
STRUTTURA E FUNZIONE DI BIOMOLECOLE · Homology Modeling L’ Homology Modeling rappresenta uno dei...
18
1 STRUTTURA E FUNZIONE DI BIOMOLECOLE Predizione della Struttura Tridimensionale della proteina umana ETHE-1 con Homology Modeling Sofia Cividini
Transcript of STRUTTURA E FUNZIONE DI BIOMOLECOLE · Homology Modeling L’ Homology Modeling rappresenta uno dei...

1
STRUTTURA E FUNZIONE DI BIOMOLECOLE
Predizione della Struttura Tridimensionale della proteina umana ETHE-1 con Homology Modeling
Sofia Cividini

2
INTRODUZIONE
Lo scopo del presente lavoro è stato quello di riuscire a predire attraverso
Homology Modeling un modello tridimensionale della struttura della proteina
umana ETHE1 che è presente nella matrice mitocondriale e di cui si conosce
soltanto la sequenza primaria. Il gene che codifica per questa proteina, quando è
mutato, è responsabile di una grave e devastante malattia metabolica infantile
detta Encefalopatia Etilmalonica. Per poter attuare questo progetto è stato quindi
necessario ricercare degli omologhi di ETHE1 di cui era già stata risolta
empiricamente la struttura tridimensionale attraverso cristallografia a raggi-X o
NMR ed utilizzare poi il programma Swiss-PDBViewer per l’ Homology Modeling
come verrà descritto in seguito.
Homology Modeling
L’ Homology Modeling rappresenta uno dei migliori metodi attualmente utilizzati
per la predizione della struttura tridimensionale (3D) di quelle proteine di cui si
conosce solamente la sequenza primaria e deriva dalla teoria della selezione
naturale. Lo scopo dell’ Homology Modeling è quindi quello di costruire un
modello 3D per una determinata proteina di struttura sconosciuta (target
sequence) in base alla similarità di sequenza con proteine (template) le cui
strutture tridimensionali sono invece già state risolte per mezzo delle comuni
tecniche cristallografiche (cristallografia a raggi-X o NMR). Per potere costruire un
modello realistico occorre che vengano rispettate due condizioni:
1. La proteina target e la/le proteine stampo devono avere una percentuale di
similarità di sequenza primaria sufficientemente alta (25-30% o più); in
questo caso si è sicuri del fatto che le due o più proteine considerate siano
degli omologhi, cioè derivino da un progenitore ancestrale comune. Le
strutture 3D delle proteine appartenenti ad una certa famiglia sono più
conservate delle loro sequenze primarie per cui, se esiste una buona
percentuale di similarità di sequenza primaria, si può di solito assumere
che esista anche una buona similarità strutturale.

3
2. E’ necessario che venga fatto un allineamento sufficientemente accurato tra
la sequenza target e le sequenze stampo considerate. L’ Homology Modeling
procede modellando il backbone della sequenza target in base a quello della
sequenza stampo, usando l’allineamento di sequenza per decidere dove
posizionare ciascun residuo. Perciò, la qualità dell’ allineamento di sequenza
è di cruciale importanza; benché esistano molti strumenti che permettono
di fare gli allineamenti in modo automatico, successivamente è sempre
necessario ricontrollare ed aggiustare manualmente l’allineamento stesso
per migliorarne ulteriormente la qualità (solitamente ci si basa sugli
allineamenti multipli forniti da ClustalW).
All’ inizio, la tecnica di routine dell’ Homology Modeling fa in modo che il
backbone della proteina target venga accomodato nella stessa maniera di quello
della proteina stampo. Questo significa che non solo le posizioni dei Cα, ma anche
gli angoli phi e psi e le strutture secondarie siano costruite in modo identico alla
proteina stampo. Successivamente, i packages di Homology Modeling più
sofisticati riescono ad aggiustare le posizioni delle catene laterali per ridurre al
minimo le collisioni e possono offrire inoltre strumenti di minimizzazione
dell’energia o di dinamica molecolare che rappresentano tutti dei tentativi di
miglioramento del modello. Anche se due proteine hanno un’ elevata identità di
sequenza ed una struttura secondaria e terziaria molto simile (folds identici), esse
non avranno comunque mai un backbone esattamente uguale, neanche in
condizioni comparabili. Per questo motivo, ci si deve aspettare che un modello
ricavato con Homology Modelling non sia proprio del tutto identico alla struttura
reale. Tutte le differenze nelle strutture del backbone proteico sono quantificate
attraverso un parametro detto rmsd, che sta per root mean square deviation e si
riferisce alle posizioni degli atomi di Cα. Un modello può essere considerato
“sufficientemente accurato” quando il suo rmsd si trova all’interno dell’ intervallo
di deviazioni osservate per le strutture sperimentali che mostrano un livello di
identità di sequenza simile al target ed alle sequenze stampo. Esempio: se noi
definiamo che un buon modello di predizione di struttura ha un valore di rmsd
<=2Å rispetto alla struttura empirica, allora la proteina stampo deve avere

4
un’identità di sequenza >=60% con la proteina target affinché si abbia una
percentuale di successo maggiore del 70%.
Errori nei modelli costruiti con Homology Modeling
Quando la similarità tra la proteina-target e le proteine-stampo diminuisce, gli
errori nel modello aumentano. Vediamo quali sono i tipi di errori che si possono
principalmente verificare:
� Errori nell’impaccamento delle catene laterali → quando le sequenze
divergono, l’impaccamento delle catene laterali nel core proteico cambia.
Gli errori nelle catene laterali sono critici se si verificano in regioni che
sono coinvolte nella funzione della proteina (sito-attivo e siti di binding di
ligandi)
� Distorsioni e shifts nelle regioni correttamente allineate → Come
conseguenza della divergenza delle sequenze, la conformazione della
catena principale cambia anche se il fold rimane lo stesso.
� Errori nelle regioni senza uno stampo → i segmenti della sequenza
target che non hanno una regione equivalente nella struttura stampo (per
esempio: inserzioni o loops) rappresentano le regioni più difficili da
modellare.
� Errori dovuti ad un errato allineamento → Questi rappresentano il
grosso degli errori che vengono commessi nell’ Homology Modeling,
specialmente quando l’identità di sequenza tra target-stampo diminuisce al
di sotto del 30%. Comunque gli errori di allineamento possono essere
minimizzati in due modi:
1. di solito è possibile usare un gran numero di sequenze per costruire un
allineamento multiplo anche se la maggior parte di queste sequenze non
ha strutture 3D note; gli allineamenti multipli sono in genere più realistici
degli allineamenti a coppie.
2. si può migliorare l’allineamento modificando iterativamente quelle regioni
nell’allineamento stesso che corrispondono agli errori predetti nel modello.
� Stampi non corretti → questo diventa un potenziale problema quando le
proteine che sono usate come stampo sono solo lontanamente correlate al
target (identità di sequenza <25%).

5
Valutazione dei modelli L’informazione che può essere ricavata da un modello di predizione di struttura
dipende dalla qualità del modello stesso. Diventa quindi essenziale stimare
l’accuratezza del modelli 3D ricavati con l’ Homology Modeling per poterli poi
correttamente interpretare. Il modello può essere valutato sia nella sua interezza
che a partire da singole regioni. Esistono molti programmi e server che
permettono di valutare un determinato modello (PROCHECK, ERRAT, BIOTECH,
AQUA, SQUID, ecc). Il primo passo nella valutazione di un modello è quello di
verificare che il modello abbia il corretto fold. Un modello avrà il fold corretto se:
� sarà stata scelta la giusta sequenza-stampo
� la sequenza-stampo sarà stata allineata approssimativamente in modo
corretto con la sequenza-target
Il fold di un modello può essere valutato in vari modi:
� attraverso un’alta similarità di sequenza con la proteina-stampo più vicina
� attraverso un’ energia basata sullo Z-score
� attraverso la conservazione nella sequenza target di residui chiave per la
funzione o la struttura della proteina stessa
Una volta che è stato accertato il fold del modello, una valutazione più dettagliata
della sua accuratezza può essere ottenuta in base alla similarità tra il target e le
sequenze-stampo. Un’ identità di sequenza superiore al 30% garantisce in linea
generale un modello di predizione relativamente buono per via delle ben note
relazioni tra similarità di struttura e di sequenza tra due proteine, della natura
geometrica del modeling che forza il modello ad essere quanto più vicino
possibile allo stampo e dell’ incapacità dell’attuale procedura di modeling di
recuperare un allineamento sbagliato.
ETHE-1
Le mutazioni che colpiscono il gene che codifica per la proteina ETHE1 sono
responsabili dell’ Encefalopatia Etilmalonica, che è una patologia metabolica
infantile estremamente grave che colpisce il cervello, il tratto gastro-intestinale ed
i vasi periferici. Nei pazienti affetti da questa sindrome si trovano alti livelli di
acido etilmalonico nei fluidi corporei e una grande diminuzione dell’ attività della

6
citocromo-c ossidasi nel muscolo scheletrico; le severe conseguenze che derivano
dal mal funzionamento di questo enzima indicano quindi che ETHE1 gioca un
ruolo molto importante nel mantenimento dell’omeostasi mitocondriale e nel
metabolismo energetico. Il gene per ETHE1 è costituito da 7 esoni e si trova sul
cromosoma 19q13. La proteina ETHE1 contiene una sequenza di 24 amino acidi
in N-Te che mostra una grande similarità con i leader peptides delle proteine
destinate al mitocondrio. Infatti, è stato dimostrato che questa proteina viene
indirizzata verso questo organello dove rimane a livello della matrice in seguito al
taglio energia-dipendente del peptide segnale in N-terminale.
Swiss-PDBViewer
Swiss-PdbViewew è un programma liberamente disponibile che permette di
analizzare diverse proteine nello stesso tempo. Queste proteine possono essere
sovrapposte allo scopo di dedurre gli allineamenti strutturali e di comparare i loro
siti attivi o altre parti di rilievo. Inoltre, si possono facilmente ottenere mutazioni
negli amino acidi, legami-H, angoli e distanze tra gli angoli. La cosa di principale
rilievo è che Swiss-PdbViewer è collegato a Swiss-Model che è un server per
Homology Modeling automatico e che è stato sviluppato all’interno dello Swiss
Institute of Bioinformatics (SIB) in collaborazione con la GlaxoSmithKline ed lo
Structural Bioinformatics Group di Basilea. Il server per l’ Homology Modeling
SWISS-MODEL restituisce un file PDB pronto per DeepView, con il modello e
ciascun stampo in un differente layer. Inoltre, con DeepView possiamo vedere gli
allineamenti delle sequenze, aggiustare le posizioni dei gaps, vedere quali sono le
regioni dell’allineamento sfavorite energeticamente, trovare e fissare i clashes
delle catene laterali.

7
RISULTATI
Attraverso il sito dell’ NCBI, è stata ricavata la sequenza primaria della proteina
ETHE1 umana che è stata quindi utilizzata per la ricerca degli omologhi di cui era
già nota la struttura 3D.
A questo scopo è stato usato BlastP con l’ opzione choose database pdb come si
può vedere nella figura 1. I risultati di BlastP mostrano che esiste un
allineamento significativo con l’enzima Gliossilasi 2 (GLO2) di Homo sapiens con
uno score di 70 ed un E-value pari a 3e-13; la percentuale dei residui identici è
del 27%. Questo enzima è costituita da 260 aa.
La Gliossilasi 2 appartiene alla superfamiglia delle metallo-β-Lattamasi ed è il
secondo dei due enzimi nel sistema delle gliossilasi; è un tiolestere che catalizza
l’idrolisi dell’ S-D-lactoilglutatione a dare glutatione ed acido D-lattico. Questo
enzima è costituito da due domini : il primo dominio si ripiega a formare una
struttura a sandwich costituita da 4 β-strands, simile a quella che è stata vista
nelle metallo-β-Lattamasi, mentre nel secondo dominio predominano le α-eliche.
>gi|14198377|gb|AAH08250.1| ETHE1 protein [Homo sapiens] MAEAVLRVARRQLSQRGGSGAPILLRQMFEPVSCTFTYLLGDRESREAVLIDPVLETAPRDAQLIKELGL RLLYAVNTHCHADHITGSGLLRSLLPGCQSVISRLSGAQADLHIEDGDSIRFGRFALETRASPGHTPGCV TFVLNDHSMAFTGDALLIRGCGRTDFQQGCAKTLYHSVHEKIFTLPGDCLIYPAHDYHGFTVSTVEEERT LNPRLTLSCEEFVKIMGNLNLPKPQQIDFAVPANMRCGVQTPTA
Figura 1

8
Il sito attivo contiene un sito di binding per due atomi di Zn ed un sito di binding
per il substrato. Sono state risolte due strutture 3D per la Gliossilasi 2 umana:
1)1QH3 che contiene acetato e cacodilato nel sito attivo (risoluzione 1.9 Å)
2)1QH5 che contiene invece S-(N-hydroxy-N-bromophenylcarbamoyl)glutathione
(GBP) (risoluzione 1.45 Å).
1QH3 e 1QH5 rappresentano gli ID di PDB (Protein Data Bank) per queste
strutture e sono stati utilizzati per scaricare i corrispondenti file.pdb contenenti
tutte le coordinate strutturali necessarie per visualizzare la struttura 3D della
Gliossilasi 2 con il programma Swiss-PDBViewer.
ZN ZINC ION Zn 2+
GTT GLUTATHIONE C10 H18 N3 O6 S +
GBP S-(N-HYDROXY-N-
BROMOPHENYLCARBAMOYL)GLUTATHIONE C17 H23 N4 O8 Br S

9

10
Length = 260 Score = 70.1 bits (170), Expect = 3e-13 Identities = 50/183 (27%), Positives = 88/183 (48%), Gaps = 12/183 (6%) Query: 36 FTYLLGDRESREAVLIDPVLETAPRDAQLIKELGLRLLYAVNTHCHADHITGSGLLRSLL 95 + YL+ D E++EA ++DPV DA ++ G++L + TH H DH G+ L L Sbjct: 13 YMYLVIDDETKEAAIVDPVQPQKVVDAA--RKHGVKLTTVLTTHHHWDHAGGNEKLVKLE 70 Query: 96 PGCQSVISRLSGAQADLHIEDGDSIRFGRFALETRASPGHTPGCVTFVL-----NDHSMA 150 G + I +++ G ++ A+P HT G + + + ++ Sbjct: 71 SGLKVYGGDDRIGALTHKITHLSTLQVGSLNVKCLATPCHTSGHICYFVSKPGGSEPPAV 130 Query: 151 FTGDALLIRGCGRTDFQQGCAKTLYHSVHEKIFTLPGDCLIYPAHDYHGFTVSTVEEERT 210 FTGD L + GCG+ F +G A + ++ E + LP D +Y H+Y T++ ++ R Sbjct: 131 FTGDTLFVAGCGK--FYEGTADEMCKALLEVLGRLPPDTRVYCGHEY---TINNLKFARH 185 Query: 211 LNP 213 + P Sbjct: 186 VEP 188

11
Clickando sul quadratino S a fianco della Gliossilasi 2, Blast ci dice che ha
trovato 4 strutture correlate all’interno di questo gruppo di sequenze identiche. I
files scaricati dalla Protein Data Bank ( 1QH5.pdb o 1QH3.pdb ) contengono due
monomeri A e B. Vedi Figura 2.
________________________________________________________________________________
In questo lavoro è stata utilizzata la struttura tridimensionale corrispondente al
codice PDB 1QH5 e contenente due atomi di Zn ed il composto S-(N-hydroxy-N-
bromophenylcarbamoyl)glutathione, mentre per la costruzione del modello
predittivo della struttura 3D di ETHE1, è stato usato SwissModel di Swiss-
PDBViewer.
Analizziamo ora nel dettaglio i vari passaggi che sono stati fatti per arrivare al
modello 3D della nostra proteina target ETHE1:
� Come prima cosa siamo andati in Preferences ed abbiamo selezionato
l’opzione SwissModel; in questo modo ci è apparsa una finestra
(SwissModel Setting) attraverso la quale abbiamo potuto inserire il nostro
nome e l’indirizzo e-mail a cui far arrivare successivamente il modello
tridimensionale ottenuto da SwissModel.
� Dopo questo primo passaggio, per mezzo dell’opzione “Load Raw Sequence
to Model” presente nel menu-item di SwissModel, abbiamo caricato nel
workspace il file.txt con la sequenza amino acidica di ETHE1 in formato
FASTA. Il risultato che appare è una lunga e perfetta α-elica.
� Quindi, attraverso “Open PDB File” presente nel menu-item di File,
abbiamo caricato nel workspace anche il file 1qh5.pdb che contiene le
coordinate necessarie per la costruzione della struttura tridimensionale
della GLO2. Possiamo vedere che compaiono due monomeri A e B la cui
sequenza primaria è perfettamente identica (come abbiamo potuto
verificare dal Control Panel nel passaggio seguente).
12
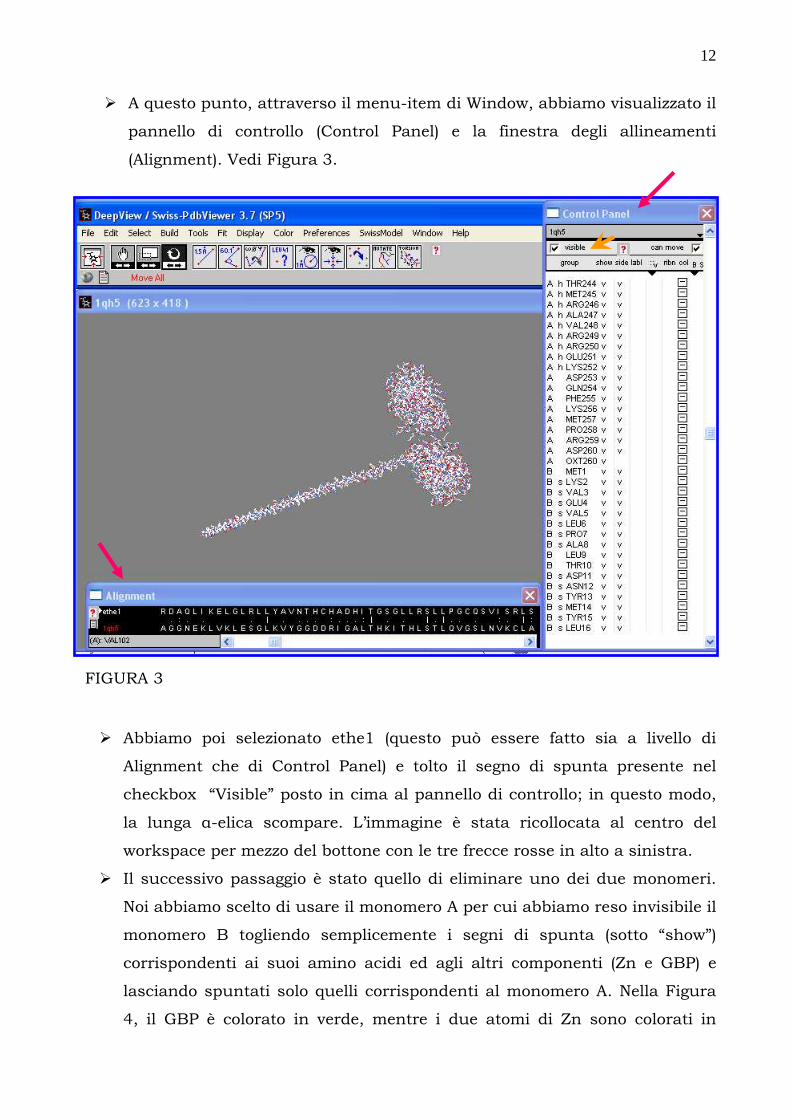
� A questo punto, attraverso il menu-item di Window, abbiamo visualizzato il
pannello di controllo (Control Panel) e la finestra degli allineamenti
(Alignment). Vedi Figura 3.
� Abbiamo poi selezionato ethe1 (questo può essere fatto sia a livello di
Alignment che di Control Panel) e tolto il segno di spunta presente nel
checkbox “Visible” posto in cima al pannello di controllo; in questo modo,
la lunga α-elica scompare. L’immagine è stata ricollocata al centro del
workspace per mezzo del bottone con le tre frecce rosse in alto a sinistra.
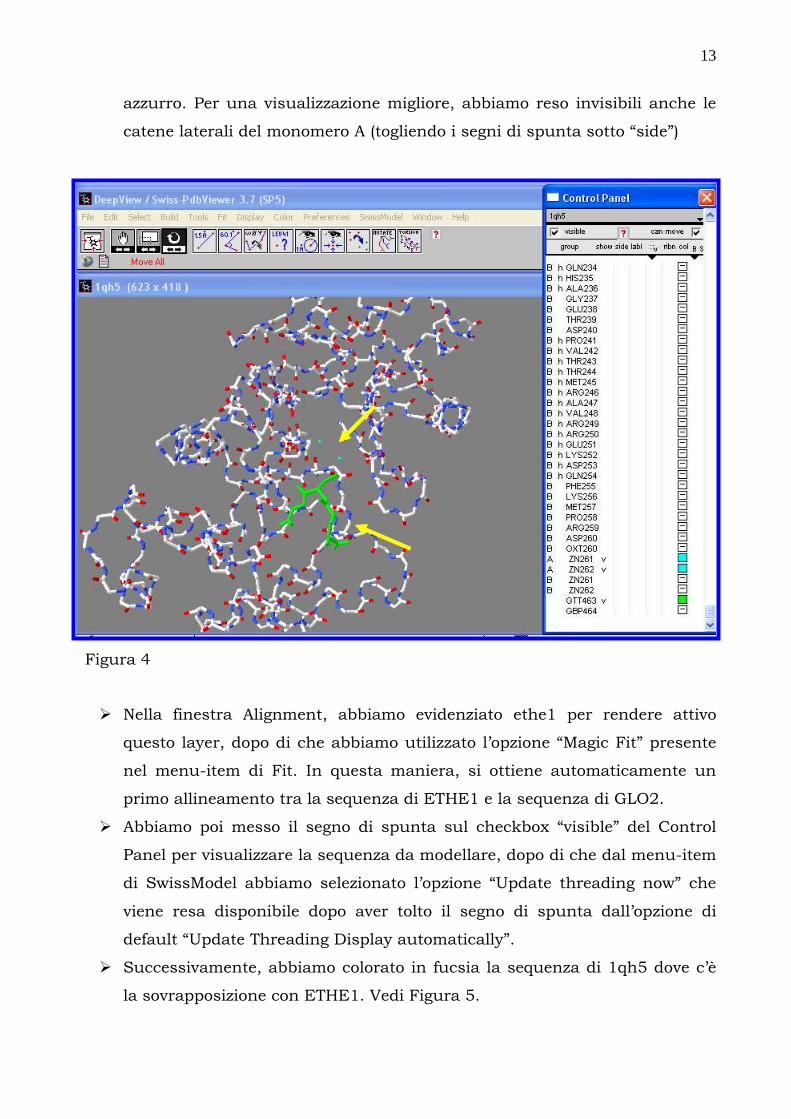
� Il successivo passaggio è stato quello di eliminare uno dei due monomeri.
Noi abbiamo scelto di usare il monomero A per cui abbiamo reso invisibile il
monomero B togliendo semplicemente i segni di spunta (sotto “show”)
corrispondenti ai suoi amino acidi ed agli altri componenti (Zn e GBP) e
lasciando spuntati solo quelli corrispondenti al monomero A. Nella Figura
4, il GBP è colorato in verde, mentre i due atomi di Zn sono colorati in
FIGURA 3
13
azzurro. Per una visualizzazione migliore, abbiamo reso invisibili anche le
catene laterali del monomero A (togliendo i segni di spunta sotto “side”)
� Nella finestra Alignment, abbiamo evidenziato ethe1 per rendere attivo
questo layer, dopo di che abbiamo utilizzato l’opzione “Magic Fit” presente
nel menu-item di Fit. In questa maniera, si ottiene automaticamente un
primo allineamento tra la sequenza di ETHE1 e la sequenza di GLO2.
� Abbiamo poi messo il segno di spunta sul checkbox “visible” del Control
Panel per visualizzare la sequenza da modellare, dopo di che dal menu-item
di SwissModel abbiamo selezionato l’opzione “Update threading now” che
viene resa disponibile dopo aver tolto il segno di spunta dall’opzione di
default “Update Threading Display automatically”.
� Successivamente, abbiamo colorato in fucsia la sequenza di 1qh5 dove c’è
la sovrapposizione con ETHE1. Vedi Figura 5.
Figura 4

14
� Il passaggio seguente è quello più importante e cioè abbiamo cercato di
migliorare manualmente l’allineamento delle due sequenze. A questo scopo,
abbiamo allineato con ClustalW la sequenza di ETHE1 e la sequenza di
GLO2 e siamo andati ad introdurre dei piccoli gaps nelle posizioni che
venivano mostrate nell’output di ClustalW sotto riportato. Abbiamo fatto
anche un allineamento multiplo con ClustalW utilizzando altre 8 sequenze
amino acidiche aventi un elevato grado di similarità con ETHE1, ma di fatto
veniva introdotto un enorme gap (24 posizioni) che rompeva la continuità
dell’allineamento tra ETHE1 e GLO2 nella zona antecedente l’estremità C-
Te (dati non mostrati). Quindi, non lo abbiamo considerato per correggere
l’allineamento delle nostre due sequenze. Siamo andati a confrontare il
valore dell’energia dell’allineamento fatto automaticamente con Magic Fit
rispetto al quello dell’allineamento fatto manualmente sulla base dei
risultati ottenuti con ClustalW e si è notato un buon miglioramento; si
passa da E=77,1 ad E=54,8 anche se i valori rimangono comunque in un
range positivo.
FIGURA 5

15
CLUSTAL W (1.82) multiple sequence alignment gi|2494849|sp|Q16775|GLO2_HUMA -----------------------MKVEVLPALTDNYMYLVIDDETKEAAI 27 gi|14198377|gb|AAH08250.1| MAEAVLRVARRQLSQRGGSGAPILLRQMFEPVSCTFTYLLGDRESREAVL 50 : ::: .:: .: **: * *::**.: gi|2494849|sp|Q16775|GLO2_HUMA VDPVQP--QKVVDAARKHGVKLTTVLTTHHHWDHAGGNEKLVKLESGLKV 75 gi|14198377|gb|AAH08250.1| IDPVLETAPRDAQLIKELGLRLLYAVNTHCHADHITGSGLLRSLLPGCQS 100 :*** : .: :: *::* .:.** * ** *. * .* .* :
gi|2494849|sp|Q16775|GLO2_HUMA YGGDDRIGALTHKITHLSTLQVGSLNVKCLATPCHTSGHICYFVSKPGGS 125 gi|14198377|gb|AAH08250.1| VISRLSGAQADLHIEDGDSIRFGRFALETRASPGHTPGCVTFVLN----- 145 . . :* . .:::.* : :: *:* **.* : :.:.
gi|2494849|sp|Q16775|GLO2_HUMA EPPAVFTGDTLFVAGCG--KFYEGTADEMCKALLEVLGRLPPDTRVYCGH 173 gi|14198377|gb|AAH08250.1| DHSMAFTGDALLIRGCGRTDFQQGCAKTLYHSVHEKIFTLPGDCLIYPAH 195 : . .****:*:: *** .* :* *. : ::: * : ** * :* .* gi|2494849|sp|Q16775|GLO2_HUMA EYTINNLKFARHVEPGNAAIREKLAWAKEKYSIGEPTVPSTLAEEFTYNP 223 gi|14198377|gb|AAH08250.1| DYHG--FTVSTVEEERTLNPRLTLSCEEFVKIMGNLNLPKPQQIDFAVPA 243 :* :..: * . * .*: : :*: .:*.. :*: . gi|2494849|sp|Q16775|GLO2_HUMA FMRVREKTVQQHAGETDPVTTMRAVRREKDQFKMPRD 260 gi|14198377|gb|AAH08250.1| NMRCGVQTPTA-------------------------- 254 ** :*
16 � Abbiamo provato anche ad adoperare l’opzione “smooth” settando il fattore di
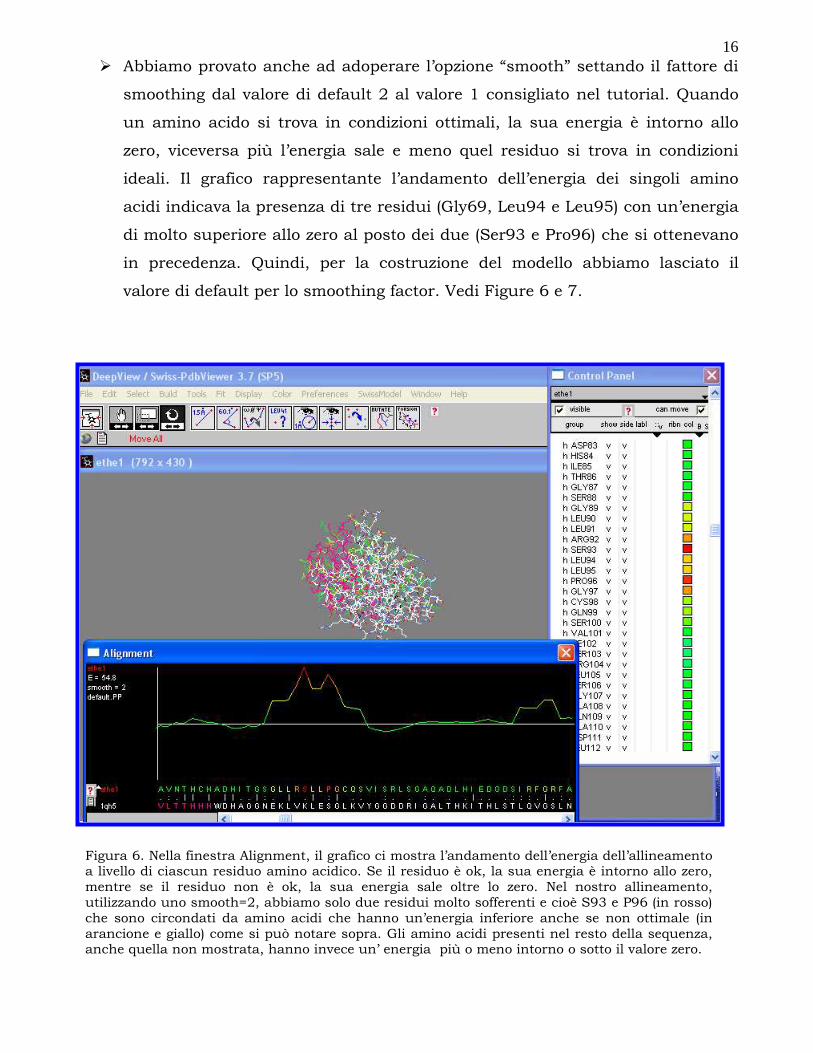
smoothing dal valore di default 2 al valore 1 consigliato nel tutorial. Quando
un amino acido si trova in condizioni ottimali, la sua energia è intorno allo
zero, viceversa più l’energia sale e meno quel residuo si trova in condizioni
ideali. Il grafico rappresentante l’andamento dell’energia dei singoli amino
acidi indicava la presenza di tre residui (Gly69, Leu94 e Leu95) con un’energia
di molto superiore allo zero al posto dei due (Ser93 e Pro96) che si ottenevano
in precedenza. Quindi, per la costruzione del modello abbiamo lasciato il
valore di default per lo smoothing factor. Vedi Figure 6 e 7.
Figura 6. Nella finestra Alignment, il grafico ci mostra l’andamento dell’energia dell’allineamento a livello di ciascun residuo amino acidico. Se il residuo è ok, la sua energia è intorno allo zero, mentre se il residuo non è ok, la sua energia sale oltre lo zero. Nel nostro allineamento, utilizzando uno smooth=2, abbiamo solo due residui molto sofferenti e cioè S93 e P96 (in rosso) che sono circondati da amino acidi che hanno un’energia inferiore anche se non ottimale (in arancione e giallo) come si può notare sopra. Gli amino acidi presenti nel resto della sequenza, anche quella non mostrata, hanno invece un’ energia più o meno intorno o sotto il valore zero.
17
� Siamo anche andati a controllare gli amino acidi che davano clashes
attraverso l’opzione “aa making clashes” presente nel menu-item Select. Dopo
di che abbiamo tentato di eliminare i clashes attraverso l’opzione “Fix Selected
Sidechains” presente nel menu-item Tools, ma questa operazione era troppo
gravosa in termini di tempo, per poter essere fatta su ciascuno dei suddetti
amino acidi. Per questo motivo purtroppo non si è potuto rifinire il modello più
di tanto.
� A questo punto, abbiamo utilizzato l’opzione “Submit modelling request”
presente nel menu-item di SwissModel ed abbiamo inviato il nostro progetto al
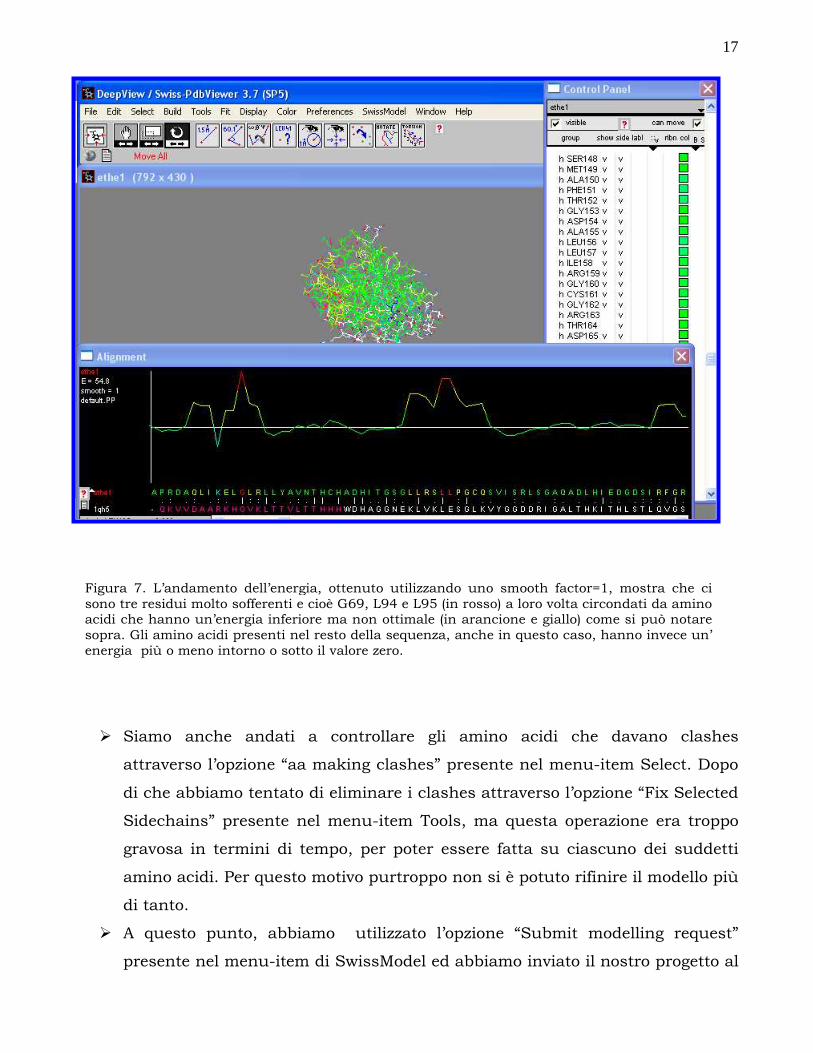
Figura 7. L’andamento dell’energia, ottenuto utilizzando uno smooth factor=1, mostra che ci sono tre residui molto sofferenti e cioè G69, L94 e L95 (in rosso) a loro volta circondati da amino acidi che hanno un’energia inferiore ma non ottimale (in arancione e giallo) come si può notare sopra. Gli amino acidi presenti nel resto della sequenza, anche in questo caso, hanno invece un’ energia più o meno intorno o sotto il valore zero.

18 server SwissModel. Successivamente, il progetto analizzato ci è stato inviato
attraverso e-mail.
� Nella Figura 8, possiamo vedere l’ipotetica struttura tridimensionale di ETHE1
predetta attraverso il nostro modello. Nel file in Power Point “ FOTO: ETHE 1”
(allegato in visione) riportiamo la stessa struttura 3D vista da diverse
angolazioni.
Nel modello tridimensionale che abbiamo ottenuto, possiamo notare la presenza del
dominio costituito da β-strand, nonché la presenza del dominio costituito da α-eliche
come avevamo già riscontrato esistere nella struttura 3D della Gliossilasi 2 risolta
sperimentalmente attraverso cristallografia a raggi-X. Questo ci dà un’indicazione
che il nostro modello sia abbastanza corretto, anche se non eccessivamente rifinito,
poiché sussiste una buona sovrapposizione tra la struttura predetta e quella reale,
tenendo anche conto che la similarità a livello della sequenza primaria tra le due
proteine non è eccessivamente alta (27%).
Figura 8 STRUTTURA 3D DI ETHE1 PREDETTA ATTRAVERSO HOMOLOGY MODELING


















