
UNIVERSITA DEGLI STUDI DI PADOVA` FACOLTA DI...
66
UNIVERSIT ` A DEGLI STUDI DI PADOVA FACOLT ` A DI INGEGNERIA DIPARTIMENTO DI INGEGNERIA DELL’INFORMAZIONE RELAZIONE LOG AVERAGE SCORING PER ALLINEAMENTI PROFILO CONTRO PROFILO DI PROTEINE Relatore: Ch.mo Prof. Carlo Ferrari Correlatore: Ch.mo Prof. Silvio C.E. Tosatto Laureanda: Alessandra Mantovan A.A. 2004/2005 Padova, 18 luglio 2005
-
Upload
nguyenthuan -
Category
Documents
-
view
214 -
download
0
Transcript of UNIVERSITA DEGLI STUDI DI PADOVA` FACOLTA DI...

UNIVERSITA DEGLI STUDI DI PADOVA
FACOLTA DI INGEGNERIA
DIPARTIMENTO DI INGEGNERIA DELL’INFORMAZIONE
RELAZIONE
LOG AVERAGE SCORING PER ALLINEAMENTI
PROFILO CONTRO PROFILO DI PROTEINE
Relatore: Ch.mo Prof. Carlo FerrariCorrelatore: Ch.mo Prof. Silvio C.E. Tosatto
Laureanda: Alessandra Mantovan
A.A. 2004/2005Padova, 18 luglio 2005

Ringraziamenti
Rivolgo un sentito ringraziamento al prof. Carlo Ferrari e al prof. Silvio CarloErmanno Tosatto per la disponibilita alla realizzazione di questo Progetto e per lafiducia accordatami.
Ringrazio inoltre tutte le persone che ho conosciuto presso Centro di RicercaInterdipartimentale di Biotecnologie Innovative di Padova ed in particolare coloro iquali mi hanno affiancato durante il lavoro.
Ringrazio tutta la mia famiglia e infine in modo particolare Enrico per il sostegnoe l’affetto che mi ha dimostrato durante tutto il mio percorso universitario.

Indice
Introduzione 3
1 Proteine e loro struttura 41.1 Struttura primaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.2 Struttura secondaria . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.3 Struttura terziaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101.4 Struttura quaternaria . . . . . . . . . . . . . . . . . . . . . . . . . . . 111.5 Ripiegamento delle proteine o folding . . . . . . . . . . . . . . . . . 12
2 Metodi sperimentali 132.1 Determinazione della struttura proteica . . . . . . . . . . . . . . . . . 13
2.1.1 Cristallografia ai raggi X . . . . . . . . . . . . . . . . . . . . . 132.1.2 Spettroscopia a risonanza magnetica nucleare (NMR) . . . . . 142.1.3 Conclusioni . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.2 Protein Data Bank (PDB) . . . . . . . . . . . . . . . . . . . . . . . . 16
3 Allineamento di sequenze proteiche 193.1 Similarita e omologia . . . . . . . . . . . . . . . . . . . . . . . . . . . 193.2 Allineamento . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.3 Matrici di sostituzione . . . . . . . . . . . . . . . . . . . . . . . . . . 20
3.3.1 Matrici PAM . . . . . . . . . . . . . . . . . . . . . . . . . . . 213.3.2 Matrici BLOSUM . . . . . . . . . . . . . . . . . . . . . . . . . 233.3.3 Confronto tra matrici PAM e matrici BLOSUM . . . . . . . . 23
3.4 Algoritmi di programmazione dinamica . . . . . . . . . . . . . . . . . 233.4.1 Algoritmo di Needleman-Wunsch . . . . . . . . . . . . . . . . 243.4.2 Algoritmo di Smith-Waterman . . . . . . . . . . . . . . . . . . 25
3.5 Algoritmi euristici . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263.5.1 FASTA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263.5.2 BLAST . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
1

INDICE 2
4 Metodi computazionali 294.1 Similarita strutturale . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.1.1 Classificazione strutturale . . . . . . . . . . . . . . . . . . . . 314.2 Predizione della struttura tridimensionale . . . . . . . . . . . . . . . . 34
4.2.1 Homology modeling . . . . . . . . . . . . . . . . . . . . . . . . 344.2.2 Fold recognition . . . . . . . . . . . . . . . . . . . . . . . . . . 364.2.3 Ab initio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
5 Allineamenti profilo contro profilo 405.1 Introduzione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 405.2 Allineamento di profili . . . . . . . . . . . . . . . . . . . . . . . . . . 415.3 Analisi di metodi per l’allineamento di profili . . . . . . . . . . . . . . 43
5.3.1 Descrizione del processo di analisi . . . . . . . . . . . . . . . . 435.3.2 Risultati . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 465.3.3 Conclusioni . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
5.4 Log average scoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . 485.5 Sperimentazione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
6 Software utilizzato e modifiche apportate 556.1 Introduzione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 556.2 Descrizione della libreria . . . . . . . . . . . . . . . . . . . . . . . . . 556.3 Modifiche e fuzionalita aggiunte . . . . . . . . . . . . . . . . . . . . . 58
Conclusioni 61

Introduzione
Il lavoro svolto in questo progetto riguarda il protein folding, ossia la predizionedella struttura tridimensionale di una proteina attraverso metodi computazionali.
Le proteine sono molecole di fondamentale importanza in quasi tutti i proces-si biologici. Esse possono ad esempio agire da catalizzatori, trasportare o con-servare altre molecole, trasmettere impulsi nervosi e controllare la crescita e ildifferenziamento.
Tutte le funzioni che una proteina puo svolgere derivano dalla sua strutturatridimensionale; ecco perche conoscere il ripiegamento (fold) di una proteina e difondamentale importanza.
Nel presente documento verranno esposti gli strumenti e gli algoritmi utilizzatiper la predizione della struttura proteica.
I metodi sviluppati per questo scopo vengono generalmente suddivisi in tre ca-tegorie: homology modeling, fold recognition e ab initio. Ognuno di questi cercadi costruire un modello di una proteina a stuttura non nota a partire da strutturedeterminate sperimentalmente utilizzando approcci diversi.
E importante a questo punto dare alcune definizioni: la proteina della quale sivuole determinare il ripiegamento e detta target ; mentre la struttura nota presacome “stampo” per la costruzione del modello e definita template o templato.
In questa relazione si porra particolare attenzione all’allineamento di sequenzeproteiche, il quale risulta essere uno dei passi fondamentali di quasi tutti i metodidi predizione della struttura proteica.
Nel Capitolo 1 vengono elencate le caratteristiche e le funzioni principali delleproteine. Il Capitolo 2 riguarda i metodi sperimentali attualmente utilizzati perdeterminare la struttura tridimensionale di molecole proteiche.
Nel Capitolo 3 vengono descritti gli strumenti e gli algoritmi usati nel calcolol’allineamento tra le sequenze del target e del template. Il Capitolo 4 espone qualisono gli approcci utilizzati dai metodi computazionali che predicono la strutturaproteica.
Nel Capitolo 5 vengono analizzati e confrontati i vari programmi che sono statisviluppati negli ultimi anni per l’allineamento profilo contro profilo. Infine il capitolo6 e interamente dedicato all’implementazione delle modifiche del progetto; oltrealle funzioni piu propriamente legate al metodo log average si e anche provvedutoall’applicazione di principi di ingegneria del software che rendessero la libreria, nelcomplesso, piu funzionale.
3

Capitolo 1
Proteine e loro struttura
Le proteine sono macromolecole composte da carbonio, idrogeno, ossigeno, azo-to e, in qualche caso, da zolfo; esse svolgono funzioni biologiche di fondamentaleimportanza:
• catalizzano specifiche reazioni (enzimi);
• trasportano molecole o ioni da un organo all’altro;
• possono essere utilizzate come sostanze nutrienti;
• permettono a cellule ed organismi di contrarsi;
• forniscono protezione e supporto alle strutture biologiche;
• difendono l’organismo dall’invasione di specie estranee;
• regolano le attivita cellulari e fisiologiche.
Queste funzioni dipendono direttamente dalla loro struttura tridimensionale.Le proteine sono costituite combinando venti tipi diversi di α-amminoacidi e
differiscono tra loro per il numero, la composizione e la sequenza degli amminoacidi.La loro struttura e solitamente suddivisa in quattro livelli: primaria, secondaria,terziaria e quaternaria.
1.1 Struttura primaria
La struttura primaria di una proteina e data dalla sequenza di amminoacidi.Un amminoacido e l’unita strutturale di base per la formazione delle proteine; e
un monomero costituito da un atomo di carbonio centrale, detto carbonio α, a cuisono legati un gruppo amminico (-NH2), un gruppo carbossilico (-COOH), un atomodi idrogeno e un gruppo R (o catena laterale) specifico per ciascun amminoacido(Fig.1.1).
In natura esistono 20 diversi amminoacidi (Fig.1.2), specificati dal codice ge-netico, che si legano l’uno all’altro attraverso una reazione di condensazione, nella
4

CAPITOLO 1. PROTEINE E LORO STRUTTURA 5
Figura 1.1: Struttura di un amminoacido.
quale il gruppo carbossilico del primo amminoacido reagisce con quello amminicodel secondo perdendo una molecola d’acqua, il legame cosı formato e detto legamepeptidico; esso ha la proprieta di essere cineticamete stabile.
In base alle proprieta chimiche della catena laterale, gli amminoacidi possonoessere classificati in alifatici non polari, polari non carichi, carichi positivamente onegativamente, e aromatici.
Quando un certo numero di amminoacidi si uniscono per mezzo di legami pep-tidici si forma una catena polipeptidica, nella quale ogni unita amminoacidica vienechiamata residuo. La catena polipeptidica e polare, infatti alle sue estremita so-no presenti gruppi diversi: da una parte vi e un gruppo amminico, dall’altra unocarbossilico. Per convenzione l’inizio della catena polipeptidica e il terminale ammi-nico, la sequenza di amminoacidi si scrive a partire da questo residuo. Una catenapolipeptidica consiste di una parte che si ripete regolarmente, detta catena princi-pale o scheletro covalente, e di una parte variabile, che comprende le catene lateralidegli amminoacidi. La catena principale e formata da un gran numero di potenzialiformatori di legami idrogeno1; ogni residuo contiene gruppi accettori (gruppo car-bonilico) e donatori (gruppo NH), che interagendo tra loro, e con i gruppi funzionalidelle catene laterali, formano strutture particolarmente stabili. In alcune proteine(proteine extracellulari) sono presenti legami trasversali, il piu comune e il legamedisolfuro, derivante dall’ossidazione di due residui di cisteina.
Le catene polipeptidiche contengono, in generale, da 50 a 2000 residui amminoa-cidici.
Il legame peptidico ha caratteristica di doppio legame; si ha la formazione di unastruttura rigida che impedisce la libera rotazione attorno al legame stesso, questocomporta una restrinzione delle possibili conformazioni dello scheletro covalente efa si che i sei atomi Cα, C, O, N, H e Cα si trovino sullo stesso piano, determinandola planarita del legame.
1Il legame idrogeno e un particolare tipo di legame secondario che si instaura tra molecolecontenenti atomi di idrogeno legati ad atomi elettronegativi come F, O ed N.

CAPITOLO 1. PROTEINE E LORO STRUTTURA 6
Figura 1.2: Struttura dei 20 amminoacidi
La caratteristica di doppio legame puo essere riconosciuta anche dalla lunghezzadel legame tra il gruppo CO e quello NH, che e pari a 1,32 A2, e risulta essereintermedia tra quella di un singolo legame C-N (1,49 A) e di un doppio legame C=N(1,27 A).
Al contrario i legami C-Cα e N-Cα sono singoli puri, quindi due unita peptidicherigide adiacenti hanno un certo grado di liberta di rotazione attorno a questi legami;questo permette alla proteina di ripiegarsi in modi diversi. L’angolo di rotazioneattorno al legame N-Cα e detto φ, quello tra il legame Cα-C e detto Ψ; una rotazionein senso orario ha valore valore positivo (Fig.1.3).
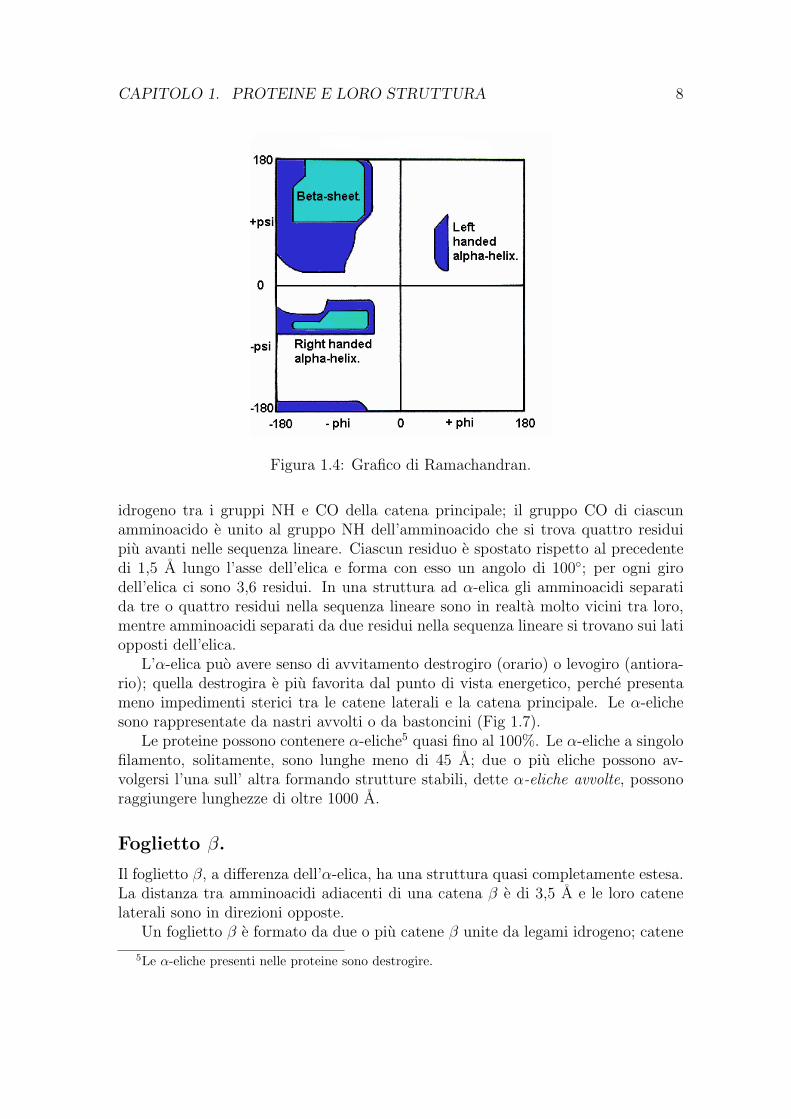
E importante evidenziare che non tutte le combinazioni degli angoli φ e Ψ sonopossibili, G.N.Ramachandran ha osservato che molte di esse sono impedite da inter-ferenze steriche tra gli atomi 3; l’insieme dei valori permessi puo essere rappresentatodal grafico in Fig. 1.4, detto grafico di Ramachandran.
Il legame peptidico puo assumere due configurazioni: quando gli atomi dei car-boni α si trovano in posizioni opposte rispetto al legame peptidico si ha una confi-gurazione trans, quando si trovano dalla stessa parte si ha, una configurazione cis;nelle proteine tutti i legami sono di tipo trans, in questo modo sono minimizzate leinterferenze steriche tra i gruppi uniti agli atomi dei carboni α.
2Un Angstrom (A) e pari a 10−10 m.3L’esclusione per ingombro sterico e un principio molto importante per l’organizzazione di
strutture chimiche e si ha quando due atomi si trovano nella stessa posizione allo stesso tempo.

CAPITOLO 1. PROTEINE E LORO STRUTTURA 7
La rigidita dell’unita peptidica, unita al ristretto numero di valori che gli angoliφ e Ψ possono assumere, limita il numero di strutture permesse nella forma nonripiegata; ne consegue che una proteina tendera a ripiegarsi spontaneamente inun’unica struttura.
Figura 1.3: Angoli di rotazione nel legame peptidico.
1.2 Struttura secondaria
La struttura secondaria riguarda la disposizione spaziale dello scheletro covalentedella catena polipepdica.
Nel 1951 Linus Pauling e Robert Corey proposero due strutture polipeptidicheperiodiche: α-elica e foglietto β 4; successivamente furono identificate altre strutturenon periodiche che concorrono alla formazione della struttura finale delle proteine,come ad esempio i ripiegameti β e l’ansa Ω. La scoperta di queste strutture fu pos-sibile grazie allo studio di tutte le conformazioni peptidiche stericamente permesse;Pauling e Corey osservarono che la maggior parte di esse presentavano la capacitadi formare legami idrogeno tra i gruppi NH e CO dello scheletro covalente.
α-elica.
L’α-elica ha una struttura a bastoncino; la sua parte interna e formata dalla catenaprincipale strettamente avvolta; le catene laterali si estendono verso l’esterno conun andamento a spirale (Fig. 1.5). L’α-elica e stabilizzata dalla presenza di legami
4β perche e stata la seconda struttura scoperta; la prima fu l’α-elica.
CAPITOLO 1. PROTEINE E LORO STRUTTURA 8
Figura 1.4: Grafico di Ramachandran.
idrogeno tra i gruppi NH e CO della catena principale; il gruppo CO di ciascunamminoacido e unito al gruppo NH dell’amminoacido che si trova quattro residuipiu avanti nelle sequenza lineare. Ciascun residuo e spostato rispetto al precedentedi 1,5 A lungo l’asse dell’elica e forma con esso un angolo di 100; per ogni girodell’elica ci sono 3,6 residui. In una struttura ad α-elica gli amminoacidi separatida tre o quattro residui nella sequenza lineare sono in realta molto vicini tra loro,mentre amminoacidi separati da due residui nella sequenza lineare si trovano sui latiopposti dell’elica.
L’α-elica puo avere senso di avvitamento destrogiro (orario) o levogiro (antiora-rio); quella destrogira e piu favorita dal punto di vista energetico, perche presentameno impedimenti sterici tra le catene laterali e la catena principale. Le α-elichesono rappresentate da nastri avvolti o da bastoncini (Fig 1.7).
Le proteine possono contenere α-eliche5 quasi fino al 100%. Le α-eliche a singolofilamento, solitamente, sono lunghe meno di 45 A; due o piu eliche possono av-volgersi l’una sull’ altra formando strutture stabili, dette α-eliche avvolte, possonoraggiungere lunghezze di oltre 1000 A.
Foglietto β.
Il foglietto β, a differenza dell’α-elica, ha una struttura quasi completamente estesa.La distanza tra amminoacidi adiacenti di una catena β e di 3,5 A e le loro catenelaterali sono in direzioni opposte.
Un foglietto β e formato da due o piu catene β unite da legami idrogeno; catene
5Le α-eliche presenti nelle proteine sono destrogire.

CAPITOLO 1. PROTEINE E LORO STRUTTURA 9
Figura 1.5: Struttura di un’α-elica.
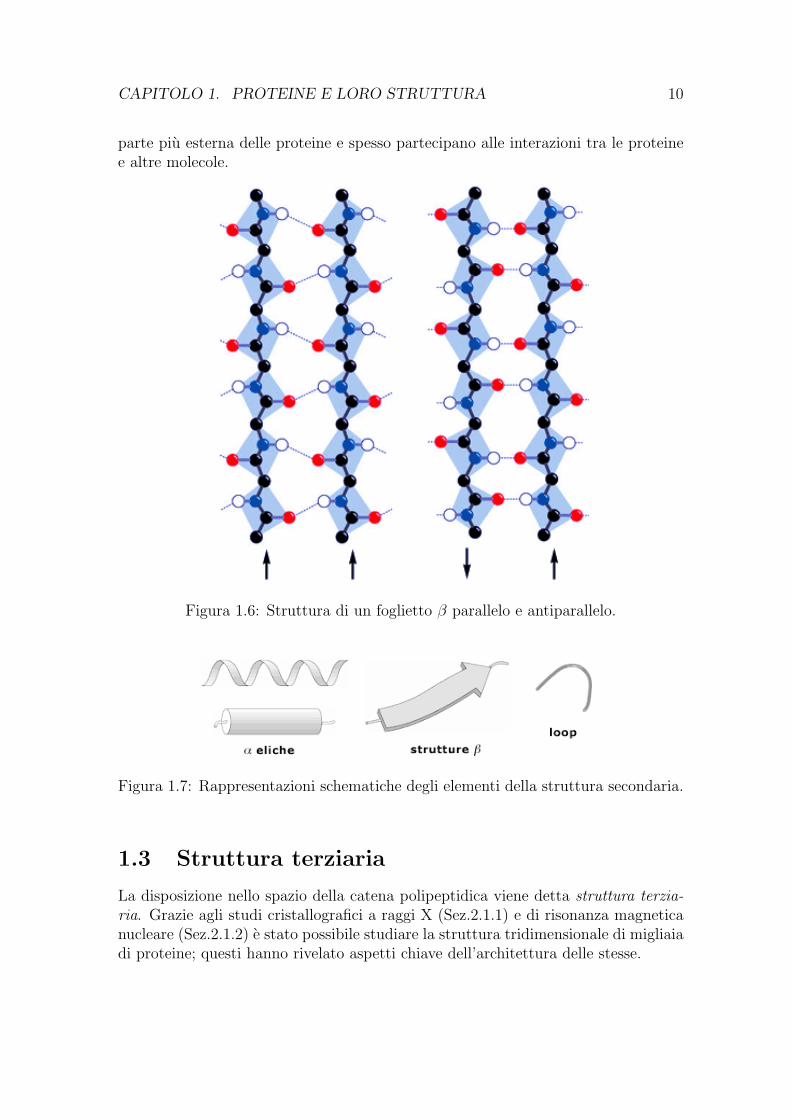
adiacenti possono avere la stessa direzione (foglietto β parallelo) oppure opposta(foglietto β antiparallelo). La struttura antiparallela presenta legami idrogeno trai gruppi NH e CO degli amminoacidi di una catena e quelli CO e NH di residuidi catene adiacenti. Nella struttura parallela, invece, la disposizione del legamiidrogeno e un po’ piu complicata: il gruppo NH di ogni amminoacido forma unlegame idrogeno con il gruppo CO di un amminoacido nella catena adiacente, mentreil suo gruppo CO forma un legame idrogeno con il gruppo NH di un amminoacidoposto due residui piu avanti lungo la catena β (Fig. 1.6).
Generalmente i foglietti β contengono 4 o 5 catene, possono essere paralleli, anti-paralleli o misti; essi sono rappresentati tramite frecce larghe rivolte nella direzionedel carbossiterminale (Fig 1.7).
Ripiegamento β e ansa Ω.
Le proteine, in genere, hanno una forma globulare compatta con frequenti cambia-menti di direzione della catena principale. L’inversione di direzione e dovuta allapresenza di strutture, come i ripiegameti β (o inversione a U), nei quali il gruppoCO del residuo i 6 forma un legame idrogeno con il gruppo NH del residuo i + 3 sta-bilizzando i bruschi cambiamenti di direzione delle catene polipepetidiche. Esistonoanche strutture piu complesse, come le anse Ω (o anse), il cui nome deriva dallaforma che assumono. Queste strutture non sono periodiche, si trovano sempre sulla
6i indica: iniziale della struttura.
CAPITOLO 1. PROTEINE E LORO STRUTTURA 10
parte piu esterna delle proteine e spesso partecipano alle interazioni tra le proteinee altre molecole.
Figura 1.6: Struttura di un foglietto β parallelo e antiparallelo.
Figura 1.7: Rappresentazioni schematiche degli elementi della struttura secondaria.
1.3 Struttura terziaria
La disposizione nello spazio della catena polipeptidica viene detta struttura terzia-ria. Grazie agli studi cristallografici a raggi X (Sez.2.1.1) e di risonanza magneticanucleare (Sez.2.1.2) e stato possibile studiare la struttura tridimensionale di migliaiadi proteine; questi hanno rivelato aspetti chiave dell’architettura delle stesse.

CAPITOLO 1. PROTEINE E LORO STRUTTURA 11
Si e constatato che in ambiente acquoso il ripiegamento di una proteina e guidatodalla forte tendenza a escludere dall’acqua i residui idrofobici.La catena polipepti-dica si ripiega spontantemente in modo che le catene laterali idrofobiche si trovinoall’interno della molecola e quelle polari, provviste di carica, si trovino sulla su-perficie; fanno eccezione le proteine che attraversano le membrane biologiche, chesolitamente presentano struttura opposta rispetto alle proteine che svolgono la lorofunzione in ambiente acquoso.
I gruppi NH o CO della catena principale non impegnati in legami peptidicihanno forte preferenza per l’acqua. Quando questi gruppi sono appaiati per mezzo dilegami idrogeno, come avviene ad esempio nelle α-eliche e nelle strutture β, allora unsegmento della catena principale puo trovarsi all’interno della molecola in ambienteidrofobico. Nelle molecole proteiche sono presenti, oltre ai legami idrogeno, le forzedi van der Waals che si formano tra le catene laterali idrocarburiche strettamenteaddensate e contribuiscono a rendere la proteina stabile.
Struttura super-secondaria e domini. Nelle proteine si possono spesso indivi-duare piccole sotto-strutture formate dalla combinazione di diversi elementi di strut-tura secondaria, tanto da ipotizzare l’esistenza di un’altro livello organizzativo dettostruttura super-secondaria o motivo. I motivi piu comuni sono α-ripiegamento-αβ-ripiegamento-β, chiave greca e β-α-β (Fig. 1.8).
Figura 1.8: Tipi di motivi.
Alcune catene polipeptidiche si ripiegano in regioni compatte, dette domini (α,β, α/β, α+β), unite tra loro da un segmento flessibile della catena polipeptidica;esse hanno dimensioni variabili da 30 a 400 amminoacidi. E importante notare chele proteine possono avere domini comuni, ma avere struttura terziaria complessivadiversa.
La combinazione di motivi puo dar luogo alla formazione di specifici domini.
1.4 Struttura quaternaria
Le proteine di grandi dimensioni possono essere formate da piu di una catena poli-peptidica, denominata subunita; per questo tipo di proteine possiamo parlare anchedi struttura quaternaria, che corrisponde dalla disposizione spaziale delle subunita.Le forze che contengono insieme subunita sono le stesse che stabilizzano la struttura

CAPITOLO 1. PROTEINE E LORO STRUTTURA 12
terziaria di proteine: forze di van der Waals, ponti salini e legami idrogeno. La strut-tura quaternaria piu semplice e costituita da due subunita uguali; esistono anchestrutture piu complesse, formate da tipi diversi di subunita e in quantita variabile.
1.5 Ripiegamento delle proteine o folding
Una proteina per poter svolgere la propria funzione biologica deve essere strutturatanella cosiddetta conformazione o struttura nativa. La conformazione nativa e quellastruttura 3D stabile e funzionale, caratterizzata da un minimo di energia potenzialee da quella particolare conformazione che consente alla proteina di svolgere la suafunzione. Il processo che dalla biosintesi del peptide porta alla formazione dellaproteina strutturata nella forma nativa viene detto folding.
Una proteina esposta a condizioni particolari di temperatura, pH, all’ azione diagenti denaturanti (quali urea o guanidina) o detergenti, perde in parte o completa-mente la sua struttura e la propria funzionalita; questo processo e detto denatura-zione della proteina. Le proteine denaturate tendono a riacquistare spontaneamentela forma nativa (refolding o rinaturazione) in seguito al ripristino delle condizioniiniziali ottimali. Il fenomeno del refolding dimostra che la sequenza amminoacidicadi una proteina contiene tutta l’informazione necessaria alla proteina per ripiegarein una struttura tridimensionale stabile e biologicamente attiva. Questa relazionetra la stuttura primaria e la conformazione 3D di una proteina fu dimostrata daChristian Anfisen.
Nonostante l’intensa ricerca condotta negli ultimi anni, il meccanismo del fol-ding di proteine non e ancora adeguatamente compreso; e stato evidenziato che ilfolding e un processo multi-stadio con formazione di intermedi di folding, cioe statiparzialmente strutturati con caratteristiche della proteina nativa.
Cyrus Levinthal, prendendo in considerazione una proteina di 100 residui, hacalcolato che se ogni residuo puo assumere 3 diverse conformazioni, il numero totaledelle strutture e pari a 3100; se ogni residuo impiega 10−13 s per passare da unastruttura all’altra, il tempo totale richiesto risulterebbe essere di 1,6 × 1027× 10−13
anni. Il ripiegamento delle proteine e quindi un processo che avviene medianteprogressive stabilizzazioni degli intermedi e non mediante una scelta casuale.
La formazione di elementi della struttura secondaria avviene probabilmente perprima; successivamente α-eliche e foglietti β si combinano tra loro formando cosı lastruttura tridimensionale della proteina.

Capitolo 2
Metodi sperimentali
La funzione di una proteina, come detto in precedenza, deriva dalla sua struttura.In questo capitolo descriveremo le tecniche attualmente utilizzate per la deter-
minazione della struttura di una macromolecola, cioe per identificare la posizionedei suoi atomi.
Una volta ottenute le coordinate degli atomi della macromolecola consideratavengono memorizzate in files di formato standard detto formato PDB e depositatenella banca dati PDB (Sez.2.2).
2.1 Determinazione della struttura proteica
2.1.1 Cristallografia ai raggi X
La cristallografia ai raggi X e la tecnica che permette di ottenere la migliore vi-sualizzazione della struttura proteica; essa infatti e in grado di determinare l’esattaposizione tridimensionale della maggior parte degli atomi.
L’uso dei raggi X consente di ottenere la migliore risoluzione perche la lorolunghezza d’onda corrisponde alla lunghezza di un legame covalente.
Per effettuare un’analisi cristallografica ai raggi X sono necessari tre componenti:un cristallo della proteina che si vuole analizzare, una sorgente di raggi X e unrivelatore.
La cristallizzazione della proteina e la parte piu delicata e piu lunga di tuttoil processo, in quanto richiede il preciso orientamento di tutte le molecole. Alcuneproteine cristallizzano facilmente, mentre altre lo fanno solo dopo molti tentativi pertrovare le condizioni giuste (temperatura, concentrazioni, presenza di soluti e cofat-tori). La cristallizzazione infatti e spesso considerata un’arte. Una volta ottenuti icristalli e necessario farli colpire da una sorgente di raggi X.
Un fascio di raggi X con una lunghezza d’onda di 1,54 A puo essere prodottoaccelerando elettroni contro un bersaglio di rame. Un sottile fascio di raggi X vienepoi diretto contro il cristallo della proteina: una parte attraversa il cristallo, l’altraviene deviata (o diffratta) in varie direzioni.
13

CAPITOLO 2. METODI SPERIMENTALI 14
I raggi diffratti, detti anche onde diffratte, possono essere rivelati da una pellicolaradiografica o da un rivelatore elettronico.
Ciascun atomo contribuisce a deviare un raggio. L’ampiezza delle onde prodot-te e proporzionale al numero di elettroni dell’atomo. Queste onde si ricombinanoin base alla disposizione degli atomi: si rinforzano l’una con l’altra a livello dellapellicola, o del rivelatore, se sono in fase in quel punto, si cancellano se sono fuorifase.
Il cristallo viene posizionato con un preciso orientamento rispetto al fascio diraggi X e alla pellicola; successivamente viene fatto ruotare in modo che il fascio diraggi X lo possa colpire da diverse direzioni. La fotografia ai raggi X che si ottiene ecomposta da macchie, dette riflessioni, disposte regolarmente. L’intensita di questemacchie viene misurata e risulta essere proporzionale all’intensita del raggio deviato.L’intensita e la posizione delle macchie costituiscono i dati sperimentali dell’analisicristallografica ai raggi X.
Il passo successivo consiste nella costruzione di un’immagine della proteina apartire da questi dati.
Nella microscopia ottica o in quella elettronica i raggi diffratti vengono messi afuoco da lenti per formare direttamente un’immagine. Nel nostro caso, invece, lastruttura della proteina viene ricostruita utilizzando la trasformata di Fourier, inquanto non esistono lenti in grado di mettere a fuoco i raggi X.
Questa operazione fornisce per ogni macchia un’onda di densita elettronica la cuiampiezza e proporzionale alla radice quadrata dell’intensita osservata nella macchia.La fase puo essere dedotta dai profili di diffrazione noti, prodotti da atomi pesantiusati come riferimento (uranio o mercurio) posti in siti specifici della proteina.
Utilizzando queste informazioni viene calcolata una mappa di densita di elettroni.In questa mappa la densita degli elettroni e rappresentata mediante linee di livello.Questa viene poi interpretata per costruire il modello tridimensionale della proteina.
Un fattore molto importante di cui si deve tener conto e la risoluzione, dallaquale dipende la fedelta dell’immagine. Se la risoluzione e abbastanza buona ipicchi presenti nella mappa di densita elettronica corrisondono ai nuclei atomici.La risoluzione finale di un’analisi ai raggi X e limitata dal grado di perfezione delcristallo; per le proteine la risoluzione limite e in genere di 2 A.
2.1.2 Spettroscopia a risonanza magnetica nucleare (NMR)
La spettroscopia a risonanza magnetica nucleare (NMR) e in grado di rivelare lastruttura atomica di macromolecole in soluzione.
Questa tecnica si basa sul fatto che alcuni nuclei atomici possiedono un momentomagnetico1 detto spin. L’esempio piu semplice e dato dal nucleo di idrogeno (1H),cioe un protone. Il moto rotatorio del protone genera un momento magnetico. Seviene applicato un campo magnetico esterno gli spin possono assumere due orien-tamenti: parallelo o antiparallo rispetto al campo esterno, detti stati di spin (α e
1Solo un numero limitato di isotopi ha questa proprieta, ad esempio 13C, 15N e 31P.

CAPITOLO 2. METODI SPERIMENTALI 15
β). La differenza di energia tra i due stati e proporzionale alla forza del campomangnetico applicato.
Quando un nucleo assorbe una radiazione elettromagnetica di frequenza appro-priata (radiofrequenza o RF ) passa dallo stato α a quello piu eccitato β, con unafrequenza pari alla differenza di energia tra i due. Si ottiene cosı la frequenza dirisonanza.
Lo spettro di risonanza di una molecola si puo ottenere in due modi: variandoil campo magnetico e mantenendo costante la frequenza della radiazione elettro-magnetica, oppure tenendo il campo magnetico costante e variando la radiazioneelettromagnetica.
Queste proprieta possono essere sfruttate per analizzare l’ambiente chimico intor-no al nucleo di idrogeno. Il flusso di elettroni attorno a un nucleo magnetico generaun piccolo campo magnetico locale che si oppone a quello applicato. Il grado dispostamento dipende dalla densita elettronica circostante, quindi nuclei in ambientidiversi risuoneranno con frequenze di radiazione diverse.
La frequenza con cui i nuclei di un campione perturbato assorbono una radiazioneelettromagnetica puo essere facilmente misurata. Le diverse frequenze sono detteanche spostamenti chimici ; esse sono espresse in unita frazionarie δ (parti per milioneo ppm) relative a un composto standard che viene aggiunto al campione. Nellemolecole proteiche generalmente lo spostamento chimico e compreso tra 0 e 9 ppm.
La maggior parte dei protoni delle proteine possono essere indentificati usandoquesta tecnica denominata NMR monodimensionale.
Maggiori informazioni si possono ottenere esaminando gli effetti che spin di pro-toni divesi hanno sui loro vicini. Attraverso la magnetizzazione transitoria, indottain un campione da un impulso di frequenza radio, e possibile alterare lo spin delnucleo vicino. Utilizzando la spettroscopia NOESY (Nuclear Overhauser Enhance-ment) si ottiene uno spettro bidimensionale che fornisce molte informazioni. Questotipo di spettroscopia rivela graficamente coppie di protoni che si trovano in strettavicinanza.
Alla base di questa tecnica vi e l’effetto nucleare Overhauser (NOE), esso met-te in relazione l’interazione tra nuclei e la distanza che li separa. In particolare:un’interazione e proporzionale all’inverso della sesta potenza della loro distanza.
La magnetizzazione viene trasferita da un nucleo eccitato a uno non eccitato sela distanza e minore di 5 A. In questo modo si ottiene un sistema di valutazionedella localizzazione relativa degli atomi nella struttura della proteina.
La diagonale di uno spettro bidimensionale NOESY corrisponde a uno spettromonodimensionale di spostamento chimico. I picchi fuori dalla diagonale identificanocoppie di protoni la cui distanza e molto piccola. Queste relazioni di prossimitapossono essere utilizzate per ricostruire la struttura tridimensionale della proteina.
Il calcolo delle strutture si basa sul fatto che i protoni che in uno spettro NOESYdistano meno di 5 A, devono essere molto vicini anche nella struttura tridimensio-nale. E necessario generare un gruppo di strutture correlate al fine di avere un altonumero di relazioni, ed essere quindi in grado di specificare interamente e con buonaapprossimazione la struttura della proteina.

CAPITOLO 2. METODI SPERIMENTALI 16
Le osservazioni sperimentali, generalmente, vengono fatte su un gran numerodi molecole in soluzione che possono assumere differenti struttura in un dato mo-mento. Gli esperimenti NMR pruducono gruppi di 20-30 strutture simili consistenticon i dati sperimentali raccolti; esse rappresentano le possibili conformazioni che laproteina in soluzione puo assumere.
Attualmente la spettroscopia NMR puo determinare la struttura di piccole pro-teine.
2.1.3 Conclusioni
La cristallografia ai raggi X e la risonanza magnetica nucleare presentano entrambedei vantaggi e degli svantaggi.
I modelli ottenuti con la spettroscopia NMR sono meno accurati di quelli ottenuticon la cristallografia ai raggi X. Il limite principale di questa tecnica e la dimensionedella proteina che puo essere oggetto di studio. Essa pero non richiede la produzionedi cristalli ed e in grado di generare un quadro della dinamica di una proteina.
La diffrazione ai raggi X offre dati molto piu precisi (la risoluzione e ormai permolte strutture al di sotto dall’Angstrom), ma necessita la produzione cristalli dibuona qualita. Inoltre non sempre le strutture costrette in cristalli rappresentanoimmagini fedeli di proteine nella loro conformazione attiva.
2.2 Protein Data Bank (PDB)
La prima banca dati di strutture di interesse biologico e la banca dati di struttureproteiche PDB (Protein Data Bank), istituita nel 1971 presso il Brookhaven NationalLaboratory (BNL). All’inizio conteneva solo sette strutture. A partire dagli anni ’80vi fu una crescita esponenziale del numero di strutture depositate; questo fu possibilegrazie al miglioramento delle tecniche utilizzate per la determinazione della strutturaproteica.
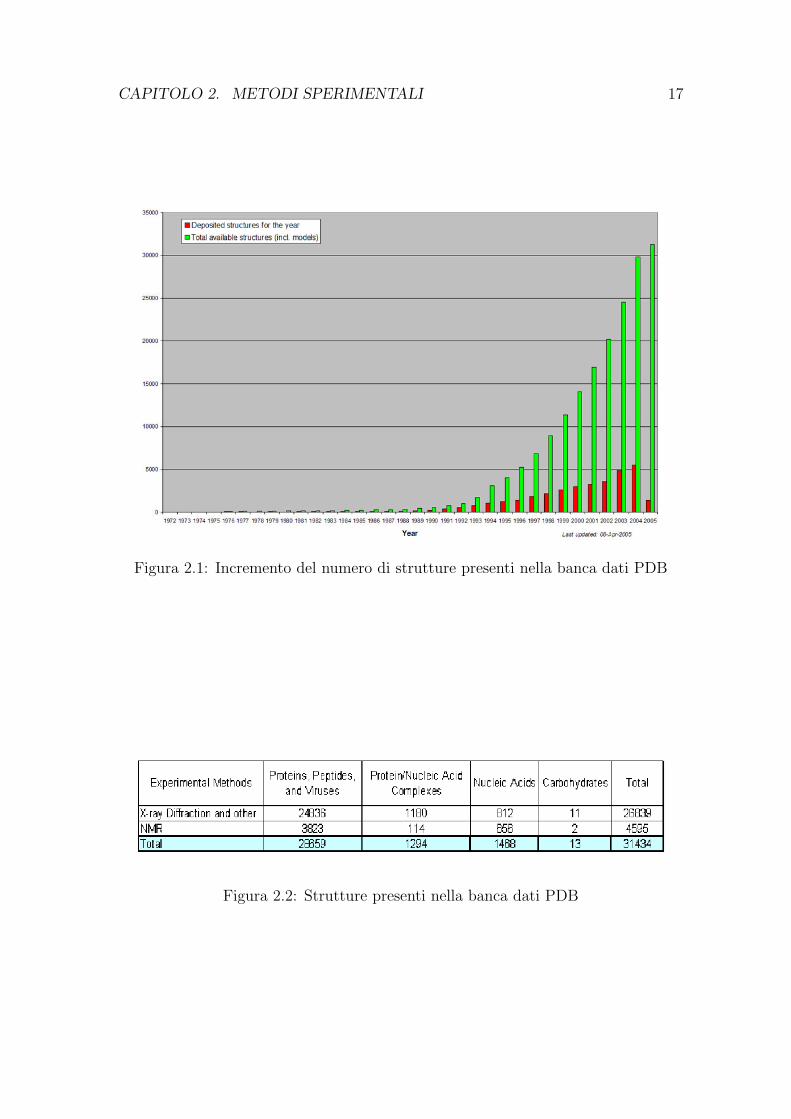
La banca dati PDB e attualmente gestita dal Research Collaboratory for Struc-tural Bioinformatics (RCSB), (giugno 2005) e raccoglie 31434 (Tab.2.2) struttureche comprendono:
• strutture proteiche (complessi proteici, capsidi virali, cofattori, substrati, ini-bitori o altri ligandi);
• complessi di proteine con acidi nucleici;
• strutture di acidi nucleici (DNA, RNA);
• strutture di carboidrati;
Il numero di strutture ”uniche” e in realta molto minore rispetto al numero diquelle depositate. In alcuni casi infatti la banca dati contiene piu versioni dellestruttura di una proteina. Questo accade quando sono state ottenute le coordinate
CAPITOLO 2. METODI SPERIMENTALI 17
Figura 2.1: Incremento del numero di strutture presenti nella banca dati PDB
Figura 2.2: Strutture presenti nella banca dati PDB

CAPITOLO 2. METODI SPERIMENTALI 18
con risoluzione migliore o quando la proteina e stata cristallizzata in condizionidiverse. Inoltre alcune proteine sono state risolte sia per diffrazione ai raggi X, siatramite NMR. Altre sono presenti in forma libera e nella forma co-cristallizata conaltri ligandi. Questa struttura “disordinata” e dovuta all’evouzione storica
Ogni file della banca dati e identificato da 4 caratteri: un numero e tre caratterialfanumerici. Il numero identifica la versione del file e i caratteri restanti sono scelti,dove possibile, in modo da ricordare i nomi delle strutture descritte dal file stesso.
Ogni file PDB e essenzialmente diviso in due parti. La prima contiene la de-scrizione della molecola in esso descritta, i dettagli sperimentali della risoluzionedella sua struttura, la risoluzione, la lista dei residui della proteina e dei nucliotididell’acido nucleico, le referenze e cosı via. La seconda parte contiene le coordinateatomiche. In ogni riga e riportata la parola ATOM seguita da numero progressivoe nome dell’atomo descritto, nome dei residui in codice di tre lettere e numero del-l’amminoacido. Di seguito sono poi riportati i valori delle coordinate cartesiane x,y, z in Angstrom misurate da un’origine arbitraria, e infine alcuni dati di interesseper i cristallografi.
Esistono altre banche dati di strutture proteiche organizzate in modo migliore,derivanti dal PDB, quali ad esempio SCOP [SCOP, 2005], CATH [CATH, 2005] eFSSP [FSSP, 2005](Sez.4.1.1).

Capitolo 3
Allineamento di sequenzeproteiche
I metodi di predizione della struttura di proteine richiedono, in generale, la genera-zione di un allineamento tra target e template, al fine di valutarne la similarita.
Le sequenze proteiche vengono rappresentate da strighe di caratteri appartenentiad un alfabeto di dimensione 20. Ogni carattere dell’alfabeto rappresenta uno dei20 diversi amminoacidi.
Nel confronto di sequenze proteiche e necessario tenere conto che proteine diversepossono derivare da una stessa proteina in seguito a processi di sostituzione, permezzo dei quali vengono cambiati alcuni residui della sequenza. Oppure processidi inserzione e delezione i quali comportanto l’aggiunta o la rimozione di residui.Inserzioni e delezioni vengono anche dette gaps.
In questo capitolo saranno date le definizioni di omologia e di similarita, inoltresi descriveranno gli algoritmi utilizzati per l’allineamento di sequenze proteiche e imodelli utilizzati per attribuire ad essi un punteggio.
3.1 Similarita e omologia
Dal punto di vista biologico la similarita tra due sequenze esprime una relazione diomologia, tuttavia i termini similarita e omologia hanno significati distinti.
Due sequenze si definiscono omologhe se condividono una stessa origine filogene-tica, da cui sono poi evolute differenziandosi l’una dall’altra. Se la relazione filoge-netica che le lega non e troppo lontana le due sequenze rimangono significativamentesimili tra loro. L’omologia e quindi una caratteristica qualitativa.
Due sequenze di dicono, invece, simili quando e presente una somiglianza, in-dipendentemente dai motivi che l’hanno determinata. La similarita deriva spessodall’omologia, ma puo anche essere dovuta al caso. Essa puo essere espressa in termi-ni quantitativi, in quanto fa riferimento al grado di similitudine che viene misuratotra due sequenze precedentemente allineate.
19

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 20
3.2 Allineamento
Come detto precedentemente la determinazione del grado di similarita tra due o piusequenze richiede che esse vengano allineate.
L’allineamento tra due sequenze consiste nella determinazione di una relazio-ne tra i residui della prima sequenza con quelli della seconda in modo da rende-re massimo il grado di similarita, o analogamente rendere minimo il numero didifferenze.
Per valutare il grado di similarita e necessario definire un modello che permettadi assegnare un punteggio all’allineamento. Il modello piu semplice attribuisce unpunteggio solo ai match, ossia ad una coppia di amminoacidi uguali.
Spesso e necessario considerare la possibilita che il migliore allineamento com-porti l’inserimento di gap nelle sequenze. Questo deve essere penalizzato nel calcolodel punteggio da attribuire all’allineamento ottimo.
Il costo associato a un gap di lunghezza g puo essere ottenuto mediante un linearscore
γ(g) = −gd (3.1)
oppure un affine score
γ(g) = −d− (g − 1)e (3.2)
Il termine d, detto gap-open penality, penalizza l’apertura di un gap, invece e, dettagap-extention penality, e il costo associato alla lunghezza del gap. La gap-extentionpenality e in generale piu piccola rispetto alla gap-open penality.
Il numero possibile di allineamenti tra due sequenze di lunghezza n e m, esclu-dendo l’inserimento di gaps, e pari a n+m-1.
Esistono vari modi per allineare due sequenze. Il metodo piu semplice, ma ancheil piu costoso in termini di tempo, consiste nel far scorrere una sequenza sull’altravalutando ad ogni spostamento il numero di appaiamenti. L’allineamento miglioresara quello con il massimo numero di match. Questo richiede di effettuare un numerodi confronti pari al prodotto della lunghezza delle due sequenze che si voglionoallineare.
Un metodo di questo tipo richiede un tempo troppo elevato, per questo motivoe preferibile l’utilizzo di vie alternative, quali algoritmi dinamici ed euristici.
La scelta dell’algoritmo dipende anche dalle finalita che l’allineamento si propone.In seguito verranno illustrati i principali algoritmi attualmete utilizzati.
3.3 Matrici di sostituzione
Negli allineamenti di sequenze di amminoacidi, in genere, vengono utilizzati criteri disimilarita che non si limitano a verificare l’identita assoluta di due amminoacidi. Sitiene conto anche del fatto che gli amminoacidi possono essere piu o meno simili traloro, e spesso nel corso dell’evoluzione prendono il posto l’uno dell’altro in proteineomologhe, senza alterare la funzionalita della proteina stessa.

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 21
Per far fronte a questa esigenza sono state introdotte delle matrici, dette matricidi sostituzione, che congono 210 valori o punteggi, uno per ogni possibile coppiadi amminoacidi. In generale queste matrici hanno dimensione 20 × 20 e riportanodue volte il punteggio che lega due diversi amminoacidi. Ad esempio la relazionealanina-argina (A-R) e riportata anche come argina-alanina (R-A).
Utilizzando le matrici di sostituzione per l’allineamento di due sequenze e quindipossibile assegnare un punteggio ad ogni coppia di amminoacidi, invece di conside-rare solo il numero di identita.
Nell’allineamento di sequenze proteiche, in genere, sono utilizzate le matrici PAMe le matrici BLOSUM. Di seguito illustreremo il loro significato e il modo con cuiesse sono calcolate.
3.3.1 Matrici PAM
La matrice di sostituzione PAM fu proposta da Margareth Dayhoff nel 1978 sullabase di uno studio di tipo evolutivo, nel quale furono cosiderate 71 famiglie diproteine omologhe con un grado di similarita superiore all’85%.
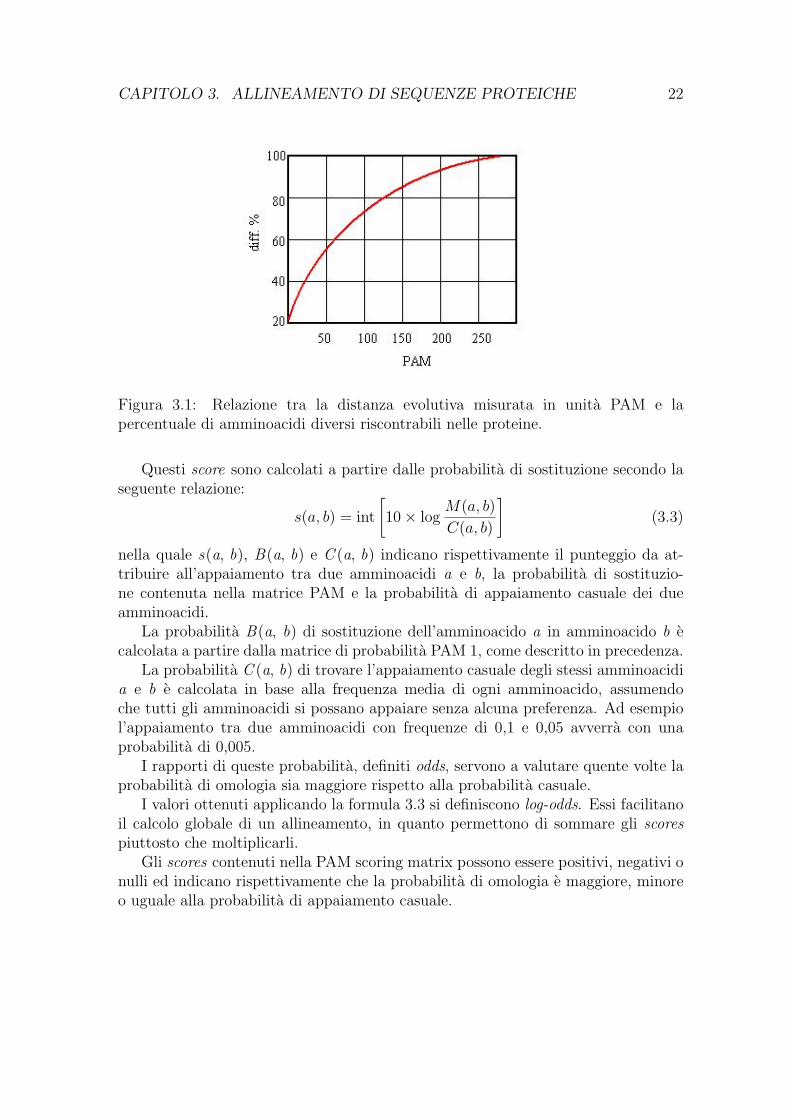
L’assunzione di partenza e che dall’analisi di sequenze correlate e possibile cal-colare la probabilita con cui ogni amminoacido subisce una mutazione, detta anchePercent Accepted Mutation (PAM)1, e metterla in relazione con la distanza evolutiva.
Mediante un calcolo statistico fu determinata la probabilita di sostituzione diogni coppia di amminoacidi, a una distanza evolutiva di 1 PAM, considerando quindila mutazione di un amminoacido ogni cento. Data la cosı piccola distanza evolutivala matrice PAM 1 risulta molto simile a una matrice identita. Essa viene utilizzataper calcolare matrici PAM di ordine superiore, cosiderando PAM 1 come un singolopasso evolutivo.
L’indice PAM indica il numero di passi evolutivi che si prevede per le proteinein analisi. PAM 100 sta quindi ad indicare che sono stati compiuti 100 passi evo-lutivi, ognuno con le sue probabilita, e non che il 100% degli amminoacidi vengonosostituiti. All’aumetare dell’indice PAM ci si allontana sempre piu dalla matrice diidentita.
Molte sostituzioni possono cadere sulla stessa posizione, di conseguenza i residuicon bassa probabilita di sostituzione resteranno invariati per molti passi evolutivi.
I valori della matrice, a una data distanza PAM, corrispondono alla probabilitache due dati amminoacidi siano omologhi.
L’indice della matrice PAM utilizzata e molto importante, ed e diverso a secondadegli scopi. Se si vogliono confrontare sequenze vicine e opportuno usare una matricePAM a basso indice, in modo da penalizzare in modo rilevante qualsiasi sostituzione.Invece se le due sequenze considerate sono distanti e preferibile utilizzare matriciPAM con indici alti al fine di valorizzare le sostituzioni conservative.
Le matrici sopra descritte sono le matrici PAM di probabilita di sostituzione. Daesse derivano le matrici PAM scoring matrix e contengono i punteggi (o score) daattribuire agli amminoacidi appaiati in un allineamento di sequenze proteiche.
1Il termine PAM indica una sostituzione amminoacidica accettata dalla selezione naturale.
CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 22
Figura 3.1: Relazione tra la distanza evolutiva misurata in unita PAM e lapercentuale di amminoacidi diversi riscontrabili nelle proteine.
Questi score sono calcolati a partire dalle probabilita di sostituzione secondo laseguente relazione:
s(a, b) = int
[10× log
M(a, b)
C(a, b)
](3.3)
nella quale s(a, b), B(a, b) e C (a, b) indicano rispettivamente il punteggio da at-tribuire all’appaiamento tra due amminoacidi a e b, la probabilita di sostituzio-ne contenuta nella matrice PAM e la probabilita di appaiamento casuale dei dueamminoacidi.
La probabilita B(a, b) di sostituzione dell’amminoacido a in amminoacido b ecalcolata a partire dalla matrice di probabilita PAM 1, come descritto in precedenza.
La probabilita C (a, b) di trovare l’appaiamento casuale degli stessi amminoacidia e b e calcolata in base alla frequenza media di ogni amminoacido, assumendoche tutti gli amminoacidi si possano appaiare senza alcuna preferenza. Ad esempiol’appaiamento tra due amminoacidi con frequenze di 0,1 e 0,05 avverra con unaprobabilita di 0,005.
I rapporti di queste probabilita, definiti odds, servono a valutare quente volte laprobabilita di omologia sia maggiore rispetto alla probabilita casuale.
I valori ottenuti applicando la formula 3.3 si definiscono log-odds. Essi facilitanoil calcolo globale di un allineamento, in quanto permettono di sommare gli scorespiuttosto che moltiplicarli.
Gli scores contenuti nella PAM scoring matrix possono essere positivi, negativi onulli ed indicano rispettivamente che la probabilita di omologia e maggiore, minoreo uguale alla probabilita di appaiamento casuale.

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 23
3.3.2 Matrici BLOSUM
Le matrici BLOSUM furono introdotte da [Henikoff and Henikoff, 1993]. Questesi basano sull’utilizzo di una banca dati, denominata BLOCKS, contenente unacollezione di allineamenti multipli di segmenti proteici senza gap.
Ogni blocco di allineamenti contiene sequenze con numero di amminoacidi in-dentici maggiore di una certa pecentuale P, la quale e compresa tra il 30% ed il 95%.Da questi blocchi si ricava la frequenza relativa di sostituzione degli amminoacidi.I valori delle frequenze vengono poi utilizzati per calcolare una matrice di log-oddsscores applicando gli stessi criteri usati per le matrici PAM.
La formula utilizzata per il calcolo dei log-odds e la seguente:
s(a, b) = int
[k × log
B(a, b)
C(a, b)
](3.4)
Il termine C(a, b) e la probabilita di appaiamento casuale considerando la fre-quenza media di ogni amminoacido un evento indipendente. La probabilita B(a, b)e la probabilita osservata nei BLOCKS. Infine k e una costante che in genere e paria 2/log2 oppure 3/log2.
3.3.3 Confronto tra matrici PAM e matrici BLOSUM
E importante notare che le matrici PAM e BLOSUM, nonostante sembrino moltosimili tra loro, presentato in realta sostanziali differenze. Esse partono prima ditutto da due presupposti diversi.
Nelle matrici PAM si assume che le distanze evolutive derivino da mutazionisuccessive e indipendenti, e che si possano quindi sommare. Le matrici BLOSUMinvece non fanno assunzioni, esse si basano su allineamenti reali.
Per quanto riguarda le sostituzioni, le matrici PAM tendono a premiare sosti-tuzioni amminoacidiche derivanti da mutazioni di una singola base, penalizzandoquelle che comportano la sostituzione di motivi strutturali degli amminoacidi, ilcontrario di quanto fanno le BLOSUM.
Un altro aspetto da valutare e l’indice. Nelle matrici BLOSUM rappresentala percentuale di identita minima all’intero blocco; al suo aumentare ci si avvici-nera sempre di piu alla matrice identita e il grado di similarita tra le proteine saramaggiore. Invece nelle PAM l’indice indica la distanza evolutiva.
In generale molti ritengono che le matrici BLOSUM siano piu adatte delle PAMper effettuare ricerche di similarita di sequenza.
3.4 Algoritmi di programmazione dinamica
Gli algoritmi di programmazione dinamica principalmente utilizzati per l’allinea-mento di sequenze proteiche sono l’algoritmo di Needleman-Wunsch e quello di

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 24
Smith-Waterman, i quaali permettono rispettivamente di trovare un allineamen-to globale e locale. Esiste anche un altro tipo di algoritmo di allineamento “misto”detto freeshift.
Di seguito verrano riportate le specifiche dei primi due algoritmi citati.
3.4.1 Algoritmo di Needleman-Wunsch
L’algoritmo di Needleman-Wunsch viene utilizzato per ottenere un allineamentoglobale ottimo tra due sequenze.
L’idea che sta alla base della costruzione dell’allineamento ottimo e di utilizzaresoluzioni ottime trovate per sottosequenze di lunghezza minore.
L’algoritmo prevede la costruzione di una matrice F di indici i e j e di dimen-sione (n+1) × (m+1); n ed m rappresentano la lunghezza delle due sequenze che sivogliono allineare.
Il valore F (i, j ) e il punteggio dell’allineamento migliore tra le sottosequenzex 1..i e y1..j
2. E cosı possibile calcolare F (i, j ) ricorsivamente. Come prima cosa enecessario inizializzare F (0, 0) = 0. La costruzione della matrice avviene dalla cellain alto a sinistra alla cella in basso a destra.
Noti i valori di F (i - 1, j - 1), F (i - 1, j ) e F (i, j - 1) e possibile calcolare F (i, j );si possono distinguere tre casi:
• xi = yj, allora F (i, j ) = F (i - 1, j - 1) + s(xi, yj);
• xi e allineato con un gap, allora F (i, j ) = F (i - 1, j ) - d ;
• yj e allineato con un gap, allora F (i, j ) = F (i, j - 1) - d.
Il miglior punteggio (i, j ) e il massimo tra i valori ottenuti nei tre casi sopra citati.E possibile scrivere la seguente relazione di ricorrenza:
F (i, j) = max
F (i− 1, j − 1) + s(xi, yj)F (i− 1, j)− dF (i, j − 1)− d
(3.5)
Nella fase di calcolo della matrice e necessario mantenere un’informazione aggiuntiva,ad esempio un puntatore, che memorizzi da quale cella il valore di F (i, j ) e derivato.
Per poter calcolare il valore di F (i, j ) come descritto dalla 3.5 e necessario in-zializzare la prima riga e la prima colonna della matrice con un valore “speciale”.Infatti quando j = 0 i valori di F (i, j - 1 ) e F (i - 1, j - 1 ) non sono definiti. Quandoi = 0, invece, non risultano definiti i valori di F (i - 1, j ) e F (i - 1, j -1 ).
Nella prima riga e nella prima colonna vengono quindi inseriti i seguenti valori:
F (i, 0) = −id
2x 1..i e la sottosequenza che va dal primo all’i-esimo carattere della sequenza x.; y1..j e lasottosequenza che va dal primo al j-esimo carattere della sequenza y.

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 25
F (0, j) = −jd
Il valore contenuto della cella F (n, m) della matrice stessa rappresenta il pun-teggio o score migliore per l’allineamento di x 1..n e y1..m, corrisponde quindi alloscore dell’allineamento globale ottimo tra le sequenze x ed y.
Una volta ottenuto il valore F (n, m) bisogna ricostruire l’allineamento ottimo.Questo viene fatto attraverso una procedura definita traceback. Essa consiste nelricostruire l’allineamento inverso a partire dall’ultima cella della matrice, utilizzandoi puntatori memorizzati durante la costruzione della matrice. Ad ogni passo delprocesso traceback si effettua uno spostamento dalla cella corrente (i, j ) ad una dellecelle (i-1, j-1 ), (i, j-1 ) o (i-1, j ) dalla quale e derivato il valore di F (i, j ). In basea questo si aggiunge un carattere ad ognuna delle due stringhe che rappresentanol’allineamento secondo le seguenti regole:
• se (i, j ) deriva da (i-1, j-1 ) =⇒ vengono inseriti i caratteri xi, yj
• se (i, j ) deriva da (i, j-1 ) =⇒ vengono inseriti i caratteri ‘-’3, yj
• se (i, j ) deriva da (i-1, j ) =⇒ vengono inseriti i caratteri xi, ‘-’
Questo processo termina quando i = j = 0.
3.4.2 Algoritmo di Smith-Waterman
Un’altra comune situazione e quella di voler trovare l’allineamento migliore tra duesottosequenze x ed y. Questo succede quando, ad esempio, si vuole verificare seuna coppia di proteine hanno un dominio comune. L’algoritmo di Smith-Watermanpermette di fare questo. E in grado infatti di determinare un allineamento ottimolocale.
Questo algoritmo e molto simile a quello di Needleman-Wunsh, presenta peroalcune sostanziali differenze.
Il valore F (i, j ) viene calcolato in questo modo:
F (i, j) = max
0F (i− 1, j − 1) + s(xi, yj)F (i− 1, j)− dF (i, j − 1)− d
(3.6)
Si preferisce quindi inserire il valore 0 nel caso in cui i valori derivanti da tutte le“opzioni” siano negativi. Questo viene fatto perche se nella costruzione dell’allinea-mento ottimo si trova ad un certo punto uno score negativo, e preferibile abbando-nare l’allineamento corrente e iniziarne un altro. Il valore 0 indica quindi l’inizio diun nuovo allineamento.
Di conseguenza i valori di F (i, 0 ) e F (0, j ) sono inizializzati a 0, a differenza diquanto accade per l’algoritmo di Needleman-Wunsh.
3Il carattere ‘-’ corrisponde all’inserimento di un gap

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 26
Un’altra differenza riguarda l’identificazione di quale cella della matrice F con-tiene il valore dell’allineamento locale ottimo cercato. In questo caso e necessariocercare il valore massimo all’interno della matrice F. Fatto questo si applica il pro-cesso traceback a partire dalla cella associata al valore massimo appena torvato.Questo processo termina quando si trova una cella contenente il valore 0 che indical’inizio dell’allineamento.
3.5 Algoritmi euristici
Gli algoritmi di programmazione dinamica, dei quali si e parlato in precedenza, sonoideali per allineare tra loro due sequenze in modo esatto.
L’utilizzo di questo tipo di algoritmi per la ricerca di similarita in banche dati disequenze biologiche richiede tempi di esecuzione troppo lunghi, sono percio necessarialgoritmi piu veloci.
Le ricerche di similarita nelle banche dati consistono nel ripetere automaticamen-te la procedura di allineamento di una data sequenza, definita query, con ognunadelle sequenze presenti nella banca dati (subject), al fine di individuare quella chepresenta il massimo punteggio di allineamento.
Per far fronte all’esigenza di diminuire i tempi di calcolo sono stati sviluppatiprogrammi, quali FASTA e BLAST, in grado di effettuare velocemente ricerche disimilarita perche basati su algoritmi di tipo euristico. Questi ultimi tengogo contodi assunzioni non certe, ma estremamente probabili. La ricerca e resa cosı piu velocea scapito della certezza di avere veramente trovato la soluzione migliore.
3.5.1 FASTA
Il programma FASTA, sviluppato da Lipman e Pearson nel 1985, e stato il primo arispondere all’esigenza di effettuare ricerche di similarita in banche dati.
FASTA si basa su una strategia di indicizzazione di parole. Un parametro im-portante e ktup che indica la lunghezza delle parole da indicizzare dette k-tuples.Per le proteine questo valore in generale e pari a 2. E possibile ottenere 202 parolediverse di due amminoacidi.
L’indicizzazione consiste nella creazione di un elenco di tutte le posizioni in cuicompare ogni possibile parola all’interno della sequenza. Le elaborazioni effettuatedal programma possono essere divise in quattro fasi distinte.
Fase 1. Nella fase 1 vengono considerate le due sequenze di amminoacidi da alli-neare e viene posto ktup pari a 2. Il programma inizia a leggere la sequenza query,una parola alla volta. Per ogni parola individuata viene aggiornato l’indice aggiun-gendo la posizione in cui essa e stata trovata all’interno della query. L’indice e quindicompilato in tempi rapidi, con un numero di operazioni proporzionale alla lunghezzadella sequenza.

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 27
Alla fine dell’indicizzazione delle parole della query il programma analizza unaalla volta, parola per parola, tutte le sequenze subject presenti nella banca dati.
In questa operazione l’indice viene solamente consultato al fine di verificare se, edeventualmete dove, ogni parola della sequenza subject e stato trovata nella sequenzaquery. Quando viene trovata una parola comune alle due sequenze il progammamemorizza la corrispondente diagonale della matrice di allineamento. In questomodo e possibile unire eventuali parole adiacenti per formare segmenti di identitapiu consistenti.
Il progamma scende di una riga nella matrice di allineamento per ogni paroladella sequenza subject, memorizzando le diagonali corrispondenti alla parola co-mune trovata. Se alla parola successiva della sequenza subject viene trovata unacorrispondenza sulla stessa diagonale, la diagonale viene allungata.
Fase 2. Nella fase 2 i punteggi delle regioni corrispondenti ai segmenti piu lunghisono ricalcolati utilizzando una matrice di sostituzione. Si ottengono cosı le bestinitial regions i cui punteggi sono indicati con il termine Init1.
Con questi valori viene stilata una graduatoria delle migliori similarita trova-te nella banca dati per decidere quali e quante sequenze utilizzare per continuarel’analisi delle fasi successive.
Fase 3. La terza fase genera regioni di similarita di maggiori dimensioni, detteInitN. Questo viene fatto congiungendo, dove possibile, le best initial regions eapplicando opportune penalita per ogni gap.
Fase 4. La quarta e ultima fase del programma effettua un allineamento accuratoutilizzando una variante dell’algoritmo Smith-Waterman che considera solo i per-corsi di allineamento entro una stretta banda (circa 20 amminoacidi) fiancheggiantele regioni precedentemente trovate. In questo modo viene calcolato il punteggioottimizzato, inidicato dal programma con Opt.
3.5.2 BLAST
BLAST (Basic Local Alignment Search Tool) e anch’esso un programma utilizzatoper la ricerca rapida di similarita in banche dati biologiche. E stato sviluppato esuccessivamente migliorato da Altschul e da altri ricercatori del NCBI negli anni ’90.Si basa anch’esso sull’indicizzazione di parole.
L’algoritmo puo essere diviso in tre fasi.
Fase 1. In questa fase viene creato un elenco di parole leggendo ad una ad unatutte le parole di una certa lunghezza W della sequenza query.
Facendo uso di una matrice di sostituzione, per ogni parola della query vienegenerata una lista di parole affini. Queste sono dette W-mers e producono unoscore maggiore di una determinata soglia T quando sono allineati con la paroladella sequenza query. Tutti questi sono inseriti nell’elenco.

CAPITOLO 3. ALLINEAMENTO DI SEQUENZE PROTEICHE 28
Fase 2. Nella seconda parte dell’algoritmo si analizzano tutte le sequenze dellabanca dati cercando i W-mers corrispondenti alle parole della lista prodotta dal-l’analisi della sequenza query. Ogni corrispondenza trovata, cioe un hit potrebberappresentare una parte di un possibile allineamento piu esteso.
Fase 3. La terza e ultima fase dell’algoritmo consiste nel verificare per ogni hit see quanto sia possibile estenderlo. Si cerca di estendere l’allineamento in entrambele direzioni, escludendo la possibilita di inserire dei gap.
Nel valutare la possibilita di estendere un hit si tiene conto di un un parametro X.Questo parametro viene utilizzato per stabilire quanto il programma debba insisterenel cercare di estendere gli hit nel caso in cui si incontrino scores negativi. Si ottienecosı un segmento di allineamento locale che non puo essere ulteriormente esteso,definito High-scoring Segment Pair (HSP). S rappresenta una soglia di score; se unHSP supera questa soglia viene preso in cosiderazione.
Negli ultimi anni sono stati sviluppati diversi progarammi basati su BLAST perfar fronte a particolari esigenze. Uno di questi e PSI-BLAST (Postition SpecificIterated BLAST) [Altschul et al., 1997]. Per mezzo di questo programma e possibilegenerare i profili che caratterizzano le sequenze conservate. Questo e possibile inquanto utilizza una ricerca iterativa; le sequenze trovate a ogni ciclo sono usate percostruire un modello di punteggio per il ciclo successivo.

Capitolo 4
Metodi computazionali
Nella prima parte di questo capitolo viene data la definizione di similaria strutturale.Successivamente viene descritto il funzionamento di alcuni programmi utilizzati perconfrontare strutture proteiche e le banche dati che derivano da essi.
La seconda parte riguarda i vari metodi e i diversi approcci che possono essereutilizzati per determinare la struttura tridimensionale di una proteina con metodicomputazionali.
4.1 Similarita strutturale
Nella Sez.3.1 e stata data la definizione di similarita tra sequenze di proteine. Inalcuni casi le sequenze che si confrontano possono essere cosı diverse che diventadifficile mettere in evidenza la loro omologia. I cambiamenti nella struttura proteicainvece si conservano maggiormente. In generale infatti l’evoluzione non altera ilripiegamento della proteina.
Oltre alle sequenze puo essere quindi interessante confrontare le strutture pro-teiche. Questo permette di evidenziare un’evoluzione divergente avvenuta tra dueproteine che hanno una bassa similarita di sequenza.
Per confrontare due strutture proteiche e necessario sovrapporle effettuando larotazione e la traslazione di una delle due strutture sull’altra. Proteine diverse nonhanno lo stesso numero e tipo di residui nelle stesse posizioni; presentano quindidelezioni e inserzioni l’una rispetto all’altra.
Per effettuare una sovrapposizione globale tra due strutture si possono utilizzarele catene dei carboni alfa appartenenti agli elementi di struttura secondaria delledue proteine considerate. In generale delezioni e inserzioni sono localizzate nei loops,che possono essere esclusi dalla sovrapposizione. Spesso succede che catene lateralidi residui identici si trovano in conformazioni diverse. Queste vengono usate inconfronti strutturali che coinvolgono porzioni molto piccole di strutture proteiche.
Una volta ottenuto l’allineamento strutturale e necessario assegnare ad esso unpunteggio che ne valuti la qualita.
Un allineamento struttuale puo essere valutato in base alla deviazione quadratica
29

CAPITOLO 4. METODI COMPUTAZIONALI 30
media (root mean square deviation o r.m.s.d), al numero di atomi accoppiati nellasovrapposizione e alla della similarita dei residui sovrapposti.
L’r.m.s.d. (4.1) e la distanza media tra gli atomi di tutte le coppie che hannopartecipato all’allineamento strutturale; piu bassa e l’r.m.s.d. tanto migliore saral’allineamento calcolato.
r.m.s.d. =
√√√√ N∑i=1
D2i /N (4.1)
Nella formula 4.1 D rappresenta la distanza tra due coppie di atomi appaiati edN e il numero di coppie considerate.
Esistono anche criteri alternativi che tengono conto del numero di atomi o diresidui accoppiati1.
In generale si cerca di massimizzare il numero di atomi appaiati e di minimiz-zare la r.m.s.d. corrispondente. A parita di r.m.s.d sara migliore l’allineamentostrutturale con il maggior numero di atomi accoppiati.
Per confrontare le strutture tridimensionali delle proteine sono stati implemen-tati vari metodi. I piu noti sono DALI [DALI, 2005], SSAP [FSSP, 2005] e CE[Shindyalov and Bourne, 1998].
DALI
Il programma DALI [DALI, 2005] confronta le mappe di contatti tra carboni alfadelle due proteine considerate.
Ogni proteina viene descritta da una matrice di distanze, nella quale l’elemento(i, j ) corrisponde alla distanza tra i caboni alfa compresi tra l’i -esimo e il j -esimoresiduo. L’utilizzo di questa matrice semplifica e rende i calcoli piu veloci: permettedi rappresentare in due dimesioni le informazioni essenziali relative alle strutturetridimensionali delle proteine.
Il confronto di porzioni di proteine si riduce quindi al confronto di sotto matricidi distanze.
Per prima cosa si cercano sottomatrici simili di dimensione 6 × 6; queste vengonoselezionate sulla base della qualita di ogni accoppiamento di sottomatrici.
Il programma DALI viene utizzato per la costruzione della banca dati FSSP[FSSP, 2005](Sez.4.1.1).
CE
Il programma CE [Shindyalov and Bourne, 1998](Combinational Extension) con-fronta coppie di strutture proteiche basandosi sull’identificazione di frammenti al-lineati detti Aligned Fragment Pairs (AFP). Gli AFP corrispondono a coppie diframmenti (uno su ognuna delle due strutture confrontate) con una buona similaritalocale.
1In alcuni casi viene considerato anche il punteggio di similarita di residui accoppiati.

CAPITOLO 4. METODI COMPUTAZIONALI 31
L’allineamento ottimo si ottiene estendendo l’allineamento ottenuto a partireda ogni AFP. Questo avviene per mezzo di una procedura rapida ed efficiente checonsente il confronto tra una proteina e una banca dati di strutture.
SSAP
Il programma SSAP [Orengo and Taylor, 1996] utilizza la doppia programmazionedinamica.
Per prima cosa si costruiscono due matrici di distanze intramolecolari, una perogni proteina da confrontare. Ogni elemento (i, j ) della matrice corrisponde aun vettore che unisce l’i -esimo e il j -esimo carbonio alfa. Successivamente vienecalcolata una matrice di differenze per ogni coppia di righe delle due matrici iniziali.
L’allineamento migliore tra le due strutture si ottiene identificando il migliorpercorso all’interno di una matrice di punteggi sommati.
Il programma SSAP viene utizzato nella costruzione della banca dati CATH[CATH, 2005] (Sez.4.1.1).
4.1.1 Classificazione strutturale
Esistono diverse banche dati derivate da PDB organizzate in modo migliore tenendoconto della classificazione strutturale delle proteine. Esse inoltre non presentanoridondanza.
Queste banche sono ad esempio SCOP [Murzin et al., 1995], CATH e FSSP chedescriviamo di seguito.
SCOP
In SCOP [Murzin et al., 1995] (Structural Classification Of Protein) le struttureproteiche sono organizzate gerarchicamente seguendo criteri evolutivi e di simila-rita strutturale. La classificazione si basa su singoli domini piuttosto che su intereproteine.
Le proteine presenti in SCOP sono divise in quattro categorie:
FAMILY: proteine che hanno una similarita maggiore del 30% , oppure si-milarita maggiore del 15% e presentano similarita con strutture e/o funzioninote
SUPERFAMILY: famiglie di proteine che contengono alcune sequenze e fun-zioni/strutture simili, dovute probabilmente a origini evolutive comuni
COMMON FOLD: superfamiglie che hanno similarita strutturale, cioe hannodei fold2 in comune
CLASS: classe di struttura secondaria (alpha, beta, alpha/beta, alpha+beta)

CAPITOLO 4. METODI COMPUTAZIONALI 32
Figura 4.1: Strutture presenti in SCOP
La classificazione dei domini viene fatta manualmente con il controllo di curatoriesperti. Questo particolare rende SCOP uno strumento affidabile e valido.
CATH
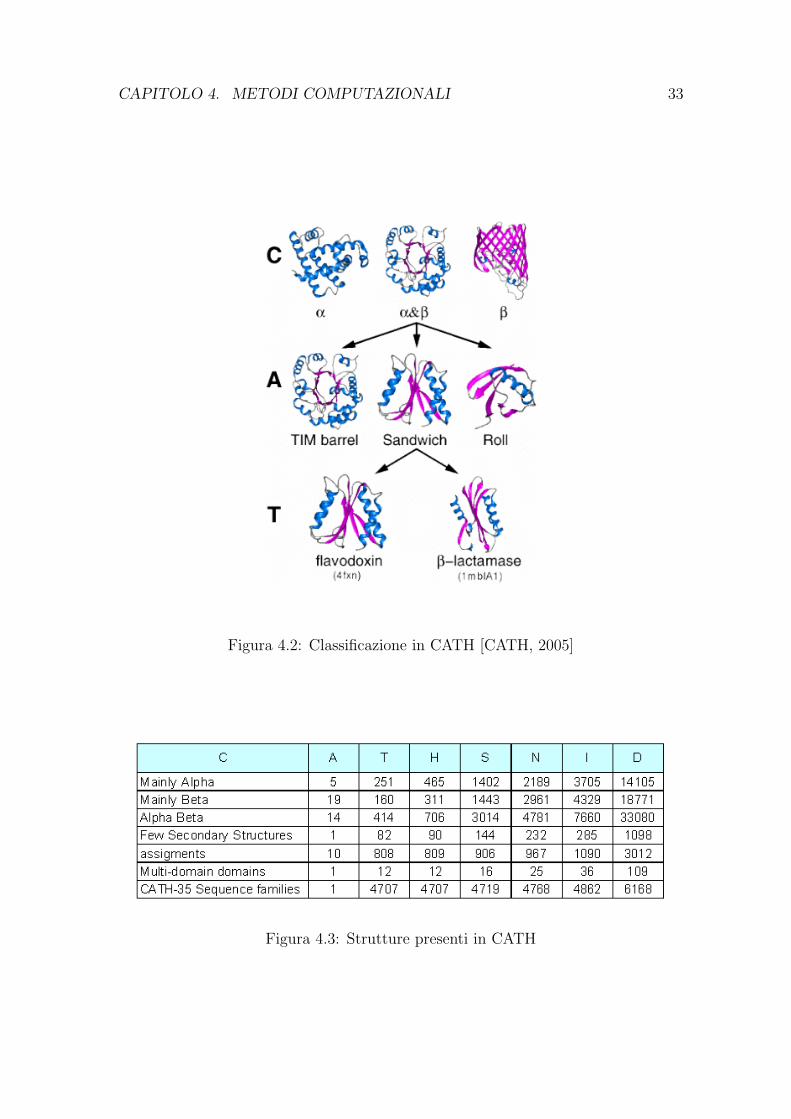
CATH (Class Architecture Topology Homologous superfamily) presenta una classifi-cazione strutturale simile a quella offerta da SCOP:
Class
Architecture
Topology
Homologous superfamily
Sequence family: identita di sequenza maggiore del 35%
Near identical structures: identita di sequenza maggiore del 95%
Identical structures: identita di sequenza pari al 100%
CATH si basa su confronti di strutture effettuati con il programma SSAP. Laclassificazione avviene cosı in modo automatico, ad eccezione della categoria Archi-tecture che viene assegnata manualmente.
Il nome della banca dati deriva dai livelli di gerarchia in essa presenti (Fig.4.2).
FSSP
La classificazione in FSSP (Fold classification based on Structure Structure alignmentof Protein) viene fatta totalmente in modo automatico basandosi sul programma perla ricerca di similarita strutturale DALI [DALI, 2005].
2ripiegamento
CAPITOLO 4. METODI COMPUTAZIONALI 33
Figura 4.2: Classificazione in CATH [CATH, 2005]
Figura 4.3: Strutture presenti in CATH

CAPITOLO 4. METODI COMPUTAZIONALI 34
Il programma DALI viene applicato a tutte le singole catene delle proteinedepositate nel PDB. Il risultati ottenuti vengono depositati nella banca dati FSSP.
Ogni file della FSSP include l’allineamento con le proteine di struttura simile eriporta i residui che sono equivalenti nelle strutture.
4.2 Predizione della struttura tridimensionale
I metodi per predire la struttura tridimensionale delle proteine attualmente utiliz-zati possono essere suddivisi in tre categorie principali:
- homology modeling : effettua delle ricerche in database e costruisce un modelloda struttura omologa nota;
- fold recognition: tenta di riconoscere strutture simili, cercando il foldingmigliore;
- ab initio: costruisce modelli basati sulle caratteristiche fisico-chimiche dellasequenza.
L’homology modeling si applica quando e possibile identificare una proteina distruttura nota la cui sequenza sia simile a quella della proteina target.Si puo utilizzare il modello strutturale del templato come sistema di riferimento percostruire il modello della proteina con struttura ignota. In generale il modello chesi ottiene e piuttosto affidabile.
Nel caso in cui non si trovino nel PDB proteine con sequenza simile alla pro-teina di cui si vuole modellare la struttura, ci si puo affidare alla fold recognition.Questa cerca di identificare il fold tridimensionale piu compatibile con la sequenzaaminoacidica della proteina della quale si vuole modellare la struttura.
Nei metodi ab initio, il ripiegamento di una proteina viene simulato in silicio, conmetodi computazionali basati su potenziali che simulano le vere forze di interazionetra gli atomi della proteina con il solvente. Tali metodi esulano dallo scopo di questatrattazione e non verranno pertanto descritti in questa sede.
4.2.1 Homology modeling
L’homology modeling sviluppa un modello tridimensionale della proteina a partiredalla sequenza. Questo viene fatto tenendo conto di strutture gia note di proteineomologhe al target.
L’idea di base e che proteine con un buon livello di similarita di sequenza risultanoessere molto simili anche nella struttura.
E importante sottolineare che un insieme di proteine che sono supposte omologhepresentano strutture tridimensionali conservate in misura maggiore rispetto alle lorostrutture primarie. Grazie a questa osservazione e stato possibile generare modellidi proteine omologhe, ma con somiglianze di sequenza molto basse.

CAPITOLO 4. METODI COMPUTAZIONALI 35
Nella maggior parte dei casi proteine che mostrano una identita di sequenzasuperiore al 30% conservano una similarita di struttura; in questo modo e possibileutilizzarne una per costruire il modello dell’altra.
Tanto maggiore e l’identita di sequenza tra due proteine tanto piu risultera altala similarita tra le loro strutture tridimensionali.
Questo equivale a dire che all’aumentare dell’identita di sequenza tra due pro-teine l’r.m.s.d. (4.1), calcolata tra i carboni α3 delle loro strutture tridimensionali,diminuisce. Di conseguenza aumenta l’affidabilita ottenibile dal modello.
In particolare e stato osservato che proteine che condividono un’identita di se-quenza maggiore o uguale al 50% mantengono circa il 90% dei residui in posizionistrutturalmente conservate.
In ogni modello saranno presenti regioni piu conservate rispetto ad altre. Infatticonfrontando proteine che svolgono le stesse funzioni, con un’identita di sequenzacrescente, si e osservato che le regioni funzionalmente importanti sono maggiormenteconservate rispetto ad altre; sia in sequenza che in struttura. L’r.m.s.d. infatti risultaessere molto bassa nei siti attivi, mentre cresce di molto nelle regioni lontane da essi.
La predizione della struttura tridimensionale di proteine mediante l’homologymodeling si basa sui seguenti passi fondamentali:
1. Identificazione di omologhi
2. Allineamento di sequenze
3. Costruzione di un pre-modello (regioni conservate)
4. Modellazione dei loop (regioni divergenti)
5. Posizionamento delle catene laterali
6. Refinement
Il primo passo per la costruzione di un modello consiste nell’identificare gli omo-loghi o templates ; ovvero individuare una o piu proteine a struttura nota che pre-sentano una buona identita di sequenza (> 30%) con il target. Questo viene fattomediante una ricerca in banca dati; generalmente viene usato PSI-BLAST. Se sitrovano piu templates e preferibile utilizzare quello piu simile. A parita di simi-larita invece si sceglie la struttura ottenuta tramite cristallografia ai raggi X conrisoluzione migliore.
Fatto questo e necessario allineare al meglio le due sequenze, correggendo l’alli-neamento a mano se necessario. L’allineamento e un’operazione critica, in quantoda esso deriva la qualita del modello che si sta per costruire.
Per identita di sequenza maggiori o uguali al 70% l’allineamento e privo di com-plicazioni e puo essere affidato a procedure automatiche; per identita minori, invecepotrebbero essere necessario un intervento manuale di correzione.
3A volte l’r.m.s.d. viene calcolata su tutti gli atomi della struttura.

CAPITOLO 4. METODI COMPUTAZIONALI 36
Una buona regola inoltre e identificare le regioni strutturalmente conservate equelle variabili. Le regioni conservate in genere sono elementi della struttura secon-daria; quelle variabili corrispondono ai loops, che ospitano la maggior parte delleinserzioni, delezioni e sostituzioni del modello in costruzione.
A questo punto e possibile costruire un pre-modello, nel quale la sequenza dellaproteina target e allineata con le regioni conservate. La struttura del templatoe utilizzata come stampo per la costruzione del modello seguendo l’allineamentotrovato.
Le regioni strutturalmente variabili, che connettono gli elementi della struttu-ra secondaria conservata, non possono essere direttamente utilizzate: e necessariopredirle. La modellazione di queste regioni e piuttosto complessa.
Per prima cosa si cercano template per i loops in un insieme composto da piu pro-teine a struttura nota. Successivamente vengono selezionanti i template per i loopsche presentino strutture pre-loop (frammento N-terminale) e post-loop (frammentoC-terminale) simili alle strutture del template prescelto.
A questo punto viene fatta la modellazione delle catene laterali della preteinatarget. Si tende in generale a mantenere le coordinate delle catene laterali del tem-plate se il residuo sconosciuto e identico o molto simile a quello conosciuto. In casocontrario si utilizzano librerie di rotameri che suggeriscono gli angoli di torsione piuprobabili e piu adeguati alle torsioni del backbone.
L’ultimo passo della modellazione per omologia consiste nel refinement. At-traverso questa operazione si riducono, dove possibile, eventuali errori commessidurante il modeling che comportano collisioni tra gli atomi. Il refinement vieneeffettuato manualmente oppure utilizzando programmi disponibili quali CHARMM,GROMOS, DISCOVER o AMBER.
Alla fine della modellazione gli atomi della proteina non dovranno collidere tradi loro e la struttura trovata deve corrispondere a quella a minor energia possibile.
4.2.2 Fold recognition
La fold recognition viene utilizzata quando il target non mostra una similarita disequenza significativa con proteine a struttura nota.
L’idea di base e cercare di rappresentare la struttura ignota con dei fold conosciutie valutare quale potrebbe essere quello “giusto”, considerando che le proteine innatura assumono un numero finito di strutture possibili compreso tra le 1000 e le3000 unita circa.
Bisogna ricordare inoltre che proteine con una non rilevante similarita di sequen-za presentano spesso lo stesso ripiegamento. Alcuni dei diversi fold conosciuti sonopresenti in piu proteine diverse, altri invece sono stati visti solo in casi singoli.
Ogni metodo di fold recognition si basa sulla presenza delle seguenti componenti:
• una banca dati di fold (fold library) derivata da PDB;
• una funzione di scoring per valutare il fit tra sequenza e fold ;

CAPITOLO 4. METODI COMPUTAZIONALI 37
• un algoritmo per la ricerca dell’allineamento ottimo tra sequenza e struttura;
• un metodo di selezione o ranking.
La fold library e un sottoinsieme dei fold presenti nel PDB, il suo contenuto ericavato da dabase organizzati secondo una classificazione strutturale delle proteinequali ad esempio SCOP [SCOP, 2005], CATH [CATH, 2005] o FSSP [FSSP, 2005](Sez.4.1.1).
Le funzioni di scoring vengono usate dall’algoritmo di allineamento per valuta-re l’allineamento target-template migliore. Queste funzioni possono essere di tipouniposizionale e multiposizionale a seconda che i punteggi siano calcolati indipen-dentemente in ogni posizione o in piu di una posizione simultaneamente.
La scelta dell’algoritmo per ottenere l’allineamento ottimo dipende dal tipo difunzione di compatibilita usata dal metodo. Se e stata utilizzata una funzione ditipo unidimensionale si puo utilizzare direttamente un algoritmo di programmazionedinamica (Sez.3.4.1, 3.4.2). In caso contrario si deve prima trasformare la funzionemultidimensionale in una unidimensionale oppure utilizzare altri algoritmi.
Dopo aver assegnato un punteggio ad ogni all’allineamento del target con tutti itemplate considerati, si passa alla fase di ranking.
Il metodo di selezione piu semplice e l’ordinamento in ordine decrescente dipunteggio. Considerando un modello di selezione di tipo probabilistico e possibiledeterminare quanto il punteggio dell’allineamento sia abbastanza elevato per essereconsiderato significativo. Una funzione utilizzata per il ranking e ad esempio lo Z-score; esso corrisponde alla deviazione standard tra un punteggio e la media di tuttii punteggi.
Un’altro metodo utilizzato e GenThreader [Jones, 1999]; e piu sofisticato e basatosu reti neurali.
I metodi di fold recognition utilizzano principalmente tre tipi di appocci:
• profili e metodi di mapping (1D-1D o 1D-3D);
• threading (3D-3D);
• metodi misti.
I diversi tipi di appocci citati differiscono tra loro per almeno uno dei componentifondamentali dei quali abbiamo discusso precedentemente.
Per capire meglio come funziona la predizione di tipo fold recognition descriviamoil metodo dei profili 1D-3D [] e il metodo Rosetta [Baker et al., 2000].
Il metodo detto dei profili 1D-3D, sviluppato da David Eisenberg, consiste nelcatalogare tutti i fold in termini di intorno per ogni posizione di ogni fold stesso.
L’intorno di ogni posizione viene identificato con la struttura secondaria (α, β ecoil), l’accessibilita al solvente (sepolto, parzialmente sepolto o esposto) e il tipo deiresidui circostanti (polari o apolari).

CAPITOLO 4. METODI COMPUTAZIONALI 38
I fold vengono poi descritti con una sequenza (1D) di simboli. Ogni posizione erappresentata da un simbolo per ogni classe4 in cui viene descritto l’intorno.
Successivamente vengono calcolati dei punteggi derivanti dalle frequenze osser-vate in una banca dati, che associano ogni tipo di residuo a determinate preferenzeper le varie classi definite.
Confrontando le preferenze dei residui della sequenza proteica con i dati riguar-danti le posizioni della struttura e possibile valutare l’allineamento tra la sequenzae il fold.
Utilizzando il metodo dei profili 1D-3D, anche quando una coppia di proteinepresenta una similarita di sequenza molto bassa, si e in grado di assegnare punteggielevati a coppie 1D-3D di proteine correlate da un punto di vista evolutivo. Que-sto e possibile perche l’intorno di una posizione nella struttura proteica risulta piuconservato rispetto alla sequenza della proteina stessa.
Il metodo Rosetta sviluppato da [Baker et al., 2000] non si basa su una bancadati di fold e puo essere suddiviso in tre parti.
Nella prima parte la sequenza della proteina a struttura non nota viene suddivisain frammenti ognuno dei quali contiene da 3 a 9 residui. Poi viene fatta una ricercaper identita o similarita di sequenza tra tutti i frammenti e la banca dati di proteinea struttura nota. Per ogni frammento viene costruito un elenco di possibili strutturetridimensionali.
La seconda parte considera la distribuzione delle conformazioni dei frammen-ti della banca dati di strutture. In questo modo vengono costruiti piu modellialternativi combinando le strutture dei frammenti identificati nella prima parte.
Infine si valutano le varie conformazione con diverse funzioni di scoring.
4.2.3 Ab initio
La predizione della struttura terziaria ab initio consiste nella simulazione in silicio delripiegamento di una proteina. Questo viene fatto utilizzando metodi computazionalibasati su potenziali che simulano le vere forze di interazione tra gli atomi dellaproteina con il solvente.
I metodi ab initio partono dal presupposto che la struttura nativa di una pro-teina corrisponde ad un sistema in equilibrio termodinamico ad un minimo di ener-gia libera. Anfisen [Anfisen, 1973] mostro che tutta l’informazione necessaria peril ripiegamento delle proteine e contenuta nella struttura primaria ed esso derivaesclusivamente da fenomeni fisici.
Lo stato nativo di una proteina corrisponde quindi a un’unica conformazione, conla minima somma di energia potenziale intramolecolare, entropia conformazionale eenergia libera del solvente.
Il calcolo della struttura tridimensionale ab initio si basa sulla presenza di:
• un modello per rappresentare la geometria della proteina;
4Il termine classe sta ad indicare una possibile combinazione dei tre elementi che costituisconol’intorno, ad esempio: α, esposto, polare. A questa classe corrisponde il simbolo 1.

CAPITOLO 4. METODI COMPUTAZIONALI 39
• una funzione energetica;
• un metodo di ricerca.
La rappresentazione della geometria e importante per il calcolo dell’energia po-tenziale del modello. Dato l’elevato numero di atomi di una proteina e dei possibilistati che la catena polipeptidica puo assumere vengono fatte alcune semplificazioni.Generalmente viene ridotto il numero di atomi usati per rappresentare la strutturadella proteina. Per fare questo si utilizza un modello atomico “virtuale” nel qualeun residuo e rappresentato da uno o piu atomi. Altri modelli invece identificano unoo piu residui con un solo atomo.
La tendenza generale e di costruire il modello considerando frammenti della ca-tena polipeptidica costituiti da almeno tre e fino a un massimo di nove residui. Cosıfacendo la procedura viene semplificata proporzionalmente al numero di frammentiutilizzati. La costruzione di modelli e inoltre facilitata dalla possibilita di estrarrestrutture di frammenti noti dalla banca dati PDB.
I potenziali considerati nel calcolo dell’energia minima sono divisi in due ca-tegorie: quelli calcolati considerando le forze fisiche che determinano la strutturadella proteina, e quelli ricavati empiricamente dalle strutture presenti nella bancadati PDB. Le funzioni di energia utilizzate devono essere in grado di discriminare laconformazione nativa dalle altre ad energia superiore.
L’ultimo elemento fondamentale di un metodo ab initio e un algoritmo effi-ciente ed affidabile per la ricerca dei minimi energetici nello spazio delle possibiliconformazioni. Generalmente viene utilizzato il metodo Monte Carlo o delle suevarianti.

Capitolo 5
Allineamenti profilo contro profilo
In questo capitolo daremo la definzione di profilo e desciveremo il processo che portaall’allineamento di due profili.
Sulla base di un’analisi condotta da [Wang and Dunbrack JR., 2004] si cercheradi capire quale sia la combinazione migliore di algoritmi, funzioni e parametri perottenere dei buoni allineamenti profilo-profilo.
5.1 Introduzione
L’allineamento di profili e uno strumento potente per la creazione di allineamenti disequenze molto accurati.
Un profilo e una matrice di dimensione 20 × L: 20 rappresenta il numero diamminoacidi presenti in natura, ed L corrisponde alla lunghezza della sequenzatarget o query. Se si tiene conto dell’informazione relativa ai gaps allora il profiloavra dimensione 21 × L.
I profili furono proposti da [Gribskov et al., 1987] nel 1987 come mezzi per laricerca in database.
I valori contenuti in un profilo corrispondono alla frequenza con cui ogni ammi-noacido e presente in ogni colonna dell’allineamento multiplo.
Fino a questo momento abbiamo sempre fatto riferimento ad allineamenti tracoppie di sequenze. Per conoscere le caratteristiche di una determinata famigliadi proteine in generale vengono utilizzati gli allineamenti multipli tra proteine afunzione omologa. Le sequenze da multiallineare si ottengono in genere dalla ricercain banca dati mediante sistemi di ricerca di similarita come BLAST e FASTA.
In un allineamento multiplo si prendono in considerazione le colonne di residui,piuttosto che le proteine a cui appartengono. L’allineamento multiplo si ottienedall’allineamento progressivo di coppie di sequenze.
I profili sono quindi utilizzati per riassumere le informazioni ottenute da unmultiallineamento.
Per creare un allineamento profilo contro profilo e necessario seguire alcuni passifondamentali:
40

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 41
1. scegliere quali sequenze includere nell’allineamento multiplo utilizzato per lacostruzione dei profili
2. definire un modello per assegnare un “peso” a sequenze simili derivanti dal-l’allineamento multiplo
3. definire un metodo per ricavare la frequenza degli amminoacidi a partiredall’allineamento pesato
4. scegliere una funzione di scoring per assegnare un punteggio agli allineamentitra ogni coppia di colonne dei due profili
5. decidere come assegnare un costo ai gaps presenti nei profili
6. definire se e come includere informazioni strutturali gia disponibili
Un accurato allineamento tra una sequenza target e una struttura utilizzata cometemplato viene determinato utilizzando altre sequenze di proteine ad esse correlate.L’informazione relativa a queste sequenze viene rappresentata mediante profili.
L’uso dei profili e incrementato con lo sviluppo del programma PSI-BLAST daparte di [Altschul et al., 1997]. PSI-BLAST utilizza un approccio euristico per laricerca rapida in banche dati di sequenze proteiche che presentino un buon punteggiodi allineamento con la sequenza query. A partire da queste sequenze e da una matricedi sostituzione viene costruito un profilo che contiene i punteggi di tipo log-odds diogni amminoacido della sequenza query.
Il profilo cosı ottenuto viene poi utilizzato per effettuare un’altra ricerca nellastessa banca dati, ma questa volta per valutare l’allineamento vengono utilizzati ipunteggi contenuti nel profilo.
Le sequenze che presentano un punteggio alto vengono utilizzate per costruireun nuovo profilo. Il processo viene poi nuovamente iterato.
[Pietrokovski, 1996] propose una generalizzazione della ricerca profilo-sequenzain database. Secondo [Pietrokovski, 1996] e possibile creare un database di profili apartire da una banca dati di sequenze ed utilizzarlo per effettuare le ricerche.
5.2 Allineamento di profili
Sono stati implementati diversi metodi per l’allineamento profilo contro profilo iquali differiscono tra loro per alcuni aspetti. In ogni caso i passi seguiti per ottenerel’allineamento di due profili e lo stesso (Fig.5.1). La prima sostanziale differenzariguarda la generazione dei profili iniziali. Generalmente viene utilizzato PSI-BLASTper la ricerca in database non ridondanti. Alcuni utilizzano gli allineamenti multiplidi sequenza generati da PSI-BLAST per costruire i profili in forma di frequenzaoppure nella forma log-odds ; altri invece utilizzano direttamente i profili log-oddsgenerati da PSI-BLAST.
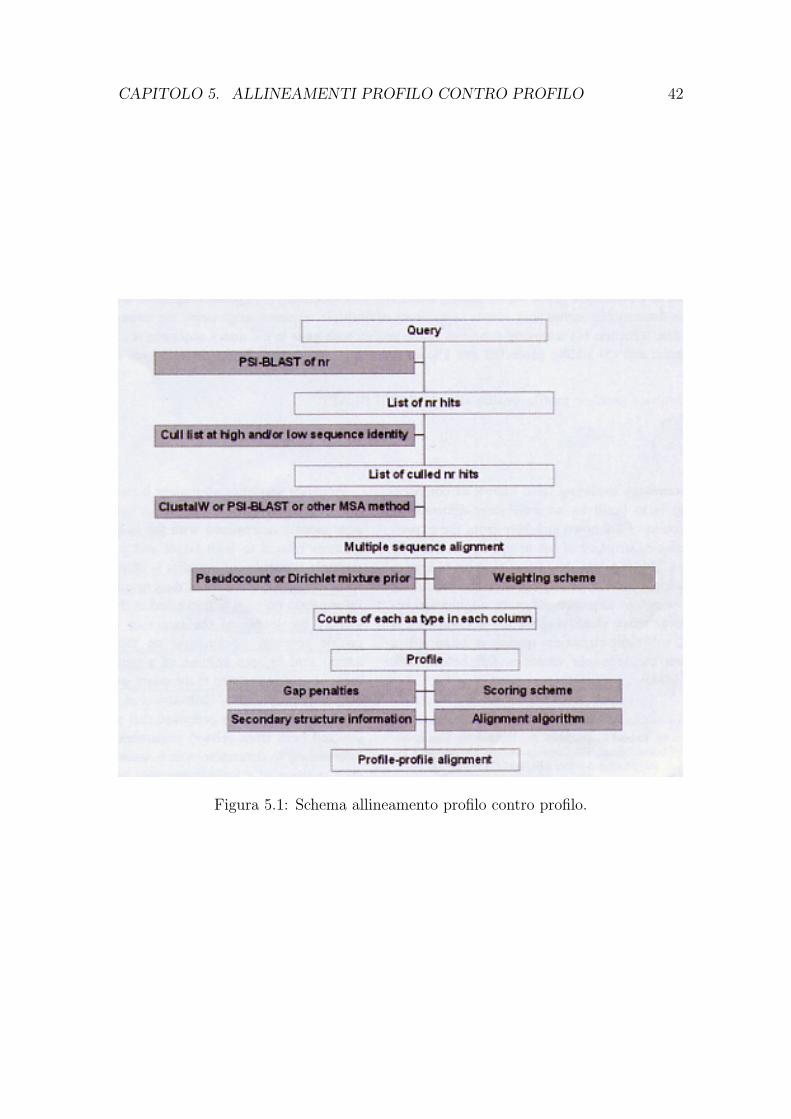
CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 42
Figura 5.1: Schema allineamento profilo contro profilo.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 43
Nella fase di costruzione dei profili e necessario scegliere quali sequenze inclu-dere e in quale modo. I profili possono inoltre differire per come i gaps vengonorappresentati in ciascuna colonna e nella sequenza query.
Inoltre nei metodi messi a confronto e diverso il metodo di scoring utilizzato pervalutare l’allineamento di una colonna del profilo query con una colonna del profilodel template.
La maggior parte dei metodi utilizza l’algoritmo di Smith-Waterman. Se i pun-teggi ottenuti dall’allineamento hanno media negativa e score massimo positivo al-lora questo algoritmo e in grado di identificare regioni comuni senza allineare regioninon correlate. Alcune delle funzioni di scoring utilizzate restituiscono pero punteggipositivi e richiedono quindi uno shift.
Una terza differenza riguarda il costo associato ai gaps. E indispensabilie tenerconto della possibilita che nell’allineamento ottimo, locale o globale, siano presentidei gaps. Il costo a loro associato viene ottimizzato in base alla funzione di scoringutilizzata.
L’allineamento profilo contro profilo ottenuto viene utilizzato per la ricerca inbanche dati di profili e per la costruzione allineamenti sequenza-struttura accurati.
5.3 Analisi di metodi per l’allineamento di profili
In questa sezione descriveremo l’analisi condotta da [Wang and Dunbrack JR., 2004]e i risultati ottenuti.
5.3.1 Descrizione del processo di analisi
Test set
Nella prima fase dell’analisi e stato ottenuto un insieme non ridondante (in terminidi identita) di sequenze dalla versione 1.48 di SCOP. L’identita tra ogni coppia disequenze appatenenti all’insieme ricavato non supera il 40%.
Utilizzando i programmi CE [Shindyalov and Bourne, 1998] (Sez.4.1) e DALI[DALI, 2005] (Sez.4.1) vengono creati due set di tutte le possibili coppie di allinea-menti strutturali tra famiglie e superfamiglie (in base alla definizione data in SCOP)all’interno di scop148 40.
A partire da questi allineamenti si costruisce un data set attraverso una proce-dura che qui non descriviamo.
Il data set risultante e composto di 3441 coppie di allineamenti, che includono1627 sequenze appartenenti a 374 famiglie e 128 superfamiglie. L’identita tra lesequenze e compresa tra lo 0% e il 35%. Questo gruppo di allineamenti e detto setA.
Al fine di valutare la selettivita e la sensitivita della ricerca in database vienecostruito un set S. Questo viene creato a partire dal set A secondo il seguente criterio:non piu di due sequenze dell’insieme devono appartenere ad una stessa famiglia enon piu di dieci sequenze alla stessa superfamiglia.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 44
Il set S risulta quindi composto da 665 sequenze costituite da 441.560 coppie diallineamenti. Questi ultimi comprendono 3320 coppie di true positive, le restantisono false positive.
Generazione di profili
Nella generazione dei profili e stato utilizzato il programma PSI-BLAST. Per primacosa viene creato un database non ridondante di sequenze. L’insieme dei multialli-neamenti ottenuto dopo 5 consecutive applicazioni di PSI-BLAST viene chiamatoLastRound profiles.
Successivante viene creato un’altro set di allineamenti multipli dal quale vengo-no eliminate non solo le sequenze ridondanti, ma anche quelle che presentano unaidentita con la sequenza query inferiore al 15%. Questo set viene detto CutLowIdentprofiles.
Weighting schemes
Un passo importante nella costruzione dei profili a partire da un allineamentomultiplo e dare un “peso” ad ogni sequenza dell’allineamento.
In questo test sono stati considerati tre tipi diversi di weighting schemes :
• Henikoff [Henikoff and Henikoff, 1994] utilizzato da PSI-BLAST
• PSIC [Sunyaev et al., 1999] utilizzato in COMPASS
• SeqDivergence [Rychlewski et al., 2000]
Funzioni di scoring
Uno degli obiettivi dell’analisi di [Wang and Dunbrack JR., 2004] e determinare inquale modo la scelta della funzione di scoring incida sull’accuratezza dell’allinea-mento di sequenze e nella sensitivita e selettivita della ricerca in database. Sonostate prese in considerazione le funzioni di scoring riportate di seguito.
Sum of pairs
Questo tipo di funzione esegue la somma dei prodotti delle frequenze di ogni coppiadi colonne dei due profili considerati. Queste corrispondono a tutte le possibilicombinazioni tra l’amminoacido a e b.
Esistono due varianti: CrossProduct e LogAverage.
S1,2 =20∑
a=1
20∑b=1
Q1aQ
2bsab (CrossProduct) (5.1)
La funzione 5.1 moltiplica ogni prodotto per il corrispondente sab che rappresenta ilvalore dei log-odds della matrice di sostituzione BLOSUM62.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 45
S1,2 = ln20∑
a=1
20∑b=1
Q1aQ
2bqab (LogAverage) (5.2)
La funzione LogAverage [von Osen and Zimmer, 2001] [von Osen and Zimmer, 2003]moltiplica i prodotti per qab. In questo caso qab rappresenta il valore contenuto nel-la matrice BLOSUM62 delle frequenze. Questa viene ricavata dalla matrice nellaforma standard log-odds. Il punteggio finale e il risultato del calcolo del logaritmodella somma dei prodotti.
Dot product
I puteggi calcolati con questo tipo di funzioni corrispondono alla somma dei prodottidelle frequenze (DotPFreq) o dei log-odds (DotPOdds) per ogni coppia di colonnedei profili.
S1,2 =20∑
a=1
Q1aQ
2a (DotPFreq) S1,2 =
20∑a=1
w1aw
2a (DotPOdds) (5.3)
dove w1a e w2
a rappresentano i log-odds ricavati dai profili.
Pearson’s correlation
Questa funzione viene utilizzata nel metodo LAMA [Pietrokovski, 1996] per il con-fronto di due profili.
S1,2 =
∑20a=1(w
1a − 〈w1
a〉)(w2a − 〈w2
a〉)√∑20a=1(w
1a − 〈w1
a〉)2∑20
a=1(w2a − 〈w2
a〉)2(Correl) (5.4)
Jensen-Shannon function
La funzione Jensen-Shannon e stata introdotta da [Yona and Levitt, 2002]. Questasi basa sul calcolo di un divergence score (D) e di un significance score (S). Ilpunteggio finale risultante e dato dalla sombinazione dei due tipi di score.
S1,2 =1
2(1−D)(1 + S) (JensenShannon) (5.5)
Symmetric log-odds multinomial score
Questa funzione di scoring e utilizzata in COMPASS [Sadreyev and Grishin, 2003]
S1,2 = c1
20∑a=1
n1aw
2a + c2
20∑a=1
n2aw
1a (LogOddsMultin) (5.6)
n1a n2
a rappresentano l’effettivo conteggio degli amminoacidi della colonna 1 e 2. c1
e c2 invece sono dei parametri che servono a bilanciare il contributo dei due terminidella formula.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 46
Parametri di valutazione
Per determinare la qualita degli allineamenti sono stati utilizzati i sequenti parametriQModeler, QDeveloper e QCombined.
QModeler e la frazione delle posizioni correttamente allineate nell’allineamentoprofilo-profilo; QDeveloper invece rappresenta la frazione di posizioni correttamenteallineate nell’allineamento strutturale.
QModeler =nC
nA
QDeveloper =nC
nS
(5.7)
dove nC , nA, nS sono rispettivamente il numero delle coppie allineate in en-trambi gli allineamenti (strutturale e di sequenza), nell’allineamento di sequenza enell’allineamento strutturale.
QModeler penalizza allineamenti di sequenze troppo lunghi (nA nS). QDeveloper
al contrario penalizza allineamenti di sequenze troppo corti (nA nS). Consideran-do entrambi questi parametri si ha una visione delle buone e cattive caratteristichedel metodo.
[Yona and Levitt, 2002] introdussero inoltre il parametro QCombined il quale pe-nalizza sia sequenze di allineamento troppo lunghe sia quelle troppo corte.
QCombined =nC
nT
(5.8)
nT e il numero totale di coppie di allineamento “uniche” derivanti dall’allineamentodi sequenza oppure da quello strutturale. Questo vuol dire che se una coppia diresidui sono allineati in tutti e due i tipi di allineamenti il patametro nT ne terraconto una sola volta.
Informazioni sulla struttura secondaria
L’obiettivo principale dell’allineamento profilo contro profilo e quello di determina-re relazioni evolutive, anche remote, tra proteine. [Wang and Dunbrack JR., 2004]hanno determinato per questo motivo una matrice di sostituzione asimmetrica trastruttura secondaria predetta e struttura secondaria nota. Una matrice di sostitu-zione simile e stata usata da [Ginalski et al., 2003] nel server ORFEUS.
5.3.2 Risultati
Nel confronto effettuato sono state valutate accuratezza, sensitivita e specificita diricerca delle funzioni di scoring descritte nella Sez.5.3.1.
Per eseguire il confronto sono stati ottimizzati i parametri gap-open, gap-extentione zero-shift1 per ogni funzione separatamente. Per fare questo si e tenuto conto delvalore di QCombined.
1Shift necessario perche i punteggi ottenuti dall’allineamento hanno media negativa e scoremassimo positivo.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 47
In un primo momento e stato utilizzato il weighting scheme di Henikoff, l’infor-mazione riguardante la struttura secondaria non e stata considerata e non e stataprevista la presenza di gaps nei profili iniziali.
Considerando il parametro QCombined i sette tipi di scoring danno risultati com-parabili in tutti i livelli di identita di sequenza presi in esame. Fanno eccezione lefunzioni LogAverage e LogOddsMultin le quali presentano una minore accuratezzaper valori bassi di identita. Se invece consideriamo i parametri QModeler e QDeveloper
si puo vedere che per valori paragonabili di QCombined le funzioni si comportano inmaniera diversa. Alcune, come ad esempio Correl, DotPOdds, JensenShannon eLogOddsMultin, producono allineamenti piu corti e maggiormente accurati in termi-ni di QModeler. Correl, LogOddsMultin hanno i valori piu bassi di QDeveloper. Altrefunzioni hanno invece bassi valori diQModeler e alti QDeveloper. CrossProduct ha ilvalore piu alto di QDeveloper. Questi risultati indicano che la scelta di quale sia lamigliore funzione di scoring da utilizzare nell’allineamento profilo contro profilo disequenze proteiche dipende dai criteri utilizzati.
Se consideriamo la sensitivita e la specificita, si e visto che le abilita di ricercadelle varie funzioni differiscono in maniera significativa. Le funzioni LogOddsMultin,LogAverage e Correl sono le migliori da questo punto di vista. Ad esempio Correltrova 850 true positive prima di trovare il centesimo false positive.
Successivamente si e cercato di capire quali siano le altre informazioni di cui sideve o meno tener conto per migliorare i risultati ottenuti dagli allineamenti. Conquesto scopo sono state inoltre analizzate diverse combinazioni dei parametri citatiin questa sezione.
Gli allineamenti profilo-profilo sono utilizzati nella predizione della struttura ter-ziaria di una proteina per allineare due sequenze, delle quali soltanto una ha strutturanota. Per questo motivo si e cercato di capire, se e in quale misura, il confrontotra la struttura secondaria predetta della sequenza query e quella di una sequenzacon struttura secondaria nota, possa migliorare gli allineamenti ottenuti. Questroviene fatto introducendo una matrice di sostituzione per la struttura secondaria. Ilprocesso di calcolo di questa matrice non verra descritto.
Il confronto tra le strutture secondarie incide negli allineamenti finali in misuradiversa a seconda della funzione di scoring utilizzata come da Tab.5.1.
CrossProduct 20%LogAverage 30%DotPFreq 35%DOtPOdds 20%Correl 30%JensenShannon 25%LogOddsMultin 30%
Tabella 5.1: Percentuale di incidenza della matrice di sostituzione relativa allastruttura secondaria per le varie funzioni di scoring.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 48
Per le funzioni di scoring considerate l’utilizzo della matrice di sostituzionerelativa alla struttura secondaria migliora, ma non di molto, l’accuratezza degliallineamenti.
5.3.3 Conclusioni
Dall’ analisi effettuata da [Gribskov et al., 1987] di cui abbiamo parlato in questasezione e emerso che
• utilizzare un weighting scheme di tipo PSIC
• eliminare nei profili le posizioni relative alla presenza di gaps nella sequenzaquery
• costruire i profili a partire dal LastRound PSI-BLAST
• utilizzate l’informazione riguardante la struttura secondaria
migliora l’accuratezza dell’allineamento e la selettivita e specificita della ricerca.Considerando quanto appena detto per effettuare l’allineamento profilo-profilo, e
emerso che in termini QCombined si e riscontrato un miglioramento, stimato tra 20%e il 27%, dell’accuratezza per sequenze con bassa identita.
Le funzioni LogAverage e LogOddsMultin hanno prestazioni migliori rispetto allealtre per quanto riguarda la specificita e l’accuratezza; DotPOdds invece e quellapeggiore.
Le funzioni analizzate hanno prestazioni molto simili in quanto i valori che esserestituiscono sono stati normalizzati. Tenendo conto di questo e stata considerata la“nuova” funzione Combined : il punteggio da essa restituito e dato dalla somma ditutti i punteggi restitui dalle 7 funzioni. Questa porta ad una maggiore sensitivitae selettivita rispetto ad ogni altra singola funzione considerata.
5.4 Log average scoring
Parte del lavoro svolto ha interessato l’aggiunta di funzionalita ad una libreria icui dettagli vengono descritti nel Cap.6. In questa era gia possibile calcolare l’alli-neamento di tipo sequenza-sequenza e sequenza-profilo. Abbiamo deciso quindi diaggiungere la possibilita di effettuare anche un allineamento profilo contro profilo,in quanto si e visto essere un ottimo strumento per ottenere allineamenti accurati.
L’implementazione ha preso in considerazione l’ultima parte dello schema rap-presentato in Fig.5.1. I profili vengono generati da un multiallineamento dato. So-no stati considerati tutti e tre i possibili algoritmi dinamici: Needleman-Wunsh,Smith-Waterman e la variante Freeshift.
Tenendo conto dei contronti fatti tra tutte le funzioni di scoring e di tutti ipossibili accorgimenti per migliorare l’allineamento profilo contro profilo abbiamodeciso di utilizzare la funzione LogAverage (5.2).

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 49
Questo tipo di scoring non da i migliori risultati in tutti i casi e in riferimentoa tutti i parametri considerati, ma ha mediamente un buon funzionamento. Inparticolare e una delle funzioni migliori se si considera il rapporto tra true positivee false positive.
Riportiamo di seguito il codice del metodo scoring della classe ScoringP2P cheimplementa la funzione di LogAverage.
Metodo scoring della classe ScoringP2P:
double ScoringP2P::scoring(int i, int j)
Vec<Vec<int> > score = sub.score;
double s = 0.00;
double tmp1 = 0.00;
double tmp2 = 0.00;
for ( int amino = 0; amino < AminoAcid_CODE_SIZE-1; amino++)
AminoAcidCode Amino = static_cast<AminoAcidCode>(amino);
char aminoacid1 = aminoAcidOneLetterTranslator(Amino);
double freq1 = p1.getAminoFrequencyFromCode(Amino,(i-1));
for ( int amino1 = 0; amino < AminoAcid_CODE_SIZE-1; amino++)
AminoAcidCode Amino1 = static_cast<AminoAcidCode>(amino1);
char aminoacid2 = aminoAcidOneLetterTranslator(Amino1);
double freq2 = p2.getAminoFrequencyFromCode(Amino1,(j-1));
double tmp = (score[aminoacid1][aminoacid2])*(freq2);
tmp1 = tmp1 + tmp;
tmp2 = tmp2 + (freq1 * tmp1);
s = log(tmp2+1);
return s;
Spieghiamo ora il suo funzionamento.Il metodo scoring riceve come parametri due numeri interi i e j che rappresen-
tano gli indici della cella della matrice dei punteggi considerata.La variabile score e una matrice bidimensionale di interi alla quale vengono asse-
gnati i valori della matrice di sostituzione considerata (in questo caso BLOSUM62);sub e una variabile di classe di tipo Blosum (vedi Cap.6).
Nelle righe successive sono definite tre variabili di tipo intero: s memorizza il ri-sultato restituito dal metodo scoring; tmp1 e tmp2 invece servono per memorizzarerisultati parziali all’interno dei successi cicli for.
Le istuzioni all’interno dei cicli effettuano le seguenti operazioni per ogni coppiadi amminoacidi.
All’interno dei cicli per ogni amminoacido della coppia considerata viene creatauna variabile di tipo AminoAcidCode contenente il codice numerico associato a quel

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 50
preciso amminoacido. Questo codice viene successivamente tradotto nel corrispon-dente carattere dell’alfabeto che lo identifica. Fatto questo viene estrapolata dalprofilo la frequenza corrispondente a quell’amminoacido nella posizione data dagliindici i e j. Le variabili p1 e p2 rappresentano i due profili utilizzati per l’allinea-mento. Le successive istruzioni implementano i calcoli necessari per ottenere unoscore di tipo LogAverage.
In base al tipo di allineamento che si vuole ottenere (globale, locale o freeshift)si utilizzeranno rispettivamente le classi NWAlign, SWAlign e FSAlign.
Per maggiori dettagli sulla libreria e sulla sua struttura vedere Cap.6.
5.5 Sperimentazione
L’allineamento profilo contro profilo di proteine utilizzando una funzione di scoringdi tipo log average e stato testato con quattro casi con diversa difficolta.
Nella sperimentazione e stata utilizzata la matrice BLOSUM62, una gap-openpenality di 1.474 e una gap-extention penality di 0.037.
L’allineamento e stato fatto con l’algoritmo di programmazione dinamica free-shift. Questo e una variante dell’algoritmo di Needleman-Wunsch (Sez.3.4.1). Ilcalcolo della matrice dei punteggi non cambia. La scelta della cella della matricecontenente il valore associato all’allineamento migliore avviene diversamente. Il va-lore massimo viene cercato cerca tra quelli contenuti nell’ultima riga e colonna. Ilprocesso di traceback ha inizio dalla cella della matrice associata al valore trovato.
L’allineamento di profili effettuato non prevede la presenza di gaps nei profili.Gli allineamenti sono poi stati confrontati con dei risultati dati di allineamen-
ti profilo contro profilo che tengono conto anche dell’informazione associata allastruttura secondaria della proteine. Nonostante questa funzionalita non sia anco-ra presente nella libreria utilizzata i risultati ottenuti risultano simili e si possonoquindi considerare attendibili.
Nella pagine seguenti riportiamo l’allineamento ottenuto pre i quattro targetconsiderati.
Per ogni caso viene indicato il grado di difficolta ad esso associato. La classifica-zione in FR/H, CM/H e CM/Easy si basa sulla suddivisione in catagorie effettuatadal CASP.
Il CASP (Critical Assessment of techniques for protein Structure Prediction) euna competizione che riguarda la predizione della struttura proteica con metodicomputazionali2
2http://PredictionCenter.llnl.gov/casp6/Casp6.html.

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 51
CASO 1
Categoria: FR/H
Target: T0213
Template: 1T62A_2
-freeshift
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
Read matrix from file: blosum62.dat
Sequence 1 (Profile1):
MQPNDITFFQRFQDDILAGRKTITIRDESESHFKTGDVLRVGRFEDDGYFCTIEVTATSTVTLDTLT
EKHAEQENMTLTELKKVIADIYPGQTQFYVIEFKCL
Sequence 2 (Profile2):
MLKNVEVFWQNFLDKHELDMLMPDVWMFGDGSSEMGNRLGQLVVSGRKTATCSSLDIYKMEEEQLPK
AGQYDIILDGQSQPLAIIRTTKVEIMPMNKVSESFAQAEGLDYWYEEHARFFKEELAPYQLQFYPDM
LLVCQSFEVVDLYTH
gap penalty: 1.475
gap extension: 0.037
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
FreeShift alignment:
Score = 21.1111
An optimal alignment:
MQPNDITFFQRFQD---DILAGRKTITIRDESESHFKTGDVLR---VGRFEDDGYFCTIEVTATSTV
TLDTLTEK-----------HAEQENMTLT-------ELKKVIADIYPGQTQFYVIEFKCL
-------MLKNVEVFWQNFLDKHELDMLMPDVWMFGDGSSEMGNRLGQLVVSGRKTATCSSLDIYKM
EEEQLPKAGQYDIILDGQSQPLAIIRTTKVEIMPMNKVSESFAQAEGLDYWYEEHARFFK

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 52
CASO 2
Categoria: CM/H
Target: T0234
Template: 1NRGA_2
-freeshift
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
Read matrix from file: blosum62.dat
Sequence 1 (Profile1):
MSQLEKAQAEYAGFIQEFQSAIISTISEQGIPNGSYAPFVIDDAKNIYIYVSGLAVHTKNIEANPLV
NVLFVDDEAKTNQIFARRRLSFDCTATLIERESQKWNQVVDQFQERFGQIIEVLRGLADFRIFQLTP
KEGRFVIGFGAAYHISGDRLDTLVPITGDKG
Sequence 2 (Profile2):
EETHLTSLDPVKQFAAWFEEAVQCPDIGEANAMCLATCTRDGKPSARMLLLKGFGKDGFRFFTNFES
RKGKELDSNPFASLVFYWEPLNRQVRVEGPVKKLPEEEAECYFHSRPKSSQIGAVVSHQSSVIPDRE
YLRKKNEELEQLYQDQEVPKPKSWGGYVLYPQVMEFWQGQTNRLHDRIVFRRGLPTGDSPLGPMTHR
GEEDWLYERLAP
gap penalty: 1.475
gap extension: 0.037
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
FreeShift alignment:
Score = 59.2779
An optimal alignment:
----------MSQLEK--AQAEYAGFIQEFQSAIISTISEQGIPNG-----SYAPFVIDDAKNIYIY
VSGLAVHTKNIEA---NPLVNVLFVDDEAKTNQIFARRRLSFDCTATLIERESQKWNQVVD------
-QFQERFGQIIEVLRGLADFRIFQLTPKEGRFVIGFGAAYHISGDRLDTLVPITGDKG
EETHLTSLDPVKQFAAWFEEAVQCPDIGEANAMCLATCTRDGKPSARMLLLKGFGKDGFRFFTNFES
RKGKELDSNPFASLVFYWEPLNRQVRVEGPVKKLPEE-----EAECYFHSRPKSSQIGAVVSHQSSV
IPDREYLRKKNEELEQLYQDQEVPKPKSWGGYVLYPQVMEFWQGQTNRLHDRIVFRRG

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 53
CASO 3
Categoria: CM/Easy
Target: T0240
Template: 1QXX_A_2
-freeshift
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
Read matrix from file: blosum62.dat
Sequence 1 (Profile1):
ASGPRALSRNQPQYPARAQALRIEGQVKVKFDVTPDGRVDNVQILSAKPANMFEREVKNAMRRWRYE
PGKPGSGIVVNILFKINGTTEIQ
Sequence 2 (Profile2):
PARAQALRIEGQVKVKFDVTPDGRVDNVQILSAKPANMFEREVKNAMRRWRYEPGKPGSGIVVNILF
KINGTTEIQ
gap penalty: 1.475
gap extension: 0.037
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
FreeShift alignment:
Score = 38.8226
An optimal alignment:
ASGPRALSRNQPQYPARAQALRIEGQVKVKFDVTPDGRVDNVQILSAKPANMFEREVKNAMRRWRYE
PGKPGSGIVVNILFKINGTTEIQ
--------------PARAQALRIEGQVKVKFDVTPDGRVDNVQILSAKPANMFEREVKNAMRRWRYE
PGKPGSGIVVNILFKINGTTEIQ

CAPITOLO 5. ALLINEAMENTI PROFILO CONTRO PROFILO 54
CASO 4
Categoria: CM/H
Target: T0267
Template: 1GHE_B_2
-freeshift
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
Read matrix from file: blosum62.dat
Sequence 1 (Profile1):
MVRIRRAGLEDLPGVARVLVDTWRATYRGVVPEAFLEGLSYEGQAERWAQRLKTPTWPGRLFVAESE
SGEVVGFAAFGPDRASGFPGYTAELWAIYVLPTWQRKGLGRALFHEGARLLQAEGYGRMLVWVLKEN
PKGRGFYEHLGGVLLGEREIELGGAKLWEVAYGFDLGGHKW
Sequence 2 (Profile2):
HAQLRRVTAESFAHYRHGLAQLLFETVHGGASVGFADLDQQAYAWCDGLKADIAAGSLLLWVVAEDD
NVLASAQLSLCQKPNGLNRAEVQKLVLPSARGRGLGRQLDEVEQVAVKHKRGLLHLDTEAGSVAEAF
YSALAYTRVGELPGYCATPDGRLHPTAIYFKTL
gap penalty: 1.475
gap extension: 0.037
-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-x-
Suboptimal FreeShift alignment:
Score = 84.4928
An optimal alignment:
MVRIRRA-----GLEDLPGVARVLVDTWRATYRGVVPEAFLEGLSYEGQAERWAQRLKTPTWPGRLF
VAESESGEVVGFAAFGPDRASGFPGYTAELWAIYVLPTWQRKGLGRALFHEGARLLQAEGYGRMLVW
VLKENPKGRGFYEHLGGVLLGEREIELGGAKLWEVAYGFDLGGH
-----HAQLRRVTAESFAH-YRHGLAQLLFE-TVHGGASVGFADLDQQAYAWCDGLKADIAAGSLLL
WVVAEDDNVLASAQLSLCQK-PNGLNRA-EVQKLVLPSA--RGRGLGRQLDEVEQVAVKHKRGLLHL
DTEAGSVAEAFYSALAYTRVGELPGYCATPDGRLHPTAIYFKTL

Capitolo 6
Software utilizzato e modificheapportate
6.1 Introduzione
La parte implementativa relativa a questo progetto si e svolta utilizzando una libreriadi nome Victor sviluppata, tra gli altri, dal gruppo Biocomputing del Centro diRicerche Interdipartimentale di Biotecnologie Innovative di Padova, del quale faparte il prof. Silvio C. E. Tosatto.
Victor permette di predire la struttura tridimensionale della proteine utilizzandoapprocci e metodologie diverse1. Questa libreria si compone di varie parti che sonostate aggiunte successivamente nel corso degli anni.
La parte della libreria presa in considerazione in questo progetto e denominataAlign; e usata per effettuare allineamenti di vario tipo, quali sequenza-sequenza eprofilo-sequenza, dando la possibilita di usare tutti gli algoritmi di programmazionedinamica citati nella Sez.3.4.
Il lavoro iniziale prevedeva di aggiungere alla libreria le funzionalita necessarieper effettuare allineamenti profilo contro profilo di sequenze proteiche utilizzandouna funzione di scoring di tipo log average.
Da un’analisi accurata riguardante la struttura di Align e emersa l’esigenza dieffettuare delle modifiche anche per permettere sviluppi futuri, quali ad esempiol’allineamento profilo contro profilo che tenga conto della struttura secondaria.
La necessita di modificare la libreria e dovuta principalmente al numero troppoelevato di classi con funzionalita diverse tra loro ma con codice in gran parte uguale.
In questo capitolo descriveremo la struttura della libreria prima e dopo il lavorosvolto in questo progetto.
6.2 Descrizione della libreria
La struttura iniziale di Align e rappresentata in Fig. 6.1
1Informazioni riguardanti la libreria si possono trovare in [Tosatto, 2003].
55

CAPITOLO 6. SOFTWARE UTILIZZATO E MODIFICHE APPORTATE 56
Di seguito ci limiteremo a descrivere il significato delle classi interessate dallemodifiche apportate nell’organizzazione di Align e di quelle che definiscono struttureutilizzate per effettuare l’allineamento profilo-profilo.
La classe Align e una classe astratta base per ogni tipo di allineamento. Daessa derivano AlignSimple utilizzata per allineamenti di tipo sequenza-sequenza eAlignProfile per allineamenti profilo-sequenza.
Come detto prima e possibile utilizzare gli algoritmi di Needleman-Wunsch,Smith-Waterman e Freeshift che vengono identificati rispettivamente dalla coppia dilettere NW, SW e FS. Queste coppie di caratteri all’interno del nome di una clas-se indicano che per effettuare l’allineamento viene utilizzato l’algoritmo associato aquella determinata coppia.
Figura 6.1: Diagramma UML iniziale.
Le classi NWAlign, SWAlign e FSAlign sono utilizzate per effettuare l’allinea-mento sequenza contro sequenza ciascuna utilizzando un diverso algoritmo: se sivuole tener conto dell’informazione riguardante la struttura secondaria ci sono leclassi SecNWAlign, SecSWAlign e SecFSAlign. NWAlignThread, SWAlignThread eFSAlignThread considerano invece uno score di tipo energetico.
L’allineamento profilo-sequenza viene fatto utilizzando le classi ProfileNWAlign,ProfileSWAlign e ProfileFSAlign; se si vuole cosiderare uno score energetico siusano ProfileNWAlignThread, ProfileSWAlignThread e ProfileFSAlignThread.

CAPITOLO 6. SOFTWARE UTILIZZATO E MODIFICHE APPORTATE 57
Tutte le classi di cui abbiamo parlato sopra hanno un metodo pCalculateMatrix
che viene utilizzato per calcolare la matrice dei punteggi. Questo varia in baseall’algoritmo di allineamento utilizzato, in quanto diversi sono i modi con cui glialgoritmi calcolano la matrice. Se si confronta invece il metodo pCalculateMatrix
di classi di “tipo” NW (o SW o FS) che utilizzano funzioni di scoring diverse, ledifferenze riscontrate nel codice sono minime.
Riportiamo un’esempio dove vengono messe in evidenza le sole parti diverse.
Metodo pCalculateMatrix della classe NWAlign
for (int i = 1; i <= static_cast<int>(n); i++)
for (int j = 1; j <= static_cast<int>(m); j++)
int s = score[seq1[i-1]][seq2[j-1]];
int extI , extJ;
Metodo pCalculateMatrix della classe ProfileNWAlign
for (int i = 1; i <= static_cast<int>(n); i++)
for (int j = 1; j <= static_cast<int>(m); j++)
int s = 0;
int extI , extJ;
double tmp2 = 0.00;
for ( int amino = 0; amino < AminoAcid_CODE_SIZE-1; amino++)
AminoAcidCode Amino = static_cast<AminoAcidCode>(amino);
char aminoacid = aminoAcidOneLetterTranslator(Amino);
double freq = profile.getAminoFrequencyFromCode(Amino,(i-1));
double tmp = (score[seq2[j-1]][aminoacid])*(freq);
tmp2 = tmp2 + tmp;
s = static_cast<int>(ceil(tmp2));
Tenendo conto di queste piccole differenze si e deciso di apportare alcune modi-fiche delle quali parleremo successivamente.
Altre classi utilizzate sono Profile per la costruzione di profili a partire da unmultiallineamento, Blosum che implementa una matrice di sostituzione e Thread
utilizzata per effettuare uno scoring energetico. Infine Traceback e una struttura

CAPITOLO 6. SOFTWARE UTILIZZATO E MODIFICHE APPORTATE 58
dati che memorizza gli indici delle celle della matrice che verrano utilizzate perricostruire l’allineamento.
6.3 Modifiche e fuzionalita aggiunte
Partendo dalle considerazioni fatte nella Sez.6.2 si e pensato di modificare la struttu-ra della libreria nel modo seguente. Le classi che introducevano “ridondanza” sonostate tolte.
Dopo le modifiche apportate la struttura di Align e come da Fig.6.2.
Figura 6.2: Diagramma UML attuale.
La classe Align e rimasta sostanzialmente la stessa per quanto riguarda le suefunzionalita. Si sono dovuti modificare alcuni metodi e variabili. In particolare sonostate aggiunte variabili puntatore di tipo ScoringScheme e AlignmentData, dellequali parleremo tra poco.
Align ha come figlie NWAlign, SWAlign e FSAlign.Tutte le funzionalita riguardanti i vari tipi di allineamento e scoring sono rag-
gruppate in un’unica classe per ogni diverso algoritmo. Questo e possibile graziealle variabili di tipo puntatore ereditate da Align.

CAPITOLO 6. SOFTWARE UTILIZZATO E MODIFICHE APPORTATE 59
La parte di codice all’interno del metodo pCalculateMatrix che presentavadifferenze e stata sostituita chiamando il metodo scoring del puntatore
double s = scheme->scoring(i, j);
In questo modo verra richiamato il metodo di scoring specifico che si vuoleutilizzare.
La classe astratta ScoringScheme ha un metodo denominato scoring che rap-presenta appunto un tipo di scoring. E stata poi creata una classe figlia per ognidiverso tipo di scoring, in particolare:ScoringS2S: scoring sequenza-sequenzaScoringS2Ssec: scoring sequenza-sequenza considerando la struttura secondariaScoringS2SThread: scoring sequenza-sequenza di tipo energeticoScoringP2S: scoring profilo-sequenzaScoringP2SThread: scoring profilo-sequenza di tipo energetico
Queste modifiche hanno fatto nascere la necessita di introdurre la classe astrattaAlignmentData che gestisce la ricostruzione e la stampa a video dell’allineamentotrovato.
In generale l’allineamento richiede l’uso di due variabili di tipo stringa che rap-presentano le due sequenze considerate. Nel caso in cui si consideri anche la strutturasecondaria sono necessarie ulteriori due variabili di tipo stringa per rappresentarla.A questo scopo sono state create le classi:
• SecSequenceData che gestisce la ricostruzione e la stampa a video dell’alli-neamento quando viene considerata anche la struttura secondaria
• SequenceData che gestisce tutti i casi di ricostruzione e stampa degli allinea-menti che non tengono conto della struttura secondaria
La stampa e la ricostruzione prima veniva gestita all’interno di ogni “tipo” diclasse quindi non esisteva il problema del diverso numero di variabili utilizzate,infatti in ognuna di esse erano presenti tutte e sole le variabili necessarie.
Se non fosse stata introdotta la classe AlignementData in NWAlign, SWAlign eFSAlign ci sarebbero state due variabili di classe di tipo stringa utilizzate solo inpochi casi.
Se si vuole effettuare un’allineamento bisogna seguire il seguente ordine:
• creare un oggetto di tipo AlignmentData
• creare un oggetto di tipo ScoringScheme
• creare un oggetto di tipo Align
Ad esempio se vogliamo effettuare l’allineamento sequenza-sequenza utilizzandol’algoritmo Needleman-Wunsh, sara necessario scrivere all’interno del programma laseguente porzione di codice:

CAPITOLO 6. SOFTWARE UTILIZZATO E MODIFICHE APPORTATE 60
SequenceData ad(2,seq1, seq2);
ScoringS2S sc(sub,&ad);
NWAlign2 nwAlign(&sc,&ad, gapPenalty, gapExtension);
cout << "A Needleman-Wunsch optimal alignment:" << endl;
nwAlign.domatch(cout);
sAddLine();
Questo tipo di organizzazione risulta essere migliore rispetto al precedente. Inquesto modo infatti se si vuole utilizzare una diversa funzione di scoring basteraaggiungere una classe figlia di ScoringScheme che la implementi.
Dopo aver fatto questo tipo di modifiche e stata aggiunta la classe ScoringP2P
come figlia di ScoringScheme, la quale implementa il log average scoring per alli-neamenti profilo contro profilo di proteine.

Conclusioni
In questa relazione sono stati presi in cosiderazione i vari metodi utilizzati per ilprotein folding.
Abbiamo visto che il punto di partenza per la costruzione di un modello accu-rato della proteina a struttura non nota e avere un buon allineamento tra target etemplate.
Dall’applicazione a casi reali del metodo di allineamento profilo contro profilo ditipo log average e emersa le potenzialita di questo strumento.
Tenendo conto all’analisi svolta da [Wang and Dunbrack JR., 2004], di cui abbia-mo parlato in precedenza, sono emersi quali saranno gli sviluppi futuri. Si e pensatoinfatti di aggiungere la possibilita di effettuare allineamenti profilo-profilo conside-rando anche la struttura secondaria, per migliorare ulteriormente l’accuratezza degliallineamenti.
Inoltre facendo riferimento allo schema di Fig.5.1 e emersa la possibilita di ag-giungere la parte riguardante il weighting schemes, implementando il metodo PSIC,il quale e risultato essere il migliore.
La realizzazione di questi possibili sviluppi e sicuramente facilitata dalla nuovastruttura di Align, la quale si presta maggiormente all’aggiunta di nuove funzionalitasenza l’introduzione di ridondanza.
61

Bibliografia
[Altschul et al., 1997] Altschul, S., T.L. Madden, A. S., Zhang, J., Zhang, Z., Miller,W., and Lipman, D. (1997). Gapped BLAST and PSI-BLAST: a new generationof protein database search programs. Nucleic Acid Res, (25):3389–3402.
[Anfisen, 1973] Anfisen, C. (1973). Principles that govern the folding of proteinchains. Science, (181):223–230.
[Baker et al., 2000] Baker, D., Bonneau, R., Tsai, J., and Ruczinski, I. (2000). Abinitio structure prediction using rosetta. CASP-4 Manual, pages A–118.
[Berg et al., 2003] Berg, J. M., Tymoczko, J. L., and Stryer, L. (2003). Biochimica,chapter 3, 4[96-102]. Zanichelli.
[Bourne and Weissig, 2003] Bourne, P. E. and Weissig, H. (2003). Structuralbioinformatics. Wiley-liss.
[CATH, 2005] CATH (2005). http://www.cathbiochem.ucl.ac.uk/bsm/cath/.
[DALI, 2005] DALI (2005). http://www.ebi.ac.uk/dali/.
[FSSP, 2005] FSSP (2005). http://www.ebi.ac.uk/dali/fssp/.
[Ginalski et al., 2003] Ginalski, K., Pas, J., Wyrwicz, L., von Grotthuss, M., Buj-nicki, J., and Rychlewski, L. (2003). ORFEUS: Detection of distant homologyusing sequence profiles and predicted secondary structure. Nucleic Acid Res,(31):3804–3807.
[Gribskov et al., 1987] Gribskov, M., McLanchlan, A., and Eisenberg, D. (1987).Profile analisys: Detection of distantly related proteins. Proc. Natl. Acad. Sci.,(84):4355–4358.
[Henikoff and Henikoff, 1993] Henikoff, S. and Henikoff, J. (1993). Performanceevaluation of amino acid substitution matrices. Protein, (17):49–61.
[Henikoff and Henikoff, 1994] Henikoff, S. and Henikoff, J. (1994). Performanceevaluation of amino acid substitution matrices. J. Mol. Biol, (233):123–138.
[Holm and Sander, 1994] Holm, L. and Sander, C. (1994). The FSSP database ofstructurally aligned protein fold families. Nucl Acid Res, (22):3600–3609.
62

BIBLIOGRAFIA 63
[Jones, 1999] Jones, D. (1999). GenTHREADER: An efficient and reliable proteinfold recognition method for genomic sequences. J Mol Bio, (287):797–815.
[Murzin et al., 1995] Murzin, A., Brenner, S., Hubbard, T., and Chothia, C. (1995).SCOP: A structural classification of proteins database for the investigation ofsequences and structures. J Mol Biol, (247):536–540.
[Orengo et al., 1997] Orengo, C., Michie, A., Jones, S., Jones, D., Swindells, M., andThornton, J. (1997). CATH - a hierarchic classification of protein domain struc-tures database provides insights into protein structure/function relationships.Structure, (5):1093–1108.
[Orengo et al., 1999] Orengo, C., Pearl, F., Bray, J., Todd, A., Martin, A., Conte,L. L., and Thornton, J. (1999). The CATH database provides insights into proteinstructure/function relationships. Nucl Acid Res, (27):275–279.
[Orengo and Taylor, 1996] Orengo, C. and Taylor, W. (1996). SSAP: sequen-tial structure alignment program for protein structure comparison. Meth Enz,(266):1617–635.
[PDB, 2005] PDB (2005). http://www.rcsb.org/pdb/.
[Pietrokovski, 1996] Pietrokovski, S. (1996). Searching databases of conservedsequence regions by aligning protein multiple-alignments. Nucleic Acid Res.
[Rychlewski et al., 2000] Rychlewski, L., Jaroszewski, L. L. W., and Godzik, A.(2000). Comparison of sequence profiles. strategies for structural predictions usingsequence information. Protein Sci, (9):232–241.
[Sadreyev and Grishin, 2003] Sadreyev, R. and Grishin, N. (2003). COMPASS: Atool for comparison of multiple protein alignments with assessment of statisticalsignificance. J. Mol. Biol, (326):317–336.
[SCOP, 2005] SCOP (2005). http://scop.mrc-lmb.cam.ac.uk/scop/.
[Shindyalov and Bourne, 1998] Shindyalov, I. and Bourne, P. (1998). Protein struc-ture alignment by incrementel combinatorial extension (CE) of the optimal path.Prot Eng, (11):739–747.
[Sunyaev et al., 1999] Sunyaev, S., Eisenhaber, F., Rodchenkov, I., Eisenhaber, B.,Tumanyan, V., and Kuznetsov, E. (1999). PSIC: Profile extraction from sequencealignmnets with position-specific counts of independent observation. Protein Eng.,(12):387–394.
[Tosatto, 2003] Tosatto, S. C. E. (2003). Protein structure prediction: improvingand automating knowledge-based approaches. PhD thesis.
[Valle, 2003] Valle, G. (2003). Introduzione alla bioinformatica. Zanichelli.

BIBLIOGRAFIA 64
[von Osen and Zimmer, 2003] von Osen, N. and Zimmer, I. S. R. (2003). Profile-profile alignment: A powerful tool for protein structure prediction. Pac. Symp.Biocomput, pages 252–263.
[von Osen and Zimmer, 2001] von Osen, N. and Zimmer, R. (2001). Improvingprofile-profile alignment via log average scoring. In Gascuel, O. and Moret, B.,editors, Algorithms in bioinformatics, First International Workshop, pages 11–26.Springer-Verlag, Berlin.
[Wang and Dunbrack JR., 2004] Wang, G. and Dunbrack JR., R. L. (2004). Scoringprofile-to-profile sequence alignments. Protein Science, (13):1612–1626.
[Yona and Levitt, 2002] Yona, G. and Levitt, M. (2002). Within the twilight zone:A sensitive profiles-profiles comparison tool based on information theory. J. Mol.Biol, (315):1257–1275.


















