Allegato 1. Metodi analitici per contaminanti inorganici
98
Protocolli analitici (sostanze organiche) 1. Acido perfluoroottanoico (PFOA) e perfluoroottansolfonato (PFOS) nel biota acquatico. Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria, Reparto di Chimica Tossicologica 2. Acido perfluoroottanoico (PFOA) e perfluoroottansolfonato (PFOS) in campioni di pesce. Istituto di Ricerche Farmacologiche Mario Negri, Dipartimento Ambiente e Salute 3. Pesticidi organo clorurati nei prodotti ittici. Istituto Zooprofilattico Sperimentale delle Regioni Lazio e Toscana, Dipartimento di Chimica e Sostanze Biologicamente Attive 4. Polibromodifenil eteri (PBDE), policlorobifenili (PCB), policlorodibenzodiossine (PCDD) e policlorodibenzofurani (PCDF) in campioni di pesce e di molluschi bivalvi. Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria, Reparto di Chimica Tossicologica 5. Policlorobifenili (PCB), policlorodibenzodiossine (PCDD), policlorodibenzofurani (PCDF), polibromodibenzodiossine (PBDD) e polibromodibenzofurani (PBDF) in campioni di omogeneizzato di pesce. Istituto di Ricerche Farmacologiche Mario Negri, Dipartimento Ambiente e Salute 6. Policlorobifenili (PCB) in campioni di pesce e di molluschi bivalvi. CNR. Istituto per la Dinamica dei Processi Ambientali. Sede Venezia 7. Policlorobifenili idrossilati (HO-PCB) in campioni di siero ematico. Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria, Reparto di Chimica Tossicologica
Transcript of Allegato 1. Metodi analitici per contaminanti inorganici

Protocolli analitici (sostanze organiche) 1. Acido perfluoroottanoico (PFOA) e perfluoroottansolfonato (PFOS) nel biota
acquatico. Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria, Reparto di Chimica Tossicologica
2. Acido perfluoroottanoico (PFOA) e perfluoroottansolfonato (PFOS) in
campioni di pesce. Istituto di Ricerche Farmacologiche Mario Negri, Dipartimento Ambiente e Salute
3. Pesticidi organo clorurati nei prodotti ittici.
Istituto Zooprofilattico Sperimentale delle Regioni Lazio e Toscana, Dipartimento di Chimica e Sostanze Biologicamente Attive
4. Polibromodifenil eteri (PBDE), policlorobifenili (PCB), policlorodibenzodiossine
(PCDD) e policlorodibenzofurani (PCDF) in campioni di pesce e di molluschi bivalvi. Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria, Reparto di Chimica Tossicologica
5. Policlorobifenili (PCB), policlorodibenzodiossine (PCDD),
policlorodibenzofurani (PCDF), polibromodibenzodiossine (PBDD) e polibromodibenzofurani (PBDF) in campioni di omogeneizzato di pesce. Istituto di Ricerche Farmacologiche Mario Negri, Dipartimento Ambiente e Salute
6. Policlorobifenili (PCB) in campioni di pesce e di molluschi bivalvi.
CNR. Istituto per la Dinamica dei Processi Ambientali. Sede Venezia 7. Policlorobifenili idrossilati (HO-PCB) in campioni di siero ematico.
Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria, Reparto di Chimica Tossicologica

1. Acido perfluoroottanoico (PFOA) e
perfluoroottansolfonato (PFOS) nel biota acquatico
Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione
Primaria, Reparto di Chimica Tossicologica
Avvertenze
Il metodo analitico in oggetto è un adattamento derivante dalla letteratura internazionale, dalle linee guida proposte nel workshop preparatorio al III studio interlaboratorio per l’analisi dei composti alchilici perfluorurati (Università Vrije, Amsterdam 1920/03/2007) e dalla esperienza di reparto derivante dalla partecipazione al I studio interlaboratorio (marzo 2005). Tale metodo deve essere ancora testato e validato. Al riguardo, si fa presente quanto segue:
� I risultati del I (2005) e II (2006) studio interlaboratorio per l’analisi dei composti organici perfluorurati nel biota acquatico e nel sangue umano hanno rilevato un inadeguato accordo tra i risultati analitici tale da non consentire la definizione e la disponibilità di metodi e materiali di riferimento;
� prodotti consumabili o strumenti identificati commercialmente nel testo, e portati come esempi, possono essere sostituiti con prodotti o strumentazione equivalenti di differente origine;
� i contaminanti in oggetto sono tossici e idonee misure protettive devono essere presenti nel laboratorio per evitare contaminazioni ambientali e rischi occupazionali;
Acronimi CRM materiale di riferimento certificato

FR fattore di risposta FRR fattore di risposta relativo LOQ limite di quantificazione N noise, o rumore di fondo (N = 4 SDN) PFOA acido perfluoroottanoico PFOS perfluoroottano solfonato SE standard esterno SI standard interno MRM multiple reaction monitoring SRM single reaction monitoring HPLC high performance liquid cromatography Introduzione Il composto acido perfluoroottanoico (PFOA, C7F15COOH) e il composto anionico perfluoroottano solfonato (PFOS, C8F17SO3–) sono ampiamente dispersi nell’ambiente e bio-accumulati a diversi livelli della catena alimentare e sono il prodotto di degradazione ambientale di una più ampia famiglia di composti organici fluorurati ampiamente utilizzati in diversi prodotti industriali. Le caratteristiche chimico-fisiche di questi composti sono notevolmente diverse da quelle che caratterizzano gli inquinanti organici persistenti clorurati. In particolar modo le caratteristiche ioniche rendono questi composti solubili in acqua. Da ciò ne consegue un comportamento metabolico non vincolato al compartimento lipidico. 1.1 Scopo e applicazione 1.1.1 Il metodo è proposto per la quantificazione di PFOA e PFOS nelle matrici biologiche solide.

1.2 Principio del metodo 1.2.1 La Figura 1.1 sintetizza le fasi di preparazione, estrazione, purificazione e quantificazione utilizzate nel presente metodo. 1.2.2 Il metodo implica l’uso di standard interni (SI) marcati con quattro 13C vicinali compreso il C funzionalizzato (C1). 1.2.3 L’estratto è soggetto a purificazione SPE per mezzo di scambiatore anionico debole (Oasis® WAX 150 mg). 1.2.4 La quantificazione può essere ottenuta o per confronto con una retta di calibrazione (anche non in matrice) o per singolo punto da standard esterni. La seconda procedura è attuabile solo dopo attenta verifica del campo di linearità. 1.2.5 La procedura per la stima dei recuperi può essere eseguita o fortificando matrici prive dei contaminanti in esame o direttamente dalle fortificazioni con analiti marcati del campione in esame. Nella seconda evenienza la retta di calibrazione deve essere costruita in matrice per ovviare al possibile effetto matrice che renderebbe non accurata la risposta strumentale dello standard di quantificazione. 1.2.6 La separazione cromatografia è ottenuta con cromatografia liquida ad alta prestazione (HPLC). La quantificazione strumentale è affidata a spettrometri di massa con configurazione a doppio quadrupolo con cella collisionale con cui è possibile effettuare acquisizioni SRM (single reaction monitoring) e MRM (multiple reaction monitoring). 1.2.7 A ciascun batch di campioni è abbinata l’analisi di un bianco procedurale per verificare l’eventuale contributo al fondo (background) procedurale. 1.3 Reattivi impiegati 1.3.1 Reagenti e materiale cromatografico
� Argon 5.0; � azoto per gas cromatografia, ultrapuro; � Oasis® WAX – 6 cc, 150 mg, 30 um (Waters, USA); � potassio idrossido (85 %); � sodio idrossido; � sodio acetato (98.5 %) ; � acido acetico (99.9 %).

1.3.2 Solventi
� Acetone, grado tecnico; � n-esano per LC, purezza > 98.0 %; � metanolo per HPLC, purezza ≥ 99.9 %.
1.3.3 Standard
� PFOA, standard naturale, purezza > 99 % (lineare) (Wellington Laboratories) ;
� PFOS (sale sodico), standard naturale, purezza > 99 % (lineare) (Wellington Laboratories);
� PFOA, standard 13C4-marcato, purezza > 99 % (lineare) (Wellington Laboratories);
� PFOS (sale sodico), standard 13C4-marcato, purezza > 99 % (lineare) (Wellington Laboratories);
� PFDA, standard 13C2-marcato, purezza > 99 % (lineare) (Wellington Laboratories).
1.4 Interferenze e cause d’errore 1.4.1 Solo un laboratorio adeguatamente costruito ed equipaggiato può ritenersi idoneo all’esecuzione delle analisi d’interesse. All’interno del medesimo, dovrà essere mantenuta la massima pulizia, eventualmente in pressione positiva per evitare apporti di contaminazione atmosferica dall’esterno. Gli operatori, tutti esperti, dovranno attenersi rigorosamente ai protocolli di “buone pratiche” e alle procedure operative standard dedicate d’accreditamento in vigore nel laboratorio. 1.4.2 Le contaminazioni derivanti dalla presenza di materiale teflonato o simile sono tra le maggiori cause di interferenza analitica riscontrabile in queste procedure. Sono quindi da evitare setti teflonati o alternativamente è possibile porre del foglio di alluminio sotto il setto direttamente a contatto con le soluzioni di lavoro. 1.4.3 Le contaminazioni derivanti dall’apparecchiatura HPLC non possono essere completamente eliminate ma devono essere continuamente monitorate e pertanto l’applicazione di una accurata procedura per il bianco analitico risulta di fondamentale importanza. 1.4.4 Poiché la vetreria, i solventi, i reagenti, e ogni tipo di strumentazione utilizzata durante l’analisi possono essere fonte di interferenti, è necessario dimostrare che tutti i materiali impiegati ne siano virtualmente privi.

1.4.5 La vetreria, tutta di Pyrex, viene preventivamente lavata con detersivo e acqua, risciacquata accuratamente con acqua distillata, e poi tenuta in stufa a 170 °C per 12–24 h (la vetreria tarata, di Classe A ove opportuno, non viene sottoposta al trattamento termico). Prima dell’uso la vetreria è ulteriormente e ripetutamente risciacquata con acetone seguito da metanolo. Tutti i contenitori in polipropilene utilizzati per il campionamento, la conservazione e l’analisi dei campioni devono essere preventivamente lavati con metanolo. 1.4.6 Tutti i solventi utilizzati in quantità relativamente rilevanti, che subiscono sensibili processi di concentrazione durante il percorso dell’analisi, devono essere sottoposti a un controllo di congruità analitica preventiva per verificarne l’eventuale contributo al background procedurale. Per tale accertamento, volumi operativi dei solventi d’interesse, presi singolarmente, vengono concentrati a piccolo volume secondo quanto si verifica nell’applicazione del metodo e sottoposti a rilevamento strumentale. Qualora questo background procedurale sia troppo elevato, occorre purificare i solventi non idonei, o effettuarne la sostituzione. 1.4.7 Tutti gli standard acquisiti da ditte specializzate devono essere certificati all’origine in merito alle identità dei composti ed isomeri forniti, al grado della loro purezza, e al loro titolo effettivo. In genere, è opportuno verificare tali parametri direttamente mediante rilevamento strumentale: qualora si rilevi un’incongruenza sensibile tra il dichiarato e il risultato del rilevamento, il prodotto dovrà essere sostituito. Delle soluzioni di riferimento derivate dagli standard commerciali se ne deve accertare la stabilità nel tempo. 1.4.8 Le soluzioni acquose dei composti organici perfluorurati non possono essere poste in contenitori di vetro in quanto è possibile l’adsorbimento di questi composti da parte dei materiali vetrosi. 1.4.9 Le miscele tecniche dei composti in esame sono miscele di isomeri lineari e ramificati (gruppi diperfluorometilici geminali e perfluorometilici). Gli standard (Wellington) attualmente in commercio sono certificati al 98 % di purezza sull’isomero lineare. I fattori di risposta strumentale sono diversi tra isomeri di diversa ramificazione da ciò ne deriva la difficoltà intrinseca di una quantificazione accurata. Dovendo valutare la presenza totale dei composti in esame è opportuno integrare completamente il segnale cromatografico. Aggiustamenti della cromatografia, tali da rendere coeluenti i vari isomeri, devono essere ben vagliati in relazione al rischio di incrementare l’effetto matrice. 1.5 Conservazione del campione 1.5.1 Il campione è conservato a –20 ºC fino all’analisi.

1.6 Procedura analitica 1.6.1 Preparazione del campione. Il dimensionamento del campione da sottoporre all’analisi è funzione dei livelli di contaminazione presunti. In fase preliminare è possibile utilizzare 1 g di campione omogeneizzato ed eventualmente modificare il volume di ripresa finale per ottenere un LOQ analitico accettabile. Sulla base dei livelli di contaminazione presunti è possibile stimare la quantità di SI da addizionare al campione prima dell’estrazione. Il campione omogeneizzato è pesato e addizionato con gli SI. Il campione così preparato è lasciato riposare per 16 h a 4 °C prima dell’estrazione. 1.6.2 Estrazione. Un grammo del campione viene addizionato a 10 mL di soluzione metanolica di potassio idrossido 0.05 N in una Falcon di polipropilene e agitato per 16 h a temperatura ambiente. Un mL dell’estratto viene diluito con 100 mL di acqua in opportuno contenitore di polipropilene prima della fase di purificazione. 1.6.3 Purificazione. L’estratto è purificato mediante cartuccia SPE Oasis WAX. Questa viene condizionata prima con 5 mL soluzione metanolica di sodio idrossido 5 mM, poi con 4 mL di acqua. Dopo caricamento del campione, vengono raccolte tre frazioni: frazione 1 (4 mL di tampone acetato 25 mM (pH 4), frazione 2 (4 mL metanolo), frazione 3 (4 mL soluzione metanolica di sodio idrossido 5 mM). La velocità di eluizione della cartuccia è di circa 1 mL ogni 10 secondi. La frazione 3 concentrata a volume opportuno sotto leggero flusso di azoto a 40 °C. Nel caso si volesse procedere ad una quantificazione dei recuperi degli analiti marcati è necessario aggiungere un volume noto della soluzione metanolica di PFDA (standard d’iniezione). 1.6.4 Determinazione strumentale. Il PFOA e il PFOS vengono determinati mediante LC-MS con triplo quadrupolo utilizzando le condizioni operative descritte nell’Allegato 1.1. 1.6.5 Calcoli e risultati. La quantificazione degli analiti di interesse viene eseguita applicando l’Equazione 1, avendo determinato i fattori di risposta (FR) e i fattori di risposta relativi (FRR). Questi vengono stimati dalle aree dei segnali relativi alle masse.
SI
SI
AN
ANAN A
Q
FRR
AX ×= (1)
dove XAN è la quantità assoluta dell’analita nel campione; AAN è l’area del segnale analitico nel campione; QSI è la quantità assoluta dello SI 13C-marcato;

ASI è l’area corrispondente al segnale dello SI 13C-marcato; FRRAN è il fattore di risposta relativo calcolato mediante l’Equazione (2):
SI
ANAN FR
FRFRR = (2)
dove FRAN e FRSI sono rispettivamente i fattori di risposta determinati per l’analita e per lo standard interno nello standard di quantificazione. I risultati analitici relativi a PFOA e PFOS sono generalmente espressi in ng/g ww (peso fresco). 1.7 Verifica della qualità dei dati 1.7.1 Il laboratorio che usa il presente metodo è invitato a operare seguendo un programma di assicurazione di qualità i cui requisiti sono riportati di seguito. 1.7.2 RIPRODUCIBILITÀ INTRA-LABORATORIO DEL METODO. Questo controllo è eseguito con misurazioni indipendenti, ripetute su una medesima matrice omogenea predisposta dal laboratorio (blank fortificato). Devianze accettate sui valori singoli o cumulativi delle concentrazione degli analiti, <│±20 %│. Valori al difuori di tale intervallo comportano una specifica valutazione di congruenza e una conseguente azione a livello operativo. 1.7.3 ACCURATEZZA INTRA-LABORATORIO DEL METODO. Questo controllo è eseguito con misurazioni ripetute su appropriati materiali di riferimento (CRM) o, in loro mancanza, su blanks propriamente fortificati nel laboratorio. Devianze accettate sui valori singoli o cumulativi delle concentrazione degli analiti, <│±30 %│. Valori non congruenti a tale intervallo comportano una specifica valutazione di congruenza e una conseguente azione a livello operativo. 1.7.4 RECUPERO DI STANDARD INTERNI MARCATI. Sono accettabili recuperi compresi nel range 50–120 %. Valori al di fuori di tale intervallo comportano una specifica valutazione di congruenza, ed eventualmente il rigetto e la ripetizione dell’analisi. 1.7.5 LIMITE DI QUANTIFICAZIONE DEL METODO (LOQ). La determinazione del LOQ viene eseguita sul campione reale, calcolando per ciascun analita la concentrazione che produce una risposta strumentale pari a quattro volte il noise (N), con N ≈ 4 SDN (SDN, deviazione standard associata al noise). 1.7.6 CONTAMINAZIONE LEGATA AD INTERFERENZE NEI BIANCHI PROCEDURALI. A ciascun batch di campioni è abbinato un bianco procedurale il cui contributo potrebbe influenzare il valore di un segnale analitico (X). Il seguente sistema di

contrassegni o flags viene utilizzato nel laboratorio per qualificare e gestire il contributo di XB nei registri interni ed eventualmente nei rapporti di prova:
� XB / X ≤ 0.05: l’interferenza è considerate “trascurabile” → nessuna correzione;
� 0.05 < XB / X ≤ 0.25: l’interferenza è considerata “modesta” → nessuna correzione (flag, o contrassegno su X, “*”);
� 0.25 < XB / X ≤ 0.75: l’interferenza è considerata “sensibile” → correzione richiesta: X[valore corretto] = X – XB (flag “**”);
� 0.75 < XB / X ≤ 1.25: l’interferenza è considerate “marcata” → il segnale analitico è paragonabile al LOQ e sostituito da “<LOQ” (flag “***”);
� 1.25 < XB / X: l’interferenza è considerata “eccessiva” e non correggibile → il valore di X è sostituito da ND (“non determinabile”).
1.7.7 RIPETIBILITÀ DEI TEMPI DI RITENZIONE. Livelli di devianza accettati, <│±2 %│. 1.7.8 RIPETIBILITÀ DEI FATTORI DI RISPOSTA RELATIVI (RRF). Livelli di devianza accettati per (RFANALYTE) × (RF13C-IS)–1, <│±15 %│ (con segnali > LOQ). 1.7.9 EFFICIENZA DI RECUPERO DEGLI STANDARD INTERNI 13C-MARCATI. Intervallo d’accettabilità, 50–120 %. Valori al difuori di tale intervallo comportano una specifica valutazione di congruenza, ed eventualmente il rigetto e la ripetizione dell’analisi. 1.7.10 CONTROLLO DELLA LINEARITÀ DI RISPOSTA. La linearità di risposta è controllata regolarmente con soluzioni standard di PFOA e PFOS su un intervallo di circa due ordini di grandezza, che includono i valori LOQ. Il LOQ strumentale non dovrebbe essere superiore a 2–3 pg iniettati in colonna per ottenere prestazioni analitiche compatibili ad un LOQ analitico nell’ordine di 1 ppb (ng/g) 1.7.11 RIPRODUCIBILITÀ INTRA-LABORATORIO NEL TEMPO. Questo controllo è basato sulla ripetizione di determinazioni eseguite a intervalli di tempo (es., semestrale) su una matrice omogenea stabile idonea. Devianze accettate sui valori singoli o cumulativi delle concentrazione degli analiti, a differenti momenti temporali, maggiori di │±30 %│ comportano una specifica valutazione di congruenza e una conseguente azione a livello operativo. 1.7.12 RIPRODUCIBILITÀ E ACCURATEZZA INTER-LABORATORIO. Valutazione basata sulle determinazioni dei congeneri d’interesse, indipendenti e ripetute su matrici omogenee e CRM distribuite da un ente organizzatore. I criteri di valutazione/accettazione in funzione del sistema di controllo utilizzato dall’ente organizzatore.

Omogeneizzazione campione
Spike del campione con SI
1 g del campione più 10 mL metanolo/KOH 0.05 N
1 mL dell’estratto diluito in 100 mL di H2O
Condizionamento Oasis WAX (vedi testo)
Caricamento campione su OASIS WAX
Frazione 1: lavaggio con 4 mL tampone acetato 25 mM (pH 4)
Frazione 2: lavaggio con 4 mL metanolo
Frazione 3 (raccolta analiti): 4 mL metanolo/NaOH 5 mM
(evaporazione) Iniezione (10 µl) in HPLC/MS/MS
Figura 1.1 Schema della procedura analitica adottata dal presente metodo.

ALLEGATO 1.1 Condizioni operative suggerite per la determinazione strumentale degli analiti di interesse LC-MS HPLC – Agilent HP1100 o equivalente. Colonna cromatografica proposta – Thermo Betasil C18 (2.1 mm i.d., 50 mm length, 5 µm ps) o equivalente. Possibile uso di precolonna. Fasi mobili – 2 mM acetato d’ammonio in soluzione acquosa (fase A), metanolo (fase B). È proponibile anche l’uso di fasi addizionate ad acido formico o acetico all’1%. Iniettare 10 µl; flusso 300 µl/min; gradiente fase B: 10 % per 30 s e al 75 % a 7 min, al 100 % a 10 s, isocratica fino a 12 min. A 20 min si ritorna al 10 %.
Spettrometro a doppio quadrupolo con cella collisionale (Waters Premiere o equivalente). Analisi eseguite in Multiple Reaction Monitoring (MRM) in acq negativa.
MRM
Analita [M-H]– Prodotto
PFOA 413 369 PFOS 499 80/99
PFOA 13C4 417 372 PFOS 13C4 503 80/99
ALLEGATO 1.2
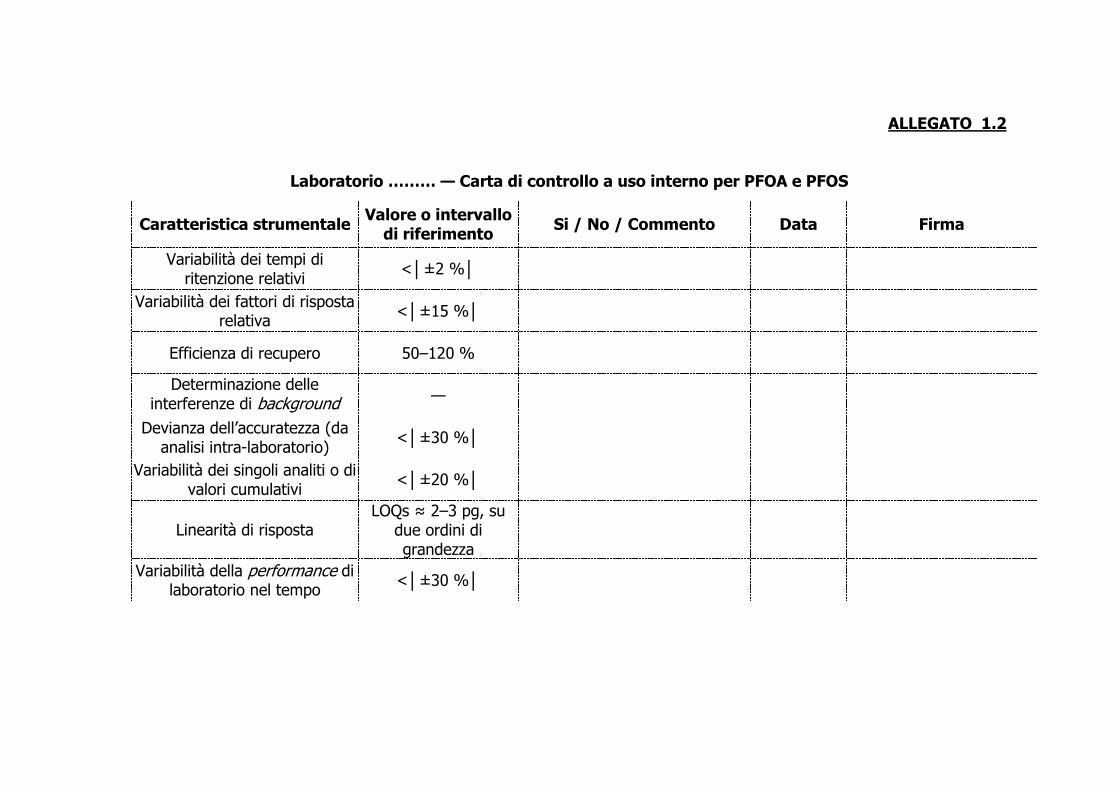
Laboratorio ……… — Carta di controllo a uso interno per PFOA e PFOS
Caratteristica strumentale Valore o intervallo di riferimento
Si / No / Commento Data Firma
Variabilità dei tempi di ritenzione relativi
<│±2 %│
Variabilità dei fattori di risposta relativa <│±15 %│
Efficienza di recupero 50–120 %
Determinazione delle interferenze di background —
Devianza dell’accuratezza (da analisi intra-laboratorio)
<│±30 %│
Variabilità dei singoli analiti o di valori cumulativi
<│±20 %│
Linearità di risposta LOQs ≈ 2–3 pg, su
due ordini di grandezza
Variabilità della performance di laboratorio nel tempo <│±30 %│

2. Acido perfluoroottanoico (PFOA) e
perfluoroottansolfonato (PFOS) in campioni di pesce
Istituto di Ricerche Farmacologiche Mario Negri, Dipartimento Ambiente e Salute
Acronimi ESI electrospray ionization HPLC high performance liquid chromatography, o cromatografia liquida ad
alta prestazione MS spettrometria di massa PFOA acido perfluoroottanoico PFOS perfluoroottansulfonato SI standard interno SPE solid phase extraction SRM selected reaction monitoring 2.1 Scopo e applicazione 2.1.1 Il presente metodo è utilizzabile per la quantificazione dell’acido perfluoroottanoico (PFOA, C7F15COOH) e del perfluoroottano solfonato (PFOS, C8F17SO3
–) mediante cromatografia liquida ad alte prestazioni abbinata a spetrtrometria di massa (HPLC-MS/MS).

2.2 Principio del metodo 2.2.1 Il rilevamento degli analiti di interesse è basato sull’impiego di standard interni (SI) marcati con 13C. Gli SI vengono aggiunti al campione prima di qualsiasi operazione analitica. 2.2.2 Dopo estrazione con solvente, i campioni sono sottoposti a una procedura di purificazione mediante cartuccia SPE e quantificati mediante HPLC-MS/MS per la determinazione di PFOS a PFOA. 2.2.3 A ciascun batch di campioni è abbinata l’analisi di un bianco procedurale per verificare l’eventuale contributo dovuto al fondo ambientale e procedurale. 2.3 Strumentazione 2.3.1 Strumentazione per l’estrazione del campione
� Estrattore automatico Aspec GX-274 (Gilson); � contenitori Falcon di polipropilene da 50 mL; � cartucce per estrazione SPE di tipo Evolute ABN (50 mg); � provette coniche da 6 mL.
2.3.2 Strumentazione per la determinazione strumentale del campione
� HPLC Perkin-Elmer Series 200 (autocampionatore e due pompe analitiche); � Spettrometro di massa Applied Biosystems API 3000, con sorgente
electrospray ionization ESI.

2.4 Reagenti e standard 2.4.1 Solventi
• Acetonitrile (Riedel-de-Haen, per LC-MS); • acqua ultrapura MilliQ per HPLC (Millipore).
2.4.2 Standard
• Perfluoro-n-octanoic acid (PFOA), 50 µg/mL in metanolo (Wellington Laboratories);
• perfluoro-n-[1,2,3,4-13C4]octanoic acid (PFOA-13C4), 50 µg/mL in metanolo (Wellington Laboratories);
• sodio perfluoro-1-octansolfonato (L-PFOS), 50 µg/mL in metanolo (Wellington Laboratories);
• sodio perfluoro-1-[1,2,3,4-13C4]octansolfonato (L-PFOS-13C4), 50 µg/mL in metanolo (Wellington Laboratories).
2.5 Conservazione del campione 2.5.1 I campioni sono stati conservati –20 °C fino all’analisi. 2.6 Procedura analitica 2.6.1 Estrazione del campione. Dopo scongelamento, vengono pesati circa 5 g di omogenato di pesce e trasferiti in Falcon da 50 mL. Al campione vengono aggiunti 40 mL di MilliQ e il tutto viene accuratamente miscelato. Dopo aver portato il pH a 8.5 tramite aggiunta di 70 µL di idrossido di ammonio, vengono aggiunti gli SI di PFOS e PFOA (1 ng totale). Il campione viene quindi mescolato su Vortex per permettere la diffusione dello standard interno e successivamente centrifugato per 10 minuti a 2800 rpm. Il surnatante viene quindi trasferito in una Falcon da 50 mL e sottoposto a purificazione. 2.6.2 Purificazione. La purificazione avviene tramite utilizzo di cartucce SPE del tipo EVOLUTE ABN mediante un estrattore automatico. La metodica prevede i seguenti passaggi:
� condizionamento della cartuccia SPE con 3 mL di metanolo, seguiti da 3 mL di acqua MilliQ;
� essiccazione della cartuccia tramite flusso di azoto; � caricamento del campione a velocità di 30 mL/min; � eluizione della cartuccia con 3 mL di metanolo.

Dopo eluizione, il solvente viene evaporato e il campione ripreso in 100 µL di una miscela di acetonitrile al 20 % in acqua. 50 µL del campione così diluito vengono trasferiti in vial di vetro con microinserti in plastica per l’analisi strumentale. 6.2 determinazione strumentale. Gli standard di PFOA e PFOS e i relativi standard marcati con 13C sono diluiti alle concentrazioni di lavoro (10 ng/µL, 100 pg/µL e 10 pg/µL) in acetonitrile. In fase di sviluppo del metodo per ogni sostanza è stata eseguita una ottimizzazione delle condizioni analitiche in MS e in MS/MS, mediante infusione continua di una soluzione standard (1 ng/µL). Tale procedura ha permesso di stabilire le migliori condizioni di ionizzazione e di frammentazione degli ioni pseudo-molecolari (Tabella 1). Le condizioni gascromatografiche di analisi (Allegato 1) sono state ottimizzate usando standard analitici alla concentrazione di 10 pg/µL. I tempi di ritenzione di PFOA e PFOS sono risultati rispettivamente: 7.7 min e 9.4 min. La sensibilità strumentale ottenuta è dell’ordine di pochi pg di sostanza iniettata in colonna.

Tabella 2.1. Condizioni di ionizzazione (ioni negativi) e di frammentazione per le sostanze indicate. Sostanza Orifice
voltage
(V)
Ring
voltage
(V)
Ione
precursore
(m/z)
Ione prodotto I
(m/z) ed
energia di collisione
(eV)
Ione prodotto II
(m/z) ed energia di
collisione (eV)
PFOA –20 –100 413 169 (26) 369 (16)
PFOA-13C4 –20 –100 417 172 (26) 372 (16)
PFOS –40 –180 499 80 (86) 99 (68)
PFOS-13C4 –40 –180 503 80 (86) 99 (68)

ALLEGATO 2.1 Condizioni operative utilizzate per la determinazione strumentale degli analiti di interesse
• colonna: XTerra MS C18 2.1x100 mm, 3.5 µm; • loop di iniezione: 20 µL; • eluente A: acido acetico 0.05 % in acqua; • eluente B: acetonitrile; • flusso: 200 µL/min; • gradiente: dal 20 al 70 % di B in 10 min, dal 70 al 100 % di B in 2 min, 100 % di B per 2 min, ritorno al 20 % di B in 2
min, 20 % di B per 7 min (riequilibrio della colonna).

3. Pesticidi organoclorurati nei prodotti ittici
Istituto Zooprofilattico Sperimentale delle Regioni Lazio e Toscana, Dipartimento di
Chimica e Sostanze Biologicamente Attive
Acronimi DCB decaclorobifenile 2,4’-DDD 1,1-dicloro-2-(2-clorofenil)-2-(4-clorofenil)-2,2-dicloroetano, noto
anche come o,p-DDD 4,4’-DDD 1,1-bis(4-clorofenil)-2,2-dicloroetano, noto anche come p,p’-DDD 2,4’-DDE 1,1-dicloro-2-(2-clorofenil)-2-(4-clorofenil)-etene, noto anche
come o,p-DDE 4,4’-DDE 1,1-dicloro-2,2-bis(4-clorofenil)-etene, noto anche come p,p’-
DDE 2,4’-DDT 1,1,1-tricloro-2-(2-clorofenil)-2-(4-clorofenil)-etano, noto anche
come o,p-DDT 4,4’-DDT 1,1,1-tricloro-2,2-bis(4-clorofenil)-etano, noto anche come p,p’-
DDT GC gas cromatografia MS spettrometria di massa OCP pesticidi organoclorurati SPE estrazione su fase solida

3.1 Scopo e applicazione 3.1.1 Il metodo è proposto per la quantificazione dei pesticidi organoclorurati in campioni di pesce mediante gascromatografia con rivelazione a selezione di massa (GC-MS/MS). 3.1.2 Il metodo consente la determinazione dei pesticidi clorurati indicati in Tabella 3.1. 3.2 Principio del metodo 3.2.1 Il campione è sottoposto ad estrazione con etere di petrolio per ottenere la sua componente lipidica. Da questa i pesticidi organoclorurati vengono estratti con acetonitrile. 3.2.2 L’estratto è purificato mediante cartucce SPE Florisil e gli analiti sono quantificati mediante gascromatografia con rivelazione a selezione di massa (GC-MS/MS). 3.3 Strumentazione
� Evaporatore rotante; � bilancia di precisione ± 1 mg; � bilancia analitica ± 0.01 mg; � sistema per l’eluizione sotto vuoto di cartucce SPE; � centrifuga refrigerata; � sistema riscaldato per l’evaporazione sotto corrente di N2; � gascromatografo Thermo-Finnigan mod. Trace GC dotato di rivelatore a
selezione di massa a trappola ionica (MS/ITD) mod. PolarisQ ed autocampionatore Thermo-Finnigan AS 3000;
� normale vetreria da laboratorio; � beute da 250 mL con tappo smeriglio; � bagno ad ultrasuoni; � palloncini da evaporazione a fondo conico (capacità almeno 250 mL); � provettoni “Falcon” in polipropilene con tappo a vite da 50 mL; � vials a tappo a vite da circa 12 mL con tappo teflonato; � vials a tappo a vite da circa 2 mL per gascromatografia;
3.4 Reagenti e standard 3.4.1 Reagenti e materiale cromatografico
� SPE-Florisil 2000 mg 12 mL; � colonna capillare con fase RTX – 5sil MS (RESTEK) 30 m × 0.25 mm × 0.25 µm
df, o di analoga selettività a basso spurgo per GC/MS.

3.4.2 Solventi
� Etere di petrolio per pesticidi, p. eb. 40–60 C; � sodio solfato anidro; � acetonitrile per pesticidi; � n-esano per pesticidi; � acetone per pesticidi; � soluzione di estrazione: 400 mL di acetonitrile per pesticidi vengono miscelati
con 100 mL di n-esano in un imbuto separatore. Una volta separate le due fasi, la fase sottostante (acetonitrile) viene recuperata e conservata in un contenitore idoneo.
3.4.3 Standard certificati
� Standard di pesticidi clorurati certificati 10 ng/µL (miscela); � standard di pesticidi clorurati marcati certificati 100 ng/µL e 50 ng/µL
(singoli); � standard di decaclorobifenile (DCB) certificato 100 ng/µL.
3.4.4 Soluzioni standard 3.4.4.1 Soluzione di PCL mix a 100 µg/mL: 100 µL della miscela di pesticidi clorurati 10 ng/µL vengono diluiti fino al volume di 10 mL con n-esano. 3.4.4.2 Soluzione di PCL mix a 1 µg/mL: 100 µL della miscela di pesticidi clorurati 10 mg/kg vengono diluiti fino al volume di 10 mL con n-esano. 3.4.4.3 Soluzione di PCL marcati a 100 µg/mL: 10 µL della soluzione pesticida marcato a 100 ng/µl e 200 µl della soluzione pesticida marcato a 20 ng/µL vengono diluiti fino al volume di 10 mL con n-esano. 3.4.4.4 Soluzione di PCL marcati a 1 µg/mL: 100 µL della soluzione pesticida marcato a 100 µg/mL vengono diluiti fino al volume di 10 mL con n-esano. Preparare la soluzione di pesticida marcato per ogni analista, utilizzando il marcato corrispondente la soluzione deve essere preparata ogni volta che si effettua l’analisi. 3.4.4.5 Soluzione di standard siringa DCB 100 µg/mL: 100 µL della soluzione DCB a 100 µg/mL vengono diluiti fino al volume di 10 mL con n-esano; 3.4.4.6 Soluzione di standard siringa DCB 1 µg/mL: 100 µL della soluzione DCB a 100 µg/mL vengono diluiti fino al volume di 10 mL con n-esano. 3.5 Conservazione del campione 3.5.1 Il campione omogeneizzato è congelato a –20 °C, fino all’analisi.

3.6 Procedura analitica 3.6.1 Estrazione della componente lipidica. Approssimativamente 30 g di campione opportunamente omogeneizzato vengono pesati in una beuta con tappo smeriglio e impastati con 50-100 g di sodio solfato anidro (la quantità di sodio solfato varia a seconda della percentuale di acqua presente nel campione). Dopo l’aggiunta di 150 mL di etere di petrolio, il campione viene sonicato per 15 minuti e lasciato con il solvente per 12 ore. Il surnatante è filtrato su sodio solfato e il solvente evaporato a pressione ridotta. 3.6.2 Estrazione dei pesticidi organoclorurati dal grasso. Circa 1 g di grasso, estratto come descritto al Punto 6.1, viene pesato in una Falcon da 50 mL, addizionato con 100 µL di miscela di standard marcati 1 µg/mL e mescolato con 30 mL di acetonitrile. Il campione così preparato viene posto in stufa a 60 °C per 20 minuti, agitato nuovamente, e posto in congelatore a –20 °C per almeno 1 ora. Dopo centrifugazione a 4000 rpm a 4 °C per 3 minuti, il surnatante viene raccolto in un pallone da evaporazione a fondo conico precedentemente pesato e il residuo viene estratto nuovamente con 30 mL di acetonitrile. Sul campione è ripetuta la fase di riscaldamento in stufa a 60°C e il congelamento a –20 °C. Dopo centrifugazione a 4000 rpm a 4 °C per 3 minuti, il surnatante è riunito al precedente. Le fasi estratte (volume totale 60 mL) vengono portate a secco in evaporatore rotante a 40 °C e sottoposte a purificazione. 3.6.3 Purificazione dei campioni. 20 mL di estratto vengono purificate sulle cartucce SPE Florisil 2000 mg 12 mL. Le cartucce vengono lavate con 20 mL di acetonitrile saturo in n-esano, asciugate per 10 minuti, e condizionate con 5 mL di acetonitrile saturo di n-esano. Caricato l’estratto su cartuccia, questa viene asciugata per 5 minuti sotto leggero vuoto e gli analiti vengono eluiti con 12 mL di una miscela n-esano:acetone (1:9). L’eluato è portato a secco sotto leggero flusso di azoto in termostato a 30 °C, ripreso con 900 µL di n-esano e 100 µL di DCB 1 µg/mL.
3.6.4 Determinazione strumentale. Gli analiti di interesse sono determinati mediante HRGC-MS/MS (trappola ionica) in modalità selective reaction monitoring (SRM) utilizzando le condizioni operative descritte nell’Allegato 1. Gli analiti di interesse e i corrispondenti composti marcati sono quantificati utilizzando le masse riportate in Tabella 3.1. Il rilevamento quantitativo viene eseguito mediante retta di taratura in matrice.

Tabella 3.1. Pesticidi organoclorurati ricercati nello studio.
Pesticida No. CAS
α-HCH 319-84-6 β-HCH 319-85-7 γ-HCH 58-89-9 HCB 118-74-1
eptacloro 76-44-8 aldrin 309-00-2
dieldrin 60-57-1 endrin 72-20-8
cis-eptacloro epossido 280044-83-9 trans-eptacloro epossido 1024-5703
cis-clordano 57-74-9 trans-clordano 57-74-2
2,4’-DDD 53-19-0 4,4’-DDD 72-54-8 2,4’-DDE 3424-82-6 4,4’-DDE 72-55-9 2,4’-DDT 789-02-6 4,4’-DDT 50-29-3
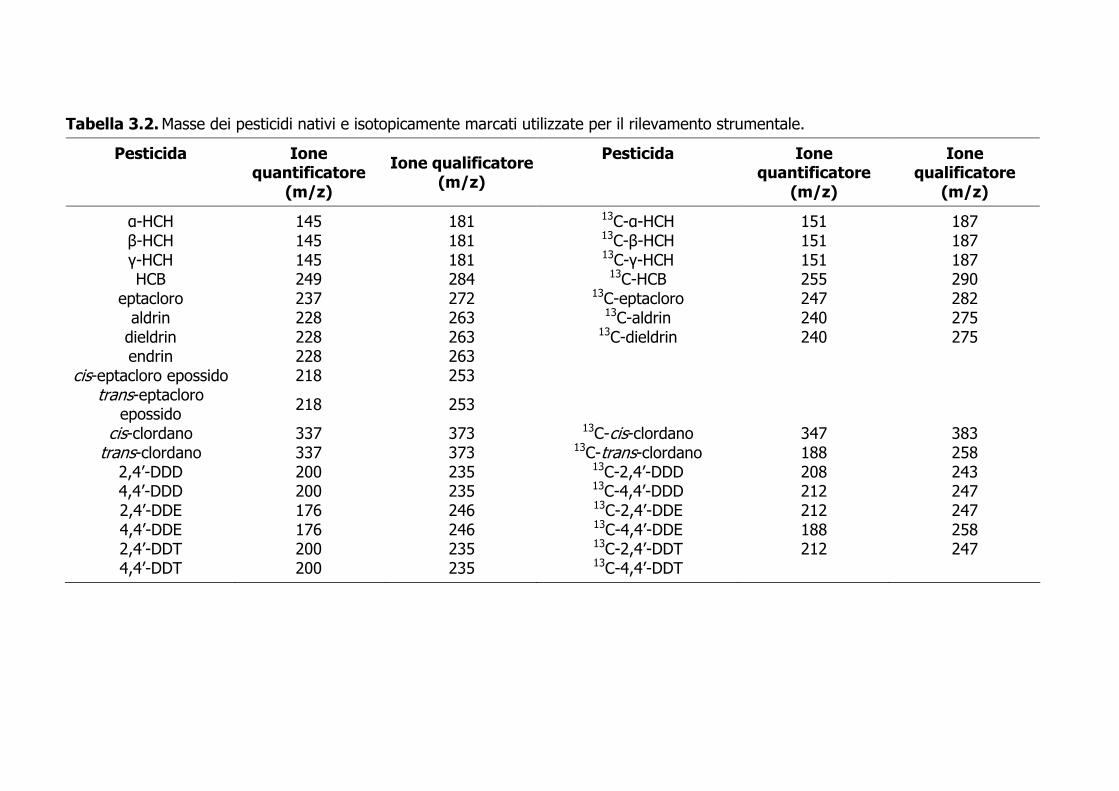
Tabella 3.2. Masse dei pesticidi nativi e isotopicamente marcati utilizzate per il rilevamento strumentale.
Pesticida Ione quantificatore
(m/z)
Ione qualificatore (m/z)
Pesticida Ione quantificatore
(m/z)
Ione qualificatore
(m/z)
α-HCH 145 181 13C-α-HCH 151 187 β-HCH 145 181 13C-β-HCH 151 187 γ-HCH 145 181 13C-γ-HCH 151 187 HCB 249 284 13C-HCB 255 290
eptacloro 237 272 13C-eptacloro 247 282 aldrin 228 263 13C-aldrin 240 275
dieldrin 228 263 13C-dieldrin 240 275 endrin 228 263
cis-eptacloro epossido 218 253 trans-eptacloro
epossido 218 253
cis-clordano 337 373 13C-cis-clordano 347 383 trans-clordano 337 373 13C-trans-clordano 188 258
2,4’-DDD 200 235 13C-2,4’-DDD 208 243 4,4’-DDD 200 235 13C-4,4’-DDD 212 247 2,4’-DDE 176 246 13C-2,4’-DDE 212 247 4,4’-DDE 176 246 13C-4,4’-DDE 188 258 2,4’-DDT 200 235 13C-2,4’-DDT 212 247 4,4’-DDT 200 235 13C-4,4’-DDT

ALLEGATO 3.1
Condizioni operative suggerite per la determinazione strumentale degli analiti di interesse Le condizioni di analisi gascromatografica sono ancora in fase di ottimizzazione.
� colonna gas cromatografia: RTX – 5sil MS (RESTEK) 30 m × 0.25 mm × 0.25 µm df, o di analoga selettività a basso spurgo per GC/MS;
� iniettore splitless con l’inlet alla temperatura di 40 °C; � programmata GC: 70 °C per 1 min, 10 °C/min fino a 180 °C, 180 °C per 3
min, 4 °C/min fino 220 °C, 220 °C per 3 min, 5 °C/min fino 270 °C, 270 °C per 10 min, 10 °C/min fino 300 °C, 300 °C per 2 min.

4. Polibromodifenil eteri (PBDE), policlorobifenili (PCB),
policlorodibenzodiossine (PCDD) e policlorodibenzofurani
(PCDF) in campioni di pesce e di molluschi bivalvi
Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione
Primaria, Reparto di Chimica Tossicologica
Avvertenze Il metodo proposto è basato su esperienze dirette (Bayarri et al., 2001; De Felip, 2003; di Domenico et al., 1997, 1998, 2002; Ingelido, 2004, 2007) e in accordo con il Metodo 1613 della US EPA (1994) sviluppato per la determinazione di PCDD e PCDF mediante HRGC-HRMS. Al riguardo, si fa presente quanto segue:
� a causa della complessità intrinseca delle analisi di cui trattasi, l’applicazione del metodo si basa sulla disponibilità di infrastrutture e strumentazione specialistiche e personale esperto o adeguatamente addestrato;
� prodotti consumabili o strumenti identificati commercialmente nel testo, e portati come esempi, possono essere sostituiti con prodotti o strumentazione equivalenti di differente origine;
� la tecnica della “diluizione isotopica” utilizzata nel metodo è descritta nel dettaglio nel Metodo 1613 della US EPA (1994): a esso si rinvia per adeguata informazione;
� i contaminanti in oggetto sono altamente tossici e idonee misure protettive devono essere presenti nel laboratorio per evitare contaminazioni ambientali e rischi occupazionali;
� a causa della fotolabilità osservata per alcuni dei PBDE in oggetto, particolari precauzioni devono essere prese dal laboratorio per evitare l’esposizione alla luce delle soluzioni standard e dei campioni.

Acronimi CRM materiale di riferimento certificato DL-PCB policlorobifenili diossina-simili EI electron impact FR fattore di risposta FRR fattore di risposta relativo GC gascromatografia HR alta risoluzione LOQ limite di quantificazione LR bassa risoluzione MS spettrometria di massa N noise, o rumore di fondo (N = 4 SDN) NDL-PCB policlorobifenili non diossina-simili PBDE polibromodifenileteri PCDD policlorodibenzodiossine PCDF policlorodibenzofurani SI standard interno SIM single ion monitoring TEF fattori di tossicità equivalente

Introduzione La classe dei polibromodifenileteri (PBDE), strutturalmente non molto dissimile da quella dei policlorobifenili (PCB) e dei polibromobifenili (PBB), comprende 209 congeneri che si differenziano per numero e posizione degli atomi di bromo. Ciascun congenere è numerato secondo il sistema IUPAC utilizzato per la numerazione dei PCB. In commercio esistono tre formulazioni, denominate Penta-BDE, Octa-BDE e Deca-BDE, prodotte per bromurazione dei difenileteri in presenza di un catalizzatore. Ciascuna di esse è costituita da miscele di prodotti con grado di bromurazione variabile. La famiglia dei policlorobifenili (PCB) è costituita da 209 congeneri, il cui substrato di base è il bifenile; esso può alloggiare da uno a 10 atomi di cloro. In relazione ai meccanismi propri dell’azione tossica dei PCB, si tende a dividere tali composti in due categorie (comunque coesistenti nelle miscele commerciali e nelle matrici ambientali): i 12 PCB diossina-simili (DL-PCB), con quattro posizioni orto (2, 2’, 6, 6’) libere (DL-PCB non-orto-sostituiti) o monoclorosostituite (DL-PCB mono-orto-sostituiti), e i PCB non diossina-simili (NDL-PCB). Policlorodibenzodiossine (PCDD) e policlorodibenzofurani (PCDF) sono termini che indicano due famiglie chimiche caratterizzate da elevata molteplicità: esistono 75 PCDD e 135 PCDF, per un totale di 210 congeneri suddivisi in otto gruppi omologhi per ciascuna famiglia, con grado di clorosostituzione da uno a otto (WHO, 1989; IARC, 1997). Con l’eccezione degli ottacloroderivati, tutti gli altri gruppi omologhi sono costituiti da più congeneri (isomeri posizionali). Per il loro potenziale tossicologico, in genere sono d’interesse analitico solo quei congeneri clorosostituiti alle posizioni C2, C3, C7, e C8, per un totale di 17 congeneri. PCDD+PCDF, insieme o separatamente, vengono comunemente chiamati “diossine”; il termine “diossina” è di massima riservato al congenere 2,3,7,8-T4CDD.

4.1 Scopo e applicazione 4.1.1 Il metodo è proposto per la quantificazione di PBDE, NDL-PCB, DL-PCB, e PCDD+PCDF in campioni omogeneizzati di pesce e di molluschi bivalvi mediante gascromatografia ad alta risoluzione-spettrometria di massa a bassa risoluzione (HRGC-LRMS) e gascromatografia ad alta risoluzione-spettrometria di massa ad alta risoluzione (HRGC-HRMS). 4.1.2 Il metodo consente la determinazione di a) 11 congeneri di PBDE appartenenti ai gruppi omologhi tetra- (T4), penta- (P5), esa- (H6), epta- (H7), otta- (O8), nona- (O9) e deca- (O10) bromosostituiti; b) 30 congeneri di NDL-PCB appartenenti ai gruppi omologhi tri- (T3), tetra- (T4), penta- (P5), esa- (H6), epta- (H7), e otta- (O8) clorosostituiti; c) 12 congeneri di DL-PCB appartenti ai gruppi omologhi tetra- (T4), penta- (P5), ed esa- (H6) clorosostituiti e comprendenti 8 congeneri mono-orto sostituiti e 4 congeneri a struttura coplanare; d) 17 congeneri tossici di PCDD+PCDF appartenenti ai gruppi omologhi tetra- (T4), penta- (P5), esa- (H6), epta- (H7), e otta- (O8) clorosostituiti. L’elenco dei singoli congeneri previsti dal presente metodo è riportato nelle Tabelle 4.1–4.4 4.2 Principio del metodo 4.2.1 La Figura 4.1 sintetizza le fasi di preparazione, estrazione, purificazione e quantificazione utilizzate nel presente metodo. 4.2.2 In accordo con il Metodo 1613 della US EPA (1994), il rilevamento degli analiti di interesse è basato sull’impiego estensivo di standard interni (SI), o traccianti, completamente marcati con 13C. Gli IS vengono aggiunti all’omogeneizzato prima di qualsiasi operazione analitica, ed eventualmente di passaggi analitici intermedi ritenuti critici. 4.2.3 La componente lipidica è estratta in Soxhlet con n-esano, così da rimuovere gli analiti altamente lipofili. 4.2.4 L’estratto è soggetto a una procedura di purificazione comprendente un trattamento con acido solforico concentrato per distruggere il carico organico co-estratto con gli analiti, seguito da passaggi su colonne cromatografiche in sequenza che permettono di concentrare gli analiti in forma purificata. 4.2.5 Dopo purificazione, il campione è quantificato contro standard esterni (SE, tanti quanti sono gli analiti da dosare) mediante HRGC-LRMS (SIM) per la determinazione di PBDE, di NDL-PCB, e di mono-orto DL-PCB e mediante HRGC-HRMS (SIM) per la determinazione di non-orto DL-PCB e PCDD+PCDF.

4.2.6 Si fa presente che, a causa dell’ampio intervallo di pesi molecolari presentati dai PBDE (fra 480 e 970 Da), la spettrometria di massa a elevata risoluzione rappresenta il sistema di rilevamento di elezione, garantendo elevata selettività e sensibilità anche per i congeneri esa–deca-sostituiti. Tuttavia, si ritiene che la determinazione congenere-specifica possa essere effettuata con successo anche in bassa risoluzione. Al riguardo, si nota che i più recenti spettrometri di massa quadrupolari coprono un range di massa fino a 1050 Da, così offrendo una potenziale alternativa all’uso di spettrometri ad alta risoluzione anche nella determinazione di PBDE con elevato grado di bromurazione. 4.2.7 Si evidenzia che il rilevamento strumentale dei PBDE è complicato dalla proprietà intrinseche di queste sostanze che presentano alti punti di ebollizione e moderata stabilità chimico-fisica (il congenere 209, per esempio, inizia a degradarsi a temperature prossime a 300 °C). Particolare attenzione deve essere posta, dunque, nella scelta del sistema d’iniezione e della colonna gascromatografica (Björklund, 2004). 4.2.8 A ciascun batch di campioni è abbinata l’analisi di un bianco procedurale per verificare l’eventuale contributo al fondo (background) procedurale. 4.2.9 Parallelamente alla quantificazione degli analiti di interesse una frazione dell’estratto, pari al 5 % in peso, viene utilizzata per la determinazione del contenuto lipidico. 4.3 Strumentazione 4.3.1 Strumentazione per l’estrazione del campione
� Estrattore Soxhlet in vetro da 250–1000 mL. 4.3.2 Strumentazione per la concentrazione del campione
� Evaporatore rotante; � apparato per la concentrazione sotto corrente di azoto.
4.3.3 Strumentazione per la purificazione del campione
� Colonna in vetro (Ø 2.5 cm) per Extrelut; � sistema di purificazione automatizzato (Power-PrepTM, Fluid Managemnet
Systems); � colonna in vetro (Ø 0.5 cm) per allumina; � palloni di vetro da 50, 100 e 250 mL.

4.3.4 Strumentazione per la determinazione strumentale del campione
� HRGC-HRMS, Waters VG Autospec; � HRGC-LRMS, Trace GC Ultra, Thermo Electron Corporation.
4.4 Reagenti e standard 4.4.1 Reagenti e materiale cromatografico. Le sostanze identificate con “*”
vengono scaldate a 110 °C per 12–24 h prima dell’uso.
� Acido solforico per analisi, concentrato (96 %); � azoto per gas cromatografia, ultrapuro; � bicarbonato di sodio, purezza ≥ 99.8 %; � Celite 545, granulometria 0.01–0.04 mm; Merck (Darmstadt, Germany);* � elio per gas cromatografia, ultrapuro; � Extrelut NT; Merck (Darmstadt, Germany); � gel di silice, per cromatografia su colonna, 70–230 mesh; diametro medio
dei pori 60 Å;* � ossido d’alluminio 90, per cromatografia su colonna, 70–230 mesh. Si
raccoglie per setacciatura la frazione di granulometria pari a circa 120–230 mesh, e si attiva in muffola a 500 °C per 12–24 h prima dell’uso. L’allumina si utilizza appena raffreddata; in alternativa, essa viene conservata protetta dall’umidità (l’efficienza della filtrazione cromatografica su allumina è particolarmente sensibile alle condizioni operative e richiede adeguata standardizzazione);
� kit di tre colonne (allumina, carbone e silice) per Power-Prep™; � sodio cloruro (per eventuale aggiunta al sodio solfato anidro per
aumentarne la permeabilità), purezza > 99.0 %;* � sodio solfato anidro per analisi, purezza ≥ 99.0 %*.
4.4.2 Solventi
� Acetone per HPLC, purezza > 98.0 %, acqua ≤ 0.1 %; � acetone grado tecnico; � diclorometano per HPLC, purezza 99.8 %; � n-esano grado tecnico; � n-esano per LC, purezza > 98.0 %; � etil acetato per HPLC, purezza 99.8 %; � iso-ottano per analisi strumentale (UV, IR, o HPLC), purezza 99.5 %; � metanolo per HPLC, purezza ≥ 99.9 %; � n-nonano per HPLC, purezza 99.7 %; � n-pentano per analisi, purezza ≥ 99.0 %; � n-tetradecano standard per GC, purezza ≥ 99.5 %; � toluene per analisi strumentale (UV, IR, o HPLC), purezza 99.5 %;

� tetracloruro di carbonio, purezza ≥ 99.5 %. 4.4.3 Standard
� Clordano 13C-marcato impiegato come standard d’iniezione nella determinazione di NDL-PCB, mono-orto DL-PCB, e PBDE, purezza 99 %;
� congeneri PBDE, standard naturali, purezza > 99 % (Wellington Laboratories);
� congeneri PBDE, standard 13C-marcati; purezza > 99 %; (Wellington Laboratories);
� congeneri PCB, standard naturali, purezza > 99 % (Cambridge Isotopic Laboratory (CIL));
� congeneri PCB, standard 13C-marcati, purezza > 99 % (Cambridge Isotopic Laboratory (CIL));
� congeneri PCDD+PCDF, standard naturali, purezza > 99 % (Cambridge Isotope Laboratories (CIL));
� congeneri PCDD+PCDF, standard completamente 13C-marcati, purezza > 99 %; (Cambridge Isotope Laboratories (CIL)). Questo gruppo di composti include tutti gli SI propriamente utilizzati come tali più due congeneri impiegati come standard d’iniezione (13C-1,2,3,7,8,9-H6CDF e 13C-1,2,3,6,7,8-H6CDD).
4.4.4 Campione controllo qualità (CQ). Il campione CQ più idoneo è rappresentato da un materiale di riferimento certificato (CRM) simile alla matrice analizzata, contenente gli analiti di interesse a concentrazioni note. In mancanza di CRM, il campione CQ potrebbe essere costituito da una soluzione standard degli analiti di interesse a concentrazione nota di origine diversa rispetto a quella utilizzata per la quantificazione. 4.5 Interferenze e cause d’errore 4.5.1 A causa delle minute quantità di analiti che devono essere identificate e quantificate, solo un laboratorio adeguatamente costruito ed equipaggiato può ritenersi idoneo all’esecuzione delle analisi d’interesse. All’interno del medesimo, dovrà essere mantenuta la massima pulizia, eventualmente in pressione positiva per evitare apporti di contaminazione atmosferica dall’esterno. Gli operatori, tutti esperti, dovranno attenersi rigorosamente ai protocolli di “buone pratiche” e alle procedure operative standard dedicate d’accreditamento in vigore nel laboratorio. 4.5.2 Poiché la vetreria, i solventi, i reagenti, e ogni tipo di strumentazione utilizzata durante l’analisi possono portare alla presenza di sostanze interferenti, è necessario dimostrare che tutti i materiali impiegati ne siano virtualmente privi.

4.5.3 La vetreria, tutta di Pyrex, viene preventivamente lavata con detersivo e acqua, risciacquata accuratamente con acqua distillata, e poi tenuta in stufa a 170 °C per 12–24 h (la vetreria tarata, di Classe A ove opportuno, non viene sottoposta al trattamento termico). Prima dell’uso, la vetreria è ulteriormente e ripetutamente risciacquata con acetone seguito da n-esano, e fatta asciugare in zona protetta. L’ultimo risciacquo della vetreria con n-esano viene raccolto quantitativamente e concentrato a piccolo volume — da 1/100 a 1/500 del volume di partenza — per verificarvi mediante rilevamento HRGC-LRMS(SIM) e HRGC-HRMS(SIM) l’eventuale presenza d’interferenze pregiudiziali sui segnali degli analiti d’interesse. Qualora questo background procedurale sia troppo elevato, occorre sottoporre la vetreria di cui trattasi a nuovo lavaggio oppure sostituirla. Dopo l’uso, se opportuno, la vetreria impiegata viene recuperata dopo decontaminazione con lavaggi ripetuti con miscela equivolumetrica di acetone e n-esano (eventualmente entrambi di grado tecnico), seguita dalla procedura di lavaggio precedentemente descritta. In una valutazione costo-beneficio, parte della vetreria usata può essere eliminata secondo le prassi di scarico dei rifiuti in atto nel laboratorio. 4.5.4 Tutti i solventi utilizzati in quantità relativamente rilevanti, che subiscono sensibili processi di concentrazione durante il percorso dell’analisi, devono essere sottoposti a un controllo di congruità analitica preventiva per verificarne l’eventuale contributo al background procedurale. Per tale accertamento, volumi operativi dei solventi d’interesse, presi singolarmente, vengono concentrati a piccolo volume secondo quanto si verifica nell’applicazione del metodo e sottoposti a rilevamento HRGC-LRMS(SIM) e HRGC-HRMS(SIM). Qualora questo background procedurale sia troppo elevato, occorre purificare il o i solventi inidonei, o effettuarne la sostituzione. 4.5.5 Tutti gli standard acquisiti da ditte specializzate devono essere certificati all’origine in merito alle identità dei congeneri forniti, al grado della loro purezza, e al loro titolo effettivo. In genere, è opportuno verificare tali parametri direttamente mediante rilevamento HRGC-LRMS(SIM) e HRGC-HRMS(SIM): qualora si rilevi un’incongruenza sensibile tra il dichiarato e il risultato del rilevamento, il prodotto dovrà essere sostituito. Le soluzioni di riferimento derivate dagli standard commerciali utilizzano solventi relativamente poco volatili (n-nonano, iso-ottano, o toluene) per le diluizioni: di esse se ne accerta la stabilità nel tempo. 4.5.6 I lipidi naturalmente presenti nel campione e co-estratti insieme agli analiti di interesse possono interferire con la loro determinazione. Pertanto, il contenuto lipidico deve essere ridotto o eliminato utilizzando la procedura di purificazione riportata.

4.6 Conservazione del campione 4.6.1 Il campione omogeneizzato è congelato a –20 °C, fino all’analisi. 4.7 Procedura analitica 4.7.1 Preparazione del campione 4.7.1.1 La dimensione del campione da sottoporre all’analisi è funzione dei livelli di contaminazione presunti e del contenuto lipidico della specie in esame. La Tabella 4.5 mostra per alcune specie di pesce e di molluschi la percentuale media di lipidi presenti nella parte edibile. 4.7.1.2 Sulla basse dei livelli di contaminazione presunti, è stata stimata per ciascuna classe di analiti la quantità di SI da addizionare al campione prima dell’estrazione (Tabella 4.6). Si fa presente che le quantità riportate sono indicative e possono variare in funzione della contaminazione presunta del campione. 4.7.1.3 Il campione omogeneizzato è pesato, mescolato con sodio solfato, in genere aggiunto in quantità doppia rispetto al campione, e addizionato con gli SI. Il campione così preparato è lasciato riposare per 12–24 h prima dell’estrazione. 4.7.2 Estrazione 4.7.2.1 L’estrazione è condotta in Soxhlet per 18–24 ore con n-esano (circa 150 ricicli). L’estratto è concentrato a pressione ridotta e pesato per prelevare una quantità pari al 5 % in peso da destinare alla determinazione del contenuto lipidico. Questo viene calcolato lasciando l’aliquota prelevata sotto leggero flusso di azoto fino a portarla a costanza di peso. 4.7.3 Purificazione 4.7.3.1 L’estratto esanico viene concentrato fino al volume di 5–10 mL ed è eluito con 350 mL di n-esano su colonna di Extrelut (12 g) impregnato con 30 mL di acido solforico concentrato (Figura 4.2) (De Felip et al., 1990, 1999). Se il carico organico è ancora visibilmente presente dopo il trattamento acido, il campione concentrato a piccolo volume viene eluito con 200 mL di n-esano o n-pentano su una colonna multistrato (Figura 4.2). L’eluato ottenuto dall’Extrelut o dalla colonna multistrato viene concentrato fino al volume di 4–5 mL e sottoposto a frazionamento mediante Power-PrepTM. Questo apparato, capace di prestazioni elevate, consente d’eseguire un cleanup automatizzato con un kit commerciale di tre colonne sequenziali impaccate, nell’ordine, con gel di silice, allumina, e

carbone. Il campione è eluito sequenzialmente con approssimativamente 80 mL di n-esano, 150 mL di una miscela 50:1 di n-esano-diclorometano (Frazione I, contenente i NDL-PCB e i mono-orto-DL-PCB), 190 mL di una miscela 1:1 di n-esano-diclorometano (Frazione II, contenente i PBDE), e in ultimo 70 mL di toluene a flusso invertito (Frazione III, contenente PCDD+PCDF e i non-orto-DL-PCB). Aggiunto 1 µL di n-tetradecano, le frazioni di interesse sono concentrate sotto azoto fin quasi a secchezza e poi riprese con 50–100 µL di iso-ottano contenente gli standard d’iniezione (clordano 13C-marcato per le Frazioni I e II, e 13C-1,2,3,7,8,9-H6CDF+13C-1,2,3,6,7,8-H6CDD per la Frazione III). 4.7.3.2 In alternativa all’uso del Power-PrepTM e qualora non fosse necessaria la determinazione dei PBDE, il frazionamento del campione può avvenire tramite allumina attivata (Figura 4.2). L’eluato, cautamente concentrato a piccolo volume (0.2–0.5 mL) viene frazionato usando come eluente, nell’ordine, circa 20 mL di n-pentano (Frazione 0), 20 mL di una miscela equivolumetrica di n-pentano-tetracloruro di carbonio (Frazione I, contenente i NDL-PCB), e 10 mL di diclorometano per PCDD+PCDF e DL-PCB (Frazione II) (De Felip et al., 1990, 1999). 4.7.4 Determinazione strumentale 4.7.4.1 I PBDE, i NDL-PCB, e i mono-orto DL-PCB sono determinati tramite HRGC-LRMS (EI, 35 eV) in modalità SIM utilizzando le condizioni operative descritte nell’Allegato 1. 4.7.4.2 I 17 congeneri di PCDD+PCDF e i 4 congeneri di non-orto-DL-PCB sono determinati mediante HRGC-HRMS VG Autospec (EI, 35 eV), operante alla risoluzione di 10000 in modalità SIM (le condizioni operative sono riportate nell’Allegato 1). 4.7.4.3 I congeneri nativi e quelli isotopicamente marcati sono quantificati utilizzando le due masse più intense dello spettro di massa nel caso dei PBDE (Tabella 4.7, Figura 4.3) e le due masse più intense di ciascun multipletto molecolare per PCB e PCDD+PCDF (Tabelle 4.8–10, Figura 4.4). 4.7.4.4 La quantificazione congenere-specifica viene eseguita impiegando una miscela dei congeneri naturali d’interesse e di quelli isotopicamente marcati a concentrazione nota (standard di quantificazione). Se una determinazione risulta interferita, essa può essere ripetuta dopo un ulteriore purificazione dell’estratto mediante Power-Prep™. 4.7.5 Calcoli e risultati 4.7.5.1 La quantificazione dei congeneri di interesse viene eseguita applicando l’Equazione 1, avendo determinato nello standard di quantificazione i fattori di

risposta (FR) e i fattori di risposta relativi (FRR). Questi vengono stimati dalle aree dei segnali relativi alle due masse più abbondanti dei congeneri di PBDE (Tabella 4.7) e, per PCB e PCDD+PCDF, mediante le aree dei segnali più intensi presenti nei multipletti corrispondenti agli ioni molecolari dei vari omologhi.
SI
SI
AN
ANAN A
Q
FRR
AX ×= (1)
dove XAN è la quantità assoluta dell’analita nel campione; AAN è l’area del segnale analitico nel campione; QSI è la quantità assoluta dello SI 13C-marcato; ASI e l’area corrispondente al segnale dello IS 13C-marcato; FRRAN è il fattore di risposta relativo calcolato mediante l’Equazione (2):
SI
ANAN FR
FRFRR = (2)
dove FRAN e FRSI sono rispettivamente i fattori di risposta determinati per l’analita e per lo standard interno nello standard di quantificazione utilizzando l’equazione (3):
i
ii Q
AFR = (3)
dove Ai è l’area del segnale analitico dell’analita o dello SI 13C-marcato nello standard di quantificazione; Qi è la quantità assoluta dell’analita o dello SI 13C-marcato nello standard di quantificazione; 4.7.5.2 La determinazione del recupero (%) dei congeneri marcati viene effettuata mediante l’Equazione (4):
100(%) cuperoRe ×=SI
SI
Q
X (4)
dove QSI è la quantità assoluta di standard interno aggiunta al campione;

XSI è la quantità assoluta di standard interno determinata nel campione utilizzando l’Equazione (5):
ING
ING
SI
SISI A
Q
FRR
AX ×= (5)
dove ASI è l’area del segnale relativo allo standard interno nel campione; FRRSI è il fattore di risposta relativo dello standard interno rispetto allo standard di iniezione determinato nella soluzione calibrante; QING è la quantità assoluta di standard di iniezione nel campione; AING è l’area del segnale relativo allo standard di iniezione nel campione. 4.7.5.3 I risultati analitici relativi a PBDE e NDL-PCB sono generalmente espressi in ng/g ww (peso fresco). Al fine di comparare il risultato analitico con altri dati di letteratura, può essere opportuno riportare la concentrazione stimata anche in termini di ng/g grasso, previa determinazione del contenuto di grasso presente nel campione di partenza (cfr. Punto 4.7.2). 4.7.5.4 Come previsto dalla normativa attualmente vigente (Regolamento 1881/2006/CE e 1883/2006/CE), i risultati analitici relativi a PCDD+PCDF e DL-PCB sono espressi in pg/g ww (peso fresco), e poi convertiti in unità TE, o TEQ, mediante opportuno sistema TEF (Tabelle 11 e 12). 4.7.5.5 I referti cumulativi in unità TE riportati per PCDD+PCDF, e DL-PCB vengono calcolati con approccio upper bound: Nel calcolo, quei congeneri non determinabili poiché al disotto del limite di determinazione (<LD), sono di fatto inseriti ponendone la concentrazione uguale al valore del limite (LD × 1). In accordo con la normativa vigente (Regolamento 1183/06/CE), in prossimità del limite di legge la differenza fra i valori espressi in upper e lower bound non deve superare il 20 %. 4.7.5.6 Salvo esigenza specifica, i referti cumulativi riportati per PBDE e NDL-PCB vengono calcolati con approccio upper bound: nel calcolo, quei congeneri non determinabili poiché al disotto del limite di determinazione (<LD) sono di fatto inseriti ponendone la concentrazione uguale al valore del limite (LD × 1). 4.8 Verifica della qualità dei dati 4.8.1 La verifica della qualità dei dati è condotta attraverso un programma di assicurazione di qualità i cui requisiti sono riportati di seguito.

4.8.2 RIPRODUCIBILITÀ INTRA-LABORATORIO DEL METODO Questo controllo è eseguito con misurazioni indipendenti, congenere-specifiche, ripetute su una medesima matrice omogenea predisposta dal laboratorio (blank fortificato). Devianze accettate sui singoli congeneri o sui valori cumulativi (analitici o TEQ), <|±20 %|. Valori al difuori di tale intervallo comportano una specifica valutazione di congruenza e una conseguente azione a livello operativo. 4.8.3 ACCURATEZZA INTRA-LABORATORIO DEL METODO Questo controllo è eseguito con misurazioni congenere-specifiche ripetute su appropriati materiali di riferimento (CRM) o, in loro mancanza, su blanks propriamente fortificati nel laboratorio. Devianze accettate sui singoli congeneri o sui valori cumulativi (analitici o TEQ), <|±30 %|. Valori al difuori di tale intervallo comportano una specifica valutazione di congruenza e una conseguente azione a livello operativo. 4.8.4 RECUPERO DI STANDARD INTERNI MARCATI Sono accettabili recuperi compresi nel range 60–120 %. Valori al di fuori di tale intervallo comportano una specifica valutazione di congruenza, ed eventualmente il rigetto e la ripetizione dell’analisi. 4.8.5 LIMITE DI DETERMINAZIONE DEL METODO (LD) La determinazione del LD viene eseguita sul campione reale, calcolando per ciascun congenere la concentrazione che produce una risposta strumentale pari a quattro volte il noise (N), con N ≈ 3–4 SDN (SDN, deviazione standard associata al noise). 4.8.6 CONTAMINAZIONE LEGATA AD INTERFERENZE NEI BIANCHI PROCEDURALI A ciascun batch di campioni è abbinato un bianco procedurale il cui contributo potrebbe influenzare il valore di un segnale analitico (X). Il seguente sistema di contrassegni o flags viene utilizzato nel laboratorio per qualificare e gestire il contributo di XB nei registri interni ed eventualmente nei rapporti di prova:
� XB / X ≤ 0.05: l’interferenza è considerate “trascurabile” → nessuna correzione;
� 0.05 < XB / X ≤ 0.25: l’interferenza è considerata “modesta” → nessuna correzione (flag, o contrassegno su X, “*”);
� 0.25 < XB / X ≤ 0.75: l’interferenza è considerata “sensibile” → correzione richiesta: X[valore corretto] = X – XB (flag “**”);
� 0.75 < XB / X ≤ 1.25: l’interferenza è considerate “marcata” → il segnale analitico è paragonabile al LD e sostituito da “<LD” (flag “***”);
� 1.25 < XB / X: l’interferenza è considerata “eccessiva” e non correggibile → il valore di X è sostituito da ND (“non determinabile”).
4.8.7 RIPETIBILITÀ DEI TEMPI DI RITENZIONE RELATIVI Livelli di devianza accettati, <|±0.1 %|.

4.8.8 RISOLUZIONE GAS CROMATOGRAFICA I picchi dei congeneri d’interesse devono essere risolti: sovrapposizione di uno o più componenti adiacenti, <10 %. 4.8.9 RIPETIBILITÀ DEI FATTORI DI RISPOSTA RELATIVI (RRF) Livelli di devianza accettati per (RFANALYTE) × (RF13C-IS)–1, <|±15 %| (con segnali >LD). 4.8.10 CORRETTEZZA DEL RAPPORTO DELLE MASSE ISOTOPICHE CRITICHE Le masse ioniche (es., M and M+2) campionate con tecnica SIM per identificazione/quantificazione dovrebbero fornire il previsto rapporto (teorico) (es., [M] × [M+2]–1) o un rapporto simile a quello dello standard. Livelli accettati di devianza dai livelli attesi, <|±20 %|. 4.8.11 QUANTIFICAZIONI PARALLELE BASATE SU DUE MASSE ISOTOPICHE Quantificazioni con due diverse masse isotopiche (es., M and M+2) dovrebbero differire <|±20 %|. 4.8.12 CONTROLLO DELLA LINEARITÀ DI RISPOSTA La linearità di risposta è controllata regolarmente con soluzioni standard di PBDE, PCDD+PCDF e di PCB su un intervallo di circa due ordini di grandezza, che includono i valori LD. Gli accertamenti sono eseguiti utilizzando generalmente i congeneri più importanti (es., 2,3,7,8-T4CDD, 1,2,3,7,8-P5CDD, 2,3,4,7,8-P5CDF). Per i congeneri più tossici (TEF più elevati), i LD strumentali devono essere compatibili con una quantità iniettata di circa 50 fg per i congeneri prioritari. 4.8.13 RIPRODUCIBILITÀ INTRA-LABORATORIO NEL TEMPO Questo controllo è basato sulla ripetizione di determinazioni eseguite a intervalli di tempo (es., semestrale) su una matrice omogenea stabile idonea. Devianze sui singoli congeneri o sui valori cumulativi (analitici o TEQ), a differenti momenti temporali, maggiori di |±30 %| comportano una specifica valutazione di congruenza e una conseguente azione a livello operativo. 4.8.14 RIPRODUCIBILITÀ E ACCURATEZZA INTER-LABORATORIO Valutazione basata sulle determinazioni dei congeneri d’interesse, indipendenti e ripetute su matrici omogenee e CRM distribuite da un ente organizzatore. I criteri di valutazione/accettazione in funzione del sistema di controllo utilizzato dall’ente organizzatore. 4.9 Esempi di stima dell’incertezza 4.9.1 La valutazione dell’affidabilità analitica si basa principalmente sulle numerose condizioni per l’assicurazione di qualità contenute nei protocolli analitici (cfr. Punti 4.8.2–4.8.14). Una stima dell’incertezza sulla singola determinazione per i congeneri individuali degli analiti di interesse o per i risultati cumulativi (analitici o TEQ nel caso di PCDD+PCDF e DL-PCB) può essere comunque eseguita in accordo con le indicazioni fornita da EURACHEM/CITAC (2000).

4.9.2 Come detto, severe condizioni per l’assicurazione di qualità devono essere osservate nell’esecuzione dei protocolli d’interesse, e queste condizioni già forniscono una base per conseguire una diminuzione dell’incertezza. A tale scopo, è di particolare efficacia l’impiego di numerosi SI 13C-marcati, aggiunti alla matrice per l’analisi all’inizio del percorso procedurale ed eventualmente anche durante fasi intermedie del medesimo. Giova precisare che, negli esempi che seguono, i campi di misurazione pei i vari congeneri sono, come appropriato, al disopra degli specifici LD — per la massa di quantificazione o più intensa di ciascun cluster molecolare, si suggerisce: S ≈ 4 N ed N ≈ 3–4 SDN (dove S rappresenta l’intensità del segnale dell’analita ed N (noise) il rumore effettivo del fondo (background) in sua prossimità) — condizione che consente di avere una più regolare e sostanzialmente piatta ripetibilità di misurazione strumentale. 4.9.3 A titolo di esempio si riporta la stima dell’incertezza applicata per PCDD+PCDF. Una trattazione analoga può essere condotta per stimare l’incertezza per le altre classi di composti prese in considerazione dal presente metodo. 4.9.4 STIMA DELL’INCERTEZZA DEI SINGOLI CONGENERI DI PCDD+PCDF La relazioni-chiave cui può farsi riferimento per la stima di cui trattasi sono (cfr. Punto 4.7.5):
SI
SI
AN
ANAN A
Q
FRR
AX ×=
SI
ANAN FR
FRFRR =
L’incertezza standard combinata uc(y) per modelli contenenti moltiplicazioni e/o divisioni può essere espressa come
⋅⋅⋅++=22
)()()(
q
qu
p
puyyuc
dove u(p)/p, u(q)/q, etc., sono le incertezze sui parametri espresse come deviazioni standard relative (RSD), o nella forma normalizzata
⋅⋅⋅+
+
=22
)()()(q
qu
p
pu
y
yuc

Ai fini dell’esempio in considerazione, e con riferimento ai pertinenti protocolli, possono assumersi le seguenti incertezze sui parametri d’interesse, espresse come quantità RSD:
� %10)( ±=ANARSD , valore medio per congenere;
� %5)( ±=ANFRRRSD , devianza media;
� %5.2)( ±=SIQRSD , devianza massima basata sulle operazioni di
fortificazione (spiking); � %10)( ±=SIARSD , valore medio per PCDD+PCDF.
Introducendo i valori sopra riportati nell’equazione normalizzata, si ottiene la seguente stima conservativa dell’incertezza:
15.0)1.0()025.0()05.0()1.0()( 2222 =+++≤
AN
ANc
X
Xu
Utilizzando un fattore di copertura k = 2 per una distribuzione normale di misure e un livello di fiducia P = 95 %, l’incertezza estesa è stimata <│±30 %│. 4.9.5 STIMA DELL’INCERTEZZA DEI CONGENERI COMBINATI DI PCDD+PCDF L’incertezza standard combinata uc(y) per modelli contenenti somme e/o sottrazioni può essere espressa come
...)()(,...)),(( 22 ++= qupuqpyuc
dove u(p), u(q), etc., sono le incertezze sui parametri espresse come deviazioni standard (SD). Quando i livelli dei congeneri di PCDD+PCDF sono sommati, è agevole osservare come l’incertezza finale totale (espressa come RSDT) sia inferiore alle singole RSD di congenere. Indipendentemente dalla marcata variabilità relativa dei congeneri rilevabile nelle misurazioni reali, si assuma che i 17 PCDD+PCDF abbiano simili concentrazioni e SD (es., 100 ± 20 pg/g, ovvero RSD = │±20 %│). La quantità totale sarà 1700 pg/g, mentre l’incertezza totale sarà:
82)20(17,...)),(( 2 ≅+=qpXu ANc
Il risultato finale sarà riportato come 1700 ± 82 pg/g: il valore 82 è approssimativamente il 5 % (RSDT) del risultato cumulativo. In diversi casi, alcuni congeneri predominano sugli altri a causa delle loro maggiori concentrazioni

analitiche e/o TEQ. Peraltro, i valori piccoli di alcuni TEF (es., quelli per O8CDD e O8CDF) possono rendere trascurabili i contributi dei corrispondenti congeneri ai valori TEQ cumulativi e alle relative incertezze. In questo caso, l’incertezza totale sarà determinata primariamente dai congeneri predominanti fino a rendere trascurabili i contributi meno importanti. Riferimenti bibliografici Bayarri S., Turrio Baldassarri L., Iacovella N., Ferrara F., di Domenico A. (2001). PCDDs, PCDFs, PCBs, and DDE in edile marine species from Adriatic sea. Chemosphere 43, 601−610. Björklund J., Tollbäck P., Hiärne C., Dyremark E., Östman C. (2004). Influence of the injection techinique and the column system on gas chromatographic determination of polybrominated diphenyl ethers. Journal Chromatograpy A 1041, 201−210. De Felip E., di Domenico A., Turrio L., Volpi F., Merli F. (1990). Analytical approaches and criteria to define environmental contamination from an accidental polychlorobiphenyl (PCB) spillage. Toxicological and Environmental Chemistry 29, 37–46. De Felip E., di Domenico A., Falleni M., Ferri F., Iacovella N., Menale G., Tafani P., Tommasino G., Turrio Baldassarri L. (1994). Polychlorodibenzodioxin and polychlorodibenzofuran levels in dielectric fluids containing polychlorobiphenyls. Toxicological and Environmental Chemistry 46, 239−260. De Felip E., Miniero R. (1999). Procedimenti analitici adottati per il rilevamento di microcontaminanti in sedimenti lagunari. ISTISAN 99/28. A report of the Istituto Superiore di Sanità, Rome (Italy). De Felip E., Päpke O., Ingelido A.M., Hermann T., Cardelli M., Porpora M.G., di Domenico A. (2003). PBDE levels in Italian nulliparous women of reproductive age. Organohalogenon Compounds 61, 287−290. di Domenico A., Turrio Baldassarri L., Ziemacki G., De Felip E., Ferri F., Iacovella N., La Rocca C., Rodriguez F., Volpi F., D’Agostino O., Sansoni R (1997). Selected carcinogenic organic microcontaminants and heavy metals in the Venice lagoon. II. Contamination levels of biota samples. Organohalogen Compounds 34, 61–66. di Domenico A., Turrio Baldassarri L., Ziemacki G., De Felip E., La Rocca C., Ferrari G., Cardelli M., Volpi F., Ferri F., Iacovella N., Lupi C., Rodriguez F., D’Agostino O.,

Sansoni R., Settimo G. (1998). Priority microcontaminats in biota samples from the Venice lagoon: a selection of concentration data and elements of risk analysis. Organohalogen Compounds 39, 199–204. di Domenico A., Biagini G., De Felip E., Dellatte E., Ferri F., Ingelido A.M., Miniero R. (2002). Laguna Veneta: Scenari di rischio per la salute umana associati alla introduzione e alla presenza di microcontaminanti chimici ad alto potenziale tossico (PR-27/IS) — Rapporto Finale, Sezione II: Sottoprogramma “Latte Umano”. Istituto Superiore di Sanità (Roma). EURACHEM/CITAC (2000). Quantifying uncertainty in analytical measurement. CITAC Guide CG 4, second edition, Paragraphs 7–8. QUAM:2000.1. Co-operation on International Traceability in Analyrical Chemistry (Pymble). IARC (1997). Polychlorinated dibenzo-para-dioxins and polychlorinated dibenzofurans. Monographs on the Evaluation of Carcinogenic Risks to Humans 69, International Agency for Research on Cancer, World Health Organization (Lyon). Ingelido A.M., di Domenico A., Ballard T., De Felip E., Dellatte E., Ferri F., Fulgenzi A.R., Herrmann T., Iacovella N., Miniero R., Papke O., Porpora M.G. (2004). Levels of polybrominated diphenyl ethers in milk from Italian women living in Rome and Venice. Organohalogenon Compounds 66, 2689−2694. Ingelido A.M., di Domenico A., Ballard T., De Felip E., Dellatte E., Ferri F., Fulgenzi A.R., Herrmann T., Iacovella N., Miniero R., Papke O., Porpora M.G. (2007). Polychlorobiphenyl (PCBs) and polybromominated diphenyl ethers (PBDEs) in milk from Italian women living in Rome and Venice. Chemosphere 67, S301−S306. NATO/CCMS (1988). International toxicity equivalency factor (I-TEF) method of risk assessment for complex mixtures of dioxins and related compounds. Report Number 176, Committee on the Challenges of Modern Society, North Atlantic Treaty Organization. Unione Europea (2006). Raccomandazione della Commissione del 6 Febbraio 2006 relativa alla riduzione della presenza di diossine, furani e PCB nei mangimi e negli alimenti. Gazzetta Ufficiale dell’Unione Europea del 14 febbraio 2006. US EPA (1994). Method 1613B. Tetra- through octachlorinated dioxins and furans by isotope dilution HRGC-HRMS. Engineering and Analysis Division (4303), Office of Water, US Environmental Protection Agency (Washington). Van den Berg M., Birnbaum L., Bosveld A.T., Brunstrom B., Cook P., Feeley M., Giesy J.P., Hanberg A., Hasegawa R., Kennedy S.W., Kubiak T., Larsen J.C., van Leeuwen F.X., Liem A.K., Nolt C., Peterson R.E., Poellinger L., Safe S., Schrenk D.,

Tillitt D., Tysklind M., Younes M., Waern F., & Zacharewski T. (1998). Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environmental Health and Perspectives 106, 775−92. WHO (1989). Polychlorinated dibenzo-p-dioxins and dibenzofurans. Environmental Health Criteria 88. International Programme on Chemical Safety, World Health Organization (Geneva). WHO (1995). Second workshop on reliable evaluation of low level contamination of food. WHO/GEMS/Food-EURO, May 26–27, Kulmbach (Germany).

Tabella 4.1. Congeneri di PBDE rilevanti ai fini degli accertamenti d’interesse.
Congenere No. CAS
2,4,4’-T3BDE [28] 2,2’,4,4’-T4BDE [47] 5436-43-1 2,2’,3,4,4’-P5BDE [85] 182346-21-0 2,2’,4,4’,5-P5BDE [99] 60348-60-9 2,2’,4,4’,6-P5BDE [100] 189084-64-8 2,2’,4,4’,5,5’-H6BDE [153] 68631-49-2 2,2’,4,4’,5,6’-H6BDE [154] 207122-15-4 2,2’,3,4,4’,5’,6’-H7BDE [183] 2,2’,3,3’,4,4’,6,6’-O8BDE [197] 2,2’,3,4,4’,5,5’,6-N9BDE [206] D10BDE [209] 1163-19-5
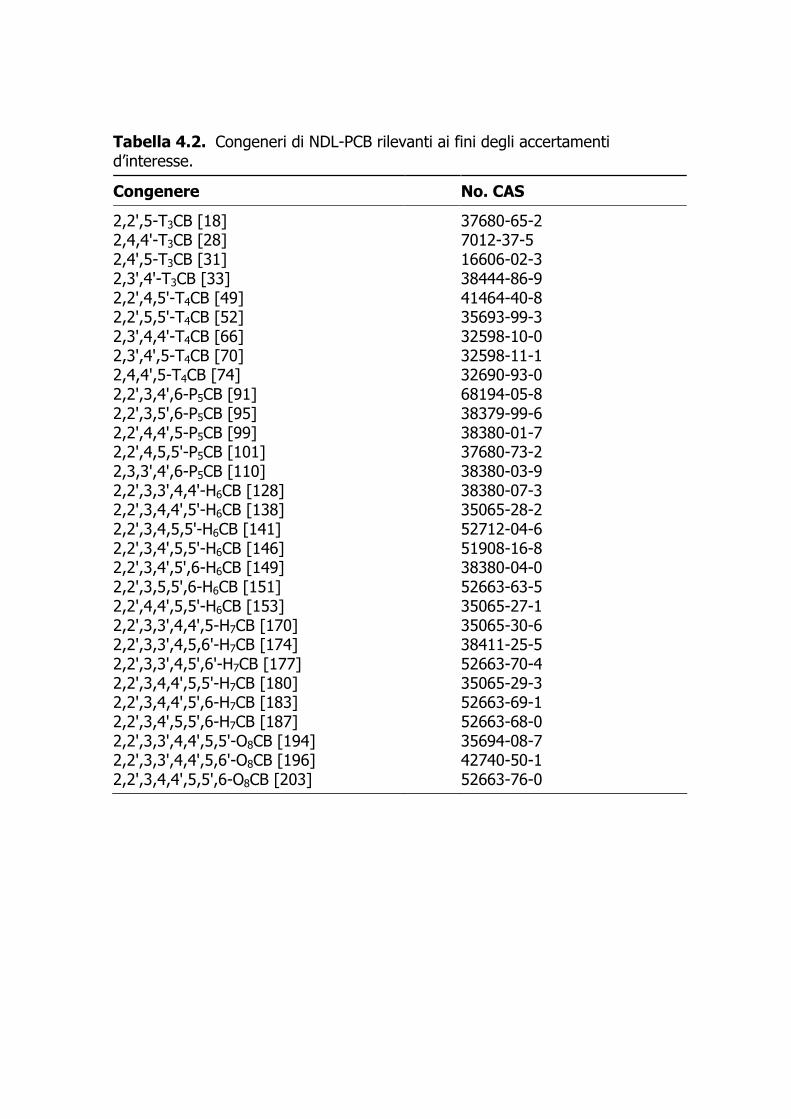
Tabella 4.2. Congeneri di NDL-PCB rilevanti ai fini degli accertamenti d’interesse.
Congenere No. CAS
2,2',5-T3CB [18] 37680-65-2 2,4,4'-T3CB [28] 7012-37-5 2,4',5-T3CB [31] 16606-02-3 2,3',4'-T3CB [33] 38444-86-9 2,2',4,5'-T4CB [49] 41464-40-8 2,2',5,5'-T4CB [52] 35693-99-3 2,3',4,4'-T4CB [66] 32598-10-0 2,3',4',5-T4CB [70] 32598-11-1 2,4,4',5-T4CB [74] 32690-93-0 2,2',3,4',6-P5CB [91] 68194-05-8 2,2',3,5',6-P5CB [95] 38379-99-6 2,2',4,4',5-P5CB [99] 38380-01-7 2,2',4,5,5'-P5CB [101] 37680-73-2 2,3,3',4',6-P5CB [110] 38380-03-9 2,2',3,3',4,4'-H6CB [128] 38380-07-3 2,2',3,4,4',5'-H6CB [138] 35065-28-2 2,2',3,4,5,5'-H6CB [141] 52712-04-6 2,2',3,4',5,5'-H6CB [146] 51908-16-8 2,2',3,4',5',6-H6CB [149] 38380-04-0 2,2',3,5,5',6-H6CB [151] 52663-63-5 2,2',4,4',5,5'-H6CB [153] 35065-27-1 2,2',3,3',4,4',5-H7CB [170] 35065-30-6 2,2',3,3',4,5,6'-H7CB [174] 38411-25-5 2,2',3,3',4,5',6'-H7CB [177] 52663-70-4 2,2',3,4,4',5,5'-H7CB [180] 35065-29-3 2,2',3,4,4',5',6-H7CB [183] 52663-69-1 2,2',3,4',5,5',6-H7CB [187] 52663-68-0 2,2',3,3',4,4',5,5'-O8CB [194] 35694-08-7 2,2',3,3',4,4',5,6'-O8CB [196] 42740-50-1 2,2',3,4,4',5,5',6-O8CB [203] 52663-76-0

Tabella 4.3. Congeneri di DL-PCB rilevanti ai fini degli accertamenti d’interesse.
Congenere No. CAS
3,3’,4,4’-T4CB [77] (a) 32598-13-3 3,3’,4’,5-T4CB [81] (a) 70362-50-4 2,3,3',4,4'-P5CB [105] (b) 32598-14-4 2,3,4,4',5-P5CB [114] (b) 74472-37-0 2,3',4,4',5-P5CB [118] (b) 31508-00-6 2,3',4,4',5'-P5CB [123] (b) 65510-44-3 3,3’,4,4’,5-H5CB [126] (a) 57465-28-8 2,3,3',4,4',5-H6CB [156] (b) 38380-08-4 2,3,3',4,4',5'-H6CB [157] (b) 69782-90-7 2,3',4,4',5,5'-H6CB [167] (b) 52663-72-6 3,3’,4,4’,5,5’-H6CB [169] (a) 32774-16-6 2,3,3',4,4',5,5'-H7CB [189] (b) 39635-31-9
(a) DL-PCB non-orto sostituiti (coplanari). (b) DL-PCB mono-orto sostituiti.

Tabella 4.4. Congeneri di PCDD+PCDF rilevanti ai fini degli accertamenti d’interesse.
Congenere No. CAS Congenere No. CAS
2,3,7,8-T4CDD 1746-01-6 2,3,7,8-T4CDF 51207-31-9 1,2,3,7,8-P5CDD 40321-76-4 1,2,3,7,8-P5CDF 57117-41-6 2,3,4,7,8-P5CDF 57117-31-4 1,2,3,4,7,8-H6CDD 39227-28-6 1,2,3,4,7,8-H6CDF 70648-26-9 1,2,3,6,7,8-H6CDD 57653-85-7 1,2,3,6,7,8-H6CDF 57117-44-9 1,2,3,7,8,9-H6CDD 19408-74-3 1,2,3,7,8,9-H6CDF 72918-21-9 2,3,4,6,7,8-H6CDF 60851-34-5 1,2,3,4,6,7,8-H7CDD 35822-46-9 1,2,3,4,6,7,8-H7CDF 67562-39-4 1,2,3,4,7,8,9-H7CDF 55673-89-7 1,2,3,4,6,7,8,9-O8CDD 3268-87-9 1,2,3,4,6,7,8,9-O8CDF 39001-02-0

Tabella 4.5. Contenuto lipidico medio presente nella parte edibile di alcune specie di pesce e di molluschi.
SPECIE Lipidi (%)
Sardine fresche 15.4 Orata fresca d'allevamento, filetti 8.4 Tonno, fresco 8.1 Spigola d'allevamento, filetti 6.8 Sarda fresca 4.5 Trota iridea d'allevamento, filetti 4.1 Orata fresca, filetti 3.8
Alto contenuto
lipidico (> 4 %
)
Halibut 3.5 Trota 3.0 Cozza o mitilo 2.7 Acciuga o alice, fresca 2.6 Vongola 2.5 Trota, surgelata 2.3 Calamaro, fresco 1.7 Calamaro, surgelato 1.5 Spigola 1.5 Sogliola, fresca 1.4 Sogliola, surgelata 1.3
Medio contenuto
lipidico
(1 – 4 %
)
Orata, surgelata 1.2 Merluzzo o nasello, surgelato
0.6
Merluzzo o nasello, surgelato, filetti
0.6
Basso
contenuto
lipidico (> 1
Merluzzo o nasello, Crudo
0.3

Tabella 4.6. Quantità per singolo congenere 13C-sostituito da utilizzare come SI prima dell’estrazione del campione omogeneizzato.
PBDE 13C-sostituito
(ng)
NDL-PCB 13C-sostituito
(ng)
DL-PCB 13C-sostituito
(ng)
PCDD+PCDF 13C-sostituito (ng)
2.5 – 12 (a) 10 (a) 0.4 (a) 0.05 – 0.35 (a)
(a) Le quantità riportate sono indicative e calcolate considerando di estrarre circa 1 g di grasso.
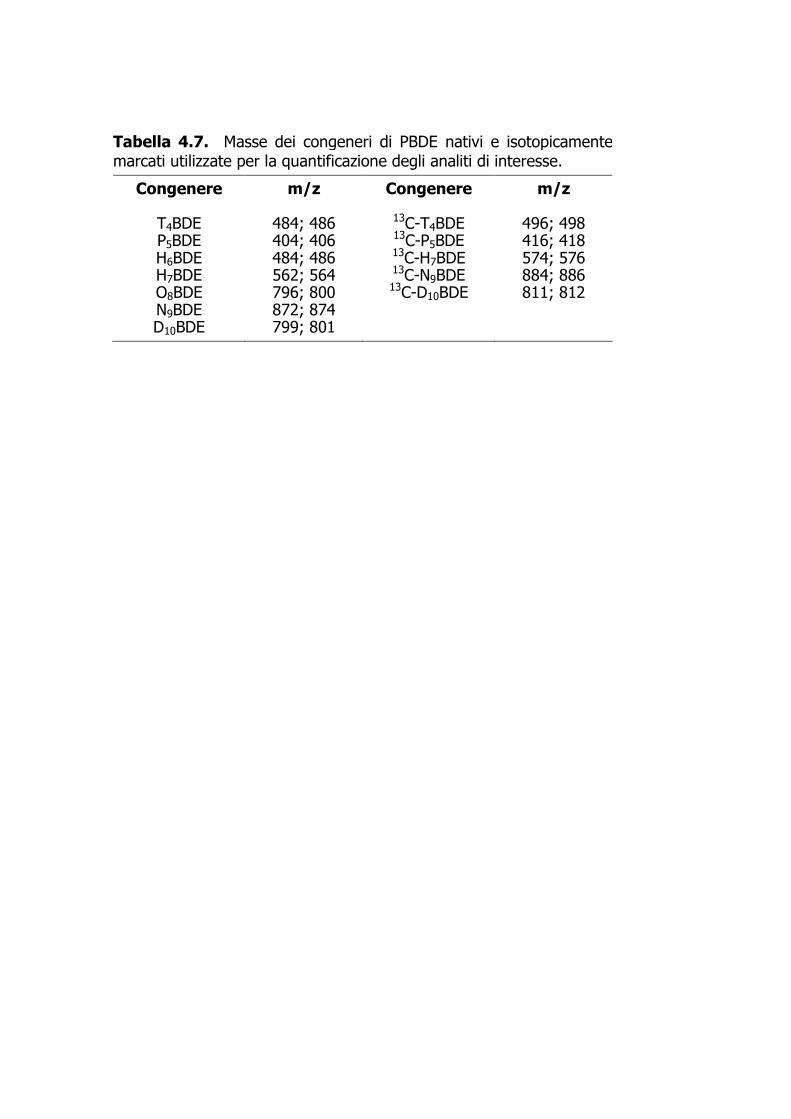
Tabella 4.7. Masse dei congeneri di PBDE nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Congenere m/z Congenere m/z
T4BDE 484; 486 13C-T4BDE 496; 498 P5BDE 404; 406 13C-P5BDE 416; 418 H6BDE 484; 486 13C-H7BDE 574; 576 H7BDE 562; 564 13C-N9BDE 884; 886 O8BDE 796; 800 13C-D10BDE 811; 812 N9BDE 872; 874 D10BDE 799; 801
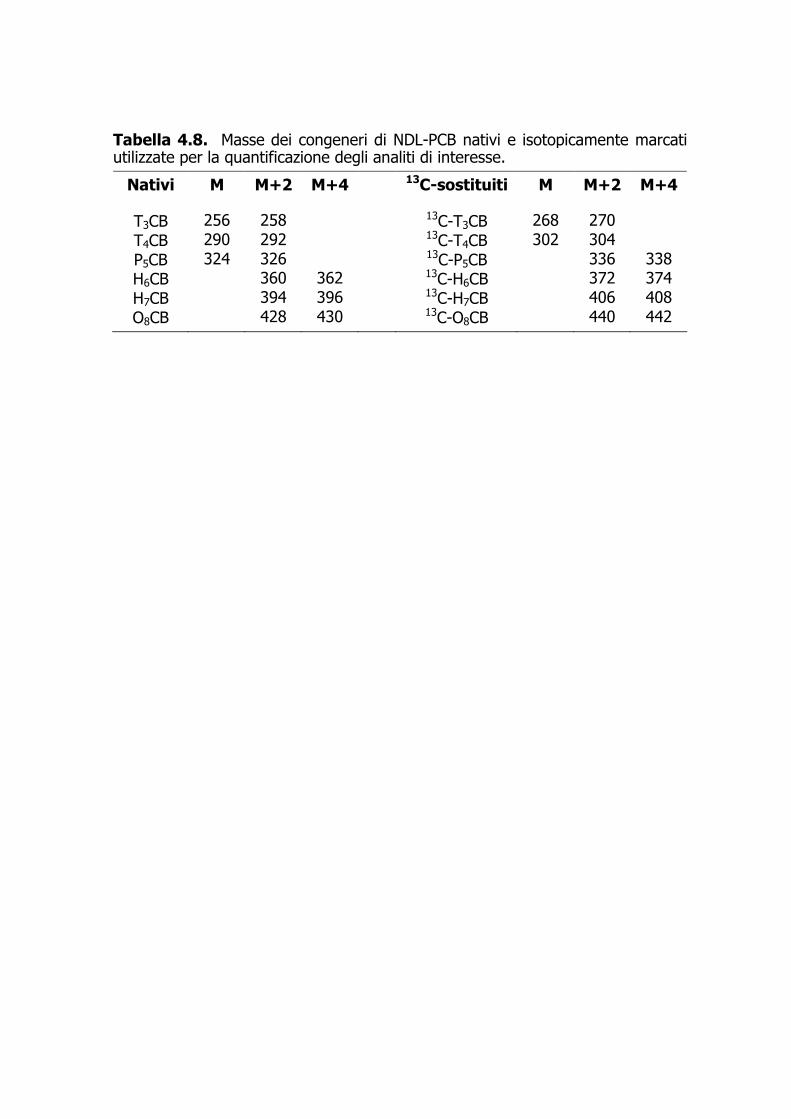
Tabella 4.8. Masse dei congeneri di NDL-PCB nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Nativi M M+2 M+4 13C-sostituiti M M+2 M+4
T3CB 256 258 13C-T3CB 268 270 T4CB 290 292 13C-T4CB 302 304 P5CB 324 326 13C-P5CB 336 338 H6CB 360 362 13C-H6CB 372 374 H7CB 394 396 13C-H7CB 406 408 O8CB 428 430 13C-O8CB 440 442
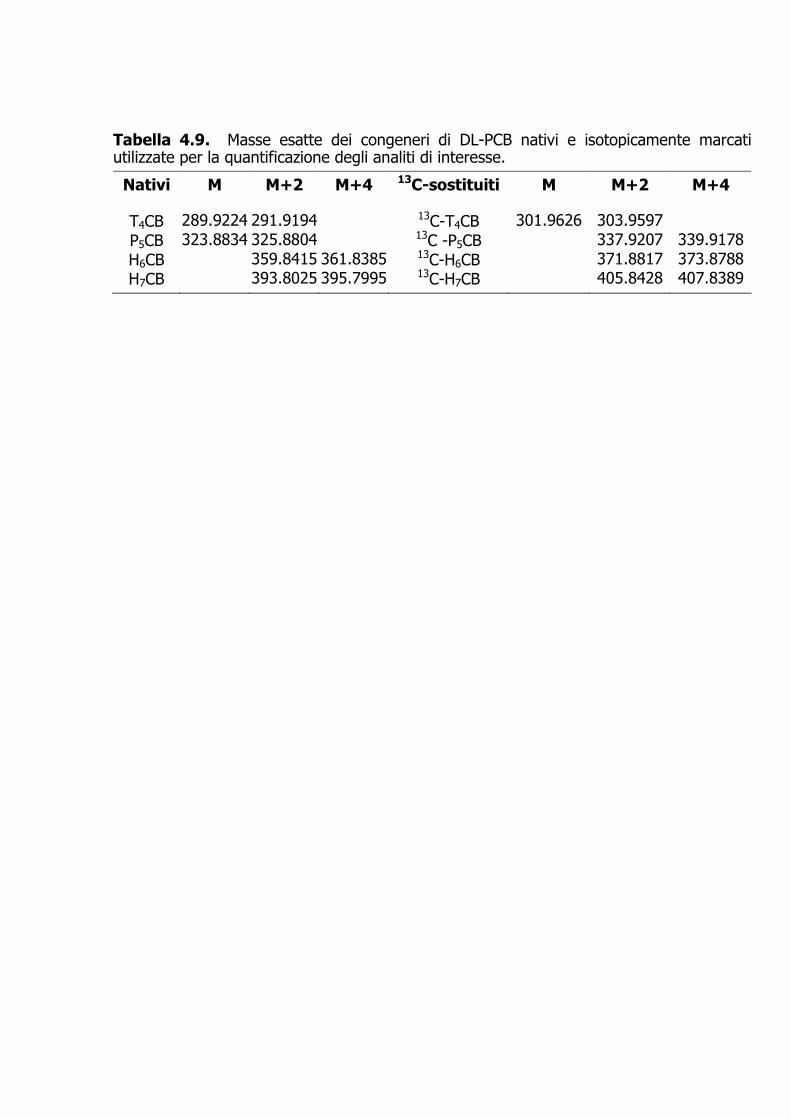
Tabella 4.9. Masse esatte dei congeneri di DL-PCB nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Nativi M M+2 M+4 13C-sostituiti M M+2 M+4
T4CB 289.9224 291.9194 13C-T4CB 301.9626 303.9597 P5CB 323.8834 325.8804 13C -P5CB 337.9207 339.9178 H6CB 359.8415 361.8385 13C-H6CB 371.8817 373.8788 H7CB 393.8025 395.7995 13C-H7CB 405.8428 407.8389
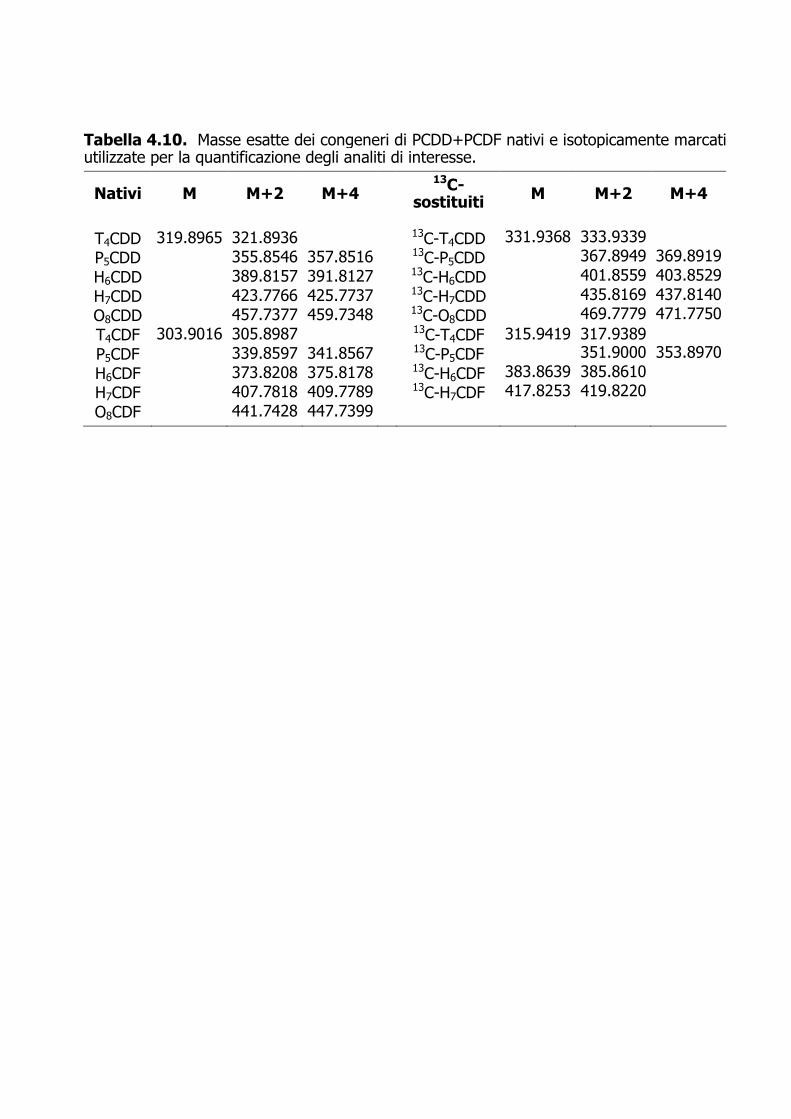
Tabella 4.10. Masse esatte dei congeneri di PCDD+PCDF nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Nativi M M+2 M+4 13C-
sostituiti M M+2 M+4
T4CDD 319.8965 321.8936 13C-T4CDD 331.9368 333.9339 P5CDD 355.8546 357.8516 13C-P5CDD 367.8949 369.8919 H6CDD 389.8157 391.8127 13C-H6CDD 401.8559 403.8529 H7CDD 423.7766 425.7737 13C-H7CDD 435.8169 437.8140 O8CDD 457.7377 459.7348 13C-O8CDD 469.7779 471.7750 T4CDF 303.9016 305.8987 13C-T4CDF 315.9419 317.9389 P5CDF 339.8597 341.8567 13C-P5CDF 351.9000 353.8970 H6CDF 373.8208 375.8178 13C-H6CDF 383.8639 385.8610 H7CDF 407.7818 409.7789 13C-H7CDF 417.8253 419.8220 O8CDF 441.7428 447.7399
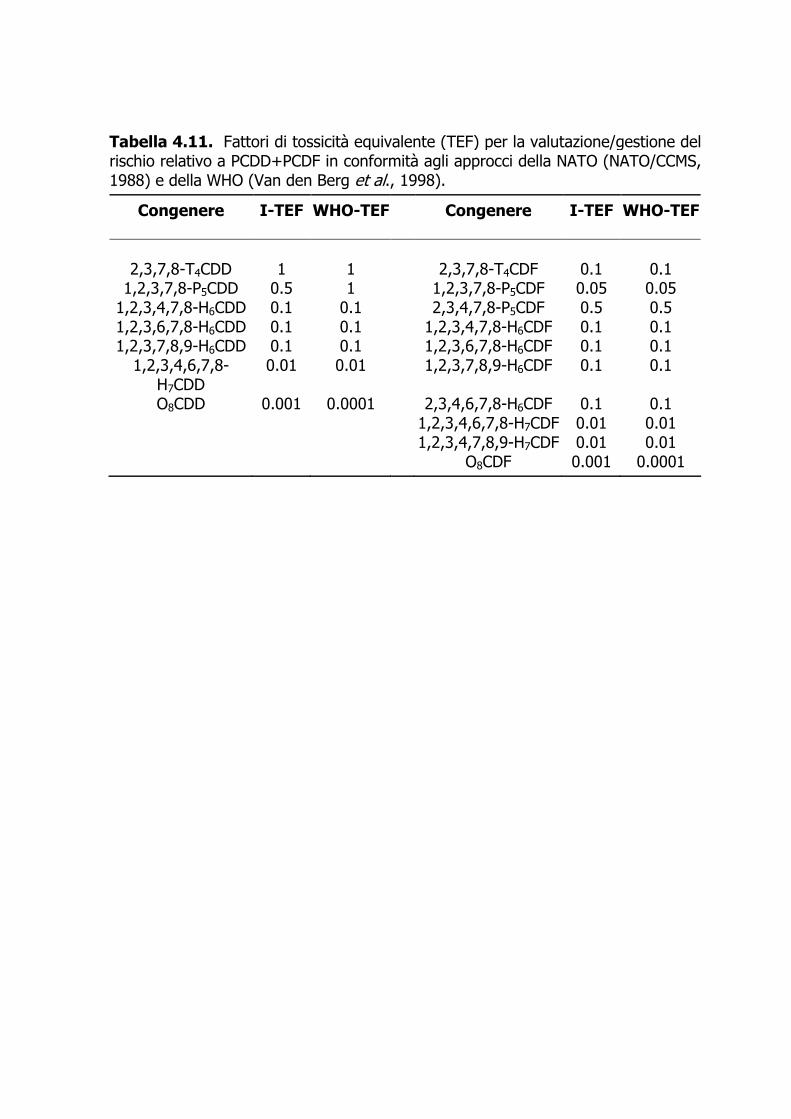
Tabella 4.11. Fattori di tossicità equivalente (TEF) per la valutazione/gestione del rischio relativo a PCDD+PCDF in conformità agli approcci della NATO (NATO/CCMS, 1988) e della WHO (Van den Berg et al., 1998).
Congenere I-TEF WHO-TEF Congenere I-TEF WHO-TEF
2,3,7,8-T4CDD 1 1 2,3,7,8-T4CDF 0.1 0.1 1,2,3,7,8-P5CDD 0.5 1 1,2,3,7,8-P5CDF 0.05 0.05
1,2,3,4,7,8-H6CDD 0.1 0.1 2,3,4,7,8-P5CDF 0.5 0.5 1,2,3,6,7,8-H6CDD 0.1 0.1 1,2,3,4,7,8-H6CDF 0.1 0.1 1,2,3,7,8,9-H6CDD 0.1 0.1 1,2,3,6,7,8-H6CDF 0.1 0.1
1,2,3,4,6,7,8-H7CDD
0.01 0.01 1,2,3,7,8,9-H6CDF 0.1 0.1
O8CDD 0.001 0.0001 2,3,4,6,7,8-H6CDF 0.1 0.1 1,2,3,4,6,7,8-H7CDF 0.01 0.01 1,2,3,4,7,8,9-H7CDF 0.01 0.01 O8CDF 0.001 0.0001

Tabella 4.12. Fattori di tossicità equivalente (TEF) per la valutazione/gestione del rischio relativo ai DL-PCB in conformità agli approcci della WHO (Van den Berg et al., 1998). Congenere WHO-TEF 3,3’,4,4’-T4CB [77] 0.0001 3,3’,4’,5-T4CB [81] 0.0001 3,3’,4,4’,5-H5CB [126] 0.1 3,3’,4,4’,5,5’-H6CB [169] 0.01
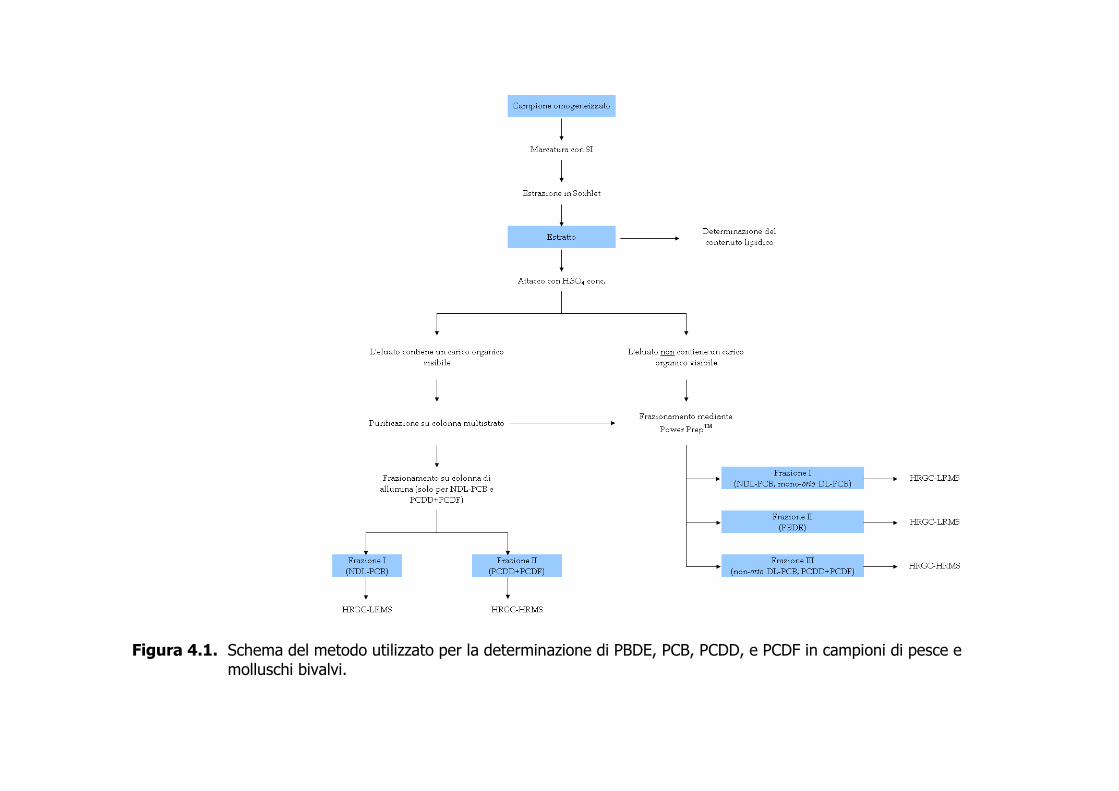
Figura 4.1. Schema del metodo utilizzato per la determinazione di PBDE, PCB, PCDD, e PCDF in campioni di pesce e
molluschi bivalvi.
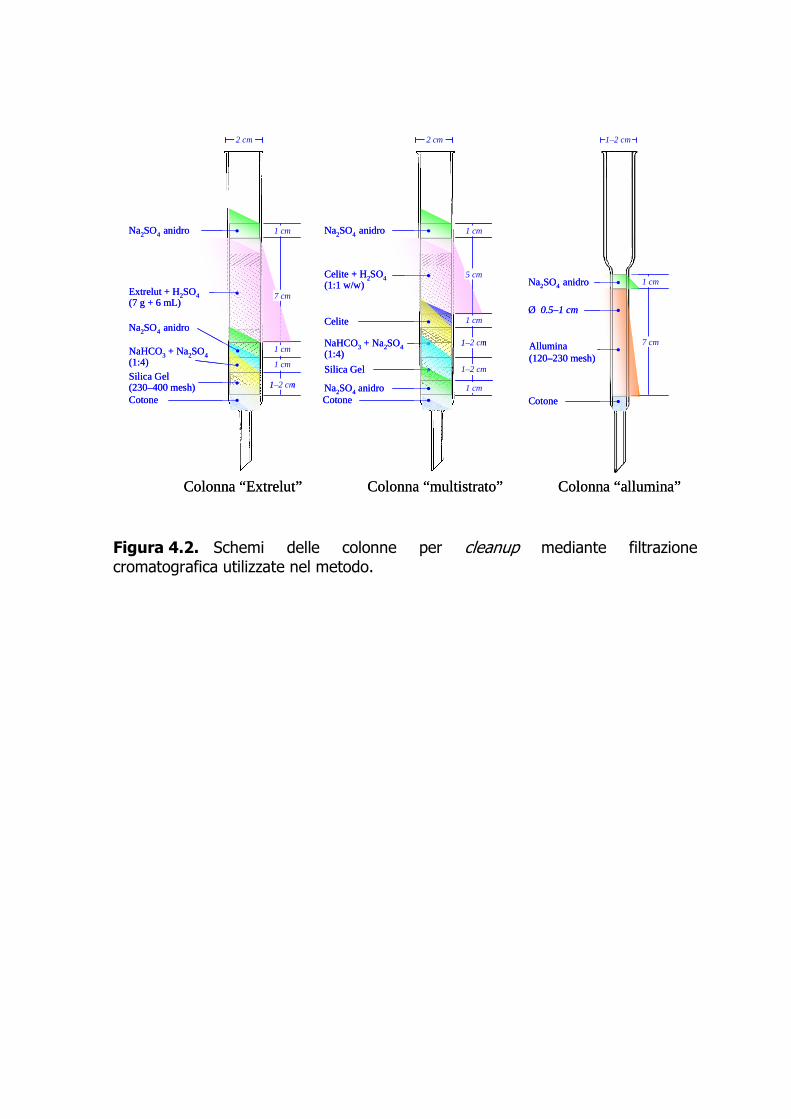
1–2 cm
1–2 cm
Na2SO4 anidro
Extrelut + H2SO4(7 g + 6 mL)
NaHCO3 + Na2SO4(1:4)Silica Gel(230–400 mesh)Cotone
1 cm
1 cm
1 cm
7 cm
Na2SO4 anidro
Colonna “Extrelut”
2 cm
Colonna “multistrato”
Na2SO4 anidro
Celite + H2SO4(1:1 w/w)
Celite
NaHCO3 + Na2SO4(1:4)
Silica Gel
Na2SO4 anidro Cotone
1 cm
1 cm
1–2 cm
1–2 cm
1 cm
5 cm
2 cm
Na2SO4 anidro
Cotone
1 cm
7 cm
Colonna “allumina”
Allumina(120–230 mesh)
Ø 0.5–1 cm
1–2 cm
1–2 cm
Na2SO4 anidro
Extrelut + H2SO4(7 g + 6 mL)
NaHCO3 + Na2SO4(1:4)Silica Gel(230–400 mesh)Cotone
1 cm
1 cm
1 cm
7 cm
Na2SO4 anidro
Colonna “Extrelut”
2 cm2 cm
Colonna “multistrato”
Na2SO4 anidro
Celite + H2SO4(1:1 w/w)
Celite
NaHCO3 + Na2SO4(1:4)
Silica Gel
Na2SO4 anidro Cotone
1 cm
1 cm
1–2 cm
1–2 cm
1 cm
5 cm
2 cm2 cm
Na2SO4 anidro
Cotone
1 cm
7 cm
Colonna “allumina”
Allumina(120–230 mesh)
Ø 0.5–1 cm
Figura 4.2. Schemi delle colonne per cleanup mediante filtrazione cromatografica utilizzate nel metodo.
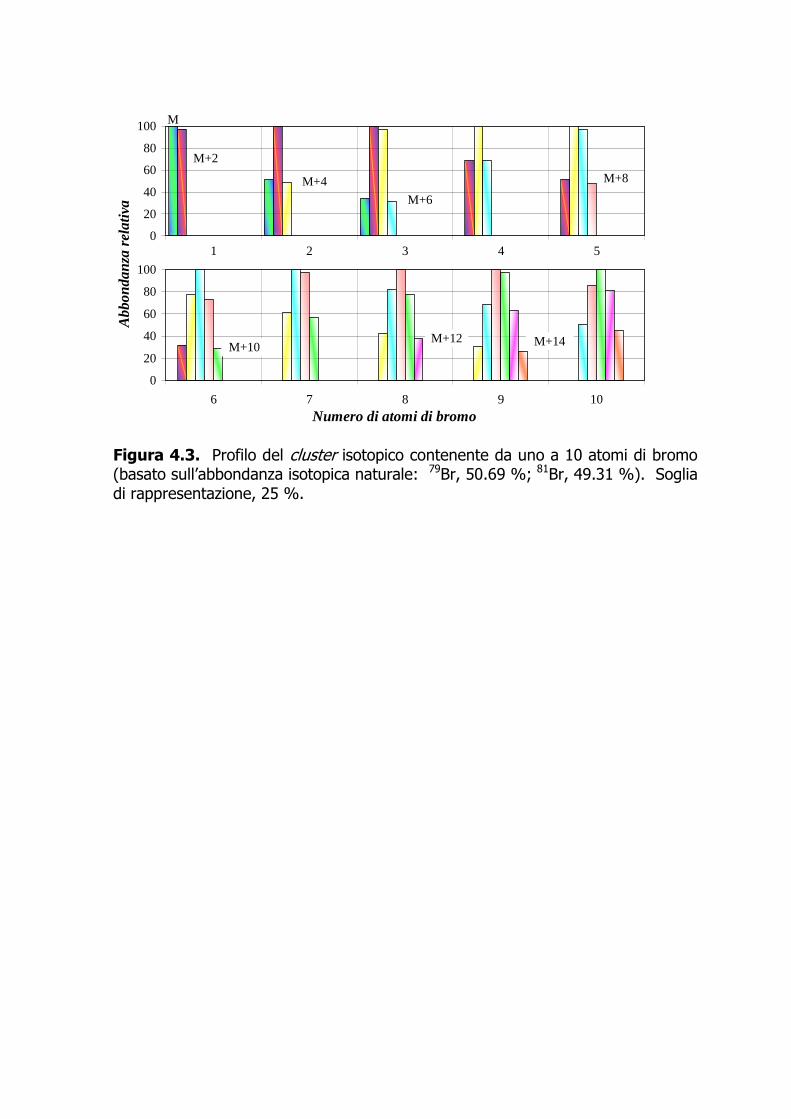
Abb
onda
nza
rela
tiva
Numero di atomi di bromo
0
20
40
60
80
100
1 2 3 4 5
M+2
M+4
M+6
M+8
0
20
40
60
80
100
6 7 8 9 10
M+10 M+14M+12
M
Figura 4.3. Profilo del cluster isotopico contenente da uno a 10 atomi di bromo (basato sull’abbondanza isotopica naturale: 79Br, 50.69 %; 81Br, 49.31 %). Soglia di rappresentazione, 25 %.
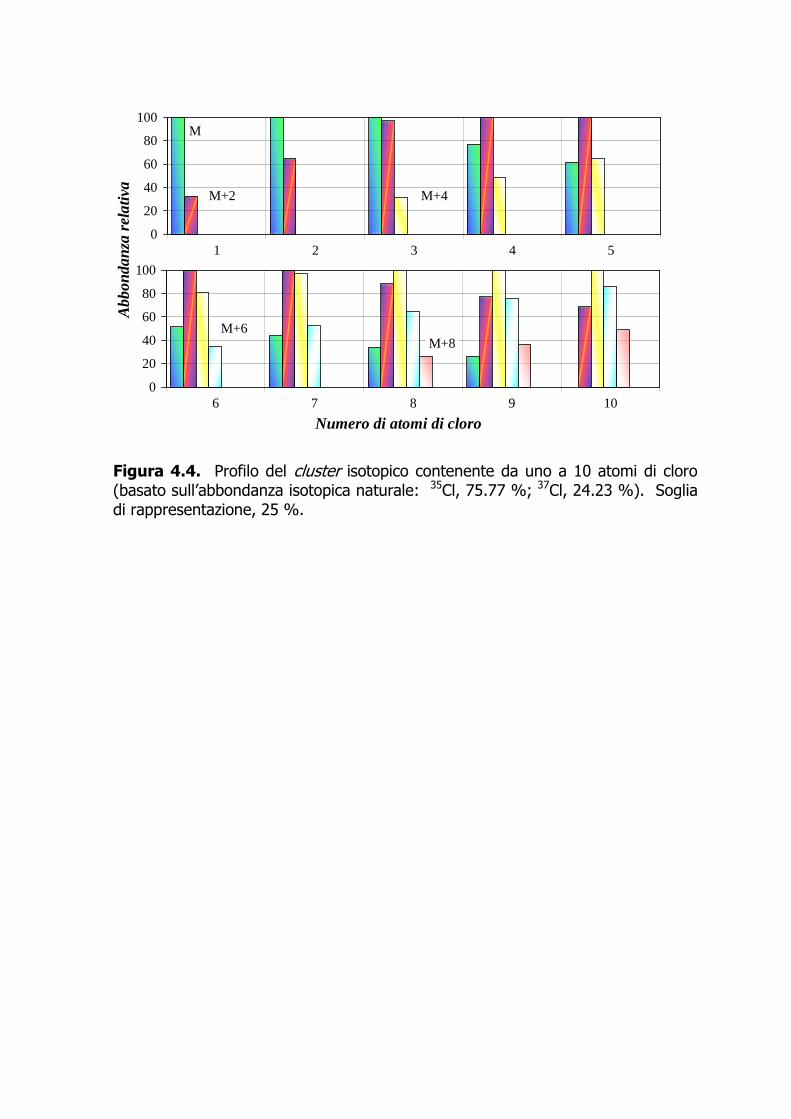
Abb
onda
nza
rela
tiva
Numero di atomi di cloro
0
20
40
60
80
100
1 2 3 4 5
M
M+2 M+4
0
20
40
60
80
100
6 7 8 9 10
M+6M+8
Figura 4.4. Profilo del cluster isotopico contenente da uno a 10 atomi di cloro (basato sull’abbondanza isotopica naturale: 35Cl, 75.77 %; 37Cl, 24.23 %). Soglia di rappresentazione, 25 %.

ALLEGATO 4.1 Condizioni operative suggerite per la determinazione strumentale degli analiti di interesse HRGC-LRMS (EI, 35 eV)
� colonna gas cromatografia: HT-5 (5% phenyl (equiv) polysiloxane-carborane, 25 m × 0.22 mm Øi × 0.1 µm) o equivalente;
� gas di trasporto: He; � iniettore PTV oppure on-column; � programmata GC per la determinazione di congeneri di PBDE: 80 °C per 1
min, 30 °C/min fino a 240 °C, 10 °C/min fino 340 °C, 340 °C per 4 min; � programmata GC per la determinazione di congeneri di NDL-PCBe di mono-
orto-DL-PCB: 80 °C per 1 min, 30 °C/min fino a 150 °C, 5 °C/min fino 255 °C, 120 °C/min fino a 340 °C per 3 min.
HRGC-HRMS, Waters VG Autospec; colonna gas cromatografica HT-5 (5% phenyl (equiv) polysiloxane-carborane, 25 m × 0.22 mm Øi × 0.1 µm) o equivalente; gas di trasporto, He; iniettore PTV oppure on-column; programmata GC, 80 °C per 1 min, 30 °C/min fino a 240 °C, 10 °C/min fino 340 °C, 340 °C per 4 min.

5. Policlorobifenili (PCB), policlorodibenzodiossine (PCDD),
policlorodibenzofurani (PCDF), polibromodibenzodiossine
(PBDD) e polibromodibenzofurani (PBDF) in campioni di
omogeneizzato di pesce
Istituto di Ricerche Farmacologiche Mario Negri, Dipartimento Ambiente e Salute
Acronimi DL-PCB policlorobifenili diossina-simili GC gascromatografia HR alta risoluzione LOQ limite di quantificazione LR bassa risoluzione MS spettrometria di massa NCI negative chemical ionization NDL-PCB policlorobifenili non diossina-simili PBDD polibromodibenzodiossine PBDE polibromodifenileteri PCDD policlorodibenzodiossine PBDF polibromodibenzofurani PCDF policlorodibenzofurani

SI standard interno SIM single ion monitoring TEF toxicity equivalency factor, o fattori di tossicità equivalente TEQ equivalenti di tossicità di 2,3,7,8-T4CDD NCI negative chemical ionization EI electron impact 5.1 Scopo e applicazione 5.1.1 Il presente metodo è impiegato per la quantificazione, mediante gascromatografia ad alta risoluzione-spettrometria di massa a bassa risoluzione (HRGC-LRMS) e gascromatografia ad alta risoluzione-spettrometria di massa ad alta risoluzione (HRGC-HRMS), di policlorobifenili non diossina-simili (NDL-PCB), policlorobifenili diossina-simili (DL-PCB), policlorodibenzodiossine (PCDD), policlorodibenzofurani (PCDF), polibromodibenzodiossine (PBDD), e polibromodibenzofurani (PBDF) in campioni di omogeneizzato di pesce. 5.1.2 Il metodo consente la determinazione di a) 16 congeneri di NDL-PCB appartenenti ai gruppi omologhi tri- (T3), tetra- (T4), penta- (P5), esa- (H6) e epta- (H7) clorosostituiti; b) 10 congeneri di DL-PCB appartenenti ai gruppi omologhi tetra- (T4), penta- (P5), ed esa- (H6) clorosostituiti e comprendenti 8 congeneri mono-orto sostituiti e 4 congeneri non orto-sostituiti; c) 17 congeneri tossici di PCDD+PCDF appartenenti ai gruppi omologhi tetra- (T4), penta- (P5), esa- (H6), epta- (H7), e otta- (O8) clorosostituiti; d) 10 congeneri di PBDD+PBDF appartenenti ai gruppi omologhi tetra- (T4), penta- (P5), esa- (H6) e epta- (H7) bromosostituiti. L’elenco dei singoli congeneri analizzati è riportato nelle Tabelle 5.1–4. 5.2 Principio del metodo 5.2.1 In accordo con il Metodo 1613 della US EPA (1994), il rilevamento degli analiti di interesse è basato sull’impiego di standard interni (SI), o traccianti, marcati con 13C. Tali SI vengono aggiunti al campione dopo la liofilizzazione, prima dell’estrazione con solvente.

5.2.2 L’estrazione avviene tramite utilizzo di estrattori Soxhlet con una miscela esano:acetone 9:1 per 12 ore. 5.2.3 La procedura di purificazione comprende un trattamento con acido solforico concentrato, seguito da passaggi su colonne cromatografiche che permettono di concentrare gli analiti in forma purificata. 5.2.4 Dopo purificazione, il campione viene quantificato mediante HRGC-LRMS (SIM, NCI) per la determinazione di PCB e PBDD+PBDF, e mediante HRGC-HRMS (SIM, EI+) per la determinazione di PCDD+PCDF. 5.2.5 A ciascun batch di campioni è abbinata l’analisi di un bianco procedurale per verificare l’eventuale contributo dovuto al fondo ambientale e procedurale. 5.3 Strumentazione 5.3.1 STRUMENTAZIONE PER LA PURIFICAZIONE DEL CAMPIONE
• colonna in vetro (Ø 2.5 cm) per Extrelut; • colonna in vetro per allumina (Ø 0.5 cm, h 7 cm); • palloni di vetro da 250 mL.
5.3.2 STRUMENTAZIONE PER LA DETERMINAZIONE STRUMENTALE DEL CAMPIONE
• HRGC-HRMS, TRACE GC 2000, Mat 95 XP; • HRGC-LRMS, GC 6890, Agilent 5973N MSD.
5.4 Reagenti e standard 5.4.1 SOLVENTI, REAGENTI E MATERIALE CROMATOGRAFICO
• n-Esano (Carlo Erba, >98 %, per pesticidi); • acido solforico concentrato (Carlo Erba, 98 %, per microanalisi); • isottano (Carlo Erba, >98 %, per pesticidi); • carbonio tetracloruro (Carlo Erba, 99.8 %); • diclorometano (Carlo Erba, >99.8 %, per pesticidi); • Extrelut NT (Merck); • ossido di allumina (Merck, attivato neutro per cromatografia su colonna); • sodio solfato anidro (Merck, 99 %); • lana di vetro (Supelco).

5.4.2 STANDARD
• Standard interno PCB: DL-PCB 12 congeneri 13C12 (10 pg/µL), NDL-PCB 6 congeneri 13C12 (10 pg/µL), PCB 209 (10 pg/µL);
• standard interno PCDD+PCDF: 16 congeneri 13C12-2,3,7,8 cloro-sostituiti (10 pg/µL);
• standard d’iniezione: 2,3,7,8-Cl37-TCDD (10 pg/µL); • standard interno PBDD+PBDF: [5 congeneri 13C12-2,3,7,8 bromo-sostituiti
(100 pg/µL).
5.5 Conservazione del campione 5.5.1 I campioni sono stati conservati in freezer a –20 °C fino all’analisi. 5.6 Procedura analitica 5.6.1 PREPARAZIONE DEL CAMPIONE 5.6.1.1 Ogni campione viene scongelato, riposto in una falcon da 50 mL, pesato prima della liofilizzazione e ricongelato. La quantità pesata è di circa 50 g. 5.6.1.2 La liofilizzazione ha inizio quando il campione è congelato. La durata di questo processo è di circa 3 giorni. 5.6.1.3 Il campione viene quindi ripesato, per avere il peso secco e riposto in un ditale in fibra di vetro per l’estrazione in Soxhlet. A questo punto vengono aggiunti gli SI di PCB (1 ng di ciascun congenere di DL-PCB e dei sei NDL-PCB indicatori), delle PCDD e PCDF (200 pg di ciascun congenere 2,3,7,8-cloro sostituito) e delle PBDD e PBDF (400 pg di 5 congeneri 2,3,7,8 bromo-sostituito). L’estrazione viene effettuata su circa 4 grammi di peso secco.
5.6.2 ESTRAZIONE 5.6.2.1 L’estrazione avviene riponendo il ditale all’interno del Soxhlet e preparando un pallone da 100 mL, in cui viene aggiunto l’anti-bumping e la miscela di n-esano-acetone 9:1. Il tempo di estrazione è di circa 8 ore. A questo punto, i campioni vengono travasati in palloni da 250 mL (precedentemente pesati) e portati a secco al rotavapor, per eliminare tutto il solvente ed isolare il grasso estratto. I palloni vengono quindi ripesati per la determinazione della quantità di grasso presente nel campione.

5.6.3 PURIFICAZIONE 5.6.3.1 Una prima purificazione viene effettuata tramite colonna Extrelut (15 g) dopo trattamento con acido solforico concentrato (15 mL) per almeno 2 ore fino a completa distruzione del carico organico. Successivamente è caricato su colonna Extrelut (15 g) dove è lasciato adsorbire per una notte. Successivamente è eluito con 120 mL di n-esano. 5.6.3.2 L’eluato, cautamente concentrato a piccolo volume, viene frazionato su colonna di ossido di alluminio (0.20 g ) usando come eluente, nell’ordine, circa 9 mL di n-esano (Frazione 0), 6 mL di CCl4 (Frazione I, contenente i PCB), 7 mL di CH2Cl2 (Frazione II, contenente PCB, PCDD+PCDF e PBDD+PBDF). 5.6.3.3 Aggiunti 200 pg.di standard d’iniezione 2,3,7,8-37Cl-T4CDD alla Frazione II, le Frazioni I e II vengono portate a secco e riprese con 50 µL d’isottano. 25 µL della Frazione II vengono utilizzati per quantificare PCDD+PCDF e PBDD+PBDF, i restanti 25 µL vengono uniti alla Frazione I e impiegati per la quantificazione dei PCB. 5.6.4 DETERMINAZIONE STRUMENTALE 5.6.4.1 Tutti i congeneri dei PCB e PBDD e PBDF vengono determinati mediante HRGC-LRMS (NCI) in modalità SIM utilizzando le condizioni operative descritte nell’Allegato 5.1. 5.6.4.2 I 17 congeneri di PCDD e PCDF vengono determinati mediante HRGC-HRMS, TRACE GC 2000, Mat 95 XP (EI+, 48 eV), operante alla risoluzione di 10000 in modalità SIM (le condizioni operative sono riportate nell’Allegato 5.1). 5.6.4.3 I congeneri nativi e quelli isotopicamente marcati sono identificati utilizzando le due masse più intense dello spettro di massa nel caso dei PBDD+PBDF (Tabella 5.8), le due masse più intense di ciascun cluster molecolare per PCB e PCDD+PCDF (Tabelle 5.5–7) e quantificati sulla base del rapporto dell’analita sullo SI relativo alla traccia più intensa. Per tali analiti, il rapporto delle aree relative alle due masse esatte di ciascun multipletto molecolare deve essere ± 20 % del corrispondente valore teorico. 5.6.4.4 La quantificazione congenere-specifica viene eseguita impiegando una miscela dei congeneri naturali d’interesse e di quelli isotopicamente marcati a concentrazione nota (standard di quantificazione). 5.6.4.5 Per i 17 congeneri di PCDD+PCDF è stata inoltre calcolata la percentuale di recupero, tramite il rapporto tra SI e SE (2,3,7,8-37Cl).

5.6.4.6 I risultati analitici relativi a PCDD+PCDF e DL-PCB sono convertiti in unità TE, o TEQ, mediante opportuno sistema TEF (Tabelle 5.9 e 5.10).

Tabella 5.1. Congeneri di NDL-PCB rilevati con il presente metodo.
Congenere No. CAS
2,4,4'-T3CB [28] 7012-37-5 2,2',5,5'-T4CB [52] 35693-99-3 2,2',3,5',6-P5CB [95] 38379-99-6 2,2',4,4',5-P5CB [99] 38380-01-7 2,2',4,5,5'-P5CB [101] 37680-73-2 2,3,3',4',6-P5CB [110] 38380-03-9 2,2',3,4,4',5'-H6CB [138] 35065-28-2 2,2',3,4',5,5'-H6CB [146] 51908-16-8 2,2',3,4',5',6-H6CB [149] 38380-04-0 2,2',3,5,5',6-H6CB [151] 52663-63-5 2,2',4,4',5,5'-H6CB [153] 35065-27-1 2,2',3,3',4,4',5-H7CB [170] 35065-30-6 2,2',3,3',4,5',6'-H7CB [177] 52663-70-4 2,2',3,4,4',5,5'-H7CB [180] 35065-29-3 2,2',3,4,4',5',6-H7CB [183] 52663-69-1 2,2',3,4',5,5',6-H7CB [187] 52663-68-0

Tabella 5.2. Congeneri di DL-PCB rilevati dal presente metodo.
Congenere No. CAS
3,3’,4,4’-T4CB [77]
a 32598-13-3 3,3’,4’,5-T4CB [81]
a 70362-50-4 2,3,3',4,4'-P5CB [105]
b 32598-14-4 2,3,4,4',5-P5CB [114]
b 74472-37-0 2,3',4,4',5-P5CB [118]
b 31508-00-6 2,3',4,4',5'-P5CB [123]
b 65510-44-3 3,3’,4,4’,5-H5CB [126]
a 57465-28-8 2,3,3',4,4',5-H6CB [156]
b 38380-08-4 2,3,3',4,4',5'-H6CB [157]
b 69782-90-7 2,3',4,4',5,5'-H6CB [167]
b 52663-72-6 3,3’,4,4’,5,5’-H6CB [169]
a 32774-16-6 2,3,3',4,4',5,5'-H7CB [189]
b 39635-31-9
(a) DL-PCB non-orto sostituiti (complanari).
(b) DL-PCB mono-orto sostituiti.

Tabella 5.3. Congeneri di PCDD+PCDF rilevati con il presente metodo.
Congenere No. CAS Congenere No. CAS
2,3,7,8-T4CDD 1746-01-6 2,3,7,8-T4CDF 51207-31-9 1,2,3,7,8-P5CDD 40321-76-4 1,2,3,7,8-P5CDF 57117-41-6 2,3,4,7,8-P5CDF 57117-31-4 1,2,3,4,7,8-H6CDD 39227-28-6 1,2,3,4,7,8-H6CDF 70648-26-9 1,2,3,6,7,8-H6CDD 57653-85-7 1,2,3,6,7,8-H6CDF 57117-44-9 1,2,3,7,8,9-H6CDD 19408-74-3 1,2,3,7,8,9-H6CDF 72918-21-9 2,3,4,6,7,8-H6CDF 60851-34-5 1,2,3,4,6,7,8-H7CDD 35822-46-9 1,2,3,4,6,7,8-H7CDF 67562-39-4 1,2,3,4,7,8,9-H7CDF 55673-89-7 1,2,3,4,6,7,8,9-O8CDD 3268-87-9 1,2,3,4,6,7,8,9-O8CDF 39001-02-0
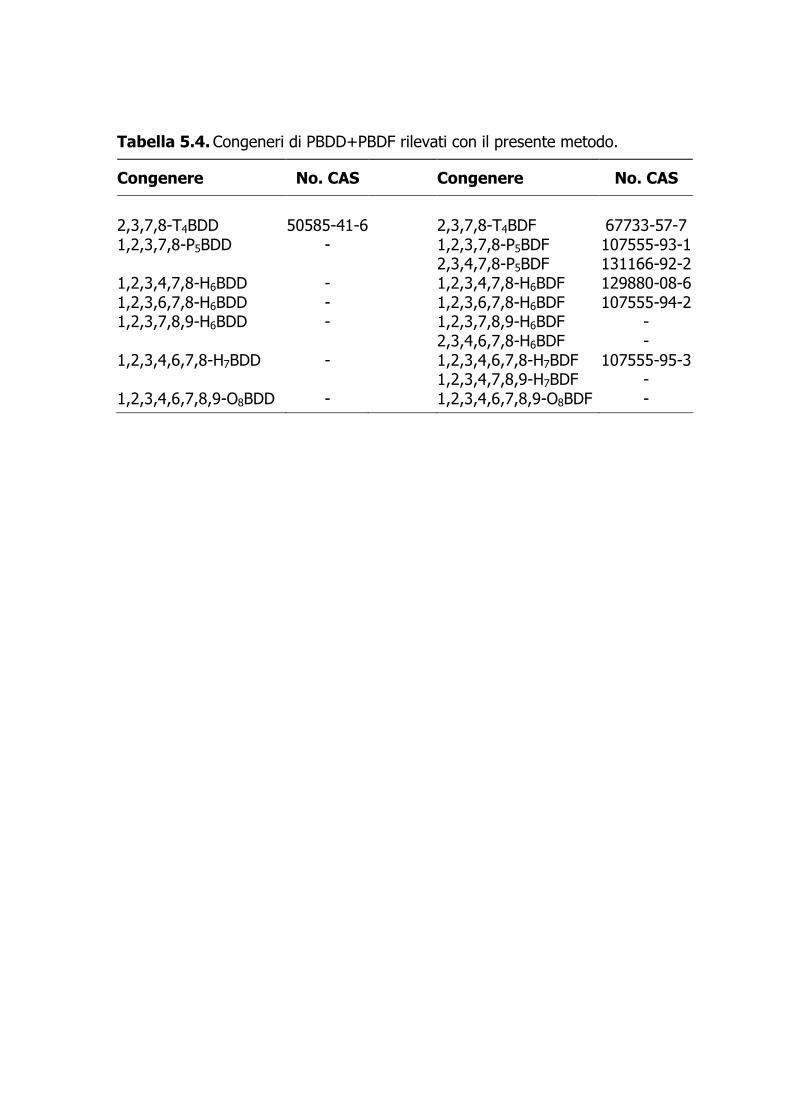
Tabella 5.4. Congeneri di PBDD+PBDF rilevati con il presente metodo.
Congenere No. CAS Congenere No. CAS
2,3,7,8-T4BDD 50585-41-6 2,3,7,8-T4BDF 67733-57-7 1,2,3,7,8-P5BDD - 1,2,3,7,8-P5BDF 107555-93-1 2,3,4,7,8-P5BDF 131166-92-2 1,2,3,4,7,8-H6BDD - 1,2,3,4,7,8-H6BDF 129880-08-6 1,2,3,6,7,8-H6BDD - 1,2,3,6,7,8-H6BDF 107555-94-2 1,2,3,7,8,9-H6BDD - 1,2,3,7,8,9-H6BDF - 2,3,4,6,7,8-H6BDF - 1,2,3,4,6,7,8-H7BDD - 1,2,3,4,6,7,8-H7BDF 107555-95-3 1,2,3,4,7,8,9-H7BDF - 1,2,3,4,6,7,8,9-O8BDD - 1,2,3,4,6,7,8,9-O8BDF -
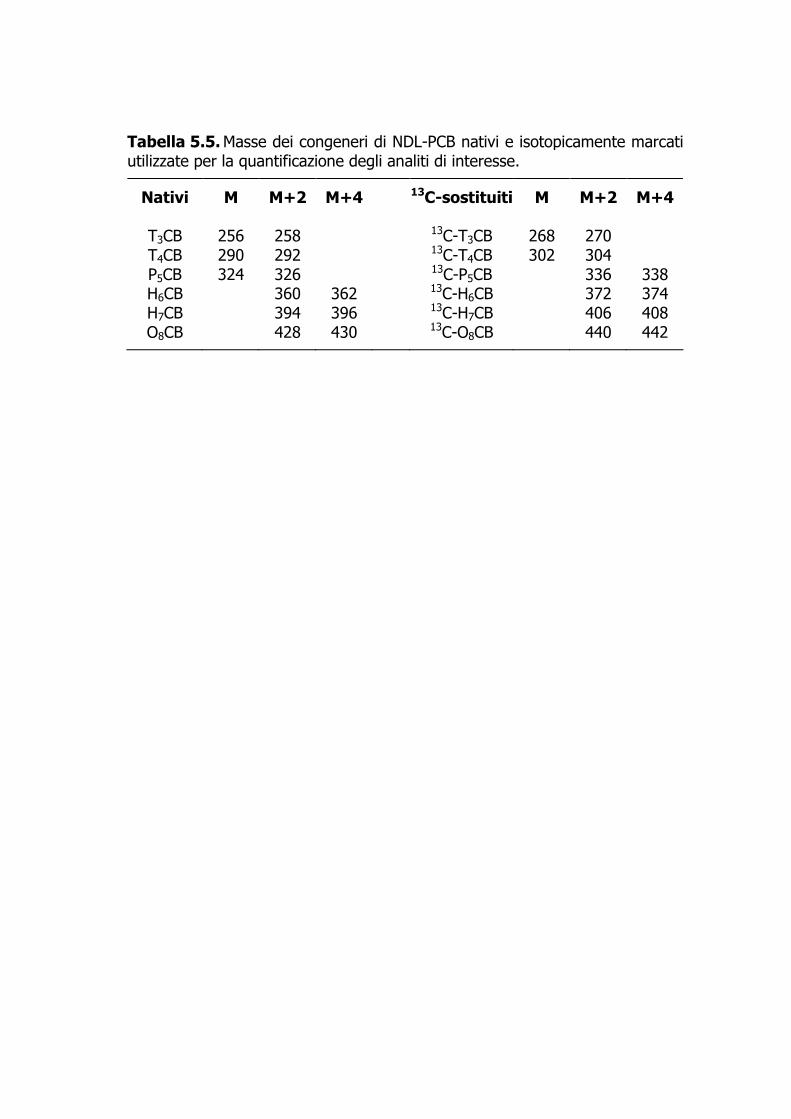
Tabella 5.5. Masse dei congeneri di NDL-PCB nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Nativi M M+2 M+4 13C-sostituiti M M+2 M+4
T3CB 256 258 13C-T3CB 268 270 T4CB 290 292 13C-T4CB 302 304 P5CB 324 326 13C-P5CB 336 338 H6CB 360 362 13C-H6CB 372 374 H7CB 394 396 13C-H7CB 406 408 O8CB 428 430 13C-O8CB 440 442

Tabella 5.6. Masse dei congeneri di DL-PCB nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Nativi M M+2 M+4 13C-
sostituiti M M+2 M+4
T4CB 290 292 13C-T4CB 302 304 P5CB 324 326 13C -P5CB 338 340 H6CB 360 362 13C-H6CB 372 374 H7CB 394 396 13C-H7CB 406 408
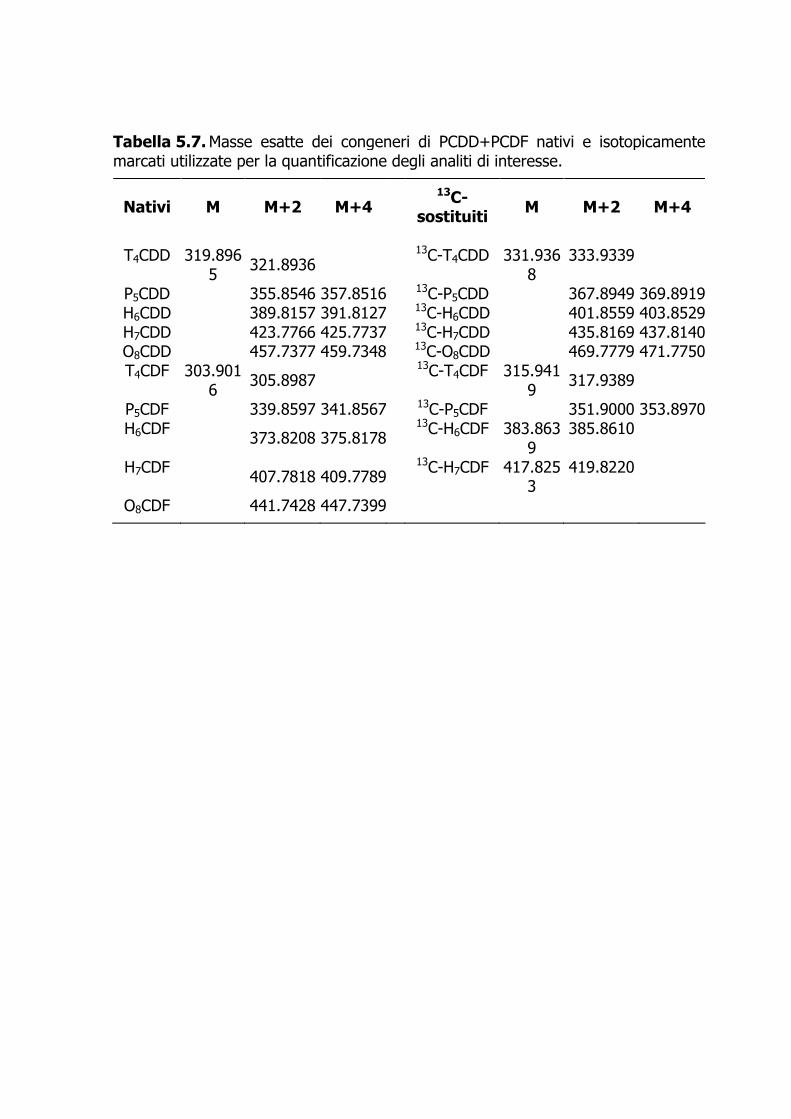
Tabella 5.7. Masse esatte dei congeneri di PCDD+PCDF nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Nativi M M+2 M+4 13C-
sostituiti M M+2 M+4
T4CDD 319.896
5 321.8936 13C-T4CDD 331.936
8 333.9339
P5CDD 355.8546 357.8516 13C-P5CDD 367.8949 369.8919H6CDD 389.8157 391.8127 13C-H6CDD 401.8559 403.8529H7CDD 423.7766 425.7737 13C-H7CDD 435.8169 437.8140O8CDD 457.7377 459.7348 13C-O8CDD 469.7779 471.7750T4CDF 303.901
6 305.8987 13C-T4CDF 315.941
9 317.9389
P5CDF 339.8597 341.8567 13C-P5CDF 351.9000 353.8970H6CDF 373.8208 375.8178 13C-H6CDF 383.863
9 385.8610
H7CDF 407.7818 409.7789 13C-H7CDF 417.8253
419.8220
O8CDF 441.7428 447.7399

Tabella 5.8. Masse dei congeneri di PBDD+PBDF nativi e isotopicamente marcati utilizzate per la quantificazione degli analiti di interesse.
Congenere M+2 M+4 M+6 M+8 Congenere M+2 M+4 M+6 M+8
T4BDD 498 500 13C-P5BDD 589 591 P5BDD 578 580 13C-T4BDF 496 498 H6BDD 657 659 13C-P5BDF 574 576
13C-H6BDF 653 655 T4BDF 484 486 13C-H7BDF 731 733 P5BDF 562 564 H6BDF 641 643 H7BDF 719 721
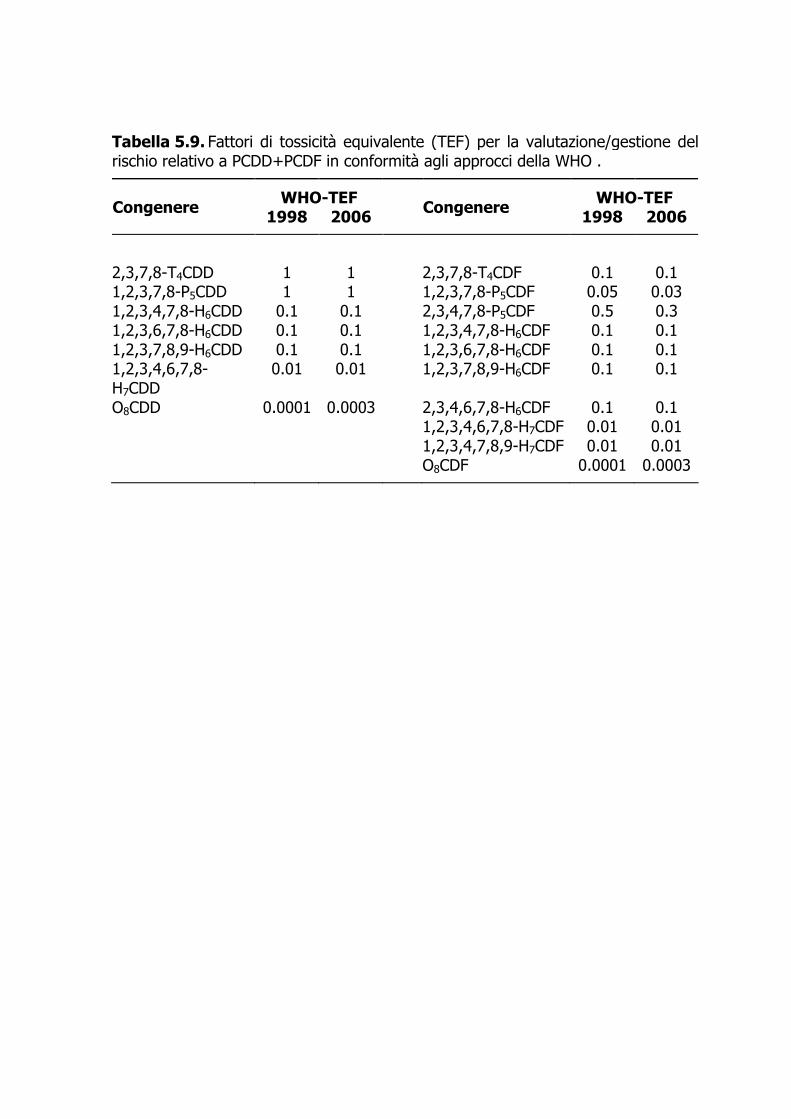
Tabella 5.9. Fattori di tossicità equivalente (TEF) per la valutazione/gestione del rischio relativo a PCDD+PCDF in conformità agli approcci della WHO .
WHO-TEF WHO-TEF Congenere
1998 2006 Congenere
1998 2006
2,3,7,8-T4CDD 1 1 2,3,7,8-T4CDF 0.1 0.1 1,2,3,7,8-P5CDD 1 1 1,2,3,7,8-P5CDF 0.05 0.03 1,2,3,4,7,8-H6CDD 0.1 0.1 2,3,4,7,8-P5CDF 0.5 0.3 1,2,3,6,7,8-H6CDD 0.1 0.1 1,2,3,4,7,8-H6CDF 0.1 0.1 1,2,3,7,8,9-H6CDD 0.1 0.1 1,2,3,6,7,8-H6CDF 0.1 0.1 1,2,3,4,6,7,8-H7CDD
0.01 0.01 1,2,3,7,8,9-H6CDF 0.1 0.1
O8CDD 0.0001 0.0003 2,3,4,6,7,8-H6CDF 0.1 0.1 1,2,3,4,6,7,8-H7CDF 0.01 0.01
1,2,3,4,7,8,9-H7CDF 0.01 0.01 O8CDF 0.0001 0.0003

Tabella 5.10. Fattori di tossicità equivalente (TEF) per la valutazione/gestione del rischio relativo ai PCB in conformità agli approcci della WHO.
Congenere WHO-TEF-1998 WHO-TEF-2006
3,3’,4,4’-T4CB [77] 0.0001 0.0001 3,3’,4’,5-T4CB [81] 0.0001 0.0003 3,3’,4,4’,5-H5CB [126] 0.1 0.1 3,3’,4,4’,5,5’-H6CB [169] 0.01 0.03 2,3,3',4,4'-pentaCB (PCB 105) 0.0001 0.00003 2,3,4,4',5-pentaCB (PCB 114) 0.0005 0.00003 2,3',4,4',5-pentaCB (PCB 118) 0.0001 0.00003 2',3,4,4',5-pentaCB (PCB 123) 0.0001 0.00003 2,3,3',4,4',5-hexaCB (PCB 156) 0.0005 0.00003 2,3,3',4,4',5'-hexaCB (PCB 157) 0.0005 0.00003 2,3',4,4',5,5'-hexaCB (PCB 167) 0.00001 0.00003 2,3,3',4,4',5,5'-heptaCB (PCB189)
0.0001 0.00003

ALLEGATO 5.1
Condizioni operative utilizzate per la determinazione strumentale degli analiti di interesse HRGC-LRMS (NCI) PER ANALISI DI PCB
• colonna gas cromatografica: HT-8 (60 m × 0.25 mm Øi × 0.25 µm); • gas di trasporto: He; • iniettore standard per colonne capillari; • programmata GC per la determinazione di congeneri di PCB: 185 °C per 2
min, 7.5 °C/min fino a 190 °C, 2 °C/min fino 280 °C, per 10 min. HRGC-LRMS (NCI) PER ANALISI DI PBDD+PBDF
• colonna gas cromatografica: HP DB-5MS (30 m × 0.25 mm Øi × 0.10 µm); • gas di trasporto: He; • iniettore standard per colonne capillari; • programmata GC per la determinazione di congeneri di PBDD/F: 100 °C per
0.25 min, 20 °C/min fino a 220 °C, 12 °C/min fino 280 °C, 20 °C/min fino a 320 °C, 320 °C per 8 minuti.
HRGC-HRMS, TRACE GC 2000 , Mat 95 XP (EI+) PER ANALISI DI PCDD+PCDF
• colonna gas cromatografica: BPX-DXN (SGE) (60 m × 0,20 mm Øi × 0.25µm);
• gas di trasporto: He; • iniettore: splitless; • programmata GC per la determinazione di PCDD/F:160 °C per 1 min, 2.5
°C/min fino a 300 °C, 300 °C per 6 min.

6. Determinazione di policlorobifenili (PCB) in campioni di
pesce e di molluschi bivalvi
CNR, Istituto per la Dinamica dei Processi Ambientali, Sede Venezia
Acronimi DL-PCB policlorobifenili diossina-simili FR fattore di risposta FRR fattore di risposta relativo GC gascromatografia GPC gel permeation chromatography, o cromatografia di gel permeazione HR alta risoluzione MID multiple ion detection MS spettrometria di massa NDL-PCB policlorobifenili non diossina-simili PSE pressurized solvent extraction, o estrazione con solvente
pressurizzato SI standard interno

Introduzione I policlorobifenili (PCB) sono una famiglia di composti aventi come struttura base quella del bifenile; questo può legarsi ad atomi di cloro in numero variabile da 1 a 10 dando luogo alla formazione di 209 possibili congeneri. Essi sono stati commercializzati come miscele di isomeri con varie denominazioni (Aroclor, Phenoclor, Kanechlor, Clophen, Fenclor, ecc.) a seconda della ditta produttrice. Facendo riferimento al principale produttore negli USA (Monsanto) le miscele commercializzate prendevano il nome di Aroclor seguito da un numero (ad es. Aroclor 1242, Aroclor 1254, Aroclor 1260), dove i numeri 42, 54 e 60 indicavano la % di cloro; ad una percentuale maggiore corrisponde una maggior presenza di congeneri a più alto grado di clorurazione. Queste miscele non contengono comunque tutti i 209 congeneri, ma solo una parte. In relazione ai meccanismi propri dell’azione tossica dei PCB, si tende a dividere tali composti in due categorie (comunque coesistenti nelle miscele commerciali e nelle matrici ambientali): i 12 PCB diossina-simili (DL-PCB), con quattro posizioni orto (2, 2’, 6, 6’) libere o mono-cloro-sostituite, e i PCB non diossina-simili (NDL-PCB). 6.1 Scopo e applicazione
6.1.1 Il metodo è utilizzato per la quantificazione di NDL-PCB e DL-PCB, in campioni omogeneizzati di pesce e di molluschi bivalvi mediante gascromatografia ad alta risoluzione-spettrometria di massa ad alta risoluzione (HRGC-HRMS). 6.1.2 Il metodo consente la determinazione di: a) 132 congeneri di NDL-PCB appartenenti ai gruppi omologhi mono- (M1), bi- (B2), tri- (T3), tetra- (T4), penta- (P5), esa- (H6), epta- (H7), otta- (O8), nona- (N9) e deca- (D10) clorosostituiti o come singoli congeneri o come somma di due o tre isomeri; b) 12 congeneri di DL-PCB appartenti ai gruppi omologhi tetra- (T4), penta- (P5), esa- (H6) ed epta- (H7) clorosostituiti e comprendenti 8 congeneri mono-orto sostituiti e 4 congeneri a struttura coplanare. L’elenco dei congeneri considerati nel presente metodo è riportato nella Tabella 6.1. 6.2 Principio del metodo 6.2.1 Il metodo analitico utilizzato fa riferimento al Metodo 1668 della US EPA (1999): “Method 1668, Revision A: Chlorinated Biphenyl Congeners in Water, Soil, Sediment, and Tissue by HRGC/HRMS”. La Figura 6.1 sintetizza le fasi di preparazione, estrazione, purificazione e quantificazione utilizzate nel presente metodo. 6.2.2 La determinazione degli analiti di interesse è basata sull’impiego di standard interni (SI) completamente marcati con 13C. Gli SI vengono aggiunti

all’omogeneizzato all’inizio della procedura analitica per la determinazione quantitativa; prima dell’analisi gascromatografica vengono aggiunti alcuni PCB come standard d’iniezione per la determinazione del recupero degli SI marcati. 6.2.3 La componente lipidica dell’omogeneizzato di pesce è estratta per estrazione con solvente pressurizzato (PSE) utilizzando un’apposita apparecchiatura ed una miscela n-esano-diclorometano 1:1 (v:v), così da rimuovere gli analiti altamente lipofili. 6.2.4 L’estratto è soggetto a una procedura di purificazione utilizzando un sistema Power-PrepTM, equipaggiato con una colonna di silice ABN (acida, basica, neutra) ad alta capacità, e un sistema basato sulla cromatografia di gel permeazione (GPC). 6.2.5 Dopo purificazione, i congeneri vengono quantificati in base a fattori di risposta (FR) mediante HRGC-HRMS, utilizzando nell’acquisizione il metodo multiple ion detection (MID). 6.2.6 All’analisi dei campioni suddivisi in gruppi è abbinata quella di un bianco procedurale per verificare il contributo al fondo (background) procedurale. 6.3 Strumentazione 6.3.1 STRUMENTAZIONE PER L’ESTRAZIONE DEL CAMPIONE
� Estrattore PSE One (Applied Separation, USA). 6.3.2 STRUMENTAZIONE PER LA CONCENTRAZIONE DEL CAMPIONE
� Apparato per la concentrazione del campione con flusso d’azoto, TurboVap II (Caliper LifeScience, USA).
6.3.3 STRUMENTAZIONE PER LA PURIFICAZIONE DEL CAMPIONE
� Sistema di purificazione automatizzato Power-PrepTM (Fluid Managemnet Systems, USA);
� sistema di purificazione automatizzato LS PPPGPC1 (LabService Analytica, Italia);
� palloni e beute di vetro.

6.3.4 STRUMENTAZIONE PER L’ANALISI STRUMENTALE
� HRGC-HRMS: spettometro di massa ad alta risoluzione MAT95XP (ThermoFisher Scientific, USA), accoppiato ad un gascromatografo Agilent (USA), mod. 6890.
6.4 Reagenti e standard 6.4.1 REAGENTI E MATERIALE CROMATOGRAFICO
� Elio Ricerche – SIAD, titolo 99.9995 %; � azoto Ricerche – SIAD, titolo 99.999 %; � colonna di silice ABN ad alta capacità per sistema Power-PrepTM; � colonna di stirene-divinilbenzene SX-3 (Bio-Rad Laboratories, USA); � colonna gas cromatografica: HP-5MS ((5%-Phenyl)-methylpolysiloxane, 60
m × 0.25 mm ∅i × 0.25 µm; Agilent Technologies, USA); � sodio solfato anidro, per analisi, (Fluka, Italia); � Spe-edTM Matrix (Applied Separation, USA); � Ottawa sand, Applied Separation, USA.
6.4.2 SOLVENTI
� Diclorometano, Pesticide Residue Analysis (Super Purity Solvent, Romil, UK);
� n-esano, Pesticide Residue Analysis (Super Purity Solvent, Romil, UK); � iso-ottano, Pestiscan (Lab-Scan, Polonia); � n-tetradecano (Aldrich, Italia).
6.4.3 STANDARD
� Miscele di congeneri dei PCB presenti negli Aroclors 1242, 1254 e 1260 (C-CS-01, C-CS-02, C-CS-03, C-CS-04, C-CS-05: 10 µg/mL); AccuStandard, USA;
� miscela di congeneri “Quebec Ministry of Environment congener mix”; Accustandard, USA;
� PCB 30, PCB 65, PCB 96, PCB 166: 10 ng/µL; Dr Ehrenstorfer GmbH, Germania;
� PCB 126-CS, PCB 169-CS: 100 g/mL; Cambridge Isotopic Laboratory , USA;
� soluzioni di congeneri marcati, Mono-Deca PCB Mixture (EC-4189), Co-Planar PCB Mixture (EC-4187), Mono-Ortho PCB Mixture (EC-4188): 1 µg/mL; PCB 178: 40 µg/mL; Cambridge Isotopic Laboratory, USA;
� CLP GPC Calibration Mix; Dr Ehrenstorfer GmbH, Germania.

6.4.4 SOLUZIONI STANDARD
� Soluzione di standard interni (SI): (solvente, iso-ottano) preparata miscelando le soluzioni di congeneri marcati Mono-Deca PCB Mixture, Co-Planar PCB Mixture, e Mono-Ortho PCB Mixture; viene utilizzata per l’aggiunta dei PCB 13C-marcati ai campioni all’inizio del processo analitico;
� soluzione standard d’iniezione (SING): (solvente, iso-ottano) preparata miscelando le soluzioni di congeneri PCB 30, PCB 65, PCB 96, PCB 166 e la soluzione del congenere 13C-marcato PCB 178; viene aggiunta alla fine del processo analitico per la determinazione del recupero dei PCB 13C-marcati;
� soluzione standard di quantificazione (SQ): (solvente, iso-ottano) preparata miscelando le soluzioni C-CS-01, C-CS-02, C-CS-03, C-CS-04, C-CS-05, le soluzioni dei congeneri PCB 126, PCB 169, PCB 30, PCB 65, PCB 96, PCB 166 e le soluzioni di congeneri 13C-marcati Mono-Deca PCB Mixture, Co-Planar PCB Mixture, Mono-Ortho PCB Mixture e PCB 178; viene utilizzata per il calcolo dei fattori di risposta relativi dei congeneri;
� soluzione standard di controllo (SC): preparata miscelando la soluzione “Quebec Ministry of Environment congener mix” e le soluzioni di congeneri marcati Mono-Deca PCB Mixture , Co-Planar PCB Mixture, Mono-Ortho PCB Mixture; viene utilizzata per controllare nel tempo la soluzione SFR.
6.5 Interferenze e contaminazione 6.5.1 La vetreria, tutta di Pyrex, viene preventivamente lavata con detersivo e acqua, e poi tenuta in stufa a 170 °C per 12–24 h. Prima dell’uso, la vetreria è ulteriormente e ripetutamente risciacquata con diclorometano seguito da n-esano, e fatta asciugare in zona protetta. L’ultimo risciacquo della vetreria con n-esano viene raccolto quantitativamente e concentrato a piccolo volume per verificarvi mediante rilevamento HRGC-HRMS l’eventuale presenza d’interferenze pregiudiziali sui segnali degli analiti d’interesse. Qualora questo background procedurale sia troppo elevato, occorre sottoporre la vetreria a nuovo lavaggio oppure sostituirla. Dopo l’uso, se opportuno, la vetreria impiegata viene recuperata dopo decontaminazione con lavaggi ripetuti con miscela equivolumetrica di acetone e n-esano (eventualmente entrambi di grado tecnico), seguita dalla procedura di lavaggio precedentemente descritta. 6.5.2 Tutti i solventi utilizzati in quantità relativamente rilevanti, che subiscono sensibili processi di concentrazione durante il percorso dell’analisi, devono essere sottoposti a un controllo di “idoneità analitica” preventiva per verificarne l’eventuale contributo al background procedurale. Per tale accertamento, volumi operativi dei solventi d’interesse, presi singolarmente, vengono concentrati a piccolo volume secondo quanto si verifica nell’applicazione del metodo e sottoposti a rilevamento HRGC-HRMS.

6.5.3 Il sodio solfato anidro viene purificato secondo la seguente procedura:
� riscaldamento in stufa a 150 °C per 24 ore; � esecuzione di 2 trattamenti di purificazione per estrazione con
diclorometano in bagno ad ultrasuoni (6 min); � esecuzione di 2 trattamenti di purificazione per estrazione con esano in
bagno ad ultrasuoni (6 min). Il sodio solfato viene quindi conservato in n-esano fino al momento dell’utilizzo. 6.5.4 La “Ottawa sand” viene purificata con una miscela esano-diclorometano 1:1 v/v eseguendo 3 estrazioni con l’estrattore PSE One (100 °C, 100 bar, 5 min); la Spe-edTM Matrix viene purificata con una miscela esano/diclorometano 1:1 v/v eseguendo 7 estrazioni con l’estrattore PSE One (100 °C, 100 bar, 5 min). 6.5.5 I lipidi naturalmente presenti nel campione e co-estratti insieme agli analiti di interesse possono interferire con la loro determinazione. Pertanto, il contenuto lipidico deve essere ridotto o eliminato utilizzando la procedura di purificazione riportata. 6.6 Conservazione del campione 6.6.1 Il campione omogeneizzato è congelato a –20 °C, fino all’analisi. 6.7 Procedura analitica 6.7.1 PREPARAZIONE DEL CAMPIONE Circa 10 g di campione, lasciati andare a temperatura ambiente, vengono mescolati con una spatola di acciaio; un’aliquota di 0.5–1.5 g viene prelevata ponendola in un pesafiltri e miscelata con circa 10 g di solfato di sodio anidro, precedentemente purificato. Il campione così preparato è lasciato riposare in un essiccatore per 12–24 h prima dell’estrazione. 6.7.2 ESTRAZIONE L’estrazione è condotta utilizzando l’estrattore PSE One. Nel serbatoio di estrazione vengono immessi partendo dal basso:
� un sottile strato di Spe-edTM Matrix; � il campione, miscelato con il solfato sodico anidro purificato, a cui vengono
aggiunti gli SI; � un sottile strato di Spe-edTM Matrix; � uno strato di “Ottawa sand”, fino a riempimento;

� un apposito filtro di cellulosa. L’estrazione viene condotta utilizzando il seguente programma:
Solvente n-esano/diclorometano 50:50 Temperatura 100 °C Pressione 100 bar Statica 5 minuti Cicli 3 Pausa No Flushing Program Solvent/gas/repeat flush: 20 sec / 2 min / 0
Circa 40 mL di estratto vengono raccolti in un’apposita vial e sottoposti a purificazione. 6.7.3 PURIFICAZIONE 6.7.3.1 L’estratto presenta sul fondo stratificata una piccola quantità di acqua (circa 1 mL). Dopo decantazione le due fasi vengono separate e la fase acquosa estratta con 3 aliquote di 3 mL di n-esano, che vengono aggiunte alla fase organica. Si aggiunge solfato di sodio anidro purificato, si agita e si lascia decantare. Si filtra, lavando la vial ed il solfato con 3 aliquote di 5 mL di esano. L’estratto viene ridotto di volume (5 mL) per evaporazione in flusso d’azoto con il TurboVap II e caricato nel sistema di purificazione Power-PrepTM equipaggiato con una colonna ABN ad alta capacità. La procedura utilizzata è la seguente:
� condizionamento della colonna con 250 mL di n-esano (flusso: 10 mL/min); � caricamento del campione (flusso: 5 mL/min); � eluizione con 250 mL di n-esano (flusso 5 mL/min);vengono raccolti 200
mL, scartando i primi 50 mL. 6.7.3.2 All’estratto, ridotto per evaporazione in flusso di azoto al volume di 0.5 mL, vengono aggiunti 4.5 mL di diclorometano e il volume viene nuovamente ridotto a 0.5 mL. La soluzione in diclorometano viene caricata sul sistema GPC e si eluisce con 150 mL di diclorometano; i primi 95 mL vengono scartati ed i secondi 55 mL raccolti. Dopo aggiunta di 2 µL di n-tetradecano, si porta con flusso d’azoto fin quasi a secchezza e poi si riprende con 100 µL della soluzione SING. Il sistema per purificazione GPC viene periodicamente tarato utilizzando la soluzione CLP GPC Calibration Mix. 6.7.4 DETERMINAZIONE STRUMENTALE 6.7.4.1 I NDL-PCB e i DL-PCB sono determinati tramite HRGC-HRMS (EI, 45 eV) operante in alta risoluzione (10000) in modalità MID ed utilizzando le seguenti condizioni gascromatografiche:

� gas di trasporto: He; flusso: 1.2 mL/min (constant flow); � iniezione in modalità splitless (purge time: 1 min; purge flow; 50 mL/min); � temperatura dell’iniettore: 300 °C; temperature della linea di trasferimento:
300 °C: programma di temperatura del forno: 120 °C (1 min), 20 °C/min fino a 150 °C (8 min), 4 °C/min fino a 235 °C (10 min), 12 °C/min fino a 290 °C (34 min).
6.7.4.2 Il rilevamento quantitativo viene eseguito impiegando la soluzione SQ. 6.7.5 CALCOLI E RISULTATI 6.7.5.1 La quantità assoluta del congenere considerato, Qxs, viene calcolata applicando l’equazione 1, avendo determinato i fattori di risposta relativi (FRRx); questi vengono stimati dalle aree dei segnali relativi alle due masse più abbondanti dei congeneri dei PCB. Il rapporto relativo alle due masse più abbondanti viene anche utilizzato per confermare qualitativamente la presenza del congenere nel campione.
x
m
xx
xxxs FRR
Q
SS
AAQ ×
++=
)21()21(
(1)
dove A1x e A2x sono le aree relative ai due segnali più intensi del congenere considerato nel campione; S1m e S2m sono le aree relative ai due segnali più intensi del PCB 13C-marcato aggiunto come standard interno al campione; Qm è la quantità assoluta del PCB 13C-marcato aggiunto come standard d’iniezione al campione; Il fattore di risposta, FRRx, del congenere considerato viene calcolato mediante l’Equazione (2):
x
m
xx
xxx C
C
ss
aaFRR ×
++=
)21()21(
(2)
dove a1x e a2x sono le aree relative ai due segnali più intensi del congenere considerato nella soluzione standard SFR; s1m e s2m sono le aree relative ai due segnali più intensi del PCB 13C-marcato presente nella soluzione standard SQ;

Cx e Cm sono le concentrazioni del congenere considerato e dello PCB 13C-marcato presente nella soluzione SQ. 6.7.5.2 I risultati analitici relativi alla concentrazione dei NDL-PCB e DL-PCB nel campione (Cxs) sono espressi in pg/g w/w (peso fresco) e vengono calcolati utilizzando la seguente Equazione:
xsxs
s
QC =
p (3)
dove ps è la massa utilizzata del campione di pesce. 6.7.5.3 La determinazione del recupero (%) dei congeneri marcati viene effettuata calcolando la concentrazione rispetto ai PCB di riferimento presenti nella soluzione SR mediante l’Equazione (4):
×concentrazione trovatarecupero (%) = 100
concentrazione aggiunta (4)

Tabella 6.1. Elenco dei PCB considerati.
omologo PCB IUPAC
omologo PCB IUPAC
omologo PCB IUPAC
1 104 179
2 103 176
1
3 100 178
10, 4 93, 95 175
7, 9 91 187
6 92 183
8,5 101, 84, 90 185
14 99 174
12, 13 119 177
2
15 83 171
19 97 173
18 87, 115, 117 172
17 85 180
24, 27 110 193
16, 32 82 191
34 124 170, 190
29 109
7
189
26 123 202
25 118 201
31,28 114 197
20, 33 122 200
3
22 105 199
54
5
126 203, 196
53 136 195
45 154 194
51 151
8
205
46 135, 144 208
69 147 207
52, 73 149
9
206
49 134 10 209
47, 48, 75 131, 130 44 146
59, 42 153 71, 41, 64 132
40 141 67 137 63 165 74 164, 138, 163 70 158 66 129
56, 60 128, 167 81 156
4
77
6
157 169
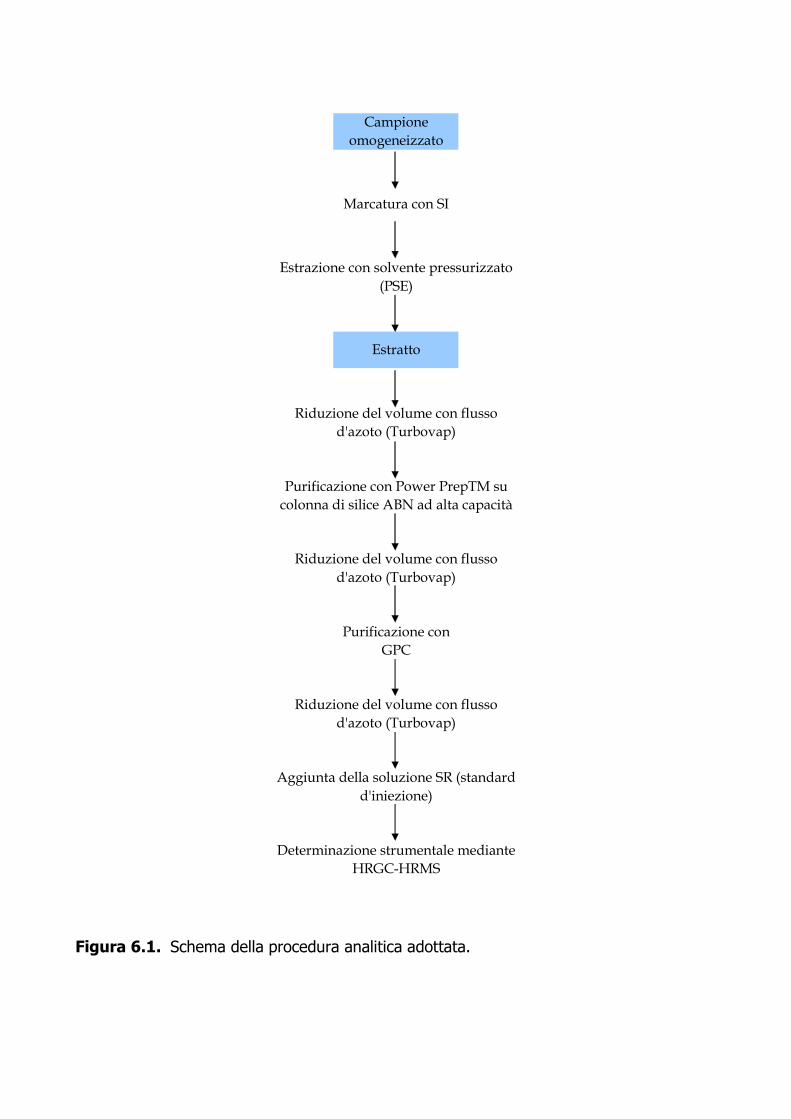
Riduzione del volume con flusso
d'azoto (Turbovap)
Riduzione del volume con flusso
d'azoto (Turbovap)
Purificazione con
GPC
Purificazione con Power PrepTM su
colonna di silice ABN ad alta capacità
Riduzione del volume con flusso
d'azoto (Turbovap)
Aggiunta della soluzione SR (standard
d'iniezione)
Determinazione strumentale mediante
HRGC-HRMS
Campione
omogeneizzato
Marcatura con SI
Estratto
Estrazione con solvente pressurizzato
(PSE)
Figura 6.1. Schema della procedura analitica adottata.

7. Policlorobifenili idrossilati (HO-PCB) in campioni di siero
ematico
Istituto Superiore di Sanità, Dipartimento Ambiente e Connessa Prevenzione Primaria,
Reparto di Chimica Tossicologica
Avvertenze Il metodo proposto è stato inizialmente sviluppato per la determinazione di policlorobifenili idrossilati (HO-PCB) in campioni di siero umano con produzione di dati accurati e ripetibili come mostrano i risultati ottenuti riportati nell’Allegato 7.1. Acronimi GC gascromatografia HO-PCB policlorobifenili idrossilati HR alta risoluzione LR bassa risoluzione MS spettrometria di massa MTBE metil ter-butiletere SI standard interno NCI negative-ion chemical ionization

7.1 Scopo e applicazione 7.1.1 Il metodo è proposto per la quantificazione di HO-PCB in campioni di biota acquatico mediante gascromatografia ad alta risoluzione-spettrometria di massa a bassa risoluzione (HRGC-LRMS) con tecnica di ionizzazione chimica negativa (NCI). 7.1.2 Il metodo consente la determinazione dei HO-PCB elencati in Tabella 1. 7.2 Principio del metodo 7.2.1 Il rilevamento degli analiti di interesse è basato sull’impiego di standard interni (SI), completamente marcati con 13C. Gli SI vengono aggiunti al campione prima di qualsiasi operazione analitica, ed eventualmente di passaggi analitici intermedi ritenuti critici (l’elenco degli SI utilizzati è riportato in Tabella 1). La componente lipidica del siero è estratta con una miscela 1:1 di n-esano-metil ter-butiletere (MTBE), e gli HO-PCB sono quantificati come metossi-derivati (MeO-PCB) mediante rilevamento con HRGC-LRMS dopo derivatizzazione con una soluzione eterea di diazometano. 7.3 Strumentazione
� HRGC-LRMS Trace GC Ultra, Thermo Electron Corporation. 7.4 Reagenti e standard 7.4.1 REAGENTI E MATERIALE CROMATOGRAFICO
� Acido cloridrico 6 M; � n-esano; � metil ter-butiletere (MBTE); � potassio idrossido 5 N; � alcool iso-propilico; � etere etilico; � acido solforico concentrato; � n-tetradecano; � N-metil-N-nitroso urea; � acqua distillata.
7.4.2 STANDARD
� congeneri HO-PCB, standard naturali, purezza > 99 % (Wellington Laboratories); � congeneri HO-PCB, standard 13C-marcati, purezza > 99 % (Wellington
Laboratories); � Clordano 13C-marcato impiegato come standard d’iniezione.

7.5 Procedura analitica 7.5.1 PREPARAZIONE DEL CAMPIONE 2 g di siero di pesce, trasferiti in provetta da centrifuga e addizionati con una soluzione a titolo noto contenente gli SI 13C-marcati 4-HO-PCB 120 e 4-HO-PCB 172, sono agitati su vortex e conservati in frigorifero per una notte. Successivamente la denaturazione delle proteine contenute nel campione è realizzata aggiungendo 200 µL di acido cloridrico 6 M e 1.2 mL di alcool iso-propilico, e lasciando il campione così preparato in agitazione per 10 min in un bagno a ultrasuoni. 7.5.2 ESTRAZIONE Il campione è sottoposto a due estrazioni sequenziali in centrifuga (3500 giri, 10 min) utilizzando due porzioni di una miscela 1:1 n-esano-MTBE (2 mL a porzione). Gli estratti sono riuniti, portati a quasi secchezza, e ripresi con 1 mL di n-esano. 7.5.3 PURIFICAZIONE L’estratto esanico in provetta da centrifuga è addizionato con 2 mL di H2SO4 concentrato. La miscela è agitata vigorosamente e sottoposta a centrifugazione (3500 giri, 10 min). La fase esanica surnatante è trasferita quantitativamente in vial a fondo conico da 8 mL e addizionata con una soluzione a titolo noto contenente gli SI 13C-marcati 4-HO-PCB 159 e 4-HO-PCB 187. 7.5.4 DERIVATIZZAZIONE DEGLI HO-PCB Dopo l’aggiunta degli SI (cfr. 7.5.3), il campione è lasciato reagire per l’intera notte alla temperatura di 4 °C con un eccesso di soluzione eterea di diazometano (derivatizzante), preparata di fresco come descritto nell’Allegato 7.2. In genere sono sufficienti 500 µL di soluzione derivatizzante per trasformare quantitativamente i possibili HO-PCB presenti nei corrispondenti metossi-derivati. Tuttavia, se dopo l’aggiunta della soluzione derivatizzante (di colore giallo), il campione non diventa giallo o il colore non perdura per almeno 2 min, è necessario aggiungere altra soluzione derivatizzante fino al verificarsi della condizione. Successivamente l’eccesso di diazometano è eliminato evaporando sotto leggero flusso di azoto fino a scomparsa del colore giallo. Dopo l’aggiunta di 1 µL di n-tetradecano, il campione è portato a quasi secchezza sotto leggero flusso d’ azoto, ripreso con 100 µL di clordano 13C-marcato, e trasferito in vial per auto-campionatore. 7.5.5 DETERMINAZIONE STRUMENTALE Gli HO-PCB sono determinati come MeO-PCB mediante HRGS-LRMS (NCI, ionizzazione chimica negativa con metano) secondo le condizioni riportate nell’Allegato 7.3.

Tabella 7.1. Congeneri di HO-PCB rilevanti ai fini degli accertamenti d’interesse e congeneri 13C-marcati utilizzati come SI.
Congeneri nativi Congeneri 13C-marcati
4-OH-PCB 107 2,3,3',4',5-Pentacloro-4-bifenilolo 13C-4-HO-PCB 120 2',3,4',5,5'-Pentacloro-4-[13C12]-bifenilolo 3-OH-PCB 138 2,2',3',4,4',5-Esacloro-3- bifenilolo 13C-4-HO-PCB 159 2',3,3',4',5,5'-Esacloro-4-[13C12]-bifenilolo 4-OH-PCB 146 2,2',3,4',5,5'- Esacloro-4- bifenilolo 13C-4-HO-PCB 172 2,2',3,3',4',5,5'-Eptacloro-4-[13C12]-bifenilolo 4-OH PCB 172 2,2',3,3',4',5,5'-Eptacloro -4- bifenilolo 13C-4-HO-PCB 187 2,2',3,4',5,5',6-Eptacloro-4-[13C12]-bifenilolo 4-OH-PCB 187 2,2',3,4',5,5',6-Eptacloro-4- bifenilolo

ALLEGATO 7.1
Risultati ottenuti nello sviluppo del metodo di determinazione di policlorobifenili idrossilati (HO-PCB) in campioni di siero umano
SIERO UMANO Lo studio dell’esattezza e della precisione (ripetibilità e riproducibilità intra-laboratorio, in termini di deviazione standard e CV%) del metodo descritto nel presente allegato è stato effettuato analizzando cinque sottocampioni di siero umano (≈ 5 g ognuno, identificati come P1, P2, P3, P4, P5), ottenuti da uno stesso campione omogeneo. Come “bianco” è stata utilizzata acqua distillata (≈ 5 g) priva degli analiti d’interesse. In assenza di un appropriato materiale di riferimento, lo studio dell’accuratezza è stato condotto aggiungendo quantità note di standard 13C-marcati (risultatati mostrati in Tabella A.7.1). I dati ottenuti nello studio di ripetibilità del metodo sono riportati in Tabella A.7.2.

Tabella A.7.1 Studio dell’accuratezza del metodo applicato a campioni di siero umano: rese di recupero (%) degli standard 13C-marcati.
13C-4-HO-PCB 120 13C-4-HO-PCB 159 13C-4-HO-PCB 172 13C-4-HO-PCB 187
P1 77 105 84 106 P2 85 130 89 107 P3 71 123 72 102 P4 77 119 77 101 P5 49 68 51 68
Bianco 62 112 64 92
Tabella A.7.2 Studio di ripetibilità del metodo applicato a campioni di siero umano: dati di concentrazione espressi in ng/g fw.
4-HO-PCB 107 3-HO-PCB 138 4-HO-PCB 146 4-HO-PCB 172 4-HO-PCB 187
P1 0.065 0.043 0.018 0.011 0.068 P2 0.059 0.039 0.017 0.013 0.065 P3 0.055 0.033 0.016 0.012 0.058 P4 0.062 0.036 0.018 0.014 0.063 P5 0.057 0.036 0.017 0.014 0.06
<X> 0.06 0.037 0.017 0.013 0.063 CV% 6 10 4 12 6

ALLEGATO 7.2 Preparazione della soluzione etera di diazometano Ad apparecchio aperto porre nella parte interna del contenitore in vetro: � 133 mg di N-metil-N-nitroso urea � 500 µL di acqua distillata e nella parte esterna: 3 mL di etere etilico Chiudere il contenitore e aggiungere con una siringa attraverso il setto forabile posto sul tappo del contenitore: � 600 µL di una soluzione di potassio idrossido 5 N (possibilmente preparata di fresco: per 10 mL di soluzione pesare 2.8 g di potassio idrossido, ovvero circa 25 pasticche) Lasciare reagire ≈ 1 ora Ipotizzando una resa del 100 % si ottiene ≈ 1 mmole di diazometano; ipotizzando invece (come indicato nella ricetta originale per un tempo di reazione di 45 min) una resa del 60 % si ottengono 0.6 mmoli di diazometano in 3 mL di etere etilico, dunque: 0.6 mmoli/3mL = 0.2 M = 2*105 pmoli/µL Si ricorda che la procedura di preparazione della soluzione di diazometano così come la reazione di derivatizzazione va eseguita rigorosamente sotto cappa.

ALLEGATO 7.3 Condizioni operative suggerite per la determinazione strumentale degli analiti di interesse HRGC-LRMS (NCI)
� colonna gas cromatografia: HT8-PCB 60 m x 0.25 mm � gas di trasporto: metano; � iniettore: PTV; � programmata GC per: 70 °C per 1 min, 30 °C/min fino a 230 °C, 3°C/min fino
280 °C, 20 °C/min fino a 330 °C per 4 min.