
Un Sistema per la Gestione dell’Informazione nella Attivit...
111
UNIVERSIT ` A DEGLI STUDI DI VERONA Facolt` a di Scienze MM.FF.NN. Corso di Laurea in Informatica Tesi di Laurea Un Sistema per la Gestione dell’Informazione nella Attivit` a di Ricerca su Fenotipi Complessi in un Laboratorio di Genetica Umana Relatore: Prof. Carlo Combi Correlatore: Prof. Pier Franco Pignatti Laureando: Luciano Xumerle ANNO ACCADEMICO 2002/2003
Transcript of Un Sistema per la Gestione dell’Informazione nella Attivit...

UNIVERSITA DEGLI STUDI DI VERONA
Facolta di Scienze MM.FF.NN.
Corso di Laurea in Informatica
Tesi di Laurea
Un Sistema per laGestione dell’Informazione nella
Attivita di Ricerca suFenotipi Complessi in un
Laboratorio di Genetica Umana
Relatore:
Prof. Carlo Combi
Correlatore:
Prof. Pier Franco Pignatti
Laureando:
Luciano Xumerle
ANNO ACCADEMICO 2002/2003


a Francesca

Ringraziamenti
Ringrazio il Dott. Giovanni Malerba, per il Suo prezioso contributo
durante la stesura di questa tesi.
Ai miei genitori va un ringraziamento per tutto quello che hanno fatto
durante i miei studi universitari.
Infine voglio ringraziare tutti i miei amici di Universita e di Laboratorio.

Riassunto
In questa tesi di Laurea viene presentato un sistema informatico per la
memorizzazione e gestione di informazioni utilizzate nelle attivita di ricerca
su malattie complesse del laboratorio di genetica umana della sezione di
Biologia e Genetica presso l’Universita degli Studi di Verona. Le informazioni
sono memorizzate in una base di dati relazionale che viene interrogata ed
aggiornata in modo automatico attraverso strumenti software sviluppati
appositamente. Il software necessario per il funzionamento e lo sviluppo
del sistema che viene proposto e distribuito con licenze Open Source.
E dimostrato l’uso del sistema per la gestione dell’informazione nei
progetti relativi allo studio genetico di asma ed di osteoporosi. Il
sistema proposto e facilmente configurabile e puo essere arricchito di nuove
funzionalita.

Indice
1 Introduzione 1
1.1 Scopo del lavoro . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2 Fondamenti 3
2.1 Geni e Trasmissione dei Caratteri . . . . . . . . . . . . . . . . 3
2.2 Cromosomi e geni . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.3 Fenotipo e Genotipo . . . . . . . . . . . . . . . . . . . . . . . 5
2.4 Malattia e genetica . . . . . . . . . . . . . . . . . . . . . . . . 6
2.5 Il difetto molecolare . . . . . . . . . . . . . . . . . . . . . . . . 7
2.6 Alberi genealogici . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.7 Come si studiano le malattie complesse . . . . . . . . . . . . . 9
2.8 Sistemi di basi di dati per la genetica . . . . . . . . . . . . . . 11
2.8.1 Il laboratorio di genetica . . . . . . . . . . . . . . . . . 13
2.9 Esempi di sistemi per il trattamento di informazioni clinichee genetiche . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.9.1 Un sistema per la gestione di dati clinici e genetici inrete . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
I

INDICE
2.9.2 MFG Tools . . . . . . . . . . . . . . . . . . . . . . . . 19
2.9.3 GenoDB . . . . . . . . . . . . . . . . . . . . . . . . . . 21
3 Progettazione della base di dati per le informazioni clinichee genetiche 23
3.1 Lo schema Entita-Relazione (E-R) . . . . . . . . . . . . . . . 24
3.2 Lo schema relazionale . . . . . . . . . . . . . . . . . . . . . . 29
3.2.1 La parte per l’amministrazione del sistema . . . . . . . 29
3.2.2 La parte per gli individui . . . . . . . . . . . . . . . . . 32
3.2.3 La parte per i genotipi . . . . . . . . . . . . . . . . . . 34
3.3 Lo schema fisico . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.4 La generazione automatica delle query . . . . . . . . . . . . . 40
4 Architettura del sistema 47
4.1 Strumenti utilizzati per lo sviluppo . . . . . . . . . . . . . . . 48
4.1.1 Il sistema di basi di dati relazionale ed il linguaggio SQL 48
4.1.2 Il linguaggio Perl . . . . . . . . . . . . . . . . . . . . . 49
4.1.3 CGI e mod perl . . . . . . . . . . . . . . . . . . . . . 51
4.1.4 L’architettura del sistema . . . . . . . . . . . . . . . . 55
4.2 Lo sviluppo dei programmi e delle librerie . . . . . . . . . . . 55
4.2.1 PDB::pgc . . . . . . . . . . . . . . . . . . . . . . . . . 56
4.2.2 PDB::bio . . . . . . . . . . . . . . . . . . . . . . . . . 57
4.2.3 PDB::profilo . . . . . . . . . . . . . . . . . . . . . . . 60
4.2.4 PDB::SQL . . . . . . . . . . . . . . . . . . . . . . . . 63
II

INDICE
4.2.5 Gli altri moduli ed i programmi . . . . . . . . . . . . . 64
5 L’utilizzo del sistema 66
5.1 Descrizione del sistema . . . . . . . . . . . . . . . . . . . . . . 67
5.1.1 L’utilizzo dei programmi . . . . . . . . . . . . . . . . . 69
5.2 Applicazione del sistema: Asma . . . . . . . . . . . . . . . . . 84
5.3 Applicazione del sistema: Osteoporosi . . . . . . . . . . . . . . 87
5.4 Sviluppi futuri . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
6 Conclusioni 93
Bibliografia 96
III

Capitolo 1
Introduzione
Negli ultimi anni abbiamo assistito a una esplosione della tecnologia sia in
ambito molecolare che computazionale che ha permesso l’identificazione di
geni coinvolti nelle malattie ereditarie umane [Ris00].
Le malattie complesse (o multifattoriali) sono le malattie dove sia fattori
ambientali che genetici giocano un ruolo nella determinazione del rischio di
sviluppare la patologia (ad esempio, asma e osteoporosi) [GS01]. Il modello di
trasmissione ed i meccanismi molecolari associati nell’insorgenza e sviluppo di
tali patologie sono spesso sconosciuti e la ricerca di fattori genetici coinvolti
richiede la disponibilita di molte informazioni cliniche e genetiche per un
numero elevato di individui. La memorizzazione e gestione dei dati diventa
un elemento chiave per la successiva consultazione delle informazioni. Il
lavoro del laboratorio di genetica e volto alla caratterizzazione genetica degli
individui e, successivamente, all’analisi statistica sui dati raccolti mentre
i dati clinici degli individui ed i campioni di DNA (estratto da sangue
periferico) sono spesso forniti da collaboratori esterni.
1

1 – Introduzione
1.1 Scopo del lavoro
Scopo della tesi e documentare il lavoro svolto al fine di progettare e
implementare un sistema che, per mezzo di un sistema di base di dati ed
una serie di strumenti software, permette di gestire in maniera efficiente
ed affidabile le informazioni cliniche e genetiche necessarie nelle attivita di
ricerca sulle malattie complesse. Gli utenti registrati del sistema hanno
a disposizione una interfaccia Web per l’inserimento e la modifica dei
dati genetici progettata con lo scopo di facilitare alcune operazioni come
l’eliminazione degli errori di battitura e l’immissione dei dati [RBK94]. La
stessa interfaccia Web consente di estrarre le informazioni presenti nella base
di dati in formati utili per i programmi di analisi statistica comunemente
utilizzati.
Nella prima parte del lavoro si presentano alcuni concetti tipici della
genetica medica utili alla comprensione dei termini utilizzati in questa tesi
di laurea. Successivamente si presentano tre sistemi per il trattamento
di informazioni cliniche e genetiche utilizzati da altri laboratori. Un
capitolo e dedicato ad una dettagliata descrizione della base di dati, delle
informazioni memorizzate e delle procedure orientate alla creazione delle
interrogazioni. Dopo aver dato una panoramica degli strumenti utilizzati
per l’implementazione del nostro sistema, vengono presentate le librerie
ed i programmi che abbiamo sviluppato. Per concludere, viene mostrato
il funzionamento programmi e l’applicazione del sistema a due malattie
complesse: asma e osteoporosi.
Il lavoro e stato svolto nel laboratorio di Genetica Matematica, della
sezione di Biologia e Genetica, Dipartimento Materno Infantile e di Biologia
2

1 – Introduzione
e Genetica dell’Universita degli Studi di Verona.
3

Capitolo 2
Fondamenti
Il lavoro descritto in questa tesi di laurea consiste nella progettazione e
sviluppo di una serie di strumenti informatici dedicati a compiti precisi
quali la gestione dei dati genetici ottenuti in laboratorio. Per facilitare
la comprensione di alcuni termini segue una breve introduzione su alcuni
concetti tipici della genetica.
2.1 Geni e Trasmissione dei Caratteri
Il fatto che certe caratteristiche siano trasmesse dai genitori ai figli e noto
fin dai tempi antichi. I primi studi sull’ereditarieta dei caratteri singoli
risalgono alla meta del 1700 quando un botanico tedesco di nome Joseph
Gottlieb Kolreuter (1733-1806) descrisse che incrociando 2 linee distinte
di pomodori ne otteneva una terza ibrida dove i genitori contribuivano
in parti identiche alle caratteristiche della linea ibrida, concludendo che
4

2 – Fondamenti
la trasmissione di un carattere potesse essere associato alla trasmissione
di singoli fattori [SGML92]. Gregorio Mendel (1822-1884) fu il primo a
riconoscere l’esistenza dei fattori ereditari ed a dimostrare i meccanismi
fondamentali dell’ereditarieta, servendosi di esperimenti su piante (Pisum
sativum, Lathyrus odoratus) [SGML92]. Nel 1866 postulo che dei fattori
discreti, o geni, venissero trasmessi da una generazione alla successiva (leggi
della segregazione e dell’assortimento). La genetica Mendeliana, utilizzata
nella agricoltura e zootecnia, riveste anche una grande importanza nello
studio della genetica umana.
Molte malattie nell’uomo sono causate da singoli geni.
2.2 Cromosomi e geni
I cromosomi rappresentano il materiale genetico di base per ogni organismo
vivente conosciuto [SGML92].
L’uomo, come molti altri organismi, e diploide, ossia ogni cellula porta
2 copie di ogni cromosoma. Le due copie sono dette cromosomi omologhi.
L’uomo possiede 22 coppie di cromosomi omologhi (numerati da 1 a 22 in
base alla loro lunghezza) e 2 cromosomi detti sessuali per un totale di 46
cromosomi. Per ogni cromosoma il figlio riceve un omologo dal padre e uno
dalla madre. Per i cromosomi sessuali il figlio ricevera uno dei 2 cromosomi X
dalla madre e il cromosoma X (sara femmina) o Y (sara maschio) dal padre.
Nei cromosomi sono contenuti i geni: ogni individuo porta quindi 2
copie dello stesso gene. Le due copie del gene costituiscono il genotipo
dell’individuo per il dato gene. Per ogni gene possono esistere molte varianti
5

2 – Fondamenti
dette alleli che costituiscono il pool genico della popolazione (l’insieme di
tutti i geni di una specie, ognuno con tutti i suoi alleli) [SGML92]. Ogni
individuo, essendo diploide, puo portare al massimo 2 alleli diversi tra quelli
presenti nella popolazione. L’individuo e detto omozigote se entrambi gli
alleli sono identici, mentre e eterozigote se i 2 alleli sono diversi fra loro.
2.3 Fenotipo e Genotipo
Il fenotipo di un individuo riferito ad un carattere e la forma che viene
mostrata (osservabile) e puo essere espressa sia in termini qualitativi (ad
esempio, affetto/non affetto) che quantitativi (ad esempio, altezza, peso).
La relazione tra genotipo e fenotipo puo essere piu o meno stretta. Nel
caso di una relazione diretta (carattere mendeliano) il genotipo determina
il fenotipo in accordo con un dato modello mentre per altri caratteri la
relazione e piu velata (malattie complesse) oppure inesistente (esiste la sola
componente ambientale). L’esempio del gruppo sanguigno AB0 puo essere di
aiuto per chiarire la relazione tra genotipo e fenotipo nel caso di un carattere
mendeliano. Con questo esempio si puo inoltre introdurre il concetto di
dominanza, codominanza e recessivita di un allele rispetto ad un altro [GS02].
Esistono 6 possibili genotipi (3 alleli: A,B,0). La tabella 2.3 riporta i
genotipi con il corrispondente fenotipo.
6

2 – Fondamenti
GENOTIPO FENOTIPO
AA A
A0 A
BB B
B0 B
AB AB
00 0
L’allele A domina su 0 (il genotipo A0 e associato al fenotipo A). Gli
alleli A e B sono codominanti (fenotipo AB). I genotipi AA e A0 sono
fenotipicamente indistinguibili.
2.4 Malattia e genetica
Le malattie in relazione alla loro origine possono essere classificate in 3
categorie principali [GS02]:
1. patologie ad esclusiva origine genetica o malattie Mendeliane:
seguono una segregazione (trasmissione del fenotipo o del genotipo
attraverso individui imparentati) Mendeliana (ad esempio la Fibrosi
Cistica).
2. patologie ambientali: traumi ed infezioni (ad esempio un incidente
stradale).
3. malattie multifattoriali o poligeniche: gruppo estremamente
variabile ed eterogeneo in relazione al numero di geni e dei fattori
ambientali (per esempio il diabete, il lupus, l’asma).
7

2 – Fondamenti
Questa classificazione e utile da un punto di vista didattico, ma appare
sempre piu inadeguata e semplicistica. Cio che si osserva e che il contributo
delle componenti geniche e ambientali e variabile nelle diverse malattie
[SR99].
Si parte dalle malattie mendeliane o semplici (come la fibrosi cistica o
l’emofilia) - causate dalla mutazione di un gene - per passare alle malattie
complesse o multifattoriali che risultano dalla interazione di piu geni tra loro
e di questi geni con fattori ambientali piu o meno influenti - in questo caso
i fattori genetici conferiscono solo una suscettibilita o predisposizione allo
sviluppo del disturbo.
2.5 Il difetto molecolare
Uno degli obiettivi della genetica umana e di identificare la mutazione
(alterazione) genetica che e associabile ad una patologia. L’alterazione
puo verificarsi a livello cromosomico (duplicazioni, delezioni, inversioni di
frammenti) o molecolare. Il difetto molecolare e spesso rilevato grazie a
tecniche di biologia molecolare che abitualmente si utilizzano nei laboratori
di genetica umana [BLC+01].
Il DNA (acido desossiribonucleico) e la molecola che contiene l’insieme dei
caratteri ereditari da cui dipendono l’organizzazione delle molecole proteiche
e la regolarita delle reazioni che avvengono nelle cellule. Il DNA e una lunga
catena di basi nucleotidiche (A adenina, T timina, C citosina, G guanina).
I cromosomi sono costituiti dal DNA e da una parte proteica con funzione
strutturale e regolativa. Alterazioni nella sequenza dei nucleotidi rispetto
8

2 – Fondamenti
ad una sequenza di riferimento indicano potenziali variazioni associabili ad
alterazione nel fenotipo.
Le variazioni piu comuni si riferiscono a cambiamenti di basi (lettere) in
posizioni fisse (Fig. 2.1),
Figura 2.1. Cambiamento di una base in posizione fissa: C diventa G.
oppure a delezioni/inserzioni di una o piu basi (Fig. 2.2), oppure a
Figura 2.2. Una delezione di basi. Viene perso il frammento GTGTTG.
variazioni nel numero di ripetizioni (Fig. 2.3) di un particolare motivo.
Queste variazioni sono utilizzate come marcatori molecolari per seguire la
trasmissione da genitori a figli della regione cromosomica che le contiene.
Quindi due individui possono presentare un diverso numero di ripetizioni:
questa differenza puo essere usata per distinguere il primo individuo dal
secondo.
Esistono esempi associati a patologie per tutti i tipi di variazione descritti.
9

2 – Fondamenti
Figura 2.3. Variazione nel numero di ripetizioni. Il frammentoTATATATA (1 ripetizione 4x del dinucleotide TA) diventaTATATATATATATATA (2 ripetizioni 4x del dinucleotide TA).
2.6 Alberi genealogici
Un albero genealogico (pedigree) e dato da un gruppo di individui con una
chiara relazione familiare di ognuno con gli altri [BLC+01].
I pedigree vengono rappresentati principalmente in 2 modi. Uno e quello
classico del genetista (Fig. 2.4a) e l’altro e quello del matematico (Fig.
2.4b) che organizza gli individui codificando le informazioni di relazione e
memorizzandole in un file [Lan97]. Gli individui per i quali i genitori non
sono noti sono detti fondatori.
2.7 Come si studiano le malattie complesse
Nelle malattie complesse il fattore genetico contribuisce nella suscettibilita
alla malattia ma non e di per se il fattore determinante.
Il passaggio dallo studio delle malattie monogeniche, dove l’alterazione del
gene e la causa della malattia, alle malattie multifattoriali, dove l’alterazione
e associata ad un rischio, ha visto un cambiamento nelle metodologie
applicate allo studio per la ricerca dei fattori genetici che sono associati ad
10

2 – Fondamenti
(a) Genetico (b) Matematico
Figura 2.4. I due modi di rappresentazione di un pedigree:(a) Il quadrato indica il maschio, il cerchio la femmina;(b) padre, madre: x indica genitore ignoto;(b) sesso: 1 indica il maschio, 2 la femmina.
una aumentata suscettibilita alla malattia.
Tradizionalmente gli studi genetici si possono suddividere in 2 categorie:
studi di associazione e studi familiari (studi di linkage).
Negli studi di associazione si misura la frequenza di un dato marcatore
genetico e lo si compara tra 2 o piu classi di individui (ad esempio i casi
e controlli) non imparentati tra loro. Una differenza significativa della
frequenza del marcatore e indice che uno degli alleli del marcatore e associato
alla patologia.
Con gli studi familiari si studia la segregazione congiunta (cosegregazione)
di un marcatore genetico con la malattia in accordo con il modello genetico
proposto (dominante, recessivo, e cosı via). Per marcatore si puo intendere
11

2 – Fondamenti
qualsiasi variazione osservabile che possa permettere la discriminazione di
un allele da un altro. Se il marcatore cosegrega con la malattia significa che
nella regione cromosomica dove si trova il marcatore si trova anche il gene
associato alla malattia.
C’e un’accesa discussione su quale possa essere il metodo piu appropriato
per ricercare i fattori genetici associati alle malattie comuni. Entrambi
i metodi hanno in comune delle forti richieste in termini di individui da
reclutare nello studio e di marcatori genetici da analizzare. Alcuni studi
distinti intrapresi dal nostro laboratorio hanno l’obiettivo di identificare
fattori genetici coinvolti nell’asma infantile, nelle alterazioni del metabolismo
osseo e nelle malattie cardiovascolari.
Lo studio per l’asma infantile ha richiesto l’analisi di circa 200 famiglie
(∼ 850 individui) dove ogni individuo e stato caratterizzato geneticamente
(genotipizzato) con 400 marcatori disposti lungo i cromosomi. Uno studio
analogo e stato attivato per l’identificazione di fattori genetici coinvolti
nell’osteoporosi (∼ 1700 individui). Lo studio relativo alle malattie
cardiovascolari sta coinvolgendo l’analisi di 1500 individui.
2.8 Sistemi di basi di dati per la genetica
Poiche le malattie complesse colpiscono una elevata percentuale di persone,
tali patologie sono studiate in campo medico con sempre maggior interesse
[GS01]. Le tecniche adottate per le malattie genetiche semplici sono
inadeguate. Per il genetista non e piu sufficiente studiare i pochi numeri
forniti da pedigree piuttosto estesi per un marcatore alla volta [Ott99].
12

2 – Fondamenti
Lo studio delle malattie multifattoriali richiede lo studio di migliaia di
individui con migliaia di marcatori genetici. Dati i grandi numeri in gioco
e la complessita dei sistemi oggetto di studio si rende necessario servirsi di
conoscenze e metodi che provengono da altre discipline quali l’informatica,
la statistica, la matematica.
La strategia usata nello studio delle malattie complesse si basa su due
possibili disegni: studio familiare e studio caso/controllo [Ott99].
Nel primo caso gli individui raccolti formano un set di famiglie. Per ogni
individuo bisogna fornire il maggior numero possibile di informazioni:
• la struttura della famiglia (pedigree)
• i caratteri fenotipici (eta, peso, test clinici, e cosı via)
• i dati genetici
Le prime informazioni raccolte per i singoli individui riguardano le relazioni
parentali con gli altri individui della famiglia, il sesso e l’eta. Di solito
i dati clinici sono raccolti per mezzo di questionari in forma cartacea
o in formati elettronici non standard [MTK+98]. Le famiglie composte
da molti individui distribuiti su piu generazioni sono le piu informative
durante gli studi linkage o di segregazione. Con lo studio di segregazione
si cerca di individuare il miglior modello di trasmissione della malattia (ad
esempio ambientale, poligenico, oligogenico, mendeliano) [Ott99]. Il linkage
rappresenta la tendenza di due geni a essere ereditati insieme in virtu della
loro vicinanza fisica su un cromosoma [GS01]. L’analisi di linkage ha sfruttato
questa caratteristica per identificare, attraverso l’uso di sequenze specifiche
13

2 – Fondamenti
di DNA (i marcatori), i geni malattia che si trasmettono in alcune grandi
famiglie [GS01].
Nel caso di studi caso/controllo, individui affetti (casi) vengono
confrontati con individui non affetti (controlli) per cercare di determinare
dei fattori di rischio che ci attendiamo essere piu presenti nei casi [Ott99].
2.8.1 Il laboratorio di genetica
Per condurre le analisi cliniche e gli studi genetici, e necessario raccogliere dei
campioni di DNA di tutti gli individui. Il laboratorio che raccoglie i campioni
puo essere diverso dal laboratorio che esegue lo studio genetico: in tal caso
il laboratorio di genetica deve assegnare ad ogni campione di DNA raccolto
il codice dell’individuo e provvedere allo stoccaggio ed alla conservazione di
tutti i campioni.
Genotipizzazione degli individui
Quando i campioni di DNA sono disponibili al laboratorio di genetica e
possibile procedere nello studio con la genotipizzazione degli individui per
diversi marcatori [MTK+98].
Lo studio di una malattia complessa richiede la genotipizzazione di
un grande numero di individui per un elevato numero di marcatori sia
per studi di scansione genomica che per studi dettagliati di linkage
disequilibrium (associazione preferenziale di alleli di diversi marcatori) in
regioni cromosomiche ristrette [Ris00]. La mole di dati genetici prodotti
14

2 – Fondamenti
cresce molto rapidamente con l’avanzare dello studio ed occorrono sistemi per
memorizzare, correggere e richiamare le informazioni necessarie [LDL+01].
La genotipizzazione dei marcatori e soggetta ad errori che dipendono da vari
fattori:
• condizioni sperimentali
• errori umani:
– puo essere difficile interpretare i risultati sperimentali
– l’operatore deve inserire i valori dei due alleli che descrivono un
singolo genotipo per tutti gli individui ed i marcatori: questa
operazione e spesso soggetta ad errori di battitura.
Memorizzazione dei dati genetici prodotti
Molti laboratori si affidano a normali “fogli elettronici” per la memorizzazione
dei dati genetici prodotti, ma tale scelta puo essere vantaggiosa solo per
piccole quantita di dati poiche non e possibile strutturare i dati e la
dimensione del foglio elettronico puo essere limitata (ad esempio, un foglio
di MS Excel ha dimensione massima di 65536 righe per 256 colonne [Inc03]).
Un’altra scelta possibile e quella di un piccolo sistema di basi di dati (ad
esempio, MS Access) che pero e spesso sottoutilizzato in quanto gli utenti
hanno una conoscenza limitata delle potenzialita del software. Una base di
dati relazionale ben progettata puo permettere di maneggiare con efficienza
ed affidabilita tutte le informazioni necessarie.
15

2 – Fondamenti
Estrazione ed analisi statistiche dei dati
I dati precedentemente memorizzati devono essere estratti in un formato
compatibile con il programma di statistica che l’operatore desidera utilizzare.
Nel caso di basi di dati relazionali l’operatore deve conoscere la struttura della
base di dati ed il linguaggio SQL per poter effettuare le interrogazioni. La
creazione di strumenti software per l’estrazione dei dati e la creazione dei
file utilizzati dai programmi di statistica permette a tutti gli operatori di
consultare le informazioni necessarie e di poter eseguire delle analisi.
Sui dati estratti vanno condotte alcune semplici analisi statistiche utili
per produrre una statistica descrittiva del campione analizzato come, ad
esempio, il calcolo della frequenza degli alleli. Tra i test abitualmente
utilizzati ricordiamo il test che compara le frequenze dei genotipi osservati
rispetto la distribuzione attesa sotto l’ipotesi del libero assortimento degli
alleli negli individui di una popolazione. Le cause di una differenza
significativa nella distribuzione - verificate attraverso il test di Hardy-
Weinberg - possono essere attribuite ad un qualsiasi fenomeno biologico che
impedisce il libero assortimento degli alleli (fenomeni selettivi dove viene
selezionato un particolare genotipo e fenomeni migratori) oppure a cause
sperimentali (una non accurata lettura dei genotipi) [SGML92] [MXP03].
E possibile inoltre condurre analisi genetiche tramite programmi
statistici sviluppati appositamente, come GeneHunter [KDRDL96] e Merlin
[ACCC02], per trovare dei marcatori associati alle malattie studiate.
16

2 – Fondamenti
2.9 Esempi di sistemi per il trattamento di
informazioni cliniche e genetiche
Consultando la letteratura sono stati individuati tre sistemi orientati alla
gestione e memorizzazione delle informazioni cliniche e genetiche necessarie
durante lo studio su una malattia complessa.
I tre sistemi propongono approcci diversi per affrontare il problema ma
sono accomunati da due fattori:
• tutte le informazioni sono memorizzate in una base di dati relazionale;
• i sistemi utilizzano software proprietario per il funzionamento.
Di seguito proponiamo una breve disamina dei tre sistemi:
• un sistema per la gestione di dati clinici e genetici in rete (sezione 2.9.1)
[MTK+98];
• MFG Tools (sezione 2.9.2) [IDoMotCK00];
• GenoDB (sezione 2.9.3) [LDL+01].
2.9.1 Un sistema per la gestione di dati clinici e
genetici in rete
Il sistema e diviso in 4 moduli ed ogni modulo e gestito da un client
[MTK+98].
17

2 – Fondamenti
L’hardware richiesto e costituito da un computer IBM compatibile (PC)
che opera da server, da alcuni PC che ospitano diversi moduli del sistema
e da un Apple Macintosh per il modulo dei genotipi. I PC utilizzati per
ospitare uno o piu moduli del sistema - a seconda delle necessita del singolo
laboratorio - operano come client, possono essere meno potenti del server ed
essere collocati in laboratori diversi.
Figura 2.5. I 4 moduli connessi al server centrale
I moduli offerti dal sistema sono 4 e permettono di gestire i dati clinici, i
dati genetici, la quantita ed il luogo di stoccaggio per i campioni di DNA di
ogni individuo, e lo sviluppo delle linee cellulari (colture di cellule in vitro)
(Fig. 2.5).
Il sistema operativo e MS Windows NT per il server e MS Windows
3.1 per i client, il sistema di basi di dati relazionale e il Borland Paradox
5.0 ed il programma per la rappresentazione grafica dei pedigree e Cyrillic
2.1. Il computer Macintosh e necessario perche il software che permette di
estrarre i dati dal sequenziatore di DNA e disponibile solo per questo tipo di
macchina. Il sequenziatore di DNA e la macchina utilizzata per estrarre delle
18

2 – Fondamenti
sequenze localizzate di DNA. Gli utenti utilizzano i pacchetti GeneScan 2.1c2
[Bioa] e GenoTyper 1.1 [Biob] - forniti con il sequenziatore - per effettuare
una gestione semi-automatica dei dati prodotti dal sequenziatore. Tutti i
programmi che permettono le operazioni di inserimento dati, di generazione
di report dalla base di dati e di formattazione dei dati estratti sono stati
sviluppati in MS Visual Basic appositamente per il sistema.
Lo schema della base di dati e diviso in 4 parti - corrispondenti ai 4 moduli
- connesse alla relazione Master Pedigree. Questa rappresenta le informazioni
principali sugli individui come gli ID univoci, la struttura della famiglia, il
probando (individuo che arriva alle osservazioni del medico e attraverso il
quale si procede poi alla raccolta delle informazioni relative ai familiari) e gli
ID alternativi che possono essere assegnati dai vari laboratori agli individui
stessi.
Le altre relazioni del sistema rappresentano le informazioni gestite dai 4
moduli disponibili:
• le relazioni che descrivono i caratteri clinici degli individui contengono
informazioni sui caratteri fenotipici, sullo stato delle famiglie (criteri
di selezione, stato della famiglia, e cosı via) e sui dati amministrativi
(pagamenti, consensi allo studio, e cosı via).
• un diverso gruppo di relazioni descrive le informazioni sui campioni di
DNA, come il frigorifero in cui si trova la provetta, la quantita residua
e l’individuo a cui e stato fatto il prelievo.
• il gruppo di relazioni che descrive la linea cellulare contiene le
informazioni sulle culture cellulari.
19

2 – Fondamenti
• Le relazioni che descrivono i dati genetici degli individui contengono
le informazioni sui marcatori (posizione sul cromosoma e, se possibile,
gene di appartenenza) ed i genotipi degli individui.
L’estrazione delle informazioni contenute nella base di dati avviene
attraverso i moduli del sistema. Inoltre e possibile esportare un file
compatibile con il programma Cyrillic 2.1 per generare un pedigree.
Il sistema permette di gestire tutte le informazioni necessarie allo studio di
malattie complesse (individui, struttura delle famiglie, fenotipi e genotipi) ed,
in aggiunta, informazioni relative ai campioni di DNA ed alle linee cellulari.
2.9.2 MFG Tools
MFG Tools [IDoMotCK00] e un sistema integrato che permette la gestione
di individui, fenotipi e genotipi. Funziona con il sistema operativo MS
Windows, utilizza il sistema di basi di dati relazionale MS SQL Server 7.0 per
la memorizzazione dei dati e fornisce alcuni programmi - e possibile scaricare
il codice sorgente - sviluppati con il linguaggio MS Visual Basic.
Il sistema e diviso in due parti distinte: una parte clinica, dove risiedono
tutte le informazioni relative agli individui, alla struttura delle famiglie ed
ai fenotipi, e una parte genetica dove sono memorizzati tutti i dati relativi
alle condizioni sperimentali (nome del marcatore, nome del gene, i reagenti
utilizzati durante l’esperimento, e cosı via) ed i genotipi degli individui.
Ogni operatore e il proprietario dei dati da lui inseriti e, con
20

2 – Fondamenti
l’amministratore di sistema, e il solo che puo permettere accessi in
lettura/scrittura agli altri utenti.
Lo schema della base di dati e diviso in parti distinte che rappresentano
vari gruppi di informazioni. La parte piu importante dello schema e quella
che rappresenta gli individui. Ogni individuo viene contrassegnato con un ID
univoco e per ogni ID sono disponibili le informazioni relative alla struttura
della famiglia (individuo, padre e madre), al sesso, all’eta e all’indirizzo. Sono
inoltre memorizzate informazioni riguardanti gli istituti (nome, indirizzo,
telefono, fax e cosı via) che hanno raccolto i dati. Tutte le relazioni della base
di dati sono collegate a questa parte di schema per mezzo di una relazione
che descrive gli ID univoci degli individui.
Di solito gli istituti che raccolgono gli individui raccolgono anche
le informazioni cliniche che costituiranno il set di caratteri fenotipici:
quest’ultimi vanno a costituire un’altra parte della base di dati.
Un’altra parte dello schema descrive tutte le informazioni necessarie per
conoscere la locazione esatta di ogni campione di DNA raccolto (laboratorio,
frigorifero e tipo di frigorifero), lo stato del campione, le date del prelievo,
l’esecutore del prelievo, la categoria del campione e l’ID dell’individuo a cui
e stato fatto il prelievo.
La parte piu estesa dello schema e riferita alla memorizzazione dei dati
genetici (condizioni sperimentali, genotipi, marcatori).
Quando tutti i dati sono disponibili, il sistema permette di verificare la
correttezza della segregazione dei genotipi all’interno delle famiglie (controllo
della segregazione mendeliana).
21

2 – Fondamenti
Il sistema fornisce metodi per l’esportazione dei dati memorizzati nella
base di dati su file di testo compatibili con i programmi di elaborazione
statistica.
MFG Tools permette di gestire tutte le informazioni necessarie allo studio
di malattie complesse (individui, struttura delle famiglie, fenotipi e genotipi)
ed, in aggiunta, informazioni relative ai campioni di DNA.
2.9.3 GenoDB
Il sistema GenoDB [LDL+01] propone un soluzione per gestire
genotipizzazioni condotte su larga scala. Non e progettato per gestire le
informazioni cliniche degli individui.
GenoDB gira su macchine con sistema operativo MS Windows NT
ed e una applicazione sviluppata per il database MS Access. Il sistema
e finalizzato all’automazione del processo di genotipizzazione. Tutte le
operazioni sono svolte attraverso delle maschere di MS Access fornite da
GenoDB e possono essere svolte in serie o separatamente. I dati prodotti dal
sequenziatore di DNA vengono elaborati da uno o piu tecnici specializzati
di laboratorio in modo semi-automatico con i programmi GENESCAN 3.1
[Bioa] e GENOTYPER 2.1 [Biob].
Lo schema della base di dati rappresenta solo i genotipi e la struttura delle
famiglie ed e costituito da poche relazioni. La relazione pedigree contiene
gli attributi che indicano la struttura della famiglia. Una relazione DNA
connette ogni individuo con i suoi campioni di DNA e ogni campione di DNA
con i genotipi della relazione genotipi. La relazione genotipi rappresenta le
22

2 – Fondamenti
informazioni sui genotipi utilizzando tre coppie di attributi per memorizzare
le informazioni sui genotipi:
1. genotipi prodotti dalla macchina;
2. genotipi proposti dai tecnici di laboratorio;
3. genotipi verificati e corretti dalla lettura di 1 e 2 che possono essere
utilizzati nelle analisi successive.
Il sistema permette di gestire tutte le operazioni coinvolte nelle fasi di
genotipizzazione. La procedura per il riconoscimento degli errori permette di
usare solo genotipi corretti nelle analisi. Il sistema memorizza anche le fasi
intermedie delle genotipizzazioni e permette di ripristinare i vecchi dati nel
caso di aggiornamenti errati.
GenoDB permette di gestire gli individui, la struttura delle famiglie e la
parte genetica ma non prevede la memorizzazione dei fenotipi. L’estrazione
delle informazioni prevede una formattazione dei dati compatibile con quella
utilizzata dal programma PedCheck che esegue il controllo della segregazione
mendeliana [OW98].
23

Capitolo 3
Progettazione della base di dati
per le informazioni cliniche e
genetiche
Il sistema da noi sviluppato si serve del sistema di basi di dati PostgreSQL
per la memorizzazione delle informazioni relative agli individui, alla struttura
delle famiglie, ai fenotipi ed ai genotipi. In aggiunta a queste informazioni
nella base di dati sono memorizzati anche i dati degli utenti che accedono
al servizio: cosı facendo possiamo regolamentare i permessi su ogni singolo
attributo di una tabella ed aumentare la sicurezza del sistema poiche un
utente puo accedere ai dati solo attraverso i programmi da noi sviluppati.
La progettazione della base di dati e iniziata con lo sviluppo di uno schema
Entita Relazione (E-R) che ha aiutato a visualizzare l’organizzazione delle
informazioni contenute nella base di dati senza badare alla memorizzazione
fisica dei dati [ACPT99].
24

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
Successivamente lo schema E-R e stato tradotto verso un modello logico
che aiuta ad individuare graficamente i legami esistenti tra le varie tabelle
della base di dati [ACPT99].
Come ultimo passo della progettazione si e aggiunta una definizione dei
parametri fisici di memorizzazione dei dati al modello logico - nel nostro caso
si tratta di un modello relazionale - per definire lo schema fisico che poi
viene creato attraverso i costrutti del linguaggio SQL per lo specifico DBMS
adottato [ACPT99].
La numerosita dei dati contenuti nella base di dati ed il tipo di
risultato richiesto dai programmi di elaborazione statistica rendono laboriosa
l’estrazione delle informazioni. Per facilitare questa operazione, abbiamo
sviluppato degli strumenti che permettono agli utenti di ottenere le
informazioni in modo automatico e senza conoscere la struttura della base di
dati, continuamente soggetta ad aggiunte e modifiche da parte degli utenti.
3.1 Lo schema Entita-Relazione (E-R)
Lo schema E-R vuole dare una descrizione grafica autoesplicativa della realta
di interesse senza scendere nei dettagli implementativi. I costrutti che
vengono utilizzati durante lo sviluppo dello schema sono le entita, le relazioni
e gli attributi [ACPT99].
Una entita, indicata graficamente con un rettangolo, rappresenta classi
di oggetti (persone, luoghi, etc.) che possiedono proprieta comuni ed una
esistenza autonoma mentre una relazione, indicata graficamente con un
rombo, rappresenta un legame logico, con dei vincoli di cardinalita minima
25

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
e massima, verso le entita alle quali e connessa.
Figura 3.1. Esempio di due entita ed una relazione. La relazionememorizza presenta un vincolo di cardinalita minima uguale a zero perl’entita admin user in quanto non e detto che un utente abbia inseritodei messaggi e un vincolo di cardinalita minima e massima uguale ad unoper l’entita admin usr msg in quanto ogni messaggio esiste solo se vieneinserito da un utente. L’entita admin usr msg ha un identificatore formatodall’attributo id memo piu l’identificatore esterno login che e, a sua volta,identificatore dell’entita admin user.
L’attributo descrive delle proprieta elementari delle entita o delle relazioni
come, ad esempio, il nome, il cognome, la data di nascita e l’eta di una
entita individuo ed e rappresentato graficamente come un ellisse. Gli attributi
che sono utilizzati per identificare univocamente ogni occorrenza dell’entita
hanno il nome sottolineato (Fig. 3.1).
I nomi che abbiano assegnato alle entita hanno dei prefissi - “fml”, “phnt”,
“chr”, “web” o “admin” - che vengono usati per aiutare l’amministratore a
comprenderne la funzione. Le entita che memorizzano le modifiche fatte sui
dati genetici dagli utenti hanno il nome che termina con il suffisso “ log”.
Per non appesantire eccessivamente lo schema E-R (Fig. 3.2),
mostriamo soltanto gli identificatori delle entita in quanto tutti gli attributi
presenti all’interno delle entita saranno indicati successivamente durante la
presentazione dello schema logico (Fig. 3.3).
Le entita aventi nome con prefisso “admin” descrivono le modalita di
26

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
Figura 3.2. Lo schema E-R 27

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
accesso alla base di dati per ogni singolo utente. L’entita admin user contiene
le informazioni sul nome dell’utente, la password, l’indirizzo e-mail, un campo
per decidere se l’utente e o no amministratore e la login (il nome utente)
come identificatore. A questa sono relazionate le entita web locus dove sono
memorizzati i singoli marcatori (la relazione descrive i permessi di lettura
e scrittura sui singoli marcatori), admin user msg dove l’utente puo salvare
delle note e admin log dove vengono memorizzati gli accessi degli utenti al
sistema. Abbiamo preferito gestire direttamente l’autenticazione degli utenti
nella base di dati, sebbene ogni RDBMS ne fornisca una propria, per poter
avere un controllo piu fine sulle operazioni che si possono fare. Vogliamo
regolamentare i permessi di lettura e scrittura di ogni singolo marcatore
mentre i permessi per gli utenti della base di dati sono sempre riferiti ad
una tabella. Inoltre, poiche la login e la password fornite agli utenti del
sistema permettono solo di accedere ai programmi ma non direttamente alla
base di dati, l’utente che viene utilizzato dai programmi per accedere alla
base di dati e noto solo agli amministratori del sistema con un conseguente
incremento della sicurezza.
Le entita che hanno prefisso “fml” descrivono gli individui, la struttura
della famiglia ed alcuni parametri clinici. Le informazioni principali
di un individuo sono contenute nell’entita fml individuo e consistono
dell’identificatore id, del nome e cognome, del numero della famiglia di
appartenenza (se presente e unico per ogni famiglia), del numero univoco
dell’individuo (ogni individuo ha un numero che lo identifica all’interno
di una famiglia), dei numeri univoci nella famiglia del padre e della
madre dell’individuo, del probando (1=si, 0=no), del sesso (1=maschio,
2=femmina), dell’eta, della data di nascita e di una stima della quantita di
DNA disponibile. L’entita fml individuo e in relazione con le altre entita
28

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
di prefisso “fml”: fml num e fml conv all. In fml num sono presenti i
nomi dei laboratori che hanno raccolto gli individui, il numero identificativo
utilizzato per ogni famiglia, il numero univoco generale che intendiamo
assegnare ad ogni famiglia ed un valore che indica quali famiglie utilizzare o
no durante l’estrazione delle informazioni. L’entita fml conv all descrive le
corrispondenze tra gli identificatori che utilizziamo nella base di dati e quelli
adottati dal laboratorio che compie le genotipizzazioni.
Le entita di prefisso “web” contengono tutte le informazioni necessarie
per la gestione dei dati genetici che vengono inseriti dagli utenti attraverso
l’interfaccia Web da noi creata. Per i marcatori, descritti nell’entita web locus
e identificati dal nome del marcatore, rappresentiamo il nome del gene e del
cromosoma, il numero degli alleli, gli alleli presenti, l’operatore che inserisce
i dati, la data di ultima modifica, le condizioni sperimentali (primer forward,
primer reverse, condizioni della PCR, enzima e condizioni di digestione),
un memo e le modifiche operate sui dati. Con la PCR (Polymerase Chain
Reaction) si ottiene una grande quantita (amplificazione) di DNA di un
frammento selezionato tramite la reazione di restrizione enzimatica a partire
dal DNA stesso del campione che si sta analizzando. L’analisi del DNA
attraverso restrizione enzimatica e una tecnica che permette di distinguere un
frammento di DNA in relazione alla presenza di un certo pattern di nucleotidi
in una data posizione (ad esempio, se il pattern e presente, l’enzima taglia
il frammento) [SR99]. Gli alleli di ogni marcatore ed il loro ordine sono
descritti nell’entita web allele e sono identificati dal nome del marcatore e dal
nome dell’allele. La relazione web genotipi presente tra le entita web locus e
fml conv all descrive i due alleli che definiscono il genotipo di un individuo
per un marcatore. Tutte le modifiche apportate ai loci ed ai genotipi sono
memorizzate nelle tabelle di log web genotipi log e web locus log.
29

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
3.2 Lo schema relazionale
Il passaggio da un modello concettuale - lo schema E-R - ad un modello
logico - il modello relazionale - non si realizza con una semplice traduzione
poiche possono essere fatte delle ristrutturazioni per soddisfare due esigenze:
“semplificare” la traduzione e “ottimizzare” il progetto [ACPT99]. L’esigenza
di ottimizzare il progetto deriva dal fatto che, mentre lo schema E-R ha
come obiettivo la rappresentazione naturale dei dati, la progettazione logica
e orientata all’effettiva realizzazione dell’applicazione ed all’ottimizzazione
delle prestazioni. Semplificare lo schema permette di utilizzare query meno
“complesse” e di velocizzare il sistema. Nel passaggio verso lo schema
relazionale vanno tradotte per prime le entita con delle relazioni, poi le entita
con gli identificativi esterni ed infine le associazioni tra le entita.
3.2.1 La parte per l’amministrazione del sistema
La traduzione relativa alla parte per l’amministrazione del sistema non
necessita di particolari ristrutturazioni e da il seguente risultato:
admin user(login, passwd, nome, email, admin)
admin table(login, tab, locus, read, write)
admin log(identif, login, time, ip add)
admin user msg(id memo,:::::::utente, data, memo, priorita, is admin)
dove con nome attributo indichiamo la PRIMARY KEY della relazione e
con:::::::::::::::::nome attributo le FOREIGN KEY. Un vincolo di integrita referenziale o
FOREIGN KEY crea un legame tra i valori di un attributo A1 di una relazione
R1 ed i valori di un attributo A2 di una relazione R2, imponendo che ogni
valore non nullo assunto dall’attributo A1 nelle righe di R1 sia presente
30

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
tra i valori assunti dall’attributo A2 della relazione R2. Per applicare
il vincolo di FOREIGN KEY e necessario che l’attributo A2 della relazione
R2 sia soggetto ad un vincolo di UNIQUE o PRIMARY KEY. Un vincolo di
UNIQUE viene applicato su uno o piu attributi ed impone che i valori degli
attributi coinvolti siano presenti in una ed una sola riga della tabella. La
sola eccezione riguarda il valore nullo, che puo essere ripetuto su piu righe
[ACPT99].
La relazione admin user memorizza la login, la password ed alcune
informazioni necessarie per descrivere gli utenti:
• login: PRIMARY KEY della relazione
• passwd: definito come NOT NULL
• nome: definito come NOT NULL
• email: facoltativo
• admin: indica se login e un amministratore o no. Definito come NOT
NULL con valore di DEFAULT=’f’
admin table rappresenta i permessi concessi agli utenti: ogni record
setta i permessi di lettura e scrittura per l’attributo di una relazione della
base di dati. Ogni utente e abilitato alla lettura di tutti i campi presenti
nelle relazioni mentre i permessi di scrittura devono essere settati dagli
amministratori.
• login, tab, locus: PRIMARY KEY della relazione. L’attributo login
e un identificatore esterno riferito alla PRIMARY KEY della relazione
31

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
admin user. Gli attributi locus e tab permettono di identificare un
marcatore (attributo locus) che si trova all’interno di una relazione
(attributo tab) della base di dati.
• read: definito come NOT NULL con valore di DEFAULT=’t’. Di
default, ogni utente puo accedere a tutti i dati in lettura
• write: definito come NOT NULL con valore di DEFAULT=’f’ (false).
Di default, l’utente non puo compiere modifiche o inserimenti. Solo
l’amministratore puo settare il valore dell’attributo a ’t’ (true).
admin user msg contiene messaggi inseriti dagli utenti o dagli
amministratori. E possibile dare una priorita ad ogni messaggio inserito.
• id memo: PRIMARY KEY della relazione
•:::::::utente: e un identificatore esterno riferito alla PRIMARY KEY della
relazione admin user
• data: la data dell’inserimento definita come NOT NULL
• memo: la nota inserita dall’utente definita come NOT NULL
• priorita: l’importanza del messaggio
• is admin: indica se il messaggio riguarda l’amministrazione del sistema
o no. Definito come NOT NULL con valore di DEFAULT=’f’
admin log mantiene traccia degli accessi al sistema da parte degli utenti.
Per ogni connessione sono memorizzate la data, una stringa random utilizzata
dai programmi CGI per identificare la connessione e l’indirizzo IP dell’utente
connesso.
32

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
• identif, login: PRIMARY KEY della relazione. L’attributo identif e
la stringa random diversa per ogni connessione effettuata dagli utenti.
L’attributo login e un identificatore esterno riferito alla PRIMARY
KEY della relazione admin user.
• time: definito come NOT NULL
• ip add: indirizzo IP dell’utente definito come NOT NULL
3.2.2 La parte per gli individui
La traduzione della parte di schema che descrive gli individui e la struttura
delle famiglie da il seguente risultato:
fml num(fam num, fam, lab, cat)
fml individuo(id,::::fam∗, sbj, fth, mth, nome, sex, age, date, fake)
fml conv all(id est, geno lab,:::id, usa, note)
La relazione fml num memorizza le informazioni che riguardano le
famiglie. In questo modo assegniamo ad ogni famiglia un numero univoco
fam_num utilizzato dai programmi di elaborazione statistica.
• fam num: PRIMARY KEY della relazione. Il numero univoco
assegnato ad ogni famiglia della base di dati
• fam: definito come NOT NULL. E il numero univoco assegnato alla
famiglia dal laboratorio lab che ha raccolto i dati clinici.
• lab: il nome del laboratorio, definito come NOT NULL.
33

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
• cat: definito come NOT NULL. Consente di raggruppare le famiglie
in set diversi con uno valore scelto dall’amministratore. Ad esempio,
possiamo volere solo le famiglie in cui c’e una coppia di fratelli affetti
oppure ignorare una famiglia poiche non si ha disponibilita di DNA.
fml individuo memorizza le informazioni principali degli individui e la
struttura della famiglia di appartenenza.
• id: identificatore degli individui, definito come PRIMARY KEY della
relazione.
•::::fam∗: Il numero univoco della famiglia. E un identificatore esterno
riferito alla PRIMARY KEY della relazione fml num. Il simbolo ∗
indica che il vincolo di cardinalita minima presente nella relazione dello
schema E-R e zero.
• sbj: il numero univoco dell’individuo all’interno di una famiglia
• fth: il numero univoco del padre all’interno della famiglia
• mth: il numero univoco della madre all’interno della famiglia
• nome: il nome e cognome dell’individuo
• sex: definito come NOT NULL. Prende valori 1 o 2 (maschio o femmina
rispettivamente).
• age: l’eta dell’individuo
• date: la data di nascita
34

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
• fake: indica la quantita di DNA disponibile. Se settato a -1000
l’individuo non e considerato. Ad esempio, bisogna scegliere un solo
individuo tra i gemelli omozigoti.
fml conv all mantiene le corrispondenze tra gli ID della base di dati
ed i codici utilizzati dal laboratorio che compie le analisi genetiche. Viene
definito un vincolo di UNIQUE sugli attributi geno_lab ed id per garantire
che ogni laboratorio utilizzi una sola volta l’individuo.
• id est, geno lab: PRIMARY KEY della relazione. L’attributo id_est
e l’identificatore utilizzato dal laboratorio geno_lab
•::id: definito come NOT NULL E un identificatore esterno riferito alla
PRIMARY KEY della relazione fml num.
• usa: definito come NOT NULL con valore di default = ’t’.
L’amministratore puo decidere se rendere visibile o no un individuo
durante la genotipizzazione. Ad esempio puo capitare che il laboratorio
che esegue le genotipizzazioni abbia piu identificatori riferiti allo stesso
individuo.
• note: note dell’amministratore
3.2.3 La parte per i genotipi
Il modello relazionale che ottenuto dalla traduzione della parte di schema
E-R che descrive i genotipi diventa:
web locus(nome, gene, cromosoma,:::::::autore, ultima modifica,
35

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
numero alleli, primer frv, primer rev, condizioni pcr, enzima,
condizioni digestione, memo)
web allele(allele, locus, num)
web genotipi(id, geno lab, locus, all 1, all 2, seleziona, memo)
web locus log(nome, gene, cromosoma,::::::::autore, ultima modifica,
numero alleli, primer frv, primer rev, condizioni pcr, enzima,
condizioni digestione, memo)
web genotipi log(::id,
:::::::::geno lab, locus, utente, data, old 1, old 2,
new 1, new 2)
L’entita web locus rappresenta i marcatori e le condizioni sperimentali
utilizzate nella genotipizzazione.
• nome: PRIMARY KEY della relazione. Il nome del marcatore.
• gene: il gene in cui e localizzato il marcatore
• cromosoma: il cromosoma sul quale e localizzato il marcatore
•:::::::autore: l’utente che inserisce le informazioni sul marcatore. E un
identificatore esterno riferito alla PRIMARY KEY della relazione
admin user.
• ultima modifica: definito come NOT NULL. La data della modifica
• numero alleli: gli alleli che possono formare il genotipo
• primer frv: il primer forward (reagente utilizzato per la PCR)
36

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
• primer rev: il primer reverse (reagente utilizzato per la PCR)
• condizioni pcr: le condizioni usate nella PCR
• enzima: reagente per la reazione di restrizione enzimatica
• condizioni digestione: le condizioni sperimentali (tempo e
temperatura) per la reazione di restrizione enzimatica
• memo: le note dell’utente
web allele descrive gli alleli di ogni marcatore presente nella relazione
web locus.
• allele, locus: PRIMARY KEY della relazione. L’attributo allele
contiene i valori degli alleli. L’attributo locus e un identificatore
esterno riferito alla PRIMARY KEY della relazione web locus.
• num: il numero d’ordine degli alleli
web genotipi memorizza, per ogni individuo presente, il genotipo
corrispondente ad ogni marcatore presente.
• id, geno lab, locus: PRIMARY KEY della relazione. Gli attributi id e
geno_lab sono identificatori esterni riferiti alla PRIMARY KEY della
relazione fml conv all mentre locus e un identificatore esterno riferito
alla PRIMARY KEY della relazione web locus
• all 1: il primo allele del genotipo
• all 2: il secondo allele del genotipo
37

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
• seleziona: definito come NOT NULL con valore di default ’t’.
L’amministratore decide se utilizzare o no un individuo.
• memo: l’utente puo inserire una breve nota. Ad esempio, il genotipo e
da rifare.
Le relazioni web locus log e web genotipi log memorizzano, aggiungendo
la data dell’inserimento, tutti gli attributi presenti nelle relazioni web locus e
web genotipi allo scopo di mantenere traccia delle modifiche apportate dagli
utenti.
3.3 Lo schema fisico
Lo schema fisico descrive come sono organizzate fisicamente le informazioni
che sono memorizzate nella base di dati. Per mezzo di una rappresentazione
grafica (Fig. 3.3) - prodotta utilizzando i programmi autodoc [Tay99] e dia
[Lar99] - mostriamo, per ogni tabella, il tipo di dato degli attributi con i
relativi valori di default (se presenti) e le informazioni previste dal modello
relazionale come le PRIMARY KEY, le FOREIGN KEY ed i vincoli di UNIQUE.
Tutte le relazioni rappresentate nello schema logico hanno una tabella
corrispondente all’interno dello schema fisico. Quest’ultimo viene ampliato
con delle tabelle il cui nome ha prefisso “phnt” o “chr” contenenti
rispettivamente informazioni sui fenotipi o sui genotipi. Queste tabelle
hanno un attributo id definito sia come PRIMARY KEY che come FOREIGN KEY
(riferita alla PRIMARY KEY della relazione fml individuo) piu un numero
variabile di campi aggiuntivi. Ogni campo delle tabelle con prefisso “phnt”
memorizza un fenotipo diverso mentre, nel caso di tabelle con prefisso “chr”,
38
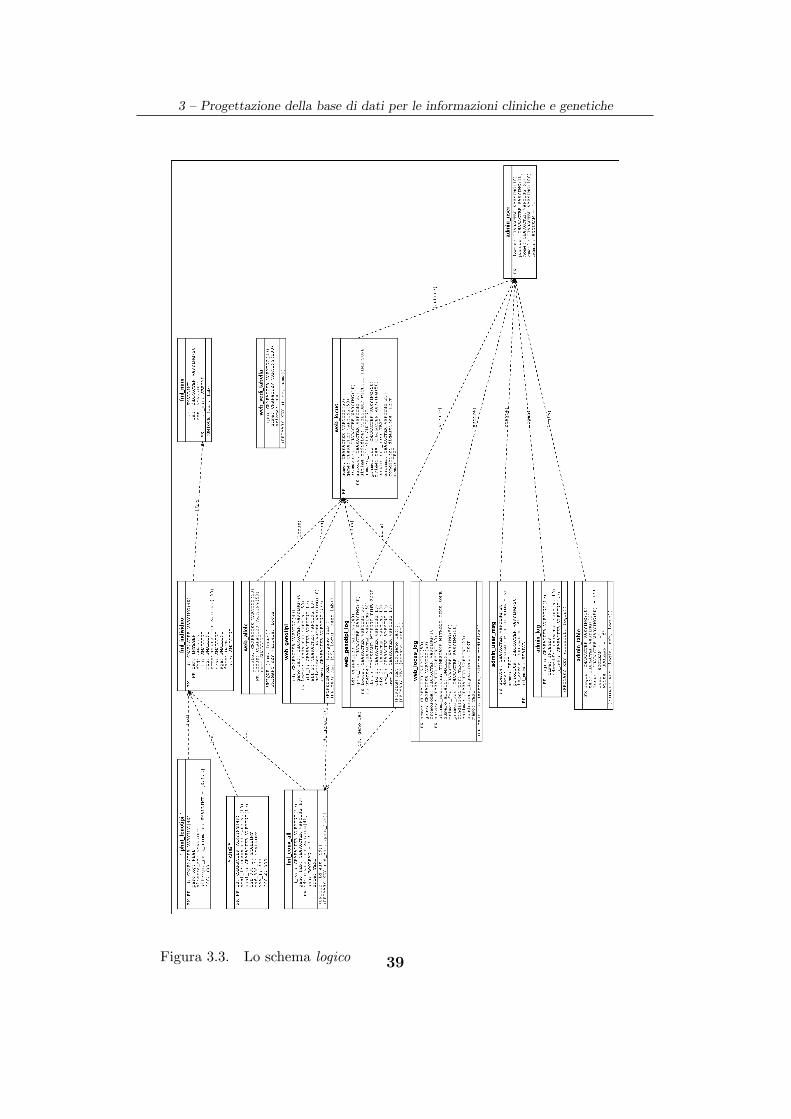
3 – Progettazione della base di dati per le informazioni cliniche e genetiche
Figura 3.3. Lo schema logico 39

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
ogni marcatore e composto da due colonne contenenti la coppia di alleli del
un genotipo (Fig. 3.3). Abbiamo deciso di strutturare le tabelle in questo
modo per una serie di motivi che andiamo ad elencare:
La rappresentazione dei dati clinici e genetici vuole essere il piu possibile
conforme al formato tabellare utilizzato dai programmi di elaborazione
statistica per velocizzare le operazioni di estrazione dei dati.
Possiamo assegnare ad ogni fenotipo o genotipo il tipo di dato che
meglio lo rappresenta, in quanto, i fenotipi possono essere qualitativi
(es. affetto/non affetto, si/no, 0/1/2) o quantitativi (es. indicano il
valore numerico, intero o reale, di un test clinico) mentre i due alleli
che descrivono il singolo genotipo possono assumere valori numerici o
alfanumerici.
La maggior difficolta richiesta per l’aggiornamento dei dati sulle tabelle
e trascurabile poiche si tratta di un operazione poco frequente in
quanto:
• i fenotipi vengono forniti da laboratori esterni e, di solito, non
necessitano di modifiche;
• i genotipi dei marcatori che sono memorizzati in queste tabelle
provengono dai genotipi inseriti via Web: questi vengono spostati
solo quando il laboratorio termina la genotipizzazione di tutti gli
individui per un marcatore e ne verifica la correttezza.
All’interno delle tabelle utilizziamo i valori nulli per descrivere l’assenza di
informazione poiche non e sempre possibile avere i dati per tutti gli individui
(ad esempio, per la terminazione del DNA).
40

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
3.4 La generazione automatica delle query
Le query (o interrogazioni) del sistema, che estraggono i dati contenuti nella
base di dati in formati compatibili con i programmi di elaborazione statistica
adottati dagli utenti, vengono generate automaticamente utilizzando i metodi
e le funzioni contenuti nei moduli perl (Sezione 4.2) che abbiamo creato
appositamente.
Per semplificare la creazione della query abbiamo creato una “vista”
fenotipi che contiene tutte le informazioni relative agli individui, alla
struttura della famiglia ed ai fenotipi presenti. Una “vista” crea una tabella
virtuale che contiene l’output generato da una SELECT. L’interrogazione
fatta per ottenere la vista fenotipi e solitamente complessa per due motivi:
primo, il numero di fenotipi coinvolti puo essere elevato, secondo, e possibile
ricavare dei nuovi fenotipi partendo da quelli gia disponibili. Ad esempio, se si
vuole ottenere un fenotipo qualitativo da uno quantitativo, si puo definire una
clausola CASE che utilizzi un valore soglia per distinguere tra un individuo
affetto o non affetto.
L’elenco di tutte le tabelle in cui sono presenti gli attributi selezionabili
dagli utenti e memorizzato in una tabella web vedi tabella che ha il seguente
schema relazionale:
web vedi tabella(tipo, nome, azione)
Il significato degli attributi nome ed azione dipende da cosa viene indicato
sull’attributo tipo:
• tipo=FENOTIPI: l’attributo nome memorizza il nome della tabella
41

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
o della vista che contiene gli identificatori degli individui, la struttura
delle famiglie ed i fenotipi - nel nostro caso la vista fenotipi - mentre
azione e vuoto.
• tipo=CHR: l’attributo nome contiene il nome di una tabella della base
di dati dove sono memorizzati i genotipi. Per distinguere i due tipi di
tabella disponibili si utilizza il campo azione. Le tabelle di tipo 1 sono
quelle aventi il nome con prefisso “chr” mentre quelle di tipo 2 hanno
la struttura della tabella “web genotipi”.
• tipo=WHERE: l’attributo nome contiene un nome identificativo per
la clausola WHERE mentre azione contiene la clausola WHERE da
utilizzare all’interno della query (Es. age>20). L’utente puo scegliere
una delle clausole WHERE memorizzate per selezionare dei subset di
individui (Es. seleziono solo i maschi, solo le femmine, solo i probandi,
solo le famiglie, ecc.).
• tipo=ORDER BY: l’attributo nome memorizza gli attributi sui quali
eseguire la clausola ORDER BY (es. id, fam) mentre azione e vuoto.
• tipo=PROFILO: gli attributi nome ed azione contengono,
rispettivamente, il nome di un profilo (Sez. 4.2.3) e la stringa che
lo descrive.
La creazione della query
L’elenco di attributi selezionabili dall’utente comprende dati sugli individui,
sulla struttura della famiglia, sui fenotipi e sui genotipi. Se si escludono i
genotipi, tutte le informazioni sono selezionate attraverso la vista fenotipi.
42

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
L’algoritmo di generazione automatica delle query permette di relazionare
alla vista fenotipi le diverse tabelle di genotipi. In particolare, la tabella
web_genotipi puo avere set di identificatori provenienti da laboratori
diversi ed un numero variabile e sconosciuto di marcatori. L’algoritmo
di estrazione e stato modificato nel tempo per poter risolvere queste
problematiche poiche, in un primo momento, non era stato previsto che i
genotipi venissero inseriti direttamente dagli utenti via Web. La presenza
di identificatori diversi ha reso necessaria la presenza di una tabella
fml_conv_all contenente le corrispondenze tra gli identificatori utilizzati
nella base di dati (tabella fml_individuo) e quelli dei laboratori esterni
presenti nella tabella web_genotipi.
La prima soluzione che abbiamo sviluppato per estrarre i dati prevedeva
la creazione di un’unica query SQL formata da una serie di clausole SELECT
nidificate ed unite attraverso delle clausole di OUTER JOIN. Una clausola
di JOIN viene utilizzata quando e necessario correlare dati provenienti da
diverse tabelle della base di dati attraverso i valori assunti dagli attributi. Le
clausole di OUTER JOIN (LEFT, RIGHT e FULL) permettono di selezionare
tutte le righe in cui vi e corrispondenza tra i valori degli attributi comuni
di due diverse tabelle piu tutte le righe che fanno parte di una (LEFT o
RIGHT) o entrambe (FULL) le tabelle coinvolte e che non hanno tuple
corrispondenti nell’altra tabella (gli attributi assenti sono rimpiazzati con
opportuni valori nulli). Una clausola di INNER JOIN permette di correlare
attraverso gli attributi comuni diverse tabelle, selezionando solo le righe in
cui vi e una corrispondenza tra i valori [ACPT99].
Ad esempio, vogliamo estrarre tutti gli attributi della vista fenotipi
ed i due alleli di apoe (il gene APO-E del cromosoma 19) dalla tabella
43

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
web_genotipi:
SELECT fen.*, tab1.all_1 as apoe_1, tab1.all_2 as apoe_2
FROM (
SELECT fml_conv_all.id,
web_genotipi.all_1, web_genotipi.all_2
FROM fml_conv_all, web_genotipi
WHERE web_genotipi.id=fml_conv_all.id_est
AND web_genotipi.geno_lab=fml_conv_all.geno_lab
AND seleziona=’t’ AND locus=’apoe’
) AS tab1
RIGHT OUTER JOIN (
SELECT * FROM fenotipi
) AS fen ON tab1.id=fen.id
La select proposta puo essere nidificata a sua volta fino ad ottenere tutti
gli attributi richiesti dall’utente. Questa soluzione e stata abbandonata
a causa dell’elevato numero di tabelle e marcatori presenti nella base di
dati. Abbiamo osservato che eseguendo delle query con numerose SELECT
nidificate le prestazioni della base di dati degradavano considerevolmente
fino a portare, nel caso peggiore, alla prematura terminazione del processo di
selezione. Ad esempio, nella base di dati asma abbiamo provato ad estrarre
i dati, prodotti da una scansione genomica, dei 23 cromosomi ed il sistema
ha interrotto l’esecuzione della query. Il motivo di tale comportamento e
legato al fatto che il sistema di basi di dati PostgreSQL deve utilizzare grosse
quantita di memoria per eseguire le query piu complesse [Mom03] e, nel
nostro caso, la quantita disponibile non e sufficiente.
44

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
Per evitare il problema abbiamo adottato un approccio diverso che
consiste nel creare automaticamente una tabella temporanea nella quale
inserire tutti i dati relativi ai genotipi. Successivamente otteniamo la
SELECT finale come INNER JOIN tra la tabelle temporanea e la tabella
dei fenotipi. Il nome assegnato dal sistema alla tabella temporanea e della
forma nomeutente_genotipi per evitare che piu utenti utilizzino la stessa
tabella. Supponendo che l’utente collegato sia ciano, il nome della tabella
temporanea ciano_genotipi e la tabella che memorizza gli individui ed i
fenotipi indicata in wev vedi tabella sia fenotipi, l’algoritmo che genera la
tabella e:
Se esiste la tabella ciano_genotipi allora:
DROP TABLE ciano_genotipi
Creazione della tabella temporanea con il comando:
SELECT id INTO ciano_genotipi FROM fenotipi
Creazione di una listaattributi contenente tutti gli attributi presenti
nel profilo ed i nomi di tutti i marcatori presenti in web_genotipi
poiche non sono conosciuti a priori.
Per ogni attributo di listaattributi verifichiamo se tale attributo e stato
selezionato dall’utente e se e un genotipo (non appartiene alla tabella
fenotipi). Se si:
aggiungiamo i campi alla tabella temporanea:
ALTER TABLE ciano_genotipi ADD nome_genotipo_1 text
ALTER TABLE ciano_genotipi ADD nome_genotipo_2 text
Gli attributi aggiunti hanno tipo text poiche ogni dato disponibile
45

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
in PostgreSQL puo essere memorizzato senza modifiche in un
campo testuale.
A seconda del tipo di tabella che memorizza il genotipo settiamo
i valori del genotipo. Per le tabelle di tipo 1 si ha:
UPDATE fenotipi_temp SET
apoe_1=chr19.apoe_1, apoe_2=chr19.apoe_2
FROM chr19 WHERE fenotipi_temp.id = chr19.id
Per le tabelle do tipo 2 si ha:
UPDATE fenotipi_temp SET
apoe_1=tt.all_1, apoe_2=tt.all_2 FROM
(
SELECT fml_conv_all.id,
web_genotipi.all_1, web_genotipi.all_2
FROM web_genotipi, fml_conv_all
WHERE web_genotipi.geno_lab=fml_conv_all.geno_lab
AND web_genotipi.id=fml_conv_all.id_est
AND seleziona = ’t’ AND locus =’apoe’
) AS tt
WHERE tt.id=fenotipi_temp.id
Dopo aver processato tutti gli attributi selezioniamo tutti i campi
richiesti dall’utente attraverso un INNER JOIN tra le tabelle fenotipi
e ciano_genotipi utilizzando le clausole WHERE ed ORDER BY
scelte dall’utente:
46

3 – Progettazione della base di dati per le informazioni cliniche e genetiche
SELECT att1, att2, ..., attn
FROM fenotipi, ciano_genotipi
WHERE fenotipi.id = ciano_genotipi.id
AND <clausola_where> ORDER BY <clausola_order_by>
I valori di <clausola_where> e <clausola_order_by> sono estratti da
web vedi tabella.
Rimuoviamo la tabella temporanea che contiene i genotipi:
DROP TABLE ciano_genotipi
L’algoritmo proposto permette di selezionare gli attributi richiesti
attraverso una sequenza di comandi SQL ma non con un unica query.
Tuttavia la possibilita di selezionare un numero imprecisato di attributi in
tempi ragionevolmente brevi e senza appesantire il sistema rende la soluzione
ottimale per i nostri scopi. Inoltre l’algoritmo proposto viene eseguito
automaticamente utilizzando procedure da noi sviluppate (Sez. 4.2.4) e in
maniera del tutto trasparente all’utente.
47

Capitolo 4
Architettura del sistema
Lo studio genetico delle malattie complesse richiede che vengano raccolte
molte informazioni genetiche e cliniche per un elevato numero di individui.
Un sistema di memorizzazione informatico studiato appositamente per
il personale specializzato di laboratorio puo aiutare a gestire tutte le
informazioni disponibili in modo efficiente.
Abbiamo sviluppato un sistema che cerca di ottimizzare e rendere
automatiche le operazioni di uso frequente. Diversi software curano
l’inserimento, la verifica, l’estrazione dei dati e la generazione di una statistica
descrittiva delle variabili genetiche. L’inserimento e la gestione dei dati
genetici avvengono via Web e sono regolamentati attraverso la verifica di
un nome utente ed una password che l’amministratore assegna a tutti gli
operatori autorizzati. Al contrario, l’inserimento di dati, che non interessano
le operazioni di genotipizzazione come la struttura delle famiglie, i fenotipi
e la gestione degli utenti, avviene per mezzo di programmi utilizzabili solo
dagli amministratori di sistema.
48

4 – Architettura del sistema
4.1 Strumenti utilizzati per lo sviluppo
Il sistema e attualmente installato su computer PC compatibili ed utilizza
esclusivamente software Open Source - il codice sorgente e disponibile e
liberamente distribuito - per il funzionamento e la realizzazione. Il sistema
operativo e Debian GNU Linux con Web server Apache - e installato su piu
del 60% dei siti Web [Fou02] - e sistema di basi di dati relazionale PostgreSQL
[pos]. Il linguaggio scelto per lo sviluppo dei programmi e il Perl [Wal].
4.1.1 Il sistema di basi di dati relazionale ed il
linguaggio SQL
PostgreSQL e un sistema di basi di dati relazionale o RDBMS (abbreviazione
di relational database management system) che supporta praticamente tutti
i costrutti sintattici di SQL (Sructured Query Language) tra cui le subselect,
le transazioni ed i vincoli relazionali [pos]. Un RDBMS e un sistema per
la gestione di dati memorizzati su relazioni. Una relazione e essenzialmente
una tabella composta da un insieme di righe aventi lo stesso set di attributi
(o colonne).
Il linguaggio SQL viene utilizzato per gestire l’interazione con l’RDBMS
attraverso tre gruppi di comandi [ACPT99] [DB00]:
• DDL (Data Definition Language): creazione degli oggetti dello
schema
• DCL (Data Control Language): controllo degli utenti e delle
autorizzazioni
49

4 – Architettura del sistema
• DML (Data Manipulation Language): manipolazione dell’istanza
della base di dati (interrogazioni e aggiornamenti)
4.1.2 Il linguaggio Perl
Il Perl (Practical Extraction and Report Language) e un linguaggio
interpretato nato per il trattamento di file di testo che fornisce una sintassi
molto simile al linguaggio C e che viene spesso associato alla realizzazione
dei programmi CGI [Wal02]. Un programma CGI puo essere scritto in
diversi linguaggi (ad esempio, Perl, C, bash). L’esecuzione di un programma
CGI (Figura 4.1) genera un file HTML (HyperText Markup Language,
il linguaggio utilizzato per la creazione delle pagine Web) che cambia in
funzione dei parametri di ingresso ricevuti. Un parametro di ingresso per un
CGI e una stringa di testo della forma chiave=valore [GGB00].
Figura 4.1. L’interfaccia CGI. L’operatore, per mezzo di un browserWeb, effettua una richiesta al Web server che, attraverso l’interfaccia CGI,esegue il programma CGI corrispondente. Quando il programma CGItermina l’elaborazione, restituisce un documento HTML al Web server che,a sua volta, lo invia al browser.
Il Perl permette di trattare i file di testo facilmente e con poche righe di
codice poiche dispone di costrutti per il “pattern matching” molto sofisticati
50

4 – Architettura del sistema
e di molte funzioni per il trattamento di stringhe e di array [Wal02]. Queste
caratteristiche permettono di trattare efficacemente alcune delle operazioni
che sono state implementate nel nostro sistema:
• generare i file HTML utilizzati nell’interfaccia Web
• creare e modificare file CSV (Es. cambiare l’ordine delle colonne). Un
file di tipo CSV e un file di testo che rappresenta una tabella: i campi
contenuti nelle righe del file sono divisi per mezzo di un separatore
(tabulatore, punto e virgola, spazio, etc.) scelto dall’utente [DB00].
• importare/esportare file CSV nel/dal sistema di basi di dati
• fare statistiche descrittive
Il Perl dispone di un elevato numero di moduli [Per] che permettono di
facilitare la scrittura dei CGI, di effettuare le connessioni al sistema di basi di
dati attraverso una interfaccia standardizzata e di importare/esportare vari
formati di documento (MS Excel, RTF, PDF, etc.).
Tutto il nostro software - CGI, script per l’amministrazione del sistema e
moduli - e scritto in Perl e si basa su alcuni moduli che sono quindi necessari
per il corretto funzionamento:
• CGI [Lin02]: preparazione delle pagine Web. Fornisce metodi per
la creazione di Web FORMs (permettono all’utente di mandare o
richiedere informazioni al Web server attraverso dei campi compilabili),
la lettura (parsing) del loro contenuto e la generazione automatica di
codice HTML
51

4 – Architettura del sistema
• DBI [Bun02]: accesso al sistema di basi di dati. Definisce una serie di
metodi, variabili e convenzioni che forniscono un’interfaccia consistente
tra uno o piu sistemi di basi di dati e l’applicazione.
• XML::Simple [McL02]: gestione dei file in formato XML (eXtensible
Markup Language). fornisce dei metodi per la creazione e la lettura di
documenti XML. XML e un meta linguaggio, cioe un linguaggio per
costruire altri linguaggi, utilizzato per la rappresentazione elettronica
di documenti [W3C02b].
• Spreadsheet::WriteExcel [McN02]: creazione di file in formato MS
Excel. Il modulo puo essere usato per creare automaticamente un file
di MS Excel utile per scambiare le informazioni con gli utenti che
utilizzano MS Office.
4.1.3 CGI e mod perl
Durante lo sviluppo il sistema ha subito diverse modifiche orientate a
migliorarne le prestazioni. Inizialmente abbiamo utilizzato l’interfaccia CGI
per generare in tempo reale delle pagine Web dinamiche in base alle richieste
degli utenti.
L’interfaccia CGI permette al Web server di eseguire un programma CGI
per compiere delle operazioni (ad esempio, accedere ad un sistema di basi di
dati ed impaginare i dati richiesti) e produrre un documento HTML con le
informazioni che sono state richieste. Tuttavia l’interfaccia CGI puo risultare
inadatta sia per le prestazioni che per la sicurezza del sistema in quanto:
• Prestazioni:
52

4 – Architettura del sistema
– ogni richiesta verso un programma CGI provoca la creazione di
un nuovo processo con conseguente carico di lavoro aggiuntivo sul
server.
– il Web server non e in grado di mantenere in memoria le pagine
gia richieste e prodotte da un CGI.
– quando si utilizza un RDBMS per la gestione delle informazioni,
ogni richiesta ad un programma CGI apre una nuova connessione
al RDBMS che viene chiusa al termine dell’esecuzione del processo.
L’apertura e chiusura della connessione provoca un ulteriore carico
di lavoro per il server ed allunga i tempi di attesa dell’utente.
• Sicurezza: puo essere pericoloso servirsi di un programma CGI che
utilizza dei programmi esterni per compiere delle operazioni: se il
programma esterno (eseguito con i permessi del Web server) processa
dati ricevuti da una FORM HTML, allora un utente potrebbe riuscire
ad eseguire dei comandi di sistema non previsti.
Sono disponibili delle tecnologie, alternative all’utilizzo dell’interfaccia
CGI, nate con lo scopo di evitare queste limitazioni:
• Active Server Pages (ASP) [Cor02]: e una tecnologia sviluppata da
Microsoft che viene integrata all’interno del Web server (ad esempio,
l’IIS - Internet Information Services) e permette di scrivere “codice”
all’interno delle pagine HTML invece di scrivere programmi separati.
ASP supporta diversi linguaggi di programmazione tra cui il piu
utilizzato e MS Visual Basic.
• PHP [Fou01]: e un linguaggio simile al Perl. L’interprete PHP
53

4 – Architettura del sistema
e integrato, come modulo, all’interno del Web server e supporta la
scrittura di “codice PHP” all’interno delle pagine HTML.
• Java servlets [SM02a]: sono state create da Sun. Sono realizzate
in Java e, per poter funzionare, devono essere compilate. Le classi
prodotte dalla compilazione sono caricate dinamicamente dal Web
server e hanno una funzionalita simile a quella dei CGI in quanto sono
programmi che generano documenti HTML.
• JavaServer Pages (JSP) [SM02b]: sono create da Sun. Permettono
di utilizzare il linguaggio Java all’interno del codice HTML ed hanno
una tecnologia molto simile a quella adottata dalle ASP.
• mod perl [Mac96]: un modulo che integra l’interprete Perl all’interno
del Web server Apache. Il modulo consente di avere contenuti dinamici
via Web creati attraverso programmi Perl evitando il costo richiesto
dall’esecuzione dell’interprete esterno [Gro97].
Tra le alternative possibili, mod perl permette, oltre a migliorare le
prestazioni offerte dal sistema, di mantenere il Perl come linguaggio di
programmazione. Inoltre permette di riutilizzare i programmi CGI emulando
l’interfaccia CGI per mezzo del modulo Apache::Registry o del modulo
Apache::PerlRun. I due moduli si differenziano in quanto i programmi CGI
eseguiti utilizzando il modulo Apache::Registry mantengono il codice e le
variabili nella memoria del Web server per le richieste successive, mentre nel
caso di Apache::PerlRun il CGI viene reinterpretato ad ogni richiesta e le
variabili non sono memorizzate nel Web server [Mac96].
Nel nostro sistema, abbiamo adottato mod perl ed il modulo
Apache::PerlRun per emulare l’interfaccia CGI. Abbiamo ottenuto
54

4 – Architettura del sistema
prestazioni comparabili a quelle ottenute con il modulo Apache::Registry
senza mantenere nella memoria del Web server le pesanti strutture dati
degli individui (le informazioni cliniche e genetiche) e senza chiudere la
connessione al sistema di basi di dati. Se configurato opportunamente, il
Web server Apache puo caricare in memoria alcuni moduli Perl durante la
fase di avvio per evitare ai programmi CGI di compiere questa operazione
ad ogni esecuzione [Mac96] [Gro97]. I moduli Perl che carichiamo all’avvio
del server Apache sono:
• Apache::PerlRun per l’esecuzione dei CGI
• Apache::DBI per la connessione persistente al sistema di basi di dati.
Apache::DBI si serve del modulo standard DBI per la gestione del
sistema di basi di dati e maschera le operazioni di apertura e chiusura
delle connessioni in modo da riutilizzare connessioni gia aperte (se
presenti). Non e necessario modificare i programmi CGI che utilizzano
il modulo DBI in quanto tutte le operazioni sul sistema di basi di dati
sono intercettate automaticamente dal modulo Apache::DBI [Mer01].
• i moduli Perl necessari al funzionamento dei nostri programmi (ad
esempio CGI.pm e DBI.pm).
In conclusione, la tecnologia offerta da mod perl ha permesso di migliorare
visibilmente le prestazioni del sistema e di riutilizzare gli script CGI gia
sviluppati grazie alla presenza dell’interprete Perl all’interno di Apache,
all’utilizzo dei moduli Apache::PerlRun e Apache::DBI ed alla possibilita
di precaricare in memoria i moduli Perl necessari ai nostri programmi.
55

4 – Architettura del sistema
4.1.4 L’architettura del sistema
L’architettura proposta consente di utilizzare una singola macchina che
funzioni contemporaneamente come Web Server e come Database Server
oppure con due macchine connesse in rete ed adibite a gestire il Web Server
ed il Database Server separatamente. (Figura 4.2). La seconda soluzione
Figura 4.2. L’architettura del sistema con due computer.L’operatore, per mezzo di un browser Web, effettua una richiesta HTTP alWeb server che, a sua volta, esegue il programma Perl corrispondente. Ilprogramma Perl si connette al sistema di basi di dati remoto utilizzandoil modulo Apache::DBI, compie le elaborazioni richieste ed invia il codiceHTML risultante al Web server che lo reindirizza al browser.
aumenta la sicurezza dei dati in quanto il sistema di basi di dati puo essere
installato su una macchina non connessa direttamente alla rete.
4.2 Lo sviluppo dei programmi e delle
librerie
Il sistema che presentiamo si serve di alcuni moduli Perl da noi sviluppati
per avere a disposizione metodi e funzioni che permettano l’esecuzione
56

4 – Architettura del sistema
delle operazioni piu frequenti. I moduli vengono usati all’interno di tutti
i programmi (CGI e script) e permettono di sviluppare velocemente nuove
applicazioni riutilizzando funzioni e metodi gia presenti e testati.
Di seguito saranno esaminati i moduli da noi sviluppati soffermandoci
maggiormente nell’analisi di quei moduli che gestiscono gli individui, i
loro dati (struttura delle famiglie, fenotipi e genotipi) e le metodologie di
inserimento ed estrazione dei dati nella di basi di dati.
4.2.1 PDB::pgc
Il modulo PDB::pgc viene utilizzato per istanziare un oggetto dedicato alla
gestione di un sistema di basi di dati PostgreSQL. PDB::pgc si serve del
modulo DBI per la connessione e la gestione del sistema di basi di dati e
fornisce dei metodi che semplificano le operazioni piu ricorrenti. I metodi
implementati gestiscono l’apertura e la chiusura di connessioni, l’esecuzione
di comandi SQL e la ricerca di tabelle o di attributi all’interno della base di
dati:
• new(nome database, user, password): istanzia l’oggetto della classe e
crea la connessione alla base di dati desiderata.
• close(): chiude la connessione
• getDBH: restituisce il puntatore all’oggetto della classe DBI che
mantiene la connessione alla base di dati.
• doSQL(comando SQL): esegue un comando SQL che non restituisce
dei risultati (ad esempio, INSERT, CREATE, ALTER)
57

4 – Architettura del sistema
• executeSQL(query SQL): esegue una query SQL e restituisce il
puntatore ad un oggetto che contiene i risultati della query
• isPresent(nome tabella): verifica se una relazione e presente nella base
di dati
• isAttPresent(nome tabella, nome attributo): verifica se un attributo
e presente in una relazione della base di dati
• getAttType(nome tabella, nome attributo): restituisce il tipo di dato
associato all’attributo di una relazione
Tutti i metodi possono gestire gli errori dovuti all’assenza della
connessione al sistema di basi di dati, all’errata sintassi delle istruzioni SQL
o alla presenza di valori non compatibili con il tipo di dato previsto dalla
base di dati.
4.2.2 PDB::bio
Il modulo PDB::bio istanzia un oggetto che contiene e gestisce tutti i dati
relativi agli individui (struttura della famiglia, fenotipi e genotipi). Tramite
i metodi contenuti nel modulo e possibile creare i file di testo formattati che
sono utilizzati dai programmi di analisi statistica, modificare il formato dei
dati e prelevare le informazioni sugli individui da un file o da una basi di
dati.
Il metodo new istanzia un oggetto vuoto della classe PDB::bio che
puo essere popolato usando il metodo fromCSV (nomefile, separatore)
se si importano i dati degli individui da un file CSV, o il metodo
58

4 – Architettura del sistema
fromSQL(database, query) se si effettua una interrogazione verso una base
di dati.
I metodi getHead e getHeadIDpos restituiscono rispettivamente la lista dei
nomi degli attributi (presenti sul file CSV o selezionati dalla base di dati) e
la posizione dell’attributo id (l’identificatore degli individui) all’interno della
lista.
I metodi getListaID, getOrderID e getUnrelatedID restituiscono un array
che contiene gli identificatori degli individui e che puo essere non ordinato,
ordinato o contenere solo gli individui non imparentati (o unrelated).
Il metodo setColonneGenotipi(valore nullo, [0,1,2], [0,1]) scorre la lista
di tutti gli attributi disponibili ed individua i marcatori cercando le coppie
di attributi che formano un genotipo. I due attributi che rappresentano
un genotipo hanno un nome del tipo <nome_locus>_1 e <nome_locus>_2.
Ad esempio, i due attributi che rappresentano le mutazioni del gene cftr
sono cftr_1 e cftr_2. Il tipo di formattazione dei genotipi che si utilizza
dipende dal programma di statistica adottato e puo essere quello abituale a
due colonne - coppie di alleli - o quello ad una sola colonna. La codifica dei
genotipi con una singola colonna puo essere fatta in due modi:
• una semplice concatenazione delle coppie di alleli che formano il
genotipo: dati gli alleli A e G, i genotipi possibili sono AA, AG e
GG
• una numerazione predefinita di ogni possibile genotipo: dati gli alleli
A e G, i genotipi possibili sono 1, 2 e 3 (stanno ad indicare AG, AG e
GG rispettivamente)
59

4 – Architettura del sistema
Per distinguere il tipo di genotipo attraverso il nome dell’attributo abbiamo
stabilito una convenzione:
• le due colonne che contengono gli alleli di un marcatore hanno nome
<nome_locus>_1 e <nome_locus>_2
• la colonna che contiene genotipi codificati numericamente ha nome
<nome_locus>_num.
• la colonna che contiene genotipi codificati come concatenazione degli
alleli ha nome <nome_locus>_con
A seconda dei parametri di ingresso passati a setColonneGenotipi e
possibile ottenere diversi risultati. Il valore del parametro [0,1,2] permette
di decidere come visualizzare i genotipi codificati con una sola colonna:
• 0: la colonna dei genotipi indicati con un numero
• 1: la colonna dei genotipi dove i due alleli sono concatenati
• 2: le due colonne mostrate in 0 e 1
Tramite il parametro [0,1] si stabilisce se visualizzare o no le due colonne
contenenti gli alleli: con 0 visualizziamo gli alleli piu i genotipi codificati in
una colonna mentre con 1 soltanto i genotipi codificati in una colonna.
Il metodo getFrequenze esegue delle elaborazioni sui dati al fine di
generare una statistica di base sugli individui presenti. L’output restituito e
una stringa contenente le frequenze alleliche e genotipiche di tutti i marcatori
presenti. Se necessario, e possibile calcolare l’equilibrio di Hardy-Weinberg
60

4 – Architettura del sistema
(hwe) [GT92] sull’insieme di individui unrelated (non imparentati). Il calcolo
di hwe e eseguito dal programma hwe [Ott95] utilizzando le frequenze degli
alleli e dei genotipi osservati sugli individui non imparentati presenti nella
base di dati.
Altri metodi della classe permettono di eseguire il confronto tra un
attributo che compare in due oggetti di tipo PDB::bio per trovare le differenze
(getAttDiff ) e di esportare su un file CSV o MS EXCEL i dati contenuti
nell’oggetto (toCSV e toXLS ).
4.2.3 PDB::profilo
Il modulo PDB::profilo fornisce delle funzioni che permettono la gestione
dei profili degli utenti. Un profilo e una astrazione della base di dati che
permette di mostrare all’utente un gruppo di tabelle fittizie indipendenti
dalla struttura fisica della base di dati.
Ad esempio, in una base di dati dove siano presenti le tabelle:
fml_individuo(id, fam, sbj, fth, mth, sex, age)
chr7(id, cftr_1, cftr_2)
chr19(id, apoe_1, apoe_2)
E possibile creare un profilo che mostri le seguenti tabelle fittizie:
fenotipi(id, sex, age)
genotipi(id, cftr_1, cftr_2, apoe_1, apoe_2)
Abbiamo definito la stringa che descrive un profilo come segue:
61

4 – Architettura del sistema
• una singola tabella fittizia (<TABELLA>) viene rappresentata come una
lista di stringhe separate dai caratteri -#-:
<TABELLA> := <nome_tab>-#-<rel>;<att>-#-...-#-<rel>;<att>
La prima stringa contiene il nome della tabella fittizia <nome_tab>
mentre le successive rappresentano gli attributi. Per identificare il
singolo attributo utilizziamo la sintassi <rel>;<att> dove indichiamo il
nome della tabella (<rel>) della base di dati in cui si trova l’attributo
(<att>) stesso. Le stringhe relative alle tabelle fittizie “fenotipi” e
“genotipi” dell’esempio sono:
fenotipi-#-fml_individuo;id-#-fml_individuo;sex-#-fml_individuo;age
genotipi-#-chr7;cftr_1-#-chr7;cftr_2-#-chr19;apoe_1-#-chr19;apoe_2
• un profilo e formato da una lista di stringhe <TABELLA> separate dai
caratteri -@-:
<PROFILO> := <TABELLA>-@-<TABELLA>[email protected]@-<TABELLA>
Il profilo risultante per le due tabelle dell’esempio e:
fenotipi-#-...-#-fml_individuo;age-@-genotipi-#-...-#-chr19;apoe_2
Sebbene le tabelle fittizie che fanno parte si un profilo, siano paragonabili
a delle viste, l’utilizzo dei profili semplifica il lavoro dell’amministratore del
sistema: la creazione e la gestione di un profilo avviene attraverso un file di
testo dove sono presenti tutti gli attributi che si vogliono estrarre dalle tabelle
della base di dati. Ogni riga del file di testo indica una diversa informazione:
62

4 – Architettura del sistema
• una riga di tipo --<stringa> indica l’inizio di una nuova tabella fittizia
di nome <stringa>.
• una riga di tipo <relazione>;<attributo> indica il campo e la tabella
di appartenenza all’interno della base di dati.
Un tipico file di testo utilizzato per generare un profilo ha questo formato:
--Fenotipi
fenotipi;fml_num
fenotipi;sbj
fenotipi;fth
fenotipi;mth
.....
--Cromosoma 6
chr6;lt
.....
--Cromosoma 7
chr7;tg
chr7;t
.....
.....
Utilizzando un editor di testo e possibile aggiungere, togliere e riordinare in
modo semplice l’elenco di attributi presenti nelle relazioni della base di dati
senza intervenire sulla base di dati (come accadrebbe servendosi delle viste).
Le funzioni profilo2file e file2profilo permettono di convertire in modo
automatico una stringa che definisce un profilo in un file e viceversa. Inoltre
63

4 – Architettura del sistema
e disponibile una funzione importProfilo per estrarre un profilo dalla base di
dati e creare una struttura dati che viene utilizzata dai programmi CGI per
visualizzare le tabelle e creare le interrogazioni.
4.2.4 PDB::SQL
Il modulo PDB::SQL fornisce i metodi usati per creare la query SQL che
seleziona gli attributi richiesti dall’utente.
L’algoritmo prevede la creazione di una tabella temporanea dove vengono
aggiunti in modo automatico tutti gli attributi richiesti tra quelli presenti
nelle tabelle della base di dati. I metodi del modulo PDB::SQL permettono
di generare la sequenza di comandi SQL che gestisce la creazione e
l’aggiornamento della tabella temporanea e l’estrazione delle informazioni
desiderate. Ogni istanza della classe contiene il nome della tabella
temporanea creata all’interno della base di dati e una lista dei comandi SQL
necessari alla sua creazione.
La classe fornisce un metodo new per istanziare l’oggetto e una serie
di metodi che permettono l’accesso ai dati della classe: getSQLcommand
restituisce il puntatore alla lista dei comandi SQL che generano la tabella
temporanea, showTemp mostra il nome della tabella temporanea, dbh
restituisce l’oggetto DBI che mantiene la connessione alla base di dati e
doTable crea la tabella temporanea utilizzando i comandi SQL contenuti
nella lista ottenuta con il metodo getSQLcommand.
Oltre ad i metodi dedicati alla gestione delle strutture dati interne ci sono i
metodi finalizzati alla creazione dei comandi SQL. Il metodo createTempTable
64

4 – Architettura del sistema
crea la tabella temporanea alla quale vengono aggiunti tutti i genotipi
richiesti attraverso il metodo addAttribute.
Nella sezione 3.4 sono presentati lo schema della base di dati ed una
descrizione dettagliata dell’algoritmo di creazione della tabella temporanea.
4.2.5 Gli altri moduli ed i programmi
Durante la realizzazione del sistema da noi sviluppato abbiamo creato altri
moduli in aggiunta a quelli precedentemente descritti.
Il modulo PDB::myCGI.pm fornisce funzioni per la generazione
automatica del codice HTML contenuto in tutte le pagine Web prodotte
dal sistema come, ad esempio, l’intestazione e la coda delle singole pagine
ed la URI del file CSS che si vuole utilizzare. Un file CSS (Cascading Style
Sheets) permette di definire lo stile (font, colori, spazi, etc) di una pagina
HTML [W3C02a].
Le procedure per creare la maschera in cui inserire “nome utente” e
“password” e per decidere se l’utente puo accedere al sistema sono definite
nel modulo PDB::Utente.pm. Ogni utente puo memorizzare brevi note
all’interno di una tabella della base di dati che viene gestita attraverso le
funzioni del modulo PDB::memo.pm.
Il modulo PDB::myUTL.pm contiene delle funzioni per gestire stringhe ed
array di stringhe. In particolare fornisce una funzione getIDorder che e molto
usata all’interno dei nostri programmi e permette di ordinare un array di
identificatori utilizzando un ordinamento basato sulla semantica della stringa
piuttosto che sull’ordine lessico-grafico. Ad esempio, gli identificatori Vr4.02
65

4 – Architettura del sistema
e Vr41.03 hanno un ordine lessico-grafico Vr4.02 > Vr25.03 mentre hanno
un ordinamento semantico Vr4.02 < Vr25.03. Infatti, gli identificatori
assegnati agli individui sono creati in modo da essere informativi sulla
provenienza di un individuo presente nella base di dati. Ad esempio, un
identificatore Vr4.02 indica l’individuo 2 della famiglia 4 del set di individui
di V r.
Per facilitare lo scambio di dati tra la base di dati ed un file CSV e per
aggiornare in modo automatico una relazione della base di dati con i dati
contenuti in un file (ad esempio, l’inserimento ed aggiornamento delle tabelle
dei fenotipi) utilizziamo le funzioni del modulo PDB::csv.pm. La funzione
csv2pg del modulo permette di importare un file CSV nella base di dati
cercando di assegnare il tipo di dato piu corretto a tutte le colonne del file.
I programmi per l’amministrazione del sistema ed i CGI utilizzano
i moduli che abbiamo presentato per implementare le operazioni basilari
dell’elaborazione. Per i programmi CGI, oltre ai moduli che permettono
di gestire la connessione alla base di dati e l’elaborazione dell’informazione,
utilizziamo i moduli che consentono la creazione del codice HTML prodotto
come risultato. Le operazioni che vengono svolte dai programmi e dai CGI
sono presentate nella sezione 5.1.1.
66

Capitolo 5
L’utilizzo del sistema
Il sistema da noi sviluppato fornisce una serie di strumenti atti alla gestione di
una base di dati contenente dati relativi allo studio delle malattie complesse.
Gli strumenti sono rivolti a due categorie di utilizzatori: l’amministratore di
sistema e gli utenti.
L’amministratore ha il compito di creare lo schema iniziale della base
di dati (tramite uno script SQL), di popolare le tabelle con i dati clinici
degli individui e di garantire il corretto funzionamento del sistema mentre gli
utenti registrati possono inserire i marcatori ed i genotipi degli individui ed
estrarre le informazioni dalla base di dati.
Il sistema viene utilizzato per gestire le basi di dati di due malattie
complesse: l’asma e l’osteoporosi. Lo studio sull’asma consta di informazioni
cliniche e genetiche per un totale di circa 850 individui, mentre per
l’osteoporosi abbiamo informazioni per circa 1700 individui.
67

5 – L’utilizzo del sistema
5.1 Descrizione del sistema
Lo studio delle malattie complesse richiede una prima fase in cui vanno
raccolti informazioni sugli individui, i loro dati clinici ed i loro campioni
di DNA che saranno poi utilizzati per la genotipizzazione. Per condurre uno
studio comparativo occorre raccogliere un gruppo di individui affetti ed un
gruppo di individui sani, mentre per le analisi di linkage occorrono gruppi di
famiglie che presentino individui affetti.
Di solito, i dati degli individui ed i loro caratteri clinici vengono forniti da
laboratori esterni sotto forma di fogli elettronici e necessitano di un lavoro
aggiuntivo per poter essere importati nella base di dati. La sequenza delle
operazioni necessarie per inizializzare la base di dati ed importare i dati
forniti consiste in:
• convertire i fogli elettronici in file di testo CSV. Questa fase, per forza
di cose, richiede un lavoro manuale ed una verifica delle informazioni
disponibili e del processo di conversione.
• assegnare gli identificatori univoci agli individui: ogni identificatore e
composto dall’unione di due o tre informazioni distinte che permettono
di stabilire la provenienza dell’individuo, la famiglia di appartenenza
(se esiste) ed il numero univoco dell’individuo all’interno della famiglia
(individui imparentati) o del gruppo di individui (non imparentati):
– una sigla identifica il laboratorio, la citta o il gruppo a cui
l’individuo e associato
– un codice numerico che identifica la famiglia all’interno del set
68

5 – L’utilizzo del sistema
– il numero univoco dell’individuo all’interno della famiglia
La concatenazione delle tre informazioni genera identificatori della
forma: <stringa><numero famiglia>.<numero individuo> nel caso
di individui imparentati e <stringa><numero individuo> altrimenti.
Ad esempio, un codice Vr2.5 indica l’individuo 5 della famiglia 2
del gruppo di individui di Verona (Vr). L’identificatore viene creato
in modo automatico per mezzo di un programma perl che abbiamo
sviluppato appositamente.
• creare la base di dati e lo schema. Abbiamo sviluppato uno script SQL
che crea tutte le tabelle principali della base di dati e tutti i vincoli
relazionali.
• importare i file CSV nella base di dati. L’operazione viene fatta
utilizzando un nostro programma che crea in automatico una tabella
(con il nome corrispondente al file CSV) nella base di dati cercando di
assegnare a tutti i campi il tipo di dato piu appropriato (Es. int2, int4,
float4, varchar, date, etc.).
• aggiornare la tabelle della base di dati che rappresentano gli individui
(tabelle con prefisso fml) utilizzando i dati importati dai file CSV.
• creare ed aggiornare le tabelle con prefisso “phnt” dove vanno
memorizzati i fenotipi che si hanno a disposizione
• aggiornare le tabelle per l’amministrazione del sistema con l’elenco degli
utenti che sono abilitati all’utilizzo della base di dati
• aggiungere alla tabella fml_conv_all gli identificatori del laboratorio
che compie le genotipizzazioni. Questi identificatori sono forniti dal
69

5 – L’utilizzo del sistema
laboratorio attraverso un foglio elettronico e sono importati nella base
di dati seguendo i passaggi visti nel caso dei fogli elettronici degli
individui.
• creare le tabelle con prefisso chr qualora fossero disponibili dei
marcatori genotipizzati
• creare il profilo che viene utilizzato per l’estrazione dei dati
Terminata la fase di inizializzazione della base di dati, gli utenti possono
iniziare ad inserire i marcatori ed i genotipi degli individui. L’amministratore,
a questo punto, deve solo compiere operazioni di backup (automatizzate) e
di riorganizzazione dei genotipi che vengono completati dal laboratorio (Es.
creazione o aggiornamento di una tabella di tipo “chr”). Nel caso in cui
vengano forniti nuovi gruppi di individui e necessario aggiornare le tabelle
che contengono i dati delle famiglie ed i fenotipi.
5.1.1 L’utilizzo dei programmi
I programmi che abbiamo sviluppato permettono di compiere le operazioni
piu ricorrenti che si incontrano durante la creazione e gestione di una base
di dati per la memorizzazione dei dati sulle malattie complesse:
• creazione e gestione degli utenti che accedono alla base di dati
• inserimento di nuovi individui
• inserimento dei dati clinici
• inserimento, verifica e modifica dei dati genetici
70

5 – L’utilizzo del sistema
• estrazione dei dati nei formati richiesti dai programmi di statistica
• calcolo di una statistica descrittiva sui dati genetici (frequenze alleliche,
genotipiche e test HWE)
I dati modificabili dall’utente riguardano solo la parte genetica poiche le
informazioni riguardanti la struttura della famiglia ed i dati clinici sono
fornite da laboratori esterni e rimangono generalmente invariate nel tempo.
Il sistema utilizza un file XML per salvare le informazioni necessarie per
eseguire la connessione verso la base di dati come:
• IP address del database server
• la login e password per accedere alla base di dati, sconosciuta agli utenti
che utilizzano il nostro sistema
• l’elenco delle patologie studiate. Ogni patologia corrisponde ad una
base di dati
• gli IP address che hanno il permesso di accedere al sistema
In questo modo, i programmi possono accedere ad un qualunque sistema
di basi di dati con una semplice modifica dei parametri contenuti nel file
XML.
L’utilizzo dei programmi CGI
I programmi utilizzabili via Web sono presentati attraverso alcune schermate
che andiamo a visualizzare di seguito. Non esiste un aiuto in linea per
71

5 – L’utilizzo del sistema
l’interfaccia Web in quanto gli utenti che accedono al servizio conoscono la
genetica ed il significato dei campi che devono andare a modificare all’interno
delle pagine.
Un utente che vuole accedere al sistema deve essere autenticato (Fig.
5.1): l’autenticazione avviene in in due fasi che verificano 1) se l’indirizzo
IP dell’utente che vuole accedere al servizio e compreso nell’elenco di quelli
abilitati e 2) se la login e la password memorizzati corrispondono a quelle
inserite dall’utente.
Figura 5.1. La schermata di login.DMIBG: Dipartimento Materno Infantile e di Biologia e Genetica.
La prima schermata presentata all’utente mostra un menu con le
operazioni disponibili (Fig. 5.2):
• inserimento e modifica dei genotipi
• visualizzazione ed estrazione dei dati
• inserimento e modifica dei memo
72

5 – L’utilizzo del sistema
• inserimento di nuovi marcatori
• torna alla pagina di login e scelta di una base di dati
Figura 5.2. Menu di scelta
In figura 5.3 e possibile vedere la schermata utilizzata per inserire un
nuovo marcatore nella base di dati. L’utente deve semplicemente inserire il
nome del marcatore, gli alleli ed il set di individui che intende genotipizzare.
Attualmente e possibile inserire solamente marcatori biallelici, ma una nuova
versione permettera di inserire marcatori con piu alleli.
La schermata per l’inserimento e la modifica dei genotipi (Fig. 5.4) mostra
tutti i marcatori disponibili e le operazioni disponibili su di essi:
• editare i genotipi per un marcatore seguendo il link che ne mostra il
nome
• visualizzare e modificare le informazioni del marcatore premendo il
bottone INFO corrispondente al marcatore
73

5 – L’utilizzo del sistema
Figura 5.3. Pagina per l’inserimento marcatori
• visualizzare una statistica descrittiva per i dati di un marcatore tramite
il link “HWE”.
Selezionando il nome del marcatore viene presentata una pagina che
permette l’inserimento e la modifica dei genotipi (Fig. 5.5). L’utente
puo settare il genotipo degli individui selezionando uno dei bottoni
corrispondenti. I genotipi selezionabili sono creati dinamicamente partendo
dalla lista degli alleli che sono presenti nella tabella web_allele della base
di dati. Questo metodo di inserimento dei genotipi preclude all’operatore
qualunque errore di battitura e velocizza notevolmente il processo di
inserimento in quanto tutte le operazioni sono fatte con il mouse in modo
intuitivo. Gli errori si possono verificare solo per false letture del genotipo o
per la selezione del bottone sbagliato. Tramite le frecce o i link presenti in
fondo alla pagina e possibile selezionare tutti gli individui presenti. Esiste
la possibilita di cambiare il numero di individui visualizzati su ogni pagina
(default=8), di selezionare un marcatore diverso per le modifiche (bottone
’Cambia locus’), di visualizzare la statistica descrittiva e di tornare alla
74

5 – L’utilizzo del sistema
Figura 5.4. Menu dei marcatori
pagina con l’elenco dei marcatori disponibili per l’inserimento dei genotipi.
La pagina che mostra le informazioni dei marcatori (Fig 5.6) contiene
il nome del marcatore, il gene, il cromosoma, i dati di laboratorio
sull’esperimento, un memo per la memorizzazione di informazioni aggiuntive
e la login dell’utente che ha inserito i dati del marcatore. Se l’utente che
accede alla pagina ha il permesso per modificare i dati del marcatore allora
puo utilizzare una FORM HTML per eseguire l’operazione di aggiornamento.
La pagina relativa alla statistica descrittiva di un marcatore mostra le
frequenze di tutti gli alleli nel set, le frequenze dei genotipi e l’output
75

5 – L’utilizzo del sistema
Figura 5.5. Modifica dei genotipi di un marcatore
del programma hwe [Ott95] che esegue una comparazione tra le frequenze
genotipiche osservate del set e quelle attese.
La pagina che permette di leggere i memo degli utenti (Fig. 5.8) contiene
delle note inserite dagli utenti o dall’amministratore.
E possibile accedere alla pagina di modifica (Fig. 5.9) o rimozione di
un messaggio se questo e stato creato dall’utente corrente. La rimozione di
un messaggio avviene immediatamente dopo la richiesta e senza avvisi da
parte del sistema. Ogni utente puo decidere di inserire un nuovo messaggio
tramite la stessa FORM che permette la modifica del messaggio (Fig. 5.9).
La login dell’utente e la data di inserimento sono inserite automaticamente
dal sistema.
76

5 – L’utilizzo del sistema
Figura 5.6. Informazioni dei marcatori
La pagina per la selezione dei dati (Fig. 5.10) visualizza una FORM che
contiene tutti gli attributi selezionabili dall’utente con le opzioni consentite.
Per velocizzare la scelta degli attributi e possibile selezionare una intera
tabella marcando il bottone Seleziona tabella.
Nelle Figure 5.10 e 5.11 e possibile osservare la disposizione degli attributi
al variare del profilo scelto dall’utente.
Scorrendo la pagina vengono visualizzate le opzioni applicabili alla
selezione degli attributi precedentemente scelti. Ogni FORM compilata puo
essere salvata con un nome per consentire all’utente di riselezionare gli stessi
attributi in un secondo momento (Fig. 5.12); il salvataggio evita un dispendio
di tempo nel caso di selezioni utilizzate frequentemente che coinvolgono molti
attributi.
77

5 – L’utilizzo del sistema
Figura 5.7. Statistica descrittiva di un marcatore
L’utente puo scegliere quale gruppo di individui utilizzare marcando uno
dei bottoni indicati nella sezione Estrazione dei soggetti (Fig. 5.12): dove
e disponibile un gruppo di clausole WHERE predefinite che permettono di
eseguire le selezioni piu frequenti (Es. selezionare solo le famiglie, selezionare
solo i probandi, etc.).
La scelta della modalita di visualizzazione (Fig. 5.12) che viene utilizzata
per mostrare i risultati permette di decidere il separatore che viene utilizzato
nel file CSV (punto e virgola, spazio, tabulatore o virgola) ed un carattere
particolare che rappresenta il valore nullo (carattere vuoto, x, 0, -9999).
Abbiamo rivolto particolare attenzione alla modalita di estrazione dei
78

5 – L’utilizzo del sistema
Figura 5.8. Lettura dei memo
genotipi in quanto i diversi programmi di elaborazione statistica utilizzano
metodologie diverse per la loro codifica. Selezionando opportunamente
i bottoni della FORM e possibile ottenere le diverse rappresentazioni
disponibili per i genotipi (Sez. 4.2.2 pag. 57).
Quando l’utente ha settato tutti i parametri desiderati puo scegliere uno
dei formati disponibili per la selezione: se visualizzare o scaricare il file CSV
risultante (Fig. 5.13) o se scaricare un foglio di MS Excel zippato (utile per
importare i dati nei programmi statistici di MS Windows).
79

5 – L’utilizzo del sistema
Figura 5.9. Modifica o inserimento dei memo
I programmi per l’amministrazione del sistema
I programmi utilizzati dall’amministratore in genere sono degli script
costruiti utilizzando i metodi e le funzioni disponibili sui moduli da noi
sviluppati. Il nome di tutti i programmi ha un prefisso DB- per meglio
identificarli. Di seguito, presentiamo una panoramica che mostra quali
operazioni sono permesse.
DB-utenti-crea: crea un utente all’interno della base di dati
DB-utenti-permessi: setta il permesso di lettura o scrittura su un
attributo di una tabella.
DB-comando: Si connette al sistema di basi di dati, esegue la query
SQL data in input e restituisce il file CSV corrispondente.
80
5 – L’utilizzo del sistema
Figura 5.10. Selezione dei campi con profilo ”default”
DB-dbest: Consente di estrarre automaticamente dalla base di dati
una tabella contenente tutti i fenotipi (la vista fenotipi) piu tutti i
genotipi che si trovano su una tabella della base di dati.
DB-csv2pg: Permette di creare nella base di dati una tabella
equivalente ad un file CSV. Lo script riesce riconoscere il tipo di dato
piu appropriato per le colonne processando tutti i campi del file (int2,
int4, float4, varchar(), etc.).
DB-csv2xls: Converte un file CSV in un file di MS Excel.
DB-tabella: Estrae una tabella dalla base di dati in formato CSV.
81
5 – L’utilizzo del sistema
Figura 5.11. Selezione dei campi con profilo ”vania”
DB-profilo-getcampi: Produce un file corrispondente ad un profilo
che contiene la lista di tutti i campi presenti nella base di dati.
Le tabelle da processare sono quelle memorizzate sulla tabelle
web_vedi_tabella.
DB-profilo-getfile: prende in input la stringa di un profilo e produce
il file corrispondente.
DB-profilo-getprofilo: produce la stringa di un profilo utilizzando il
file di testo corrispondente.
DB-confronta: confronta gli attributi comuni di due tabelle della base
di dati e mostra le differenze.
82

5 – L’utilizzo del sistema
Figura 5.12. Opzioni utilizzabili durante la selezione
DB-frequenze-hwe: Produce la statistica descrittiva per un
marcatore della base di dati.
DB-settaGeneticaDaGenitori: La presenza di un allele con
frequenza molto maggiore degli altri in un marcatore porta ad avere
molti individui omozigoti. L’individuo omozigote puo trasmettere un
solo allele al figlio (ha due alleli uguali per quel marcatore) per cui,
se entrambi i genitori sono omozigoti, non e necessario genotipizzare i
figli per conoscere il genotipo. Lo script permette di processare un file
CSV contenente la struttura della famiglia ed un marcatore e di definire
automaticamente il genotipo dei figli che hanno i genitori omozigoti.
83
5 – L’utilizzo del sistema
Figura 5.13. Risultato della selezione
DB-backup: crea il backup di una base di dati. Il nome del file
ha sempre la data per poter mantenere una cronologia ed evitare
sovrascritture di vecchi backup.
DB-backup-script: crea un archivio contenente tutti gli script, i
moduli ed i CGI sviluppati. Il nome del file ha sempre la data per poter
mantenere una cronologia ed evitare sovrascritture di vecchi backup.
84

5 – L’utilizzo del sistema
5.2 Applicazione del sistema: Asma
L’asma viene definita come una malattia respiratoria cronica di tipo
infiammatorio dovuto ad ampie variazioni, per brevi periodi di tempo, della
resistenza al flusso nelle vie aeree intrapolmonari. Dal punto di vista clinico
e caratterizzata da crisi ricorrenti di tosse, dispnea, respiro sibilante e senso
di costrizione toracica. Il fenotipo asma viene individuato in vari modi, quali
i sintomi, o misure obiettive, quali l’iperreattivita bronchiale (BHR), i livelli
sierici di IgE o entrambi. Di solito e associata ad allergia (da ’allos’ che
significa diverso e da ’ergon’, effetto), ma puo anche essere causata da fattori
emotivi, fisici, chimici e genetici. Quando si parla di allergia si intende la
reattivita spontanea ed esagerata dell’organismo dell’individuo allergico a
particolari sostanze, che risultano invece innocue nell’80% della popolazione.
Gli allergeni hanno la particolarita di sviluppare il sistema immunitario
mediante la produzione di una classe di anticorpi: le immunoglobuline di tipo
E (IgE). Segue che gli individui allergici presentano un numero maggiore di
IgE. Gli allergeni piu comuni sono quelli stagionali primaverili, i pollini, e
quelli contenuti nelle polveri di casa (gli acari) [HCH+99].
I probandi raccolti per il nostro studio hanno asma allergico e provengono
da due laboratori distinti (Verona e Bolzano). La raccolta dei dati clinici ha
fornito, per tutti gli individui, informazioni su BHR, IgE e sulle allergie piu
comuni (polveri, animali piante, etc.).
La dimensione dei dati memorizzati all’interno della base di dati e:
• individui ∼ 850
• fenotipi ∼ 20
85

5 – L’utilizzo del sistema
• geni e cromosomi candidati ∼ 120 marcatori. Ad esempio:
– gene cd14 su cromosoma 5
– gene cftr su cromosoma 7
– gene il11 su cromosoma 19
– marcatori sui cromosomi 7, 12, 14 e chr19
• scansione genomica ∼ 400 marcatori
Le informazioni raccolte sono memorizzate in una base di dati di nome
asma: le tabelle presenti sono quelle gia viste nella presentazione del modello
relazionale (Sez. 3.2 pag 29), piu le tabelle che memorizzano fenotipi e
genotipi.
Abbiamo creato quattro tabelle di tipo “phnt”:
phnt asma(id, asma)
phnt ige(id, ige)
phnt bhr(id, bhr orig, val, cat)
phnt prick(id, prick, spt, epiteli, dfdp, gram, erbe, alberi)
phnt rinite(id, rinite)
Le tabelle rappresentano i fenotipi “asma”, “BHR”, “IgE” e i valori
qualitativi di alcuni tipi di allergia. Per ottenere il fenotipo qualitativo di
BHR ed IgE abbiamo creato le funzioni getbhr e getige.
La funzione getbhr seleziona il valore qualitativo di BHR utilizzando tre
parametri: il valore fornito di BHR, il tipo di valore e la soglia. Abbiamo
86

5 – L’utilizzo del sistema
bisogno del tipo di valore in quanto i laboratori che hanno fornito la
clinica utilizzano dei valori non numerici per indicare i casi piu gravi (con
tipovalore = 1) o evidentemente sani (con tipovalore = 2). I valori numerici
esatti hanno sempre tipovalore = 10.
CREATE FUNCTION "getbhr" (real,smallint,smallint)
RETURNS integer AS
’select CASE
WHEN ($2 = 1) THEN 1
WHEN ($2 = 2) THEN 2
WHEN ($2 = 10) THEN
CASE WHEN ($1 < $3) THEN 2
ELSE 1 END
ELSE 0 END;’
LANGUAGE ’sql’;
La funzione getige seleziona il valore qualitativo delle IgE utilizzando
l’eta ed il valore clinico di un individuo. Per valori di Ige > 200 l’individuo
e marcato affetto senza ulteriori verifiche.
CREATE FUNCTION "getige" (smallint,real) RETURNS integer AS
’select CASE
WHEN $2 < 0 OR $2 = NULL THEN 0
WHEN $2 >= 200 THEN 2
ELSE CASE
WHEN $1 = 0 AND $2 >= 20 THEN 2
WHEN $1 = 1 AND $2 >= 50 THEN 2
WHEN $1 = 2 AND $2 >= 60 THEN 2
87

5 – L’utilizzo del sistema
WHEN $1 = 3 AND $2 >= 85 THEN 2
WHEN ($1 = 4 OR $1 = 5) AND $2 >= 130 THEN 2
WHEN ($1 = 6 OR $1 = 7) AND $2 >= 160 THEN 2
WHEN ($1 = 8 OR $1 = 9) AND $2 >= 180 THEN 2
ELSE 1 END
END;’
LANGUAGE ’sql’;
Il nostro laboratorio ha genotipizzato gli individui disponibili per dei
marcatori presenti in diversi cromosomi. Ogni tabella di tipo “chr”
che abbiamo creato memorizza i marcatori che appartengono ad singolo
cromosoma. I cromosomi per i quali abbiamo dei marcatori genotipizzati
sono il 5, 6, 7, 11, 12, 14, 16, 19 e sono memorizzati nelle tabelle chr5, chr6,
chr7, chr11, chr12, chr14, chr16 e chr19 rispettivamente.
Inoltre, e stato incaricato un laboratorio specializzato per eseguire una
scansione genomica. I risultati della scansione ci sono pervenuti in formato
MS Excel e di testo CSV. Per ogni cromosoma abbiamo importato il file
CSV corrispondente nella base di dati assegnando il prefisso chrsg (chrsg01,
chrsg02,...,chrsg22, chrsgx ) ad ognuna delle tabelle create. Il prefisso indica
le tabelle di tipo “chr” relative alla scansione genomica (sg).
5.3 Applicazione del sistema: Osteoporosi
L’osteoporosi, letteralmente osso poroso, e una malattia caratterizzata
da una diminuzione della componente proteica e minerale dell’osso, con
conseguente alterazione della microstruttura dello scheletro. Questa
88

5 – L’utilizzo del sistema
condizione comporta una maggiore suscettibilita alle fratture, anche per
microtraumi [sum98][PTEF02].
L’osteoporosi e condizionata da numerosi fattori e da concause non
completamente conosciute. Alcuni fattori di rischio sono genetici: tra questi,
il piu rilevante e il sesso femminile. L’osteoporosi interessa la donna con un
rapporto 8 a 2 nei confronti dell’uomo; la donna ha in effetti un apparato
scheletrico meno robusto di quello maschile, e esposta alla perdita accelerata
di minerale dopo la menopausa, ed inoltre vive piu a lungo (l’eta infatti puo
essere considerata di per se un fattore di rischio). Altri fattori di rischio
riconosciuti per ambedue i sessi sono la taglia corporea ridotta, la razza ed il
rischio legato alla storia familiare. Alcuni fattori di rischio sono dipendenti
da abitudini di vita e nutrizionali, e sono quindi modificabili, oppure sono in
rapporto a patologie concomitanti o ad assunzioni di determinati farmaci.
Tra le abitudini che possono predisporre all’osteoporosi vanno incluse il
fumo (anticipa l’eta della menopausa), l’eccessivo consumo di alcolici (causa
di malnutrizione e di compromissione dell’equilibrio) e la vita sedentaria
(riducendosi le forze muscolari applicate all’osso, lo scheletro riduce la sua
mineralizzazione come risposta di adattamento). Tra i fattori nutrizionali si
possono includere le diete sbilanciate povere di sali minerali, l’assunzione di
calcio non adeguata alle richieste, la dieta prevalentemente carnea (l’eccesso
di proteine induce perdita di calcio con le urine) [sum98][PTEF02].
La diagnosi di osteoporosi si basa inizialmente su esami clinici. Per
la diagnosi definitiva si ricorre alla densitometria (o mineralometria) ossea
computerizzata, un esame strumentale che misura la densita minerale ossea
(BMD o Bone Mineral Density). Per convenzione internazionale, si ha
osteoporosi quando la BMD e inferiore di oltre 2.5 volte rispetto al valore
89

5 – L’utilizzo del sistema
medio di un giovane adulto (Tscore < −2.5DS). L’esame riporta sempre
la BMD e, quasi sempre, lo scostamento rispetto alla media dei soggetti
della stessa eta del paziente (Z score) o dei soggetti giovani (T score)
[sum98][PTEF02].
Gli individui raccolti per il nostro studio hanno La raccolta dei dati clinici
ha fornito, per tutti gli individui, informazioni su BHR, IgE e sulle allergie
piu comuni (polveri, animali piante, etc.).
La dimensione dei dati memorizzati all’interno della base di dati e:
• individui ∼ 1700
• fenotipi ∼ 70
• geni e cromosomi candidati ∼ 15 marcatori. Ad esempio:
– gene APO-E su cromosoma 19
Le informazioni raccolte sono memorizzate in una base di dati di nome
osteo. Oltre alle tabelle standard gia viste nella presentazione del modello
relazionale (Sez. 3.2 pag. 29), abbiamo le tabelle che memorizzano fenotipi
e genotipi.
Il laboratorio che ha raccolto gli individui ci ha fornito i dati degli
individui ed dei fenotipi tramite un unico file di MS Excel contenente
tutti i campi. La tabella phnt fenotipi contiene i fenotipi disponibili che
riguardano i dati clinici degli individui ed i risultati della densitometria sugli
individui.
I tecnici specializzati di laboratorio utilizzano i codici riportati sui
90

5 – L’utilizzo del sistema
campioni di DNA per identificare gli individui. I codici sono diversi da quelli
standard della base di dati e sono memorizzati nella tabella fml_conv_all
per mantenere una corrispondenza tra i codici degli individui.
I marcatori che abbiamo genotipizzato per gli individui sono localizzati
sui cromosomi 6, 7, 11, 17 e 19 e sono memorizzati sulle tabelle chr6, chr7,
chr11, chr17 e chr19 rispettivamente.
Gli individui presenti nella base di dati osteo sono divisi in tre gruppi
per poter condurre diverse tipologie di studio. Abbiamo un gruppo di
famiglie, uno di maschi unrelated (non imparentati) ed un gruppo di femmine
unrelated. Le procedure di estrazione dati consentono di estrarre solo gli
individui del gruppo desiderato per mezzo delle clausole WHERE che abbiamo
definito.
91

5 – L’utilizzo del sistema
5.4 Sviluppi futuri
Gli studi condotti nel laboratorio della sezione di Biologia e Genetica sono
molto diversificati e, per poter garantire un sistema adatto alle esigenze
dei ricercatori, sono necessari continui aggiornamenti atti a migliorare le
prestazioni, offrire nuove funzionalita o rimuovere eventuali errori.
Per esempio, gli studi longitudinali richiedono che i principali caratteri
fenotipici correlati con una patologia vengano rilevati piu volte ad intervalli
di tempo successivi al fine di individuare fattori genetici legati con una
manifestazione piu o meno grave della patologia stessa. Modificare il
sistema per consentire la memorizzazione ed il successivo utilizzo di queste
informazioni richiedera, da una parte, l’aggiunta di nuove tabelle nella base di
dati e, dall’altra, un nuovo sistema di estrazione ed analisi delle informazioni.
Attualmente stiamo lavorando allo sviluppo di un programma per la
categorizzazione automatica degli alleli [MSX+02]: questo deve processare
i file generati dal sequenziatore di DNA e, attraverso metodi probabilistici,
definire dei valori di aggiustamento per gli alleli. Inoltre si vuole verificare
la correttezza dei risultati utilizzando le informazioni sulla struttura delle
famiglie: se la segregazione degli alleli all’interno di una famiglia e errata, e
molto probabile (se si scartano ipotesi di false paternita) che uno o piu valori
di aggiustamento, che sono stati assegnati dal programma agli alleli, siano
errati.
Nello stato attuale del sistema non e possibile eseguire alcune operazioni
via Web. Ad esempio, manca una interfaccia che permetta l’aggiunta e
la rimozione degli utenti e la gestione delle operazioni a loro consentite
92

5 – L’utilizzo del sistema
all’interno del sistema. Inoltre, l’interfaccia che gestisce l’estrazione dei dati
dalla base di dati dovrebbe permettere agli utenti di compiere direttamente
alcune operazioni aggiuntive tipo il check mendeliano della segregazione o
una analisi statistica sui risultati prodotti. Per ottenere questo risultato
stiamo valutando la possibilita di utilizzare un programma per la statistica
computazionale come R [GI97], uno strumento programmabile che dispone
di un suo linguaggio specifico.
Una delle difficolta che si incontrano durante lo sviluppo di programmi
CGI deriva dalla necessita di scrivere codice perl che, oltre a produrre dati
richiesti, produca il codice HTML. Per rendere piu agevole la realizzazione
ed il mantenimento dei programmi CGI esistono dei moduli creati
appositamente con lo scopo di separare la parte algoritmica vera e propria
dal codice HTML tra cui: CGI::Application [Erl00] e HTML::Template
[Tre00] [Erl01]. Il modulo CGI::Application si appoggia al modulo standard
CGI e consente di sviluppare uno script CGI minimale che utilizza un
modulo contenente tutte le funzioni necessarie per la creazione di ogni
pagina generata [Erl00]. Per ogni pagina Web esiste un template che
viene processato con i metodi di HTML::Template per generare la pagina
definitiva che presenta i dati desiderati. Il sistema proposto consentirebbe di
modificare l’aspetto grafico delle pagine senza andare a toccare gli algoritmi
e di modificare l’algoritmo senza preoccuparsi dell’aspetto grafico.
93

Capitolo 6
Conclusioni
Il sistema che abbiamo proposto permette di trattare efficientemente tutte
le informazioni necessarie durante lo studio di una malattia complessa ed
e disegnato per integrare dati clinici e genetici in modo efficiente. Le
informazioni possono essere rappresentate in una varieta di formati (file
di testo CSV o di MS Excel) compatibili con i programmi di elaborazione
statistica.
Il sistema e modulare, espandibile ed adattabile allo studio di piu
patologie. E costituito da una serie di strumenti software creati per gestire
una base di dati relazionale per facilitare le interrogazioni complesse ed
il controllo sui dati memorizzati. Dopo una prima fase di preparazione
della base di dati (fenotipi e struttura delle famiglie), tutte le operazioni
piu frequenti sono svolte per mezzo di una interfaccia Web che permette di
utilizzare una interfaccia grafica sviluppata ad hoc su qualunque piattaforma
hardware e software che disponga di un Web browser e di una connessione
ad Internet.
94

6 – Conclusioni
Il confronto tra il nostro sistema e gli strumenti presentati in precedenza
(2.9.1, 2.9.2 e 2.9.3) mette in risalto alcune differenze. In primo luogo,
possiamo osservare che, mentre noi utilizziamo esclusivamente software Open
Source per lo sviluppo dei nostri programmi, tutti gli altri sistemi si servono
di software proprietario. Nel nostro caso la scelta di utilizzare software Open
Source non ha ristretto le possibilita di sviluppo portando, al contrario, alcuni
benefici:
1. Costi: tutto il software e la documentazione necessaria sono disponibili
liberamente in Internet e non richiedono licenze (spesso costose) con il
vantaggio di poter stanziare la quasi totalita dei fondi sulla ricerca di
laboratorio.
2. Flessibilita: Il sistema consente l’aggiunta di nuove caratteristiche
senza modificare la struttura attuale in quanto - a meno di modifiche
allo schema della base di dati - e sufficiente implementare le funzioni
necessarie in un modulo che puo essere richiamato all’interno dei
programmi (amministrazione del sistema e CGI ).
3. Disponibilita del software: E sufficiente installare una distribuzione
GNU/Linux per poter far funzionare correttamente il nostro
sistema. Attualmente stiamo utilizzando una distribuzione Debian
GNU/Linux 3.0 ma i pacchetti che abbiamo installato sono
disponibili anche su altre distribuzioni come, ad esempio, Mandrake
(http://www.mandrake.com) o Red-Hat (http://www.redhat.com).
4. Mantenimento: e sempre possibile aggiornare gli strumenti necessari
al funzionamento del sistema nel momento in cui sono disponibili nuove
versioni dei pacchetti o della distribuzione completa.
95

6 – Conclusioni
5. Portabilita: il sistema puo essere installato su qualunque Sistema
Operativo (e piattaforma Hardware) per il quale siano disponibili il
Web server Apache, il RDBMS PostgreSQL ed il linguaggio Perl.
Alcune operazioni che si possono compiere utilizzando gli altri sistemi che
abbiamo visto sono mancanti in quanto o sono in fase di realizzazione (ad
esempio, la categorizzazione automatica degli alleli [MSX+02]) o non sono
strettamente necessarie per il nostro laboratorio (ad esempio, una interfaccia
Web per inserire i fenotipi). Al contrario, alcune funzionalita del sistema che
abbiamo proposto non trovano corrispondenze, come:
• la possibilita di definire “profili” personalizzati per tutti gli utenti del
nostro sistema;
• l’estrazione dei dati in diversi formati compatibili con i programmi di
elaborazione statistica;
• la generazione di una statistica descrittiva in cui sono descritte le
frequenze alleliche e genotipiche degli individui ed il risultato del test
di Hardy-Weinberg.
I due punti su cui abbiamo focalizzato maggiormente il nostro lavoro
sono le interfacce per l’inserimento delle informazioni sui genotipi e per
l’estrazione dei dati. Abbiamo cercato di fornire degli strumenti che fossero
allo stesso tempo efficienti e facili da utilizzare nel lavoro quotidiano del
genetista umano.
96

Bibliografia
[ACCC02] G.R. Abecasis, S.S. Cherny, W.O. Cookson e L.R. Cardon.
“Merlin-rapid analysis of dense genetic maps using sparse
gene flow trees”. Nat Genet , vol. 30: pp. 97–101 (2002).
[ACPT99] P. Atzeni, S. Ceri, S. Paraboschi e R. Torlone. Basi di dati:
concetti, linguaggi e architetture. McGraw-Hill Italia (1999).
[Bioa] Applied Biosystems. “Genescan analysis software”.
http://www.appliedbiosystems.com/.
[Biob] Applied Biosystems. “Genotyper software”.
http://www.appliedbiosystems.com/.
[BLC+01] Franca Dagna Bricarelli, Faustina Lalatta, Romeo Carrozzo,
Mario Lituania, Simona Cavani e Umberto Nicolini. Filo
diretto con le malattie genetiche, volume 2 . UTET (2001).
[Bun02] Tim Bunce. “Dbi - database independent interface for perl”
(1994-2002).
[Cor02] Microsoft Corporation. “Active server pages (asp)” (2002).
http://www.asp.net/.
97

BIBLIOGRAFIA
[DB00] Alligator Descartes e Tim Bunce. Programming the Perl
DBI . O’Reilly & Associates, Inc. (Feb 2000).
[Erl00] Jesse Erlbaum. “Cgi::application” (2000).
http://www.cpan.org/authors/id/J/JE/JERLBAUM/.
[Erl01] Jesse Erlbaum. “Using cgi::application” (may 2001).
http://www.perl.com/lpt/a/2001/06/05/cgi.html.
[Fou01] Apache Software Foundation. “Php” (2001-2001).
http://www.php.net/.
[Fou02] The Apache Software Foundation. “Apache” (1999-2002).
http://httpd.apache.org/.
[GGB00] Scott Guelich, Shishir Gundavaram e Gunther Birznieks.
CGI Programming with Perl . O’Reilly & Associates (2000).
[GI97] Robert Gentleman e Ross Ihaka. “The r project for statistical
computing” (1997).
http://www.r-project.org/.
[Gro97] John Groenveld. “Apache and perl, an alternative
architecture to oracle webserver” (Apr 1997).
http://www.cse.psu.edu/∼groenvel/ECO97/.
[GS01] GlaxoSmithKline e SIGU. Incontri 2 - Farmaci e genoma.
GlaxoSmithKline e Societa Italiana di Genetica Umana
(2001).
[GS02] GlaxoSmithKline e SIGU. Incontri 3 - Dalla Ricerca
Genetica alla pratica clinica. GlaxoSmithKline e Societa
Italiana di Genetica Umana (2002).
98

BIBLIOGRAFIA
[GT92] S.W. Guo e E.A. Thompson. “Performing the exact test of
hardy-weinberg proportion for multiple alleles”. Biometrics ,
vol. 48: pp. 361–372 (1992).
[HCH+99] S.T. Holgate, W. Cookson, P.G. Holt, C. Macaubas, P.A.
Stumbles, P.D. Sly, D.B. Corry, F. Kheradmand, Turner H.,
Kinet J.P. e Barnes P.J. Allergy and Asthma. Nature Suppl
(1999).
[IDoMotCK00] platforms Bioinformatics I. Department of Medicine of the
CAU Kiel. “Mfg tools, genotyping lab software” (2000).
http://www.mucosa.de/.
[Inc03] The MathWorks Inc. “Excel link for use with matlab”
(2003).
http://www.mathworks.com/access/helpdesk/
help/pdf doc/exlink/exlink.pdf.
[KDRDL96] L. Kruglyak, M.J. Daly, M.P. Reeve-Daly e E.S. Lander.
“Parametric and nonparametric linkage analysis: A unified
multipoint approach and nonparametric linkage analysis: A
unified multipoint approach”. American Journal of Human
Genetics , vol. 58: pp. 1347–1363 (jun 1996).
[Lan97] Kenneth Lange. Mathematical and Statistical Methods for
Genetic Analysis . Springer Verlag (1997).
[Lar99] Alexander Larsson. “Dia - a diagram drawing program”
(1999).
[LDL+01] Jin-Long Li, Hongyi Deng, Dong-Bing Lai, Fuhua Xu,
Jian Chen, Guimin Gao, Robert R. Recker e Hong-Wen
99

BIBLIOGRAFIA
Deng. “Toward high-throughput genotyping: dynamic and
automatic software for manipulating large-scale genotype
data using fluorescently labeled dinucleotide markers”.
Genome Res., vol. 11(7): pp. 1304–1314 (jul 2001).
[Lin02] D. Stein Lincoln. “Cgi - simple common gateway interface
class” (1995-2002).
[Mac96] Doug MacEachern. “mod perl” (1996).
http://perl.apache.org/.
[McL02] Grant McLean. “Xml::simple - easy api to read/write xml”
(1999-2002).
[McN02] John McNamara. “Spreadsheet::writeexcel - write to a cross-
platform excel binary file” (2000-2002).
[Mer01] Edmund Mergl. “Apache::dbi - initiate a persistent database
connection” (2001).
[Mom03] Bruce Momjian. “Postgresql hardware performance tuning”
(2003).
http://www.ca.postgresql.org/docs/
momjian/hw performance/.
[MSX+02] Giovanni Malerba, Antonella Sangalli, Luciano Xumerle,
Monica Mottes e Pier Franco Pignatti. “Studio di procedure
per l’aggiustamento delle genotipizzazioni condotte su un
elevato numero di famiglie”. Rap. tecn., V Congresso
Nazionale S.I.G.U. (2002).
100

BIBLIOGRAFIA
[MTK+98] F.J. McMahon, C.J. Thomas, R.J. Koskela, T.S. Breschel,
T.C. Hightower, N. Rohrer, C. Savino, M.G. McInnis,
S.G. Simpson e J.R. DePaulo. “Integrating clinical and
laboratory data in genetic studies of complex phenotypes: a
network-based data management system”. Am J Med Genet ,
vol. 81(3): pp. 248–256 (1998).
[MXP03] Giovanni Malerba, Luciano Xumerle e Pier Franco Pignatti.
“Il test dell’equilibrio di hardy-weinberg in situazioni
scomode”. Rap. tecn., VI Congresso Nazionale S.I.G.U.
(2003).
[Ott95] Jurg Ott. “Hwe program” (1995).
http://linkage.rockefeller.edu/soft/linkutil/.
[Ott99] Jurg Ott. Analysis of Human Genetic Linkage. Johns
Hopkins Univ Pr (1999).
[OW98] Jeffrey R. O’Connell e Daniel E. Weeks. “Pedcheck:
A program for identification of genotype incompatibilities
in linkage analysis”. Am J. Hum. Genet., vol. 63(1):
pp. 259–266 (jul 1998).
[Per] “Perl modules”.
http://www.cpan.org/modules/.
[pos] “Postgresql - a sophisticated object-relational dbms”.
http://www.postgresql.org/.
[PTEF02] Munro Peacock, Charles H. Turner, Michael J. Econs e
Tatiana Foroud. “Genetics of osteoporosis”. Endocrine
Reviews , vol. 3(23): pp. 303–326 (jun 2002).
101

BIBLIOGRAFIA
[RBK94] G.A. Rohrer, D.W. Behrens e J.W. Keele. “A simplified
procedure for entry of raw genotypic data”. J Comput Biol ,
vol. 1(2): pp. 111–119 (1994).
[Ris00] N.J. Risch. “Searching for genetic determinants in the new
millennium”. Nature, (405): pp. 847–856 (jun 2000).
[SGML92] David T. Suzuki, Anthony J. F. Griffiths, Jeffrey H. Miller e
Richard C. Lewontin. Genetica, principi di analisi formale.
Zanichelli (1992).
[SM02a] Inc. Sun Microsystems. “Java servlet technology” (1995-
2002).
http://java.sun.com/products/servlet/.
[SM02b] Inc. Sun Microsystems. “Javaserver pages (jsp)” (1995-
2002).
http://java.sun.com/products/jsp/.
[SR99] Tom Strachan e Andrew P. Read. Human Molecular
Genetics, 2nd Edition. BIOS Scientific Publishers (1999).
[sum98] Executive summary. “Osteoporosis: review of the
evidence for prevention, diagnosis and treatment and
cost-effectiveness analysis”. Osteoporosis Int., vol. 8(4):
pp. S3–S6 (jun 1998).
[Tay99] Rod Taylor. “Postgres auto-doc” (1999).
[Tre00] Sam Tregar. “Html::template” (2000).
http://html-template.sourceforge.net.
102

BIBLIOGRAFIA
[W3C02a] W3C. “Cascading style sheets (css)” (1996-2002).
http://www.w3.org/Style/CSS/.
[W3C02b] W3C. “Extensible markup language (xml)” (1996-2002).
http://www.w3.org/XML/.
[Wal] Larry Wall. “perl - practical extraction and report
language”.
http://www.perl.org/.
[Wal02] Larry Wall. “Embperl” (1993-2002).
http://www.perldoc.com/.
103


















