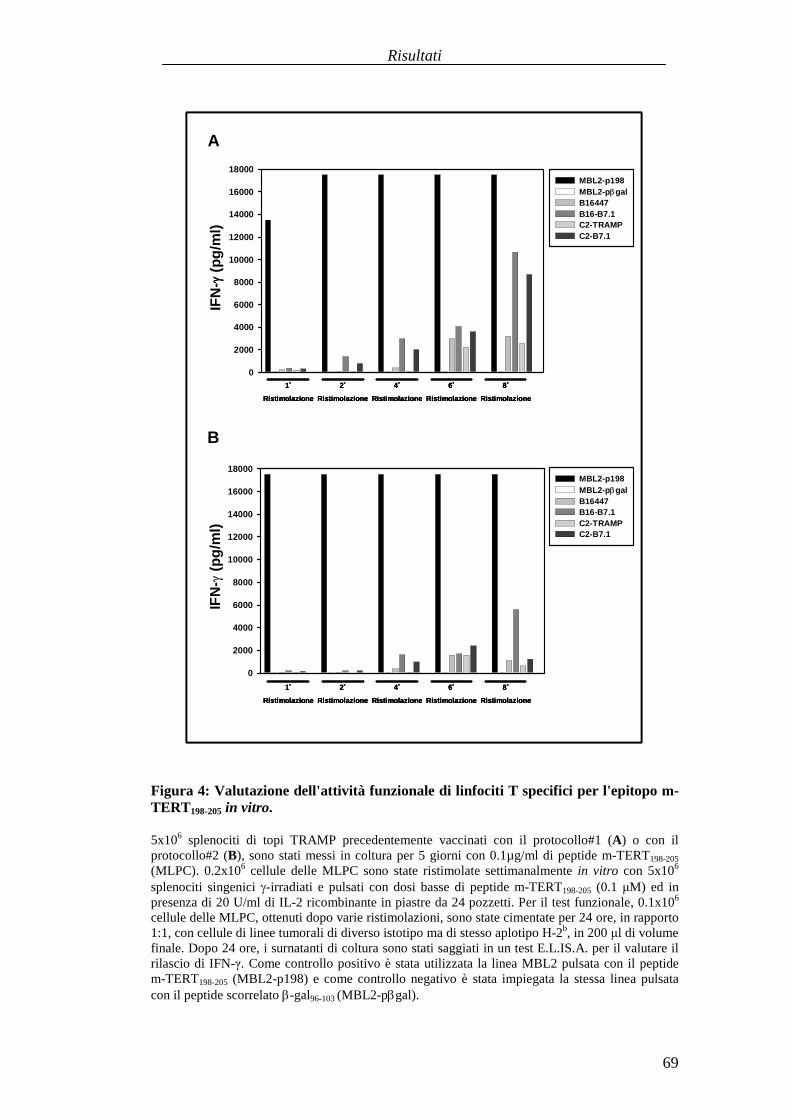
Risultati
69
Figura 4: Valutazione dell'attività funzionale di linfociti T specifici per l'epitopo m-TERT198-205 in vitro.
5x106 splenociti di topi TRAMP precedentemente vaccinati con il protocollo#1 (A) o con ilprotocollo#2(B), sonostati messi in colturaper 5 giorni con 0.1µg/ml di peptidem-TERT198-205
(MLPC). 0.2x106 cellule delle MLPC sono state ristimolate settimanalmente in vitro con 5x106
splenociti singenici γ-irradiati e pulsati con dosi bassedi peptide m-TERT198-205 (0.1 µM) ed inpresenzadi 20 U/ml di IL-2 ricombinantein piastreda 24 pozzetti. Peril testfunzionale, 0.1x106
cellule delle MLPC, ottenutidopovarie ristimolazioni, sonostatecimentate per24 ore,in rapporto1:1, concellule di linee tumorali di diversoistotipo madi stessoaplotipo H-2b, in 200µl di volumefinale.Dopo 24 ore, i surnatantidi colturasonostati saggiati in un testE.L.IS.A. per il valutareilrilascio di IFN-γ. Come controllo positivo è statautilizzatala lineaMBL2 pulsatacon il peptidem-TERT198-205 (MBL2-p198) e come controllo negativo è stata impiegatala stessalinea pulsatacon il peptide scorrelatoβ-gal96-103(MBL2-pβgal).
A
B
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
IFN
-γ(p
g/m
l)
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
IFN
-γ(p
g/m
l)
MBL2-p198MBL2-pβgalB16447B16-B7.1C2-TRAMPC2-B7.1
MBL2-p198MBL2-pβgalB16447B16-B7.1C2-TRAMPC2-B7.1
A
B
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
IFN
-γ(p
g/m
l)
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
1°
Ristimolazione
6°
Ristimolazione
4°
Ristimolazione
8°
Ristimolazione
2°
Ristimolazione
IFN
-γ(p
g/m
l)
MBL2-p198MBL2-pβgalB16447B16-B7.1C2-TRAMPC2-B7.1
MBL2-p198MBL2-pβgalB16447B16-B7.1C2-TRAMPC2-B7.1

Risultati
70
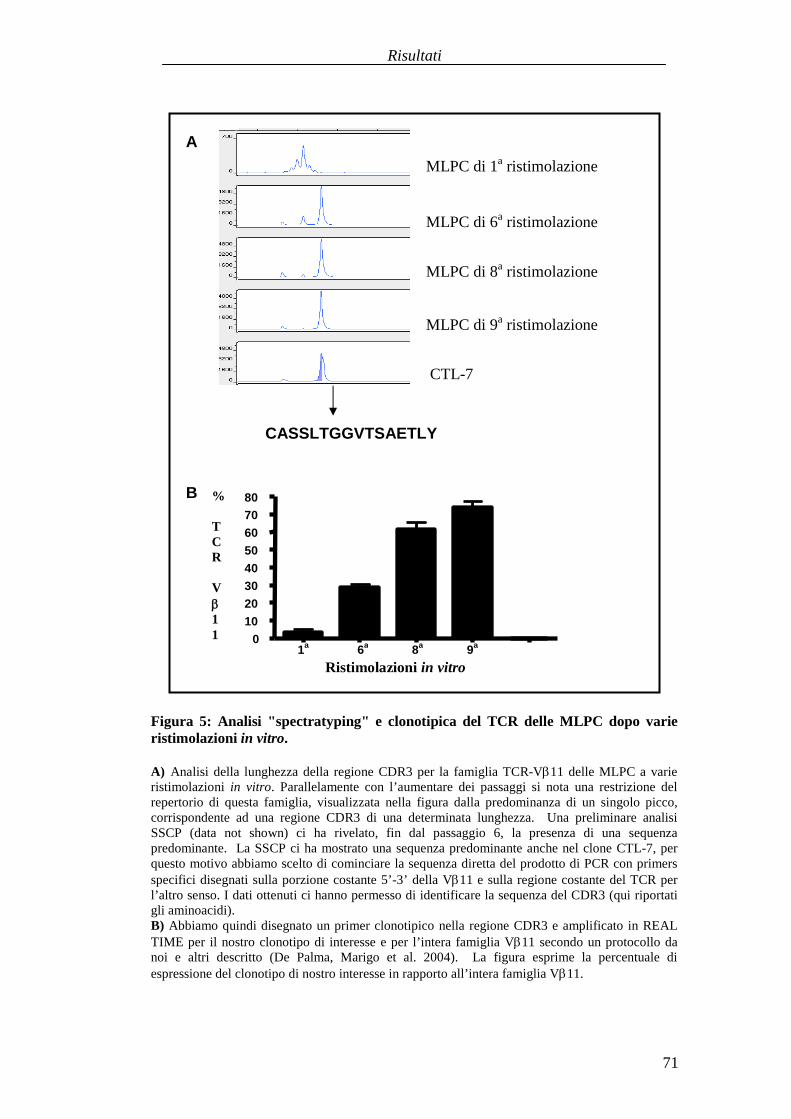
Risultati
71
Figura 5: Analisi "spectratyping" e clonotipica del TCR delle MLPC dopo varieristimolazioni in vitro.
A) Analisi della lunghezza della regione CDR3 per la famiglia TCR-Vβ11 delle MLPC a varieristimolazioni in vitro. Parallelamentecon l’aumentare dei passaggi si nota una restrizione delrepertorio di questa famiglia, visualizzata nella figura dalla predominanzadi un singolo picco,corrispondente ad una regione CDR3 di una determinata lunghezza. Una preliminare analisiSSCP (data not shown) ci ha rivelato, fin dal passaggio6, la presenzadi una sequenzapredominante. La SSCP ci ha mostrato una sequenza predominanteanchenel cloneCTL-7, perquesto motivo abbiamo scelto di cominciarela sequenzadiretta del prodotto di PCRcon primersspecifici disegnati sulla porzionecostante 5’-3’ della Vβ11 e sulla regionecostantedel TCR perl’altro senso. I dati ottenuti ci hannopermessodi identificarela sequenzadel CDR3 (qui riportatigli aminoacidi).B) Abbiamo quindi disegnatoun primer clonotipico nella regioneCDR3 e amplificato in REALTIME per il nostro clonotipo di interesse e per l’ intera famiglia Vβ11 secondoun protocollo danoi e altri descritto (De Palma, Marigo et al. 2004). La figura esprime la percentuale diespressionedel clonotipodi nostrointeressein rapporto all’i nterafamiglia Vβ11.
MLPC di 1a ristimolazione
CTL-7
MLPC di 6a ristimolazione
MLPC di 8a ristimolazione
MLPC di 9a ristimolazione
CASSLTGGVTSAETLY
1a 6a 8a 9a01020304050607080
A
B
Ristimolazioni in vitro
%
TCR
Vβ11

Risultati
72

Risultati
73
Valutazione dell'effetto terapeutico del trasferimento adottivo della
popolazione policlonale di linfociti T citotossici (CTL) specifici per
l'epitopo m-TERT198-205 in animali portatori di tumore.
Una volta completata la caratterizzazione funzionale dei CTL derivati dagli
animali vaccinatia solo DNA plasmidicoe specifici per l'epitopo m-TERT198-205,
lo studio è proseguitonel valutare la loro efficacia in vivo nel contrastare la
crescitatumorale in animali portatori di neoplasia.Abbiamo studiato l'effetto
terapeuticosia in un modello di metastatizzazione polmonare che in modelli di
crescitatumoraleindottadall'inoculosottocutaneodi lineecellulari.
Nel casodel modellodi metastizzazione, abbiamo inoculato endovena la lineadi
melanomaB16 in animaliC57BL/6Je, 3 giorni dopol'inoculo, abbiamotrasferito
ad un gruppo di animali (n=5) 5x106 CTL m-TERT198-205-specifici, mentre un
gruppo di ugual numero di animali non è stato trattato. A entrambi i gruppi
sperimentali sono statesomministrate 60000 IU/topo/die di IL-2 ricombinante,
tramite inoculo intraperitoneale(i.p.). La somministrazione di IL-2 è stata
eseguitaanchenei duesuccessivi giorni dal trasferimento adottivo dei CTL. Gli
animali sono stati sacrificati dopo 15 giorni dall'i noculo del tumore e l'effetto
terapeuticoè stato valutato mediante conta in doppio cieco del numero di
metastasi polmonari: il gruppo di animali trattati con il trasferimento adottivo di
CTL specifici per l'epitopom-TERT198-205 presentavanoun numerodi metastasi
significativamente inferiore rispetto agli animali non trattati (p=0.0004), come
verificatoin dueesperimentiindipendenti(Figura6).
Nel casodel modello di tumore trapiantabile, abbiamo deciso di utilizzare due
tumori di diversaistologia: la linea di melanomaB16 (Figura 7) e la linea di
carcinomaprostatico C2-TRAMP (Figura8). L'impianto sperimentalehaprevisto
l'inoculo sottocutaneo, nel fiancodestrodell'animale, in topi C57BL/6J di 0.2x106
cellule di melanomaB16 o di 1x106 cellule di carcinomaprostatico C2-TRAMP.
Quando il tumore presentavadimensionidi 10 mm2, all'incirca dopo 14 giorni
dall'inoculo del tumore,gli animali sonostati sottopostial trasferimentoadottivo
di CTL specifici per l'epitopo m-TERT198-205 (m-TERT CTL, n=5) o CTL
specifici per l'epitopo β-gal96-103 (β-gal CTL, n=5) tramite inoculo endovenoso
(i.v). Alcune ore prima dell'inoculo dei CTL, gli animali sonostati sottoposti a
mielo-linfo-deplezione tramite γ-irradiazione "total-body" (500 cGy) per

Risultati
74
eliminare le popolazioni in grado di ostacolare la proliferazione dei linfociti
trasferiti, comedescrittoprecedentementenell'introduzione. Alcune ore dopo il
trasferimento dei CTL, gli animali sono stati immunizzati tramite inoculo
intramuscolo (i.m) di 5x108 PFU/topo di un vettore adenovirale ricombinante
codificantem-TERT o β-gal. A entrambi i gruppisonostateinoltre somministrate
60000 IU/topo/diedi IL-2 ricombinante,tramite inoculo intraperitoneale (i.p.). La
somministrazione di IL-2 è stata eseguita anche nei due giorni successivi al
trasferimentoadottivo dei CTL. Le dimensioni del tumore sonostatemonitorate
ogni duegiorni e gli animalisonostatisacrificati quandoil tumoreharaggiuntole
dimensioni di 100mm2.
Abbiamodimostrato che, in entrambii modelli , il trasferimentoadottivo è statoin
grado di rallentare significativamentela crescita tumorale rispetto al gruppo di
controllo. Infatti, gli animali trattati con i CTL specifici per l'epitopom-TERT198-
205 presentanoun incremento significativo della sopravvivenza rispetto agli
animali trattati con i CTL specifici per l'epitopoβ-gal96-103. In conclusione, questi
dati dimostranol'efficacia dall'immunoterapia adottiva nel trattamento di tumori
ed evidenziano come l'antigene TERT sia potenzialmente utili zzabile come
bersagliodi interventi immunoterapici anti-tumorali rivolti a neoplasiedi diversa
istologia.
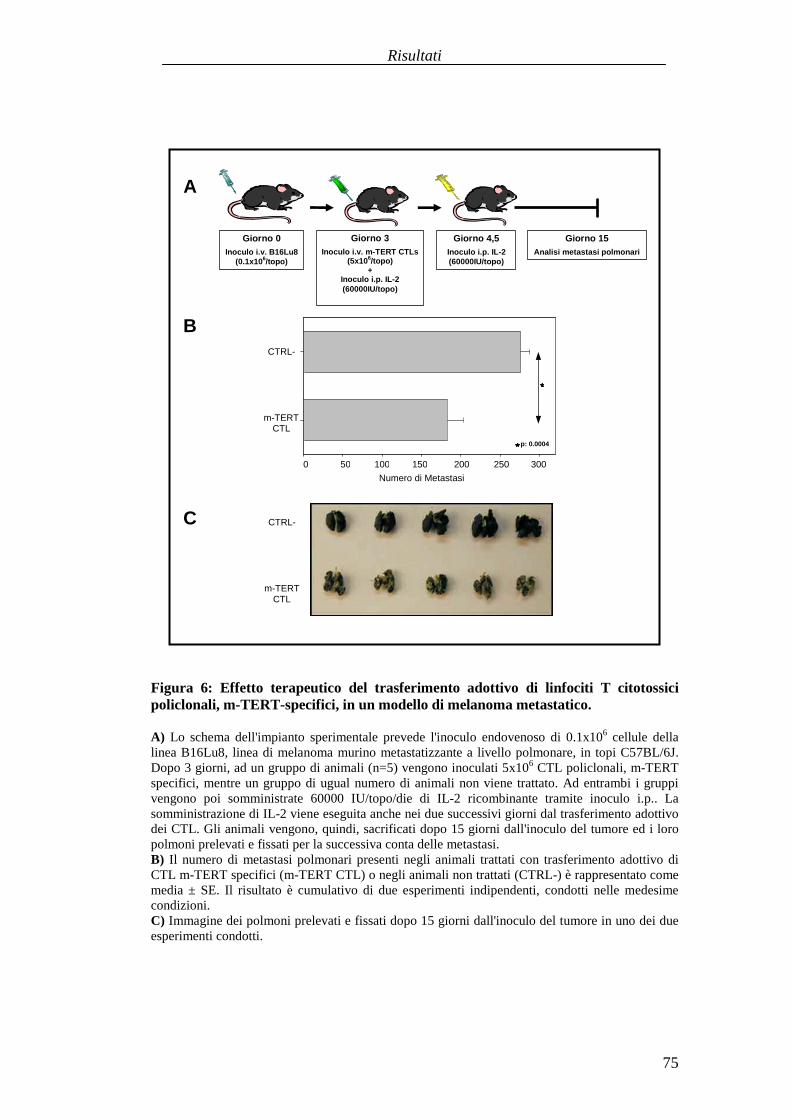
Risultati
75
Figura 6: Effetto terapeutico del trasferimento adottivo di linfociti T citotossicipoliclonali, m-TERT-specifici, in un modello di melanoma metastatico.
A) Lo schemadell'impianto sperimentale prevede l'inoculo endovenosodi 0.1x106 cellule dellalineaB16Lu8, lineadi melanoma murino metastatizzantea livello polmonare, in topi C57BL/6J.Dopo3 giorni, ad un gruppodi animali (n=5) vengono inoculati 5x106 CTL policlonali, m-TERTspecifici, mentre un gruppo di ugual numerodi animali non viene trattato. Ad entrambi i gruppivengono poi somministrate 60000 IU/topo/die di IL-2 ricombinante tramite inoculo i.p.. Lasomministrazionedi IL-2 viene eseguitaanchenei duesuccessivi giorni dal trasferimento adottivodei CTL. Gli animali vengono, quindi, sacrificati dopo 15 giorni dall'i noculo del tumoreed i loropolmoni prelevatie fissati perla successivaconta delle metastasi.B) Il numerodi metastasipolmonari presentinegli animali trattati con trasferimentoadottivo diCTL m-TERT specifici (m-TERT CTL) o negli animali nontrattati (CTRL-) è rappresentatocomemedia ± SE. Il risultatoè cumulativo di due esperimenti indipendenti, condotti nelle medesimecondizioni.C) Immaginedei polmoni prelevati e fissati dopo15 giorni dall'inoculo del tumorein unodei dueesperimenti condotti.
Giorno 0Inoculo i.v. B16Lu8
(0.1x106/topo)
Giorno 4,5Inoculo i.p. IL-2 (60000IU/topo)
Giorno 15Analisi metastasi polmonari
A
0 50 100 150 200 250 300
CTRL-
m-TERTCTL
Numero di Metastasi
p: 0.0004
B
CTRL-
m-TERTCTL
C
Giorno 3Inoculo i.v. m-TERT CTLs
(5x106/topo)+
Inoculo i.p. IL-2 (60000IU/topo)

Risultati
76
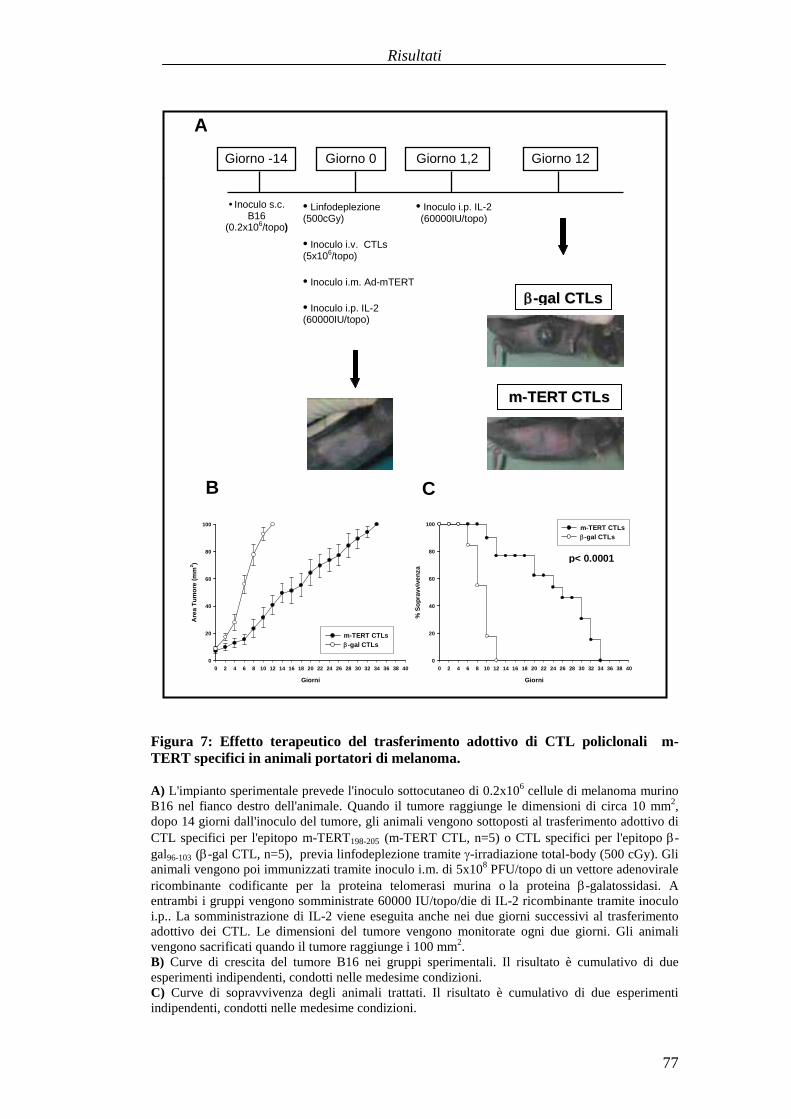
Risultati
77
Figura 7: Effetto terapeutico del trasferimento adottivo di CTL policlonali m-TERT specifici in animali portatori di melanoma.
A) L'impianto sperimentale prevedel'inoculo sottocutaneodi 0.2x106 cellule di melanomamurinoB16 nel fianco destro dell'animale.Quandoil tumore raggiunge le dimensioni di circa 10 mm2,dopo14 giorni dall'inoculodel tumore, gli animali vengonosottopostial trasferimento adottivo diCTL specifici per l'epitopom-TERT198-205 (m-TERT CTL, n=5) o CTL specifici per l'epitopo β-gal96-103 (β-gal CTL, n=5), previa linfodeplezionetramite γ-irradiazione total-body(500cGy). Glianimali vengono poi immunizzati tramiteinoculoi.m. di 5x108 PFU/topodi un vettore adenoviralericombinante codificante per la proteina telomerasi murina o la proteina β-galatossidasi. Aentrambi i gruppi vengonosomministrate60000IU/topo/die di IL-2 ricombinantetramiteinoculoi.p.. La somministrazionedi IL-2 vieneeseguitaanche nei duegiorni successivi al trasferimentoadottivo dei CTL. Le dimensioni del tumore vengono monitorate ogni due giorni. Gli animalivengonosacrificati quandoil tumore raggiunge i 100mm2.B) Curve di crescita del tumore B16 nei gruppi sperimentali. Il risultato è cumulativo di dueesperimenti indipendenti, condottinelle medesimecondizioni.C) Curve di sopravvivenzadegli animali trattati. Il risultato è cumulativo di due esperimentiindipendenti, condotti nelle medesimecondizioni.
• Inoculo s.c.B16
(0.2x106/topo)
• Linfodeplezione(500cGy)
• Inoculo i.v. CTLs(5x106/topo)
• Inoculo i.m. Ad-mTERT
• Inoculo i.p. IL-2 (60000IU/topo)
• Inoculo i.p. IL-2 (60000IU/topo)
Giorno 12Giorno -14 Giorno 0 Giorno 1,2
ββ--ggaall CCTTLLss
mm--TTEERRTT CCTTLLss
B C
Giorni
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40
Are
aT
um
ore
(mm
2 )
0
20
40
60
80
100
Giorni
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40
%S
op
ravv
iven
za
0
20
40
60
80
100
p< 0.0001
A
m-TERT CTLsβ-gal CTLs
m-TERT CTLsβ-gal CTLs

Risultati
78
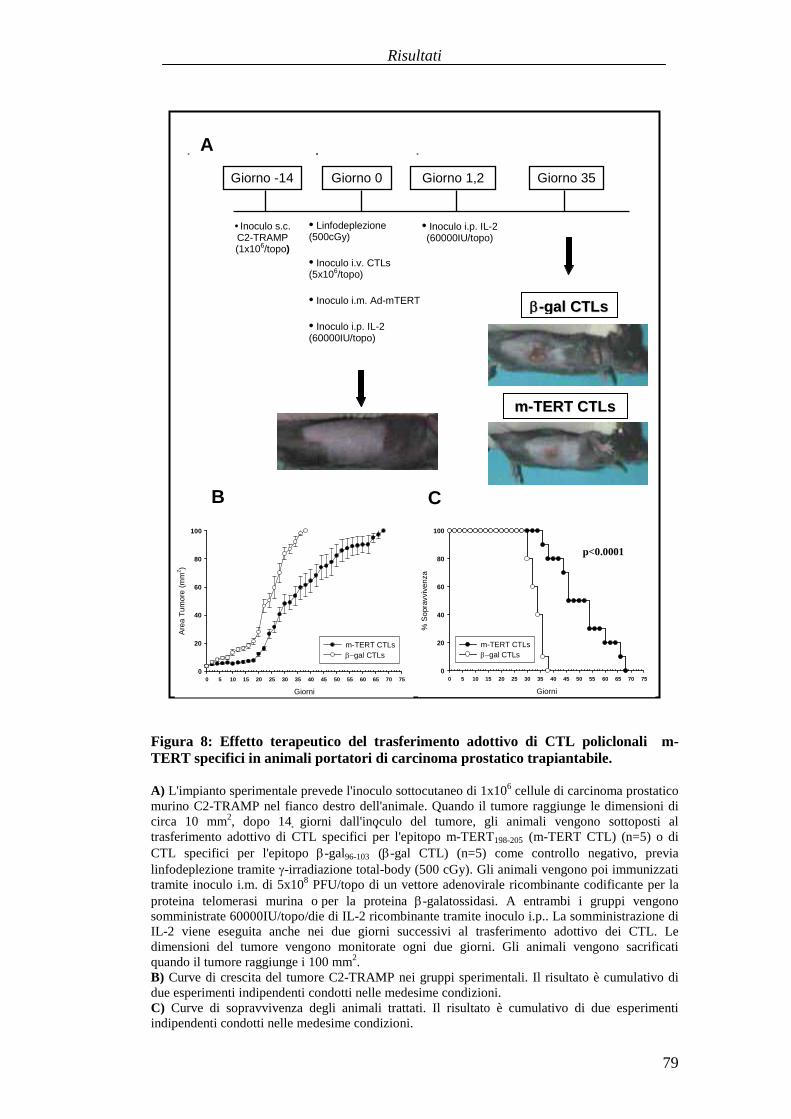
Risultati
79
Figura 8: Effetto terapeutico del trasferimento adottivo di CTL policlonali m-TERT specifici in animali portatori di carcinoma prostatico trapiantabile.
A) L'impianto sperimentaleprevedel'inoculo sottocutaneodi 1x106 cellule di carcinomaprostaticomurino C2-TRAMP nel fianco destrodell'animale.Quandoil tumoreraggiungele dimensioni dicirca 10 mm2, dopo 14 giorni dall'inoculo del tumore, gli animali vengono sottoposti altrasferimento adottivo di CTL specifici per l'epitopo m-TERT198-205 (m-TERT CTL) (n=5) o diCTL specifici per l'epitopo β-gal96-103 (β-gal CTL) (n=5) come controllo negativo, previalinfodeplezionetramiteγ-irradiazionetotal-body(500cGy). Gli animali vengonopoi immunizzatitramite inoculo i.m. di 5x108 PFU/topo di un vettoreadenovirale ricombinante codificanteper laproteina telomerasi murina o per la proteina β-galatossidasi. A entrambi i gruppi vengonosomministrate 60000IU/topo/die di IL-2 ricombinante tramite inoculo i.p.. La somministrazionediIL-2 viene eseguita anche nei due giorni successivi al trasferimento adottivo dei CTL. Ledimensioni del tumore vengono monitorate ogni due giorni. Gli animali vengono sacrificatiquandoil tumore raggiungei 100mm2.B) Curve di crescita del tumoreC2-TRAMP nei gruppi sperimentali. Il risultato è cumulativo didueesperimenti indipendenti condotti nelle medesimecondizioni.C) Curve di sopravvivenzadegli animali trattati. Il risultato è cumulativo di due esperimentiindipendenticondotti nellemedesimecondizioni.
Giorno 35Giorno -14 Giorno 0 Giorno 1,2
A
• Inoculo s.c.C2-TRAMP(1x106/topo)
• Linfodeplezione(500cGy)
• Inoculo i.v. CTLs(5x106/topo)
• Inoculo i.m. Ad-mTERT
• Inoculo i.p. IL-2 (60000IU/topo)
• Inoculo i.p. IL-2 (60000IU/topo)
B C
ββ--ggaall CCTTLLss
mm--TTEERRTT CCTTLLss
Giorni
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75
Are
aT
umor
e(m
m2)
0
20
40
60
80
100
m-TERT CTLsβ−gal CTLs
Giorni
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75
%S
opra
vviv
enza
0
20
40
60
80
100
m-TERT CTLsβ−gal CTLs
p<0.0001

Risultati
80

Risultati
81
Identificazione e caratterizzazione funzionale di un clone CTL ad
alta affinità specifico per l'epitopo m-TERT198-205 .
Considerando l'eterogeneità della popolazione CTL ottenuta mediante
sitmolazioniripetutein vitro, il nostro studioèproseguito con l'isolamento di CTL
ad alta affinità per l'epitopom-TERT198-205, tramite"limiting-dilution". Abbiamo
ottenutouno spettro di cloni CTL con TCR a diverseavidità per il complessom-
TERT198-205-MHC. Tra questi, il clone 7 (CTL-7) è risultato il più interessante.
Questoclone,infatti, è risultatoavereun TCR costituito dalla catenaVβ-11 ed è
stato in gradodi riconoscereun ampiospettro di lineetumorali di diversaistologia
(carcinoma prostatico, carcinomadi colon,melanomae sarcoma) sia tramite saggi
di citotossicità (rilasciodi 51Cr) chesaggiE.L.I.S.A. perrilasciodi IFN-γ.
Un datointeressanteè statoil fatto cheil livello di riconoscimentodel CTL-7 dei
diversibersagli cellulari è risultatougualea quello del bersaglio cellularecaricato
con il peptide m-TERT198-205. Inaspettatamente, le linee derivate da tumorali
ematologici,quali linfomi (MBL2) e timomi (EL-4), non sonostate riconosciute.
Abbiamo inoltre isolato ancheun clone a bassaaffinità: il clone 13 (CTL-13).
Questo clone presentava caratteristicheopposte al clone CTL-7: infatti, non è
stato in gradodi riconoscerebersaglicellulari ad eccezionedi quelli caricati con il
peptide m-TERT198-205 (MBL2-p198) e in misura blanda bersagli cellulari
transfettati per l'espressionedella molecola costimolatoria B7.1/CD80(Figura9).
Questa diversa funzionalità tra il CTL-7 e il CTL-13 è in linea con l'avidità
funzionaledei dueCTL per il complessocostituito dalle molecole di primaclasse
e il peptidem-TERT198-205. Infatti, in un testdi citotossicitàper rilascio di 51Cr in
cui il bersaglio cellulare (MBL2) è stato caricato con concentrazioni scalari di
peptide m-TERT198-205, il CTL-7 presentavalivelli di lisi maggiori rispetto al
CTL-13 aparitàdi concentrazionedi peptidecaricato (Figura9).
La caratterizzazionedei cloni specifici per l'epitopo m-TERT198-205 è proseguita
valutando l'efficacia terapeutica in vivo. Abbiamo util izzato il modello di
metastatizzazionea livello polmonareinoculandoendovenala linea di melanoma
murino B16. Dopo 3 giorni dall'inoculo del tumore,gli animali hanno ricevuto
5x106 cellule del clone CTL-7, del clone CTL-13 o del clone CTL-β-gal,
quest'ultimo usato comecontrollo negativo.I vari gruppi sonostati trattati con
60000IU/topo/die di IL-2 ricombinantetramite inoculo i.p., come descritto in
precedenza.Gli animali sono stati sacrificati dopo 15 giorni dall'inoculo del

Risultati
82
tumore e l'effetto terapeuticoè statovalutato come diminuzionedel numero di
metastasi:il gruppodi animali trattaticonil cloneCTL-7 presentavanoun numero
di metastasi(n=18)significativamenteinferiorerispetto agli animali del gruppodi
controllo (n=77,p<0.001).Anchegli animali trattati con il cloneCTL-13 a bassa
affinità presentavanouna significativa (n=52, p=0.006)diminuzione del numero
di metastasirispetto al gruppodi controllo ma inferiore rispetto a quella indotta
dal cloneCTL-7 (Figura10).
Questidati dimostranocomel'isolamentoe l'espansionein vitro di cloni CTL con
TCR ad alta affinità per il complesso MHC+peptide antigenico, sia unastrategia
efficace per potenziare l'effetto terapeutico dell'immunoterapia adottiva nel
trattamento dei tumori metastatici.
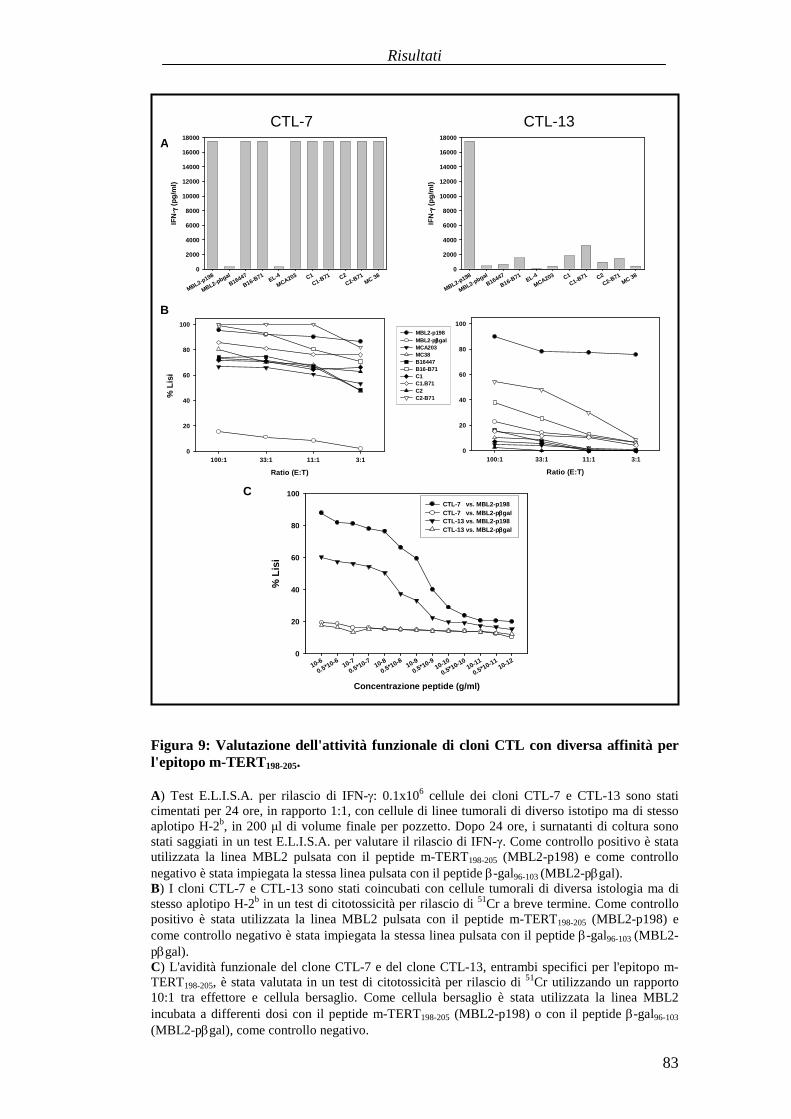
Risultati
83
Figura 9: Valutazione dell'attività funzionale di cloni CTL con diversa affinità perl'epitopo m-TERT198-205.
A) Test E.L.I.S.A. per rilascio di IFN-γ: 0.1x106 cellule dei cloni CTL-7 e CTL-13 sono staticimentatiper24 ore, in rapporto 1:1, con cellule di linee tumorali di diversoistotipo ma di stessoaplotipo H-2b, in 200 µl di volume finale per pozzetto. Dopo 24 ore, i surnatanti di colturasonostati saggiati in un test E.L.I.S.A. per valutare il rilasciodi IFN-γ. Comecontrollo positivo è statautilizzata la linea MBL2 pulsata con il peptide m-TERT198-205 (MBL2-p198) e come controllonegativo è stataimpiegata la stessalinea pulsata conil peptideβ-gal96-103(MBL2-pβgal).B) I cloni CTL-7 e CTL-13 sonostati coincubaticon cellule tumorali di diversa istologia ma distesso aplotipo H-2b in un testdi citotossicitàper rilascio di 51Cr a brevetermine. Comecontrollopositivo è stata utilizzata la linea MBL2 pulsatacon il peptidem-TERT198-205 (MBL2-p198) ecome controllo negativo è stataimpiegatala stessalineapulsatacon il peptideβ-gal96-103 (MBL2-pβgal).C) L'avidità funzionale del clone CTL-7 e del clone CTL-13, entrambi specifici per l'epitopo m-TERT198-205, è stata valutata in un testdi citotossicità per rilasciodi 51Cr utilizzando un rapporto10:1 tra effettore e cellula bersaglio. Come cellula bersaglio è stata utilizzata la linea MBL2incubataa differenti dosi con il peptide m-TERT198-205 (MBL2-p198) o con il peptideβ-gal96-103
(MBL2-pβgal), come controllo negativo.
Concentrazione peptide (g/ml)
10-6
0.5*10-6 10-7
0.5*10-7 10-8
0.5*10-8 10-9
0.5*10-910-10
0.5*10-1010-11
0.5*10-1110-12
%L
isi
0
20
40
60
80
100CTL-7 vs. MBL2-p198CTL-7 vs. MBL2-pβgalCTL-13 vs. MBL2-p198CTL-13 vs. MBL2-pβgal
C
MBL2-p198
MBL2-pbgal
B16447
B16-B71EL-4
MCA203 C1C1-B71 C2
C2-B71MC 38
IFN
- γ(p
g/m
l)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
MBL2-p198
MBL2-pbgal
B16447
B16-B71EL-4
MCA203 C1C1-B71 C2
C2-B71MC 38
IFN
- γ(p
g/m
l)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000A
CTL-7 CTL-13
Ratio (E:T)
100:1 33:1 11:1 3:1
%L
isi
0
20
40
60
80
100
B
Ratio (E:T)
100:1 33:1 11:1 3:1
%L
isiC
r51
0
20
40
60
80
100MBL2-p198MBL2-pβgalMCA203MC38B16447B16-B71C1C1.B71C2C2-B71

Risultati
84
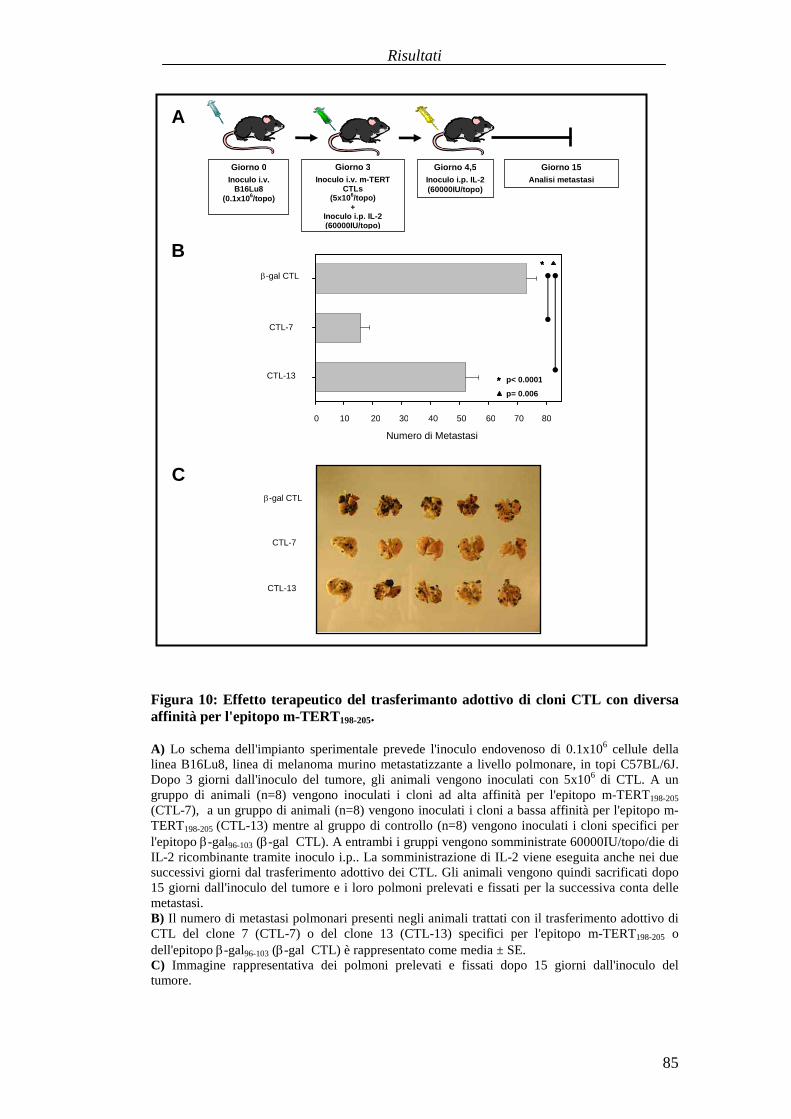
Risultati
85
Figura 10: Effetto terapeutico del trasferimanto adottivo di cloni CTL con diversaaffinità per l'epitopo m-TERT198-205.
A) Lo schemadell'impianto sperimentale prevede l'inoculo endovenosodi 0.1x106 cellule dellalineaB16Lu8, lineadi melanoma murino metastatizzantea livello polmonare, in topi C57BL/6J.Dopo 3 giorni dall'inoculo del tumore, gli animali vengono inoculati con 5x106 di CTL. A ungruppo di animali (n=8) vengono inoculati i cloni ad alta affinità per l'epitopo m-TERT198-205
(CTL-7), a un gruppodi animali (n=8) vengono inoculati i cloni a bassaaffinità per l'epitopo m-TERT198-205 (CTL-13) mentre al gruppo di controllo (n=8) vengonoinoculati i cloni specifici perl'epitopo β-gal96-103 (β-gal CTL). A entrambi i gruppivengono somministrate60000IU/topo/die diIL-2 ricombinante tramiteinoculo i.p.. La somministrazionedi IL-2 viene eseguitaanchenei duesuccessivi giorni dal trasferimento adottivo dei CTL. Gli animali vengono quindi sacrificatidopo15 giorni dall'inoculo del tumoree i loro polmoni prelevati e fissati per la successiva contadellemetastasi.B) Il numerodi metastasipolmonari presentinegli animali trattati con il trasferimentoadottivodiCTL del clone 7 (CTL-7) o del clone 13 (CTL-13) specifi ci per l'epitopo m-TERT198-205 odell'epitopoβ-gal96-103 (β-gal CTL) è rappresentatocomemedia± SE.C) Immagine rappresentativa dei polmoni prelevati e fissati dopo 15 giorni dall'inoculo deltumore.
C
Giorno 0Inoculo i.v.
B16Lu8(0.1x106/topo)
Giorno 4,5Inoculo i.p. IL-2 (60000IU/topo)
Giorno 15Analisi metastasi
Giorno 3Inoculo i.v. m-TERT
CTLs(5x106/topo)
+Inoculo i.p. IL-2 (60000IU/topo)
B
A
β-gal CTL
CTL-7
CTL-13
0 10 20 30 40 50 60 70 80
p< 0.0001
p= 0.006
Numero di Metastasi
β-gal CTL
CTL-7
CTL-13

Risultati
86

Risultati
87
Valutazione dell'efficacia terapeutica delle due strategie vaccinali a
DNA verso l'antigene TERT in un modello murino di carcinogenesi
prostatica spontanea.
La nostraricercasi è ancheproposta di valutare l'efficacia terapeutica dei due
protocolli di vaccinazione, decritti precedentemente, in un modello di
carcinogenesiprostaticaspontanea.
Abbiamoutilizzatocomemodellodi studio il ceppomurinoTRAMP (Transgenic
adenocarcinoma mouseprostate, (Greenberg, DeMayo et al. 1995). Il ceppodi
topi TRAMP è derivato dai topi C57BL/6Jed è caratterizzato dallo sviluppo,tra
le 8-12 settimane di vita, di una neoplasia intraepiteliale prostatica che
progrediscein adenocarcinomainvasivo nel 100%dei soggetti di 24-30 settimane.
Il transgeneresponsabiledell’insorgenza del tumore (Tag di SV40) è sotto il
controllo del promotoredella probasina(PB) per cui, nonostantesia presentein
tutte le cellule dell’animale,vieneespressonelle solecellule epiteliali prostatiche
a seguitodell’esposizionealla probasina, enzima prodotto durantel’età puberale
del topo, che innescala tumorigenesi. L’istologia e la storia naturaledel tumore
murino sono molto simili al tumore umano (Kaplan-Lefko, Chen et al. 2003).
Tutti gli animali sono stati selezionati per la presenza del trasgenePB-Tag
medianteanalisi in PCRdaDNA estrattoda leucociti del sangueperiferico. Solo
gli animali risultati positivi allo screening sono stati adoperati per gli incroci.
Dopo 23 generazioni, siamo riusciti ad ottenere una linea di topi in cui il
transgeneè presente in doppiacopia (omozigoti). Questi topi sonostati quindi
incrociati con animali C57BL/6J inbred per otteneregli animali eterozigoti che
sono stati utili zzati nei protocolli sperimentali che saranno descritti. L’analisi
anatomo-patologica condotta nei nostri animali eterozigoti ha confermato
l’i stogenesiprostatica del tumore TRAMP, così come riportato in letteratura
(Figura11A).
Prima di intraprendereil percorsovaccinale, abbiamo affrontato due aspettidi
primaria importanza,ovvero la valutazione del sistema immunitario dei topi
TRAMP in fasi diversedella progressionetumorale e la verifica della presenza
dell'antigene telomersi a livello del sito tumorale.
Al fine di valutare lo stato immunologico degli animali portatori di tumore di
diversostadio, sonostati sacrificatigruppi (n=3) di topi TRAMP diversi per età
(8, 24, 36 e 48 settimanedi vita) edestensionedi malattia, e gli splenociti ottenuti

Risultati
88
dagli animali sonostati analizzatiper la rispostaalla stimolazione in vitro con
alloantigeni, medianteallestimentodi colture leucocitarie miste (MLC). Dopo 5
giorni, le colture sonostatesaggiatecontro bersagli allogenici e di controllo. I
risultati hanno dimostrato che la capacità delle colture di produrre IFN-γ in
risposta allo stimolo allogenico è conservata durante la progressione della
neoplasia.Si è poi procedutoa valutarela risposta spontaneaversol’antigenem-
TERT. Con gli stessi splenociti sono state allestite MLPC utili zzando come
stimolo il peptide m-TERT198-205. Le colturecellulari sonostatesuccessivamente
ristimolate con 105 cellule singenichepulsatecon il peptide di interesse (m-
TERT198-205) o con il peptidescorrelato( β-gal96-103). Dopo24 oredi incubazione
il surnatante della colturaè statosottopostoa saggio E.L.I.S.A. perquantificareil
rilascio di IFN-γ. Abbiamoosservatounarisposta assente nel gruppodi animali
TRAMP di 8, 24 e 36; solo animali TRAMP di 48 settimanepresentavano una
debolerisposta per l'antigene(Figura11B). Poichéquesta reattivi tà è assente nei
topi TRAMP di 8 settimanedi età,in cui il processotumoralesi è appenaavviato,
mentre è presentein topi TRAMP di 48 settimane di vita, in cui il tumore è
avanzato,è possibile ipotizzareche il sistema immune sia espostoall’antigene
TERT durante la progressione tumorale, ma che tuttavia non è in grado di
contrastare la crescitaneoplastica.L'indagine è proseguita per valutare lo stato
immunologico degli animali portatori di tumore a seguito dell'intervento
vaccinale. Conseguentemente,animali TRAMP diversi per età (8, 24, 36 e 48
settimane di vita) sono stati vaccinati con un vettore adenovirale ricombinante
(5x108 PFU/topo) codificante la proteina telomerasi. Dopo 14 giorni dalla
vaccinazionegli animali sonostati sacrificati e gli splenociti sonostati stimolati
per 24 ore con 10 U/ml di IL-2 ricombinante, in presenzadi 1µg/ml del peptide
m-TERT198-205 (p-198) o del peptideβ-gal96-103 (p-βgal), in un saggio ELISPOT
per rilascio di IFN-γ. Come si evincedalla figura 11C,solo gli animali di 8 e 24
settimanedi età presentanoun'espansionedi linfociti T in grado di riconoscere
significativamentel'epitopom-TERT198-205 rispetto all'epitopo scorrelato β-gal96-
103, mentregli animali di 36 e 48 settimanedi età non mostrano una risposta
analoga.L'analisi è proseguitaanchedopo stimolazione in vitro, utili zzando gli
splenociti per allestire MLPC con il peptide m-TERT198-205. Dopo 5 giorni di
coltura, le MLPC sonostatetestatesia in un saggiodi citotossicità per rilascio di51Cr che in un test E.L.I.S.A. per il rilascio di IFN-γ, utilizzando comebersagli

Risultati
89
cellulari la lineaMBL2 pulsatacon il peptide m-TERT198-205 e la medesimalinea
cellulare pulsata con il peptidescorrelato β-gal96-103. Entrambi i test funzionali
hanno evidenziatocomesolo le MLPC degli animali TRAMP di 8 e 24 settimane
di etàpresentavanounasignificativacapacità di riconoscerel'epitopom-TERT198-
205 mentre le MLPC degli animaliTRAMP di 36 e 48 settimanenon contengono
linfociti reattivi verso questoepitopo(Figura11C). Limitatamentealle condizioni
utilizzate, possiamo concludere che la finestra temporale efficace per la
vaccinazione è limitata alla 36 settimana di vita poichè, oltre questo limi te
temporale,i topi TRAMP non sonoin gradodi montareunarispostaimmuneper
l'antigene TERT. Al momento, possiamo solo ipotizzare i motivi di questa
inefficienza quali, ad esempio, fenomeni di tolleranza/soppressione indotti dal
tumore; risulta evidente,comunque,che esisteun ampio arco temporale in cui
intervenire somministrando ripetutamente il vaccino, per mantenere una buona
rispostaprolungatanel tempoanchenellefasi di malattiaavanzata.
Per dimostrarela presenzadell'antigeneTERT nella prostata, durantelo sviluppo
della neoplasia, abbiamo sfruttato un anticorpo policlonale specifi co per
l'isoforma murina di telomerasiin un'analisi immunoistochimica condotta dalla
Dr.ssaManuela Iezzi dell'Universitàdi Chieti. Abbiamo potuto dimostrareche, se
pur il tessuto prostatico normale presentaun'espressionebasaledi telomerasi,la
proteina risulta iperespressa nelle fasi di iperplasia e soprattutto di
adenocarcinoma (Figura 12). Questodato avvalora il significato dell'antigene
telomerasi comeTAA, in quantola sua espressione differenziale ci permette di
distinguere tessuto normale da tessuto tumorale. Verificato la presenza
dell'antigene a livello del tumore prostatico, abbiamo paragonato l'eff icacia
terapeuticadi due protocolli di vaccinazione per l'antigene TERT in animali
TRAMP: il primo basatosull' immunizzazionetramite inoculo di DNA plasmidico
e l'altro basatosu un regime "prime-boost". Gli animali TRAMP sono stati
vaccinatia partiredalla 6a settimanadi vita tramite un doppioinoculo di plasmide
intervallato di 15 giorni (protocollo#1)o tramite l'inoculo del plasmide seguito
dalla somministrazionedi un vettoreadenovirale ricombinante (protocollo#2), e
un doppio richiamo con DNA plasmidico dopo 10 settimane dall'ultima
somministrazione del vaccino. Per verificare l’efficacia dei suddetti protocolli
vaccinaliabbiamo sacrificatogli animalialla 24a settimanadi vita. Agli animali è
stata prelevatala prostataper un’indagine istologica volta a valutare lo stadio

Risultati
90
della progressione tumorale. Tale indagineha mostrato che la vaccinazione a
DNA plasmidico haun impattoterapeutico: l'istologiadel tessuto prostatico degli
animali vaccinati con il plasmide vuoto ha svelato, infatti, quadri diffusi di
iperplasiaprostatica definita da cellule iperproliferanti che hanno persola loro
polarizzazione,mentreil tessutoprostatico degli animali vaccinati con l'antigene
TERT presentavanoaree di iperplasia meno estese. Questo può esser
schematizzatodalla distribuzionepercentuale di areeneoplastiche: in particolare,
la prostatadegli animali vaccinati con TERT è costituita da un 35% di tessuto
sano e daun 8.75%di adenocarcinomamentre le prostate degli animali vaccinati
con il plasmide vuoto presentano un 6.17% di tessuto sano e un 24.30% di
adenocarcinoma (p<0.05). Al contrario la vaccinazione a "prime-boost" non ha
presentatoalcun effetto terapeutico:la percentualedi adenocarcinomapresente a
livello delle prostatedegli animali vaccinati controtelomerasi è risultata del tutto
simile a quella presentea livello delle prostate degli animali di controllo (Figura
13).
Dato che la vaccinazionea "prime-boost" è risultata inefficace dal puntodi vista
terapeutico,abbiamodecisodi approfondire unicamente la vaccinazionea DNA
plasmidico.In primo luogoabbiamosvolto un'indagineimmunoistochimica, volta
a valutare il differente infiltrato leucocitario nella sede tumorale tra animali
vaccinati con il plasmidevuoto e quelli vaccinati con il plasmide codificante
TERT. Questa indagineimmunoistochimica ha evidenziato che le prostate degli
animali vaccinati con TERT presentavanoun infilt rato leucocitario nel sito
tumorale, costituito in prevalenzada cellule fagocitiche (macrofagi) a cui si
accompagnavanoelementidella risposta immune adattativa (linfociti T CD4+ e
CD8+), maggiore rispetto alle prostatedegli animali vaccinati con il plasmide
vuoto. La presenzadi un infiltrato di linfociti T indica chequestecellule sonoin
grado di arrivarenella sedetumoraledopo immunizzazioneattiva.
Infine, abbiamo dimostratocomela vaccinazione controTERT determina anche
un effetto terapeuticoin termini di aumento di sopravvivenza. Gli animali
TRAMP sonostati vaccinatia patiredalla 6a settimanadi vita con un protocollo
che prevedeva un doppio inoculo di DNA plasmidico intervallato di 15 giorni e
successivi richiami ogni 10 settimaneper l'intera vita dell'animale. Abbiamo
accertatoche la vaccinazioneè in gradodi determinareun incremento medio di
circa 11 settimanedi sopravvivenzarispetto agli animali vaccinati con il plasmide

Risultati
91
di controllo. Tale incrementodella sopravvivenzainoltre è risultato indipendente
dal costrutto plasmidicoutilizzatonella vaccinazione. Infatti, un gruppodi topi
TRAMP è stato vaccinatocon il costrutto plasmidico pVlJnsA vuoto (n=8) o
codificanteTERT (n=8) mentreun gruppodi topi TRAMP è stato vaccinatocon il
costruttoplasmidico VICAL vuoto(n=8) o codificantem-TERT (n=8). Possiamo,
quindi, concludere che l'utilizzo della vaccinazione a DNA contro telomerasi
risulta avereun effetto preventivonella progressione del carcinoma prostatico e
potrebberappresentareun buoncandidatoper futuri approcci immunoterapeutici
applicati all'uomo(Figura14).

Risultati
92

Risultati
93
Figura 11: Analisi della capacità di topi TRAMP, in stadi diversi della progressionetumorale, di montare una risposta immunitaria nei confronti dell'epitopo m-TERT198-205.
A) Immagine anatomo-patologica macroscopica rappresentativa di sezioni dell'apparatourogenitale di topi TRAMP di diverseetàcorrispondenti a fasi progressivedella malattia.B) Splenociti di topi TRAMP di diverseetà sono stati messi in colturaper5 giorni con 0.1µg/mldipeptidem-TERT198-205 (MLPC) o con 3x106 di splenociti allogenici irradiati (MLC). Per il testfunzionale0.1x106 cellule di MLPC sonostatecimentate per 24 ore, in rapporto 1:1, con la lineatumoraleMBL2 pulsatao con il peptideβ-gal96-103 o con il peptide m-TERT198-205, in 200 µl divolumefinale, mentre 0.1x106 cellule delle MLC sono statecimentate nelle medesimecondizionio conun bersaglio cellularesingenico(MBL2) o conun bersaglio cellulareallogenico(CT-26). Inseguito, i surnatanti di coltura sonostati saggiati in un test E.L.I.S.A. per valutare il rilascio diIFN-γ. I valori sono espressicomedifferenza della concentrazione di IFN-γ rilasciatodalle MLPC/MLC incubate con il bersaglio specifico e quello rilasciato dopo incubazione con il bersaglioscorrelato.C) Topi TRAMP di diverse età (n=3) sono stati vaccinati tramite inoculo di un adenovirusricombinante codificante m-TERT. Dopo 14 giorni dalla vaccinazione gli animali sono statisacrificati. 1x106 splenociti di topi TRAMP, precedentementevaccinati, sonostati stimolati per24ore con 10 U/ml di IL-2 ricombinante ed in presenzadi 1µg/ml o del peptide m-TERT198-205 (p-198) o del peptideβ-gal96-103 come controllo negativo, in un saggioELISPOT per il rilascio diIFN-γ. I dati sonorappresentaticomemedia ± SEdi animali della stessaetà. Successivamenteglisplenociti di ciascun animale sonostati messi in coltura per 5 giorni con 0.1µg/ml di peptidem-TERT198-205 (MLPC). Dopo 5 giorni le colture sono state saggiate sia in un testE.L.I.S.A. per ilrilascio di IFN-γ sia in un test di rilasciodi 51Cr utilizzandocome bersaglio in entrambi i casi lalinea cellulareMBL-2 pulsatacon il peptidem-TERT198-205o conil peptide di controllo β-gal96-103.I risultati in entrambii testsonoriportati comedifferenzao di rilascio di IFN-γ o di percentuale dilisi tra le MBL2 pulsatecon il peptidem-TERT198-205 e le MBL2 pulsateconil peptideβ-gal96-103. Idati sonorappresentaticomemedia ± SE di animali della stessaetà.
B
A C
Num
ero
diS
pots
0
50
100
150
200
8a settimana
24a settimana
36a settimana
48a settimana
p-198 p- β gal
Ratio (E:T)
100:1 33:1 11:1 3:1
%di
Lisi
0
20
40
60
80
100
8a settimana24a settimana36a settimana48a settimana
IFN
- γ(p
g/m
l)
0
1000
2000
3000
4000
5000
6000
7000
8a
settimana24a
settimana36a
settimana48a
settimana
TRAMP 48a settimana
TRAMP 36a settimana
TRAMP 24a settimana
TRAMP 8a settimana
IFN
-γ(p
g/m
l)
1000
2000
10000
12000
14000
16000
18000
8a
settimana24a
settimana36a
settimana48a
settimana
m-TERT198-205 -responsività
Allo-responsività

Risultati
94
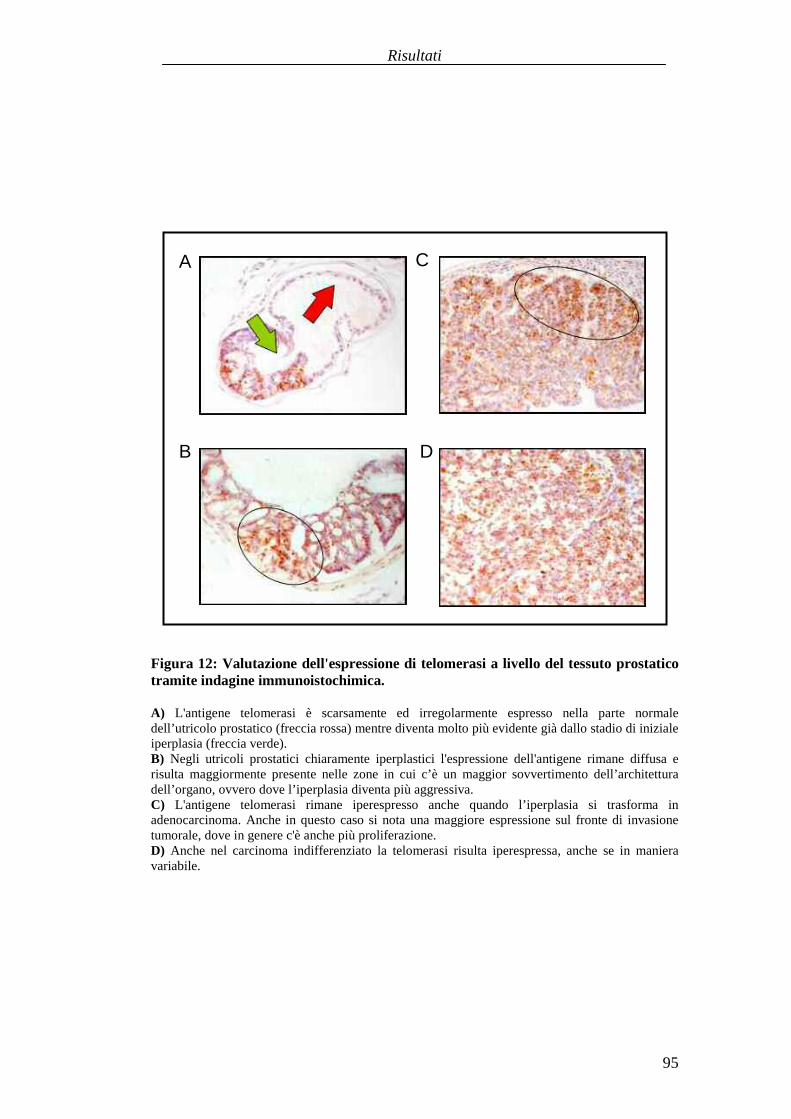
Risultati
95
Figura 12: Valutazione dell'espressione di telomerasi a livello del tessuto prostaticotramite indagine immunoistochimica.
A) L'antigene telomerasi è scarsamente ed irregolarmente espresso nella parte normaledell’utricolo prostatico (freccia rossa) mentrediventa molto più evidentegià dallo stadiodi inizialeiperplasia(frecciaverde).B) Negli utricoli prostatici chiaramenteiperplastici l'espressione dell'antigene rimanediffusa erisulta maggiormente presente nelle zone in cui c’è un maggior sovvertimento dell’ architetturadell’organo,ovverodovel’iperplasia diventa più aggressiva.C) L'antigene telomerasi rimane iperespresso anche quando l’ iperplasia si trasforma inadenocarcinoma.Anche in questo casosi nota una maggiore espressionesul fronte di invasionetumorale,dovein genere c'èanchepiù proliferazione.D) Anche nel carcinomaindifferenziatola telomerasirisulta iperespressa,anche se in manieravariabile.
A C
B D

Risultati
96
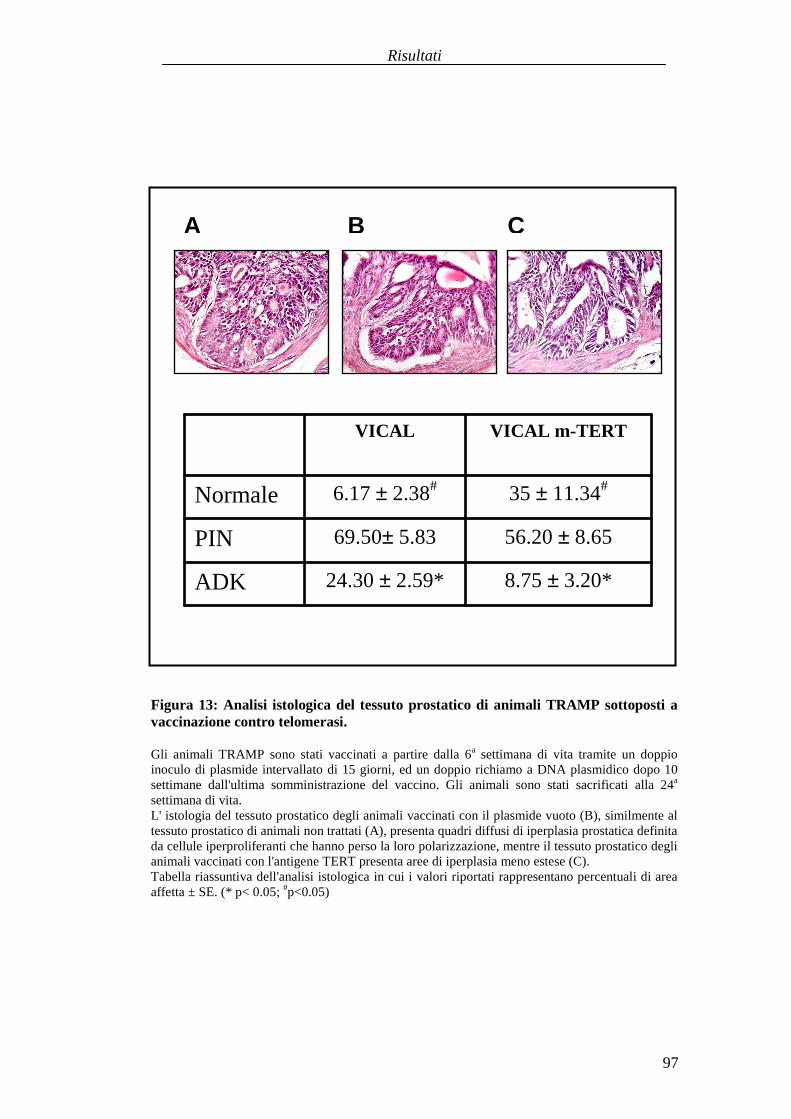
Risultati
97
Figura 13: Analisi istologica del tessuto prostatico di animali TRAMP sottoposti avaccinazione contro telomerasi.
Gli animali TRAMP sonostati vaccinati a partire dalla 6a settimanadi vita tramite un doppioinoculodi plasmide intervallatodi 15 giorni, ed un doppiorichiamo a DNA plasmidico dopo10settimane dall'ultima somministrazione del vaccino. Gli animali sono stati sacrificati alla 24a
settimanadi vita.L' istologia del tessuto prostatico degli animali vaccinati con il plasmidevuoto (B), similmentealtessutoprostatico di animali non trattati(A), presentaquadridiffusi di iperplasiaprostaticadefinitada celluleiperproliferanti chehannopersola loro polarizzazione, mentre il tessutoprostatico deglianimali vaccinati conl'antigeneTERT presenta areedi iperplasiameno estese(C).Tabellariassuntiva dell'analisiistologicain cui i valori riportati rappresentanopercentuali di areaaffetta ± SE. (* p< 0.05;#p<0.05)
A CB
8.75± 3.20*24.30± 2.59*ADK
56.20± 8.6569.50± 5.83PIN
35 ± 11.34#6.17 ± 2.38#Normale
VICAL m-TERTVICAL

Risultati
98
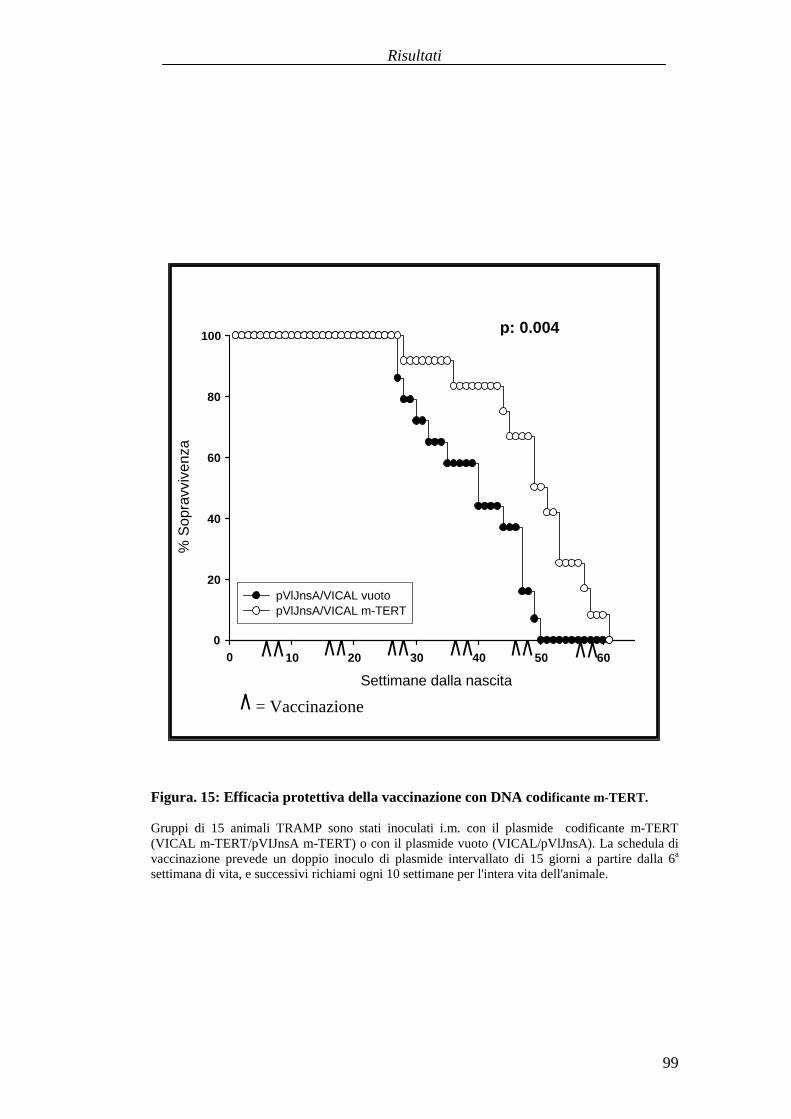
Risultati
99
Figura. 15: Efficacia protettiva della vaccinazione con DNA codificante m-TERT.
Gruppi di 15 animali TRAMP sono stati inoculati i.m. con il plasmide codificante m-TERT(VICAL m-TERT/pVIJnsA m-TERT) o con il plasmide vuoto (VICAL/ pVlJnsA). La schedula divaccinazione prevedeun doppio inoculo di plasmide intervallato di 15 giorni a partire dalla 6a
settimanadi vita,e successivirichiami ogni 10 settimane per l'interavita dell'animale.
= Vaccinazione
p: 0.004
Settimane dalla nascita
0 10 20 30 40 50 60
% Sopravvivenza
0
20
40
60
80
100
pVlJnsA/VICAL vuotopVlJnsA/VICAL m-TERT
= Vaccinazione
p: 0.004
Settimane dalla nascita
0 10 20 30 40 50 60
%S
opra
vviv
enza
0
20
40
60
80
100
pVlJnsA/VICAL vuotopVlJnsA/VICAL m-TERT

Risultati
100

Discussione
101
DISCUSSIONE
Per esser consideratoun candidatoideale per approccidi immunoterapia anti-
tumorale,un TAA devepresentaredellecaratteristichefondamentali: in primis, in
basealla sua espressione,deveesserin grado di discriminare il tessutonormale
dal tessutoneoplastico;in secondoluogo,deve esser in grado di attivareeffettori
immunitari. L'enzima ribonucleoproteico telomerasi (TERT) responsabile
dell'elongazionedei telomeri alle estremità dei cromosomi, riassumeentrambe
questecaratteristiche: infatti, comerecentementedescritto in letteratura,h-TERT
è sottopostaa processazionecitosolica,similmentea quantoaccade alle proteine
endogene,ed i suoi peptidi presentatia livello della membranaplasmatica in
associazionealle molecoleMHC, sono riconosciuti da linfociti T (Vonderheide,
Hahn et al. 1999). Inoltre, TERT è espressain circa l'85% delle cellule tumorali,
indipendentementedalla loro origine e istologia, mentre è attiva solamente in
alcunepopolazioni cellulari endogene,quali cellule staminali, cellule germinative
e linfociti attivati (Gross, Graff-Dubois et al. 2004). Tuttavia i livelli di
espressionedi TERT in queste popolazionicellulari risultanoinferiori di circa100
volte separagonate aquelli dellecelluletumorali. In questo lavorodi tesiabbiamo
dimostratoche anche l'isoforma murina di TERT, similmente a quella umana,
genera peptidi immunogenici a seguito della processazione endogena. In
particolare, siamoriusciti ad identificareil peptide m-TERT198-205 (VGRNFTNL)
come epitopo immunodominanteper aplotipo H-2b (ristretto per l'allele Kb).
Infatti, gli splenociti di animali con background C57BL/6J, vaccinati contro
TERT, sono in grado di riconoscere unicamente questo peptide. Abbiamo
dimostratocome, a livello dellaprostatadei topi TRAMP, l'espressionedi TERT
permettadi distinguereil tessutoprostatico normale dal tessuto tumorale. Infatti,
TERT risulta espressa nella prostatanormale ma a livelli decisamente inferiori
rispettoagli stadi di neoplasiaintraepiteliale (PIN) o di carcinoma.
L’utilizzo del modello murino TRAMP nei nostri studi trova il suo razionale
nell’elevata omologia con la neoplasia umana, in termini di progressione di
malattia e condivisioned’antigeniprostata-specifici. Questoagevolala traslazione
dei risultati ottenuti dagli studi preclinici di immunoterapia, illustrati in questa
tesi, all'ambito clinico. I nostri risultati dimostrano che i topi TRAMP sono
immunocompetenti, indipendentementedall’età e dall’estensionedi malattia.
Anzi, esisteuna seppurdebole reattività spontanea contro TERT che sembra

Discussione
102
essere in relazione con l'avanzamentodello stato della patologia. Infatti, la
reattività immunologica verso l’epitopo m-TERT198-205 è assente nei topi di 8
settimaned’età (ancoraprivi di tumoreinfi ltrante)ed apparein concomitanzacon
la progressionedella malattia.E’ importantenotareche, nonostante la presenza di
tale reattività, la crescita progressivadel tumore in tutti gli animali dimostra
inequivocabilmente l’inefficacia della popolazione di linfociti m-TERT198-205 -
reattivi nel contrastarela progressione del tumore. Una spiegazione di questa
mancatarisposta è sicuramenteda ricercarenei fenomeni di soppressioneindotti
dal microambiente tumorale. Da un recente studio pubblicato dal nostro
laboratorio (Bronte,Kasicet al. 2005), risulta chei linfociti intratumorali dei topi
TRAMP presentano un fenotipo di attivazione abortiva (CD44int, CD62L-,
CD25int), paragonabile a quellodescritto nei linfociti T umanimelanoma specifici
(Lee, Yee et al. 1999). Questofenotipo è indotto dall'espressionedi alti li velli
degli enzimi ARG e NOS, presentinell’epitelio prostatico neoplastico ma non in
quello normale (Bronte, Kasic et al. 2005). A rafforzare il ruolo del
microambientetumorale nel condizionarel'attivazionedegli effettori immunitari,
è stata l'osservazioneche solo animali privi di malattia o in presenzadi una
neoplasiaai primi stadi (animali di 24 settimane di età) sono responsivi alla
vaccinazione, essendocapaci di montare una risposta immunitaria effi cace e
specificaper l'antigenesomministrato,mentreanimali portatori di tumori avanzati
non sonoin gradodi innescarealcunarisposta immunitaria. La ricercadel miglior
protocollo di vaccinazioneha evidenziato come il vaccino con solo DNA
plasmidico, seguito dall'elettroporazionedel sito di inoculo, sia più efficace
(rispettoall'utili zzodi DNA plasmidicoin combinazionecon la somministrazione
di vettori adenoviraliricombinanti)nel selezionarelinfociti T CD8+ funzionali nel
riconoscereil complesso MHC/m-TERT198-205 presentea livello della membrana
plasmaticadelle cellule tumorali. Inoltre, la vaccinazionea soloDNA plasmidico
risulta avere una capacitàprofilattica, inducendo un significativo rallentamento
della progressioneneoplasticanel modelloTRAMP, doveal contrario il regime a
"prime-boost" non ha dimostrato alcun effetto terapeutico. A sottolineare
l'efficaciadella vaccinazionea DNA è anchela capacità di richiamare,a livello
del sito tumorale, un numero maggiore di effettori immunitari. A questo si
aggiunga che i topi TRAMP vaccinati con due somministrazioni di plasmide
codificantem-TERT, intervallati di quindici giorni l’una dall’al tra, e successivi

Discussione
103
richiami dopo10 settimane,hannodimostrato un significativo aumento in termini
di sopravvivenzarispetto agli animali vaccinati con il plasmidevuoto. Questo
effettoterapeutico è risultatoindipendente dal costrutto plasmidico ma dipendente
unicamentedalla sequenzaantigenica.Sulla scorta dei dati ottenuti la strategia
vaccinalefutura prevederàdi potenziare l'effetto terapeutico della vaccinazione
associandola alla somministrazionedi inibitori dell'attivi tà di ARG e NOS che,
come descritto in precedenza, sono responsabili del fenotipo abortivo dei TIL.
Studi già pubblicati dal nostrogruppo,infatti, hanno dimostrato chel’i mpiegodi
inibitori dei due enzimi in combinazioneconsenteun recuperofunzionale di
questi linfociti in vitro (Bronte,Kasicet al. 2005). La somministrazionedi questi
inibitori potrà avvenire in modo continuativo, o comunqueper lunghi tempi,
tramite l'utili zzo di una dieta medicatacontenente i farmaci, in modo da non
utilizzaremetodicheinvasive.
Gli studi in vitro ci hannopermesso di isolaredalle milze dei topi vaccinati con
DNA plasmidico codificanteTERT,popolazioni di CTL policlonali specifi cheper
l'epitopo m-TERT198-205 capaci di riconoscere in modo efficace varie linee
tumorali di diversa istologia ma con stessoaplotipo. Inoltre, siamo riusciti a
caratterizzareper intero la sequenzadella catena Vα e Vβ del TCR di alcuni di
questi linfociti. Questo ci consentiràdi trasdurre linfociti T di topo con un
recettoread alta affinità per TERT e valutare l'impatto terapeutico in animali
portatori di neoplasieTERT-positive.
Originale rispetto a quanto è riportato in letteraturaè stato, infatti, lo studio
eseguitosull'affinità dei CTL m-TERT specific i e soprattutto sul loro impiegoin
approcci di trasferimentocellulare adottivo in animali portatori di neoplasie di
diversa istologia. Abbiamo, infatti, dimostrato che il trasferimento di CTL
policlonali specifici per l'epitopo m-TERT198-205, associatoadun regime di mielo-
linfo-deplezione,è in gradodi rallentarein modosignificativo la crescitatumorale
in duemodelli di tumoretrapiantabile(melanomae carcinomaprostatico),quando
paragonatoall'inoculodi CTL specificiperun antigeneirrilevante.Inoltre,usando
un modello di metastatizzazionepolmonarebasato sull'inoculo di una linea
cellularedi melanoma,abbiamodimostratochel'inoculo di CTL specifici perm-
TERT198-205 causaunasignificativa diminuzionedel numero di metastasirispetto
ai controlli. L'effetto terapeuticorisultapotenziato quandovengonoinoculati CTL
ad alta affinità per l'epitopom-TERT198-205. Per potenziare ulteriormentel'effetto

Discussione
104
del trasferimento adottivo abbiamo di recente sfruttato l'util izzo di aAPC
sintetiche caricate con complessi MHC-peptide e molecole costimolatorie
(CD80), dimostrando l'incrementodell'effetto terapeutico anchequandovengono
inoculati linfociti T con TCR a bassaavidità (Zoso et al., 2008, inviato per la
pubblicazione).Al tra strategiacheabbiamo percorsoin questoultimo periodoè la
somministrazione ripetuta dei CTL. Abbiamo, infatti, utili zzato nel modello
TRAMP un regime di trattamentoche prevede tre somministrazioni di CTL a
partire dalla 6a settimana di vita, intervallate di 5 settimane l'una dall'altra.
Interessanteè poi la possibilità di applicarele metodichedescritte in questolavoro
nel modellomurino trasgenicoHLA-A*0201 (topi HHD), per isolare linfociti T
con TCR specifico per il complessoumano costituito dalle molecole HLA-A2 e
peptidi specifici per l'isoforma umana di TERT. Inoltre sarà possibile
somministrarei CTL specifici per h-TERT in topi doppi knock-out per RAG-2 e
per la catenaγ comuneal recettore di diversecitochineportatoridi tumori umani.
Questitopi sonodeficitari per le popolazionilinfocitarieT, B cellule NK e NKT e
consentono, conseguentemente,l'attecchimento di tumori umani, escludendo il
rischio di rigetto dovuto al xenotrapianto o qualunquealtro coinvolgimento del
sistemaimmunitario dell'animaleospite nell'azioneantitumorale. Questo modello
rappresentala piattaformaidealeper traslare gli studi dal camposperimentale a
quello clinico.

Bibliografia
105
BIBLIOGRAFIA
Acuto, O. and F. Michel (2003). "CD28-mediated co-stimulation: a quantitativesupport for TCR signalling."Nat RevImmunol3(12): 939-51.
Appay, V., P. R. Dunbar, et al. (2002). "Memory CD8+ T cells vary indifferentiation phenotypein differentpersistentvirus infections."Nat Med8(4): 379-85.
Bach, E. A., M. Aguet, et al. (1997)."The IFN gamma receptor:a paradigm forcytokine receptor signaling."AnnuRevImmunol15: 563-91.
Bacich, D. J., J. T. Pinto, et al. (2001). "Cloning, expression, genomiclocalization, and enzymatic activities of the mousehomologof prostate-specific membrane antigen/NAALADase/folate hydrolase." MammGenome12(2): 117-23.
Baniyash, M. (2004). "TCR zeta-chain downregulation: curtailing an excessiveinflammatoryimmuneresponse."Nat RevImmunol4(9): 675-87.
Bernhardt,S.L., M. K. Gjertsen,et al. (2006)."Telomerasepeptide vaccination ofpatientswith non-resectablepancreaticcancer:A doseescalating phaseI/IIstudy." Br J Cancer95(11): 1474-82.
Bronte, V., E. Apolloni, et al. (2000). "Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressingCD8(+) T cells." Blood96(12): 3838-46.
Bronte, V., S. Cingarlini, et al. (2003). "Effective genetic vaccination with awidely shared endogenousretroviral tumor antigen requires CD40stimulationduringtumorrejectionphase."J Immunol171(12): 6396-405.
Bronte, V., T. Kasic, et al. (2005). "Boosting antitumor responsesof Tlymphocytes infil trating human prostate cancers." J Exp Med 201(8):1257-68.
Bronte, V., P. Serafini, et al. (2003). "L-arginine metabolism in myeloid cellscontrols T-lymphocytefunctions."TrendsImmunol24(6): 302-6.
Bronte, V. and P. Zanovello (2005). "Regulation of immune responsesby L-argininemetabolism." Nat RevImmunol5(8): 641-54.
Bui, J. D. and R. D. Schreiber (2007). "Cancer immunosurveill ance,immunoediting and inflammation: independent or interdependentprocesses?"CurrOpin Immunol19(2): 203-8.
Burnet, F. M. (1970). "The conceptof immunological surveill ance."Prog ExpTumorRes13: 1-27.
Burnet,M. (1964)."ImmunologicalFactorsin theProcessof Carcinogenesis."BrMedBull 20: 154-8.
Burstein, N. A. and L. W. Law (1971). "Neonatal thymectomy and non-viralmammarytumoursin mice."Nature231(5303):450-2.
Calarota, S. A. and D. B. Weiner (2004). "Enhancement of humanimmunodeficiency virus type 1-DNA vaccine potency throughincorporation of T-helper1 molecularadjuvants."Immunol Rev 199: 84-99.
Cheever, M. A., P. D. Greenberg, et al. (1980). "Specifi city of adoptivechemoimmunotherapy of established syngeneic tumors." J Immunol125(2): 711-4.
Choudhury,A., S. Mosolits,et al. (2006)."Clinical results of vaccine therapy forcancer: learningfrom history for improving the future." Adv CancerRes95: 147-202.

Bibliografia
106
Cohen, C. J., Z. Zheng, et al. (2005). "Recognition of fresh humantumor byhuman peripheral blood lymphocytes transducedwith a bicistronicretroviral vector encodinga murine anti-p53 TCR." J Immunol 175(9):5799-808.
Cohen, E. P. (2001). "DNA-basedvaccines for the treatment of cancer--anexperimental model." TrendsMol Med 7(4): 175-9.
Colombo, M. P. and S. Piconese(2007). "Regulatory-T-cell inhibition versusdepletion: the right choice in cancerimmunotherapy."Nat Rev Cancer7(11): 880-7.
Comoli, P., R. De Palma,et al. (2004)."Adoptive transferof allogeneic Epstein-Barr virus (EBV)-specificcytotoxic T cellswith in vitro antitumor activit yboosts LMP2-specific immune responsein a patient with EBV-relatednasopharyngealcarcinoma."Ann Oncol 15(1): 113-7.
Cong,Y. S., W. E. Wright, et al. (2002)."Human telomeraseand its regulation."Microbiol Mol Biol Rev66(3): 407-25, tableof contents.
Cortez-Gonzalez,X. andM. Zanetti(2007)."Telomeraseimmunity from benchtobedside:roundone."J TranslMed 5: 12.
De, A. K., C. L. Mi ller-Graziano,et al. (2005)."Selective activation of peripheralblood T cell subsetsby endotoxin infusion in healthy human subjectscorrespondsto differential chemokineactivation." J Immunol 175(9):6155-62.
De Palma, R., I. Marigo, et al. (2004)."Therapeutic effectivenessof recombinantcancervaccinesis associatedwith a prevalent T-cell receptor alphausageby melanoma-specific CD8+ T lymphocytes." Cancer Res64(21): 8068-76.
Dighe, A. S.,E. Richards, et al. (1994)."Enhanced in vivo growthandresistanceto rejection of tumor cells expressing dominant negative IFN gammareceptors." Immunity1(6): 447-56.
Doe, B., M. Selby, et al. (1996). "Induction of cytotoxic T lymphocytes byintramuscular immunization with plasmid DNA is facili tated by bonemarrow-derived cells." ProcNatl AcadSci U S A 93(16): 8578-83.
Dudley, M. E., J. R. Wunderlich, et al. (2002). "Cancer regression andautoimmunity in patients after clonal repopulation with antitumorlymphocytes." Science298(5594):850-4.
Dudley, M. E., J. R. Wunderlich, et al. (2002). "A phase I study ofnonmyeloablative chemotherapyand adoptive transfer of autologoustumor antigen-specific T lymphocytes in patients with metastaticmelanoma."J Immunother(1997)25(3): 243-51.
Dunn, G. P., A. T. Bruce, et al. (2002). "Cancer immunoediting: fromimmunosurveillanceto tumorescape."Nat Immunol3(11): 991-8.
Dunn, G. P., C. M. Koebel, et al. (2006). "Interferons, immunity and cancerimmunoediting." Nat RevImmunol6(11): 836-48.
Dunn, G. P., L. J. Old, et al. (2004). "The immunobiology of cancerimmunosurveillanceandimmunoediting."Immunity 21(2): 137-48.
Eng, M. H., L. G. Charles,et al. (1999). "Early castration reduces prostaticcarcinogenesisin transgenicmice."Urology54(6): 1112-9.
Engel,A. M., I. M. Svane,et al. (1997)."MCA sarcomasinduced in scid mice aremore immunogenic than MCA sarcomas induced in congenic,immunocompetentmice."ScandJ Immunol45(5): 463-70.

Bibliografia
107
Fetsch, P. A., F. M. Marincola, et al. (1999). "The new melanomamarkers:MART-1 and Melan-A (the NIH experience)." Am J Surg Pathol23(5):607-10.
Feunou,P., L. Poulin, et al. (2003). "CD4+CD25+and CD4+CD25- T cells actrespectively as inducer and effector T suppressorcells in superantigen-inducedtolerance."J Immunol171(7): 3475-84.
Filaci, G., M. Fravega,et al. (2006)."Frequencyof telomerase-specific CD8+ Tlymphocytesin patientswith cancer."Blood 107(4): 1505-12.
Finn, O. J. (2003)."Cancervaccines: between the ideaand the reality." Nat RevImmunol3(8): 630-41.
Foster,B. A., J.R. Gingrich, et al. (1997)."Characterization of prostatic epithelialcell lines derivedfrom transgenicadenocarcinoma of the mouseprostate(TRAMP) model." CancerRes57(16): 3325-30.
Gabrilovich, D. (2004). "Mechanismsand functional significance of tumour-induceddendritic-cell defects."Nat RevImmunol4(12): 941-52.
Gattinoni, L., C. A. Klebanoff,et al. (2005)."Acquisition of full effector functionin vitro paradoxically impairsthe in vivo antitumor efficacy of adoptivelytransferredCD8+T cells." J Clin Invest115(6): 1616-26.
Gattinoni, L., D. J. Powell, Jr., et al. (2006). "Adoptive immunotherapy forcancer: buildingonsuccess." Nat RevImmunol 6(5): 383-93.
Gelbard, A., C. T. Garnett, et al. (2006). "Combination chemotherapy andradiation of human squamouscell carcinoma of the head and neckaugmentsCTL-mediatedlysis." Clin CancerRes12(6): 1897-905.
Gingrich, J. R., R. J. Barrios, et al. (1996). "Metastatic prostate cancer in atransgenicmouse." CancerRes56(18): 4096-102.
Grabert,R. C., L. P. Cousens,et al. (2006)."Human T cells armedwith Her2/neubispecific antibodies divide, are cytotoxic, and secrete cytokines withrepeatedstimulation." Clin CancerRes12(2): 569-76.
Grant, G. A. and J. F. Miller (1965). "Effect of neonatal thymectomy on theinductionof sarcomatain C57BL mice."Nature205(976):1124-5.
Greenberg,N. M., F. DeMayo, et al. (1995). "Prostate cancer in a transgenicmouse."ProcNatl AcadSci U S A 92(8): 3439-43.
Gross, D. A., S. Graff-Dubois, et al. (2004)."High vaccination efficiencyof low-affin ity epitopes in antitumorimmunotherapy." J Clin Invest 113(3): 425-33.
Grover, A., G. J. Kim, et al. (2006). "Intralymphatic dendritic cell vaccinationinducestumor antigen-specific, skin-homingT lymphocytes."Clin CancerRes12(19): 5801-8.
Hahn, W. C. and M. Meyerson (2001). "Telomerase activation, cellularimmortalizationandcancer."Ann Med 33(2): 123-9.
Harlin, H., T. V. Kuna, et al. (2006)."Tumor progression despite massive influxof activatedCD8(+)T cells in a patientwith malignantmelanomaascites."CancerImmunol Immunother55(10): 1185-97.
Hastie, N. D., M. Dempster, et al. (1990). "Telomere reduction in humancolorectalcarcinomaandwith ageing."Nature346(6287):866-8.
Hendriks, J.,L. A. Gravestein, et al. (2000). "CD27 is required for generation andlong-termmaintenanceof T cell immunity." Nat Immunol1(5): 433-40.
Hildeman,D. A., T. Mitchell, et al. (2003). "Control of Bcl-2 expression byreactive oxygenspecies."ProcNatl AcadSci U S A 100(25): 15035-40.

Bibliografia
108
Hsu, F. J., C. Benike, et al. (1996). "Vaccination of patients with B-celllymphomausing autologousantigen-pulseddendritic cells." Nat Med 2(1):52-8.
Hu, H. M., H. Winter, et al. (2000). "Divergent roles for CD4+ T cells in thepriming and effector/memoryphasesof adoptive immunotherapy." JImmunol 165(8): 4246-53.
Huang,X., M. Bennett,et al. (2004). "Anti -tumor effects andlack of side effectsin mice of an immunotoxindirectedagainst human and mouseprostate-specificmembraneantigen." Prostate61(1): 1-11.
Hubert,R. S., I. Vivanco,et al. (1999)."STEAP: a prostate-specific cell-surfaceantigenhighly expressedin humanprostate tumors."ProcNatl AcadSci US A 96(25): 14523-8.
Kaech,S. M., J. T. Tan, et al. (2003). "Selective expression of the interleukin 7receptor identifies effector CD8 T cells that give rise to long-livedmemory cells." Nat Immunol4(12): 1191-8.
Kagamu, H., J. E. Touhalisky, et al. (1996). "Isolation basedon L-selectinexpressionof immuneeffectorT cellsderived from tumor-draining lymphnodes."CancerRes56(19): 4338-42.
Kanegane, H., K. Nomura,et al. (2002). "Biological aspects of Epstein-Barr virus(EBV)-infected lymphocytes in chronic active EBV infection andassociatedmalignancies."Crit RevOncolHematol 44(3): 239-49.
Kaplan-Lefko, P. J., T. M. Chen,et al. (2003). "Pathobiology of autochthonousprostatecancer in a pre-clinical transgenic mousemodel." Prostate 55(3):219-37.
Kaplan, D. H., V. Shankaran,et al. (1998). "Demonstration of an interferongamma-dependenttumor surveillancesystemin immunocompetent mice."ProcNatl AcadSci U S A 95(13): 7556-61.
Kawakami, Y., M. I. Nishimura, et al. (1993). "T-cell recognition of humanmelanomaantigens." J ImmunotherEmphasisTumor Immunol 14(2): 88-93.
Kershaw,M. H., M. W. Teng, et al. (2005). "Supernatural T cells: geneticmodification of T cells for cancertherapy." Nat Rev Immunol 5(12): 928-40.
Khong, H. T. andN. P. Restifo(2002)."Natural selection of tumorvariants in thegenerationof "tumor escape"phenotypes."Nat Immunol3(11): 999-1005.
Kieper, W. C., A. Troy, et al. (2005). "Recent immunestatus determines thesourceof antigens that drive homeostaticT cell expansion." J Immunol174(6): 3158-63.
Kim, N. W., M. A. Piatyszek,et al. (1994). "Specific association of humantelomeraseactivity with immortal cells and cancer." Science266(5193):2011-5.
Klebanoff, C. A., H. T. Khong, et al. (2005). "Sinks, suppressorsand antigenpresenters: how lymphodepletion enhances T cell-mediated tumorimmunotherapy." TrendsImmunol26(2): 111-7.
Klein, G. (1966)."Tumorantigens." AnnuRevMicrobiol 20: 223-52.Kobayashi,Y., T. Higashi, et al. (2000). "Expression of MAGE, GAGE and
BAGE genes in human liver diseases:utilit y as molecular markersforhepatocellular carcinoma."J Hepatol32(4): 612-7.
Koya,R. C., N. Kasahara, et al. (2003). "Potent maturation of monocyte-deriveddendritic cells afterCD40L lentiviral genedelivery." J Immunother (1997)26(5): 451-60.

Bibliografia
109
Krieg, A. M. (2002). "CpG motifs in bacterial DNA and their immuneeffects."AnnuRevImmunol20: 709-60.
Kuball, J., F. W. Schmitz, et al. (2005). "Cooperation of humantumor-reactiveCD4+ and CD8+ T cells after redirection of their specificity by a high-affin ity p53A2.1-specificTCR." Immunity 22(1): 117-29.
Lanzavecchia,A. and F. Sallusto (2002). "Progressive differentiation andselectionof the fi ttest in the immuneresponse."Nat Rev Immunol 2(12):982-7.
Larin, S. S., G. P. Georgiev,et al. (2004). "Genetransferapproachesin cancerimmunotherapy." GeneTher11 Suppl 1: S18-25.
Lee, P. P., C. Yee, et al. (1999). "Characterization of circulating T cells specificfor tumor-associated antigensin melanoma patients." Nat Med 5(6): 677-85.
Lipponen,P. K., M. J. Eskelinen,et al. (1992)."Tumour infilt rating lymphocytesas an independent prognosticfactor in transitional cell bladdercancer."Eur J Cancer29A(1): 69-75.
Loll ini, P.L., F. Cavallo,et al. (2006)."Vaccines for tumourprevention." Nat RevCancer6(3): 204-16.
Ludlow, J. W. (1993). "InteractionsbetweenSV40 large-tumor antigen and thegrowthsuppressorproteinspRB andp53."FasebJ 7(10): 866-71.
Masutomi, K., S. Kaneko, et al. (2002). "Identification of serum anti-humantelomerase reverse transcriptase (hTERT) auto-antibodies duringprogressionto hepatocellularcarcinoma."Oncogene21(38): 5946-50.
Mavroudis, D., I. Bolonakis, et al. (2006). "A phaseI study of the optimizedcryptic peptide TERT(572y) in patients with advanced malignancies."Oncology70(4): 306-14.
Mazzoni, A., V. Bronte, et al. (2002). "Myeloid suppressorlines inhibit T cellresponsesby anNO-dependentmechanism."J Immunol168(2): 689-95.
Meyerson, M., C. M. Counter, et al. (1997). "hEST2, the putative humantelomerasecatalytic subunit gene, is up-regulated in tumor cells andduring immortalization." Cell 90(4): 785-95.
Mihm, M. C., Jr., C. G. Clemente,et al. (1996)."Tumor infilt rating lymphocytesin lymph node melanoma metastases: a histopathologic prognosticindicatorandan expressionof local immuneresponse."Lab Invest74(1):43-7.
Mocellin, S., S. Mandruzzato,et al. (2004). "Cancer vaccines: pessimism incheck." Nat Med 10(12): 1278-9; author reply 1279-80.
Montgomery,D. L., J. W. Shiver, et al. (1993). "Heterologousand homologousprotectionagainst influenzaA by DNA vaccination: optimization of DNAvectors."DNA Cell Biol 12(9): 777-83.
Moore,A. C. andA. V. Hill (2004)."Progressin DNA-basedheterologousprime-boostimmunization strategiesfor malaria."ImmunolRev199: 126-43.
Morgan,R. A., M. E. Dudley, et al. (2006). "Cancer regressionin patients aftertransferof genetically engineeredlymphocytes."Science314(5796):126-9.
Muranski, P., A. Boni, et al. (2006). "Increased intensity lymphodepletion andadoptive immunotherapy--how far can we go?" Nat Clin Pract Oncol3(12): 668-81.
Nagorsen,D., M. Panelli,et al. (2003)."Biasedepitopeselection by recombinantvaccinia-virus (rVV)-infectedmatureor immaturedendritic cells." GeneTher10(20): 1754-65.

Bibliografia
110
Nair, S. K., A. Heiser,et al. (2000). "Induction of cytotoxic T cell responsesandtumor immunity against unrelated tumors using telomerase reversetranscriptaseRNA transfecteddendritic cells." Nat Med 6(9): 1011-7.
Naito, Y., K. Saito, et al. (1998)."CD8+ T cells infi ltratedwithin cancercell nestsas a prognostic factor in humancolorectal cancer." Cancer Res 58(16):3491-4.
Old, L. J. andE. A. Boyse(1964). "Immunology of Experimental Tumors."AnnuRevMed 15: 167-86.
Onizuka, S., I. Tawara,et al. (1999)."Tumor rejection by in vivo administrationof anti-CD25(interleukin-2 receptoralpha)monoclonalantibody."CancerRes59(13): 3128-33.
Parkhurst,M. R., J. P. Riley, et al. (2004). "Immunization of patients with thehTERT:540-548 peptideinducespeptide-reactive T lymphocytesthat donot recognizetumorsendogenouslyexpressing telomerase."Clin CancerRes10(14): 4688-98.
Pavlenko, M., A. K. Roos,et al. (2004)."A phaseI trial of DNA vaccination witha plasmidexpressingprostate-specific antigen in patientswith hormone-refractory prostatecancer."Br J Cancer91(4): 688-94.
Peng, Y., Y. Laouar, et al. (2004). "TGF-beta regulates in vivo expansionofFoxp3-expressing CD4+CD25+ regulatory T cells responsible forprotectionagainstdiabetes."ProcNatl AcadSci U S A 101(13): 4572-7.
Pertmer,T. M., M. D. Eisenbraun,et al. (1995). "Genegun-based nucleic acidimmunization: elicitation of humoral and cytotoxic T lymphocyteresponsesfollowing epidermaldelivery of nanogramquantities of DNA."Vaccine13(15): 1427-30.
Powell, D. J., Jr., M. E. Dudley, et al. (2005)."Transition of late-stageeffector Tcells to CD27+ CD28+tumor-reactiveeffector memory T cells in humansafter adoptivecell transfertherapy." Blood 105(1): 241-50.
Quaglino, E., M. Iezzi, et al. (2004). "Electroporated DNA vaccine clearsawaymultifocal mammary carcinomasin her-2/neu transgenicmice." CancerRes64(8): 2858-64.
Quaglino, E., S. Rolla, et al. (2004). "Concordant morphologic and geneexpression data show that a vaccine halts HER-2/neu preneoplasticlesions." J Clin Invest113(5): 709-17.
Radi, R. (2004)."Nitric oxide,oxidants,andprotein tyrosinenitration." ProcNatlAcadSci U S A 101(12): 4003-8.
Reiter, R. E., Z. Gu, et al. (1998). "Prostate stem cell antigen: a cell surfacemarker overexpressedin prostate cancer."Proc Natl Acad Sci U S A95(4): 1735-40.
Reits, E. A., J. W. Hodge, et al. (2006). "Radiation modulates the peptiderepertoire, enhancesMHC class I expression, and induces successfulantitumorimmunotherapy."J ExpMed203(5): 1259-71.
Romero, P., J. C. Cerottini, et al. (2004). "Monitoring tumor antigenspecific T-cell responsesin cancerpatientsandphaseI clinical trialsof peptide-basedvaccination." CancerImmunolImmunother53(3): 249-55.
Rosenberg,S. A., J. J. Mule, et al. (1985). "Regressionof establishedpulmonarymetastases and subcutaneous tumor mediated by the systemicadministration of high-doserecombinant interleukin 2." J Exp Med 161(5):1169-88.

Bibliografia
111
Rosenberg,S. A., J. R. Yannelli, et al. (1994). "Treatment of patients withmetastatic melanomawith autologoustumor-infilt rating lymphocytesandinterleukin2." J Natl CancerInst 86(15): 1159-66.
Rubinstein,N., M. Alvarez,et al. (2004)."Targeted inhibition of galectin-1 geneexpression in tumor cells resultsin heightenedT cell-mediated rejection;A potential mechanism of tumor-immune privilege." Cancer Cell 5(3):241-51.
Sasada, T., M. Kimura,et al. (2003)."CD4+CD25+regulatory T cells in patientswith gastrointestinal malignancies:possible involvementof regulatory Tcells in diseaseprogression."Cancer98(5): 1089-99.
Schopfer,F. J.,P. R. Baker,et al. (2003)."NO-dependent protein nitration: a cellsignaling eventor an oxidativeinflammatory response?" TrendsBiochemSci 28(12): 646-54.
Schuler,W., I. J. Weiler, et al. (1986)."Rearrangement of antigen receptor genesis defectivein micewith severecombinedimmunedeficiency." Cell 46(7):963-72.
Shankaran, V., H. Ikeda, et al. (2001). "IFNgamma and lymphocytes preventprimary tumour developmentandshapetumour immunogenicity." Nature410(6832):1107-11.
Shay, J. W. and W. E. Wright (2002). "Telomerase: a target for cancertherapeutics."CancerCell 2(4): 257-65.
Sheil,A. G. (1986)."Canceraftertransplantation." World J Surg10(3): 389-96.Speiser,D. E., R. Miranda,et al. (1997)."Self antigensexpressedby solid tumors
Do not efficiently stimulatenaive or activated T cells: implications forimmunotherapy." J Exp Med186(5): 645-53.
Steinbrink,K., H. Jonuleit, et al. (1999). "Interleukin-10-treated human dendriticcells induce a melanoma-antigen-specific anergy in CD8(+) T cellsresulting in a failureto lysetumorcells."Blood93(5): 1634-42.
Stennett,L. S., A. I. Riker, et al. (2004). "Expression of gp100 and CDK2 inmelanomacells is not co-regulatedby a sharedpromoter region."PigmentCell Res 17(5): 525-32.
Street,S. E., E. Cretney, et al. (2001)."Perforin and interferon-gamma activitiesindependently control tumor initiation, growth, and metastasis."Blood97(1): 192-7.
Stutman, O. (1974). "Tumor development after 3-methylcholanthrene inimmunologically deficientathymic-nudemice."Science183(124):534-6.
Stutman,O. (1975). "Immunodepression and malignancy." Adv Cancer Res22:261-422.
Stutman,O. (1979)."Chemicalcarcinogenesisin nudemice: comparisonbetweennudemicefrom homozygousmatingsandheterozygousmatingsandeffectof ageandcarcinogendose."J Natl Cancer Inst 62(2): 353-8.
Su, Z., J. Dannull, et al. (2005). "TelomerasemRNA-transfected dendritic cellsstimulate antigen-specific CD8+ and CD4+ T cell responsesin patientswith metastatic prostatecancer."J Immunol174(6): 3798-807.
Sussman,J. J., R. Parihar,et al. (2004). "Prolongedculture of vaccine-primedlymphocytesresults in decreasedantitumorkill ing and changein cytokinesecretion."CancerRes64(24): 9124-30.
Sutmuller,R. P., L. M. vanDuivenvoorde,et al. (2001)."Synergismof cytotoxicT lymphocyte-associated antigen4 blockadeand depletion of CD25(+)regulatoryT cells in antitumor therapyreveals alternative pathways for

Bibliografia
112
suppression of autoreactivecytotoxic T lymphocyte responses."J ExpMed194(6): 823-32.
Suzuki, E., V. Kapoor, et al. (2004)."Solubletype II transforming growth factor-beta receptor inhibits establishedmurine malignantmesothelioma tumorgrowth by augmentinghostantitumorimmunity." Clin CancerRes 10(17):5907-18.
Takeda, K., M. J. Smyth,et al. (2002). "Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveill anceagainst tumordevelopment."J Exp Med 195(2): 161-9.
Terabe, M. and J. A. Berzofsky (2004). "Immunoregulatory T cells in tumorimmunity."CurrOpin Immunol16(2): 157-62.
Terabe, M., S. Matsui, et al. (2000). "NKT cell-mediated repression of tumorimmunosurveillance by IL-13 and the IL-4R-STAT6 pathway." NatImmunol 1(6): 515-20.
Thornburg, C., D. Boczkowski, et al. (2000). "Induction of cytotoxic Tlymphocyteswith dendritic cells transfected with human papillomavirusE6 and E7 RNA: implications for cervical cancer immunotherapy." JImmunother(1997)23(4): 412-8.
Ulmer, J. B., J. J. Donnelly, et al. (1993). "Heterologous protection againstinfluenza by injection of DNA encoding a viral protein." Science259(5102):1745-9.
Uyttenhove, C., L. Pilotte, et al. (2003). "Evidence for a tumoral immuneresistancemechanismbasedon tryptophandegradation by indoleamine2,3-dioxygenase."Nat Med9(10): 1269-74.
Vandeputte,M. and P. De Somer (1965). "Influence of thymectomy on viraloncogenesisin rats."Nature206(983): 520-1.
Vilella, R., D. Benitez, et al. (2003). "Treatment of patients with progressiveunresectable metastatic melanoma with a heterologous polyvalentmelanomawholecell vaccine."Int J Cancer106(4): 626-31.
Vonderheide,R. H., S. M. Domchek,et al. (2004)."Vaccination of cancer patientsagainst telomeraseinduces functional antitumor CD8+ T lymphocytes."Clin CancerRes10(3): 828-39.
Vonderheide,R. H., W. C. Hahn,et al. (1999)."The telomerasecatalytic subunitis a widely expressedtumor-associatedantigenrecognized by cytotoxic Tlymphocytes." Immunity 10(6): 673-9.
Wang, L. X., W. X. Huang,et al. (2004). "Adoptive immunotherapyof cancerwith polyclonal, 108-fold hyperexpanded, CD4+ and CD8+ T cells." JTranslMed 2(1): 41.
Widera, G., M. Austin, et al. (2000). "Increased DNA vaccine delivery andimmunogenicity by electroporationin vivo." J Immunol164(9): 4635-40.
Wittke, F., R. Hoffmann, et al. (1999). "Interleukin 10 (IL-10): animmunosuppressivefactor and independentpredictor in patients withmetastaticrenalcell carcinoma." Br J Cancer 79(7-8): 1182-4.
Wolf, A. M., D. Wolf, et al. (2003). "Increase of regulatory T cells in theperipheral bloodof cancerpatients."Clin CancerRes 9(2): 606-12.
Wolff, J.A., R. W. Malone,et al. (1990)."Direct genetransferinto mousemusclein vivo." Science247(4949Pt 1): 1465-8.
Workman,P., A. Balmain,et al. (1988)."UKCCCR guidelinesfor thewelfare ofanimalsin experimentalneoplasia."LabAnim 22(3): 195-201.

Bibliografia
113
Wrzesinski, C., C. M. Paulos,et al. (2007)."Hematopoietic stem cells promote theexpansion andfunction of adoptivelytransferredantitumorCD8 T cells." JClin Invest 117(2): 492-501.
Xia, Y., L. J. Roman,et al. (1998). "Inducible nitric-oxide synthasegeneratessuperoxidefrom thereductasedomain."J Biol Chem273(35): 22635-9.
Xia, Y. and J. L. Zweier (1997). "Superoxideandperoxynitrite generation frominduciblenitric oxide synthase in macrophages." ProcNatl Acad Sci U SA 94(13): 6954-8.
Yang, D., G. E. Holt, et al. (2001)."Murine six-transmembraneepithelial antigenof the prostate, prostatestemcell antigen,andprostate-specific membraneantigen:prostate-specificcell-surfaceantigenshighly expressedin prostatecancer of transgenicadenocarcinomamouseprostate mice." Cancer Res61(15): 5857-60.
Zakian,V. A. (1997)."Life andcancerwithouttelomerase." Cell 91(1): 1-3.

114

Abbreviazioni
115
ABBREVIAZIONI
2β-Me...................................................................................2-β-Mercaptoetanolo
Ab...........................................................................................................Anticorpo
ACT ....................................................................Trasferimentoadottivo cellulare
Ag.............................................................................................................Antigene
APC ........................................................................Cellula presentante l’antigene
ARG..........................................................................................................Arginasi
β-gal ................................................................................................β-galattosidasi
CD ................................................................................Cluster di differenziazione
CTL ...................................................................................LinfocitaT citotossico
CpG ...........................................................Dinucleotidi citosina-fosfato-guanina
DC ..............................................................................................Celluladendritica
DNA-PK ............................................................Protein chinasiDNA-dipendente
EDTA....................................................................Acido etilendiaminotetracetico
E.L.I.S.A...................................................Enzyme-linked Immunosorbent Assay
FACS ...........................................................FluorescenceActivatedCell Sorting
FBS ..........................................................................................Sierofetalebovino
FITC ............................................................................Fluoresceinaisotiocianato
G-CSF...............................Fattorestimolantela crescitadi coloniedi granulociti
GM-CSF .......Fattorestimolantela crescitadi coloniedi granulociti emacrofagi
GvHD................................................................Reazionedi rigetto versol’ospite
H2O2....................................................................................Perossidodi idrogeno
H&E.......................................................................................Ematossilinaeosina
HEPES......................................Acido 4-(2-idrossietil)-piperazin-1etansulfonico
IDO ...........................................................................Indolamina2,3-diossigenasi
IFN ........................................................................................................Interferone
IL ......................................................................................................Interleuchina
i.p. ................................................................................................. Intraperitoneale
K.O. .......................................................................................................Knockout
LAK ...............................................................CelluleKi ller attivatedalinfochine
L-Arg ................................................................................................... L-Arginina
mAb .................................................................................Anticorpomonoclonale
MCA .............................................................................................Metilcolantrene
MHC ....................................................Complessomaggioredi istocompatibil ità

Abbreviazioni
116
MLC..............................................................................Colturaleucocitariamista
MLPC ..............................................Colturaleucocitariastimolataconil peptide
MDSC .....................................................................Cellulemieloidi soppressorie
Nf-kB.........................................................................................Nuclearfactor kB
NK ........................................................................................Cellulekiller naturali
NKT .................................................................................CelluleT naturalkill er
NO ...................................................................................................Ossidonitrico
NOS ............................................................................Sintetasidell’ossidonitrico
O2-...........................................................................................Anionesuperossido
ONOO- ..............................................................................................Perossinitriti
PAA .............................................................................Antigeniprostataassociati
PBS..............................................Phosfatebuffersaline,soluzionesalinafosfato
PCR ................................................................Reazioneacatenadella polimerasi
PE ................................................................................Phycoerythrin, Ficoeritrina
PSCA.............................................................................Prostatestemcell antigen
PSMA ............................................................Prostatespecific membraneantigen
RAG2 ................................................................Recombination activatinggene2
RNOS ...............................................................Specie reattive dell’ossidonitrico
ROS ..........................................................................Specie reattive dell’ossigeno
s.c.............................................................................................................Sottocute
SCID............................................................Severe-combinedimmunodeficiency
STEAP .....................................Six transmembraneephitelial antigenof prostate
Treg.....................................................................................................T regolatorie
TAA...........................................................................Antigeniassociati al tumore
TAP ...............................................................Transporterassociatedwith antigen
TCR...............................................................Recettoreclonotipicodei linfociti T
TERT ...................................................................................................Telomerasi
TGF-β ...............................................................Fattoredi crescita trasformanteβ
TIL ...........................................................................Linfociti infilt ranti il tumore
Th ..............................................................................................Linfociti T helper
TRAMP .....................Modello transgenicomurinodel carcinomadellaprostata
w.t. ..................................................................................Wild type,tipo parentale

117
PUBBLICAZIONI
• BronteV., Cingarlini S., Marigo I., De SantoC., Gallina G., Dolcetti L.,Ugel S., PeranzoniE., MandruzzatoS., Zanovello P. Leukocyte infiltrationin cancer creates an unfavorable environment for antitumor immuneresponses: a novel target for therapeutic intervention.ImmunolInvest.2006;35(3-4): 327-57.
• Peranzoni E., Marigo I., Dolcetti L., Ugel S., SondaN., Taschin E.,Mantelli B., Bronte V. and ZanovelloP. Role of arginine metabolism inimmunity and immunopathology.Immunobiology2008; 212(9-10): 795-812.

118

Ringraziamenti
Un grazie a Enzo per avermi permesso di vivere quest'entusiasmante avventura e per avermi
retto per tre anni, un grazie altresì sentito alla Prof.ssa Zanovello per esser stata sempre
disponibile e presente.
Un grazie a tutti coloro che hanno collaborato per la realizzazione di questo lavoro: il gruppo
IRBM (Elisa, Carmela, Francesco, Barbara) il gruppo di Chieti (Manu, Marianna) e il Dr. De
Palma.
Un grazie a quelle canaglie del laboratorio che ormai son diventate una seconda famiglia:
grazie ILA per aver badato a me meglio di una mamma (ed è ora che lo diventi!), grazie Gigi
per non avermi mai picchiato, grazie Sonda per la compagnia nelle serate alcoliche, grazie Zilio
per le PCR, grazie Mantelli per non avermi mai urtato in pieno, grazie Circe per esser riuscita
a legger quei tetrameri, grazie Audry per il corso di inglese...ah quasi dimenticavo...grazie (ed è
un grazie speciale) Peranzoni per non esser mai d'accordo con me ma soprattutto per la tua
infinita pazienza nel sopportarmi...e si che non peso tanto!
Un grazie particolare a Sara amica sincera e leale (hai visto il tuo virgulto come è stato bravo!)
sempre presente se pur distante ma come dice De Gregori due buoni compagni di viaggio, anche
se scelgono imbarchi diversi, saranno sempre due marinai!
Un grazie alla colonia TRAMP per aver sfornato in maniera continuativa le bestiole su cui mi
son sbizzarito.
Un grazie ai due fratelli Paolo ed Elisabetta presenze insostituibili nella mia vita, un grazie
alla due nonne sempre affettuose, un grazie agli zii e ai cugini sempre presenti, un grazie
anche ai due cagnetti Birbo e Perla.
Un grazie speciale a mia mamma per avermi fatto testardo, un grazie speciale al mio papà per
avermi fatto idealista e sognatore ( e soprattutto interista). Vi voglio un gran bene e non posso
far altro che dedicarvi questo lavoro che rappresenta la realizzazione di un traguardo in cui ho
messo in gioco tutto me stesso tra sacrifici e fatiche ma sempre con tanta abnegazione e
volontà e sempre con il sorriso....ce l'ho fatta!

120


















