
Polymorphisms of the prion protein gene in Italian patients with Creutzfeldt-Jakob disease
5
Hum Genet (1994) 94:375-379 Springer-Verlag 1994 Mirella Salvatore - Maurizio Genuardi Rosella Petraroli Carlo Masullo Marco D'Alessandro Maurizio Pocchiari Polymorphisms of the Prion Protein gene in italian patients with Creutzfeldt-Jakob disease Received: 20 December 1993 / Revised: 13 March 1994 Abstract Creutzfeldt-Jakob disease (CJD) is a transmis- sible neurodegenerative disorder characterized by the ac- cumulation of the amyloid protein PrP in the CNS. Two coding polymorphisms of the PrP gene (PRNP) are a me- thionine (Met) to valine (Val) change at codon 129, and a deletion in the octapeptide coding region. In the United Kingdom, homozygosity at codon 129 appears to be asso- ciated with a predisposition to develop CJD. However, in Japan, where allelic frequencies and genotype distribution are significantly different, such an association has not been demonstrated. To determine whether such deletion(s) or codon 129 polymorphisms of PRNP predispose to the de- velopment of CJD in Italian patients, 31 sporadic CJD pa- tients with no known PRNP mutations, and 186 unrelated control subjects were studied. Genotypic frequencies at codon 129 in these Italian CJD patients revealed a signif- icant excess of methionine alleles, and a different geno- type distribution in comparison with the normal Italian population. Deletions of a 24-bp segment located in the PrP octapeptide coding region were found in two control subjects, but in none of the sporadic CJD patients. These data suggest that Met homozygosity at codon 129 may contribute, with other enviromental or endogenous fac- tors, to CJD development. M. Salvatore (5:~). R. Petraroli - M. D'Alessandro M. Pocchiari Laboratory of Virology, Istituto Superiore di Sanith, Viale Regina Elena 299, 1-00161 Rome, Italy M. Genuardi Istituto di Genetica, Universith Cattolica del Sacro Cuore, Rome, Italy C. Masullo Istituto di Neurologia, Universith Cattolica del Sacro Cuore, Rome, Italy Introduction The pathological hallmark of transmissible spongiform encephalopathies [i.e., Creutzfeldt-Jakob disease (CJD) and Gerstmann-Straussler Syndrome (GSS) in man, and scrapie and bovine spongiform encephalopathy in animals] is the accumulation of a partially protease-resistant protein (PrP- res) in the brain of affected individuals (Brunori et al. 1988). This protein is critically involved in the develop- ment of the disease(s) (Bueler et al. 1993), and derives from a posttranslational modification of its protease sensi- tive cellular isoform (PrP-sen) (Caughey et al. 1989) whose function is still unknown. Although the two isoforms are encoded by the same chromosomal gene, PrP-res is found only in the brain of affected individuals, while normal PrP-sen is found both in affected and unaffected individu- als (Oesch et al. 1985). In mice, polymorphisms within the PrP murine homologous gene control the incubation period of experimental scrapie (Westaway et al. 1987). In man, missense point mutations or insertions of additional octapeptide repeats within the PrP gene (PRNP) have been linked to the clinical development of familial CJD and GSS (Dlouhy et al. 1992; Doh-ura et al. 1989; Gold- farb et al. 1991a, b, 1992; Goldgaber et al. 1989; Hsiao et al. 1989, 1992; Owen et al. 1989; Pocchiari et al. 1993; Poulter et al. 1992; Ripoll et al. 1993). Moreover, some specific mutations are associated with the phenotypic ex- pression of the disease (Brown et al. 1991). In the human gene, two polymorphisms leading to a change in the amino acid sequence have been described. The first is an ATG to GTG polymorphic change at codon ! 29 of PRNP that results in the substitution of valine (Val) for the more common methionine (Met). It has been re- ported that most CJD patients in the UK carry a homo- zygous genotype at codon 129, suggesting that it confers a genetic predisposition to the development of sporadic CJD in the UK (Palmer et al. 1991). Moreover, homozy- gosity seems to enhance susceptibility to iatrogenic CJD of peripheral and central origin (Collinge et al. 1991; Preece 1993; Brown et al. 1994; Deslys et al. 1994).
-
Upload
mirella-salvatore -
Category
Documents
-
view
213 -
download
0
Transcript of Polymorphisms of the prion protein gene in Italian patients with Creutzfeldt-Jakob disease

Hum Genet (1994) 94:375-379 �9 Springer-Verlag 1994
Mirella Salvatore - Maurizio Genuardi �9 Rosella Petraroli Carlo Masullo �9 Marco D'Alessandro Maurizio Pocchiari
Polymorphisms of the Prion Protein gene in italian patients with Creutzfeldt-Jakob disease
Received: 20 December 1993 / Revised: 13 March 1994
Abs t rac t Creutzfeldt-Jakob disease (CJD) is a transmis- sible neurodegenerative disorder characterized by the ac- cumulation of the amyloid protein PrP in the CNS. Two coding polymorphisms of the PrP gene (PRNP) are a me- thionine (Met) to valine (Val) change at codon 129, and a deletion in the octapeptide coding region. In the United Kingdom, homozygosity at codon 129 appears to be asso- ciated with a predisposition to develop CJD. However, in Japan, where allelic frequencies and genotype distribution are significantly different, such an association has not been demonstrated. To determine whether such deletion(s) or codon 129 polymorphisms of PRNP predispose to the de- velopment of CJD in Italian patients, 31 sporadic CJD pa- tients with no known PRNP mutations, and 186 unrelated control subjects were studied. Genotypic frequencies at codon 129 in these Italian CJD patients revealed a signif- icant excess of methionine alleles, and a different geno- type distribution in comparison with the normal Italian population. Deletions of a 24-bp segment located in the PrP octapeptide coding region were found in two control subjects, but in none of the sporadic CJD patients. These data suggest that Met homozygosity at codon 129 may contribute, with other enviromental or endogenous fac- tors, to CJD development.
M. Salvatore (5:~). R. Petraroli - M. D'Alessandro �9 M. Pocchiari Laboratory of Virology, Istituto Superiore di Sanith, Viale Regina Elena 299, 1-00161 Rome, Italy
M. Genuardi Istituto di Genetica, Universith Cattolica del Sacro Cuore, Rome, Italy
C. Masullo Istituto di Neurologia, Universith Cattolica del Sacro Cuore, Rome, Italy
Introduction
The pathological hallmark of transmissible spongiform encephalopathies [i.e., Creutzfeldt-Jakob disease (CJD) and Gerstmann-Straussler Syndrome (GSS) in man, and scrapie and bovine spongiform encephalopathy in animals] is the accumulation of a partially protease-resistant protein (PrP- res) in the brain of affected individuals (Brunori et al. 1988). This protein is critically involved in the develop- ment of the disease(s) (Bueler et al. 1993), and derives from a posttranslational modification of its protease sensi- tive cellular isoform (PrP-sen) (Caughey et al. 1989) whose function is still unknown. Although the two isoforms are encoded by the same chromosomal gene, PrP-res is found only in the brain of affected individuals, while normal PrP-sen is found both in affected and unaffected individu- als (Oesch et al. 1985). In mice, polymorphisms within the PrP murine homologous gene control the incubation period of experimental scrapie (Westaway et al. 1987). In man, missense point mutations or insertions of additional octapeptide repeats within the PrP gene (PRNP) have been linked to the clinical development of familial CJD and GSS (Dlouhy et al. 1992; Doh-ura et al. 1989; Gold- farb et al. 1991a, b, 1992; Goldgaber et al. 1989; Hsiao et al. 1989, 1992; Owen et al. 1989; Pocchiari et al. 1993; Poulter et al. 1992; Ripoll et al. 1993). Moreover, some specific mutations are associated with the phenotypic ex- pression of the disease (Brown et al. 1991).
In the human gene, two polymorphisms leading to a change in the amino acid sequence have been described. The first is an ATG to GTG polymorphic change at codon ! 29 of PRNP that results in the substitution of valine (Val) for the more common methionine (Met). It has been re- ported that most CJD patients in the UK carry a homo- zygous genotype at codon 129, suggesting that it confers a genetic predisposition to the development of sporadic CJD in the UK (Palmer et al. 1991). Moreover, homozy- gosity seems to enhance susceptibility to iatrogenic CJD of peripheral and central origin (Collinge et al. 1991; Preece 1993; Brown et al. 1994; Deslys et al. 1994).

376
The other PRNP polymorphism is represented by a deletion within the region encoding for the octapeptide re- peats (Vnencak-Jones and Phillips 1992). Its role, if any, in the clinical appearance and development of the disease has yet to be clarified.
To understand better the role of these PRNP polymor- phisms on CJD predisposition in another Caucasian popu- lation, we studied codon 129 and deletion polymorphism distribution in an Italian control population and in Italian CJD patients.
BsmAI, and Mspl, respectively) using buffers and reaction condi- tions as recommended by the manufacturers. Since the A to T transversion at codon 210 does not create or abolish any suitable restriction site, screening for this mutation was performed by si- multaneously creating and abolishing Hphl restriction sites at dif- ferent positions during amplification (Pocchiari et al. 1993).
The presence of deletions or insertions within the PRNP gene was tested by directly electrophoresing PCR amplified fragments on agarose gels. Samples positive for the deletion were treated with the restriction enzymes NcoI (which cuts at codons 76 and 165) and Pvull (which cuts at codon 117) in order to locate the deletion with respect to these restriction sites.
Subjects and methods
Thirty-one unrelated CJD patients of Italian ancestry and 186 un- related healthy Italian control subjects were examined. Patients, from all over Italy, were referred to the Italian National survey center for CJD. In 27 patients, the clinical diagnosis of CJD was confirmed by neuropathological examination of the brain (24 cases) and/or by Western blot detection of PrP-res from cortical samples of frozen brains (20 cases) using a polyclonal antibody prepared against PrP-res of scrapie-infected hamsters (Xi et al. 1992). In two of the remaining four patients, a definite diagnosis of CJD was based on clinical criteria (Brown et al. 1986). In the other two patients, still living, a probable diagnosis of CJD was made based on the presence of dementia, myoclonus, typical EEG and the exclusion of other neurologic diseases. Control specimens were randomly selected from DNA samples of unrelated adult in- dividuals of both sexes collected from all over Italy. DNA was ex- tracted from frozen autopsy brain tissue (patients) or from blood (patients and controls) according to standard procedures (Sam- brook et al. 1985) and the PRNP open reading frame was amplified by the polymerase chain reaction (PCR) in an automated thermal cycler (Perkin Elmer-Cetus) as previously described (Goldgaber et al. 1989).
Codon 129 polymorphism was studied by digesting amplified DNAs with MaeIl restriction endonuclease (patients and controls), and confirmed in CJD patients by digestion with NspI (Fig. I). To exclude the presence of any of the previously described mutations at codons 102, 117, 178, 198, 200, and 217 within the PRNP of CJD patients, amplified DNAs were screened by digestion with the appropriate restriction enzymes (Ddel, Pvull, Tth l l l l , Hi?hi,
Results
Genotype frequencies at codon 129 in 186 unrelated Ital- ian controls were 45.16% Met/Met, 40,32% Met/Val and 14.52% Val/Val giving an allele frequency of 0.653:0.348 Met:Val. This distribution follows the Hardy-Weinberg equi l ibr ium (Z e = 2.249; P > 0.1 ) and does not differ from that previously reported for the Italian population (Z2= 0.034; P > 0.9; Medori and Tritscbler 1993) nor from that reported for other Caucasian populations, such as the British (Z 2 = 3.097; P > 0.2; Owen et al. 1990), French (Z 2 = 3.687; P > 0.1; Deslys et al. 1994) and American (Z 2 = 2.939; P > 0.2; Brown et al. 1994) populations,
None of the 31 sporadic CJD patients carried any of the known PrP mutations. Twenty-five of them were Met homozygous, five were heterozygous and one was Val homozygous (Table 1). There was a significant excess of the Met allele in CJD patients [Z 2 = 12.44 (with Yates cor- rection for continuity), P = 0.0004] and the genotype dis- tribution of CJD patients was significantly different from that found in controls (Z 2 = 13.50; P = 0.001 ),
Statistical analysis was also significant when per- formed only on 26 CJD patients with a definite diagnosis of CJD [Z 2 = 8.48 (with Yates correction for continuity), P = 0.004 for comparison of alleles; Z2 = 9.35; P = 0.009 for comparison of genotypes].
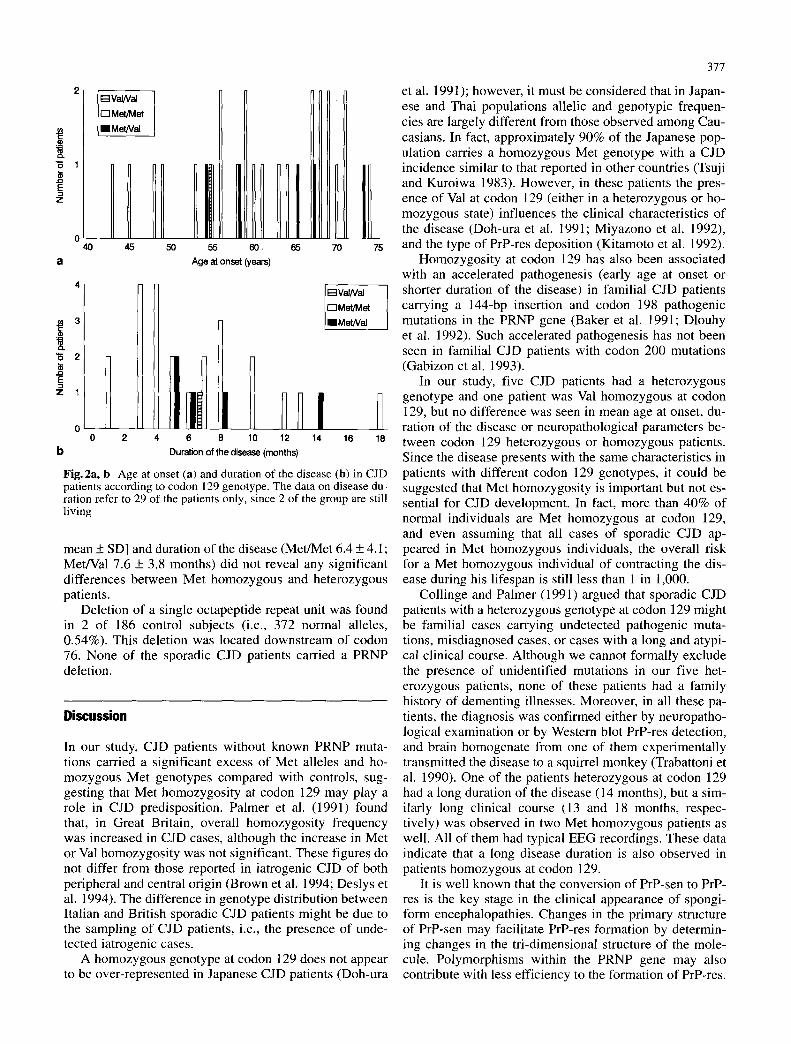
No difference was seen in the distribution of age at on- set (Fig. 2a), duration of the disease (Fig. 2b), or severity of neuropathological f indings (data not shown) among pa- tients carrying Met/Met, Met/Val or Val/Val genotypes. Moreover, parametric (unpaired t-test) and nonparametric (Wilcoxon test) statistical analysis on the mean age at on- set [Met/Met 60.9 _+ 8.7 (61.4 _+ 8.6 for 23 patients with a definite diagnosis of CJD); Met/Val 63.4 _+ 7.5 years,
Fig. 1 MaelI (top) and Nspl (bottom) restriction nuclease diges- tion of the 803-bp amplified PRNP in 18 Creutzfeldt-Jakob disease (CJD) patients. Maell cuts when Val 129 is present, while Nspl cuts when Met 129 is present. Lane 1 molecular weight standard DNA pBR322 digested with Avail + EcoR1; lanes 4,5 and 10 Met/Val; lane 9 Val/Val; remaining lanes Met/Met patients. Dig- its on the left indicate the length (bp) of molecular weight standard DNA
Table 1 Genotype frequencies at codon 129 in CJD patients and controls
n Codon 129
Met/Met Val/Val Met/Val
Definite CJD 29 23 (79.31) I (3.45) 5 (17.24) Probable CJD 2 v - -
TotalCJD 31 25 (80.64) 1 (3.23) 5(16.13)
Controls 186 84 (45.16) 75 (40.32) 27 (14.52)
"5
E
"6
.Q
Z
0 40
0 --
0 2
BValNaJ
/-i Met/Met
BBMetJVal
45 50 55 60 65 70 75 Age at onset (years)
4 6
EBVaINal
r-IMet/Met
IlMetNal
8 10 12 14 16 18 Duration of the disease (months)
Fig.2a, b Age at onset (a) and duration of the disease (b) in CJD patients according to codon 129 genotype. The data on disease du- ration refer to 29 of the patients only, since 2 of the group are still living
mean + SD] and duration of the disease (Met/Met 6.4 _+ 4.1; Met/Val 7.6 _+ 3.8 months) did not reveal any significant differences between Met homozygous and heterozygous patients.
Deletion of a single octapeptide repeat unit was found in 2 of 186 control subjects (i.e., 372 normal alleles, 0.54%). This deletion was located downstream of codon 76. None of the sporadic CJD patients carried a PRNP deletion.
D i s c u s s i o n
In our study, CJD patients without known PRNP muta- tions carried a significant excess of Met alleles and ho- mozygous Met genotypes compared with controls, sug- gesting that Met homozygosity at codon 129 may play a role in CJD predisposition. Palmer et al. (1991) found that, in Great Britain, overall homozygosity frequency was increased in CJD cases, although the increase in Met or Val homozygosity was not significant. These figures do not differ from those reported in iatrogenic CJD of both peripheral and central origin (Brown et al. 1994; Deslys et al. 1994). The difference in genotype distribution between Italian and British sporadic CJD patients might be due to the sampling of CJD patients, i.e., the presence of unde- tected iatrogenic cases.
A homozygous genotype at codon 129 does not appear to be over-represented in Japanese CJD patients (Doh-ura
377
et al. 1991); however, it must be considered that in Japan- ese and Thai populations allelic and genotypic frequen- cies are largely different from those observed among Cau- casians. In fact, approximately 90% of the Japanese pop- ulation carries a homozygous Met genotype with a CJD incidence similar to that reported in other countries (Tsuji and Kuroiwa 1983). However, in these patients the pres- ence of Val at codon 129 (either in a heterozygous or ho- mozygous state) influences the clinical characteristics of the disease (Doh-ura et al. 1991; Miyazono et al. 1992), and the type of PrP-res deposition (Kitamoto et al. 1992).
Homozygosity at codon 129 has also been associated with an accelerated pathogenesis (early age at onset or shorter duration of the disease) in familial CJD patients carrying a 144-bp insertion and codon 198 pathogenic mutations in the PRNP gene (Baker et al. 1991; Dlouhy et al. 1992). Such accelerated pathogenesis has not been seen in familial CJD patients with codon 200 mutations (Gabizon et al. 1993).
In our study, five CJD patients had a heterozygous genotype and one patient was Val homozygous at codon 129, but no difference was seen in mean age at onset, du- ration of the disease or neuropathological parameters be- tween codon 129 heterozygous or homozygous patients. Since the disease presents with the same characteristics in patients with different codon 129 genotypes, it could be suggested that Met homozygosity is important but not es- sential for CJD development. In fact, more than 40% of normal individuals are Met homozygous at codon 129, and even assuming that all cases of sporadic CJD ap- peared in Met homozygous individuals, the overall risk for a Met homozygous individual of contracting the dis- ease during his lifespan is still less than 1 in 1,000.
Collinge and Palmer (1991) argued that sporadic CJD patients with a heterozygous genotype at codon 129 might be familial cases carrying undetected pathogenic muta- tions, misdiagnosed cases, or cases with a long and atypi- cal clinical course. Although we cannot formally exclude the presence of unidentified mutations in our five het- erozygous patients, none of these patients had a family history of dementing illnesses. Moreover, in all these pa- tients, the diagnosis was confirmed either by neuropatho- logical examination or by Western blot PrP-res detection, and brain homogenate from one of them experimentally transmitted the disease to a squirrel monkey (Trabattoni et al. 1990). One of the patients heterozygous at codon 129 had a long duration of the disease (14 months), but a sim- ilarly long clinical course (13 and 18 months, respec- tively) was observed in two Met homozygous patients as well. All of them had typical EEG recordings. These data indicate that a long disease duration is also observed in patients homozygous at codon 129.
It is well known that the conversion of PrP-sen to PrP- res is the key stage in the clinical appearance of spongi- form encephalopathies. Changes in the primary structure of PrP-sen may facilitate PrP-res formation by determin- ing changes in the tri-dimensional structure of the mole- cule. Polymorphisms within the PRNP gene may also contribute with less efficiency to the formation of PrP-res.

378
According to the prion hypothesis, Met (or Val) homozy- gosity favors the formation of dimers between PrP-sen and PrP-res, and hence the transformation of PrP-sen into PrP-res (Prusiner 1993). Alternatively, if PrP-sen acts as cellular receptor for an exogenous agent that causes the disease, homozygosi ty might confer an increased affinity with the infectious agent (Diringer 1992).
By analogy with insertions in the octapeptide coding region that have been linked to the development of fa- milial CJD (Owen et al. 1989) and GSS (Goldfarb et al. 1992), a deletion in the same region might be expected to alter the protein conformation (Puckett et al. 1991), thus enhancing the formation of PrP-res and the development of the disease. However, a deletion located downstream of codon 76 was identified in 2 out of 186 control subjects as previously reported (Laplanche et al. 1990, 1991 ; Puckett et al. 1991; Vnencak-Jones and Phillips 1992) but in none of the sporadic CJD patients. Similar downstream dele- tions, however, have been detected in a patient with un- classified dementia (Dietrich et al. 1992), in two cases of iatrogenic CJD (Brown et al. 1994) and in a CJD patient with a codon 178 mutation on the same allele (Bosque et al. 1992). In these patients the deletion did not appear to influence the phenotypic expression of the disease. Differ- ent deletions located upstream of codon 76 were observed on the non-mutated allele of a familial CJD patient carry- ing a codon 210 mutation, which probably delays the age at onset of the disease (Pocchiari et al. 1993), in unaf- fected members of the same family and, in a homozygous state, in a 33-year-old woman with unclassified dementia (Masullo et al. 1994). These findings indicate that dele- tion of a single repeat coding region is a low-frequency polymorphism, but its role, if any, in the manifestation of CJD has yet to be ascertained.
In conclusion, we have found an association between codon 129 Met homozygosi ty and sporadic CJD. Al- though it is not essential in determining the disease, ho- mozygosity at codon 129 of PRNP may contribute with other host or environmental factors to the development of sporadic CJD.
Acknowledgments We thank Prof. G. Macchi lk)r neuropatho- logic examination of our cases, Dr. P. Tanci for the selection of DNA samples of healthy Italian individuals, Drs. F. Cardone and Y.G. Xi for Western blot detection of PrP, Mr. A. Sbriccoli for technical support and Ms D. Bryant for editorial assistance. We also thank Prof. G.B. Rossi, whose recent death was a great loss for us all, for his continuous support of our work and Italian neuro- logists for referrals of CJD cases. This work was partially supported by CNR PF lnvecchiamento (no. 942409) and by the National registry of Creutzfeldt-Jakob disease, granted by the Italian Min- istry of Health - lstituto Superiore di Sanita'.
References
Baker HF, Poulter M, Crow TJ, Frith CD, Lofthouse R, Ridley RM (1991) Aminoacid polymorphism in human prion protein and age at death in inherited prion disease. Lancet 337:1289
Bosque PJ, Vnencak-Jones CL, Johnson MD, Whitlock JA, McLean MJ (1992) A PrP gene codon 178 base substitution and a 24-bp interstitial deletion in familial Creutzfeldt-Jakob disease. Neurology 42 : 1864 1870
Brown P, Cathala F, Castaigne P, Gajdusek DC (1986) Creutz- feldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 20: 597-602
Brown P, Goldfarb LG, Gibbs CJ, Gajdusek DC (1991) The phe- notypic expression of different mutations in transmissible fa- milial Creutzfeldt-Jakob disease. Eur J Epidemiol 7 : 469-476
Brown P, Cervenakova L, Goldfarb LG, McCombie WR, Ruben- stein R, Will RG, Pocchiari M, Martinez-kage JF, Scalici C, Masullo C, Graupera G, Ligan J, Gajdusek DC (1994) latro- genie Creutzfeldt-Jakob disease: an example of the interplay between ancient genes and modern medicine. Neurology 44: Brunori G, Silvestrini C, Pocchiari M (1988) The scrapie agent and the prion hypothesis. Trends Biochem Sci 13:309 313
Bueler H, Aguzzi A, Saller A, Greiner RA, Autenried P, Aguet M, Weissmann C (1993) Mice devoid of PrP are resistant to scrapie. Cell 73 :1339-1347
Caughey B, Race R, Ernst D, Buchmeier M, Chesebro B (1989) Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol 63 : 175 18 l
Collinge J, Palmer M (1991) CJD discrepancy. Nature 353:802 Collinge J, Pahner MS, Dryden AJ (1991) Genetic predisposition
to iatrogenic Creutzfeldt-Jakob disease. Lancet 337:1441 1442
Deslys J-P, Marc~ D, Dormont D (1994) Similar genetic suscepti- bility in iatrogenic and sporadic Creutzfeldt-Jakob disease. J Gen Virol 75 :23 -25
Dietrich JF, Knopman DS, List JF, Olson K, Frey WH I1, Emory CR, Sung JH, Haase AT (1992) Deletion in the prion protein gene in a demented patient. Hum Mol Gen 1 : 443-444
Diringer H (1992) Hidden amiloydoses. Exp Clin lmmunogenet 9: 212-229
Dlouhy SR, Hsiao K, Farlow MR, Foroud T, Conneally PM, John- son P, Prusiner SB, Hodes ME, Gheni B (1992) Linkage of the Indiana kindred of Gerstmann-Straussler-Sheinker disease to the prion protein gene. Nature Genet I :64-67
Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y (1989) Pro -> leu change at position 102 of prion protein is the most com- mon but not the sole mutation related to Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 163:974-979
Doh-Ura K, Kitamoto T, Sakaki Y, Tateishi J (1991) CJD discrep- ancy. Nature 353 : 801-802
Gabizon R, Rosenmann H, Meiner Z, Kahana 1, Kahana E, Shugart Y, On J, Prusiner SB (1993) Mutation and polymorphism of the prion protein gene in Libyan Jews with Creutzfeldt-Jakob disease (CJD). Am J Hum Genet 53 : 828 835
Goldfarb LG, Haltia M, Brown P, Nieto A, Kovanen J, McCombie WR, Trapp S, Gajdusek DC (1991a) New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creutzfeldt- Jakob kindred. Lancet 337 : 425
Goldfarb LG. Brown P, McCombie WR, Goldgaber D, Swergold GD, Wills PR, Cervenacowt L, Baron H, Gibbs CJ Jr, Gajdusek DC ( 1991 b) Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Nail Acad Sci USA 88 :10926 10930
Goldfarb LG, Brown P, Vrbovska A, Baron H, McCombie WR, Cathala F, Gibbs CJ Jr, Gaidusek DC (1992) An insert muta- tion in the chromosome 20 amyloid precursor gene in a Gerst- mann-Straussler-Scheinker family. J Neurol Sci 11 I : 189-194
Goldgabcr D, Goldfarb LG, Brown P, Asher DM, Brown WT, Lin S, Teener JW. Feinstone SM, Rubenstein R, Kacsak R J, Boellaard JW, Ga:jdusek CD (1989) Mutations in familial Creutzfeldt-,lakob disease and Gerstmann-Straussler-Schein- ker 's syndrome. Exp Neurol 106:204-206
Hsiao K, Baker HF, Crow TY, Poulter M, Owen F, Terwilliger JD, Westaway W. Ott ,I, Prusiner SB (1989) Linkage of a prion protein missense w~riant to Gerstmann-Straussler syndrome. Nature 338 : 342-345

379
Hsiao K, Dlouhy SR, Farlow MR, Cass C, Costa M Da, Conneally PM, Hodes ME, Ghetti B, Prusiner SB (1992) Mutant prion proteins in Gerstmann-Straussler-Sheinker disease with neu- rofibrillary tangles. Nature Genet 1:68-71
Kitamoto T, Doh-Ura K, Muramoto T, Miyazono M, Tateishi J (1992) The primary structure of the prion protein influences the distribution of abnormal Prion protein in the central nervous system. Am J Pathol 141 : 1-7
Laplanche JL, Chatelain J, Launay JM, Gazengel C, Vidaud M (1990) Deletion in prion protein gene in a Moroccan family. Nucleic Acids Res 18 : 6745
Laplanche JL, Chatelein J, Thomas S, Brown P, Chatala F (1991) Analyse du gene PrP dans une famille d'origine tunisienne at- teinte de Maladie de Creutzfeldt-Jakob. Rev Neurol 147 : 825- 827
Masullo C, Salvatore M, Genuardi M, Pocchiari M, Macchi M (1994) Progressive dementia in a young patient with homozy- gosity del PrP gene. Ann NY Acad Sci 724: 358-360
Medori R, Tritschler H-J (1993) Prion protein gene analysis in three kindreds with fatal familial insomnia (FFI): codon 178 mutation and codon 129 polymorphism. Am J Hum Genet 53 : 822-827
Miyazono M, Kitamoto T, Doh-Ura K, lwaki T, Tateishi J (1992) Creutzfeldt-Jakob disease with codon 129 polymorphism (Va- line): a comparative study of patients with codon 102 point mu- tation or without mutations Acta Neuropathol 84 : 349-354
Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hodd LE, Prusiner SB, Weissman C (1985) A cellular gene encodes scrapie PrP27-30 protein. Cell 40 : 735-746
Owen F, Poulter M, Lofthouse R, Collinge J, Crow T J, Risby D, Baker HF, Ridley RM, Hsiao K, Prusiner SB (1989) Insertion in prion protein gene in familial Creutzfeldt Jakob. Lancet 1 : 51-52
Owen F, Poulter M, Collinge J, Crow TJ (1990) Codon 129 changes in the prion protein gene in Caucasians. Am J Hum Genet 46:1215-1216
Palmer MS, Dryden AJ, Hughes JT, Collinge J (1991) Homozy- gous prion protein predisposes to sporadic Creutzfeldt-Jakob disease. Nature 352 : 340-342
Pocchiari M, Salvatore M, Cutruzzolzi F, Genuardi M, Travaglini- Allocatelli C, Masullo C, Macchi G, Alem~i G, Galgani S, Xi YG, Petraroli R, Silvestrini MC, Brunori M (1993) A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. Ann Neurol 34 : 802-807
Poulter M, Baker KF, Frith CD, Leach M, Lofthouse R, Ridley RM, Shah T, Owen F, Collinge J, Brown J, Hardy J, Mullan M J, Harding AE, Bennet C, Doshi R, Crow TJ (1992) Inherited Prion Disease with 144 base pair gene insertion. 1. Genealogi- cal and molecular studies. Brain 115:675-685
Preece M (1993) Human pituitary growth hormone and Creutz- feldt-Jakob disease. Horm Res 39 : 95-98
Prusiner SB (1993) Genetic and infectious priori diseases. Arch Neurol 50:1129-1153
Puckett C, Concannon P, Casey C, Hood L (1991) Genomic struc- ture of the human prion protein gene. Am J Hum Gen 49: 320-329
Ripoll L, Laplance JL, Salzmann M, Jouvet A, Planques B, Dus- saucy M, Chatelain J, Beaudry P, Launay JM (1993) A new point mutation in the prion protein gene at codon 210 in Creutzfeldt-Jakob disease. Neurology 43 : 1934-1938
Sambrook J, Fritsch EF, Maniatis T (1985) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Trabattoni G, Lechi A, Bettoni L, Macchi G, Brown P (1990) Con- sideration on a group of 13 patients with Creutzfeldt-Jakob dis- ease in the region of Parma (Italy). Eur J Epidemiol 6:238-243
Tsuji S, Kuroiwa Y (1983) Creutzfeldt-Jakob disease in Japan. Neurology 33 : 1503-1506
Vnencak-Jones C, Phillips JA III (1992) Identification of hetero- geneous PrP gene deletions in controls by detection of allele- specific heteroduplexes (DASH). Am J Hum Gen 50:871-872
Westaway D, Goodman PA, Mirenda CA, McKinley MP, Carlson GA, Prusiner SB (1987) Distinct prion proteins in short and long scrapie incubation period mice. Cell 51 : 651-662
Xi YG, Ingrosso L, Ladogana A, Masullo C, Pocchiari M (1992) Amphotericin B treatment dissociates in vivo replication of the scrapie agent from PrP accumulation. Nature 356:598-601


















