







Le lingue
Pagine
Legale


UTS Materiali e Nuove Tecnologie, Centro Ricerche Casaccia , Roma - Italy
Fabrizio CleriENEA, Unità Materiali e Nuove Tecnologie
Centro Ricerche Casaccia, Roma
La simulazione al calcolatore come strumentodi progettazione di materiali avanzati
Staff Team :
Fabrizio Cleri (ENEA Casaccia)
Gregorio D’Agostino (ENEA Casaccia)
Pier Giuseppe Gabrielli (ENEA Casaccia)
M ichele Gusso (ENEA Brindisi)
Principali collaborazioni in corso:
Luciano Colombo (Cagliari)
Giovanni Ciccotti (Roma “La Sapienza”)
Antonio Di Carlo (Roma Tre)
Patrizia Trovalusci (Roma “La Sapienza”)
Pawel Keblinski (Rennselaer, NY)
Susan B. Sinnott (U. Florida, Gainesville)
C. Delerue (ISEN Lille)
I nostri collaboratori:
M anuela Volpe
Sara Letardi
Vittorio Sansalone
M ariella Ippolito
Andrea M oriani (ENEA Frascati)
Alessandro M attoni (Cagliari)
Alessandra Satta (*)
Enrico Pisanu (*)
M anlio M essina (*)
Sonia Costantini (*)
Luigi Brambilla (*)
UTS Materiali e Nuove Tecnologie, Centro Ricerche Casaccia , Roma - Italy
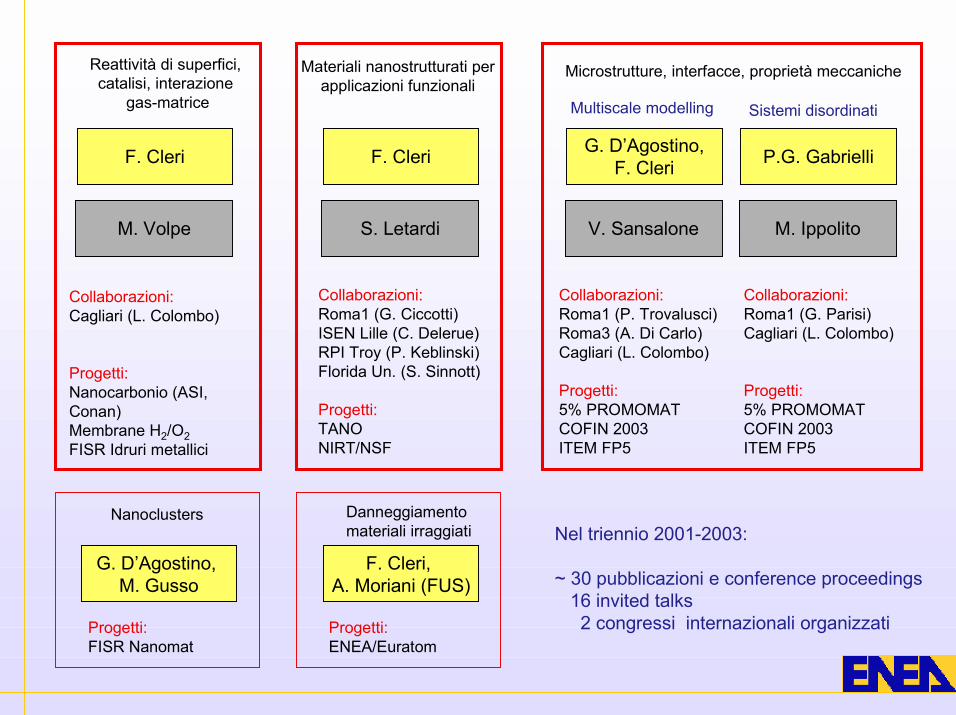
F. Cleri
Reattività di superfici,catalisi, interazione
gas-matrice
Materiali nanostrutturati perapplicazioni funzionali
Microstrutture, interfacce, proprietà meccaniche
M. Volpe
G. D’Agostino,F. Cleri
V. Sansalone
P.G. Gabrielli
M. Ippolito
F. Cleri
S. Letardi
Collaborazioni:Cagliari (L. Colombo)
Progetti:Nanocarbonio (ASI,Conan)Membrane H2/O2FISR Idruri metallici
Collaborazioni:Roma1 (G. Ciccotti)ISEN Lille (C. Delerue)RPI Troy (P. Keblinski)Florida Un. (S. Sinnott)
Progetti:TANONIRT/NSF
Nanoclusters
G. D’Agostino, M. Gusso
Progetti:FISR Nanomat
Danneggiamentomateriali irraggiati
F. Cleri, A. Moriani (FUS)
Progetti:ENEA/Euratom
Sistemi disordinatiMultiscale modelling
Collaborazioni:Roma1 (P. Trovalusci)Roma3 (A. Di Carlo)Cagliari (L. Colombo)
Progetti:5% PROMOMATCOFIN 2003ITEM FP5
Collaborazioni:Roma1 (G. Parisi)Cagliari (L. Colombo)
Progetti:5% PROMOMATCOFIN 2003ITEM FP5
Nel triennio 2001-2003:
~ 30 pubblicazioni e conference proceedings 16 invited talks 2 congressi internazionali organizzati

RISORSE HARDWARE/SOFTWARE (Il “laboratorio di fisica computazionale”…?)
-Cluster di PC Linux (5 Athlon 1-1.8 Ghz + 1+1 twin-AMD 2.8 Ghz) + alcuni Mac G4/G5 OsX
-Sistema operativo Linux Debian Woody 3.1, GNU Fortran/C/C++, OpenOffice-Grafica di base: Xmgr, GNUplot, PGPlot + pacchetti sviluppati in casa-Grafica avanzata: JMOL, gOpenMol
-Principali software di sviluppo:-CAST (“Casaccia Atomistic Simulation Tools”) comprende tutto il necessario per dinamica molecolare classica e tight binding, Monte Carlo (HELLF), in diversi ensemble termodinamici e con diversi potenziali di interazione-ABINIT, codice di simulazione di struttura elettronica e dinamica molecolare da principi primi, sviluppato da UL Bruxelles-fhi98md, fhi98PP, codice di simulazione di struttura elettronica da principi primi, e modulo per la generazione di pseudopotenziali a partire da funzioni d’onda all electron

DINAMICA MOLECOLARE - Modelli su scala atomica dei materiali
Gli atomi sono descritti esplicitamente come masse puntiformi che interagiscono medianteforze su scala atomica.L’evoluzione del sistema si ottiene integrando le equazioni newtoniane del moto per ciascunatomo, nel campo di potenziale determinato dall’interazione complessiva tra gli atomi.
Le forze interatomiche si possono ottenere da unpotenziale empirico derivabile analiticamente…Gli elettroni sono cioè “congelati” nello statofondamentale, rendendo il calcolo molto efficiente.Si possono così studiare sistemi di dimensioni finoa qualche frazione di micrometro.
Oppure, il legame esplicito tra atomi può esseredescritto mediante gli elettroni di legame. In questocaso le forze hanno una base quanto-meccanica.Questo approccio è computazionalmente molto piùpesante ed è quindi limitato a sistemi composti alpiù da qualche decina-centinaia di atomi. (J. Schiotz et al., Nature 391, 561 (1998))

METODI “MONTE CARLO” : Coarse-graining dei gradi di libertà
Simulazione Grand-Canonical Monte Carlo delfisisorbimento di H2 in una matrice di nanotubi dicarbonio idrogenati.(M. Volpe, F. Cleri, Chem. Phys. Lett. 231, 133 (2003))
I metodi Monte Carlo sono basati sullastima di integrali multi-dimensionalimediante tecniche di campionamentostocastico.
L’integrale da calcolare può essere unafunzione termodinamica, un funzionale dienergia totale, il kernel di una equazioneintegrale (ad es., equazione diBoltzmann), o altro ancora.
Le variabili indipendenti sono solo unsottoinsieme limitato di tutte le variabilicanoniche del sistema.
Campionando le variabili in base a unafunzione di distribuzione data, si calcolail valore corrispondente dell’integrando ese ne ottiene così una media statistica.

SOMMARIO:
• La Scienza dei Materiali Computazionale
• Il paradigma della modellistica multiscala
• Modellistica multiscala di microstrutture
• Modelli di nanomateriali per elettronica molecolare

“Computational Materials Science”
Un innovativo approccio sistematico alla Scienza dei Materiali che combina:
• Ricerca interdisciplinare• Esperimenti di laboratorio critici• Sviluppo di metodi e tecniche computazionali
allo scopo di ridurre il gap tra il “concetto” e il “prodotto finale”.
I benefici di un tale approccio combinato sono (almeno) i seguenti:
- scoraggiare progressivamente il costoso approccio “trial-and-error” e la serendipitàdell’atteggiamento “Edisoniano” tradizionale
- aumentare la probabilità che i nuovi materiali così sintetizzati possiederanno leproprietà desiderate quando trasferiti dalla scala di laboratorio
- diminuire la tendenza a perseguire soluzioni già “compromesse” e/o troppoconservative che risultano da tecnologie preesistenti e meno affidabili su base“fondamentale”…

MOTIVAZIONI: PERCHE’ LE SIMULAZIONI AL CALCOLATORE?
IL PUNTO DI VISTA DEL “TEORICO”:
Le simulazioni numeriche sono alternative (non “sostitutive”) agli esperimenti,nel senso che forniscono una conoscenza fondamentale sintetica derivante dauna formulazione strettamente teorica e astratta di un problema.
IL PUNTO DI VISTA DELLO “SPERIMENTALE”:
Le simulazioni sono complementari agli esperimenti, ne supportanol’interpretazione, e rappresentano una specie di microscopio (o “omniscopio”)ideale mediante il quale ottenere il più completo dettaglio dei sistemi e processidi interesse.

Il paradigma “multiscala” nella Scienza dei Materiali Computazionale
ElettroniNucleiAtomi
FrammentimolecolariAngoli di legameForce fields
Interazioni di superficieOrientazioneImpaccamento reticolare
CostituentiFasiDanneggiamento
Previsioni quantitative Previsioni qualitative
Meccanica quantistica Nano-meccanica Meso-meccanica Micromeccanica
Fibra
Matrice
Tempo (s) 10-15 10-12 10-9 10-6 10-3 100
Lunghezza (m) 10-12 10-9 10-6 10-3 100

Studio di difetti isolati:bordi grano, dislocazioni,
microcricche in Si
Interazione tra difetti:creazione di giunzioni triple,dislocazioni vs. bordi grano,microcricche vs. giunz. triple
Evoluzione dimicrostrutture su
scala atomica
Evoluzione di microstrutturesu scala mesoscopica, sistemi
disordinati
Modelli di materiali compositi suscala macroscopica, elementi finiti,
continui alla Cosserat
Codici industriali,Abaqus, Nastran,
FEMLab….

Modellistica multiscala dell’evoluzione di microstrutture
Lamellar austenite-bainite microstructurein heat-treated (700 °C) carbon steel.
Natural pyroclastic granite from theAppennine mountains (central Italy).
Polycrystalline Silicon produced by100 ns laser-pulse recrystallization ofamorphous Si film.

INTERAZIONE FRA DIFETTI: GEOMETRIA vs. TOPOLOGIA
Ruolo delle giunzioni triple nell’evoluzione microstrutturale
Le giunzioni triple sono i siti dove avvengono le modificazioni topologiche dellamicrostruttura, cioè variazioni della connettività e del numero dei difetti:
Tutti gli altri eventi evolutivi (ad es. migrazione e slittamento dei bordi grano, diffusioneatomica, deformazione plastica nei grani, propagazione di cricche) contribuiscono allemodificazioni geometriche changes, cioè variazioni localizzate di dilatazione/contrazioneche preservano la auto-similarità topologica della microstruttura.
T1 T2 anti-T2
(F. Cleri., G.D’Agostino, A.Satta and L.Colombo, Computational Materials Science 24 (2002) 21)

I Twin boundaries sui piani {111} esibiscono simmetria mirror su scala atomica con energiadi eccesso quasi 0: sono difetti di crescita comuni in Si, Ge, C …
Il twinning multiplo si verifica quando unostesso cristallo forma dei twin su piani {111}non-equivalenti: si forma un terzo bordo granoche, per simmetria fcc, può essere solo un{221} o un {114} symmetric-tilt (anche dettotwin secondario).
I tre bordi grano si congiungono ad una linea(un punto in 2D), cioè formano una giunzionetripla.
Nella nomenclatura CSL il {111} è Σ3 e tantoil {221} che {114} sono Σ9 : quindi la giunzionetripla è detta una Σ3-Σ3-Σ9 ...
DINAMICA MOLECOLARE DI DIFETTI ISOLATI: GIUNZIONI TRIPLE
Scelta di un sistema modello rappresentativo: Twin boundaries in Si

Si ottiene la struttura atomica dei due bordi grano Σ3 e Σ9 isolati. Porzioni dei due bordigrano vengono tagliati e ruotati in modo da costruire una giunzione tripla
Il legame direzionale in Si è descritto con il potenziale Stillinger-Weber.
La struttura risultante è equilibrata a T~500 K e raffreddata a T=0 K mediante MD.
Condizioni al bordo a trazione costante sono impiegate per immergere il sistema in untricristallo Σ3-Σ3-Σ9 infinito.
(see: F. Cleri, Phys. Rev. B65 (2002) 014137)
Σ Σ Σ Σ 3
Σ Σ Σ Σ 3
Σ Σ Σ Σ 9
HREM of a multiple twin in Si

Canpo di stress su scalaatomica intorno alla giunzionetripla ΣΣΣΣ3-ΣΣΣΣ3-ΣΣΣΣ9
Un lobo di stress compressivo (bianco)si oppone a un lobo di stress tensile(nero) formando un dipolo di stress deldiametro di circa 3 nm.
La giunzione tripla funziona come unconcentratore di stress (propriocome una microcricca, unadislocazione….
Ad una analisi più accurata la TJassomiglia molto ad unadisclinazione deformata.
zzσ (kBar)
(S. Costantini, P.Alippi, L. Colombo and F.Cleri, Phys. Rev. B 63, 045602 (2000) )

MICROMECCANICA DELLA FRATTURA: Interazione fra una giunzione tripla e una microcricca
(A. Satta, L. Colombo, E. Pisanu, F. Cleri, J. Phys. Cond. Matt. 14 (2002) 13030)

La configurazione iniziale del substrato è ottenuta con i seguenti passi:
• Si definiscono dei centri dinucleazione: un volume cilindricodi raggio dato attorno a ogni nucleo.
(2) I cilindri sono “congelati” mentreil resto del sistema viene fuso adelevata temperatura.
(3) I semi cristallini cilindrici vengonoquindi ruotati a caso entro il fuso.
(4) Da questa configurazione, latemperatura del sistema viene poiabbassata disotto del punto difusione. I semi cristallini possono oracrescere fino al completo esaurimentodel liquido.
UN ESEMPIO DI SINTESI DI UNA NANOSTRUTTURA COMPATTA
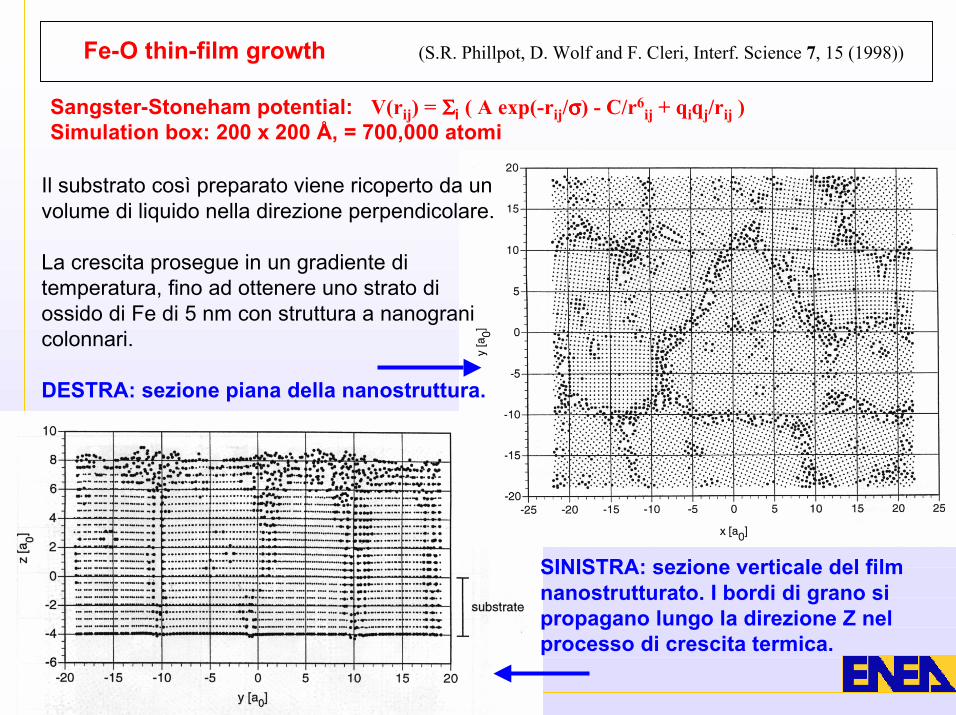
SINISTRA: sezione verticale del filmnanostrutturato. I bordi di grano sipropagano lungo la direzione Z nelprocesso di crescita termica.
Fe-O thin-film growth (S.R. Phillpot, D. Wolf and F. Cleri, Interf. Science 7, 15 (1998))
Sangster-Stoneham potential: V(rij) = ΣΣΣΣi ( A exp(-rij/σσσσ) - C/r6ij + qiqj/rij )
Simulation box: 200 x 200 Å, = 700,000 atomi
Il substrato così preparato viene ricoperto da unvolume di liquido nella direzione perpendicolare.
La crescita prosegue in un gradiente ditemperatura, fino ad ottenere uno strato diossido di Fe di 5 nm con struttura a nanogranicolonnari.
DESTRA: sezione piana della nanostruttura.

Simulazione Monte Carlo di sistemi dinamici dissipativi
Sviluppo di una microstruttura lamellare in un gradiente (verticale) di orientazione atemperatura costante. I soli gradi di libertà descritti esplicitamente sono i vertici dove siincontrano 3 bordi di grano. La dinamica stocastica fa evolvere l’insieme dei bordi di grano inmodo da minimizzare l’energia dissipata.
0
0.2
0.4
0.
6
0
.8
1.0
1
.2
⟨ θ ⟩
(rad)
(F. Cleri, Physica A 282, 339 (2000); F.Cleri & G. D’Agostino, J. Mat. Res. 17,117 (2002))

MODELLO SU RETICOLO DI UN COMPOSITO MICROFESSURATO
A livello microscopico il composito è descritto come una matrice continua in cui sonoimmerse una distribuzione di fibre rigide, e una distribuzione stazionaria di microcricche.
I due sottoreticoli interagenti sono descritti da un modulo :- un reticolo di particelle rigide connesse da N travi linear-elastiche, che rappresentano la matrice con le fibre- un reticolo di M fessure ellittiche interagenti che rappresenta le microcricche
ta
i
tb
i
zk
j
zh
jr h
l
r al
(P. Trovalusci and R. Masiani, Int. J. Solids & Str. 36, 2091 (1999) P. Trovalusci and G. Augusti, J. Phys. IV, 8, 383 (2000) )
MODULO
Questo modello su reticolo “parla” direttamentecon le simulazioni su scala atomica.
INPUT: interazioni microscopiche fra matrice,fibre e cricche
OUTPUT: funzionale energia per un elementofinito del modello continuo con gradi di libertàinterni.

MODELLO CONTINUO EQUIVALENTE
L’ energia in un dominio continuo C dotato di microstruttura descritto dai campiomogenei u(x), R(x) and d(x) è:
(G. Capriz, Continua with Microstructure, Springer-Verlag 1989) ΠC u,R,d( ) = S ⋅ ∇u + 1
2 S ⋅ ∇R + z ⋅ d + P ⋅ ∇d( )dVC∫
Ciascun elemento di volume rappresentativo (RV) del continuo è descritto a scalamicroscopica da un modulo del reticolo discreto.
LHRV
q
Ad esempio, una trave dimateriale composito di lunghezzaL e sezione H soggetta ad uncarico assiale uniforme q
Modellistica di materiali nanostrutturatiper applicazioni funzionali

BENZO-TIOLO/TIOLATO SU CLUSTER DI Au
In queste simulazioni di struttura elettronica mediante la teoria del Funzionale Densità(DFT) viene accuratamente descritta la chimica dei legami molecola-metallo, la strutturaenergetica dei livelli molecolari, il rilassamento ionico, lo shift dei livelli molecolari…
In questa simulazione si osserva, fra l’altro, l’evidenza della stereo-specificità del legametiolo S-H, passando da tiolo a tiolato, con chemisorbimento di H sul cluster.
(S. Letardi e F. Cleri, J. Chem. Phys., submitted)

Building block: di-benzene di-tiolatosu un cluster planare di Au(3).Unità elementare da replicare: DBDT su untrimero di Au.Alternando con trimeri vuoti si possonosimulare diversi coverage della superficie.
DINAMICA MOLECOLARE CLASSICA
Simulazione di self-assembled monolayers (SAM) sulla superficie Au (111).Codice di dinamica molecolare DLProtein (sviluppato dal Dipartimento di Fisica,Università di Roma “La Sapienza”, sulla base del codice DLPoly).
Simulazioni a temperatura finite di monolayers con diverse ricoperture dellasuperficie.Termostato di Nosé-Hoover per il controllo della temperatura.All-atom (Amber) force field. Dati per C-S-Au derivati dalle simulazioni ab-initio.

FULL Au (111) SURFACE COVERAGE

HALF-COVERAGETendenza a formare aggregati di Van der Waals

Una nanogiunzione tra due nanotubi di carbonio ottenuta medianteirraggiamento con fascio di elettroni e fusione localizzata.
(P. Ajayan et al., PRL 89, 2002)

In questo caso usiamo la dinamica molecolare tight-binding (TBMD) per sudiare lastruttua atomica ed elettronica di giunzioni modello tra nanotubi di carbonio (5,5):
Hamiltoniana di Wang-Ho per le interazioni C-C (Xu et al., J. Phys. Cond. Matt. 1992).
Otteniamo modelli di strutture irraggiate con un modello di dinamica di non-equilibrioimpiegando il potenziale empirico di Tersoff (Jang & Sinnott, Florida).Quindi le strutture così ottenute sono “passate” alla TBMD per il calcolo elettronico.
Sulla matrice degli autovalori della H si svolge la analisi di localizzazione studiando il“participation ratio”.

Modello TBMD di una giunzioneirraggiata da fascio elettronico.
La regione centrale è fortementedisordinata (praticamente amorfa), conun elevata concentrazione di difetti dicoordinazione e topologici.
La DOS contiene molte bande di statifortemente localizzati.
In particolare, tra 0. e 0.5 eV gli statielettronici sono totalmente localizzati.
(F. Cleri et al., Phys. Rev. Lett., submitted)

Gli stati elettronici sonolocalizzati e quantizzati nellaregione di giunzione deinanotubi.
La regione di giunzione formacioè un QUANTUM DOT.
LOCALIZZAZIONE + QUANTIZZAZIONE …
Plot della |Ψ(z)|2 lungo l’asse dei nanotubi per alcuni stati vicino ad EF
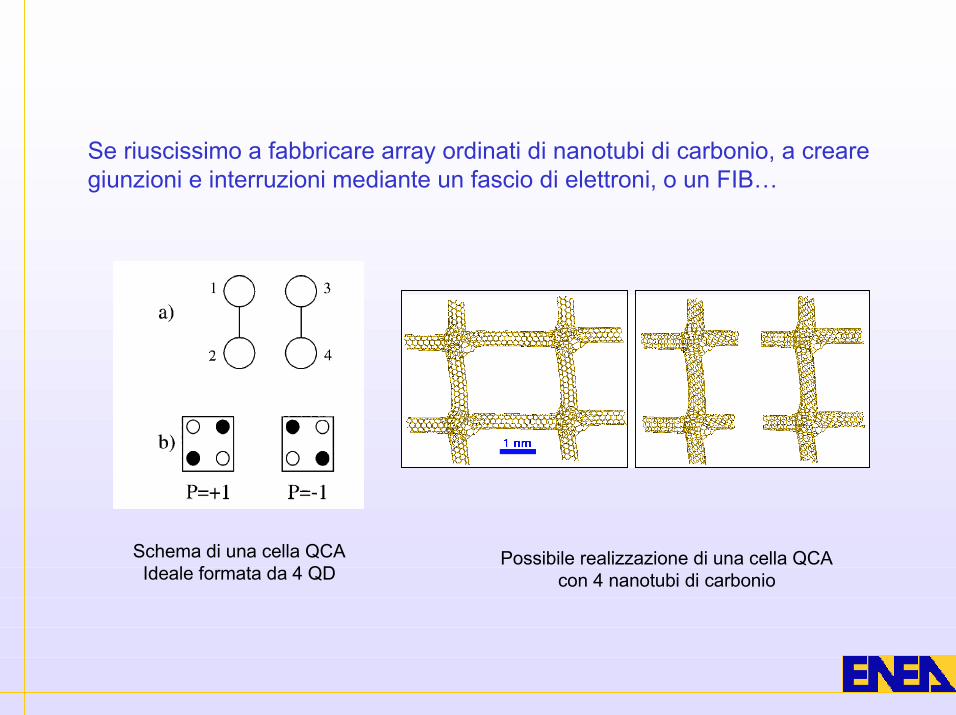
Schema di una cella QCAIdeale formata da 4 QD
Possibile realizzazione di una cella QCAcon 4 nanotubi di carbonio
Se riuscissimo a fabbricare array ordinati di nanotubi di carbonio, a crearegiunzioni e interruzioni mediante un fascio di elettroni, o un FIB…

In conclusione…
Le simulazioni al calcolatore offrono una opportunità unicaalle nanotecnologie: per la prima volta è infatti possibileconfrontare modelli teorici e dati sperimentali in scala 1:1(almeno per le lunghezze…).
Sussistono limitazioni intrinseche di scala per le simulazioniatomistiche, che non potranno essere superate neanche conun eccezionale aumento della potenza di calcolo.Viene in aiuto il paradigma multiscala, il quale però richiede undeciso cambiamento di prospettiva.
Peraltro, l’improvement dei metodi teorici e degli algoritminumerici, unito allo sviluppo di tecniche di intelligenza artificiale, potrà rendere solubili problemi oggi ancora lontanidallo stadio di fattibilità tecnologica (e.g., materiali ibridi).
Top Related